
Submitted:
01 August 2024
Posted:
02 August 2024
You are already at the latest version
Abstract
One of the bottlenecks to bringing new therapies to the clinic has been a lack of vectors for delivering novel therapeutics in a targeted manner. Cell penetrating peptides (CPPs) have received a lot of attention and have been the subject of numerous developments since their identification nearly 3 decades ago. Known for their transduction abilities, they have generally been considered inert vectors. In this review we present a schema for their classification, highlight what is known about their mechanism of transduction, and outline the existing literature, as well as our own experience, vis a vis the intrinsic anti-inflammatory properties that certain CPPs exhibit. Given the inflammatory responses associated with viral vectors, CPPs represent a viable alternative to these vectors; further, the anti-inflammatory properties of CPPs, mostly through inhibition of the NF-κB pathway, are encouraging. Much work in relevant animal models, toxicity studies in large animal models, and ultimately human trials are needed before their potential is fully realized.
Keywords:
Cell Penetrating Peptides
; Inflammation
; NF-κB
; Protein Transduction Domains
1. Introduction
Among the most promising forefronts for novel vectors has been the development of protein transduction domains, more commonly referred to as cell penetrating peptides (CPPs). CPPs are small molecules, ranging from 5 to 30 amino acids in length, which function to transduce the cell membrane either alone or attached to a variety of cargos [1]. CPPs are unique in their ability to cross the cell membrane without requiring the use of a receptor, a function first identified in 1988 in HIV’s trans-activator of transcription protein (Tat) [2,3]. Notably, the entire protein is not responsible for cell transduction; the identification of a short, 11-amino-acid, primarily cationic sequence within Tat [3] which proved to be responsible for crossing the membrane has paved the way for the research and development of CPPs as we know them today.
Following the discovery of Tat, other non-cell specific CPPs have been discovered. This includes Antp, also called penetratin, discovered from Drosophila melanogaster [4]. CPPs have traditionally been used mostly for their ability to cross cell membrane barriers and deliver large or hydrophobic cargos, as first demonstrated through the conjugation of proteins such as RNase A, β-galactosidase, and horseradish peroxidase to Tat; in this case, transduction was robustly observed without any cell-type specificity [5]. Such transduction has also been observed in vivo, with protein functionality being preserved after transduction into all tissues including brain tissue [6]. Since then, CPPs have been used in a variety of applications, including targeted drug delivery [7,8,9], the delivery of RNA interference agents such as siRNA [10,11,12,13,14], enhancing the delivery of viral vectors [15] and radioactive agents [16,17], along with many more applications [18,19,20,21,22].
While CPPs remain an exciting frontier in terms of targeted delivery and enhancing cell transduction, they have generally been considered as inert vectors without any intrinsic biological properties. Although studies on both targeted and non-targeted delivery of potentially therapeutic or diagnostic molecules abound in the literature, intrinsic biological properties of CPP have received little attention. Therefore, this review will focus on the existing literature on the intrinsic biological properties of CPPs, beyond transduction, which could potentially be harnessed to treat, and prevent disease more effectively.
2. Mechanisms and Characteristics of CPPs
2.1. Classification of CPPs
CPPs can be categorized in a variety of ways but are generally divided into non-cell-specific peptides and cell-specific peptides. Non-specific peptides can be further divided based on their structures and associated charges; these peptides are often classified as being cationic, hydrophobic, or amphipathic (Figure 1). Tat (YGRKKRRQRRR) and penetratin (YGRKKRRQRRR) are key examples of cationic peptides, rich in both lysine and arginine. Following these examples, synthetic, non-naturally occurring peptides have been synthesized using simply homopolymers of arginine [23] or lysine [24] residues. In fact, Wender et al. found that peptides consisting of only L-arginine or D-arginine were over 20 times better at cell transduction than Tat alone, suggesting that Tat’s cationic properties play a major role in its transduction abilities [23]. Such peptides, however, seem to have a clearly defined length; poly-arginine peptides have been found to be ineffective with less than 6 residues [25] and poly-lysine polymers show a decrease in transduction with greater than 12 amino acids [26].
Though far less common than the other types of CPPs, hydrophobic CPPs have also been successfully used for membrane transduction. These peptides are often created by the conjugation of hydrophobic signal peptide sequences, which usually function to allow the secretion of proteins from inside the cell membrane; modifying these sequences can reverse the direction of peptide travel and lead to membrane transduction instead [1]. Modifications to these peptides’ structure can serve to enhance their stability in solution and transduction ability. One such peptide called TP10, an analog of transportan [27], has been successfully modified to enhance its three-dimensional structure and therefore transduction abilities [28]. The stabilization of three-dimensional structure, specifically helical structures, has been shown to enhance membrane transduction and delivery efficiency [29]. However, hydrophobic CPPs are less stable in solution and present many challenges when it comes to their synthesis and use, such as their tendency to aggregate due to their hydrophobic nature [30].
Amphipathic CPPs, the final type of non-specific cell penetrating peptides, are hybrid peptides created by attaching a hydrophilic sequence to a hydrophobic CPP. For instance, LAH4-L1 is an amphipathic CPP modulated with a nuclear localization signal (NLS) from Simian virus 40; this peptide was able to affect gene expression up to 10-fold compared to baseline levels of expression [31]. Amphipathic peptides can also be created from a mixture of evenly distributed hydrophobic amino acids and other hydrophilic amino acids [32], allowing them to directly interact with the lipid bilayer for internalization [33].
2.2. CPP Transduction
Despite many years of research, the exact mechanism of how CPPs transduce the membrane remains elusive, and likely varies between CPP subtypes as well as their concentrations. Furthermore, CPP internalization has been observed in both energy dependent and energy independent manners. Which pathway a given peptide uses will depend on both the properties of the cell membrane as well as the properties of the peptide and/or its cargo [34].
Initially, membrane transduction by CPPs was observed to be solely an energy independent process. Transduction with transportan was observed to occur at a range of temperatures from 0-37 degrees, and was not prevented by endocytosis inhibition at high concentrations [35]. Tat and penetratin were also observed to enter the cell by energy independent methods [4,36,37]. In an attempt to explain this phenomenon, Grasso et al. [38] describe in detail how membrane mechanics are affected by different CPPs. Binding of various CPPs can rearrange lipid molecules, leading to changes in membrane stiffness and eventual peptide internalization. They also suggest that highly charged CPPs may attract more water to the lipid membrane, further destabilizing it and allowing for peptide entry [37,38]. Indeed, it has been observed that changing the shape of a cellular membrane can affect transduction ability [39]. The changes in the cell membrane which result from CPP association could force the formation of inverted micelles containing the peptide, which then facilitates release into the cytoplasm [40].
Although energy-independent processes for CPP internalization have been widely observed, this process can also occur through endocytic (energy-dependent) pathways, particularly at lower peptide concentrations. Because endocytic pathways create endosomes, CPPs must be able to both induce endocytic uptake and escape the endosome once internalized. Although many endocytic pathways exist, macropinocytosis seems to be the prevailing mechanism for CPP transduction [34]. For instance, Ichimizu et al. [41] describe how macropinocytosis facilitates the entry of a peptide called palmitoyl-cyclic-(D-Arg)12/HSA. In this study, this peptide interacted with several key macropinocytosis markers, including CXCR4 and pathways such as the PKC and mTOR pathways. Endosomal escape of palmitoyl-cyclic-(D-Arg)12/HSA was also observed to be inhibited by the presence of heparin, suggesting that electrostatic interactions play a key role in endosomal escape. Interestingly, this finding suggests that the methods CPPs use to transduce cells by changing phospholipid membrane dynamics can also contribute to endosomal escape.
Other endocytic pathways besides macropinocytosis can be responsible for CPP internalization; for instance, clathrin-mediated endocytosis (CME) provides a logical method for CPP transduction. CME occurs after an endocytosis-inducing ligand binds to a receptor [34]. Therefore, it stands to reason that CPPs could possibly act as an agonist to a CME-associated receptor, activating endocytosis. This phenomenon has been observed with multiple CPPs, including Tat: Richard et al. observed that chlorpromazine, a known inhibitor of CME, inhibited transduction of Tat in HeLa cells, while filipin III and nystatin, both inhibitors of caveolin-dependent endocytosis, had little to no effect on Tat’s transduction abilities [42]. CME dependent transduction has also been observed with other CPPs. For instance, siRNA knockdown of syndecan-4, a surface proteoglycan associated with CME, has been shown to prevent the uptake of an 8 oligomer of arginine [43]. Other studies have also shown chlorpromazine to be an effective inhibitor of CME-associated CPP transduction in various cell types and with various CPPs [44,45,46,47,48,49]. It does appear, however, that the primary pathway (CME, macropinocytosis, direct transduction, etc.) a peptide uses to transduce the membrane is also dependent on concentration; at lower concentrations, especially when peptides are attached to large cargo, endocytosis is the driving factor behind peptide internalization, while at higher concentrations, peptides appear to transduce the membrane independently of endocytosis [34].
2.3. Cell-Specific CPPs
Although non-cell-specific CPPs have played a vital role in CPP research and present exciting new ways to cross the plasma membrane, they show limited clinical usefulness for targeted cell delivery. Cell-specific CPPs provide several key advantages over non-specific CPPs; namely, cell-specific CPPs require lower concentrations and avoid off-target interactions and potential side effects. The identification of such CPPs has largely been accomplished through the use of phage display libraries [1], a technique first identified by Smith et al. [50]. This process has been used successfully many times to identify a variety of cell-specific CPPS, including CPPs to target cardiomyocytes [51], dendritic cells [52], B lymphocytes [53], pancreatic islet cells [54], and more, and does not require a priori knowledge of internalization or binding partners before a cell-specific CPP can be identified.
CPPs with specific biological activity present a new forefront in CPP research. While many studies have been performed to assess the toxicity of such peptides [55,56,57,58,59,60], less research exists on the intrinsic biological properties of these peptides. Cell-specific CPPs with inherent therapeutic properties present a new wave of drug development for highly targeted, direct-to-tissue therapies.
3. Biological Effects and Therapeutic Implications
Cell penetrating peptides have been the subject of intense research since the serendipitous discovery of the Tat protein to have intrinsic transduction ability. These studies have focused on identifying new naturally occurring or synthetic CPPs, their mechanism(s) of transduction, and their applications in both the diagnostic and therapeutic arenas. To date, CPPs have largely been considered inert, small peptides with robust transduction abilities but little intrinsic biological activity beyond transduction. A handful of studies have looked at the anti-inflammatory properties of CPPs, with a larger number pertaining to using CPPs to deliver anti-inflammatory peptides/cargoes in myriad different diseases which have inflammation as an underlying pathophysiological hallmark.. A few studies have looked at the inherent anti-inflammatory nature of known cationic peptides, while others study peptides derived from molecular pathways associated with Toll-like receptors (TLRs), or the NF-κB pathways which were found to also have cell transduction properties.
As stated above, some of the earliest CPPs were cationic, and non-cell specific, like Tat, Antennapedia homeodomain-derived peptide, and synthetic homopolymers of arginine (nona-arginine). These three were compared in a head-to-head fashion in vitro in a human cervical cancer and human melanoma cell line. All three inhibited TNF-α mediated signal transduction by inducing internalization of TNF-α receptors via clathrin mediated endocytosis [61]. The full-length Tat protein had a similar effect, and apoptosis caused by Smac protein (an inhibitor of “Inhibitors of Apoptosis”, or IAPs) was abrogated by Antennapedia [61]. Another in vitro study investigated the effects of various concentrations of an arginine homopolymer (octa-arginine) with incubations at various lengths of time on a U-937 macrophage cell line. There was an initial burst of super-oxide production at 30 mins, but not at later time-points, and no increase in production of pro-inflammatory cytokines like TNF-α, IL-1β, or IL-6 [62]. In another study, antennapedia-derived CPP penetratin decreased transcriptional activity of NF-κB in TNF-α stimulated L929 fibroblasts and lipopolysaccharide-activated RAW 264.7 macrophages [63]. Moreover, in an in vivo rat model of acute pancreatitis, pre-treatment with 2 mg/Kg of penetratin attenuated the severity of pancreatitis by inhibiting IκB degradation and nuclear import of NF-κB dimers, inhibiting expression of several downstream proinflammatory genes [63].
In contrast to the above studies that explored the anti-inflammatory nature and pathways involved of known, well-established CPPs, the following studies have aimed to design peptides with cell penetrating properties and targeting different inflammatory pathways or have aimed to identify anti-inflammatory peptides that serendipitously have cell transduction properties. In this regard, the NF-κB signaling pathway, and modulation thereof, has been the subject of numerous studies due to its central role in a number of inflammatory pathologies, ranging from but not limited to atherosclerosis, diabetes and rheumatoid arthritis. There are highly conserved DNA-binding domains across all NF-κB members. Investigators have searched for peptides that would bind to these domains, thereby inhibiting NK-κB transcriptional activities. One such peptide, AIP6, inhibited DNA binding and transcriptional activity of the p65 NF-κB subunit in stimulated macrophages in vitro, and inhibited zymosan-induced inflammation in vivo in mice [64]. In other studies, another bifunctional peptide, cSN50.1, was designed to inhibit nuclear transport of stress-mediated transcription factors [65]. This peptide mimics the NF-κB/p50 nuclear localizing sequence, with further studies showing it selectively targets importin α5 [66] and mitigates atherosclerosis, fatty liver, blood glucose and lipid levels in a mouse model of familial hypercholesterolemia [67]. This peptide has a hydrophobic region (AAVALLPAVLLALLAP) and a nuclear localizing region (CVQRKRQKLMPC); its ability to cross nuclear membranes translated into a similar ability to cross plasma membranes.
Anti-inflammatory cell penetrating peptides also have applications for the nervous system. Gliomas are intracranial tumors with high mortality and limited options for treatment due to the blood brain barrier which prevents adequate drug concentrations being achieved in tumor tissue. Additionally, NF-κB activation has been correlated with tumor drug resistance. To counter both of these issues, a peptide was specifically designed to have both cell penetrating and NF-κB inhibition capabilities and was conjugated to pegylated liposomes loaded with doxorubicin for delivery to glioma tumors in vivo. This approach decreased tumor size and significantly prolonged survival in nude mice bearing intracranial gliomas [68]. Lastly, a heparin-binding sequence derived from the heparin-binding epidermal growth factor transduced RAW 264.7 macrophages within ten minutes. Pretreatment with this peptide reduced LPS-induced production of nitric oxide, inducible nitric oxide synthase, and cytokines TNF-α and IL-6 in a dose-dependent manner by blocking the phosphorylation and degradation of IκBα leading to inhibition of nuclear translocation of p65 subunit of NF-κB. It also decreased infiltration by polymorphonuclear cells in an in vivo lung inflammation model [69].
Eosinophil mediated inflammation plays an important role in several common allergic lung pathologies, including asthma. Eosinophil granules have several proteins, including eosinophil cationic protein (ECP), that is thought to play a central role in airway hyper-responsiveness. A 10-amino acid peptide (NYRWRCKNQN) derived from ECP and named CPPecp has a heparan-sulfate binding core. Several glycosaminoglycans present on cell surfaces bind to ligands to mediate myriad immune responses; therefore, it was hypothesized that CPPecp binding to these cell surface receptors will block this interraction and modulate immune responses in various models of lung inflammation. Indeed, this turned out to be true in BEAS-2B, a lung epithelial cell line, where CPPecp decreased ECP mRNA expression, eotaxin secretion and p-STAT6 activation [70]. In vivo studies also reduced mite-induced airway inflammation by decreasing neutrophil and eosinophil count in broncho-alveolar lavage fluid, and decreased IL-5, IL-13, IL-17, and eotaxin expression in lung tissue [70]. Further studies identified CPPecp’s anti-inflammatory activity through inhibition of the NLRP3 inflammasome [71].
Acute lung injury (ALI) is a life-threatening, rapidly progressing inflammatory condition driven by TLR4 activation. In an attempt to mitigate the effects of ALI, a decoy CPP designed to inhibit binding of TLR4 to MyD88 was created and termed TM6 (RQIKIWFQNRRMKWKKENFLRDTWCNFQFY). In a mouse model of lipopolysaccharide (LPS) induced lung injury, treatment with TM6 alleviated negative histological changes, inhibited myeloperoxidase activity, and lowered TNF-α, IL-1β, and IL6 levels in lung tissue [72]. Inflammation via TLR4 activation also plays a key pathophysiological role in insulin resistance in type II diabetes mellitus. The death domain of MyD88 has been found to interact with TLR4. Several studies have identified a 10-residue amino acid (M10-RRRLSLFLNV) derived from the death domain of MyD88 to have cell transduction abilities (likely due to its arginine rich residues), which also functioned to inhibit LPS-induced nuclear translocation of NF-κB, decreased TNF-α and IL6 levels, lowered blood glucose levels, and improved glucose intolerance in db/db mice [73].
CIBG-552 is an anti-tumor peptide with cell penetrating activity developed from the screening of an Ala-library derived from de LALF32–51 region [74]. It has been shown to exert anti-angiogenic and anti-inflammatory effects through inhibition of NF-κB and HIF-1 pathways via its interaction with COMMD1 [75]. Interestingly, this peptide has been tested in a Phase I clinical trial of 25 patients with advanced, refractory solid tumors [76]. Patients received varying doses of subcutaneous injections of CIBG-522 3x/week for two weeks, in which the maximum tolerated dose was found to be 4.7 mg. Stable disease was reported in 5 patients, and 7 out of 10 assessed patients showed a significant change in the ratio of CD4/CD8 cells in response to therapy [76].
As far as the transduction abilities of CPPs are concerned, numerous studies have utilized these properties to deliver small molecules or other decoy peptides inhibiting various inflammatory pathways like NF-κB [77,78,79,80,81,82,83,84], the TLR4 pathway [64,85,86,87] and STAT-6 [88,89], to name a few. This list is by no means exhaustive, and we apologize to the authors whose valuable work was inevitably left out due to space limitations.
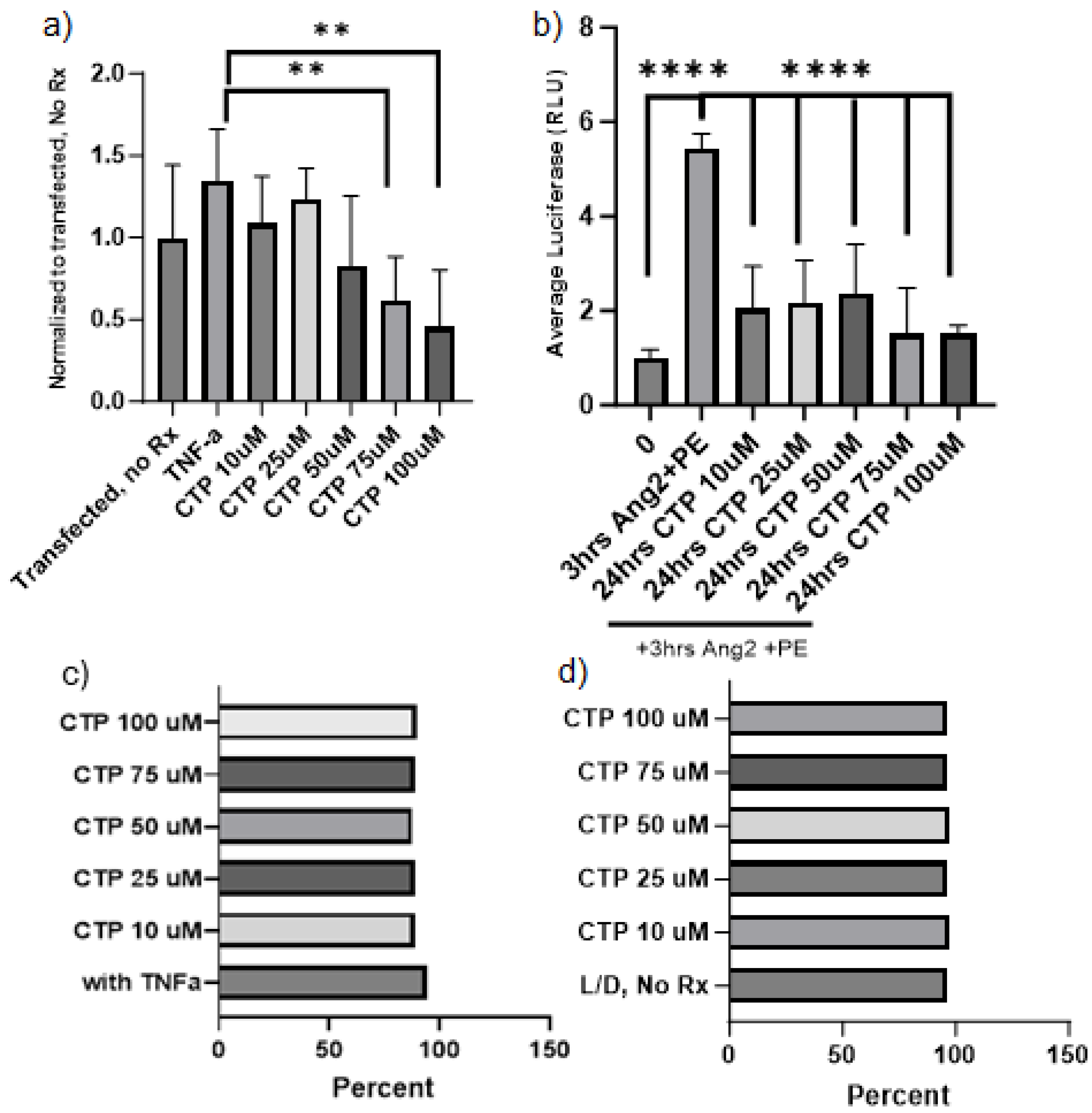
Our lab has had a long-standing interest in cell-type specific CPPs [90]. Our work using a combinatorial in vitro and in vivo phage display methodology [51] identified a cardiac targeting peptide (CTP) that is a cardiomyocyte-specific CPP which transduces heart tissue in as little as 15 minutes after a peripheral intravenous injection in mice [91]. This work has been confirmed by at least three different independent investigators from around the world [92]. Our recent work involved conjugating the N-terminus of CTP to amiodarone, a well-established anti-arrhythmic in routine clinical use, via a disulfide bond [93]. The conjugate was injected intraperitoneally into guinea pigs at 1/10th the molar dose of amiodarone. After 5-7 days of daily injections, the animals were euthanized, and hearts placed in a Langendorff perfusion system to measure heart rates, calcium currents, and action potential durations. To our surprise, CTP alone without any cargo displayed several salutary effects on calcium handling and increased heart rates while decreasing action potential durations [8]. RNA-sequencing from these heart extracts revealed that CTP increased alpha-adrenergic receptor expression and decreased beta-adrenergic receptor expression, explaining the changes in heart rate. It also upregulated calcium handling genes (SERCA2a) and surprisingly down-regulated several NF-κB pathway genes, as well as TNF-α [8]. To further study the physiological effects of CTP, we used a human cardiomyocyte cell line transfected with a reporter plasmid expressing luciferase gene under an NF-κB promoter. Forty-eight hours later, cells were treated with varying concentrations of CTP for 24 hours, and then challenged with TNF-α (20ng/mL) for 4 hours prior to running a luciferase assay in a luminometer. Cell viability in response to above manipulations was also assessed by FACS using live-dead stain. CTP inhibited TNF-α mediated NF-κB activation in a dose-dependent manner, with significant inhibition seen with CTP at concentrations of 75µM and above (p<0.01; Figure 2). Furthermore, challenging cardiomyocytes with angiotensin/phenylephrine for 3hrs resulted in a significant increase in NF- κB response. This increase was prevented if cells were pre-treated with CTP prior to addition of angiotensin/phenylephrine. Studies looking at the molecular pathways leading to this NF-κB inhibition, as well as expression of downstream cytokines are ongoing.
4. Conclusions
The potential of CPPs as novel vectors has been an area of intense study for the last three decades. They have generally been considered to be inert vectors for delivery of other diagnostic or therapeutic drugs ranging from small molecules, other peptides of therapeutic potential, to oligonucleotides for gene therapy as well as most recently RNA-interference therapeutics. Little attention has been paid to the possibility of these CPPs having inherent biological properties beyond transduction. Many of the earliest traditional non-specific CPPs like Tat and penetratin, and more recently our cardiomyocyte-targeting peptide CTP, have shown immense potential as anti-inflammatory agents, which can serve as a stand-alone therapeutic or add to the therapeutic potential of these CPPs. This is of particular significance, as the more traditional viral vectors are limited by pre-existing immunity [94], or development of rapid immunity to these vectors on first exposure [95]. Beyond developing immunity, these viral vectors also suffer from inciting an acute inflammatory response that can be life-threatening [96] including myocarditis [97] and/or a sepsis-like syndrome [98]. In this review we make a case for devoting more research to the less obvious, but perhaps equally important biological effects as transduction that these CPPs exhibit. At a minimum evolving data indicates that these peptides do not evoke an immune response, a highly desirable characteristic of a vector. However, far more longitudinal in vivo studies in large animal models are necessary to confirm these findings.
Funding
M.Z. is supported by NIH grant R01HL153407 awarded to her.
Conflicts of Interest
The authors declare no conflict of interest as pertains to this work.
References
- Taylor, R.E.; Zahid, M. Cell Penetrating Peptides, Novel Vectors for Gene Therapy. Pharmaceutics 2020, 12. [Google Scholar] [CrossRef] [PubMed]
- Green, M.; Loewenstein, P.M. Autonomous functional domains of chemically synthesized human immunodeficiency virus tat trans-activator protein. Cell 1988, 55, 1179–1188. [Google Scholar] [CrossRef] [PubMed]
- Frankel, A.D.; Pabo, C.O. Cellular uptake of the tat protein from human immunodeficiency virus. Cell 1988, 55, 1189–1193. [Google Scholar] [CrossRef] [PubMed]
- Derossi, D.; Joliot, A.H.; Chassaing, G.; Prochiantz, A. The third helix of the Antennapedia homeodomain translocates through biological membranes. J Biol Chem 1994, 269, 10444–10450. [Google Scholar] [CrossRef] [PubMed]
- Fawell, S.; Seery, J.; Daikh, Y.; Moore, C.; Chen, L.L.; Pepinsky, B.; Barsoum, J. Tat-mediated delivery of heterologous proteins into cells. Proc Natl Acad Sci U S A 1994, 91, 664–668. [Google Scholar] [CrossRef] [PubMed]
- Schwarze, S.R.; Ho, A.; Vocero-Akbani, A.; Dowdy, S.F. In vivo protein transduction: delivery of a biologically active protein into the mouse. Science 1999, 285, 1569–1572. [Google Scholar] [CrossRef]
- de la Torre, B.G.; Hornillos, V.; Luque-Ortega, J.R.; Abengozar, M.A.; Amat-Guerri, F.; Acuna, A.U.; Rivas, L.; Andreu, D. A BODIPY-embedding miltefosine analog linked to cell-penetrating Tat(48-60) peptide favors intracellular delivery and visualization of the antiparasitic drug. Amino Acids 2014, 46, 1047–1058. [Google Scholar] [CrossRef]
- Zahid, M.; Weber, B.; Yurko, R.; Islam, K.; Agrawal, V.; Lopuszynski, J.; Yagi, H.; Salama, G. Cardiomyocyte-Targeting Peptide to Deliver Amiodarone. Pharmaceutics 2023, 15. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Yang, X.Z.; Du, X.; Wang, J.W.; Zhang, R.; Zhao, J.; Wang, F.J.; Dong, Y.; Li, P.F. Enhancing tumor-specific intracellular delivering efficiency of cell-penetrating peptide by fusion with a peptide targeting to EGFR. Amino Acids 2015, 47, 997–1006. [Google Scholar] [CrossRef]
- Vogel, E.; Santos, D.; Huygens, C.; Peeters, P.; Van den Brande, S.; Wynant, N.; Vanden Broeck, J. The Study of Cell-Penetrating Peptides to Deliver dsRNA and siRNA by Feeding in the Desert Locust, Schistocerca gregaria. Insects 2023, 14. [Google Scholar] [CrossRef]
- Furukawa, K.; Tanaka, M.; Oba, M. siRNA delivery using amphipathic cell-penetrating peptides into human hepatoma cells. Bioorg Med Chem 2020, 28, 115402. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Duan, X.; Pan, H.; Akk, A.; Sandell, L.J.; Wickline, S.A.; Rai, M.F.; Pham, C.T.N. Development of a peptide-siRNA nanocomplex targeting NF- kappaB for efficient cartilage delivery. Sci Rep 2019, 9, 442. [Google Scholar] [CrossRef]
- Falato, L.; Gestin, M.; Langel, U. Cell-Penetrating Peptides Delivering siRNAs: An Overview. Methods Mol Biol 2021, 2282, 329–352. [Google Scholar] [CrossRef] [PubMed]
- Rathnayake, P.V.; Gunathunge, B.G.; Wimalasiri, P.N.; Karunaratne, D.N.; Ranatunga, R.J. Trends in the Binding of Cell Penetrating Peptides to siRNA: A Molecular Docking Study. J Biophys 2017, 2017, 1059216. [Google Scholar] [CrossRef]
- Vanova, J.; Hejtmankova, A.; Zackova Suchanova, J.; Sauerova, P.; Forstova, J.; Hubalek Kalbacova, M.; Spanielova, H. Influence of cell-penetrating peptides on the activity and stability of virus-based nanoparticles. Int J Pharm 2020, 576, 119008. [Google Scholar] [CrossRef] [PubMed]
- Polyakov, V.; Sharma, V.; Dahlheimer, J.L.; Pica, C.M.; Luker, G.D.; Piwnica-Worms, D. Novel Tat-peptide chelates for direct transduction of technetium-99m and rhenium into human cells for imaging and radiotherapy. Bioconjug Chem 2000, 11, 762–771. [Google Scholar] [CrossRef]
- Dong, P.; Cai, H.; Chen, L.; Li, Y.; Yuan, C.; Wu, X.; Shen, G.; Zhou, H.; Zhang, W.; Li, L. Biodistribution and evaluation of (131) I-labeled neuropilin-binding peptide for targeted tumor imaging. Contrast Media Mol Imaging 2016, 11, 467–474. [Google Scholar] [CrossRef]
- Avula, U.M.; Yoon, H.K.; Lee, C.H.; Kaur, K.; Ramirez, R.J.; Takemoto, Y.; Ennis, S.R.; Morady, F.; Herron, T.; Berenfeld, O.; et al. Cell-selective arrhythmia ablation for photomodulation of heart rhythm. Sci Transl Med 2015, 7, 311ra172. [Google Scholar] [CrossRef]
- Yang, J.; Firdaus, F.; Azuar, A.; Khalil, Z.G.; Marasini, N.; Capon, R.J.; Hussein, W.M.; Toth, I.; Skwarczynski, M. Cell-Penetrating Peptides-Based Liposomal Delivery System Enhanced Immunogenicity of Peptide-Based Vaccine against Group A Streptococcus. Vaccines (Basel) 2021, 9. [Google Scholar] [CrossRef]
- Yang, Y.Y.; Zhang, W.; Liu, H.; Jiang, J.J.; Wang, W.J.; Jia, Z.Y. Cell-Penetrating Peptide-Modified Graphene Oxide Nanoparticles Loaded with Rictor siRNA for the Treatment of Triple-Negative Breast Cancer. Drug Des Devel Ther 2021, 15, 4961–4972. [Google Scholar] [CrossRef]
- Chu, X.; Wu, B.; Fan, H.; Hou, J.; Hao, J.; Hu, J.; Wang, B.; Liu, G.; Li, C.; Meng, S. PTD-fused p53 as a potential antiviral agent directly suppresses HBV transcription and expression. Antiviral Res 2016, 127, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Arap, W.; Pasqualini, R.; Ruoslahti, E. Cancer treatment by targeted drug delivery to tumor vasculature in a mouse model. Science 1998, 279, 377–380. [Google Scholar] [CrossRef]
- Wender, P.A.; Mitchell, D.J.; Pattabiraman, K.; Pelkey, E.T.; Steinman, L.; Rothbard, J.B. The design, synthesis, and evaluation of molecules that enable or enhance cellular uptake: peptoid molecular transporters. Proc Natl Acad Sci U S A 2000, 97, 13003–13008. [Google Scholar] [CrossRef] [PubMed]
- Mai, J.C.; Shen, H.; Watkins, S.C.; Cheng, T.; Robbins, P.D. Efficiency of protein transduction is cell type-dependent and is enhanced by dextran sulfate. J Biol Chem 2002, 277, 30208–30218. [Google Scholar] [CrossRef]
- Mitchell, D.J.; Kim, D.T.; Steinman, L.; Fathman, C.G.; Rothbard, J.B. Polyarginine enters cells more efficiently than other polycationic homopolymers. J Pept Res 2000, 56, 318–325. [Google Scholar] [CrossRef]
- Mi, Z.; Mai, J.; Lu, X.; Robbins, P.D. Characterization of a class of cationic peptides able to facilitate efficient protein transduction in vitro and in vivo. Mol Ther 2000, 2, 339–347. [Google Scholar] [CrossRef] [PubMed]
- Soomets, U.; Lindgren, M.; Gallet, X.; Hallbrink, M.; Elmquist, A.; Balaspiri, L.; Zorko, M.; Pooga, M.; Brasseur, R.; Langel, U. Deletion analogues of transportan. Biochim Biophys Acta 2000, 1467, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Tsuchiya, K.; Horikoshi, K.; Fujita, M.; Hirano, M.; Miyamoto, M.; Yokoo, H.; Demizu, Y. Development of Hydrophobic Cell-Penetrating Stapled Peptides as Drug Carriers. Int J Mol Sci 2023, 24. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, H.; Oba, M.; Misawa, T.; Tanaka, M.; Hattori, T.; Naito, M.; Kurihara, M.; Demizu, Y. A Helix-Stabilized Cell-Penetrating Peptide as an Intracellular Delivery Tool. Chembiochem 2016, 17, 137–140. [Google Scholar] [CrossRef]
- Mueller, L.K.; Baumruck, A.C.; Zhdanova, H.; Tietze, A.A. Challenges and Perspectives in Chemical Synthesis of Highly Hydrophobic Peptides. Front Bioeng Biotechnol 2020, 8, 162. [Google Scholar] [CrossRef]
- Xu, Y.; Liang, W.; Qiu, Y.; Cespi, M.; Palmieri, G.F.; Mason, A.J.; Lam, J.K. Incorporation of a Nuclear Localization Signal in pH Responsive LAH4-L1 Peptide Enhances Transfection and Nuclear Uptake of Plasmid DNA. Mol Pharm 2016, 13, 3141–3152. [Google Scholar] [CrossRef]
- Sajid, M.I.; Moazzam, M.; Stueber, R.; Park, S.E.; Cho, Y.; Malik, N.U.A.; Tiwari, R.K. Applications of amphipathic and cationic cyclic cell-penetrating peptides: Significant therapeutic delivery tool. Peptides 2021, 141, 170542. [Google Scholar] [CrossRef]
- Alves, I.D.; Goasdoue, N.; Correia, I.; Aubry, S.; Galanth, C.; Sagan, S.; Lavielle, S.; Chassaing, G. Membrane interaction and perturbation mechanisms induced by two cationic cell penetrating peptides with distinct charge distribution. Biochim Biophys Acta 2008, 1780, 948–959. [Google Scholar] [CrossRef] [PubMed]
- Ruseska, I.; Zimmer, A. Internalization mechanisms of cell-penetrating peptides. Beilstein J Nanotechnol 2020, 11, 101–123. [Google Scholar] [CrossRef] [PubMed]
- Pooga, M.; Hallbrink, M.; Zorko, M.; Langel, U. Cell penetration by transportan. FASEB J 1998, 12, 67–77. [Google Scholar] [CrossRef]
- Green, M.; Ishino, M.; Loewenstein, P.M. Mutational analysis of HIV-1 Tat minimal domain peptides: identification of trans-dominant mutants that suppress HIV-LTR-driven gene expression. Cell 1989, 58, 215–223. [Google Scholar] [CrossRef]
- Zorko, M.; Langel, U. Cell-Penetrating Peptides. Methods Mol Biol 2022, 2383, 3–32. [Google Scholar] [CrossRef]
- Grasso, G.; Muscat, S.; Rebella, M.; Morbiducci, U.; Audenino, A.; Danani, A.; Deriu, M.A. Cell penetrating peptide modulation of membrane biomechanics by Molecular dynamics. J Biomech 2018, 73, 137–144. [Google Scholar] [CrossRef]
- Sakamoto, K.; Morishita, T.; Aburai, K.; Ito, D.; Imura, T.; Sakai, K.; Abe, M.; Nakase, I.; Futaki, S.; Sakai, H. Direct entry of cell-penetrating peptide can be controlled by maneuvering the membrane curvature. Sci Rep 2021, 11, 31. [Google Scholar] [CrossRef] [PubMed]
- Alves, I.D.; Jiao, C.Y.; Aubry, S.; Aussedat, B.; Burlina, F.; Chassaing, G.; Sagan, S. Cell biology meets biophysics to unveil the different mechanisms of penetratin internalization in cells. Biochim Biophys Acta 2010, 1798, 2231–2239. [Google Scholar] [CrossRef]
- Ichimizu, S.; Watanabe, H.; Maeda, H.; Hamasaki, K.; Ikegami, K.; Chuang, V.T.G.; Kinoshita, R.; Nishida, K.; Shimizu, T.; Ishima, Y.; et al. Cell-penetrating mechanism of intracellular targeting albumin: Contribution of macropinocytosis induction and endosomal escape. J Control Release 2019, 304, 156–163. [Google Scholar] [CrossRef] [PubMed]
- Richard, J.P.; Melikov, K.; Brooks, H.; Prevot, P.; Lebleu, B.; Chernomordik, L.V. Cellular uptake of unconjugated TAT peptide involves clathrin-dependent endocytosis and heparan sulfate receptors. J Biol Chem 2005, 280, 15300–15306. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, Y.; Takeuchi, T.; Kuwata, K.; Chiba, J.; Hatanaka, Y.; Nakase, I.; Futaki, S. Syndecan-4 Is a Receptor for Clathrin-Mediated Endocytosis of Arginine-Rich Cell-Penetrating Peptides. Bioconjug Chem 2016, 27, 1119–1130. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wang, C.; Lu, M.; Xing, H.; Yang, T.; Cai, C.; Zhao, X.; Wei, M.; Yu, J.; Ding, P. Intracellular distribution and internalization pathways of guanidinylated bioresponsive poly(amido amine)s in gene delivery. Asian J Pharm Sci 2018, 13, 360–372. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, M.; de la Torre, B.G.; Andreu, D.; Santos, N.C. Kinetic uptake profiles of cell penetrating peptides in lymphocytes and monocytes. Biochim Biophys Acta 2013, 1830, 4554–4563. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Ma, J.; Yang, Y.; Zeng, F.; Liu, C. Highly Efficient Delivery of Functional Cargoes by a Novel Cell-Penetrating Peptide Derived from SP140-Like Protein. Bioconjug Chem 2016, 27, 1373–1381. [Google Scholar] [CrossRef]
- Chao, T.Y.; Raines, R.T. Mechanism of ribonuclease A endocytosis: analogies to cell-penetrating peptides. Biochemistry 2011, 50, 8374–8382. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.J.; Tian, D.M.; Fu, L.; Jin, B.; Liu, Y.; Xu, Y.S.; Ye, Y.B.; Wang, X.B.; Xu, X.J.; Tang, C.; et al. Elastin-Derived VGVAPG Fragment Decorated Cell-Penetrating Peptide with Improved Gene Delivery Efficacy. Pharmaceutics 2023, 15. [Google Scholar] [CrossRef]
- Zhu, M.; Liu, H.; Cao, W.; Fang, Y.; Chen, Z.; Qi, X.; Luo, D.; Chen, C. Transcytosis mechanisms of cell-penetrating peptides: Cation-independent CC12 and cationic penetratin. J Pept Sci 2022, 28, e3408. [Google Scholar] [CrossRef]
- Smith, G.P. Filamentous fusion phage: novel expression vectors that display cloned antigens on the virion surface. Science 1985, 228, 1315–1317. [Google Scholar] [CrossRef]
- Zahid, M.; Phillips, B.E.; Albers, S.M.; Giannoukakis, N.; Watkins, S.C.; Robbins, P.D. Identification of a cardiac specific protein transduction domain by in vivo biopanning using a M13 phage peptide display library in mice. PLoS One 2010, 5, e12252. [Google Scholar] [CrossRef] [PubMed]
- Chamarthy, S.P.; Jia, L.; Kovacs, J.R.; Anderson, K.R.; Shen, H.; Firestine, S.M.; Meng, W.S. Gene delivery to dendritic cells facilitated by a tumor necrosis factor alpha-competing peptide. Mol Immunol 2004, 41, 741–749. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, K.; Adachi, Y.; Komoike, Y.; Kamada, Y.; Koyama, R.; Fukuda, Y.; Kadotani, A.; Asami, T.; Sakamoto, J.I. Novel DOCK2-selective inhibitory peptide that suppresses B-cell line migration. Biochem Biophys Res Commun 2017, 483, 183–190. [Google Scholar] [CrossRef] [PubMed]
- Rehman, K.K.; Bertera, S.; Bottino, R.; Balamurugan, A.N.; Mai, J.C.; Mi, Z.; Trucco, M.; Robbins, P.D. Protection of islets by in situ peptide-mediated transduction of the Ikappa B kinase inhibitor Nemo-binding domain peptide. J Biol Chem 2003, 278, 9862–9868. [Google Scholar] [CrossRef] [PubMed]
- Sahagun, D.A.; Lopuszynski, J.B.; Feldman, K.S.; Pogodzinski, N.; Zahid, M. Toxicity Studies of Cardiac-Targeting Peptide Reveal a Robust Safety Profile. Pharmaceutics 2024, 16. [Google Scholar] [CrossRef] [PubMed]
- Kilk, K.; Mahlapuu, R.; Soomets, U.; Langel, U. Analysis of in vitro toxicity of five cell-penetrating peptides by metabolic profiling. Toxicology 2009, 265, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Suhorutsenko, J.; Oskolkov, N.; Arukuusk, P.; Kurrikoff, K.; Eriste, E.; Copolovici, D.M.; Langel, U. Cell-penetrating peptides, PepFects, show no evidence of toxicity and immunogenicity in vitro and in vivo. Bioconjug Chem 2011, 22, 2255–2262. [Google Scholar] [CrossRef]
- Saar, K.; Lindgren, M.; Hansen, M.; Eiriksdottir, E.; Jiang, Y.; Rosenthal-Aizman, K.; Sassian, M.; Langel, U. Cell-penetrating peptides: a comparative membrane toxicity study. Anal Biochem 2005, 345, 55–65. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.G.; Sayers, E.J.; He, L.; Narayan, R.; Williams, T.L.; Mills, E.M.; Allemann, R.K.; Luk, L.Y.P.; Jones, A.T.; Tsai, Y.H. Cell-penetrating peptide sequence and modification dependent uptake and subcellular distribution of green florescent protein in different cell lines. Sci Rep 2019, 9, 6298. [Google Scholar] [CrossRef]
- Hoffmann, K.; Milech, N.; Juraja, S.M.; Cunningham, P.T.; Stone, S.R.; Francis, R.W.; Anastasas, M.; Hall, C.M.; Heinrich, T.; Bogdawa, H.M.; et al. A platform for discovery of functional cell-penetrating peptides for efficient multi-cargo intracellular delivery. Sci Rep 2018, 8, 12538. [Google Scholar] [CrossRef]
- Fotin-Mleczek, M.; Welte, S.; Mader, O.; Duchardt, F.; Fischer, R.; Hufnagel, H.; Scheurich, P.; Brock, R. Cationic cell-penetrating peptides interfere with TNF signalling by induction of TNF receptor internalization. J Cell Sci 2005, 118, 3339–3351. [Google Scholar] [CrossRef] [PubMed]
- Kuo, J.H.; Jan, M.S.; Lin, Y.L.; Lin, C. Interactions between octaarginine and U-937 human macrophages: global gene expression profiling, superoxide anion content, and cytokine production. J Control Release 2009, 139, 197–204. [Google Scholar] [CrossRef] [PubMed]
- Letoha, T.; Kusz, E.; Papai, G.; Szabolcs, A.; Kaszaki, J.; Varga, I.; Takacs, T.; Penke, B.; Duda, E. In vitro and in vivo nuclear factor-kappaB inhibitory effects of the cell-penetrating penetratin peptide. Mol Pharmacol 2006, 69, 2027–2036. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.F.; Xu, X.; Fan, X.; Zhang, C.; Wei, Q.; Wang, X.; Guo, W.; Xing, W.; Yu, J.; Yan, J.L.; et al. A cell-penetrating peptide suppresses inflammation by inhibiting NF-kappaB signaling. Mol Ther 2011, 19, 1849–1857. [Google Scholar] [CrossRef] [PubMed]
- DiGiandomenico, A.; Veach, R.A.; Zienkiewicz, J.; Moore, D.J.; Wylezinski, L.S.; Hutchens, M.A.; Hawiger, J. The “genomic storm” induced by bacterial endotoxin is calmed by a nuclear transport modifier that attenuates localized and systemic inflammation. PLoS One 2014, 9, e110183. [Google Scholar] [CrossRef] [PubMed]
- Zienkiewicz, J.; Armitage, A.; Hawiger, J. Targeting nuclear import shuttles, importins/karyopherins alpha by a peptide mimicking the NFkappaB1/p50 nuclear localization sequence. J Am Heart Assoc 2013, 2, e000386. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Major, A.S.; Zienkiewicz, J.; Gabriel, C.L.; Veach, R.A.; Moore, D.J.; Collins, R.D.; Hawiger, J. Nuclear transport modulation reduces hypercholesterolemia, atherosclerosis, and fatty liver. J Am Heart Assoc 2013, 2, e000093. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, L.; Hu, Y.; Jiang, K.; Li, Z.; Lin, Y.Z.; Wei, G.; Lu, W. Cell-permeable NF-kappaB inhibitor-conjugated liposomes for treatment of glioma. J Control Release 2018, 289, 102–113. [Google Scholar] [CrossRef]
- Lee, J.Y.; Seo, Y.N.; Park, H.J.; Park, Y.J.; Chung, C.P. The cell-penetrating peptide domain from human heparin-binding epidermal growth factor-like growth factor (HB-EGF) has anti-inflammatory activity in vitro and in vivo. Biochem Biophys Res Commun 2012, 419, 597–604. [Google Scholar] [CrossRef]
- Fu, L.S.; Wu, Y.R.; Fang, S.L.; Tsai, J.J.; Lin, H.K.; Chen, Y.J.; Chen, T.Y.; Chang, M.D. Cell Penetrating Peptide Derived from Human Eosinophil Cationic Protein Decreases Airway Allergic Inflammation. Sci Rep 2017, 7, 12352. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.J.; Liao, E.C.; Sheu, M.L.; Chang, D.T.; Tsai, J.J. Cell-penetrating peptide derived from human eosinophil cationic protein inhibits mite allergen Der p 2 induced inflammasome activation. PLoS One 2015, 10, e0121393. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Tian, Y.; Qu, S.; Cao, Y.; Li, S.; Zhang, W.; Zhang, Z.; Zhang, N.; Fu, Y. Protective effect of TM6 on LPS-induced acute lung injury in mice. Sci Rep 2017, 7, 572. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.; Kumari, T.; Gupta, A.; Akhtar, S.; Verma, R.D.; Ghosh, J.K. Identification of a 10-mer peptide from the death domain of MyD88 which attenuates inflammation and insulin resistance and improves glucose metabolism. Biochem J 2024, 481, 191–218. [Google Scholar] [CrossRef] [PubMed]
- Vallespi, M.G.; Fernandez, J.R.; Torrens, I.; Garcia, I.; Garay, H.; Mendoza, O.; Granadillo, M.; Falcon, V.; Acevedo, B.; Ubieta, R.; et al. Identification of a novel antitumor peptide based on the screening of an Ala-library derived from the LALF(32-51) region. J Pept Sci 2010, 16, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Daghero, H.; Fernandez Masso, J.R.; Astrada, S.; Guerra Vallespi, M.; Bollati-Fogolin, M. The Anticancer Peptide CIGB-552 Exerts Anti-Inflammatory and Anti-Angiogenic Effects through COMMD1. Molecules 2020, 26. [Google Scholar] [CrossRef] [PubMed]
- Vallespi, M.G.; Mestre, B.; Marrero, M.A.; Uranga, R.; Rey, D.; Lugiollo, M.; Betancourt, M.; Silva, K.; Corrales, D.; Lamadrid, Y.; et al. A first-in-class, first-in-human, phase I trial of CIGB-552, a synthetic peptide targeting COMMD1 to inhibit the oncogenic activity of NF-kappaB in patients with advanced solid tumors. Int J Cancer 2021, 149, 1313–1321. [Google Scholar] [CrossRef] [PubMed]
- Orange, J.S.; May, M.J. Cell penetrating peptide inhibitors of nuclear factor-kappa B. Cell Mol Life Sci 2008, 65, 3564–3591. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Qiao, Y.; Xue, L.; Xu, S.; Zhang, N. Targeted and MMP-2/9 responsive peptides for the treatment of rheumatoid arthritis. Int J Pharm 2019, 569, 118625. [Google Scholar] [CrossRef] [PubMed]
- Kanemaru, Y.; Momiki, Y.; Matsuura, S.; Horikawa, T.; Gohda, J.; Inoue, J.; Okamoto, Y.; Fujita, M.; Otsuka, M. An artificial copper complex incorporating a cell-penetrating peptide inhibits nuclear factor-kappaB (NF-kappaB) activation. Chem Pharm Bull (Tokyo) 2011, 59, 1555–1558. [Google Scholar] [CrossRef]
- Lai, J.; Yao, Y.; Zhang, Y.; Liu, Y.; Lu, C.; Meng, C.; Xia, D.; Li, Y.; Cao, K.; Gao, X.; et al. Cell-Penetrating Peptide Conjugated Au Nanoclusters Selectively Suppress Refractory Lymphoma Cells via Targeting Both Canonical and Noncanonical NF-kappaB Signaling Pathways. Bioconjug Chem 2023, 34, 228–237. [Google Scholar] [CrossRef]
- Davoudi, Z.; Akbarzadeh, A.; Rahmatiyamchi, M.; Movassaghpour, A.A.; Alipour, M.; Nejati-Koshki, K.; Sadeghi, Z.; Dariushnejad, H.; Zarghami, N. Molecular target therapy of AKT and NF-kB signaling pathways and multidrug resistance by specific cell penetrating inhibitor peptides in HL-60 cells. Asian Pac J Cancer Prev 2014, 15, 4353–4358. [Google Scholar] [CrossRef]
- Hong, S.; Yum, S.; Yoo, H.J.; Kang, S.; Yoon, J.H.; Min, D.; Kim, Y.M.; Jung, Y. Colon-targeted cell-permeable NFkappaB inhibitory peptide is orally active against experimental colitis. Mol Pharm 2012, 9, 1310–1319. [Google Scholar] [CrossRef]
- Urata, M.; Kokabu, S.; Matsubara, T.; Sugiyama, G.; Nakatomi, C.; Takeuchi, H.; Hirata-Tsuchiya, S.; Aoki, K.; Tamura, Y.; Moriyama, Y.; et al. A peptide that blocks the interaction of NF-kappaB p65 subunit with Smad4 enhances BMP2-induced osteogenesis. J Cell Physiol 2018, 233, 7356–7366. [Google Scholar] [CrossRef] [PubMed]
- von Bismarck, P.; Winoto-Morbach, S.; Herzberg, M.; Uhlig, U.; Schutze, S.; Lucius, R.; Krause, M.F. IKK NBD peptide inhibits LPS induced pulmonary inflammation and alters sphingolipid metabolism in a murine model. Pulm Pharmacol Ther 2012, 25, 228–235. [Google Scholar] [CrossRef] [PubMed]
- Kwon, H.K.; Patra, M.C.; Shin, H.J.; Gui, X.; Achek, A.; Panneerselvam, S.; Kim, D.J.; Song, S.J.; Hong, R.; Kim, K.S.; et al. A cell-penetrating peptide blocks Toll-like receptor-mediated downstream signaling and ameliorates autoimmune and inflammatory diseases in mice. Exp Mol Med 2019, 51, 1–19. [Google Scholar] [CrossRef]
- Dong, H.; Zhang, Y.; Song, L.; Kim, D.S.; Wu, H.; Yang, L.; Li, S.; Morgan, K.A.; Adams, D.B.; Wang, H. Cell-Permeable Peptide Blocks TLR4 Signaling and Improves Islet Allograft Survival. Cell Transplant 2016, 25, 1319–1329. [Google Scholar] [CrossRef] [PubMed]
- Nilsen, K.E.; Zhang, B.; Skjesol, A.; Ryan, L.; Vagle, H.; Boe, M.H.; Orning, P.; Kim, H.; Bakke, S.S.; Elamurugan, K.; et al. Peptide derived from SLAMF1 prevents TLR4-mediated inflammation in vitro and in vivo. Life Sci Alliance 2023, 6. [Google Scholar] [CrossRef]
- McCusker, C.T.; Wang, Y.; Shan, J.; Kinyanjui, M.W.; Villeneuve, A.; Michael, H.; Fixman, E.D. Inhibition of experimental allergic airways disease by local application of a cell-penetrating dominant-negative STAT-6 peptide. J Immunol 2007, 179, 2556–2564. [Google Scholar] [CrossRef]
- Wang, Y.; Li, Y.; Shan, J.; Fixman, E.; McCusker, C. Effective treatment of experimental ragweed-induced asthma with STAT-6-IP, a topically delivered cell-penetrating peptide. Clin Exp Allergy 2011, 41, 1622–1630. [Google Scholar] [CrossRef]
- Zahid, M.; Lu, X.; Mi, Z.; Robbins, P.D. Cationic and tissue-specific protein transduction domains identification, characterization, and therapeutic application. Adv Genet 2010, 69, 83–95. [Google Scholar] [CrossRef]
- Zahid, M.; Feldman, K.S.; Garcia-Borrero, G.; Feinstein, T.N.; Pogodzinski, N.; Xu, X.; Yurko, R.; Czachowski, M.; Wu, Y.L.; Mason, N.S.; et al. Cardiac Targeting Peptide, a Novel Cardiac Vector: Studies in Bio-Distribution, Imaging Application, and Mechanism of Transduction. Biomolecules 2018, 8. [Google Scholar] [CrossRef] [PubMed]
- Sahagun, D.; Zahid, M. Cardiac-Targeting Peptide: From Discovery to Applications. Biomolecules 2023, 13. [Google Scholar] [CrossRef] [PubMed]
- Yurko, R.; Islam, K.; Weber, B.; Salama, G.; Zahid, M. Conjugation of amiodarone to a novel cardiomyocyte cell penetrating peptide for potential targeted delivery to the heart. Front Chem 2023, 11, 1220573. [Google Scholar] [CrossRef] [PubMed]
- Manno, C.S.; Pierce, G.F.; Arruda, V.R.; Glader, B.; Ragni, M.; Rasko, J.J.; Ozelo, M.C.; Hoots, K.; Blatt, P.; Konkle, B.; et al. Successful transduction of liver in hemophilia by AAV-Factor IX and limitations imposed by the host immune response. Nat Med 2006, 12, 342–347. [Google Scholar] [CrossRef] [PubMed]
- Duan, D. Lethal immunotoxicity in high-dose systemic AAV therapy. Mol Ther 2023, 31, 3123–3126. [Google Scholar] [CrossRef] [PubMed]
- Hinderer, C.; Katz, N.; Buza, E.L.; Dyer, C.; Goode, T.; Bell, P.; Richman, L.K.; Wilson, J.M. Severe Toxicity in Nonhuman Primates and Piglets Following High-Dose Intravenous Administration of an Adeno-Associated Virus Vector Expressing Human SMN. Hum Gene Ther 2018, 29, 285–298. [Google Scholar] [CrossRef] [PubMed]
- Silver, E.; Argiro, A.; Hong, K.; Adler, E. Gene therapy vector-related myocarditis. Int J Cardiol 2024, 398, 131617. [Google Scholar] [CrossRef]
- Morales, L.; Gambhir, Y.; Bennett, J.; Stedman, H.H. Broader Implications of Progressive Liver Dysfunction and Lethal Sepsis in Two Boys following Systemic High-Dose AAV. Mol Ther 2020, 28, 1753–1755. [Google Scholar] [CrossRef]
Figure 1.
Classification for CPPs.
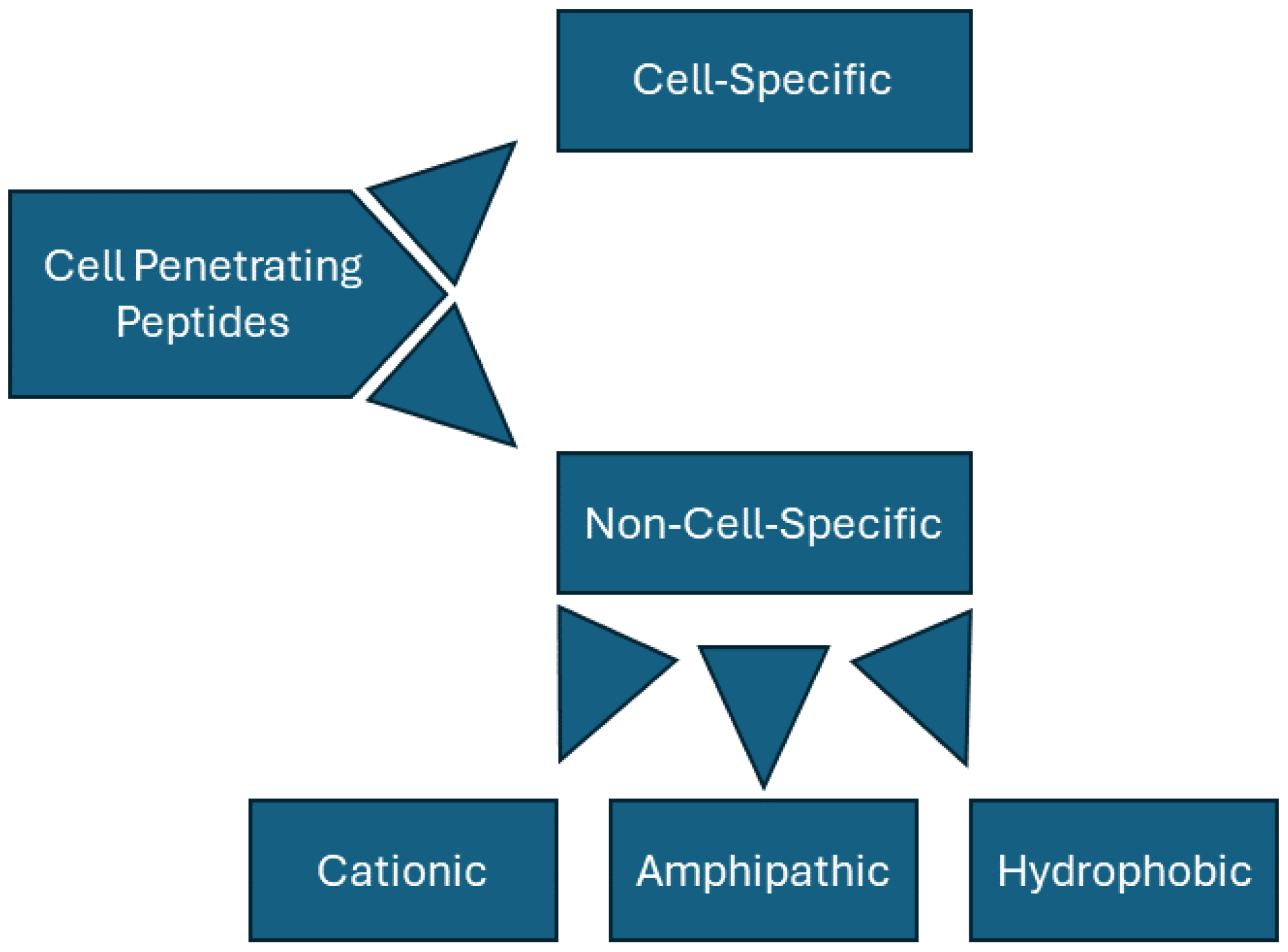
Figure 2.
Inhibition of TNF-α mediated NF-κB activation by CTP in a human cardiomyocyte cell line (a). Incubating human cardiomyocytes in angiotensin/phenylephrine for 3hrs resulted in a significant increase in NF-kB activation, which was prevented by pre-treatment with CTP (b). Neither one of these observed effects were due to changes in cell viability or number which did not change significantly in response to CTP treatment after transfection with NF-κB reporter plasmid and TNF-α challenge (c) or after daily treatments with varying concentrations of CTP (d) as assessed by FACS using live-dead stain.
Figure 2.
Inhibition of TNF-α mediated NF-κB activation by CTP in a human cardiomyocyte cell line (a). Incubating human cardiomyocytes in angiotensin/phenylephrine for 3hrs resulted in a significant increase in NF-kB activation, which was prevented by pre-treatment with CTP (b). Neither one of these observed effects were due to changes in cell viability or number which did not change significantly in response to CTP treatment after transfection with NF-κB reporter plasmid and TNF-α challenge (c) or after daily treatments with varying concentrations of CTP (d) as assessed by FACS using live-dead stain.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



