
Submitted:
30 July 2024
Posted:
31 July 2024
You are already at the latest version
Abstract
Ferroptosis and dysregulation of iron metabolism are increasingly recognized as contributors to the onset and development of cardiovascular diseases (CVD), including hypertension, cardiomyopathy, atherosclerosis, pulmonary hypertension, myocardial ischemia/reperfusion injury, heart failure, and cardiovascular manifestations of Coronavirus disease 19 (COVID-19). In-flammation plays a central role in these conditions, prompting exploration into the inflammatory and immunoregulatory molecular mechanisms un-derlying ferroptosis and its contribution to CVD progression. In particular, emerging evidence suggests that interleukin (IL)-37 is a protective cytokine capable of activating the nuclear factor erythroid 2-related factor 2 (Nrf2) pathway, suppressing macrophage ferroptosis, and activating the nuclear factor erythroid 2-related factor 2 (Nrf2) pathway to attenuate atherosclerosis progression in murine models. Despite this, a comprehensive review detailing IL-37 and its protective role against ferroptosis in CVD is absent in the current literature. Thus, this review consolidates the current state of knowledge on IL-37, summarizing its regulatory functions and modulation of ferroptosis in diseases such as atherosclerosis, myocardial infarction, aneurysm, stroke, and other CVD. We also discuss experimental approaches and propose that tar-geting IL-37 to regulate ferroptosis holds promise as a therapeutic strategy in preventing and treating diverse CVD.
Keywords:
Interleukin-37
; ferroptosis
; macrophages
; atherosclerosis
; cardiovascular diseases
; inflammation
1. Introduction
Ferroptosis is a novel regulated non-apoptotic cell death characterized by iron deregulation associated with oxidative stress (OS) and lipid peroxidation accumulation. This results in cell membrane damage and subsequent cell death due to oxidative damage [1,2,3,4,5]. Oxidative damage and cell death may produce inflammation and inflammation, which can produce ferroptosis, establishing a positive feedback loop. Both ferroptosis and inflammation processes are potential therapeutic targets in several pathological processes, including cardiovascular diseases (CVD) [6].
In the context of inflammation, macrophages play a central role in the progression of CVD, such as coronary artery disease, peripheral artery disease, and aortic atherosclerosis. Interleukin (IL)-37, a member of the IL-1 family, is a cytokine known for its ability to downregulate pro-inflammatory cytokines and inhibit innate immunity [7]. IL-37 is constitutively expressed by immune cells, including macrophages and suppressive cells such as B and T regulatory cells, and its expression is upregulated in response to pro-inflammatory stimuli [8,9]. Remarkably, IL-37 decreases macrophage ferroptosis by activating the Nrf2 pathway, thereby attenuating the progression of atherosclerosis in a murine model [10].
Despite the growing evidence, there has been no comprehensive review of IL-37’s protective effect on ferroptosis in CVD. Then, we review the current state of knowledge on the regulatory cytokine IL-37 and its immunomodulatory role in ferroptosis within the context of CVD. We discuss experimental approaches and provide insights into therapeutic strategies involving IL-37 via ferroptosis in CVD, highlighting its potential as a therapeutic strategy for preventing and treating these diseases.
2. Ferroptosis and CVDs
Excessive iron concentrations promote the generation of reactive oxygen species (ROS) via iron-dependent Fenton and Haber-Weiss reactions. While ROS production is regulated by enzymatic and no enzymatic antioxidants, depletion of these antioxidants can lead to overproduction of ROS, OS, and oxidative damage. Under these conditions, lipoxygenase (LOX) enzymes and ROS overproduction generate lipid radicals during the lipid peroxidation process. Normally, lipid radicals are degraded by glutathione peroxidase 4 (GPX4); however, GPX4 dysfunction results in the accumulation of lipid radicals, which damage biomolecules such as proteins, DNA, and lipids. Since cell membranes are composed of phospholipids, lipid peroxidation disrupts membrane integrity and fluidity, leading to cell death associated with oxidative damage and ferroptosis (Figure 1) [1,2]. Although cell death related to oxidative damage has been studied for years, the term “ferroptosis”, describing a form of programmed cell death distinct from apoptosis or autophagy, was coined in 2012 by Dixon et al. [11]. This group discovered that erastin decreases cysteine absorption, preventing glutathione (GSH) production, an essential cofactor of GPX4 to reduce the accumulation of lipid peroxides. The reduction of GSH, coupled with increased iron levels, significantly increases ROS production and, consequently, lipid peroxides. Oxidative damage from this process can be observed by microscopy as increased plasma bilayer membrane density, reduced mitochondrial size, disappearance of mitochondrial cristae, mitochondrial condensation, swelling, and membrane rupture, all characteristics of cell death due to ferroptosis.[12]
Ferroptosis differs from other types of programmed cell death, such as apoptosis, as it does not involve DNA fragmentation or caspase activation. Instead, cells undergoing ferroptosis exhibit necrotic morphology, including a lack of chromatin condensation, increased membrane density, and rupture of the outer cell membrane [13]. Rupture is related to the formation of membrane nanopores, which promotes cell bursting. [14] Consequently, ferroptosis differs in morphology, biochemistry, and genetics from other forms of programmed death. Moreover, ferroptosis is inhibited by molecules such as iron-chelating agents or antioxidants rather than caspase inhibitors [14]
Dixon et al. also demonstrated that ferroptosis induced by erastin or 3R-RSL3 (RSL3) augments intracellular iron [11]. During Fenton and Haber Weiss reactions, iron directly generates excessive ROS, promoting lipid peroxidation. Additionally, iron has been shown to increase the activity of LOXs enzymes responsible for lipid peroxidation [15]. Since iron is closely involved in ferroptosis induction, different genes or proteins involved in iron homeostasis, including import, exportation, storage, and regeneration, affect ferroptosis sensitivity. [16] For instance, gastric acid reduces Fe3+ to Fe2+ in the digestive system, which is then absorbed in the duodenum and jejunum. It should be noted that Fe2+ is oxidized to Fe3+ by the action of ceruloplasmin in the cell membrane, which combines Fe3+ with transferrin (TF) to form TF-Fe3+. TF-Fe3+ subsequently forms a complex with transferrin receptor 1 to be endocytosed by the cell. Then, Fe3+ is reconverted to Fe2+ by six-membrane epithelial cells (Step 3). Fe2+ is released into the cytoplasm for use in the cytosol and mitochondria. The iron that is not used in the cytoplasm is stored in ferritin or secreted by ferroportin 1 (FPN1). Note, that this is just a look at the biochemistry of iron, and discussing its full metabolism is beyond the scope of this review. However, changes in iron metabolism directly promote ferroptosis processes, which can contribute to the development and progression of diseases such as atherosclerosis, characterized by lipid metabolism disorders, endothelial damage, oxidative stress, inflammation, and immune dysfunction. Indeed, high iron levels have been associated with increased atherosclerosis, promoting OS and inflammatory responses related to ferroptosis. In vivo studies have shown that Fer-1, a ferroptosis inhibitor, reduces atherosclerotic lesions and lipid peroxidation induced by high-fat diets [17]. Thus, this work demonstrates the relationship between ferroptosis and CVD, such as atherosclerosis [17].
During ferroptosis, the lipid peroxidation of esterified polyunsaturated fatty acids (PUFA) is more prominent compared to desaturated PUFA due to the presence of bis-allylic hydrogen atoms that are susceptible to lipid peroxidation [18]. Esterified PUFA, such as arachidonic acid (AA) and adrenic acid (ADA), are prime substrates for lipid peroxidation. During their oxidation, these PUFA are catalyzed into their acyl-CoA esters by acyl-CoA synthetase long-chain family member 4 (ACSL4) and reacylated into lysophospholipids by lysophosphatidylcholine acyltransferase 3 (LPCAT3). These lysophospholipids are then oxidized by LOX, resulting in cell membrane rupture and ferroptotic cell death (Figure 1) [16]. The products of lipid peroxidation, such as lipid hydroperoxides and aldehydes like 4-hydroxynonenal, increase during ferroptosis, destabilizing the cell membrane and leading to pore formation, which further promotes ferroptosis [14]. Interestingly, supplementing cells with AA and AdA PUFA promotes ferroptosis, while monounsaturated fatty acids (MUFA), such as oleic acid, suppress it by reducing lipid peroxidation [19,20]. MUFAs displace PUFAs from plasma membrane phospholipids, decreasing ferroptosis. Additionally, a high-fat diet (HFD) promotes systemic accumulation of lipids and their metabolites, repressing ferroptosis [20]. The latter suggests that different lipid components of the diet can regulate ferroptosis n vascular smooth muscle cells [17].
3. Inflammation and Ferroptosis
Inflammation is an immunological process that helps to reduce pathogens during host defense and facilitates the elimination or healing of tissue damage [21]. During tissue injury, cytokines and chemokines initiate and maintain an inflammatory process, promoting ROS production, which results in oxidative stress, lipid peroxidation, and oxidative damage. Inflammation is associated with various diseases, such as chronic diseases, such as cardiovascular disease, neurodegenerative pathologies, auto-immune disorders, obesity, type 2 diabetes, endocrine diseases, osteoporosis, cancer, colitis, Crohn’s disease, and metabolic dysfunction-associated steatohepatitis (MASH). [22,23]
Interestingly, in these diseases, blocking ferroptosis alleviates clinical symptoms of colitis but promotes colon tumorigenesis, indicating a dual role of ferroptosis in intestinal diseases [24,25]. In addition, the use of ferroptosis inhibitors represses hepatic lipid peroxidation, resulting in reduced MASH severity [26]. Because reducing ferroptosis alleviates inflammatory diseases, it has been suggested that ferroptosis plays a key role in the pathophysiology of inflammation and may be a potential therapeutic target. Indeed, numerous experiments have confirmed the role of ferroptosis in inflammation, with strong evidence indicating that ferroptosis accelerates inflammation by its immunogenicity [27]. Ferroptosis also triggers inflammation by releasing damage-associated molecular pattern (DAMP), which are immunogenic. Once released from cells, DAMPs promote a non-infectious inflammatory response by binding to pattern recognition receptors (PRR) [27]. For example, high mobility group box 1 (HMGB1) is a typical DAMP released by ferroptotic cells, which, upon binding to its PRR drives inflammation by activating macrophages to produce pro-inflammatory cytokines [28].
During inflammation, DAMP production also increases AA production by phospholipase A2 (PLA2). AA is then metabolized by cyclooxygenase-2 (COX2) into inflammatory mediators such as bioactive prostaglandins, which activate macrophages and other inflammatory cells, including neutrophils and T and B lymphocytes. ROS production significantly increases in this inflammatory response, reacting with lipids, especially PUFAs. PUFAs are essential regulators of crucial cellular processes in the context of inflammation because they induce lipid remodeling in immunological cells. This lipid remodeling occurs by incorporating phospholipids such as phosphatidylethanolamines (PE), which are transformed into PUFA-PE by two enzymes: ACSL4 and LPCAT3 [29]. Together the latter enzymes, proteins such as aldokete reductase family 1 member C1 (AKR1C1), ChaC glutathione-specific gammaglutamylcyclotransferase 1 (CHAC1), ferritin heavy chain 1 (FTH1), and prostaglandin-endoperoxide synthase 2 (PTGS2) are associated with lipid metabolism, glutathione metabolism, and iron storage, with their deregulation leading to cellular ferroptosis. [30]. For instance, AKR1C1 prevents ferroptosis by reducing lipid peroxidation end products to non-toxic lipid-derived alcohols [31]. CHAC1 expression is induced by cystine starvation-triggered ferroptosis [32] and FTH1 is generally upregulated during ferroptosis [33].
Ferroptotic cells can serve as donors of AA for the transcellular biosynthesis of eicosanoids and promote LOX activity through the massive release of oxidized lipid mediators. When AA is released from phospholipids by PLA2 and phospholipase C (PLC), it serves as a precursor for bioactive proinflammatory mediators, such as prostaglandins, interleukin (IL)-1, IL-6, and tumor necrosis factor (TNF), which promote inflammatory cascades. These cytokines and interferon (IFN)-γ are implicated in tissue iron storage, especially in ferritin synthesis regulation. Evidence suggests that an abnormal inflammatory response may contribute to the pathogenesis of iron metabolism disorders and directly affect the redox system balance. For instance, it has been demonstrated that nuclear receptor coactivator 4 (NCOA4) mediates ferritinophagy, degrading the iron storage protein ferritin by autophagolysosomes, resulting in intracellular iron overload, triggering oxidative stress, and exacerbating inflammation [34,35,36,37]. Collectively, these findings indicate that inflammation molecules, such as AA, induce ferroptosis by activating different signaling pathways and molecules related to inflammation.
Several signaling pathways and regulatory mechanisms are tightly linked to ferroptosis-related inflammation. For instance, through the Janus kinase – Signal transducer and activator of transcription (JAK-STAT) cell signaling pathway), when IL-6, TNF, and IL-1β bind to their specific receptors to phosphorylate and to activate JAKs, which in turn phosphorylate STATs and induce their dimerization and nuclear translocation. The latter induces the transcription of target genes such as STAT3, which increase hepcidin expression, inhibiting iron exportation, resulting in ferroptosis [38,39].
Another signaling pathway related to ferroptosis-related inflammation is nuclear factor-κB (NF-κB) pathway. NF-kB is crucial for the onset of various chronic diseases through the up-regulation of the transcription of pro-inflammatory genes, which leads to acute or subacute levels of IL-1, TNF and IL-6. These cytokines are associated with risk of cardiovascular diseases, neurodegenerative diseases, endocrine-metabolic alterations and autoimmune disorders [23]. NF-kB is also by DAMPs such as HMGB1. HMGB1, which is released by ferroptotic cells, downregulates the transcription of target genes related to antioxidant system, enhancing oxidative stress. NF-κB is also involved in iron metabolism during inflammatory processes, promoting secretion of lipocalin 2 (LCN2) to carry extracellular iron and transfer it into cells via their specific receptor. Furthermore, NF-kB can be activated by ROS, which is one of the mainstays in ferroptosis, generating inflammatory mediators such as TNF-α, CXC Chemokine Ligand 1 (CXCL1), C-X-C Motif Chemokine Ligand 8 (CXCL8), and Colony Stimulating Factor 2 (CSF2) [40].
The Mitogen-activated protein kinase (MAPK) pathway is another cell signaling pathway influenced by the intracellular iron overload and excessive lipid peroxidation accumulation, causing phosphorylation of ERK1/2 and increased phosphorylation of c-FOS and p38, thereby increasing oxidative stress, and the latter is involved in the synthesis of IL-1β, IL-6, and IL-18 and at the same time with decreased expression of SLC7A11 and GPX4 and which may mediate neuroinflamation by the release of iron from the labile iron reserve during of ferritinophagy improvement [41]. c-FOS and p38 MAPK proteins are a group of Ser/Thr kinases that are activated by MAPK kinases in response to extracellular stress, infections, ischemia, DNA damage, oxidative stress, and cytokines [42,43]. These kinases can phosphorylate many substrates in the cytoplasm and in the nucleus and play pivotal roles in cell adaptation to stress, inflammation, and tumor formation. p38MAPKs are also involved in ischemia-reperfusion injury, heart failure, arrhythmia, Alzheimer’s epilepsy, and tumorigenesis. Moreover, the inhibition of p38MAPK may exert a systemic anti-inflammatory effect and target numerous diseases with inflammatory components, such as atherosclerosis [44]
Ferroptosis also induces the cyclic GMP-AMP Synthase-Stimulator of Interferon Genes (cGAS-STING) signaling pathway, generating ROS, sensitizing cells to ferroptosis, and facilitating inflammatory infiltrate in tissues when erastin triggers mitochondrial OS, increasing the mitochondrial translocation of STING [45,46]. STING is a PRR, which ligands are cyclic dinucleotides (CDNs) synthesized by microorganisms or by the cytoplasmic cGAS. STING can bind to host or pathogen-derived double-stranded (ds)DNA, either nuclear DNA (nDNA) or mitochondrial DNA (mtDNA), activating this cell signaling pathway [47]. Interestingly, the ferroptosis-induced mitochondria damage could be the source of dsDNA to activate the cGAS-STING pathway.It has been shown that an increase of lipid peroxidation could activate this signaling pathway through the STING carbonylation, resulting in the inhibition of innate antiviral immune responses and reduced recognition of DAMPs or PAMPs in peritoneal macrophages [48]. Furthermore, the cCAS-STING signaling pathway appears to be involved in the inflammasome pathway, where the NLPR3 could be the initiator, further inducing OS, lipid peroxidation, and ferroptosis.
Heme mediates another inflammatory mechanism. Heme is a potent prooxidant and proinflammatory molecule and prototypical alarmin that triggers NLRP3 activation in macrophages [49,50]
Additionally, endothelial cells respond to different alarmins by NLRP3 inflammasome activation and subsequent release of IL-1β, and this mechanism has been shown to play a significant role in diverse pathological conditions, including atherosclerosis [51] In this sense, heme-derived ROS strongly affect endothelial cells, signaling by nitric oxide dismutase (NOD), leucine-rich repeats (LRR), and pyrin domain-containing proteins (NLRP3) [52], that induce an increased IL-1β secretion and exocytosis of Weibel-Palade bodies [53]. Activated endothelial cells upregulate cell adhesion molecules such as vascular cell adhesion molecule-1 (VCAM-1), intercellular adhesion molecule 1, (ICAM-1), and selectins, triggering the recruitment of proinflammatory neutrophils and the induction of antioxidant proteins, including ferritin (bearing ferroxidase activity), and heme oxygenase 1 (HO-1). HO-1 hampers inflammation by inhibiting NF-κB, and reduces hemolysis and vasoocclusion in murine model [54]. Heme itself is a DAMP protein that can bind to Toll-like receptor (TLR) 4 with subsequent activation of NF-κB signaling [la anterior]. Following heme-mediated TLR4 activation, endothelial cells are actively damaged by complement deposition in a P-selectin-dependent manner. Also, heme is toxic for mouse macrophages and can initiate a necrotic process mediated by TNF that is induced downstream of TLR4/MyD88 activation, acting as an autocrine signal via TNFR1 and ultimately resulting in macrophage death [55]. These events result in proinflammatory environment linked to tissue injury, immune infiltration, and vascular dysfunction [55]. Thus, different cell signaling pathways associated with inflammation such as MAPKs kinases, cGAS-STING, NFkB, among others, are related to the activation of ferroptosis.
4. Macrophages and Ferroptosis
Ferroptosis in macrophages is observed in advanced atherosclerotic plaques, contributing to the formation of necrotic core and plaque destabilization, thereby aggravating advanced atherosclerosis [10,11]. Elevated uric acid levels have been shown to enhance macrophage ferroptosis, accelerating the progression of atherosclerosis [12]. Upon uptake of oxidized low-density lipoproteins (LDL), macrophages release proinflammatory cytokines, including IL-1, IL-3, IL-6, IL-8, and IL-18, TNF, as well as growth factors. Particularly, IL-1β and TNF exhibit atherogenic effects by promoting the expression of surface molecules such as intercellular adhesion molecule-1 (ICAM-1), vascular cell adhesion molecule-1 (VCAM)-1), CD40, and selectins on endothelial cells, smooth muscle cells, and macrophages. [56]
Emerging evidence suggests that macrophage polarization and ferroptosis can influence each other at the cell-autonomous level or through communication with other cells in a context-dependent manner [57]. Moreover, it has been shown that M1 cells are resistant to ferroptosis due to the loss of arachidonate 15-lipoxygenase (ALOX15) activity [57]. In the context of atherosclerosis, macrophages contribute to tissue repair and the proliferation of vascular smooth muscle cells, thereby enhancing plaque stability [13].
However, in a study by Marques et al., macrophages exposed to high levels of oxLDL and proinflammatory cytokines increased heme oxygenase (Hmox1), H-ferritin (Fth1), hepcidin (Hamp) and FPN1 mRNA levels, although only the latter did not increase its protein levels, leading to macrophage iron retention due to decrease export and increase storage. In addition, the macrophage iron overload could play a crucial role in plaque instability in atherosclerosis [58].
Macrophages can obtain iron from two sources, phagocytized red blood cells (RBC) and extracellular ferric iron (Fe3+). The first depends upon the activity of the Hmox1 to degrade heme and produce ferrous iron (Fe2+), while the second employs the coupling of transferrin (TF) with its receptor (TRF1) as previous explained [59]. Hepcidin is a key regulator for iron homeostasis and is upregulated by iron and inflammation [60]. Hamp targets and degrades FPN1, which leads to a decrease of the iron export, and increase the iron accumulation. This makes the macrophages susceptible to ferroptosis. Furthermore, Hamp also enhances oxLDL uptake and reduces the cholesterol excretion in macrophages via autocrine Hamp formation, induced by iron excess, high levels of oxLDL, aggravating atherosclerosis through a vicious cycle [61].
“Efferocytosis,” an efficient mechanism for debris clearance by macrophages, has been identified as crucial in preventing secondary necrosis and in stimulating the release of anti-inflammatory cytokines such as IL-35 and IL-37 [13]. Given the mechanisms of inflammation described above, it is not difficult to understand how ferroptosis plays a pro-inflammatory role in atherosclerosis.
5. Protective Role of Interleukin-37 in Cardiovascular Diseases
Cytokines are small proteins involved in the development and pathogenesis of inflammatory and/or autoimmune diseases through their secretion [62]. The involvement of cytokines in cardiomyocyte apoptosis highlights their crucial role in modulating the delicate balance between cell survival and death in the heart.
Interleukin-37 (IL-37) is a recently identified member of the interleukin-1 (IL-1) family and is a pivotal anti-inflammatory cytokine involved in regulating inflammation [63,64,65]. IL-37 has been identified in diverse human tissues, including the skin, tonsils, esophagus, placenta, as well as in melanoma, breast, prostate, and colon tumors [66]. IL-37 can suppress innate immune response by reducing the production of pro-inflammatory cytokines induced by Toll-like receptors (TLR) [67]. Moretti et al. [68] reported that IL-37 can significantly inhibit the activation of Th2/Th17 cells in mice with allergic aspergillosis, showing that IL-37 might also affect adaptive immunity.
Previous studies have shown that Smad3, the main protein for receptors of the transforming growth factor beta (TGF-B) superfamily, is involved in the immunosuppressive and anti-inflammatory properties of IL-37 both in vitro and in vivo. It has been shown that blocking Smad3 activation or Smad3knockdown decreases the anti-inflammatory activity of IL-37 [66,67,68]. However, the effects of the IL-37 Smad3 signaling pathway and sequential inflammatory factors during the development of inflammatory cardiovascular diseases remain largely unknown. A strong suggestion pointing out that Smad3 is a crucial protein in CVD, such as atherosclerosis, because this disease is a chronic inflammatory disease characterized by the generation and release of pro-inflammatory cytokines. Thus, IL-37, an emerging anti-inflammatory cytokine, exhibits intriguing potential in the context of atherosclerosis. This regulatory cytokine has a promising role in atherosclerotic diseases as a potent inflammation inhibitor by shifting cytokine equilibrium away from excessive inflammation.
Regulatory T cells (Tregs) mediate immunomodulation and protect against atherosclerosis. Enhancing the anti-inflammatory response could reduce the amount of pro-inflammatory mediators, and this may be considered one of the main targets for therapy development. The inhibitory cytokines secreted by Tregs mainly include interleukin-10 (IL-10), and transforming growth factor-beta (TGF-β) and interleukin-35 (IL-35) [69]
Lotfy, Hassan et al. [70] revealed that IL-37 was increased in patients with chronic lower limb atherosclerotic ischemia compared to non-atherosclerotic controls. Also, the expression levels of circulating IL-37 correlated with the disease severity of chronic lower limb ischemia. Interestingly, supplementation with rIL-37 augmented levels of released IL-10 and TGF-β in supernatants of T cells co-cultured with Tregs in the enrolled patients. Authors suggest a role for IL-37 in mediating anti-inflammatory functions in the atherosclerotic process, potentially involving enhancement of Treg inhibitory function and anti-inflammatory cytokine secretion with a particularly marked direct response in severe disease [70]. Although previous studies found some relation between IL-37 and acute coronary syndrome (ACS), the specific correlation and the prediction of a patient’s prognosis still need to be established.
By the other hand, Ji Q, Zeng Q, Huang Y, et al. demonstrated that the levels of certain anti-inflammatory cytokines were significantly decreased in patients with ACS [71]. This study also indicated that IL-37 levels were markedly increased in foam-like cells of atherosclerotic coronary and carotid artery plaques, with a negative correlation to left ventricular ejection fraction (LVEF) in ACS patients [71]. However, the precise role of altered IL-37 levels in the progression of atherosclerosis and the onset of ACS remains uncertain.
Although it is required to perform mechanistic studies in which the administration of exogenous IL-37 (e.g., recombinant human IL-37 and a model animal with IL-37 transgenic mice) clarifies the role of IL-37 in atherosclerosis. Chai M, et al. [72] found that IL-37 has a significant role in atherosclerosis. These authors used apoE-deficient diabetic mice to assess the IL-37 effects on vascular calcification and atherosclerosis progression. IL-37-treated mice exhibited significantly reduced calcification areas, as detected by von Kossa and Alizarin Red staining, and decreased expression of Bone Morphogenetic Protein 2 B(MP-2), TNF, IL-18, and IL-10 in atherosclerotic lesions. This group also observed smaller plaque sizes and lower plaque vulnerability scores in the aortic root. Upregulation of IL-10 expression and the concomitant decrease in TNF and IL-18 production were direct anti-inflammatory mechanisms through which IL-37 ameliorated atherosclerosis and coronary artery calcification [72,73].
In patients with atherosclerosis, higher IL-37 concentrations were detected in calcified samples, primarily originating from macrophages and vascular smooth muscle cells. Patients with coronary artery calcification also showed significantly higher plasma IL-37 levels. Correlation analysis revealed that IL-37 was positively associated with age, fasting glucose, alkaline phosphatase, IL-6, TNF, and C-reactive protein [74]
Interestingly, IL-37 is also involved in regulating cholesterol homeostasis and reducing plasma cholesterol, fatty acids, and triglycerides levels [75]. Recently, it has been reported that the IL-37 rs2708961, rs2723187, and rs2708947 polymorphisms are associated with a lower risk of hypercholesterolemia (HC) in the Mexican population. These polymorphisms were also associated with cardiovascular risk factors. Some IL-37 polymorphisms were associated with cardiometabolic factors in individuals with and without HC [75]. Additionally, Yin et al. reported an association between the IL-37 rs3811047 polymorphism and coronary artery disease, along with IL-37 decreased mRNA expression levels [76]. On the other hand, Xu et al. [77] noted that IL-37 mitigated the toxic effects of inflammatory mediators on myocardial cells. IL-37 also ameliorated myocardial infarction. Wu B, et al. [78] demonstrated that IL-37 protected mouse cardiomyocytes from apoptosis under ischemia/reperfusion (I/R) conditions and suppressed the production of pro-inflammatory cytokines, chemokines, and neutrophil infiltration. This contributed to reduced cardiomyocyte apoptosis and ROS generation. The regulatory role of IL-37 was demonstrated through the inhibition of Toll-like receptor (TLR)-4 expression and NF-kB activation after I/R, while increasing anti-inflammatory IL-10 level [78].
In another study Law et al. [79] discussed the clinical implications of IL-37 in cardiovascular manifestations of COVID-19 in a recent review. Elevated circulating IL-37 levels were reported in COVID-19-infected patients, with higher IL-37 levels associated with shorter hospitalization periods. This suggests that IL-37 may provide protective effects during COVID-19 infection, highlighting its potential protective role in CVD.
5. Interleukin-37 and Ferroptosis in Cardiovascular Diseases
IL-37 is also implicated in decreasing macrophage ferroptosis. Recent research has investigated the relationship between ferroptosis and the protective effect of IL-37 on macrophage ferroptosis stimulated with high glucose (HG)/ox-LDL in diabetic ApoE-/- mice. Xu, Jinmei et al. identified that IL-37 treatment significantly decreased plaque area, improved blood lipid levels, reduced serum levels of inflammatory mediators, including IL-1β and IL-18, and increased GPX4 and nuclear factor erythroid 2-related factor 2 (Nrf2) in the aorta of diabetic mice [17]. Additionally, in vitro experiments revealed that IL-37 inhibited HG/ox-LDL-induced ferroptosis in macrophages, as evidenced by improved cell membrane oxidation, reduced malondialdehyde production, and increased GPX4 expression. In the same study, the Nrf2 pathway was promoted, inhibiting ferroptosis in the macrophages (Figure 2) [10]. The transcription factor Nrf2 is crucial to maintain cellular homeostasis against OS through the transcription of antioxidant enzymes genes, inducing cell protection. Its activation upregulates the expression of downstream anti-ferroptotic genes such as GPX4, SLC7A11, and FPN1 and downregulates the ACSL4. Moreover, numerous studies have shown the beneficial results of the Nrf2 pathway via alleviating ferroptosis in myocardial infarction, colitis, ischemia-reperfusion injury, and ischemic stroke [80,81,82]. Whether IL-37 promotes the dissociation, directly or indirectly, between Nrf2 and its regulator, the Kelch like ECH-associated protein 1 (KEAP1) is still unknown, which deserves further investigation.
While the role of IL-37 in atherosclerosis has been established, its involvement in macrophage ferroptosis and its potential as a therapeutic target is still an emerging area of interest. As mechanisms regulating ferroptosis become further elucidated, ferroptosis holds clear potential for therapeutic benefit in several CVD
6. Conclusions and Conclusion Remarks
Inflammation produces ROS and OS, which ultimately promotes ferroptosis cell death. We reviewed that ferroptosis is a mechanistically and morphologically distinct form of regulated cell death that has provided insights into the pathophysiology of multiple disease entities. Elucidating the regulatory pathways related to ferroptosis can provide new opportunities for understanding the pathophysiology of diseases, including CVDs, by identifying novel targets and strategies for target ferroptosis for therapeutic advantage. Regulation of ferroptosis via IL-37 could be an effective intervention strategy for preventing and treating inflammatory diseases. In our review, we discussed this possibility, suggesting the high possibility of reducing ferroptosis cell death in CVDs by decreasing inflammation via IL-37.
Author Contributions
Conceptualization, G.F-C and A.C-G; Investigation, G.F-C, A.C.-G., L.M.A-G; B.U.F-B; A.R-B and.; Writing—Original Draft Preparation, G.F.-C and A.C.-G.; Writing—Review and Editing, G.F-C., A.C.-G. and L.M.A-G.; Visualization, L.M.A-G.; Supervision, G.F-C. All authors have read and agreed to the published version of the manuscript.
Funding
Open access funding for this article was supported by Instituto Nacional de Cardiología Ignacio Chávez.
Conflicts of Interest
The authors declare no conflicts of interest
References
- Xie, L.-H.; Fefelova, N.; Pamarthi, S.H.; Gwathmey, J.K. Molecular Mechanisms of Ferroptosis and Relevance to Cardiovascular Disease. Cells 2022, 11, 2726. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Li, X.; Wang, S.; Miao, R.; Zhong, J. Targeting Iron Metabolism and Ferroptosis as Novel Therapeutic Approaches in Cardiovascular Diseases. Nutrients 2023, 15, 591. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Fan, H.; Wang, S.; Tang, G.; Zhai, C.; Shen, L. Ferroptosis: A Novel Therapeutic Target for Ischemia-Reperfusion Injury. Front. Cell Dev. Biol. 2021, 9, 688605. [Google Scholar] [CrossRef]
- Cantrell, A.C.B.; Zeng, H.; Chen, J.-X. The therapeutic potential of targeting ferroptosis in the treatment of mitochondrial cardiomyopathies and heart failure. J. Cardiovasc. Pharmacol. 2023, 83, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.; Wang, K.; Li, Y.; Ye, Y.; Chen, Y.; Wang, J.; Yao, C. Construction of a Novel Ferroptosis-Related Gene Signature of Atherosclerosis. Front. Cell Dev. Biol. 2022, 9, 800833. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Ardehali, H.; Min, J.; Wang, F. The molecular and metabolic landscape of iron and ferroptosis in cardiovascular disease. Nat. Rev. Cardiol. 2023, 20, 7–23. [Google Scholar] [CrossRef] [PubMed]
- Nold, M.F.; A Nold-Petry, C.; A Zepp, J.; E Palmer, B.; Bufler, P.; A Dinarello, C. IL-37 is a fundamental inhibitor of innate immunity. Nat. Immunol. 2010, 11, 1014–1022. [Google Scholar] [CrossRef]
- Fonseca-Camarillo, G.; Furuzawa-Carballeda, J.; Yamamoto-Furusho, J.K. Interleukin 35 (IL-35) and IL-37: Intestinal and peripheral expression by T and B regulatory cells in patients with Inflammatory Bowel Disease. Cytokine 2015, 75, 389–402. [Google Scholar] [CrossRef]
- Law, C.C.; Puranik, R.; Fan, J.; Fei, J.; Hambly, B.D.; Bao, S. Clinical Implications of IL-32, IL-34 and IL-37 in Atherosclerosis: Speculative Role in Cardiovascular Manifestations of COVID-19. Front. Cardiovasc. Med. 2021, 8, 630767. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Han, X.; Xia, N.; Zhao, Q.; Cheng, Z. IL-37 suppresses macrophage ferroptosis to attenuate diabetic atherosclerosis via the NRF2 pathway. Exp. Ther. Med. 2023, 25, 1–10. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Chen, X.; Kang, R.; Kroemer, G. Ferroptosis: molecular mechanisms and health implications. Cell Res. 2021, 31, 107–125. [Google Scholar] [CrossRef] [PubMed]
- Hassannia, B.; Vandenabeele, P.; Berghe, T.V. Targeting Ferroptosis to Iron Out Cancer. Cancer Cell 2019, 35, 830–849. [Google Scholar] [CrossRef] [PubMed]
- Pedrera, L.; Espiritu, R.A.; Ros, U.; Weber, J.; Schmitt, A.; Stroh, J.; Hailfinger, S.; von Karstedt, S.; García-Sáez, A.J. Ferroptotic pores induce Ca2+ fluxes and ESCRT-III activation to modulate cell death kinetics. Cell Death Differ. 2021, 28, 1644–1657. [Google Scholar] [CrossRef]
- Jiang, X.; Stockwell, B.R.; Conrad, M. Ferroptosis: mechanisms, biology and role in disease. Nat. Rev. Mol. Cell Biol. 2021, 22, 266–282. [Google Scholar] [CrossRef]
- Doll, S.; Conrad, M. Iron and ferroptosis: A still ill-defined liaison. IUBMB Life 2017, 69, 423–434. [Google Scholar] [CrossRef] [PubMed]
- You, J.; Ouyang, S.; Xie, Z.; Zhi, C.; Yu, J.; Tan, X.; Li, P.; Lin, X.; Ma, W.; Liu, Z.; et al. The suppression of hyperlipid diet-induced ferroptosis of vascular smooth muscle cells protests against atherosclerosis independent of p53/SCL7A11/GPX4 axis. J. Cell. Physiol. 2023, 238, 1891–1908. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.S.; Stockwell, B.R. Ferroptosis: Death by Lipid Peroxidation. Trends Cell Biol. 2016, 26, 165–176. [Google Scholar] [CrossRef]
- Tesfay, L.; Paul, B.T.; Konstorum, A.; Deng, Z.; Cox, A.O.; Lee, J.; Furdui, C.M.; Hegde, P.; Torti, F.M.; Torti, S.V. Stearoyl-CoA Desaturase 1 Protects Ovarian Cancer Cells from Ferroptotic Cell Death. Cancer Res. 2019, 79, 5355–5366. [Google Scholar] [CrossRef]
- Zhang, X.; Ma, Y.; Lv, G.; Wang, H. Ferroptosis as a therapeutic target for inflammation-related intestinal diseases. Front. Pharmacol. 2023, 14, 1095366. [Google Scholar] [CrossRef]
- Medzhitov, R. Origin and physiological roles of inflammation. Nature 2008, 454, 428–435. [Google Scholar] [CrossRef] [PubMed]
- Ocansey, D.K.W.; Yuan, J.; Wei, Z.; Mao, F.; Zhang, Z. Role of ferroptosis in the pathogenesis and as a therapeutic target of inflammatory bowel disease (Review). Int. J. Mol. Med. 2023, 51, 53. [Google Scholar] [CrossRef] [PubMed]
- Fioranelli, M.; Roccia, M.G.; Flavin, D.; Cota, L. Regulation of Inflammatory Reaction in Health and Disease. Int. J. Mol. Sci. 2021, 22, 5277. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wang, J.; Li, J.; Zhu, J.; Wang, R.; Xi, Q.; Wu, H.; Shi, T.; Chen, W. Astragalus polysaccharide prevents ferroptosis in a murine model of experimental colitis and human Caco-2 cells via inhibiting NRF2/HO-1 pathway. Eur. J. Pharmacol. 2021, 911, 174518. [Google Scholar] [CrossRef]
- Xu, C.; Liu, Z.; Xiao, J. Ferroptosis: A Double-Edged Sword in Gastrointestinal Disease. Int. J. Mol. Sci. 2021, 22, 12403. [Google Scholar] [CrossRef]
- Qi, J.; Kim, J.-W.; Zhou, Z.; Lim, C.-W.; Kim, B. Ferroptosis Affects the Progression of Nonalcoholic Steatohepatitis via the Modulation of Lipid Peroxidation–Mediated Cell Death in Mice. Am. J. Pathol. 2019, 190, 68–81. [Google Scholar] [CrossRef] [PubMed]
- Gong, T.; Liu, L.; Jiang, W.; Zhou, R. DAMP-sensing receptors in sterile inflammation and inflammatory diseases. Nat. Rev. Immunol. 2019, 20, 95–112. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Kang, R.; Tang, D. The mechanism of HMGB1 secretion and release. Exp. Mol. Med. 2022, 54, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Agborbesong, E.; Li, L.X.; Li, L.; Li, X. Molecular Mechanisms of Epigenetic Regulation, Inflammation, and Cell Death in ADPKD. Front. Mol. Biosci. 2022, 9, 922428. [Google Scholar] [CrossRef] [PubMed]
- Stockwell, B.R.; Angeli, J.P.F.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascón, S.; Hatzios, S.K.; Kagan, V.E.; et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef]
- Gagliardi, M.; Cotella, D.; Santoro, C.; Corà, D.; Barlev, N.A.; Piacentini, M.; Corazzari, M. Aldo-keto reductases protect metastatic melanoma from ER stress-independent ferroptosis. Cell Death Dis. 2019, 10, 1–15. [Google Scholar] [CrossRef]
- Chen, M.-S.; Wang, S.-F.; Hsu, C.-Y.; Yin, P.-H.; Yeh, T.-S.; Lee, H.-C.; Tseng, L.-M. CHAC1 degradation of glutathione enhances cystine-starvation-induced necroptosis and ferroptosis in human triple negative breast cancer cells via the GCN2-eIF2α-ATF4 pathway. Oncotarget 2017, 8, 114588–114602. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Tao, J.; Yang, Y.; Tan, S.; Liu, H.; Jiang, J.; Zheng, F.; Wu, B. Ferroptosis involves in intestinal epithelial cell death in ulcerative colitis. Cell Death Dis. 2020, 11, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Çolakoğlu, M.; Tunçer, S.; Banerjee, S. Emerging cellular functions of the lipid metabolizing enzyme 15-Lipoxygenase-1. Cell Prolif. 2018, 51, e12472. [Google Scholar] [CrossRef] [PubMed]
- Uauy, R.; Mena, P.; Rojas, C. Essential fatty acid metabolism in the micropremie. Clin. Perinatol. 2000, 27, 71–93. [Google Scholar] [CrossRef] [PubMed]
- I Sperling, R.; I Benincaso, A.; Knoell, C.T.; Larkin, J.K.; Austen, K.F.; Robinson, D.R. Dietary omega-3 polyunsaturated fatty acids inhibit phosphoinositide formation and chemotaxis in neutrophils. J. Clin. Investig. 1993, 91, 651–660. [Google Scholar] [CrossRef] [PubMed]
- H.W. de Jonge, D.H. Dekkers, J.M. Lamers, Polyunsaturated fatty acids and signalling via phospholipase C-beta and A2 in myocardium, Mol. Cell. Biochem. 157, (1996) 199–210.
- Zhang Z, Tang J, Song J, Xie M, Liu Y, Dong Z, Liu X, Li X, Zhang M, Chen Y, Shi H, Zhong J. Elabela alleviates ferroptosis, myocardial remodeling, fibrosis and heart dysfunction in hypertensive mice by modulating the IL-6/STAT3/GPX4 signaling. Free Radic Biol Med. 2022; 181:130–42.
- Ren F, Yang Y, Wu K, Zhao T, Shi Y, Song M, et al. The effects of dandelion polysaccharides on iron me-tabolism by regulating hepcidin via JAK/STAT signaling pathway. Oxid Med Cell Longev. 2021; 2021:7184760.
- Alam Z, Devalaraja S, Li M, To TKJ, Folkert IW, MitchellVelasquez E, Dang MT, Young P, Wilbur CJ, Sil-verman MA, Li X, Chen YH, Hernandez PT, Bhattacharyya A, Bhattacharya M, Levine MH, Haldar M. Counter regulation of spic by NF-κB and STAT signaling controls inflammation and iron metabolism in macrophages. Cell Rep. 2020;31: 107825.
- Liu N, Liang Y, Wei T, Zou L, Huang X, Kong L, et al. The role of ferroptosis mediated by NRF2/ERK-regulated ferritinophagy in CdTe QDs induced inflammation in macrophage. J Hazard Mater. 2022;436:129043.
- Sanz-Ezquerro, J.J.; Cuenda, A. p38 Signalling Pathway. Int. J. Mol. Sci. 2021, 22, 1003. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Liu, Y.; Guo, Y.; Liu, C.; Yang, Y.; Fan, X.; Yang, H.; Liu, Y.; Ma, T. Function and inhibition of P38 MAP kinase signaling: Targeting multiple inflammation diseases. Biochem. Pharmacol. 2024, 220, 115973. [Google Scholar] [CrossRef]
- Gluba-Brzózka, A.; Franczyk, B.; Rysz-Górzyńska, M.; Ławiński, J.; Rysz, J. Emerging Anti-Atherosclerotic Therapies. Int. J. Mol. Sci. 2021, 22, 12109. [Google Scholar] [CrossRef]
- Liang, J.-L.; Jin, X.-K.; Zhang, S.-M.; Huang, Q.-X.; Ji, P.; Deng, X.-C.; Cheng, S.-X.; Chen, W.-H.; Zhang, X.-Z. Specific activation of cGAS-STING pathway by nanotherapeutics-mediated ferroptosis evoked endogenous signaling for boosting systemic tumor immunotherapy. Sci. Bull. 2023, 68, 622–636. [Google Scholar] [CrossRef]
- Li C, Liu J, Hou W, Kang R, Tang D. STING1 promotes ferroptosis through MFN1/ 2-dependent mitochondrial fusion. Front Cell Dev Biol. 2021;9:698679–698679.
- Couillin, I.; Riteau, N. STING Signaling and Sterile Inflammation. Front. Immunol. 2021, 12. [Google Scholar] [CrossRef] [PubMed]
- Jia, M.; Qin, D.; Zhao, C.; Chai, L.; Yu, Z.; Wang, W.; Tong, L.; Lv, L.; Wang, Y.; Rehwinkel, J.; et al. Redox homeostasis maintained by GPX4 facilitates STING activation. Nat. Immunol. 2020, 21, 727–735. [Google Scholar] [CrossRef]
- Jeney, V.; Eaton, J.W.; Balla, G.; Balla, J. Natural History of the Bruise: Formation, Elimination, and Biological Effects of Oxidized Hemoglobin. Oxidative Med. Cell. Longev. 2013, 2013, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Soares, M.P.; Bozza, M.T. Red alert: labile heme is an alarmin. Curr. Opin. Immunol. 2016, 38, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wang, L.; Pitzer, A.L.; Li, X.; Li, P.-L.; Zhang, Y. Contribution of redox-dependent activation of endothelial Nlrp3 inflammasomes to hyperglycemia-induced endothelial dysfunction. J. Mol. Med. 2016, 94, 1335–1347. [Google Scholar] [CrossRef]
- Erdei, J.; Tóth, A.; Balogh, E.; Nyakundi, B.B.; Bányai, E.; Ryffel, B.; Paragh, G.; Cordero, M.D.; Jeney, V. Induction of NLRP3 Inflammasome Activation by Heme in Human Endothelial Cells. Oxid. Med. Cell. Longev. 2018, 2018, 4310816. [Google Scholar] [CrossRef]
- Schillemans, M.; Karampini, E.; Kat, M.; Bierings, R. Exocytosis of Weibel–Palade bodies: how to unpack a vascular emergency kit. J. Thromb. Haemost. 2019, 17, 6–18. [Google Scholar] [CrossRef] [PubMed]
- Belcher, J.D.; Chen, C.; Nguyen, J.; Milbauer, L.; Abdulla, F.; Alayash, A.I.; Smith, A.; Nath, K.A.; Hebbel, R.P.; Vercellotti, G.M. Heme triggers TLR4 signaling leading to endothelial cell activation and vaso-occlusion in murine sickle cell disease. Blood 2014, 123, 377–390. [Google Scholar] [CrossRef]
- Martins, R.; Knapp, S. Heme and hemolysis in innate immunity: adding insult to injury. Curr. Opin. Immunol. 2018, 50, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Fang, Z.-M.; Yi, X.; Wei, X.; Jiang, D.-S. The interaction between ferroptosis and inflammatory signaling pathways. Cell Death Dis. 2023, 14, 1–13. [Google Scholar] [CrossRef]
- Kapralov, A.A.; Yang, Q.; Dar, H.H.; Tyurina, Y.Y.; Anthonymuthu, T.S.; Kim, R.; St Croix, C.M.; Mikulska-Ruminska, K.; Liu, B.; Shrivastava, I.H.; et al. Redox lipid reprogramming commands susceptibility of macrophages and microglia to ferroptotic death. Nat. Chem. Biol. 2020, 16, 278–290. [Google Scholar] [CrossRef] [PubMed]
- Marques, L.; Negre-Salvayre, A.; Costa, L.; Canonne-Hergaux, F. Iron gene expression profile in atherogenic Mox macrophages. Biochim. et Biophys. Acta (BBA) - Mol. Basis Dis. 2016, 1862, 1137–1146. [Google Scholar] [CrossRef]
- Ma, J.; Zhang, H.; Chen, Y.; Liu, X.; Tian, J.; Shen, W. The Role of Macrophage Iron Overload and Ferroptosis in Atherosclerosis. Biomolecules 2022, 12, 1702. [Google Scholar] [CrossRef]
- Cornelissen, A.; Guo, L.; Sakamoto, A.; Virmani, R.; Finn, A.V. New insights into the role of iron in inflammation and atherosclerosis. EBioMedicine 2019, 47, 598–606. [Google Scholar] [CrossRef] [PubMed]
- Xiao, L.; Luo, G.; Guo, X.; Jiang, C.; Zeng, H.; Zhou, F.; Li, Y.; Yu, J.; Yao, P. Macrophage iron retention aggravates atherosclerosis: Evidence for the role of autocrine formation of hepcidin in plaque macrophages. Biochim. et Biophys. Acta (BBA) - Mol. Cell Biol. Lipids 2019, 1865, 158531. [Google Scholar] [CrossRef] [PubMed]
- Lai Y, Dong C: Therapeutic antibodies that target inflammatory cytokines in autoimmune diseases. Int Immunol, 2016; 28: 181–88.
- Dinarello CA, Nold-Petry C, Nold M et al: Suppression of innate inflammation and immunity by interleukin-37. Eur J Immunol, 2016; 46: 1067–81.
- Xu, W.-D.; Zhao, Y.; Liu, Y. Insights into IL-37, the role in autoimmune diseases. Autoimmun. Rev. 2015, 14, 1170–1175. [Google Scholar] [CrossRef] [PubMed]
- Quirk, S.; Agrawal, D.K. Immunobiology of IL-37: mechanism of action and clinical perspectives. Expert Rev. Clin. Immunol. 2014, 10, 1703–1709. [Google Scholar] [CrossRef] [PubMed]
- Kumar S, Hanning CR, Brigham-Burke MR, Rieman DJ, Lehr R, Khandekar S, et al. Interleukin-1F7B (IL-1H4/IL-1 F7) is processed by caspase-1 and mature IL-1F7B binds to the IL-18 receptor but does not induce IFN-gamma production. Cytokine, 2002;18:61–71.
- Tetè, S.; Tripodi, D.; Rosati, M.; Conti, F.; Maccauro, G.; Saggini, A.; Cianchetti, E.; Caraffa, A.; Antinolfi, P.; Toniato, E.; et al. IL-37 (IL-1F7) the Newest Anti-Inflammatory Cytokine Which Suppresses Immune Responses and Inflammation. Int. J. Immunopathol. Pharmacol. 2012, 25, 31–38. [Google Scholar] [CrossRef]
- Moretti, S.; Bozza, S.; Oikonomou, V.; Renga, G.; Casagrande, A.; Iannitti, R.G.; Puccetti, M.; Garlanda, C.; Kim, S.; Li, S.; et al. IL-37 Inhibits Inflammasome Activation and Disease Severity in Murine Aspergillosis. PLOS Pathog. 2014, 10, e1004462. [Google Scholar] [CrossRef]
- Kuan, R.; Agrawal, D.K.; Thankam, F.G. Treg cells in atherosclerosis. Mol. Biol. Rep. 2021, 48, 4897–4910. [Google Scholar] [CrossRef]
- Lotfy, H.; Moaaz, M.; Moaaz, M. The novel role of IL-37 to enhance the anti-inflammatory response of regulatory T cells in patients with peripheral atherosclerosis. Vascular 2020, 28, 629–642. [Google Scholar] [CrossRef]
- Ji, Q.; Zeng, Q.; Huang, Y.; Shi, Y.; Lin, Y.; Lu, Z.; Meng, K.; Wu, B.; Yu, K.; Chai, M.; et al. Elevated Plasma IL-37, IL-18, and IL-18BP Concentrations in Patients with Acute Coronary Syndrome. Mediat. Inflamm. 2014, 2014, 165742. [Google Scholar] [CrossRef] [PubMed]
- Chai, M.; Ji, Q.; Zhang, H.; Zhou, Y.; Yang, Q.; Zhou, Y.; Guo, G.; Liu, W.; Han, W.; Yang, L.; et al. The Protective Effect of Interleukin-37 on Vascular Calcification and Atherosclerosis in Apolipoprotein E-Deficient Mice with Diabetes. J. Interf. Cytokine Res. 2015, 35, 530–539. [Google Scholar] [CrossRef] [PubMed]
- Bello, R.O.; Chin, V.K.; Isnadi, M.F.A.R.; Majid, R.A.; Abdullah, M.A.; Lee, T.Y.; Zakaria, Z.A.; Hussain, M.K.; Basir, R. The Role, Involvement and Function(s) of Interleukin-35 and Interleukin-37 in Disease Pathogenesis. Int. J. Mol. Sci. 2018, 19, 1149. [Google Scholar] [CrossRef] [PubMed]
- Yu, K.; Min, X.; Lin, Y.; Huang, Y.; Huang, S.; Liu, L.; Peng, Y.; Meng, K.; Li, D.; Ji, Q.; et al. Increased IL-37 concentrations in patients with arterial calcification. Clin. Chim. Acta 2016, 461, 19–24. [Google Scholar] [CrossRef]
- López-Bautista, F.; Posadas-Sánchez, R.; Vázquez-Vázquez, C.; Fragoso, J.M.; Rodríguez-Pérez, J.M.; Vargas-Alarcón, G. IL-37 Gene and Cholesterol Metabolism: Association of Polymorphisms with the Presence of Hypercholesterolemia and Cardiovascular Risk Factors. The GEA Mexican Study. Biomolecules 2020, 10, 1409. [Google Scholar] [CrossRef] [PubMed]
- Yin, D.; Naji, D.H.; Xia, Y.; Li, S.; Bai, Y.; Jiang, G.; Zhao, Y.; Wang, X.; Huang, Y.; Chen, S.; et al. Genomic Variant in IL-37 Confers A Significant Risk of Coronary Artery Disease. Sci. Rep. 2017, 7, 42175. [Google Scholar] [CrossRef] [PubMed]
- Xu, D., Wang A., Jiang F., Hu J., Zhang X. Effects of interleukin-37 on cardiac function after myocardial infarction in mice. Int. J. Clin. Exp. Pathol. 2015;8:5247–5251.
- Wu, B.; Meng, K.; Ji, Q.; Cheng, M.; Yu, K.; Zhao, X.; Tony, H.; Liu, Y.; Zhou, Y.; Chang, C.; et al. Interleukin-37 ameliorates myocardial ischaemia/reperfusion injury in mice. Clin. Exp. Immunol. 2014, 176, 438–451. [Google Scholar] [CrossRef]
- Law, C.C.; Puranik, R.; Fan, J.; Fei, J.; Hambly, B.D.; Bao, S. Clinical Implications of IL-32, IL-34 and IL-37 in Atherosclerosis: Speculative Role in Cardiovascular Manifestations of COVID-19. Front. Cardiovasc. Med. 2021, 8, 630767. [Google Scholar] [CrossRef]
- Tian, H.; Xiong, Y.; Zhang, Y.; Leng, Y.; Tao, J.; Li, L.; Qiu, Z.; Xia, Z. Activation of NRF2/FPN1 pathway attenuates myocardial ischemia–reperfusion injury in diabetic rats by regulating iron homeostasis and ferroptosis. Cell Stress Chaperon- 2022, 27, 149–164. [Google Scholar] [CrossRef]
- Ru, Y.; Luo, Y.; Liu, D.; Huang, Q.; Zhou, X.; Linghu, M.; Luo, X.; Lv, Z.; Wu, Y.; Zhang, H.; et al. Isorhamnetin alleviates ferroptosis-mediated colitis by activating the NRF2/HO-1 pathway and chelating iron. Int. Immunopharmacol. 2024, 135, 112318. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.; Chu, W.; Zhang, H.; Peng, Y. Nrf2 and Ferroptosis: A New Research Direction for Ischemic Stroke. Cell. Mol. Neurobiol. 2023, 43, 3885–3896. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Ferroptosis mechanisms and the system Xc- – GSH – GXP4 pathway. Iron metabolism: The transferrin receptor (TRF1) mediates the endocytosis of the ferric iron (Fe3+)-transferrin complex, then the ferric iron is reduced to ferrous iron (Fe++) by the metalloreductase STEAP3 which leads to the entrance to the cytosol by the divalent metal transporter (DMT1). In the cytosol, iron can form the labile iron pool (LIP), couple with other enzymes, be stored by ferritin or exported by ferroportin (FPN). Lipid metabolism: The enzymes ACSL4 and LPCAT3/5 catalyze the formation of membrane phospholipids-containing polyunsaturated fatty acid chains (PUFA-PL). Reactive oxygen species: Superoxide anion (●O2-) is produced in mitochondria, and then is reduced to peroxide hydrogen (H2O2) by superoxide dismutase (SOD). Lipid peroxidation: The non-enzymatic pathway initiates with the production of hydroxyl radicals (●OH) via Fenton reactions, a ferrous iron-catalyzed H2O2 oxidation process. The ●OH react with PUFAs that leads to lipid radicals and propagate the lipid peroxidation. The lipoxygenase (LOX) is essential for the enzymatic pathway by catalyzing PUFAs into lipid hydroperoxide (PL-OOH). Malondialdehyde (MDA) and 4-hydroxynenanol (4HNE) are the final metabolites of lipid peroxidation. System Xc—–GSH–GPX4 pathway: System Xc— regulate the cystine importation and glutamate exportation, cystine is reduced to cysteine by thioredoxin reductase (TXNRD) so it can be used to form glutathione (GSH). The enzyme glutathione peroxidase 4 (GPX4) utilizes GSH to reduce PL-OOH into lipid alcohols (PL-OH) and inhibit lipid peroxidation. Falta definer PL y PL-OO.
Figure 1.
Ferroptosis mechanisms and the system Xc- – GSH – GXP4 pathway. Iron metabolism: The transferrin receptor (TRF1) mediates the endocytosis of the ferric iron (Fe3+)-transferrin complex, then the ferric iron is reduced to ferrous iron (Fe++) by the metalloreductase STEAP3 which leads to the entrance to the cytosol by the divalent metal transporter (DMT1). In the cytosol, iron can form the labile iron pool (LIP), couple with other enzymes, be stored by ferritin or exported by ferroportin (FPN). Lipid metabolism: The enzymes ACSL4 and LPCAT3/5 catalyze the formation of membrane phospholipids-containing polyunsaturated fatty acid chains (PUFA-PL). Reactive oxygen species: Superoxide anion (●O2-) is produced in mitochondria, and then is reduced to peroxide hydrogen (H2O2) by superoxide dismutase (SOD). Lipid peroxidation: The non-enzymatic pathway initiates with the production of hydroxyl radicals (●OH) via Fenton reactions, a ferrous iron-catalyzed H2O2 oxidation process. The ●OH react with PUFAs that leads to lipid radicals and propagate the lipid peroxidation. The lipoxygenase (LOX) is essential for the enzymatic pathway by catalyzing PUFAs into lipid hydroperoxide (PL-OOH). Malondialdehyde (MDA) and 4-hydroxynenanol (4HNE) are the final metabolites of lipid peroxidation. System Xc—–GSH–GPX4 pathway: System Xc— regulate the cystine importation and glutamate exportation, cystine is reduced to cysteine by thioredoxin reductase (TXNRD) so it can be used to form glutathione (GSH). The enzyme glutathione peroxidase 4 (GPX4) utilizes GSH to reduce PL-OOH into lipid alcohols (PL-OH) and inhibit lipid peroxidation. Falta definer PL y PL-OO.
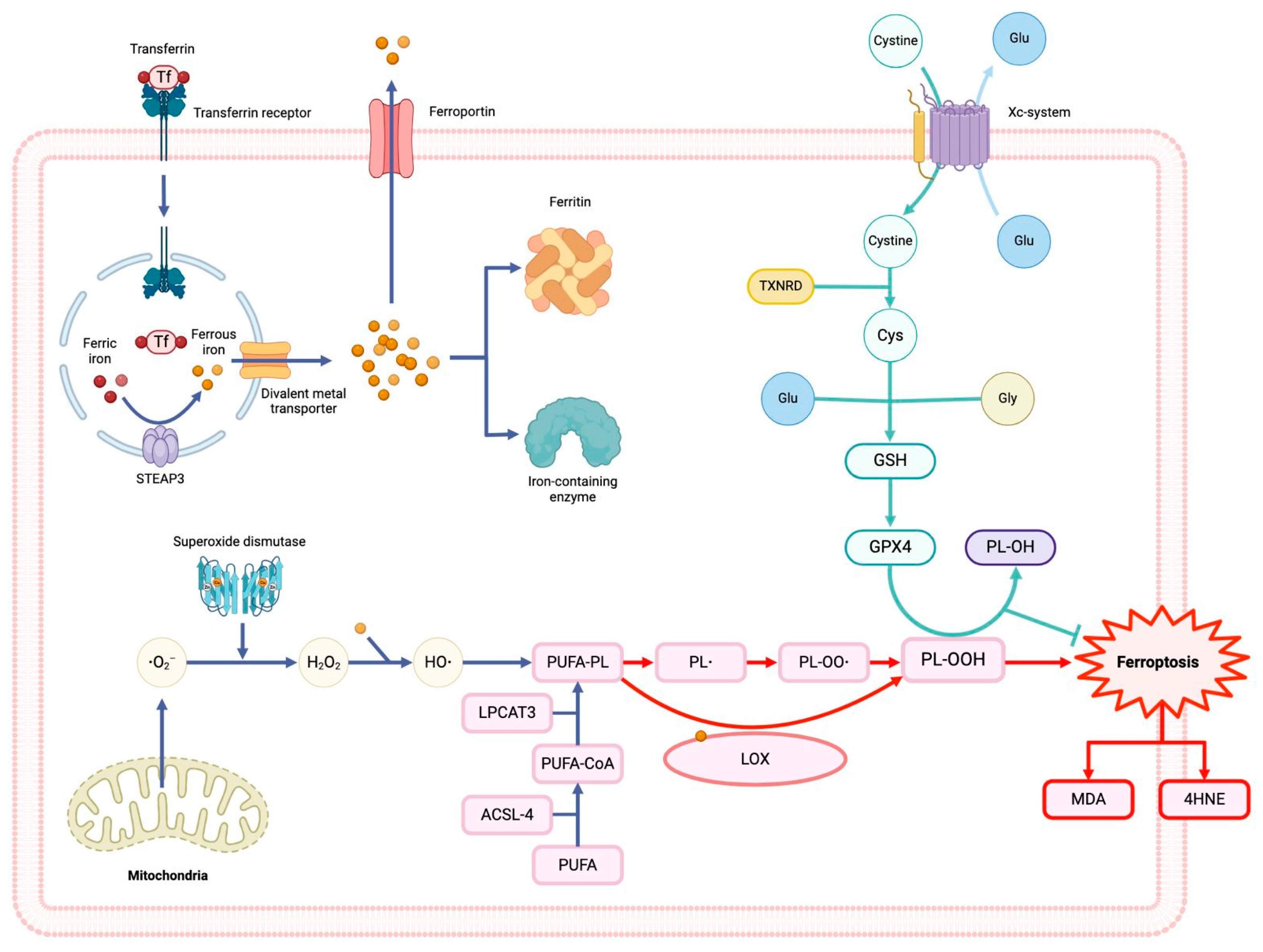
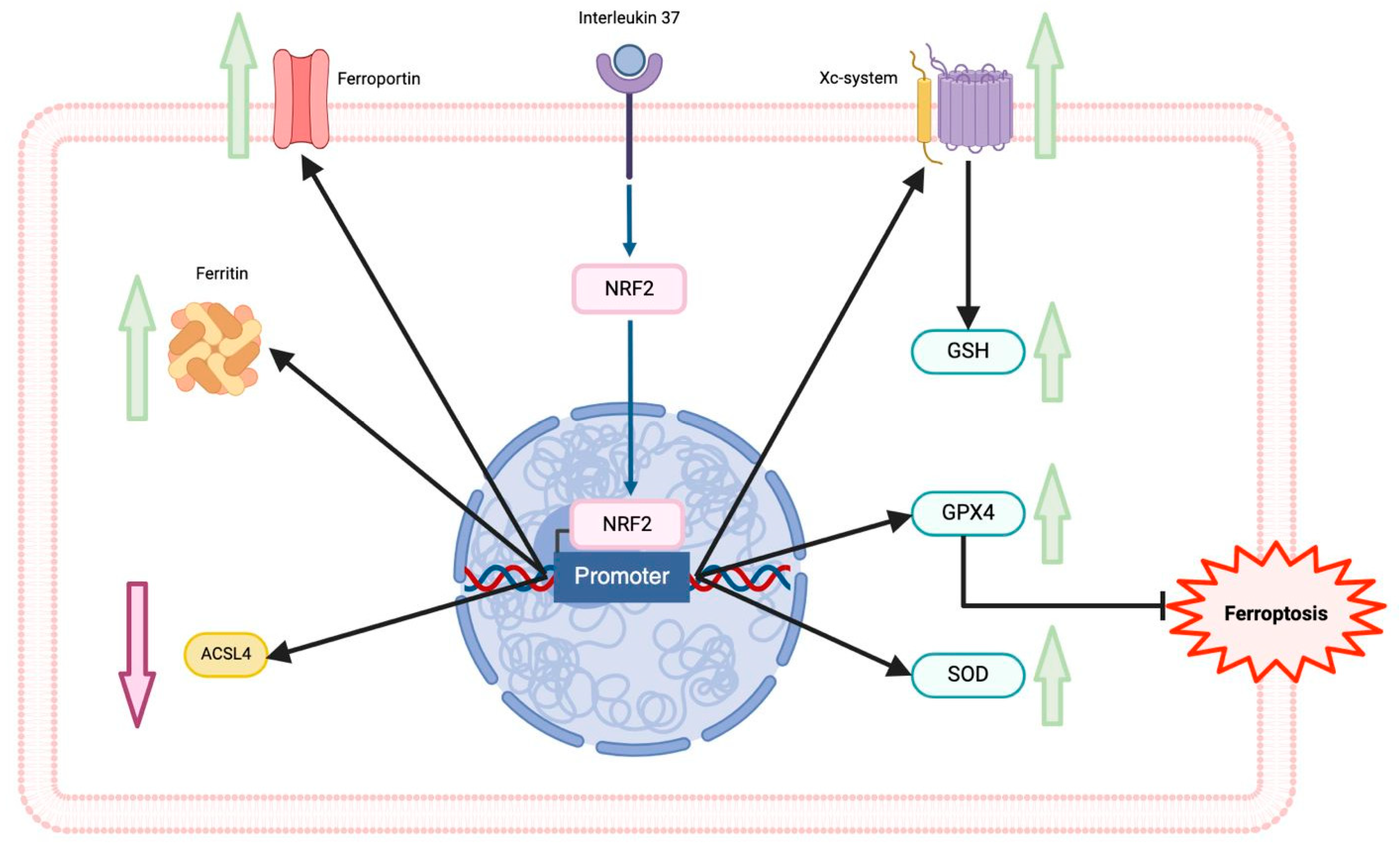
Figure 2.
Interleukin 37 (IL-37) and nuclear factor erythroid 2-related factor 2 (Nrf2 pathway. The master regulator of antioxidant response Nrf2 is activated under significant oxidative stress, and IL-37, then it is translocated to induce its target genes in the nucleus. The Nrf2 pathway upregulates proteins of the iron metabolism, superoxide dismutase, and the system Xc—–GSH–GPX4 pathway and downregulates ACSL4. IL-37 can induce the activation and translocation of Nrf2 to the nucleus. Glutathione (GSH), Glutathione peroxidase 4 (GPX4), acyl-CoA synthetase long chain family member 4 (ACSL4).
Figure 2.
Interleukin 37 (IL-37) and nuclear factor erythroid 2-related factor 2 (Nrf2 pathway. The master regulator of antioxidant response Nrf2 is activated under significant oxidative stress, and IL-37, then it is translocated to induce its target genes in the nucleus. The Nrf2 pathway upregulates proteins of the iron metabolism, superoxide dismutase, and the system Xc—–GSH–GPX4 pathway and downregulates ACSL4. IL-37 can induce the activation and translocation of Nrf2 to the nucleus. Glutathione (GSH), Glutathione peroxidase 4 (GPX4), acyl-CoA synthetase long chain family member 4 (ACSL4).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



