
Submitted:
31 July 2024
Posted:
31 July 2024
You are already at the latest version
Abstract
Lung cancer, including both non-small cell lung cancer and small cell lung cancer, remains the leading cause of cancer-related mortality worldwide representing 18% of the total cancer deaths in 2020. Many patients are identified already at an advanced stage with metastatic disease and have a worsening prognosis. Recent advances in the genetic understanding of lung cancer have opened new avenues for personalized treatments and targeted therapies. This review examines the latest discoveries in the genetics of lung cancer, discusses key biomarkers, and analyzes current clinical therapies based on this genetic information. It will conclude with a discussion of future prospects and potential research directions.
Keywords:
lung cancer
; genetic discoveries
; target therapy
; personalized treatments
1. Introduction
Lung cancer is a neoplasm that originates from the cells of the respiratory system, including bronchi, bronchioles, and alveoli. It usually presents as a mass that can grow to obstruct airflow or cause bleeding in the lungs or bronchi. Lung cancer is one of the leading causes of cancer mortality globally, and its high incidence and often unfavorable prognosis make it a significant public health issue. According to the latest statistics, lung cancer is responsible for approximately 2.5 million new cases and 1.8 million deaths per year, accounting for nearly 18% of all cancer deaths worldwide. This high mortality rate is largely due to the often late diagnosis and the aggressive nature of the disease [1,2]. In the United States, lung cancer is the most common cause of cancer death among men and has surpassed breast cancer as the leading cause of cancer death among women. In Italy, approximately 35,000-40,000 new cases are reported annually per 100,000 inhabitants, with a mortality rate of 81 per 100,000 among men and 12 per 100,000 among women. Its incidence is continuously increasing in industrialized countries due to associated risk factors, including tobacco smoking, exposure to toxic substances such as asbestos and radon, and air pollution. Extensive case studies have demonstrated the relationship between lung cancer and smoking, and it is estimated that heavy smokers (more than 40 cigarettes per day) have a 60 times higher risk of developing the disease compared to non-smokers. This risk decreases with the number of years since smoking cessation [3,4,5]. Non-smokers are also at risk from secondhand cigarette smoke. Other causes include smog and air pollution produced by the combustion of petroleum derivatives, as well as processes involving the use of specific metals (nickel, chromium) and radioactive substances. Many occupational substances are recognized as lung carcinogens, although they are less significant than tobacco. Workers in tar, railways, and refineries are particularly at risk. Chemical substances tend to remain in the lungs for a long time due to their stability and difficulty in being eliminated.
Among the most implicated inorganic compounds is asbestos, followed by others with lesser frequency, including arsenic, chromium, nickel, and cadmium [8]. To date, the histological classification and staging of lung cancer by the World Health Organization (WHO) is constantly being updated and serves as the foundation for therapeutic advances, facilitating the development of targeted immunotherapies and ensuring precise diagnoses. Epidemiological data on cancer provide essential information for the prevention, diagnosis, and management of the disease, supporting health measures. In the latest update, various advanced diagnostic methods have allowed for more accurate pathological and genetic classification of lung tumors, offering improved therapeutic options. In general, lung cancer is primarily distinguished into two categories: non-small cell lung carcinoma (NSCLC) and small cell lung carcinoma (SCLC). NSCLC accounts for about 85% of cases and includes subtypes such as adenocarcinoma, squamous cell carcinoma (SQ), and large cell carcinoma (LC). Although less common, SCLC is known for its rapid growth and spread, accounting for about 10-15% of cases [9,10,11] Figure 1.
1.1. Lung Adenocarcinoma
Lung adenocarcinoma is a type of NSCLC that originates from the glandular cells of the lungs. It is the most common type of lung cancer, accounting for about 40% of NSCLC cases [2]. This tumor tends to develop in the peripheral areas of the lungs and is often diagnosed at an advanced stage, as it can be asymptomatic in the early stages [12]. The causes of pulmonary adenocarcinoma are multifactorial; cigarette smoking is the main risk factor, but this type of carcinoma can also affect non-smokers, with a higher incidence in women and young people [6,13]. Other risk factors include exposure to chemicals such as asbestos, radon, and air pollution, as well as a genetic predisposition [6,7,8]. The diagnosis of pulmonary adenocarcinoma requires a combination of clinical, radiological, and histological examinations. Imaging techniques such as chest X-rays, computed tomography (CT), and magnetic resonance imaging (MRI) are used to identify and evaluate the extent of the tumor [14,15]. Pulmonary biopsy, which takes a sample of tumor tissue for analysis, is essential to confirm the diagnosis and determine the specific characteristics of the tumor, including the presence of genetic mutations that may influence therapeutic choice [14,35].
The treatment of pulmonary adenocarcinoma depends on the stage of the disease and the general condition of the patient. Options include surgery, chemotherapy, radiotherapy, and targeted therapies. In cases where the tumor is localized and resectable, surgery offers the best chance for a cure [15,16]. Chemotherapy and radiotherapy are often used as adjuvant treatments to reduce the risk of recurrence [17,18]. Targeted therapies and immunotherapy are revolutionizing lung cancer treatment, offering new hope, especially for patients with specific genetic mutations [19,20]. The prognosis for patients with pulmonary adenocarcinoma varies greatly depending on the stage at which it is diagnosed. In cases of early diagnosis, long-term survival rates are significantly better. However, many patients are diagnosed at advanced stages, when treatment is more complicated and survival prospects are reduced [21].
1.2. Squamous Cell Carcinoma
Squamous cell carcinoma (SCC) is a type of NSCLC that originates from the squamous cells lining the airways of the lungs. It accounts for approximately 25-30% of all NSCLC cases and is strongly associated with tobacco smoking, being more common in men and long-term smokers [2]. This type of carcinoma often develops in the central airways and can cause bronchial obstructions. The main risk factor for pulmonary squamous cell carcinoma is cigarette smoking, responsible for the majority of cases [21]. However, exposure to chemicals such as asbestos, radon, and air pollution, as well as a family history of lung cancer, can also increase the risk [6,8]. Pulmonary squamous cell carcinoma is characterized by slower growth compared to other types of lung cancer, but it can still metastasize to other parts of the body [23]. Symptoms of pulmonary squamous cell carcinoma can vary depending on the location and extent of the tumor [21]. The most common symptoms include persistent cough, often with bloody sputum, breathing difficulties, chest pain, and recurrent lung infections such as pneumonia. Other symptoms may include weight loss, fatigue, and bone pain, especially if the cancer has spread to other areas of the body [24].
The diagnosis of pulmonary squamous cell carcinoma involves a combination of clinical, radiological, and histological examinations. Imaging, such as chest X-ray and computed tomography (CT), is used to visualize the tumor mass and assess its extent [23]. Bronchoscopy, which allows direct examination of the airways and tissue sampling, is often used to confirm the diagnosis through histopathological analysis of the sample [23,25]. Other tests, such as open lung biopsy or needle biopsy, may be necessary to obtain a definitive diagnosis [25,35]. The treatment of pulmonary squamous cell carcinoma depends on the stage of the disease and the patient's overall condition. In cases where the tumor is localized, surgery to remove the affected part of the lung may offer the best chance for a cure [16,19]. Radiotherapy and chemotherapy are often used as adjuvant or neoadjuvant treatments to reduce tumor size before surgery or to eliminate any residual cancer cells after surgery. In advanced cases where surgery is not possible, chemotherapy and radiotherapy remain the main treatments [19]. Recently, targeted therapies and immunotherapy have shown promising results, offering new hope, especially for patients with specific genetic mutations or particular immune profiles [19,20]. The prognosis for patients with pulmonary squamous cell carcinoma varies significantly depending on the stage at which it is diagnosed [27]. Early diagnosis significantly improves the chances of successful treatment and long-term survival. However, since many patients are diagnosed at advanced stages due to the insidious nature of the initial symptoms, the prognosis generally remains reserved [25].
1.3. Large Cell Lung Carcinoma
Large cell lung carcinoma (LCLC) is a type of NSCLC characterized by the presence of large and anaplastic tumor cells that do not fall into the histological categories of squamous cell carcinoma or adenocarcinoma [29,30]. It accounts for about 10-15% of NSCLC cases and can manifest in any part of the lung, although it tends to develop more frequently in peripheral areas [9,28]. The primary causes of large-cell lung carcinoma are similar to those of other types of lung cancer, with cigarette smoking being the main risk factor [31]. Exposure to carcinogens such as asbestos, radon, and air pollutants, as well as genetic predisposition, can also increase the risk of developing LCLC [6,7,8]. Despite its prevalent association with smoking, this type of carcinoma can also occur in non-smokers [6]. Large cell lung carcinoma is known for its rapid growth and ability to metastasize to other parts of the body, such as lymph nodes, liver, bones, and brain [32,33]. Symptoms of LCLC are often similar to those of other types of lung cancer and can include persistent cough, shortness of breath, chest pain, hemoptysis, and unintentional weight loss. Due to its aggressive nature, symptoms can progress quickly, often leading to a diagnosis at an advanced stage [33].
The diagnosis of large-cell lung carcinoma requires a thorough clinical evaluation and a series of diagnostic tests. Imaging studies such as chest X-ray, computed tomography (CT), and magnetic resonance imaging (MRI) are used to identify the presence of the tumor and assess its extent [34]. Definitive diagnosis is obtained through a biopsy, which involves the removal of a tissue sample for histopathological analysis [35]. In some cases, bronchoscopy or CT-guided fine needle biopsy is used to obtain tissue samples. The treatment of large-cell lung carcinoma depends on the stage of the disease at the time of diagnosis and the overall condition of the patient. In the early stages, surgery is the main treatment option and may include the removal of part of the lung (lobectomy) or the entire lung (pneumonectomy) [16,19]. However, due to the aggressive nature of LCLC, many patients are diagnosed at advanced stages when surgery is no longer feasible. In these cases, chemotherapy and radiotherapy are the primary treatment options. Recently, targeted therapies and immunotherapy have shown potential in the treatment of large-cell lung carcinoma, offering new hope for patients, especially those with specific genetic alterations [19,20]. The prognosis for patients with large cell lung carcinoma is generally unfavorable, particularly because diagnosis often occurs at an advanced stage of the disease. Long-term survival depends on various factors, including the stage of the tumor at the time of diagnosis, the patient's age and health conditions, as well as the response to therapies [32,33]
1.4. Small Cell Lung Cancer
Small cell lung cancer (SCLC) is a type of lung carcinoma that accounts for about 10-15% of all lung cancers. It is characterized by rapid growth and an early tendency to metastasize. This type of cancer is closely associated with tobacco smoking, with over 95% of cases diagnosed among smokers or former smokers [37]. The most common clinical manifestations at the time of diagnosis include the presence of central tumor masses, involvement of the mediastinum, and spread outside the chest in 75-80% of cases. Patients may present with symptoms such as cough, wheezing, difficulty breathing, coughing up blood, weight loss, pain, fatigue, and paraneoplastic syndromes [37,39]. SCLC is distinguished by the presence of small, round or oval tumor cells with scant cytoplasm and prominent nuclei. It is highly aggressive and tends to spread rapidly to regional lymph nodes and other parts of the body, including the liver, bones, and brain 38.. At the molecular level, SCLC is often associated with mutations in the TP53 and RB1 genes, which are crucial in regulating the cell cycle and preventing tumor growth. The loss of function of these genes contributes to the uncontrolled proliferation of tumor cells [37]. The diagnosis of SCLC is based on a multimodal approach that includes clinical evaluation, radiological imaging, and lung tissue biopsy. Computed tomography (CT) and positron emission tomography (PET) are crucial tools for staging and assessing the extent of the disease. Diagnostic confirmation is obtained through histopathological examination, which reveals the typical cytological characteristics of SCLC [40,41,42]. The treatment of SCLC depends on the stage of the disease at the time of diagnosis. Traditionally, SCLC is classified into two main stages: limited disease and extensive disease. For limited disease, the standard treatment consists of a combination of chemotherapy (often with etoposide and platinum) and thoracic radiotherapy [42,43]. This approach can potentially be curative in some patients. For extensive disease, the main treatment is systemic chemotherapy, as radiotherapy has a limited role. Recently, immunotherapy with immune checkpoint inhibitors, such as atezolizumab, has shown promising results when combined with standard chemotherapy [43,44].
1.5. Other Less Common Types of Lung Cancer: Pulmonary Carcinoid Tumors
Pulmonary carcinoid tumors are rare neuroendocrine cell neoplasms of the lung, representing about 1-2% of all lung tumors [9]. They are divided into typical carcinoids (TC) and atypical carcinoids (AC), with slower growth compared to other lung carcinomas [9,45]. The cause of pulmonary carcinoid tumors is not completely known. Unlike other lung tumors, they are not strongly associated with tobacco smoking. Genetic mutations, such as in the MEN1 and TP53 genes, may be involved [46]. Symptoms vary and can include persistent cough, hemoptysis, bronchial obstruction, and hormone secretion syndromes, such as carcinoid syndrome. Some tumors are asymptomatic [45]. Diagnosis is based on radiological imaging (CT, MRI) and histopathological confirmation through biopsy. Octreotide scintigraphy can identify somatostatin receptors on tumor cells [47]. Surgery is the main treatment for typical carcinoids, while for atypical carcinoids, chemotherapy or radiotherapy may also be necessary [48]. For advanced cases, immunotherapy and somatostatin analogs are considered [49]. The prognosis is generally favorable for typical carcinoids with a 5-year survival rate of 80-90%. Atypical carcinoids have a worse prognosis, with a 5-year survival rate of 50-70% [48].
This comprehensive review seeks to provide insights into the genetics of lung cancer and analyzes current clinical therapies and potential future perspectives based on genetic information.
2. Genetic Landscape of Lung Cancer
Lung cancer is a highly complex and heterogeneous disease with genomic diversity in each histological subtype, a complicated mutation spectrum, and a genetic regulatory mechanism that has not yet been elucidated [50]. It is increasingly evident that the genetic alterations in lung cancers are numerous but also complex and varied in terms of their timing and mechanisms. New findings highlight molecular lesions unique to each of the two primary lung cancer subtypes, as well as those shared by both. Some genes are implicated in both subtypes, while others are specific to one subtype and may influence its differentiation [51].
Identifying the genetic mutations that drive lung cancer progression is important to better understand the underlying mechanisms and to develop personalized therapies that target tumor cells and inhibit pathways that promote tumor growth and progression. The development and implementation of high-throughput genomic technologies, enabling the identification of key genetic mutations that drive cancer progression at a lower cost and with faster turnaround times than previously possible, has made it possible to have a more complete view of the mutations involved in lung cancer [52,53].
2.1. Main Driver Genes
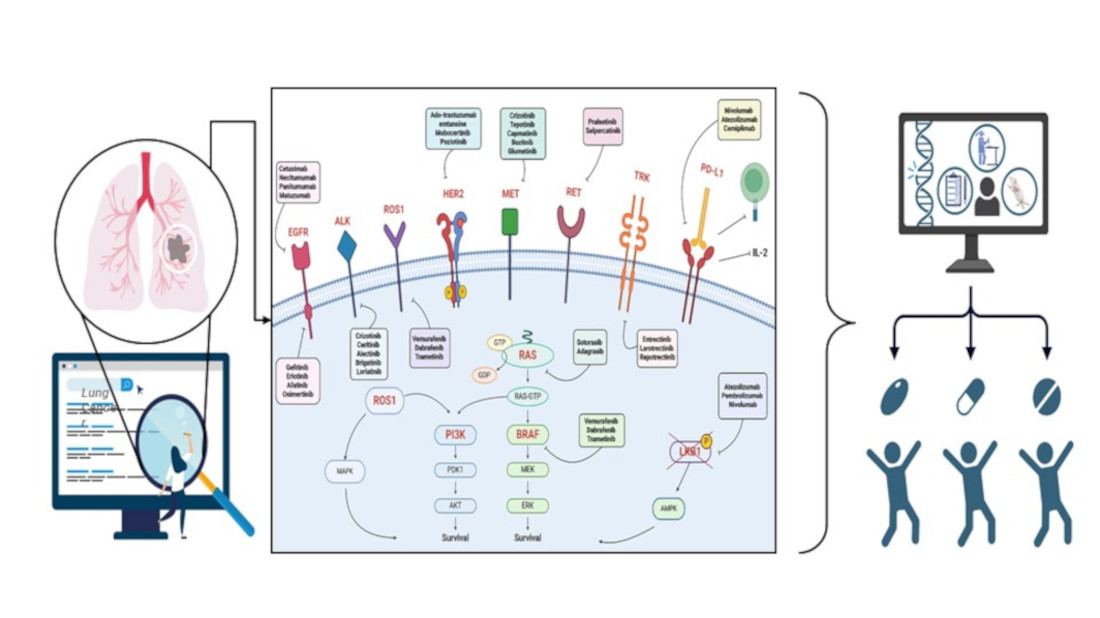
The main druggable genetic alterations in non-small cell lung cancer involved EGFR (epidermal growth factor receptor), KRAS (Kirsten rat sarcoma viral oncogene homolog), ALK (anaplastic lymphoma kinase), BRAF (V-raf murine sarcoma oncogene homolog B1) and ROS1 (c-ros oncogene 1) genes.
EGFR is a transmembrane protein with intrinsic tyrosine kinase activity, crucial for regulating cell proliferation, survival, and differentiation. Activating mutations in the EGFR gene were first identified in non-small cell lung cancers (NSCLC) in 2004, marking one of the most significant advancements in understanding the molecular biology of these tumors [54]. EGFR gene mutations in lung tumors are primarily found in exon 19 (deletions) and exon 21 (L858R substitution) [55]. Specifically, these mutations lead to constitutive activation of the receptor's tyrosine kinase, independent of ligand presence, promoting growth, survival, and cell proliferation through downstream signaling pathways such as PI3K/AKT and RAS/RAF/MEK/ERK. EGFR mutations are more common in women, non-smokers, patients with adenocarcinoma histology, and Asian patients [56,67]. In these populations, EGFR mutation prevalence can reach up to 50%, whereas in Caucasian patients it ranges around 10-15% [55,56]. The discovery of activating mutations of the epidermal growth factor receptor (EGFR) in patients with lung adenocarcinoma led to the development of a new family of biological agents, called tyrosine kinase inhibitors (TKIs), that have revolutionized the clinical management of LC patients.
Mutations in the KRAS gene are among the most common genetic alterations in lung cancers, particularly in NSCLC [65,66]. KRAS is a proto-oncogene that encodes a GTPase protein, which is involved in signal transduction that influences cell proliferation, differentiation, and survival. Under normal conditions, the KRAS protein alternates between an active (GTP-bound) state and an inactive (GDP-bound) state [64,66]. Mutations, however, result in a constitutively active protein that continuously stimulates downstream signaling pathways, such as the MAPK and PI3K-AKT pathways, promoting uncontrolled growth of cancer cells [65,66]. KRAS mutations are present in approximately 20-25% of NSCLC cases, with a higher prevalence in patients with adenocarcinoma compared to those with other histological subtypes, suggesting a strong correlation with tobacco exposure [67]. The most common types of mutations in lung cancers are primarily concentrated in codons 12, 13, and 61, with G12C, G12D, and G12V being the most frequent among these [68]. Each specific mutation can differently influence tumor biology and treatment response, generally leading to an unfavorable prognosis for the patient [69].
The ALK gene encodes a receptor tyrosine kinase that is involved in the regulation of cell growth. Originally identified in anaplastic large cell lymphomas [57], ALK alterations are now also recognized in various solid tumors, including lung cancers [58]. ALK mutations in lung cancers are often the result of genetic rearrangements, leading to the formation of fusion proteins with oncogenic activity [58]. In lung cancers, the most common rearrangement involves the fusion of the ALK gene with EML4 (echinoderm microtubule-associated protein-like 4). This rearrangement, known as EML4-ALK, was first identified in 2007 and results in the production of a fusion protein with constitutive tyrosine kinase activity, promoting cell proliferation and tumor survival [58,59]. This translocation accounts for about 5% of lung carcinomas, and among the multiple genetic alterations involved in the development of these tumors, it has emerged as an important biomarker [63]. In addition to the known rearrangement with EML4, other ALK fusion partners have been identified, including KIF5B, TFG, and KLC1 [60,61,62]. Although these rearrangements are less common, they still contribute to the pathogenesis of lung cancer through similar mechanisms of aberrant activation of the ALK pathway. ALK-positive NSCLC exhibits highly aggressive behavior and is often diagnosed at advanced stages compared to wild-type patients.
Despite BRAF gene mutations in lung carcinoma being identified before EML4-ALK translocations, there have been few clinical studies completed on this type of lung carcinoma with BRAF mutation. To date, BRAF gene mutations are recognized as one of the genetic factors contributing to the development of various types of cancer, including lung cancer [71]. BRAF is a gene that encodes a serine/threonine kinase protein, which is part of the MAPK/ERK signaling pathway involved in the regulation of cell growth, differentiation, and survival [72]. Mutations in this gene can lead to abnormal cell signaling and, consequently, uncontrolled proliferation of cancer cells. Specifically, conformational changes occur in the BRAF protein, causing constitutive activation of the MAPK/ERK signaling pathway [73]. This persistent activation stimulates cell proliferation, inhibits apoptosis, and promotes the survival of cancer cells. Several BRAF mutations have been identified, the most common being the substitution from valine to glutamate at codon 600 (V600E), which accounts for over 90% of BRAF mutations in melanoma [74]. This mutation, in particular, creates a form of BRAF that is independent of upstream regulatory signals, leading to incessant kinase activity. BRAF gene mutations are present in a minority of lung cancer cases, with an estimated prevalence of about 1-3% in NSCLC. These mutations have been identified mainly in adenocarcinoma subtypes [75]. BRAF gene mutations can be classified into three main classes. Class I BRAF mutations are commonly identified in human tumors and represent about 50% of BRAF mutations in lung tumors [75,76]. They affect the V600 amino acid, including V600D/E/K/R, and act as active monomers independent of RAS, resulting in marked activation of BRAF kinase activity and constant activation of the MAPK pathway [77]. Class II and Class III mutations, on the other hand, are non-V600 mutations and represent 50-80% of BRAF mutations in lung tumors [75]. Class II mutations are mainly found in the activation segment (such as K601, L597) or in the P-loop (such as G464, G469) [78,79], and have intermediate to high kinase activity. Class III mutations have been found in the P-loop (G466), the catalytic loop (N581), and the DFG motif (D594, G596) [78,79], and are characterized by absent or low kinase activity. In terms of prognosis, class I BRAF mutations generally show a slightly better prognosis compared to class II and III mutations. The latter are linked to more aggressive behavior, a less favorable clinical course, and earlier disease progression following initial chemotherapy. BRAF mutations globally exhibit a significant predominance among males (61%) and individuals who have a history of smoking (81%), with varying frequencies across different mutational classes.
The ROS1 gene encodes a receptor tyrosine kinase involved in the regulation of cell growth and intracellular signaling. Initially, it was found to be implicated in the development of human glioblastoma [80]. and was later recognized in other malignant neoplasms, including lung, ovarian, and gastric tumors [81,82,83]. ROS1 gene rearrangements are described in 1-2% of patients with NSCLC and were first identified in 2007 [80,81]. The majority of recorded patients are young, light smokers, or non-smokers. These rearrangements are more frequent in adenocarcinoma but have also been reported in large-cell carcinoma cases [84,85]. ROS1 mutations are primarily represented by gene translocations, where a part of the ROS1 gene fuses with another gene, creating a fusion oncogene. Among the most common translocations are fusions between ROS1 and the genes CD74, SLC34A2, TPM3, and SDC4 [81,84,86,87]. These fusions result in the production of potent oncogenic drivers that promote cell proliferation, activation, and cell cycle progression by activating downstream signaling pathways, accelerating the development and progression of lung carcinoma due to the upregulation of JAK/STAT, PI3K/AKT, and MAPK/ERK signaling pathways [88]. Despite the low frequency of ROS1 gene mutations, their identification is clinically significant as patients harboring this mutation tend to respond well to specific tyrosine kinase inhibitors [89]. This ensures that the therapeutic choice for the patient is targeted and precise.
2.2. Emerging Biomarkers
Lung cancer is a heterogeneous disease that requires precise patient stratification to optimize treatment options. In recent years, specific biomarkers such as PD-L1 (Programmed death-ligand 1), MET (Mesenchymal Epithelial Transition), RET, NTRK, PIK3CA, HER2 (human epidermal growth factor receptor) and STK11 have gained attention as crucial tools for the diagnosis, prognosis, and therapeutic management of lung cancer [70].
Programmed death-ligand 1 (PD-L1), also known as CD274, is considered an immune checkpoint that facilitates the suppression of the antitumor immune response. PD-L1 can be present on the surface of various cells, including macrophages, antigen-presenting cells, B and T lymphocytes, epithelial cells, muscle cells, and endothelial cells [90]. The factor inducing its expression is interferon-gamma (IFN-γ), released by CD8 T cells [91]. The receptor of PD-L1 (PD-1) is primarily expressed by activated cytotoxic T cells [90]. When PD-L1 binds to the PD-1 receptor on activated T cells, immune system suppression occurs [90,92]. This interaction prevents autoimmune responses in peripheral tissues during inflammations, contributing to maintaining immunological tolerance [93]. The ligand-receptor complex triggers two reactions that inhibit the immune response: the first is the inhibition of interleukin 2 (IL-2) synthesis [94], and the second is related to the inhibition of the T cell receptor, known as the "stop signal," which can modify the duration of contact between T cells and target cells or antigen-presenting cells [95]. The PD-L1-PD-1 interaction is exploited in carcinogenesis to evade the immune system [96]. Elevated levels of PD-L1 have been detected on the surface of various tumor cell types, including NSCLC. This mechanism allows tumor cells to escape the immune response; PD-L1 acts as a pro-tumorigenic factor by activating proliferative and survival signaling pathways, also promoting tumor progression [97]. In lung tumors, PD-L1 expression can be regulated by various mechanisms, including genetic mutations, gene amplifications, and transcriptional regulation induced by inflammation or hypoxia. PD-L1 overexpression has been associated with a worse prognosis and greater tumor aggressiveness [97]. Mutations in the PD-L1 gene in lung tumors are relatively rare compared to other mechanisms of overexpression. However, some mutations can significantly impact PD-L1 function and the immune response. Specifically, point mutations in PD-L1 have been found that can influence its ability to interact with PD-1, altering the antitumor immune response. Some studies have identified that these mutations lead to a loss of function or stabilization of the protein [98]. Additionally, amplifications of the 9p24.1 locus, where PD-L1 resides, have been observed in a fraction of lung tumors. These amplifications are often correlated with high PD-L1 expression and have been associated with an unfavorable response to PD-1/PD-L1 inhibitor therapies [99,100]. Finally, although rare, gene translocations involving PD-L1 can lead to the fusion of PD-L1 with other genes, influencing its expression and function. These translocations can generate chimeric isoforms with new functional properties for tumor cells [101]. Due to the clinical relevance in the context of immunotherapy observed in lung tumors, the molecular bases of PD-L1 regulation and its role in the tumor microenvironment are still being explored. The aim is to understand hidden molecular mechanisms to identify new therapeutic targets and treatment combinations that can improve patient outcomes [102].
The MET gene encodes a tyrosine kinase receptor known as hepatocyte growth factor receptor (HGFR). This receptor plays a crucial role in regulating cell proliferation, motility, morphogenesis, and survival [103]. Upon binding to its ligand HGF, MET undergoes dimerization and auto-phosphorylation, activating important intracellular signaling pathways such as PI3K/AKT and MAPK/ERK [104]. Alterations in the MET gene, including mutations, amplifications, and fusions, are implicated in the pathogenesis of various cancers, including lung cancers [103,104,105]. Point mutations in the tyrosine kinase domain of MET can lead to constitutive activation of the receptor, promoting the growth and survival of tumor cells [106]. Among these, the most studied is the MET exon 14 skipping mutation (METex14), which prevents receptor degradation and results in prolonged MET signaling [107]. MET exon 14 skipping mutations occur in approximately 3-4% of NSCLC cases and are generally not associated with other driver mutations [108,109,110]. Amplification of the MET gene, detected in 1-6% of NSCLC patients, leads to an increased number of gene copies and overexpression of the MET receptor with aberrant signaling. This amplification is often observed in cases of acquired resistance to treatments with epidermal growth factor receptor inhibitors [110,111,112,113,114]. Recently, rearrangements of the MET gene, including KIF5B-MET fusion, have been identified and although rare, have shown significance in NSCLC. These fusions are present in approximately 0.2% - 0.3% of NSCLC cases and are considered potential driver mutations targetable by specific therapies [115,116,117]. In conclusion, MET gene alterations are associated with poor prognosis and resistance to standard treatments in lung cancers. Therefore, detecting these alterations is critically important for the therapeutic management of patients [118].
The proto-oncogene RET, first identified in 1985 [119], encodes a receptor tyrosine kinase that activates downstream signaling pathways (RAS/MAPK/ERK, PI3K/AKT, and phospholipase C-γ), thereby promoting cellular proliferation, migration, and differentiation [120]. Genetic alterations, including chromosomal rearrangements and point mutations, lead to aberrant RET activation, contributing to tumorigenesis. Specifically, chromosomal rearrangements of RET have been found in approximately 1%-2% of NSCLC patients [87,121]. These alterations result in the formation of chimeric proteins with constitutive oncogenic activity, stimulating downstream signaling pathways that promote tumor cell proliferation and survival [122]. The most commonly identified rearrangement in NSCLC is the KIF5B-RET fusion [87,123,124], although other fusion partners such as CCDC6, NCOA4, TRIM33, and CUX1 have also been identified [125]. Patients with RET fusion-positive NSCLC represent a distinct molecular subgroup with specific clinical and pathological characteristics. RET gene fusions appear to be mutually exclusive with other mutations, including those in EGFR, KRAS, ALK, HER2, and BRAF genes, suggesting independent oncogenic driver roles. Furthermore, initial reports indicate that RET rearrangements are more frequent among younger (< 60 years old), female, non-smoking patients with adenocarcinoma histology [87,123,124,126,127]. In addition to rearrangements, point mutations in the RET gene have also been identified in lung tumors, albeit less commonly than fusions. Similar to rearrangements, these mutations can lead to RET receptor activation, contributing to oncogenesis [22]. Therefore, RET gene mutations represent a significant class of oncogenic alterations in lung tumors, and understanding their molecular mechanisms is facilitating the development of new targeted inhibitors, advancing therapies for RET-positive patients.
Mutations in NTRK genes are emerging as significant molecular markers in lung cancers, influencing diagnosis, prognosis, and therapeutic options [128]. The NTRK1, NTRK2, and NTRK3 genes encode the TRKA, TRKB, and TRKC receptors, respectively. These transmembrane proteins belong to the TRK receptor family and play a crucial role in cellular signaling and neural growth by activating pathways such as PIK3/PLCγ/MAPK [129,130]. NTRK gene fusions are rare but frequent oncogenic alterations, present in up to 1% of all solid tumors [131]. In NSCLC, the frequency of these fusions is approximately 0.1-0.2%, occurring when the 3' sequence of the NTRK gene fuses with the 5' sequence of a fusion partner gene [132]. The resulting fusion protein is aberrantly expressed, constitutively activating the receptor's kinase domain, which leads to persistent activation of cellular signaling pathways necessary for oncogenesis [131,133]. NTRK gene fusions typically exclude other canonical oncogenic mutations, suggesting they act as sole oncogenic drivers in the development and maintenance of their host tumors [134]. Most patients with lung cancers harboring NTRK gene fusions exhibit clinical characteristics similar to those with ALK, RET, or ROS1 fusions [132], and are often found in a younger population with minimal or no smoking history. However, studies have also identified NTRK gene fusions in patients of various ages and with a previous smoking history [132,135]. Additionally, many patients with TRK fusion-positive lung carcinoma have developed metastases in the central nervous system [136]. Generally, NTRK gene mutations in lung cancers can be associated with variable prognoses. Some studies suggest that patients with these mutations may benefit from targeted therapies with NTRK inhibitors, as these inhibitors block aberrant tyrosine kinase activity, thereby reducing tumor growth and improving clinical response [137]. Despite current knowledge, further studies are necessary to evaluate the exact incidence of NTRK gene mutations in different populations and to better understand the clinical implications of these mutations. These mutations represent a promising field for the personalization of therapy in NSCLC patients.
The PIK3CA gene encodes a catalytic subunit of phosphatidylinositol-3-kinase (PI3K), a class of enzymes involved in numerous cellular processes, including cell growth, proliferation, survival, and metabolism. Mutations in the PIK3CA gene have been implicated in various types of cancer [138], including lung cancer, with a frequency of 2-4% in NSCLC cases [138,139]. PIK3CA is often mutated or amplified due to a missense variant that mainly affects the helical binding domain (exon 9, E545K or E542K) or the catalytic subunit (exon 20, H1047R or H1047L) [138,140,141]. These alterations lead to constitutive and PI3K/AKT/mTOR pathway-independent PI3K enzymatic activation, resulting in uncontrolled proliferation and survival of cancer cells with consequent drug resistance [142]. PIK3CA gene mutations in lung adenocarcinomas have not been reported as mutually exclusive. On the contrary, co-occurrence with alterations in EGFR, BRAF, ALK, and more frequently, KRAS genes has been observed. This observation raises the question of whether PIK3CA mutation alone can be a sufficient oncogenic driver for NSCLC tumor formation [143,144]. PIK3CA mutations have been reported to be associated with smoking exposure. Specifically, patients with PIK3CA mutation exhibit greater smoking exposure than patients with EML4-ALK translocation or EGFR mutation, and less smoking exposure than patients with smoking-associated aberrations such as KRAS [145,146]. Additionally, PIK3CA mutation has been observed to occur more frequently in patients with various prior malignancies compared to NSCLC [146]. The presence of PIK3CA mutations in lung cancer has several clinical implications, including worse prognosis due to increased tumor aggressiveness and metastatic potential [142,145]. Furthermore, PIK3CA/EGFR co-mutation has been associated with reduced efficacy of EGFR inhibitors, thus necessitating alternative or combinatorial therapeutic strategies [147]. This underscores the need for further studies to ensure a deeper understanding of molecular mechanisms to improve NSCLC treatment.
Mutations in the HER2 gene in lung tumors are a topic of growing interest in oncological research. HER2, also known as ERBB2, is an oncogene that encodes a tyrosine kinase receptor involved in the regulation of cell growth, differentiation, migration, and apoptosis [148]. Although HER2 alterations are well documented in various types of cancers, such as breast carcinoma, their role in lung tumors has only recently started to emerge [149]. HER2 mutations are relatively rare, found in approximately 1-3% of NSCLC, and are observed more frequently in adenocarcinomas compared to other NSCLC subtypes [22,148,149,150]. HER2 alterations in lung tumors can primarily manifest through two mechanisms: gene amplification and point mutations. Amplification of the HER2 gene leads to overexpression of the HER2 protein on the cell surface, promoting uncontrolled cell proliferation and tumor cell survival [151,152]. The point mutations described to date are insertions within a small stretch of exon 20 with A775_G776insYVMA insertion/duplication at the COOH-terminal end of the αC-helix; these can increase the receptor's enzymatic activity independently of ligand presence, leading to constant proliferative signaling [151,152,153,154]. HER2 mutations in lung tumors are predominantly observed in female, non-smoking patients and are often associated with an unfavorable prognosis [150,153,155]. Patients with these mutations tend to present with more aggressive tumors and a less favorable response to standard therapies [154,155]. For this reason, it is necessary to further investigate HER2 mutations which, although rare, are emerging as mutations of particular interest, especially for the development and progression of adenocarcinoma. Further studies might indeed highlight HER2 as a promising and relevant therapeutic target.
Mutations in the STK11 gene, also known as LKB1, represent a tumor suppressor gene implicated in the autosomal dominant disorder that predisposes individuals to cancer known as Peutz-Jeghers syndrome (PJS) [156]. Located on chromosome 19p13.3, the STK11 gene encodes a protein that functions as a kinase involved in the regulation of energy metabolism, cell growth, apoptosis, and cell polarity. Disruption of these processes is implicated in carcinogenesis. STK11 mutations have been identified as significant genetic events in various cancer types, notably within the context of NSCLC [157,158]. STK11 mutations are recorded in approximately 20-30% of NSCLC cases, with a higher prevalence in adenocarcinoma subtypes compared to squamous cell carcinomas, and they deactivate the LBK1 protein [159]. LKB1 is a key regulator of the AMPK (AMP-activated protein kinase) pathway, a crucial pathway for cellular responses to energy metabolism and stress. It is one of the few known serine/threonine kinases to be inactivated, which implicates dysfunction of the AMPK pathway, leading to dysregulated cell growth and survival under energy-stress conditions [160]. This contributes to the uncontrolled proliferation of cancer cells. STK11 mutations are often associated with a more aggressive tumor phenotype and a poorer prognosis [161]. Additionally, STK11 mutations can influence therapeutic responses. For instance, tumors harboring STK11 mutations tend to exhibit intrinsic resistance to immune checkpoint inhibitors, such as anti-PD-1/PD-L1 antibodies [161,162,163]. This resistance may be due to the decreased presence of tumor-infiltrating lymphocytes (TILs) and a less inflammatory tumor microenvironment, defined as a "cold" immunosuppressive microenvironment [165,166]. Despite the complexity and critical nature of STK11 mutations, ongoing research continues to explore the interactions between STK11 and other molecular pathways to identify additional therapeutic targets and optimize treatment combinations.
3. Therapies Based on Genetic Information
Lung cancer represents one of the leading causes of mortality worldwide. For many years, therapeutic options were limited to surgery, chemotherapy, and radiation therapy, which often had significant side effects and limited efficacy in advanced cases. With the advancement of genetic and molecular research, target therapies have emerged as new therapeutic strategies [167]. Targeted therapies are treatments that specifically act on certain molecules involved in the growth and proliferation of tumor cells. Unlike traditional chemotherapy and radiation therapy, which indiscriminately target all rapidly dividing cells, targeted therapies block specific molecular signals that promote tumor growth [168]. This approach not only improves treatment efficacy but also reduces side effects.
3.1. Targeted Therapy
Targeted therapies for EGFR gene mutations in lung cancers represent one of the most significant and well-studied innovations in the fight against NSCLC [169] including TKIs and monoclonal antibodies. [169,170]. The concept behind TKIs is the use of small molecules capable of disrupting downstream signaling pathways that promote cell proliferation and survival. This effect is achieved either by directly inhibiting the kinase’s catalytic activity by interfering with ATP and substrate binding or by inhibiting receptor activation by blocking its dimerization [171]. Among these, the first-generation drugs targeting the EGFR tyrosine kinase domain were gefitinib and erlotinib [172,173]. Gefitinib binds to the ATP-binding site of the receptor, preventing autophosphorylation and receptor activation. It is used as a first-line therapy in patients with NSCLC and has shown significant improvements in progression-free survival (PFS) compared to traditional chemotherapy in patients with EGFR mutations [174,175]. Erlotinib inhibits EGFR tyrosine kinase like gefitinib, by binding to the ATP-binding region of the receptor and blocking autophosphorylation. It is used both as a first-line therapy and in patients with advanced or metastatic disease [174]. Also, Erlotinib has demonstrated significant improvement in PFS compared to chemotherapy in patients with EGFR mutations [176]. Among second-generation drugs, afatinib, for example, is an irreversible inhibitor of EGFR, HER2, and HER4 tyrosine kinases. Its ability to block multiple members of the ErbB family makes it effective against some forms of resistance developed with first-generation inhibitors [177,178]. Finally, among third-generation drugs, osimertinib, an irreversible inhibitor, is used for treating patients with activating EGFR mutations and the resistance mutation T790M, common in acquired resistance to first- and second-generation TKIs [179]. This has shown significant improvement in PFS and overall survival (OS) compared to first-generation TKIs, and it is particularly effective in patients with brain metastases [180]. This has made it a first-choice drug for patients with advanced disease [179]. A different approach to prevent EGFR activation and signaling is through monoclonal antibodies against TRK or their ligands. These are designed to interrupt the receptor signaling pathway upstream by neutralizing or blocking the ligand, receptor internalization, and via a cytotoxic action. Among those currently available are cetuximab, necitumumab, panitumumab, and matuzumab [181,182].
Most tumors harboring KRAS mutations have often been considered difficult to treat pharmacologically due to the frequency and heterogeneity of these gene mutations. However, two direct inhibitors of KRAS, Sotorasib and Adagrasib, have recently been identified as reliable and promising for treating patients with metastatic KRAS-positive lung cancer. Sotorasib is the first FDA-approved drug that irreversibly and selectively inactivates KRAS G12C, blocking the signaling pathway that promotes tumor growth. Phase 1 and 2 clinical trials demonstrated significant tumor reduction in patients with G12C KRAS-positive NSCLC, with an objective response rate (ORR) of about 37% and a median PFS of 6.8 months [183,184,185,186]. Adagrasib is another specific inhibitor for KRAS G12C currently in advanced clinical development. Like sotorasib, adagrasib binds irreversibly to the mutated protein, blocking cell proliferation. Preliminary clinical trial data showed a response rate similar to sotorasib, with an ORR of 45% in pretreated patients. The median PFS for adagrasib was found to be around 7 months. Additionally, it has shown an effective antitumor effect against brain metastases [187,188,189]. Besides specific inhibitors for KRAS G12C, research is expanding towards other KRAS mutations and associated signaling pathways. Indeed, combination therapies and the development of new drugs promise to further expand therapeutic options, improving survival and quality of life for patients with KRAS-mutated NSCLC.
Therapies for lung cancer with ALK gene mutations currently focus mainly on TKIs. Crizotinib was the first ALK inhibitor approved for the first- and second-line treatment of patients with ALK-positive NSCLC [190]. It showed significant improvements in PFS compared to traditional chemotherapy [191,192]. However, many patients develop resistance to crizotinib, requiring additional therapeutic options [193]. Indeed, ceritinib, a second-generation ALK inhibitor, has demonstrated efficacy in treating ALK-positive NSCLC in patients who have acquired resistance to crizotinib [194,195]. and in patients with advanced or metastatic cancer [196,197]. To date, numerous clinical trials are currently investigating additional ALK inhibitors, such as alectinib, brigatinib, lorlatinib, and therapeutic combinations to further improve outcomes in patients with ALK-positive NSCLC. The goal is to prolong PFS, enhance control of brain metastases, and increase overall survival.
In 2011, the United States approved Vemurafenib (PLX4032) as the first drug against cancer with BRAF mutation [198]. Introduced in 2008, Vemurafenib is a selective inhibitor of the oncogenic kinase B-Raf, targeting melanoma with the BRAF V600E mutation. This drug blocks the RAF/MEK/ERK pathway by inhibiting the activity of the V600E BRAF mutation [199]. In 2015, patients with BRAF-positive NSCLC treated with Vemurafenib showed a PFS of 7.3 months and an ORR of 42% [200]. Consistent with these findings, a phase 2 study in 2017 demonstrated that Vemurafenib improved PFS in previously untreated patients with V600 BRAF-driven NSCLC 201.. Dabrafenib, on the other hand, is a competitive adenosine triphosphate (ATP) inhibitor of BRAF specific for V600 mutations [202], while Trametinib is a MEK inhibitor, a downstream protein in the same signaling pathway [203]. These two drugs have been combined and have shown remarkable efficacy in patients with BRAF V600E-positive NSCLC [204]. Clinical studies have reported that the Dabrafenib-Trametinib combination results in an ORR of 64% and a median PFS of 10.9 months [205]; the high efficacy of this combination has led to the approval of this therapy as the standard of care for this subgroup of patients [206]. In addition to BRAF and MEK inhibitors, further studies are underway to develop new BRAF inhibitors and to explore the efficacy of new combinations with other therapies, which could further enhance treatment response and overcome emerging resistances.
The main targeted treatments approved for lung cancer with ROS1 rearrangement include drugs such as Crizotinib, a tyrosine kinase inhibitor initially approved for ALK-positive patients, which has shown significant efficacy in ROS1-positive patients as well [190,207]. Clinical trials have reported an ORR of 70-80% and a PFS of approximately 19 months. Despite its efficacy, it has been observed that some patients develop resistance to this drug, leading to tumor recurrence. Resistance to Crizotinib arises due to acquired secondary point mutations [209,210], establishing the urgent need to develop new therapies for carriers of this mutation. Entrectinib, a second-generation drug, was subsequently developed in 2016 and immediately showed promising results in clinical studies, recording an overall ORR of 77%, with a median duration of response of 24.6 months [212]. Additionally, it demonstrated efficacy in treating brain metastases [213], making it a viable alternative for the treatment of ROS1-positive patients. Along with Entrectinib, Lorlatinib, a third-generation TKI, has shown excellent results in terms of ORR, median PFS, and treatment of brain metastases [214,215,216]. Numerous clinical trials are ongoing to test the efficacy and safety of combined therapies for ROS1-positive tumors, aiming to address emerging resistances, improve survival, and enhance the quality of life for patients. A schematic representation of the available therapies for the well-known lung cancer-involved genes is shown in Table 1.
Despite the availability and use of assigned drugs for common biomarkers of lung cancer, research in this field continues unabated. Owing to the growing understanding of the genetic landscape of lung cancer, new clinical studies are ongoing to assess the efficacy of experimental drugs on less common but equally significant biomarkers (Table 2). This new wave of targeted therapies represents a breakthrough in the fight against lung cancer, offering personalized treatment with fewer side effects compared to traditional ones.
3.2. Immunotherapy
The application of immune checkpoint inhibitors (ICIs) has dramatically changed the therapeutic landscape for lung cancer, particularly NSCLC. ICIs like nivolumab, pembrolizumab, and cemiplimab, which target the PD-1 receptor, and atezolizumab, durvalumab, and avelumab, which target PD-L1, have shown significant efficacy. Ipilimumab, targeting CTLA-4, has also been explored in combination therapies. Nivolumab enhances effector T cell populations while suppressing PD-1 receptor expression, thus mitigating immune response inhibition [217]. Pembrolizumab binds to the PD-1 receptor, preventing interactions with PD-L1 and PD-L2, and has demonstrated durable antitumor activity and high five-year overall survival rates, especially in patients with high PD-L1 tumor proportion scores [218]. Cemiplimab, approved for use with chemotherapy, offers new treatment strategies for advanced NSCLC, regardless of PD-L1 expression levels, showing significant improvements in survival metrics compared to chemotherapy alone [219]. Atezolizumab, a humanized IgG1 monoclonal antibody, has been effective as a first-line treatment for locally advanced or metastatic NSCLC with high PD-L1 expression, providing a significant survival benefit over traditional chemotherapy [220]. Durvalumab, showing promise in treating both extensive-stage SCLC and NSCLC, has benefited from biomarkers like blood tumor mutational burden to predict clinical advantages [221]. Ipilimumab, although less common as a monotherapy, has shown effectiveness in combination therapies [222]. These agents have proven particularly effective when combined with chemotherapy [223], which addresses tumor and microenvironment heterogeneity and underscores the importance of predictive biomarkers for therapeutic decisions to enhance patient outcomes.
Unfortunately, despite the described clinical benefits, only about 20-60% of treated patients respond to immunotherapy [224]. In addition, immune-related adverse events (irAEs) remain a significant challenge with ICIs, affecting multiple organ systems with varying severity. Common irAEs include thyroid disease, diabetes, pneumonitis, and various autoimmune conditions [225]. Understanding and predicting these toxicities through biomarkers are critical for optimizing patient management and ensuring the safe administration of ICIs. Biomarkers like PD-L1 expression, tumor mutation burden (TMB), and microsatellite instability-high/mismatch repair-deficient (MSI-H/dMMR) status are shown to be effective in forecasting anti-tumor effectiveness and potential toxicities of ICIs [226,227,228].
Programmed death-ligand 1 (PD-L1) plays a crucial role in the immune evasion mechanisms of cancer cells and has emerged as a significant therapeutic target and biomarker in NSCLC. The expression level of PD-L1 significantly influences the efficacy of ICIs) such as nivolumab, pembrolizumab [229,230] High PD-L1 expression (≥ 50%) has been established as a criterion for administering pembrolizumab monotherapy, now considered the first-line treatment for metastatic NSCLC [230]. Detecting PD-L1 expression in lung cancer involves several methodologies, primarily focusing on immunohistochemistry (IHC) [231] and liquid biopsy techniques [232]. IHC is a widely used method where tissue samples, such as those obtained from surgical resections or biopsies, are stained with specific antibodies against PD-L1. Liquid biopsy, particularly the analysis of circulating tumor cells (CTCs), is another emerging method. Techniques like EpCAM-coated magnetic beads and microfluidic devices have been used to isolate CTCs from blood samples, with subsequent PD-L1 evaluation through immunofluorescent staining [233]. This method has shown promise, with PD-L1 expression detected in a significant proportion of CTCs from SCLC patients. Advanced techniques like radiogenomics, which integrate radiologic imaging with genomic data through artificial intelligence, are also being developed to non-invasively predict PD-L1 expression and other actionable mutations [234]. A multi-label multi-task deep learning system has shown high accuracy in predicting PD-L1 status from CT images, offering a non-invasive alternative to traditional testing methods [235]. Lastly, the prevalence and clinical significance of PD-L1 expression have been studied in various populations, where PD-L1 positivity was associated with specific genetic mutations and histologic types, underscoring the importance of comprehensive molecular profiling in guiding treatment strategies [236] .Collectively, these methods highlight the diverse and evolving landscape of PD-L1 detection in lung cancer, each with its unique advantages and applications in clinical practice. While PD-L1 expression significantly influences treatment outcomes, the variability in response rates and the challenges in accurate assessment necessitate ongoing research and refinement of detection methodologies. Further studies are required to standardize non-invasive PD-L1 testing and validate its predictive power in clinical settings.
TMB, quantified by the number of mutations per megabase of DNA, has gained prominence as a biomarker, with elevated TMB levels indicating a higher likelihood of producing immunogenic neo-antigens capable of eliciting a T cell response [237]. The FDA's approval of pembrolizumab for tumors with TMB exceeding 10 mutations per megabase, based on findings from the KEYNOTE-158 trial, has sparked debate due to the intricate nature of TMB as a biomarker [238]. In the context of SCLC, the BIOLUMA trial suggested that high TMB might predict responses to the combination of nivolumab and ipilimumab [239]. For NSCLC, blood-based TMB (b-TMB) has demonstrated a robust correlation with tissue TMB (t-TMB), supporting its use as a reliable biomarker for monitoring treatment response during concurrent chemoradiotherapy.[240] Yet, TMB's prognostic value in early-stage NSCLC is less clear, as a meta-analysis found no significant differences in OS or disease-free survival (DFS) between high and low TMB groups [241]. Real-world evidence from over 8,000 patients spanning 24 cancer types treated with ICIs indicated that higher TMB levels were linked to improved OS, reinforcing TMB's clinical validity as a predictor of ICI efficacy [242]. Detecting TMB expression involves several sophisticated methods to ensure accuracy and reliability, including whole-exome sequencing (WES) and gene-targeted sequencing [243]. TMB testing is typically performed using next-generation sequencing approaches, which range from whole genome sequencing to WES and large targeted gene panels. WES of tumor versus germline DNA is considered a reference standard, although various commercial gene panel assays and laboratory-developed tests are used [244] but, differences in gene coverage, in types of mutations included and in computational pipelines to estimate TMB can lead to variability among different laboratories, so their standardization is needed [245]. In the coming years, the assessment of tumor mutational burden may become fundamental in immuno-oncology. However, its application in routine clinical practice requires further optimization.
Deficient DNA mismatch repair (dMMR) refers to the failure of the cellular mechanism responsible for correcting errors that occur during DNA replication. This system involves several key proteins, including MutL protein homolog 1 (MLH1), postmeiotic segregation increased 2 (PMS2), MutS homolog 2 (MSH2), and MutS homolog 6 (MSH6), which work together to recognize and repair mismatches in the DNA sequence [246]. This complex is crucial for maintaining genomic stability by identifying and repairing base-to-base mismatches and insertion-deletion loops that arise during DNA synthesis [246]. dMMR and microsatellite instability-high (MSI-H) are emerging as pivotal biomarkers for the efficacy of ICIs across various cancers, but implications in lung cancer remain less well-defined [247]. Tumors exhibiting MSI-H are predisposed to robust responses to ICIs, attributed to their elevated mutational burden that promotes neoantigen formation and subsequent immune detection [248]. However, not all dMMR/MSI-H lung cancer patients experience benefit from ICIs [249]. This resistance mechanism may be explained by the fact that MMR-deficient cells can evade apoptosis, a critical pathway for the efficacy of many anticancer therapies, including ICIs [250]. Various advanced techniques have been developed to enhance the sensitivity and specificity to dMMR detection, such as polymerase chain reaction and next-generation sequencing, in colorectal and endometrial cancers [251]. Advancements in deep learning (DL) now enable the prediction of microsatellite instability and deficient mismatch repair status directly from digitized hematoxylin and eosin histopathology slides with high accuracy [252]. These techniques, characterized by high sensitivity, specificity, and simplified protocols, can be adapted for the detection of dMMR to evaluate the efficacy of immune checkpoint inhibitors, ensuring accurate and reliable results in clinical diagnostics.
Peripheral blood for circulating tumor DNA (ctDNA) is emerging as a promising prognostic and predictive biomarker for immune checkpoint inhibitor therapy . Sequential ctDNA studies have demonstrated the ability to detect patients who respond to ICI therapy, including those with NSCLC. A study conducted by Anagnostou et al. demonstrated the correlation between ctDNA response and survival outcomes in NSCLC patients treated with pembrolizumab is significant. Patients with lower ctDNA levels or those whose ctDNA levels decrease during treatment tend to have better survival outcomes [253]. Another study showed that high ctDNA plasma levels are associated with early PD in NSCLC treated with nivolumab [254]. Moreover, a machine learning model was developed that uses longitudinal data of circulating tumor DNA (ctDNA) dynamics to predict survival in NSCLC patients treated in combination with immune checkpoint inhibitors and chemotherapy (chemo-ICI) [255]. In addition to measuring the overall ctDNA burden, further characterization of ctDNA can reveal tumor-specific factors, such as tumor mutational burden and microsatellite instability, that influence responses or resistance to immunotherapy [256]. Current methods to measure ctDNA in liquid biopsies involve a variety of advanced techniques aimed at enhancing the precision and utility of cancer diagnostics and monitoring, like RT-PCR, digital PCR (dPCR), mass spectrometry, Next-Generation Sequencing (NGS) and hybrid sequencing (NanoString) [257]. However, it is necessary to adopt specific practical recommendations for ctDNA application to be used routinely in clinical practice to genotype advanced tumors and to be able to choose targeted therapy [258].
The response of non-small cell lung cancer with driver mutations to immune checkpoint inhibitors (ICIs) has been extensively studied due to the varying outcomes seen across different genetic profiles. ICIs have significantly changed the treatment landscape for NSCLC, though their effectiveness in patients with specific molecular alterations remains inconsistent [259]. For instance, patients with EGFR mutations often show poor responses to ICIs. This is likely due to the low immunogenicity of tumors with single driver mutations and the intricate interactions between oncogenic pathways and immune evasion mechanisms [260]. Conversely, NSCLC patients with KRAS mutations generally respond more favorably to ICIs, likely due to higher immunogenicity and PD-L1 expression in these tumors [261]. Research has demonstrated that combining ICIs with chemotherapy (CT-IO) can improve outcomes, especially in patients with novel driver alterations such as MET exon 14 skipping, BRAF mutations, RET rearrangements, and HER2 mutations, compared to ICI monotherapy [262], but with other driver mutations like PIK3CA and STK11 is limited [263]. The integration of molecular diagnostics, such as next-generation sequencing (NGS), is essential for identifying actionable mutations and guiding personalized treatment strategies [264]. Despite these advancements, the optimal sequencing and combination of ICIs, chemotherapy, and targeted therapies are still under investigation to maximize patient outcomes, particularly in never-smoking patients who are more likely to harbor actionable driver mutations [265]. Overall, while ICIs have transformed NSCLC treatment, their efficacy in patients with specific driver alterations varies, highlighting the need for further research to refine therapeutic approaches and improve clinical outcomes.
Several single nucleotide polymorphisms (SNPs) have been identified as influential in determining the efficacy of nivolumab and pembrolizumab across various cancers. In NSCLC patients treated with nivolumab, the CD274 (PD-L1) rs2282055 and rs4143815 SNPs correlated with better ORR and PFS[266]. Specifically, the G allele of rs2282055 and the C/C and C/G genotypes of rs4143815 were linked to significantly better clinical responses compared to alternative alleles [266]. However, another study on NSCLC patients treated with nivolumab found the PDCD1 (PD1) 804C>T SNP was associated with reduced odds of any grade treatment-related toxicities, though this result lacked validation in a subsequent cohort, casting uncertainty on its clinical relevance in predicting toxicity [267]. Another SNP-candidate study performed on a Caucasian cohort of 44 patients tried to validate the predictive role of rs822336, rs2282055 and rs4143815 PD-L1 SNPs in advanced NSCLC patients treated with ICIs. T/T genotype in rs2282055 was slightly associated with longer PFS (P = 0.08) and OS (P = 0.09) as compared to G/T and G/G genotypes. In contrast, patients carrying C/C genotype in rs822336 were significantly associated with better ORR (P = 0.004), PFS (P = 0.003) and OS (P = 0.002) as compared to those carrying G/C and G/G genotypes [268]. Genetic factors linked to autoimmune diseases, particularly HLA genes, have been associated with various conditions, highlighting the complex genetic basis of autoimmune disorders. For instance, the HLA class II molecule HLA-DRA has been identified as a marker for immuno-hot tumors, predicting therapeutic responses to anti-PD-1 immunotherapy in NSCLC [269]. Additionally, in patients with advanced solid tumors, including lung cancer, treated with bempegaldesleukin plus nivolumab, certain KIR/KIR-ligand genotypes, such as inhibitory KIR2DL2 and its ligand HLA-C1, were associated with greater tumor shrinkage and improved PFS, suggesting that these genetic factors may also influence responses to nivolumab-based therapies [270].
The integration of biomarkers such as PD-L1 expression, TMB, dMMR/MSI-H status, ctDNA, driver mutations and SNPs is essential for predicting responses to ICIs in lung cancer. The genetic biomarkers described are just some of those that possibly can be used to predict the efficacy and side effects of ICIs. Research is still ongoing, but many others that are not genetic ones but are particularly promising have already been reported in the literature, such as tumor-infiltrating lymphocytes (TILs) [271], serum pro-inflammatory cytokines [272] and gut microbiome [273]. Advanced detection methods and comprehensive molecular profiling are necessary to refine these biomarkers and improve patient outcomes. Further research is required to standardize non-invasive testing and validate the predictive power of these biomarkers in clinical settings. By enhancing our understanding of these biomarkers and their measurement techniques, personalized cancer therapy can be more effectively tailored to individual patient needs, optimizing treatment efficacy and minimizing adverse effects.
3.3. Emerging Therapy
The introduction of CRISPR-Cas9 gene editing technology represents a significant advancement in the field of molecular biology and genetic editing. This enables interventions on specific genomic alterations that have the potential to influence lung cancer treatment outcomes, with the potential to substantially improve patient outcomes. The ability to precisely modify the DNA of lung cancer cells using CRISPR-Cas9 offers new opportunities for targeted therapies, overcoming drug resistance, and potentially curing the disease.
Despite the significant progress in CRISPR-Cas9 delivery strategies, numerous challenges remain to be addressed. It is crucial to optimize delivery systems to ensure that the CRISPR-Cas9 complex effectively reaches target cells. Additionally, improving specificity is essential to minimize off-target effects and ensure treatment safety. Equally important is addressing immunogenicity issues, which could limit treatment efficacy, and managing the regulatory and ethical considerations associated with the use of this technology [274].
In this context, ongoing and future clinical studies play a critical role, providing essential data on the feasibility, safety, and therapeutic potential of CRISPR-Cas9-based treatments. These studies will not only facilitate regulatory approval but also provide valuable insights that will guide the optimization of delivery strategies and the selection of target genes.
4. Challenges and Future Prospects
Lung cancer represents one of the leading causes of mortality worldwide, with a generally poor prognosis due to late diagnosis and the aggressive nature of the disease. In recent decades, oncological research has made significant progress, paving the way for new therapeutic strategies based on the personalization of treatments. This approach aims to optimize the effectiveness of therapies and reduce side effects by considering the specific characteristics of the tumor and the patient.
The understanding of the molecular bases of lung cancer has revolutionized clinical oncology. Genetic studies have identified numerous mutations and genetic alterations that drive the growth and proliferation of tumor cells. The identification of such mutations through molecular tests has become crucial for treatment selection. In this context, targeted therapies and immunotherapy have represented a significant breakthrough in the management of lung cancer. However, many patients have developed acquired resistance to treatments. Acquired resistance occurs when tumor cells, initially sensitive to treatment, evolve mechanisms that render them refractory. This phenomenon can manifest through various mechanisms, including secondary mutations, activation of alternative signaling pathways, and phenotypic modifications of the tumor. To date, this represents a crucial obstacle but also the starting point for personalized medicine. Precision medicine indeed goes beyond the mere identification of genetic mutations, also including the analysis of the tumor microenvironment, gene expression profiles, and other biomarkers. Advanced techniques such as liquid biopsy, which allows for the analysis of circulating tumor DNA in the blood, are emerging as promising tools for monitoring disease progression and treatment efficacy in real-time. The personalization of therapeutic choices requires a multidisciplinary approach involving oncologists, pathologists, radiologists, and geneticists. Every patient is unique, and therapeutic decisions must consider various factors, including age, general health status, comorbidities, and personal preferences. Continuous dialogue between the medical team and the patient is essential to ensure that therapeutic choices are aligned with care objectives and quality of life. In addition, another obstacle to overcome is the limited access to innovative therapies due to economic and logistical factors. For this reason, it is essential to fund ongoing research to identify new therapeutic targets, improve understanding of the interactions between cancer and the immune system, and ensure access to treatment for all types of patients.
In the future, the integration of omics data (genomics, proteomics, metabolomics) and the use of artificial intelligence to analyze large volumes of clinical data could further revolutionize the personalization of care. The development of more accurate predictive models will allow for the anticipation of treatment responses and the dynamic adaptation of therapeutic strategies.
5. Conclusions
This review highlights the intricate and complex nature of treatment based on genetic data in lung cancer patients, underscoring the importance of evaluating and monitoring the cancer genetic landscape. Genetic discoveries have transformed the approach to lung cancer treatment, leading to targeted, personalized therapies that improve patient survival and quality of life. However, challenges such as therapeutic resistance and the need for more precise customization require further research and clinical innovation. The future of lung cancer therapy lies in the integration of advanced genomics with clinical practice to develop increasingly effective treatments.
Author Contributions
EF and FC designed the review; AR, DM, FM, ME, MP, VBS, DR and FM searched databases, collected full-text papers, extracted relevant information and drafted the article main text; all authors contributed to revising the article, and gave final approval of the version to be published.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
The authors thank the NextGenerationEU of PNRR “Piano Nazionale di Ripresa e Resilienza” Missione 4 Componente 2 – M4C2a - Project THE – Tuscany Health Ecosystem – SPOKE 6 – CUP B63C22000680007 (MILESTONE n. 6.3.2: Innovative tools for target genes, disease pathways and therapeutic strategies discovery) (E.F.); the EU funding within the NextGenerationEU with Italian Ministry of University and Research (MUR) for “Fondo per il Programma Nazionale di Ricerca e Progetti di Rilevante Interesse Nazionale (PRIN)” - PRIN 2022 – Project 2022ARXHR2_002 - CUP B53D23007910006 (E.F. and F.C.); the EU funding within the “NextGenerationEU of Piano Nazionale di Ripresa e Resilienza (PNRR) - Missione 4 “Istruzione e Ricerca” - Componente 2 – M4C2a Investimento 1.1, “Fondo per il Programma Nazionale di Ricerca e Progetti di Rilevante Interesse Nazionale (PRIN)” - PRIN 2022 PNRR – Project P2022NLEBP - CUP B53D23025110001 (E.F.); the “INAIL (BRiC - 2022) Piano Attività di Ricerca 2022-2024” (E.F); Italian Ministry of University and Research for PNRR-National Center for Gene Therapy and Drugs based on RNA Technology - CN00000041 (F.M.).
Conflicts of Interest
The authors declare no conflicts of interest.
References
- S. H.; F. J., Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin. 2021;71(3):209-249. [CrossRef]
- S. RL.; M. KD., Cancer statistics, 2020. CA Cancer J Clin. 2020, 70(1):7-30. [CrossRef]
- GBD 2015 Tobacco Collaborators; Smoking prevalence and attributable disease burden in 195 countries and territories, 1990-2015: a systematic analysis from the Global Burden of Disease Study 2015 Lancet. 2017 7;390(10103):1644. [CrossRef]
- P. HA.; I.B., The association between smoking quantity and lung cancer in men and women. Chest. 2013, 143(1):123-129. [CrossRef]
- D. R.; P. R., Mortality in relation to smoking: 50 years' observations on male British doctors. BMJ. 2004, 26;328(7455):1519. [CrossRef]
- S. JM; A.E., Lung cancer in never smokers: clinical epidemiology and environmental risk factors. Clin Cancer Res. 2009, 15;15(18):5626-45. [CrossRef]
- S. D.; Z. P., Occupational exposure and lung cancer. J Thorac Dis. 2013, 5 Suppl 4(Suppl 4):S440-5. [CrossRef]
- L. JH.; B. JD J., Lung cancer in radon-exposed miners and estimation of risk from indoor exposure. J Natl Cancer Inst. 1995, 7;87(11):817-27. [CrossRef]
- T. WD.; B. E., Introduction to The 2015 World Health Organization Classification of Tumors of the Lung, Pleura, Thymus, and Heart. J Thorac Oncol. 2015, 10(9):1240-1242. [CrossRef]
- E. DS.; W. DE., Non-Small Cell Lung Cancer, Version 3.2022, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw. 2022, 20(5):497-530. [CrossRef]
- T. WD.; B. E., International association for the study of lung cancer/american thoracic society/european respiratory society international multidisciplinary classification of lung adenocarcinoma. J Thorac Oncol. 2011, 6(2):244-85. [CrossRef]
- N. M.; M. A., Small adenocarcinoma of the lung. Histologic characteristics and prognosis. Cancer. 1995, 15;75(12):2844-52. [CrossRef]
- C. S.; Z. G, Lung cancer in never smokers--a review. Eur J Cancer. 2012, 48(9):1299-311. [CrossRef]
- R. R.; C. S., Lung cancer staging: a concise update. Eur Respir J. 2018, 17;51(5):1800190. [CrossRef]
- M. N.; S. B. Lung adenocarcinoma presenting as intramedullary spinal cord metastasis: Case report and review of literature. J Clin Neurosci. 2018, 52:124-131. [CrossRef]
- P. F.; R. S., State of the art and new perspectives in surgical treatment of lung cancer: a narrative review. Transl Cancer Res. 2022, 11(10):3869-3875. [CrossRef]
- L. X.; W. J., Optimal Initial Time Point of Local Radiotherapy for Unresectable Lung Adenocarcinoma: A Retrospective Analysis on Overall Arrangement of Local Radiotherapy in Advanced Lung Adenocarcinoma. Front Oncol. 2022, 10;12:793190. [CrossRef]
- L. D.; N. N., Customized Adjuvant Chemotherapy Based on Biomarker Examination May Improve Survival of Patients Completely Resected for Non-small-cell Lung Cancer. Anticancer Res. 2017, 37(5):2501-2507. [CrossRef]
- E. GJ.; W. DE., Non-Small Cell Lung Cancer, Version 4.2024, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw. 2024, 22(4):249-274. [CrossRef]
- B. JE.; V. K., Targeting Metabolism to Improve the Tumor Microenvironment for Cancer Immunotherapy. Mol Cell. 2020, 18;78(6):1019-1033. [CrossRef]
- J. N.; X. X., Exploring the survival prognosis of lung adenocarcinoma based on the cancer genome atlas database using artificial neural network. Medicine (Baltimore). 2019, 98(20):e15642. [CrossRef]
- Cancer Genome Atlas Research Network; Comprehensive genomic characterization of squamous cell lung cancers. Nature. 2012, 8;491(7423):288. [CrossRef]
- T. WD.; B. E., The 2015 World Health Organization Classification of Lung Tumors: Impact of Genetic, Clinical and Radiologic Advances Since the 2004 Classification. J Thorac Oncol. 2015, 10(9):1243-1260. [CrossRef]
- S. BR.; G. DP., Squamous Cell Lung Cancer. 2024, 14. In: StatPearls [Internet]. Treasure Island (FL): StatPearls.
- S. GA.; G. AV., Diagnosis and management of lung cancer, 3rd ed: American College of Chest Physicians evidence-based clinical practice guidelines. Chest. 2013, 143(5 Suppl):e211S-e250S. [CrossRef]
- G. C., A. A., Recent issues in first-line treatment of advanced non-small-cell lung cancer: Results of an International Expert Panel Meeting of the Italian Association of Thoracic Oncology. Lung Cancer. 2010, 68(3):319-31. [CrossRef]
- Z. K., C. H., Treatment options and prognosis of patients with lung squamous cell cancer in situ: a comparative study of lung adenocarcinoma in situ and stage IA lung squamous cell cancer. Transl Lung Cancer Res. 2023, 30;12(6):1276-1292. [CrossRef]
- S. LM.; Large-cell carcinoma of the lung: a diagnostic category redefined by immunohistochemistry and genomics. Curr Opin Pulm Med. 2014, 20(4):324-31. [CrossRef]
- C. A.; The fine structure of large cell undifferentiated carcinoma of the lung. Evidence for its relation to squamous cell carcinomas and adenocarcinomas. Hum Pathol. 1978, 9(2):143-56. [CrossRef]
- A. KS.; T. LD., Larg cell carcinoma of the lung. Ultrastructural differentiation and clinicopathologic correlations. Cancer. 1985, 1;56(7):1618-23. [CrossRef]
- M. JE.; S. SD., Cigarette smoking and large cell carcinoma of the lung. Cancer Epidemiol Biomarkers Prev. 1997, 6(7):477-80. [PubMed]
- X. L.; J. Y., Clinical characteristics and prognosis of pulmonary large cell carcinoma: A population-based retrospective study using SEER data. Thorac Cancer. 2020, 11(6):1522-1532. [CrossRef]
- T. Q.; Z. L., Clinical characteristics and treatments of large cell lung carcinoma: a retrospective study using SEER data. Transl Cancer Res. 2020, 9(3):1455-1464. [CrossRef]
- P. G.; B. M., Large cell carcinoma of the lung: a tumor in search of an author. A clinically oriented critical reappraisal. Lung Cancer. 2015, 87(3):226-31. [CrossRef] [PubMed]
- D. M,; B. L., Diagnostic procedures for non-small-cell lung cancer (NSCLC): recommendations of the European Expert Group. Thorax. 2016, 71(2):177-84. [CrossRef]
- A.; H. K., Prospective study of adjuvant chemotherapy for pulmonary large cell neuroendocrine carcinoma. Ann Thorac Surg. 2006, 82(5):1802-7. [CrossRef]
- G,, AF.; B, PA. , Small-cell lung cancer: what we know, what we need to know and the path forward. Nat Rev Cancer. 2017, 17(12):725-737. [CrossRef]
- K. GP.; L. BW, NCCN Guidelines Insights: Small Cell Lung Cancer, Version 2.2018. J Natl Compr Canc Netw. 2018, 16(10):1171-1182. [CrossRef]
- W. S,; Z. S, Current Diagnosis and Management of Small-Cell Lung Cancer. Mayo Clin Proc. 2019, 94(8):1599-1622. [CrossRef]
- D. AC,; F. M., Small-cell lung cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up☆. Ann Oncol. 2021, 32(7):839-853. [CrossRef]
- F. M.; D.R. D., Small-cell lung cancer (SCLC): ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2013, 24 Suppl 6:vi99-105. [CrossRef]
- K. K.; V. S., Small Cell Lung Carcinoma: Current Diagnosis, Biomarkers, and Treatment Options with Future Perspectives. Biomedicines. 2023, 13;11(7):1982. [CrossRef]
- D. AC.; F.M, Small-cell lung cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up☆. Ann Oncol. 2021 Jul;32(7):839-853. [CrossRef]
- L. SV.; R. M., Updated Overall Survival and PD-L1 Subgroup Analysis of Patients With Extensive-Stage Small-Cell Lung Cancer Treated With Atezolizumab, Carboplatin, and Etoposide (IMpower133). J Clin Oncol. 2021, 20;39(6):619-630. [CrossRef]
- C. ME; B. E.,. Pulmonary neuroendocrine (carcinoid) tumors: European Neuroendocrine Tumor Society expert consensus and recommendations for best practice for typical and atypical pulmonary carcinoids. Ann Oncol. 2015, 26(8):1604-20. [CrossRef]
- S. M.; B. S., Gene Expression Profiling of Lung Atypical Carcinoids and Large Cell Neuroendocrine Carcinomas Identifies Three Transcriptomic Subtypes with Specific Genomic Alterations. J Thorac Oncol. 2019, 14(9):1651-1661. [CrossRef]
- H. AE.; M. AM, Neuroendocrine Tumors of the Lung: Current Challenges and Advances in the Diagnosis and Management of Well-Differentiated Disease. J Thorac Oncol. 2017, 12(3):425-436. [CrossRef]
- G. M.;,M. JM., Prognostic factors in neuroendocrine lung tumors: a Spanish Multicenter Study. Spanish Multicenter Study of Neuroendocrine Tumors of the Lung of the Spanish Society of Pneumonology and Thoracic Surgery (EMETNE-SEPAR). Ann Thorac Surg. 2000, 70(1):258-63. [CrossRef]
- V. SD.;, N. A, Emerging Immunotherapeutic and Diagnostic Modalities in Carcinoid Tumors. Molecules. 2023, 22;28(5):2047. [CrossRef]
- Z. Y.; W. DC., Genome analyses identify the genetic modification of lung cancer subtypes. Semin Cancer Biol. 2017, 42:20-30. [CrossRef]
- S. Y.; F. KM., Molecular genetics of lung cancer. Annu Rev Med. 2003, 54:73-87. [CrossRef]
- C. MB.; C. PP., Clinical potential of gene mutations in lung cancer. Clin Transl Med. 2015, 4(1):33. [CrossRef]
- B. P.; H. P., Genetics of lung-cancer susceptibility. Lancet Oncol. 2011, 12(4):399-408. [CrossRef]
- L. TJ.; B. DW., Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004, 20;350(21):2129-39. [CrossRef]
- K. T.; Y. Y., Prognostic implication of EGFR, KRAS, and TP53 gene mutations in a large cohort of Japanese patients with surgically treated lung adenocarcinoma. J Thorac Oncol. 2009, 4(1):22-9. [CrossRef]
- M. T.; Y. Y. Mutations of the epidermal growth factor receptor gene and related genes as determinants of epidermal growth factor receptor tyrosine kinase inhibitors sensitivity in lung cancer. Cancer Sci. 2007, 98(12):1817-24. [CrossRef]
- M. SW.; K. MN., Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin's lymphoma. Science. 1994, 20;267(5196):316-7. [CrossRef]
- S. M.; C. YL., Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature. 2007, 2;448(7153):561-6. [CrossRef]
- S. M.; T. S., A mouse model for EML4-ALK-positive lung cancer. Proc Natl Acad Sci U S A. 2008, 16;105(50):19893-7. [CrossRef]
- T. K., C. YL., KIF5B-ALK, a novel fusion oncokinase identified by an immunohistochemistry-based diagnostic system for ALK-positive lung cancer. Clin Cancer Res. 2009, 1;15(9):3143-9. [CrossRef]
- D. X.; S. Y., ALK-rearrangement in non-small-cell lung cancer (NSCLC). Thorac Cancer. 2018, 9(4):423-430. [CrossRef]
- T. Y., S. M., KLC1-ALK: a novel fusion in lung cancer identified using a formalin-fixed paraffin-embedded tissue only. PLoS One. 2012, 7(2):e31323. [CrossRef]
- C. PL; M. P., Prevalence and natural history of ALK positive non-small-cell lung cancer and the clinical impact of targeted therapy with ALK inhibitors. Clin Epidemiol. 2014, 20;6:423-32. [CrossRef]
- C. AD.; D. CJ., Ras family signaling: therapeutic targeting. Cancer Biol Ther. 2002, 1(6):599-606. [CrossRef]
- P. W.; G. N., New driver mutations in non-small-cell lung cancer. Lancet Oncol. 2011, 12(2):175-80. [CrossRef]
- P. IA.; L. PD., A comprehensive survey of Ras mutations in cancer. Cancer Res. 2012, 15;72(10):2457-67. [CrossRef]
- W. X.; R. B., Association between Smoking History and Tumor Mutation Burden in Advanced Non-Small Cell Lung Cancer. Cancer Res. 2021, 1;81(9):2566-2573. [CrossRef]
- T. J.; The clinical relevance of KRAS gene mutation in non-small-cell lung cancer. Curr Opin Oncol. 2014, 26(2):138-44. [CrossRef]
- G. A.; R. P., Current therapy of KRAS-mutant lung cancer. Cancer Metastasis Rev. 2020, 39(4):1159-1177. [CrossRef]
- B. EH., A. TC., Update on emerging biomarkers in lung cancer. J Thorac Dis. 2019, 11(Suppl 1):S81-S88. [CrossRef]
- J. H.; W. Z., Mutations in BRAF and KRAS converge on activation of the mitogen-activated protein kinase pathway in lung cancer mouse models. Cancer Res. 2007, 15;67(10):4933-9. [CrossRef]
- R. MJ.; C. MH., Mitogen-activated protein kinase pathways. Curr Opin Cell Biol. 1997, 9(2):180-6. [CrossRef]
- W. PT; G. MJ., Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell. 2004, 19;116(6):855-67. [CrossRef]
- D. H., B. GR., Mutations of the BRAF gene in human cancer. Nature. 2002, 27;417(6892):949-54. [CrossRef]
- P. PK.; A. ME., Clinical characteristics of patients with lung adenocarcinomas harboring BRAF mutations. J Clin Oncol. 2011, 20;29(15):2046-51. [CrossRef]
- F. KT.; M. G., BRAF, a target in melanoma: implications for solid tumor drug development. Cancer. 2010, 1;116(21):4902-13. [CrossRef]
- W. PT.; G. MJ., Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell. 2004, 19;116(6):855-67. [CrossRef]
- Y. Z.; T. NM., BRAF Mutants Evade ERK-Dependent Feedback by Different Mechanisms that Determine Their Sensitivity to Pharmacologic Inhibition. Cancer Cell. 2015, 14;28(3):370-83. [CrossRef]
- Y. Z.; Y. R., Tumours with class 3 BRAF mutants are sensitive to the inhibition of activated RAS. Nature. 2017, 10;548(7666):234-238.
- D. KD., L. AT., Identifying and targeting ROS1 gene fusions in non-small cell lung cancer. Clin Cancer Res. 2012, 1;18(17):4570-9. [CrossRef]
- R. K.; G. A., Global survey of phosphotyrosine signaling identifies oncogenic kinases in lung cancer. Cell. 2007, 14;131(6):1190-203. [CrossRef]
- B. AH.; A. SL., Chromosome 3 anomalies investigated by genome wide SNP analysis of benign, low malignant potential and low grade ovarian serous tumours. PLoS One. 2011, 6(12):e28250. [CrossRef]
- L. J.; L. SE., Identification of ROS1 rearrangement in gastric adenocarcinoma. Cancer. 2013, 1;119(9):1627-35. [CrossRef]
- L. JJ.; S. AT., Recent Advances in Targeting ROS1 in Lung Cancer. J Thorac Oncol. 2017, 12(11):1611-1625. [CrossRef]
- B. K.; S. AT., ROS1 rearrangements define a unique molecular class of lung cancers. J Clin Oncol. 2012, 10;30(8):863-70. [CrossRef]
- U. A.; D. B. M., ROS1 fusions in cancer: a review. Future Oncol. 2016, 12(16):1911-28. [CrossRef]
- T. K.;, S. M., RET, ROS1 and ALK fusions in lung cancer. Nat Med. 2012, 12;18(3):378-81. [CrossRef]
- R. R. Jr; ROS1 protein-tyrosine kinase inhibitors in the treatment of ROS1 fusion protein-driven non-small cell lung cancers. Pharmacol Res. 2017, 121:202-212. [CrossRef]
- S. A.; C. F., Targeted therapies in non-small cell lung cancer: a focus on ALK/ROS1 tyrosine kinase inhibitors. Expert Rev Anticancer Ther. 2018, 18(1):71-80. [CrossRef]
- P. ER.; V. P., Comparison of Different Antibody Clones for Immunohistochemistry Detection of Programmed Cell Death Ligand 1 (PD-L1) on Non-Small Cell Lung Carcinoma. Appl Immunohistochem Mol Morphol. 2018, 26(2):83-93. [CrossRef]
- L. G.; F. X., Prognostic significance of PD-L1 expression and tumor infiltrating lymphocyte in surgically resectable non-small cell lung cancer. Oncotarget. 2017, 12;8(48):83986-83994. [CrossRef]
- S. M,; S. S., Clinical and pathologic features of lung cancer expressing programmed cell death ligand 1 (PD-L1). Lung Cancer. 2016, 98:69-75. [CrossRef]
- S. X.; W. S., PD-L1 expression in lung adenosquamous carcinomas compared with the more common variants of non-small cell lung cancer. Sci Rep. 2017, 7;7:46209. [CrossRef]
- S. KA.; F. LJ., PD-1 inhibits T-cell receptor induced phosphorylation of the ZAP70/CD3zeta signalosome and downstream signaling to PKCtheta. FEBS Lett. 2004, 10;574(1-3):37-41. [CrossRef]
- P. DM.L; The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012, 22;12(4):252-64. [CrossRef]
- D. P.; X. Y., Intrinsic PD-L1 Signaling in Cancer Initiation, Development and Treatment: Beyond Immune Evasion. Front Oncol. 2018, 19;8:386. [CrossRef]
- G. A.; S. DS., Interferon Receptor Signaling Pathways Regulating PD-L1 and PD-L2 Expression. Cell Rep. 2019, 10;29(11):3766. [CrossRef]
- R. NA., H. MD., Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015, 3;348(6230):124-8. [CrossRef]
- C. S.; P. L., CD274 (PDL1) and JAK2 genomic amplifications in pulmonary squamous-cell and adenocarcinoma patients. Histopathology. 2018, 72(2):259-269. [CrossRef]
- I. Y.; Y. K., Clinical significance of PD-L1 and PD-L2 copy number gains in non-small-cell lung cancer. Oncotarget. 2016, 31;7(22):32113-28. [CrossRef]
- K. K.; S. Y., Aberrant PD-L1 expression through 3'-UTR disruption in multiple cancers. Nature. 2016, 16;534(7607):402-6. [CrossRef]
- C. L.; Z. XZ., PD-L1 as a predictive factor for non-small-cell lung cancer prognosis. Lancet Oncol. 2024, ;25(6):e233. [CrossRef]
- T. JH.; Y. SF., MET Amplification and Exon 14 Splice Site Mutation Define Unique Molecular Subgroups of Non-Small Cell Lung Carcinoma with Poor Prognosis. Clin Cancer Res. 2016, 15;22(12):3048-56. [CrossRef]
- B. C.; B. W.,. Met, metastasis, motility and more. Nat Rev Mol Cell Biol. 2003, ;4(12):915-25. [CrossRef]
- S. L.; D. FM., Germline and somatic mutations in the tyrosine kinase domain of the MET proto-oncogene in papillary renal carcinomas. Nat Genet. 1997, 16(1):68-73. [CrossRef]
- S. M.; M. A., screening and molecular characterization of MET alterations in non-small cell lung cancer. Clin Transl Oncol. 2018, 20(7):881-888. [CrossRef]
- R. T.; L. Y., The race to target MET exon 14 skipping alterations in non-small cell lung cancer: The Why, the How, the Who, the Unknown, and the Inevitable. Lung Cancer. 2017, 103:27-37. [CrossRef]
- A. MM.; O. GR., MET Exon 14 Mutations in Non-Small-Cell Lung Cancer Are Associated With Advanced Age and Stage-Dependent MET Genomic Amplification and c-Met Overexpression. J Clin Oncol. 2016, 1;34(7):721-30. [CrossRef]
- S. AB.; F. GM., Characterization of 298 Patients with Lung Cancer Harboring MET Exon 14 Skipping Alterations. J Thorac Oncol. 2016, 11(9):1493-502. [CrossRef]
- D. A.; MET Exon 14 Alterations in Lung Cancer: Exon Skipping Extends Half-Life. Clin Cancer Res. 2016, 15;22(12):2832-4. [CrossRef]
- O. K.; S. H., Met gene copy number predicts the prognosis for completely resected non-small cell lung cancer. Cancer Sci. 2008, 99(11):2280-5. [CrossRef]
- R. S.; M. A., MET Signaling Pathways, Resistance Mechanisms, and Opportunities for Target Therapies. Int J Mol Sci. 2022, 11;23(22):13898. [CrossRef]
- C. F.; M. A., Increased MET gene copy number negatively affects survival of surgically resected non-small-cell lung cancer patients. J Clin Oncol. 2009, 1;27(10):1667-74. [CrossRef]
- S. HU.; S. AM., MET amplification status in therapy-naïve adeno- and squamous cell carcinomas of the lung. Clin Cancer Res. 2015, 15;21(4):907-15. [CrossRef]
- C. M.; S. I, KIF5B-MET fusion variant in non-small cell lung cancer. Pulmonology. 2022, 28(4):315-316. [CrossRef]
- S. D.; W. W., Identification of MET fusions as novel therapeutic targets sensitive to MET inhibitors in lung cancer. J Transl Med. 2023, 25;21(1):150. [CrossRef]
- S. D.; X. X., MET fusions are targetable genomic variants in the treatment of advanced malignancies. Cell Commun Signal. 2024, 9;22(1):20. [CrossRef]
- L. X., J. Y., Next-Generation Sequencing of Pulmonary Sarcomatoid Carcinoma Reveals High Frequency of Actionable MET Gene Mutations. J Clin Oncol. 2016, 10;34(8):794-802. [CrossRef]
- T. M.; R. J., Activation of a novel human transforming gene, ret, by DNA rearrangement. Cell. 1985, 42(2):581-8. [CrossRef]
- I. CF.; Structure and physiology of the RET receptor tyrosine kinase. Cold Spring Harb Perspect Biol. 2013, 1;5(2):a009134. [CrossRef]
- S. JS.; J. YS., transcriptional landscape and mutational profile of lung adenocarcinoma. Genome Res. 2012, 22(11):2109-19. [CrossRef]
- T. M.; RET receptor signaling: Function in development, metabolic disease, and cancer. Proc Jpn Acad Ser B Phys Biol Sci. 2022, 98(3):112-125. [CrossRef]
- K. T.; I. H., KIF5B-RET fusions in lung adenocarcinoma. Nat Med. 2012, 12;18(3):375-7. [CrossRef]
- J. YS., L. WC., A transforming KIF5B and RET gene fusion in lung adenocarcinoma revealed from whole-genome and transcriptome sequencing. Genome Res. 2012, 22(3):436-45. [CrossRef]
- K. T.; N. T., Beyond ALK-RET, ROS1 and other oncogene fusions in lung cancer. Transl Lung Cancer Res. 2015, 4(2):156-64. [CrossRef]
- L. D.; C. M., Identification of new ALK and RET gene fusions from colorectal and lung cancer biopsies. Nat Med. 2012, 12;18(3):382-4. [CrossRef]
- W. R.; H. H., RET fusions define a unique molecular and clinicopathologic subtype of non-small-cell lung cancer. J Clin Oncol. 2012, 10;30(35):4352-9. [CrossRef]
- R. M.; C. G. M., NTRK gene fusion testing and management in lung cancer. Cancer Treat Rev. 2024, 127:102733. [CrossRef]
- A. A.; S. A., Tropomyosin receptor kinase (TRK) biology and the role of NTRK gene fusions in cancer. Ann Oncol. 2019, 1;30(Suppl_8):viii5-viii15. [CrossRef]
- A. JC.; W. SH., Neurotrophin signaling: many exciting surprises! Cell Mol Life Sci. 2006, 63(13):1523-37. [CrossRef]
- R. EY.; G. DA., TRK Fusions Are Enriched in Cancers with Uncommon Histologies and the Absence of Canonical Driver Mutations. Clin Cancer Res. 2020, 1;26(7):1624-1632. [CrossRef]
- M. CA.; B. DC., A review of NTRK fusions in cancer. Ann Med Surg (Lond). 2022, 13;79:103893. [CrossRef]
- V. A.; L. AT., TRKing down an old oncogene in a new era of targeted therapy. Cancer Discov. 2015, 5(1):25-34. [CrossRef]
- W. CB.; K. MG., Genomic context of NTRK1/2/3 fusion-positive tumours from a large real-world population. NPJ Precis Oncol. 2021, 5(1):86. [CrossRef]
- H. G.; S. FC., NTRK fusions in lung cancer: From biology to therapy. Lung Cancer. 2021, 161:108-113. [CrossRef]
- T. AC.; I. M., Brain Metastases in Lung Cancers with Emerging Targetable Fusion Drivers. Int J Mol Sci. 2020, 21(4):1416. [CrossRef]
- L. F.; W. Y., NTRK Fusion in Non-Small Cell Lung Cancer: Diagnosis, Therapy, and TRK Inhibitor Resistance. Front Oncol. 2022, 12:864666. [CrossRef]
- K. S.; B. AG., Phosphatidylinositol 3-kinase mutations identified in human cancer are oncogenic. Proc Natl Acad Sci U S A. 2005, 102(3):802-7. [CrossRef]
- O. GR., B. A., New targetable oncogenes in non-small-cell lung cancer. J Clin Oncol. 2013, 31(8):1097-104. [CrossRef]
- S. Y., V. VE., Oncogenic mutations of PIK3CA in human cancers. Cell Cycle. 2004, 3(10):1221-4. [CrossRef]
- H. CH.; M. D., The structure of a human p110alpha/p85alpha complex elucidates the effects of oncogenic PI3Kalpha mutations. Science. 2007, 318(5857):1744-8. [CrossRef]
- J. F.; W. JJ., PIK3CA mutation H1047R is associated with response to PI3K/AKT/mTOR signaling pathway inhibitors in early-phase clinical trials. Cancer Res. 2013, 73(1):276-84. [CrossRef]
- K. O.; S. H., PIK3CA mutation status in Japanese lung cancer patients. Lung Cancer. 2006, 54(2):209-15. [CrossRef]
- C. JE.; A. ME, Paik PK, Lau C, Riely GJ, Pietanza MC, Zakowski MF, Rusch V, Sima CS, Ladanyi M, Kris MG. Coexistence of PIK3CA and other oncogene mutations in lung adenocarcinoma-rationale for comprehensive mutation profiling. Mol Cancer Ther. 2012, 11(2):485-91. [CrossRef]
- W. Y.; W. Y Clinical Significance of PIK3CA Gene in Non-Small-Cell Lung Cancer: A Systematic Review and Meta-Analysis. Biomed Res Int. 2020, 17;2020:3608241. [CrossRef]
- S. M.; B. M., mutations in non-small cell lung cancer (NSCLC): genetic heterogeneity, prognostic impact and incidence of prior malignancies. Oncotarget. 2015, 6(2):1315-26. [CrossRef]
- W. L.; H. H., PIK3CA mutations frequently coexist with EGFR/KRAS mutations in non-small cell lung cancer and suggest poor prognosis in EGFR/KRAS wildtype subgroup. PLoS One. 2014, 9(2):e88291. [CrossRef]
- D. JM.; J. CB., Neu differentiation factor induces ErbB2 down-regulation and apoptosis of ErbB2-overexpressing breast tumor cells. Cancer Res. 1997, 57(17):3804-11.
- A. ME.; C. JE., Prevalence, clinicopathologic associations, and molecular spectrum of ERBB2 (HER2) tyrosine kinase mutations in lung adenocarcinomas. Clin Cancer Res. 2012, 18(18):4910-8. [CrossRef]
- H. FR.; V. M., Evaluation of HER-2/neu gene amplification and protein expression in non-small cell lung carcinomas. Br J Cancer. 2002, 86(9):1449-56. [CrossRef]
- L. BT.; R. DS., HER2 Amplification and HER2 Mutation Are Distinct Molecular Targets in Lung Cancers. J Thorac Oncol. 2016, 11(3):414-9. [CrossRef]
- K. V.; G. M., Effect of HER2/neu expression on survival in non-small-cell lung cancer. Clin Lung Cancer. 2001, 2(3):216-9. [CrossRef]
- A. ME.; C. JE., Nafa K, Roy-Chowdhuri S, Lau C, Zaidinski M, Paik PK, Zakowski MF, Kris MG, Ladanyi M. Prevalence, clinicopathologic associations, and molecular spectrum of ERBB2 (HER2) tyrosine kinase mutations in lung adenocarcinomas. Clin Cancer Res. 2012, 18(18):4910-8. [CrossRef]
- M. J.; P. S., Lung cancer that harbors an HER2 mutation: epidemiologic characteristics and therapeutic perspectives. J Clin Oncol. 2013, 31(16):1997-2003. [CrossRef]
- T. K.; S. K., Prognostic and predictive implications of HER2/ERBB2/neu gene mutations in lung cancers. Lung Cancer. 2011, 74(1):139-44. [CrossRef]
- H. A.; M. D., A serine/threonine kinase gene defective in Peutz-Jeghers syndrome. Nature. 1998, 391(6663):184-7. [CrossRef]
- S. M.; P. P., Inactivation of LKB1/STK11 is a common event in adenocarcinomas of the lung. Cancer Res. 2002, 62(13):3659-62.
- C.J.; M. PP., Novel and natural knockout lung cancer cell lines for the LKB1/STK11 tumor suppressor gene. Oncogene. 2004, 23(22):4037-40. [CrossRef]
- J. H.; R. MR., LKB1 modulates lung cancer differentiation and metastasis. Nature. 2007, 448(7155):807-10. [CrossRef]
- S. DB.; S. RJ., The LKB1-AMPK pathway: metabolism and growth control in tumour suppression. Nat Rev Cancer. 2009, 9(8):563-75. [CrossRef]
- S. NJ.; K. AB., STK11 (LKB1) mutations in metastatic NSCLC: Prognostic value in the real world. PLoS One. 2020, 15(9):e0238358. [CrossRef]
- J. M.; W.S, Somatic STK11/LKB1 mutations to confer resistance to immune checkpoint inhibitors as monotherapy or in combination in advanced NSCLC.. JCO. 2018, 36, 3028-3028. [CrossRef]
- S. F.; G. ME., STK11/LKB1 Mutations and PD-1 Inhibitor Resistance in KRAS-Mutant Lung Adenocarcinoma. Cancer Discov. 2018, 8(7):822-835. [CrossRef]
- S. F.; A. K. C., Association of STK11/LKB1 genomic alterations with lack of benefit from the addition of pembrolizumab to platinum doublet chemotherapy in non-squamous non-small cell lung cancer.. JCO. 2019, 37, 102-102. [CrossRef]
- M. B.; H. S., The Importance of STK11/LKB1 Assessment in Non-Small Cell Lung Carcinomas. Diagnostics (Basel). 2021, 11(2):196. [CrossRef]
- K. S.; A. EA., STK11/LKB1 Deficiency Promotes Neutrophil Recruitment and Proinflammatory Cytokine Production to Suppress T-cell Activity in the Lung Tumor Microenvironment. Cancer Res. 2016, 76(5):999-1008. [CrossRef]
- D. S.; S. J., Mutation incidence and coincidence in non small-cell lung cancer: meta-analyses by ethnicity and histology (mutMap). Ann Oncol. 2013, 24(9):2371-6. [CrossRef]
- M. M.; I. A., or chemotherapy for non-small-cell lung cancer with mutated EGFR. N Engl J Med. 2010, 362(25):2380-8. [CrossRef]
- Z. E.; B. J., Receptor tyrosine kinase signalling as a target for cancer intervention strategies. Endocr Relat Cancer. 2001, 8(3):161-73. [CrossRef]
- M. U.; M. R., Targeted therapy in advanced non-small cell lung cancer: current advances and future trends. J Hematol Oncol. 2021, 14(1):108. [CrossRef]
- P. C.; F. M., Tyrosine Kinase Inhibitors in Cancer: Breakthrough and Challenges of Targeted Therapy. Cancers (Basel). 2020, 12(3):731. [CrossRef]
- H. RS.; F. M., Gefitinib--a novel targeted approach to treating cancer. Nat Rev Cancer. 2004, 4(12):956-65. [CrossRef]
- D. J.; M. JD., Erlotinib hydrochloride. Nat Rev Drug Discov. 2005, 4(1):13-4. [CrossRef]
- J. DM.; Y. BY., 19 deletion mutations of epidermal growth factor receptor are associated with prolonged survival in non-small cell lung cancer patients treated with gefitinib or erlotinib. Clin Cancer Res. 2006, 12(13):3908-14. [CrossRef]
- H. YH.; T. JS., The impact of different first-line EGFR-TKIs on the clinical outcome of sequential osimertinib treatment in advanced NSCLC with secondary T790M. Sci Rep. 2021, 11(1):17646. [CrossRef]
- P. T.; T. J., Real world data of efficacy and safety of erlotinib as first-line TKI treatment in EGFR mutation-positive advanced non-small cell lung cancer: Results from the EGFR-2013-CPHG study. Respir Med Res. 2021, 80:100795. [CrossRef]
- S. LV.; Y. JC., Phase III study of afatinib or cisplatin plus pemetrexed in patients with metastatic lung adenocarcinoma with EGFR mutations. J Clin Oncol. 2013, 31(27):3327-34. [CrossRef]
- W. YL.; Z. C., Afatinib versus cisplatin plus gemcitabine for first-line treatment of Asian patients with advanced non-small-cell lung cancer harbouring EGFR mutations (LUX-Lung 6): an open-label, randomised phase 3 trial. Lancet Oncol. 2014, 15(2):213-22. [CrossRef]
- G. SL.; Osimertinib: First Global Approval. Drugs. 2016, 76(2):263-73. [CrossRef]
- Z. Y.; L. S., Overall survival benefit of osimertinib and clinical value of upfront cranial local therapy in untreated EGFR-mutant nonsmall cell lung cancer with brain metastasis. Int J Cancer. 2022, 150(8):1318-1328. [CrossRef]
- K. S.; H. CT., Analysis of potential predictive markers of cetuximab benefit in BMS099, a phase III study of cetuximab and first-line taxane/carboplatin in advanced non-small-cell lung cancer. J Clin Oncol. 2010, 28(6):918-27. [CrossRef]
- P. R.; P. JR., Cetuximab plus chemotherapy in patients with advanced non-small-cell lung cancer (FLEX): an open-label randomised phase III trial. Lancet. 2009, 373(9674):1525-31. [CrossRef]
- H. D. S.; B. Y. J., 1257O Durability of clinical benefit and biomarkers in patients (pts) with advanced non-small cell lung cancer (NSCLC) treated with AMG 510 (sotorasib). Ann Oncol. 2020, 31, S812. [CrossRef]
- H. DS.; F. MG., KRASG12C Inhibition with Sotorasib in Advanced Solid Tumors. N Engl J Med. 2020, 383(13):1207-1217. [CrossRef]
- R. M.; S. A., 1416TiP CodeBreak 200: A phase III multicenter study of sotorasib (AMG 510), a KRAS (G12C) inhibitor, versus docetaxel in patients with previously treated advanced non-small cell lung cancer (NSCLC) harboring KRAS p. G12C mutation. Ann Oncol. 2020, 31, S894-S895. [CrossRef]
- S. F.; L. BT., Sotorasib for Lung Cancers with KRAS p.G12C Mutation. N Engl J Med. 2021, 384(25):2371-2381. [CrossRef]
- F. JB.; F. JP., Identification of the Clinical Development Candidate MRTX849, a Covalent KRASG12C Inhibitor for the Treatment of Cancer. J Med Chem. 2020, 63(13):6679-6693. [CrossRef]
- H. J.; E. LD., The KRASG12C Inhibitor MRTX849 Provides Insight toward Therapeutic Susceptibility of KRAS-Mutant Cancers in Mouse Models and Patients. Cancer Discov. 2020, 10(1):54-71. [CrossRef]
- R. G. J.; O. S. I., 99O_PR KRYSTAL-1: Activity and preliminary pharmacodynamic (PD) analysis of adagrasib (MRTX849) in patients (Pts) with advanced non–small cell lung cancer (NSCLC) harboring KRASG12C mutation. J Thorac Oncol. 2021, 16(4), S751-S752. [CrossRef]
- F. PM.; R. CM, Crizotinib in the treatment of non-small-cell lung cancer. Expert Opin Pharmacother. 2012, 13(8):1195-201. [CrossRef]
- C. JJ.; T. M., Structure based drug design of crizotinib (PF-02341066), a potent and selective dual inhibitor of mesenchymal-epithelial transition factor (c-MET) kinase and anaplastic lymphoma kinase (ALK). J Med Chem. 2011, 54(18):6342-63. [CrossRef]
- H. Z.; X. Q, Efficacy and safety of crizotinib plus bevacizumab in ALK/ROS-1/c-MET positive non-small cell lung cancer: an open-label, single-arm, prospective observational study. Am J Transl Res. 2021, 13(3):1526-1534.
- L. C.; L. C., Genetic correlation of crizotinib efficacy and resistance in ALK- rearranged non-small-cell lung cancer. Lung Cancer. 2022, 171:18-25. [CrossRef]
- G. JF.; T. DS., Progression-Free and Overall Survival in ALK-Positive NSCLC Patients Treated with Sequential Crizotinib and Ceritinib. Clin Cancer Res. 2015, 21(12):2745-52. [CrossRef]
- T. RK.; Overcoming drug resistance in ALK-rearranged lung cancer. N Engl J Med. 2014, 370(13):1250-1. [CrossRef]
- F. E.; K. D., Efficacy and safety of ceritinib in patients (pts) with advanced anaplastic lymphoma kinase (ALK)-rearranged (ALK+) non-small cell lung cancer (NSCLC): an update of ASCEND-1. Annal Oncol. 2014, 25: iv456. [CrossRef]
- S. BJ.; M. T., First-line crizotinib versus chemotherapy in ALK-positive lung cancer. N Engl J Med. 2015, 373(16):1582. [CrossRef]
- B. G.; T. J., Vemurafenib: the first drug approved for BRAF-mutant cancer. Nat Rev Drug Discov. 2012, 11(11):873-86. [CrossRef]
- T. J.; L. JT., Discovery of a selective inhibitor of oncogenic B-Raf kinase with potent antimelanoma activity. Proc Natl Acad Sci U S A. 2008, 105(8):3041-6. [CrossRef]
- H. DM.; P. I., Vemurafenib in Multiple Nonmelanoma Cancers with BRAF V600 Mutations. N Engl J Med. 2018, 379(16):1585. [CrossRef]
- S. V.; G. R., Efficacy of Vemurafenib in Patients With Non-Small-Cell Lung Cancer With BRAF V600 Mutation: An Open-Label, Single-Arm Cohort of the Histology-Independent VE-BASKET Study. JCO Precis Oncol. 2019, 3:PO.18.00266. [CrossRef]
- K. AJ.; A. MR., Dabrafenib; preclinical characterization, increased efficacy when combined with trametinib, while BRAF/MEK tool combination reduced skin lesions. PLoS One. 2013, 8(7):e67583. [CrossRef]
- Z. R.; Trametinib. Recent Results Cancer Res. 2014, 201:241-8. [CrossRef]
- P. D.; K. TM., Dabrafenib in patients with BRAF(V600E)-positive advanced non-small-cell lung cancer: a single-arm, multicentre, open-label, phase 2 trial. Lancet Oncol. 2016, 17(5):642-50. [CrossRef]
- P. D.; S. EF., Dabrafenib plus trametinib in patients with previously untreated BRAFV600E-mutant metastatic non-small-cell lung cancer: an open-label, phase 2 trial. Lancet Oncol. 2017, 18(10):1307-1316. [CrossRef]
- B. MI.; N. J., FDA Approval Summary: Dabrafenib in Combination with Trametinib for BRAFV600E Mutation-Positive Low-Grade Glioma. Clin Cancer Res. 2024, 30(2):263-268. [CrossRef]
- S. AT.; O. SH., Crizotinib in ROS1-rearranged non-small-cell lung cancer. N Engl J Med. 2014, 371(21):1963-71. [CrossRef]
- V. HG.; N. TQ., Crizotinib in the Treatment of Advanced Non-Small-Cell Lung Cancer with ROS1 Rearrangement or MET Alteration: A Systematic Review and Meta-Analysis. Target Oncol. 2020, 15(5):589-598. [CrossRef]
- G. JF.; T. D., Patterns of Metastatic Spread and Mechanisms of Resistance to Crizotinib in ROS1-Positive Non-Small-Cell Lung Cancer. JCO Precis Oncol. 2017, 2017:PO.17.00063. [CrossRef]
- A. MM.; K. R., Acquired resistance to crizotinib from a mutation in CD74-ROS1. N Engl J Med. 2013, 368(25):2395-401. [CrossRef]
- M. M,; A. E., Discovery of Entrectinib: A New 3-Aminoindazole As a Potent Anaplastic Lymphoma Kinase (ALK), c-ros Oncogene 1 Kinase (ROS1), and Pan-Tropomyosin Receptor Kinases (Pan-TRKs) inhibitor. J Med Chem. 2016, 59(7):3392-408. [CrossRef]
- D. A.; S. S., Entrectinib in ROS1 fusion-positive non-small-cell lung cancer: integrated analysis of three phase 1-2 trials. Lancet Oncol. 2020, 21(2):261-270. [CrossRef]
- A. E.; M. M., Entrectinib, a Pan-TRK, ROS1, and ALK Inhibitor with Activity in Multiple Molecularly Defined Cancer Indications. Mol Cancer Ther. 2016, 15(4):628-39. [CrossRef]
- Z. HY.; L. Q., PF-06463922 is a potent and selective next-generation ROS1/ALK inhibitor capable of blocking crizotinib-resistant ROS1 mutations. Proc Natl Acad Sci U S A. 2015, 112(11):3493-8. [CrossRef]
- F. E.; B. T., MA07. 11 safety and efficacy of lorlatinib (PF-06463922) in patients with advanced ALK+ or ROS1+ non-small-cell lung cancer (NSCLC). J Thorac Oncol. 2017, 12.1: S383-S384. [CrossRef]
- S.AT.; S. BJ., Lorlatinib in advanced ROS1-positive non-small-cell lung cancer: a multicentre, open-label, single-arm, phase 1-2 trial. Lancet Oncol. 2019, 20(12):1691-1701. [CrossRef]
- S.R.; Cho BC, Brahmer JR, Soo RA. Nivolumab in NSCLC: latest evidence and clinical potential. Ther Adv Med Oncol. 2015, (2):85-96. [CrossRef]
- R. D., Pembrolizumab versus Chemotherapy for PD-L1-Positive Non-Small-Cell Lung Cancer. N Engl J Med. 2016, 375(19):1823-1833. [CrossRef]
- S. A.; K.S., Cemiplimab monotherapy for first-line treatment of advanced non-small-cell lung cancer with PD-L1 of at least 50%: a multicentre, open-label, global, phase 3, randomised, controlled trial. Lancet. 2021, 397(10274):592-604. [CrossRef]
- L.ES.; S.A., Analysis of Tumor Mutational Burden, Progression-Free Survival, and Local-Regional Control in Patents with Locally Advanced Non-Small Cell Lung Cancer Treated With Chemoradiation and Durvalumab. JAMA Netw Open. 2023, 6(1):e2249591. [CrossRef]
- B. JR.; L. JS., Five-Year Survival Outcomes With Nivolumab Plus Ipilimumab Versus Chemotherapy as First-Line Treatment for Metastatic Non-Small-Cell Lung Cancer in CheckMate 227. J Clin Oncol. 2023, 41(6):1200-1212. [CrossRef]
- W.Y.; H. H., Immune checkpoint inhibitors alone vs immune checkpoint inhibitors-combined chemotherapy for NSCLC patients with high PD-L1 expression: a network meta-analysis. Br J Cancer. 2022, 127(5):948-956. [CrossRef]
- S.Y.; K. F., Immune Checkpoint Inhibitors in Cancer Therapy. Curr Oncol. 2022, 29(5):3044-3060. [CrossRef]
- M. F.; S. L., Adverse effects of immune-checkpoint inhibitors: epidemiology, management and surveillance. Nat Rev Clin Oncol. 2019, 16(9):563-580. [CrossRef]
- D. AA.; P. VG., The role of PD-L1 expression as a predictive biomarker: an analysis of all US Food and Drug Administration (FDA) approvals of immune checkpoint inhibitors. J Immunother Cancer. 2019, 7(1):278. [CrossRef]
- R. B.; W. X., Association of High Tumor Mutation Burden in Non-Small Cell Lung Cancers With Increased Immune Infiltration and Improved Clinical Outcomes of PD-L1 Blockade Across PD-L1 Expression Levels. JAMA. Oncol. 2022, 8(11):1702. [CrossRef]
- M. A.; L. DT, Efficacy of Pembrolizumab in Patients With Noncolorectal High Microsatellite Instability/Mismatch Repair-Deficient Cancer: Results From the Phase II KEYNOTE-158 Study. J Clin Oncol. 2020, 38(1):1-10. [CrossRef]
- G. A.; M. DF., Nivolumab for the treatment of cancer. Expert Opin Investig Drugs. 2015, 24(2):253-60. [CrossRef]
- R. M.; R. D., Pembrolizumab Versus Platinum-Based Chemotherapy for Advanced Non-Small-Cell Lung Cancer With PD-L1 Tumor Proportion Score of 50% or Greater. J Clin Oncol. 2019, 37(7):537-546. [CrossRef]
- A. M.; R. S., Al-Bozom IA. PD-L1 immunostaining: what pathologists need to know. Diagn Pathol. 2022, 17(1):50. [CrossRef]
- E. Z.; C. LE., Circulating tumour cells and PD-L1-positive small extracellular vesicles: the liquid biopsy combination for prognostic information in patients with metastatic non-small cell lung cancer. Br J Cancer. 2024 Jan;130(1):63-72. [CrossRef]
- A. TA.; The Role of Circulating Tumor Cells as a Liquid Biopsy for Cancer: Advances, Biology, Technical Challenges, and Clinical Relevance. Cancers (Basel). 2024, 16(7):1377. [CrossRef] [PubMed]
- S. J.; M. J., Radiogenomic System for Non-Invasive Identification of Multiple Actionable Mutations and PD-L1 Expression in Non-Small Cell Lung Cancer Based on CT Images. Cancers (Basel). 2022, 14(19):4823. [CrossRef]
- W.C.; M. J., Non-Invasive Measurement Using Deep Learning Algorithm Based on Multi-Source Features Fusion to Predict PD-L1 Expression and Survival in NSCLC. Front Immunol. 2022, 13:828560. [CrossRef]
- K. A.; D. M., The role of genetic polymorphism within PD-L1 gene in cancer. Review. Exp Mol Pathol. 2020, 116:104494. [CrossRef]
- Y.M.; H.A., Tumor Mutational Burden and Response Rate to PD-1 Inhibition. N Engl J Med. 2017, 377(25):2500-2501. [CrossRef]
- M. L.; F. LA., FDA Approval Summary: Pembrolizumab for the Treatment of Tumor Mutational Burden-High Solid Tumors. Clin Cancer Res. 2021, 27(17):4685-4689. [CrossRef] [PubMed]
- R. N. F.; J. G., 1028P BIOLUMA: A phase II trial of nivolumab in combination with ipilimumab to evaluate efficacy and safety in lung cancer and to evaluate biomarkers predictive for response – results from the NSCLC cohort. Annal Oncol. 2022, 33:S1025. [CrossRef]
- F. F.; P. R., Measuring tumor mutation burden in non-small cell lung cancer: tissue versus liquid biopsy. Transl Lung Cancer Res. 2018, 7(6):668-677. [CrossRef]
- W. D.; G. S., The prognostic value of TMB in early-stage non-small cell lung cancer: a systematic review and meta-analysis. Ther Adv Med Oncol. 2023, 15:17588359231195199. [CrossRef]
- F. L.; G. S., Tumor mutational burden quantification from targeted gene panels: major advancements and challenges. J Immunother Cancer. 2019, 7(1):183. [CrossRef]
- B. R.; L. JW., Implementing TMB measurement in clinical practice: considerations on assay requirements. ESMO Open. 2019, 4(1):e000442. [CrossRef]
- P. GF.; A. S., TMBcalc: a computational pipeline for identifying pan-cancer Tumor Mutational Burden gene signatures. Front Genet. 2024, 15:1285305. [CrossRef]
- F. L.; G. S., Tumor mutational burden quantification from targeted gene panels: major advancements and challenges. J Immunother Cancer. 2019, 7(1):183. [CrossRef]
- J. T.; B. CR., Structure and function of the components of the human DNA mismatch repair system. Int J Cancer. 2006, 119(9):2030-5. [CrossRef]
- L. DT.; U. JN., PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N Engl J Med. 2015, 372(26):2509-20. [CrossRef]
- B. M.;, L. DT., DNA mismatch repair in cancer. Pharmacol Ther. 2018, 189:45-62. [CrossRef]
- A. V.; M. G., Mechanisms of Immune Escape and Resistance to Checkpoint Inhibitor Therapies in Mismatch Repair Deficient Metastatic Colorectal Cancers. Cancers (Basel). 2021, 13(11):2638. [CrossRef]
- B. M.; L. DT., DNA mismatch repair in cancer. Pharmacol Ther. 2018, 189:45-62. [CrossRef]
- A. V.; M. G., Mechanisms of Immune Escape and Resistance to Checkpoint Inhibitor Therapies in Mismatch Repair Deficient Metastatic Colorectal Cancers. Cancers (Basel). 2021, 13(11):2638. [CrossRef]
- D. F.; C. KB., Comparison of microsatellite instability detection by immunohistochemistry and molecular techniques in colorectal and endometrial cancer. Sci Rep. 2021, 11(1):12880. [CrossRef]
- E. A.; L. G. N., Deep learning for the detection of microsatellite instability from histology images in colorectal cancer: a systematic literature review. ImmunoInformatics. 2021, 3-4:100008.
- A.V.; H. C., ctDNA response after pembrolizumab in non-small cell lung cancer: phase 2 adaptive trial results. Nat Med. 2023, 29(10):2559-2569. [CrossRef]
- P. F.; A.P., "Circulating tumor DNA (ctDNA) as predictive biomarker in NSCLC patients treated with nivolumab." Annal Oncol. 2017, 28:( ii10-ii11). [CrossRef]
- M. EJ.; L. Y., Circulating Tumor DNA Dynamics Predict Benefit from Consolidation Immunotherapy in Locally Advanced Non-Small Cell Lung Cancer. Nat Cancer. 2020, 1(2):176-183. [CrossRef]
- S. JC.; B. Y., Current and Future Clinical Applications of ctDNA in Immuno-Oncology. Cancer Res. 2022, 82(3):349-358. [CrossRef]
- K. H.; P. KU., Clinical Circulating Tumor DNA Testing for Precision Oncology. Cancer Res Treat. 2023, 55(2):351-366. [CrossRef]
- C. SA.; Liu MC, Aleshin A. Practical recommendations for using ctDNA in clinical decision making. Nature. 2023, 619(7969):259-268. [CrossRef]
- K. D.; T. A., Response to Checkpoint Inhibition in Non-Small Cell Lung Cancer with Molecular Driver Alterations. Oncol Res Treat. 2020, 43(6):289-298. [CrossRef]
- B. D., D. A. Immunotherapy in EGFR mutant non-small cell lung cancer: when, who and how? Transl Lung Cancer Res. 2019, 5):710-714. [CrossRef]
- L. C.; Z. S., The superior efficacy of anti-PD-1/PD-L1 immunotherapy in KRAS-mutant non-small cell lung cancer that correlates with an inflammatory phenotype and increased immunogenicity. Cancer Lett. 2020, ;470:95-105. [CrossRef]
- V. NI.; P. K, Le X. Efficacy of immunotherapy in oncogene-driven non-small-cell lung cancer. Ther Adv Med Oncol. 2023, 15:17588359231161409. [CrossRef]
- T. S.; Q. C., Immune Checkpoint Inhibitors in Non-Small Cell Lung Cancer: Progress, Challenges, and Prospects. Cells. 2022, 11(3):320. [CrossRef]
- M. PC.; K. MI., Targeted Next-Generation Sequencing Reveals Exceptionally High Rates of Molecular Driver Mutations in Never-Smokers With Lung Adenocarcinoma. Oncologist. 2022, 27(6):476-486. [CrossRef]
- C. A.; R. JW., Brahmer JR. Checkpoint Blockade in Lung Cancer With Driver Mutation: Choose the Road Wisely. Am Soc Clin Oncol Educ Book. 2020, 40:372-384. [CrossRef]
- N. T.; O. H., Clinical Impact of Single Nucleotide Polymorphism in PD-L1 on Response to Nivolumab for Advanced Non-Small-Cell Lung Cancer Patients. Sci Rep. 2017, 7:45124. [CrossRef]
- B.E. A.; S. B., Association of single nucleotide polymorphisms with efficacy in nivolumab-treated NSCLC patients. Annal Oncol. 2017, 28: v473. [CrossRef]
- P.G.; L. L., rs822336 binding to C/EBPβ and NFIC modulates induction of PD-L1 expression and predicts anti-PD-1/PD-L1 therapy in advanced NSCLC. Mol Cancer. 2024, 23(1):63. [CrossRef]
- H. L. M.; M. A,, KIR-HLA gene diversities and susceptibility to lung cancer. Sci Rep. 2022, 12(1):17237. [CrossRef]
- M. J.; J. G., HLA class II molecule HLA-DRA identifies immuno-hot tumors and predicts the therapeutic response to anti-PD-1 immunotherapy in NSCLC. BMC Cancer. 2022, 22(1):738. [CrossRef]
- H. S.; S. R., Assessing the Host Immune Response, TILs in Invasive Breast Carcinoma and Ductal Carcinoma In Situ, Metastatic Tumor Deposits and Areas for Further Research. Adv Anat Pathol. 2017, (5):235-251. [CrossRef]
- K. D.; K. K.,Systemic inflammation and pro-inflammatory cytokine profile predict response to checkpoint inhibitor treatment in NSCLC: a prospective study. Sci Rep. 2021, 11(1):10919. [CrossRef]
- D.S.; H. T., The Gut Microbiome from a Biomarker to a Novel Therapeutic Strategy for Immunotherapy Response in Patients with Lung Cancer. Curr Oncol. 2023, 30(11):9406-9427. [CrossRef]
- S.D.; Revolutionizing Lung Cancer Treatment: Innovative CRISPR-Cas9 Delivery Strategies. AAPS PharmSciTech. 2024, 25(5):129. [CrossRef]
Figure 1.
Classification of Lung Cancers. Lung cancer It is divided into two main categories: about 85% of non-small cell lung carcinoma (NSCLC) including the subtypes of adenocarcinoma, squamous cell carcinoma (SQ) and large cell carcinoma (LC) and about 10-15% of small cell lung carcinoma (SCLC). Only 1-2% of lung tumors are pulmonary carcinoid.
Figure 1.
Classification of Lung Cancers. Lung cancer It is divided into two main categories: about 85% of non-small cell lung carcinoma (NSCLC) including the subtypes of adenocarcinoma, squamous cell carcinoma (SQ) and large cell carcinoma (LC) and about 10-15% of small cell lung carcinoma (SCLC). Only 1-2% of lung tumors are pulmonary carcinoid.
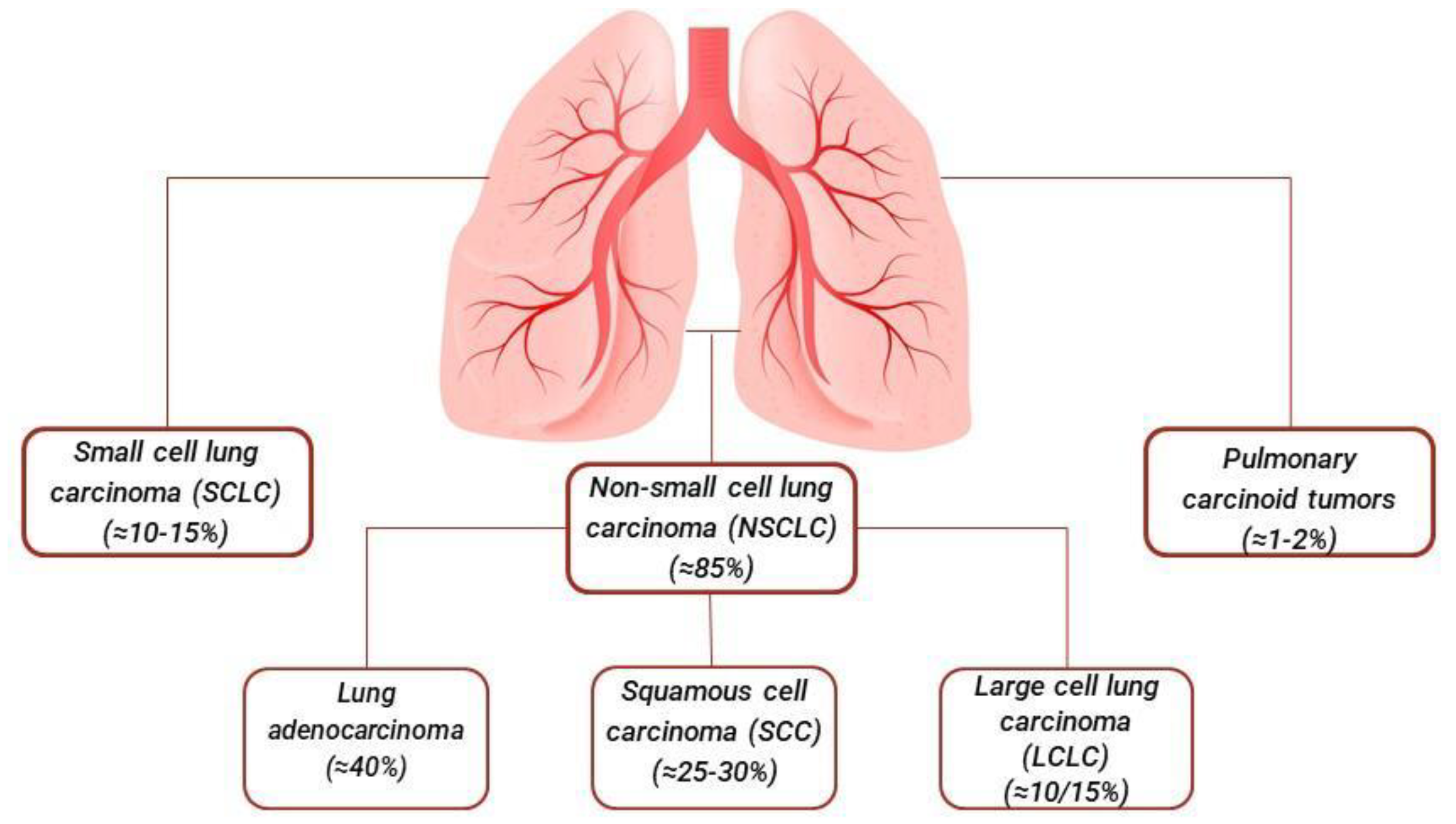

Table 1.
Well-known genes involved in lung cancer and current therapies.
| Gene | Molecular alteration |
Locus | Mutational Hotspot | Drug |
|---|---|---|---|---|
| EGFR | Mutation | 7p11.2 | Deletion in exon 19 Substitution in exon 21 (L858R) |
Gefitinib Erlotinib Afatinib Osimertinib |
| KRAS | Mutation | 12p12.1 | Substitution in exon 12 (G12C), Substitution in exon 13 (G12D) Substitution in exon 61 (G12V) |
Sotorasib Adagrasib |
| ALK | Variable chromosome rearrangement | 2p23.2-p23.1 | Fusion EML4-ALK, KIF5B-ALK, TFG-ALK, KLC1-ALK | Crizotinib Ceritinib Alectinib Brigatinib Lorlatinib |
| BRAF | Mutation | 7q34 | Sostitution V600E, V600D, V600K, V600R K601N/E, L597V, G464V, G469V/R/A, G466V/A,N581S , D594N/G, G596R |
Vemurafenib Dabrafenib Trametinib |
| ROS1 | Variable chromosome rearrangement |
6q22.1 | Fusion CD74-ROS1, SLC34A2-ROS1, TPM3-ROS1, SDC4-ROS1 | Vemurafenib Dabrafenib Trametinib |
Table 2.
Emerging genes involved in lung cancer and current therapies.
| Gene | Molecular alteration |
Locus | Mutational Hotspot | Drug |
|---|---|---|---|---|
| PD-L1 | Mutation Amplification |
9p24.1 | Substitution G1268C | Nivolumab Pembrolizumab Cemiplimab Durvalumab Atezolizumab Avelumab |
| MET | Amplification Mutation Chromosome rearrangement |
7q31.2 | METex14 Fusion KIF5B-MET |
Crizotinib Tepotinib Capmatinib Bozitnib Glumetinib |
| RET | Variabel chromosome rearrangement Mutation |
10q11.21 | Fusion KIF5B-RET, CCDC6-RET, NCOA4-RET, TRIM33-RET, CUX1-RET | Pralsetinib Selpercatinib |
| NTRK | Gene fusions | 13q31.1 13q31.2 Xq27.3 3q26.1 |
Fusion SQSTM1-NTRK1, RFWD2-NTRK1, CD74-NTRK1, TRIM 24 – NTRK2 | Entrectinib Larotrectinib Repotrectinib |
| PIK3CA | Mutation Amplification |
3q26.32 | Substitution in exon 9 (E545K/E542K) Substitution in exon 20 (H1047R/H1047L) |
Gedatolisib Idelalisib |
| HER2 | Mutation Amplification Overexpression |
17q12 | Insertion in exon 20 (A775_G776insYVMA) | Ado-trastuzumab emtansine Mobocertinib Poziotinib |
| STK11 | Mutation Inactivation |
19p13.3 | Deletion of 19p13.3 | Atezolizumab Pembrolizumab Nivolumab |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



