Submitted:
30 July 2024
Posted:
01 August 2024
You are already at the latest version
Abstract
Mammalian species possess sophisticated innate immune mechanisms that collectively combat a wide array of viral pathogens. Among these, the well-characterized interferon (IFN) response has been extensively researched; however, the role of antiviral RNA interference (RNAi) in mammals is emerging as an area of significant interest. Previous research has noted that Dicer, an enzyme crucial for processing double-stranded RNA (dsRNA), exhibits reduced activity in vitro, and that the IFN response may overshadow or inhibit the antiviral functions of RNAi in mammalian cells. Consequently, the functional relevance of RNAi in antiviral defense within mammalian somatic cells remains an open question. The evolution of antiviral systems in human populations reflects their substantial advantages, paralleling the evolutionary pressure on genomes encoding such defense mechanisms. While the well-studied protein-guided immune responses in mammals are essential for survival in viral environments, small RNA-mediated antiviral systems, which utilize complementary base pairing to silence non self-genetic material, also play a crucial role. In mammals, evidence suggests that microRNAs (miRNAs) regulate genes integral to antiviral responses, and emerging data indicate that small interfering RNAs (siRNAs), PIWI-interacting RNAs (piRNAs), and transfer RNAs (tRNAs) can directly target virus-derived nucleic acids. This review aims to highlight some of the recent progress in understanding mammalian antiviral RNAi mechanisms.
Keywords:
RNAi
; antiviral response
; interferon response
; immunity
A Brief Idea about RNAi Pathway
RNA interference (RNAi) is a process that leads to the silencing of gene expression after transcription, a phenomenon initially noticed in pigmented petunia flowers in 1990 [1]. This gene-silencing effect was found to be induced by double-stranded RNA (dsRNA), as demonstrated by Fire et al.,1998 through their work with the nematode Caenorhabditis elegans [3]. Their discovery revealed that the presence of dsRNA significantly decreased the levels of corresponding mRNA, thus effectively silencing the gene [4].
The RNAi pathway is controlled by Dicer, an enzyme belonging to the ribonuclease III (RNase III) family, which plays a critical role in the generation of small RNA molecules from longer RNA precursors (Figure 1). Dicer processes these long dsRNA molecules into small interfering RNAs (siRNAs) and converts precursor microRNAs (pre-miRNAs) into mature microRNAs (miRNAs) by cleaving hairpin structures [5]. These small RNA fragments are then incorporated into the RNA-induced silencing complex (RISC), where they guide the Argonaute protein (AGO) to recognize and bind to complementary mRNA sequences [6]. Argonaute, an essential component of RISC, facilitates the degradation or translational repression of the target mRNA, thereby silencing the gene.
Mammalian Dicer’s Long dsRNA Inefficiency in Processing
The molecular properties of Dicer, the primary enzyme involved in double-stranded RNA interference (dsRNAi), have a major effect on the efficacy of dsRNAi in mammalian cells. This multi-domain structure enzyme consists of an ATPase site-containing DExD/H helicase domain at the N-terminus, an RNAse III domain arranged in tandem, a Piwi Argonaute Zwille (PAZ) domain, a domain of unknown function (DUF283), and a C-terminal double-stranded RNA-binding domain [17]. While each RNAse III domain cleaves one strand of the RNA duplex, the PAZ domain is in charge of binding the 3′ 2-nucleotide overhangs at the ends of dsRNA substrates. Human Dicer (hDcr) is less effective at processing lengthy dsRNA into siRNAs than it is at processing pre-miRNA into miRNAs, according to in vitro investigations [18,19]. Modifying Dicer by either deleting or partially proteolyzing the helicase domain enhances the rate at which it cleaves dsRNA, with only a modest effect on pre-miRNA cleavage [18]. Furthermore, a deletion mutant of hDcr, which lacks almost the entire helicase domain, has shown a greater ability to process endogenously transcribed long dsRNA and long hairpin RNAs into siRNAs, thereby enabling dsRNAi activity in engineered cells [20]. In mouse oocytes, which actively engage in dsRNAi, a truncated isoform of Dicer known as DicerO, which lacks the N-terminal helicase domain, efficiently processes endogenous or ectopically expressed long hairpin RNAs [21]. These observations collectively imply that the helicase domain of Dicer restricts its catalytic activity for long dsRNA and suggest that the inclusion of this domain in the mature enzyme might be controlled by alternative transcription mechanisms [17]. However, outside of mouse germ cells, the expression of DicerO has not been detected, and in humans, truncated Dicer isoforms have been observed only in certain cancer cell lines [22].
Another theory suggests that the activity of proteins linked to the digestive system may be the cause of the inhibitory modulated by the helicase domain rather than alternative transcription. According to structural research, co-factors like PACT (Protein Activator of PKR) and TRBP can cause the Dicer helicase domain to shift conformation, potentially simulating the effects of the domain’s deletion [24]. This conformational change facilitated by TRBP and PACT suggests they play a role in modulating Dicer’s ability to process long dsRNA in vivo [19,25]. During replication, RNA viruses produce double-stranded RNA viral replicative intermediates (vRI-dsRNA), which are cleaved by Dicer to generate 21–23 nucleotide vsiRNAs with 2-nucleotide 3’ overhangs [2]. These vsiRNAs then enter Argonaute protein 2 (AGO2), the sole AGO protein in mammals with slicing activity, to play a downstream role in antiviral immunity [26,27]. The discovery of an isoform of Dicer, termed antiviral Dicer (aviD), which lacks exons 7 and 8, resulting in the absence of the Hel2i subdomain was seen to have enhanced antiviral RNAi capability and protects stem cells from Zika virus (ZIKV) and severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infections by processing viral dsRNAs into siRNAs more effectively [28,29].
IFN Response vs. dsRNAi
In a study conducted by Maillard et al., 2016, the antagonistic relationship between the IFN system and dsRNAi was examined in somatic cells that were genetically modified to lack MAVS or IFNAR [30]. The results revealed that introducing dsRNA into these cells led to the accumulation of siRNAs in a Dicer-dependent manner, and subsequently, sequence-specific gene silencing that required Ago2 was observed [30]. Further research by Van der Veen et al., 2018 indicated that the IFN system actively suppresses dsRNAi, at least partially, by inducing the expression of LGP2 [31]. This protein binds to Dicer, thereby inhibiting the processing of long dsRNA into siRNAs. Notably, in this study, the binding of LGP2 to Dicer did not affect the biogenesis of two housekeeping miRNAs, even though LGP2 is also known to interact with TRBP (HIV TAR RNA-binding protein), a co-factor of Dicer, and inhibit the processing of certain TRBP-bound miRNAs [32,33]. It remains to be clarified whether LGP2 further inhibits dsRNAi through its interaction with TRBP.
The rationale behind the inhibition of dsRNAi in somatic cells during an IFN response is not fully understood. Insights might be drawn from Girardi et al., 2015, who observed that mammalian cells stably expressing Drosophila dcr-2 to artificially enhance dsRNAi exhibited a reduced IFN response upon treatment with poly(I:C), a synthetic dsRNA analog [34]. Both viral infection and poly(I:C) treatment have been shown to induce poly-ADP-ribosylation of Ago2 and other components of the RISC, which inhibits RISC activity and thereby alleviates miRNA-mediated repression of some interferon-stimulated genes (ISGs) [35]. This suggests that inhibiting Dicer and RISC might be crucial for the effective activation of the IFN pathway, potentially by preserving dsRNA substrates necessary for RIG-I-like receptor (RLR) activation [17]. Moreover, maintaining dsRNA in infected cells could ensure that the activity of antiviral proteins encoded by ISGs is not compromised. For instance, the protein kinase PKR requires dsRNA longer than 30 nucleotides to dimerize and become active, leading to translational repression [36]. If Dicer cleaves long dsRNA, it might deprive the cell of necessary substrates for PKR activation or cause an accumulation of 21-22 nucleotide siRNA duplexes that could bind to PKR monomers and prevent their dimerization, thereby blocking PKR activation [17].
During the coevolution of viruses and their hosts, a range of viral suppressors of RNA interference (VSRs) have emerged, developed by various viruses to counteract the host’s antiviral responses and enhance their ability to invade. These VSRs include proteins such as NoV B2 from Nodamura virus, NS2A from Dengue virus 2, Semliki Forest virus (SFV) capsid protein, and Rubella Virus capsid protein [2,37,38]. Notably, among these suppressors, NoV B2 and DENV2 NS2A are unique in their ability to function as dsRNA-binding proteins with IFN-independent VSR activity. In contrast, other VSRs, such as IAV NS1, exhibit a dual role by interacting with dsRNA to both suppress RNA interference and simultaneously antagonize the interferon response [39]. Lately, Fang et al., 2021 had designed VSR-targeting peptides (VTPs) that disrupt the function of human enterovirus 71 (HEV71) 3A, thereby unleashing the antiviral RNAi response to reduce viral replication, highlighting a promising therapeutic strategy [40].
In plants, it has been shown that endogenous miRNAs and siRNAs are exported in exosome-like extracellular vesicles (EVs) to suppress virulence gene expression in fungal pathogens [41]. The first report of systemic antiviral RNAi in insects by Goic et al., 2013 showed that vsiRNAs produced during the RNAi pathway play a role in systemic immunity [42]. Subsequent research by Tassetto et al., 2017 detected vsiRNAs in exosome-like vesicles, mediating systemic antiviral RNAi in fruit flies [43]. While much research has focused on cell-autonomous immune regulation via antiviral RNAi, recent work from Wang, J. and Li, Y. (2024) has demonstrated that vsiRNAs in EVs can enter the bloodstream and target complementary viral RNAs in the cytoplasm, thereby restricting viral replication, indicating that mammalian antiviral RNAi functions in both cell-autonomous and non-cell-autonomous host defenses [44]. Despite the significant progress in understanding antiviral RNAi, several fundamental questions remain. These include the inconsistent detection of vsiRNAs, the efficiency of full-length Dicer cleavage, and the failure to observe increased viral replication in Dicer-deficient mammalian cells [45]. Addressing these questions is crucial for advancing research on antiviral RNAi.
Absence of Stable Mammalian vsiRNAs Detection During Viral Infection
In mammals, the production of viral small interfering RNAs (vsiRNAs) during viral infections is a significant marker of the antiviral RNA interference (RNAi) response. Studies have demonstrated that vsiRNAs can be generated using mutant viruses deficient in VSRs, such as B2-deficient NoV, NS1-deficient influenza A virus (IAV), 3A-deficient human enterovirus 71 (HEV71), and NS2A-deficient Dengue virus 2 (DENV2) when tested in mammalian cells [2,46]. These vsiRNAs have also been identified in experiments involving wild-type viruses, including encephalomyocarditis virus (EMCV), Zika virus (ZIKV), Sindbis virus (SINV), and NoV, in various cell types such as undifferentiated or somatic cells [47]. However, despite these observations, numerous studies, including those analyzing a range of wild-type viruses—such as negative-stranded Vesicular Stomatitis Virus (VSV) and IAV, as well as positive-stranded viruses like Poliovirus (PV), Hepatitis C virus (HCV), DENV, SINV, coxsackievirus B3 (CVB3), and EV71—have not detected vsiRNAs across several mammalian cell lines using deep sequencing techniques [2,48]. This lack of detection might be attributed to the low abundance of vsiRNAs within the total RNA pool, as well as the potential absence of typical vsiRNA characteristics, such as the expected enrichment for 22-nucleotide sequences [45].
Several factors could account for the inability of some studies to detect vsiRNAs in mammalian cells infected with various viruses. One reason could be that earlier research did not perform deep sequencing to a sufficient depth to identify vsiRNAs [49]. Another possible explanation is that RNase L cleavage products may obscure the 22-nucleotide peak characteristic of vsiRNAs [2]. For example, Girardi et al., 2013 infected HEK293 and Vero cells with SINV and suggested that the degradation-like viral small RNAs originated from RNase L cleavage [50].Many earlier studies used different cell lines for vsiRNA detection, and the RNase L pathway might be overly activated in ex vivo conditions [50]. In contrast, under in vivo conditions, RNase L is effectively regulated, allowing for the easy detection of vsiRNAs in studies involving ZIKV and SINV [47].
Full-Length Dicer also Processes dsRNA
Insects possess two distinct Dicer proteins, Dicer-1 and Dicer-2, where Dicer-1 is involved in miRNA production, and Dicer-2 is responsible for generating siRNAs [16]. For a long time, it was believed that mammals only had a single type of Dicer that could process both miRNAs and siRNAs [2]. However, an isoform of Dicer distinct from the full-length Dicer was discovered in mouse oocytes by Flemr et al., 2013 due to a loss in the N-terminal helicase domain [21]. When compared to the full-length Dicer, this isoform showed improved effectiveness in cleaving long hairpin dsRNA substrates, suggesting that these oocytes may have a higher capacity for antiviral RNAi [21]. Given this, Kennedy et al., 2015 created a mutant form of human Dicer that does not have the amino-terminal helicase domain (N1 hDcr), and they produced it in NoDice/ΔPKR cells together with 257-bp dsRNA and empty vector, wild-type hDcr, or N1 hDcr. [51]. Their results demonstrated that expressing N1 hDcr significantly increased the production of short RNA reads from 257-bp dsRNA from 0.25% to 23.9%, compared to NoDice/ΔPKR cells expressing the empty vector. In contrast, expressing wild-type hDcr increased short RNA reads to 7.04% [51]. Despite the fact that the N1 hDcr mutant generated 3.39 times more short RNA reads than wild-type hDcr, the wild-type hDcr was still capable of processing long dsRNA into siRNAs. The findings of Wang, J. and Li, Y. (2024) align with this, showing that Dicer efficiently processes IAV-derived dsRNA into vsiRNAs, even when IFN is activated by IAV infection in mammalian somatic cells [2,52]. It’s interesting to note that Poirier et al., 2021 compared the in vitro cleavage efficiency of synthetic dsRNA substrates by aviD and full-length Dicer and discovered that, presumably, aviD still possessed some dicing activity because its cleavage efficiency was roughly twice that of full-length Dicer [28]. They noted that aviD showed increased resilience to LGP2 inhibition [28]. Future studies should investigate whether Dicer’s capacity to cleave dsRNA in vivo is influenced by other cofactors, such as PKR-associated activator (PACT), protein kinase RNA-activated (PKR), and adenosine deaminases acting on RNA 1 (ADAR1) [53,54].
The significant role of antiviral RNA interference (RNAi) in managing viral infections in mammals is underscored by the theoretical consequences of depleting Dicer, a pivotal protein in this defense mechanism. If Dicer, which is essential for antiviral RNAi, is knocked down in mammalian cells, it is expected that viral replication would increase. This hypothesis finds support in the work of Xu et al., 2019, who documented enhanced Zika virus replication in human neural progenitor cells with reduced Dicer levels [12]. In contrast, Cullen et al., 2014 did not observe a corresponding increase in viral replication when Dicer-knockout human somatic cells were exposed to various viruses [55]. Findings from Witteveldt et al., 2019 showed that mouse embryonic stem cells lacking Dicer displayed heightened resistance to several viral infections [56]. Further research has demonstrated that mammalian cells deficient in Dicer accumulate endogenous double-stranded RNAs (dsRNAs), including Alu RNAs—predominantly found in the human genome—and B2 RNAs—prevalent in the mouse genome [57]. Typically, in healthy mammalian cells, ADAR1 enzyme modifies these endogenous dsRNAs by converting adenosine to inosine, a process that aids in their processing by Dicer and prevents their detection by other dsRNA-sensing proteins [58]. However, in the absence of Dicer, these accumulated dsRNAs can be erroneously identified by dsRNA-sensing innate immune response proteins such as PKR and MDA5, which then activate downstream interferon signaling pathways. This activation results in the production of interferon and the expression of interferon-stimulated genes (ISGs), including IFNβ, thus instigating an antiviral state within the cells [59,60]. Therefore, Dicer deletion not only leads to the buildup of endogenous dsRNAs and erroneous activation of dsRNA-sensing proteins but also triggers an interferon response and ISG activation, collectively contributing to an antiviral state that hampers viral replication [2]. A deficiency in Dicer can result in reduced levels of miRNAs and disruption in the regulation of miRNA-targeted genes, which affects essential biological processes such as cell differentiation and apoptosis. For instance, research by Witteveldt et al., 2019 found that the absence of miR-673 in Dicer knockout embryonic stem cells led to elevated levels of mitochondrial antiviral signaling protein (MAVS) and subsequent activation of the interferon response [57].
Utilizing Dicer-knockout mammalian cells as a model for studying antiviral RNA interference (RNAi) presents limitations, making it an unsuitable approach for accurately evaluating this mechanism. Instead, several alternative methodologies offer more effective means to assess antiviral RNAi functionality. One such method involves pre-inoculating organisms with viruses that lack VSRs in vivo or employing virus replicons devoid of VSRs in vitro to activate antiviral RNAi pathways. This can be followed by evaluating viral replication rescue through a recombinant virus carrying virus fragments as a reporter system, a technique illustrated in studies by Zhang et al. (2021) and Qiu et al. (2017) [52,61]. Another approach is to isolate viral small interfering RNAs (vsiRNAs) from infected mammalian tissues using an anti-pan Ago antibody for immunoprecipitation. These vsiRNAs can then be subjected to an in vitro slicing assay with synthetic RNA substrates to test whether Argonaute (Ago) proteins, when loaded with vsiRNAs, are capable of cleaving complementary single-stranded RNAs [14]. While AGO2 deficiency results in embryonic lethality in mammals, indicating its crucial role in RNAi, the zebrafish model also reveals limitations due to impaired AGO2 cleavage capacity [2,62]. Thus, exploring these alternative methods provides more accurate and feasible strategies for studying antiviral RNAi and its functional mechanisms.
Conclusion
Recent investigations into mammalian antiviral responses have uncovered that distinct positive- and negative-strand RNA viruses can induce antiviral RNA interference (RNAi) across a variety of cell types, suggesting a broader applicability of this defence mechanism beyond specific viruses or cellular environments. Notably, several of these viruses produce double-stranded RNA-binding VSRs that are crucial for their infectivity. This evidence implies that antiviral RNAi in mammals is not restricted to certain viral strains or cell types. Recent findings have demonstrated that in human cancer cells, antiviral RNAi operates independently of IFN pathways, with VSR-B2-mediated suppression of RNAi enhancing the therapeutic efficacy of an oncolytic vesicular stomatitis virus variant against cancer cells. However, a contrasting study observed that the generation of abundant vsiRNAs in human 293T cells did not correlate with a reduction in influenza A virus (IAV) replication, as had been previously reported. This discrepancy indicates that while primary mouse embryonic fibroblasts (MEFs) show effective suppression of IAV replication through antiviral RNAi, such effects are not evident in 293T cells, despite antiviral RNAi’s broad activity against viruses like Norovirus (NoV) and Enterovirus 71 (HEV71) in various models. Although primary Ago2D597A MEFs exhibit greater susceptibility to infections by the RNA viruses studied compared to wild type MEFs, this increased susceptibility is not mirrored in immortalized AGO2-knockout MEFs, which lack IFN-I signalling, underscoring the necessity of developing novel infection models to better characterize antiviral RNAi functions without interfering with cellular microRNA activities.
The identification of a novel antiviral immune mechanism in mammals offers significant opportunities for advancing our understanding of mammalian immunology. Antiviral RNAi represents a genetic pathway for clearing viral infections without necessitating cell death and is activated immediately upon infection with a specificity programmed in RNA form. Despite these insights, several crucial questions remain unanswered, including whether antiviral RNAi is active and essential in adult mammals, which exhibit more robust IFN-dependent antiviral responses compared to cultured cells or neonates. The precise role of AGO2 in mediating antiviral defence—whether through RNA slicing, mRNA degradation, or translational repression—remains unclear, as does the extent of antiviral RNAi’s prevalence in mammals relative to plants and insects. Unravelling the mechanisms behind mammalian antiviral responses has led to significant advances in antiviral therapies, including plant-derived compounds, IFN treatments, CCR5 antagonists, and monoclonal antibodies. Clarifying the role of small RNA-mediated antiviral immune systems in mammals holds promise for additional therapeutic successes. Recent progress in the development of mRNA-based therapies and vaccines, accelerated by the SARS-CoV-2 pandemic, has demonstrated the potential of small RNAs for future therapeutic applications, overcoming prior limitations in nucleic acid medicines.
References
- Napoli, C.; Lemieux, C.; Jorgensen, R. Introduction of a chimeric Chalcone synthase gene into petunia results in reversible Co-suppression of homologous genes in trans. Plant Cell 1990, 2, 279–289. [Google Scholar] [CrossRef]
- Wang, J.; Li, Y. Current advances in antiviral RNA interference in mammals. The FEBS journal 2024, 291, 208–216. [Google Scholar] [CrossRef]
- Fire, A.; Xu, S.; Montgomery, M.K.; Kostas, S.A.; Driver, S.E.; Mello, C.C. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 1998, 391, 806–811. [Google Scholar] [CrossRef] [PubMed]
- Berkhout, B. RNAi-mediated antiviral immunity in mammals. Curr Opin Virol 2018, 32, 9–14. [Google Scholar] [CrossRef]
- Song, M.S.; Rossi, J.J. Molecular mechanisms of Dicer: endonuclease and enzymatic activity. The Biochemical journal 2017, 474, 1603–1618. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, K. Are Argonaute-Associated Tiny RNAs Junk, Inferior miRNAs, or a New Type of Functional RNAs? . Frontiers in molecular biosciences 2021, 8, 795356. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, T.; Ui-Tei, K. Mutual regulation of RNA silencing and the IFN response as an antiviral defense system in mammalian cells. Int J Mol Sci 2020, 21, 1348. [Google Scholar] [CrossRef]
- Hur, S. Double-stranded RNA sensors and modulators in innate immunity. Annu Rev Immunol 2019, 37, 349–375. [Google Scholar] [CrossRef] [PubMed]
- Tsai, K.; Courtney, D.G.; Kennedy, E.M.; Cullen, B.R. Influenza a virus-derived siRNAs increase in the absence of NS1 yet fail to inhibit virus replication. RNA 2018, 24, 1172–1182. [Google Scholar] [CrossRef]
- tenOever, B.R. Questioning antiviral RNAi in mammals. Nat Microbiol 2017, 2, 17052. [Google Scholar] [CrossRef]
- Zhang, Y.; Xu, Y.; Dai, Y.; Li, Z.; Wang, J.; Ye, Z.; Ren, Y.; Wang, H.; Li, W.X.; Lu, J.; et al. Efficient dicer processing of virus-derived double-stranded RNAs and its modulation by RIG-I-like receptor LGP2. PLoS Pathog 2021, 17, e1009790. [Google Scholar] [CrossRef]
- Xu, Y.P.; Qiu, Y.; Zhang, B.; Chen, G.; Chen, Q.; Wang, M.; Mo, F.; Xu, J.; Wu, J.; Zhang, R.R.; et al. Zika virus infection induces RNAi-mediated antiviral immunity in human neural progenitors and brain organoids. Cell Res 2019, 29, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Y.; Xu, Y.P.; Wang, M.; Miao, M.; Zhou, H.; Xu, J.; Kong, J.; Zheng, D.; Li, R.T.; Zhang, R.R.; et al. Flavivirus induces and antagonizes antiviral RNA interference in both mammals and mosquitoes. Sci Adv 2020, 6, eaax7989. [Google Scholar] [CrossRef]
- Han, Q.; Chen, G.; Wang, J.; Jee, D.; Li, W.X.; Lai, E.C.; Ding, S.W. Mechanism and function of antiviral RNA interference in mice. mBio 2020, 11, e03278–19. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, T.; Nakano, Y.; Onomoto, K.; Yoneyama, M.; Ui-Tei, K. LGP2 virus sensor enhances apoptosis by upregulating apoptosis regulatory genes through TRBP-bound miRNAs during viral infection. Nucleic Acids Res 2020, 48, 1494–1507. [Google Scholar] [CrossRef] [PubMed]
- Schuster, S.; Miesen, P.; van Rij, R.P. Antiviral RNAi in insects and mammals: parallels and differences. Viruses 2019, 11, 448. [Google Scholar] [CrossRef] [PubMed]
- Maillard, P.V.; van der Veen, A.G.; Poirier, E.Z.; Reis e Sousa, C. Slicing and dicing viruses: antiviral RNA interference in mammals. The EMBO journal 2019, 38, e100941. [Google Scholar] [CrossRef] [PubMed]
- Ma, E.; MacRae, I.J.; Kirsch, J.F.; Doudna, J.A. Autoinhibition of human dicer by its internal helicase domain. J Mol Biol 2008, 380, 237–243. [Google Scholar] [CrossRef] [PubMed]
- Chakravarthy, S.; Sternberg, S.H.; Kellenberger, C.A.; Doudna, J.A. Substrate-specific kinetics of Dicer-catalyzed RNA processing. J Mol Biol 2010, 404, 392–402. [Google Scholar] [CrossRef]
- Kennedy, E.M.; Whisnant, A.W.; Kornepati, A.V.R.; Marshall, J.B.; Bogerd, H.P.; Cullen, B.R. Production of functional small interfering RNAs by an amino-terminal deletion mutant of human Dicer. Proc Natl Acad Sci USA 2015, 112, E6945–E6954. [Google Scholar] [CrossRef]
- Flemr, M.; Malik, R.; Franke, V.; Nejepinska, J.; Sedlacek, R.; Vlahovicek, K.; Svoboda, P. A retrotransposon-driven dicer isoform directs endogenous small interfering RNA production in mouse oocytes. Cell 2013, 155, 807–816. [Google Scholar] [CrossRef]
- Cantini, L.P.; Andino, L.M.; Attaway, C.C.; Butler, B.; Dumitriu, A.; Blackshaw, A.; Jakymiw, A. Identification and characterization of Dicer1e, a Dicer1 protein variant, in oral cancer cells. Mol Cancer 2014, 13, 190. [Google Scholar] [CrossRef] [PubMed]
- Taylor, D.W.; Ma, E.; Shigematsu, H.; Cianfrocco, M.A.; Noland, C.L.; Nagayama, K.; Nogales, E.; Doudna, J.A.; Wang, H.-W. Substrate-specific structural rearrangements of human Dicer. Nat Struct Mol Biol 2013, 20, 662–670. [Google Scholar] [CrossRef]
- Ota, H.; Sakurai, M.; Gupta, R.; Valente, L.; Wulff, B.E.; Ariyoshi, K.; Iizasa, H.; Davuluri, R.V.; Nishikura, K. ADAR1 forms a complex with Dicer to promote microRNA processing and RNA-induced gene silencing. Cell 2013, 153, 575–589. [Google Scholar] [CrossRef]
- Takahashi, T.; Heaton, S.M.; Parrish, N.F. Mammalian antiviral systems directed by small RNA. PLoS Pathog 2021, 17, e1010091. [Google Scholar] [CrossRef]
- Adiliaghdam, F.; Basavappa, M.; Saunders, T.L.; Harjanto, D.; Prior, J.T.; Cronkite, D.A.; Papavasiliou, N.; Jeffrey, K.L. A requirement for Argonaute 4 in mammalian antiviral defense. Cell Rep 2020, 30, 1690–1701e4. [Google Scholar] [CrossRef] [PubMed]
- Poirier, E.Z.; Buck, M.D.; Chakravarty, P.; Carvalho, J.; Frederico, B.; Cardoso, A.; Healy, L.; Ulferts, R.; Beale, R.; Reis e Sousa, C. An isoform of dicer protects mammalian stem cells against multiple RNA viruses. Science 2021, 373, 231–236. [PubMed]
- Jeffrey, K.L. Upping the ante on mammalian antiviral RNA interference. Cell Host Microbe 2021, 29, 1333–1335. [Google Scholar] [CrossRef] [PubMed]
- Maillard, P.V.; Van der Veen, A.G.; Deddouche Grass, S.; Rogers, N.C.; Merits, A.; Reis e Sousa, C. Inactivation of the type I interferon pathway reveals long double-stranded RNA-mediated RNA interference in mammalian cells. EMBO J 2016, 35, 2505–2518. [Google Scholar] [CrossRef]
- Van der Veen, A.G.; Maillard, P.V.; Schmidt, J.M.; Lee, S.A.; Deddouche Grass, S.; Borg, A.; Kjær, S.; Snijders, A.P.; Reis e Sousa, C. The RIG-I-like receptor LGP2 inhibits Dicer-dependent processing of long double-stranded RNA and blocks RNA interference in mammalian cells. e: EMBO J 2018, 37, 2018. [Google Scholar]
- Takahashi, T.; Nakano, Y.; Onomoto, K.; Murakami, F.; Komori, C.; Suzuki, Y.; Yoneyama, M.; Ui-Tei, K. LGP2 virus sensor regulates gene expression network mediated by TRBP-bound microRNAs. Nucleic Acids Res 2018, 42, D68–14. [Google Scholar] [CrossRef]
- Komuro, A.; Homma, Y.; Negoro, T.; Barber, G.N.; Horvath, C.M. The TAR-RNA binding protein is required for immunoresponses triggered by Cardiovirus infection. Biochem Biophys Res Commun 2016, 480, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Girardi, E.; Lefèvre, M.; Chane-Woon-Ming, B.; Paro, S.; Claydon, B.; Imler, J.-L.; Meignin, C.; Pfeffer, S. Cross-species comparative analysis of Dicer proteins during Sindbis virus infection. Sci Rep 2015, 5, 10693–12. [Google Scholar] [CrossRef] [PubMed]
- Seo, G.J.; Kincaid, R.P.; Phanaksri, T.; Burke, J.M.; Pare, J.M.; Cox, J.E.; Hsiang, T.-Y.; Krug, R.M.; Sullivan, C.S. Reciprocal inhibition between intracellular antiviral signaling and the RNAi machinery in mammalian cells. Cell Host Microbe 2013, 14, 435–445. [Google Scholar] [CrossRef] [PubMed]
- Husain, B.; Mukerji, I.; Cole, J.L. Analysis of high-affinity binding of protein kinase R to double-stranded RNA. Biochemistry 2012, 51, 8764–8770. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Kong, J.; Lyu, B.; Wang, X.; Qian, Q.; Zhou, X.; Qiu, Y. The capsid protein of rubella virus antagonizes RNA interference in mammalian cells. Viruses 2021, 13, 154. [Google Scholar] [CrossRef] [PubMed]
- Qian, Q.; Zhou, H.; Shu, T.; Mu, J.; Fang, Y.; Xu, J.; Li, T.; Kong, J.; Qiu, Y.; Zhou, X. The capsid protein of Semliki Forest virus antagonizes RNA interference in mammalian cells. J Virol 2020, 94, e01233–19. [Google Scholar] [CrossRef] [PubMed]
- Li, W.-X.; Ding, S.-W. Mammalian viral suppressors of RNA interference. Trends Biochem Sci 2022, 47, 978–988. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Liu, Z.; Qiu, Y.; Kong, J.; Fu, Y.; Liu, Y.; Wang, C.; Quan, J.; Wang, Q.; Xu, W.; et al. Inhibition of viral suppressor of RNAi proteins by designer peptides protects from enteroviral infection in vivo. Immunity 2021, 54, 2231–2244e6. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Rechavi, O. Plant and animal small RNA communications between cells and organisms. Nat Rev Mol Cell Biol 2022, 23, 185–203. [Google Scholar] [CrossRef]
- Goic, B.; Vodovar, N.; Mondotte, J.A.; Monot, C.; Frangeul, L.; Blanc, H.; Gausson, V.; Vera-Otarola, J.; Cristofari, G.; Saleh, M.C. RNA-mediated interference and reverse transcription control the persistence of RNA viruses in the insect model drosophila. Nat Immunol 2013, 14, 396–403. [Google Scholar] [CrossRef]
- Tassetto, M.; Kunitomi, M.; Andino, R. Circulating immune cells mediate a systemic RNAi-based adaptive antiviral response in drosophila. Cell 2017, 169, 314–325.e13. [Google Scholar] [CrossRef]
- Schierhorn, K.L.; Sanchez-David, R.Y.; Maillard, P.V. Mammalian antiviral RNAi is on the move. EMBO J 2022, 41, e111210. [Google Scholar] [CrossRef]
- Ding, S.W.; Han, Q.; Wang, J.; Li, W.X. Antiviral RNA interference in mammals. Curr Opin Immunol 1210 2018, 54, 109–114. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Wang, J.; Xu, Y.; Li, Z.; Wang, B.; Li, Y. The interaction of influenza a NS1 and cellular TRBP protein modulates the function of RNA interference machinery. Front Microbiol 2022, 13, 859420. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Li, Z.; Ye, Z.; Xu, Y.; Wang, B.; Wang, C.; Dai, Y.; Lu, J.; Lu, B.; Zhang, W.; et al. The activation of antiviral RNA interference not only exists in neural progenitor cells but also in somatic cells in mammals. Emerg Microbes Infect 2020, 9, 1580–1589. [Google Scholar] [CrossRef] [PubMed]
- Schuster, S.; Overheul, G.J.; Bauer, L.; van Kuppeveld, F.J.M.; van Rij, R.P. No evidence for viral small RNA production and antiviral function of Argonaute 2 in human cells. Sci Rep 2019, 9, 13752. [Google Scholar] [CrossRef]
- Parameswaran, P.; Sklan, E.; Wilkins, C.; Burgon, T.; Samuel, M.A.; Lu, R.; Ansel, K.M.; Heissmeyer, V.; Einav, S.; Jackson, W.; et al. Six RNA viruses and forty-one hosts: viral small RNAs and modulation of small RNA repertoires in vertebrate and invertebrate systems. PLoS Pathog 2010, 6, e1000764. [Google Scholar] [CrossRef]
- Girardi, E.; Chane-Woon-Ming, B.; Messmer, M.; Kaukinen, P.; Pfeffer, S. Identification of RNase L-dependent, 3′-end-modified, viral small RNAs in Sindbis virus-infected mammalian cells. MBio 2013, 4, e00698–13. [Google Scholar] [CrossRef]
- Kennedy, E.M.; Whisnant, A.W.; Kornepati, A.V.; Marshall, J.B.; Bogerd, H.P.; Cullen, B.R. Production of functional small interfering RNAs by an amino-terminal deletion mutant of human dicer. Proc Natl Acad Sci USA 2015, 112, E6945–E6954. [Google Scholar] [CrossRef]
- Zhang, Y.; Xu, Y.; Dai, Y.; Li, Z.; Wang, J.; Ye, Z.; Ren, Y.; Wang, H.; Li, W.X.; Lu, J.; et al. Efficient dicer processing of virus-derived double-stranded RNAs and its modulation by RIG-I-like receptor LGP2. PLoS Pathog 2021, 17, e1009790.
- Guo, Y.-L.; Gurung, C.; Fendereski, M.; Huang, F. Dicer and PKR as novel regulators of embryonic stem cell fate and antiviral innate immunity. J Immunol 2022, 208, 2259–2266. [Google Scholar] [CrossRef]
- Girardi, E.; Messmer, M.; Lopez, P.; Fender, A.; Chicher, J.; Chane-Woon-Ming, B.; Hammann, P.; Pfeffer, S. Proteomics-based determination of double stranded RNA interactome reveals known and new factors involved in Sindbis virus infection. bioRxiv 2022. [CrossRef]
- Cullen, B.R. Viruses and RNA interference: issues and controversies. J Virol 2014, 88, 12934–12936. [Google Scholar] [CrossRef]
- Witteveldt, J.; Knol, L.I.; Macias, S. MicroRNA-deficient mouse embryonic stem cells acquire a functional interferon response. Elife 2019, 8, e44171. [Google Scholar] [CrossRef]
- Gurung, C.; Fendereski, M.; Sapkota, K.; Guo, J.; Huang, F.; Guo, Y.L. Dicer represses the interferon response and the double-stranded RNA-activated protein kinase pathway in mouse embryonic stem cells. J Biol Chem 2021, 296, 100264. [Google Scholar] [CrossRef] [PubMed]
- Shiromoto, Y.; Sakurai, M.; Qu, H.; Kossenkov, A.V.; Nishikura, K. Processing of Alu small RNAs by DICER/ADAR1 complexes and their RNAi targets. RNA 2020, 26, 1801–1814. [Google Scholar] [CrossRef] [PubMed]
- Sadeq, S.; Al-Hashimi, S.; Cusack, C.M.; Werner, A. Endogenous double-stranded RNA. Noncoding RNA 2021, 7, 15. [Google Scholar] [CrossRef]
- Kim, S.; Ku, Y.; Ku, J.; Kim, Y. Evidence of aberrant immune response by endogenous double-stranded RNAs: attack from within. Bioessays 2019, 41, e1900023. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Y.; Xu, Y.; Zhang, Y.; Zhou, H.; Deng, Y.Q.; Li, X.F.; Miao, M.; Zhang, Q.; Zhong, B.; Hu, Y.; et al. Human virus-derived small RNAs can confer antiviral immunity in mammals. Immunity 2017, 46, 992–1004e5. [Google Scholar] [CrossRef]
- Chen, G.R.; Sive, H.; Bartel, D.P. A seed mismatch enhances Argonaute2-catalyzed cleavage and partially rescues severely impaired cleavage found in fish. Mol Cell 2017, 68, 1095–1107e5. [Google Scholar] [CrossRef]
- Price, B.D.; Eckerle, L.D.; Ball, L.A.; Johnson, K.L. Nodamura virus RNA replication in Saccharomyces cerevisiae: heterologous gene expression allows replication-dependent colony formation. Journal of virology 2005, 79, 495–502. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, C.S.; Ganem, D. A virus-encoded inhibitor that blocks RNA interference in mammalian cells. Journal of virology 2005, 79, 7371–7379. [Google Scholar] [CrossRef] [PubMed]
- Aliyari, R.; Wu, Q.; Li, H.W.; Wang, X.H.; Li, F.; Green, L.D.; Han, C.S.; Li, W.X.; Ding, S.W. (2008). Mechanism of induction and suppression of antiviral immunity directed by virus-derived small RNAs in Drosophila. Cell host.; microbe, 4, 387–397.
- Li, Y.; Lu, J.; Han, Y.; Fan, X.; Ding, S.W. RNA interference functions as an antiviral immunity mechanism in mammals. Science (New York, N.Y.) 2013, 342, 231–234. [Google Scholar]
- Bogerd, H.P.; Skalsky, R.L.; Kennedy, E.M.; Furuse, Y.; Whisnant, A.W.; Flores, O.; Schultz, K.L.; Putnam, N.; Barrows, N.J.; Sherry, B.; Scholle, F.; Garcia-Blanco, M.A.; Griffin, D.E.; Cullen, B.R. Replication of many human viruses is refractory to inhibition by endogenous cellular microRNAs. Journal of virology 2014, 88, 8065–8076. [Google Scholar] [CrossRef]
- Bartel, D.P. Metazoan MicroRNAs. Cell 2018, 173, 20–51. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
This image explains the processes occurring within the nucleus and cytoplasm of a cell during both miRNA biogenesis and viral replication. Within the nucleus, miRNA genes are transcribed into primary miRNAs (pri-miRNAs), which are then processed by the Drosha enzyme into pre-miRNAs. These pre-miRNAs are exported to the cytoplasm via Exportin 5, where they are further processed into mature miRNAs that can regulate gene expression. The nucleus is shown hosting viral replication, where viral RNA (vRNA) is transcribed and replicated, forming double-stranded RNA (dsRNA) intermediates and single-stranded RNA (ssRNA) that can form secondary structures. These viral RNAs can be shuttled to the cytoplasm to continue the infection cycle. Image Source: https://www.ncbi.nlm.nih.gov/probe/docs/techrnai/.
Figure 1.
This image explains the processes occurring within the nucleus and cytoplasm of a cell during both miRNA biogenesis and viral replication. Within the nucleus, miRNA genes are transcribed into primary miRNAs (pri-miRNAs), which are then processed by the Drosha enzyme into pre-miRNAs. These pre-miRNAs are exported to the cytoplasm via Exportin 5, where they are further processed into mature miRNAs that can regulate gene expression. The nucleus is shown hosting viral replication, where viral RNA (vRNA) is transcribed and replicated, forming double-stranded RNA (dsRNA) intermediates and single-stranded RNA (ssRNA) that can form secondary structures. These viral RNAs can be shuttled to the cytoplasm to continue the infection cycle. Image Source: https://www.ncbi.nlm.nih.gov/probe/docs/techrnai/.
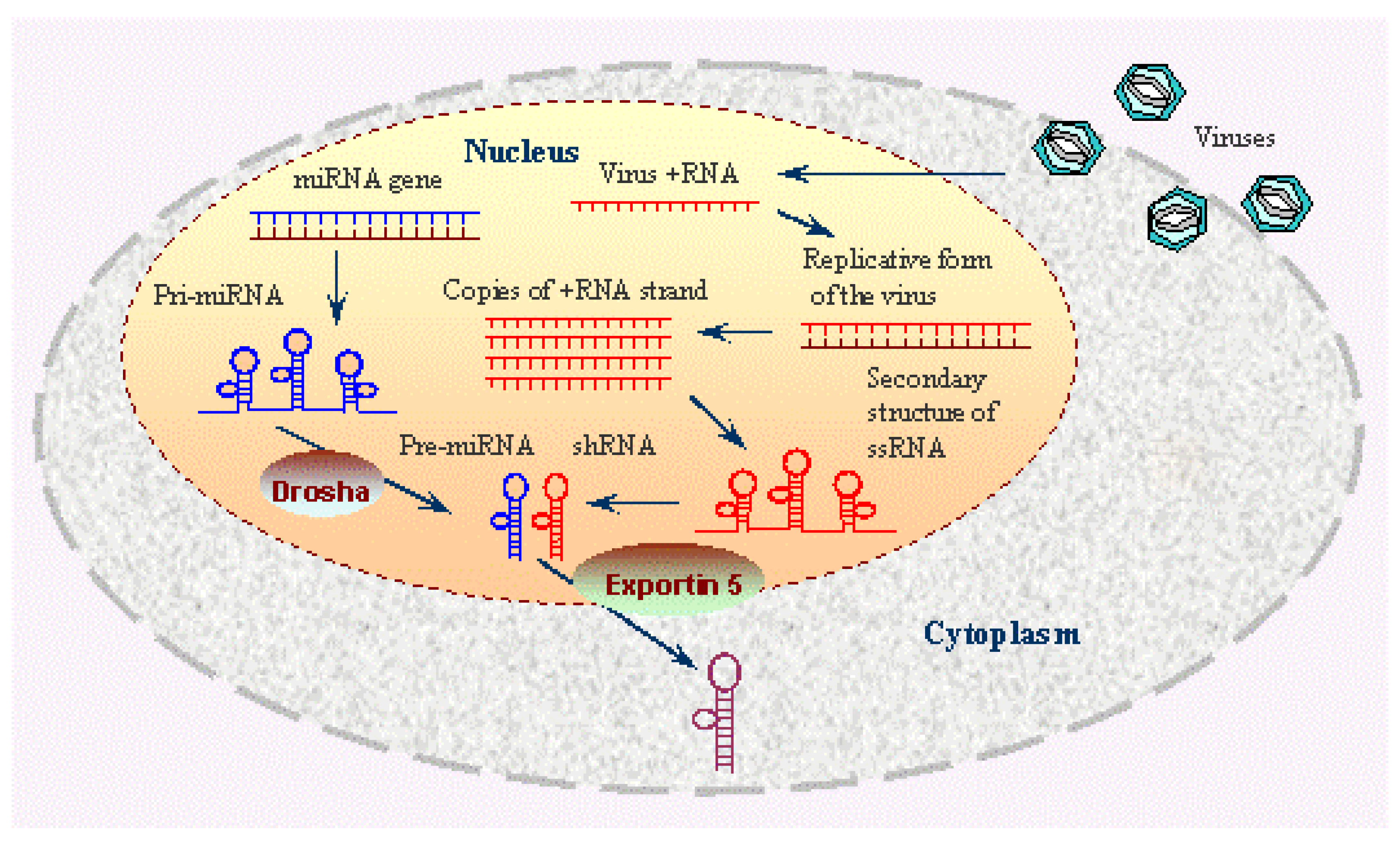
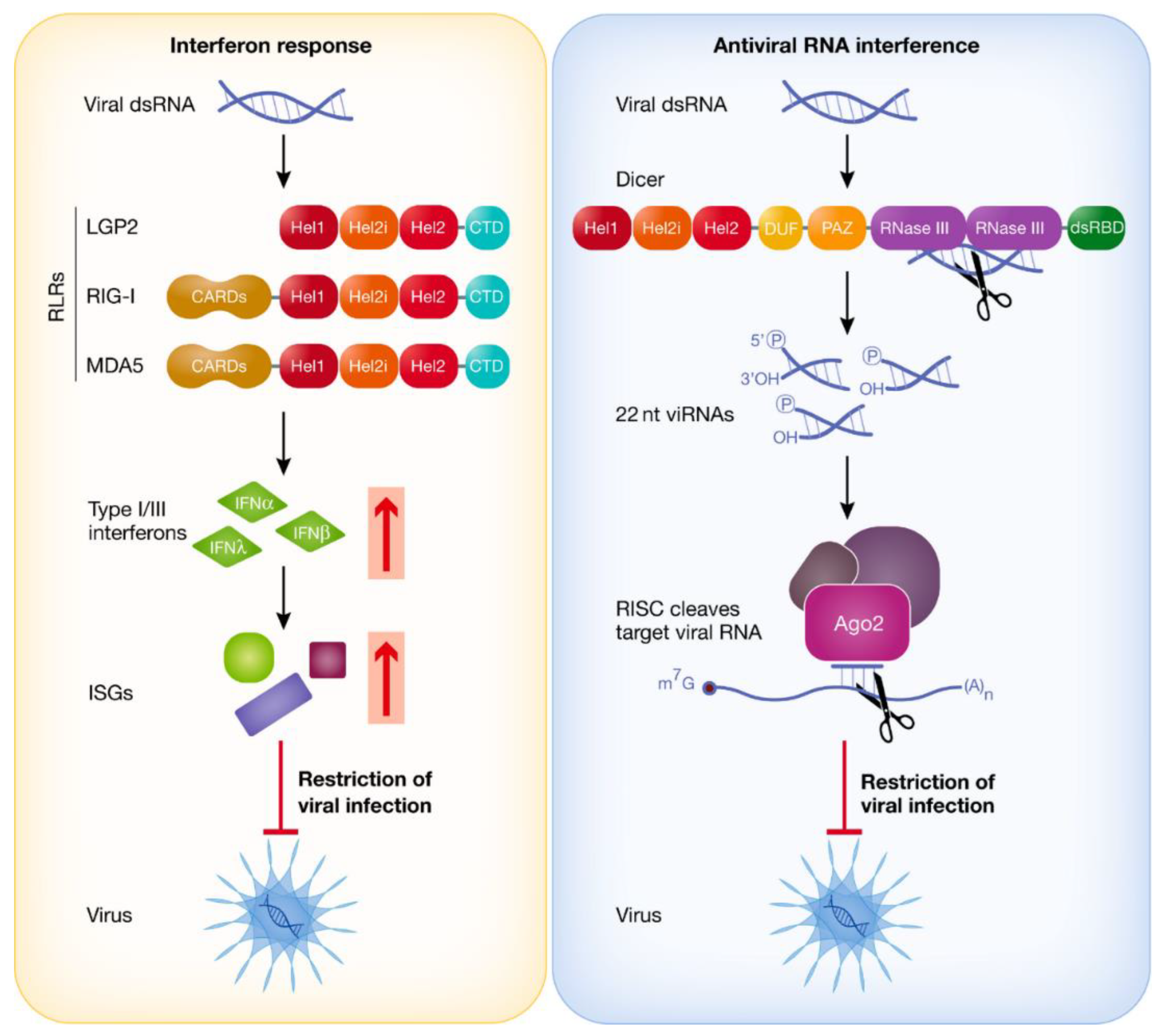
Figure 2.
This image illustrates the dual mechanisms by which mammalian cells restrict viral infections: the IFN response and antiviral RNA interference (RNAi). On the left, the IFN response is depicted, where viral double-stranded RNA (dsRNA) is detected by RIG-I-like receptors (RLRs) such as LGP2, RIG-I, and MDA5, leading to the activation of type I and III interferons (IFN-α, IFN-λ, IFN-β). These interferons then induce the expression of interferon-stimulated genes (ISGs) that collectively inhibit viral replication. On the right, the antiviral RNAi pathway is shown, starting with the processing of viral dsRNA by Dicer into 22-nucleotide viral small interfering RNAs (viRNAs), which are then incorporated into the RISC. The RISC, guided by viRNAs, cleaves the target viral RNA, leading to the degradation of viral RNA and the inhibition of viral replication. Image Source: [17].
Figure 2.
This image illustrates the dual mechanisms by which mammalian cells restrict viral infections: the IFN response and antiviral RNA interference (RNAi). On the left, the IFN response is depicted, where viral double-stranded RNA (dsRNA) is detected by RIG-I-like receptors (RLRs) such as LGP2, RIG-I, and MDA5, leading to the activation of type I and III interferons (IFN-α, IFN-λ, IFN-β). These interferons then induce the expression of interferon-stimulated genes (ISGs) that collectively inhibit viral replication. On the right, the antiviral RNAi pathway is shown, starting with the processing of viral dsRNA by Dicer into 22-nucleotide viral small interfering RNAs (viRNAs), which are then incorporated into the RISC. The RISC, guided by viRNAs, cleaves the target viral RNA, leading to the degradation of viral RNA and the inhibition of viral replication. Image Source: [17].
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



