
Submitted:
30 July 2024
Posted:
31 July 2024
You are already at the latest version
Abstract
Ubiquitin-specific protease 2 (USP2) maintains mitochondrial integrity in culture myoblasts. In this study, we investigated molecular mechanisms underlying the protective role of USP2 on mitochondria. Knockout (KO) of the Usp2 gene or chemical inhibition of USP2 induced robust accumulation of mitochondrial reactive oxygen species (ROS), accompanied by defects in mitochondrial membrane potential, in C2C12 myoblasts. ROS removal by N-acetyl-L-cysteine restored the mitochondrial dysfunction induced by USP2 deficiency. Comprehensive RT-qPCR screening and following protein analysis indicated that both genetic and chemical inhibition of USP2 elicited a decrease of uncoupling protein 2 (UCP2) at mRNA and protein levels. Accordingly, the introduction of a Ucp2-expressing construct effectively recovered mitochondrial membrane potentials, entailing an increment of the intracellular ATP level in Usp2KO C2C12 cells. In contrast, USP2 deficiency also decreased peroxisome proliferator-activated receptor coactivator 1 (PGC1 protein in C2C12 cells, while it upregulated Ppargc1a mRNA. Overexpression studies indicate that USP2 potentially stabilizes PGC1 in an isopeptidase-dependent manner. Given that PGC1 is an inducer of UCP2 in C2C12 cells, USP2 might ameliorate mitochondrial ROS by maintaining the PGC1–UCP2 axis in myoblasts.
Keywords:
ubiquitin specific protease 2
; myoblasts
; mitochondria
; ROS
; uncoupling protein 2
; PGC1alpha
1. Introduction
Loss of muscle mass has a critical influence on quality of life. For instance, age-dependent loss of muscle mass, sarcopenia, is an atypical geriatric disease with adverse outcomes: frailty, dysmobility, and mortality [1]. Maintenance of muscle progenitor cells, including satellite cells and myoblasts, impedes the progression of sarcopenia [2]. Oxidative stress is believed to damage muscle progenitor cells. For instance, hydrogen peroxide causes apoptosis and cytoplasmic distribution of p21 in myoblasts rather than in myotubes [3]. Moreover, excessive reactive oxygen species (ROS) severely inhibit muscle regeneration by inhibiting myocyte differentiation from progenitor cells [4,5]. Notably, satellite cells from elderly people exhibited higher intracellular ROS compared to younger people, resulting in impaired mitochondrial activity [6]. Therefore, managing intracellular ROS levels of muscle progenitor cells might be an effective therapeutic intervention for muscular atrophy.
Mitochondria are one of the primary sources of ROS under both physiological and pathological conditions [7]. Under physiological conditions, mitochondria generate moderate levels of ROS, mainly from the respiratory chain complexes I and III [8]. Pathological conditions, such as hyperglycemia and cardiomyopathy, perturbate the mitochondrial respiratory chain and culminate in excessive ROS production [9,10]. Superoxide is produced in the mitochondria and is converted to hydrogen peroxide by superoxide dismutase (SOD) [11]. Hydrogen peroxide is eventually quenched by catalase, the glutathione peroxidase (Gpx)-glutathione reductase (GR) system, or the peroxiredoxin (Prdx)-thioredoxin (Trx)-thioredoxin reductase (TrxR) system [12]. Uncoupling proteins (UCPs) in the inner mitochondrial membrane are also thought to minimize mitochondrial ROS generation [13]. UCPs are carrier proteins that induce proton leak and attenuation of mitochondrial membrane potential, resulting in suppression of ROS production [14]. Of the three canonical UCPs, UCP2 protects against ROS in various cells, including pancreatic β-cells, cardiomyocytes, and macrophages [14,15,16]. Although UCP2 is ubiquitously expressed in multiple tissues, including skeletal muscle and adipose tissue in normal conditions [17], the expression of the Ucp2 gene is also under the control of transcription factors and co-activators, including peroxisomal activators-activated receptors (PPARs) and PPARγ coactivator 1α (PGC1α) [18].
Emerging evidence indicates that excess accumulation of mitochondrial ROS severely encumbers mitochondrial function. Pathological content of mitochondrial ROS induced changes in mitochondrial morphology, oxidative phosphorylation, and mitochondrial transcription factor A level in renal proximal tubule epithelial cells [19]. Moreover, hypoxia-induced mitochondrial ROS interrupts mitochondrial gene expression and ATP synthesis in pig sperm [20]. Mitochondrial ROS also provokes mitochondrial DNA mutation, which is associated with tissue aging [21]. Although adequate levels of ROS function as the redox signal to maintain the viability of stem cells, prevention of excessive accumulation of ROS is necessary to sustain an efficient number of stem cells for regeneration [22]. To support this idea, Minet and Gaster reported that senescent culture human satellite cells displayed more prominent ROS accompanying an increase of mitochondrial oxidative phosphorylation [23]. The author hypothesized that accumulated ROS consequently causes mitochondrial damage at a later time. Therefore, preventing mitochondrial damage caused by ROS might be a potential therapeutic approach for regenerative medicine and geriatrics.
Ubiquitination and deubiquitination, which are counteractive protein modifications, are catalyzed by ubiquitin-ligase and deubiquitinating enzymes (DUBs), respectively [24,25]. Ubiquitin-specific protease (USP) is the largest subfamily of DUBs, which consists of 58 members in vertebrates [26]. USP2 is a widely expressed USP with several molecular targets, including receptors, intracellular signal adaptors, and transcriptional regulators [27]. To date, we and others have demonstrated that USP2 controls energy homeostasis at local and systemic levels [28,29,30,31]. For example, USP2 in ventromedial hypothalamic neurons attenuates aberrant increases in blood glucose by mitigating glycogenolysis in the liver, whereas hepatic USP2 regulates gluconeogenesis and diurnal glucose metabolism in the liver [28,31]. Additionally, USP2 directly or indirectly maintains mitochondrial ATP synthesis of myoblasts, sperm, and neural cells [31,32,33]. With regards to myoblasts, chemical and genetic ablation of USP2 rapidly induces ROS accumulation accompanying defects in mitochondrial ATP synthesis [33]. Therefore, USP2 is likely to sustain mitochondrial respiration in myoblasts via the removal of ROS. In this study, we aimed to verify the involvement of ROS in the USP2-influenced mitochondrial dysfunction of myoblasts. To this end, we used C2C12 myoblastic cells derived from the regenerative muscle of C3H mice [34], which are widely used for myoblast research [33,35,36,37]. We also investigated the molecular mechanisms underlying ROS accumulation in USP2-deficient myoblasts.
2. Results
2.1. Mitochondrial ROS Is Involed in Mitochondrial Dysfunction in Usp2KO C2C12 Cells
We previously demonstrated that knockout of the Usp2 gene causes mitochondrial dysfunction in C2C12 myoblasts [33]. Since mitochondrial ROS markedly accumulated in Usp2KO C2C12 cells, we speculated that ROS accumulation might cause impaired ATP synthesis in Usp2KO C2C12 cells [33]. To verify this possibility, we first alleviated ROS accumulation in Usp2KO C2C12 cells by treating them with N-acetyl-L-cysteine (NAC) for 8 h. Figure 1A,B show mitochondrial ROS, which is visualized with MitoSOX Red in Usp2KO and control C2C12 cells. As previously reported [33], Usp2KO C2C12 cells displayed an intense MitoSOX signal compared to the vehicle-treated control C2C12 cells. NAC treatment reduced ~27% of the fluorescent signals in Usp2KO cells. Thus, NAC treatment effectively sequestered ROS accumulation in Usp2KO C2C12 cells.
We next assessed whether the NAC treatment weaken the Usp2 deficiency-elicited mitochondrial dysfunction. Mitochondrial membrane potential, verified by tetramethylrhodamine methyl ester (TMRM) staining, was significantly lower in Usp2KO C2C12 cells than in control C2C12 cells (Figure 1C). NAC treatment partially recovered the mitochondrial membrane potential in Usp2KO C2C12 cells. Correspondingly, NAC treatment remarkably recovered the decrease of intracellular ATP in Usp2KO C2C12 cells (Figure 1D). Therefore, ROS damages the mitochondria of Usp2KO C2C12 cells, resulting in impaired ATP production.
2.2. Mitochondrial ROS Is Involved in Mitochondrial Dysfunction in ML364-Treated C2C12 Cells
ML364 is the most popular USP2 chemical inhibitor and has been used for USP2 blockade in vivo and in vitro [31,38]. Similar to the Usp2 gene knockout, ML364 treatment caused mitochondrial dysfunction accompanied by robust ROS accumulation in C2C12 cells [33]. Thus, we also investigated the effects of NAC on the ML364-elicited mitochondrial dysfunction. Figure 2A,B show that treatment with 10 μM ML364 for 8 h substantially increased mitochondrial ROS. Since this ML364 treatment did not increase lactate dehydrogenase (LDH) content in the culture supernatant of C2C12 cells, this condition is unlikely to be toxic to C2C12 cells (Figure S1). Although NAC did not completely abrogate ROS accumulation in ML364-treated cells, NAC significantly abated mitochondrial ROS levels (Figure 2A,B). Correspondingly, NAC significantly attenuated the suppressive effects of ML364 on mitochondrial membrane potential (Figure 2C). Moreover, NAC partially restored the ML364-induced decrease of intracellular ATP (Figure 2D). Hence, ROS contributes to the ML364-elicited mitochondrial damage in C2C12 cells.
2.3. USP2 Maintains UCP2 Expression at mRNA and Protein Levels in C2C12 Cells
Since the mitochondrial respiratory complexes did not alter activity in Usp2KO C2C12 cells [33], impairment of the respiratory chain is unlikely to be a major cause of excess accumulation of mitochondrial ROS in Usp2KO C2C12 cells. Thus, we assumed that USP2 deficiency causes expressional defects of certain antioxidative molecules. To explore them, we performed comprehensive reverse transcription– quantitative polymerase chain reaction (RT-qPCR) screening of 22 genes, which can be postulated to repress ROS accumulation (Figure 3A). Expression levels of 2 genes were significantly (Prdx2, Ucp2; P < 0.01) lower in Usp2KO C2C12 cells than control C2C12 cells, whereas those of 3 genes were higher in Usp2KO cells (Gsta4, Hmox1, Sod1; P < 0.01) (Figure 3A, left). ML364 treatment influenced the expression of a larger number of genes: 4 genes (Gsta4, Hmox1, Nfe2l2, Prdx6) and 4 genes (Glrx, Prdx1, Selenos, Ucp2) were significantly (P < 0.01) up- and down-regulated, respectively (Figure 3A, right). In this screening, Ucp2 was the only gene for which the expression level was more than 2-fold downregulated by both genetic and chemical inhibition of USP2 (Figure 3B). Further RT-qPCR analysis using more replicates confirmed a greater than 50% decrement of Ucp2 mRNA by Usp2 deficiency in C2C12 cells (Figure 3C). In accordance, ML364 also repressed Ucp2 mRNA to a similar degree (Figure 3D). We also elucidated UCP2 protein levels in Usp2KO cells. As with mRNA levels, Usp2KO C2C12 cells displayed ~40 % lower UCP2 protein than control C2C12 cells (Figure 3E). Similarly, ML364 strongly decreased the UCP2 protein level in C2C12 cells (Figure 3F).
We also checked the activities of canonical antioxidative enzymes in Usp2KO C2C12 cells and found activities of SOD, Gpx, GR, and TrxR were not distinguishable between control and Usp2KO C2C12 cells (Figure 3G). Similarly, neither was modulated by ML364 (10 μM, 8 h) treatment (Figure 3H). Therefore, USP2 did not sustain the overall activities of SOD, Gpx, GR, and TrxR in C2C12 myoblasts.
2.4. Decrement of UCP2 Seems to Participate in USP2 Deficiency–Elicited Mitochondrial Dysfunction
To evaluate whether UCP2 contributes to the USP2-dependent mitochondria integrity, we introduced a lentivirus encoding the Ucp2 gene to Usp2KO or control C2C12 cells. Abundant Ucp2 mRNA was observed in Ucp2-introduced Usp2KO and control C2C12 cells (Figure 4A). Moreover, UCP2 protein levels between Ucp2-introduced Usp2KO and C2C12 cells were comparable (Figure 4B). Although the introduction of the Ucp2-expressing construct failed to abolish excess accumulation of mitochondrial ROS in Usp2KO C2C12 cells, overexpression of Ucp2 significantly decreased ROS in these cells (~35%; Figure 4C). Ucp2-infected Usp2KO C2C12 cells exhibited an apparent increase in mitochondrial membrane potential (Figure 4D). Additionally, transfection of Ucp2 abrogated the decrease of intracellular ATP in Usp2KO C2C12 cells (Figure 4E). Collectively, USP2 maintains mitochondria integrity via UCP2 expression.
2.5. USP2 Stabilizes PGC1α in an Isopeptidase-Dependent Manner
PGC1α is known to potentiate Ucp2 mRNA in C2C12 myotubes [39]. Thus, we hypothesized that USP2 controls PGC1α protein levels. The abundance of Ppargc1a mRNA, which encodes PGC1α, was more than 10-fold higher in Usp2KO cells than in control C2C12 cells (Figure 5A). In sharp contrast, the nuclear PGC1 protein level was relatively low in Usp2KO C2C12 cells (Figure 5B). Accordingly, an overexpression study using 293FT cells demonstrated that the introduction of USP2 increased the accumulation of Halo-tagged PGC1α in the nucleus (Figure 5C). These results suggest that USP2 increases the PGC1α protein via a post-transcriptional process.
Since USP2 stabilizes several proteins by deubiquitination [40], we tested whether USP2 retards the degradation of the PGC1α protein. Similar to the results of Figure 5C, PGC1α protein content was remarkably lower in mock-transfected 293FT cells than in Usp2-transfected cells before cycloheximide application (Figure 5D). After cycloheximide addition, PGC1α protein in mock-transfected 293FT cells was rapidly degraded, decreasing to less than 5% at 5 h (Figure 5D). However, degradation of PGC1α was slower in Usp2-transfected cells: more than 70% of PGC1α was retained at 5 h, and ~37% was still visible even at 10 h.
Next, we evaluated the ubiquitin protease activity of USP2 in the control of PGC1α stability. Since the substitution of the 276th amino acid residue of USP2 from cysteine to alanine ablates ubiquitin isopeptidase activity, we assessed the effects of the C276A mutant on the PGC1α level. In contrast to a wild-type USP2 protein, the C276A mutant failed to sustain PGC1α in 293FT cells in the presence of cycloheximide (Figure 5E). Given that protein levels of USP2 or C276A were comparable in USP2- and C276A-transfected cells, USP2 modulates PGC1α levels via ubiquitin isopeptidase activity.
Finally, we examined whether USP2 directly digests the polyubiquitin chain on PGC1α. As shown in Figure 5F, an immunoprecipitation–western blot analysis reveals that overexpression of GFP-tagged USP2 slightly affected polyubiquitination of PGC1α (see lanes for immunoprecipitants). In contrast, GFP-tagged USP2 substantially decreased overall polyubiquitination of the nuclear protein (see lanes for inputs). However, the amount of PGC1α in the immunopecipitant was smaller in GFP-expressing control cells than USP2-GFP–transfected cells. Thus, the ratios of band intensities between the polyubiquitinated PGC1α smear to total PGC1α in the immunoprecipitant were smaller in USP2-GFP expressing cells than in GFP-expressing control cells. Therefore, USP2 seems to directly digest the polyubiquitin chain on PGC1α.
3. Discussion
As previously reported [33], genetic and chemical inhibition of USP2 provoked the accumulation of mitochondrial ROS in C2C12 myocytes. Similarly, our preliminary experiments showed that inhibition of USP2 also potentiated mitochondrial ROS production in myotubes. Thus, USP2 seems to prevent ROS production in mitochondria in the muscle cell lineage. Given that the chemical blockade of USP2 brought about ROS accumulation in hypothalamic neurons, leading to activation of the efferent sympathetic nerves [31], the antioxidative roles of USP2 might be common to several types of cells.
In this study, NAC treatment restored mitochondrial ATP synthesis in both Usp2KO and ML364-treated C2C12 cells, indicating that oxidative stress contributes to impaired oxidative phosphorylation in USP2-deficient cells. In addition to defects of ATP synthesis, USP2-deficient C2C12 cells also showed blunted mitochondria [33]. To date, USP2 has been shown to stabilize mitofusin 2 (MFN2) in cardiomyocytes [41]. MFN2, a GTPase at the outer mitochondrial membrane, is a prerequisite for mitochondrial fusion [42]. Mitochondria dynamics, consisting of fission and fusion, are a counterbalance mechanism that accommodates mitochondria to energy requirements [43]. Thus, the decrease of MFN2 might also aggravate mitochondrial function in USP2-deficient myoblasts.
Some USPs confer resistance against oxidative stress via the induction of antioxidative enzymes. For instance, USP18 induces SOD and catalase to reduce malondialdehyde in pulmonary endothelial cells [44]. Alternatively, USP30 participates in the autophagy of peroxisome (so-called “pexophagy”), indicating its role in intracellular ROS levels [45]. To explore molecules responsible for the USP2-dependent antioxidation, we performed mRNA screening of 22 antioxidative molecules. Since only UCP2 was influenced by both genetic and chemical inhibition of USP2, molecular mechanisms underlying the USP2-dependent antioxidation seem to be relatively selective. However, we only checked the expression of 22 molecules at the mRNA level. Therefore, USP2 might control these molecules at post-transcriptional or post-translational levels, although USP2 did not modulate the overall activities of SOD, GR, Gpx, and TrxR. Additionally, USP2 might regulate other antioxidative or ROS-generating molecules, which were not examined in this study. Previously, USP2 was preferably localized to peroxisomes in hepatocytes, suggesting that USP2 might control ROS generation through pexophagy [46]. Further investigation might reveal additional factors participating in the USP2-dependent antioxidation.
Of 22 genes, USP2 preserves Ucp2 expression in C2C12 cells. UCP2 dampens ROS generation and subsequent metabolic defects, including diabetes mellitus, atherosclerosis, and cardiomyopathy [14,47]. Thus, impaired expression of Ucp2 may cause ROS accumulation in USP2-deficient C2C12 cells. This theory was supported by introducing a Ucp2-expressing construct conferring resistance against mitochondrial dysfunction in Usp2KO C2C12 cells. Unlike the restricted expression of UCP1 and UCP3 in brown adipose tissue and skeletal muscle, UCP2 is widely distributed across various tissues or cells, including muscle, adipose tissue, brain, pancreatic islet, and macrophages [17]. Considering that tissue distribution of USP2 is also relatively ubiquitous [48], USP2 might prevent oxidative stress in various tissues by the sustenance of UCP2.
Lack of USP2 dramatically repressed UCP2 at the mRNA level, implying that USP2 stimulates the transcription of Ucp2 genes. In an early study, Spiegelman and colleagues demonstrated that overexpression of PGC1α increased Ucp2 mRNA levels in mature C2C12 cells, suggesting that PGC1α transcriptionally augments Ucp2 expression [39]. Since USP2 retarded degradation of PGC1α, USP2 might maintain UCP2 levels due to the stabilization of PGC1α. Indeed, overexpression of the Usp2 gene decreased poly-ubiquitin chains on PGC1α, suggesting that USP2 is a direct stabilizing enzyme for PGC1α by deubiquitination. A recent paper reported that PGC1α repressed the accumulation of mitochondrial ROS and subsequent DNA damage in myoblasts, preventing sarcopenia [49]. Therefore, USP2 might impede sarcopenia by maintenance of PGC1α.
Although USP2 deficiency caused a remarkable decrement of Ucp2 mRNA in parallel with the disappearance of the PGC1α protein, we cannot exclude the possibility that USP2 promotes Ucp2 expression via other transcriptional regulators. It has been suggested that PPARs, FOXA1, and SMAD4 regulate Ucp2 expression [50]. Additionally, the promoter region of the human UCP2 gene has several cis-regulatory elements, including the specific protein-1 (Sp1) binding site, the sterol regulatory elements, and the thyroid hormone response elements, indicating that Sp1, sterol binding proteins (SRBPs), and thyroid receptor (TR) can control Ucp2 expression [18]. Notably, PGC1α potentiates the expression of SREBP-1c and SREBP2 in β-cell-like INS-E1 cells, implying that PGC1α can indirectly maintain Ucp2 expression via SREBP induction [51].
As with the current study, perturbation of USP2 interrupts mitochondrial ATP synthesis in myoblasts, leading to defects in proliferation and differentiation [33,52]. In contrast, Usp2KO mice typically develop without any defects in skeletal muscle [53]. The difference between cellular and animal models might be attributed to the degree of oxidative stress. Most progenitors of myocytes in tissues remain quiescent under a physiological condition, while cultured myoblasts continuously proliferate. To obtain sufficient ATP for proliferation, hematopoietic stem cells in the fetal liver promote oxidative phosphorylation and subsequent ROS generation compared with those in bone marrow [54]. Similarly, cultured myoblasts might expedite oxidative phosphorylation compared to myocyte progenitors in muscle, bringing about the accumulation of mitochondrial ROS. Furthermore, cultured cells are usually maintained at atmospheric oxygen levels, while oxygen concentrations in tissues range between 2-9% [55]. Since 1-4 % of the oxygen consumed by the mitochondria is deflected to produce ROS, an excess of ROS is produced in cultured cells compared to cells within the body under normal physiological conditions [55]. Therefore, cultured myoblasts might be liable to express oxidative mitochondrial damage owing to USP2 deficiency. Given this, USP2-dependent mitochondrial defects in myoblasts are likely to become more apparent in the skeletal muscle of individuals suffering from oxidative stress, such as diabetes mellitus.
4. Materials and Methods
4.1. Cells
Mouse myoblastic C2C12 cells were obtained from the RIKEN Bioresource Center (Tsukuba, Japan). Usp2KO C2C12 clone #1 has previously been documented [33]. Human embryonic kidney 293FT cells were purchased from Thermo Fisher Scientific (Waltham, MA, USA). All cells were cultured in 4.5 g/L glucose-containing Dulbecco’s modified Eagle medium supplemented with 10% FCS. In some experiments, C2C12 cells and their derivatives were treated with 10 μM ML364 (MedChem Express, Monmouth Junction, NJ, USA) for 8 h. For the removal of mitochondrial ROS, cells were incubated in the presence of 5 mM NAC (Fujifilm Wako, Osaka, Japan) for 8 h. Cells were treated with 100 μg/mL cycloheximide (Fujifilm Wako) to prevent nascent protein synthesis. To inhibit the proteasome, 10 μM MG132 (TCI, Tokyo, Japan) was supplemented for 5 h.
4.2. Plasmid Constructs and Transfection
Construction of phUSP2-EGFP and phUSP2-HA plasmids was performed as previously described [29]. pEGFP-N2 (Clontech, Mountain View, CA, USA) and pcDNA3-HAC (Provided by Dr. Hiroyuki Takatsu, Kyoto University) were used as “empty plasmid” controls. pFN21A-HaloTag-hPGC1α and pFN21A-HaloTag-CMV Flexi plasmids were purchased from Promega (Madison, WI, USA). HA-ubiquitin was obtained from Addgene (Watertown, MA, USA). A point mutation, which substitutes cysteine to alanine at the 276th amino acid of human USP2A (NP_004196), was performed using a KOD-Plus-Mutagenesis kit (Toyobo, Osaka, Japan). The plasmids were transfected into 293FT cells using Fugene HD reagent (Promega).
4.3. Lentivirus Constructs and Infection
The coding region of mouse Ucp2 (Accession no. BC012697) was cloned into a pDONR221 vector (Thermo Fisher Scientific) by a Gateway BP reaction and subsequently converted to pLenti6/V5-DEST (Thermo Fisher Scientific) by a Gateway LR reaction. The DNA sequence of the pLenti6-mUcp2-DEST construct was checked using Hokkaido System Science (Sapporo, Japan). Recombinant lentiviral particles were produced in 293FT cells using a ViraPower Lentiviral Packaging Mix (Thermo Fisher Scientific). Lentivirus particles derived from the transfected cells were concentrated using Lenti-X Concentrator (Takara Bio, Otsu, Japan) and used to infect C2C12 cells. Infected cells were selected using 1–2 μg/mL Blasticidin S (InvivoGen, Hong Kong, China).
4.3. Mitochondrial ROS Accumulation
Mitochondrial ROS was visualized by treatment with MitoSOX Red superoxide indicator (Thermo Fisher Scientific). The cells were stained with 5 μM MitoSOX Red reagent for 20 min. After washing with PBS twice, the cells were monitored using a FACS Verse (BD Biosciences, Franklin Lakes, NJ, USA) or by BZ-H4A microscopy (Keyence, Osaka, Japan). The nuclei were stained with 5 ng/mL Hoechst33342 (Thermo Fisher Scientific). Quantitative analysis of flow cytometry was performed using FACS Diva (BD Biosciences).
4.4. Mitochondrial Membrane Potential
The electric potential of the mitochondrial inner membrane was visualized using TMRM (Setaresh Biotech, Eugene, OR, USA). The cells were incubated in the presence of 20 nM TMRM for 30 min. After washing with PBS twice, the cells were subjected to analysis using FACS Verse flow cytometry.
4.5. Intracellular ATP Content
Intracellular ATP content was measured using an ATP measurement solution (Toyo B-Net, Tokyo, Japan). After adding equal volume of the ATP measurement solution to the culture medium, the cells were vigorously mixed for 1 min, and subsequently incubated at room temperature in a dark place. Chemiluminescence of the cell lysate was then measured using a NIVO multimode microplate reader (Perkin Elmer, Waltham, MA, USA).
4.6. Cellular Toxicity
The cellular toxicity of ML364 was validated by monitoring LDH content in the culture supernatant. LDH content was measured using a Cytotoxicity LDH assay kit (Dojindo, Kumamoto, Japan) according to the manufacturer’s instructions. Absorbance at 490 nm was measured using an iMark Microplate Absorbance Reader (Bio-Rad, Hercules, CA, USA).
4.7. RT-qPCR Analysis
Total RNA was extracted with RNAiso Plus reagent (Takara Bio). cDNA was synthesized using M-MLV reverse transcriptase (Nippon Gene, Tokyo, Japan). A quantitative PCR was performed using the KAPA SYBR Fast qPCR kit (KAPA Biosystems, Wilmington, MA, USA), GeneAce SYBR qPCR Mix α, or GeneAce SYBR qPCR Mix II (Nippon Gene) using an ECO qPCR system (illumina, San Diego, CA, USA). Dual-labeled probes were synthesized using Sigma Genosys (Ishikari, Japan). The sequences of primers and the probe for qPCR are listed in Table S1. Heatmaps were created using Heatmapper (http://www.heatmapper.ca).
4.8. Antioxidative Enzyme Activities
The activities of SOD, GR, Gpx, and TrxR were assessed using commercially available kits (Cayman Chemical, Ann Arbor, MI, USA) and following the manuals’ instructions. Absorbance was measured using a Multiskan Sky High microplate spectrophotometer (Thermo Fisher Scientific).
4.9. Western Blot Analysis
Western blot analysis was conducted as previously described [56]. Total cell lysates were obtained using RIPA buffer [25 mM Tris-HCl, 150 mM NaCl, 1% Nonidet P-40, 1 % sodium deoxycholate, 0.1 %SDS]. Nuclear protein was extracted using a Nuclear Extraction kit (Active Motif, Carlsbad, CA, USA). After electrophoresis in 7.5, 10 or 15% SuperSep acrylamide gels (Fujifilm Wako), the protein was transferred onto an immobilon-P membrane (Merck Millipore (Billerica, MA, USA)). After blocking with Blocking One solution (Nacalai Tesque, Kyoto, Japan), the membrane was reacted with 1,000-2,000-fold diluted primary antibodies against USP2 (#AP2131a, Abgent, San Diego, CA, USA), UCP2 (#CSB-PA299698, Cusabio, Houston, TX, USA), PGC1 (#ab54481; Abcam, Cambridge, UK), Halo-tag (#G921A, Promega), HA-tag (#51064-2-AP; Proteintech, Rosemont, IL, USA), GFP (#598, MBL, Tokyo, Japan), β-actin (#010-27841; Fujifilm Wako), GAPDH (#016-25523, Fujifilm Wako), and γ-tubulin (#sc-17787; Santa Cruz Biotechnology, Dallas, TX, USA) at 4°C overnight. A 5,000-fold diluted horseradish-conjugated anti-rabbit (#7074; Cell Signaling Technology, Danvers, MA, USA) or anti-mouse (#7076; Cell Signaling Technology) immunoglobulin were used as secondary antibodies. Primary and secondary antibodies were diluted with Can-Get-Signal enhancer solution (for USP2, UCP2, PGC1, Halo-tag, HA-Tag, and GFP) or Tris-buffered saline with Tween 20 (TBS-T: 50 mM Tris-HCl, 140 mM NaCl, 2.7 mM KCl, 0.5% Tween 20, and pH 7.6) supplemented with 10% Blocking One solution (for β-actin and γ-tubulin). The immune complex was visualized using Chemilumi One Super reagent (Nacalai Tesque) and scanned using a Gene Gnome 5 (Syngene, Cambridge, UK) or an EZ-capture system (Atto, Tokyo, Japan). Quantification analysis was performed using GeneTools software (Syngene) or CS Analyzer (Atto).
4.10. Immune Precipitation
Immune precipitation of Halo-tagged PGC1α was performed using Halo-Trap Magnetic Particles M-270 (Chromotek, Planegg-Martinsried, Germany) according to the manual’s instructions.
4.11. Statistical Analysis
Statistical analyses were performed using a Student t-test (for two groups) or one-way analysis of variance followed by a Tukey’s post hoc test (for more than three groups) using KaleidaGraph software (Synergy Software, Reading, PA, USA).
5. Conclusions
USP2 prevents mitochondrial dysfunction by mitigating oxidative stress in culture myoblasts. Mechanistically, the protective roles of USP2 may be attributed to the maintenance of UCP2 expression. Since USP2 stabilizes PGC1α by deubiquitination, USP2 might eliminate mitochondrial ROS by activating the PGC1α—UCP2 axis.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Figure S1: Effects of ML364 on LDH content of the C2C12-cultured medium; Table S1: RT-qPCR primers and probes used in this study.
Author Contributions
Conceptualization, H.K.; methodology, M.F., M.H., H.K., and Y.H. ; investigation, M.F., H.K., and M.H.; data curation, H.K., M.F., and M.H.; writing—original draft preparation, H.K.; writing—review and editing, M.F. and M.H.; visualization, H.K. and M.H. ; supervision, O.I.; funding acquisition, H.K.
Funding
This research was supported by JSPS KAKENHI Grant Number JP21K06001.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
All data generated or analyzed during this study are included in this manuscript and additional supporting files, which can be downloaded at: https://www.dropbox.com/scl/fo/rrzmoxr1b3rj2smini7eo/AGb86qDAec7Wvkasek0kLic?rlkey=tv8svj3lbe0hhn8kdoc42zcnf&st=xky3rfu0&dl=0
Acknowledgments
This manuscript was proofread by Uni-Edit.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Cruz-Jentoft, A.J.; Sayer, A.A. Sarcopenia. Lancet 2019, 393, 2636–2646. [Google Scholar] [CrossRef]
- García-Prat, L.; Martínez-Vicente, M.; Perdiguero, E.; Ortet, L.; Rodríguez-Ubreva, J.; Rebollo, E.; Ruiz-Bonilla, V.; Gutarra, S.; Ballestar, E.; Serrano, A.L.; et al. Autophagy Maintains Stemness by Preventing Senescence. Nature 2016, 529, 37–42. [Google Scholar] [CrossRef] [PubMed]
- Haramizu, S.; Asano, S.; Butler, D.C.; Stanton, D.A.; Hajira, A.; Mohamed, J.S.; Alway, S.E. Dietary Resveratrol Confers Apoptotic Resistance to Oxidative Stress in Myoblasts. J. Nutr. Biochem. 2017, 50, 103–115. [Google Scholar] [CrossRef]
- Wang, S.; Zhao, X.; Liu, Q.; Wang, Y.; Li, S.; Xu, S. Selenoprotein K Protects Skeletal Muscle from Damage and Is Required for Satellite Cells-Mediated Myogenic Differentiation. Redox Biol. 2022, 50, 102255. [Google Scholar] [CrossRef]
- Le Moal, E.; Pialoux, V.; Juban, G.; Groussard, C.; Zouhal, H.; Chazaud, B.; Mounier, R. Redox Control of Skeletal Muscle Regeneration. Antioxidants Redox Signal. 2017, 27, 276–310. [Google Scholar] [CrossRef] [PubMed]
- Di Filippo, E.S.; Mancinelli, R.; Pietrangelo, T.; La Rovere, R.M.L.; Quattrocelli, M.; Sampaolesi, M.; Fulle, S. Myomir Dysregulation and Reactive Oxygen Species in Aged Human Satellite Cells. Biochem. Biophys. Res. Commun. 2016, 473, 462–470. [Google Scholar] [CrossRef]
- Brand, M.D.; Affourtit, C.; Esteves, T.C.; Green, K.; Lambert, A.J.; Miwa, S.; Pakay, J.L.; Parker, N. Mitochondrial Superoxide: Production, Biological Effects, and Activation of Uncoupling Proteins. Free Radic. Biol. Med. 2004, 37, 755–767. [Google Scholar] [CrossRef] [PubMed]
- Willems, P.H.G.M.; Rossignol, R.; Dieteren, C.E.J.; Murphy, M.P.; Koopman, W.J.H. Redox Homeostasis and Mitochondrial Dynamics. Cell Metab. 2015, 22, 207–218. [Google Scholar] [CrossRef]
- Yu, T.; Robotham, J.L.; Yoon, Y. Increased Production of Reactive Oxygen Species in Hyperglycemic Conditions Requires Dynamic Change of Mitochondrial Morphology. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 2653–2658. [Google Scholar] [CrossRef]
- Tahrir, F.G.; Langford, D.; Amini, S.; Mohseni Ahooyi, T.; Khalili, K. Mitochondrial Quality Control in Cardiac Cells: Mechanisms and Role in Cardiac Cell Injury and Disease. J. Cell. Physiol. 2019, 234, 8122–8133. [Google Scholar] [CrossRef]
- Wang, Y.; Branicky, R.; Noë, A.; Hekimi, S. Superoxide Dismutases: Dual Roles in Controlling ROS Damage and Regulating ROS Signaling. J. Cell Biol. 2018, 217, 1915–1928. [Google Scholar] [CrossRef]
- Molavian, H.; Madani Tonekaboni, A.; Kohandel, M.; Sivaloganathan, S. The Synergetic Coupling among the Cellular Antioxidants Glutathione Peroxidase/Peroxiredoxin and Other Antioxidants and Its Effect on the Concentration of H2O2. Sci. Rep. 2015, 5, 1–8. [Google Scholar] [CrossRef]
- Mailloux, R.J.; Harper, M.E. Uncoupling Proteins and the Control of Mitochondrial Reactive Oxygen Species Production. Free Radic. Biol. Med. 2011, 51, 1106–1115. [Google Scholar] [CrossRef] [PubMed]
- Cadenas, S. Mitochondrial Uncoupling, ROS Generation and Cardioprotection. Biochim. Biophys. Acta - Bioenerg. 2018, 1859, 940–950. [Google Scholar] [CrossRef] [PubMed]
- Arsenijevic, D.; Onuma, H.; Pecqueur, C.; Raimbault, S.; Manning, B.S.; Miroux, B.; Couplan, E.; Alves-Guerra, M.C.; Goubern, M.; Surwit, R.; et al. Disruption of the Uncoupling Protein-2 Gene in Mice Reveals a Role in Immunity and Reactive Oxygen Species Production. Nat. Genet. 2000, 26, 435–439. [Google Scholar] [CrossRef]
- Li, L.X.; Jørgensen, I.H.; Grill, I.H.; Skorpen, F.; Egeberg, K. Uncoupling Protein-2 Participates in Cellular Defense against Oxidative Stress in Clonal β-Cells. Biochem. Biophys. Res. Commun. 2001, 282, 273–277. [Google Scholar] [CrossRef]
- Erlanson-Albertsson, C. The Role of Uncoupling Proteins in the Regulation of Metabolism. Acta Physiol. Scand. 2003, 178, 405–412. [Google Scholar] [CrossRef]
- Donadelli, M.; Dando, I.; Fiorini, C.; Palmieri, M. UCP2, a Mitochondrial Protein Regulated at Multiple Levels. Cell. Mol. Life Sci. 2014, 71, 1171–1190. [Google Scholar] [CrossRef]
- Zhao, M.; Wang, Y.; Li, L.; Liu, S.; Wang, C.; Yuan, Y.; Yang, G.; Chen, Y.; Cheng, J.; Lu, Y.; et al. Mitochondrial ROS Promote Mitochondrial Dysfunction and Inflammation in Ischemic Acute Kidney Injury by Disrupting TFAM-Mediated MtDNA Maintenance. Theranostics 2021, 11, 1845–1863. [Google Scholar] [CrossRef]
- Zhu, Z.; Kawai, T.; Umehara, T.; Hoque, S.A.M.; Zeng, W.; Shimada, M. Negative Effects of ROS Generated during Linear Sperm Motility on Gene Expression and ATP Generation in Boar Sperm Mitochondria. Free Radic. Biol. Med. 2019, 141, 159–171. [Google Scholar] [CrossRef]
- Quan, Y.; Xin, Y.; Tian, G.; Zhou, J.; Liu, X. Mitochondrial ROS-Modulated MtDNA: A Potential Target for Cardiac Aging. Oxid. Med. Cell. Longev. 2020, 2020. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarty, R.P.; Chandel, N.S. Mitochondria as Signaling Organelles Control Mammalian Stem Cell Fate. Cell Stem Cell 2021, 28, 394–408. [Google Scholar] [CrossRef] [PubMed]
- Minet, A.D.; Gaster, M. Cultured Senescent Myoblasts Derived from Human Vastus Lateralis Exhibit Normal Mitochondrial ATP Synthesis Capacities with Correlating Concomitant ROS Production While Whole Cell ATP Production Is Decreased. Biogerontology 2012, 13, 277–285. [Google Scholar] [CrossRef] [PubMed]
- Wolberger, C. Mechanisms for Regulating Deubiquitinating Enzymes. Protein Sci. 2014, 23, 344–353. [Google Scholar] [CrossRef] [PubMed]
- Pickart, C.M.; Eddins, M.J. Ubiquitin: Structures, Functions, Mechanisms. Biochim. Biophys. Acta - Mol. Cell Res. 2004, 1695, 55–72. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, H. Ubiquitin-Specific Proteases (USPs) and Metabolic Disorders. Int. J. Mol. Sci. 2023, 24. [Google Scholar] [CrossRef]
- Kitamura, H.; Hashimoto, M. USP2-Related Cellular Signaling and Consequent Pathophysiological Outcomes. Int. J. Mol. Sci. 2021, 22. [Google Scholar] [CrossRef] [PubMed]
- Molusky, M.M.; Li, S.; Ma, D.; Yu, L.; Lin, J.D. Ubiquitin-Specific Protease 2 Regulates Hepatic Gluconeogenesis and Diurnal Glucose Metabolism through 11&beta-Hydroxysteroid Dehydrogenase 1. Diabetes 2012, 61, 1025–1035. [Google Scholar] [CrossRef]
- Kitamura, H.; Kimura, S.; Shimamoto, Y.; Okabe, J.; Ito, M.; Miyamoto, T.; Naoe, Y.; Kikuguchi, C.; Meek, B.; Toda, C.; et al. Ubiquitin-Specific Protease 2-69 in Macrophages Potentially Modulates Metainflammation. FASEB J. 2013, 27, 4940–4953. [Google Scholar] [CrossRef]
- Saito, N.; Kimura, S.; Miyamoto, T.; Fukushima, S.; Amagasa, M.; Shimamoto, Y.; Nishioka, C.; Okamoto, S.; Toda, C.; Washio, K.; et al. Macrophage Ubiquitin-Specific Protease 2 Modifies Insulin Sensitivity in Obese Mice. Biochem. Biophys. Reports 2017, 9, 322–329. [Google Scholar] [CrossRef]
- Hashimoto, M.; Fujimoto, M.; Konno, K.; Lee, M.L.; Yamada, Y.; Yamashita, K.; Toda, C.; Tomura, M.; Watanabe, M.; Inanami, O.; et al. Ubiquitin-Specific Protease 2 in the Ventromedial Hypothalamus Modifies Blood Glucose Levels by Controlling Sympathetic Nervous Activation. J. Neurosci. 2022, 42, 4607–4618. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, M.; Kimura, S.; Kanno, C.; Yanagawa, Y.; Watanabe, T.; Okabe, J.; Takahashi, E.; Nagano, M.; Kitamura, H. Macrophage Ubiquitin-Specific Protease 2 Contributes to Motility, Hyperactivation, Capacitation, and in Vitro Fertilization Activity of Mouse Sperm. Cell. Mol. Life Sci. 2021, 78, 2929–2948. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, M.; Saito, N.; Ohta, H.; Yamamoto, K.; Tashiro, A.; Nakazawa, K.; Inanami, O.; Kitamura, H. Inhibition of Ubiquitin-Specific Protease 2 Causes Accumulation of Reactive Oxygen Species, Mitochondria Dysfunction, and Intracellular ATP Decrement in C2C12 Myoblasts. Physiol. Rep. 2019, 7, 1–14. [Google Scholar] [CrossRef] [PubMed]
- YAFFE, D.; SAXEL, O. Serial Passaging and Differentiation of Myogenic Cells Isolated from Dystrophic Mouse Muscle. Nature 1977, 270, 725–727. [Google Scholar] [CrossRef] [PubMed]
- Sin, J.; Andres, A.M.; Taylo, R.D.J.R.; Weston, T.; Hiraumi, Y.; Stotland, A.; Kim, B.J.; Huang, C.; Doran, K.S.; Gottlieb, R.A. Mitophagy Is Required for Mitochondrial Biogenesis and Myogenic Differentiation of C2C12 Myoblasts. Autophagy 2016, 12, 369–380. [Google Scholar] [CrossRef] [PubMed]
- Forterre, A.; Jalabert, A.; Berger, E.; Baudet, M.; Chikh, K.; Errazuriz, E.; De Larichaudy, J.; Chanon, S.; Weiss-Gayet, M.; Hesse, A.M.; et al. Proteomic Analysis of C2C12 Myoblast and Myotube Exosome-like Vesicles: A New Paradigm for Myoblast-Myotube Cross Talk? PLoS One 2014, 9. [Google Scholar] [CrossRef]
- Calzia, D.; Ottaggio, L.; Cora, A.; Chiappori, G.; Cuccarolo, P.; Cappelli, E.; Izzotti, A.; Tavella, S.; Degan, P. Characterization of C2C12 Cells in Simulated Microgravity: Possible Use for Myoblast Regeneration. J. Cell. Physiol. 2020, 235, 3508–3518. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.I.; Pragani, R.; Fox, J.T.; Shen, M.; Parmar, K.; Gaudiano, E.F.; Liu, L.; Tanega, C.; McGee, L.; Hall, M.D.; et al. Small Molecule Inhibition of the Ubiquitin-Specific Protease USP2 Accelerates Cyclin D1 Degradation and Leads to Cell Cycle Arrest in Colorectal Cancer and Mantle Cell Lymphoma Models. J. Biol. Chem. 2016, 291, 24628–24640. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Puigserver, P.; Andersson, U.; Zhang, C.; Adelmant, G.; Mootha, V.; Troy, A.; Cinti, S.; Lowell, B.; Scarpulla, R.C.; et al. Mechanisms Controlling Mitochondrial Biogenesis and Respiration through the Thermogenic Coactivator PGC-1. Cell 1999, 98, 115–124. [Google Scholar] [CrossRef]
- Yi, J.; Tavana, O.; Li, H.; Wang, D.; Baer, R.J.; Gu, W. Targeting USP2 Regulation of VPRBP-Mediated Degradation of P53 and PD-L1 for Cancer Therapy. Nat. Commun. 2023, 14. [Google Scholar] [CrossRef]
- Fu, D.; Luo, J.; Wu, Y.; Zhang, L.; Li, L.; Chen, H.; Wen, T.; Fu, Y.; Xiong, W. Angiotensin II-Induced Calcium Overload Affects Mitochondrial Functions in Cardiac Hypertrophy by Targeting the USP2/MFN2 Axis. Mol. Cell. Endocrinol. 2023, 571, 111938. [Google Scholar] [CrossRef] [PubMed]
- Santel, A.; Fuller, M.T. Control of Mitochondrial Morphology by a Human Mitofusin. J. Cell Sci. 2001, 114, 867–874. [Google Scholar] [CrossRef]
- Pernas, L.; Scorrano, L. Mito-Morphosis: Mitochondrial Fusion, Fission, and Cristae Remodeling as Key Mediators of Cellular Function. Annu. Rev. Physiol. 2016, 78, 505–531. [Google Scholar] [CrossRef]
- Jiang, Z.; Shen, J.; Ding, J.; Yuan, Y.; Gao, L.; Yang, Z.; Zhao, X. USP18 Mitigates Lipopolysaccharide-Induced Oxidative Stress and Inflammation in Human Pulmonary Microvascular Endothelial Cells through the TLR4/NF-ΚB/ROS Signaling. Toxicol. Vitr. 2021, 75, 105181. [Google Scholar] [CrossRef]
- Marcassa, E.; Kallinos, A.; Jardine, J.; Rusilowicz-Jones, E.V.; Martinez, A.; Kuehl, S.; Islinger, M.; Clague, M.J.; Urbé, S. Dual Role of USP 30 in Controlling Basal Pexophagy and Mitophagy. EMBO Rep. 2018, 19, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Molusky, M.M.; Ma, D.; Buelow, K.; Yin, L.; Lin, J.D. Peroxisomal Localization and Circadian Regulation of Ubiquitin-Specific Protease 2. PLoS One 2012, 7, 1–10. [Google Scholar] [CrossRef]
- Nègre-Salvayre, A.; Hirtz, C.; Carrera, G.; Cazenave, R.; Troly, M.; Salvayre, R.; Pénicaud, L.; Casteilla, L. A Role for Uncoupling Protein-2 as a Regulator of Mitochondrial Hydrogen Peroxide Generation. FASEB J. 1997, 11, 809–815. [Google Scholar] [CrossRef]
- Gousseva, N.; Baker, R.T. Gene Structure, Alternate Splicing, Tissue Distribution, Cellular Localization, and Developmental Expression Pattern of Mouse Deubiquitinating Enzyme Isoforms Usp2-45 and Usp2-69. Gene Expr. 2003, 11, 163–179. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Tao, L.; Wang, X.; Wang, B.; Qin, W.; Song, L. PGC-1α Participates in Regulating Mitochondrial Function in Aged Sarcopenia through Effects on the Sestrin2-Mediated MTORC1 Pathway. Exp. Gerontol. 2024, 190, 112428. [Google Scholar] [CrossRef]
- Oishi, Y.; Manabe, I.; Tobe, K.; Ohsugi, M.; Kubota, T.; Fujiu, K.; Maemura, K.; Kubota, N.; Kadowaki, T.; Nagai, R. SUMOylation of Krüppel-like Transcription Factor 5 Acts as a Molecular Switch in Transcriptional Programs of Lipid Metabolism Involving PPAR-δ. Nat. Med. 2008, 14, 656–666. [Google Scholar] [CrossRef]
- Oberkofler, H.; Klein, K.; Felder, T.K.; Krempler, F.; Patsch, W. Role of Peroxisome Proliferator-Activated Receptor-γ Coactivator-1α in the Transcriptional Regulation of the Human Uncoupling Protein 2 Gene in INS-1E Cells. Endocrinology 2006, 147, 966–976. [Google Scholar] [CrossRef] [PubMed]
- Park, K.C.; Kim, J.H.; Choi, E.J.; Min, S.W.; Rhee, S.; Baek, S.H.; Chung, S.S.; Bang, O.; Park, D.; Chiba, T.; et al. Antagonistic Regulation of Myogenesis by Two Deubiquitinating Enzymes, UBP45 and UBP69. Proc. Natl. Acad. Sci. U. S. A. 2002, 99, 9733–9738. [Google Scholar] [CrossRef] [PubMed]
- Bedard, N.; Yang, Y.; Gregory, M.; Cyr, D.G.; Suzuki, J.; Yu, X.; Chian, R.C.; Hermo, L.; O’Flaherty, C.; Smith, C.E.; et al. Mice Lacking the USP2 Deubiquitinating Enzyme Have Severe Male Subfertility Associated with Defects in Fertilization and Sperm Motility. Biol. Reprod. 2011, 85, 594–604. [Google Scholar] [CrossRef] [PubMed]
- Manesia, J.K.; Xu, Z.; Broekaert, D.; Boon, R.; van Vliet, A.; Eelen, G.; Vanwelden, T.; Stegen, S.; Van Gastel, N.; Pascual-Montano, A.; et al. Highly Proliferative Primitive Fetal Liver Hematopoietic Stem Cells Are Fueled by Oxidative Metabolic Pathways. Stem Cell Res. 2015, 15, 715–721. [Google Scholar] [CrossRef] [PubMed]
- Jagannathan, L.; Cuddapah, S.; Costa, M. Oxidative Stress Under Ambient and Physiological Oxygen Tension in Tissue Culture. Curr. Pharmacol. Reports 2016, 2, 64–72. [Google Scholar] [CrossRef]
- Kitamura, H.; Ishino, T.; Shimamoto, Y.; Okabe, J.; Miyamoto, T.; Takahashi, E.; Miyoshi, I. Ubiquitin-Specific Protease 2 Modulates the Lipopolysaccharide-Elicited Expression of Proinflammatory Cytokines in Macrophage-like HL-60 Cells. Mediators Inflamm. 2017, 2017. [Google Scholar] [CrossRef]
Figure 1.
ROS is involved in the Usp2 deficiency–elicited mitochondrial dysfunction in C2C12 myoblasts. Control and Usp2KO C2C12 cells were treated with 5 mM NAC or a vehicle (2 mM DMSO) for 8 h. (A, B) Accumulation of mitochondrial reactive oxygen species (ROS). Mitochondrial ROS (mtROS) was visualized using MitoSOX Red. Representative microscopic images of three experiments were shown (A). The nuclei were stained with Hoechst33342. The scale bar represents 50 μm. The mean intensity of the MitoSOX Red-derived fluorescent signal was measured using a flow cytometer (B). (C) Mitochondrial membrane potential was visualized using TMRM staining. (D) Intracellular ATP content. All values are relative to the mean of the vehicle-treated control C2C12 cells (B–D). Data are means of 6 wells (B–D). *P < 0.05 vs. control; #P < 0.05 vs. vehicle-treated group.
Figure 1.
ROS is involved in the Usp2 deficiency–elicited mitochondrial dysfunction in C2C12 myoblasts. Control and Usp2KO C2C12 cells were treated with 5 mM NAC or a vehicle (2 mM DMSO) for 8 h. (A, B) Accumulation of mitochondrial reactive oxygen species (ROS). Mitochondrial ROS (mtROS) was visualized using MitoSOX Red. Representative microscopic images of three experiments were shown (A). The nuclei were stained with Hoechst33342. The scale bar represents 50 μm. The mean intensity of the MitoSOX Red-derived fluorescent signal was measured using a flow cytometer (B). (C) Mitochondrial membrane potential was visualized using TMRM staining. (D) Intracellular ATP content. All values are relative to the mean of the vehicle-treated control C2C12 cells (B–D). Data are means of 6 wells (B–D). *P < 0.05 vs. control; #P < 0.05 vs. vehicle-treated group.
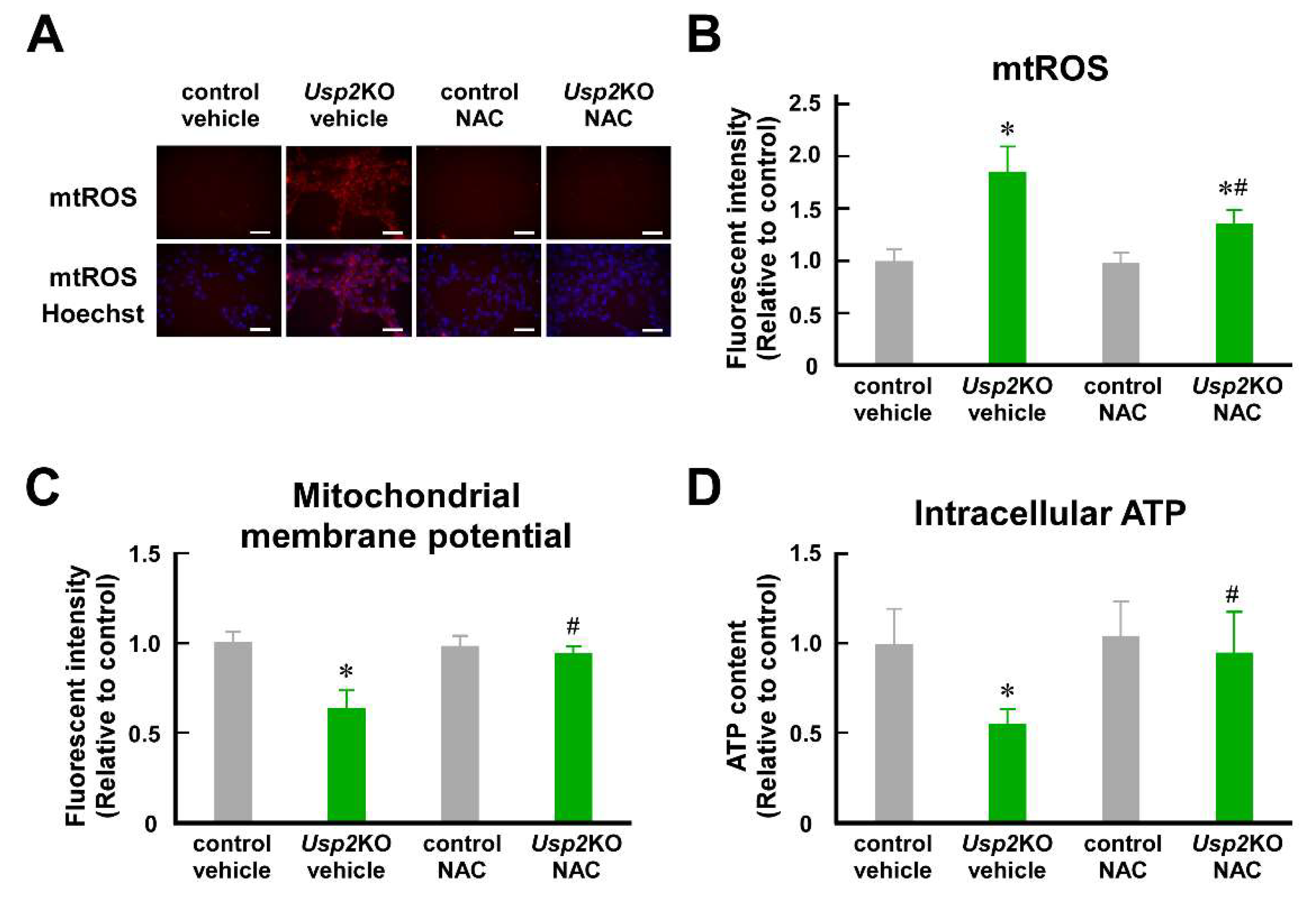
Figure 2.
ROS is involved in the ML364-elicited mitochondrial dysfunction in C2C12 myoblasts. C2C12 cells were treated with 10 μM ML364 or a vehicle (2 mM DMSO) for 8 h. Five mM NAC or the vehicle (2 mM DMSO) were added at the same time as ML364 or the vehicle application. (A–D) Mitochondrial reactive oxygen species (ROS) (A, B), mitochondrial membrane potential (C), and intracellular ATP content (D) were analyzed as per Figure 1. The scale bar represents 50 μm (A). All values are relative to the mean of the vehicle-treated C2C12 cells (B–D). Data are means of 6 wells (B–D). *P < 0.05 vs. ML364-untreated group; #P < 0.05 vs. NAC-untreated group.
Figure 2.
ROS is involved in the ML364-elicited mitochondrial dysfunction in C2C12 myoblasts. C2C12 cells were treated with 10 μM ML364 or a vehicle (2 mM DMSO) for 8 h. Five mM NAC or the vehicle (2 mM DMSO) were added at the same time as ML364 or the vehicle application. (A–D) Mitochondrial reactive oxygen species (ROS) (A, B), mitochondrial membrane potential (C), and intracellular ATP content (D) were analyzed as per Figure 1. The scale bar represents 50 μm (A). All values are relative to the mean of the vehicle-treated C2C12 cells (B–D). Data are means of 6 wells (B–D). *P < 0.05 vs. ML364-untreated group; #P < 0.05 vs. NAC-untreated group.
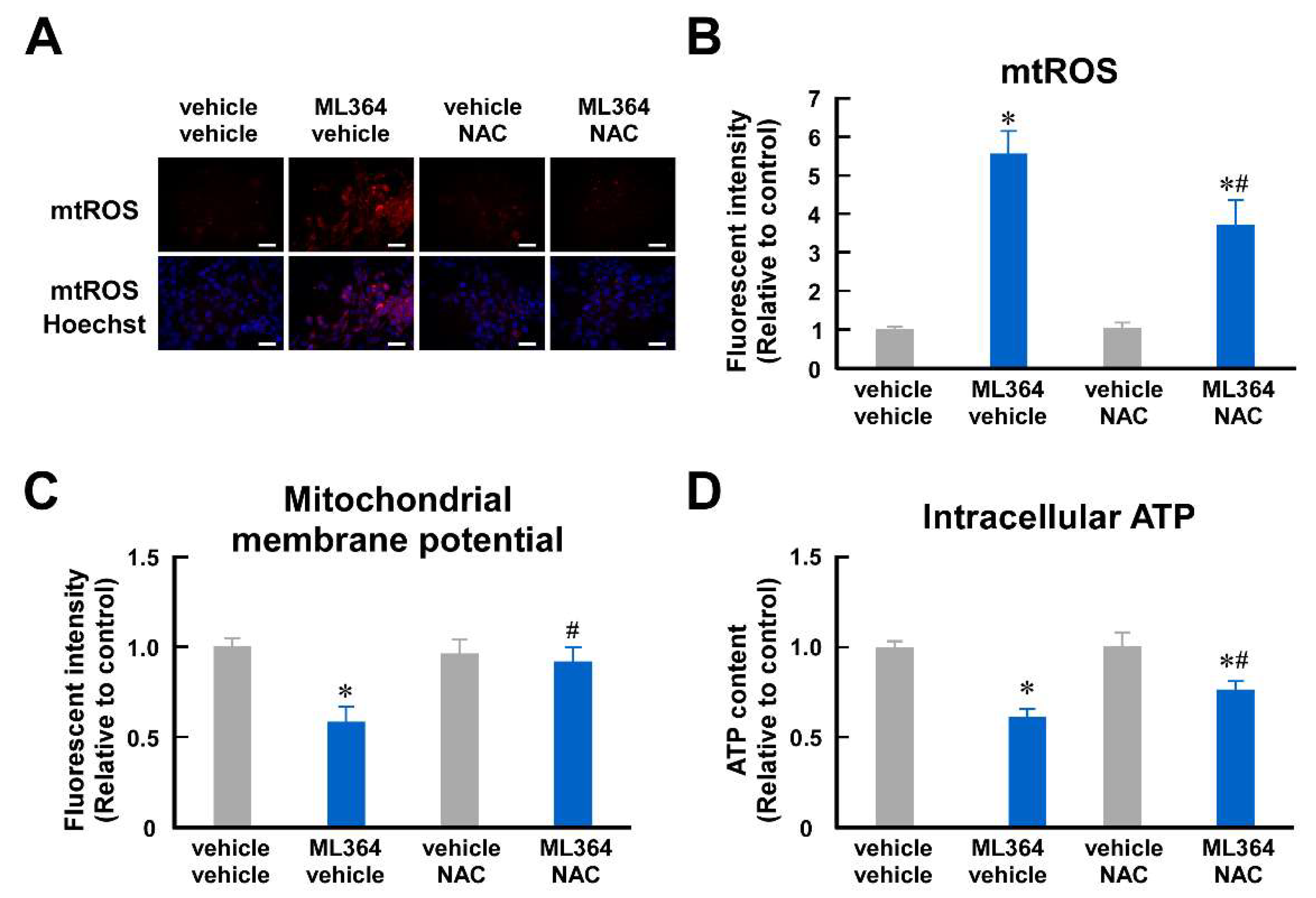
Figure 3.
USP2 maintains UCP2 expression in C2C12 myoblasts. (A) RT-qPCR screening of canonical antioxidative genes. Two comparisons were carried out: control and Usp2KO C2C12 cells (left) and treatment with vehicle and ML364 (10 μM, 8 h) (right). Heat maps represent the expression ratio to control (left) or vehicle-treated cells (right). (B) Venn diagram of genes for which expression was affected by genetic or chemical deprivation of USP2. (C, D) The abundance of Ucp2 mRNA in control and Usp2KO C2C12 cells (C) and vehicle- or ML364-treated C2C12 cells (D). Expression values are normalized to the Hprt-1 expression level. (E, F) UCP2 protein levels. Western blot analysis was performed using total cellular lysate of control and Usp2KO C2C12 cells (E) and vehicle- or ML364-treated C2C12 cells (F). Representative images are shown (top). Band intensity of UCP2 was normalized with that of β-actin. (G, H) Activities of SOD, GR, Gpx, and TrxR in Usp2KO C2C12 cells (G) and ML364-treated C2C12 cells (H). All values are relative to means of the control C2C12 cells (C, E, G) or vehicle-treated C2C12 cells (D, F, H). Data are means of 6 (C-E, G, H) or 3 (F) wells. *P < 0.05 vs. control (C, E) or vehicle-treated groups (D, F).
Figure 3.
USP2 maintains UCP2 expression in C2C12 myoblasts. (A) RT-qPCR screening of canonical antioxidative genes. Two comparisons were carried out: control and Usp2KO C2C12 cells (left) and treatment with vehicle and ML364 (10 μM, 8 h) (right). Heat maps represent the expression ratio to control (left) or vehicle-treated cells (right). (B) Venn diagram of genes for which expression was affected by genetic or chemical deprivation of USP2. (C, D) The abundance of Ucp2 mRNA in control and Usp2KO C2C12 cells (C) and vehicle- or ML364-treated C2C12 cells (D). Expression values are normalized to the Hprt-1 expression level. (E, F) UCP2 protein levels. Western blot analysis was performed using total cellular lysate of control and Usp2KO C2C12 cells (E) and vehicle- or ML364-treated C2C12 cells (F). Representative images are shown (top). Band intensity of UCP2 was normalized with that of β-actin. (G, H) Activities of SOD, GR, Gpx, and TrxR in Usp2KO C2C12 cells (G) and ML364-treated C2C12 cells (H). All values are relative to means of the control C2C12 cells (C, E, G) or vehicle-treated C2C12 cells (D, F, H). Data are means of 6 (C-E, G, H) or 3 (F) wells. *P < 0.05 vs. control (C, E) or vehicle-treated groups (D, F).
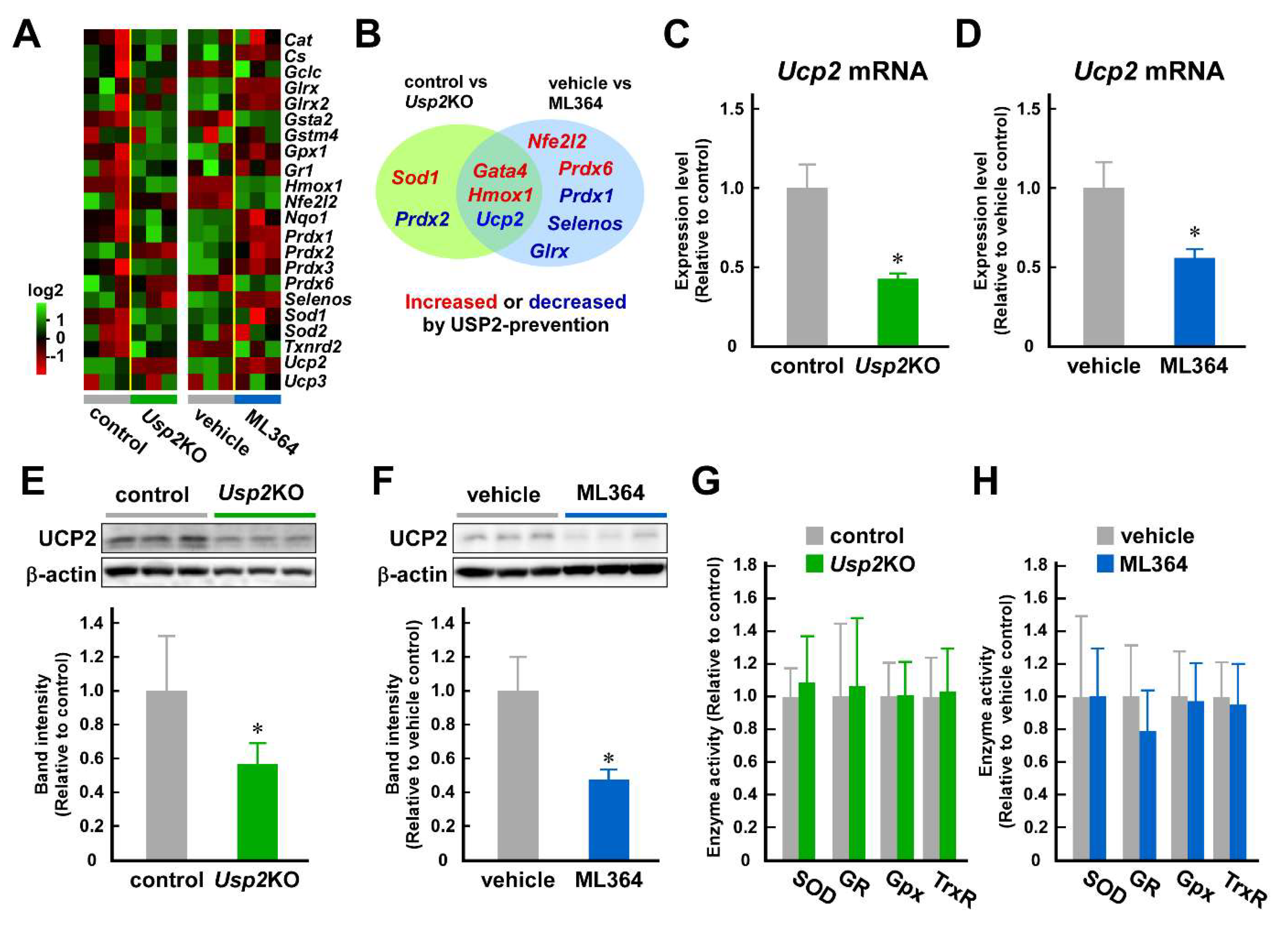
Figure 4.
The introduction of UCP2 partially recovers the Usp2KO-elicited mitochondrial dysfunction. Control or Usp2KO C2C12 cells were infected with Ucp2-expressing or mock lentiviral particles. (A, B) mRNA (A) and protein (B) levels of UCP2. A representative image is shown (B, left). Expression values were normalized with Hprt-1 mRNA levels (A) or GAPDH levels (B). (C, D) Mitochondrial ROS and mitochondrial membrane potential were measured by flow cytometry. (E) Intracellular ATP content. All values are relative to means of mock-transfected control C2C12 cells. Data are means of 4 (A), 3 (B), or 6 (C–E) wells. *P < 0.05 vs. control C2C12 cells; #P < 0.05 vs. mock-transfected group.
Figure 4.
The introduction of UCP2 partially recovers the Usp2KO-elicited mitochondrial dysfunction. Control or Usp2KO C2C12 cells were infected with Ucp2-expressing or mock lentiviral particles. (A, B) mRNA (A) and protein (B) levels of UCP2. A representative image is shown (B, left). Expression values were normalized with Hprt-1 mRNA levels (A) or GAPDH levels (B). (C, D) Mitochondrial ROS and mitochondrial membrane potential were measured by flow cytometry. (E) Intracellular ATP content. All values are relative to means of mock-transfected control C2C12 cells. Data are means of 4 (A), 3 (B), or 6 (C–E) wells. *P < 0.05 vs. control C2C12 cells; #P < 0.05 vs. mock-transfected group.

Figure 5.
USP2 prevents degradation of PGC1α. (A) Expression levels of the Ppargc1a gene in control and Usp2KO C2C12 cells. Ppargc1a expression was normalized to that of Hprt1. (B) Nuclear content of PGC1 in control and Usp2KO C2C12 cells. PGC1 levels were normalized by γ-tubulin levels (bottom). (C) Co-transfection analysis of USP2 and PGC1α. Halo-tagged PGC1α and HA-tagged USP2 or empty plasmids were transfected into 293FT cells. The nuclear fraction was subjected to western blot analysis two days after transfection. (D) Effects of USP2 on the stability of PGC1α. Two days after transfection with Halo-tagged PGC1α, HA-tagged ubiquitin (Ub) and USP2, or empty (mock) plasmids into 293FT cells, cycloheximide was added to reach 100 μg/mL. The nuclear fraction was extracted at the indicated times. Data are represented as % intensities of cycloheximide-untreated cells. (E) Roles of isopeptidase activity of USP2 on PGC1α stability. Halo-tagged PGC1α, HA-tagged ubiquitin (Ub) and USP2, isopeptidase-mutated USP2 (C276A), or empty plasmid (mock) were transfected. Cells were treated with 100 μg/mL cycloheximide for 5 h. (F) Effects of USP2 on polyubiquitination of PGC1α. The nuclear extract of 293FT cells, which were transfected with Halo-tagged PGC1α and HA-tagged ubiquitin plasmids in combination with GFP-tagged USP2 or GFP (mock) plasmids, were treated with 10 μM MG132 for 5 h, and then subjected to immunoprecipitation against Halo-tag. Western blot images of input samples are also shown. (G) The ratio of intensities between polyubiquitinated Halo-tagged PGC1α smear and Halo-tagged PGC1α in immunoprecipitant, obtained using an anti-Halo-tag antibody. For a loading control, γ-tubulin was detected (B-E). A representative image of 3–6 replicates is shown (B–F). Data are means of 3 (B, G) or 4 (A, D) wells. Data were obtained from two membranes (D, G). Differences in band intensities between the membranes were normalized to the sum of the band intensities of each replicate (D, G). The logarithmic transformation was perfomed for statistical analysis (G). *P < 0.05 vs. mock-transfected control (A,G). P-value is shown (B).
Figure 5.
USP2 prevents degradation of PGC1α. (A) Expression levels of the Ppargc1a gene in control and Usp2KO C2C12 cells. Ppargc1a expression was normalized to that of Hprt1. (B) Nuclear content of PGC1 in control and Usp2KO C2C12 cells. PGC1 levels were normalized by γ-tubulin levels (bottom). (C) Co-transfection analysis of USP2 and PGC1α. Halo-tagged PGC1α and HA-tagged USP2 or empty plasmids were transfected into 293FT cells. The nuclear fraction was subjected to western blot analysis two days after transfection. (D) Effects of USP2 on the stability of PGC1α. Two days after transfection with Halo-tagged PGC1α, HA-tagged ubiquitin (Ub) and USP2, or empty (mock) plasmids into 293FT cells, cycloheximide was added to reach 100 μg/mL. The nuclear fraction was extracted at the indicated times. Data are represented as % intensities of cycloheximide-untreated cells. (E) Roles of isopeptidase activity of USP2 on PGC1α stability. Halo-tagged PGC1α, HA-tagged ubiquitin (Ub) and USP2, isopeptidase-mutated USP2 (C276A), or empty plasmid (mock) were transfected. Cells were treated with 100 μg/mL cycloheximide for 5 h. (F) Effects of USP2 on polyubiquitination of PGC1α. The nuclear extract of 293FT cells, which were transfected with Halo-tagged PGC1α and HA-tagged ubiquitin plasmids in combination with GFP-tagged USP2 or GFP (mock) plasmids, were treated with 10 μM MG132 for 5 h, and then subjected to immunoprecipitation against Halo-tag. Western blot images of input samples are also shown. (G) The ratio of intensities between polyubiquitinated Halo-tagged PGC1α smear and Halo-tagged PGC1α in immunoprecipitant, obtained using an anti-Halo-tag antibody. For a loading control, γ-tubulin was detected (B-E). A representative image of 3–6 replicates is shown (B–F). Data are means of 3 (B, G) or 4 (A, D) wells. Data were obtained from two membranes (D, G). Differences in band intensities between the membranes were normalized to the sum of the band intensities of each replicate (D, G). The logarithmic transformation was perfomed for statistical analysis (G). *P < 0.05 vs. mock-transfected control (A,G). P-value is shown (B).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



