
Submitted:
31 July 2024
Posted:
31 July 2024
You are already at the latest version
Abstract
This study presents the first genome assembly of the freshwater saprobe fungus Neonectria lugdunensis and a comprehensive phylogenomics analysis of the Nectriaceae family, examining genomic traits according to fungal lifestyles. The Nectriaceae family, one of the largest in Hypocreales, includes fungi with significant ecological roles and economic importance as plant pathogens, endophytes, and saprobes. The phylogenomics analysis identified 2684 single-copy orthologs, providing a robust evolutionary framework for the Nectriaceae family. We analyzed the genomic characteristics of 17 Nectriaceae genomes, focusing on their carbohydrate-active enzymes (CAZymes), biosynthetic gene clusters (BGCs), and adaptations to environmental temperatures. Our results highlight the adaptation mechanisms of N. lugdunensis, emphasizing its capabilities for plant-litter degradation and enzyme activity in varying temperatures. Comparative genomics of different Nectriaceae lifestyles revealed significant differences in genome size, gene content, repetitive elements, and secondary metabolite production. Endophytes exhibited larger genomes, more effector proteins, and BGCs, while plant pathogens had higher thermo-adapted protein counts, suggesting greater resilience to global warming. In contrast, the freshwater saprobe shows less adaptation to warmer temperatures and is important for conservation goals. This study underscores the importance of understanding fungal genomic adaptations to predict ecosystem impacts and conservation targets in the face of climate change.
Keywords:
Nectriaceae family
; Fungal lifestyles
; Global warming
; Genomic Adaptations
1. Introduction
The Nectriaceae family was introduced in 1865 to accommodate the hypocrealean species having ascomata that are generally yellow, orange-red to purple and usually change colour in potassium hydroxide and lactic acid [1,2]. Nectrioid fungi were mainly characterized by slightly bright-colored and generally superficial perithecia containing ascospores with articulated cells, and their various phialidic anamorph. Some genera affiliated with Nectriaceae have straight asci and conidia, while those of other genera are strongly curved [1].
The reexamination of nectrioid fungi has been carried out continuously since 1950 by several taxonomists [3,4,5,6,7,8,9,10,11,12].
The genus Neonectria is characterized by well-developed or minute stromata, subglobose to broadly pyriform and gregarious perithecia that are laterally or not collapsing when dry, 2- or 3-layered perithecial wall, smooth to warted perithecial surface, cylindrical to clavate asci with or without an apical ring, and uni- or multiseptate ascospores with spinulose or striate surface [13].
Nectriaceae is one of the largest families of Hypocreales (Sordariomycetes, Ascomycota) [14], with 167 genera described in Mycobank (09.02.2024, Mycobank). They are frequently found on both living and decaying woody materials, soil, fruiting bodies of other fungi, and insects and occur on various substrates in tropical and subtropical regions around the world [8]. Several of them are documented as endophytes, or opportunistic plant-pathogens, pathogens of crops and humans [15]. They have potential to degrade resistant plant material [16], and are used in industrial and commercial applications [17].
The genera Fusarium and Neonectria are the most studied taxa in Nectriaceae due to their relevant economic importance as plant-pathogen. The genus Fusarium is one of the largest genera in the family Nectriaceae, and more than 1,000 species have been reported in 2022 [14]. Fusarium was included in the top 10 globally most important genera of plant pathogenic fungi based on scientific and economic importance [18]. The genus Neonectria has 57 species reported in Mycobank (09.02.2024, Mycobank). Neonectria species are known for infection of the branch and trunk which causes lesions known as cankers, they are normally associated with beech and fruit trees bark canker [19]. The most known Neonectria plant-pathogens are N. coccinea (European beech bark disease); N. faginata (American beech bark disease); N. ditissima (hardwood canker disease) [20,21].
Neonectria is also a common genus of endophytic fungi [19,22]. Fungal endophytes of plants are widespread and important for host plant health [23]. They spend all or part of their life, residing in healthy plant tissue, often in non-pathogenic mutualistic relationships, often protecting the plant against biotic and abiotic stress and promoting plant growth [22,24,25]. Their presence and abundance are often associated with the development stage of the plant, season, and environment [25,26]. When in association with the roots, fungal endophytes play important roles in ecosystem processes and nutrient cycling, with beneficial symbiotic relationships with many plants [27,28]. Endophytic fungi can be latent pathogens [29,30], mutualists, for example mycorrhizal fungi [31], and/or saprobes [32,33], but should be detected within the tissues of healthy host plants [34,35].
The Nectriaceae family also includes fungi with saprobe capabilities, in soil and freshwater [17,36]. The Freshwater fungi are believed to be originated from soil fungi approximately 1.5 billion years ago due to a similar microbial machinery found in both groups and evolutionary studies [37,38]. In soil, Nectriaceae saprobes have a cosmopolitan distribution around the world and colonize newly dead, organic plant material [39]. Freshwater fungi are characterized as having partial or complete lifecycle in freshwater environments [40], where they act primarily in the decomposition of leaf litter, particularly aquatic hyphomycetes [41]. The aquatic hyphomycetes are characterized by tetraradiate, branched, and sigmoid conidial shapes that facilitate the attachment on the surface of aquatic leaf litter [42,43]. However, in aquatic Nectriaceae the typical conidial shape can be clove--shaped with or without minute outgrows [43,44]. Freshwater ascomycetes possess distinctive adaptations that enable their survival in freshwater environments for example their unique lignocellulose enzymes that work by softening the leaves and wood, which is particularly fundamental for breaking down wood underwater [37].
According to many studies, in order to adapt to colder environments, certain freshwater fungi have developed unique mechanisms, such as cold-active enzymes and antifreeze proteins [45,46,47,48,49,50], these characteristics help the freshwater fungi to survive in extreme environmental conditions like arctic and subarctic streams and as well in freshwater streams. However, the escalating temperatures caused by global warming have emphasized the need to comprehend and forecast the reaction of microbial life and ecosystems to severe drought and warm weather occurrences. These events are one of the major environmental stresses experienced by microorganisms such as fungi [51] and can affect the activity of their enzymes and specialized metabolites. For example, the production of specialized metabolites in lichens is affected by light, UV radiation, altitude, temperature fluctuations, and seasonality [52,53,54]. In addition, global warming may have significant consequences in human health, crop production, and the well-being of forests and other habitats [53]. Therefore, research mechanisms to identify temperature adaptations and the possible impacts of climate change on fungal species have been implemented not only to predict ecosystem impact but also to target conservation goals [47,55,56].
In this manner, we present the first genome of the saprobe freshwater fungus Neonectria lugdunensis and an up-to-date phylogenomics tree of the Nectriaceae family observing the divergences in genome traits according to fungal lifestyle. We emphasized their machinery for the breakdown of plant-litter and their enzyme capability to resist and be active in colder and warmer temperatures, which is an important mechanism to identify not only the environment adaptation for temperature fluctuations but also may indicate possible target species for conservation goals in times of climate change-related impacts.
2. Materials and Methods
The Neonectria lugdunensis strain was obtained from the internal collection of DSMZ (Leibniz Institute DSMZ, German Collection of Microorganisms and Cell Cultures) with the assigned identifier DSM 113088. Originally this strain was derived from ascospores of the perithecial morph on a twig in Slovakia (GenBank accession number of ITS- sequence DQ247777) by Ludmila Marvanová and deposited in the Czech Collection of Microorganisms under CCM F-13783. The fungus was cultivated on agar plates containing 2% malt extract (Feelwell, Germany) using Oxoid brand agar (Germany) at a temperature of 16°C within a designated cooling facility for DNA extraction. To confirm the taxonomic assignation of the strain cultures, we employed Sanger sequencing of the Beta Tubulin marker gene. This was accomplished using the pair primers T1 (5′-AAC-ATG-CGT-GAG-ATT-GTA-AGT-3′) and T2 (5′-TAG-TGA-CCC-TTG-GCC-CAG-TTG-3′). The obtained sequences were manually curated using the Sequencher tool v5.4.6 (http://www.genecodes.com). Subsequently, a BLAST analysis was conducted against the National Center for Biotechnology Information (NCBI) nucleotide database (https://www.ncbi.nlm.nih.gov/).
After taxonomy confirmation of the strain, the biomass was transferred to 1 litter flask each with potato-glucose liquid medium (Carl Roth, Germany), and transferred to a 16°C cooling room over a shaker (Bottmingen, Switzerland) at 120 RPM. The samples were kept in a shaker until enough biomass (14 grams) for genome extraction (see Supplementary File). The genomic DNA with fragment sizes greater than fifteen kilobases, verified with an Agilent Femto Pulse (Agilent, United States), were sent to Hifi genome sequencing at Macrogen, Netherlands. The raw sequence files were obtained from Macrogen, using Flye v2.9 [57], and the quality was accessed with Busco v5.2.2 [58] using the ascomycota_odb10 database.
For the comparative genomics of the Nectriaceae family, we have obtained 17 genomes of Nectriaceae from the NCBI database (see Table 1). We aim to select representative genomes of each genus at random but taking into consideration the lifestyle of each fungal species, and we added all genomes available for the Neonectria genus. We also added 2 outgroup genomes for phylogenomics analysis belonging to the Ophiocordycipitaceae family, phylogenetically closely related to Nectriaceae [37], and the newly assembled genome of Neonectria lugdunensis. The genomes were verified according to their genome completeness to assure values higher than 95%, according to the results obtained with BUSCO and the ascomycota_odb10 database.
An all-vs-all Genome-to-genome alignment and comparison analysis was performed using the DNADIFF program from MUMMER3 [59]. All genomes were aligned and compared against each other to obtain the average nucleotide identity (ANI) and genomes with similarity higher than 95% were considered of the same species [60]
The integrative method of RepeatModeler2 [61] was employed to generate a comprehensive denovo species-specific repeat library for all genomes. The identification of new repeats by RepeatModeler v2.05 resulted in a library of consensus sequences for each species combined with repeat annotation carried out using RepeatMasker v4.1.5 [62], via sequence comparison against the Dfam library, generating transposable elements results, while soft masking the genome. The soft-masked genome was used to predict the tRNA sequences determined by tRNAscan-SE 2.0.12 [63] with default parameters for eukaryotic organisms.
The soft-masked genomes from NCBI (17 Nectriaceae and 2 outgroup taxa) together with the newly assembled genome of Neonectria lugdunensis, were then submitted to the Braker3 v3.0.3 [64] pipeline to perform ab initio gene prediction with parameters “--esmode” and “--fungus”, using therefore, the software GeneMarker-ES v4.71_lic [65], to produce denovo hints to train Augustus v3.5.0 [66] which after that, predicted the aminoacid sequences of each genome.
The produced aminoacid sequences were used in Orthofinder v3.0.0 [67]with the “-og” option to predict only the Single-Orthologs common to all species of this study, generating then sequence clusters. Each cluster was aligned using MAFFT v7.520 [68] with the option “--auto” and trimmed using Trimal v1.4.rev15 [69] with the option “-automated1” to remove poor alignment regions. The phylogenetic clusters were than merged into a unique phylogenetic tree using IQTREE2 v2.2.2.7 [70] with the options –bb 1000 -m TEST. The -m TEST parameter stands for ModelFinder [71], which searches for the best evolutive model for each gene cluster, respecting the evolutionary specificities of each gene.
To investigate the genetic potential of the Nectriaceae fungi for secondary metabolite production, the AntiSMASH fungal cluster predictor (v7.0.1) [72] was implemented. The presence of genes and clusters associated with the biosynthesis of secondary metabolites was assessed in every genome. Fasta files containing genome sequences were utilized as inputs, with the default search parameter set to "relaxed." Every additional feature was enabled, including Cluster-border prediction by utilizing transcription factor binding sites (CASSIS).
The predicted aminoacid sequences from the Braker3 pipeline were used for secretome prediction using a combination of tools. To predict where the provided protein sequence possesses a signal peptide, at its N-terminus, that directs the protein towards secretion, we use the consensus sequences obtained from the tools Signalp 6.0 [73] and Targetp2 [74]. In addition, the sequences were also filtered out using DeepTMHMM [75]to remove transmembrane domain signals.
Secreted, not secreted and effector proteins were then annotated for Carbohydrate-active enzyme (CAZymes) with run_dbCAN4 (https://github.com/linnabrown/run_dbcan), a standard alone tool of dbCAN3 web server (https://bcb.unl.edu/dbCAN2/), using the tools HMMER v3.3.2 [76], DIAMOND v2.1.8 [77], and dbCAN_sub with the dbCAN3 [78] database v12. The analysis was performed against all 6 available classes of carbohydrate-active enzymes (CAZys): carbohydrate-binding module (CBM), glycoside hydrolases (GHs), polysaccharide lyases (PLs), auxiliary activities (AAs), carbohydrate esterases (CEs), and glycosyl transferases (GTs). Only sequences that were identified as CAZy enzymes with all 2 or more tools (HMMER, DIAMOND, and dbCAN_sub) were considered for the results, as recommended by dbcan3.
Putative secreted effector proteins were identified from the secreted proteins pool using EffectorP v3.0 [79] where proteins were predicted to have or not interact with plants at the apoplast and cytoplasmic level, through the tool, we can observe the secreted effector proteins associated with plant infection by Fungi including the main local of infection of the protein.
To better understand the adaption of the freshwater hyphomycetes to the environment, proteins with psychrophilic and thermophilic characteristics were predicted using the machine learning tool ThermoProt [80], for the secretome, and effector proteins.
3. Results
The newly assembled genome of Neonectria lugdunensis, has 44.78 Mbp and 97.6% genome completeness with N50 of 44.7 Mbp, showing similar characteristics from many genomes available in NCBI from Nectriaceae as shown in this study (Table 1). We detected 4.38% of repetitive elements in N. lugdunensis, 2.04% were retroelements, and 0.33% were DNA transposons. Only 207 sequences of tRNA were identified in N. lugdunensis, which is the smallest amount in comparison with the freshwater saprobe fungi Aquanectria penicillioides (222) and the soil saprobe Thelonectria discophora (225).
The N. lugdunensis has fewer predicted genes (11480) than the freshwater saprobe Aquanectria penicillioides with 12575 although higher than the soil saprobe Thelonectria discophora with 10364 genes. This result is also reflected in the number of secreted proteins of N. lugdunensis being smaller (797) than freshwater A. penicillioides (820) and higher than the soil saprobe Thelonectria discophora (655) and also reflected the number of effector genes (229 in N. lugdunensis, 254 in A. penicillioides, and 191 in soil saprobe T. discophora) (Table S1). Regarding BGCs, N. lugdunensis, had higher amounts among saprobe fungi with 48 predicted clusters, followed by the other freshwater A. penicillioides with 40, and the soil saprobe T. discophora with 33. (Table S2). N. lugdunensis was the only freshwater fungi in this study to have a BGC for NRP-Metallophore.
The species Ilyonectria robusta (current name according to Mycobank: Ramularia robusta) is known to be a plant pathogen and endophyte. However, the genome strain was isolated as an endophyte (https://mycocosm.jgi.doe.gov/Ilyrob1/Ilyrob1.home.html) and therefore presented in this study as such (Table S1)
We obtained 2684 Single-copy ortholog genes yielded a robust, well-resolved, and comprehensive phylogeny of the Nectriaceae family (Figure 1). The internodes in the tree received a strong bootstrap value higher than 95%, characterizing a strongly supported tree and not particularly associated with the lifestyle of the fungi.
The species Neonectria galligena is nowadays a known synonym of Neonectria ditissima and has been described as being the same species since 1995 [8] although, in the NCBI database, both names are represented with different genomes. The genome-to-genome analysis through ANI confirmed that they are the same species (ANI value of 98.91%), but the enzymatic profile suggests being from different strains, presenting different phenotypes, and therefore the synonymy was kept in the study. All other genome comparisons using ANI show values smaller than 95% between species genomes.
Our results show that the genome length of endophyte organisms was on average bigger (49.24 Mbp) than plant-pathogens (48.04 Mbp) and saprobes (46.71 Mbp), despite the largest genome in the study being Dactylonectria alcacerensis which is a plant pathogen with 61.76 Mbp (Figure S1). This value was also reflected in the average number of aminoacid sequences predicted where endophytes (12971) were the highest followers by plant-pathogens (12798) and saprobes (11473) (Figure S2). However, we observed a change in the GC content where the highest average in the plant pathogens than saprobe fungi and endophytes (51.54, 51.42, 51.07, respectively) (Figure S3).
The plant pathogenic fungi in this study presented more tRNA on average than saprobe and endophyte (218.42, 218.00, 212.67 respectively). This is contradictory to what was found in other studies [81,82] (Figure S4).
We observed a large variation in repetitive elements according to the fungi lifestyle (Figure S5), where on average the saprobe (7.35) has more repetitive elements than plant pathogens (5.28) and endophytes (3.13). When observing the repetitive elements sequences with origins in retroelements, we found statistically significant differences between endophytes (0.69) and plant pathogens (2.60) (Figure 2). The difference in retroelements between saprobe, the largest on average with 3.67, and plant pathogens, is not significant, as well as saprobe and endophytes. Regarding the repetitive elements classified as DNA transposons (Figure S6), we observed a shift where saprobes have more repetitive DNA transposons on average (0.75) than endophytes (0.68) and plant pathogens (0.53) (Figure S6).
The number of genes present in endophytes was also higher than in plant-pathogens and saprobes (Table S1), The number of secreted CAZy enzymes was higher in plant pathogens than in endophytes and saprobe fungi, (953.33, 907, and 757.33, respectively) (Figure S7). The secreted CAZy enzymes with cold properties are higher in endophytes (382.67) than in plant pathogens (359.92) and saprobes (313.33) (Figure S8). On the other hand, the secreted CAZy enzymes with thermo resistance were higher in plant pathogens (217.17) than endophytes (196.33) and statistically significant among plant pathogens and saprobes (146.00) (Figure 3).
The average number of effector proteins secreted by endophytes (278.33) is higher than in plant pathogens (268.17) and saprobes (224.67) (Figure S9). The temperature adaptation for effector genes shows statistical significance between endophytes and saprobes for cold and thermos adaptations, where the effector genes adapted to cold was higher in endophytes (166.33), followed by plant pathogens (145.42) and saprobes (126.67) (Figure 4). The endophytes also show higher potentiation in adapting to warmer temperatures, than plant pathogens and saprobes (65.00, 59.08, and 44.67, respectively) (Figure 5).
The endophytes have shown to have more effector genes on average with apoplastic characteristics (237.00) than plant pathogens (223.92) and saprobes (190.67) (Figure S10). On another hand, the effector genes with cytoplasmatic characteristics are shown to be statistically significant between endophytes (99.93 on average) and saprobes (71.00 on average), despite that, plant pathogens (101.58) had higher amounts than endophytes and saprobes (Figure S11).
Regarding Biosynthetic Gene Clusters, endophytes were shown to produce on average more biosynthetic metabolites based on cluster results than plant pathogens and saprobes (54, 53.25, 40.33, respectively) (Figure S12).
We observed also the formation of clusters based on fungal lifestyle when considering their genome products associated with plants (enzymes and metabolites) (Figure 6). In the central region of the figure, we have saprobe organisms surrounded by plant pathogens above and below them, while two endophytes are located on the right side and Mariannaeae in the extreme north of the graph.
4. Discussion
The endophytes exist within plant tissues without causing plant apparent harm to the host, whereas plant pathogens infect and cause illness in plants. Both of these groups work closely with living plant cells, overcoming the plant's immunological defenses and adapting to its metabolic environment [83,84]. These interactions need a complex set of genes that create enzymes, toxins, and other compounds that aid the invasion of plant cells, suppressing the plant immune system responses, and manipulating plant metabolism. As a result, their genomes frequently contain genes for complicated secondary metabolite production, a variety of transporters, and genes that confer resistance to plant defense mechanisms [85,86]. On the other hand, saprobe fungi, as decomposers of organic material, require a different set of enzymatic tools focused primarily on breaking down cellulose, lignin, and other plant structural components. While this still requires a diverse set of enzymes, the interaction with non-living material is less complex than manipulating and responding to living cells, and therefore, is expected that saprobes have fewer genes than endophytes and plant pathogens [87,88,89].
The repetitive elements are microsatellite regions, or more often transposable elements (transposons) which increase their number in the genome mainly due to cellular stress, which is not a characteristic of endophyte environments [90,91,92]. This corroborates our findings for fewer repetitive elements in endophyte genomes likely influenced by their lifestyle since being endophytic is a very cost-effective strategy. When establishing themselves within plants, they are shielded from abiotic and biotic challenges being less subject to stress, which is a factor for the multiplication of transposable elements [93].
Our study confirms the results of Queiroz, 2020 [94] where the number of repetitive elements was a key element in distinguishing between pathogenic and endophytic fungi, despite non-statistical significance. Several studies found Transposable elements abundant in plant-pathogenic fungi [95,96,97]. In our study, this case can be even better observed with retrotransposons, where the different occurrence between endophytes and plant pathogens was statistically significant. The amount of retroelements identified in endophytes is fewer than in saprobe and statistically significantly less than in plant pathogens.
The transposable elements contribute significantly to genomic plasticity, which is crucial for adapting to host defenses. For instance, in the case of the plant pathogen Verticillium dahliae, TEs exert an influence on gene expression variations essential for pathogenicity enabling the fungus to adapt rapidly to host-induced stresses or resistances [98]. Meanwhile, saprobe fungi are not known for presenting large amounts of TEs [96,99,100], since they are less subject to adaptation to host living cells and may reside in less stressful environments [101,102,103,104], although, the higher number of TEs for saprobes in our study could indicate potential adaptation to more dynamic or challenging environmental conditions [96].
The secretome of fungi, which includes CAZy enzymes, plays a pivotal role in the interaction between fungi and plants. These enzymes are crucial not only for breaking down the complex polysaccharides found in plant cell walls, which aids in nutrient acquisition and infection processes but also for helping fungi evade plant immune responses, facilitating their survival and propagation in various environments [104,105,106]. The CAZymes of classes CE, GH, and PL are often referred to as plant-litter degrading enzymes because they play crucial roles in the degradation of plant biomass by fungi and bacteria [107,108].
In our study, saprobe fungi produced less secreted CAZy enzymes than endophytes and plant pathogens. According to Zhao (2013) [109], saprobe fungi are expected to only degrade plant-litter complex polysaccharides from dead material without having to interact with the plant-living cell defense, and for that, typically require a narrower range of enzymes. In contrast, endophytes and pathogens interact directly with living plant tissues, requiring a broader array of enzymes not only to breakdown living plant tissues but also evade or suppress plant immune responses, and successfully infect their hosts. These interactions demand more specialized enzymatic functions, such as those that degrade pectin, hemicelluloses, and other plant cell wall components under varying physiological conditions as to successfully establish their mutualistic relationships with their hosts [89,110–1142.
The CAZy enzymes when secreted have several functions other than the degradation of plant cell-wall. They are also involved in promoting attachment, invasion, colonization, and nutrient acquisition from the hosts and that’s why numerous studies suggest that endophytes and plant pathogens can manufacture the same leaf-degrading enzymes as closely related saprobic fungi [89,108,110,113,114,115,116,117,118].
Pathogens and endophytes must overcome various layers of plant protection to successfully infect the plant [119,120]. The first immunological response of the plant is through receptor-like kinases that can detect pathogen-associated molecular patterns called microbe-associated molecular patterns (MAMPs) which activate the MAMP-triggered immunity or pathogen-triggered immunity, which is effective against a wide range of microorganisms [121,122,123,124]. However, microorganisms can bypass this mechanism by producing effector proteins that alter cellular processes in the host, leading to effector-triggered susceptibility [122,125,126,127,128]. In addition, the plant has a second layer of receptors called resistant proteins or R proteins which also recognize patterns in effector proteins in the host generating a resistance response that promotes the death of infected cells containing the infection [122,129,130,131,132,133]. Hence, effector proteins have distinct properties to act in the host extracellular space (apoplast) and intracellular space (cytoplasm) during infection. The cytoplasmic effectors have a higher proportion of positively charged amino acids, whereas apoplastic effectors are enriched for cysteine residues [79]. Therefore, it is expected that the more interactions needed between the fungi and plants to overcome their immune system, the more equipped the plant pathogen with effector genes should be the fungi.
However, the cell defense system is not the only obstacle to the survival and adaptation of fungi. The environmental conditions play a major role in protein synthesis, sporulation, and diversity of their communities. Currently, the world is undergoing a significant and fast-paced climate change caused by human activities, specifically global warming. This has resulted in a rise in temperatures, which are presently approximately 1.5 °C higher than they were during the pre-industrial period [134]. This even raises concerns about the adaptation of fungal species with pathogenic capabilities, such as those that infect plants, since is expected an increase in plant diseases in crops under projected climate change scenarios [134,135,136]
In our study, the pathogenic fungi from the Nectriaceae family have more secreted enzymes adapted to higher temperatures than endophytes and saprobes which indicates a higher potential to adapt to global warming situations. The potential for growth and infection of Nectriaceae fungi, are observed in many of the most important economical plant-pathogens from Nectriaceae family belonging to the genera Neonectria and Fusarium [137,138,139,140,141,142,143]. In our study plant-pathogens presented less cold-adapted and more thermo-adapted proteins indicating a potential to perform better in higher temperatures, which reinforces the findings of [139] showing that phytopathogens from Nectriaceae order are organisms that may prevail in global warming increasing their distribution and impact.
On the other hand, in some cases, the temperature can be a comprehensive factor that impacts cellular metabolism and functions. This encompasses essential biochemical parameters, such as the rates at which reactions occur, molecule binding, and the flexibility of cell membranes [134,144]. As example, the freshwater saprobe fungal communities are susceptible to being affected by climate change in species composition and abundance and therefore, impact freshwater ecosystem function [145,146,147,148]. They are richer in cold regions or environments and have genome machinery more adapted to colder temperatures [47,149]. Therefore, the increase in the temperature from global warming can cause a reduction in their activity and could negatively impact energy, carbon, and nutrient cycling, threatening the delivery of ecosystem services to higher organisms [150,151,152,153].
The newly assembled genome of Neonectria lugdunensis presented in the study, was first isolated from a submerged decaying twig in a stream bed (personal communication L. Marvanová) and is commonly reported in aquatic ecology studies [154] but has also been identified in soil [155]. The N. lugdunensis has been shown to be resistant to long drought at 25°C and with the potential to endure even higher temperatures for a limited time, as shown in [153]. Other freshwater fungi have better adaptation to colder temperatures showing growth peaks at temperatures between 15°C and 25°C [146,153]
The low amount of thermo-adapted protein observed in the saprobe fungi from this study is a concerning situation when the increase in world temperature can also represent a loss in saprobe biodiversity, growth, and survival. The temperature is one of the most important factors influencing the structure of saprobe communities, which may lead to severe consequences in the ecological process of decomposition [156]. Therefore, monitoring their biodiversity and the implementation of conservation efforts are vital to preserve the biodiversity of saprobe fungi.
The protein synthesis is a central cellular process that is partly regulated by the availability of tRNA molecules. The tRNAs can pre-present in the genome and spread in multiple families in multiple copies or single genes [81]. They are vital for the breakdown of organic matter and the synthesis of proteins and metabolites that can interact with living organisms. In endophytes and plant pathogens, they are essential for the organism virulence and evasion of host defense and are evolved with pathogenicity [82,157,158]. Therefore, the number of copies of tRNA and their variability may be utilized to predict the efficiency with which genes may be translated. This allows for the estimation of protein synthesis rates, cell responsiveness to external influences, and eventually, evolutionary adaptations to novel environments and has been considered a determinant for lifestyle transitions among basidiomycetes fungi [81,159]. In our data, tRNA did show to be significant in differentiating fungal lifestyle.
The fungi have developed distinct mechanisms for producing CAZyme and secondary metabolites to suit their lifestyles [160,161,162,163]. The secondary metabolites are primarily encoded by biosynthetic gene clusters (BGCs), that are jointly controlled and located very close together in a certain genome region, and as a result, the BGCs in charge of a given specialized metabolite are either "silent" or upregulated [52,164,165,166].
These metabolites play a crucial role in several key adaptive processes associated with ecological interactions and stress responses within their environment and are not directly associated with the growth or reproduction of the fungi [164,167,168,169] being often linked to communication and the defense and/or attack against their surrounding organisms or hosts [52], therefore expected to be present in higher quantities in endophytes and phytopathogens, as presented in this study.
Many secondary metabolites act as pathogenicity factors and have detrimental impacts on host health [163] largely due to the production of mycotoxins – toxic secondary metabolites encoded by BGCs. Mycotoxins can act as virulence factors weakening or killing host plants, and aiding colonization [170,171,172]. The Nectriaceae fungi have been noted for their biological activities, for parasitism on plants, fungi, and insects, and as producers of antibiotics and/or mycotoxins [173], although not much research was implemented in the study of the mycotoxins of the genus Neonectria during plant infection.
The non-ribosomal peptide synthetases (NRPS) and polyketide synthases (PKS) are two types of large, modular enzyme complexes (megasynthases) involved in the biosynthesis of non-ribosomal peptides and polyketides, respectively. The NRPS creates molecules by joining amino acids. In contrast, PKS creates molecules by joining acyl groups to form large polypeptide chains responsible for many different catalytic domains, each performing a different chemical reaction. This allows them to create highly complex and diverse molecules [54]. In plant-associated fungi, NRPS and PKS have distinct functions in the synthesis of phytotoxins, mycotoxins, and antibiotics [174]. According to Yoder (2001) [175] numerous virulence factors were identified in NRPS clusters, demonstrating them as necessary mechanisms in fungal pathogenesis. They were found in higher concentrations in plant pathogenic and endophytes in this study and lower amounts in freshwater saprobes.
Terpene cyclases are enzymes found in plants and microorganisms that help to form monoterpenes, sesquiterpenes, and diterpenes by converting prenyl diphosphate chains. Terpenoids come from isopentenyl diphosphate (IPP), which prenyl transferases modify to create geranyl diphosphate (GPP), farnesyl diphosphate (FPP), and geranyl diphosphate (GGPP). These compounds serve as starting points for making monoterpenes (from GPP), sesquiterpenes (from FPP), and diterpenes (from GGPP) with the help of terpene cyclases enzymes [176,177,178]. Currently, over 80,000 terpenoids are known [179]. Our study found the highest amount of terpene clusters in plant pathogenic fungi. The terpene is known in pathogenic fungi for producing many mycotoxins of the class sesquiterpenoids which play a vital role in fungal virulence [180]. The terpenes can also have effects on fungal growth and protective effect against oxidative stress and UV radiation by the production of carotenoids [178,181,182]. Different environmental factors including light and temperature were shown to change the production level and composition of carotenoids [183].
The ribosomally synthesized and post-translationally modified peptides (RiPPs) were found without many variations in this study. are an increasingly important group of natural products known for their powerful biological activities [184]. They are produced through a straightforward process. The initial step involves the synthesis of a precursor peptide by the ribosome, which consists of leader, core, and follower amino acid sequences. Subsequently, the core sequence undergoes specific post-translational modifications, guided by the leader and follower sequences. The final bioactive RiPP is released after these sequences are removed [185].
Metal ions play a vital role in numerous enzymatic processes, but their excess can be detrimental to the growth and development of various organisms [186]. Research has shown that siderophores, originally known for their iron-binding capacity, can bind to a variety of metals. This multi-faceted functionality has prompted the broader classification of these compounds as "metallophores," referring to secondary metabolites capable of binding a range of metal(loid) cations [187,188]. Metallophores are low-molecular-weight organic ligands that facilitate the delivery of essential metal ions to an organism, while the organism regulates the production and release of these ligands based on its metal ion requirements [187]. In our study, we detected the Siderophore bind with transporter nickel (NI-Siderophore) in only three species (C. pteridis, I. robusta, and F. nematophilium), while Metallophore (NRP- Metallophore) was found in 11 species in this study with one cluster per organism.
The BGC indole is a volatile compound that has been associated with effective fungicides to face continuous fungal infections [189,190] and was not identified on the saprobes genomes in this study showing that it might be a crucial cluster for the fungi infection and virulence in plants.
The phosphate compounds produced in BGCs are chemicals with phosphorus-carbon bonds with the general chemical formula C−PO(OH)2 or C−PO(OR)2 where R is an alkyl or aryl functional group [191]. They have a wide range of bioactivities, such as antibiotics, antivirals, pesticides, and antiparasitics, where approximately 15% of all phosphonate natural products are commercialized [192]. In our study, it was found in 5 plant pathogens and 1 endophyte showing to be an additional feature in fungal infection.
Isocyanides, also known as isonitriles, are a group of microbial secondary metabolites that have been extensively studied due to their wide range of pharmacological applications such as antifungal, antibacterial, antitumor, and antiprotozoal bioactivity [193,194,195,196]. In addition, they play an important role in the pathogenesis of insect, plant, and human diseases [196,197,198] showing to be an additional accessory for fungal infection found in 10 plant pathogen fungi and 2 endophytes in this study. These compounds are distinguished by the highly reactive isocyanide functional group (R≡N+-C−), which originates from the conversion of specific amino acids within the compound [199].
For the endophytes, mycotoxins can also cause herbivory limitation: Ergot alkaloids produced by endophytes limit herbivory, protecting the plant and increasing the fitness of both the plant and the fungus [200]. In addition, the endophyte mycotoxins can also prevent their predation by insects, aiding in the competition against these pests [201], as a consequence protecting the plant and the fungi itself from predation. Biosynthetic gene clusters such as betalactone are known to be an antiviral heterocyclic compound contributing to biocontrol activity and in addition, acting in insect immune suppression, [202,203,204]. This might be important for endophytes and plant pathogens to avoid predation of the leaf and competition by other organisms.
5. Conclusions
We found that endophytes and plant pathogen fungi carry more effector genes and genes dedicated to secondary metabolism than saprobes, likely due to their higher interaction with the defense mechanisms of living cells of plants.
Despite not finding many variables with statistical significance, except the number of retroelements and thermo-effector genes, we can still differentiate fungal lifestyle based on the combination of characteristics associated with plant interaction of enzymes and metabolites shown in this study. This suggests that we can determine the lifestyle of the fungi based on this data and therefore, machine learning algorithms could be applied efficiently for lifestyle prediction in larger datasets. In addition, the low abundance of effector genes adapted to higher temperatures found in the freshwater saprobes in this study may suggest that they would be important organisms for targeting preservation goals due to global warming events. In contrast, the higher amounts of thermo-adapted effector genes in plant pathogens suggest a greater capacity to enhance their bioactivity as temperatures rise, and therefore a potential to escalate the economic impact of these fungi when proliferating in agricultural systems.
A lot still needs to be explored for Nectriaceae: Within the genomes, the laboratory, and the field, such as the secondary metabolites during fungal infection of plants and their adaptations to different temperatures due to global warming. In the next years, many plant pathologists may study effector proteins and metabolites that contribute to effector functions in plant cell-wall to better understand the mechanisms of infection of fungi and reduce their economic impact. In the same direction, freshwater fungi show a lot of potential to be explored in enzymes adapted to cold temperatures and secondary metabolites that can have biotechnological importance in different areas.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Figure S1 to S12.
Author Contributions
Conceptualization, C.B. and D.V.R.; methodology, D.V.R.; software, D.V.R.; validation, D.V.R.; formal analysis, D.V.R. and M.I.; investigation, D.V.R., and M.I.; resources, C.B.; data curation, D.V.R.; writing—original draft preparation, D.V.R. and C.B; writing—review and editing, C.B. and M.I.; visualization, D.V.R.; supervision, C.B.; project administration, C.B.; funding acquisition, C.B. All authors have read and agreed to the published version of the manuscript.
Funding
This study was funded by the German Research Foundation (DFG), Project-447009466/BA 3924 within the DFG priority program SPP 1991Taxon-Omics.
Data Availability Statement
Data are contained within the article, Supplementary Materials, the tables S1 and S2 with the number of features annotated are available at https://doi.org/10.5281/zenodo.12808644 and genome sequences used in this research are deposited in NCBI’s GenBank (you can find their accession numbers in Table 1).
Acknowledgments
In The authors thank C. Berg and C. Plagge for their expert assistance in the laboratory.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Zeng, Z.Q.; Zhuang, W.Y. The Genera Rugonectria and Thelonectria (Hypocreales, Nectriaceae) in China. MycoKeys 55: 101-120 2019, 55, 101–120. [Google Scholar] [CrossRef] [PubMed]
- Rossman, A.Y.; Seifert, K.A.; Samuels, G.J.; Minnis, A.M.; Schroers, H.J.; Lombard, L.; Crous, P.W.; Põldmaa, K.; Cannon, P.F.; Summerbell, R.C.; et al. Genera in Bionectriaceae, Hypocreaceae, and Nectriaceae (Hypocreales) Proposed for Acceptance or Rejection. IMA Fungus 2013, 4, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Booth, C. Studies of Pyrenomycetes Nectria (Part 1). Mycol Pap 1959, 73, 1–115. [Google Scholar]
- Rogerson, C.T. The Hypocrealean Fungi (Ascomycetes, Hypocreales). Mycologia 1970, 62, 865–910. [Google Scholar] [CrossRef] [PubMed]
- Samuels, G. A Revision of the Fungi Formerly Classified as Nectria Subgenus Hyponectria. Mem N Y Bot Gard 1976, 26, 1–126. [Google Scholar]
- Seifert, K. A Monograph of Stilbella and Some Allied Hyphomycetes. Stud Mycol 1985, 27, 1–235. [Google Scholar]
- GJ Samuels, D.B. Variation in Nectria Radicicola and Its Anamorph Cylindrocarpon Destructans. Mycol Res 1990, 94, 433–442. [Google Scholar] [CrossRef]
- AY Rossman, G.S.C.R.R.L. Genera of Bionectriaceae, Hypocreaceae, and Nectriaceae (Hypocreales, Ascomycetes). Stud Mycol 1999, 42, 1–248. [Google Scholar]
- E Lieckfeldt, K.S. An Evaluation of the Use of ITS Sequences in the Taxonomy of the Hypocreales. Stud Mycol 2000, 45, 35–44. [Google Scholar]
- Rossman, A. Towards Monophyletic Genera in the Holomorphic Hypocreales. Stud Mycol 2000, 45, 27–34. [Google Scholar]
- Schroers, H.-J. A Monograph of Bionectria (Ascomycota, Hypocreales, Bionectriaceae) and Its Clonostachys Anamorphs. Stud Mycol 2001, 46, 1–214. [Google Scholar]
- Hirooka, Y.; Kobayashi, T. Taxonomic Studies of Nectrioid Fungi in Japan. I: The Genus Neonectria. Mycoscience 2007 48:1 2007, 48, 53–62. [Google Scholar] [CrossRef]
- Luo, J.; Zhuang, W.Y. Three New Species of Neonectria (Nectriaceae, Hypocreales) with Notes on Their Phylogenetic Positions. Mycologia 2010, 102, 142–152. [Google Scholar] [CrossRef] [PubMed]
- Goh, J.; Oh, Y.; Park, Y.-H.; Mun, H.Y.; Park, S.; Cheon, W. The Korean Journal of Mycology Isolation and Characterization of Previously Undescribed Seventeen Fungal Species Belonging to the Order Hypocreales in Korea. The Korean Journal of Mycology 2022, 50, 1–29. [Google Scholar] [CrossRef]
- Zeng, Z.Q.; Zhuang, W.Y. New Species of Nectriaceae (Hypocreales) from China. Journal of Fungi 2022, Vol. 8, Page 1075 2022, 8, 1075. [Google Scholar] [CrossRef] [PubMed]
- Habtewold, J.Z.; Helgason, B.L.; Yanni, S.F.; Janzen, H.H.; Ellert, B.H.; Gregorich, E.G. Litter Composition Has Stronger Influence on the Structure of Soil Fungal than Bacterial Communities. Eur J Soil Biol 2020, 98, 103190. [Google Scholar] [CrossRef]
- Lombard, L.; van der Merwe, N.A.; Groenewald, J.Z.; Crous, P.W. Generic Concepts in Nectriaceae. Stud Mycol 2015, 80, 189–245. [Google Scholar] [CrossRef] [PubMed]
- Dean, R.; Van Kan, J.A.L.; Pretorius, Z.A.; Hammond-Kosack, K.E.; Di Pietro, A.; Spanu, P.D.; Rudd, J.J.; Dickman, M.; Kahmann, R.; Ellis, J.; et al. The Top 10 Fungal Pathogens in Molecular Plant Pathology. Mol Plant Pathol 2012, 13, 414–430. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Xu, H.; Liu, L.; Li, H.; Gao, Z. Draft Genome Sequence of Neonectria Sp. DH2 Isolated from Meconopsis Grandis Prain in Tibet. 3 Biotech 2020, 10, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Impacts of the International Code of Nomenclature for Algae, Fungi and Plants (Melbourne Code) on the Scientific Names of Plant Pathogenic Fungi. APS Features. APSnet Feature Articles 2013. [Google Scholar] [CrossRef]
- Bennett, J.W.; Klich, M. Mycotoxins. Clin Microbiol Rev 2003, 16, 497–516. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.R.; Hu, X.P.; Jiang, C.J.; Qi, J.; Wu, Y.C.; Li, W.; Zeng, Y.J.; Li, C.F.; Liu, S.X. Diversity and Antimicrobial Activity of Endophytic Fungi Isolated from Cephalotaxus Hainanensis Li, a Well-known Medicinal Plant in China. Lett Appl Microbiol 2015, 61, 484–490. [Google Scholar] [CrossRef] [PubMed]
- Sofian, F.F.; Suzuki, T.; Supratman, U.; Harneti, D.; Maharani, R.; Salam, S.; Abdullah, F.F.; Koseki, T.; Tanaka, K.; Kimura, K. ichi; et al. Cochlioquinone Derivatives Produced by Coculture of Endophytes, Clonostachys Rosea and Nectria Pseudotrichia. Fitoterapia 2021, 155, 105056. [Google Scholar] [CrossRef] [PubMed]
- JIANG, S.; QIAN, D.; YANG, N.; TAO, J.; DUAN, J. Biodiversity and Antimicrobial Activity of Endophytic Fungi in Angelica Sinensis. Chin Herb Med 2013, 5, 264–271. [Google Scholar] [CrossRef]
- Verma, A.; Shameem, N.; Jatav, H.S.; Sathyanarayana, E.; Parray, J.A.; Poczai, P.; Sayyed, R.Z. Fungal Endophytes to Combat Biotic and Abiotic Stresses for Climate-Smart and Sustainable Agriculture. Front Plant Sci 2022, 13, 953836. [Google Scholar] [CrossRef] [PubMed]
- Li, H.Y.; Wei, D.Q.; Shen, M.; Zhou, Z.P. Endophytes and Their Role in Phytoremediation. Fungal Divers 2012, 54, 11–18. [Google Scholar] [CrossRef]
- Yu, Z. Whole RDNA Analysis Reveals Novel and Endophytic Fungi in Bletilla Ochracea (Orchidaceae). 2008.
- Christensen, M. A View of Fungal Ecology. Mycologia 1989, 81, 1–19. [Google Scholar] [CrossRef]
- Jumpponen, A. Dark Septate Endophytes - Are They Mycorrhizal? Mycorrhiza 2001, 11, 207–211. [Google Scholar] [CrossRef]
- Photita, W.; Lumyong, S.; Lumyong, P.; Mckenzie, E.H.C.; Hyde, K.D.; Photita, W.; Lumyong, S.; Lumyong, P.; Hyde, M.E.H.C. Fungal Diversity Are Some Endophytes of Musa Acuminata Latent Pathogens?
- Bernstein, N. Bernstein, N. Effects of Salinity on Root Growth. Plant Roots: The Hidden Half, Fourth Edition 2013, 595–612. [CrossRef]
- Gardes, M. An Orchid-Fungus Marriage: Physical Promiscuity, Conflict and Cheating. New Phytol 2002, 154, 4–7. [Google Scholar] [CrossRef]
- Promputtha, I.; Lumyong, S.; Dhanasekaran, V.; McKenzie, E.H.C.; Hyde, K.D.; Jeewon, R. A Phylogenetic Evaluation of Whether Endophytes Become Saprotrophs at Host Senescence. Microb Ecol 2007, 53, 579–590. [Google Scholar] [CrossRef] [PubMed]
- Schulz, B.; Boyle, C. The Endophytic Continuum. Mycol Res 2005, 109, 661–686. [Google Scholar] [CrossRef]
- Yu, Z. Whole RDNA Analysis Reveals Novel and Endophytic Fungi in Bletilla Ochracea (Orchidaceae). 2008.
- Morrison, E.S.; Thomas, P.; Ogram, A.; Kahveci, T.; Turner, B.L.; Chanton, J.P. Characterization of Bacterial and Fungal Communities Reveals Novel Consortia in Tropical Oligotrophic Peatlands. Microb Ecol 2021, 82, 188–201. [Google Scholar] [CrossRef] [PubMed]
- Bao, D.F.; Hyde, K.D.; Maharachchikumbura, S.S.N.; Perera, R.H.; Thiyagaraja, V.; Hongsanan, S.; Wanasinghe, D.N.; Shen, H.W.; Tian, X.G.; Yang, L.Q.; et al. Taxonomy, Phylogeny and Evolution of Freshwater Hypocreomycetidae (Sordariomycetes). Fungal Diversity 2023 121:1 2023, 121, 1–94. [Google Scholar] [CrossRef]
- Shearer, C.A.; Raja, H.A.; Miller, A.N.; Nelson, P.; Tanaka, K.; Hirayama, K.; Marvanová, L.; Hyde, K.D.; Zhang, Y. The Molecular Phylogeny of Freshwater Dothideomycetes. Stud Mycol 2009, 64, 145–153. [Google Scholar] [CrossRef] [PubMed]
- Salgado-Salazar, C.; Rossman, A.Y.; Chaverri, P. Not as Ubiquitous as We Thought: Taxonomic Crypsis, Hidden Diversity and Cryptic Speciation in the Cosmopolitan Fungus Thelonectria Discophora (Nectriaceae, Hypocreales, Ascomycota). PLoS One 2013, 8, e76737. [Google Scholar] [CrossRef] [PubMed]
- Shearer, C.A.; Descals, E.; Kohlmeyer, B.; Kohlmeyer, J.; Marvanová, L.; Padgett, D.; Porter, D.; Raja, H.A.; Schmit, J.P.; Thorton, H.A.; et al. Fungal Biodiversity in Aquatic Habitats. Biodivers Conserv 2007, 16, 49–67. [Google Scholar] [CrossRef]
- Bärlocher, F. Research on Aquatic Hyphomycetes: Historical Background and Overview. 1992, 1–15. [CrossRef]
- Webster, J. Experiments with Spores of Aquatic Hyphomycetes: I. Sedimentation, and Impaction on Smooth Surfaces. Ann Bot 1959, 23, 595–611. [Google Scholar] [CrossRef]
- Dix, N.J.; Webster, J. Fungal Ecology. 1995. [CrossRef]
- Webster, J.; Shearer, C.A.; Spooner, B.M. Mollisia Casaresiae (Ascomycetes) the Teleomorph of Casaresia Sphagnorum, an Aquatic Fungus. Nova Hedwigia 1993, 57, 3–4. [Google Scholar]
- Vasconcelos Rissi, D.; Ijaz, M.; Baschien, C. Comparative Genome Analysis of the Freshwater Fungus Filosporella fistucella. [CrossRef]
- Brown, A.D. Compatible Solutes and Extreme Water Stress in Eukaryotic Micro-Organisms. Adv Microb Physiol 1978, 17, 181–242. [Google Scholar] [CrossRef] [PubMed]
- Robinson, C.H. Cold Adaptation in Arctic and Antarctic Fungi. New Phytologist 2001, 151, 341–353. [Google Scholar] [CrossRef]
- Mycorrhizal Fungi: Their Habitats and Nutritional Strategies. Environmental and Microbial Relationships 2007, 229–256. [CrossRef]
- Hassan, N.; Rafiq, M.; Hayat, M.; Shah, A.A.; Hasan, F. Psychrophilic and Psychrotrophic Fungi: A Comprehensive Review. Reviews in Environmental Science and Bio/Technology 2016 15:2 2016, 15, 147–172. [Google Scholar] [CrossRef]
- Weinstein, R.N.; Montiel, P.O.; Johnstone, K. Influence of Growth Temperature on Lipid and Soluble Carbohydrate Synthesis by Fungi Isolated from Fellfield Soil in the Maritime Antarctic. Mycologia 2000, 92, 222–229. [Google Scholar] [CrossRef]
- Sui, Y.; Wisniewski, M.; Droby, S.; Norelli, J.; Liu, J. Recent Advances and Current Status of the Use of Heat Treatments in Postharvest Disease Management Systems: Is It Time to Turn up the Heat? Trends Food Sci Technol 2016, 51, 34–40. [Google Scholar] [CrossRef]
- Zhgun, A.A. Fungal BGCs for Production of Secondary Metabolites: Main Types, Central Roles in Strain Improvement, and Regulation According to the Piano Principle. International Journal of Molecular Sciences 2023, Vol. 24, Page 11184 2023, 24, 11184. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Pei, R.; Zhang, Y.; Tu, Y.; He, B. Light Regulation of Secondary Metabolism in Fungi. J Biol Eng 2023, 17, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Yogabaanu, U.; Weber, J.F.F.; Convey, P.; Rizman-Idid, M.; Alias, S.A. Antimicrobial Properties and the Influence of Temperature on Secondary Metabolite Production in Cold Environment Soil Fungi. Polar Sci 2017, 14, 60–67. [Google Scholar] [CrossRef]
- Parain, E.C.; Rohr, R.P.; Gray, S.M.; Bersier, L.F. Increased Temperature Disrupts the Biodiversity–Ecosystem Functioning Relationship. 2019, 193, 227–239. [Google Scholar] [CrossRef] [PubMed]
- Morera, A.; Martínez de Aragón, J.; Bonet, J.A.; Liang, J.; de-Miguel, S. Performance of Statistical and Machine Learning-Based Methods for Predicting Biogeographical Patterns of Fungal Productivity in Forest Ecosystems. For Ecosyst 2021, 8, 1–14. [Google Scholar] [CrossRef]
- Freire, B.; Ladra, S.; Parama, J.R. Memory-Efficient Assembly Using Flye. IEEE/ACM Trans Comput Biol Bioinform 2021, 1–1. [Google Scholar] [CrossRef] [PubMed]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E. V.; Zdobnov, E.M. BUSCO: Assessing Genome Assembly and Annotation Completeness with Single-Copy Orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [PubMed]
- Kurtz, S.; Phillippy, A.; Delcher, A.L.; Smoot, M.; Shumway, M.; Antonescu, C.; Salzberg, S.L. Versatile and Open Software for Comparing Large Genomes. Genome Biol 2004, 5, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Lachance, M.A.; Lee, D.K.; Hsiang, T. Delineating Yeast Species with Genome Average Nucleotide Identity: A Calibration of ANI with Haplontic, Heterothallic Metschnikowia Species. Antonie van Leeuwenhoek, International Journal of General and Molecular Microbiology 2020, 113, 2097–2106. [Google Scholar] [CrossRef] [PubMed]
- Flynn, J.M.; Hubley, R.; Goubert, C.; Rosen, J.; Clark, A.G.; Feschotte, C.; Smit, A.F. RepeatModeler2 for Automated Genomic Discovery of Transposable Element Families. Proc Natl Acad Sci U S A 2020, 117, 9451–9457. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, D. RepeatMasker. https://home.liebertpub.com/bsi 2004, 1, 36–39. [Google Scholar] [CrossRef]
- Chan, P.P.; Lin, B.Y.; Mak, A.J.; Lowe, T.M. TRNAscan-SE 2.0: Improved Detection and Functional Classification of Transfer RNA Genes. Nucleic Acids Res 2021, 49, 9077–9096. [Google Scholar] [CrossRef] [PubMed]
- Gabriel, L.; Brůna, T.; Hoff, K.J.; Ebel, M.; Lomsadze, A.; Borodovsky, M.; Stanke, M. BRAKER3: Fully Automated Genome Annotation Using RNA-Seq and Protein Evidence with GeneMark-ETP, AUGUSTUS, and TSEBRA. Genome Res 2024. [Google Scholar] [CrossRef] [PubMed]
- Brůna, T.; Lomsadze, A.; Borodovsky, M. GeneMark-EP+: Eukaryotic Gene Prediction with Self-Training in the Space of Genes and Proteins. NAR Genom Bioinform 2020, 2. [Google Scholar] [CrossRef] [PubMed]
- Stanke, M.; Keller, O.; Gunduz, I.; Hayes, A.; Waack, S.; Morgenstern, B. AUGUSTUS: Ab Initio Prediction of Alternative Transcripts. Nucleic Acids Res 2006, 34, W435–W439. [Google Scholar] [CrossRef] [PubMed]
- Emms, D.M.; Kelly, S. OrthoFinder: Phylogenetic Orthology Inference for Comparative Genomics. Genome Biol 2019, 20, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol Biol Evol 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Capella-Gutiérrez, S.; Silla-Martínez, J.M.; Gabaldón, T. TrimAl: A Tool for Automated Alignment Trimming in Large-Scale Phylogenetic Analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef] [PubMed]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; Von Haeseler, A.; Lanfear, R.; Teeling, E. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol Biol Evol 2020, 37, 1530–1534. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; Von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast Model Selection for Accurate Phylogenetic Estimates. Nature Methods 2017 14:6 2017, 14, 587–589. [Google Scholar] [CrossRef] [PubMed]
- Blin, K.; Shaw, S.; Augustijn, H.E.; Reitz, Z.L.; Biermann, F.; Alanjary, M.; Fetter, A.; Terlouw, B.R.; Metcalf, W.W.; Helfrich, E.J.N.; et al. AntiSMASH 7.0: New and Improved Predictions for Detection, Regulation, Chemical Structures and Visualisation. Nucleic Acids Res 2023, 51, W46–W50. [Google Scholar] [CrossRef] [PubMed]
- Teufel, F.; Almagro Armenteros, J.J.; Johansen, A.R.; Gíslason, M.H.; Pihl, S.I.; Tsirigos, K.D.; Winther, O.; Brunak, S.; von Heijne, G.; Nielsen, H. SignalP 6.0 Predicts All Five Types of Signal Peptides Using Protein Language Models. Nature Biotechnology 2022 40:7 2022, 40, 1023–1025. [Google Scholar] [CrossRef] [PubMed]
- Emanuelsson, O.; Nielsen, H.; Brunak, S.; Von Heijne, G. Predicting Subcellular Localization of Proteins Based on Their N-Terminal Amino Acid Sequence. J Mol Biol 2000, 300, 1005–1016. [Google Scholar] [CrossRef] [PubMed]
- Hallgren, J.; Tsirigos, K.D.; Damgaard Pedersen, M.; Juan, J.; Armenteros, A.; Marcatili, P.; Nielsen, H.; Krogh, A.; Winther, O. DeepTMHMM Predicts Alpha and Beta Transmembrane Proteins Using Deep Neural Networks. bioRxiv, 2022. [Google Scholar] [CrossRef]
- Finn, R.D.; Clements, J.; Eddy, S.R. HMMER Web Server: Interactive Sequence Similarity Searching. Nucleic Acids Res 2011, 39, W29–W37. [Google Scholar] [CrossRef] [PubMed]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and Sensitive Protein Alignment Using DIAMOND. Nature Methods 2014 12:1 2014, 12, 59–60. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Ge, Q.; Yan, Y.; Zhang, X.; Huang, L.; Yin, Y. DbCAN3: Automated Carbohydrate-Active Enzyme and Substrate Annotation. Nucleic Acids Res 2023, 51, W115–W121. [Google Scholar] [CrossRef] [PubMed]
- Sperschneider, J.; Dodds, P.N. EffectorP 3.0: Prediction of Apoplastic and Cytoplasmic Effectors in Fungi and Oomycetes. Molecular Plant-Microbe Interactions 2022, 35, 146–156. [Google Scholar] [CrossRef] [PubMed]
- Erickson, E.; Gado, J.E.; Avilán, L.; Bratti, F.; Brizendine, R.K.; Cox, P.A.; Gill, R.; Graham, R.; Kim, D.J.; König, G.; et al. Sourcing Thermotolerant Poly(Ethylene Terephthalate) Hydrolase Scaffolds from Natural Diversity. Nature Communications 2022 13:1 2022, 13, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Guerreiro, M.A.; Yurkov, A.; Nowrousian, M.; Stukenbrock, E.H. Lifestyle Transitions in Basidiomycetous Fungi Are Reflected by TRNA Composition and Translation Efficiency of Metabolic Genes. bioRxiv, 2023. [Google Scholar] [CrossRef]
- Fijarczyk, A.; Hessenauer, P.; Hamelin, R.C.; Landry, C.R. Lifestyles Shape Genome Size and Gene Content in Fungal Pathogens. bioRxiv, 2022. [Google Scholar] [CrossRef]
- Raffaele, S.; Kamoun, S. Genome Evolution in Filamentous Plant Pathogens: Why Bigger Can Be Better. Nature Reviews Microbiology 2012 10:6 2012, 10, 417–430. [Google Scholar] [CrossRef] [PubMed]
- Talhinhas, P.; Carvalho, R.; Loureiro, J. The Use of Flow Cytometry for Fungal Nuclear DNA Quantification. Cytometry Part A 2021, 99, 343–347. [Google Scholar] [CrossRef] [PubMed]
- Ting, A.S.Y.; Chaverri, P.; Edrada-Ebel, R.A. Editorial: Endophytes and Their Biotechnological Applications. Front Bioeng Biotechnol 2021, 9, 795174. [Google Scholar] [CrossRef] [PubMed]
- Gouda, S.; Das, G.; Sen, S.K.; Shin, H.S.; Patra, J.K. Endophytes: A Treasure House of Bioactive Compounds of Medicinal Importance. Front Microbiol 2016, 7, 219261. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Plaza, C.; Ochoa-Hueso, R.; Trivedi, C.; Wang, J.; Trivedi, P.; Zhou, G.; Piñeiro, J.; Martins, C.S.C.; Singh, B.K.; et al. Litter and Soil Biodiversity Jointly Drive Ecosystem Functions. Glob Chang Biol 2023, 29, 6276–6285. [Google Scholar] [CrossRef]
- Mirabile, G.; Ferraro, V.; Mancuso, F.P.; Pecoraro, L.; Cirlincione, F. Biodiversity of Fungi in Freshwater Ecosystems of Italy. Journal of Fungi 2023, Vol. 9, Page 993 2023, 9, 993. [Google Scholar] [CrossRef]
- Bhunjun, C.S.; Phukhamsakda, C.; Hyde, K.D.; McKenzie, E.H.C.; Saxena, R.K.; Li, Q. Do All Fungi Have Ancestors with Endophytic Lifestyles? Fungal Diversity 2023 125:1 2023, 125, 73–98. [Google Scholar] [CrossRef]
- Gorbunova, V.; Seluanov, A.; Mita, P.; McKerrow, W.; Fenyö, D.; Boeke, J.D.; Linker, S.B.; Gage, F.H.; Kreiling, J.A.; Petrashen, A.P.; et al. The Role of Retrotransposable Elements in Ageing and Age-Associated Diseases. Nature 2021 596:7870 2021, 596, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Latzel, V.; Puy, J.; Thieme, M.; Bucher, E.; Götzenberger, L.; de Bello, F. Phenotypic Diversity Influenced by a Transposable Element Increases Productivity and Resistance to Competitors in Plant Populations. Journal of Ecology 2023, 111, 2376–2387. [Google Scholar] [CrossRef]
- Liu, C.; Li, B.; Dong, Y.; Lin, H. Endophyte Colonization Enhanced Cadmium Phytoremediation by Improving Endosphere and Rhizosphere Microecology Characteristics. J Hazard Mater 2022, 434, 128829. [Google Scholar] [CrossRef] [PubMed]
- Mascarin, G.M.; Jaronski, S.T. The Production and Uses of Beauveria Bassiana as a Microbial Insecticide. World Journal of Microbiology and Biotechnology 2016 32:11 2016, 32, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Queiroz, C.B. de; Santana, M.F. Prediction of the Secretomes of Endophytic and Nonendophytic Fungi Reveals Similarities in Host Plant Infection and Colonization Strategies. Mycologia 2020, 112, 491–503. [Google Scholar] [CrossRef] [PubMed]
- Oggenfuss, U.; Croll, D. Recent Transposable Element Bursts Are Associated with the Proximity to Genes in a Fungal Plant Pathogen. PLoS Pathog 2023, 19, e1011130. [Google Scholar] [CrossRef] [PubMed]
- Muszewska, A.; Steczkiewicz, K.; Stepniewska-Dziubinska, M.; Ginalski, K. Transposable Elements Contribute to Fungal Genes and Impact Fungal Lifestyle. Scientific Reports 2019 9:1 2019, 9, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Grandaubert, J.; Lowe, R.G.T.; Soyer, J.L.; Schoch, C.L.; Van De Wouw, A.P.; Fudal, I.; Robbertse, B.; Lapalu, N.; Links, M.G.; Ollivier, B.; et al. Transposable Element-Assisted Evolution and Adaptation to Host Plant within the Leptosphaeria Maculans-Leptosphaeria Biglobosa Species Complex of Fungal Pathogens. BMC Genomics 2014, 15, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Torres, D.E.; Thomma, B.P.H.J.; Seidl, M.F. Transposable Elements Contribute to Genome Dynamics and Gene Expression Variation in the Fungal Plant Pathogen Verticillium Dahliae. Genome Biol Evol 2021, 13. [Google Scholar] [CrossRef] [PubMed]
- Seidl, M.F.; Faino, L.; Shi-Kunne, X.; Van Den Berg, G.C.M.; Bolton, M.D.; Thomma, B.P.H.J. The Genome of the Saprophytic Fungus Verticillium Tricorpus Reveals a Complex Effector Repertoire Resembling That of Its Pathogenic Relatives. 2015, 28, 362–373. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, P.N.; Brookes, L.M.; Edwards, H.M.; Fisher, M.C.; Jervis, P.; Kappel, D.; Sewell, T.R.; Shelton, J.M.G.; Skelly, E.; Rhodes, J.L. Cross-Disciplinary Genomics Approaches to Studying Emerging Fungal Infections. Life 2020, Vol. 10, Page 315 2020, 10, 315. [Google Scholar] [CrossRef] [PubMed]
- Bennett, E.A.; Coleman, L.E.; Tsui, C.; Pittard, W.S.; Devine, S.E. Natural Genetic Variation Caused by Transposable Elements in Humans. Genetics 2004, 168, 933–951. [Google Scholar] [CrossRef] [PubMed]
- Castanera, R.; López-Varas, L.; Borgognone, A.; LaButti, K.; Lapidus, A.; Schmutz, J.; Grimwood, J.; Pérez, G.; Pisabarro, A.G.; Grigoriev, I. V.; et al. Transposable Elements versus the Fungal Genome: Impact on Whole-Genome Architecture and Transcriptional Profiles. PLoS Genet 2016, 12, e1006108. [Google Scholar] [CrossRef] [PubMed]
- Biscotti, M.A.; Olmo, E.; Heslop-Harrison, J.S. (Pat) Repetitive DNA in Eukaryotic Genomes. Chromosome Research 2015, 23, 415–420. [Google Scholar] [CrossRef] [PubMed]
- Gazis, R.; Kuo, A.; Riley, R.; LaButti, K.; Lipzen, A.; Lin, J.; Amirebrahimi, M.; Hesse, C.N.; Spatafora, J.W.; Henrissat, B.; et al. The Genome of Xylona Heveae Provides a Window into Fungal Endophytism. Fungal Biol 2016, 120, 26–42. [Google Scholar] [CrossRef] [PubMed]
- Vincent, D.; Rafiqi, M.; Job, D. The Multiple Facets of Plant–Fungal Interactions Revealed Through Plant and Fungal Secretomics. Front Plant Sci 2020, 10, 487828. [Google Scholar] [CrossRef] [PubMed]
- Cantarel, B.I.; Coutinho, P.M.; Rancurel, C.; Bernard, T.; Lombard, V.; Henrissat, B. The Carbohydrate-Active EnZymes Database (CAZy): An Expert Resource for Glycogenomics. Nucleic Acids Res 2009, 37, D233–D238. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Liu, H.; Wang, C.; Xu, J.R. Correction to Comparative Analysis of Fungal Genomes Reveals Different Plant Cell Wall Degrading Capacity in Fungi [BMC Genomics 14(2013) 274]. BMC Genomics 2014, 15, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Ospina-Giraldo, M.D.; Griffith, J.G.; Laird, E.W.; Mingora, C. The CAZyome of Phytophthora Spp.: A Comprehensive Analysis of the Gene Complement Coding for Carbohydrate-Active Enzymes in Species of the Genus Phytophthora. BMC Genomics 2010, 11, 1–16. [Google Scholar] [CrossRef]
- Zhao, Z.; Liu, H.; Wang, C.; Xu, J.R. Comparative Analysis of Fungal Genomes Reveals Different Plant Cell Wall Degrading Capacity in Fungi. BMC Genomics 2013, 14, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Czislowski, E.; Zeil-rolfe, I.; Aitken, E.A.B. Effector Profiles of Endophytic Fusarium Associated with Asymptomatic Banana (Musa Sp.) Hosts. International Journal of Molecular Sciences 2021, Vol. 22, Page 2508 2021, 22, 2508. [Google Scholar] [CrossRef] [PubMed]
- Promputtha, I.; Hyde, K.D.; McKenzie, E.H.C.; Peberdy, J.F.; Lumyong, S. Can Leaf Degrading Enzymes Provide Evidence That Endophytic Fungi Becoming Saprobes? Fungal Divers 2010, 41, 89–99. [Google Scholar] [CrossRef]
- Sarkar, S.; Dey, A.; Kumar, V.; Batiha, G.E.S.; El-Esawi, M.A.; Tomczyk, M.; Ray, P. Fungal Endophyte: An Interactive Endosymbiont With the Capability of Modulating Host Physiology in Myriad Ways. Front Plant Sci 2021, 12, 701800. [Google Scholar] [CrossRef] [PubMed]
- Pointing, S.B.; Parungao, M.M.; Hyde, K.D. Production of Wood-Decay Enzymes, Mass Loss and Lignin Solubilization in Wood by Tropical Xylariaceae. Mycol Res 2003, 107, 231–235. [Google Scholar] [CrossRef] [PubMed]
- Pointing, S.B.; Pelling, A.L.; Smith, G.J.D.; Hyde, K.D.; Reddy, C.A. Screening of Basidiomycetes and Xylariaceous Fungi for Lignin Peroxidase and Laccase Gene-Specific Sequences. Mycol Res 2005, 109, 115–124. [Google Scholar] [CrossRef]
- Koide, K.; Osono, T.; Takeda, H. Fungal Succession and Decomposition of Camellia Japonica Leaf Litter. Ecol Res 2005, 20, 599–609. [Google Scholar] [CrossRef]
- Bucher, V.V.C.; Hyde, K.D.; Pointing, S.B.; Reddy, C.A.; Reddy, S.B. Production of Wood Decay Enzymes, Mass Loss and Lignin Solubilization in Wood by Marine Ascomycetes and Their Anamorphs.
- Purahong, W.; Hyde, K.D. Effects of Fungal Endophytes on Grass and Non-Grass Litter Decomposition Rates. Fungal Divers 2011, 47, 1–7. [Google Scholar] [CrossRef]
- Barrett, K.; Jensen, K.; Meyer, A.S.; Frisvad, J.C.; Lange, L. Fungal Secretome Profile Categorization of CAZymes by Function and Family Corresponds to Fungal Phylogeny and Taxonomy: Example Aspergillus and Penicillium. Scientific Reports 2020 10:1 2020, 10, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.M.; Zhang, Y. Plant Immunity: Danger Perception and Signaling. Cell 2020, 181, 978–989. [Google Scholar] [CrossRef] [PubMed]
- Zúñiga, E.; Romero, J.; Ollero-Lara, A.; Lovera, M.; Arquero, O.; Miarnau, X.; Torguet, L.; Trapero, A.; Luque, J. Inoculum and Infection Dynamics of Polystigma Amygdalinum in Almond Orchards in Spain. Plant Dis 2020, 104, 1239–1246. [Google Scholar] [CrossRef] [PubMed]
- Ali, G.S.; Reddy, A.S.N. PAMP-Triggered Immunity. Plant Signal Behav 2008, 3, 423–426. [Google Scholar] [CrossRef]
- Chellappan, B.V.; El-Ganainy, S.M.; Alrajeh, H.S.; Al-Sheikh, H. In Silico Characterization of the Secretome of the Fungal Pathogen Thielaviopsis Punctulata, the Causal Agent of Date Palm Black Scorch Disease. Journal of Fungi 2023, 9, 303. [Google Scholar] [CrossRef] [PubMed]
- Boller, T.; Felix, G. A Renaissance of Elicitors: Perception of Microbe-Associated Molecular Patterns and Danger Signals by Pattern-Recognition Receptors. Annu Rev Plant Biol 2009, 60, 379–407. [Google Scholar] [CrossRef] [PubMed]
- Mattoo, A.J.; Nonzom, S. Endophytic Fungi: Understanding Complex Cross-Talks. Symbiosis 2021 83:3 2021, 83, 237–264. [Google Scholar] [CrossRef]
- Lo Presti, L.; Lanver, D.; Schweizer, G.; Tanaka, S.; Liang, L.; Tollot, M.; Zuccaro, A.; Reissmann, S.; Kahmann, R. Fungal Effectors and Plant Susceptibility. Annu Rev Plant Biol 2015, 66, 513–545. [Google Scholar] [CrossRef] [PubMed]
- Pradhan, A.; Ghosh, S.; Sahoo, D.; Jha, G. Fungal Effectors, the Double Edge Sword of Phytopathogens. Current Genetics 2020 67:1 2020, 67, 27–40. [Google Scholar] [CrossRef] [PubMed]
- Todd, J.N.A.; Carreón-Anguiano, K.G.; Islas-Flores, I.; Canto-Canché, B. Fungal Effectoromics: A World in Constant Evolution. International Journal of Molecular Sciences 2022, Vol. 23, Page 13433 2022, 23, 13433. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Wang, Y.; McDowell, J. Focus on Effector-Triggered Susceptibility. 2017; 31, 5. [Google Scholar] [CrossRef]
- Hammond-Kosack, K.E.; Jones, J.D.G. Plant Disease Resistance Genes. Annu Rev Plant Biol 1997, 48, 575–607. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Cheng, Y. Advances in Fungal Elicitor-Triggered Plant Immunity. International Journal of Molecular Sciences 2022, Vol. 23, Page 12003 2022, 23, 12003. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Chen, Y.; Li, B.; Zhang, Z.; Qin, G.; Chen, T.; Tian, S. Molecular Mechanisms Underlying Multi-Level Defense Responses of Horticultural Crops to Fungal Pathogens. Hortic Res 2022, 9. [Google Scholar] [CrossRef] [PubMed]
- Petit-Houdenot, Y.; Fudal, I. Complex Interactions between Fungal Avirulence Genes and Their Corresponding Plant Resistance Genes and Consequences for Disease Resistance Management. Front Plant Sci 2017, 8, 250670. [Google Scholar] [CrossRef] [PubMed]
- Balint-Kurti, P. The Plant Hypersensitive Response: Concepts, Control and Consequences. Mol Plant Pathol 2019, 20, 1163–1178. [Google Scholar] [CrossRef] [PubMed]
- Casadevall, A. Global Warming Could Drive the Emergence of New Fungal Pathogens. Nature Microbiology 2023 8:12 2023, 8, 2217–2219. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.K.; Delgado-Baquerizo, M.; Egidi, E.; Guirado, E.; Leach, J.E.; Liu, H.; Trivedi, P. Climate Change Impacts on Plant Pathogens, Food Security and Paths Forward. Nature Reviews Microbiology 2023 21:10 2023, 21, 640–656. [Google Scholar] [CrossRef] [PubMed]
- Seidel, D.; Wurster, S.; Jenks, J.D.; Sati, H.; Gangneux, J.P.; Egger, M.; Alastruey-Izquierdo, A.; Ford, N.P.; Chowdhary, A.; Sprute, R.; et al. Impact of Climate Change and Natural Disasters on Fungal Infections. Lancet Microbe 2024, 5, e594–e605. [Google Scholar] [CrossRef]
- Stauder, C.M.; Garnas, J.R.; Morrison, E.W.; Salgado-Salazar, C.; Kasson, M.T. Characterization of Mating Type Genes in Heterothallic Neonectria Species, with Emphasis on N. Coccinea, N. Ditissima, and N. Faginata. Mycologia 2020, 112, 880–894. [Google Scholar] [CrossRef] [PubMed]
- Latorre, B.A.; Rioja, M.E.; Lillo, C.; Muñoz, M. The Effect of Temperature and Wetness Duration on Infection and a Warning System for European Canker (Nectria Galligena) of Apple in Chile. Crop Protection 2002, 21, 285–291. [Google Scholar] [CrossRef]
- Langer, G.J.; Bußkamp, J. Fungi Associated With Woody Tissues of European Beech and Their Impact on Tree Health. Front Microbiol 2021, 12, 702467. [Google Scholar] [CrossRef] [PubMed]
- Purahong, W.; Tanunchai, B.; Wahdan, S.F.M.; Buscot, F.; Schulze, E.D. Molecular Screening of Microorganisms Associated with Discolored Wood in Dead European Beech Trees Suffered from Extreme Drought Event Using next Generation Sequencing. Plants 2021, 10, 2092. [Google Scholar] [CrossRef] [PubMed]
- Morrison, E.W.; Kasson, M.T.; Heath, J.J.; Garnas, J.R. Pathogen and Endophyte Assemblages Co-Vary With Beech Bark Disease Progression, Tree Decline, and Regional Climate. Frontiers in Forests and Global Change 2021, 4, 673099. [Google Scholar] [CrossRef]
- Saremi, H.; Burgess, L.W.; Backhouse, D. Temperature Effects on the Relative Abundance of Fusarium Species in a Model Plant–Soil Ecosystem. Soil Biol Biochem 1999, 31, 941–947. [Google Scholar] [CrossRef]
- Suárez-Estrella, F.; Vargas-García, M.C.; Elorrieta, M.A.; López, M.J.; Moreno, J. Temperature Effect on Fusarium Oxysporum f.Sp. Melonis Survival during Horticultural Waste Composting. J Appl Microbiol 2003, 94, 475–482. [Google Scholar] [CrossRef] [PubMed]
- Knapp, B.D.; Huang, K.C. The Effects of Temperature on Cellular Physiology. Annu Rev Biophys 2022, 51, 499–526. [Google Scholar] [CrossRef] [PubMed]
- Bärlocher, F.; Seena, S.; Wilson, K.P.; Dudley Williams, D. Raised Water Temperature Lowers Diversity of Hyporheic Aquatic Hyphomycetes. Freshw Biol 2008, 53, 368–379. [Google Scholar] [CrossRef]
- Dang, C.K.; Schindler, M.; Chauvet, E.; Gessner, M.O. Temperature Oscillation Coupled with Fungal Community Shifts Can Modulate Warming Effects on Litter Decomposition. Ecology 2009, 90, 122–131. [Google Scholar] [CrossRef] [PubMed]
- Větrovský, T.; Kohout, P.; Kopecký, M.; Machac, A.; Man, M.; Bahnmann, B.D.; Brabcová, V.; Choi, J.; Meszárošová, L.; Human, Z.R.; et al. A Meta-Analysis of Global Fungal Distribution Reveals Climate-Driven Patterns. Nature Communications 2019 10:1 2019, 10, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Author, C.; Zhao, W. Endophytic Fungi in Green Manure Crops; Friends or Foe? Mycosphere 2023, 14, 7019. [Google Scholar] [CrossRef]
- Fenoy, E.; Pradhan, A.; Pascoal, C.; Rubio-Ríos, J.; Batista, D.; Moyano-López, F.J.; Cássio, F.; Casas, J.J. Elevated Temperature May Reduce Functional but Not Taxonomic Diversity of Fungal Assemblages on Decomposing Leaf Litter in Streams. Glob Chang Biol 2022, 28, 115–127. [Google Scholar] [CrossRef] [PubMed]
- Follstad Shah, J.J.; Kominoski, J.S.; Ardón, M.; Dodds, W.K.; Gessner, M.O.; Griffiths, N.A.; Hawkins, C.P.; Johnson, S.L.; Lecerf, A.; LeRoy, C.J.; et al. Global Synthesis of the Temperature Sensitivity of Leaf Litter Breakdown in Streams and Rivers. Glob Chang Biol 2017, 23, 3064–3075. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Sun, J.; Xie, P.; Guo, C.; Zhu, K.; Tian, K. Climate Warming Enhances Microbial Network Complexity by Increasing Bacterial Diversity and Fungal Interaction Strength in Litter Decomposition. Science of The Total Environment 2024, 908, 168444. [Google Scholar] [CrossRef] [PubMed]
- Boyero, L.; Pearson, R.G.; Gessner, M.O.; Barmuta, L.A.; Ferreira, V.; Graça, M.A.S.; Dudgeon, D.; Boulton, A.J.; Callisto, M.; Chauvet, E.; et al. A Global Experiment Suggests Climate Warming Will Not Accelerate Litter Decomposition in Streams but Might Reduce Carbon Sequestration. Ecol Lett 2011, 14, 289–294. [Google Scholar] [CrossRef] [PubMed]
- Simões, S.; Gonçalves, A.L.; Jones, T.H.; Sousa, J.P.; Canhoto, C. Air Temperature More than Drought Duration Affects Litter Decomposition under Flow Intermittency. Science of The Total Environment 2022, 829, 154666. [Google Scholar] [CrossRef] [PubMed]
- Shearer, C.A.; Webster, J. Aquatic Hyphomycete Communities in the River Teign. IV. Twig Colonization. Mycol Res 1991, 95, 413–420. [Google Scholar] [CrossRef]
- Cobo-Díaz, J.F.; Baroncelli, R.; Le Floch, G.; Picot, A. A Novel Metabarcoding Approach to Investigate Fusarium Species Composition in Soil and Plant Samples. FEMS Microbiol Ecol 2019, 95, 84. [Google Scholar] [CrossRef] [PubMed]
- Barros, J.; Seena, S. Fungi in Freshwaters: Prioritising Aquatic Hyphomycetes in Conservation Goals. Water 2022, Vol. 14, Page 605 2022, 14, 605. [Google Scholar] [CrossRef]
- Chen, H.; Raffaele, S.; Dong, S. Silent Control: Microbial Plant Pathogens Evade Host Immunity without Coding Sequence Changes. FEMS Microbiol Rev 2021, 45, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Hinsch, J.; Galuszka, P.; Tudzynski, P. Functional Characterization of the First Filamentous Fungal TRNA-Isopentenyltransferase and Its Role in the Virulence of Claviceps Purpurea. New Phytologist 2016, 211, 980–992. [Google Scholar] [CrossRef] [PubMed]
- dos Reis, M.; Savva, R.; Wernisch, L. Solving the Riddle of Codon Usage Preferences: A Test for Translational Selection. Nucleic Acids Res 2004, 32, 5036–5044. [Google Scholar] [CrossRef] [PubMed]
- Kohler, A.; Kuo, A.; Nagy, L.G.; Morin, E.; Barry, K.W.; Buscot, F.; Canbäck, B.; Choi, C.; Cichocki, N.; Clum, A.; et al. Convergent Losses of Decay Mechanisms and Rapid Turnover of Symbiosis Genes in Mycorrhizal Mutualists. Nature Genetics 2015 47:4 2015, 47, 410–415. [Google Scholar] [CrossRef] [PubMed]
- Collemare, J.; Billard, A.; Böhnert, H.U.; Lebrun, M.H. Biosynthesis of Secondary Metabolites in the Rice Blast Fungus Magnaporthe Grisea: The Role of Hybrid PKS-NRPS in Pathogenicity. Mycol Res 2008, 112, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Liang, P.; Liu, S.; Xu, F.; Jiang, S.; Yan, J.; He, Q.; Liu, W.; Lin, C.; Zheng, F.; Wang, X.; et al. Powdery Mildews Are Characterized by Contracted Carbohydrate Metabolism and Diverse Effectors to Adapt to Obligate Biotrophic Lifestyle. Front Microbiol 2018, 9, 423656. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wu, J.; Yan, J.; Guo, M.; Xu, L.; Hou, L.; Zou, Q. Comparative Genome Analysis of Plant Ascomycete Fungal Pathogens with Different Lifestyles Reveals Distinctive Virulence Strategies. BMC Genomics 2022, 23, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Rokas, A.; Mead, M.E.; Steenwyk, J.L.; Raja, H.A.; Oberlies, N.H. Biosynthetic Gene Clusters and the Evolution of Fungal Chemodiversity. Nat Prod Rep 2020, 37, 868–878. [Google Scholar] [CrossRef] [PubMed]
- Mózsik, L.; Iacovelli, R.; Bovenberg, R.A.L.; Driessen, A.J.M. Transcriptional Activation of Biosynthetic Gene Clusters in Filamentous Fungi. Front Bioeng Biotechnol 2022, 10, 901037. [Google Scholar] [CrossRef] [PubMed]
- García-Estrada, C.; Domínguez-Santos, R.; Kosalková, K.; Martín, J.F. Transcription Factors Controlling Primary and Secondary Metabolism in Filamentous Fungi: The β-Lactam Paradigm. Fermentation 2018, Vol. 4, Page 47 2018, 4, 47. [Google Scholar] [CrossRef]
- Roberts, D.W.; St. Leger, R.J. Metarhizium Spp., Cosmopolitan Insect-Pathogenic Fungi: Mycological Aspects. Adv Appl Microbiol 2004, 54, 1–70. [Google Scholar] [CrossRef] [PubMed]
- Keller, N.P. Translating Biosynthetic Gene Clusters into Fungal Armor and Weaponry. Nature Chemical Biology 2015 11:9 2015, 11, 671–677. [Google Scholar] [CrossRef] [PubMed]
- Gluck-Thaler, E.; Haridas, S.; Binder, M.; Grigoriev, I. V.; Crous, P.W.; Spatafora, J.W.; Bushley, K.; Slot, J.C. The Architecture of Metabolism Maximizes Biosynthetic Diversity in the Largest Class of Fungi. Mol Biol Evol 2020, 37, 2838–2856. [Google Scholar] [CrossRef] [PubMed]
- Blacutt, A.A.; Gold, S.E.; Voss, K.A.; Gao, M.; Glenn, A.E. Fusarium Verticillioides: Advancements in Understanding the Toxicity, Virulence, and Niche Adaptations of a Model Mycotoxigenic Pathogen of Maize. Phytopathology 2018, 108, 312–326. [Google Scholar] [CrossRef] [PubMed]
- Mehmood, M.A.; Rauf, A.; Ashfaq, M.; Ahmad, F. Exploring Biological Control Strategies for Managing Fusarium Mycotoxins. Nanohybrid Fungicides: New Frontiers in Plant Pathology 2024, 257–293. [CrossRef]
- Yazid, S.N.E.; Jinap, S.; Ismail, S.I.; Magan, N.; Samsudin, N.I.P. Phytopathogenic Organisms and Mycotoxigenic Fungi: Why Do We Control One and Neglect the Other? A Biological Control Perspective in Malaysia. Compr Rev Food Sci Food Saf 2020, 19, 643–669. [Google Scholar] [CrossRef] [PubMed]
- Rossman, A. Morphological and Molecular Perspectives on Systematics of the Hypocreales. Mycologia 1996, 88, 1–19. [Google Scholar] [CrossRef]
- Yang, Y.; Liu, X.; Cai, J.; Chen, Y.; Li, B.; Guo, Z.; Huang, G. Genomic Characteristics and Comparative Genomics Analysis of the Endophytic Fungus Sarocladium Brachiariae. BMC Genomics 2019, 20, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Yoder, O.C.; Turgeon, B.G. Fungal Genomics and Pathogenicity. Curr Opin Plant Biol 2001, 4, 315–321. [Google Scholar] [CrossRef] [PubMed]
- Christianson, D.W. Structural Biology and Chemistry of the Terpenoid Cyclases. Chem Rev 2006, 106, 3412–3442. [Google Scholar] [CrossRef] [PubMed]
- Davis, E.M.; Croteau, R. Cyclization Enzymes in the Biosynthesis of Monoterpenes, Sesquiterpenes, and Diterpenes. 2000, 53–95. [CrossRef]
- Crutcher, F.K.; Parich, A.; Schuhmacher, R.; Mukherjee, P.K.; Zeilinger, S.; Kenerley, C.M. A Putative Terpene Cyclase, Vir4, Is Responsible for the Biosynthesis of Volatile Terpene Compounds in the Biocontrol Fungus Trichoderma Virens. Fungal Genetics and Biology 2013, 56, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Buckingham, J.; Cooper, C.M.; Purchase, R. Natural Products Desk Reference. Natural Products Desk Reference 2015, 1–222. [Google Scholar] [CrossRef]
- Schmidt-Dannert, C. Biosynthesis of Terpenoid Natural Products in Fungi. Adv Biochem Eng Biotechnol 2014, 148, 19–61. [Google Scholar] [CrossRef]
- Akiyama, K.; Matsuzaki, K.I.; Hayashi, H. Plant Sesquiterpenes Induce Hyphal Branching in Arbuscular Mycorrhizal Fungi. Nature 2005 435:7043 2005, 435, 824–827. [Google Scholar] [CrossRef] [PubMed]
- Kai, M.; Effmert, U.; Berg, G.; Piechulla, B. Volatiles of Bacterial Antagonists Inhibit Mycelial Growth of the Plant Pathogen Rhizoctonia Solani. Arch Microbiol 2007, 187, 351–360. [Google Scholar] [CrossRef] [PubMed]
- Foppen, F.H.; Gribanovski-Sassu, O. Lipids Produced by Epicoccum Nigrum in Submerged Culture. Biochemical Journal 1968, 106, 97–100. [Google Scholar] [CrossRef] [PubMed]
- Ozaki, T.; Minami, A.; Oikawa, H. Recent Advances in the Biosynthesis of Ribosomally Synthesized and Posttranslationally Modified Peptides of Fungal Origin. The Journal of Antibiotics 2022 76:1 2022, 76, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Vignolle, G.A.; Mach, R.L.; Mach-Aigner, A.R.; Derntl, C. Novel Approach in Whole Genome Mining and Transcriptome Analysis Reveal Conserved RiPPs in Trichoderma Spp. BMC Genomics 2020, 21, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Deicke, M.; Mohr, J.F.; Roy, S.; Herzsprung, P.; Bellenger, J.P.; Wichard, T. Metallophore Profiling of Nitrogen-Fixing Frankia Spp. to Understand Metal Management in the Rhizosphere of Actinorhizal Plants. Metallomics 2019, 11, 810–821. [Google Scholar] [CrossRef] [PubMed]
- Kraemer, S.M.; Duckworth, O.W.; Harrington, J.M.; Schenkeveld, W.D.C. Metallophores and Trace Metal Biogeochemistry. Aquat Geochem 2015, 21, 159–195. [Google Scholar] [CrossRef]
- Kuzyk, S.B.; Hughes, E.; Yurkov, V. Discovery of Siderophore and Metallophore Production in the Aerobic Anoxygenic Phototrophs. Microorganisms 2021, 9, 959. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Aty, A.S. Fungicidal Activity of Indole Derivatives against Some Plant Pathogenic Fungi. J Pestic Sci 2010, 35, 431–440. [Google Scholar] [CrossRef]
- Shen, Q.; Liu, L.; Wang, L.; Wang, Q. Indole Primes Plant Defense against Necrotrophic Fungal Pathogen Infection. PLoS One 2018, 13, e0207607. [Google Scholar] [CrossRef] [PubMed]
- Svara, J.; Weferling, N.; Hofmann, T. Phosphorus Compounds, Organic. In Ullmann’s Encyclopedia of Industrial Chemistry; 2006. [Google Scholar] [CrossRef]
- Metcalf, W.W.; Van Der Donk, W.A. Biosynthesis of Phosphonic and Phosphinic Acid Natural Products. Annu Rev Biochem 2009, 78, 65–94. [Google Scholar] [CrossRef] [PubMed]
- Lim, F.Y.; Won, T.H.; Raffa, N.; Baccile, J.A.; Wisecaver, J.; Keller, N.P.; Rokas, A.; Schroeder, F.C. Fungal Isocyanide Synthases and Xanthocillin Biosynthesis in Aspergillus Fumigatus. mBio 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Won, T.H.; Bok, J.W.; Nadig, N.; Venkatesh, N.; Nickles, G.; Greco, C.; Lim, F.Y.; González, J.B.; Turgeon, B.G.; Keller, N.P.; et al. Copper Starvation Induces Antimicrobial Isocyanide Integrated into Two Distinct Biosynthetic Pathways in Fungi. Nature Communications 2022 13:1 2022, 13, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Massarotti, A.; Brunelli, F.; Aprile, S.; Giustiniano, M.; Tron, G.C. Medicinal Chemistry of Isocyanides. Chem Rev 2021, 121, 10742–10788. [Google Scholar] [CrossRef] [PubMed]
- Raffa, N.; Won, T.H.; Sukowaty, A.; Candor, K.; Cui, C.; Halder, S.; Dai, M.; Landero-Figueroa, J.A.; Schroeder, F.C.; Keller, N.P. Dual-Purpose Isocyanides Produced by Aspergillus Fumigatus Contribute to Cellular Copper Sufficiency and Exhibit Antimicrobial Activity. Proc Natl Acad Sci U S A 2021, 118, e2015224118. [Google Scholar] [CrossRef] [PubMed]
- Crawford, J.M.; Portmann, C.; Zhang, X.; Roeffaers, M.B.J.; Clardy, J. Small Molecule Perimeter Defense in Entomopathogenic Bacteria. Proc Natl Acad Sci U S A 2012, 109, 10821–10826. [Google Scholar] [CrossRef]
- Harris, N.C.; Sato, M.; Herman, N.A.; Twigg, F.; Cai, W.; Liu, J.; Zhu, X.; Downey, J.; Khalaf, R.; Martin, J.; et al. Biosynthesis of Isonitrile Lipopeptides by Conserved Nonribosomal Peptide Synthetase Gene Clusters in Actinobacteria. Proc Natl Acad Sci U S A 2017, 114, 7025–7030. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.Y.; Chen, J.; Tang, Y.; Zhou, J.; Guo, Y.; Chang, W. chen Current Understanding toward Isonitrile Group Biosynthesis and Mechanism. Chin J Chem 2021, 39, 463–472. [Google Scholar] [CrossRef] [PubMed]
- Sweany, R.R.; Breunig, M.; Opoku, J.; Clay, K.; Spatafora, J.W.; Drott, M.T.; Baldwin, T.T.; Fountain, J.C. Why Do Plant-Pathogenic Fungi Produce Mycotoxins? Potential Roles for Mycotoxins in the Plant Ecosystem. Phytopathology 2022, 112, 2044–2051. [Google Scholar] [CrossRef] [PubMed]
- Drott, M.T.; Lazzaro, B.P.; Brown, D.L.; Carbone, I.; Milgroom, M.G. Balancing Selection for Aflatoxin in Aspergillus Flavus Is Maintained through Interference Competition with, and Fungivory by Insects. Proceedings of the Royal Society B: Biological Sciences 2017, 284. [Google Scholar] [CrossRef]
- Fenteany, G.; Standaert, R.F.; Reichard, G.A.; Corey, E.J.; Schreiber, S.L. A Beta-Lactone Related to Lactacystin Induces Neurite Outgrowth in a Neuroblastoma Cell Line and Inhibits Cell Cycle Progression in an Osteosarcoma Cell Line. Proceedings of the National Academy of Sciences 1994, 91, 3358–3362. [Google Scholar] [CrossRef] [PubMed]
- Meesil, W.; Muangpat, P.; Sitthisak, S.; Rattanarojpong, T.; Chantratita, N.; Machado, R.A.R.; Shi, Y.M.; Bode, H.B.; Vitta, A.; Thanwisai, A. Genome Mining Reveals Novel Biosynthetic Gene Clusters in Entomopathogenic Bacteria. Scientific Reports 2023 13:1 2023, 13, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Ji, S.; Tian, Y.; Li, J.; Xu, G.; Zhang, Y.; Chen, S.; Chen, Y.; Tang, X. Complete Genome Sequence of Bacillus Cereus Z4, a Biocontrol Agent against Tobacco Black Shank, Isolated from the Western Pacific Ocean. Mar Genomics 2023, 72, 101071. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Phylogenomics tree of Nectriaceae genomes. Phylogenomics tree of Nectriaceae family using 2684 single-copy orthologs genes. The plant pathogen fungi N. galligena current name is N. ditissima. The species in red represent plant pathogens, while in green endophytes and brown saprobes. (*) are saprobe freshwater fungi.
Figure 1.
Phylogenomics tree of Nectriaceae genomes. Phylogenomics tree of Nectriaceae family using 2684 single-copy orthologs genes. The plant pathogen fungi N. galligena current name is N. ditissima. The species in red represent plant pathogens, while in green endophytes and brown saprobes. (*) are saprobe freshwater fungi.
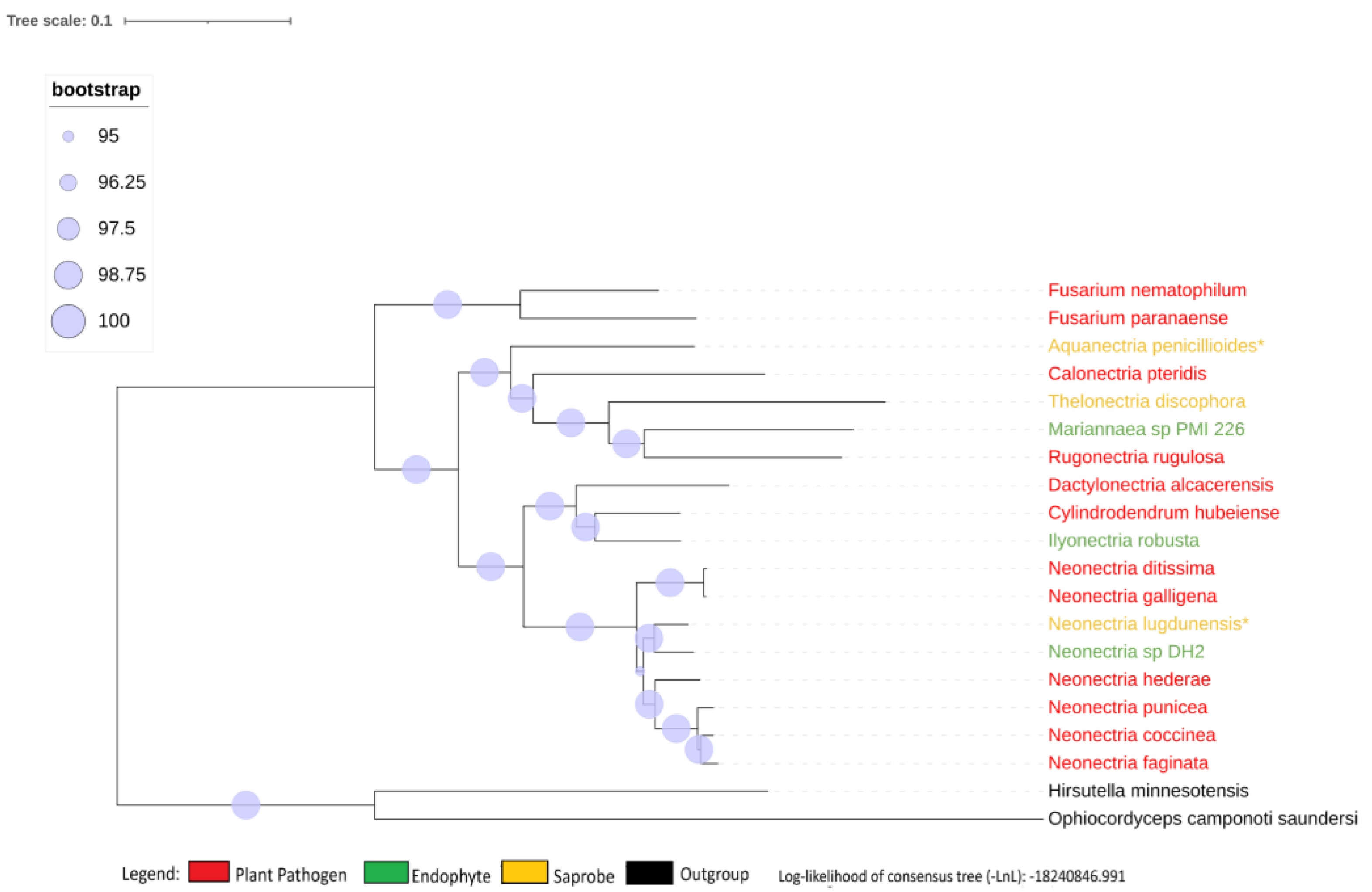
Figure 2.
Comparative Analysis of Retroelement Counts Among Different Fungal Lifestyles. Distribution of retroelement across three fungal lifestyles: Endophyte, Plant Pathogen, and Saprobes. Endophyte: Represented in green, the retroelement counts for endophytes are low, with an average count of 0.69. The box plot shows minimal variation with most values clustering around the average. Plant Pathogen: Represented by red, plant pathogens exhibit a higher average retroelement count of 2.60. The distribution shows greater variability compared to endophytes, with several outliers indicating significantly higher counts. Saprobe: Represented by brown color, has the highest average retroelement count of 3.67. This group shows larger variability in retroelement counts.
Figure 2.
Comparative Analysis of Retroelement Counts Among Different Fungal Lifestyles. Distribution of retroelement across three fungal lifestyles: Endophyte, Plant Pathogen, and Saprobes. Endophyte: Represented in green, the retroelement counts for endophytes are low, with an average count of 0.69. The box plot shows minimal variation with most values clustering around the average. Plant Pathogen: Represented by red, plant pathogens exhibit a higher average retroelement count of 2.60. The distribution shows greater variability compared to endophytes, with several outliers indicating significantly higher counts. Saprobe: Represented by brown color, has the highest average retroelement count of 3.67. This group shows larger variability in retroelement counts.
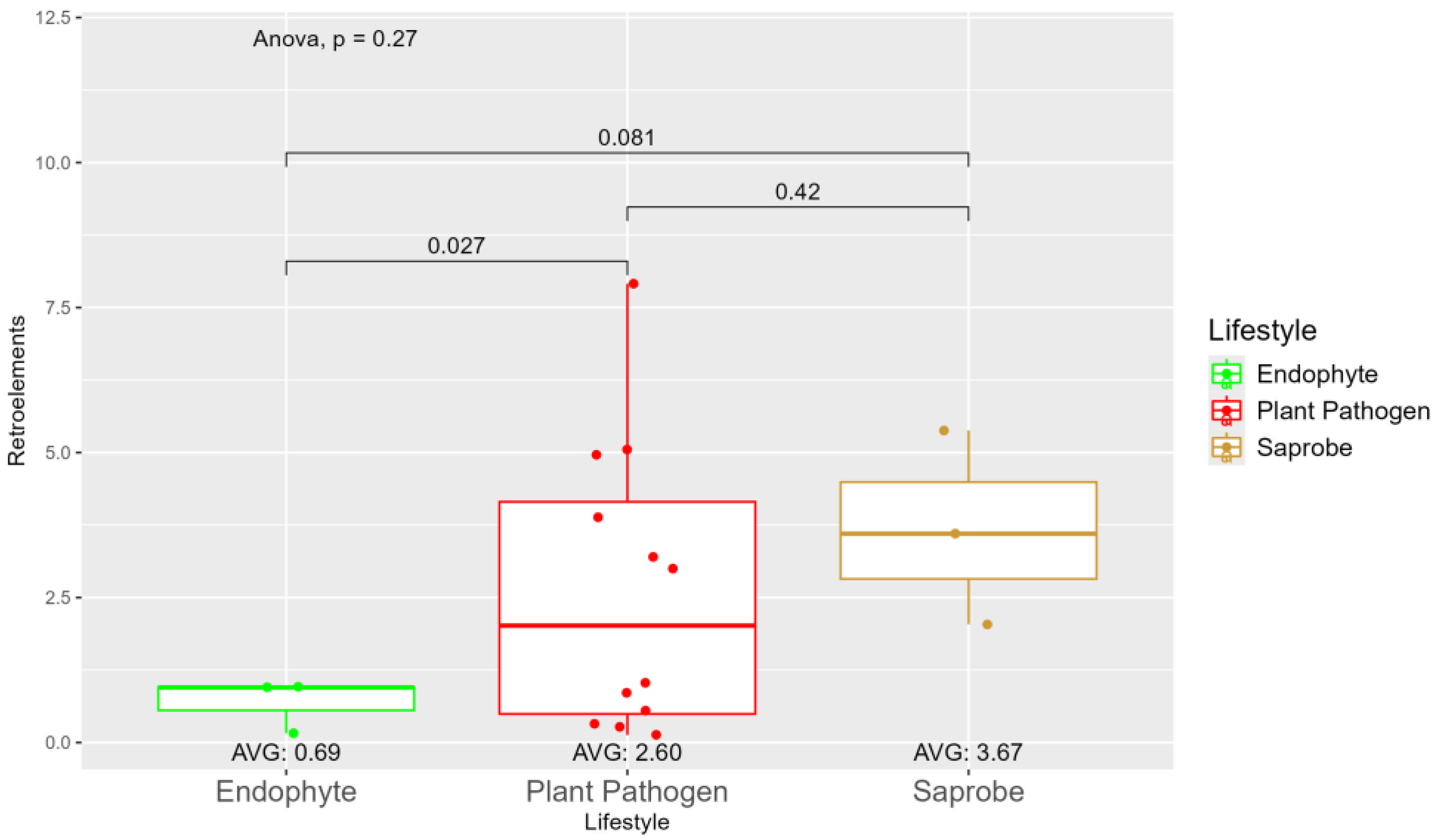
Figure 3.
Comparative Analysis of Secreted Thermo Effector Counts Among Different Fungal Lifestyles. Comparative Analysis of Secreted Thermo Effector Counts Among Different Fungal Lifestyles. Endophyte: Represented by green, the secreted thermo effector counts for endophytes have an average count of 196.33. The box plot shows moderate variation with values clustering around the average. Plant Pathogen: Represented by red, plant pathogens exhibit a higher average secreted thermo effector count of 217.17. The distribution shows greater variability compared to endophytes, with several outliers indicating significantly higher counts. Saprobe: Represented by brown, saprobes have the lowest average secreted thermo effector count of 147.00. This group shows minimal variability in secreted thermo effector counts. The comparison between Plant Pathogens and Saprobes shows a significant difference (p = 0.0074).
Figure 3.
Comparative Analysis of Secreted Thermo Effector Counts Among Different Fungal Lifestyles. Comparative Analysis of Secreted Thermo Effector Counts Among Different Fungal Lifestyles. Endophyte: Represented by green, the secreted thermo effector counts for endophytes have an average count of 196.33. The box plot shows moderate variation with values clustering around the average. Plant Pathogen: Represented by red, plant pathogens exhibit a higher average secreted thermo effector count of 217.17. The distribution shows greater variability compared to endophytes, with several outliers indicating significantly higher counts. Saprobe: Represented by brown, saprobes have the lowest average secreted thermo effector count of 147.00. This group shows minimal variability in secreted thermo effector counts. The comparison between Plant Pathogens and Saprobes shows a significant difference (p = 0.0074).
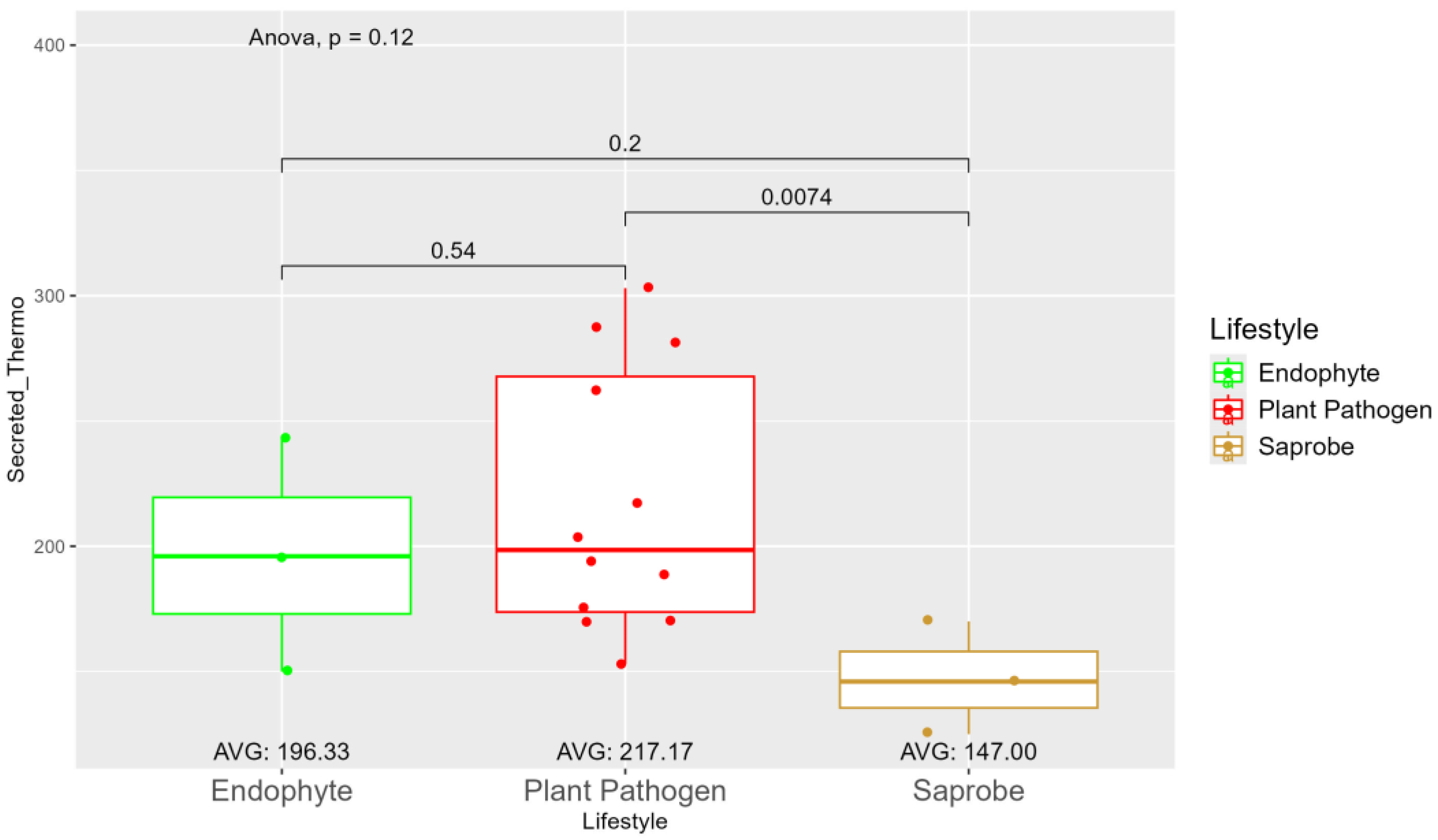
Figure 4.
Comparative Analysis of Secreted Cold Effector Counts Among Different Fungal Lifestyles. Difference in effector genes adapted to colder temperatures in endophytes, plant pathogens, and saprobes fungi. Endophyte: Represented by green, the cold effector counts for endophytes have an average count of 166.33. The box plot shows moderate variation with values clustering around the average. Plant Pathogen: Represented by red, plant pathogens exhibit a lower average cold effector count of 145.42 than endophytes. The distribution shows greater variability compared to endophytes, with several outliers indicating higher counts. Saprobe: Represented by brown, saprobes have the lowest average cold effector count of 126.67. This group shows minimal variability in cold effector counts. Notably, the comparison between Plant Pathogens and Saprobes shows a significant difference (p = 0.044).
Figure 4.
Comparative Analysis of Secreted Cold Effector Counts Among Different Fungal Lifestyles. Difference in effector genes adapted to colder temperatures in endophytes, plant pathogens, and saprobes fungi. Endophyte: Represented by green, the cold effector counts for endophytes have an average count of 166.33. The box plot shows moderate variation with values clustering around the average. Plant Pathogen: Represented by red, plant pathogens exhibit a lower average cold effector count of 145.42 than endophytes. The distribution shows greater variability compared to endophytes, with several outliers indicating higher counts. Saprobe: Represented by brown, saprobes have the lowest average cold effector count of 126.67. This group shows minimal variability in cold effector counts. Notably, the comparison between Plant Pathogens and Saprobes shows a significant difference (p = 0.044).
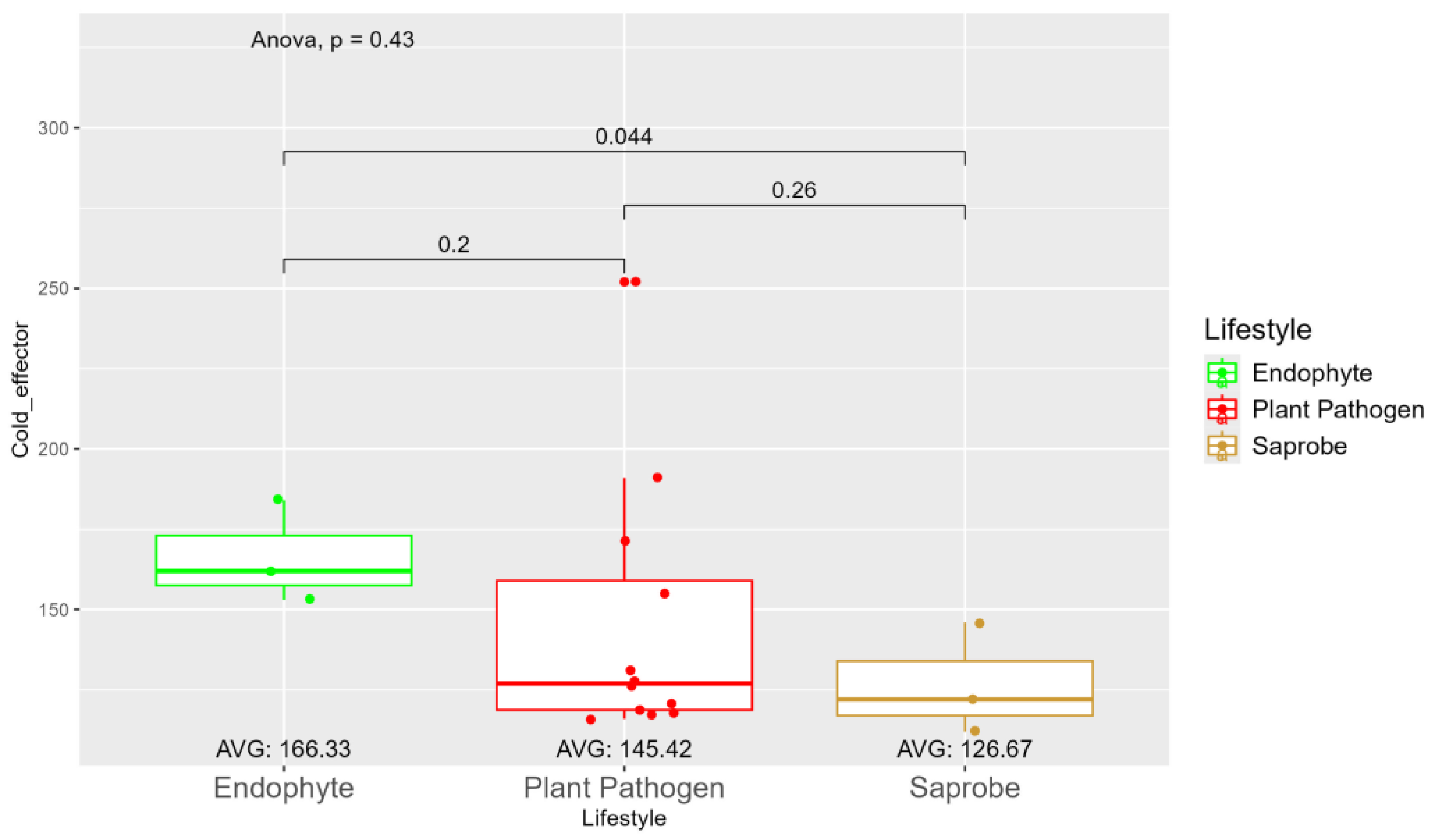
Figure 5.
Comparative Analysis of Thermo Effector Counts Among Different Fungal Lifestyles. Difference in effector genes adapted to higher temperatures in endophytes, plant pathogens, and saprobes fungi. Endophyte: Represented by green, the thermo effector counts for endophytes have an average count of 65.00. The box plot shows moderate variation with values clustering around the average. Plant Pathogen: Represented by red, plant pathogens exhibit an average thermo effector count of 59.08. The distribution shows greater variability compared to endophytes, with several outliers indicating higher counts. Saprobe: Represented by brown, saprobes have the lowest average thermo effector count of 44.67. This group shows minimal variability in thermo effector counts. Notably, the comparison between Endophytes and Plant Pathogens shows a significant difference (p = 0.039).
Figure 5.
Comparative Analysis of Thermo Effector Counts Among Different Fungal Lifestyles. Difference in effector genes adapted to higher temperatures in endophytes, plant pathogens, and saprobes fungi. Endophyte: Represented by green, the thermo effector counts for endophytes have an average count of 65.00. The box plot shows moderate variation with values clustering around the average. Plant Pathogen: Represented by red, plant pathogens exhibit an average thermo effector count of 59.08. The distribution shows greater variability compared to endophytes, with several outliers indicating higher counts. Saprobe: Represented by brown, saprobes have the lowest average thermo effector count of 44.67. This group shows minimal variability in thermo effector counts. Notably, the comparison between Endophytes and Plant Pathogens shows a significant difference (p = 0.039).
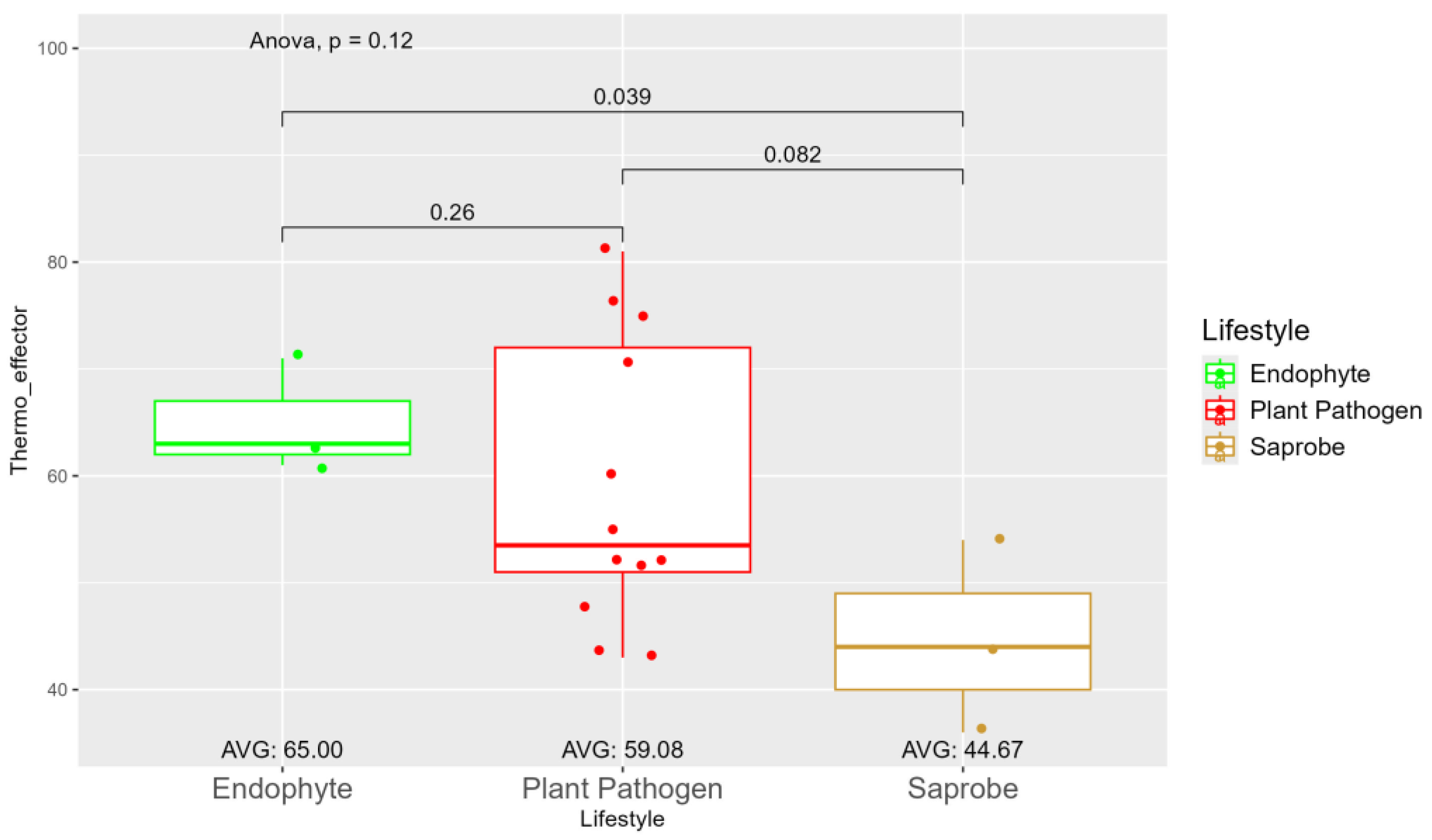
Figure 6.
Principal Component Analysis (PCA) of Fungal Species Based on Lifestyle considering secreted genes, effector protein, and total biosynthetic gene clusters.
Figure 6.
Principal Component Analysis (PCA) of Fungal Species Based on Lifestyle considering secreted genes, effector protein, and total biosynthetic gene clusters.
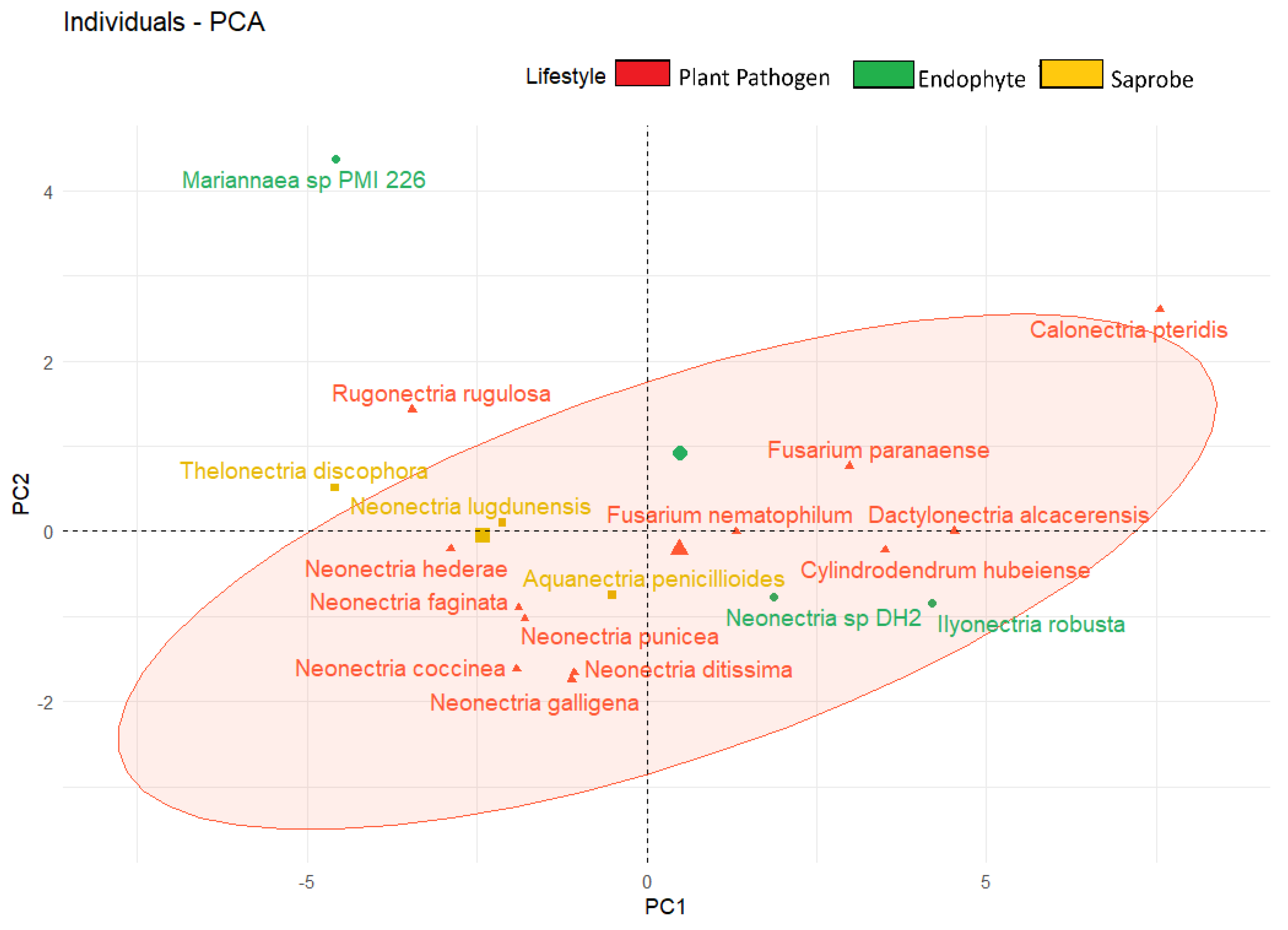

Table 1.
Basal genome information of the Nectriaceae family in this study.
| Species | Genome Length (Mbp) | Genome Compl. (%) | GC Content (%) | N50 | Assembly Accession |
|---|---|---|---|---|---|
| Ilyonectria robusta | 59.64 | 97.5% | 51.68 | 1.2Mb | GCF_021365365.1 |
| Calonectria pteridis | 58.37 | 97.7% | 50.21 | 3.2Mb | GCA_022837005.1 |
| Dactylonectria alcacerensis | 61.76 | 97.9% | 49.86 | 4.3Mb | GCA_029931735.1 |
| Fusarium paranaense | 53.40 | 98.2% | 49.21 | 859.2Kb | GCA_027886155.1 |
| Fusarium nematophilum | 50.82 | 96.4% | 53.92 | 148.30Kb | GCA_033030565.1 |
| Cylindrodendrum hubeiense | 48.81 | 96.5% | 51.81 | 85.2Kb | GCA_014621425.1 |
| Rugonectria rugulosa | 46.95 | 97.6% | 51.43 | 56.0Kb | GCA_023509875.1 |
| Neonectria sp DH2 | 45.82 | 94.8% | 52.99 | 45.8Mb | GCA_003934905.1 |
| Aquanectria penicillioides | 53.76 | 95.0% | 47.94 | 4.93Mbp | GCA_003415625.1 |
| Neonectria ditissima | 44.95 | 97.5% | 51.83 | 1.8Mb | GCA_001305505.1 |
| Neonectria lugdunensis | 44.78 | 97.6% | 52.17 | 44.7Mb | To be added during review |
| Neonectria punicea | 41.47 | 96.8% | 52.72 | 41.4Mb | GCA_003385315.1 |
| Neonectria galligena | 41.00 | 96.7% | 53.9 | 31.3Kb | GCA_013759035.1 |
| Neonectria coccinea | 42.74 | 97.3% | 51.65 | 178.8Kb | GCA_019137265.1 |
| Neonectria faginata | 42.94 | 97.4% | 52.48 | 4.4Mb | GCA_030864175.1 |
| Neonectria hederae | 43.28 | 97.5% | 49.43 | 248.9Kb | GCA_003385265.1 |
| Thelonectria discophora | 41.60 | 97.8% | 54.16 | 41.6Mb | GCA_911649645.1 |
| Mariannaea sp PMI 226 | 42.25 | 97.7% | 48.55 | 2.9Mb | GCA_024336345.1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



