
Submitted:
30 July 2024
Posted:
31 July 2024
You are already at the latest version
Abstract
This narrative review challenges the recent World Workshop Consensus (WWC) conferences failure to classify aggressive periodontitis (AP) in young adults as distinct from adult periodontitis. Deficiencies in the consensus process, results of these deficiencies, and their impact on disease classification are presented. Support for retaining localized aggressive periodontitis (LAgP) in adolescents as a unique disease, focuses on the; 1) age of onset, 2) rate of bone loss in those affected, and 3) unique microbiological etiological associations. Examples of, 1) unique clinical signs and symptoms of LAgP are presented, and 2) the microbial subgingival consortia that precedes these clinical signs and symptoms are described. Tables show, 1) decreased publications for AP since publication of WWC guidelines and 2) Bradford-Hill guidelines that support the unique etiological consortia replicated in clinically well-defined longitudinal studies. The review describes major deficiencies in the WWC as compared to other medically-related well-run consensus conferences that, 1) set pre-conference standards for 70 – 80% agreement of expert participants, and 2) published the dissenting point of view. In contrast, the WWC conference was decided by a mere majority, and never presented the dissenting view. The review concludes that averaging of mean pocket depth reduction, or attachment gain can misrepresent disease and/or success of treatment in aggressive diseases. In contrast, clinical assessment of time to recurrence of subgingival re-infection with possible translocation of oral microbes to distant sites is presented as an alternative measurement of success. Other questions and future directions are presented.
Keywords:
Aggressive Periodontitis
; Aggregatibacter actinomycetemcomitans
; Treatment success
; Consensus conferences
; microbiome consortia
; damage/response framework
1. Importance of Recognizing Subtypes of Periodontitis for Scientific Discovery
The primary focus of this narrative review is an assessment of the serious deficiencies of the Stage III Grade C classification of a form of periodontitis previously known as Localized Aggressive Periodontitis (LAgP), Periodontosis, Localized Juvenile Periodontitis, and Early Onset Periodontitis [1,2]. This paper presents information that challenges both the process and the decisions made by the World Workshop on the Classification of Periodontal and Peri-Implant Diseases and Conditions, referred to hereafter as WWC, that led to the elimination of LAgP as a unique disease entity [1,3]. The Staging and Grading method of classification adopted by the leadership of the WWC is a scheme that has been used extensively by cancer researchers and clinicians for purposes of creating a consistent system for prognosis and treatment [4]. However, classification of disease by staging and grading was never intended to serve as a substitute for a robust framework for disease diagnosis [5]. As noted above, throughout the extensive history of efforts to characterize LAgP many name changes have evolved but several features unique to this disease have remained constant including: 1) the early age of onset of disease, 2) the localized nature of the disease to incisors and first molars, and 3) the rapid rate of severe bone loss seen in the disease as compared to forms of periodontitis presenting in adults [2,6]. It has also been obvious that, unlike adult periodontitis, LAgP frequently exhibits strong familial aggregation with transmission across multiple generations [7]. LAgP also exhibits a far higher prevalence of 2.05% in African Americans ages 14-17 compared to only 0.14% in persons of European ancestry in the United States, a nearly 15-fold difference in frequency [8].
The revised classification of LAgP as Stage III Grade C ignores the distinct clinical, microbiological and genetic differences in this aggressive form of periodontitis as compared to adult forms of disease [9]. Historical revisions of nomenclature that reflect shifting opinions are valid, but in this case the change fails to reflect the most current scientific knowledge [10]. The goal of this review is to challenge the Stage/Grade classification of LAgP, including the way in which the name change occurred (i.e., “the consensus process”), the negative effects this is already having on research aimed at understanding of the etiology and progression of the disease, and the impacts on clinical diagnosis and treatment. Our emphasis in the paper will be on: 1) the unique clinical presentation of LAgP, and 2) the unique microbiological risk factors associated with LAgP. Another paper in this volume on “Pathogens” (Miguel and Shaddox) will present the unique genetic, immunological and additional clinical features of LAgP [11].
Dental biology does not exist in a vacuum. The WWC reliance on histopathological differences as the ultimate discriminator for LAgP classification is premature.
Histopathological discrimination is unlikely until biopsy material related to active disease has been collected [12]. In time, it is likely that both histopathological and genetic differences between periodontitis in older adults and LAgP in children and young adults will be uncovered [13]. However, at this moment it is already inappropriate to ignore the major clinical signs and symptoms that are unique to LAgP [14]. Thus far, etiology based on a single gene of major effect causing LAgP appears unlikely, aside from rare syndromic forms such as those caused by mutations in the cathepsin C gene [15]. Instead, it is more probable that an ensemble of genes of small to moderate effect, combined with environmental risk factors such as smoking, play a prominent role in LAgP underlie susceptibility to the disorder [13,16]. The WWC argument that LAgP is too limited in numbers to warrant a separate diagnosis doesn’t hold up because orphan diseases (affecting less than 200,000 individuals) receive specific diagnoses in medicine [17]. The 2% prevalence among the 11+ million African American adolescents in the United States noted above alone would mean that approximately 317,000 individuals could be affected by LAgP. Moreover, necrotizing diseases are also very uncommon but despite their limited numbers these diseases were still recognized with a unique diagnosis by the WWC [18,19]. The idea that the determinations of the WWC are fixed is invalid because changes in the WWC classification have occurred previously [20].
Necrotizing diseases such as Acute Necrotizing (AN) Gingivitis and AN Periodontitis provide good examples of how “impairment of the host immune system” can create a dysbiotic state. In these examples, histopathological differences are extolled [18]. We propose that patients with LAgP fit into the same category, also having an impaired host immune system but in this case at the local level, as demonstrated by polymorphonuclear leukocyte dysfunction which in itself supports the concept that this is a specific category of disease [21]. In-spite of these inconsistencies the WWC authors failed to create a separate case definition for LAgP as a distinctive disease entity [1]. There is no recognition of this in the Consensus decision despite the fact that many reports in the literature, including the foundation papers on LAgP, clearly point to specific signs and symptoms, especially in patients of African descent who have excessive bone loss at an early age [12,22].
Over the years disease diagnoses have been debated among the “lumpers” and “splitters”. These terms originated in evolutionary biology but could be used in any form of biology [23]. Most simply put, “lumpers” tend to join things together, while ‘splitters” separate things so that they can be examined individually in greater detail. In the words of Siddhartha Mukherjee, author of the bestselling book “The Emperor of all Maladies” [24], there is great value in differentiating cancer types for diagnosis and therapy. In his words, “How can you treat something you can’t name.” As one example, Dr. Mukherjee points out that targeted immunotherapy is successful because definitions of disease highlighting different types of cancers can be affected by specific targeted treatments [24]. This reasoning can also be applied to LAgP. While Staging and Grading was first introduced in the 1940’s and 1950’s by Dr. Pierre Denoix it was never intended as a diagnostic strategy [25].
Personalized/Precision Medicine was promulgated by the insightful work of Dr. Ralph Snyderman (former member of the immunology section of the National Institute of Dental and Craniofacial Research and Dean of Duke University Medical School) [26,27]. Personalized Medicine makes great efforts to define diseases precisely as opposed to lumping different forms of a disease into a single group [22]. In the recent WWC the “Consensus” decision was to eliminate LAgP as a disease entity despite its characteristically unique clinical features that separate it from other forms of periodontitis [22]. This decision has had an outsized influence on diagnosis, treatment, prevention and scholarship in the areas in microbiology, immunology, and pathobiology of periodontal disease. Failing to name a disease with features as distinctive as LAgP has already led to less research (see Table 1), limited treatment options and going forward risks further divorcing our field’s biological initiatives from that of modern medicine.
Curiously, Pediatric Dentistry has deemed it appropriate and beneficial to name a disease so clinicians know the best way for it to be treated [28]. For example, the biological processes of caries, despite its various locations in the mouth, are very similar, and all varieties are related to acid producing, aciduric microbes that affiliate themselves with tooth surfaces and demineralize the surface and subsurface to create a carious lesion [29]. Nevertheless, the caries process has been recognized as having distinct subtypes: 1) occlusal caries, 2) proximal caries, and 3) root caries [30]. Moreover, early childhood caries (ECC) has been recognized as a distinct entity [31]. These well-defined diagnoses have led to specific treatments such as; 1) sealants used to obliterate occlusal pits and fissures [32], 2) fluoride varnishes for proximal decay [33], and 3) dietary counseling for prevention of ECC [34]. These treatments have resulted in changes in caries prevalence [35]. Unfortunately, our field’s new WWC guidelines fail to make similar distinctions for a major subtype of periodontitis, and this is going to have a negative impact on treatment and prevention of the disease.
2. Consensus (the Process) Pros and Cons
A. The Process: The Periodontitis and Peri-implantitis Classification formula developed by the Consensus Conference of the World Workshop in 2017 has several flaws. The WWC report by-passes clinical phenotypes and then eliminates the distinctions between adult periodontitis and disease that occur in adolescents and children with a marked bias toward host related pathobiology as supportive evidence for their re-classification [1]. The lead authors of the final report fail to recognize relevant differences in clinical presentation and microbial dysbiosis as well as host response elements as described in the previous World Workship classification [36]. The lead WWC authors fail to distinguish between diagnosis, treatment and prognosis and may have been more focused on improving opportunities for reimbursement by third party payers.
Consensus meetings in science usually gather experts in the field with a goal of reviewing the current literature to define disease diagnoses and most effective treatments [37]. The meetings are intended to generate a report presented to the world of clinical scientists and practitioners with guidelines based on the information currently available. The resulting conclusions can have a profound influence on preventive, diagnostic and treatment strategies for years into the future. The rules and regulations concerned with the conduct and presentation of material derived from consensus conferences vary widely and sometimes appear to be determined in a haphazard manner [37,38,39]. In other words, all Consensus Conferences are not always equally responsive to their constituents. One of the foundational papers of the WWC was commissioned to assess LAgP as a distinct clinical entity [22]. In this context, Staging and Grading was considered for LAgP but with the intent of keeping LAgP as a distinct clinical entity [22]. Approximately 100 clinician scientists were invited to the two-day WWC meeting. In the first day the participants were divided into groups to discuss specific periodontal clinical entities one of which was LAgP. While a variety of topics were discussed most were centered around disease classification. Among the 100 participants from Europe, South America, Asia and North America there were approximately ten to fifteen participants assigned to discuss LAgP. The following day representatives presented highlights of the group discussions. The summation of the LAgP group discussion was presented in a manner that was opposed to consideration of LAgP as a unique disease entity.
B. The Result: After the summary presentation, a vigorous and contentious discussion followed and a vote was taken in which 51% of the participants favored dropping the LAgP classification, and 49% were in favor of keeping LAgP as a distinct entity. Subsequently, the WWC organizers eliminated LAgP as a distinct form of periodontitis and lumped these patients into the general Periodontitis group, now classified as Stage III Grade C Periodontitis. To call this consequential decision a “Consensus” was clearly inappropriate, as the decision was supported only by a small majority of votes.
This decision was strongly contested [3]. The participants’ views appeared to be largely split by the fact that many clinical studies conducted in the USA have focused on adolescents of African descent in cases with onset as teenagers or in the early 20’s while other groups (excluding some studies from Scandinavia and Brazil) paid little or no attention to these strong associations [1,40,41] . Many participants were so disturbed by this outcome that a rebuttal to that final report has been subsequently written [12,42]. In efforts to reconcile the way in which the Workshop decision was concluded, efforts were made to examine the literature to explore how well constructed, rigorous consensus conferences are normally conducted.
While there is a great deal of inconsistency in the rules and regulation of consensus conferences there are several examples that set standards of good practice. One example of a well performed consensus conference required a vote of 70-80% or higher by the experts involved to reach what could be labelled as a consensus [37,38,39]. Even with the 80% rule, consensus conference participants in the minority were required to publish the dissenting point of view so that readers could have a clear understanding as to how decisions were made, what the opposing views were, and the future directions projected [43]. This process was not used in the WWC.
C. The Impact: The effect of this approach has had an untoward effect on scholarly activity as witnessed by publications in the years following the decision. Below we have provided a survey of papers published using the search term Localized Aggressive Periodontitis in either the title or abstract as compared to search engines using terms such as Aggressive Periodontitis, Periodontitis and Stage III Grade C Periodontitis. Our search consisted of articles published in the last 12 years, broken into groups based on 6 years prior to the new WWC classification and 6 years following the Consensus publication. Pub Med, Scopus and the Web of Science databases were searched, and the data were averaged to get a general sense of work done in these areas over the designated time span. The results show an overall 18% increase in papers with “Periodontitis” in the title and/or abstract six years following the WWC and a 33% decrease in those with “Aggressive Periodontitis.” It is abundantly clear that elimination of LAgP as a case definition has had a major negative impact on numbers of publications focusing on patients who previously were classified as LAgP (see Table 1).
3. World Workshop Case Definition of Periodontitis
A further dissection of the new definition of Aggressive Periodontitis (now called Stage III Grade C Periodontitis) points out that there is no distinction within this framework that discriminates between a young adolescent with disease and older patients presenting with a similar pattern of disease [14]). In our efforts to understand Periodontal diseases many factors come into play that can be used to distinguish one form of disease from another. It appears as if data representative of cases of Localized Aggressive Periodontitis in adolescents or children who show extreme levels of disease were overlooked [44].
A. Age as a consideration for case determination: There is no dispute that subgingival bacteria initiate a host damage response in both LAgP and other forms of Periodontitis [45]. However, it appears as if factors such as age of onset, ethnicity and rate of disease progression are ignored in the recent Consensus document [19]. The Consensus summary states that differences in the pathobiology of Aggressive and Chronic forms of Periodontitis are not significant enough to establish distinct disease entities [1]. Of curiosity, age as a factor for disease is reported by one of the foundation papers but here age assessment begins with individuals 30 and above [46]. The paper concludes that …”empirical evidence-driven definitions of CAL (Clinical Attachment Level) thresholds signifying disproportionate severity of periodontitis by age are feasible [46] and that ..”age is a significant determinant of the clinical presentation of periodontitis…”, “… individuals with fewer remaining teeth have higher mean CAL and PD measures.” [46]. This is especially true for LAgP but this disease classification is not considered in the paper. The focus of those of us who study LAgP has usually been on individuals aged 20 and younger [47].
B. Rate of bone loss as a consideration for a Case Definition. The most prominent illustration of differences in the rate of disease progression occurs in comparing LAgP to Adult periodontitis (AP). Rate of disease progression is a difficult parameter to measure unless longitudinal studies are performed [48,49]. A simple examination of specific site disease progression can be determined when LAgP and Adult Periodontitis are compared, and age is introduced as a parameter. There is no denial that 6 to 8 mm of bone loss is rarely seen in a short period of 2-3 years in other forms of Periodontitis aside from those who were previously diagnosed as having LAgP [12,42]. However, even in LAgP, this very significant fact (rate of bone loss) is hidden when mean CAL is averaged over 168 sites [48] (see Section 10).
C. Etiology as a consideration for a case definition. The hazardous often challenging, shifting, and at times impaired oral environment (i.e., irradiated salivary glands, dry mouth, circadian rhythms) has resulted in a highly adaptable oral microbiome [50]. As the main entryway into the body, bacteria first reaching the oral cavity face enormous environmental obstacles and a multitude of surfaces for colonization [51]. The more we probe, the more we realize that oral bacteria are not only involved in dental disease but that these highly adaptable bacteria can and have been shown to escape their initial oral habitat and move to remote sites to exacerbate diseases at sites distant from the oral cavity [52,53].
Many infectious diseases are considered to have a microbial etiology, and thus specific infectious agents are used to define the disease of interest. The recognition of microbiological aspects of dental disease began in full force at the earliest days when microbiology emerged as a field of biology. W.D. Miller, one of the first graduates of the Pennsylvania Dental College (graduating in 1879) went to Berlin to study in the laboratory of Robert Koch. Miller developed the chemo-parasitic theory of caries, suggesting that oral bacteria fermented sugars and produced acids that demineralized enamel [54]. He also developed the focal theory of infection that suggested that bacteria or products of bacteria can travel to sites distant from the oral cavity and play a role in the development of various diseases that can affect the brain, lungs and stomach. Sometime thereafter, Kritchevsky and Seguin [55], two French microbiologists, focused on periodontal disease and its treatment. They also believed that oral disease could be implicated in systemic diseases. Over time the focal theory of infection was abandoned because treatments such as full mouth extraction designed to eliminate oral infection had no impact on the systemic diseases that these oral microbes were proposed to cause. Immunological research in the same era examined the role of oral microbes on immune responsiveness. Notable among them, were papers by Beckwith and colleagues who showed in dramatic fashion how oral bacterial plaque provoked a severe full body immunological/inflammatory response [56].
Sexually transmitted diseases (STDs) serve as an example of cases where disease is determined by the initiating infective agent [57]. For instance, Syphilis is initiated by Treponema pallidum, Gonorrhea, is due to Neisseria gonorrhoeae, Chlamydia, is due to infection by Chlamydia trachomatis. Hepatitis B, Herpes Simplex virus (HSV), Human Papillomavirus (HPV) and AIDS initiated by HIV are infections due to specific viruses [58]. In contrast, some diseases can be initiated by a variety of infectious agents and promote morbidity and mortality resulting from disparate confounding factors. For example, Kaposi’s sarcoma can be caused by; 1) Herpes virus (HHV-8), 2) AIDS – related HIV, 3) Iatrogenic transplant-associations, 4) Advanced age, most common in Eastern Europeans, Middle Eastern and Mediterranean men, and 5) Endemic Kaposi sarcoma, occurring in young people in Africa [59]. These distinctions in diseases and the resulting tissue damage are best described by the Damage Response Framework introduced by Casadevall and Pirofski [60,61]. These authors described how HIV, the primary infectious agent in AIDS, typically was not the cause of mortality or morbidity [60,61]. The disease and irreversible tissue and organ damage were due to the immunocompromised host being unable to counteract damage inflicted by otherwise harmless microorganisms [62,63,64].
4. Conclusions for the Consensus Section
The WWC organizers’ re-classification of periodontitis case definitions is confounded by the use of overlapping and inaccurate clinical definitions that, in the case of LAgP, disregarded key clinical features that set it apart from other forms of periodontitis [22]. In contrast, conditions such as necrotizing periodontal disease and endo-periodontal lesions were presented as distinct case definitions [1].
To avoid problems such as occurred in the most recent WWC, it is recommended that future Consensus Conferences include the following characteristics: 1) A statement clearly articulating the percentage of participants who agree with recommendations from the Conference in order for the recommendation to be considered a consensus opinion (i.e., 70 -80% agreement); 2) Dissenting points of view should be clearly reported with explanations for such views; 3) A criteria statement should be made at the outset of each foundational paper meeting as to what publications will be included in each foundation paper (i.e., Delphi process, longitudinal data, case defined data, etc.); and 4) The concluding statement should present realistic suggestions designed to improve case definitions in the future [43].
Most of us who disagreed with the final Consensus report were left with the question as to why a group of well-meaning expert periodontists decided to separate themselves from the mainstream of medical research and lump together what clearly is a distinct clinical entity. A group of participants and their colleagues previously challenged the decision to eliminate LAgP as a distinct diagnosis in a paper published in JADA, but this journal is not widely read by our European colleagues [12,42].
5. Localized Juvenile Periodontitis: History
In efforts to abbreviate the long history of Aggressive Periodontitis in Adolescents, it is worth mentioning the historical significance of the original recognition of what was first called Periodontosis by Dr. Bernhard Gottlieb in Vienna in 1923 [10]. In the “modern era” Dr. Paul Baer (1971) presented a prescient, more detailed clinical description of this entity, then called Localized Juvenile Periodontitis (LJP) [6]. His description depicted rapid bone loss in younger individuals and was founded on a minimum of the following five clinical features: 1) age of onset, 2) family history, 3) lack of relationship between local factors and deep pockets, 4) rapid rate of progression and, 5) effect on primary teeth. In the over five decades since this publication the name of this disease and the disease itself has often been debated [1,2,65]. Unfortunately, since Baer’s descriptive and thorough paper many twists and turns have resulted in great confusion, which has and will continue to seriously impede our efforts to understand the microbiology, pathology, clinical definition and treatment of this silent (often painless symptomatology) and uncommon condition.
In 1976, Actinobacillus actinomycetemcomitans (Aa; now Aggregatibacter) was shown to be associated with what was then called Localized Juvenile Periodontitis (LJP) [66,67]. This discovery coincided with an effort to demonstrate that specific plaque entities were responsible for specific forms of periodontal disease and led to the birth of the concept of The Specific Plaque Hypothesis (SPH) as compared to the Non-Specific Plaque Hypothesis (NSPH) [68,69]. Aa, a Gram-negative capnophilic microbe, was discovered in 1912 by Klinger [70] in a disease called Actinomycosis (lumpy jaw disease). Interestingly, Aa was not presented as a sole actor in this disease and was shown to have acted in concert with Actinomyces israelii; hence the species name “actinomycetemcomitans” or in common with Actinomyces [70]. The SPH stimulated a significant body of research that explored specific microbes and their relationship to specific clinically recognizable forms of disease [71]. They were indeed hypotheses for their time and stimulated a voluminous literature that examined the biological features of several microbes associated with both caries and periodontitis [72,73,74]. This intellectually challenging approach added significant scientific information to microbial features related to these diseases that were here-to-fore unexplored [75]. The understanding of dental diseases as well as the biology and pathogenicity of several microorganisms associated with distinct forms of dental disease that benefited from these broad hypotheses were; Streptococcus mutans (and its relationship to caries [29], Actinobacillus (now; Aggregatibacter) actinomycetemcomitans and its relationship to LJP [66,67], Bacteroides melaninogenicus (now Porphyromonas gingivalis) [73,76] (and its relationship to adult periodontitis) and Actinomyces viscosus (and its relationship to root caries) [77] to name a few of these associations [78]. While in retrospect this appears to be a naïve approach it stimulated research that led to a better understanding of glucans and dextran’s in the case of S. mutans [79], Leukotoxin and cytolethal distending toxin in the case of Aa [80], gingipains [81], hemolysins [82] and collagenases in the case of P. gingivalis etc. [83]. This choice between the SPH and the NSPH was replaced by the Ecological Plaque Hypothesis [84] which though modified over the years has highlighted how dental diseases are influenced by ecological interactions and how ecological interactions can influence and modify dental diseases.
During the early period of microbiological furor, there was also an appropriate and important emphasis placed on the host immunological response [85] . At the time, the disease process was thought of as a war between “bad” bacteria and the host responsiveness to those bacteria [86]. The battleground was the junctional epithelium, the epithelial barrier that formed a boundary between the subgingival pocket area and the gingival and periodontal complex [87]. This complex boundary tissue contained all the elements that could thwart the inward progress of the destructive bacterial elements such as toxins, enzymes, antigens and lipo- proteins, polysaccharides and teichoic acids, etc. It was proposed that a weakened barrier could lead to ingress of noxious elements, that could result in an overall aberrant host responsiveness, tissue alteration and disease at the local tissue site [88,89]. These understandings re-awakened interest in movement of bacteria from oral to distant sites [89]. Local destruction was thought to result from the direct result of bacterial by-products such as enzymes, toxins etc. or from an overactive host response initially designed to stem the tide in favor of healing [90]. However, many believed that an excessive host response could also be responsible for local tissue destruction and/or disease. The experimental gingivitis model of Harald Loe and colleagues provided a close examination of chronological association of oral bacteria in supragingival plaque and the coordinated dysregulation of the host response to accumulating bacteria [91] (Figure 1). Notable experiments and descriptions by Page and Schroeder [92], work by Taubman et al [93], Genco and Sanz [94], Taichman and colleagues [95,96] and Lehner and colleagues [85] emphasized stages in the inflammatory process that highlighted the prominence of specific cells such as polymorphonuclear leukocytes (PMNs), monocytes, macrophages and plasma cells. The importance of each of these cell types in the process of tissue damage were carefully described and particular emphasis was placed on plasma cells and their ability to destroy bone via osteoclastic activity. In parallel to this work, hemolysins, collagenases, and a host of destructive microbial factors derived from B. melaninogenicus, and then a leukotoxin derived from Aa were uncovered. The apparent emphasis and conflicts between microbiologists and immunologists became perceptible in many forms of infectious diseases [86]. More recently periodontal research has shown that shifting levels of cytokines and chemokines have been shown to be signaling molecules that encourage cellular activity [97]. The emergence of the Damage/Response Framework shifted the emphasis away from microbes or the host to the interaction of the microbe and its host in relationship to disease progression [60] (Figure 2).
6. Bradford Hill Guidelines for Distinguishing Causation from Correlation
A longitudinal model designed to study the transition from health to disease has shown that a consortium of microbes in addition to Aa are required for disease to occur (Aa is necessary but not sufficient) [47]. It is possible that the consortia can be expanded to include other microbes but this observation and those of others to this point have shown that Aa is instrumental in the disease process (see Table 2). We suggest that Aa is a social influencer in that despite its low level it has an outsized influence on microbial community geography and behavioral interactions. The most likely rationale is that Aa impairs local immune responsiveness to allow less adaptable subgingival microbes in the community to overgrow. Models and clinical studies suggest that the consortia consist of Aa and Filifactor alocis among other subgingival microbes [47,98]. It appears as if F. alocis invades the biofilm at the later stages of subgingival biofilm formation but then provokes an aggressive and damaging host response by continuing to alter the local immune capacity. It appears as if both Aa and F. alocis relationships with disease could be strain related [99]. Other studies have found a somewhat different consortia associated with Periodontitis in older patients [100]. There is a great deal of evidence that points to bacteria as initiators of cell-mediated tissue loss in periodontitis [72,73,101]. Studies of carious lesions are more dependent on environmental factors, namely carbohydrate consumption, that stimulate the growth and survival of acid producing – acid loving microbes [29,75].
Of all dental diseases, LAgP is the closest to fulfilling the Bradford Hill Guidelines for association of provocative agents and disease association [102]. LAgP, if defined in the most stringent manner, focusing on adolescents with bone loss in the molar or incisor region, typically of African descent, satisfies six of the nine aspects of association with disease causation. These associations are; 1) temporality, or, exposure to the agent prior to disease 2) strength of association, or, the level of association of the microbe as determined statistically , 3) biological gradient, or, a dose-related response of a biologically active substance to tissue damage and thus the higher the exposure the greater the disease, 4) consistency, or, reproducibility of critical experiments by others, 5) plausibility, or, the association of an agent with pathological consequences such as tissue damage caused by the microbe, 6) alteration of the disease, or experimental intervention that alters the provoking agent (either the bacteria, bacterial complex, or virulence agents) and the disease (See Table 2).
To support these associations, we have set a minimum of three independent longitudinal studies that test for these associations in categories 1 through 6. For positive proof, at least 2 to 3 longitudinal studies are needed to show a microbial consortia or virulence complex implicated in disease. Due to the dearth of longitudinal studies done using the appropriate populations, we proposed that one of the three studies could show that the presence of Aa alone could satisfy categories 1 through 6. Guidelines 7 to 9 were also assessed. Guideline 7 evaluates specificity and requires that the study demonstrates that a cause can produce an effect. Guideline 8 evaluates experimental alteration which requires demonstrating that the disease can be altered by an intervention that reduces the overall damage response. Guideline 9 discusses coherence which requires that the theory presented is in keeping with existing knowledge. Guidelines 7 to 9 have been limited to Aa and its effects because studies with the consortia in these categories have been few. While newer approaches to data integration have been used to expand the interpretation of The Bradford Hill guidelines they remain as an important way in which to associate infectious agents with the etiology of disease.
7. The Changing landscape of Infectious Diseases: The Oral Cavity as a New Focus
There has been a shift in our understanding of infectious diseases in the last thirty years.
Up until the mid-1980’s diagnostic microbiology, immunization and antibiotic therapy have proven to provide a crucially important strategy used to control many prevalent infections caused by identifiable microorganisms [103]. However, since then infections such as acquired immunodeficiency syndrome (AIDS) have led to new insights into complex diseases that result from host modification leading to lethal progression due to secondary well-defined infections. These complex secondary infections required a holistic approach to diagnosis, prevention and treatment of disease hence the evolution of the damage/response framework [60].
Data acquired over the last 60 years of research in microbiology and immunology suggest that LAgP in several ways shows similarity to a local form of AIDS [47]. This realization contrasts to typical blood borne mono-infections such as Syphilis (Treponema pallidum), Diphtheria (Corynebacterium diphtheriae) or Anthrax (Bacillus anthracis) or any blood borne mono-infection that spreads disease producing toxins [104]. In the localized immunomodulated AIDS-type scenario (LAgP/type scenario) once the barrier effect at the local site has been affected, bacteria from subgingival plaque weaken the local epithelial local barrier and can enter the connective tissue, alter it, and then invade the blood stream and translocate to distant sites [105,106].
In context of this newly emerging concept of complex/host compromised/multi-species/multi-layered infection, the oral cavity provides an ideal environment to study diagnosis, initiation, and progression of diseases that involve multi-species microbial interactions and that produce altered host responses that fail to control shifting microbial challenges [107]. These sentiments are not meant to minimize post-infection host healing efforts to repair damage caused by infectious insults. On the contrary, because of access and exposure, the oral cavity can provide an ideal environment for studying disease initiation, metabolic processes, and disease progression in these complex multi-layered infections [108].
The Damage/Response Framework appears to be more appropriate for the study of Localized Aggressive forms of periodontitis and periapical infections. Periodontitis, particularly LAgP, can be equated to a form of an acquired immunomodulated host response at the local level [109]. Aa and other microbes can be associated with disease susceptibility created by an imbalance between a dysbiotic microbiome resulting in a perturbed host homeostasis [71]. A. actinomycetemcomitans is a pathobiont that is opportunistic in its own regard but Aa also creates a perturbation of homeostasis by modulating PMNs, macrophages, Lymphocytes and other local immune functions [9,110,111,112]. The damage process can be further altered by subsets of other pathobionts such as P. gingivalis [113], and F. alocis [114] such that exacerbated local damage can occur as a result of the overwhelming challenge due to the overgrowth of otherwise commensal/opportunistic microbes (F. nucleatum, S. parasanguinis ) [47,98]. Overgrowth of these specific pathobionts and commensals in an otherwise compromised local immune response can now diminish the reparative capacity at the local site and enhance the resulting tissue damage [115]. The damage can remain localized but in certain cases an altered barrier can result in movement of pathobionts and/or commensals to sites distant from the oral cavity [116]. Several experiments have re-iterated the early work of Okell and Elliott [105] showing transient bacteremia’s emanating from oral procedures move many oral microbes to distant sites through the blood stream [106,117]. Mechanical dental procedures such as scaling and root planning as well as flossing, brushing and eating an apple can also induce transient bacteremia’s [118,119].
8. Oral Microbiology and Immunology and Infectious Diseases
A. Microbiology: The oral cavity provides an ideal place to study host/microbial interactions [107,120]. First and foremost, the oral cavity is the entry point for most external substances making it an ideal place to study host/microbial interactions. Material derived from the mouth is easily accessible for longitudinal analysis. Analysis of the interaction of cellular and acellular material can be collected and normalized for quantitative assessment. Oral collections can be done painlessly, without aggressive/invasive/destructive methodologies and with little to no interference with bodily function [107]. Finally, host influences as well as external influences such as diet, radiation, circadian rhythms, drug effects, stress, aging, trauma healing etc. can be documented in vivo [121,122].
Many experiments were done in the early days of microbiology in efforts to understand the role that oral microbes play in periodontology and cariology [123,124,125]. These landmark experiments should be put into context based on the current understanding of disease [30]. Two seminal experiments conducted in the 1960’s moved microbiology into the consciousness of the dental academic community. A series of rigorous experiments by Paul Keyes and colleagues clearly showed the caries could be passed from mother to child when he compared caries experiences of Golden Hamsters to Albino Hamsters [126]. Golden Hamsters were discovered to be susceptible to carious dental lesions, while Albino Hamsters were caries free. By shifting newly born albino pups into cages with Golden Hamster Dames he found that these pups, delivered by Cesarian section in a Germ-Free chamber, would now show caries because they were suckled by caries exposed Dames. In contrast, Albino Dames who suckled Golden Hamster pups, also delivered by Cesarian section, showed no caries. Penicillin could impede the carious process and finally it was shown that the agent provocateur was a Gram-positive coccus (later identified as Streptococcus). These experiments showed that caries was caused by oral microbes in animals fed carbohydrates, and, that the microbes could be passed from mother to child. These experiments were dramatic and demonstrated the importance of acid production and demineralization but were conducted in the absence of a fully developed microbiome. As a result, dysbiosis and homeostatic imbalance were not studied (Figure 3).
The second seminal experiment was developed by Harald Löe and colleagues in Denmark [127]. These experiments opened the door to our recognition that microbes formed on teeth in an ordered, deliberate manner. Exploration of this model showed that over time the shift from a clean tooth surface to one colonized sequentially by microbial pioneers followed by a complex mass of bacteria overtime moved from above to below the gumline. This progression led to tissue inflammation (Figure 2). These studies showed that resumption of toothbrushing and other oral hygiene methods after abstention for a three-week period caused a removal of microbial masses and a return to gingival health. This simple, elegant model permitted our colleagues to investigate plaque biofilm development and the associated effect of microbial dysbiosis on tissue inflammation. Sophisticated microbiological and histopathological studies provided a new and vital understanding of how microbes interacted and how they may have affected the underlying epithelial and connective tissue response [92,128]. However these experiments also illustrated how host inflammation provided a feedback loop for nutritional supplementation that might have accounted for microbial expansion [129]. It was also shown that not all subjects responded in the same way to the same extent relative to the microbiological challenge, and that within the same mouth different sites responded differently [97,130]. While now appreciated to be much more complicated than previously thought these experiments resulted in more questions than answers and stimulated new areas of research that have moved well beyond dentistry. The whole field of coaggregation initiated by Paul Kolenbrander and colleagues was derived from these primitive but elegant experiments [131,132].
These landmark experiments provided the impetus for studies of microbial colonization of teeth and oral soft tissue from birth to senescence which proved to be thorough and accurate and more easily studied than microbial colonization of the gut lining [133]. While feces collection is a way of studying gut colonization, microbial contact with gut epithelium can only be studied using invasive, colonoscopic methodologies [134]. The experimental model developed by Löe and colleagues has led to a more complete understanding of biofilm formation, coaggregation and host responsiveness to microbial challenges [135]. Most recently, this model has been amplified to examine chemokine and cytokine levels over the time course of the 3-week non-brushing model [97]. Unfortunately, many assumptions made about the host response to the immediate bacterial challenge in this model have only recently been documented. It is quite conceivable that microbial factors move through the subgingival epithelial barrier formed by the junctional and sulcular epithelium and that host inflammation plays an important role in microbial dysbiosis [88,136].
9. Immunology and Studies of Host Responsiveness to Infectious Agents
What we lack are sufficient time course experiments that document host cellular changes that evolve from health to early, middle, and later stages of periodontal disease development [92]. Studies of these events have occurred more directly in experiments of endodontic lesions [137]. While different in some respects, oral biologists should be encouraged to examine the similarities and differences in periodontal and endodontic lesions. Just as the experimental gingivitis model enlightened our understanding of time related events associated with microbial development in both the supragingival and subgingival environment, we still need to decipher the passage of substances from the “pocket” to and through the epithelial basement membrane to challenge the immediate area below the barrier membrane. It has taken 60 years from the inception of this classical gingivitis model to document interbacterial signaling distances and bacterial by product host cell interactions. However, we have now come to a time when technological advances have caught up to our theoretical understandings. We are now on the threshold of the merging of ideas and technological advances. DNA techniques [138], bar-coding [139] and CLASI_FISH [140] technologies now provide us with the tools required to study subgingival bacterial biogeography and cellular phenotypic responsiveness in a time related manner [141]. In this quest for a more complete understanding of microbial-host interactions we as oral biologists now could study these events in a sequential manner with easy access to microbial and host induced inflammatory response elements [142].
To re-iterate, early plaque development and initiation of gingivitis provided a straightforward path. Plaque could be collected easily for example from a tooth surrogate which when placed in the mouth could serve as an aseptic surface for microbial associations over time [143,144,145]. Microbiological, immunological and DNA technologies are now being used to catalogue biogeography. A tooth analogue can be placed at or just below the gum line to document the transition from supra to subgingival biofilm formation [146]. Documentation of subgingival plaque is more difficult. Early efforts to use mylar strips, cemental strips and polyvinyl strips have yielded useful but incomplete information [143,144]. There have been several reports that have tried to document the complexity of subgingival plaque, but since this is the focal point for spread of commensal oral microbes throughout the body more must be done.
10. Measurements as Determinants of Treatment Failure or Success
In the overall scheme of things, we have determined through both clinical observations and scientific testing that microbial plaque leads to gingival inflammation, which leads to tissue destruction, barrier alterations, pocketing, attachment loss and then bone loss [147]. While this sequence of events has been observed in human and replicated in animals the timing of these events and their route of progression can be influenced and altered by a shifting microbiota as well as host responsiveness [89]. In the presence of sophisticated biological methodologies, our clinical measurement methods have remained stagnant and reliant on a periodontal probe and an x-ray.
It is hard to consider that periodontitis is not due to a dysbiotic microbiome which results in disease defined by tissue destruction and weakened tooth support [147]. In most if not all diseases, successful treatment is typically measured by repair of altered tissue and/or alternatively in reduced recurrence or relapse of disease [48]. In most cases, treatment of periodontitis has relied on tissue repair as demonstrated by pocket depth reduction or attachment level gain [49]. However, in recent years many studies have shown that oral microbes can travel through the blood stream and these oral bacteria can exacerbate systemic diseases at distant sites such colorectal cancer and heart disease etc. [148]. In these cases, it might be prudent to look at treatment success in an alternative manner, as the prevention of disease progression, recurrence or relapse as a way of reducing initiation of diseases at distant sites.
A. An additional way to assess periodontitis treatment. success
Many of our most revered longitudinal studies have relied on averaging pocket depth reduction or attachment level gain resulting from a specific treatment. Often times, data is presented as reduction of probing pocket depth or attachment gain over a 3 month to 1 year period after completion of treatment [49]. These measurements are usually averaged over 28 teeth, each tooth having 6 measured surfaces, thus yielding up to a total of 168 probable or measurable sites per mouth. For more illustrative examples two fictitious but illustrative cases will be compared (Cases A and B). Case A represents a patient that has 2 probable pockets with each site having a 10 mm probing pocket depth on initial examination. Thus, we start with 2, 10mm pockets on the distomesial and lingual-mesial surfaces of the left and right mandibular first molar or a total of 40 mm of pocketing at 4 diseased sites (4 x 10 = 40). The remaining 164 probable sites have pockets of 3 mm. Cumulatively, these sites have a total of 492 mm (164 sites x 3 mm pockets per site = 492 mm of total probing depth). Overall, the patient presents with a total of 532 mm of pocketing in 168 probable sites ( 492 + 40 = 532 mm) over the whole mouth or an average of 3.17 mm of pocketing per site. After treatment, consisting of deep scaling and root planning, the four-10 mm pockets were reduced to 8 mm (now a total of 32 mm) while the remaining 164 sites remain at 3mm (164 x 3 = 492). After treatment the total pocketing in the mouth is 492 mm + 32 mm = 524 mm. In this scenario the average pocket depth is 524/168 = 3.12 mm per site, a reduction of only 0.05 mm (3.17mm to 3.12 mm). In Case B the patient starts at the same level as seen in Case A, with 4 – 10 mm pockets and 164 – 3 mm pockets or a total of 532 total mm of pocket probing depth over the span of 28 teeth (3.17 mm of pocketing per site) but in this case the treatment is scaling and root planning plus the addition of Metronidazole and Ampicillin (for example). The result in this case is a reduction in the 4 deep sites to 2 mm (now a total 8 mm). Therefore, the total probing pocket depth in this case is 500 mm (164 x 3mm = 492 mm + 8 mm) over the span of 28 teeth. In this scenario, the total probing pocket depth over the 28 teeth is now 500/168 = 2.98 mm per site (a reduction of 0.19 mm). Comparing Case A to Case B there is an overall probing depth reduction resulting from treatment of .14 mm (3.12 – 2.98 = 0.14mm) even though the infected site reduction went from 10 mm to 8 mm in Case A, as compared to 10 mm to 2 mm in Case B. There has been some effort recently to focus on categorization of changes in pocket depth reduction or attachment level gain in sites of low to moderate to high risk, but this has not been fully conceptualized or actuated [149].
When disease returns at specific sites it is almost always associated with a dysbiotic microbiome [74,78]. Therefore, in contrast to measuring pocket depth reduction or attachment gain that we are accustomed to doing, a disease re-occurrence reduction, or relapse to infection model is proposed. This can be applied independently or combined with standard measurement of improvement at the disease affected sites. In this relapse model we question how treatment has resulted in the reduced risk for re-occurrence of disease. This disease re-occurrence reduction model implies that a goal of treatment is to enhance the local tissue barrier effect such that spreading of the infective oral bacteria from the site below the gum-line to areas distant to the oral cavity is the goal of treatment. For example, in Case A re-infection and return of deeply infected sites might likely occur 4 weeks post treatment intervention; whereas in Case B it might take 2 years or more for re-infection and reemergence of large pocket depths to re-occur. This pocket depth comparison presents an example of how oversight of re-infection in the oral dentition could provide a superior environment for patient overall health. This re-infection model takes into account the fact that while in our dental plaque model we establish “good guys” and “bad guys” in a supra or subgingival plaque environment we are fully aware of the fact that some of the so-called “good guys” become “bad guys” when they move past the local epithelial barrier and through the blood stream such that they can then colonize heart valves, colon cells, lung or brain tissue [150]. Our proposed model moves away from the concept that infection and disease is a war between bacteria and the host and moves toward the concept that disease is the result of a damage/response ratio. The overall result appears to be dependent on coping mechanisms by the immune system that is designed to successfully reduce the consequences and spread of microbial/viral/fungal interactions from the local site to sites distant from their origin [151].
B. An illustrative Case: This case documents aggressive periodontal bone loss in a 20-year-old patient who reported to our clinic (RSDM) with extensive periodontal disease. After obtaining consent (IRB:PRO#012008035; year = 2009) we collected subgingival plaque, saliva and buccal cells for analysis. The subgingival sample taken from various healthy and diseased sites had the “b” serotype of Aa with the JP2 promoter region. The patient had only one strain of Aa in his subgingival microbiome isolated from both healthy and diseased sites, with substantially more Aa isolated from diseased sites. We tested the Aa isolates for antibiotic sensitivity and for the presence of the hbpA-1 and hbP-2 and tbp-A pseudogenes using primers reported by Haubek et al [152]. Saliva was assessed for salivary anti-S. mutans activity and buccal cells were used for detection of the lactoferrin single nucleotide polymorphism associated with anti-S. mutans activity [153]. Our primary goal was to use our laboratory data to provide information about Aa antibiotic sensitivity, which could act to supplement treatment aimed at resolving this progressive disease in this young patient. The focus on Aa was pragmatic because our previous data had suggested that Aa was necessary but subsequently proved to be insufficient on its own to cause disease [47]; conversely, assessment of the complete consortia was impractical at that time. However, several of our other laboratory assessments proved useful.
While we recovered Aa from the diseased site, we cannot attribute disease to the presence of Aa. Microbial causation can only be implied if the bacterium preceded disease at the site of disease initiation [29]. Therefore, in this case, linking Aa to disease initiation and development can only be seen as speculative. Second, based on the complex patient history, it is reasonable to conclude that confounding social, psychological, and ecological modifiers could have contributed to a diminished host response, factors that could clearly be implicated in disease progression [30]. Based on the antibiotic sensitivity testing we ruled out the use of penicillin derivatives due to Aa insensitivity, which was a clinically useful finding. Furthermore, the age of the patient, the tooth loss attributed to periodontal disease and the extent of bone loss indicated an aggressive nature of localized disease in this patient (Figure 4). We posit, based on tooth location and the patient’s history that the loss of two mandibular incisors, was due to extensive periodontal disease (Figure 4). This conclusion appears to be a realistic appraisal of tooth loss as a consequence of: 1) the dramatic level of bone loss in the existing molars, and 2) the complete lack of caries in this subject’s mouth (and the fact that the mandibular incisors lost are not typically vulnerable to caries) [154]. Testing for salivary anti-S. mutans activity gave an incomplete picture since several other oral microorganisms can also be related to caries [155]. The fact that anti-LF antibody had no effect on S. mutans suggests that factors other than LF were responsible for the anti-S. mutans activity. This finding agrees with our previous data where 20-30% of subjects tested showed factors independent of LF that also killed S. mutans [153]. The level of bone loss and lack of proximal decay reflects a pattern seen in many cases of LAgP [156,157]. Finally, point mutations in the hpbA-1 and tnp-B pseudogenes suggested that the patient was of West African descent [152].
In summary, the clinical presentation coupled with the presence of minimal plaque, the absence of proximal decay, severe periodontal disease and the presence of Aa, all present a strong argument that LAgP is a disease uniquely distinguishable from periodontitis that occurs in adults (Figure 5).
11. Future Challenges and Suggestions to the Clinical and Research Community
First, it is our hope based on the extensive data reviewed here that colleagues in our clinical and research community will re-instate LAgP as a distinct disease entity. This prospect does not in any manner attempt to overrule Staging and Grading but proposes that the WWC system should make a distinction between this unique disease and the generally accepted adult form of periodontitis for the betterment of research and practice. Second, we propose expansion of treatment goals to include reduction of re-emergence of infection as another way of assessing treatment success. This point speaks to the concern that re-infection of the local site may lead to passage of either oral commensals or pathobionts from the local periodontal site to sites distant from the oral cavity. Therefore, the goals of treatment for Periodontitis or any other dental infection are both to support healing and repair coupled with the goal of reduction of translocation of oral microbes throughout the body to limit the scope and extent of damage to diseases distant from the oral cavity.
Will these expanded goals lead to emergence of different treatment regimens for different disease categories (e.g., antibiotic use – yes or no) that are not only designed to reverse pocket depth and improve gain of attachment but also to delay time to relapse? Will this expanded approach limit the potential for local dental infections to exacerbate systemic issues at distant sites? The goal of this paper has been to raise questions such as these that expand the horizons of dental research, diagnosis and treatment so that clinicians and researchers can enthusiastically study how dental infections can be assessed and controlled better for the benefit of our patients.

Table 2.
Causation of disease by microbial consortia assessed by Bradford Hill criteria.
| Hill criteria | Example | Feasibility Yes/No? |
Impact of study and reference [ ] |
| 1.Temporal relationship | Exposure to agent precedes outcome | Yes | Longitudinal; healthy controls; age approp Aa; [41,158] Longitudinal; health controls age approp Aa + consort;[47,159] |
| 2.Strength of Association | Size of association determined statistically | Yes | Show stats Aa ; [41] Show stats Aa + consort; [47,159] |
| 3.Dose-Response | ^exposure> ^response | Yes | Measure consort vs Aa alone; [47,159] |
| 4.Consistency | Experiments reproduced |
Yes | Show consort X-sect; [160,161] Show consort longitude;[47,159] |
| 5.Plausibility | Assoc agree with pathobiological explanations |
Yes | Cdt has impact; [162] Ltx has impact; [99] Consortia passed from mother with disease to Child: local debridement improves inflammation, but consort remains; [163]. Consort metabolomics; [98] |
| 6.Experimental evidence |
Disease altered By intervention |
Yes | Tetracycline admin reduces disease; [164] Tetracycline eliminates Aa and reduces disease; [165] Amox/Metra reduces disease, no antibiotic no improve; [166] |
| 7.Alternative explanation |
Rule out other explanations | ? Open ? Open |
[41,47,159] |
| 8. Specificity | Cause produces effect | Yes | Flp and no disease; [167] Ltx and more bone loss; [168] Pga B is modified and disease is reduced; [169] |
| 9.Coherence | Theory consistent with Existing knowledge |
Yes | Ltx and infections; [170] Cdt and infections; [171] Metabolomics and consortia; [98] |
References
- Papapanou, P.N.; Sanz, M.; Buduneli, N.; Dietrich, T.; Feres, M.; Fine, D.H.; Flemmig, T.F.; Garcia, R.; Giannobile, W.V.; Graziani, F.; et al. Periodontitis: Consensus report of workgroup 2 of the 2017 World Workshop on the Classification of Periodontal and Peri-Implant Diseases and Conditions. Journal of Periodontology 2018, 89 (Suppl. 1), S173–S182. [Google Scholar] [CrossRef] [PubMed]
- Armitage, G.C. Periodontal diseases: diagnosis. Ann Periodontol 1996, 1, 37–215. [Google Scholar] [CrossRef] [PubMed]
- Caton, J.G.; Armitage, G.; Berglundh, T.; Chapple, I.L.C.; Jepsen, S.; Kornman, K.S.; Mealey, B.L.; Papapanou, P.N.; Sanz, M.; Tonetti, M.S. A new classification scheme for periodontal and peri-implant diseases and conditions - Introduction and key changes from the 1999 classification. J Clin Periodontol 2018, 45 Suppl 20, S1–S8. [Google Scholar] [CrossRef]
- Greene, F.L. Cancer staging in outcomes assessment. J Surg Oncol 2014, 110, 616–620. [Google Scholar] [CrossRef] [PubMed]
- Park, B.K.; Schneider, J.; Suh, Y.J. Survival analysis for patients with metachronous contralateral breast cancer: Insights from a retrospective study. Oncol Lett 2024, 28, 390. [Google Scholar] [CrossRef] [PubMed]
- Baer, P.N. The case for periodontosis as a clinical entity. Journal of Periodontology 1971, 42, 516–520. [Google Scholar] [CrossRef] [PubMed]
- Marazita, M.L.; Burmeister, J.A.; Gunsolley, J.C.; Koertge, T.E.; Lake, K.; Schenkein, H.A. Evidence for autosomal dominant inheritance and race-specific heterogeneity in early-onset periodontitis. Journal of Periodontology 1994, 65, 623–630. [Google Scholar] [CrossRef]
- Loe, H.; Brown, L.J. Early onset periodontitis in the United States of America. Journal of Periodontology 1991, 62, 608–616. [Google Scholar] [CrossRef]
- Fine, D.H.; Kaplan, J.B.; Kachlany, S.C.; Schreiner, H.C. How we got attached to Actinobacillus actinomycetemcomitans: A model for infectious diseases. Periodontol 2000 2006, 42, 114–157. [Google Scholar] [CrossRef]
- Fine, D.H.; Cohen, D.W.; Bimstein, E.; Bruckmann, C. A ninety-year history of periodontosis: the legacy of Professor Bernhard Gottlieb. Journal of Periodontology 2015, 86, 1–6. [Google Scholar] [CrossRef]
- Miguel, M.V.M.; Shaddox, L.M. Grade C molar-incisor pattern periodontitis in young adults: What have we learned so far? Pathogens 2024, accepted in press, 1–21. [Google Scholar] [CrossRef]
- Zambon, J.J. Authors’ response. J Am Dent Assoc 2020, 151, 160–161. [Google Scholar] [CrossRef] [PubMed]
- de Carvalho, F.M.; Tinoco, E.M.; Govil, M.; Marazita, M.L.; Vieira, A.R. Aggressive periodontitis is likely influenced by a few small effect genes. J Clin Periodontol 2009, 36, 468–473. [Google Scholar] [CrossRef] [PubMed]
- Fine, D.H.; Armitage, G.C.; Genco, R.J.; Griffen, A.L.; Diehl, S.R. Unique etiologic, demographic, and pathologic characteristics of localized aggressive periodontitis support classification as a distinct subcategory of periodontitis. J Am Dent Assoc 2019, 150, 922–931. [Google Scholar] [CrossRef] [PubMed]
- Pallos, D.; Acevedo, A.C.; Mestrinho, H.D.; Cordeiro, I.; Hart, T.C. Novel cathepsin C mutation in a Brazilian family with Papillon-Lefevre syndrome: case report and mutation update. J Dent Child (Chic) 2010, 77, 36–41. [Google Scholar] [PubMed]
- Diehl, S.R.; Wu, T.; Michalowicz, B.S.; Brooks, C.N.; Califano, J.V.; Burmeister, J.A.; Schenkein, H.A. Quantitative measures of aggressive periodontitis show substantial heritability and consistency with traditional diagnoses. Journal of Periodontology 2005, 76, 279–288. [Google Scholar] [CrossRef] [PubMed]
- Yoo, H.W. Development of orphan drugs for rare diseases. Clin Exp Pediatr 2024, 67, 315–327. [Google Scholar] [CrossRef] [PubMed]
- Wade, D.N.; Kerns, D.G. Acute necrotizing ulcerative gingivitis-periodontitis: a literature review. Mil Med 1998, 163, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Albandar, J.M. Disparities and social determinants of periodontal diseases. Periodontol 2000 2024. [Google Scholar] [CrossRef]
- Botelho, J.; Machado, V.; Proenca, L.; Mendes, J.J. The 2018 periodontitis case definition improves accuracy performance of full-mouth partial diagnostic protocols. Sci Rep 2020, 10, 7093. [Google Scholar] [CrossRef]
- Fredman, G.; Oh, S.F.; Ayilavarapu, S.; Hasturk, H.; Serhan, C.N.; Van Dyke, T.E. Impaired phagocytosis in localized aggressive periodontitis: rescue by Resolvin E1. PLoS One 2011, 6, e24422. [Google Scholar] [CrossRef] [PubMed]
- Fine, D.H.; Patil, A.G.; Loos, B.G. Classification and diagnosis of aggressive periodontitis. Journal of Periodontology 2018, 89 Suppl 1, S103–S119. [Google Scholar] [CrossRef]
- Endersby, J. Lumpers and splitters: Darwin, Hooker, and the search for order. Science 2009, 326, 1496–1499. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S. The Emperor of All Maladies: A Biography of Cancer; Scribner: New York, USA, 2010. [Google Scholar]
- Doll, R. The Pierre Denoix Memorial Lecture: nature and nurture in the control of cancer. Eur J Cancer 1999, 35, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Snyderman, R.; Phillips, J.K.; Mergenhagen, S.E. Biological activity of complement in vivo. Role of C5 in the accumulation of polymorphonuclear leukocytes in inflammatory exudates. J Exp Med 1971, 134, 1131–1143. [Google Scholar] [CrossRef] [PubMed]
- Snyderman, R. Personalized health care: from theory to practice. Biotechnol J 2012, 7, 973–979. [Google Scholar] [CrossRef] [PubMed]
- American Academy of Pediatric Dentistry Council on Clinical, A. Policy on use of a caries-risk assessment tool (CAT) for infants, children, and adolescent. Pediatr Dent 2005, 27, 25–27. [Google Scholar]
- Loesche, W.J. Role of Streptococcus mutans in human dental decay. Microbiol Rev 1986, 50, 353–380. [Google Scholar] [CrossRef]
- Marsh, P.D. Are dental diseases examples of ecological catastrophes? Microbiology (Reading) 2003, 149, 279–294. [Google Scholar] [CrossRef]
- Folayan, M.N.O.; Amalia, R.; Kemoli, A.; Sun, I.G.; Duangthip, D.; Abodunrin, O.; Virtanen, J.I.; Masumo, R.M.; Vukovic, A.; Al-Batayneh, O.B.; et al. Can the sustainable development goal 9 support an untreated early childhood caries elimination agenda? BMC Oral Health 2024, 24, 776. [Google Scholar] [CrossRef]
- Sheykholeslam, Z.; Buonocore, M.G. Bonding of resins to phosphoric acid-etched enamel surfaces of permanent and deciduous teeth. J Dent Res 1972, 51, 1572–1576. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Chen, A.Y.; Geissler, K.H.; Dick, A.W.; Kranz, A.M. Clinician characteristics associated with fluoride varnish applications during well-child visits. Am J Manag Care 2024, 30, e203–e209. [Google Scholar] [CrossRef] [PubMed]
- Lumsden, C.L.; Edelstein, B.L.; Basch, C.E.; Wolf, R.L.; Koch, P.A.; McKeague, I.; Leu, C.S.; Andrews, H. Protocol for a family-centered behavioral intervention to reduce early childhood caries: the MySmileBuddy program efficacy trial. BMC Oral Health 2021, 21, 246. [Google Scholar] [CrossRef] [PubMed]
- Lienhart, G.; Elsa, M.; Farge, P.; Schott, A.M.; Thivichon-Prince, B.; Chaneliere, M. Factors perceived by health professionals to be barriers or facilitators to caries prevention in children: a systematic review. BMC Oral Health 2023, 23, 767. [Google Scholar] [CrossRef] [PubMed]
- Armitage, G.C. A brief history of periodontics in the United States of America: Pioneers and thought-leaders of the past, and current challenges. Periodontol 2000 2020, 82, 12–25. [Google Scholar] [CrossRef] [PubMed]
- Diamond, I.R.; Grant, R.C.; Feldman, B.M.; Pencharz, P.B.; Ling, S.C.; Moore, A.M.; Wales, P.W. Defining consensus: a systematic review recommends methodologic criteria for reporting of Delphi studies. J Clin Epidemiol 2014, 67, 401–409. [Google Scholar] [CrossRef] [PubMed]
- Hutchinson, P.J.; Kolias, A.G.; Tajsic, T.; Adeleye, A.; Aklilu, A.T.; Apriawan, T.; Bajamal, A.H.; Barthelemy, E.J.; Devi, B.I.; Bhat, D.; et al. Consensus statement from the International Consensus Meeting on the Role of Decompressive Craniectomy in the Management of Traumatic Brain Injury : Consensus statement. Acta Neurochir (Wien) 2019, 161, 1261–1274. [Google Scholar] [CrossRef]
- Pelosi, L.; Aranyi, Z.; Beekman, R.; Bland, J.; Coraci, D.; Hobson-Webb, L.D.; Padua, L.; Podnar, S.; Simon, N.; van Alfen, N.; et al. Expert consensus on the combined investigation of ulnar neuropathy at the elbow using electrodiagnostic tests and nerve ultrasound. Clin Neurophysiol 2021, 132, 2274–2281. [Google Scholar] [CrossRef] [PubMed]
- Fine, D.H.; Markowitz, K.; Furgang, D.; Fairlie, K.; Ferrandiz, J.; Nasri, C.; McKiernan, M.; Gunsolley, J. Aggregatibacter actinomycetemcomitans and its relationship to initiation of localized aggressive periodontitis: longitudinal cohort study of initially healthy adolescents. J Clin Microbiol 2007, 45, 3859–3869. [Google Scholar] [CrossRef]
- Haubek, D.; Ennibi, O.K.; Poulsen, K.; Vaeth, M.; Poulsen, S.; Kilian, M. Risk of aggressive periodontitis in adolescent carriers of the JP2 clone of Aggregatibacter (Actinobacillus) actinomycetemcomitans in Morocco: a prospective longitudinal cohort study. Lancet 2008, 371, 237–242. [Google Scholar] [CrossRef]
- Fine, D.H.; Armitage, G.C.; Griffen, A.L.; Diehl, S.R. Authors’ response. J Am Dent Assoc 2020, 151, 160. [Google Scholar] [CrossRef] [PubMed]
- Clyne, B.; Sharp, M.K.; M, O.N.; Pollock, D.; Lynch, R.; Amog, K.; Ryan, M.; Smith, S.M.; Mahtani, K.; Booth, A.; et al. An international modified Delphi process supported updating the web-based “right review” tool. J Clin Epidemiol 2024, 170, 111333. [Google Scholar] [CrossRef] [PubMed]
- Albandar, J.M. Aggressive periodontitis: case definition and diagnostic criteria. Periodontol 2000 2014, 65, 13–26. [Google Scholar] [CrossRef] [PubMed]
- Cekici, A.; Kantarci, A.; Hasturk, H.; Van Dyke, T.E. Inflammatory and immune pathways in the pathogenesis of periodontal disease. Periodontol 2000 2014, 64, 57–80. [Google Scholar] [CrossRef] [PubMed]
- Billings, M.; Holtfreter, B.; Papapanou, P.N.; Mitnik, G.L.; Kocher, T.; Dye, B.A. Age-dependent distribution of periodontitis in two countries: Findings from NHANES 2009 to 2014 and SHIP-TREND 2008 to 2012. Journal of Periodontology 2018, 89 (Suppl. 1), S140–S158. [Google Scholar] [CrossRef] [PubMed]
- Fine, D.H.; Markowitz, K.; Fairlie, K.; Tischio-Bereski, D.; Ferrendiz, J.; Furgang, D.; Paster, B.J.; Dewhirst, F.E. A consortium of Aggregatibacter actinomycetemcomitans, Streptococcus parasanguinis, and Filifactor alocis is present in sites prior to bone loss in a longitudinal study of localized aggressive periodontitis. J Clin Microbiol 2013, 51, 2850–2861. [Google Scholar] [CrossRef] [PubMed]
- Needleman, I.; Garcia, R.; Gkranias, N.; Kirkwood, K.L.; Kocher, T.; Iorio, A.D.; Moreno, F.; Petrie, A. Mean annual attachment, bone level, and tooth loss: A systematic review. Journal of Periodontology 2018, 89 (Suppl. 1), S120–S139. [Google Scholar] [CrossRef] [PubMed]
- Leow, N.M.; Moreno, F.; Marletta, D.; Hussain, S.B.; Buti, J.; Almond, N.; Needleman, I. Recurrence and progression of periodontitis and methods of management in long-term care: A systematic review and meta-analysis. J Clin Periodontol 2022, 49 (Suppl. 24), 291–313. [Google Scholar] [CrossRef] [PubMed]
- Bik, E.M.; Long, C.D.; Armitage, G.C.; Loomer, P.; Emerson, J.; Mongodin, E.F.; Nelson, K.E.; Gill, S.R.; Fraser-Liggett, C.M.; Relman, D.A. Bacterial diversity in the oral cavity of 10 healthy individuals. The ISME journal 2010, 4, 962–974. [Google Scholar] [CrossRef]
- Proctor, D.M.; Shelef, K.M.; Gonzalez, A.; Davis, C.L.; Dethlefsen, L.; Burns, A.R.; Loomer, P.M.; Armitage, G.C.; Ryder, M.I.; Millman, M.E.; et al. Microbial biogeography and ecology of the mouth and implications for periodontal diseases. Periodontol 2000 2019, 82, 26–41. [Google Scholar] [CrossRef]
- Rubinstein, M.R.; Baik, J.E.; Lagana, S.M.; Han, R.P.; Raab, W.J.; Sahoo, D.; Dalerba, P.; Wang, T.C.; Han, Y.W. Fusobacterium nucleatum promotes colorectal cancer by inducing Wnt/beta-catenin modulator Annexin A1. EMBO Rep 2019, 20. [Google Scholar] [CrossRef]
- Han, Y.W.; Redline, R.W.; Li, M.; Yin, L.; Hill, G.B.; McCormick, T.S. Fusobacterium nucleatum Induces Premature and Term Stillbirths in Pregnant Mice: Implication of Oral Bacteria in Preterm Birth. Infect Immun 2004, 72, 2272–2279. [Google Scholar] [CrossRef]
- Miller, W.D. The Micro-organisms of the Human Mouth: The Local and General Diseases which are Caused by Them; Classics of Dentistry Library: 1890.
- Kritchevsky, B.; Seguin, P. The Pathogenesis and Treatment of Pyorrhea Alveolaris. The Dental Cosmos: a monthly record of dental science 1918, 60, 781–784. [Google Scholar]
- Beckwith, T.D.; Williams, A.; Rose, E.T. The role of bacteria in pyorrhea. Medical journal and record 1929, 129, 333–336. [Google Scholar]
- Woodward, C.; Fisher, M.A. Drug treatment of common STDs: Part II. Vaginal infections, pelvic inflammatory disease and genital warts. Am Fam Physician 1999, 60, 1716–1722. [Google Scholar] [PubMed]
- Malhotra, M.; Sood, S.; Mukherjee, A.; Muralidhar, S.; Bala, M. Genital Chlamydia trachomatis: an update. Indian J Med Res 2013, 138, 303–316. [Google Scholar]
- Radu, O.; Pantanowitz, L. Kaposi sarcoma. Arch Pathol Lab Med 2013, 137, 289–294. [Google Scholar] [CrossRef]
- Casadevall, A.; Pirofski, L.A. Host-pathogen interactions: redefining the basic concepts of virulence and pathogenicity. Infect Immun 1999, 67, 3703–3713. [Google Scholar] [CrossRef] [PubMed]
- Casadevall, A.; Pirofski, L.A. Microbiology: Ditch the term pathogen. Nature 2014, 516, 165–166. [Google Scholar] [CrossRef]
- Casadevall, A.; Pirofski, L.A. What is a pathogen? Ann Med 2002, 34, 2–4. [Google Scholar] [CrossRef]
- Pirofski, L.; Casadevall, A. The Damage-Response Framework as a Tool for the Physician-Scientist to Understand the Pathogenesis of Infectious Diseases. J Infect Dis 2018, 218, S7–S11. [Google Scholar] [CrossRef] [PubMed]
- Pirofski, L.A.; Casadevall, A. The meaning of microbial exposure, infection, colonisation, and disease in clinical practice. Lancet Infect Dis 2002, 2, 628–635. [Google Scholar] [CrossRef] [PubMed]
- Armitage, G.C. Learned and unlearned concepts in periodontal diagnostics: a 50-year perspective. Periodontol 2000 2013, 62, 20–36. [Google Scholar] [CrossRef]
- Newman, M.G.; Socransky, S.S.; Savitt, E.D.; Propas, D.A.; Crawford, A. Studies of the microbiology of periodontosis. Journal of Periodontology 1976, 47, 373–379. [Google Scholar] [CrossRef]
- Slots, J. The predominant cultivable organisms in juvenile periodontitis. Scand J Dent Res 1976, 84, 1–10. [Google Scholar] [CrossRef]
- Loesche, W.J. Chemotherapy of dental plaque infections. Oral Sci Rev 1976, 9, 65–107. [Google Scholar]
- Loesche, W.J. Clinical and microbiological aspects of chemotherapeutic agents used according to the specific plaque hypothesis. J Dent Res 1979, 58, 2404–2412. [Google Scholar] [CrossRef]
- Klinger, R. Untersuchungen uber menschliche aktinomycose. Zentralbl. Bakteriol. (Orig) 1912, 62, 191–200. [Google Scholar]
- Marsh, P.D.; Zaura, E. Dental biofilm: ecological interactions in health and disease. J Clin Periodontol 2017, 44 Suppl 18, S12–S22. [Google Scholar] [CrossRef]
- Moore, W.E.; Moore, L.H.; Ranney, R.R.; Smibert, R.M.; Burmeister, J.A.; Schenkein, H.A. The microflora of periodontal sites showing active destructive progression. J Clin Periodontol 1991, 18, 729–739. [Google Scholar] [CrossRef]
- Socransky, S.S.; Haffajee, A.D.; Cugini, M.A.; Smith, C.; Kent, R.L., Jr. Microbial complexes in subgingival plaque. J Clin Periodontol 1998, 25, 134–144. [Google Scholar] [CrossRef] [PubMed]
- Socransky, S.S.; Haffajee, A.D. Periodontal microbial ecology. Periodontol 2000 2005, 38, 135–187. [Google Scholar] [CrossRef] [PubMed]
- Marsh, P.D. In Sickness and in Health-What Does the Oral Microbiome Mean to Us? An Ecological Perspective. Adv Dent Res 2018, 29, 60–65. [Google Scholar] [CrossRef] [PubMed]
- Gibbons, R.J.; Macdonald, J.B. Hemin and vitamin K compounds as required factors for the cultivation of certain strains of Bacteroides melaninogenicus. J Bacteriol 1960, 80, 164–170. [Google Scholar] [CrossRef] [PubMed]
- Komiyama, K.; Khandelwal, R.L.; Heinrich, S.E. Glycogen synthetic and degradative activities by Actinomyces viscosus and Actinomyces naeslundii of root surface caries and noncaries sites. Caries Res 1988, 22, 217–225. [Google Scholar] [CrossRef]
- Moore, W.E.C.; Moore, L.V.H. The bacteria of periodontal disease. Periodontol 2000 1994, 5, 66–77. [Google Scholar] [CrossRef] [PubMed]
- Schachtele, C.F.; Loken, A.E.; Schmitt, M.K. Use of specifically labeled sucrose for comparison of extracellular glucan and fructan metabolism by oral streptococci. Infect Immun 1972, 5, 263–266. [Google Scholar] [CrossRef]
- Kachlany, S.C.; Fine, D.H.; Figurski, D.H. Secretion of RTX leukotoxin by Actinobacillus actinomycetemcomitans. Infect Immun 2000, 68, 6094–6100. [Google Scholar] [CrossRef] [PubMed]
- Pavloff, N.; Potempa, J.; Pike, R.N.; Prochazka, V.; Kiefer, M.C.; Travis, J.; Barr, P.J. Molecular cloning and structural characterization of the Arg-gingipain proteinase of Porphyromonas gingivalis. Biosynthesis as a proteinase-adhesin polyprotein. J Biol Chem 1995, 270, 1007–1010. [Google Scholar] [CrossRef]
- Chu, L.; Bramanti, T.E.; Ebersole, J.L.; Holt, S.C. Hemolytic activity in the periodontopathogen Porphyromonas gingivalis: kinetics of enzyme release and localization. Infect Immun 1991, 59, 1932–1940. [Google Scholar] [CrossRef]
- Gibbons, R.J.; Macdonald, J.B. Degradation of collagenous substrates by Bacteroides melaninogenicus. J Bacteriol 1961, 81, 614–621. [Google Scholar] [CrossRef] [PubMed]
- Marsh, P.D. Microbial ecology of dental plaque and its significance in health and disease. Adv Dent Res 1994, 8, 263–271. [Google Scholar] [CrossRef] [PubMed]
- Ivanyi, L.; Challacombe, S.J.; Lehner, T. The specificity of serum factors in lymphocyte transformation in periodontal disease. Clin Exp Immunol 1973, 14, 491–500. [Google Scholar] [PubMed]
- Ebersole, J.L.; Dawson, D., 3rd; Emecen-Huja, P.; Nagarajan, R.; Howard, K.; Grady, M.E.; Thompson, K.; Peyyala, R.; Al-Attar, A.; Lethbridge, K.; et al. The periodontal war: microbes and immunity. Periodontol 2000 2017, 75, 52–115. [Google Scholar] [CrossRef] [PubMed]
- Fine, D.H. Incorporating new technologies in periodontal diagnosis into training programs and patient care: a critical assessment and a plan for the future. Journal of Periodontology 1992, 63, 383–393. [Google Scholar] [CrossRef] [PubMed]
- Listgarten, M.A. Periodontal probing: what does it mean? J Clin Periodontol 1980, 7, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Cugini, C.; Ramasubbu, N.; Tsiagbe, V.K.; Fine, D.H. Dysbiosis From a Microbial and Host Perspective Relative to Oral Health and Disease. Front Microbiol 2021, 12, 617485. [Google Scholar] [CrossRef] [PubMed]
- Casadevall, A.; Pirofski, L.A. What is a host? Incorporating the microbiota into the damage-response framework. Infect Immun 2015, 83, 2–7. [Google Scholar] [CrossRef]
- Loe, H.; Silness, J. Periodontal disease in pregancy. Prevalence and severity. Acta Odontol Scand 1963, 21, 533–551. [Google Scholar] [CrossRef]
- Page, R.C.; Schroeder, H.E. Pathogenesis of inflammatory periodontal disease. A summary of current work. Lab Invest 1976, 34, 235–249. [Google Scholar]
- Taubman, M.A.; Valverde, P.; Han, X.; Kawai, T. Immune response: the key to bone resorption in periodontal disease. Journal of Periodontology 2005, 76, 2033–2041. [Google Scholar] [CrossRef] [PubMed]
- Genco, R.J.; Sanz, M. Clinical and public health implications of periodontal and systemic diseases: An overview. Periodontol 2000 2020, 83, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Taichman, N.S.; Dean, R.T.; Sanderson, C.J. Biochemical and morphological characterization of the killing of human monocytes by a leukotoxin derived from Actinobacillus actinomycetemcomitans. Infect Immun 1980, 28, 258–268. [Google Scholar] [CrossRef] [PubMed]
- Taichman, N.S.; Wilton, J.M. Leukotoxicity of an extract from Actinobacillus actinomycetemcomitans for human gingival polymorphonuclear leukocytes. Inflammation 1981, 5, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Bamashmous, S.; Kotsakis, G.A.; Kerns, K.A.; Leroux, B.G.; Zenobia, C.; Chen, D.; Trivedi, H.M.; McLean, J.S.; Darveau, R.P. Human variation in gingival inflammation. Proc Natl Acad Sci U S A 2021, 118. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Wright, C.J.; Dingming, H.; Uriarte, S.M.; Lamont, R.J. Oral community interactions of Filifactor alocis in vitro. PLoS One 2013, 8, e76271. [Google Scholar] [CrossRef] [PubMed]
- Johansson, A.; Claesson, R.; Aberg, C.H.; Haubek, D.; Lindholm, M.; Jasim, S.; Oscarsson, J. Genetic Profiling of Aggragatibacter actinomycetemcomitans Serotype B Isolated from Periodontitis Patients Living in Sweden. Pathogens 2019, 8, 153–167. [Google Scholar] [CrossRef] [PubMed]
- Haffajee, A.D.; Patel, M.; Socransky, S.S. Microbiological changes associated with four different periodontal therapies for the treatment of chronic periodontitis. Oral Microbiol Immunol 2008, 23, 148–157. [Google Scholar] [CrossRef] [PubMed]
- Socransky, S.S.; Haffajee, A.D. Microbial mechanisms in the pathogenesis of destructive periodontal diseases: a critical assessment. J Periodontal Res 1991, 26, 195–212. [Google Scholar] [CrossRef]
- Usman, M.; Hameed, Y.; Ahmad, M. Does human papillomavirus cause human colorectal cancer? Applying Bradford Hill criteria postulates. Ecancermedicalscience 2020, 14, 1107. [Google Scholar] [CrossRef]
- Blaser, M.J. Missing microbes : how the overuse of antibiotics is fueling our modern plagues, First edition. ed.; Henry Holt and Company: New York, 2014. [Google Scholar]
- Del Romero, J.; Moreno Guillen, S.; Rodriguez-Artalejo, F.J.; Ruiz-Galiana, J.; Canton, R.; De Lucas Ramos, P.; Garcia-Botella, A.; Garcia-Lledo, A.; Hernandez-Sampelayo, T.; Gomez-Pavon, J.; et al. Sexually transmitted infections in Spain: Current status. Rev Esp Quimioter 2023, 36, 444–465. [Google Scholar] [CrossRef] [PubMed]
- Okell, C.C.; Elliott, S.D. Bacteriaaemia and oral sepsis with special reference to the aetiology of subacute endocarditis. The Lancet 1935, 869–872. [Google Scholar] [CrossRef]
- Silver, J.G.; Martin, A.W.; McBride, B.C. Experimental transient bacteraemias in human subjects with varying degrees of plaque accumulation and gingival inflammation. J Clin Periodontol 1977, 4, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Proctor, D.M.; Shelef, K.M.; Gonzalez, A.; Davis, C.L.; Dethlefsen, L.; Burns, A.R.; Loomer, P.M.; Armitage, G.C.; Ryder, M.I.; Millman, M.E.; et al. Microbial biogeography and ecology of the mouth and implications for periodontal diseases. Periodontol 2000 2020, 82, 26–41. [Google Scholar] [CrossRef] [PubMed]
- Wells, P.M.; Sprockett, D.D.; Bowyer, R.C.E.; Kurushima, Y.; Relman, D.A.; Williams, F.M.K.; Steves, C.J. Influential factors of saliva microbiota composition. Sci Rep 2022, 12, 18894. [Google Scholar] [CrossRef] [PubMed]
- Fine, D.H.; Schreiner, H. Oral microbial interactions from an ecological perspective: a narrative review. Front Oral Health 2023, 4, 1229118. [Google Scholar] [CrossRef] [PubMed]
- Taichman, N.S.; Tsai, C.C.; Baehni, P.C.; Stoller, N.; McArthur, W.P. Interaction of inflammatory cells and oral microorganisms. IV. In vitro release of lysosomal constituents from polymorphonuclear leukocytes exposed to supragingival and subgingival bacterial plaque. Infect Immun 1977, 16, 1013–1023. [Google Scholar] [CrossRef] [PubMed]
- Van Dyke, T.E.; Horoszewicz, H.U.; Genco, R.J. The polymorphonuclear leukocyte (PMNL) locomotor defect in juvenile periodontitis. Study of random migration, chemokinesis and chemotaxis. Journal of Periodontology 1982, 53, 682–687. [Google Scholar] [CrossRef]
- Fives-Taylor, P.M.; Meyer, D.H.; Mintz, K.P.; Brissette, C. Virulence factors of Actinobacillus actinomycetemcomitans. Periodontol 2000 1999, 20, 136–167. [Google Scholar] [CrossRef]
- Zambon, J.J. Periodontal diseases: microbial factors. Ann Periodontol 1996, 1, 879–925. [Google Scholar] [CrossRef]
- Aruni, W.; Chioma, O.; Fletcher, H.M. Filifactor alocis: The Newly Discovered Kid on the Block with Special Talents. J Dent Res 2014, 93, 725–732. [Google Scholar] [CrossRef] [PubMed]
- Fine, D.H.; Patil, A.G.; Velusamy, S.K. Aggregatibacter actinomycetemcomitans (Aa) Under the Radar: Myths and Misunderstandings of Aa and Its Role in Aggressive Periodontitis. Front Immunol 2019, 10, 728. [Google Scholar] [CrossRef] [PubMed]
- Offenbacher, S.; Katz, V.; Fertik, G.; Collins, J.; Boyd, D.; Maynor, G.; McKaig, R.; Beck, J. Periodontal infection as a possible risk factor for preterm low birth weight. Journal of Periodontology 1996, 67, 1103–1113. [Google Scholar] [CrossRef] [PubMed]
- Fine, D.H.; Furgang, D.; McKiernan, M.; Tereski-Bischio, D.; Ricci-Nittel, D.; Zhang, P.; Araujo, M.W. An investigation of the effect of an essential oil mouthrinse on induced bacteraemia: a pilot study. J Clin Periodontol 2010, 37, 840–847. [Google Scholar] [CrossRef] [PubMed]
- Kinane, D.F.; Riggio, M.P.; Walker, K.F.; MacKenzie, D.; Shearer, B. Bacteraemia following periodontal procedures. J Clin Periodontol 2005, 32, 708–713. [Google Scholar] [CrossRef] [PubMed]
- Shanson, D. New British and American guidelines for the antibiotic prophylaxis of infective endocarditis: do the changes make sense? A critical review. Curr Opin Infect Dis 2008, 21, 191–199. [Google Scholar] [CrossRef] [PubMed]
- Costello, E.K.; Stagaman, K.; Dethlefsen, L.; Bohannan, B.J.; Relman, D.A. The application of ecological theory toward an understanding of the human microbiome. Science 2012, 336, 1255–1262. [Google Scholar] [CrossRef] [PubMed]
- Tabak, L.A.; Levine, M.J.; Mandel, I.D.; Ellison, S.A. Role of salivary mucins in the protection of the oral cavity. J Oral Pathol 1982, 11, 1–17. [Google Scholar] [CrossRef]
- Mandel, I.D. The role of saliva in maintaining oral homeostasis. J Am Dent Assoc 1989, 119, 298–304. [Google Scholar] [CrossRef]
- Rosebury, T.; Linton, R.W.; Buchbinder, L. A Comparative Study of Dental Aciduric Organisms and Lactobacillus Acidophilus. J Bacteriol 1929, 18, 395–412. [Google Scholar] [CrossRef]
- Hemmens, E.S.; Harrison, R.W. Studies on the Anaerobic Bacterial Flora of Suppurative Periodontitis. The Journal of Infectious Diseases 1942, 70, 131–146. [Google Scholar] [CrossRef]
- Rosebury, T.; Clark, A.R.; Macdonald, J.B.; O’Connell, D.C. Studies of fusospirochetal infection. III. Further studies of a guinea pig passage strain of fusospirochetal infection, including the infectivity of sterile exudate filtrates, of mixed cultures through ten transfers, and of recombined pure cultures. J Infect Dis 1950, 87, 234–248. [Google Scholar] [CrossRef]
- Keyes, P.H.; Fitzgerald, R.J. Dental caries in the Syrian hamster. IX. Arch Oral Biol 1962, 7, 267–277. [Google Scholar] [CrossRef]
- Loe, H.; Theilade, E.; Jensen, S.B. Experimental Gingivitis in Man. Journal of Periodontology 1965, 36, 177–187. [Google Scholar] [CrossRef] [PubMed]
- Curtis, M.A.; Diaz, P.I.; Van Dyke, T.E. The role of the microbiota in periodontal disease. Periodontol 2000 2020, 83, 14–25. [Google Scholar] [CrossRef] [PubMed]
- Lang, N.P.; Kiel, R.A.; Anderhalden, K. Clinical and microbiological effects of subgingival restorations with overhanging or clinically perfect margins. J Clin Periodontol 1983, 10, 563–578. [Google Scholar] [CrossRef] [PubMed]
- Theilade, E.; Wright, W.H.; Jensen, S.B.; Loe, H. Experimental gingivitis in man. II. A longitudinal clinical and bacteriological investigation. J Periodontal Res 1966, 1, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Kolenbrander, P.E.; London, J. Adhere today, here tomorrow: oral bacterial adherence. J Bacteriol 1993, 175, 3247–3252. [Google Scholar] [CrossRef]
- Kolenbrander, P.E.; Palmer, R.J., Jr.; Periasamy, S.; Jakubovics, N.S. Oral multispecies biofilm development and the key role of cell-cell distance. Nat Rev Microbiol 2010, 8, 471–480. [Google Scholar] [CrossRef]
- Socransky, S.S.; Manganiello, S.D. The oral microbiota of man from birth to senility. Journal of Periodontology 1971, 42, 485–496. [Google Scholar] [CrossRef]
- Lowe, A.M.; Yansouni, C.P.; Behr, M.A. Causality and gastrointestinal infections: Koch, Hill, and Crohn’s. Lancet Infect Dis 2008, 8, 720–726. [Google Scholar] [CrossRef] [PubMed]
- Ritz, H.L. Microbial population shifts in developing human dental plaque. Arch Oral Biol 1967, 12, 1561–1568. [Google Scholar] [CrossRef] [PubMed]
- Hujoel, P.P.; White, B.A.; Garcia, R.I.; Listgarten, M.A. The dentogingival epithelial surface area revisited. J Periodontal Res 2001, 36, 48–55. [Google Scholar] [CrossRef]
- Bergenholtz, G. Effect of bacterial products on inflammatory reactions in the dental pulp. Scand J Dent Res 1977, 85, 122–129. [Google Scholar] [PubMed]
- Bik, E.M.; Eckburg, P.B.; Gill, S.R.; Nelson, K.E.; Purdom, E.A.; Francois, F.; Perez-Perez, G.; Blaser, M.J.; Relman, D.A. Molecular analysis of the bacterial microbiota in the human stomach. Proc Natl Acad Sci U S A 2006, 103, 732–737. [Google Scholar] [CrossRef] [PubMed]
- Black, S.; Phillips, D.; Hickey, J.W.; Kennedy-Darling, J.; Venkataraaman, V.G.; Samusik, N.; Goltsev, Y.; Schurch, C.M.; Nolan, G.P. CODEX multiplexed tissue imaging with DNA-conjugated antibodies. Nat Protoc 2021, 16, 3802–3835. [Google Scholar] [CrossRef] [PubMed]
- Mark Welch, J.L.; Dewhirst, F.E.; Borisy, G.G. Biogeographty of the Oral Microbiome: The Site-Specific Hyothesis. Ann Rev Microbiol. 2019, 73, 335–358. [Google Scholar] [CrossRef]
- Lakschevitz, F.S.; Hassanpour, S.; Rubin, A.; Fine, N.; Sun, C.; Glogauer, M. Identification of neutrophil surface marker changes in health and inflammation using high-throughput screening flow cytometry. Exp Cell Res 2016, 342, 200–209. [Google Scholar] [CrossRef]
- Lamont, R.J.; Koo, H.; Hajishengallis, G. The oral microbiota: dynamic communities and host interactions. Nat Rev Microbiol 2018, 16, 745–759. [Google Scholar] [CrossRef]
- Oshrain, H.I.; Salind, A.; Mandel, I.D. A method for collection of subgingival plaque and calculus. Journal of Periodontology 1968, 39, 322–325. [Google Scholar] [CrossRef]
- Fine, D.H.; Greene, L.S. Microscopic evaluation of root surface associations in vivo. J Periodontal Res 1984, 19, 152–167. [Google Scholar] [CrossRef] [PubMed]
- Wecke, J.; Wolf, V.; Fath, S.; Bernimoulin, J.P. The occurrence of treponemes and their spherical bodies on polytetrafluoroethylene membranes. Oral Microbiol Immunol 1995, 10, 278–283. [Google Scholar] [CrossRef]
- Fine, D.H.; Markowitz, K.; Furgang, D.; Velliyagounder, K. Aggregatibacter actinomycetemcomitans as an early colonizer of oral tissues: epithelium as a reservoir? J Clin Microbiol 2010, 48, 4464–4473. [Google Scholar] [CrossRef] [PubMed]
- Hajishengallis, G.; Lamont, R.J.; Koo, H. Oral polymicrobial communities: Assembly, function, and impact on diseases. Cell Host Microbe 2023, 31, 528–538. [Google Scholar] [CrossRef] [PubMed]
- Hajishengallis, G. Interconnection of periodontal disease and comorbidities: Evidence, mechanisms, and implications. Periodontol 2000 2022, 89, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Holtfreter, B.; Kuhr, K.; Borof, K.; Tonetti, M.S.; Sanz, M.; Kornman, K.; Jepsen, S.; Aarabi, G.; Volzke, H.; Kocher, T.; et al. ACES: A new framework for the application of the 2018 periodontal status classification scheme to epidemiological survey data. J Clin Periodontol 2024, 51, 512–521. [Google Scholar] [CrossRef] [PubMed]
- Herzberg, M.C. Platelet-streptococcal interactions in endocarditis. Crit Rev Oral Biol Med 1996, 7, 222–236. [Google Scholar] [CrossRef]
- Peters, J.; Robinson, F.; Dasco, C.; Gentry, L.O. Subacute bacterial endocarditis due to Actinobacillus actinomycetemcomitans. Am J Med Sci 1983, 286, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Haubek, D.; Poulsen, K.; Kilian, M. Microevolution and patterns of dissemination of the JP2 clone of Aggregatibacter (Actinobacillus) actinomycetemcomitans. Infect Immun 2007, 75, 3080–3088. [Google Scholar] [CrossRef]
- Fine, D.H.; Toruner, G.A.; Velliyagounder, K.; Sampathkumar, V.; Godboley, D.; Furgang, D. A lactotransferrin single nucleotide polymorphism demonstrates biological activity that can reduce susceptibility to caries. Infect Immun 2013, 81, 1596–1605. [Google Scholar] [CrossRef]
- Esmaeilyfard, R.; Bonyadifard, H.; Paknahad, M. Dental Caries Detection and Classification in CBCT Images Using Deep Learning. Int Dent J 2024, 74, 328–334. [Google Scholar] [CrossRef] [PubMed]
- Pitts, N.B.; Twetman, S.; Fisher, J.; Marsh, P.D. Understanding dental caries as a non-communicable disease. Br Dent J 2021, 231, 749–753. [Google Scholar] [CrossRef]
- Sioson, P.B.; Furgang, D.; Steinberg, L.M.; Fine, D.H. Proximal caries in juvenile periodontitis patients. Journal of Periodontology 2000, 71, 710–716. [Google Scholar] [CrossRef] [PubMed]
- Zambon, J.J. Actinobacillus actinomycetemcomitans in human periodontal disease. J Clin Periodontol 1985, 12, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Hoglund Aberg, C.; Kwamin, F.; Claesson, R.; Dahlen, G.; Johansson, A.; Haubek, D. Progression of attachment loss is strongly associated with presence of the JP2 genotype of Aggregatibacter actinomycetemcomitans: a prospective cohort study of a young adolescent population. J Clin Periodontol 2014, 41, 232–241. [Google Scholar] [CrossRef] [PubMed]
- Razooqi, Z.; Tjellstrom, I.; Hoglund Aberg, C.; Kwamin, F.; Claesson, R.; Haubek, D.; Johansson, A.; Oscarsson, J. Association of Filifactor alocis and its RTX toxin gene ftxA with periodontal attachment loss, and in synergy with Aggregatibacter actinomycetemcomitans. Front Cell Infect Microbiol 2024, 14, 1376358. [Google Scholar] [CrossRef] [PubMed]
- Shaddox, L.M.; Huang, H.; Lin, T.; Hou, W.; Harrison, P.L.; Aukhil, I.; Walker, C.B.; Klepac-Ceraj, V.; Paster, B.J. Microbiological characterization in children with aggressive periodontitis. J Dent Res 2012, 91, 927–933. [Google Scholar] [CrossRef] [PubMed]
- Koo, S.S.; Fernandes, J.G.; Li, L.; Huang, H.; Aukhil, I.; Harrison, P.; Diaz, P.I.; Shaddox, L.M. Evaluation of microbiome in primary and permanent dentition in grade C periodontitis in young individuals. Journal of Periodontology 2024. [Google Scholar] [CrossRef]
- Shenker, B.J.; Walker, L.P.; Zekavat, A.; Korostoff, J.; Boesze-Battaglia, K. Aggregatibacter actinomycetemcomitans Cytolethal Distending Toxin-Induces Cell Cycle Arrest in a Glycogen Synthase Kinase (GSK)-3-Dependent Manner in Oral Keratinocytes. Int J Mol Sci 2022, 23. [Google Scholar] [CrossRef]
- Monteiro, M.F.; Altabtbaei, K.; Kumar, P.S.; Casati, M.Z.; Ruiz, K.G.S.; Sallum, E.A.; Nociti-Junior, F.H.; Casarin, R.C.V. Parents with periodontitis impact the subgingival colonization of their offspring. Sci Rep 2021, 11, 1357. [Google Scholar] [CrossRef]
- Novak, M.J.; Stamatelakys, C.; Adair, S.M. Resolution of early lesions of juvenile periodontitis with tetracycline therapy alone: long-term observations of 4 cases. Journal of Periodontology 1991, 62, 628–633. [Google Scholar] [CrossRef]
- Mandell, R.L.; Tripodi, L.S.; Savitt, E.; Goodson, J.M.; Socransky, S.S. The effect of treatment on Actinobacillus actinomycetemcomitans in localized juvenile periodontitis. Periodontology 1986, 57, 94–99. [Google Scholar] [CrossRef] [PubMed]
- Miller, K.A.; Branco-de-Almeida, L.S.; Wolf, S.; Hovencamp, N.; Treloar, T.; Harrison, P.; Aukhil, I.; Gong, Y.; Shaddox, L.M. Long-term clinical response to treatment and maintenance of localized aggressive periodontitis: a cohort study. J Clin Periodontol 2017, 44, 158–168. [Google Scholar] [CrossRef]
- Schreiner, H.C.; Sinatra, K.; Kaplan, J.B.; Furgang, D.; Kachlany, S.C.; Planet, P.J.; Perez, B.A.; Figurski, D.H.; Fine, D.H. Tight-adherence genes of Actinobacillus actinomycetemcomitans are required for virulence in a rat model. Proc Natl Acad Sci U S A 2003, 100, 7295–7300. [Google Scholar] [CrossRef] [PubMed]
- Schreiner, H.; Li, Y.; Cline, J.; Tsiagbe, V.K.; Fine, D.H. A comparison of Aggregatibacter actinomycetemcomitans (Aa) virulence traits in a rat model for periodontal disease. PLoS One 2013, 8, e69382. [Google Scholar] [CrossRef] [PubMed]
- Parthiban, C.; Varudharasu, D.; Shanmugam, M.; Gopal, P.; Ragunath, C.; Thomas, L.; Nitz, M.; Ramasubbu, N. Structural and functional analysis of de-N-acetylase PgaB from periodontopathogen Aggregatibacter actinomycetemcomitans. Mol Oral Microbiol 2017, 32, 324–340. [Google Scholar] [CrossRef] [PubMed]
- Ristow, L.C.; Welch, R.A. RTX Toxins Ambush Immunity’s First Cellular Responders. Toxins (Basel) 2019, 11. [Google Scholar] [CrossRef]
- Azzi-Martin, L.; Touffait-Calvez, V.; Everaert, M.; Jia, R.; Sifre, E.; Seeneevassen, L.; Varon, C.; Dubus, P.; Menard, A. Cytolethal Distending Toxin Modulates Cell Differentiation and Elicits Epithelial to Mesenchymal Transition. J Infect Dis 2024, 229, 1688–1701. [Google Scholar] [CrossRef]
Figure 1.
Illustration of the experimental 4 gingivitis model. Panel Upper Left: illustration of pre-experimental gingival health of patient prior to abstaining form oral hygiene. Panel Lower Left: Gingival indices indicate punctate areas of redness around marginal ginigivae especially in upper premolar and molar areas while incisors show minimal inflammation. Panel Upper Right: Plaque disclosure seven days following abstaining from oral hygiene using erythrocin staining. Note minimal levels of plaque around gingival margin of teeth. Panel Lower Right: Plaque disclosure 21 days after abstaining from oral hygiene. Note the increased intensity of erythrocin staining which illustrates increased plaque thickness and how upper anterior teeth show less staining, i.e., less plaque accumulation.
Figure 1.
Illustration of the experimental 4 gingivitis model. Panel Upper Left: illustration of pre-experimental gingival health of patient prior to abstaining form oral hygiene. Panel Lower Left: Gingival indices indicate punctate areas of redness around marginal ginigivae especially in upper premolar and molar areas while incisors show minimal inflammation. Panel Upper Right: Plaque disclosure seven days following abstaining from oral hygiene using erythrocin staining. Note minimal levels of plaque around gingival margin of teeth. Panel Lower Right: Plaque disclosure 21 days after abstaining from oral hygiene. Note the increased intensity of erythrocin staining which illustrates increased plaque thickness and how upper anterior teeth show less staining, i.e., less plaque accumulation.
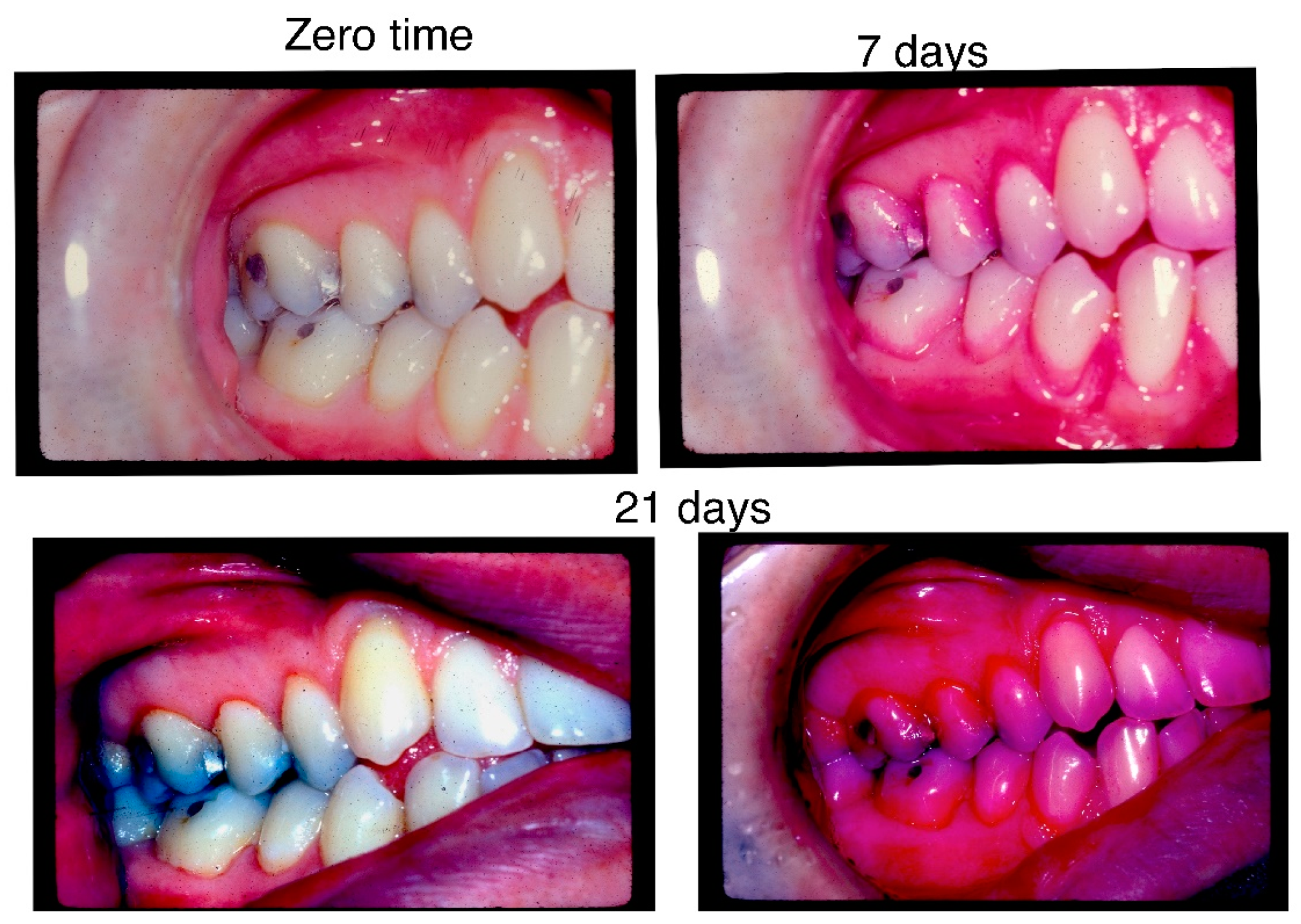
Figure 2.
Diagram of Damage/Response simplified. Solid line represents host response framework where after the period of disease the host response kicks in and disease is resolved and then the host response is tapered. Whereas, the doted line represents the muted host response through the diseased phase and then a spike in the response, representing further damage to the host in the recuperation phase.
Figure 2.
Diagram of Damage/Response simplified. Solid line represents host response framework where after the period of disease the host response kicks in and disease is resolved and then the host response is tapered. Whereas, the doted line represents the muted host response through the diseased phase and then a spike in the response, representing further damage to the host in the recuperation phase.
Figure 3.
Illustration of the Golden and Albino Hamster experiments showing transmission from mother to child. Golden Hamster Dame (caries positive) is on left and Albino hamster dame (caries negative) is on right side. Hamster pups are delivered in a sterile chamber by Cesarian section under sterile conditions and then albino pubs are put in the cage of the Golden Hamster pups are to be suckled by the Albino Dame pubs (bottom right side of illustration) while the albino pups are suckled by the Golden Hamster Dame (bottom left side of the illustration). As shown the Golden pubs have no caries now (bottom right; while the albino pubs have caries (bottom left).
Figure 3.
Illustration of the Golden and Albino Hamster experiments showing transmission from mother to child. Golden Hamster Dame (caries positive) is on left and Albino hamster dame (caries negative) is on right side. Hamster pups are delivered in a sterile chamber by Cesarian section under sterile conditions and then albino pubs are put in the cage of the Golden Hamster pups are to be suckled by the Albino Dame pubs (bottom right side of illustration) while the albino pups are suckled by the Golden Hamster Dame (bottom left side of the illustration). As shown the Golden pubs have no caries now (bottom right; while the albino pubs have caries (bottom left).
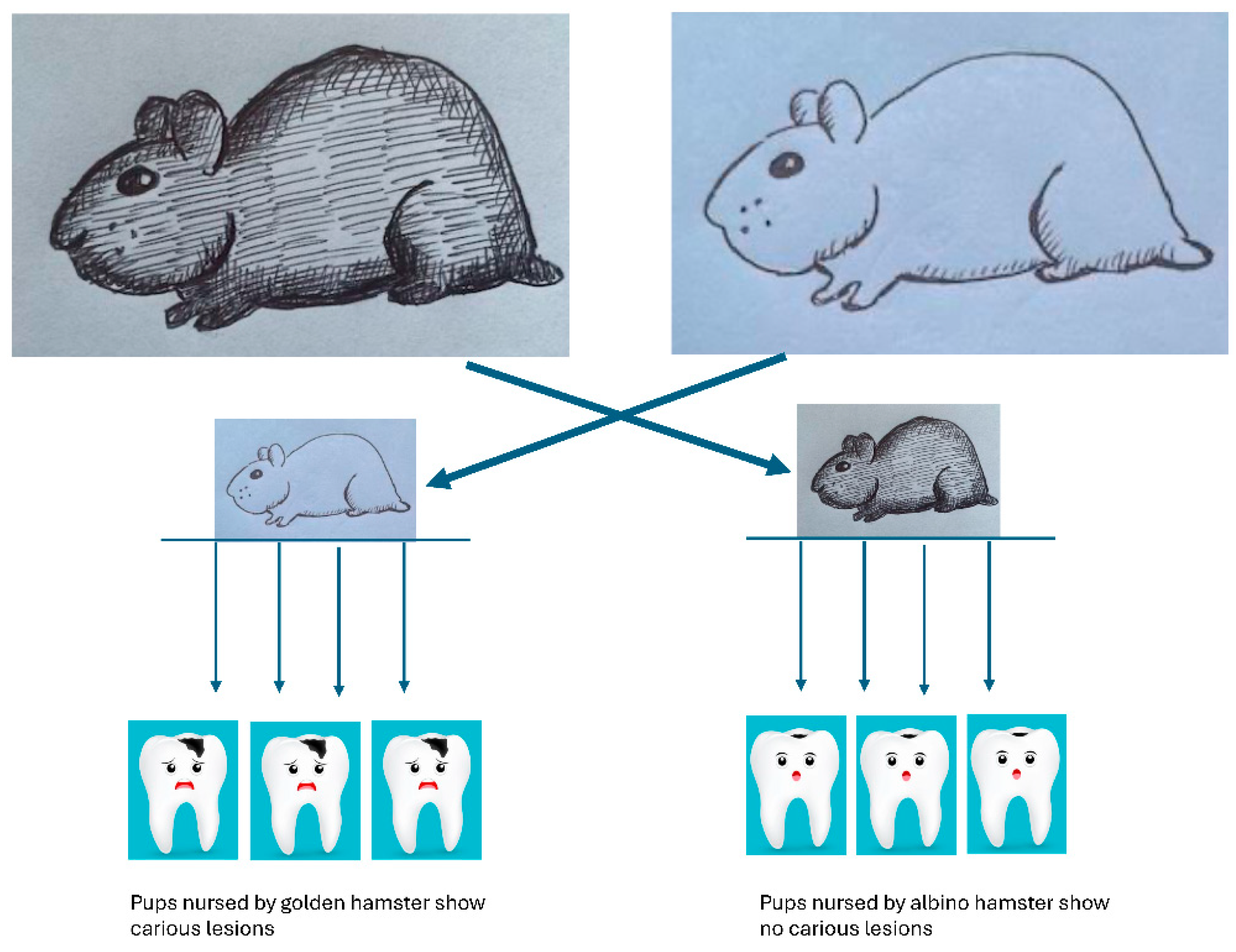
Figure 4.
Charting and radiographs of patient with significant pocket depth and bone loss. Charting shows loss of two mandibular incisors and deep pocketing in first molar region, which is confirmed by radiographic evidence. Note lack of carious lesions on radiographs.
Figure 4.
Charting and radiographs of patient with significant pocket depth and bone loss. Charting shows loss of two mandibular incisors and deep pocketing in first molar region, which is confirmed by radiographic evidence. Note lack of carious lesions on radiographs.
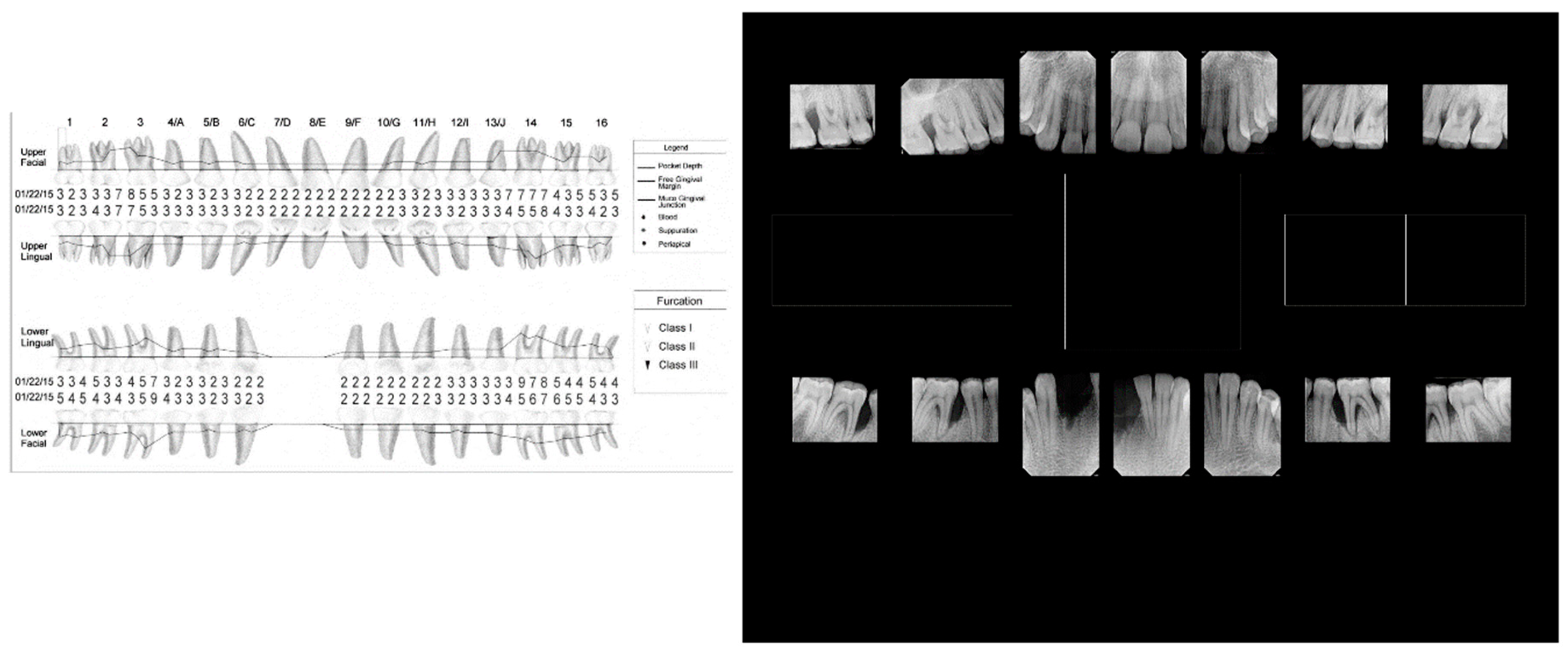
Figure 5.
Panoramic radiographs of cases of aggressive periodontitis in adolescents. Panel A (left): shows panoramic view of excessive bone loss in first molars and no carious lesions. Panel B (right). Shows more extensive disease in an adolescent with bone loss around molars and missing molars and incisors with occlusal but proximal decay is related to blow-out occlusal lesion in mandibular right second molar.
Figure 5.
Panoramic radiographs of cases of aggressive periodontitis in adolescents. Panel A (left): shows panoramic view of excessive bone loss in first molars and no carious lesions. Panel B (right). Shows more extensive disease in an adolescent with bone loss around molars and missing molars and incisors with occlusal but proximal decay is related to blow-out occlusal lesion in mandibular right second molar.
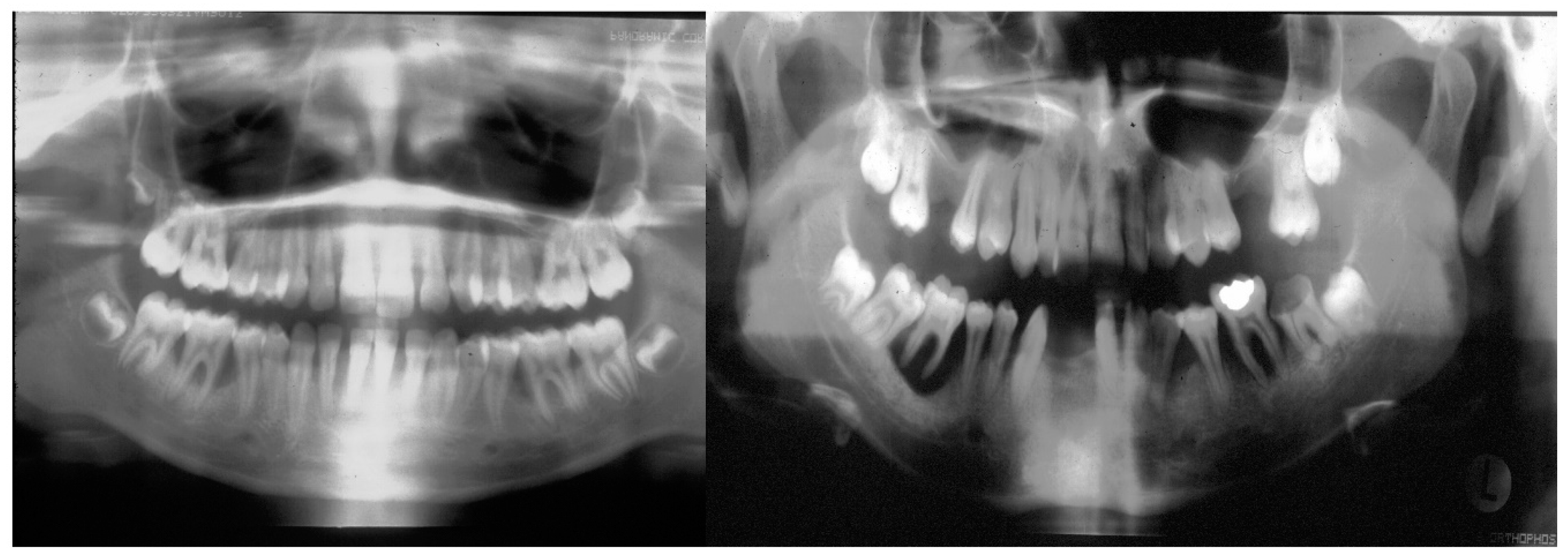
Table 1.
Numbers of Publications by Periodontitis Disease Categories from 2012-2023.
| Disease Category |
Pub Med Years |
Web of Science Years |
Pub Med Years |
|||
| 2012-17 | 2018-23 | 2012-17 | 2018-23 | 2012-17 | 2018-23 | |
| Periodontitis | 6,492 | 6,287 | 10,181 | 16,420 | 13,000 | 18,926 |
| Aggressive Periodontitis |
884 | 534 | 1.053 | 817 | 1,030 | 679 |
| Localized Aggressive Periodontitis |
179 | 114 | 134 | 102 | 118 | 80 |
| *Stage III Grade C Periodontitis |
*148 | *138 | *135 | |||
Comparisons between 6 years prior to 6 years following category changes are seen below as percentages disclosed by each search engine. Periodontitis changes varied from -3% to + 38%. Aggressive Periodontitis changes varied from -22% to -40%. Localized Aggressive Periodontitis changes varied from -24% to – 36%. *Stage III Grade C includes all forms of Aggressive Periodontitis and does not discriminate between adolescents and adults who have disease thus the # of publications are at a minimum 60% less when compared to LAgP; in addition, 20% of these publications define the new classification and did not present clinical data.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



