
Submitted:
01 August 2024
Posted:
01 August 2024
You are already at the latest version
Abstract
Structural Maintenance of Chromosomes (SMC) complexes are an evolutionary conserved protein family. In most eukaryotes, three SMC complexes have been characterized: cohesin, condensin, and SMC5-6 complexes. These complexes are involved in a plethora of functions and defects in SMC genes can lead to increased risk of chromosomal abnormalities, infertility, and cancer. To investigate th evolution of SMC complex genes in mammals, we analyzed their selective patterns in an extended phylogeny. Signals of positive selection were identified for condensin NCAPG, for two SMC5/6 complex genes (SMC5 and NSMCE4A) and for all cohesin genes with almost exclusive meiotic expression (RAD21L1, REC8, SMC1B, and STAG3). For the latter, evolutionary rates correlate with expression during female meiosis and most positively selected sites fall in intrinsically disordered regions (IDRs). Our results support growing evidence that IDRs are fast evolving and that most likely contribute to adaptation through modulation of phase separation. We suggest that the natural selection signals identified in SMC complexes may be the result of different selective pressures: a host-pathogen arms race in the condensin and SMC5/6 complexes, and an intragenomic conflict similar to that described for centromeres and telomeres for meiotic cohesin genes.
Keywords:
Structural Maintenance of Chromosomes
; meiotic cohesins
; positive selection
; host-pathogen arms race
; intragenomic conflict
1. Introduction
Structural Maintenance of Chromosomes (SMC) complexes are an evolutionary conserved protein family present from bacteria to humans [1]. In most eukaryotes, three SMC complexes have been characterized: cohesin, condensin, and SMC5-6 complexes [1]. Such complexes are involved in a plethora of functions, including mitotic and meiotic chromosome condensation, sister chromatid cohesion, accurate chromosome segregation, DNA replication and repair, genome compartmentalisation, and transcriptional regulation. All SMC complexes share structural features. Each complex is composed of three core proteins (two SMC proteins and a kleisin subunit) and peripheral subunits forming a ring-shaped structure [1,2].
The cohesin complex is most likely the best studied SMC complex. In mammalian cells, the cohesin complex comprises two SMC proteins (SMC3 and SMC1A or SMC1B), an alpha-kleisin subunit (RAD21, RAD21L, or REC8), and a stromal antigen protein (STAG1, 2, or 3) [2]. Four of these subunits (REC8, RAD21L1, SMC1B, and STAG3) have an almost exclusive meiotic expression and are therefore referred to as meiotic-specific cohesins. Hereafter, for the sake of simplicity, SMC3 (present in all cohesin complexes) and the remaining cohesin subunits (expressed preferentially in somatic cells) will be designated non-meiotic cohesins. Cohesin complexes are involved in a number of different mechanisms: from keeping sister chromatids together to contributing to the compartmentalization of chromosomes in topologically associative domains (TADs). Chromosome and nuclear compartmentalization, as well as TAD assembly, are mediated by phase separation. It has recently been reported that a fraction of cohesin associates with chromatin in a manner consistent with bridging-induced phase separation (BIPS, also known as polymer–polymer phase separation) [3,4]. BIPS uses multivalent protein‒DNA interactions bridging two distinct DNA regions and forming a DNA loop that acts as a nucleation structure for phase condensation [3,5]. In addition, during meiosis, meiotic-specific cohesins mediate Sister Chromatid Cohesion (SCC), Synaptonemal Complex (SC) assembly and synapsis, as well as telomere attachment to the nuclear envelope and telomere maintenance. The essential role of the cohesin complex in many aspects of chromosome biology is supported by the fact that defects in cohesin genes can lead to different diseases in which chromatid cohesion, DNA repair, transcriptional regulation, and genome topology are altered. Mutations in meiotic-specific cohesin genes have been associated with infertility, age-related aneuploidy, and premature ovarian failure [6]. Moreover, mutations in non-meiotic cohesin complex components and in their regulators have been associated with cancer [7,8,9]. Globally, mutations in these genes lead to disease conditions also known as cohesinophaties. Among these, Cornelia de Lange syndrome (CdLS) is the most frequent and best known entity [10,11]. CdLS is a malformative syndrome affecting many organs, in which intellectual and growth retardations are the main phenotypic manifestations [12,13]. Patients require life-long rehabilitation and about 80% of cases carry mutations in one of cohesin complex components or in one of their regulators (SMC1A, SMC3, RAD21, STAG1, STAG2, NIPBL, HDAC8) [11,12,14].
In addition to cohesin complex, most eukaryotic genomes contain two distinct condensin complexes (Condensin I and II) that differ in their non-SMC subunits, in cellular localization, and in their regulation during cell cycle [15,16,17]. In particular Condensin I localizes in the cytoplasm and gains access to chromosomes between prometaphase and telophase, when the nuclear envelope breaks down (NEBD). Conversely, Condensin II has a nuclear localization and, in mitosis, it binds stably to chromatin. Like cohesins, the condensin complex plays a key role in chromosome condensation, assembly, and segregation during mitosis and meiosis [18,19,20]. Condensins have also been associated to pathological conditions, as mutations in condensin subunits result in microcephaly due to impaired DNA decatenation [21,22].
The third member of SMC family, the SMC5/6 complex, has important functions in DNA repair by recombination, but also plays a role in influencing genome stability and dynamics in undamaged cells [23,24]. Furthermore, by preventing accumulation of toxic recombination intermediates, SMC5/6 promotes correct mitotic and meiotic chromosome segregation [23,24]. As in the case of cohesins, protein levels of SMC5/6 components decrease with age in mouse oocytes [25]. It was thus speculated that, in humans, reduced SMC5/6 availability may be associated with the increased risk of chromosomal abnormalities and infertility linked to maternal age. Moreover, mutations in NSMCE2 or NSMCE3 have been described in patients with primordial dwarfism, extreme insulin resistance, gonadal failure [26], and lung disease immunodeficiency and chromosome breakage syndrome (LICS) [27]. Finally, the complex acts as a host-restrictor factor, inhibiting the transcription of genomes of different viruses (i.e.: HBV, unintegrated HIV1, HSV1, HCMV, KSHV, and HPV) [28,29,30,31,32,33,34,35,36].
Due to their essential functions and association to pathological conditions, SMC complex proteins would be expected to evolve under strict evolutionary constraint. Nevertheless, King and colleagues [37] recently observed signatures of recurrent positive selection in the Condensin II and in mitotic cohesin complexes across Drosophila and mammals. They also suggested the presence of an evolutionary arms race driven by viral infections.
To better understand the selective events underlying the evolution of genes that encode SMC complex proteins, we analyzed the selective patterns of all the proteins that contribute to the formation of Cohesin, Condensin, and SMC5/6 complexes.
2. Materials and Methods
2.1. Sequence Retrieval and Alignment
In this study we analyzed 26 subunits of cohesin, condensin I and II, and SMC5/6 complexes (Table 1) reported as “subunits” by Haering and Gruber [2]. Mammalian homologs of human genes were included only if they represented 1-to-1 orthologs, as reported in the EnsemblCompara GeneTrees [38]. Coding sequence information for at least 46 mammalian species was retrieved from the NCBI database (http://www.ncbi.nlm.nih.gov/) and from the UCSC server (http://genome.ucsc.edu/). The list of species and the number of sequences analyzed for each gene are reported in Table 1 and Supplementary Table S1.
The RevTrans 2.0 utility was used to generate Multiple Sequence Alignments (MSAs) using MAFFT v6.240 as an aligner [39]. Phylogenetic trees were reconstructed using the phyML program with a General Time Reversible (GTR) model plus gamma-distributed rates and 4 substitution rate categories with a fixed proportion of invariable sites [40].
Because recombination can generate false positive inferences of positive selection [41,42], MSAs were screened for the presence of recombination using GARD (Genetic Algorithm Recombination Detection) [43]. GARD is a genetic algorithm implemented in the HYPHY suite (version 2.2.4) [44], which uses phylogenetic incongruence among segments in the alignment to detect the best-fit number and location of recombination breakpoints. No significant breakpoint was detected.
2.2. Evolutionary Analysis in Mammals
The average nonsynonymous substitution (dN)/synonymous substitution (dS) rate ratio and the dN-dS parameter were calculated using the Single-Likelihood Ancestor Counting (SLAC) method (10.1093/molbev/msi105). Inputs were the MSAs and phyML trees (see section 2.1).
To detect positive selection, we used the codon-based codeml program implemented in the PAML (Phylogenetic Analysis by Maximum Likelihood) suite [45]. We applied different site (NSsite) models; specifically, we compared models of gene evolution that allow (NSsite models M2a and M8) or disallow (NSsite models M1a, and M7) a class of codons to evolve with dN/dS >1. To assess statistical significance, twice the difference of the likelihood (ΔlnL) for the models (M1a vs M2a and M7 vs M8) is compared to a χ2 distribution (2 degrees of freedom for both comparisons). To assure reliability, different codon substitution models (F3x4 and F61) were used.
In order to identify specific sites subject to positive selection, we applied three different methods: 1) the Bayes Empirical Bayes (BEB) analysis (with a cutoff ≥ 0.90), which calculates the posterior probability that each codon is from the site class of positive selection (under model M8) [46]; 2) Fast Unbiased Bayesian AppRoximation (FUBAR) [47], an approximate hierarchical Bayesian method that generates an unconstrained distribution of selection parameters to estimate the posterior probability of positive diversifying selection at each site in a given alignment (with a cutoff ≥ 0.90); 3) the Fixed Effects Likelihood (FEL) [48], a maximum-likelihood (ML) approach to infer dN/dS on a per-site basis, assuming that the selection pressure for each site is constant along the entire phylogeny (with a p-value cutoff < 0.1). To be conservative and to limit false positives, only sites detected using at least two methods were considered as positive selection targets.
FEL, FUBAR, and SLAC analyses were run locally through the HYPHY suite [44].
The PAML Free Ratio (FR) model was used to estimate different value of dN/dS on the branches of the phylogeny [49]. The FR model assumes different dN/dS for each lineage and is compared with a null model with one dN/dS for the entire phylogeny. Statistical significance is assessed by comparing twice the ΔlnL of the two models with a χ2 distribution with degrees of freedom equal to the difference in model parameters.
2.3. Correlation with Meiotic Gene Expression
2.4. Prediction of Disordered Regions and Functional Motifs
Intrinsically disordered regions (IDRs) were identified by the Metapredict V2 tool [52,53]. This tool defines IDRs by applying a deep-learning algorithm based on a consensus score calculated from eight different disorder predictors [53]. Metapredict V2 was run using default parameters and IDRs were defined as consecutive disordered stretches longer than 30 residues. Prediction of functional motifs and nuclear localization signals was performed using PROSITE (https://prosite.expasy.org) [54] and NLStradamus software (http://www.moseslab.csb.utoronto.ca/NLStradamus/) [55], respectively.
PS-promoting regions were identified using the ParSe method v 2.0 [56,57]. ParSe uses sequence-based calculations of hydrophobicity, α-helix propensity, and a model of the polymer scaling exponent (νmodel) to predict regions prone to prone to undergo PS. We used a model that also includes the effects of interactions between amino acids (U π for π–π and cation–π interactions and U q for charge-based effects) trained on csat (the saturation concentration associated with protein PS) [58,59].
3. Results
3.1. Evolutionary Analysis in Mammals: SMC Complexes Evolve at Different Rates
We first aimed to comprehensively analyze the selective pressure acting on mammalian genes that encode proteins of SMC complexes. In particular, we analyzed the evolutionary history of 26 SMC genes in at least 46 mammalian species: 11 cohesins (4 of them meiosis-specific), 8 condensins, and 7 SMC5/6 genes (Table 1,Supplementary Table S1) [2].
For coding genes, the strength of selection can be quantified by comparing the rate of non-synonymous nucleotide substitutions per non-synonymous site (dN) with that of synonymous substitution per synonymous site (dS). We thus calculated the average dN/dS ratio using the single-likelihood ancestor counting (SLAC) method [48]. dN/dS values greater than 1 are consistent with positive (diversifying) selection, whereas ratios lower than 1 indicate purifying selection (selective constraint). The expected dN/dS under selective neutrality is 1.
As reported in Table 1, all genes had dN/dS values much lower than 1, indicating that, as is the case of most mammalian genes [42], purifying selection is the major force acting on SMC complexes genes. Comparison of dN/dS values among meiosis-specific and mitotic cohesin genes indicated that these latter tend to show higher evolutionary constraint. The same results were observed comparing mitotic cohesin genes with condensin and SMC5/6 genes (Table 1). To gain insight into the relative evolutionary rates of these protein in a wider genomic context, we compared the average dN/dS values of the SMC complex genes with those previously calculated in more than 9000 genes in a representative mammalian phylogeny (24 species) [60]. In this phylogeny, the average dN/dS values were calculated for only 11 SMC genes, so we carried out a correlation analyses between dN/dS values obtained for these 11 genes on the two phylogenies (Figure 1A). There was a strong correlation (Spearman test, p value= 2.2x10-16, rho= 0.95; Kendall test, p value= 4.6x10-5, tau= 0.85) between dN/dS values calculated on our phylogeny with those calculated by Ebel and colleagues [60], thus we assumed we could compare our data with those calculated on a large gene set. As evident in Figure 1, all mitotic cohesin genes displayed the lowest dN/dS values among SMC genes and their dN/dS values were well below the median for all human genes, confirming stronger evolutionary constraint. Conversely, RAD21L1 showed the fastest evolutionary rate among SMC complex genes, with a dN/dS value higher than the 98th percentile of the distribution.
To investigate the selection pattern of individual codons in SMC genes, we calculated the dN-dS parameter at each site [48]. dN-dS was preferred over the conventional dN/dS because it is not rendered to infinite for dS values equal to 0. The analysis was done on all the genes collected in 4 groups: meiotic and mitotic cohesins, condensin, and SMC5/6. All gene groups displayed a high proportion of constrained sites (dN-dS<0), in particular mitotic cohesin genes were more constrained than the other gene groups, confirming the average dN/dS analysis. The distribution of dN-dS values was significantly different across the 4 gene groups (Figure 1B).
3.2. Positive Selection Drives the Evolution of Meiosis-Specific Cohesins
While constraints on protein function and structure typically result in overall purifying selection being the primary evolutionary force acting on protein regions, diversifying selection is often limited to specific sites or domains [42]. Thus, to identified pervasive positive selection, we applied maximum-likelihood analyses implemented in the PAML (Phylogenetic Analysis by Maximum Likelihood) package [45,61]. Specifically, we used the codeml program to compare models of gene evolution that allow (NSsite models M2a and M8, positive selection model) or disallow (NSsite models M1a and M7, null models) a class of codons to evolve with dN/dS >1. The null models were rejected in favor of the positive selection models for all meiotic-specific cohesin genes (RAD21L1, REC8, SMC1B, and STAG3), for condensin NCAPG, and for two SMC5/6 complex genes (SMC5 and NSMCE4A) (Table 2, Supplementary Tables S2–4). Overall, these data indicate that a high proportion (27%) of SMC complex genes experienced positive selection.
In previous studies [37,62], King and colleagues reported signals of positive selection in all 4 mitotic cohesin genes analyzed (SMC1, SMC3, RAD21, and STAG1). This divergence between our results and those reported in the literature may be due to several factors. I) the evolutionary analyses were conducted on different phylogenies: while our data derive from analyses carried out on an extended mammalian phylogeny, King and colleagues analyzed separately the different groups of mammals (primates, murinae, cricetidae, bats, and bovidae); ii) in our analyses we applied an extremely conservative approach, in fact a gene was considered to be under positive selection only if all the M1/M2 and M7/M8 comparisons for two codon frequency models (F3x4 and F61) were significant, while King and colleagues applied only one comparison (M7 vs M8, model F3x4).
We next sought to analyze selection patterns across the whole mammalian phylogeny. To this aim, we applied the free ratio (FR) model implemented in the PAML software [49]. This model estimates a value of dN/dS for each lineage in the phylogeny and it is compared with a null model that estimates a single dN/dS for all lineages. The FR model fitted the data better than the null model for 21 genes (Supplementary Table S5), suggesting that, for these genes, the selective pressure has been acting differently across the phylogeny. To display specific lineages that carry natural selection signals, we overlaid the proportion of genes showing dN/dS>1 over the mammalian tree for each of the 3 SMC complexes separately. Most of the branches leading to superoders/orders showed at least one gene with dN/dS>1, for all SMC complex genes (Supplementary Figure 1). In particular, the branches leading to primates and laurasiatheria showed a relatively high number of selected genes. Similarly, for tip branches selection appeared strong in primates. In general, weak selection signals were detected in rodents (Supplementary Figure 1).
3.3. Analysis of Positively Selected Sites
To identify specific codons targeted by positive selection, we applied a conservative strategy and called a site as positively selected only if it was detected by at least two of the following methods: BEB, FUBAR or FEL (see Materials and Methods). The positively selected sites are reported in Table 2. Briefly, we identified 48 positively selected sites 38 of which in meiotic cohesins (10 in RAD21L and STAG3, 12 in REC8, and 6 in SMC1B), 4 in NCAPG, 4 in SMC5 and 2 in NSMCE4A. We next aimed to investigate the potential functional effects of positive selection. By looking at the positions of the positively selected sites within the proteins, we observe that most sites (~67%) are located in intrinsically disordered regions (IDRs) (Figure 2). IDRs are regions that do not adopt a stable three dimensional structure, but rather exist in a collection of structurally distinct conformers. Nevertheless, they are known to play different regulatory functions in the cell and mediate protein-protein interactions, because their lack of structure allows them to adapt their conformation to different interacting partners [63]. We thus tested whether in these genes IDRs are significantly enriched of positively selected sites. We found this to be the case for RAD21L1, REC8, STAG3 and SMC5 (binomial test; RAD21L1 p-value: 0.01; REC8 p-value: 0.013; STAG3 p-value: 1.77x10-4; SMC5 p-value: 0.023). Moreover, proteins containing IDRs are known to be essential for phase separation (PS). PS plays a role in many biological processes, including chromosome organization [64,65,66]. We thus applied the ParSe (Partition Sequence) method (10.1002/pro.4756) to identify regions that promote PS in the selected genes. PS-promoting regions were detected in the IDRs of RAD21L1 and STAG3. Interestingly, all three PS-promoting regions identified carry at least one positively selected site (Figure 2).
We then scanned protein sequences using the PROSITE tool to infer functional motifs. In summary, 13 out of 48 positively selected sites fall in a funcional motif: 9 are phosphorilation sites, 3 myristorylation sites, and 1 a glycosilation site. Notably, in NCAPG, 3 out of 4 positively selected sites are phosphorilation sites, recognized by Protein Kinase C (site 36) and Casein Kinase 2 (sites 37 and 84), of which site 37 involves the residue that is phosphorylated. Finally, since most SMC complex components have nuclear expression, we looked for nuclear localization signals in positively selected genes. In STAG3 two positively selected sites (R83 and H86) fall- in nuclear localization signals predicted by the NLStradamus software.
3.4. Meiotic Cohesin Evolutionary Rates Correlate with Expression during Female Meiosis
Because meiosis-specific Cohesin genes displayed high average dN–dS values and were found to be positively selected, we investigated the relationship between evolutionary rates and gene meiotic expression. In particular, we used genome-wide RNA-seq data for fetal mouse ovaries to retrieve information on gene expression before and during meiosis [50]. Specifically, we obtained expression level changes (fold-change) for the leptotene (E14.5) and pachytene (E16.5) stages compared to a pre-meiotic (E12.5) stage. Furthermore, we retrieved expression changes during different stages of mouse male meiosis compared to pre-meiotic stages (6 days post partum, dpp). In particular, time periods that roughly correspond to the leptotene/zygotene stage (10 dpp) and pachytene stage (14 dpp) were analyzed [51]. Finally, these values were correlated to average dN/dS. A positive correlation was obtained for the leptotene stage of female meiosis, whereas a correlation with borderline significance was observed for the pachytene stage (Figure 3). Conversely, no significant correlation was observed between dN/dS and increased meiotic expression for male meiosis. As shown in the Figure 3, meiotic cohesin genes that are up-regulated in female meiosis evolve faster than mitotic cohesins; these latter, condensin and SMC5/6 subunits show no or limited upregulation during meiosis.
4. Discussion
Large-scale three-dimensional rearrangements of chromosomal DNA drive and facilitate diverse genomic processes, from chromosome segregation to gene expression, DNA repair, and recombination. SMC complexes are involved in these fundamental processes of genome organization, they are essential for all organisms across the tree of life, and they are deeply conserved in eukaryotes [1]. The importance of these complexes is not limited to mitosis and meiosis, where in fact they are fundamental, but they participate with different functions throughout the all cell cycle [16]. The pivotal role played by the SMC components is confirmed by two other pieces of evidence: i) mutations in SMC genes determine pathological conditions, including tumor forms; ii) some of these genes are targets of natural selection as previously reported in Drosophila and in some mammalian groups [37,62]. In these studies, evolutionary analyses have only been conducted on a limited number of SMC genes. Thus, we aimed to cover this gap by analyzing the evolutionary history of all the components of the SMC complexes, including meiotic cohesins, which were never analyzed previously. Indeed, given the key role of these genes in the regulation of primary biological processes of the cell machinery, many different selective forces are expected to drive their evolution.
Our observations on the genes of the cohesin complexes are particularly interesting. In these genes, two distinct trends are highlighted. On one hand, the mitotic cohesins are highly constrained; on the other, the meiotic cohesins show signals of pervasive positive selection. Indeed, in all cohesin genes with predominantly meiotic expression we identified strong positive selection signals and the selected sites are significantly clustered within IDRs, supporting growing evidence that IDRs are fast evolving in different systems [67,68,69,70,70,71,72,73]. Protein containing IDRs are known to be essential for phase separation (PS), a process that consists in the compartmentalization of proteins and nucleic acids within the cell and plays a role in a wide range of processes, including meiotic chromosome organization, chromosome dynamics and meiotic sex chromosome inactivation (MSCI) [64,65,66,74]. A series of meiosis-specific events, including programmed DNA double-strand break formation, homologous pairing, synaptonemal complex installation, and inter-homolog crossover formation, take place to ensure successful chromosome segregation. During meiosis, cohesins and chromosomal phase separation are fundamental in these processes. In this light, we suggest that meiotic cohesins may be engaged in an intra-genomic conflict similar to the ones previously described for centromeres, telomeres, and telomere/centromere-binding proteins [75,76,77]. The centromere drive hypothesis posits that selfish centromeric DNA elements promote their preferential inclusion in the oocyte through the recruitment of kinetochore components. Similarly, we previously proposed that selfish subtelomeric DNA elements can influence the directionality of chromosome movements to the centrosome during meiosis, and that this skews their segregation; the fast evolution of telomere-binding proteins would thus serve the purpose of suppressing meiotic drive and restore equal partitioning [75]. Because cohesins can potentially influence chromosome movement during meiosis, they may also participate in the control of cheating DNA elements to ensure proper segregation. In support of this hypothesis, we detected a significant correlation between the evolutionary rate of meiotic cohesin genes and their upregulation during mouse female meiosis. We thus suggest that cohesins join centromere- and telomere-binding proteins as elements involved in intra-genomic conflicts fueled by selfish elements that promote meiotic drive. Also, MSCI is considered a driving force for genomic evolution. In particular, germline X chromosome inactivation, which occurs in the in the germ cells of XY males, has been linked to genetic conflict related to sexual antagonism [78]. Thus, an alternative, non mutually exclusive possibility is that meiotic cohesins are involved in an intra-genetic conflict related to MSCI.
The SMC5/6 complex, in addition to its physiological roles in chromosome maintenance (repair of chromosomal DNA, conformational compaction of bound DNA, DNA replication), functions as a host restriction factor against several viruses, including HBV, unintegrated HIV-1, papillomavirus (HPV), and different herpesviruses (KSHV, EBV, HSV-1) [30]. The SMC5/6 complex recognizes and binds viral episomal DNA molecules inducing their epigenetic silencing. In turn, episomal DNA viruses antagonize the function of the SMC5/6 complex by expressing viral proteins that degrade one or more SMC5/6 components. For example, the HBV HBx protein recruits cellular DNA damage-binding protein 1 (DDB1), which contains an E3 ubiquitin ligase that targets SMC5/6 for proteasomal degradation. This antagonism of the SMC5/6 complex by HBx is an evolutionarily conserved function found in divergent mammalian HBV species [62] and leads to the specific degradation of SMC5 and SMC6 components [28,29]. A similar function is reported for EBV BNRF1 and KSHV RTA [34,36].
In general, these observations suggest that components of the SMC5/6 complex are engaged in a host-pathogen genetic conflict. The latter ensues when a host restriction factor targets one or more viruses, which evolve counter-restriction mechanisms. The viral proteins mutate to escape restriction by the host factor, which in turn evolves to re-estabilish viral restriction. This cycle recurs repeatedly and results in an evolutionary arms race [79].
The arms race with viral pathogens may underlie the positive selection signal identified in the two components of the SMC5/6 complex, as both are directly involved in pathogen-host conflict: SMC5 is a HBV Hbx target for proteasomal degradation, while NSMCE4A interacts with episomal DNA template.
Mammals have two Nse4 paralogs, Nse4a and Nse4b (encoded by NSMCE4A and EID3, respectively), which share two highly conserved kleisin domains. The two proteins are equally efficient at supporting the assembly of a full SMC5/6 complex, nevertheless it has been suggested that smc5/6 containing NSE4a or NSE4b may exhibit different DNA binding substrate preferences [80]. Indeed, the Nse4a-containing SMC5/6 complex exhibits episomal restriction activity and has been recovered in HBx pull-down experiments. In contrast, the Nse4b-containing SMC5/6 complex is defective in its interaction with episomal DNA template, supporting our hypothesis that the positive selection signals identified in NSMCE4A gene (but not in EID3 gene) arise from a host-pathogen conflict.
An evolutionary conflict between hosts and pathogens could also underlie the positive selection found in NCAPG. By acting on the condensin complex, gammaherpesviruses are able to induce host chromosomal condensation to promote the replication of the viral genome. EBV is known to activate the condensin complex by NCAPG phosphorilation [81]. Specifically the viral BGLF4 kinase induces NCAPG phosphorylation at the Cdc2 target motifs, suggesting that the viral kinase might induce chromosome condensation by mimicking Cdc2. The Condensin I complex is constitutively present throughout the cell cycle and regulates the state of chromatin condensation, which is in a relaxed form during interphase and is converted into compact rod-like structures (chromosomes) over a short period of time during mitosis. The function of Condensin I must be tightly regulated during the cell cycle and this occurs through the phosphorylation of its components by different kinases. Three of the four positively selected sites in NCAPG fall into phosphorylation sites and in particular site 37 corresponds to the residue that is phosphorylated by Casein Kinase 2 (CK2). CK2 is the main kinase that phosphorylates Condensin I during interphase and reduces its supercoiling activity, in contrast to the slight stimulatory effect of mitosis-specific phosphorylation by Cdc2 [82]. We speculate that other NCAPG phosphorylation sites other than Cdc2 sites may be the targets of viral kinases determining the effects of natural selection on this gene and in particular on CK2 phoshorilation sites.
In conclusion, we suggest that the natural selection signals identified in SMC complexes may be the result of different selective pressures. Regarding the selection signals in the condensin and SMC5/6 complexes, the data suggest a host-pathogen arms race. In contrast, the evolutionary rate of meiotic cohesion genes could be the result of an intragenomic conflict similar to that described for centromeres and telomeres.
Author Contributions
Conceptualization: Cagliani Rachele and Manuela Sironi; Formal analysis: Cagliani Rachele, Diego Forni, Manuela Sironi; Investigation: Rachele Cagliani, Diego Forni, Alessandra Mozzi; Visualization: Cagliani Rachele, Diego Forni, Alessandra Mozzi; Writing –Original Draft: Cagliani Rachele, Manuela Sironi; Writing –Review & Editing: Cagliani Rachele, Manuela Sironi, Alessandra Mozzi, Diego Forni; Funding acquisition: Manuela Sironi. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Italian Ministry of Health (“Ricerca Corrente” to MS).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data that support the findings of this study are included in the article, further inquiries can be directed to the corresponding author upon reasonable request.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Uhlmann, F. SMC Complexes: From DNA to Chromosomes. Nature reviews.Molecular cell biology 2016, 17, 399–412. [Google Scholar] [CrossRef] [PubMed]
- Haering, C.H.; Gruber, S. SnapShot: SMC Protein Complexes Part I. Cell 2016, 164, 326–326.e1. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Kim, J.-J.; Ryu, J.-K. Mechanism of Phase Condensation for Chromosome Architecture and Function. Exp Mol Med 2024, 56, 809–819. [Google Scholar] [CrossRef]
- Ryu, J.-K.; Bouchoux, C.; Liu, H.W.; Kim, E.; Minamino, M.; De Groot, R.; Katan, A.J.; Bonato, A.; Marenduzzo, D.; Michieletto, D.; et al. Bridging-Induced Phase Separation Induced by Cohesin SMC Protein Complexes. Sci. Adv. 2021, 7, eabe5905. [Google Scholar] [CrossRef] [PubMed]
- Erdel, F.; Rippe, K. Formation of Chromatin Subcompartments by Phase Separation. Biophysical Journal 2018, 114, 2262–2270. [Google Scholar] [CrossRef] [PubMed]
- Beverley, R.; Snook, M.L.; Brieño-Enríquez, M.A. Meiotic Cohesin and Variants Associated With Human Reproductive Aging and Disease. Front. Cell Dev. Biol. 2021, 9, 710033. [Google Scholar] [CrossRef] [PubMed]
- Hill, V.K.; Kim, J.-S.; Waldman, T. Cohesin Mutations in Human Cancer. Biochimica et Biophysica Acta (BBA) - Reviews on Cancer 2016, 1866, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Pati, D. Role of Chromosomal Cohesion and Separation in Aneuploidy and Tumorigenesis. Cell. Mol. Life Sci. 2024, 81, 100. [Google Scholar] [CrossRef]
- Di Nardo, M.; Pallotta, M.M.; Musio, A. The Multifaceted Roles of Cohesin in Cancer. J Exp Clin Cancer Res 2022, 41, 96. [Google Scholar] [CrossRef]
- Liu, J.; Krantz, I.D. Cornelia de Lange Syndrome, Cohesin, and Beyond. Clinical genetics 2009, 76, 303–314. [Google Scholar] [CrossRef]
- Barbero, J.L. Genetic Basis of Cohesinopathies. The application of clinical genetics 2013, 6, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Kline, A.D.; Moss, J.F.; Selicorni, A.; Bisgaard, A.-M.; Deardorff, M.A.; Gillett, P.M.; Ishman, S.L.; Kerr, L.M.; Levin, A.V.; Mulder, P.A.; et al. Diagnosis and Management of Cornelia de Lange Syndrome: First International Consensus Statement. Nat Rev Genet 2018, 19, 649–666. [Google Scholar] [CrossRef]
- Kline, A.D.; Krantz, I.D.; Sommer, A.; Kliewer, M.; Jackson, L.G.; FitzPatrick, D.R.; Levin, A.V.; Selicorni, A. Cornelia de Lange Syndrome: Clinical Review, Diagnostic and Scoring Systems, and Anticipatory Guidance. American J of Med Genetics Pt A 2007, 143A, 1287–1296. [Google Scholar] [CrossRef] [PubMed]
- Yuan, B.; Neira, J.; Pehlivan, D.; Santiago-Sim, T.; Song, X.; Rosenfeld, J.; Posey, J.E.; Patel, V.; Jin, W.; Adam, M.P.; et al. Clinical Exome Sequencing Reveals Locus Heterogeneity and Phenotypic Variability of Cohesinopathies. Genetics in Medicine 2019, 21, 663–675. [Google Scholar] [CrossRef] [PubMed]
- Hirano, T. Condensin-Based Chromosome Organization from Bacteria to Vertebrates. Cell 2016, 164, 847–857. [Google Scholar] [CrossRef]
- Hoencamp, C.; Rowland, B.D. Genome Control by SMC Complexes. Nat Rev Mol Cell Biol 2023, 24, 633–650. [Google Scholar] [CrossRef]
- Ono, T.; Fang, Y.; Spector, D.L.; Hirano, T. Spatial and Temporal Regulation of Condensins I and II in Mitotic Chromosome Assembly in Human Cells. MBoC 2004, 15, 3296–3308. [Google Scholar] [CrossRef]
- Cuylen, S.; Haering, C.H. Deciphering Condensin Action during Chromosome Segregation. Trends in Cell Biology 2011, 21, 552–559. [Google Scholar] [CrossRef]
- Kinoshita, K.; Hirano, T. Dynamic Organization of Mitotic Chromosomes. Current Opinion in Cell Biology 2017, 46, 46–53. [Google Scholar] [CrossRef]
- Kakui, Y.; Uhlmann, F. SMC Complexes Orchestrate the Mitotic Chromatin Interaction Landscape. Curr Genet 2018, 64, 335–339. [Google Scholar] [CrossRef]
- Martin, C.-A.; Murray, J.E.; Carroll, P.; Leitch, A.; Mackenzie, K.J.; Halachev, M.; Fetit, A.E.; Keith, C.; Bicknell, L.S.; Fluteau, A.; et al. Mutations in Genes Encoding Condensin Complex Proteins Cause Microcephaly through Decatenation Failure at Mitosis. Genes Dev. 2016, 30, 2158–2172. [Google Scholar] [CrossRef] [PubMed]
- Pang, D.; Yu, S.; Yang, X. A Mini-Review of the Role of Condensin in Human Nervous System Diseases. Front. Mol. Neurosci. 2022, 15, 889796. [Google Scholar] [CrossRef] [PubMed]
- Peng, X.P.; Zhao, X. The Multi-Functional Smc5/6 Complex in Genome Protection and Disease. Nat Struct Mol Biol 2023, 30, 724–734. [Google Scholar] [CrossRef] [PubMed]
- Aragón, L. The Smc5/6 Complex: New and Old Functions of the Enigmatic Long-Distance Relative. Annu. Rev. Genet. 2018, 52, 89–107. [Google Scholar] [CrossRef] [PubMed]
- Hwang, G.; Sun, F.; O’Brien, M.; Eppig, J.J.; Handel, M.A.; Jordan, P.W. SMC5/6 Is Required for the Formation of Segregation-Competent Bivalent Chromosomes during Meiosis I in Mouse Oocytes. Development 2017, dev.145607. [Google Scholar] [CrossRef] [PubMed]
- Payne, F.; Colnaghi, R.; Rocha, N.; Seth, A.; Harris, J.; Carpenter, G.; Bottomley, W.E.; Wheeler, E.; Wong, S.; Saudek, V.; et al. Hypomorphism in Human NSMCE2 Linked to Primordial Dwarfism and Insulin Resistance. J. Clin. Invest. 2014, 124, 4028–4038. [Google Scholar] [CrossRef] [PubMed]
- Van Der Crabben, S.N.; Hennus, M.P.; McGregor, G.A.; Ritter, D.I.; Nagamani, S.C.S.; Wells, O.S.; Harakalova, M.; Chinn, I.K.; Alt, A.; Vondrova, L.; et al. Destabilized SMC5/6 Complex Leads to Chromosome Breakage Syndrome with Severe Lung Disease. Journal of Clinical Investigation 2016, 126, 2881–2892. [Google Scholar] [CrossRef]
- Decorsière, A.; Mueller, H.; Van Breugel, P.C.; Abdul, F.; Gerossier, L.; Beran, R.K.; Livingston, C.M.; Niu, C.; Fletcher, S.P.; Hantz, O.; et al. Hepatitis B Virus X Protein Identifies the Smc5/6 Complex as a Host Restriction Factor. Nature 2016, 531, 386–389. [Google Scholar] [CrossRef]
- Murphy, C.M.; Xu, Y.; Li, F.; Nio, K.; Reszka-Blanco, N.; Li, X.; Wu, Y.; Yu, Y.; Xiong, Y.; Su, L. Hepatitis B Virus X Protein Promotes Degradation of SMC5/6 to Enhance HBV Replication. Cell Reports 2016, 16, 2846–2854. [Google Scholar] [CrossRef]
- Irwan, I.D.; Cullen, B.R. The SMC5/6 Complex: An Emerging Antiviral Restriction Factor That Can Silence Episomal DNA. PLoS Pathog 2023, 19, e1011180. [Google Scholar] [CrossRef]
- Xu, W.; Ma, C.; Zhang, Q.; Zhao, R.; Hu, D.; Zhang, X.; Chen, J.; Liu, F.; Wu, K.; Liu, Y.; et al. PJA1 Coordinates with the SMC5/6 Complex To Restrict DNA Viruses and Episomal Genes in an Interferon-Independent Manner. J Virol 2018, 92, e00825-18. [Google Scholar] [CrossRef]
- Gibson, R.T.; Androphy, E.J. The SMC5/6 Complex Represses the Replicative Program of High-Risk Human Papillomavirus Type 31. Pathogens 2020, 9, 786. [Google Scholar] [CrossRef]
- Bentley, P.; Tan, M.J.A.; McBride, A.A.; White, E.A.; Howley, P.M. The SMC5/6 Complex Interacts with the Papillomavirus E2 Protein and Influences Maintenance of Viral Episomal DNA. J Virol 2018, 92, e00356-18. [Google Scholar] [CrossRef]
- Yiu, S.P.T.; Guo, R.; Zerbe, C.; Weekes, M.P.; Gewurz, B.E. Epstein-Barr Virus BNRF1 Destabilizes SMC5/6 Cohesin Complexes to Evade Its Restriction of Replication Compartments. Cell Reports 2022, 38, 110411. [Google Scholar] [CrossRef]
- Dupont, L.; Bloor, S.; Williamson, J.C.; Cuesta, S.M.; Shah, R.; Teixeira-Silva, A.; Naamati, A.; Greenwood, E.J.D.; Sarafianos, S.G.; Matheson, N.J.; et al. The SMC5/6 Complex Compacts and Silences Unintegrated HIV-1 DNA and Is Antagonized by Vpr. Cell Host & Microbe 2021, 29, 792–805.e6. [Google Scholar] [CrossRef]
- Han, C.; Zhang, D.; Gui, C.; Huang, L.; Chang, S.; Dong, L.; Bai, L.; Wu, S.; Lan, K. KSHV RTA Antagonizes SMC5/6 Complex-Induced Viral Chromatin Compaction by Hijacking the Ubiquitin-Proteasome System. PLoS Pathog 2022, 18, e1010744. [Google Scholar] [CrossRef] [PubMed]
- King, T.D.; Leonard, C.J.; Cooper, J.C.; Nguyen, S.; Joyce, E.F.; Phadnis, N. Recurrent Losses and Rapid Evolution of the Condensin II Complex in Insects. Molecular Biology and Evolution 2019, 36, 2195–2204. [Google Scholar] [CrossRef]
- Vilella, A.J.; Severin, J.; Ureta-Vidal, A.; Heng, L.; Durbin, R.; Birney, E. EnsemblCompara GeneTrees: Complete, Duplication-Aware Phylogenetic Trees in Vertebrates. Genome research 2009, 19, 327–335. [Google Scholar] [CrossRef] [PubMed]
- Wernersson, R. RevTrans: Multiple Alignment of Coding DNA from Aligned Amino Acid Sequences. Nucleic Acids Research 2003, 31, 3537–3539. [Google Scholar] [CrossRef]
- Guindon, S.; Delsuc, F.; Dufayard, J.F.; Gascuel, O. Estimating Maximum Likelihood Phylogenies with PhyML. Methods in molecular biology (Clifton, N.J.) 2009, 537, 113–137. [Google Scholar] [CrossRef]
- Anisimova, M.; Nielsen, R.; Yang, Z. Effect of Recombination on the Accuracy of the Likelihood Method for Detecting Positive Selection at Amino Acid Sites. Genetics 2003, 164, 1229–1236. [Google Scholar] [CrossRef] [PubMed]
- Sironi, M.; Cagliani, R.; Forni, D.; Clerici, M. Evolutionary Insights into Host-Pathogen Interactions from Mammalian Sequence Data. Nat Rev Genet 2015, 16, 224–236. [Google Scholar] [CrossRef] [PubMed]
- Pond, S.L.K.; Posada, D.; Gravenor, M.B.; Woelk, C.H.; Frost, S.D. Automated Phylogenetic Detection of Recombination Using a Genetic Algorithm. Molecular biology and evolution 2006, 23, 1891–1901. [Google Scholar] [CrossRef] [PubMed]
- Pond, S.L.K.; Frost, S.D.W.; Muse, S.V. HyPhy: Hypothesis Testing Using Phylogenies. Bioinformatics 2005, 21, 676–679. [Google Scholar] [CrossRef]
- Yang, Z. PAML 4: Phylogenetic Analysis by Maximum Likelihood. Mol Biol Evol 2007, 24, 1586–1591. [Google Scholar] [CrossRef] [PubMed]
- Anisimova, M.; Bielawski, J.P.; Yang, Z. Accuracy and Power of Bayes Prediction of Amino Acid Sites Under Positive Selection. Molecular Biology and Evolution 2002, 19, 950–958. [Google Scholar] [CrossRef] [PubMed]
- Murrell, B.; Wertheim, J.O.; Moola, S.; Weighill, T.; Scheffler, K.; Pond, S.L.K. Detecting Individual Sites Subject to Episodic Diversifying Selection. PLoS genetics 2012, 8, e1002764. [Google Scholar] [CrossRef] [PubMed]
- Kosakovsky Pond, S.L.; Frost, S.D.W. Not So Different After All: A Comparison of Methods for Detecting Amino Acid Sites Under Selection. Molecular Biology and Evolution 2005, 22, 1208–1222. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Nielsen, R. Synonymous and Nonsynonymous Rate Variation in Nuclear Genes of Mammals. J Mol Evol 1998, 46, 409–418. [Google Scholar] [CrossRef]
- Soh, Y.Q.; Junker, J.P.; Gill, M.E.; Mueller, J.L.; Oudenaarden, A. van; Page, D.C. A Gene Regulatory Program for Meiotic Prophase in the Fetal Ovary. PLoS genetics 2015, 11, e1005531. [Google Scholar] [CrossRef]
- Margolin, G.; Khil, P.P.; Kim, J.; Bellani, M.A.; Camerini-Otero, R.D. Integrated Transcriptome Analysis of Mouse Spermatogenesis. BMC genomics 2014, 15, 39–39. [Google Scholar] [CrossRef] [PubMed]
- Emenecker, R.J.; Griffith, D.; Holehouse, A.S. Metapredict V2: An Update to Metapredict, a Fast, Accurate, and Easy-to-Use Predictor of Consensus Disorder and Structure 2022.
- Emenecker, R.J.; Griffith, D.; Holehouse, A.S. Metapredict: A Fast, Accurate, and Easy-to-Use Predictor of Consensus Disorder and Structure. Biophysical Journal 2021, 120, 4312–4319. [Google Scholar] [CrossRef] [PubMed]
- Sigrist, C.J.A.; De Castro, E.; Cerutti, L.; Cuche, B.A.; Hulo, N.; Bridge, A.; Bougueleret, L.; Xenarios, I. New and Continuing Developments at PROSITE. Nucleic Acids Research 2012, 41, D344–D347. [Google Scholar] [CrossRef]
- Nguyen Ba, A.N.; Pogoutse, A.; Provart, N.; Moses, A.M. NLStradamus: A Simple Hidden Markov Model for Nuclear Localization Signal Prediction. BMC Bioinformatics 2009, 10, 202. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, A.Y.; Khaodeuanepheng, N.P.; Amarasekara, D.L.; Correia, J.J.; Lewis, K.A.; Fitzkee, N.C.; Hough, L.E.; Whitten, S.T. Intrinsically Disordered Regions That Drive Phase Separation Form a Robustly Distinct Protein Class. Journal of Biological Chemistry 2023, 299, 102801. [Google Scholar] [CrossRef] [PubMed]
- Wilson, C.; Lewis, K.A.; Fitzkee, N.C.; Hough, L.E.; Whitten, S.T. ParSe 2.0: A Web Tool to Identify Drivers of Protein Phase Separation at the Proteome Level. Protein Science 2023, 32, e4756. [Google Scholar] [CrossRef]
- Tesei, G.; Trolle, A.I.; Jonsson, N.; Betz, J.; Pesce, F.; Johansson, K.E.; Lindorff-Larsen, K. Conformational Ensembles of the Human Intrinsically Disordered Proteome: Bridging Chain Compaction with Function and Sequence Conservation 2023.
- Tesei, G.; Lindorff-Larsen, K. Improved Predictions of Phase Behaviour of Intrinsically Disordered Proteins by Tuning the Interaction Range. Open Res Europe 2023, 2, 94. [Google Scholar] [CrossRef] [PubMed]
- Ebel, E.R.; Telis, N.; Venkataram, S.; Petrov, D.A.; Enard, D. High Rate of Adaptation of Mammalian Proteins That Interact with Plasmodium and Related Parasites. PLoS genetics 2017, 13, e1007023. [Google Scholar] [CrossRef]
- Yang, Z. PAML: A Program Package for Phylogenetic Analysis by Maximum Likelihood. Computer applications in the biosciences : CABIOS 1997, 13, 555–556. [Google Scholar] [CrossRef] [PubMed]
- Abdul, F.; Filleton, F.; Gerossier, L.; Paturel, A.; Hall, J.; Strubin, M.; Etienne, L. Smc5/6 Antagonism by HBx Is an Evolutionarily Conserved Function of Hepatitis B Virus Infection in Mammals. J Virol 2018, 92, e00769-18. [Google Scholar] [CrossRef]
- Wright, P.E.; Dyson, H.J. Intrinsically Disordered Proteins in Cellular Signalling and Regulation. Nat Rev Mol Cell Biol 2015, 16, 18–29. [Google Scholar] [CrossRef] [PubMed]
- Strom, A.R.; Emelyanov, A.V.; Mir, M.; Fyodorov, D.V.; Darzacq, X.; Karpen, G.H. Phase Separation Drives Heterochromatin Domain Formation. Nature 2017, 547, 241–245. [Google Scholar] [CrossRef]
- Mirny, L.A.; Imakaev, M.; Abdennur, N. Two Major Mechanisms of Chromosome Organization. Current Opinion in Cell Biology 2019, 58, 142–152. [Google Scholar] [CrossRef]
- Larson, A.G.; Elnatan, D.; Keenen, M.M.; Trnka, M.J.; Johnston, J.B.; Burlingame, A.L.; Agard, D.A.; Redding, S.; Narlikar, G.J. Liquid Droplet Formation by HP1α Suggests a Role for Phase Separation in Heterochromatin. Nature 2017, 547, 236–240. [Google Scholar] [CrossRef]
- Holehouse, A.S.; Kragelund, B.B. The Molecular Basis for Cellular Function of Intrinsically Disordered Protein Regions. Nat Rev Mol Cell Biol 2024, 25, 187–211. [Google Scholar] [CrossRef] [PubMed]
- Afanasyeva, A.; Bockwoldt, M.; Cooney, C.R.; Heiland, I.; Gossmann, T.I. Human Long Intrinsically Disordered Protein Regions Are Frequent Targets of Positive Selection. Genome research 2018, 28, 975–982. [Google Scholar] [CrossRef]
- Brown, C.J.; Johnson, A.K.; Dunker, A.K.; Daughdrill, G.W. Evolution and Disorder. Current opinion in structural biology 2011, 21, 441–446. [Google Scholar] [CrossRef] [PubMed]
- Molteni, C.; Forni, D.; Cagliani, R.; Mozzi, A.; Clerici, M.; Sironi, M. Evolution of the Orthopoxvirus Core Genome. Virus Research 2023, 323, 198975. [Google Scholar] [CrossRef]
- Mozzi, A.; Forni, D.; Cagliani, R.; Clerici, M.; Pozzoli, U.; Sironi, M. Intrinsically Disordered Regions Are Abundant in Simplexvirus Proteomes and Display Signatures of Positive Selection. Virus evolution 2020, 6, veaa028. [Google Scholar] [CrossRef]
- Zarin, T.; Strome, B.; Nguyen Ba, A.N.; Alberti, S.; Forman-Kay, J.D.; Moses, A.M. Proteome-Wide Signatures of Function in Highly Diverged Intrinsically Disordered Regions. eLife 2019, 8, e46883. [Google Scholar] [CrossRef]
- Cagliani, R.; Forni, D.; Mozzi, A.; Fuchs, R.; Tussia-Cohen, D.; Arrigoni, F.; Pozzoli, U.; De Gioia, L.; Hagai, T.; Sironi, M. Evolution of virus-like features and intrinsically disordered regions in retrotransposon-derived mammalian genes. Molecular Biology and Evolution 2024, in press. [Google Scholar] [CrossRef]
- Zhang, R.; Liu, Y.; Gao, J. Phase Separation in Controlling Meiotic Chromosome Dynamics. In Current Topics in Developmental Biology; Elsevier, 2023; Vol. 151, pp. 69–90 ISBN 978-0-12-820156-5.
- Pontremoli, C.; Forni, D.; Cagliani, R.; Pozzoli, U.; Clerici, M.; Sironi, M. Evolutionary Rates of Mammalian Telomere-Stability Genes Correlate with Karyotype Features and Female Germline Expression. Nucleic acids research 2018, 46, 7153–7168. [Google Scholar] [CrossRef]
- Pontremoli, C.; Forni, D.; Pozzoli, U.; Clerici, M.; Cagliani, R.; Sironi, M. Kinetochore Proteins and Microtubule-destabilizing Factors Are Fast Evolving in Eutherian Mammals. Molecular Ecology 2021, 30, 1505–1515. [Google Scholar] [CrossRef]
- Henikoff, S.; Ahmad, K.; Malik, H.S. The Centromere Paradox: Stable Inheritance with Rapidly Evolving DNA. Science (New York, N.Y.) 2001, 293, 1098–1102. [Google Scholar] [CrossRef]
- Wu, C.-I.; Yujun Xu, E. Sexual Antagonism and X Inactivation – the SAXI Hypothesis. Trends in Genetics 2003, 19, 243–247. [Google Scholar] [CrossRef] [PubMed]
- Tenthorey, J.L.; Emerman, M.; Malik, H.S. Evolutionary Landscapes of Host-Virus Arms Races. Annu. Rev. Immunol. 2022, 40, 271–294. [Google Scholar] [CrossRef]
- Abdul, F.; Diman, A.; Baechler, B.; Ramakrishnan, D.; Kornyeyev, D.; Beran, R.K.; Fletcher, S.P.; Strubin, M. Smc5/6 Silences Episomal Transcription by a Three-Step Function. Nat Struct Mol Biol 2022, 29, 922–931. [Google Scholar] [CrossRef]
- Lee, C.-P.; Huang, Y.-H.; Lin, S.-F.; Chang, Y.; Chang, Y.-H.; Takada, K.; Chen, M.-R. Epstein-Barr Virus BGLF4 Kinase Induces Disassembly of the Nuclear Lamina To Facilitate Virion Production. J Virol 2008, 82, 11913–11926. [Google Scholar] [CrossRef] [PubMed]
- Takemoto, A.; Kimura, K.; Yanagisawa, J.; Yokoyama, S.; Hanaoka, F. Negative Regulation of Condensin I by CK2-Mediated Phosphorylation. EMBO J 2006, 25, 5339–5348. [Google Scholar] [CrossRef]
Figure 1.
Evolutionary rates in SMC complexes. (a) Comparison of evolutionary rates. The distribution of dN/dS values for more than 9000 genes in a representative mammalian phylogeny [60] is shown. The hatched red lines correspond to the 10th, 50th and 90th percentiles. The dN/dS values of the genes we analyzed are indicated. The inset shows the correlation between the dN/dS values we calculated and those previously reported by Ebel and coworkers for 11 SMC complex genes (NCAPD2, NCAPD3, NCAPG, NCAPH, RAD21, RAD21L, REC8, SMC1B, SMC4, STAG3). (b) Boxplot representation of dN–dS values calculated for meiotic and mitotic Cohesin, Condensin, and SMC5/6 genes. Statistical significance was assessed by Nemenyi post hoc pairwise comparison after Kruskal Wallis test. All comparisons are significant with a p-value <0.001.
Figure 1.
Evolutionary rates in SMC complexes. (a) Comparison of evolutionary rates. The distribution of dN/dS values for more than 9000 genes in a representative mammalian phylogeny [60] is shown. The hatched red lines correspond to the 10th, 50th and 90th percentiles. The dN/dS values of the genes we analyzed are indicated. The inset shows the correlation between the dN/dS values we calculated and those previously reported by Ebel and coworkers for 11 SMC complex genes (NCAPD2, NCAPD3, NCAPG, NCAPH, RAD21, RAD21L, REC8, SMC1B, SMC4, STAG3). (b) Boxplot representation of dN–dS values calculated for meiotic and mitotic Cohesin, Condensin, and SMC5/6 genes. Statistical significance was assessed by Nemenyi post hoc pairwise comparison after Kruskal Wallis test. All comparisons are significant with a p-value <0.001.
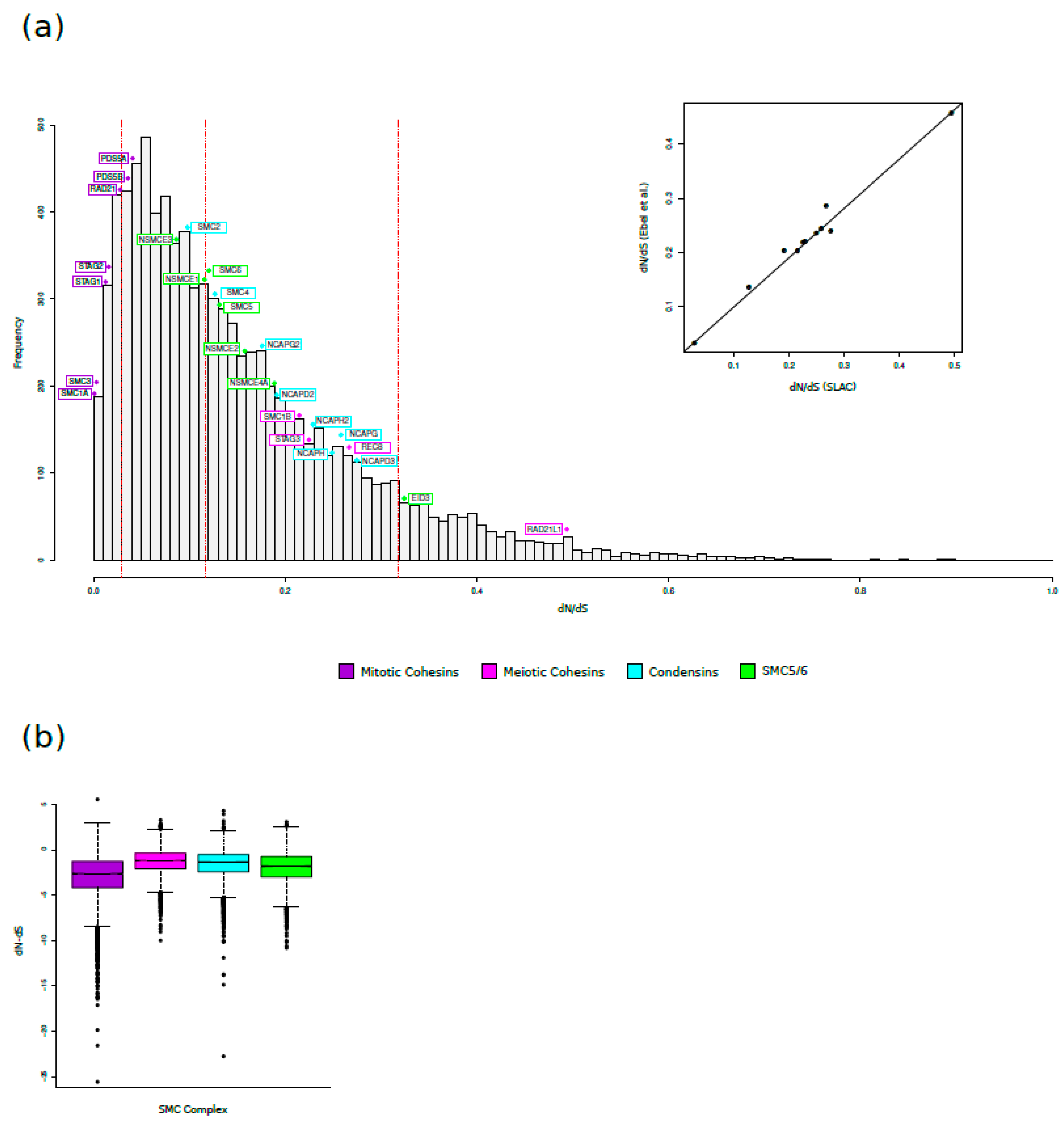
Figure 2.
Domain structures of SMC complexes. Schematic domain structures of the 7 proteins with evidence of positive selection are drawn to scale. Domains are defined using the InterPro (https://www.ebi.ac.uk/interpro/) classification. The grey shaded areas represent IDRs identified by the Metapredict tool based on human proteins. The red arrows denote positively selected sites as obtained from positive selection analysis. ParSe sequences are represented in blue.
Figure 2.
Domain structures of SMC complexes. Schematic domain structures of the 7 proteins with evidence of positive selection are drawn to scale. Domains are defined using the InterPro (https://www.ebi.ac.uk/interpro/) classification. The grey shaded areas represent IDRs identified by the Metapredict tool based on human proteins. The red arrows denote positively selected sites as obtained from positive selection analysis. ParSe sequences are represented in blue.
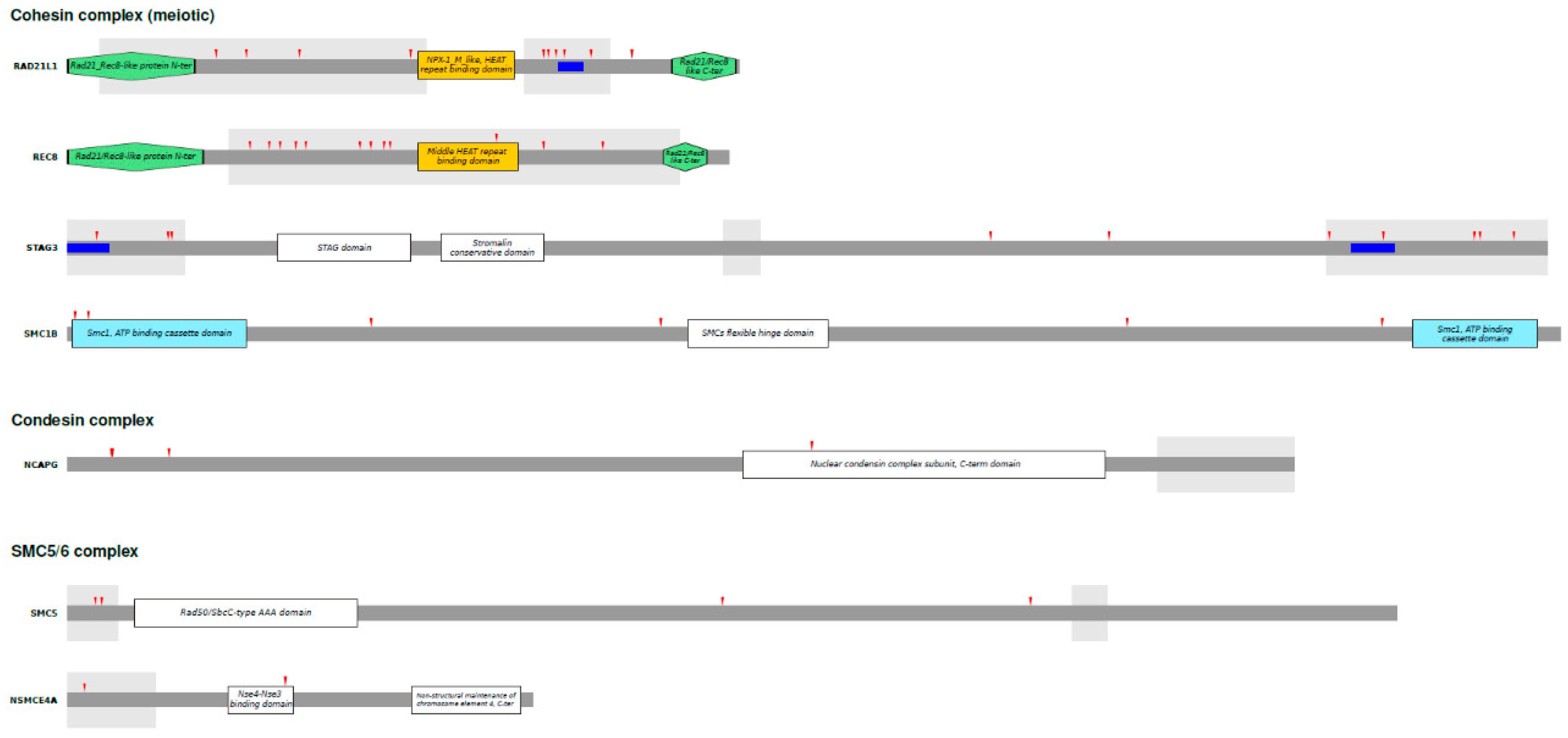
Figure 3.
Evolutionary rates and gene expression in meiosis. Average dN/dS for all SMC complex genes is plotted against the log2 fold-change (FC) of gene expression in the leptotene or pachytene stages versus the pre-meiotic stage of mouse oogenesis or spermatogenesis. Kendall's correlation coefficients are also reported.
Figure 3.
Evolutionary rates and gene expression in meiosis. Average dN/dS for all SMC complex genes is plotted against the log2 fold-change (FC) of gene expression in the leptotene or pachytene stages versus the pre-meiotic stage of mouse oogenesis or spermatogenesis. Kendall's correlation coefficients are also reported.
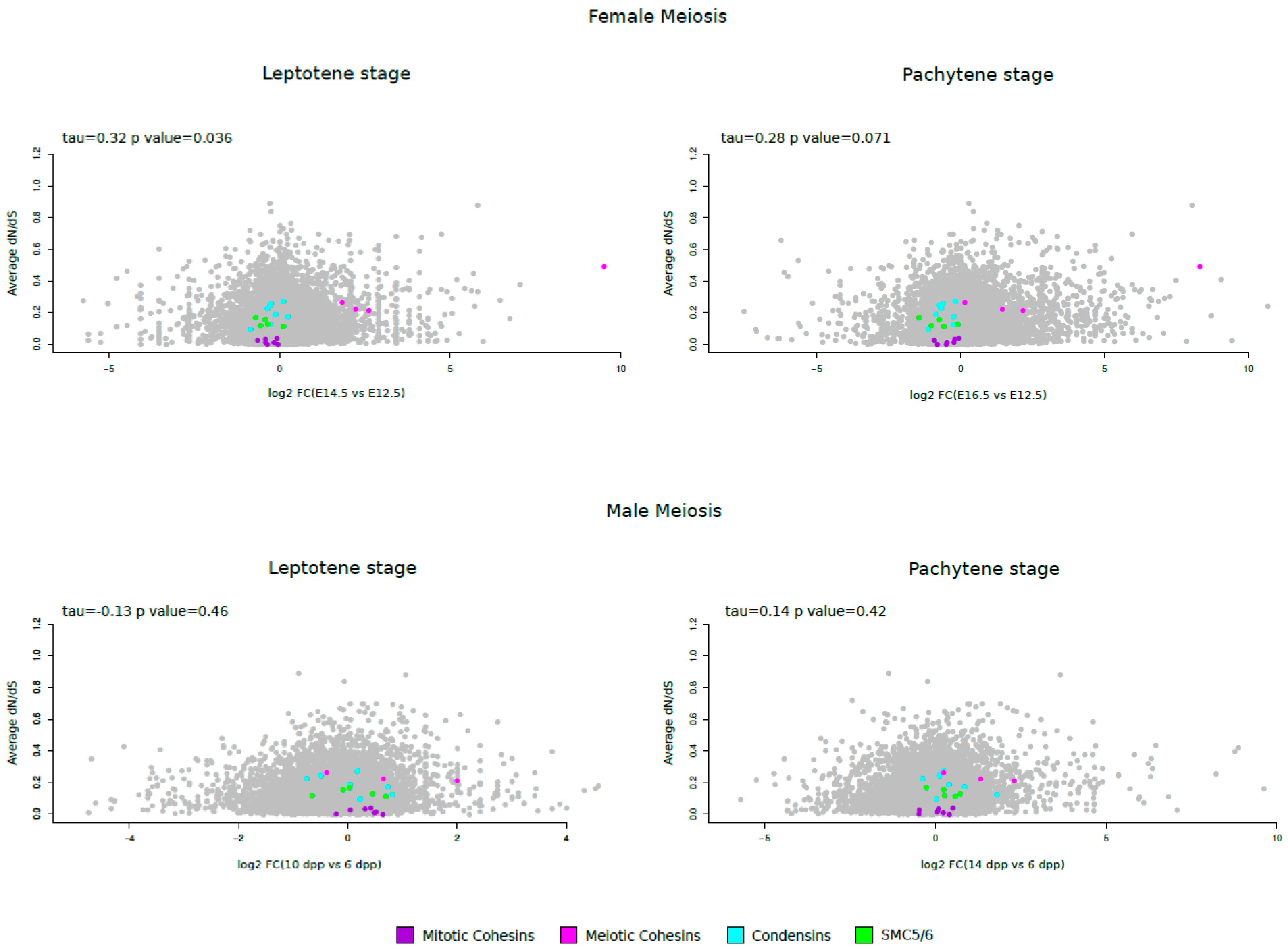

Table 1.
List of analyzed SMC complex genes.
| Gene | Alias gene symbol | Subunits | n. of species | dN/dS | |
|---|---|---|---|---|---|
| Cohesin complex | |||||
| RAD21 | SCC1 | Kleisin | 63 | 0.028 | |
| RAD21L1* | RAD21L | Kleisin | 63 | 0.494 | |
| REC8* | - | Kleisin | 63 | 0.267 | |
| SMC1A | - | SMC | 61 | 0.003 | |
| SMC1B* | - | SMC | 60 | 0.215 | |
| SMC3 | - | SMC | 63 | 0.001 | |
| PDS5A | SCC112 | HEAT-A | 57 | 0.041 | |
| PDS5B | APRIN, AS3 | HEAT-A | 60 | 0.036 | |
| STAG1 | SA1 | HEAT-B | 63 | 0.013 | |
| STAG2 | SA2 | HEAT-B | 63 | 0.016 | |
| STAG3* | SA3 | HEAT-B | 63 | 0.225 | |
| Condensin complex | |||||
| NCAPD2 | CAP-D2 | HEAT-A (I) | 62 | 0.191 | |
| NCAPD3 | CAP-D3 | HEAT-A (II) | 63 | 0.275 | |
| NCAPG | CAP-G | HEAT-B (I) | 63 | 0.258 | |
| NCAPG2 | CAP-G2 | HEAT-B (II) | 59 | 0.176 | |
| NCAPH | CAP-H | Kleisin (I) | 63 | 0.249 | |
| NCAPH2 | CAP-H2 | Kleisin (II) | 62 | 0.229 | |
| SMC2 | CAP-E | SMC | 62 | 0.098 | |
| SMC4 | CAP-C | SMC | 61 | 0.127 | |
| SMC5/6 complex | |||||
| NSMCE1 | NSE1 | Tandem-WHD E3 ligase | 60 | 0.120 | |
| NSMCE2 | NSE2 | SUMO ligase | 63 | 0.158 | |
| NSMCE3 | NSE3/MAGEG1 | Tandem-WHD | 54 | 0.087 | |
| NSMCE4A | NSE4A | Kleisin | 63 | 0.189 | |
| EID3 | NSMCE4B | Kleisin | 46 | 0.342 | |
| SMC5 | - | SMC | 63 | 0.131 | |
| SMC6 | - | SMC | 63 | 0.116 |
Table 2.
Likelihood ratio test statistics for models of variable selective pressure among sites (F3x4 and F61 codon frequency model) for SMC complexes.
Table 2.
Likelihood ratio test statistics for models of variable selective pressure among sites (F3x4 and F61 codon frequency model) for SMC complexes.
| Gene/ LRT model |
n. of species | F3x4 | F61 | Positively selected sitesc | |||
|---|---|---|---|---|---|---|---|
| -2ΔlnLa | p valueb | -2ΔlnLa | p valueb | ||||
| Cohesin Complex | |||||||
| RAD21L* | 63 | ||||||
| M1 vs M2 | 102.59 | 5.28x10-23 | 92.78 | 7.13x10-21 | 122,148,192,284,394,398, 404,411,477,433 |
||
| M7 vs M8 | 113.97 | 1.79x10-25 | 108.91 | 2.25x10-24 | |||
| REC8* | 63 | ||||||
| M1 vs M2 | 51.13 | 7.89x10-12 | 10.11 | 0.0064 | 152,168,191,199,253,264, 269, 358,400,449,178,244 |
||
| M7 vs M8 | 88.22 | 6.97x10-20 | 50.28 | 1.21x10-11 | |||
| SMC1B* | 60 | ||||||
| M1 vs M2 | 37.77 | 6.29x10-09 | 16.92 | 0.00021 | 6,18,251,491,877,1088 | ||
| M7 vs M8 | 105.04 | 1.55x10-23 | 55.29 | 9.85x10-13 | |||
| STAG3* | 62 | ||||||
| M1 vs M2 | 27.39 | 1.13x10-06 | 18.02 | 0.00012 | 24,83,86,764,862,1044, 1089,1154,1159,1197 |
||
| M7 vs M8 | 79.88 | 4.51x10-18 | 58.44 | 2.04x10-13 | |||
| Condensin Complex | |||||||
| NCAPG | 63 | ||||||
| M1 vs M2 | 46.98 | 6.29x10-11 | 48.72 | 2.63x10-11 | 36,37,84,616 | ||
| M7 vs M8 | 90.97 | 1.76x10-20 | 102.35 | 5.96x10-23 | |||
| SMC5/6 Complex | |||||||
| SMC5 | 63 | ||||||
| M1 vs M2 | 17.97 | 0.000125 | 7.91 | 0.019 | 797,38,542,33 | ||
| M7 vs M8 | 61.40 | 4.65x10-14 | 45.29 | 1.46x10-10 | |||
| NSMCE4A | 63 | ||||||
| M1 vs M2 | 33.96 | 4.22x10-08 | 22.82 | 1.11x10-05 | 14, 185 | ||
| M7 vs M8 | 45.11 | 1.60x10-10 | 35.79 | 1.69x10-08 | |||
a Twice the difference of likelihood for the two models compared; b p value of rejecting the neutral models (M8a and M7) in favor of the positive selection model (M8); c positively selected sites detected by at least two methods among BEB, FEL, and FUBAR.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



