
Submitted:
30 July 2024
Posted:
01 August 2024
You are already at the latest version
Abstract
Idiopathic granulomatous mastitis (IGM) is a rare condition characterised by chronic inflammation and granuloma formation in the breast. The aetiology of IGM is unclear. By focusing on the protein-coding regions of the genome, where most disease-related mutations often occur, whole-exome sequencing (WES) is a powerful approach for investigating rare and complex conditions, like IGM. We report WES results on paired blood and tissue samples from eight IGM patients. Samples were processed using standard genomic protocols. Somatic variants were called with two analytical pipelines: nf-core/sarek with Strelka2 and GATK4 with Mutect2. Our WES study of eight patients did not find evidence supporting a clear genetic component. The discrepancies between variant calling algorithms, along with the considerable genetic heterogeneity observed amongst the eight IGM cases, indicate that common genetic drivers are not readily identifiable. With only three genes, CHIT1, CEP170, and CTR9, recurrently altered in multiple cases, the genetic basis of IGM remains uncertain. The absence of validation for somatic variants by Sanger sequencing raises further questions about the role of genetic mutations in the disease. These observations suggest that IGM may be influenced by non-genetic factors, such as environmental triggers or immune system dysregulation, rather than being primarily a genetic disorder.
Keywords:
breast pathology
; breast abscess
; suppurative breast lesion
; tuberculous mastitis
; breast cancer
; mastitis
; idiopathic granulomatous mastitis
; somatic mutations
; pathogenic mutations
; whole-exome sequencing
1. Introduction
Idiopathic granulomatous mastitis (IGM) poses a significant clinical challenge [1,2]. It is characterised by chronic inflammation and granuloma formation in the breast, with the precise molecular mechanisms driving its pathogenesis remaining ambiguous [3,4,5]. Unlike other forms of mastitis with known risk factors of breastfeeding, infection, or autoimmunity, IGM perplexes clinicians and researchers alike due to its elusive aetiology and diverse clinical presentation [5,6,7,8].
The rarity of IGM complicates both its diagnosis and treatment [2,9]. Specific prevalence and incidence rates are not well-established in the literature due to their rarity and the diagnosis being one of exclusion [10]. Some studies indicate that the incidence may be higher in certain geographic regions and populations, but precise numbers are not widely available [2,11]. With its low incidence rate and heterogeneous clinical manifestations, establishing standardised diagnostic criteria and therapeutic guidelines proves challenging [2,11,12]. The lack of consensus regarding management also underscores the pressing need for a deeper understanding of the disease’s molecular underpinnings [10].
Advancements in genomic technologies have heralded a new era in unravelling the genetic basis of complex diseases. Whole exome sequencing (WES) has emerged as a powerful tool for comprehensively interrogating the coding regions of the genome, offering a promising avenue to explore the genetic landscape of rare disorders like IGM [13]. Studying IGM-related somatic mutations enhances our understanding of the molecular mechanisms underlying the disease’s pathogenesis. Such insights can lead to improved diagnostics through biomarker identification, enabling quicker and more accurate differentiation from other breast diseases [14]. In this study, we embark on a first-of-its-kind endeavour to identify somatic mutations associated with IGM by employing WES on matched blood and tissue samples from IGM patients.
2. Results
2.1. Patient Demographics and Clinical Characteristics
Paired blood and breast tissue samples donated by eight women diagnosed with IGM were processed for WES. The patient demographic and clinical characteristics are shown in Table 1.
The women have a median age of 33 (interquartile range 27.3-34.5) (Table 1). The other demographic variables reported were ethnicity, body mass index and education level (Table 1). Clinical characteristics reported were parity, number of children, smoking, chronic illness diagnosis, and first-degree family history of breast cancer (Table 1). All patients reported no alcohol consumption; no previous or existing diagnosis of autoimmune conditions (coeliac disease, type 1 diabetes mellitus, Graves’ disease, inflammatory bowel disease, multiple sclerosis, psoriasis, rheumatoid arthritis, or lupus erythematosus); and no previous or existing cancer diagnosis.
2.2. DNA Quality and Sequencing Metrics
Genomic DNA extraction yielded mean DNA concentration from blood samples at 12.5 ng/µL, and from tissue samples at 10.7 ng/µL (Supplementary Table S1, Supplementary Figure S1). WES libraries prepared had typical fragment size distributions with peak range of 320-337 bp for the blood samples, and 315-332 bp for the tissue samples (Supplementary Table S2). An average of almost 91 million reads per sample was obtained (Supplementary Table S3). Reads were aligned to the human GRCh38 reference genome using BWA, with a mean mapping rate of 100.0% (Supplementary Table S3). The average duplication rate was 17.2%, ensuring efficient use of sequencing capacity (Supplementary Table S3). The average coverage depth across target exonic regions was 37.5x (range 26.66-53.51x), ensuring high sensitivity for variant detection (Supplementary Table S3). 100% of target regions were covered at least 20x, indicating uniform coverage across the exome (Supplementary Table S3). The GC content of the reads was within the expected range for human exonic sequences (range 42.3-43.7%) (Supplementary Table S3). Other summary statistics for sequencing performance, coverage metrics and sequencing read quality control values are displayed in Supplementary Table S3.
2.3. Somatic variants identified from WES
Variant calling by Strelka2 in nf-core/sarek pipeline and Mutect2 in GATK4 Best Practises workflow, yielded
- Variants called from blood samples: Variants identified in blood samples (Supplementary Table S4), and
- Variants called from paired blood/tissue samples: Variants identified in the tissue sample that were not present in the corresponding blood sample (Table 2).

Table 2.
Somatic variants identified from WES of paired blood/tissue samples through Strelka2 and Mutect2 variant calling.
Table 2.
Somatic variants identified from WES of paired blood/tissue samples through Strelka2 and Mutect2 variant calling.
| Case | Somatic variants | SNVs1 | Indels2 | PTVs3 | Pathogenic4 | Pathogenic / Likely Pathogenic4 | Likely Pathogenic4 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Strelka2 | Mutect2 | Strelka2 | Mutect2 | Strelka2 | Mutect2 | Strelka2 | Mutect2 | Strelka2 | Mutect2 | Strelka2 | Mutect2 | Strelka2 | Mutect2 | |
| 1 | 57 | 224 | 56 | 219 | 1 | 1 | 1 | 13 | 0 | 0 | 0 | 0 | 0 | 0 |
| 2 | 50 | 46 | 48 | 38 | 2 | 7 | 0 | 5 | 0 | 0 | 0 | 0 | 0 | 0 |
| 3 | 57 | 72 | 56 | 69 | 1 | 2 | 1 | 4 | 0 | 0 | 0 | 0 | 0 | 0 |
| 4 | 53 | 52 | 51 | 48 | 2 | 4 | 3 | 5 | 0 | 0 | 0 | 0 | 0 | 0 |
| 5 | 56 | 33 | 54 | 28 | 2 | 5 | 2 | 6 | 0 | 0 | 0 | 0 | 0 | 0 |
| 6 | 80 | 35 | 79 | 26 | 1 | 8 | 0 | 4 | 0 | 0 | 0 | 0 | 0 | 0 |
| 7 | 65 | 39 | 65 | 32 | 0 | 6 | 0 | 5 | 0 | 0 | 0 | 0 | 0 | 0 |
| 8 | 53 | 52 | 52 | 39 | 1 | 11 | 0 | 8 | 0 | 0 | 0 | 0 | 0 | 0 |
| Median (range) | 56.5 (50-80) |
49 (23-224) |
55 (48-79) |
38.5 (26-219) |
1 (0-2) |
5.5 (1-11) |
0.5 (0-3) |
5 (4-13) |
0 (0-0) |
0 (0-0) |
0 (0-0) |
0 (0-0) |
0 (0-0) |
0 (0-0) |
1 Single nucleotide variants. 2 Insertions and deletions. 3 Protein-truncating variants. These correspond to variants annotated as nonsense mutations, or frameshift insertions or deletions by GATK4 Funcotator. 4 ClinVar annotation of pathogenicity within GATK4 Funcotator variant annotation.
Supplementary Table S4 and Table 2 show the number of somatic variants, single nucleotide variants (SNVs), insertions and deletions (indels) called by the two variant callers for blood samples, and blood/tissue paired samples respectively, for the 8 cases. Variants annotated as “Nonsense_Mutation”, “Frame_Shift_Ins”, “Frame_Shift_Del” with Funcotator were labelled protein-truncating variants (PTVs). Table 2 also shows the number of PTVs, and ClinVar pathogenicity annotations for the eight cases.
In paired blood-tissue samples, Strelka2 called more variants than Mutect2 (Table 2, Figure 1a). Medians of all variants called were 56.5.5 (range 50-80) for Strelka2 and 49 (range 23-224) for Mutect2 (Table 2). Strelka2 and Mutect2 also called more SNVs than indels (Table 2, Figure 1a).
Amongst non-synonymous mutations annotated with Funcotator, missense mutations were annotated the most (Figure 2a).
More non-synonymous mutations and more types of non-synonymous mutations were annotated from the variants called by Mutect2, than by Strelka2 (Figure 2a). Median mutations annotated per sample was 49 for variants called by Mutect2, vs 11.5 for variants called by Strelka2 (Figure 2a). Variants called by Strelka2 were annotated as missense mutations, splice sites and nonsense mutations; variants called by Mutect2 were annotated as those already mentioned, as well as frameshift insertions and deletions, in-frame insertion and deletions, and non-stop mutations (Figure 2a). None of the variants called in matched blood-tissue comparisons were pathogenic, or likely pathogenic, as per ClinVar annotation (Table 2). Less variants called in matched blood-tissue samples were annotated as PTVs in Strelka2 variant calling (median 0.5, range 0-3), compared to that of Mutect2 (median 5, range 4-13), despite the opposite comparison for total number of variants called (Table 2, Figure 1a and Figure 2b).
Further examination of PTVs from the matched blood-tissue Strelka2 and Mutect2 variant calling, found 53 genes altered across the eight cases (Figure 3).
49 genes altered were identified from variants called by Mutect2, and the remaining 4 genes were identified from variants called by Strelka2 (Figure 3). Only 3 out of the 53 genes were altered in more than 1 case (Figure 3):
- CHIT1, altered in Cases 3, 4 and 5, nonsense mutations
- CEP170, altered in Cases 4 and 5, nonsense mutations
- CTR9, altered in Cases 7 and 8, nonsense mutation and frameshift deletion, respectively
The remaining genes were only altered in single cases (Figure 3).
Functional enrichment analysis of the altered genes with enrichR did not reveal any statistically significant (p<0.05) pathways enriched (Supplementary Table S5, Supplementary Figure S2). Pathways with the lowest p-values identified include terpenoid backbone biosynthesis (p=0.0526, adjusted p=0.481), protein export (p=0.0549, adjusted p=0.481), and protein processing in the endoplasmic reticulum (p=0.0618, adjusted p=0.481) (Supplementary Table S5, Supplementary Figure S2). The genes IDI2, SEC62, DNAJB12, and DNAH1 were implicated in these pathways (Supplementary Table S5).
A median of 2 [range 1-3] overlapping variants per sample were called by both variant callers from paired blood/tissue samples (Table 3).
All overlapping variants are single nucleotide variants, none of which were annotated as PTVs, or pathogenic or likely pathogenic (Table 3). Only 1 variant from Patient 4 (missense mutation) and 1 variant from Patient 8 (splice site) were annotated as non-synonymous mutations.
Supplementary Table S4 and Figure 2b show the variants and their categorisations for the Strelka2 and Mutect2 variant calling in only the blood samples. Supplementary Table S6 details the overlapping variants per sample called by both variant callers in only the blood samples.
2.4. Validation of Somatic Variants with Sanger Sequencing
A subset of variants was selected for validation using Sanger sequencing (Supplementary Table S7). None of the selected variants were validated through Sanger sequencing (Supplementary Table S7).
3. Discussion
This study presents somatic variants identified through WES in paired blood and breast tissue samples from eight women diagnosed with IGM. WES libraries exhibited typical fragment size distributions, high mapping rates, and sufficient coverage depth across exonic regions. Somatic variant calling revealed more variants by Strelka2 compared to Mutect2 in paired samples. This flips when variants called by Mutect2 are annotated to more non-synonymous mutations and PTVs than those called by Strelka2. However, none of the variants were pathogenic per ClinVar annotation. Further examination identified 53 altered genes, with CHIT1, CEP170 and CTR9 genes altered in more than one case. Functional enrichment analysis did not show statistically significant pathways, although terpenoid backbone biosynthesis, protein export, and protein processing in the endoplasmic reticulum were implicated. Validation of variants through Sanger sequencing did not yield any validated variants.
Differences in the variability of sensitivity and specificity between different variant calling algorithms have been extensively discussed [15,16,17,18,19]. Their differences in algorithm and focus of the variant caller underscores this discrepancy between Strelka2 and Mutect2 in terms of the number of variants identified, and their limited overlap. Strelka2 uses a probabilistic model leveraging local assembly and realignment to call variants for more sensitivity in identifying low-frequency somatic mutations, especially in matched tumour-normal pairs, by using Bayesian methods to model both the tumour and normal samples [20]. Contrastingly, the haplotype-based approach in Mutect2 employs a sophisticated filtering process that incorporates various sources of evidence to distinguish between true mutations and sequencing artefacts [21]. Designed to balance sensitivity and specificity, Mutect2 minimises false positives by implementing additional artefact filters for oxidative artefacts and strand bias, on top of the standard filtering preprocessing [21]. Furthermore, nf-core/sarek Strelka2’s use of hard filters based on fixed thresholds, vs GATK4 Mutect2 use of machine learning to filter variants could provide additional explanation for the large discrepancy in called variants [20,21,22].
Both algorithms identified more SNVs than indels, which is consistent with typical findings in WES studies [23,24,25]. Despite the larger number of variants identified by Strelka2, the fewer non-synonymous mutations and PTVs annotation in Funcotator contrasted with Mutect2 variant calls in matched blood-tissue samples, emphasises the need for the multiple variant caller approach for capturing the full spectrum of genetic alterations [26,27]. The limited overlap across the different categorisations of the variants in both the matched blood-tissue calls and the blood only calls, signals further optimisation of the variant calling pipelines and validating identified variants through independent methods are necessary.
From the PTVs from the matched blood-tissue Strelka2 and Mutect2 variant calling, CHIT1, CEP170, and CTR9, were altered in more than one case. CHIT1 has been implicated in both granulomatous and non-granulomatous inflammatory conditions, including multiple sclerosis, sarcoidosis, inflammatory bowel disease, and a few fibrotic interstitial lung diseases (tuberculosis, idiopathic pulmonary fibrosis, scleroderma-associated interstitial lung diseases, chronic obstructive pulmonary diseases) [28,29,30,31,32]. Song and Shao (2024) have also proposed CHIT1 as one of 12 genes in an immune-mediated genetic prognostic risk score model when administering immunotherapy in triple negative breast cancer [33]. CEP170 and CTR9 are involved in cell cycle processes, in which dysregulation have been described to result in the secretion of inflammatory factors, impair immune-mediated processes, and increase inflammation [34,35,36,37,38]. Potentially, the alterations in CHIT1, CEP170, and CTR9 may individually, or collectively contribute to granuloma formation and chronic inflammation in the breast tissue in IGM.
Unfortunately, there is considerable genetic heterogeneity amongst the eight IGM cases since the remaining 50 genes are each altered in single cases. This variability complicates efforts to pinpoint common genetic drivers of the disease, and could suggest IGM may arise from multiple genetic pathways. Such heterogeneity is consistent with the clinical diversity observed in IGM, where patients present with a wide range of symptoms and disease severities [4,10,39]. However, the lack of statistically significant pathways identified from functional enrichment analysis of the 53 genes, with all pathways identified enriched by only one to two genes, suggests there may not be identifiable genetic drivers for IGM among these eight patients.
Both pipelines rely on rigorous variant calling and annotation processes to maximise the reliability and validity of the identified somatic variants. Unfortunately, none of the selected variants identified from WES were validated with Sanger sequencing. Despite achieving high coverage depth across all exome-targeted regions, WES is prone to inaccuracies, due to sequencing artefacts or accurately identifying variants in regions of genomic instability [40]. False positives can arise in short-read technology, particularly in regions with high GC content or repetitive sequences [41]. Sanger sequencing, with its ability to provide uniform coverage and longer read lengths, is a valuable orthogonal validation tool [42]. Other studies have also described somatic variants identified from WES that were not found in Sanger sequencing [24,25,43,44]. Discrepancies between WES and Sanger sequencing results can be attributed to their inherent differences in their error profiles or limitations in detecting variants present at low allele frequencies, especially in heterogeneous samples like those from IGM patients [45,46,47].
It must be recognised that the application of WES to the study of IGM presents its own unique set of challenges. WES mainly targets the exonic regions of the genome and is not as effective in identifying large structural variations, including deletions, duplications, inversions, and translocations [48]. Additionally, the rarity of IGM limits access to large patient cohorts for comprehensive genomic analysis with sufficient power to detect smaller effect sizes and lower impact somatic mutations [49]. Given our sample size of eight patients with matched blood and tissue samples, this study has low statistical power (0.230) to detect a significant difference in somatic mutations between the paired samples if such a difference truly exists [50]. The ideal power of at least 0.80 requires an odds ratio of approximately 7.25 to detect a statistically significant difference in somatic mutations between the paired blood and tissue samples [50].
Another difficulty lies in the disease’s inherent heterogeneity [2,4,8,10]. With a broad spectrum of clinical features, ranging from localised breast masses to diffuse inflammatory changes, identifying consistent genetic signatures associated with IGM is challenging [39]. Moreover, the multifactorial and inflammatory nature of IGM adds another layer of complexity to the study of its aetiology [8,51]. While various hypotheses, including immune dysregulation, infectious triggers, and hormonal influences, have been proposed, the precise interplay of genetic and environmental factors remains poorly understood [8,11]. Understanding these complex interactions is key to determining the underlying causes of IGM.
This study pioneers the investigation into somatic variants in IGM patients. While the matched blood-tissue WES variant calls did not identify any ClinVar annotated pathogenic variants, the detection of variants in multiple genes suggests that IGM may involve a variety of molecular mechanisms. Larger studies with more comprehensive datasets are needed to uncover significant genomic drivers and biological pathways associated with IGM. Furthermore, larger scale studies could also unearth possible associations between different variants and, the clinical manifestations and severity of IGM [2,10]. Future studies should also focus on integrating additional omics data, such as transcriptomics and proteomics, to provide a more comprehensive understanding of the disease. The challenges in validating somatic variants underscore the need for improved methodologies and protocols for variant validation. Addressing these challenges requires a multifaceted approach, including refining bioinformatics pipelines to mitigate false positives, and an integrated orthogonal validation approach to ensure the accuracy of variant calls.
4. Materials and Methods
4.1. Patient Recruitment
The study population and patient recruitment have been previously described [8]. In brief, adult female patients with IGM were recruited from five participating hospitals in Singapore between 2018 and 2020. IGM diagnoses were based on breast core biopsy histopathology for non-caseating granulomatous inflammation and absence of malignancy. Patients were also negative for Mycobacterium tuberculosis infection (acid-fast bacillus stain), and fungal infection (Grocott (methenamine) silver stain or Periodic acid–Schiff stain). Study coordinators sought written informed consent from potential IGM patients identified by clinicians and physicians. All studies were performed in accordance with the Declaration of Helsinki. This study was approved by the National Healthcare Group Domain Specific Review Board (reference number: 2017/01057) and the Agency for Science, Technology and Research Institutional Review Board (reference number: 2020-152).
4.2. Sample Collection
A subset of eight IGM patients who donated paired blood and core tissue biopsy samples for research use were included in our study (Figure 4).
Fresh blood samples were collected with DNA/RNA Shield Blood Collection tubes (catalogue number R1150; Zymo Research, Irvine, CA, USA). Fresh tissue samples were obtained with ultrasound-guided core tissue biopsies from three different regions of the affected breast area. Tissue cores were collected in 2mL-collection tubes with 300µL DNA/RNA Shield without breads (catalogue number R1100-250; Zymo Research, Irvine, CA, USA).
4.3. DNA Extraction and Sequencing
Genomic DNA was extracted from the collected whole blood and fresh tissue samples, and the WES library was prepared with standard protocols. Briefly, DNA extraction was performed with Quick-DNA Miniprep Plus Kit (catalogue number D4069; Zymo Research, Irvine, CA, USA) according to the manufacturer’s instructions. WES library was prepared with NEBNext® Ultra™ II DNA Library Prep Modules for Illumina® (catalogue number E7645L; New England Biolabs, Ipswich, MA, USA) according to the manufacturer’s instructions. Exome capture was performed with NimbleGen SeqCap EZ Exome Library Kit v3.0 (catalogue number 06465692001; Roche, Basel, Switzerland). DNA concentrations and quality were measured after extraction, shearing, pre-, and post-exome capture for quality control. Libraries were sequenced with 2 × 150 bp paired-end reads on HiSeq4000.
4.4. Quality Control and Somatic Variant Calling
Two analytical pipelines were used to identify somatic variants in the paired blood and tissue samples. The nf-core/sarek pipeline (version 3.3.0) was executed for identifying somatic mutations with singularity [52,53,54,55,56,57]. Raw sequencing reads were aligned to human GRCh38 reference genome (version from 22 July 2016, Broad Institute, from https://console.cloud.google.com/storage/browser/genomics-public-data/resources/broad/hg38/v0;tab=objects?prefix=&forceOnObjectsSortingFiltering=false) with BWA [58]. GATK4 was applied according to GATK Best Practises recommendations parameters for hard filtering and score recalibration for removing duplicates and base quality score recalibration [21,59,60]. Strelka2 was used for matched tissue-normal pair variant calling [20].
Another analytical pipeline, also applying GATK4 according to the GATK Best Practices workflow, was also used to identify somatic mutations [21,59,60]. Preprocessing sequencing reads followed the same parameters as above, but the different processes were applied individually in the same sequence, without the wrapped container. Briefly, the raw sequencing reads were also aligned to the human GRCh38 reference genome using BWA [58]. Picard tools were utilised to mark and remove duplicates [21], and GATK4 BaseRecalibrator and GATK4 ApplyBQSR for base quality score recalibration, consistent with GATK Best Practices [21,59,60]. Matched tissue-normal pair somatic variant calling was executed using GATK4 Mutect2 [21]. The called variants underwent filtering with GATK FilterMutectCalls before variant annotation [21].
Variants called with Strelka2 in the nf-core/sarek pipeline, and variants called with Mutect2 in the GATK Best Practises workflow, were annotated with GATK4 Funcotator [21]. Variant annotation was performed against Funcotator data sources v1.7.20200521s (GRCh38), encompassing the following databases:
- Catalogue of Somatic Mutations in Cancer (COSMIC) Cancer Gene Census (CGC) (data source dated 15 March 2012) [61]
- NCBI dbSNP (data source dated 18 April 2018) [64]
- Human DNA repair genes (data source dated 24 May 2018) [65]
- GENCODE (v34) [68]
- Genome Aggregation Database (gnomAD) (v3.1.2) [69]
- Human Genome Organisation (HUGO) Gene Nomenclature Committee (HGNC) Database (data source dated 30 November 2017) [70]
4.5. Validating Called Variants with Sanger Sequencing
A subset of somatic variants identified through WES and visualised with Integrative Genomics Viewer (IGV, version 2.17.0) were validated with Sanger sequencing [71]. Primers for PCR amplification of the regions containing the variants were designed using Primer3web (version 4.1.0) [72,73,74]. The PCR products were purified using AMPure XP beads (product number A63882; Beckman Coulter Life Sciences, Indianapolis, IN, USA) in a ratio of 1:1 sample to bead volume. Purified PCR products were Sanger sequenced. The sequencing data were analysed using the CLC Main Workbench (version 7.7.3, QIAGEN, Hilden, Germany) to confirm the presence or absence of the variants. The validity of somatic variants identified was determined based on sequence quality, allele frequency, and variants presence in the corresponding matched blood samples.
4.6. Gene-Set Analysis
Functional enrichment analysis of gene sets of somatic variants identified was performed with enrichR (version 3.2) using the 2019 version of Kyoto Encyclopedia of Genes and Genomes (KEGG) knowledge base [75,76,77,78,79]; comprehensive analysis and visualisation of somatic variants identified was performed with maftools (version 2.6.05) [79]; and other data visualisation was performed with ggplot2 (version 3.4.4) [80]. All analyses and visualisations were performed in R (version 4.0.4) unless otherwise stated.
4.7. Data and Sample Availability Statement
The datasets used and analysed in the current study are available from the corresponding author on reasonable request, within limitations of the study’s Institutional Review Board (IRB).
5. Conclusions
This study leads the way in advancing our understanding of the genetic complexities of IGM, through WES and comprehensive bioinformatics analysis. While numerous somatic variants have been identified, their clinical significance remains to be fully elucidated. Challenges encountered in variability across variant calling algorithms, Sanger validation, and heterogeneous genetic profiles, underscore the ongoing need for methodological refinement and larger-scale studies to comprehensively understand the genetic underpinnings of IGM. Despite these challenges, this study represents a critical step forward in decoding the genetic landscape of IGM. More large-scale investigations, robust validation studies, and integration of multi-omics approaches in future work, will contribute to unravelling the genomic intricacies of IGM.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org., Supplementary Table S1: Concentration and yield of extracted genomic DNA before WES library preparation.; Supplementary Figure S1: Yield of extracted genomic DNA before whole exome sequencing (WES) library preparation.; Supplementary Table S2: Concentration and yield from whole exome sequencing (WES) library preparation.; Supplementary Table S3: Summary statistics of sequencing performance, coverage metrics and sequencing reads quality control values.; Supplementary Table S4: Somatic variants identified from whole exome sequencing (WES) of blood samples through Strelka2 and Mutect2 variant calling.; Supplementary Table S5: EnrichR functional enrichment of genes altered in protein-truncating variants (PTVs) called by Strelka2 and Mutect2 matched blood-tissue variant calling, using 2019 version of Kyoto Encyclopedia of Genes and Genomes (KEGG) knowledge base.; Supplementary Figure S2: Visualisation of enriched pathways from Supplementary Table S5, ordered by p-value.; Supplementary Table S6: Somatic variants identified from whole exome sequencing (WES) of blood samples that overlap across Strelka2 and Mutect2 variant calling.; Supplementary Table S7: Variants identified through whole exome sequencing (WES) validation with Sanger sequencing.
Author Contributions
Conceptualisation, S.S.O., P.J.H., J.L. and M.H.; methodology, S.S.O., P.J.H., A.J.K. and J.L.; software, S.S.O., P.J.H. and J.L.; validation, A.J.K.; formal analysis, S.S.O. and J.L.; investigation, S.S.O., A.J.K. and J.L.; resources, A.J.K., J.L. and M.H.; data curation, A.J.K., B.K.T.T., Q.T.T., E.Y.T., S.M.T., T.C.P., S.H.L., E.L.S.T. and M.H.; writing—original draft preparation, S.S.O.; writing—review and editing, S.S.O., P.J.H., A.J.K., B.K.T.T., Q.T.T., E.Y.T., S.M.T., T.C.P., S.H.L., E.L.S.T., J.L. and M.H.; visualisation, S.S.O., P.J.H., A.J.K., and J.L.; supervision, P.J.H., J.L. and M.H.; project administration, S.S.O., A.J.K., J.L. and M.H.; funding acquisition, J.L. and M.H. All authors have read and agreed to the published version of the manuscript.
Funding
This study was supported by National University Health System (NUHS) Seed Fund (NUHSRO/2020/027/T1/Seed-Aug/11, awarded to M Hartman), and the Agency for Science, Technology and Research (A*STAR).
Institutional Review Board Statement
All studies were performed in accordance with the Declaration of Helsinki. This study was approved by the National Healthcare Group Domain Specific Review Board (reference number: 2017/01057) and the Agency for Science, Technology and Research Institutional Review Board (reference number: 2020-152).
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
The datasets used and analysed in the current study are available from the corresponding author on reasonable request, within limitations of the study’s Institutional Review Board (IRB).
Acknowledgments
This work was supported by the A*STAR Computational Resource Centre through the use of its high performance computing facilities. The study team thanks all the patients for their participation. The authors would like to express their gratitude to Nur Khaliesah Binte Mohamed Riza, Ganga Devi d/o Chandrasegran, Yuk Kei Wan, Dominik Stanojević, Jonathan Göke and Mile Šikić for their contribution to this study. We would also like to thank the clinical research coordinators and research assistants for their contributions in recruitment and data collection.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Al-Jarrah, A.; Taranikanti, V.; Lakhtakia, R.; Al-Jabri, A.; Sawhney, S. Idiopathic Granulomatous Mastitis: Diagnostic strategy and therapeutic implications in Omani patients. Sultan Qaboos Univ Med J 2013, 13, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Ong, S.S.; Ho, P.J.; Liow, J.J.K.; Tan, Q.T.; Goh, S.S.N.; Li, J.; Hartman, M. A meta-analysis of idiopathic granulomatous mastitis treatments for remission and recurrence prevention. Front Med (Lausanne) 2024, 11, 1346790. [Google Scholar] [CrossRef]
- Hugon-Rodin, J.; Plu-Bureau, G.; Hugol, D.; Gompel, A. Management of granulomatous mastitis: a series of 14 patients. Gynecol Endocrinol 2012, 28, 921–924. [Google Scholar] [CrossRef] [PubMed]
- Baslaim, M.M.; Khayat, H.A.; Al-Amoudi, S.A. Idiopathic granulomatous mastitis: a heterogeneous disease with variable clinical presentation. World J Surg 2007, 31, 1677–1681. [Google Scholar] [CrossRef]
- Yukawa, M.; Watatani, M.; Isono, S.; Fujiwara, Y.; Tsujie, M.; Kitani, K.; Hara, J.; Kato, H.; Takeyama, H.; Kanaizumi, H.; et al. Management of granulomatous mastitis: a series of 13 patients who were evaluated for treatment without corticosteroids. Int Surg 2015, 100, 774–782. [Google Scholar] [CrossRef] [PubMed]
- Goulabchand, R.; Hafidi, A.; Van de Perre, P.; Millet, I.; Maria, A.T.J.; Morel, J.; Quellec, A.L.; Perrochia, H.; Guilpain, P. Mastitis in Autoimmune Diseases: Review of the Literature, Diagnostic Pathway, and Pathophysiological Key Players. J Clin Med 2020, 9. [Google Scholar] [CrossRef]
- Pevzner, M.; Dahan, A. Mastitis While Breastfeeding: Prevention, the Importance of Proper Treatment, and Potential Complications. J Clin Med 2020, 9. [Google Scholar] [CrossRef]
- Ong, S.S.; Xu, J.; Sim, C.K.; Khng, A.J.; Ho, P.J.; Kwan, P.K.W.; Ravikrishnan, A.; Tan, K.B.; Tan, Q.T.; Tan, E.Y.; et al. Profiling Microbial Communities in Idiopathic Granulomatous Mastitis. Int J Mol Sci 2023, 24. [Google Scholar] [CrossRef]
- Dilaveri, C.; Degnim, A.; Lee, C.; DeSimone, D.; Moldoveanu, D.; Ghosh, K. Idiopathic Granulomatous Mastitis. Breast J 2024, 2024, 6693720. [Google Scholar] [CrossRef]
- Ong, S.S.; Sim, J.X.Y.; Chan, C.W.; Ho, P.J.; Lim, Z.L.; Li, J.; Hartman, M. Current Approaches to Diagnosing and Treating Idiopathic Granulomatous Mastitis in Asia: A Summary from In-Depth Clinician Interviews. SSRN Preprints with The Lancet https://ssrn.com/abstract=4908728. 2024. [Google Scholar]
- Altintoprak, F.; Kivilcim, T.; Ozkan, O.V. Aetiology of idiopathic granulomatous mastitis. World J Clin Cases 2014, 2, 852–858. [Google Scholar] [CrossRef]
- Wolfrum, A.; Kummel, S.; Theuerkauf, I.; Pelz, E.; Reinisch, M. Granulomatous Mastitis: A Therapeutic and Diagnostic Challenge. Breast Care (Basel) 2018, 13, 413–418. [Google Scholar] [CrossRef]
- Sathyanarayanan, A.; Manda, S.; Poojary, M.; Nagaraj, S.H. Exome Sequencing Data Analysis. In Encyclopedia of Bioinformatics and Computational Biology, Ranganathan, S., Gribskov, M., Nakai, K., Schönbach, C., Eds.; Academic Press: Oxford, 2019; pp. 164–175. [Google Scholar]
- Sprissler, R.; Perkins, B.; Johnstone, L.; Babiker, H.M.; Chalasani, P.; Lau, B.; Hammer, M.; Mahadevan, D. Rare Tumor-Normal Matched Whole Exome Sequencing Identifies Novel Genomic Pathogenic Germline and Somatic Aberrations. Cancers (Basel) 2020, 12. [Google Scholar] [CrossRef]
- Chen, Z.; Yuan, Y.; Chen, X.; Chen, J.; Lin, S.; Li, X.; Du, H. Systematic comparison of somatic variant calling performance among different sequencing depth and mutation frequency. Sci Rep 2020, 10, 3501. [Google Scholar] [CrossRef]
- Jin, J.; Chen, Z.; Liu, J.; Du, H.; Zhang, G. Towards an accurate and robust analysis pipeline for somatic mutation calling. Front Genet 2022, 13, 979928. [Google Scholar] [CrossRef]
- Cai, L.; Yuan, W.; Zhang, Z.; He, L.; Chou, K.C. In-depth comparison of somatic point mutation callers based on different tumor next-generation sequencing depth data. Sci Rep 2016, 6, 36540. [Google Scholar] [CrossRef]
- Alioto, T.S.; Buchhalter, I.; Derdak, S.; Hutter, B.; Eldridge, M.D.; Hovig, E.; Heisler, L.E.; Beck, T.A.; Simpson, J.T.; Tonon, L.; et al. A comprehensive assessment of somatic mutation detection in cancer using whole-genome sequencing. Nat Commun 2015, 6, 10001. [Google Scholar] [CrossRef]
- Kroigard, A.B.; Thomassen, M.; Laenkholm, A.V.; Kruse, T.A.; Larsen, M.J. Evaluation of Nine Somatic Variant Callers for Detection of Somatic Mutations in Exome and Targeted Deep Sequencing Data. PLoS One 2016, 11, e0151664. [Google Scholar] [CrossRef]
- Kim, S.; Scheffler, K.; Halpern, A.L.; Bekritsky, M.A.; Noh, E.; Kallberg, M.; Chen, X.; Kim, Y.; Beyter, D.; Krusche, P.; et al. Strelka2: fast and accurate calling of germline and somatic variants. Nat Methods 2018, 15, 591–594. [Google Scholar] [CrossRef] [PubMed]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- Carson, A.R.; Smith, E.N.; Matsui, H.; Braekkan, S.K.; Jepsen, K.; Hansen, J.B.; Frazer, K.A. Effective filtering strategies to improve data quality from population-based whole exome sequencing studies. BMC Bioinformatics 2014, 15, 125. [Google Scholar] [CrossRef] [PubMed]
- Boulygina, E.A.; Borisov, O.V.; Valeeva, E.V.; Semenova, E.A.; Kostryukova, E.S.; Kulemin, N.A.; Larin, A.K.; Nabiullina, R.M.; Mavliev, F.A.; Akhatov, A.M.; et al. Whole genome sequencing of elite athletes. Biol Sport 2020, 37, 295–304. [Google Scholar] [CrossRef] [PubMed]
- Shi, W.; Ng, C.K.Y.; Lim, R.S.; Jiang, T.; Kumar, S.; Li, X.; Wali, V.B.; Piscuoglio, S.; Gerstein, M.B.; Chagpar, A.B.; et al. Reliability of Whole-Exome Sequencing for Assessing Intratumor Genetic Heterogeneity. Cell Rep 2018, 25, 1446–1457. [Google Scholar] [CrossRef] [PubMed]
- Lyu, N.; Guan, L.L.; Ma, H.; Wang, X.J.; Wu, B.M.; Shang, F.H.; Wang, D.; Wen, H.; Yu, X. Failure to Identify Somatic Mutations in Monozygotic Twins Discordant for Schizophrenia by Whole Exome Sequencing. Chin Med J (Engl) 2016, 129, 690–695. [Google Scholar] [CrossRef] [PubMed]
- Xiao, W.; Ren, L.; Chen, Z.; Fang, L.T.; Zhao, Y.; Lack, J.; Guan, M.; Zhu, B.; Jaeger, E.; Kerrigan, L.; et al. Toward best practice in cancer mutation detection with whole-genome and whole-exome sequencing. Nat Biotechnol 2021, 39, 1141–1150. [Google Scholar] [CrossRef] [PubMed]
- Barbitoff, Y.A.; Abasov, R.; Tvorogova, V.E.; Glotov, A.S.; Predeus, A.V. Systematic benchmark of state-of-the-art variant calling pipelines identifies major factors affecting accuracy of coding sequence variant discovery. BMC Genomics 2022, 23, 155. [Google Scholar] [CrossRef] [PubMed]
- Belien, J.; Swinnen, S.; D’Hondt, R.; de Juan, L.V.; Dedoncker, N.; Matthys, P.; Bauer, J.; Vens, C.; Moylett, S.; Dubois, B. CHIT1 at diagnosis predicts faster disability progression and reflects early microglial activation in multiple sclerosis. Nat Commun 2024, 15, 5013. [Google Scholar] [CrossRef] [PubMed]
- Dymek, B.; Sklepkiewicz, P.; Mlacki, M.; Guner, N.C.; Nejman-Gryz, P.; Drzewicka, K.; Przysucha, N.; Rymaszewska, A.; Paplinska-Goryca, M.; Zagozdzon, A.; et al. Pharmacological Inhibition of Chitotriosidase (CHIT1) as a Novel Therapeutic Approach for Sarcoidosis. J Inflamm Res 2022, 15, 5621–5634. [Google Scholar] [CrossRef] [PubMed]
- Di Francesco, A.M.; Verrecchia, E.; Sicignano, L.L.; Massaro, M.G.; Antuzzi, D.; Covino, M.; Pasciuto, G.; Richeldi, L.; Manna, R. The Use of Chitotriosidase as a Marker of Active Sarcoidosis and in the Diagnosis of Fever of Unknown Origin (FUO). J Clin Med 2021, 10. [Google Scholar] [CrossRef]
- Mazur, M.; Zielinska, A.; Grzybowski, M.M.; Olczak, J.; Fichna, J. Chitinases and Chitinase-Like Proteins as Therapeutic Targets in Inflammatory Diseases, with a Special Focus on Inflammatory Bowel Diseases. Int J Mol Sci 2021, 22. [Google Scholar] [CrossRef]
- Cho, S.J.; Weiden, M.D.; Lee, C.G. Chitotriosidase in the Pathogenesis of Inflammation, Interstitial Lung Diseases and COPD. Allergy Asthma Immunol Res 2015, 7, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Song, X.Q.; Shao, Z.M. Identification of immune-related prognostic biomarkers in triple-negative breast cancer. Transl Cancer Res 2024, 13, 1707–1720. [Google Scholar] [CrossRef] [PubMed]
- Wiechmann, S.; Ruprecht, B.; Siekmann, T.; Zheng, R.; Frejno, M.; Kunold, E.; Bajaj, T.; Zolg, D.P.; Sieber, S.A.; Gassen, N.C.; et al. Chemical Phosphoproteomics Sheds New Light on the Targets and Modes of Action of AKT Inhibitors. ACS Chem Biol 2021, 16, 631–641. [Google Scholar] [CrossRef] [PubMed]
- Escudero-Hernandez, C.; van Beelen Granlund, A.; Bruland, T.; Sandvik, A.K.; Koch, S.; Ostvik, A.E.; Munch, A. Transcriptomic Profiling of Collagenous Colitis Identifies Hallmarks of Nondestructive Inflammatory Bowel Disease. Cell Mol Gastroenterol Hepatol 2021, 12, 665–687. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Li, B.; Liu, L.; Sun, S.; Sun, S. Centrosome dysfunction: a link between senescence and tumor immunity. Signal Transduct Target Ther 2020, 5, 107. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Lee, J.W.; Yoo, K.D.; Yoo, J.Y.; Lee, J.P.; Kim, D.K.; Chin, H.J.; Kim, Y.S.; Yang, S.H. Cln 3-requiring 9 is a negative regulator of Th17 pathway-driven inflammation in anti-glomerular basement membrane glomerulonephritis. Am J Physiol Renal Physiol 2016, 311, F505–519. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.W.; Bae, E.; Kwon, S.H.; Yu, M.Y.; Cha, R.H.; Lee, H.; Kim, D.K.; Lee, J.P.; Ye, S.K.; Yoo, J.Y.; et al. Transcriptional modulation of the T helper 17/interleukin 17 axis ameliorates renal ischemia-reperfusion injury. Nephrol Dial Transplant 2019, 34, 1481–1498. [Google Scholar] [CrossRef]
- Vinayagam, R.; Cox, J.; Webb, L. Granulomatous Mastitis: A Spectrum of Disease. Breast Care (Basel) 2009, 4, 251–254. [Google Scholar] [CrossRef] [PubMed]
- Corominas, J.; Smeekens, S.P.; Nelen, M.R.; Yntema, H.G.; Kamsteeg, E.J.; Pfundt, R.; Gilissen, C. Clinical exome sequencing-Mistakes and caveats. Hum Mutat 2022, 43, 1041–1055. [Google Scholar] [CrossRef]
- Duan, J.; Liu, H.; Zhao, L.; Yuan, X.; Wang, Y.P.; Wan, M. Detection of False-Positive Deletions from the Database of Genomic Variants. Biomed Res Int 2019, 2019, 8420547. [Google Scholar] [CrossRef]
- Arteche-Lopez, A.; Avila-Fernandez, A.; Romero, R.; Riveiro-Alvarez, R.; Lopez-Martinez, M.A.; Gimenez-Pardo, A.; Velez-Monsalve, C.; Gallego-Merlo, J.; Garcia-Vara, I.; Almoguera, B.; et al. Sanger sequencing is no longer always necessary based on a single-center validation of 1109 NGS variants in 825 clinical exomes. Sci Rep 2021, 11, 5697. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.S.; Huang, H.D.; Yeh, K.T.; Chang, J.G. Evaluation of whole exome sequencing by targeted gene sequencing and Sanger sequencing. Clin Chim Acta 2017, 471, 222–232. [Google Scholar] [CrossRef] [PubMed]
- Robinson, P.N. Whole-exome sequencing for finding de novo mutations in sporadic mental retardation. Genome Biol 2010, 11, 144. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.; Lian, T.; Lin, J.; Li, S.; Zheng, H.; Cheng, C.; Ye, J.; Jing, Z.; Wang, X.; Huang, W. Whole-exome sequencing improves genetic testing accuracy in pulmonary artery hypertension. Pulm Circ 2018, 8, 2045894018763682. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.H.; Chen, S.X.; Cheng, L.Y.; Rodriguez, A.Y.; Tang, R.; Cabrera, K.; Zhang, D.Y. Confirming putative variants at </= 5% allele frequency using allele enrichment and Sanger sequencing. Sci Rep 2021, 11, 11640. [Google Scholar] [CrossRef]
- Hamilton, A.; Tetreault, M.; Dyment, D.A.; Zou, R.; Kernohan, K.; Geraghty, M.T.; Consortium, F.C.; Care4Rare Canada, C.; Hartley, T.; Boycott, K.M. Concordance between whole-exome sequencing and clinical Sanger sequencing: implications for patient care. Mol Genet Genomic Med 2016, 4, 504–512. [Google Scholar] [CrossRef]
- Tattini, L.; D’Aurizio, R.; Magi, A. Detection of Genomic Structural Variants from Next-Generation Sequencing Data. Front Bioeng Biotechnol 2015, 3, 92. [Google Scholar] [CrossRef]
- Mahmodlou, R.; Dadkhah, N.; Abbasi, F.; Nasiri, J.; Valizadeh, R. Idiopathic granulomatous mastitis: dilemmas in diagnosis and treatment. Electron Physician 2017, 9, 5375–5379. [Google Scholar] [CrossRef]
- Pourhoseingholi, M.A.; Vahedi, M.; Rahimzadeh, M. Sample size calculation in medical studies. Gastroenterol Hepatol Bed Bench 2013, 6, 14–17. [Google Scholar]
- Maione, C.; Palumbo, V.D.; Maffongelli, A.; Damiano, G.; Buscemi, S.; Spinelli, G.; Fazzotta, S.; Gulotta, E.; Buscemi, G.; Lo Monte, A.I. Diagnostic techniques and multidisciplinary approach in idiopathic granulomatous mastitis: a revision of the literature. Acta Biomed 2019, 90, 11–15. [Google Scholar] [CrossRef]
- Hanssen, F.; Garcia, M.U.; Folkersen, L.; Pedersen, A.S.; Lescai, F.; Jodoin, S.; Miller, E.; Seybold, M.; Wacker, O.; Smith, N.; et al. Scalable and efficient DNA sequencing analysis on different compute infrastructures aiding variant discovery. NAR Genom Bioinform 2024, 6, lqae031. [Google Scholar] [CrossRef] [PubMed]
- Garcia, M.; Juhos, S.; Larsson, M.; Olason, P.I.; Martin, M.; Eisfeldt, J.; DiLorenzo, S.; Sandgren, J.; Diaz De Stahl, T.; Ewels, P.; et al. Sarek: A portable workflow for whole-genome sequencing analysis of germline and somatic variants. F1000Res 2020, 9, 63. [Google Scholar] [CrossRef]
- Ewels, P.A.; Peltzer, A.; Fillinger, S.; Patel, H.; Alneberg, J.; Wilm, A.; Garcia, M.U.; Di Tommaso, P.; Nahnsen, S. The nf-core framework for community-curated bioinformatics pipelines. Nat Biotechnol 2020, 38, 276–278. [Google Scholar] [CrossRef]
- Di Tommaso, P.; Chatzou, M.; Floden, E.W.; Barja, P.P.; Palumbo, E.; Notredame, C. Nextflow enables reproducible computational workflows. Nat Biotechnol 2017, 35, 316–319. [Google Scholar] [CrossRef]
- Kurtzer, G.M.; Sochat, V.; Bauer, M.W. Singularity: Scientific containers for mobility of compute. PLoS One 2017, 12, e0177459. [Google Scholar] [CrossRef] [PubMed]
- Maxime U Garcia, F.H., Anders Sune Pedersen, Gisela Gabernet, WackerO, SusiJo, et al. nf-core/sarek: Sarek 3.3.0 - Rapaselet. Zenodo 2023. [CrossRef]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv: Genomics (q-bio.GN). [CrossRef]
- DePristo, M.A.; Banks, E.; Poplin, R.; Garimella, K.V.; Maguire, J.R.; Hartl, C.; Philippakis, A.A.; del Angel, G.; Rivas, M.A.; Hanna, M.; et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 2011, 43, 491–498. [Google Scholar] [CrossRef] [PubMed]
- Van der Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; Del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr Protoc Bioinformatics 2013, 43, 11 10 11–11 10 33. [Google Scholar] [CrossRef]
- Sondka, Z.; Bamford, S.; Cole, C.G.; Ward, S.A.; Dunham, I.; Forbes, S.A. The COSMIC Cancer Gene Census: describing genetic dysfunction across all human cancers. Nat Rev Cancer 2018, 18, 696–705. [Google Scholar] [CrossRef]
- Landrum, M.J.; Lee, J.M.; Riley, G.R.; Jang, W.; Rubinstein, W.S.; Church, D.M.; Maglott, D.R. ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res 2014, 42, D980–985. [Google Scholar] [CrossRef]
- Harrison, S.M.; Riggs, E.R.; Maglott, D.R.; Lee, J.M.; Azzariti, D.R.; Niehaus, A.; Ramos, E.M.; Martin, C.L.; Landrum, M.J.; Rehm, H.L. Using ClinVar as a Resource to Support Variant Interpretation. Curr Protoc Hum Genet 2016, 89, 8 16 11–8 16 23. [Google Scholar] [CrossRef]
- Sherry, S.T.; Ward, M.; Sirotkin, K. dbSNP-database for single nucleotide polymorphisms and other classes of minor genetic variation. Genome Res 1999, 9, 677–679. [Google Scholar] [CrossRef] [PubMed]
- Wood, R.D.; Mitchell, M.; Lindahl, T. Human DNA repair genes, 2005. Mutat Res 2005, 577, 275–283. [Google Scholar] [CrossRef] [PubMed]
- Sijmons, R.H.; Burger, G.T. Familial cancer database: a clinical aide-memoire. Fam Cancer 2001, 1, 51–55. [Google Scholar] [CrossRef] [PubMed]
- Sijmons, R.H.; Burger, G.T. The Use of a Diagnostic Database in Clinical Oncogenetics. Hered Cancer Clin Pract. 2003, 1, 31–33. [Google Scholar] [CrossRef]
- Harrow, J.; Frankish, A.; Gonzalez, J.M.; Tapanari, E.; Diekhans, M.; Kokocinski, F.; Aken, B.L.; Barrell, D.; Zadissa, A.; Searle, S.; et al. GENCODE: the reference human genome annotation for The ENCODE Project. Genome Res 2012, 22, 1760–1774. [Google Scholar] [CrossRef] [PubMed]
- Gudmundsson, S.; Singer-Berk, M.; Watts, N.A.; Phu, W.; Goodrich, J.K.; Solomonson, M.; Genome Aggregation Database, C.; Rehm, H.L.; MacArthur, D.G.; O’Donnell-Luria, A. Variant interpretation using population databases: Lessons from gnomAD. Hum Mutat 2022, 43, 1012–1030. [Google Scholar] [CrossRef] [PubMed]
- Seal, R.L.; Braschi, B.; Gray, K.; Jones, T.E.M.; Tweedie, S.; Haim-Vilmovsky, L.; Bruford, E.A. Genenames.org: the HGNC resources in 2023. Nucleic Acids Res 2023, 51, D1003–D1009. [Google Scholar] [CrossRef] [PubMed]
- Thorvaldsdottir, H.; Robinson, J.T.; Mesirov, J.P. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform 2013, 14, 178–192. [Google Scholar] [CrossRef] [PubMed]
- Untergasser, A.; Cutcutache, I.; Koressaar, T.; Ye, J.; Faircloth, B.C.; Remm, M.; Rozen, S.G. Primer3--new capabilities and interfaces. Nucleic Acids Res 2012, 40, e115. [Google Scholar] [CrossRef]
- Koressaar, T.; Remm, M. Enhancements and modifications of primer design program Primer3. Bioinformatics 2007, 23, 1289–1291. [Google Scholar] [CrossRef]
- Koressaar, T.; Lepamets, M.; Kaplinski, L.; Raime, K.; Andreson, R.; Remm, M. Primer3_masker: integrating masking of template sequence with primer design software. Bioinformatics 2018, 34, 1937–1938. [Google Scholar] [CrossRef]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res 2016, 44, W90–97. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Bailey, A.; Kuleshov, M.V.; Clarke, D.J.B.; Evangelista, J.E.; Jenkins, S.L.; Lachmann, A.; Wojciechowicz, M.L.; Kropiwnicki, E.; Jagodnik, K.M.; et al. Gene Set Knowledge Discovery with Enrichr. Curr Protoc 2021, 1, e90. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M. Toward understanding the origin and evolution of cellular organisms. Protein Sci 2019, 28, 1947–1951. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Sato, Y.; Kawashima, M.; Ishiguro-Watanabe, M. KEGG for taxonomy-based analysis of pathways and genomes. Nucleic Acids Res 2023, 51, D587–D592. [Google Scholar] [CrossRef]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer-Verlag New York: 2016.
Figure 1.
Single nucleotide variants (SNVs) and insertion and deletion variants (indels) called by Strelka2 and Mutect2, from (a) matched blood-tissue samples; (b) blood samples.
Figure 1.
Single nucleotide variants (SNVs) and insertion and deletion variants (indels) called by Strelka2 and Mutect2, from (a) matched blood-tissue samples; (b) blood samples.
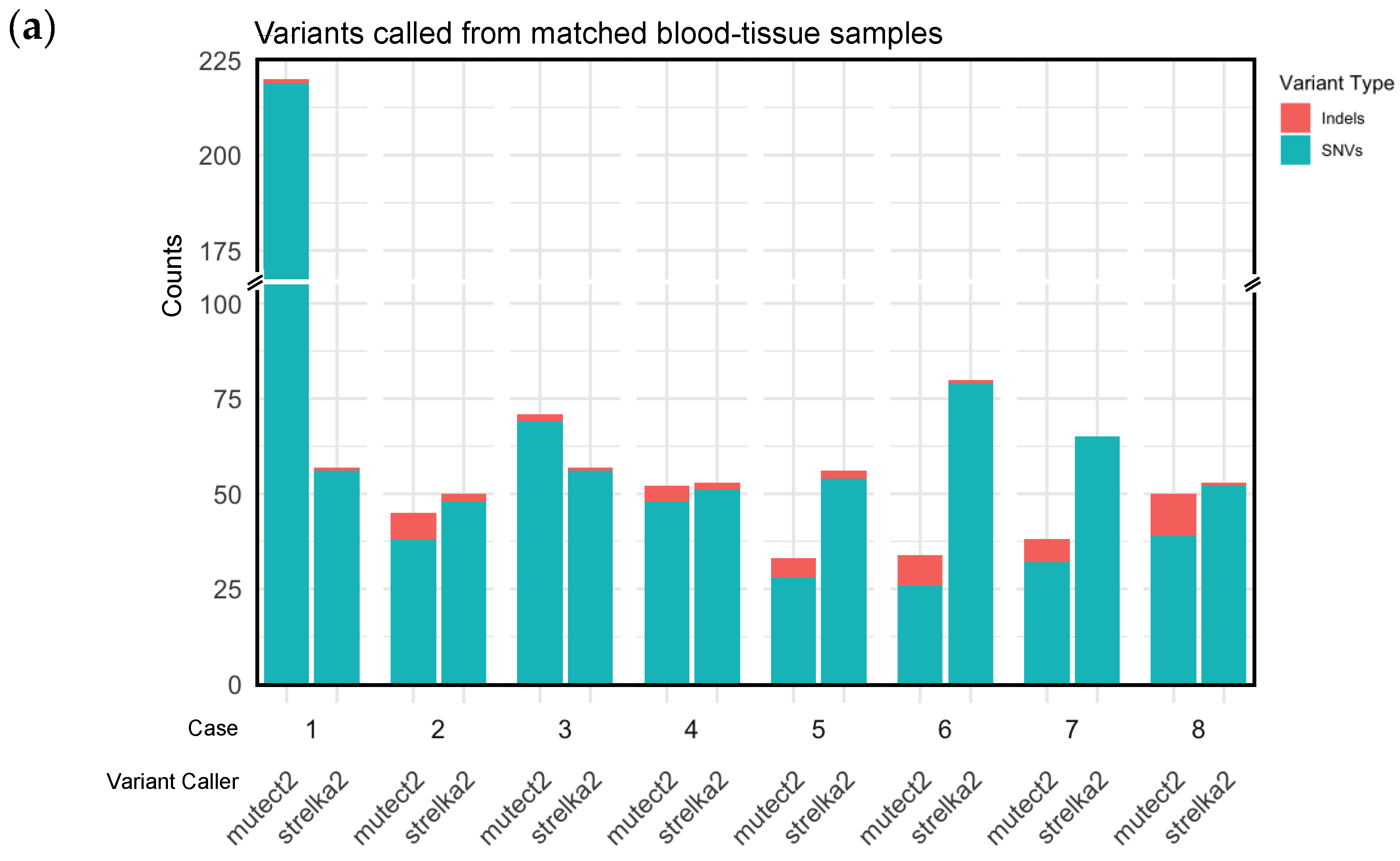
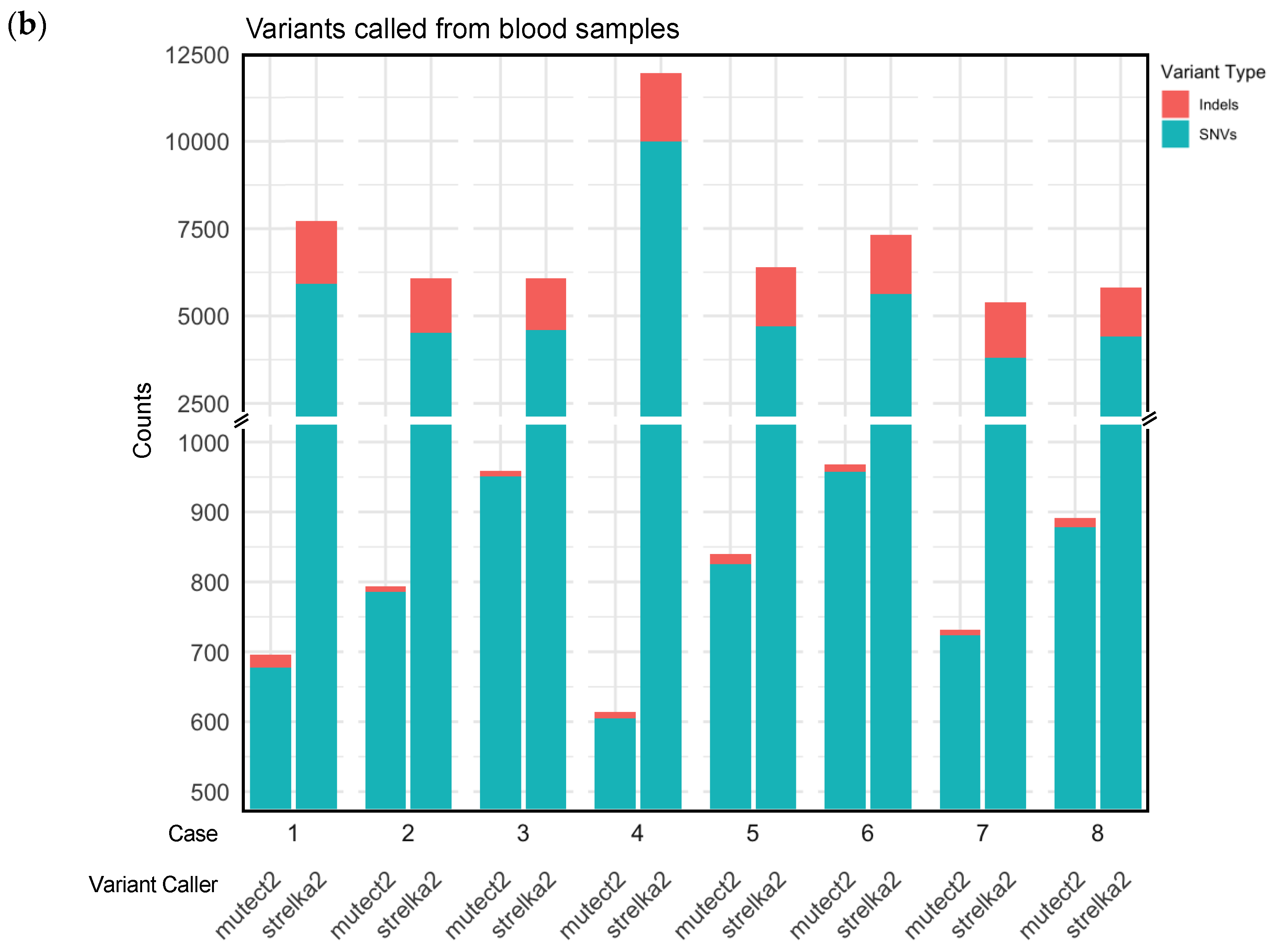
Figure 2.
Mutations annotated by Funcotator from variants called with Strelka2 and Mutect2 matched blood-tissue samples. (a) All non-sysnonymous mutations annotated from Strelka2 and Mutect2 variant calls; (b) Protein-truncating variants (nonsense mutations, and frame-shift insertions and deletions) annotated from Strelka2 and Mutect 2 variant calls.
Figure 2.
Mutations annotated by Funcotator from variants called with Strelka2 and Mutect2 matched blood-tissue samples. (a) All non-sysnonymous mutations annotated from Strelka2 and Mutect2 variant calls; (b) Protein-truncating variants (nonsense mutations, and frame-shift insertions and deletions) annotated from Strelka2 and Mutect 2 variant calls.
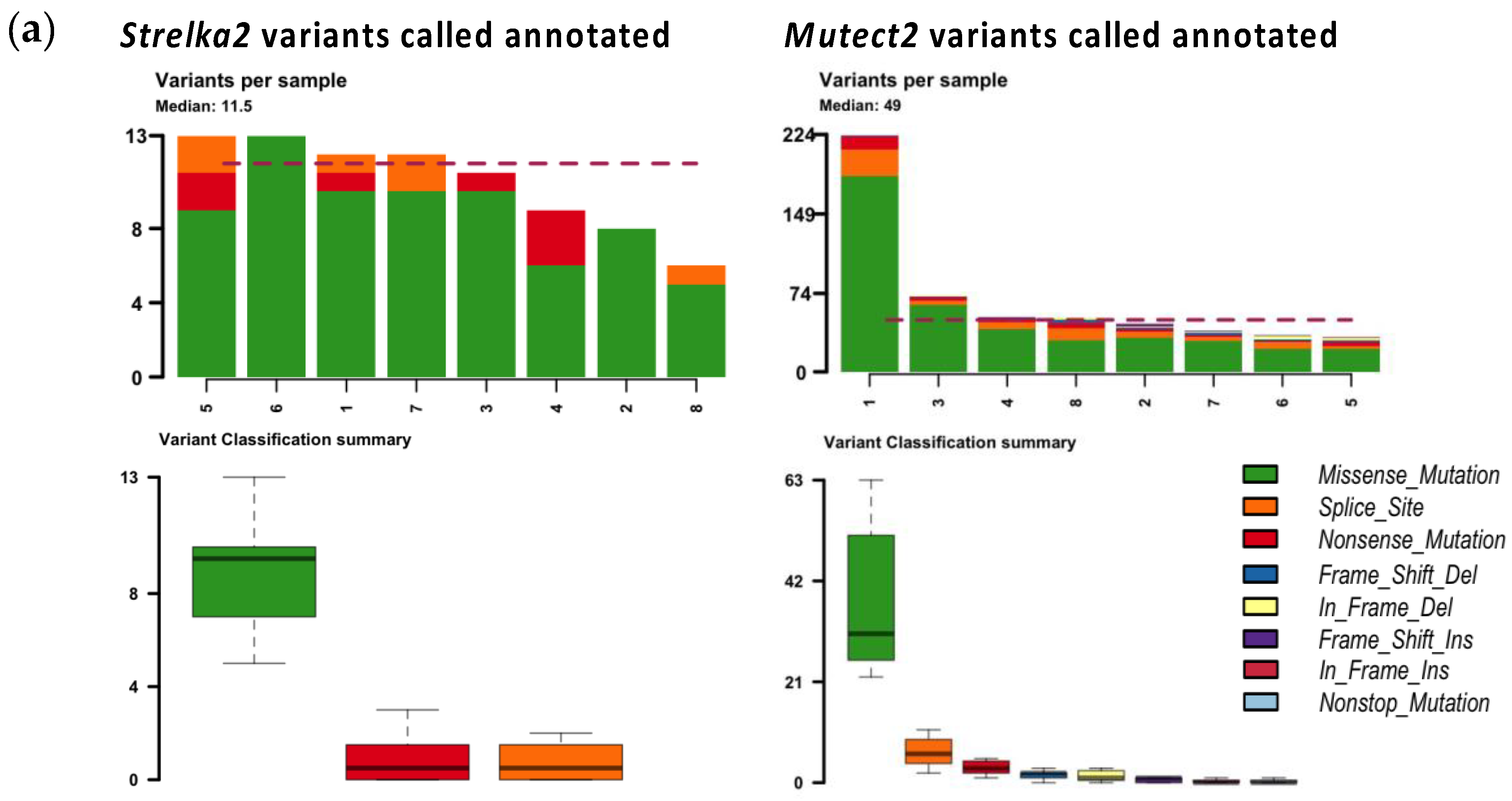
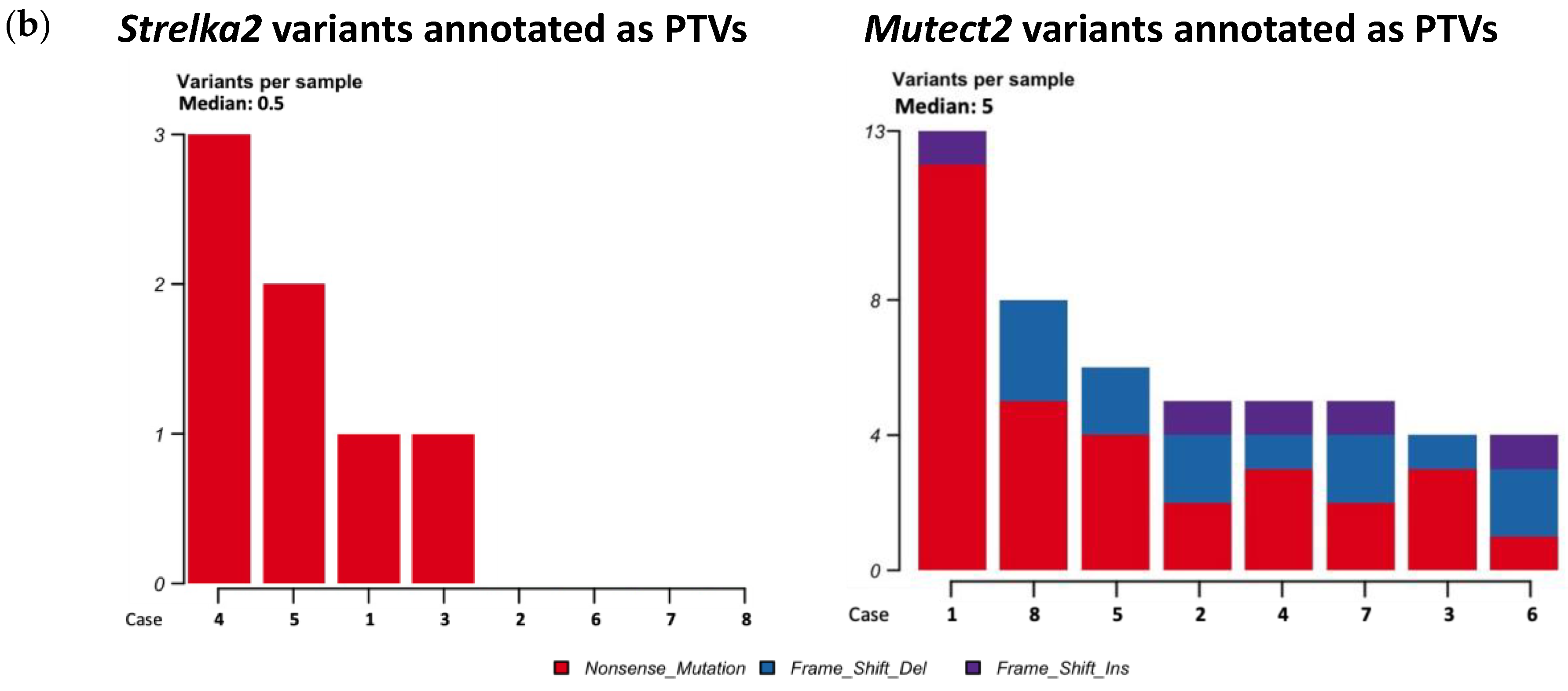
Figure 3.
Altered genes in PTVs from Strelka2 and Mutect2 matched blood-tissue variant calling.
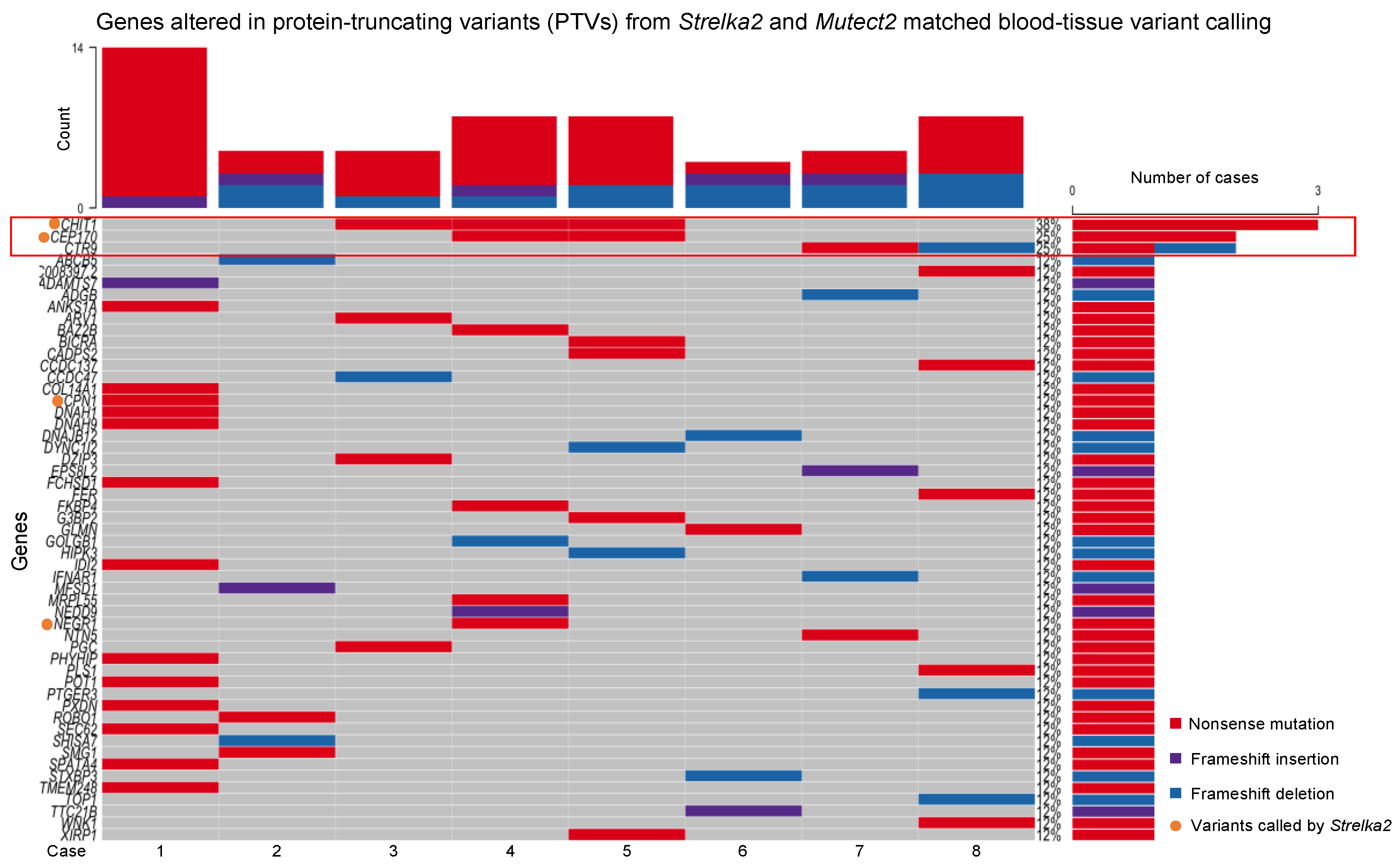
Figure 4.
Patient recruitment, inclusion, and exclusion flowchart.
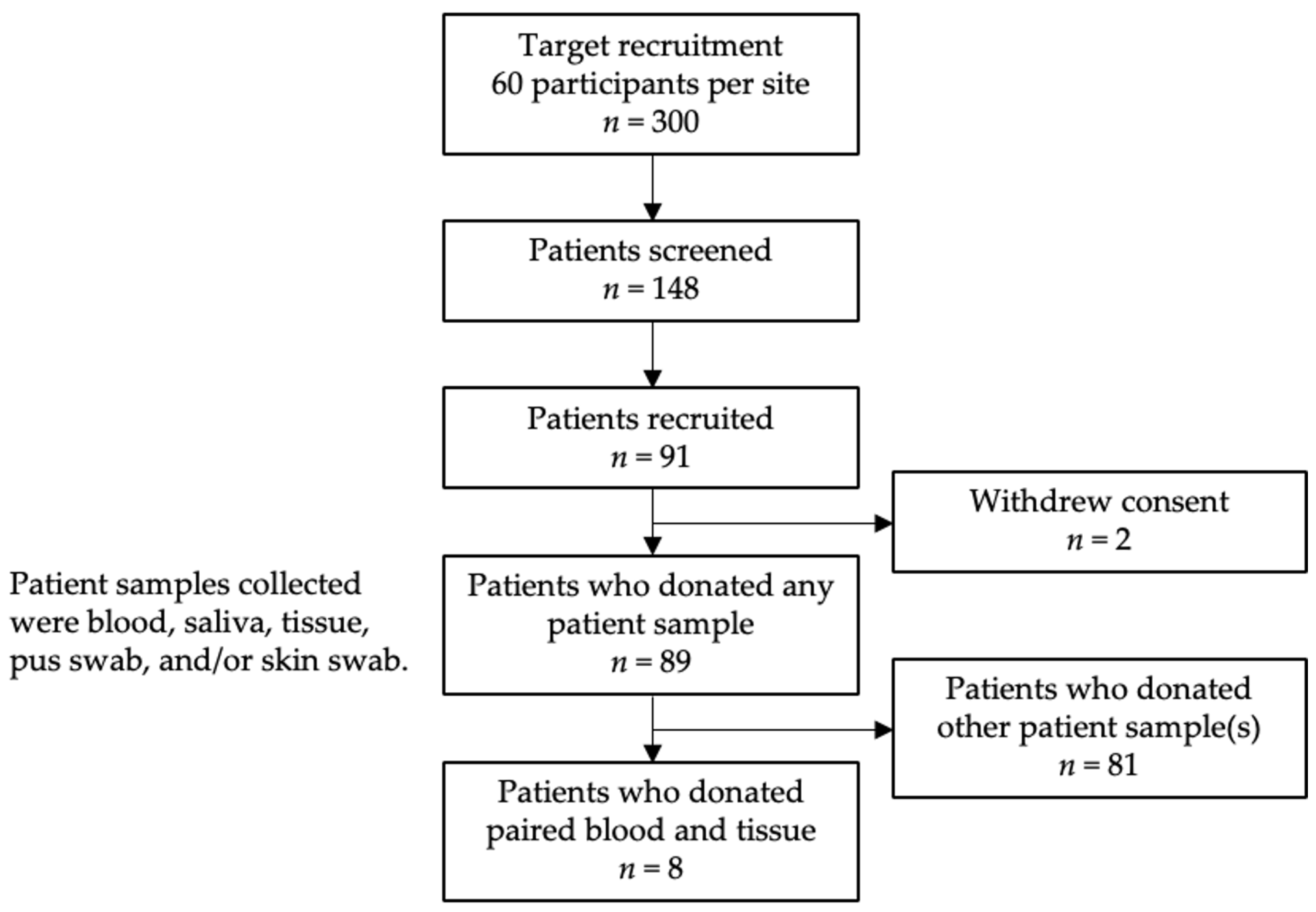
Table 1.
Description of demographic and clinical parameters of idiopathic granulomatous mastitis (IGM) patients.
Table 1.
Description of demographic and clinical parameters of idiopathic granulomatous mastitis (IGM) patients.
| Demographic and clinical parameters | IGM n = 8 |
|---|---|
| Demographics | |
| Median age at diagnosis (years, IQR) | 33.0 (27.3-34.5) |
| Ethnicity (n, %) | |
| Chinese | 4 (50) |
| Malay | 3 (38) |
| Others | 1 (12) |
| Body mass index (kg/m2, IQR) | 27.770 (23.460-33.454) |
| Education level (n, %) | |
| Up to secondary school | 3 (38) |
| Secondary school to pre-university | 3 (38) |
| Tertiary education | 2 (25) |
| Patient characteristics | |
| Parity (n, %) | |
| Yes | 6 (75) |
| No | 2 (25) |
| Number of children (n, %) | |
| No children | 2 (25) |
| 1-2 children | 5 (62) |
| More than 2 children | 1 (12) |
| Smoking (n, %) | |
| Yes | 2 (25) |
| No | 6 (75) |
| Chronic illness1 diagnosis (n, %) | |
| Yes | 2 (25) |
| No | 6 (75) |
| Family history2 of breast cancer (n, %) | |
| Yes | 1 (12) |
| No | 7 (88) |
1 Chronic illness: Heart attack, stroke, or high blood pressure. 2 First degree family history: Breast cancer in a parent, sibling, or child.
Table 3.
Somatic variants identified from WES of paired blood/tissue samples that overlap in Strelka2 and Mutect2 variant calling.
Table 3.
Somatic variants identified from WES of paired blood/tissue samples that overlap in Strelka2 and Mutect2 variant calling.
| Case | Somatic variants | SNVs1 | Indels2 | PTVs3 | Pathogenic4 | Pathogenic / Likely Pathogenic4 | Likely Pathogenic4 |
|---|---|---|---|---|---|---|---|
| 1 | 3 | 3 | 0 | 0 | 0 | 0 | 0 |
| 2 | 2 | 2 | 0 | 0 | 0 | 0 | 0 |
| 3 | 1 | 1 | 0 | 0 | 0 | 0 | 0 |
| 4 | 1 | 1 | 0 | 0 | 0 | 0 | 0 |
| 5 | 2 | 2 | 0 | 0 | 0 | 0 | 0 |
| 6 | 2 | 2 | 0 | 0 | 0 | 0 | 0 |
| 7 | 3 | 3 | 0 | 0 | 0 | 0 | 0 |
| 8 | 2 | 2 | 0 | 0 | 0 | 0 | 0 |
| Median (range) | 2 (1-3) | 2 (1-3) | 0 (0-0) | 0 (0-0) | 0 (0-0) | 0 (0-0) | 0 (0-0) |
1 Single nucleotide variants. 2 Insertions and deletions. 3 Protein-truncating variants. These correspond to variants annotated as nonsense mutations, or frameshift insertions or deletions by GATK4 Funcotator. 4 ClinVar annotation of pathogenicity within GATK4 Funcotator variant annotation.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



