
Submitted:
30 July 2024
Posted:
31 July 2024
You are already at the latest version
Abstract
Endolysin of bacteriophage T5 (EndoT5) is a Zn2+-dependent and Ca2+-activated L-alanyl-D-glutamate peptidase that hydrolyses peptidoglycans during the destruction of the cell wall of the phage host bacterium Escherichia coli. To elucidate the mechanism of calcium activation, the spatial solution structure of EndoT5 in a complex with two ions - catalytic Zn2+ and regulatory Ca2+ (EndoT5-Zn2+Ca2+, PDB ID: 8P3A) was determined by high-resolution NMR and deposited in the Protein Data Bank. We show that coordination of the Ca2+ ion fix the spatial position of polar amino acid residues D113, N115 and S117. As a result, the intramolecular mobility of extended protein loops (residues 40-70 and 111-132) is significantly reduced compared to the mobility of these loops in the structure of an enzyme containing only a catalytic a Zn2+ ion (EndoT5-Zn2+, PDB ID: 2MXZ). In the stabilized EF-like calcium-binding loop (residues 111-132), the number of van der Waals interactions with amino acid residues of the globular core of the protein increases. The binding of the Ca2+ ion is accompanied by the selection of the functional states of both extended loops of the enzyme (residues 40-70 and 111-132), because of which EndoT5-Zn2+Ca2+ acquires the structural integrity necessary for catalysis. Therefore, the mechanism of activation of L-alanyl-D-glutamate peptidase of bacteriophage T5 by Ca2+ ions involves stabilization of the enzyme molecule in a catalytically active “open” conformation. This work sheds light on the structural grounds of the ion-dependent functional proteoforms of this interesting protein.
Keywords:
calcium activation
; endolysin structure
; bacteriophage
; catalysis
; NMR
; ion-induced proteoforms
1. Introduction
Calcium ions are necessary for bacteriophage T5 at all stages of its life cycle. Their participation is required for the phage DNA penetration into the host cell and for the processes of phage-specific transcription and translation [1,2,3]. It has been established that Ca2+ promotes the association of the main capsid protein pb8p and plays a key role in the formation of a stable procapsid [4]. At the final stage of the phage life cycle, bacteriophage T5 endolysin (EndoT5) hydrolyzes cell wall peptidoglycan, promoting further cell lysis and release of phage progeny. EndoT5 is a small (137 amino acid residues) zinc-containing L-alanyl-D-glutamate peptidase, belonging to the M15 family of metallopeptidases, subfamily C. The lytic activity of EndoT5 also depends on Ca2+ ions, which can be replaced by Mn2+ ions in vitro [5]. The spatial structure of this enzyme in complex with the catalytic ion Zn2+ was previously established using high-resolution NMR spectroscopy (PDB ID: 2MZX) [6]. EndoT5 is a globular protein of the α+β class with a hydrophobic core consisting of three α-helices and four antiparallel β-strands forming a sheet. The Zn2+ ion is coordinated in the enzyme structure by conservative amino acid residues H66, D73, and H133. The binding of the Zn2+ ion in the active site of the enzyme is necessary for the formation of a stable spatial structure of the protein.
Analysis of the long disordered regulatory loop of EndoT5, formed by residues 111-130, using site-directed mutagenesis revealed that its polar residues D113, N115, and S117 functionally correspond to residues 1, 3 and 5 of the consensus sequence of the canonical EF-motif, characteristic of eukaryotic calcium-binding proteins [7]. Canonical EF-loop provides 6 ligands for Ca2+ coordination located at positions 1, 3, 5, 7, 9, and 12, with 7 oxygen atoms organized into a pentagonal bipyramid [8]. The first 6 positions of the canonical EF-loop have a consensus sequence DxDxDG [9]. However, in contrast to the classical EF-motif, the EndoT5 regulatory loop has a shortened size (11 residues instead of 12), contains substitutions in the C-terminal region, and most importantly, has a low affinity for calcium ion [7]. It was suggested that calcium ion plays the role of a structural regulator of EndoT5, leading to a change in the conformation of the protein globule. However, the specific structural basis of calcium activation of EndoT5 remained unclear, and the non-canonical features of the discovered EF-like loop did not allow us to answer the question of how calcium ion binding promotes catalysis. In this work, the 3D solution structure of EndoT5 in a complex with Zn2+ and Ca2+ ions was established for the first time (PDB ID 8P3A). A comparative analysis of this structure with the single-ion EndoT5-Zn2+ structure (PDB ID: 2MXZ) made it possible to shed light on the structural basis of the mechanism of calcium activation of cell lysis at the final stage of the lytic bacteriophage T5 development. Furthermore, the present study provides important structural grounds for better understanding of the functional ion-dependent proteoforms of the EndoT5.
2. Materials and Methods
2.1. Isolation and Purification of Endolysin
To produce labeled EndoT5, E. coli BL21(DE3) cells carrying the pT5lys plasmid obtained previously [5] were grown in minimal M9 medium containing 15NH4Cl as a nitrogen source and 13C6H12O6 as a carbon source, until reaching OD550=0.6. Then, the synthesis of the target protein was induced with 0.8 mM IPTG. After induction, the cells were grown for 5 hours and collected by centrifugation at 3000 g for 10 minutes. For protein purification, E. coli BL21(DE3) cells (1.1 g) were suspended in 10 ml of buffer A containing 25 mM Tris-HCl (pH = 8.0), 40 mM NaCl and 1 mM EDTA. The cell suspension was ultrasonicated at a power of 75 W for 1 minute (2 times for 30 s), then clarified by centrifugation at 20,000 g for 30 minutes. The supernatant (9.5 ml) was passed through a 10 ml Toyopearl DEAE 650 M column, then applied to a 5 ml phosphocellulose P11 column in buffer A. Proteins were eluted with a linear gradient of sodium chloride (0.04-0.50 M) in a volume of 80 ml. Fractions (2 ml each) were analyzed by electrophoresis in 15% PAGE. The target EndoT5 protein was eluted with 0.3 M NaCl. Desalting of the sample was carried out by dialysis against an NH3 solution in water pH=7.2. The desalted preparation was freeze-dried.
2.2. Sample Preparation
Freeze-dried labeled EndoT5 was dissolved in 0.5 ml of 8 M Urea solution in buffer B of the following composition: 50 mM CD3COONa, 1 mM ZnCl2, 1 mM CaCl2 and 0.01% NaN3 (w/v) pH = 4.1, and then dialyzed 6 times against 200 ml of buffer B. 50 μl of D2O was added to the protein sample refolded in the presence of Zn2+ and Ca2+ and transferred to an NMR tube. The enzymatic activity of the sample was monitored using a turbidimetric approach based on the decrease in optical density at 450 nm of E. coli cells pretreated with chloroform, as described [5].
2.3. NMR Spectroscopy
The samples for the NMR experiments contained 0.8 mM [15N/13C]EndoT5-Zn2+Ca2+ in 50 mM Na-acetate buffer (pH 4.1) containing 0.03% NaN3 and prepared in a mixture of H2O/D2O (9/1). All experiments were carried out on an AVANCE-III 600 spectrometer (Bruker, Germany) operating at 1H frequencies of 600 MHz, equipped with 5 mm triple resonance inverse detection probe (TXI) with Z-gradient. The sample temperature was 298 K. To reduce protein association and prevent its autolysis during the experiment, solution with pH 4.1 was chosen.
For primary processing of the retrieved data and automatic collection of signal positions of 3D 13C-NOESY and 3D 15N-NOESY spectra, the TOPSPIN 2.1 program (Bruker Biospin, Karlsruhe, Germany) was used. The assignment of 1H, 13C, 15N signals in the spectra was carried out using the CARA algorithm [10]. Semi-automatic assignment of polypeptide chain signals to a specific residue in the primary protein sequence was carried out based on the data from the 2D-1H/15N-HSQC, 3D-HNCACB, and 3D-CBCA(CO)NH spectra using the integrated AutoLinc-9.4 module [11] of the CARA platform. Subsequently, based on the HNCACB and CC(CO)NH spectra, the fragments of semi-automatic assignment were combined, and its reliability was checked. In addition, the CC(CO)NH spectrum was also used to assign 13C resonances of the side chains of aliphatic residues. The corresponding proton resonances were established based on 3D-15N-TOCSY and 3D-HCСH-TOCSY experiments.
Chemical shifts of carbonyl group carbons were determined based on the 3D-HNCO spectrum. The 1H and 13C resonances of the side chains of aromatic residues were determined based on the 2D-CBHD and 3D-HCСH-TOCSY spectra. 2D-1H/15N-HSQC, 3D-15N-TOCSY, and 3D-15N-NOESY spectra were used to assign the indole protons of the tryptophan residues 84, 91, and 114. Chemical shifts of protons are indicated relative to the signal of sodium 2,2-dimethyl-2-silapentane-5-sulfonate, according to the IUPAC recommendation [12]; chemical shifts of other nuclei were recalculated from the ratio of gyromagnetic factors [13].
To study the dynamics of the EndoT5-Zn2+Ca2+ protein backbone in the fast, subnanosecond time range, heteronuclear 15N{1H} NOE values were measured at a field strength of 14.1 T and a temperature of 298 K [14]. The 15N{1H} NOE values were calculated as the ratio of signal intensities in two-dimensional 1H/15N correlation spectra with and without saturation of amide protons for 3.5 seconds and a relaxation delay of 10 seconds. The measurement error was assessed taking into account signal-to-noise ratios [15].
2.4. Calculation of Spatial Structure
Distance constraints were obtained from the 3D-15N and 13C-NOESY spectra with magnetization mixing times of 80 ms for 15N-NOESY, 160 ms for 13C-NOESY-ali, and 100 ms for 13C-NOESY-aro. Editing and integration of the collected signals was carried out on the CARA platform [10]. The torsion angles φ and ψ of the polypeptide chain were predicted by the TALOS program based on the chemical shifts of the core chain atoms 1HN, 15N, 1H, 13C, and 13CO [16]. To identify hydrogen bonds, a 3D-HNCO sequence was used [17], modified for the 2D registration option and optimized for observing long-range constants 3hJNC', realized through hydrogen bonds [18]. It was possible to distinguish 11 reliably detected hydrogen bonds. According to the geometric criterion [19], the formation of 20 hydrogen bonds is possible in the determined structure. For each hydrogen bond, distance restrictions were introduced: two upper ones (d(O,HN) = 2.0 Å, d(O,N) = 3.0 Å) and two lower ones d(O,HN) = 1.8 Å, d(O, N) = 2.7 Å). Thus, 80 additional restrictions were introduced to take hydrogen bonds into account in subsequent calculations. Constraint parameters for the coordinated Zn2+ ion were obtained from X-ray diffraction data of the catalytic domain of phage A500 endolysin (PDB ID: 2VO9, [20]). Similar parameters for the coordinated Ca2+ ion were partially obtained from the X-ray diffraction data of calcium-loaded apo-obelin from Obelia longissima (PDB ID: 1SL7, [21]) and modified to take into account the effect of point substitutions on the activity of EndoT5 [7].
The structure was calculated using the CYANA program [22]. A standard protocol was used, including seven cycles of automated assignment of NOESY cross peaks and calculation of the structures of 100 conformations in each cycle. As a result, 20 structures with the minimum value of the objective function were selected, which were used for the final analysis. The resulting ensemble of structures is in agreement with the experimental data and shows good Ramachandran statistics (Table 1).
The MOLMOL algorithm [23] was used to visually represent the structure. Experimentally obtained NMR constraints and atomic coordinates of 20 model protein structures of EndoT5-Zn2+Ca2+ were deposited in the PDB database with accession code 8P3A. The assignment of 1H, 13C, and 15N signals has been deposited in the BioMagResBank database with accession number 34817.
2.5. Overall Structure of EndoT5-Zn2+Ca2+[M1]
3. Results and Discussion
3.1. Comparative Analysis of the Secondary Structure of Single and Dual Ion Forms of EndoT5
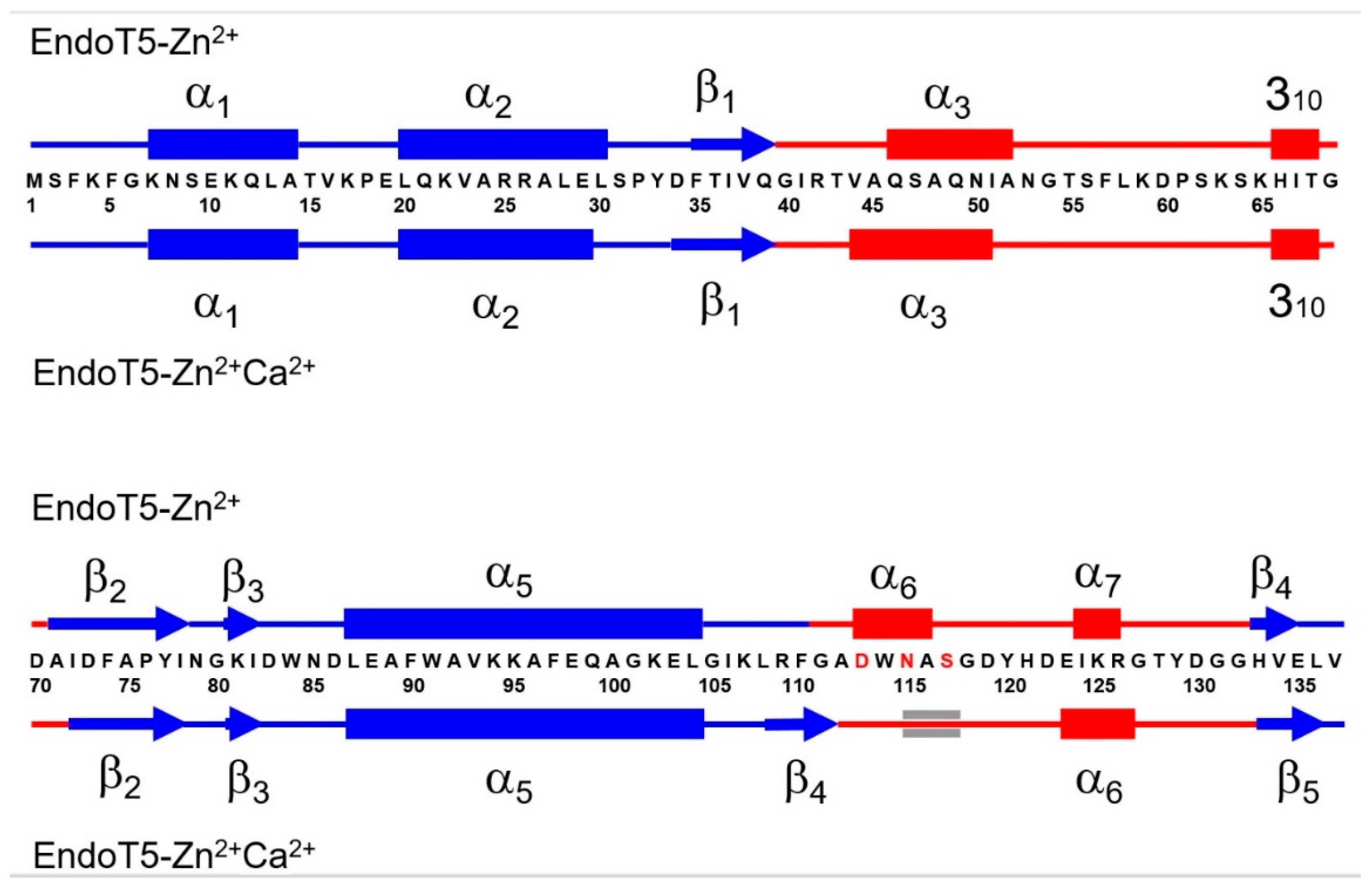
The spatial structure of EndoT5 in complex with catalytic Zn2+ and regulatory Ca2+ (EndoT5-Zn2+Ca2+, PDB ID: 8P3A) was obtained by high-resolution NMR. For comparative analysis, we used the previously solved 3D structure of EndoT5 in complex with catalytic Zn2+ ion (EndoT5-Zn2+, PDB ID 2MZX). An accurate evaluation of the secondary structure content in both forms of the protein was carried out based on the analysis of the average spatial structures of each of the ensembles obtained on the basis of NMR data. It was shown that upon binding of regulatory Ca2+, the α-helical content decreased from 49 to 36%, while the content of β-sheet structure increased from 12 to 16%. The arrangement of secondary structure elements for both forms of EndoT5 is shown in Figure 1.
It is interesting that the C-terminal part of the α3-helix of the single-ionic form EndoT5-Zn2+ (I51–A52) transforms into an irregular structure upon Ca2+ binding. In this case, two residues flanking the N-terminus of this helix, V44 and A45, on the contrary, take on an α-helical conformation. Further, the residues L108–G111 flanking the calcium-binding regulatory loop change from an irregular conformation upon Ca2+ binding to a β-structure, forming an additional β-strand that is part of the globular domain. In this case, residues D113-A116 of the beginning of the regulatory loop, which formed an α-helix in the absence of Ca2+, take on an irregular conformation, in which the configuration of only three residues - N115-S117 - is close to the parameters of an α-helix. The short C-terminal α-helix I124–R126 after Ca2+ binding includes one additional residue, E123. The secondary structure of the globular domain also undergoes a moderate transformation upon Ca2+ ion binding. The lengths of α-helical (L20-L30) and β-structural (A71-I78) segments are shortened by one residue from the C- and N-terminal ends, respectively. In contrast, β-strands F35-Q39 and H133-E135 each add one residue from the N- or C-end. In addition, as noted above, some residues form an additional β-strand L108-G111. Thus, the changes in secondary structure induced by binding of regulatory Ca2+ are observed both in the composition of extended loops and in the globular part of the macromolecule.
Translated with DeepL.com (free version)
Figure 1.
Comparative analysis of the secondary structures of EndoT5-Zn2+ and EndoT5-Zn2+Ca2+. Cylinders indicate α-helical sections, arrows indicate β-structural sections. The Ca2+ coordinating residues of the regulatory loop are shown in red. Sequence fragments forming the globular domain are shown in blue, whereas regions located in extended loops are shown in red.
Figure 1.
Comparative analysis of the secondary structures of EndoT5-Zn2+ and EndoT5-Zn2+Ca2+. Cylinders indicate α-helical sections, arrows indicate β-structural sections. The Ca2+ coordinating residues of the regulatory loop are shown in red. Sequence fragments forming the globular domain are shown in blue, whereas regions located in extended loops are shown in red.
3.2. Binding of Ca2+ Ion by the Regulatory Loop and Its Effect on Intramolecular Mobility
Details of the binding of regulatory calcium by the EF-like calcium-binding loop formed by amino acid residues 113 to 123 are presented in Figure 2. The figure clearly illustrates that polar amino acid residues D113, N115 and S117, corresponding to positions 1, 3, and 5 of the canonical EF loop, are involved in the coordination of the Ca2+ ion. The emerging coordination bonds fix the spatial position of these residues. As a result, the length of the moving sections of the regulatory loop is significantly reduced, stabilizing it. In this stable conformation of the loop, an increase in the number of van der Waals contacts of the amino acid residues with the residues of the globular core of the protein is observed.
Intramolecular movements in proteins play an important role in many biological processes, in particular enzymatic catalysis and its allosteric regulation. The presence of Ca2+-mediated allosteric regulation is well illustrated by the differences in the local RMSD of the main chain of the ensembles of EndoT5-Zn2+ and EndoT5-Zn2+Ca2+ structures (Figure 3). Despite the spatial distance of the extended loops 40-70 and 111-132 and the absence of direct contact between them, the ordering of their conformational states has a symbatic character. Therefore, the Zn2+Ca2+ form of EndoT5 manifests itself as a single conformational cooperative unit [24]. This property of the catalytically active “open” conformation of the enzyme molecule is necessary for all stages of the catalytic cycle.
A comparative analysis of 15N{1H} NOE amides of the main chains of both forms of the protein (Figure 4A and Figure 4B) shows that in the subnanosecond time range, only the backbone fragment of the extended G40–D70 loop has significant intramolecular mobility. At the same time, the regulatory Ca2+-binding loop (G111–G132) is globally immobile. However, a detailed comparison shows that after calcium binding, the 15N{1H}NOE values for amide pairs neighboring along the polypeptide chain tend to align throughout the whole structure, indicating the emergence of the dynamic conjugation of residues adjacent within the chain. These data indicate the emergence of conformational relationships in the structure of EndoT5-Zn2+ macromolecules when they bind Ca2+ ions, which is consistent with the structural data obtained by analyzing the local RMSD of the main chain of ensembles of EndoT5-Zn2+ and EndoT5-Zn2+Ca2+ structures (see Figure 3).
The coordination of the intramolecular movements of extended loops in the presence of the Ca2+ ion is necessary for the implementation of all the stages of catalysis (binding – hydrolysis – substrate release), regardless of the view on the recognition processes: whether to adhere to the theory of induced fit or the theory of conformational selection, the debate around which is still ongoing. From the point of view of Koshland’s theory of induced fit [25], intramolecular mobility provides the protein globule with the ability to interact with the substrate, and the binding of regulatory calcium with a subsequent change in the loop structure can be considered as a stage of this process and at the same time a special case of induced loop-ligand fit. If one discusses the intramolecular mobility in terms of the conformational selection [26], mobility provides variability in conformational substates in a population of macromolecules, and the calcium ion, interacting with protein globules that have a conformation with the lowest free binding energy, shifts the equilibrium towards catalytically active forms. Therefore, the binding of the Ca2+ ion is accompanied by the selection of functionally significant conformational states of the enzyme’s extended loops (40-70 and 111-132).
3.3. Structural Basis of Activation of EndoT5 by Regulatory Ca2+
Figure 5A and Figure 5B compare the structure of EndoT5 with one Zn2+ ion (PDB ID: 2MXZ) and two ions, Zn2+ and Ca2+ (PDB ID: 8P3A). Both ions in the EndoT5-Zn2+Ca2+ structure are located in close proximity, but their roles are different. The zinc ion in EndoT5 is catalytic, tightly coordinated to H66, H133, and D73, and, together with the D130 residue, is involved in the nucleophilic substitution that occurs during hydrolysis of the peptide bond in the substrate by endolysin. Calcium plays a regulatory role and is bound by the mobile EndoT5 loop, which has convergent similarity to the canonical EF loop of the eukaryotic calcium buffers and sensors. The affinity of Ca2+ for the enzyme is low: the dissociation constant determined previously is 2×10-4 M [7].
There are no analogues of the EndoT5-Zn2+Ca2+ two-ion structure in the databases yet. Among other members of the family of zinc-containing peptidases M15 of subfamily C homologous to EndoT5, there are several representatives whose structures (or parts of structures) were determined by X-ray diffraction analysis. These include peptidoglycan hydrolase ChiX from the Gram-negative bacterium Serratia marcescens (PDB IDs: 5OPZ and 5OQ1, [27]); the LysB4 endolysin from Bacillus cereus-targeting bacteriophage B4 (PDB ID: 6AKV, [28]); and catalytic domain of the Listeria bacteriophage endolysin Ply500 (PDB ID: 2VO9, [20]). All of these structures contain a zinc ion coordinated by an H-D-H triad, similar to EndoT5, but they do not contain a calcium ion or a calcium-binding loop.
Among the two-ionic structures of endolysins, we can mention the structure of the CHAP catalytic domain of the staphylococcal endolysin LysK, containing Ca2+ and Zn2+ [29]. However, the differences with EndoT5 are radical. CHAP LysK domain is a papain-type cysteine protease that hydrolyzes the D-Ala-Gly bond in staphylococcal peptidoglycan (between D-alanine of the tetra-peptide stem and the first glycine of the penta-glycine cross-bridge). In the CHAP structure (PDB ID: 4CSH, [29]), unlike EndoT5, the calcium ion is tightly bound by the canonical EF loop. The zinc ion in the LysK structure is coordinated not by the H-D-H triad, as in EndoT5, but by the C57 cysteine residue, is weakly bound, and its role is presumably to regulate the admission of the substrate to the catalytic site. Interestingly, the structure of the CHAP domain of a close LysK homologue, LysGH15 (PDB ID: 4OLK), does not contain zinc ions, whereas calcium is bound similarly to the classical EF loop [30].
Comparative analysis of EndoT5 protein globules with one and two ions shows that the mechanism of calcium activation of EndoT5 is based on the formation and stabilization of the catalytically active “open” conformation of the active site. Binding of Ca2+ by side chains D113, N115, and S117 (positions 1, 3, and 5 of the regulatory loop) locally changes the secondary structure, reduces the mobility of the loop, fixing it on the globular domain. We recently showed that tryptophan residue W114, which is strictly conserved among metallopeptidases of the M15_C subfamily, plays an important role in maintaining the affinity for regulatory Ca2+ [31]. The presence of tryptophan at position 2 of the calcium-binding loop is not typical for canonical EF motifs. However, in endolysins of the M15_C peptidase family, this residue affects the configuration of the hydrophobic core of the globule, and is possibly involved in the binding of the substrate, peptidoglycan [31]. Apparently, calcium binding by the EndoT5 regulatory loop regulates the spatial position of the conserved tryptophan residue, which mediates fixation of the loop on the globular domain, while simultaneously entering into hydrophobic interactions with the substrate. As a result, the entire protein globule of the enzyme passes into a catalytically active conformation, which is open relative to the inactive single-ion form, which is clearly visible while comparing Figure 5A and Figure 5B.
It should be noted that NMR experiments on the relaxation dispersion of populations of the conformational ensemble of enzyme molecules using the example of adenylate kinase, discussed in the review [32], showed that the conformational exchange between open and closed conformations is not exclusively a function of catalytic turnover, since it is also observed in free enzyme [33]. This was confirmed by relaxation experiments using the example of a ligand-dependent enzyme, E. coli dihydrofolate reductase [34]. The conformational dynamics of the enzyme was ligand dependent, where the binary complexes with a bound cofactor or product fluctuated into a conformation resembling a ternary complex (with ligand and substrate/product), which correlated with the dynamics of chemical shifts. Therefore, binding (or release) of a ligand can change the ratio of substates in the conformational ensemble, as well as the kinetic and thermodynamic parameters governing conformational equilibrium. From the viewpoint of the conformational selection theory, EndoT5 calcium binding should be considered not as a mechanism for inducing a conformational transition, but as a tool for shifting the equilibrium in a population of molecules towards a catalytically active open conformation with higher energy. In this case, the high value of the calcium dissociation constant reflects the height of the energy barrier between the free and calcium-bound states.
The structural basis for the activation of EndoT5 by calcium ions is similar to mechanism that underlies the work of calcium “sensors”, proteins, the binding of calcium to which causes intramolecular changes leading to the exposure of the hydrophobic regions of the protein globule to the surface of the “sensor”, thereby promoting interaction with other proteins [34]. A similar mechanism is characteristic, for example, of Ca2+-calmodulin. Here, two domains of this universal protein form a hydrophobic channel capable of binding target proteins, such as kinases, phosphatases, transcription factors, cytoskeletal components, and transporters [35]. Therefore, calcium ions mediate signal transduction in the cells, mainly of eukaryotic origin. Conformational rearrangements also underlie the work of the Extracellular Calcium-Sensing Receptor (CaR), where upon the Ca2+ binding, the open cleft of the Venus Flytrap domain closes inducing, in turn, conformational changes in both the transmembrane domain and the intracellular domains that are believed to initiate signal transduction [36].
In our case, obviously, calcium binding plays a similar structural role, promoting the enzymatic function of the protein, and not signaling. The low affinity of EndoT5 for regulatory Ca2+ indicates that the binding of the ion does not occur in the cytoplasm of the host cell of this E. coli phage, where the Ca2+ concentration is much lower than the dissociation constant, but in the periplasm serving as a calcium “depot”, where, as was shown by Jones et al. [37]), the Ca2+ concentration reaches 1 mM, and where the EndoT5 substrate, peptidoglycan, is located. Probably, like calcium sensors, binding of calcium by EndoT5 triggers conformational changes (or conformational selection) leading to the predominance of protein forms exposure of hydrophobic amino acids, primarily those located in the W114 regulatory loop involved in the interaction with the substrate. The activated enzyme destroys the layer of peptidoglycan, which is the main factor in the strength of bacterial cell walls and the last obstacle to the exit of phage progeny from the host cell. Therefore, activation of the bacteriophage T5 endolysin by periplasmic calcium is one of the triggers for cell lysis by this lytic bacteriophage.
Finally, results of this analysis should be considered from the viewpoint of the proteoform concept, according to which, a single gene can encode a diverse set of structurally and functionally distinct protein molecules [38]. The structural and functional diversification of proteoforms is achieved through various mechanisms that alter the chemical structure of gene-encoded proteins: at the DNA level via allelic variations such as single or multiple point mutations, indels, and SNPs; at the mRNA level through alternative splicing and other pre-translational events; and at the protein level via a wide array of PTMs [38,39,40,41,42,43]. Additionally, intrinsic disorder and structural alterations induced by protein function contribute to this structural and functional diversification [44].
One should also keep in mind that protein structure is a highly dynamic entity characterized by an exceptional spatio-temporal heterogeneity and containing a continuous spectrum of differently structured/disordered conformations [45]. Proteins structure can be envisioned as a complex conformational mosaic containing foldons (independently foldable protein units), inducible foldons (intrinsically disordered regions (IDRs) capable of at least partial folding triggered by the interactions with binding partners), morphing inducible foldons (IDRs with the potential to fold differently at binding to different partners), semi-foldons (regions that are always in a semi-folded form), non-foldons (IDRs that never fold), and even unfoldons (ordered regions undergoing order-to-disorder transition to make protein active). Importantly, these differently (dis)ordered regions can be found within one protein molecule, where they might have different functions. This defines the protein structure-function continuum model postulating that a given protein exists as a dynamic conformational ensemble containing multiple proteoforms of different origin (conformational/basic, inducible/modified, and functioning) characterized by a broad spectrum of structural features and possessing different functional potentials [46,47,48,49]. The existence of different proteoforms combined with the highly heterogeneous spatio-temporal organization of functional proteins challenges the oversimplified "one gene – one protein – one function" model in favor of a more nuanced "one gene – many proteins – many functions" paradigm [44,46,47,49,50,51,52,53,54].
In line with these considerations, the present study provides important structural grounds for better understanding of the ion-dependent functioning proteoforms of EndoT5. In fact, with its Ca2+-binding induced structural and dynamic changes, EndoT5 serves as an illustrative example of basic (or conformational, or intrinsic) and functioning proteoforms. Here, basic (or intrinsic, or conformational) proteoforms originate from the fact that structurally, a functional protein represents a dynamic conformational ensemble, members of which have different structures and might have different functions. Furthermore, since protein structural ensemble can be affected by protein function, interaction with specific partners, or even just placement of a protein inside its natural cellular environment that contains high concentrations of various biological macromolecules [55,56,57], has limited available volume [58], and contains restricted amounts of free water [55,59,60,61,62,63]) protein functionality itself represents a factor generating functioning proteoforms [44,50]. Figure 6 illustrates this ideas showing the Ca2+ binding-induced transformation of a fuzzy EndoT5-Zn2+ (which is a functioning basic proteoform itself, as it is rather flexible and contains a bound Zn2+ ion) into a less flexible EndoT5-Zn2+Ca2+ thereby promoting the enzymatic function of the protein. In other words, Ca2+ binding reduces structural flexibility of the basic/intrinsic/conformational proteoform and creates a less flexible functioning proteoform capable of catalytic activity.
Author Contributions
Conceptualization, D.A.P., V.P.K., and G.V.M.; methodology and investigation, D.A.P., G.V.M., V.P.K. and V.N.U.; writing—original draft preparation, G.V.M. and D.A.P.; writing—review and editing, V.N.U. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
PDB structure 2MXZ. https://doi.org/10.2210/pdb2MXZ/pdb, release date on 11 February 2015. PDB structure 8P3A. https://doi.org/10.2210/pdb8P3A/pdb, release date on 5 July 2023. The data present in the current study are available from the corresponding authors on reasonable request.
Acknowledgments
This work is dedicated to the memory of Professor Victor P. Kutyshenko who has passed away on October 18, 2021.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Bonhivers, M.; Letellier, L. Calcium controls phage T5 infection at the level of the Escherichia coli cytoplasmic membrane. FEBS Lett 1995, 374, 169–173. [Google Scholar] [CrossRef] [PubMed]
- Moyer, R.W.; Buchanan, J.M. Effect of calcium ions on synthesis of T5-specific ribonucleic acid. J Biol Chem 1970, 245, 5904–5913. [Google Scholar] [CrossRef]
- Moyer, R.W.; Buchanan, J.M. Effect of calcium ions on synthesis of T5-specific proteins. J Biol Chem 1970, 245, 5897–5903. [Google Scholar] [CrossRef]
- Huet, A.; Conway, J.F.; Letellier, L.; Boulanger, P. In vitro assembly of the T=13 procapsid of bacteriophage T5 with its scaffolding domain. J Virol 2010, 84, 9350–9358. [Google Scholar] [CrossRef]
- Mikoulinskaia, G.V.; Odinokova, I.V.; Zimin, A.A.; Lysanskaya, V.Y.; Feofanov, S.A.; Stepnaya, O.A. Identification and characterization of the metal ion-dependent L-alanoyl-D-glutamate peptidase encoded by bacteriophage T5. FEBS J 2009, 276, 7329–7342. [Google Scholar] [CrossRef]
- Prokhorov, D.A.; Mikoulinskaia, G.V.; Molochkov, N.V.; Uversky, V.N.; Kutyshenko, V.P. High-resolution NMR structure of a Zn 2+-containing form of the bacteriophage T5 L-alanyl-D-glutamate peptidase. RSC advances 2015, 5, 41041–41049. [Google Scholar] [CrossRef]
- Kovalenko, A.O.; Chernyshov, S.V.; Kutyshenko, V.P.; Molochkov, N.V.; Prokhorov, D.A.; Odinokova, I.V.; Mikoulinskaia, G.V. Investigation of the calcium-induced activation of the bacteriophage T5 peptidoglycan hydrolase promoting host cell lysis. Metallomics 2019, 11, 799–809. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, H.; Nakayama, S.; Kretsinger, R.H. Classification and evolution of EF-hand proteins. Biometals 1998, 11, 277–295. [Google Scholar] [CrossRef]
- Dominguez, D.C.; Guragain, M.; Patrauchan, M. Calcium binding proteins and calcium signaling in prokaryotes. Cell Calcium 2015, 57, 151–165. [Google Scholar] [CrossRef]
- Keller, R. The computer aided resonance assignment tutorial; CANTINA verlag: 2004.
- Masse, J.E.; Keller, R. AutoLink: automated sequential resonance assignment of biopolymers from NMR data by relative-hypothesis-prioritization-based simulated logic. J Magn Reson 2005, 174, 133–151. [Google Scholar] [CrossRef]
- Markley, J.L.; Bax, A.; Arata, Y.; Hilbers, C.W.; Kaptein, R.; Sykes, B.D.; Wright, P.E.; Wuthrich, K. Recommendations for the presentation of NMR structures of proteins and nucleic acids--IUPAC-IUBMB-IUPAB Inter-Union Task Group on the standardization of data bases of protein and nucleic acid structures determined by NMR spectroscopy. Eur J Biochem 1998, 256, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Bigam, C.G.; Yao, J.; Abildgaard, F.; Dyson, H.J.; Oldfield, E.; Markley, J.L.; Sykes, B.D. 1H, 13C and 15N chemical shift referencing in biomolecular NMR. J Biomol NMR 1995, 6, 135–140. [Google Scholar] [CrossRef]
- Farrow, N.A.; Muhandiram, R.; Singer, A.U.; Pascal, S.M.; Kay, C.M.; Gish, G.; Shoelson, S.E.; Pawson, T.; Forman-Kay, J.D.; Kay, L.E. Backbone dynamics of a free and phosphopeptide-complexed Src homology 2 domain studied by 15N NMR relaxation. Biochemistry 1994, 33, 5984–6003. [Google Scholar] [CrossRef]
- Kharchenko, V.; Nowakowski, M.; Jaremko, M.; Ejchart, A.; Jaremko, L. Dynamic (15)N(1)H NOE measurements: a tool for studying protein dynamics. J Biomol NMR 2020, 74, 707–716. [Google Scholar] [CrossRef]
- Cornilescu, G.; Delaglio, F.; Bax, A. Protein backbone angle restraints from searching a database for chemical shift and sequence homology. J Biomol NMR 1999, 13, 289–302. [Google Scholar] [CrossRef] [PubMed]
- Grzesiek, S.; Bax, A. Improved 3D triple-resonance NMR techniques applied to a 31 kDa protein. Journal of Magnetic Resonance (1969) 1992, 96, 432–440. [Google Scholar] [CrossRef]
- Shenkarev, Z.O.; Balashova, T.A.; Yakimenko, Z.A.; Ovchinnikova, T.V.; Arseniev, A.S. Peptaibol zervamicin IIb structure and dynamics refinement from transhydrogen bond J couplings. Biophys J 2004, 86, 3687–3699. [Google Scholar] [CrossRef]
- Stickle, D.F.; Presta, L.G.; Dill, K.A.; Rose, G.D. Hydrogen bonding in globular proteins. J Mol Biol 1992, 226, 1143–1159. [Google Scholar] [CrossRef]
- Korndorfer, I.P.; Kanitz, A.; Danzer, J.; Zimmer, M.; Loessner, M.J.; Skerra, A. Structural analysis of the L-alanoyl-D-glutamate endopeptidase domain of Listeria bacteriophage endolysin Ply500 reveals a new member of the LAS peptidase family. Acta Crystallogr D Biol Crystallogr 2008, 64, 644–650. [Google Scholar] [CrossRef]
- Deng, L.; Vysotski, E.S.; Markova, S.V.; Liu, Z.J.; Lee, J.; Rose, J.; Wang, B.C. All three Ca2+-binding loops of photoproteins bind calcium ions: the crystal structures of calcium-loaded apo-aequorin and apo-obelin. Protein Sci 2005, 14, 663–675. [Google Scholar] [CrossRef]
- Guntert, P. Automated NMR structure calculation with CYANA. Methods Mol Biol 2004, 278, 353–378. [Google Scholar] [CrossRef] [PubMed]
- Koradi, R.; Billeter, M.; Wuthrich, K. MOLMOL: a program for display and analysis of macromolecular structures. J Mol Graph 1996, 14, 51–55. [Google Scholar] [CrossRef] [PubMed]
- Fersht, A. Structure and mechanism in protein science: a guide to enzyme catalysis and protein folding; Macmillan: 1999.
- Koshland, D.E. Application of a Theory of Enzyme Specificity to Protein Synthesis. Proc Natl Acad Sci U S A 1958, 44, 98–104. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.J.; Kumar, S.; Ma, B.; Nussinov, R. Folding funnels, binding funnels, and protein function. Protein Sci 1999, 8, 1181–1190. [Google Scholar] [CrossRef] [PubMed]
- Owen, R.A.; Fyfe, P.K.; Lodge, A.; Biboy, J.; Vollmer, W.; Hunter, W.N.; Sargent, F. Structure and activity of ChiX: a peptidoglycan hydrolase required for chitinase secretion by Serratia marcescens. Biochem J 2018, 475, 415–428. [Google Scholar] [CrossRef]
- Hong, S.; Son, B.; Ryu, S.; Ha, N.C. Crystal Structure of LysB4, an Endolysin from Bacillus cereus-Targeting Bacteriophage B4. Mol Cells 2019, 42, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Sanz-Gaitero, M.; Keary, R.; Garcia-Doval, C.; Coffey, A.; van Raaij, M.J. Crystal structure of the lytic CHAP(K) domain of the endolysin LysK from Staphylococcus aureus bacteriophage K. Virol J 2014, 11, 133. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.; Feng, Y.; Feng, X.; Sun, C.; Lei, L.; Ding, W.; Niu, F.; Jiao, L.; Yang, M.; Li, Y.; et al. Structural and biochemical characterization reveals LysGH15 as an unprecedented "EF-hand-like" calcium-binding phage lysin. PLoS Pathog 2014, 10, e1004109. [Google Scholar] [CrossRef] [PubMed]
- Mikoulinskaia, G.V.; Prokhorov, D.A.; Chernyshov, S.V.; Sitnikova, D.S.; Arakelian, A.G.; Uversky, V.N. Conservative Tryptophan Residue in the Vicinity of an Active Site of the M15 Family l,d-Peptidases: A Key Element in the Catalysis. Int J Mol Sci 2023, 24. [Google Scholar] [CrossRef]
- Boehr, D.D.; Nussinov, R.; Wright, P.E. The role of dynamic conformational ensembles in biomolecular recognition. Nat Chem Biol 2009, 5, 789–796. [Google Scholar] [CrossRef]
- Henzler-Wildman, K.A.; Thai, V.; Lei, M.; Ott, M.; Wolf-Watz, M.; Fenn, T.; Pozharski, E.; Wilson, M.A.; Petsko, G.A.; Karplus, M.; et al. Intrinsic motions along an enzymatic reaction trajectory. Nature 2007, 450, 838–844. [Google Scholar] [CrossRef] [PubMed]
- Boehr, D.D.; McElheny, D.; Dyson, H.J.; Wright, P.E. The dynamic energy landscape of dihydrofolate reductase catalysis. Science 2006, 313, 1638–1642. [Google Scholar] [CrossRef] [PubMed]
- Luan, S.; Wang, C. Calcium Signaling Mechanisms Across Kingdoms. Annu Rev Cell Dev Biol 2021, 37, 311–340. [Google Scholar] [CrossRef]
- Gerbino, A.; Colella, M. The Different Facets of Extracellular Calcium Sensors: Old and New Concepts in Calcium-Sensing Receptor Signalling and Pharmacology. Int J Mol Sci 2018, 19. [Google Scholar] [CrossRef]
- Jones, H.E.; Holland, I.B.; Campbell, A.K. Direct measurement of free Ca(2+) shows different regulation of Ca(2+) between the periplasm and the cytosol of Escherichia coli. Cell Calcium 2002, 32, 183–192. [Google Scholar] [CrossRef]
- Smith, L.M.; Kelleher, N.L.; Consortium for Top Down, P. Proteoform: a single term describing protein complexity. Nat Methods 2013, 10, 186–187. [Google Scholar] [CrossRef]
- Uhlen, M.; Bjorling, E.; Agaton, C.; Szigyarto, C.A.; Amini, B.; Andersen, E.; Andersson, A.C.; Angelidou, P.; Asplund, A.; Asplund, C.; et al. A human protein atlas for normal and cancer tissues based on antibody proteomics. Molecular & Cellular Proteomics 2005, 4, 1920–1932. [Google Scholar] [CrossRef]
- Farrah, T.; Deutsch, E.W.; Omenn, G.S.; Sun, Z.; Watts, J.D.; Yamamoto, T.; Shteynberg, D.; Harris, M.M.; Moritz, R.L. State of the Human Proteome in 2013 as Viewed through PeptideAtlas: Comparing the Kidney, Urine, and Plasma Proteomes for the Biology- and Disease-Driven Human Proteome Project. Journal of Proteome Research 2014, 13, 60–75. [Google Scholar] [CrossRef]
- Farrah, T.; Deutsch, E.W.; Hoopmann, M.R.; Hallows, J.L.; Sun, Z.; Huang, C.Y.; Moritz, R.L. The State of the Human Proteome in 2012 as Viewed through PeptideAtlas. Journal of Proteome Research 2013, 12, 162–171. [Google Scholar] [CrossRef]
- Reddy, P.J.; Ray, S.; Srivastava, S. The Quest of the Human Proteome and the Missing Proteins: Digging Deeper. Omics-a Journal of Integrative Biology 2015, 19, 276–282. [Google Scholar] [CrossRef]
- Kim, M.S.; Pinto, S.M.; Getnet, D.; Nirujogi, R.S.; Manda, S.S.; Chaerkady, R.; Madugundu, A.K.; Kelkar, D.S.; Isserlin, R.; Jain, S.; et al. A draft map of the human proteome. Nature 2014, 509, 575–581. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. p53 Proteoforms and Intrinsic Disorder: An Illustration of the Protein Structure-Function Continuum Concept. Int J Mol Sci 2016, 17, 1874. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. Unusual biophysics of intrinsically disordered proteins. Biochim Biophys Acta 2013, 1834, 932–951. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. p53 Proteoforms and Intrinsic Disorder: An Illustration of the Protein Structure-Function Continuum Concept. Int J Mol Sci 2016, 17. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. Protein intrinsic disorder and structure-function continuum. Prog Mol Biol Transl Sci 2019, 166, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. Intrinsically disordered proteins and their “mysterious” (meta)physics. Frontiers in Physics 2019, 7, Article 10 (18 pages). [CrossRef]
- Uversky, V.N. Dancing Protein Clouds: The Strange Biology and Chaotic Physics of Intrinsically Disordered Proteins. J Biol Chem 2016, 291, 6681–6688. [Google Scholar] [CrossRef]
- Uversky, V.N. Functional roles of transiently and intrinsically disordered regions within proteins. FEBS J 2015, 282, 1182–1189. [Google Scholar] [CrossRef]
- Fonin, A.V.; Darling, A.L.; Kuznetsova, I.M.; Turoverov, K.K.; Uversky, V.N. Multi-functionality of proteins involved in GPCR and G protein signaling: making sense of structure-function continuum with intrinsic disorder-based proteoforms. Cell Mol Life Sci 2019, 76, 4461–4492. [Google Scholar] [CrossRef]
- Gupta, M.N.; Uversky, V.N. Protein structure-function continuum model: Emerging nexuses between specificity, evolution, and structure. Protein Sci 2024, 33, e4968. [Google Scholar] [CrossRef]
- Kulkarni, P.; Leite, V.B.P.; Roy, S.; Bhattacharyya, S.; Mohanty, A.; Achuthan, S.; Singh, D.; Appadurai, R.; Rangarajan, G.; Weninger, K.; et al. Intrinsically disordered proteins: Ensembles at the limits of Anfinsen's dogma. Biophys Rev (Melville) 2022, 3, 011306. [Google Scholar] [CrossRef]
- Uversky, V.N. Functional unfoldomics: Roles of intrinsic disorder in protein (multi)functionality. Adv Protein Chem Struct Biol 2024, 138, 179–210. [Google Scholar] [CrossRef] [PubMed]
- Zimmerman, S.B.; Trach, S.O. Estimation of macromolecule concentrations and excluded volume effects for the cytoplasm of Escherichia coli. J. Mol. Biol. 1991, 222, 599–620. [Google Scholar] [CrossRef]
- van den Berg, B.; Ellis, R.J.; Dobson, C.M. Effects of macromolecular crowding on protein folding and aggregation. The EMBO journal 1999, 18, 6927–6933. [Google Scholar] [CrossRef]
- Rivas, G.; Ferrone, F.; Herzfeld, J. Life in a crowded world. EMBO reports 2004, 5, 23–27. [Google Scholar] [CrossRef] [PubMed]
- Ellis, R.J.; Minton, A.P. Cell biology: join the crowd. Nature 2003, 425, 27–28. [Google Scholar] [CrossRef]
- Zimmerman, S.B.; Minton, A.P. Macromolecular crowding: biochemical, biophysical, and physiological consequences. Annu. Rev. Biophys. Biomol. Struct. 1993, 22, 27–65. [Google Scholar] [CrossRef] [PubMed]
- Fulton, A.B. How crowded is the cytoplasm? Cell 1982, 30, 345–347. [Google Scholar] [CrossRef]
- Minton, A.P. Influence of excluded volume upon macromolecular structure and associations in 'crowded' media. Curr. Opin. Biotechnol. 1997, 8, 65–69. [Google Scholar] [CrossRef]
- Ellis, R.J. Macromolecular crowding: obvious but underappreciated. Trends Biochem Sci 2001, 26, 597–604. [Google Scholar] [CrossRef]
- Minton, A.P. Protein folding: Thickening the broth. Curr. Biol. 2000, 10, R97–R99. [Google Scholar] [CrossRef] [PubMed]
Figure 2.
Calcium binding by the EndoT5 regulatory loop. The green sphere is a zinc ion, the lilac sphere is a calcium ion. The numbers indicate amino acid residues that coordinate both ions.
Figure 2.
Calcium binding by the EndoT5 regulatory loop. The green sphere is a zinc ion, the lilac sphere is a calcium ion. The numbers indicate amino acid residues that coordinate both ions.
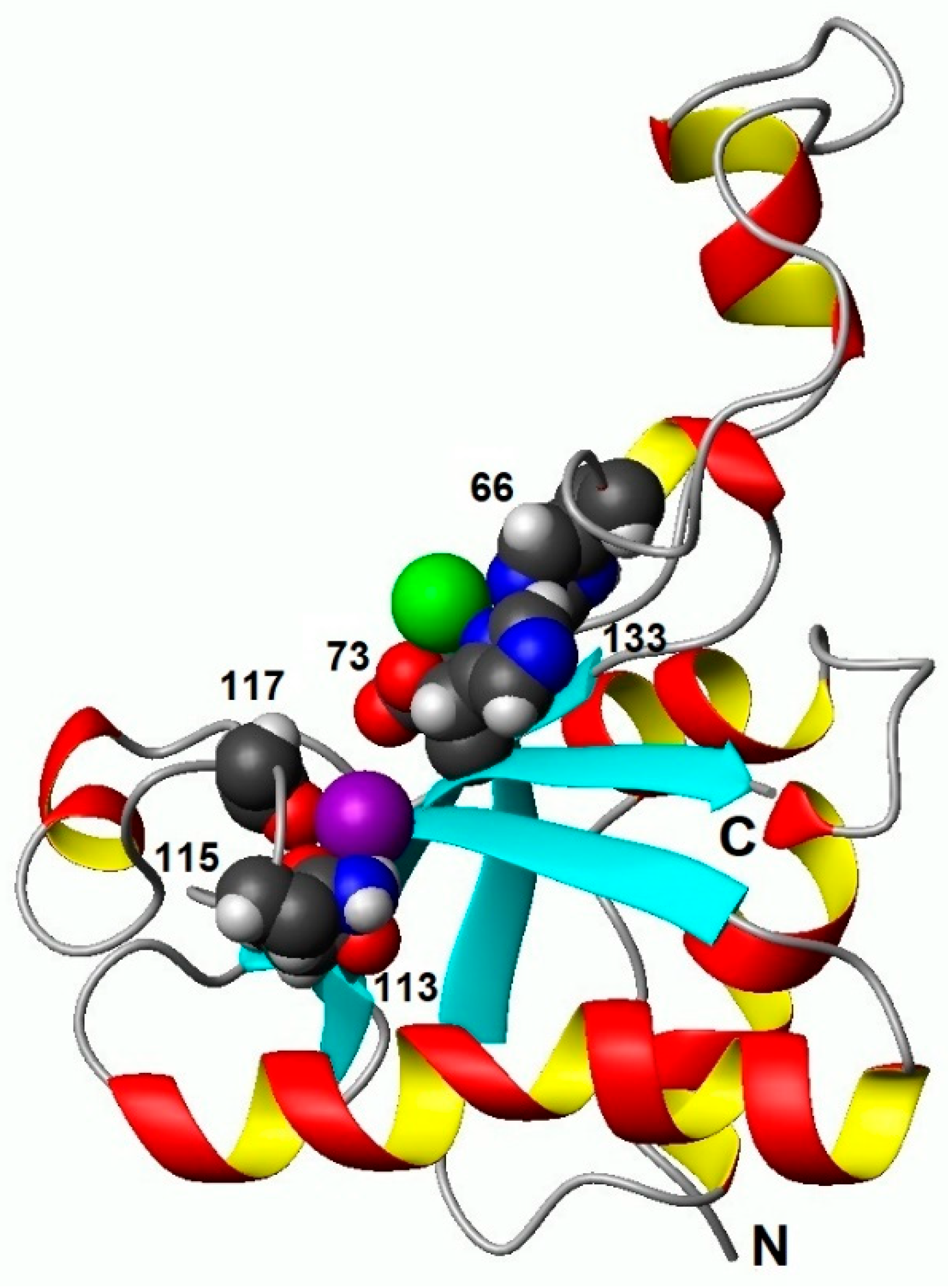
Figure 3.
Comparison of backbone RMSD (Å) for EndoT5-Zn2+ (ο) and EndoT5-Zn2+Ca2+ (•) structure ensembles. Each ensemble contains 20 structures.
Figure 3.
Comparison of backbone RMSD (Å) for EndoT5-Zn2+ (ο) and EndoT5-Zn2+Ca2+ (•) structure ensembles. Each ensemble contains 20 structures.
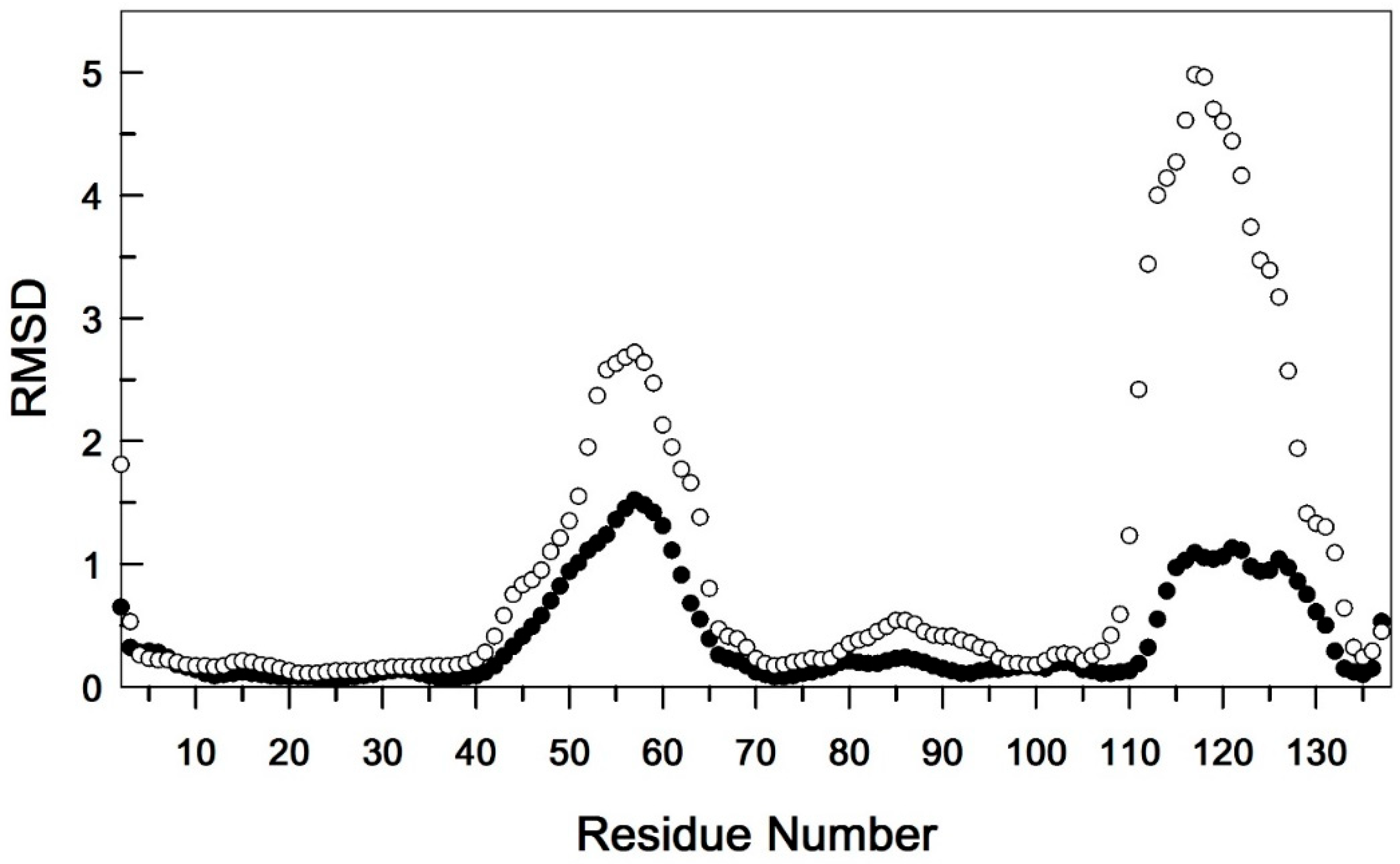
Figure 4.
Heteronuclear 15N{1H} NOE values measured for backbone 15N-1H amide pairs as a function of residue number. EndoT5-Zn2+(A) and EndoT5-Zn2+Ca2+(B). Secondary structural elements are shown at the top of the plot. Cylinders indicate α-helical sections, arrows indicate β-structural sections. Sequence fragments forming the globular domain are shown in blue, whereas regions located in extended loops are shown in red.
Figure 4.
Heteronuclear 15N{1H} NOE values measured for backbone 15N-1H amide pairs as a function of residue number. EndoT5-Zn2+(A) and EndoT5-Zn2+Ca2+(B). Secondary structural elements are shown at the top of the plot. Cylinders indicate α-helical sections, arrows indicate β-structural sections. Sequence fragments forming the globular domain are shown in blue, whereas regions located in extended loops are shown in red.
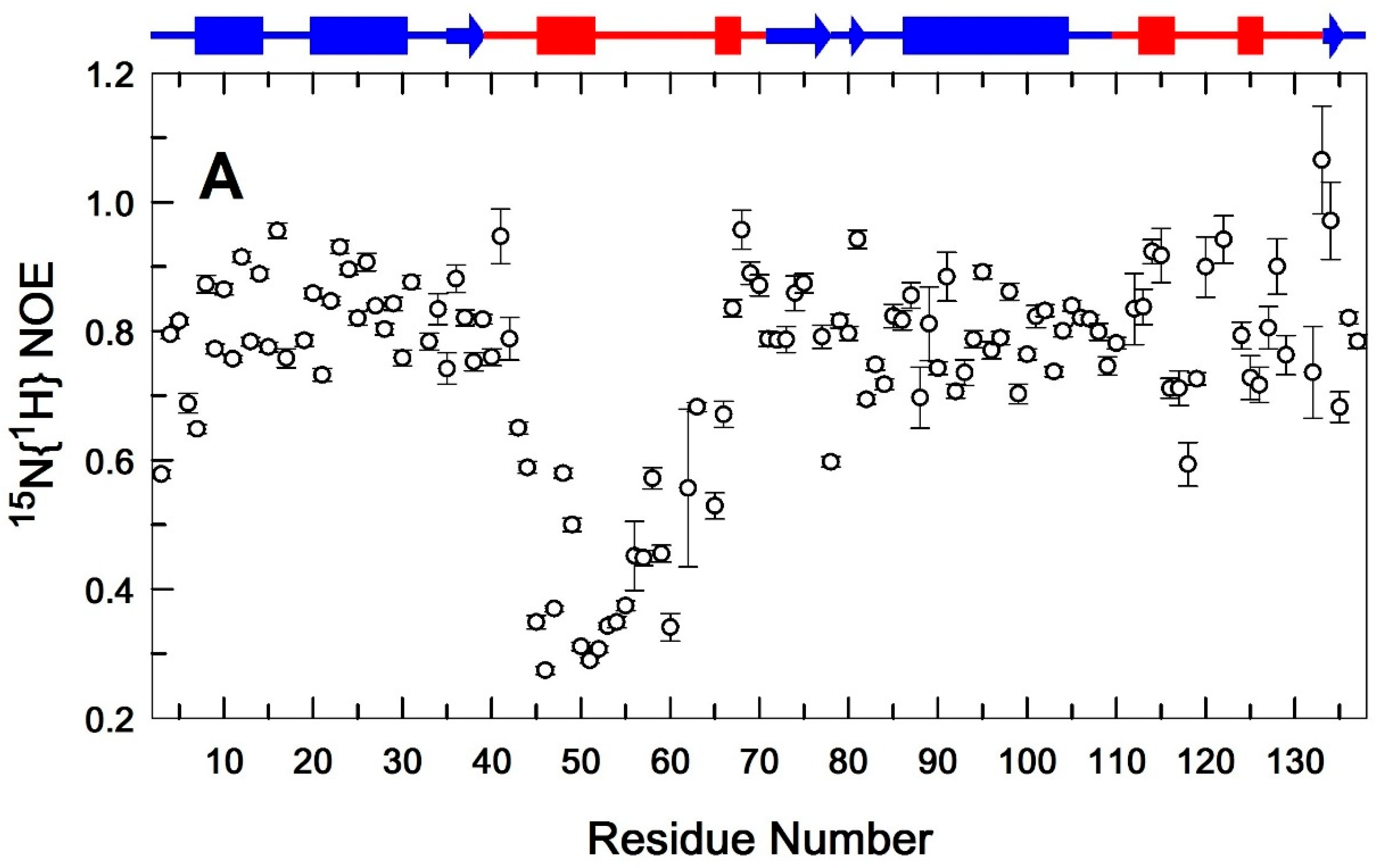
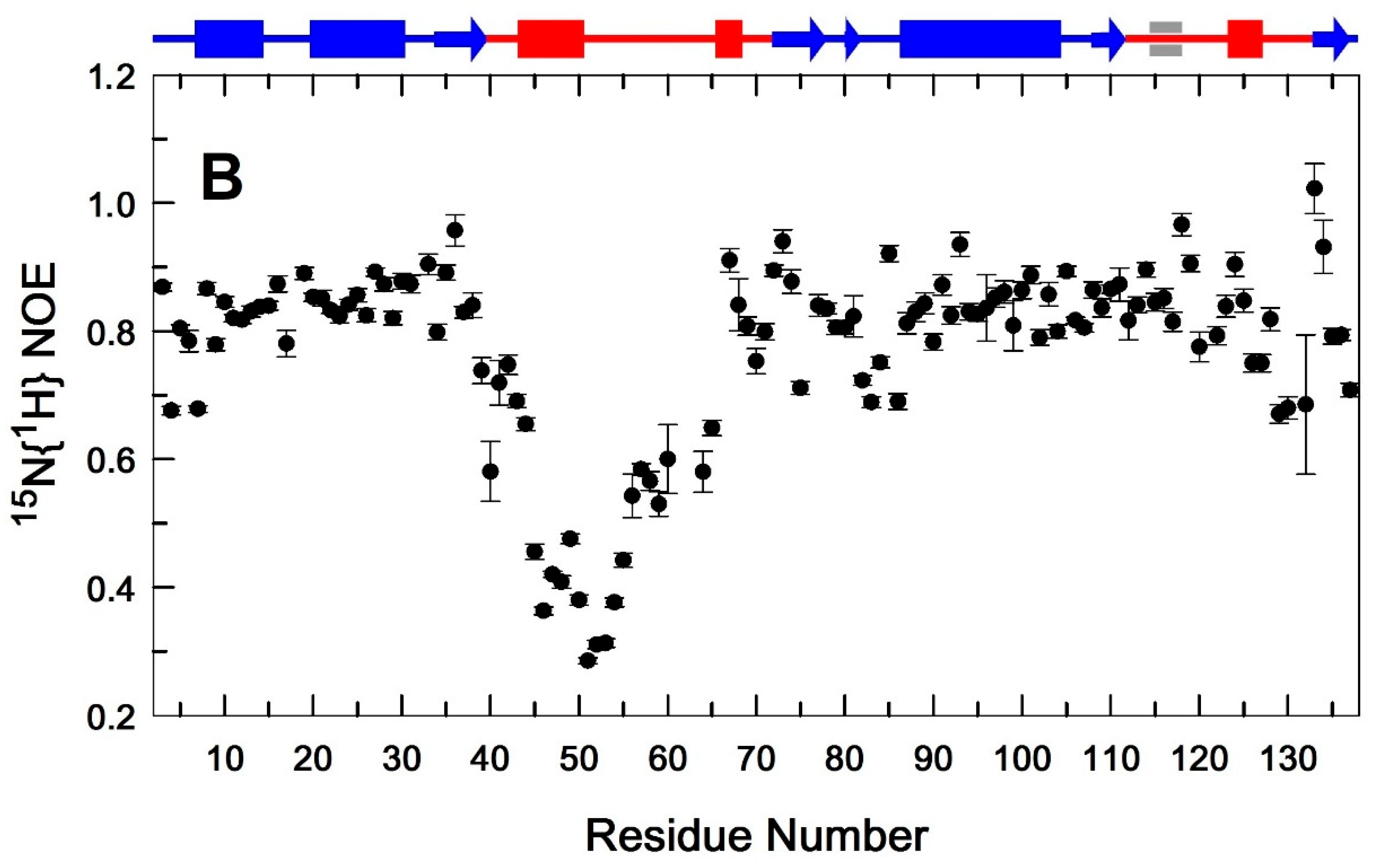
Figure 5.
Comparative analysis of two EndoT5 structures: A. Zn2+ form (2MXZ), B. Zn2+Ca2+ form (8P3A). Zn2+ ions are marked in green, Ca2+ ions in lilac. The side chains of ion-coordinating amino acid residues are detailed.
Figure 5.
Comparative analysis of two EndoT5 structures: A. Zn2+ form (2MXZ), B. Zn2+Ca2+ form (8P3A). Zn2+ ions are marked in green, Ca2+ ions in lilac. The side chains of ion-coordinating amino acid residues are detailed.
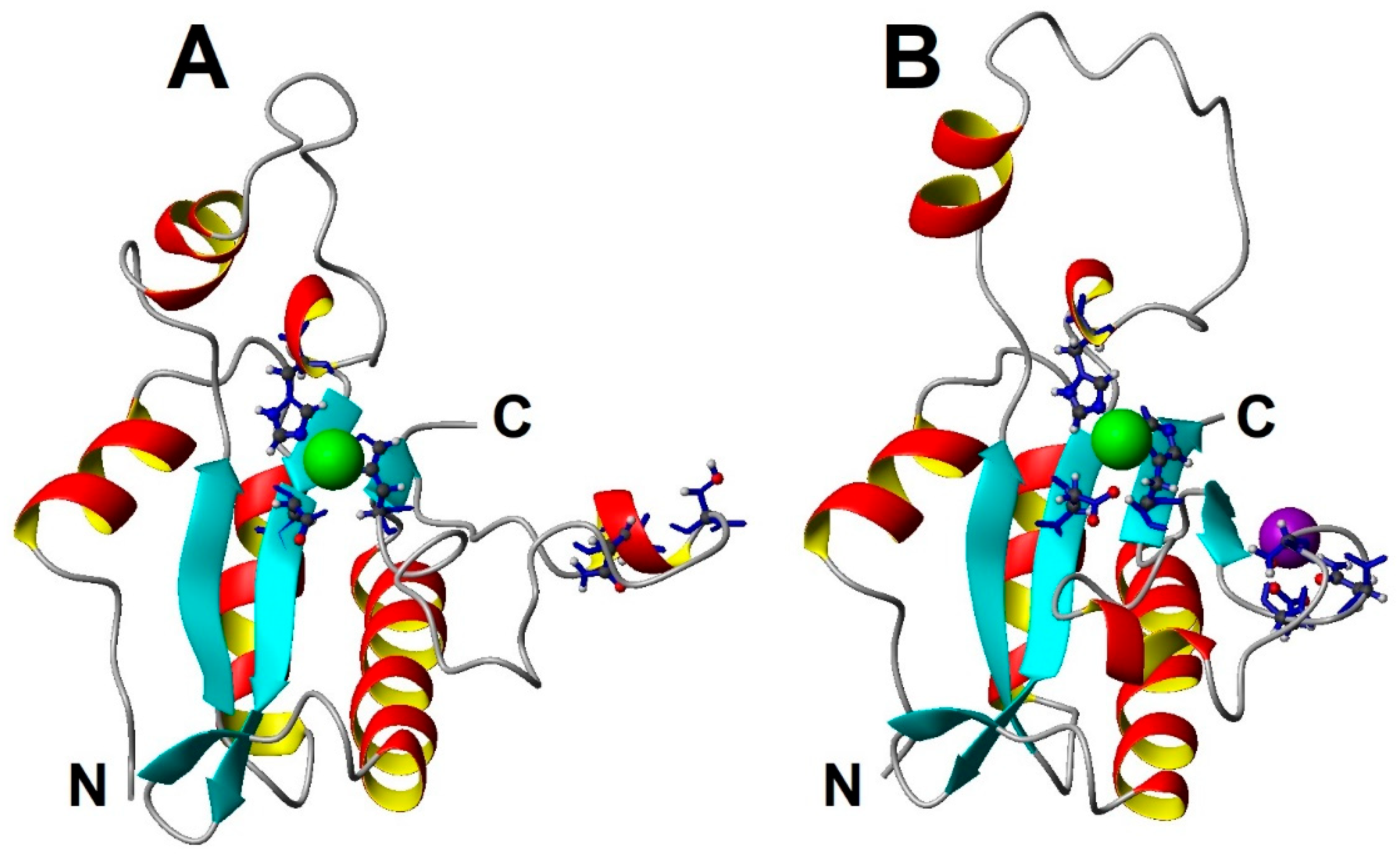
Figure 6.
Conformational ensembles of two EndoT5 forms: A. Zn2+ form (2MXZ), B. Zn2+Ca2+ form (8P3A). In both cases, an overlay of 20 lowest energy members of the corresponding NMR ensembles is presented.
Figure 6.
Conformational ensembles of two EndoT5 forms: A. Zn2+ form (2MXZ), B. Zn2+Ca2+ form (8P3A). In both cases, an overlay of 20 lowest energy members of the corresponding NMR ensembles is presented.
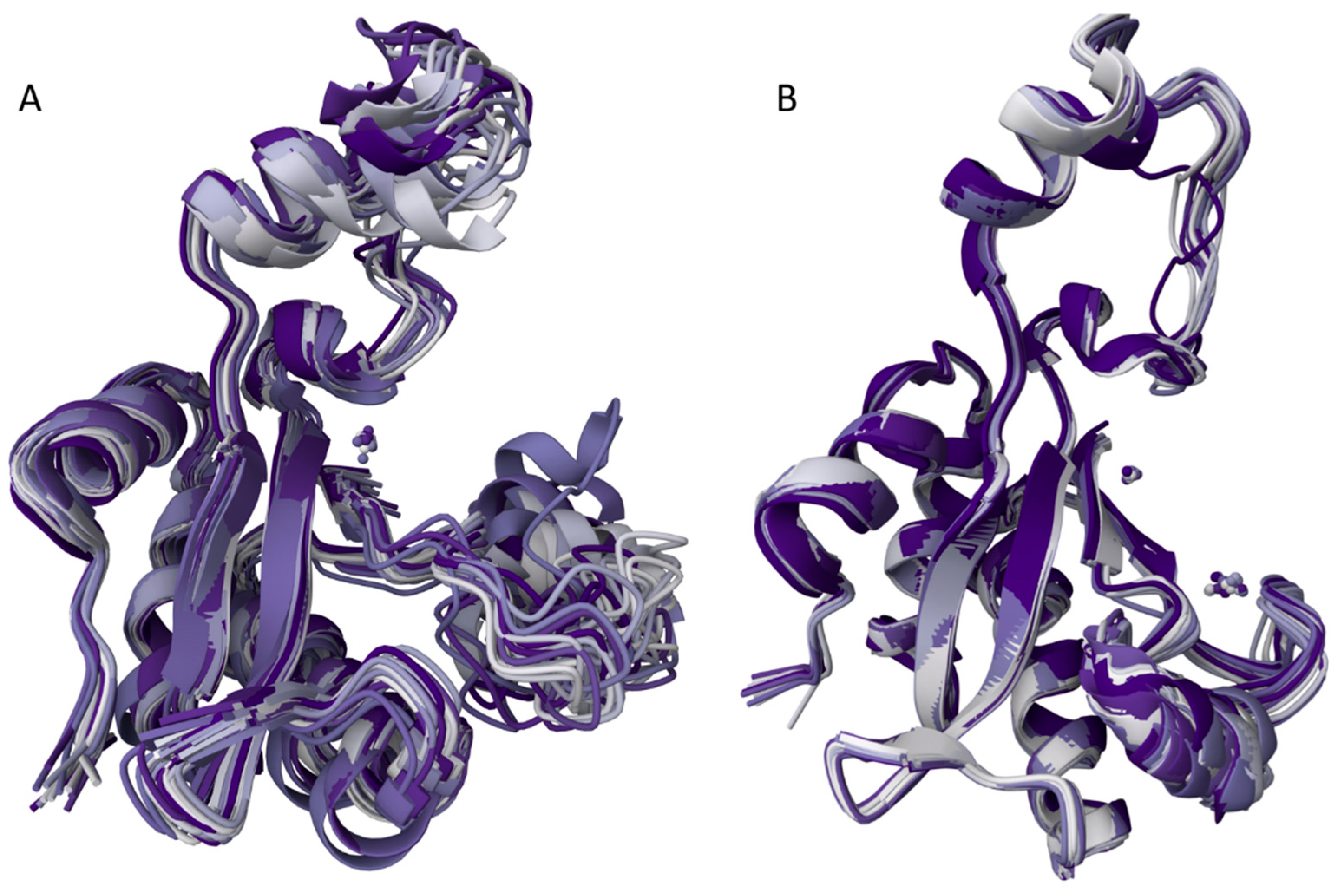

Table 1.
Statistics for calculating the structure of EndoT5-Zn2+Ca2+ a.
| Used restrictions b | |
|---|---|
| Spatial nuclear Overhauser effect (NOE) restrictions | |
| Intra-residual (i-j = 0) | 491 |
| Sequential (|i-j| = 1) | 667 |
| Mid-range (1<|i-j|≤4) | 417 |
| Long-range (|i-j|≥5) | 965 |
| Total number of NOE restrictions | 2540 |
| Restrictions on dihedral angles | |
| Torsion angle (φ/ψ) | 102/103 |
| Restrictions on hydrogen bonds (upper/lower) | 40/40 |
| Restrictions on coordination links | |
| with Zn2+ ion in the active site (upper / lower) | 6/6 |
| with Ca2+ ion in the regulatory loop (upper / lower) | 6/6 |
| Violations of the NOE restrictions | |
| at distances >0.1Å | 46 |
| at distances >0.2Å | 8 |
| to dihedral angles >5° | 0 |
| RMSD (Å) in (residues 5-137) c | |
| main chain atoms | 0.52±0.27 |
| all heavy atoms | 0.96±0.21 |
| Ramachandran Map Analysis d | |
| % of residues in the most preferred regions | 76.3 |
| % of residues in additional allowed regions | 20.9 |
| % of residues in conditionally permitted regions | 2.8 |
| % of residues in prohibited regions | 0.0 |
a Statistics obtained for 20 calculated EndoT5-Zn2+Ca2+ structures with the best objective function. b Spatial constraints derived from contacts. c Root mean square deviation (RMSD) calculated by pairwise comparison of structures in the ensemble with the average structure. d The Ramachandran map for all residues, including the first seven high mobility residues, was obtained using the PROCHECK-NMR program at www.rcsb.org/.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



