
Submitted:
31 July 2024
Posted:
01 August 2024
You are already at the latest version
Abstract
Dent disease type 1 is a rare inherited renal disorder affecting mainly young males, generally leading to end-stage renal failure and for which there is no cure. It is caused by inactivating mutations in the gene encoding ClC-5, a 2Cl-/H+ exchanger found on endosomes in the renal proximal tubule. This transporter participates in reabsorbing all filtered plasma proteins which justifies why proteinuria is commonly observed when ClC-5 is defective. In the context of Dent disease type 1, a proximal tubule dedifferentiation was shown to be accompanied by a dysfunctional cell metabolism. However, the exact mechanisms linking such alterations to chronic kidney disease are still unclear. In this review, we gather knowledge from several Dent disease type 1 models to summarize the current hypotheses generated to understand the progression of this disorder. We also highlight some urinary biomarkers for Dent disease type 1 suggested in different studies.
Keywords:
Dent disease type 1
; proximal tubule
; metabolism
; chronic kidney disease
1. Introduction
The proximal tubule is the most metabolically active segment of the nephron. It reabsorbs 100% of the filtered load of glucose and a significant part of other solutes such as sodium and calcium which originate from the Bowman’s space. This explains why proximal tubule cells contain a high number of mitochondria that provide them with ATP, an energy source that is required for active solute transepithelial reabsorption [1,2]. Another major function of the proximal tubule is the reabsorption of low-molecular-weight proteins via receptor-mediated endocytosis. When dysfunctional, this may lead to growth retardation due to the urinary loss of vitamin-bound proteins [3]. Such abnormality has been observed in patients with Dent disease type 1, a hereditary renal disorder characterized by the urinary loss of low molecular weight proteins, calcium, and other solutes such as glucose or phosphate, usually associated with a proximal tubule dysfunction. Diagnosed during childhood and being progressive, the disease leads to end-stage renal failure, therefore requiring dialysis or kidney transplantation [3,4]. Dent disease type 1 is characterized by inactivating mutations of the CLCN5 gene encoding the 2Cl-/H+ exchanger and is observed in about 65% of patients with Dent disease. Up to now, there is no specific treatment for Dent disease, therefore, the current therapy is aimed at slowing down the progression of some clinical traits (e.g. nephrocalcinosis) [3,5,6,7]. Numerous in vitro and in vivo models have been generated to decipher the cellular and molecular mechanisms underlying Dent disease type 1. They include ClC-5 knock-out (KO) [8,9,10,11], knock-in (KI) mice harboring the non-pathogenic E211G mutation [12] and the pathogenic N340K mutation [13], knock-down of the ClC-5 homolog in D. melanogaster [14], cells carrying ClC-5 mutants derived from DD type 1 patients (ciPTECs) [15,16], X. laevis oocytes and different stably or transiently transfected cell lines [11,17,18,19,20,21,22,23,24].
Although some of the studies performed using these different models of Dent disease type 1 revealed progressive proximal tubule dysfunction with parallel impairment of cell metabolism, the precise role of ClC-5 in these alterations remains yet to be understood. In this review, we highlight past and newly generated hypotheses to explain the pathogenesis of Dent disease type 1 – from the onset to the progression to chronic kidney disease – that have emerged from the different experimental models.
2. A Dysfunctional ClC-5 Leads to Proximal Tubule Dedifferentiation
Previous studies in the context of Dent disease type 1 allowed to explore the role of ClC-5 in maintaining an optimal endocytosis of proteins in proximal tubule cells. Indeed, when this transporter is dysfunctional, proteinuria appears as a consequence of reduced protein uptake in this segment of the nephron [4,8,9,25]. Due to its colocalization with the vacuolar H+ ATPase, described as the endo-lysosomal acidifying pump, it has been for a long time suspected of creating an electrical shunt for the pump to keep working [25,26]. Hence, the accumulation of Cl- in early endosomes would potentiate endosomal acidification that seems necessary for several processes, from receptor-ligand dissociation to creating an optimal environment for protein digestion in lysosomes [25,27,28,29]. Over the years, ClC-5 physical interaction with cytoskeleton proteins playing a crucial role in endocytosis mechanisms was also highlighted, suggesting that this transporter might participate in endocytosis independently from its electrical activity [30,31]. However, some pathogenic mutants of ClC-5 were described as correctly expressed but bearing impaired electrical activity [19,20,21,22,32]; while patients carrying these mutants still presented proteinuria, we can imagine the proteins they interact with will not show altered localization. Among these mutants, the artificial mutation of ClC-5 E211G converting the transporter in a pure Cl- conductance was induced in a mouse model [12,25]. These mice showed a typical Dent disease phenotype with impaired receptor-mediated endocytosis but normal endosomal acidification, revealing, for the first time in vivo, that endocytosis can be inefficient even with normal endo-lysosomal pH evolution [12,21,25,33]. More recently, endosomal pH and Cl- concentration were measured in a highly differentiated proximal tubule cell line expressing a similar ClC-5 mutant and showed an impairment of both parameters [11]. Therefore, ClC-5’s exact contribution to maintaining an efficient endocytosis is still an unresolved matter and a combination of all previously mentioned roles of this transporter could be important for optimal protein uptake.
As previous studies aiming at characterizing the real impact of ClC-5 dysfunction on receptor-mediated endocytosis relied solely on end-time endocytosis assays [8,9,12,13], the Hall team recently explored this phenomenon in ClC-5 KO mice using intravital microscopy [34]. These experiments revealed that endocytosis of filtered proteins is not abolished in these animals but rather slowed down as endo-lysosome trafficking seems to be impaired [34]. Moreover, past data from Dent disease patients and ClC-5 KO animals further validate that the endo-lysosomal apparatus does not seem to be altered and multi-ligand receptors are not down-regulated in proximal tubules but instead less recycled back at the plasma membrane [11,35,36]. In the recent pathogenic N340K mutant model, protein endocytosis was shown to be extended to later proximal tubule segments indicating a nephron plasticity in trying to optimize cell function in disease states [13]. Besides, lysosomes – the final compartments of the endo-lysosomal system but also essential organelles for optimal autophagy within the cell – displayed overall altered distribution in proximal tubules [13]. Lysosomal hydrolases responsible for protein degradation were also shown to be abnormally distributed and expressed in mouse models [13,37]. Many cathepsins need further processing when traveling along the endo-lysosomal pathway and a final activating cleavage occurs in optimally acidified lysosomes [38,39,40,41].
In addition to these observations, other major transporters necessary for the reabsorption of electrolytes and metabolites were shown to be less available at the apical plasma membrane; hence, justifying the loss of molecules typically observed when proximal tubule dysfunction occurs [3,8,9,12,13]. An epithelial-to-mesenchymal transition was also observed from transcriptomic data and tissue immunostainings supporting even more the proximal tubule dedifferentiation taking place in the context of Dent disease type 1 [13]. As a consequence of this phenomenon, proximal tubule cell apoptosis and replacement proliferation were observed and accompanied by fibrosis and inflammation ultimately leading to chronic renal failure in aging animals and some patients [13,42,43]. In mice presenting a clear proximal tubule dedifferentiation and proliferation, oxidative stress was observed which suggests that proximal tubule metabolism is also impacted during Dent disease type 1 [13,42]. Interestingly, even in models with a mutant ClC-5 retained within the endoplasmic reticulum, no unfolded protein response could be detected suggesting that this other cell stress does not contribute to the observed oxidative stress [13,16].
3. An Alteration of Proximal Tubule Cell Metabolism Is Observed in the Context of Dent Disease Type 1
Cell mitochondrial abundance depends on its energetic needs and as previously mentioned, the proximal tubule requires enough energy production to maintain its massive absorptive functions [44]. In addition to ATP production, mitochondria maintain intracellular calcium concentrations, redox homeostasis, and participate in regulating cell apoptosis [45,46,47]. Thus, in several pathological contexts affecting the proximal tubule, mitochondrial dysfunctions have been observed [48,49,50]. Regarding Dent disease type 1, the pathogenic N340K mouse model shows for the first time clear signs of mitochondrial metabolism alterations [13]. Indeed, a decreased expression of genes involved in oxidative phosphorylation and fatty acid metabolism as well as a significant urinary loss and altered renal cortex handling of mitochondrial metabolites were highlighted in this model [13]. We are currently exploring if similar phenomena are occurring in Dent patients by analyzing their urine content. Furthermore, the Clcn5 KO model studied by Wright et al. displayed altered gene expression related to proximal tubule metabolism and carbohydrate homeostasis, that could impact gluconeogenesis [51,52]. Thus, other metabolic pathways should be further explored. Indeed, a possible way for cells to show changes in the functioning of their ATP-producing TCA cycle could be by altering gluconeogenesis which is responsible for regulating the cataplerosis process [52,53]. Apart from the common glucosuria observed in Dent disease patients and animal models, no alteration in glycemia was communicated but this phenomenon could be adjusted at the systemic level. Indeed, the pathogenic N340K mouse model showed a clear reduction in their fat mass suggesting a decreased energy storage or an increased need for energy release [13].
Mitochondrial dysfunction may compromise the amounts of ATP generated and contribute to increased oxidative stress via the mitochondrial respiratory chain [54]. This would, in turn, cause damage to cellular components such as proteins, lipids, and DNA, promote mitochondrial fragmentation and activate pro-inflammatory pathways [55]. In cells and animal models for Dent disease type 1, increased antioxidant defenses were observed, with an upregulation of SOD1 and Thioredoxin encoding genes, activation of ROS metabolic processes, and in some cases even a mild increase in protein carboxylation [13,16,42,51,56]. The role of albumin in controlling oxidative stress was also suggested. Indeed, this protein could show a protective role in proximal tubules by binding to ROS. In Dent disease type 1, a decreased endocytosis of albumin could compromise this function and participate in the observed oxidative stress [57,58].
In addition to the potentially altered functioning of proximal tubule mitochondria, lipid metabolism was also shown to be impacted in the context of Dent disease type 1 [13,51]. Fatty acid β-oxidation is the main pathway used to produce energy in proximal tubule mitochondria. When β-oxidation is reduced and the cell's capacity to oxidize fatty acids is exceeded, these molecules are stored in the form of lipid droplets, which can – in excess – cause lipotoxicity and eventually impact on renal function [59]. The accumulation of these lipid droplets has been observed in models developing chronic kidney disease including in the pathogenic N340K mouse model for Dent disease type 1 and is thought to participate in ROS production, thus promoting fibrosis and inflammation ultimately leading to chronic renal failure [13,52,59,60]. On the other hand, studies in cultured cell models for Dent disease type 1 did not highlight any alteration of lipid metabolism suggesting that these mechanisms occur over a longer time in integrated models [16].
Furthermore, it was shown that plasma membrane lipid and cholesterol composition can modify not only the electrical activity of transporters but also the dynamics of vesicle trafficking [4,61,62]. Hence, as already suggested by colleagues, the altered lipid metabolism observed in animal models for Dent disease type 1 could play a role in reduced endosomal recycling and trafficking [4,13,51]. Up to now, it is difficult to pinpoint a cause for this altered lipid metabolism other than an initial mitochondrial impairment. The latter could be a consequence of a defect in clearing damaged mitochondria because of the endo-lysosomal system inefficiency (altered mitophagy). It could also be caused by a reduced uptake of metabolites necessary for cell metabolism following the decreased apical availability of their transporters (impaired recycling) [13]. Another possibility could be a direct interaction between the endo-lysosomal system and lipid droplets – that would be impaired in the context of Dent disease type 1 – as such a phenomenon was already observed in hepatocytes using high-resolution microscopy [63].
Although the exact mechanisms behind the altered proximal tubule cell metabolism are yet to be unraveled, previous transcriptomic, proteomic, and metabolomic studies allowed to shed light on potential biomarkers for Dent disease type 1 and its evolution to more severe forms.
4. Studies Revealed a Diversity of Potential Biomarkers for the Progression of Dent Disease Type 1
In 2008 the carbonic anhydrase type III (CA III) kidney-specific upregulation was described in the context of Dent disease type 1 (patient and Clcn5 KO mice) [42]. This enzyme catalyzes the production of HCO3- and H+ in skeletal muscles, adipose tissue, and, to a lesser extent, in the liver and the kidney [42,64,65]. While it does not seem to play a crucial role in pH regulation, it was shown to be induced and remains stable under oxidative stress conditions where it acts as a ROS scavenger hence protecting cells against the bad sides of such stress [65,66]. Its encoding gene Car3 was shown to be overexpressed in Dent disease type 1 models inducing a secretion of CA III from dedifferentiating proximal tubules [13,42,51]. Moreover, this protein was found slightly increased in urines from Megalin KO mice, presenting altered endo-lysosomal function and dedifferentiating tubules as well [42]. Hence, this urinary marker could be used as a reflector of the oxidative stress level within proximal tubules and their consequent dysfunction [42].
A-ketoglutarate (AKG) is responsible for the regulation of the TCA cycle and its substrates, it participates in amino acid synthesis, ATP production, the respiratory chain functioning and can modulate oxidative stress [67,68]. It also plays a role in the hydroxylation of proteins, nucleic acids, lipids, and metabolites, as well as cell proliferation and differentiation through calcium-mediated mechanisms [69,70]. In young pathogenic N340K mice, cortical and urinary AKG were increased which could play a significant role in protecting the nephron at this disease stage whereas with time, these effects would not be sufficient to contain the tissular damages observed [13]. The excretion of this metabolite could either reflect the activation of defense mechanisms or be the consequence of an uncontrolled proximal tubule dysfunction. Further studies would be needed to evaluate the potential of using AKG as a urinary biomarker of proximal tubule metabolic alteration in Dent disease type 1.
In the kidney cortex, the hormone serotonin, which is synthesized from the amino acid tryptophan, participates in the regulation of glomerular filtration, vascular resistance, and mitochondrial biogenesis to improve renal function [71,72,73]. However, it was also suggested that high levels of serotonin promote proximal tubule epithelial-to-mesenchymal transition, local inflammation, and tissue damage [72]. In a current analysis of urines from patients with Dent disease, serotonin was generally found in very low amounts (unpublished data). Moreover, young pathogenic N340K mice, not displaying chronic kidney disease, presented reduced serotonin in their urine paralleled by increased urinary tryptophan [13]. As Dent disease type 1 is characterized by altered uptake of amino acids, it could justify the reduced availability of tryptophan to produce serotonin. Hence, this marker does not seem to be playing a role in the proximal tubule dedifferentiation and fibrosis observed. Other metabolites could be more implicated in these phenomena in the context of Dent disease type 1.
In vivo, inosine is generated from the less stable adenosine nucleoside and was shown to significantly increase in body fluids in several pathological contexts [74,75,76,77]. Both molecules can bind to the same receptor A2A-R but would activate different pathways; inosine would stimulate ERK1/2-mediated proliferation and apoptosis of cells whereas adenosine would activate a cAMP pathway with opposite consequences [78,79,80]. Inosine to adenosine ratio could therefore be an important parameter to modulate the immune response and associated tissue damage [78]. In the urines from the pathogenic N340K mouse model, this ratio is increased which could participate in the tubular cell apoptosis, progressive inflammation, and fibrosis observed [13]. Further studies are required to explore this urinary ratio in patients with Dent disorder at various disease states.
In different Dent disease type 1 models, NGAL/Lipocalin 2 was shown to be overexpressed and even excreted [13,51]. In past studies, it was hypothesized that this protein is overexpressed by distal tubules as a result of the increased presence of retinoic acid there. Indeed, the latter would be less taken up by proximal tubules because of the reduced endocytosis of its binding protein RBP. Reaching the more distal tubules, it would freely diffuse through the plasma membrane to stimulate the expression of its target genes including Lcn2 [81,82]. More recently, Sakhi et al. showed a proximal tubule localization of NGAL in young animals that was switched to the distal nephron in older N340K mice presenting chronic kidney disease. Hence, authors hypothesized that proximal tubules express this protein because of the increased local oxidative stress and that the circulating NGAL activates inflammation and fibrosis pathways through its distal receptor 24p3R [13]. Anyway, these increased urinary NGAL levels were observed in different renal diseases and are thought to directly reflect renal function [83]. This biomarker could then be used in the Dent disease type 1 context to evaluate the level of renal damage patients are experiencing.
5. Current State of the Art – Hypotheses on Dent Disease Type 1 Progression
Experimental models designed for Dent disease type 1 study allowed to pinpoint several potential pathogenetic mechanisms involved in this disorder [8,9,10,11,12,13,14,15,16,17,19,20,21,22,23,24]. Some observations are clearly defined: in the proximal tubule, ClC-5 is necessary for the proper functioning of receptor-mediated endocytosis of plasma filtered proteins and when this phenomenon is altered over a long time, proximal tubules dedifferentiate, and progressive tissue damage occurs (Figure 1) [3,8,9,12,17,25,42,51,58]. However, the exact role of ClC-5 in protein uptake remains unclear. As colleagues showed that endocytosis could be disrupted even with normal pH gradients, they hypothesized that an optimal Cl- accumulation in endosomes is essential for endosomal Ca2+ transport that would itself intervene in vesicle trafficking and functioning [12,25,84,85]. New emerging tools coupled with high-resolution microscopy could help answer this question and define whether ClC-5’s contribution to efficient endocytosis lies in its electrical activity and/or its physical interaction with crucial partners.
In several models, renal oxidative stress and metabolic disorders have been observed [13,42,51]. In 2015, based on observations made in other contexts, colleagues hypothesized it could be a consequence of altered autophagy and dedifferentiating proximal tubules [58]. Indeed, excessive ROS production from damaged and non-cleared mitochondria could disrupt cell junction and consequently impact on epithelium integrity [86]. A free zona ocludens 1 would then release its binding protein, the transcription factor ZONAB, that would upregulate (e.g. Cyclin D1) or downregulate (e.g. Megalin) its target genes [87,88]. This would cause cell proliferation and dedifferentiation as observed in some models for Dent disease type 1 [13,42,58]. In the latest mouse model for this disorder, no alteration of cell junctions was observed, and the multi-ligand receptor was not downregulated at the gene level but rather, less recycled back at the plasma membrane and probably rerouted to degradative pathways [13]. In addition, no clear accumulation of damaged mitochondria has been shown so far, and the autophagy defect was also not directly defined. Even though the distribution of lysosomes is abnormal, and some markers of autophagy suggest this pathway is impacted [13], no exploration of this mechanism in dynamic conditions has been done yet (Figure 1). Up to now, we only know that metabolites that are crucial for mitochondrial function are more excreted in the context of Dent disease type 1 [13] (and unpublished data); meanwhile, genes encoding proteins contributing to cell metabolism seem to be downregulated [13,51]. Moreover, lipid metabolism also appears to be impacted showing fatty acids that are less excreted in urines from Dent disease type 1 mice and lipid droplets that accumulate (Figure 1) [4,13,89]. In other contexts, these parameters usually reflect mitochondria dysfunction [59,90].
As highlighted in the biomarker section, several metabolites and proteins are found in increased amounts in kidneys and urines from mice affected with Dent disease type 1. Many of them could reflect the cell oxidative stress status and the progression of fibrosis and inflammation in the kidneys of animals that start to show signs of renal failure [13]. Among them, NGAL/Lipocalin-2 could even play pro-inflammatory and pro-fibrotic roles itself as it could trigger its distal receptor 24p3R while being in excess in the nephron lumen (Figure 1). However, this should be explored further in animals and patient urines. Past studies could not highlight any correlation between genotype and phenotype as patients carrying the same mutation would present with different clinical traits [3]. It was suggested that environmental factors could heavily contribute to this clinical variability [82]. Besides, one way to predict the actual severity of one patient’s phenotype could be to use these non-invasive biomarkers to estimate the level of tissue damage they are experiencing. Indeed, knowing that urinary NGAL could efficiently reflect a patient’s GFR and that it starts to be over- excreted even in animals not presenting CKD yet, measuring this marker could allow us to estimate how close one is to developing end-stage renal disease [13,91].
In conclusion, as several new techniques and models are emerging within the field, some of the uncertainties on Dent disease type 1 mechanisms could become clearer in the following years. Therefore, ways of slowing disease progression could emanate from this research. Involving exploration of patient urines on a larger cohort would also validate the relevance of using non-invasive biomarkers to predict patient’s renal outcome.
Author Contributions
EDC, IBS and SL; writing—original draft preparation, EDC, IBS and SL; writing—review and editing. All authors have read and agreed to the published version of the manuscript.
Acknowledgments
Figure 1 was created with BioRender.com.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Chrysopoulou, M.; Rinschen, M.M. Metabolic Rewiring and Communication: An Integrative View of Kidney Proximal Tubule Function. Annu. Rev. Physiol. 2024, 86, 405–427. [Google Scholar] [CrossRef]
- Miguel, V.; Kramann, R. Metabolic reprogramming heterogeneity in chronic kidney disease. FEBS Open Bio 2023, 13, 1154–1163. [Google Scholar] [CrossRef]
- Mansour-Hendili, L.; Blanchard, A.; Le Pottier, N.; Roncelin, I.; Lourdel, S.; Treard, C.; González, W.; Vergara-Jaque, A.; Morin, G.; Colin, E.; et al. Mutation Update of theCLCN5Gene Responsible for Dent Disease 1. Hum. Mutat. 2015, 36, 743–752. [Google Scholar] [CrossRef]
- E Shipman, K.; A Weisz, O. Making a Dent in Dent Disease. Function 2020, 1. [Google Scholar] [CrossRef]
- Devuyst O, Thakker RV. Dent’s disease. Orphanet J Rare Dis. 2010 Oct 14;5:28.
- Ehlayel, A.M.; Copelovitch, L. Update on Dent Disease. Pediatr. Clin. North Am. 2018, 66, 169–178. [Google Scholar] [CrossRef]
- Arnous, M.G.; Arroyo, J.; Cogal, A.G.; Anglani, F.; Kang, H.G.; Sas, D.; Harris, P.C.; Lieske, J.C. The Site and Type of CLCN5 Genetic Variation Impact the Resulting Dent Disease-1 Phenotype. Kidney Int. Rep. 2023, 8, 1220–1230. [Google Scholar] [CrossRef]
- Piwon, N.; Günther, W.; Schwake, M.; Bösl, M.R.; Jentsch, T.J. ClC-5 Cl--channel disruption impairs endocytosis in a mouse model for Dent's disease. Nature 2000, 408, 369–373. [Google Scholar] [CrossRef]
- Wang, S.S.; Devuyst, O.; Courtoy, P.J.; Wang, X.-T.; Wang, H.; Wang, Y.; Thakker, R.V.; Guggino, S.; Guggino, W.B. Mice lacking renal chloride channel, CLC-5, are a model for Dent's disease, a nephrolithiasis disorder associated with defective receptor-mediated endocytosis. Hum. Mol. Genet. 2000, 9, 2937–2945. [Google Scholar] [CrossRef]
- Yadav, M.K.; Yoo, K.W.; Atala, A.; Lu, B. Lentiviral vector mediated gene therapy for type I Dent disease ameliorates Dent disease-like phenotypes for three months in ClC-5 null mice. Mol. Ther. - Methods Clin. Dev. 2022, 27, 149–166. [Google Scholar] [CrossRef]
- Shipman, K.E.; Baty, C.J.; Long, K.R.; Rbaibi, Y.; Cowan, I.A.; Gerges, M.; Marciszyn, A.L.; Kashlan, O.B.; Tan, R.J.; Edwards, A.; et al. Impaired Endosome Maturation Mediates Tubular Proteinuria in Dent Disease Cell Culture and Mouse Models. J. Am. Soc. Nephrol. 2023, 34, 619–640. [Google Scholar] [CrossRef]
- Novarino, G.; Weinert, S.; Rickheit, G.; Jentsch, T.J. Endosomal Chloride-Proton Exchange Rather Than Chloride Conductance Is Crucial for Renal Endocytosis. Science 2010, 328, 1398–1401. [Google Scholar] [CrossRef] [PubMed]
- Sakhi IB, De Combiens E, Frachon N, Durussel F, Brideau G, Nemazanyy I, Frère P, Thévenod F, Lee WK, Zeng Q, Klein C, Lourdel S, Bignon Y. A novel transgenic mouse model highlights molecular disruptions involved in the pathogenesis of Dent disease 1. Gene. 2024 Nov 30;928:148766.
- Reynolds, C.J.; Gillen, C.M.; Burke, R.; Tsering, Y.; Loucks, E.; Judd-Mole, S.; Dow, J.A.; Romero, M.F. Drosophila ClC-c Is a Homolog of Human CLC-5 and a New Model for Dent Disease Type 1. Kidney360 2024, 5, 414–426. [Google Scholar] [CrossRef] [PubMed]
- Gorvin, C.M.; Wilmer, M.J.; Piret, S.E.; Harding, B.; Heuvel, L.P.v.D.; Wrong, O.; Jat, P.S.; Lippiat, J.D.; Levtchenko, E.N.; Thakker, R.V. Receptor-mediated endocytosis and endosomal acidification is impaired in proximal tubule epithelial cells of Dent disease patients. Proc. Natl. Acad. Sci. 2013, 110, 7014–7019. [Google Scholar] [CrossRef] [PubMed]
- Durán, M.; Burballa, C.; Cantero-Recasens, G.; Butnaru, C.M.; Malhotra, V.; Ariceta, G.; Sarró, E.; Meseguer, A. Novel Dent disease 1 cellular models reveal biological processes underlying ClC-5 loss-of-function. Hum. Mol. Genet. 2021, 30, 1413–1428. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Cai, H.; Cebotaru, L.; Hryciw, D.H.; Weinman, E.J.; Donowitz, M.; Guggino, S.E.; Guggino, W.B. ClC-5: role in endocytosis in the proximal tubule. Am. J. Physiol. Physiol. 2005, 289, F850–F862. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xu, L.; Zhang, Y.; Fu, H.; Gao, L.; Guan, Y.; Gu, W.; Sun, J.; Chen, X.; Yang, F.; et al. Dent disease 1-linked novel CLCN5 mutations result in aberrant location and reduced ion currents. Int. J. Biol. Macromol. 2024, 257, 128564. [Google Scholar] [CrossRef] [PubMed]
- Grand, T.; Mordasini, D.; L'Hoste, S.; Pennaforte, T.; Genete, M.; Biyeyeme, M.-J.; Vargas-Poussou, R.; Blanchard, A.; Teulon, J.; Lourdel, S. Novel CLCN5 mutations in patients with Dent’s disease result in altered ion currents or impaired exchanger processing. Kidney Int. 2009, 76, 999–1005. [Google Scholar] [CrossRef] [PubMed]
- Grand, T.; L'Hoste, S.; Mordasini, D.; Defontaine, N.; Keck, M.; Pennaforte, T.; Genete, M.; Laghmani, K.; Teulon, J.; Lourdel, S. Heterogeneity in the processing of CLCN5 mutants related to Dent disease. Hum. Mutat. 2011, 32, 476–483. [Google Scholar] [CrossRef]
- Bignon, Y.; Alekov, A.; Frachon, N.; Lahuna, O.; Doh-Egueli, C.J.-B.; Deschênes, G.; Vargas-Poussou, R.; Lourdel, S. A novel CLCN5 pathogenic mutation supports Dent disease with normal endosomal acidification. Hum. Mutat. 2018, 39, 1139–1149. [Google Scholar] [CrossRef]
- Sakhi, I.; Bignon, Y.; Frachon, N.; Hureaux, M.; Arévalo, B.; González, W.; Vargas-Poussou, R.; Lourdel, S. Diversity of functional alterations of the ClC-5 exchanger in the region of the proton glutamate in patients with Dent disease 1. Hum. Mutat. 2021, 42, 537–550. [Google Scholar] [CrossRef]
- Satoh, N.; Yamada, H.; Yamazaki, O.; Suzuki, M.; Nakamura, M.; Suzuki, A.; Ashida, A.; Yamamoto, D.; Kaku, Y.; Sekine, T.; et al. A pure chloride channel mutant of CLC-5 causes Dent’s disease via insufficient V-ATPase activation. Pfl?gers Arch. Eur. J. Physiol. 2016, 468, 1183–1196. [Google Scholar] [CrossRef] [PubMed]
- Chang MH, Brown MR, Liu Y, Gainullin VG, Harris PC, Romero MF, Lieske JC. Cl- and H+ coupling properties and subcellular localizations of wildtype and disease-associated variants of the voltage-gated Cl-/H+ exchanger ClC-5. J Biol Chem. 2020 Feb 7;295(6):1464–73.
- Jentsch, T.J.; Pusch, M. CLC Chloride Channels and Transporters: Structure, Function, Physiology, and Disease. Physiol. Rev. 2018, 98, 1493–1590. [Google Scholar] [CrossRef] [PubMed]
- Günther, W.; Lüchow, A.; Cluzeaud, F.; Vandewalle, A.; Jentsch, T.J. ClC-5, the chloride channel mutated in Dent’s disease, colocalizes with the proton pump in endocytotically active kidney cells. Proc. Natl. Acad. Sci. 1998, 95, 8075–8080. [Google Scholar] [CrossRef] [PubMed]
- Eshbach, M.L.; Weisz, O.A. Receptor-Mediated Endocytosis in the Proximal Tubule. Annu. Rev. Physiol. 2017, 79, 425–448. [Google Scholar] [CrossRef] [PubMed]
- Beenken, A.; Cerutti, G.; Brasch, J.; Guo, Y.; Sheng, Z.; Erdjument-Bromage, H.; Aziz, Z.; Robbins-Juarez, S.Y.; Chavez, E.Y.; Ahlsen, G.; et al. Structures of LRP2 reveal a molecular machine for endocytosis. Cell 2023, 186, 821–836. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.; Homaei, A.; El-Seedi, H.R.; Akhtar, N. Cathepsins: Proteases that are vital for survival but can also be fatal. Biomed. Pharmacother. 2018, 105, 526–532. [Google Scholar] [CrossRef] [PubMed]
- Hryciw, D.H.; Wang, Y.; Devuyst, O.; Pollock, C.A.; Poronnik, P.; Guggino, W.B. Cofilin Interacts with ClC-5 and Regulates Albumin Uptake in Proximal Tubule Cell Lines. J. Biol. Chem. 2003, 278, 40169–40176. [Google Scholar] [CrossRef]
- Reed AA, Loh NY, Terryn S, Lippiat JD, Partridge C, Galvanovskis J, Williams SE, Jouret F, Wu FT, Courtoy PJ, Nesbit MA, Rorsman P, Devuyst O, Ashcroft FM, Thakker RV. CLC-5 and KIF3B interact to facilitate CLC-5 plasma membrane expression, endocytosis, and microtubular transport: relevance to pathophysiology of Dent’s disease. Am J Physiol Ren Physiol. 2010 Feb;298(2):F365-80.
- Lourdel, S.; Grand, T.; Burgos, J.; González, W.; Sepúlveda, F.V.; Teulon, J. ClC-5 mutations associated with Dent’s disease: a major role of the dimer interface. Pfl?gers Arch. Eur. J. Physiol. 2011, 463, 247–256. [Google Scholar] [CrossRef] [PubMed]
- Alekov, A.K. Mutations associated with Dent's disease affect gating and voltage dependence of the human anion/proton exchanger ClC-5. Front. Physiol. 2015, 6. [Google Scholar] [CrossRef]
- Polesel, M.; Kaminska, M.; Haenni, D.; Bugarski, M.; Schuh, C.; Jankovic, N.; Kaech, A.; Mateos, J.M.; Berquez, M.; Hall, A.M. Spatiotemporal organisation of protein processing in the kidney. Nat. Commun. 2022, 13, 1–13. [Google Scholar] [CrossRef]
- Devuyst, O.; Jouret, F.; Auzanneau, C.; Courtoy, P.J. Chloride Channels and Endocytosis: New Insights from Dent’s Disease and ClC-5 Knockout Mice. Nephron Physiol. 2005, 99, p69–p73. [Google Scholar] [CrossRef] [PubMed]
- Long, K.R.; Rbaibi, Y.; Kashlan, O.B.; Weisz, O.A. Receptor-associated protein impairs ligand binding to megalin and megalin-dependent endocytic flux in proximal tubule cells. Am. J. Physiol. Physiol. 2023, 325, F457–F464. [Google Scholar] [CrossRef] [PubMed]
- Christensen, E.I.; Devuyst, O.; Dom, G.; Nielsen, R.; Van Der Smissen, P.; Verroust, P.; Leruth, M.; Guggino, W.B.; Courtoy, P.J. Loss of chloride channel ClC-5 impairs endocytosis by defective trafficking of megalin and cubilin in kidney proximal tubules. Proc. Natl. Acad. Sci. 2003, 100, 8472–8477. [Google Scholar] [CrossRef] [PubMed]
- Braulke, T.; Geuze, H.; Slot, J.; Hasilik, A.; Vonfigura, K. On The Effects of Weak Bases and Monensin on Sorting and Processing of Lysosomal-Enzymes in Human-Cells. Eur J Cell Biol. 1987, 43, 316–321.
- Nielsen, R.; Courtoy, P.J.; Jacobsen, C.; Dom, G.; Lima, W.R.; Jadot, M.; Willnow, T.E.; Devuyst, O.; Christensen, E.I. Endocytosis provides a major alternative pathway for lysosomal biogenesis in kidney proximal tubular cells. Proc. Natl. Acad. Sci. 2007, 104, 5407–5412. [Google Scholar] [CrossRef] [PubMed]
- Cocchiaro, P.; De Pasquale, V.; Della Morte, R.; Tafuri, S.; Avallone, L.; Pizard, A.; Moles, A.; Pavone, L.M. The Multifaceted Role of the Lysosomal Protease Cathepsins in Kidney Disease. Front. Cell Dev. Biol. 2017, 5, 114. [Google Scholar] [CrossRef] [PubMed]
- Cavallo-Medved D, Moin K, Sloane B. Cathepsin B. AFCS-Nat Mol Pages [Internet]. 2011 [cited 2020 Nov 23];2011. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5541861/.
- Gailly, P.; Jouret, F.; Martin, D.; Debaix, H.; Parreira, K.; Nishita, T.; Blanchard, A.; Antignac, C.; Willnow, T.; Courtoy, P.; et al. A novel renal carbonic anhydrase type III plays a role in proximal tubule dysfunction. Kidney Int. 2008, 74, 52–61. [Google Scholar] [CrossRef] [PubMed]
- Blanchard, A.; Curis, E.; Guyon-Roger, T.; Kahila, D.; Treard, C.; Baudouin, V.; Bérard, E.; Champion, G.; Cochat, P.; Dubourg, J.; et al. Observations of a large Dent disease cohort. Kidney Int. 2016, 90, 430–439. [Google Scholar] [CrossRef] [PubMed]
- Bhargava, P.; Schnellmann, R.G. Mitochondrial energetics in the kidney. Nat. Rev. Nephrol. 2017, 13, 629–646. [Google Scholar] [CrossRef] [PubMed]
- Protasoni, M.; Zeviani, M. Mitochondrial Structure and Bioenergetics in Normal and Disease Conditions. Int. J. Mol. Sci. 2021, 22, 586. [Google Scholar] [CrossRef]
- Herst, P.M.; Rowe, M.R.; Carson, G.M.; Berridge, M.V. Functional Mitochondria in Health and Disease. Front. Endocrinol. 2017, 8, 296–296. [Google Scholar] [CrossRef]
- Devin, *!!! REPLACE !!!*; et al. Mitochondria: Ultrastructure, Dynamics, Biogenesis and Main Functions. In: Mitochondria in Obesity and Type 2 Diabetes [Internet]. Elsevier Inc 2019. Academic Press; 2019 [cited 2024 Jul 14]. p. 3–32. Available from: https://www.sciencedirect.com/science/article/abs/pii/B9780128117521000018.
- Cassina, L.; Chiaravalli, M.; Boletta, A. Increased mitochondrial fragmentation in polycystic kidney disease acts as a modifier of disease progression. FASEB J. 2020, 34, 6493–6507. [Google Scholar] [CrossRef] [PubMed]
- Festa, B.P.; Chen, Z.; Berquez, M.; Debaix, H.; Tokonami, N.; Prange, J.A.; van de Hoek, G.; Alessio, C.; Raimondi, A.; Nevo, N.; et al. Impaired autophagy bridges lysosomal storage disease and epithelial dysfunction in the kidney. Nat. Commun. 2018, 9, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Tanriover, C.; Copur, S.; Ucku, D.; Cakir, A.B.; Hasbal, N.B.; Soler, M.J.; Kanbay, M. The Mitochondrion: A Promising Target for Kidney Disease. Pharmaceutics 2023, 15, 570. [Google Scholar] [CrossRef] [PubMed]
- Wright, J.; Morales, M.M.; Sousa-Menzes, J.; Ornellas, D.; Sipes, J.; Cui, Y.; Cui, I.; Hulamm, P.; Cebotaru, V.; Cebotaru, L.; et al. Transcriptional adaptation to Clcn5 knockout in proximal tubules of mouse kidney. Physiol. Genom. 2008, 33, 341–354. [Google Scholar] [CrossRef] [PubMed]
- Faivre, A.; Verissimo, T.; Auwerx, H.; Legouis, D.; de Seigneux, S. Tubular Cell Glucose Metabolism Shift During Acute and Chronic Injuries. Front. Med. 2021, 8. [Google Scholar] [CrossRef] [PubMed]
- Owen, O.E.; Kalhan, S.C.; Hanson, R.W. The Key Role of Anaplerosis and Cataplerosis for Citric Acid Cycle Function. J. Biol. Chem. 2002, 277, 30409–30412. [Google Scholar] [CrossRef] [PubMed]
- Turrens, JF. Mitochondrial formation of reactive oxygen species. J Physiol. 2003 Oct 15;552(Pt 2):335–44.
- Ott M, Gogvadze V, Orrenius S, Zhivotovsky B. Mitochondria, oxidative stress and cell death. Apoptosis. 2007 May 1;12(5):913–22.
- Guggino, SE. Can we generate new hypotheses about Dent’s disease from gene analysis of a mouse model? Exp Physiol. 2009 Feb;94(2):191–6.
- Iglesias, J.; Abernethy, V.E.; Wang, Z.; Lieberthal, W.; Koh, J.S.; Levine, J.S. Albumin is a major serum survival factor for renal tubular cells and macrophages through scavenging of ROS. Am. J. Physiol. Physiol. 1999, 277, F711–F722. [Google Scholar] [CrossRef] [PubMed]
- Devuyst O, Luciani A. Chloride transporters and receptor-mediated endocytosis in the renal proximal tubule. J Physiol. 2015 Sep 15;593(18):4151–64.
- Kang, H.M.; Ahn, S.H.; Choi, P.; Ko, Y.-A.; Han, S.H.; Chinga, F.; Park, A.S.D.; Tao, J.; Sharma, K.; Pullman, J.; et al. Defective fatty acid oxidation in renal tubular epithelial cells has a key role in kidney fibrosis development. Nat. Med. 2015, 21, 37–46. [Google Scholar] [CrossRef]
- Hou, Y.; Tan, E.; Shi, H.; Ren, X.; Wan, X.; Wu, W.; Chen, Y.; Niu, H.; Zhu, G.; Li, J.; et al. Mitochondrial oxidative damage reprograms lipid metabolism of renal tubular epithelial cells in the diabetic kidney. Cell. Mol. Life Sci. 2024, 81, 1–19. [Google Scholar] [CrossRef]
- Levental I, Lyman E. Regulation of membrane protein structure and function by their paralipidomes. Nat Rev Mol Cell Biol. 2023 Feb;24(2):107–22.
- Cornelius, F. Modulation of Na,K-ATPase and Na-ATPase Activity by Phospholipids and Cholesterol. I. Steady-State Kinetics. Biochemistry 2001, 40, 8842–8851. [Google Scholar] [CrossRef]
- Schulze, R.J.; Krueger, E.W.; Weller, S.G.; Johnson, K.M.; Casey, C.A.; Schott, M.B.; McNiven, M.A. Direct lysosome-based autophagy of lipid droplets in hepatocytes. Proc. Natl. Acad. Sci. 2020, 117, 32443–32452. [Google Scholar] [CrossRef]
- Supuran, CT. Carbonic anhydrases--an overview. Curr Pharm Des. 2008;14(7):603–14.
- Silagi, E.S.; Batista, P.; Shapiro, I.M.; Risbud, M.V. Expression of Carbonic Anhydrase III, a Nucleus Pulposus Phenotypic Marker, is Hypoxia-responsive and Confers Protection from Oxidative Stress-induced Cell Death. Sci. Rep. 2018, 8, 4856. [Google Scholar] [CrossRef] [PubMed]
- Räisänen SR, Lehenkari P, Tasanen M, Rahkila P, Härkönen PL, Väänänen HK. Carbonic anhydrase III protects cells from hydrogen peroxide-induced apoptosis. FASEB J Off Publ Fed Am Soc Exp Biol. 1999 Mar;13(3):513–22.
- Krebs, H.; Johnson, W. The role of citric acid in intermediate metabolism in animal tissues. FEBS Lett. 1980, 117, 2–10. [Google Scholar] [CrossRef]
- Mailloux RJ, Singh R, Brewer G, Auger C, Lemire J, Appanna VD. Alpha-ketoglutarate dehydrogenase and glutamate dehydrogenase work in tandem to modulate the antioxidant alpha-ketoglutarate during oxidative stress in Pseudomonas fluorescens. J Bacteriol. 2009 Jun;191(12):3804–10.
- A McDonough, M.; Loenarz, C.; Chowdhury, R.; Clifton, I.J.; Schofield, C.J. Structural studies on human 2-oxoglutarate dependent oxygenases. Curr. Opin. Struct. Biol. 2010, 20, 659–672. [Google Scholar] [CrossRef]
- Liu, S.; He, L.; Yao, K. The Antioxidative Function of Alpha-Ketoglutarate and Its Applications. BioMed Res. Int. 2018, 2018, 1–6. [Google Scholar] [CrossRef]
- Sole, M.J.; Madapallimattam, A.; Baines, A.D. An active pathway for serotonin synthesis by renal proximal tubules. Kidney Int. 1986, 29, 689–694. [Google Scholar] [CrossRef] [PubMed]
- Erikci, A.; Ucar, G.; Yabanoglu-Ciftci, S. Role of serotonin in the regulation of renal proximal tubular epithelial cells. Ren. Fail. 2016, 38, 1141–1150. [Google Scholar] [CrossRef]
- Hurtado, K.; Scholpa, N.E.; Schnellmann, J.G.; Schnellmann, R.G. Serotonin regulation of mitochondria in kidney diseases. Pharmacol. Res. 2024, 203, 107154. [Google Scholar] [CrossRef]
- Moser, G.H.; Schrader, J.; Deussen, A. Turnover of adenosine in plasma of human and dog blood. Am. J. Physiol. Physiol. 1989, 256, C799–C806. [Google Scholar] [CrossRef]
- Viegas, T.X.; Omura, G.A.; Stoltz, R.R.; Kisicki, J. Pharmacokinetics and Pharmacodynamics of Peldesine (BCX-34), a Purine Nucleoside Phosphorylase Inhibitor, following Single and Multiple Oral Doses in Healthy Volunteers. J. Clin. Pharmacol. 2000, 40, 410–420. [Google Scholar] [CrossRef]
- Phillis, J.W.; Walter, G.A.; O'Regan, M.H.; Stair, R.E. Increases in Cerebral Cortical Perfusate Adenosine and Inosine Concentrations during Hypoxia and Ischemia. J. Cereb. Blood Flow Metab. 1987, 7, 679–686. [Google Scholar] [CrossRef] [PubMed]
- L F, F H, V K, E H, E B, R F, T S, A JN, A N. Enhanced accumulation of pericardial fluid adenosine and inosine in patients with coronary artery disease. Life Sci. 1999 Jan 1;65(10):1005–12.
- Welihinda, A.A.; Kaur, M.; Greene, K.; Zhai, Y.; Amento, E.P. The adenosine metabolite inosine is a functional agonist of the adenosine A2A receptor with a unique signaling bias. Cell. Signal. 2016, 28, 552–560. [Google Scholar] [CrossRef] [PubMed]
- Merighi, S.; Mirandola, P.; Milani, D.; Varani, K.; Gessi, S.; Klotz, K.-N.; Leung, E.; Baraldi, P.G.; Borea, P.A. Adenosine Receptors as Mediators of Both Cell Proliferation and Cell Death of Cultured Human Melanoma Cells. J. Investig. Dermatol. 2002, 119, 923–933. [Google Scholar] [CrossRef] [PubMed]
- Mabley, J.G.; Pacher, P.; Liaudet, L.; Soriano, F.G.; Haskó, G.; Marton, A.; Szabó, C.; Salzman, A.L. Inosine reduces inflammation and improves survival in a murine model of colitis. Am. J. Physiol. Liver Physiol. 2003, 284, G138–G144. [Google Scholar] [CrossRef] [PubMed]
- Garay-Rojas, E.; Harper, M.; Hraba-Renevey, S.; Kress, M. An apparent autocrine mechanism amplifies the dexamethasone- and retinoic acid-induced expression of mouse lipocalin-encoding gene 24p3. Gene 1996, 170, 173–180. [Google Scholar] [CrossRef] [PubMed]
- Maritzen, T.; Rickheit, G.; Schmitt, A.; Jentsch, T. Kidney-specific upregulation of vitamin D3 target genes in ClC-5 KO mice. Kidney Int. 2006, 70, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Buonafine, M.; Martinez-Martinez, E.; Jaisser, F. More than a simple biomarker: the role of NGAL in cardiovascular and renal diseases. Clin. Sci. 2018, 132, 909–923. [Google Scholar] [CrossRef]
- Morgan, A.J.; Platt, F.M.; Lloyd-Evans, E.; Galione, A. Molecular mechanisms of endolysosomal Ca2+ signalling in health and disease. Biochem. J. 2011, 439, 349–378. [Google Scholar] [CrossRef] [PubMed]
- Scott CC, Gruenberg J. Ion flux and the function of endosomes and lysosomes: pH is just the start: the flux of ions across endosomal membranes influences endosome function not only through regulation of the luminal pH. BioEssays News Rev Mol Cell Dev Biol. 2011 Feb;33(2):103–10.
- Yu W, Beaudry S, Negoro H, Boucher I, Tran M, Kong T, Denker BM. H2O2 activates G protein, alpha 12 to disrupt the junctional complex and enhance ischemia reperfusion injury. Proc Natl Acad Sci U A. 2012 Apr 24;109(17):6680–5.
- Lima, W.R.; Parreira, K.S.; Devuyst, O.; Caplanusi, A.; N′Kuli, F.; Marien, B.; Van Der Smissen, P.; Alves, P.M.; Verroust, P.; Christensen, E.I.; et al. ZONAB Promotes Proliferation and Represses Differentiation of Proximal Tubule Epithelial Cells. J. Am. Soc. Nephrol. 2010, 21, 478–488. [Google Scholar] [CrossRef]
- Raggi C, Luciani A, Nevo N, Antignac C, Terryn S, Devuyst O. Dedifferentiation and aberrations of the endolysosomal compartment characterize the early stage of nephropathic cystinosis. Hum Mol Genet. 2014 May 1;23(9):2266–78.
- Wright, J.; Morales, M.M.; Sousa-Menzes, J.; Ornellas, D.; Sipes, J.; Cui, Y.; Cui, I.; Hulamm, P.; Cebotaru, V.; Cebotaru, L.; et al. Transcriptional adaptation to Clcn5 knockout in proximal tubules of mouse kidney. Physiol. Genom. 2008, 33, 341–354. [Google Scholar] [CrossRef]
- Hallan, S.; Afkarian, M.; Zelnick, L.R.; Kestenbaum, B.; Sharma, S.; Saito, R.; Darshi, M.; Barding, G.; Raftery, D.; Ju, W.; et al. Metabolomics and Gene Expression Analysis Reveal Down-regulation of the Citric Acid (TCA) Cycle in Non-diabetic CKD Patients. EBioMedicine 2017, 26, 68–77. [Google Scholar] [CrossRef] [PubMed]
- Chou, K.-M.; Lee, C.-C.; Chen, C.-H.; Sun, C.-Y. Clinical Value of NGAL, L-FABP and Albuminuria in Predicting GFR Decline in Type 2 Diabetes Mellitus Patients. PLOS ONE 2013, 8, e54863. [Google Scholar] [CrossRef] [PubMed]
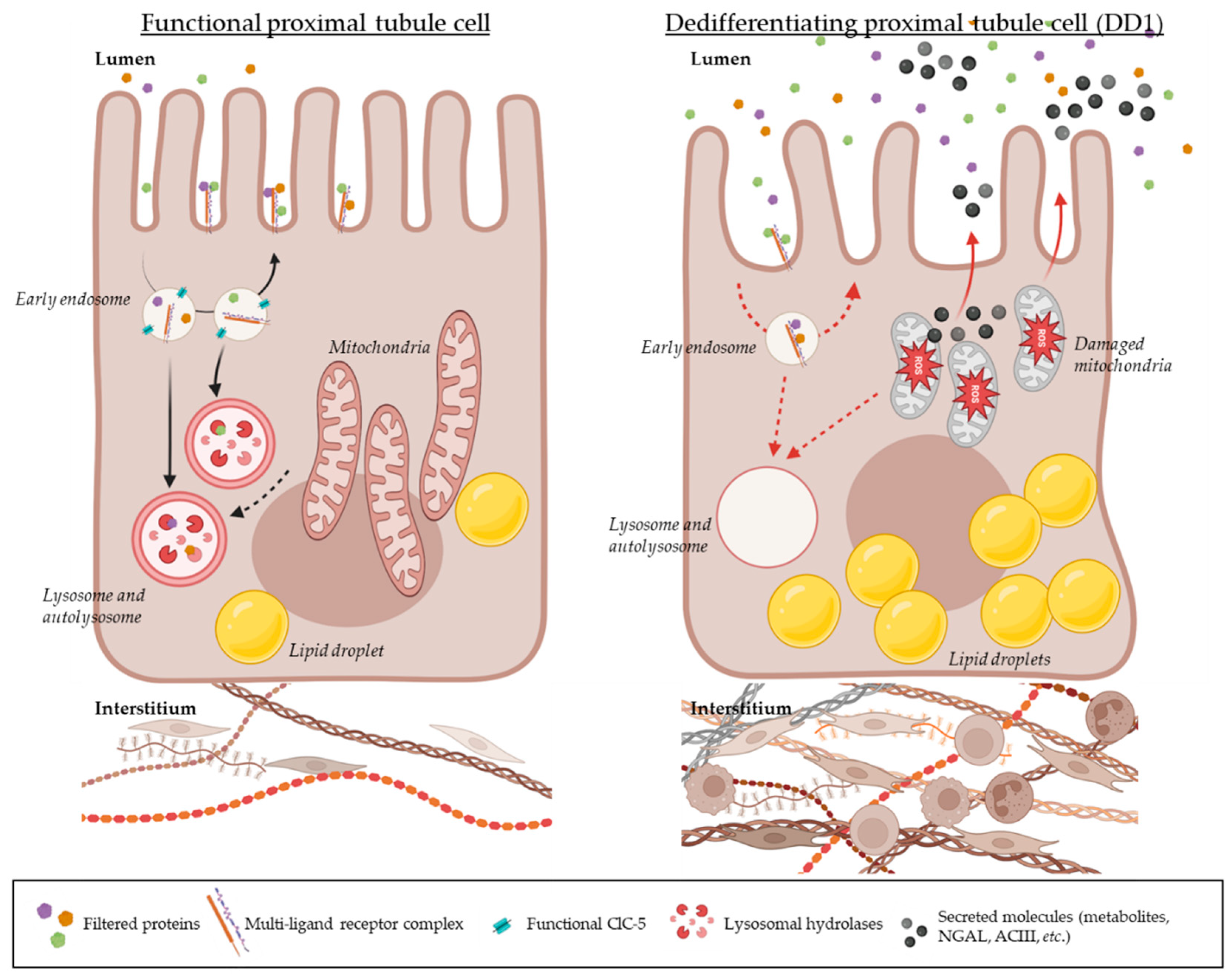
Figure 1.
Hypothesis on the pathogenesis of Dent disease type 1 (DD1). In physiological contexts (left image), the proximal tubule reabsorbs filtered plasma proteins using receptor-mediated endocytosis (through a multi-ligand receptor complex composed of Megalin, Cubilin, and Ammionless). Once attached to their receptors, proteins are internalized within the cell and reach a first endosomal compartment where a drop in pH occurs thanks to a functional ClC-5 transporter. In these early endosomes, proteins detach from their receptors and further traffic along the endo-lysosomal apparatus until reaching lysosomes; while receptors are recycled back to the plasma membrane to catch other filtered proteins. The highly acidified lysosomes enriched with functional hydrolases called cathepsins take care of digesting these proteins. According to Polesel et al., the degradation product is secreted back to the lumen, and the resulting peptides are further processed by later proximal tubule segments [34]. On the other hand, functional lysosomes also fuse with autophagosomes to ensure damaged organelle clearance for general cell health. In the context of Dent disease type 1 (right image), the absence of a functional ClC-5 on the early endosome membrane would prevent the early acidification step from occuring; hence, uptaken proteins would hardly detach from their receptors which would in turn slow down the internalization of additional proteins, the recycling of multi-ligand receptors and the further trafficking of vesicles along the endo-lysosomal pathway. Additionally, lysosomes would lack functional cathepsins as these proteases need an optimal endo-lysosomal system to be efficient. Transporters that would be internalized on early endosomes would also be less recycled back at the plasma membrane which would reduce the reabsorption of electrolytes and other molecules in proximal tubules. Stressed mitochondria lacking their essential metabolites to ensure their functions would accumulate within the cells but not effectively being cleared by autophagy. Lipids would also accumulate within the proximal tubules as they would be harder to process by the dysfunctional cells. The consecutive increased oxidative stress would activate stress pathways and the production of damage-associated biomarkers. A renal function decline would be observed as the tissue’s local inflammation and fibrosis progress (with an activation of fibroblasts in the environing interstitium and the infiltration of immune cells). The dysfunctional proximal tubule cells would slowly activate epithelial to mesenchymal transition factors and dedifferentiate.
Figure 1.
Hypothesis on the pathogenesis of Dent disease type 1 (DD1). In physiological contexts (left image), the proximal tubule reabsorbs filtered plasma proteins using receptor-mediated endocytosis (through a multi-ligand receptor complex composed of Megalin, Cubilin, and Ammionless). Once attached to their receptors, proteins are internalized within the cell and reach a first endosomal compartment where a drop in pH occurs thanks to a functional ClC-5 transporter. In these early endosomes, proteins detach from their receptors and further traffic along the endo-lysosomal apparatus until reaching lysosomes; while receptors are recycled back to the plasma membrane to catch other filtered proteins. The highly acidified lysosomes enriched with functional hydrolases called cathepsins take care of digesting these proteins. According to Polesel et al., the degradation product is secreted back to the lumen, and the resulting peptides are further processed by later proximal tubule segments [34]. On the other hand, functional lysosomes also fuse with autophagosomes to ensure damaged organelle clearance for general cell health. In the context of Dent disease type 1 (right image), the absence of a functional ClC-5 on the early endosome membrane would prevent the early acidification step from occuring; hence, uptaken proteins would hardly detach from their receptors which would in turn slow down the internalization of additional proteins, the recycling of multi-ligand receptors and the further trafficking of vesicles along the endo-lysosomal pathway. Additionally, lysosomes would lack functional cathepsins as these proteases need an optimal endo-lysosomal system to be efficient. Transporters that would be internalized on early endosomes would also be less recycled back at the plasma membrane which would reduce the reabsorption of electrolytes and other molecules in proximal tubules. Stressed mitochondria lacking their essential metabolites to ensure their functions would accumulate within the cells but not effectively being cleared by autophagy. Lipids would also accumulate within the proximal tubules as they would be harder to process by the dysfunctional cells. The consecutive increased oxidative stress would activate stress pathways and the production of damage-associated biomarkers. A renal function decline would be observed as the tissue’s local inflammation and fibrosis progress (with an activation of fibroblasts in the environing interstitium and the infiltration of immune cells). The dysfunctional proximal tubule cells would slowly activate epithelial to mesenchymal transition factors and dedifferentiate.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



