
Submitted:
30 July 2024
Posted:
02 August 2024
You are already at the latest version
Abstract
Midkine (MDK) is a multifunctional heparin-binding growth factor, and has been shown to regulate cell growth, survival and migration. It also plays important roles in several inflammatory diseases such as sepsis. However, the role of MDK in the lungs has not yet been elucidated. In the present study, we investigated the role of MDK in pulmonary inflammation experiments using a mouse lipopolysaccharide (LPS)-induced pneumonia model and human bronchial cells. Wild-type and MDK-deficient mice were injected intratracheally with LPS, and several inflammatory parameters were analyzed. In the wild-type mice, mRNA expression of MDK in lung tissues was significantly increased after intratracheal LPS injection. The MDK-deficient mice showed significantly lower counts of total cells and neutrophils, as well as lower concentrations of neutrophil chemokines, KC and MIP-2 in bronchoalveolar lavage fluid, compared to wild-type mice. Moreover, mRNA expressions of TNF-α, KC and MIP-2 in lung tissues, as well as histopathological lung inflammation score, were significantly lower in the MDK-deficient mice. Furthermore, in in vitro experiments using bronchial epithelial cells, LPS stimulation increased mRNA expression of MDK, and MDK knockdown by siRNA decreased LPS-induced TNF-α and CXCL8 upregulation. These findings suggest that deficiency of MDK attenuates LPS-induced pulmonary inflammation.
Keywords:
midkine
; pulmonary inflammation
; bronchial epithelial cells
; chemokine
; neutrophils
Introduction
Acute respiratory distress syndrome (ARDS) is a life-threatening respiratory failure triggered by several risk factors such as pneumonia and sepsis. ARDS is characterized by acute inflammatory lung injury with non-cardiogenic pulmonary edema. There are no established pharmacologic therapies for ARDS, with reported mortality rates ranging from 35% to 46%. In the United States, approximately 190,000 patients are diagnosed annually, with a hospital mortality rate of 38.5% [1]. The prevalence of ARDS is reported to be 10% among patients in intensive care units and 23% among all ventilated patients [2]. The mechanism is complex, and various types of cells and mediators have been reported to be involved. However, there are still many unclear points regarding the molecular pathophysiology of ARDS.
Midkine (MDK), discovered in 1988, is a low-molecular-weight heparin-binding protein with a molecular weight of about 13 kDa [3]. It is highly expressed mainly in epithelial tissues in the process of epithelial mesenchymal interactions, in differentiating nerve tissues, and in mesodermal tissues undergoing remodeling. Additionally, MDK plays roles in various physiological activities, including development, survival and cell migration [3,4,5]. While MDK expression in adults is limited, it has been reported to be highly expressed in cancer cells [6] and the processes of inflammation and repair [7,8], as well as the pathology of various diseases [9,10].
There have been few reports showing the role of MDK in the lung. However, it has been reported that its expression is accelerated in the respiratory tract of cystic fibrosis patients [11], and that it has antimicrobial activity against bacteria and fungi [12,13]. While these findings suggest that MDK plays an important role in pulmonary inflammation, Zhang et al. have reported the role of MDK in ARDS-associated lung fibrosis [14]. However, the specific role of MDK in lung diseases remains unclear.
In the present study, we conducted in vitro experiments using a mouse lipopolysaccharide (LPS)-induced pulmonary inflammation model and human bronchial cells to determine the role of MDK in pulmonary inflammation.
Materials and Methods
Reagents
The reagents used in this study were: Escherichia coli serotype 0111:B4 LPS (Signa-Aldrich, St. Louis, MO), Power SYBR Green PCR Master Mix (Applied Biosystems, Foster City, CA), BEAS-2B cells (ATCC, Manassas, VA), mouse TNF-α DuoSet ELISA kit (R&D Systems, Minneapolis, MN), mouse KC DuoSet ELISA kit (R&D Systems), mouse MIP-2 DuoSet ELISA kit (R&D Systems) and human IL-8/CXCL8 DuoSet ELISA kit (R&D Systems).
Animal Protocols
The Animal Research Committee of Fukushima Medical University approved all animal experiments. MDK-deficient mice (Mdk KO mice; obtained from Dr. K. Kadomatsu) used in this study had no gross abnormalities in the brain, lungs, heart, stomach, kidneys, testes or ovaries by macroscopic and microscopic observation, consistent with Nakamura et al.’s study [15]. Wild-type (WT) and Mdk KO mice were anesthetized with ketamine/xylazine, and 1 mg/kg of LPS was administered intratracheally. Bronchoalveolar lavage (BAL) was performed as previously described [16,17]. Briefly, a BD Insyte Autoguard catheter (Becton, Dickinson and Company, Franklin Lakes, NJ) was inserted into the trachea, and 0.6 mL of physiological saline was infused a total of three times. After recovery of the fluid, the mice were sacrificed, and lung tissue was excised for use in analyzing the mRNA expression of mediators. In addition, for histopathological examinations, 10% formalin (Wako Pure Chemical Industries; Osaka, Japan) was injected into the trachea and lung tissues were fixed with 25 cm H2O at 24 h after LPS administration.
Isolation of RNA
RNA was isolated with the Absolute RNA Miniprep Kit (Stratagene, La Jolla, CA). Genomic DNA was digested with DNase I (Ambion, Austin, TX), and RNA was reverse transcribed with the SuperScript III First-Strand Synthesis System (Invitrogen, Carlsbad, CA).
Time Course of Midkine Expression
mRNA levels of MDK in lung tissues were analyzed by quantitative real-time PCR using the following primers: Fwd: 5′- CTCGCCCTTCTTGCCCTCTT-3′, Rev: 5′- GCAGGGCACCTTGCAATGGA-3′ as previously described [9].
Measurement of mRNA
Quantitative PCR was performed using Power SYBR Green PCR Master Mix and an ABI PRISM 7000 (Applied Biosystems). The threshold cycle (Ct) was calculated using Cts for the target genes and GAPDH. Relative mRNA expression was expressed as fold increase over values obtained from RNA from normal lungs or human reference total RNA (Stratagene) as previously described [18,19].
Measurement of Inflammatory Cytokine Levels
The concentrations of human TNF-α and CXCL8 as well as murine keratinocyte chemoattractant (KC) and macrophage inflammatory protein (MIP)-2 were measured with ELISA kits (DuoSet ELISA development kit, R&D Systems) according to the manufacturer’s protocols.
Pathological Evaluation of Lung Sections
Pathological evaluation was performed as previously described [17]. Briefly, the lungs were excised and fixed by inflation at 25 cm of H2O with a phosphate buffer (10 mM, pH 7.4) containing 10% formalin for 24 h, and then embedded in paraffin. A 5-m-thick tissue section was prepared and stained with hematoxylin and eosin. An observer who was blinded to the animal group assignment assessed 10 randomly chosen regions per tissue sample at a magnification of 400×, and scored lung damage severity as described previously [20]. Within each field, lung injury was scored lung damage severity in each field based on two criteria: a) neutrophil infiltration or aggregation in the airspace or vessel wall, and b) alveolar wall thickness. Each criterion was evaluated on a 4-point scale (0 = no damage, 1 = mild damage, 2 = moderate damage, and 3 = severe damage). The sum of these scores was presented as the lung injury score.
Cell Culture
Human lung bronchial epithelial cells, BEAS-2B cells, were cultured in RPMI-1640 medium supplemented with 10% FBS (Gibco by Life Technologies, Grand Island, NY), 100 IU/mL penicillin and 100 μg/mL streptomycin (Sigma-Aldrich, St. Louis, MO). After reaching 80% confluence, the cells were isolated, counted and cultured in RPMI-1640 medium for 24 h. LPS (1.0 mg/mL) was added to the wells with or without knockdown of MDK, and the cells were incubated and harvested at specified time points.
Knockdown of Midkine
Small inhibitory RNAs (siRNAs) were obtained from Thermo Scientific (ON-TARGET plus SMART human MDK and ON-TARGET plus Non-Targeting siRNA, Waltham, MA). Transfection of siRNAs was performed according to the manufacturer’s protocol as described previously [19]. BEAS-2B cells were incubated in growth medium for 24 h, and a final concentration of 100 nM siRNA was applied to the cells. Lipofectamine RNAiMAX (Invitrogen) was used as transfection medium. After 24 h, the cells were washed and incubated with or without LPS for 3 h.
Statistical Analysis
The Student’s t-test or Mann-Whitney U test was used to compare two groups, while ANOVA was used to compare multiple groups. Fisher’s least significant difference test was used for post hoc analysis. For all analysis, p < 0.05 was considered statistically significant.
Results
Change in Midkine Expression in Lung Tissues after LPS Administration
To characterize changes in the expression of mRNA for MDK, WT mice were treated with LPS, and quantitative real-time PCR was performed. Twenty-four hours after LPS treatment, the mRNA expression of MDK was significantly elevated compared to baseline level (Figure 1).
Bronchoalveolar Lavage Findings in Midkine-Deficient Mice
Inflammatory Cytokine Concentration in Bronchoalveolar Lavage Fluid of Midkine-Deficient Mice
To investigate the reason of decreased neutrophil lung inflammation in the Mdk KO mice, we analyzed the concentrations of KC and MIP-2 in BAL fluid, which play critical roles in neutrophil recruitment into the lungs. The concentration of KC was significantly lower in the Mdk KO mice compared to the WT mice at 3 and 6 h after LPS treatment (Figure 3a). In addition, the concentration of MIP-2 was significantly lower in the Mdk KO mice compared to the WT mice at 3 h after LPS treatment (Figure 3b).
Inflammatory Cytokine Expression in Lung Tissues of Midkine-deficient Mice
For the next analysis, we analyzed the mRNA expression of inflammatory cytokines in lung tissues. The mRNA expressions of TNF-α, KC and MIP-2 were significantly lower in the Mdk KO mice compared to the WT mice at 3 h after LPS treatment (Figure 4a–c).
Histopathological Analysis of Midkine-Deficient Mice
For further analysis, we compared the histological findings of lung tissues between the WT and Mdk KO mice. At 24 h after LPS administration, in the Mdk KO mice, pulmonary inflammation was decreased (Figure 5) and the lung injury score was significantly lower compared to the WT mice (2.3 ± 0.4 vs 3.4 ± 0.4, p < 0.01; mean ± SEM). These results suggest that LPS-induced pulmonary inflammation was suppressed in the Mdk KO mice.
Change in Midkine mRNA Expression in Bronchial Epithelial Cells after LPS Stimulation
Because lung epithelial cells play important roles in pulmonary inflammation, we used BEAS-2B human bronchial cells to analyze the role of MDK in LPS-induced pulmonary inflammation. We stimulated the cells with LPS, and analyzed the changes in MDK mRNA expression, which revealed a significant increase at 3 h post-LPS stimulation (Figure 6).
Effect of Midkine Knockdown on LPS-induced TNF-α and CXCL8 Expressions in Bronchial Epithelial Cells
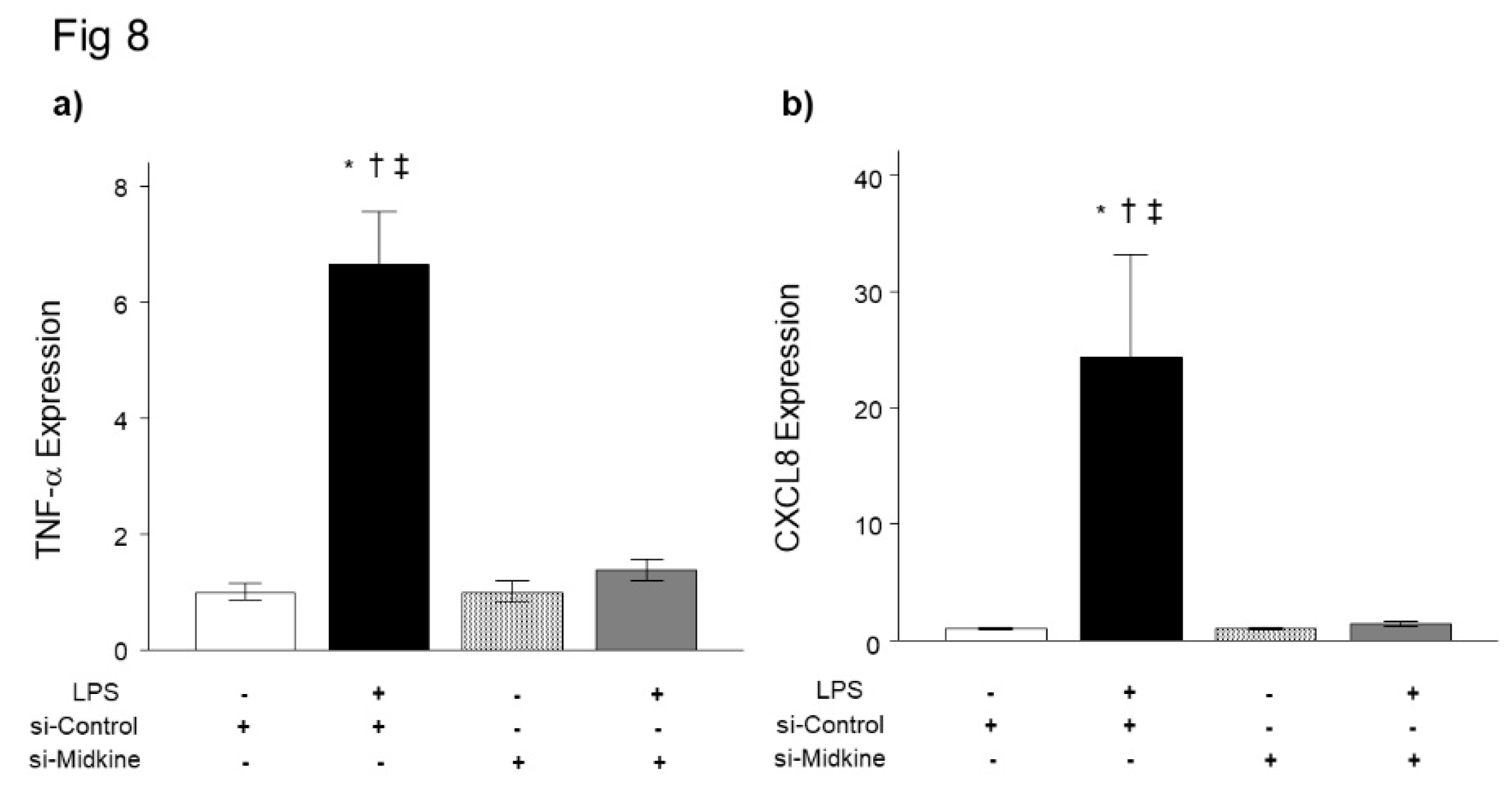
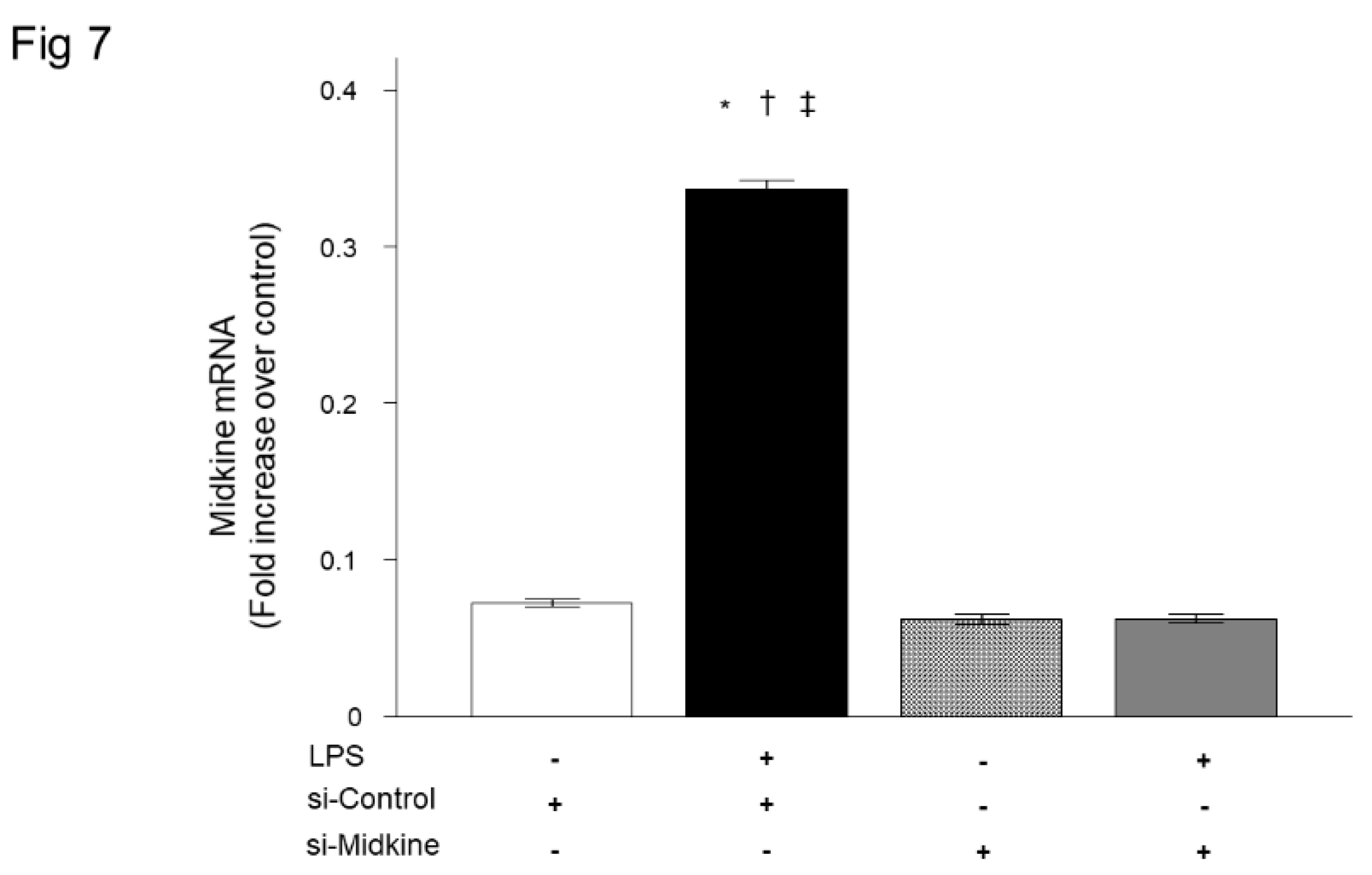
For further evaluation of the role of MDK in pulmonary inflammation, we analyzed the effect of MDK knockdown on LPS-induced inflammatory cytokine expression in bronchial epithelial cells. We first confirmed that MDK mRNA expression was decreased to the baseline level 3 h after transfection of MDK siRNA (Figure 7). Consistent with the results of Mdk KO mice, knockdown of MDK expression by MDK siRNA significantly decreased LPS-induced mRNA upregulation of TNF-α (Figure 8a) and CXCL8 (Figure 8b) 3 h after LPS stimulation.
Discussion
The present study demonstrated that MDK expression was increased in lung tissues after LPS administration in mice, and neutrophil accumulation in the lungs was significantly attenuated in Mdk KO mice compared with WT mice. In addition, expressions of inflammatory cytokines and chemokines as well as histopathological lung injury score were significantly lower in Mdk KO mice compared with WT mice. Furthermore, in in vitro experiments using bronchial epithelial cells, MDK knockdown by siRNA decreased LPS-induced upregulation of TNF-α and CXCL8. These findings suggest that MDK exerts a pro-inflammatory effect in LPS-induced pulmonary inflammation.
MDK was first discovered as a heparin-binding cytokine that is highly expressed during embryogenesis [3]. Although MDK expression is relatively low in healthy adults, it has been found to increase during inflammation, tissue repair and neoplastic transformation. Regarding inflammation, Sato et al. reported that MDK expression in renal proximal tubules was increased after renal ischemic reperfusion injury, and the numbers of neutrophils and macrophages infiltrating into the tubulointerstitium were significantly lower in Mdk KO mice than in WT mice [21]. Reduced renal damage with impaired infiltration of inflammatory cells into the renal tissue in the absence of MDK was also reported by other studies [22,23,24]. Moreover, in a mouse model of experimental autoimmune encephalomyelitis, infiltration of inflammatory cells into the spinal cord was decreased in MDK-deficient mice compared to WT mice [25]. These results indicate that MDK has a pro-inflammatory effect.
However, conflicting results have been reported as well. For example, Kojima et al. reported increases in inflammatory cell infiltration and matrix deposition in the glomerulus and the interstitium during the progression of crescentic glomerulonephritis induced by anti-glomerular basement membrane antibody in Mdk KO mice, showing that deficiency of MDK exacerbates necrotizing glomerular injuries in progressive glomerulonephritis [26]. It was also reported that knocking down endogenous MDK expression by siRNA enhanced TNF-α-induced apoptosis through activation of caspase-3 in prostatic cancer cells [27], and MDK had a protective role against cardiac ischemic/reperfusion injury [28]. Moreover, Takenaka et al. reported a protective effect of MDK in acute cardiac infarction [29], suggesting the possibility of organ-dependent effects of MDK.
In lung diseases, MDK is reported to be expressed in small airways, type II pneumocytes, and alveolar macrophages in COPD, as well as in the sputum of patients with ventilator-associated pneumonia caused by S. aureus [30]. Additionally, MDK expression has been demonstrated to be induced in the respiratory epithelium by hypoxia through hypoxia inducible factor-1α [31]. These findings suggest that MDK plays important roles in several lung diseases. In acute lung inflammation, plasma concentrations of MDK were found to be higher in patients with ARDS than in healthy volunteers [14], and were associated with pulmonary and kidney injury, as well as 28-day mortality in septic patients with ARDS [32]. Increase in serum MDK levels were also reported in patients with SARS-CoV-2 infection (COVID-19) [33].
To determine the role of MDK in acute lung inflammation, we used an LPS-induced pulmonary inflammation model. Our results showed an increase in lung inflammation in Mdk KO mice, consistent with previous studies. Xu et al. reported that MDK inhibition attenuated sepsis-induced lung injury via angiotensin-converting enzyme/angiotensin II pathway, and demonstrated the role of MDK in pulmonary endothelial cells [34]. In the present study, we showed that neutrophil recruitment was attenuated with decreased levels of chemokines in the lung of Mdk KO mice when compared to WT mice. In addition, we focused on the role of MDK in bronchial epithelial cells, and demonstrated that LPS-induced upregulation of TNF-α and CXCL8 was attenuated by MDK knockdown in bronchial epithelial cells.
In the current study, we did not address several mechanistically relevant questions of interest. First, we analyzed epithelial cells because the cells play a critical role in pulmonary inflammation and increase in MDK had already been demonstrated in the cells [12,31]. However, we cannot exclude the possibility that other types of cells are involved in the pathogenesis of pulmonary inflammation, because MDK exists in a variety of cells. MDK expression has been shown in type II pneumocytes and alveolar macrophages in COPD [12], and increased expression of MDK protein due to hypoxia has been demonstrated in neutrophils, monocytes, and endothelial cells [35]. Second, we showed attenuation of LPS-induced neutrophil chemokines upregulation in MDK inhibition. The low levels of chemokines in the lung are considered to be one of the mechanisms by which LPS-induced pulmonary inflammation was attenuated in Mdk KO mice. To the best of our knowledge, the present study is the first to report showing the role of MDK in the lungs, consistent with previous research investigating renal ischemia-reperfusion injury [21]. However, there are several other roles of MDK regarding neutrophil recruitment. MDK by itself was reported to act as a haptotactic and chemotactic agent for neutrophils [36]. In addition, MDK was shown to support neutrophil trafficking during acute inflammation by promoting adhesion via β2 integrin [37,38]. The relationship between MDK and neutrophil extracellular traps (NETosis) was also reported [39,40]. Finally, we did not analyze the role of angiotensin-converting enzyme/angiotensin II pathway in the present study. Further study is required to fully elucidate the role of MDK in acute lung inflammation.
In conclusion, the results of the current study showed an increase in MDK in response to intratracheal LPS administration and reduced pulmonary inflammatory response in MDK-deficient mice following intratracheal LPS treatment. In addition, knockdown of MDK attenuated inflammatory responses in lung epithelial cells in vitro. Taken together, we conclude that deficiency of MDK attenuates acute lung inflammation, at least in part, through inhibiting inflammatory cytokine and chemokine upregulation in the lungs.
Authors Contributions
Conceptualization, Y.T., X.W., T.N., K.K., and Y.S.; Methodology, Y.T., X.W., T.N., Y.S., R.T., N.W., M.T., K.K., and Y.S.; Formal Analysis, Y.T., and X.W; Investigation, Y.T., X.W., T.N., Y.S., R.T., N.W., and M.T.; Resources, Y.T., X.W., M.T., K.K., and Y.S.; Data Curation, Y.T., X.W., T.N., Y.S., R.T., N.W., and M.T.; Writing – Original Draft Preparation, Y.T.; Writing – Review & Editing, Y.T., X.W., T.N., Y.S., R.T., N.W., M.T., K.K., and Y.S.; Supervision, Y.S.; Funding Acquisition, Y.T., and Y.S.
Conflicts of Interest
All authors declare that there is no conflict of interest.
References
- Matthay, MA.; Zemans, RL.; Zimmerman, GA.; Yaseen M Arabi, YM.; Beitler, JR.; Mercat, A.; Margaret Herridge, M.; Adrienne G Randolph, AG.; Carolyn S Calfee, CS. Acute respiratory distress syndrome. Nat. Rev. Dis. Primers. 2019, 5, 18. [Google Scholar] [CrossRef] [PubMed]
- Bellani, G.; Laffey, JG.; Pham, T.; Fan, E.; Brochard, L.; Esteban, A.; Gattinoni, L.; van Haren, F.; Larsson, A.; McAuley, DF.; Ranieri, M.; Rubenfeld, G.; Thompson, BT.; Wrigge, H.; Slutsky, AS.; Pesenti, A.; LUNG SAFE Investigators; ESICM Trials Group. Epidemiology, patterns of care, and mortality for patients with acute respiratory distress syndrome in intensive care units in 50 countries. JAMA. 2016, 315, 788–800. [Google Scholar] [CrossRef] [PubMed]
- Kadomatsu, K.; Tomomura, M.; Muramatsu, T. cDNA cloning and sequencing of a new gene intensely expressed in early differentiation stages of embryonal carcinoma cells and in mid-gestation period of mouse embryogenesis. Biochem. Biophys. Res. Commun. 1988, 151, 1312–1318. [Google Scholar] [CrossRef] [PubMed]
- Kadomatsu, K.; Huang, RP.; Suganuma, T.; Murata, F.; Muramatsu, T. A retinoic acid responsive gene MK found in the teratocarcinoma system is expressed in spatially and temporally controlled manner during mouse embryogenesis. J. Cell Biol. 1990, 110, 607–616. [Google Scholar] [CrossRef] [PubMed]
- Matsubara, S.; Tomomura, M.; Kadomatsu, K.; Muramatsu, T. Structure of a retinoic acid-responsive gene, MK, which is transiently activated during the differentiation of embryonal carcinoma cells and the mid-gestation period of mouse embryogenesis. J. Biol. Chem. 1990, 265, 9441–9443. [Google Scholar] [CrossRef] [PubMed]
- Tsutsui, J.; Kadomatsu, K.; Matsubara, S.; Nakagawara, A.; Hamanoue, M.; Takao, S.; Shimazu, H.; Ohi, Y.; Muramatsu, T. A new family of heparin-binding growth/differentiation factors: increased midkine expression in Wilms’ tumor and other human carcinomas. Cancer Res. 1993, 53, 1281–1285. [Google Scholar] [PubMed]
- Ohta, S.; Muramatsu, H.; Senda, T.; Zou, K.; Iwata, H.; Muramatsu, T. Midkine is expressed during repair of bone fracture and promotes chondrogenesis. J. Bone Miner. Res. 1999, 14, 1132–1144. [Google Scholar] [CrossRef] [PubMed]
- Misa, K.; Tanino, Y.; Wang, X.; Nikaido, T.; Kikuchi, M.; Sato, Y.; Togawa, R.; Tanino, M.; Tanaka, S.; Kadomatsu, K.; Munakata, M. Involvement of midkine in the development of pulmonary fibrosis. Physiol. Rep. 2017, 5, e13383. [Google Scholar] [CrossRef] [PubMed]
- Muramatsu, T. Midkine and pleiotrophin: two related proteins involved in development, survival, inflammation and tumorigenesis. J. Biochem. 2002, 132, 359–371. [Google Scholar] [CrossRef]
- Kinoshita, D.; Shishido, T.; Takahashi, T.; Yokoyama, M.; Sugai, T.; Watanabe, K.; Tamura, H.; Nishiyama, S.; Takahashi, H.; Arimoto, T.; Miyamoto, T.; Watanabe, T.; Kishida, S.; Kadomatsu, K.; Abe, J.; Takeishi, Y.; Konta, T.; Kubota, I.; Watanabe, M. Growth factor midkine aggravates pulmonary arterial hypertension via surface nucleolin. Sci. Rep. 2020, 10, 10345. [Google Scholar] [CrossRef]
- Nordin, SL.; Jovic, S.; Kurut, A.; Andersson, C.; Gela, A.; Bjartell, A.; Morgelin, M.; Olin, AI.; Lund, M.; Egesten, A. High expression of midkine in the airways of patients with cystic fibrosis. Am. J. Respir. Cell. Mol. Biol. 2013, 49, 935–942. [Google Scholar] [CrossRef] [PubMed]
- Linge, HM.; Andersson, C.; Nordin, SL.; Olin, AI.; Petersson, AC.; Mörgelin, M.; Welin, A.; Bylund, J.; Bjermer, L.; Erjefält, J.; Egesten, A. Midkine is expressed and differentially processed during chronic obstructive pulmonary disease exacerbations and ventilator-associated pneumonia associated with Staphylococcus aureus infection. Mol. Med. 2013, 19, 314–323. [Google Scholar] [CrossRef] [PubMed]
- Netsu, S.; Shishido, T.; Kitahara, T.; Honda, Y.; Funayama, A.; Narumi, T.; Kadowaki, S.; Takahashi, H.; Miyamoto, T.; Arimoto, T.; Nishiyama, S.; Watanabe, T.; Woo, CH.; Takeishi, Y.; Kubota, I. Midkine exacerbates pressure overload-induced cardiac remodeling. Biochem. Biophys. Res. Commun. 2014, 443, 205–210. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Pan, Y.; Fanelli, V.; Wu, S.; Luo, AA.; Islam, D.; Han, B.; Mao, P.; Ghazarian, M.; Zeng, W.; Spieth, PM.; Wang, D.; Khang, J.; Mo, H.; Liu, X.; Uhlig, S.; Liu, M.; Laffey, J.; Slutsky, AS.; Li, Y.; Zhang, H. Mechanical Stress and the Induction of Lung Fibrosis via the Midkine Signaling Pathway. Am. J. Respir. Crit. Care. Med. 2015, 192, 315–323. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, E.; Kadomatsu, K.; Yuasa, S.; Muramatsu, H.; Mamiya, T.; Nabeshima, T.; Fan, QW.; Ishiguro, K.; Matsubara, S.; Kaname, T.; Horiba, M.; Saito, H.; Muramatsu, T. Disruption of the midkine gene (Mdk) results in altered expression of calcium binding protein in the hippocampus of infant mice and their abnormal behaviour. Genes Cells 1998, 3, 811–822. [Google Scholar] [CrossRef] [PubMed]
- Nikaido, T.; Tanino, Y.; Wang, X.; Sato, S.; Misa, K.; Fukuhara, N.; Sato, Y.; Fukuhara, A.; Uematsu, M.; Suzuki, Y.; Kojima, T.; Tanino, M.; Endo, Y.; Tsuchiya, K.; Kawamura, I.; Frevert, CW.; Munakata, M. Serum syndecan-4 as a possible biomarker in patients with acute pneumonia. J. Infect. Dis. 2015, 212, 1500–1508. [Google Scholar] [CrossRef] [PubMed]
- Tanino, Y.; Makita, H.; Miyamoto, K.; Betsuyaku, T.; Ohtsuka, Y.; Nishihira, J.; Nishimura, M. Role of macrophage migration inhibitory factor in bleomycin-induced lung injury and fibrosis in mice. Am. J. Physiol. Lung Cell Mol. Physiol. 2002, 283, L156–62. [Google Scholar] [CrossRef] [PubMed]
- Tanino, Y.; Chang, MY.; Wang, X.; Gill, SE.; Skerrett, S.; McGuire, JK.; Sato, S.; Nikaido, T.; Kojima, T.; Munakata, M.; Mongovin, S.; Park, WC.; Martin, TR.; Wight, TN.; Frevert, CW. Syndecan-4 regulates early neutrophil migration and pulmonary inflammation in response to lipopolysaccharide. Am. J. Respir. Cell Mol. Biol. 2012, 47, 196–202. [Google Scholar] [CrossRef] [PubMed]
- Tanino, Y.; Wang, X.; Nikaido, T.; Misa, K.; Sato, Y.; Togawa, R.; Kawamata, T.; Kikuchi, M.; Frevert, CW.; Tanino, M.; Kojima, T.; Shibata, Y. Syndecan-4 inhibits the development of pulmonary fibrosis by attenuating TGF-β signaling. Int. J. Mol. Sci. 2019, 20, 4989. [Google Scholar] [CrossRef]
- Tang, SE.; Wu, SY.; Chu, SJ.; Tzeng, YS.; Peng, CK.; Lan, CC.; Perng, WC.; Wu, CP.; Huang, KL. Pre-treatment with ten-minute carbon dioxide inhalation prevents lipopolysaccharide-induced lung injury in mice via down-regulation of toll-like receptor 4 expression. Int. J. Mol. Sci. 2019, 20, 6293. [Google Scholar] [CrossRef]
- Sato, W.; Kadomatsu, K.; Yuzawa, Y.; Muramatsu, H.; Hotta, N.; Matsuo, S.; Muramatsu, T. Midkine is involved in neutrophil infiltration into the tubulointerstitium in ischemic renal injury. J. Immunol. 2001, 167, 3463–3469. [Google Scholar] [CrossRef] [PubMed]
- Kosugi, T.; Yuzawa, Y.; Sato, W.; Arata-Kawai, H.; Suzuki, N.; Kato, N.; Matsuo, S.; Kadomatsu, K. Midkine is involved in tubulointerstitial inflammation associated with diabetic nephropathy. Lab. Invest. 2007, 87, 903–913. [Google Scholar] [CrossRef] [PubMed]
- Sato, W.; Takei, Y.; Yuzawa, Y.; Matsuo, S.; Kadomatsu, K.; Muramatsu, T. Midkine antisense oligodeoxyribonucleotide inhibits renal damage induced by ischemic reperfusion. Kidney Int. 2005, 67, 1330–1339. [Google Scholar] [CrossRef] [PubMed]
- Kawai, H.; Sato, W.; Yuzawa, Y.; Kosugi, T.; Matsuo, S.; Takei, Y.; Kadomatsu, K.; Muramatsu, T. Lack of the growth factor midkine enhances survival against cisplatin-induced renal damage. Am. J. Pathol. 2004, 165, 1603–1612. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Takeuchi, H.; Sonobe, Y.; Jin, S.; Mizuno, T.; Miyakawa, S.; Fujiwara, M.; Nakamura, Y.; Kato, T.; Muramatsu, H.; Muramatsu, T.; Suzumura, A. Inhibition of midkine alleviates experimental autoimmune encephalomyelitis through the expansion of regulatory t cell population. Proc. Natl. Acad. Sci. USA 2008, 105, 3915–3920. [Google Scholar] [CrossRef] [PubMed]
- Kojima, H.; Kosugi, T.; Sato, W.; Sato, Y.; Maeda, K.; Kato, N.; Kato, K.; Inaba, S.; Ishimoto, T.; Tsuboi, N.; Matsuo, S.; Maruyama, S.; Yuzawa, Y.; Kadomatsu, K. Deficiency of growth factor midkine exacerbates necrotizing glomerular injuries in progressive glomerulonephritis. Am. J. Pathol. 2013, 182, 410–419. [Google Scholar] [CrossRef] [PubMed]
- Ohuchida, T.; Okamoto, K.; Akahane, K.; Higure, A.; Todoroki, H.; Abe, Y.; Kikuchi, M.; Ikematsu, S.; Muramatsu, T.; Itoh, H. Midkine protects hepatocellular carcinoma cells against trail-mediated apoptosis through down-regulation of caspase-3 activity. Cancer 2004, 100, 2430–2436. [Google Scholar] [CrossRef] [PubMed]
- Horiba, M.; Kadomatsu, K.; Yasui, K.; Lee, JK.; Takenaka, H.; Sumida, A.; Kamiya, K.; Chen, S.; Sakuma, S.; Muramatsu, T.; Kodama, I. Midkine plays a protective role against cardiac ischemia/reperfusion injury through a reduction of apoptotic reaction. Circulation 2006, 114, 1713–1720. [Google Scholar] [CrossRef] [PubMed]
- Takenaka, H.; Horiba, M.; Ishiguro, H.; Sumida, A.; Hojo, M.; Usui, A.; Akita, T.; Sakuma, S.; Ueda, Y.; Kodama, I.; Kadomatsu, K. Midkine prevents ventricular remodeling and improves long-term survival after myocardial infarction. Am. J. Physiol. Heart Circ. Physiol. 2009, 296, H462–469. [Google Scholar] [CrossRef]
- Linge, HM.; Andersson, C.; Nordin, SL.; Olin, AI.; Petersson, AC.; Mörgelin, M.; Welin, A.; Bylund, J.; Bjermer, L.; Erjefält, J.; Egesten, A. Midkine is expressed and differentially processed during chronic obstructive pulmonary disease exacerbations and ventilator-associated pneumonia associated with staphylococcus aureus infection. Mol. Med. 2013, 19, 314–323. [Google Scholar] [CrossRef]
- Reynolds, PR.; Mucenski, ML.; Le Cras, TD.; Nichols, WC.; Whitsett, JA. Midkine is regulated by hypoxia and causes pulmonary vascular remodeling. J. Biol. Chem. 2004, 279, 37124–37132. [Google Scholar] [CrossRef] [PubMed]
- Chang, W.; Peng, F.; Sun, Q.; Meng, SS.; Qiu, HB.; Xu, JY. Plasma midkine is associated with 28-day mortality and organ function in sepsis. J. Intensive. Care Med. 2020, 35, 1290–1296. [Google Scholar] [CrossRef] [PubMed]
- Ketenci, S.; Kalayci, MU.; Dündar, B.; Duranay, R.; Aynacıoğlu, AS. Elevated serum midkine levels in severe acute respiratory syndrome coronavirus 2 (sars-cov-2) infected patients. Int. Immunopharmacol. 2022, 110, 108939. [Google Scholar] [CrossRef] [PubMed]
- Xu, JY.; Chang, W.; Sun, Q.; Peng, F.; Yang, Y. Pulmonary midkine inhibition ameliorates sepsis induced lung injury. J. Transl. Med. 2021, 19, 91. [Google Scholar] [CrossRef] [PubMed]
- Weckbach, LT.; Groesser, L.; Borgolte, J.; Pagel, JI.; Pogoda, F.; Schymeinsky, J.; Müller-Höcker, J.; Shakibaei, M.; Muramatsu, T.; Deindl, E.; Walzog, B. Midkine acts as proangiogenic cytokine in hypoxia-induced angiogenesis. Am. J. Physiol. Heart Circ. Physiol. 2012, 303, H429–438. [Google Scholar] [CrossRef] [PubMed]
- Takada, T.; Toriyama, K.; Muramatsu, H.; Song, XJ.; Torii, S.; Muramatsu, T. Midkine, a retinoic acid-inducible heparin-binding cytokine in inflammatory responses: chemotactic activity to neutrophils and association with inflammatory synovitis. J. Biochem. 1997, 122, 453–458. [Google Scholar] [CrossRef] [PubMed]
- Weckbach, LT.; Gola, A.; Winkelmann, M.; Jakob, SM.; Groesser, L.; Borgolte, J.; Pogoda, F.; Pick, R.; Pruenster, M.; Müller-Höcker, J.; Deindl, E.; Sperandio, M.; Walzog, B. The cytokine midkine supports neutrophil trafficking during acute inflammation by promoting adhesion via β2 integrins (CD11/CD18). Blood 2014, 123, 1887–1896. [Google Scholar] [CrossRef] [PubMed]
- Herter, JM.; Mayadas, TN. Midkine, a middle manager of β2 integrins. Blood 2014, 123, 1777–1779. [Google Scholar] [CrossRef] [PubMed]
- Weckbach, LT.; Grabmaier, U.; Uhl, A.; Gess, S.; Boehm, F.; Zehrer, A.; Pick, R.; Salvermoser, M.; Czermak, T.; Pircher, J.; Sorrelle, N.; Migliorini, M.; Strickland, DK.; Klingel, K.; Brinkmann, V.; Abed, UA.; Eriksson, U.; Massberg, S.; Brunner, S.; Walzog, B. Midkine drives cardiac inflammation by promoting neutrophil trafficking and netosis in myocarditis. J. Exp. Med. 2019, 216, 350–368. [Google Scholar] [CrossRef]
- Liu, G.; Ren, X.; Li, Y.; Li, H. Midkine promotes kidney injury in diabetic kidney disease by increasing neutrophil extracellular traps formation. Ann. Transl. Med. 2022, 10, 693. [Google Scholar] [CrossRef]
Figure 1.
Midkine expression in lung tissues after intratracheal LPS injection into wild-type mice. Midkine was significantly increased in lung tissues 24 h after intratracheal LPS injection in wild-type mice (n = 3–5 per time point). Statistical differences between each group and 0 h were compared using ANOVA with Fisher’s least significant difference test as a post hoc test. *, p < 0.05 vs. 0 h.
Figure 1.
Midkine expression in lung tissues after intratracheal LPS injection into wild-type mice. Midkine was significantly increased in lung tissues 24 h after intratracheal LPS injection in wild-type mice (n = 3–5 per time point). Statistical differences between each group and 0 h were compared using ANOVA with Fisher’s least significant difference test as a post hoc test. *, p < 0.05 vs. 0 h.
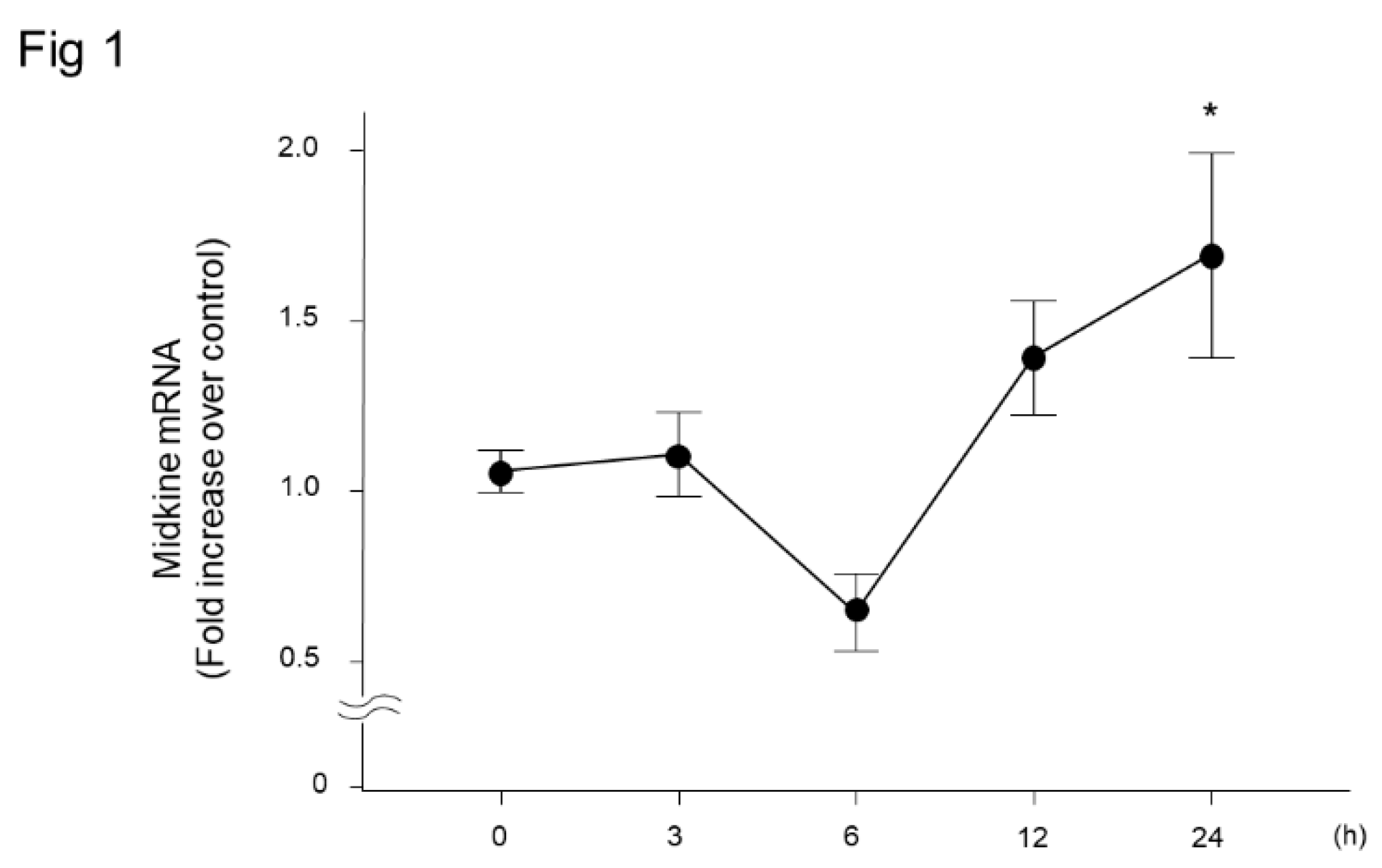
Figure 2.
BAL fluid findings after intratracheal LPS injection in midkine-deficient mice. Total cell (a) and neutrophil (b) counts in BAL fluid were significantly lower in midkine-deficient mice (KO) compared to wild-type mice (WT) 6, 12 and 24 h after LPS injection. n = 5–16/group, * p < 0.05 vs KO, mean ± SEM.
Figure 2.
BAL fluid findings after intratracheal LPS injection in midkine-deficient mice. Total cell (a) and neutrophil (b) counts in BAL fluid were significantly lower in midkine-deficient mice (KO) compared to wild-type mice (WT) 6, 12 and 24 h after LPS injection. n = 5–16/group, * p < 0.05 vs KO, mean ± SEM.
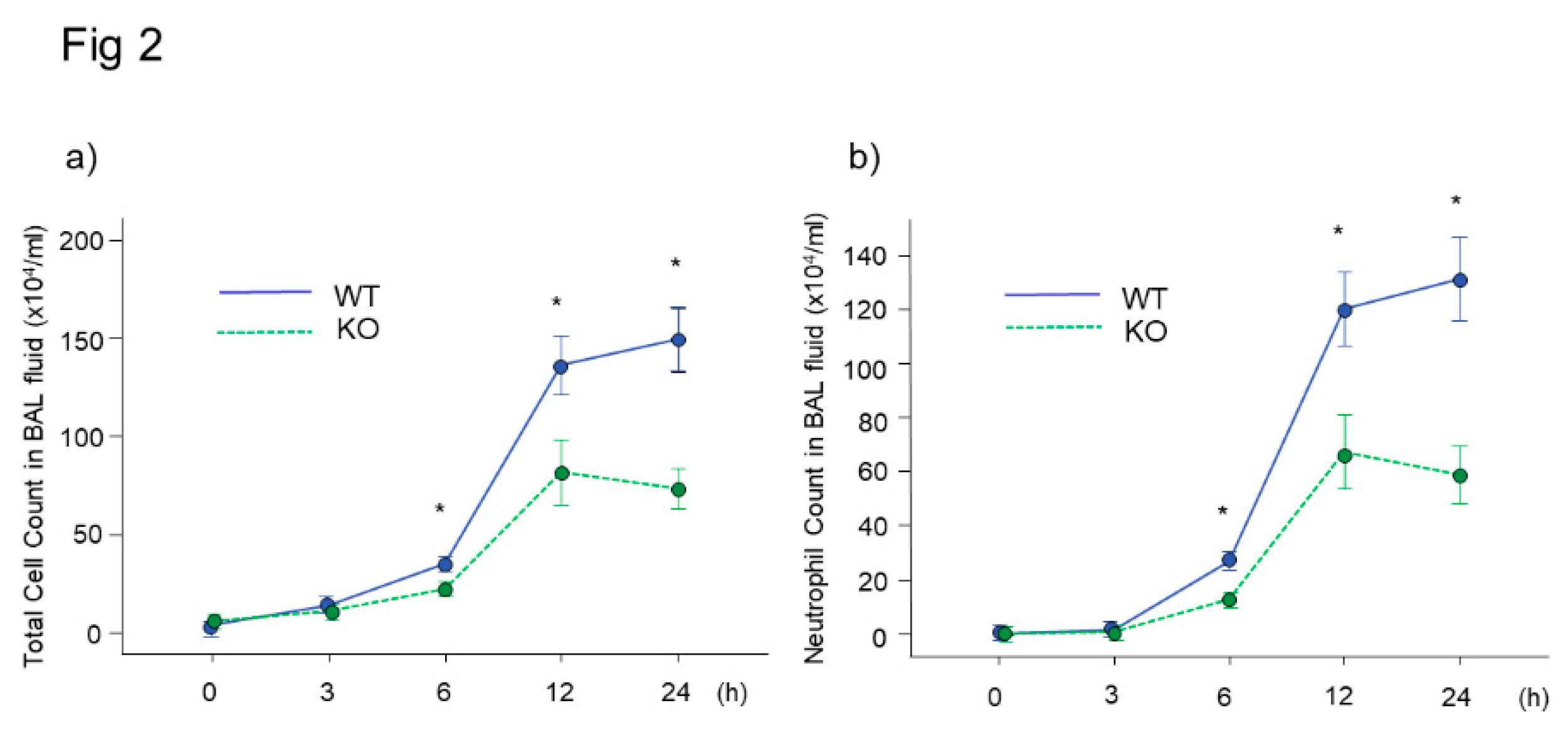
Figure 3.
Chemokine levels in BAL fluid in midkine-deficient mice. The levels of KC at 3 and 6 h and MIP-2 at 3 h were significantly lower in midkine-deficient mice (KO) compared to wild-type mice (WT). n = 5–12/group, * p < 0.05 vs KO, mean ± SEM.
Figure 3.
Chemokine levels in BAL fluid in midkine-deficient mice. The levels of KC at 3 and 6 h and MIP-2 at 3 h were significantly lower in midkine-deficient mice (KO) compared to wild-type mice (WT). n = 5–12/group, * p < 0.05 vs KO, mean ± SEM.
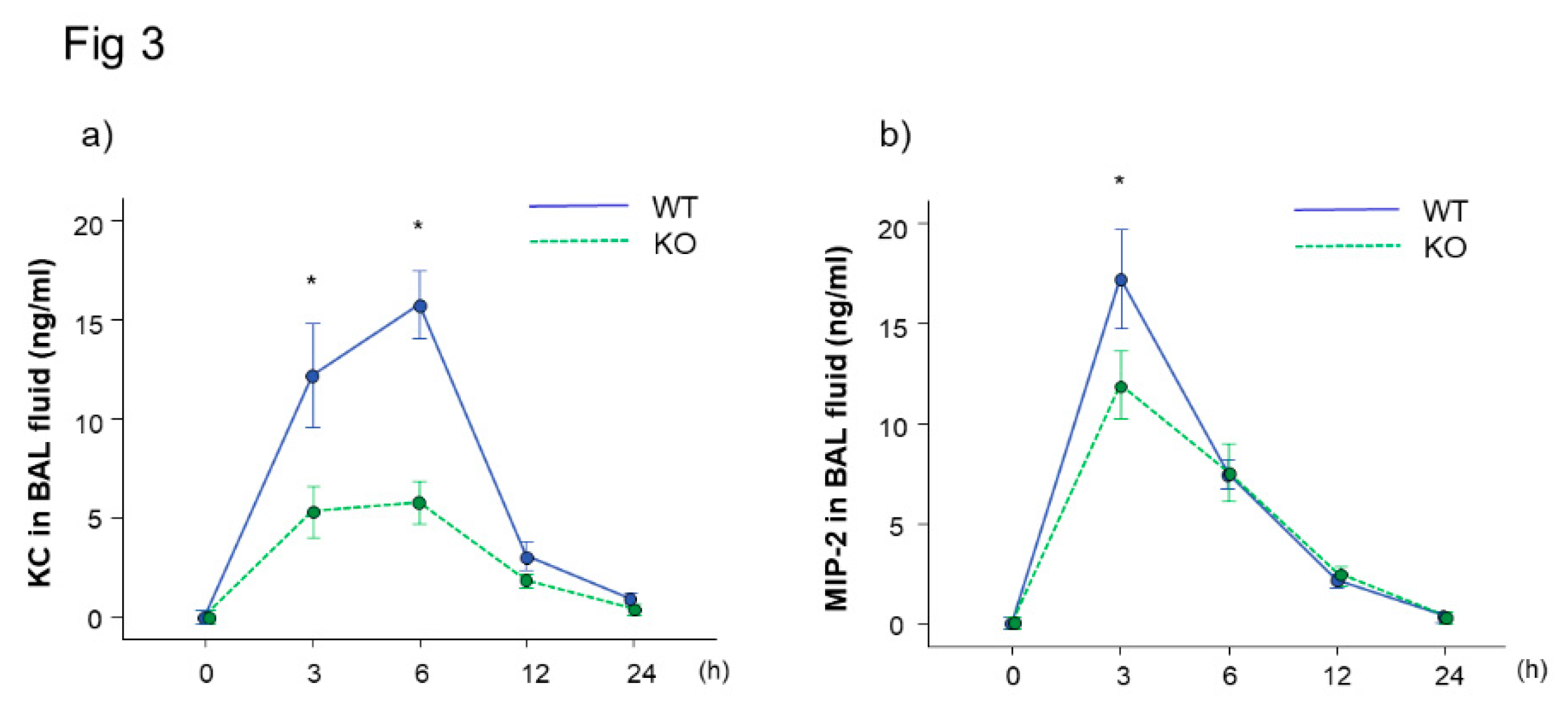
Figure 4.
mRNA expression of inflammatory cytokines in lung tissues in midkine-deficient mice. mRNA expression of TNF-α, KC and MIP-2 in lung tissues were lower in midkine-deficient mice (KO; n = 8) compared to wild-type mice (WT; n = 6) 12 h after LPS. mRNA expression was expressed as fold increase over normal. * p < 0.05 vs WT. Mean ± SEM.
Figure 4.
mRNA expression of inflammatory cytokines in lung tissues in midkine-deficient mice. mRNA expression of TNF-α, KC and MIP-2 in lung tissues were lower in midkine-deficient mice (KO; n = 8) compared to wild-type mice (WT; n = 6) 12 h after LPS. mRNA expression was expressed as fold increase over normal. * p < 0.05 vs WT. Mean ± SEM.
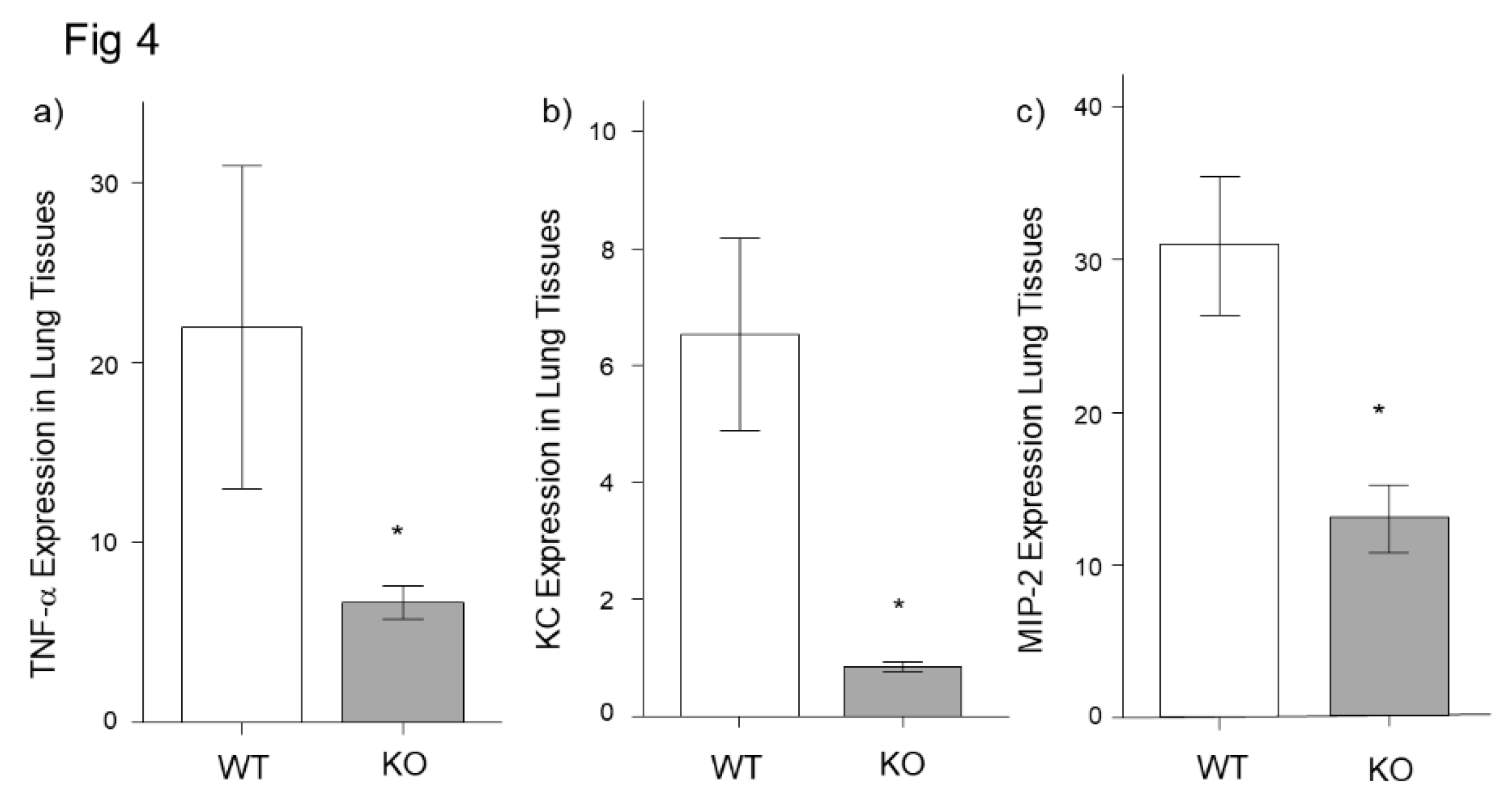
Figure 5.
Histopathological findings in midkine-deficient mice. Lung inflammation was more severe in wild-type mice (WT) compared to midkine-deficient mice (KO) 24 h after LPS. Hematoxylin and eosin stain.
Figure 5.
Histopathological findings in midkine-deficient mice. Lung inflammation was more severe in wild-type mice (WT) compared to midkine-deficient mice (KO) 24 h after LPS. Hematoxylin and eosin stain.
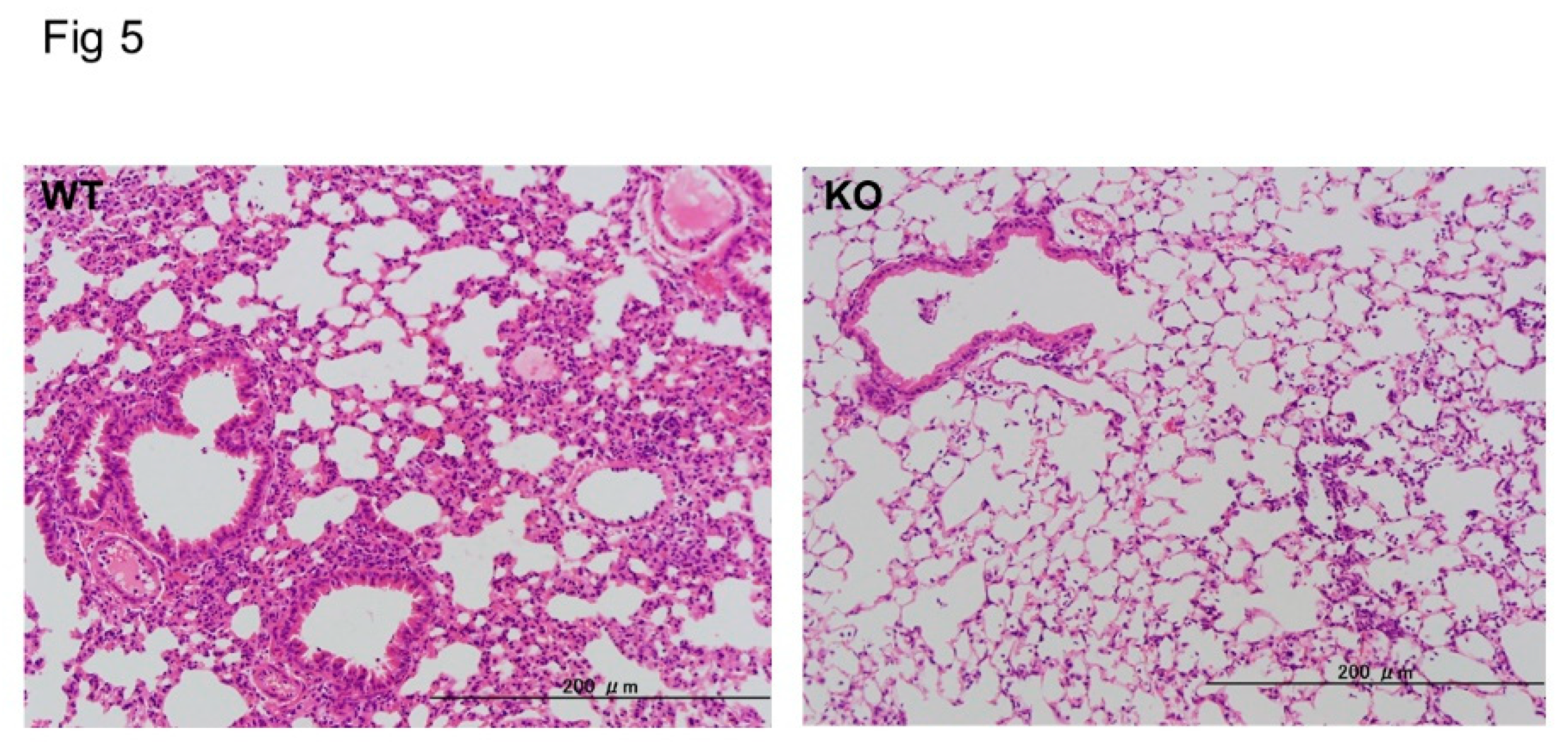
Figure 6.
Midkine expression after LPS stimulation in bronchial epithelial cells. Midkine was significantly increased at and after 3 h post LPS stimulation in BEAS-2B bronchial epithelial cells. n = 4/group, * p < 0.05 vs 0 h, mean ± SEM.
Figure 6.
Midkine expression after LPS stimulation in bronchial epithelial cells. Midkine was significantly increased at and after 3 h post LPS stimulation in BEAS-2B bronchial epithelial cells. n = 4/group, * p < 0.05 vs 0 h, mean ± SEM.
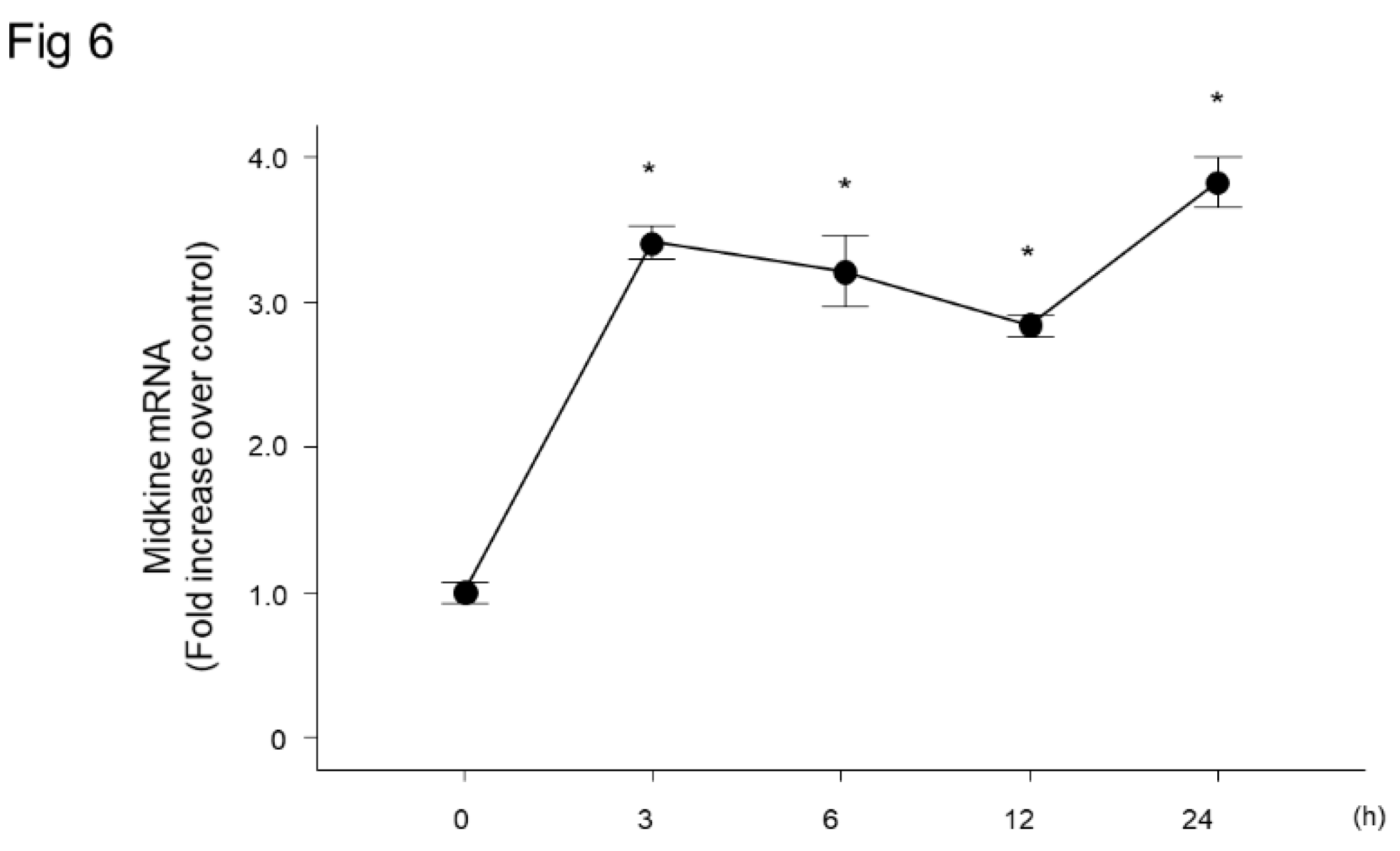
Figure 7.
Effect of midkine knockdown on mRNA expression of inflammatory cytokines after LPS in bronchial epithelial cells. LPS significantly increased mRNA expression of TNF-α (a) and CXCL8 (b) in BEAS-2B cells. Knockdown of midkine by siRNA significantly inhibited upregulation of TNF-α and CXCL8 mRNA 3 h after LPS stimulation. n = 4/group. *p < 0.05 vs si-control group, † p < 0.05 vs si-midkine group, ‡ p < 0.05 vs LPS with si-midkine group. Mean ± SEM.
Figure 7.
Effect of midkine knockdown on mRNA expression of inflammatory cytokines after LPS in bronchial epithelial cells. LPS significantly increased mRNA expression of TNF-α (a) and CXCL8 (b) in BEAS-2B cells. Knockdown of midkine by siRNA significantly inhibited upregulation of TNF-α and CXCL8 mRNA 3 h after LPS stimulation. n = 4/group. *p < 0.05 vs si-control group, † p < 0.05 vs si-midkine group, ‡ p < 0.05 vs LPS with si-midkine group. Mean ± SEM.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



