
Submitted:
08 September 2024
Posted:
09 September 2024
You are already at the latest version
Abstract
Knee joint injuries, including those affecting the anterior cruciate ligament (ACL), meniscus, and cartilage, present complex challenges in sports medicine and orthopedics due to the intricate cellular and molecular mechanisms involved in tissue damage and repair. Understanding the molecular biology underpinning these processes is crucial for developing effective therapeutic and rehabilitation strategies. This systematic review investigates the impact of mechanical loading on the cellular responses during knee joint injury repair, with a particular focus on the molecular pathways involved in tissue regeneration.
Mechanical loading plays a dual role, where controlled early loading can promote tissue repair, while excessive or inappropriate loading can exacerbate tissue damage. Fibroblasts, chondrocytes, and mesenchymal stem cells (MSCs) are central to the repair process, and their activation, proliferation, and differentiation are regulated by key molecular pathways. Upon injury, mechanotransduction pathways such as the integrin/FAK signaling axis are activated, which convert mechanical signals into biochemical responses that regulate cell adhesion, migration, and extracellular matrix (ECM) synthesis. Additionally, mechanosensitive ion channels like Piezo1 and TRPV4 modulate intracellular calcium levels, triggering downstream signaling cascades such as calmodulin/CaMKII, which regulate gene transcription and cellular responses to mechanical stress.
The YAP/TAZ pathway, a critical component of the Hippo signaling pathway, responds to mechanical stimuli and regulates cell proliferation and ECM production in fibroblasts and chondrocytes. YAP/TAZ translocate to the nucleus in response to mechanical loading, where they interact with transcription factors such as TEAD, promoting the expression of genes involved in collagen synthesis and tissue repair. In parallel, growth factors like transforming growth factor-beta (TGF-β) and fibroblast growth factor (FGF) activate the TGF-β/Smad and PI3K/Akt signaling pathways, driving MSC differentiation into fibroblasts and chondrocytes, essential for ligament and cartilage repair.
Mechanical loading also influences the inflammatory response at the injury site by modulating immune cell activity. Early mechanical loading can shift macrophages from a pro-inflammatory M1 phenotype to an anti-inflammatory M2 phenotype, mediated by growth factors such as TGF-β and interleukin-10 (IL-10). This phenotypic switch promotes tissue regeneration by enhancing ECM synthesis and resolving inflammation, which is crucial for long-term joint health. Matrix metalloproteinases (MMPs), regulated by NF-κB and MAPK pathways, play a role in ECM remodeling, where an imbalance between MMPs and their inhibitors (TIMPs) can lead to excessive matrix degradation, impeding tissue repair.
Furthermore, Wnt/β-catenin signaling is activated in response to moderate mechanical loading, promoting chondrocyte proliferation and enhancing cartilage repair by upregulating type II collagen and aggrecan synthesis. However, dysregulation of Wnt signaling under excessive mechanical stress can lead to chondrocyte hypertrophy and cartilage degradation, contributing to osteoarthritis development.
This review also explores emerging therapeutic strategies that leverage the molecular biology of knee joint repair, including biologics like platelet-rich plasma (PRP) and MSC-derived exosomes, which deliver bioactive molecules that activate critical regenerative pathways such as TGF-β/Smad and PI3K/Akt. Gene therapies targeting Wnt signaling or YAP/TAZ offer potential for enhancing tissue regeneration by modulating mechanotransduction and repair processes at the molecular level.
In conclusion, the molecular biology of cellular responses to mechanical loading is central to knee joint repair following injuries. By understanding these processes and targeting specific molecular pathways, clinicians can optimize rehabilitation protocols and develop novel therapeutic approaches that enhance tissue regeneration, prevent chronic degeneration, and restore joint function. This comprehensive synthesis highlights the importance of integrating molecular insights into treatment strategies for ACL, meniscal, and cartilage injuries to improve patient outcomes.
Keywords:
knee joint
; cell biology
; mechanical loading
Introduction
Knee joint injuries are highly prevalent, affecting both athletes and the general population, and are commonly caused by trauma, overuse, or degenerative changes associated with aging. The knee joint, a hinge-like synovial joint, is vital for load-bearing and mobility, making it susceptible to various forms of injury due to its complex anatomy and function. Injuries to the knee can range from ligamentous tears, such as those of the anterior cruciate ligament (ACL), to meniscal damage and cartilage degradation, each of which can severely impair joint function and quality of life. Among these injuries, ACL tears are particularly common, especially in high-impact sports, and they often necessitate surgical repair due to the ligament’s poor healing capacity and the biomechanical instability it causes when torn. The ACL is essential for stabilizing the knee joint during dynamic movements such as pivoting, jumping, and rapid deceleration, and its rupture significantly increases the risk of developing further joint damage, including meniscal tears and cartilage wear, which can lead to early-onset osteoarthritis.
Historically, the focus of rehabilitation following ACL and other knee injuries has been on restoring mobility, strength, and joint function through progressively increasing physical activity. However, recent advances in molecular biology and biomechanics have shed light on the crucial role of early mechanical loading in influencing the repair and regeneration of injured tissues. Mechanical loading refers to the physical forces exerted on tissues during movement and weight-bearing activities. In the context of knee rehabilitation, mechanical loading not only promotes physical strength and stability but also directly affects cellular behavior at the injury site. The timing, intensity, and type of mechanical loading are now understood to play a pivotal role in regulating cellular responses, including proliferation, differentiation, and extracellular matrix (ECM) synthesis, which are critical for effective tissue repair. Mechanical loading impacts various cell types within the knee, including fibroblasts, chondrocytes, mesenchymal stem cells (MSCs), and immune cells, each of which responds to mechanical stimuli through specific signaling pathways that drive tissue remodeling and healing.
Emerging research has identified several mechanotransduction pathways, through which cells convert mechanical signals into biochemical responses. These pathways involve key molecular players such as integrins, focal adhesion kinase (FAK), and mechanosensitive ion channels, which regulate gene expression related to tissue repair. Additionally, the YAP/TAZ and PI3K/Akt signaling pathways have been shown to be critical in fibroblast and chondrocyte responses to mechanical stress, promoting cell proliferation and ECM production. Early mechanical loading, when applied correctly, has been associated with enhanced alignment of collagen fibers, improved cartilage regeneration, and better overall tissue functionality. However, improper or excessive loading can lead to adverse outcomes, such as increased inflammation, excessive ECM degradation through the upregulation of matrix metalloproteinases (MMPs), and delayed healing.
Given these advancements in understanding how mechanical loading influences cellular processes, it is essential to integrate this knowledge into therapeutic strategies and rehabilitation protocols. This systematic review aims to explore the current understanding of how early mechanical loading affects the cellular and molecular mechanisms in knee joint injuries, with a particular focus on ACL injuries. By examining the underlying biological responses and signaling pathways activated by mechanical loading, this review seeks to provide valuable insights into optimizing rehabilitation strategies, enhancing tissue repair, and potentially improving long-term outcomes for patients suffering from knee joint injuries. Additionally, it explores novel therapeutic approaches, such as biologics and gene therapies, that could complement traditional rehabilitation by targeting specific molecular pathways involved in tissue regeneration and repair.
Types of Knee Joint Injuries
Anterior Cruciate Ligament (ACL) Injuries
The Anterior Cruciate Ligament (ACL) is a key structural component of the knee joint, responsible for maintaining stability by limiting excessive anterior translation of the tibia relative to the femur and controlling rotational movements. Its role is particularly crucial during dynamic activities such as pivoting, jumping, and sudden stops, which involve high levels of stress on the knee. This makes the ACL highly vulnerable to injury, especially in sports like soccer, basketball, and skiing, where rapid directional changes, decelerations, and high-impact landings are common. When the ACL is torn, it results in immediate joint instability, disrupting the intricate balance of forces within the knee, which not only impairs normal movement but also increases the risk of secondary injuries to other structures like the meniscus and articular cartilage. Over time, this joint instability can lead to altered knee mechanics, abnormal wear patterns, and a significantly increased risk of developing osteoarthritis, a degenerative joint condition characterized by cartilage breakdown and chronic pain. The biological response to ACL injury is multifaceted, beginning with an acute inflammatory phase that involves the release of cytokines, growth factors, and damage-associated molecular patterns (DAMPs) from the injured tissue. This inflammatory response triggers the recruitment of immune cells such as neutrophils and macrophages, which clear debris and initiate tissue repair. However, the injury also causes significant extracellular matrix (ECM) degradation, mediated by the upregulation of matrix metalloproteinases (MMPs), which degrade collagen and other ECM components. Concurrently, mechanical forces within the knee, including altered loading patterns and abnormal joint kinematics, further influence the healing process by modulating cellular behaviors such as fibroblast and chondrocyte activity, affecting ECM synthesis, collagen alignment, and overall tissue remodeling. These mechanical forces, along with the biochemical environment, play a crucial role in determining the balance between successful repair and long-term joint degeneration.
Mechanisms of Injury
ACL injuries, particularly those arising from non-contact mechanisms, account for the vast majority of cases, with estimates suggesting that approximately 70% of ACL injuries are caused by these mechanisms. These injuries are frequently observed during athletic activities that involve sudden, high-stress movements, such as pivoting on a planted foot, rapid deceleration, or improper landing from a jump. These movements place tremendous force on the knee, especially when the joint is subjected to valgus stress or rotational forces. The ACL, which is responsible for preventing excessive forward movement of the tibia and controlling rotational stability, becomes highly vulnerable under these conditions. During these rapid motions, the ligament experiences sudden, intense strain, often resulting in a partial or complete tear when the force exceeds its tensile strength. In many cases, these tears occur without any direct physical contact, underscoring the importance of biomechanics and neuromuscular control in preventing ACL injuries.
Once the ACL is ruptured, the body immediately initiates a mechanical and biochemical response to address the injury. The mechanical rupture of the ligament disrupts the knee’s structural integrity, leading to immediate instability, swelling, and pain. Simultaneously, a complex inflammatory cascade is triggered, which is crucial for initiating the healing process. The disruption of the ligament and surrounding tissues results in the release of damage-associated molecular patterns (DAMPs), which activate pattern recognition receptors (PRRs) on immune cells and other resident cells in the knee. This leads to the rapid production of pro-inflammatory cytokines, including tumor necrosis factor-alpha (TNF-α), interleukin-1 beta (IL-1β), and interleukin-6 (IL-6). These cytokines play a pivotal role in recruiting immune cells like neutrophils and macrophages to the injury site. Neutrophils are the first responders, arriving within hours of the injury, where they release reactive oxygen species (ROS) and proteolytic enzymes to clear debris and damaged tissue.
Following neutrophil infiltration, macrophages arrive at the injury site and transition from a pro-inflammatory (M1) phenotype to an anti-inflammatory (M2) phenotype as the inflammatory phase progresses. M1 macrophages continue to secrete pro-inflammatory cytokines and chemokines, amplifying the immune response and recruiting additional immune cells to the site. However, their role extends beyond inflammation, as they also secrete matrix metalloproteinases (MMPs), particularly MMP-1, MMP-3, and MMP-13, which degrade the extracellular matrix (ECM) components, such as collagen, in the injured ligament. This degradation is necessary for clearing damaged matrix to allow for repair, but excessive MMP activity can lead to further ECM destruction and impair tissue regeneration.
As inflammation continues, the macrophages shift toward the M2 phenotype, which promotes tissue repair and regeneration. This shift is mediated by anti-inflammatory cytokines like interleukin-10 (IL-10) and transforming growth factor-beta (TGF-β), which suppress the inflammatory response and stimulate fibroblast proliferation. Fibroblasts, in turn, begin synthesizing new ECM components, including type I collagen, which is essential for restoring the structural integrity of the ligament. However, the success of this repair process is influenced by mechanical forces within the knee. Altered joint mechanics following the injury, such as abnormal loading patterns due to instability, can significantly impact fibroblast function, collagen fiber alignment, and overall tissue remodeling.
The biomechanical environment of the knee post-injury plays a critical role in determining the outcome of the healing process. Early mechanical loading, when appropriately applied, has been shown to positively influence tissue repair by promoting ECM synthesis and aligning collagen fibers in a manner that restores the ligament’s mechanical properties. Mechanical forces are sensed by mechanoreceptors on cells, such as integrins and mechanosensitive ion channels like Piezo1, which convert these physical stimuli into biochemical signals through pathways such as focal adhesion kinase (FAK), YAP/TAZ, and PI3K/Akt. These pathways regulate cellular processes like proliferation, migration, and ECM production, which are critical for ligament repair. However, improper or excessive mechanical loading can exacerbate tissue damage by further disrupting the already compromised ECM and prolonging the inflammatory phase, thereby impeding repair and increasing the risk of chronic joint instability and degeneration.
Thus, the inflammatory response to ACL injury is a double-edged sword. While it is essential for initiating tissue repair and clearing damaged tissue, it must be tightly regulated to prevent excessive ECM degradation and chronic inflammation, which can delay healing and predispose the joint to long-term complications like osteoarthritis. The interplay between inflammation, mechanical forces, and tissue repair mechanisms is central to the healing process, and understanding these dynamics is crucial for developing effective therapeutic strategies to improve outcomes following ACL injuries.
Cellular and Molecular Responses to ACL Injury
The cellular and molecular responses to an ACL injury are highly complex and play a vital role in determining the effectiveness of the healing process. Following the rupture of the ACL, the initial response is marked by a robust inflammatory reaction, which is essential for clearing debris and initiating tissue repair. This phase is driven by the release of pro-inflammatory cytokines such as tumor necrosis factor-alpha (TNF-α), interleukin-1 beta (IL-1β), and interleukin-6 (IL-6), which recruit immune cells like neutrophils and macrophages to the injury site. These immune cells not only clear damaged tissues but also secrete matrix metalloproteinases (MMPs), enzymes responsible for degrading the extracellular matrix (ECM). ECM degradation is a necessary step for clearing the injured matrix and creating space for new tissue formation, but excessive activity of MMPs can lead to further tissue damage if not tightly regulated. As inflammation subsides, the remodeling phase begins, where fibroblasts are activated to synthesize new ECM components, predominantly type I collagen, which is critical for restoring the structural integrity of the ligament. Fibroblast activation is mediated by growth factors such as transforming growth factor-beta (TGF-β) and fibroblast growth factor (FGF), which activate intracellular signaling pathways like TGF-β/Smad and MAPK, promoting cell proliferation and ECM production.
In parallel, mesenchymal stem cells (MSCs) are recruited to the injury site, primarily from the bone marrow and synovium. MSC recruitment is driven by chemotactic signals such as stromal cell-derived factor-1 (SDF-1) and vascular endothelial growth factor (VEGF), which bind to their receptors on MSCs, initiating signaling cascades like the SDF-1/CXCR4 and VEGF/VEGFR pathways. Upon arrival at the injury site, MSCs differentiate into fibroblasts and other cell types necessary for tissue repair, while also exerting paracrine effects by secreting cytokines and growth factors that modulate immune responses and promote tissue regeneration. Angiogenesis, the formation of new blood vessels, is another critical process in ACL healing, driven by growth factors such as VEGF, which stimulates endothelial cell proliferation and migration through PI3K/Akt and ERK1/2 signaling pathways. The newly formed blood vessels ensure adequate oxygen and nutrient delivery to the healing tissue, supporting the metabolic demands of regenerating cells and promoting successful tissue repair. Understanding these intricate cellular and molecular responses at the injury site can greatly influence therapeutic strategies, such as the use of biologics like platelet-rich plasma (PRP) or MSC-derived exosomes to enhance repair processes, and guide rehabilitation protocols to optimize mechanical loading that promotes tissue regeneration while minimizing further damage. Integrating this molecular knowledge into treatment plans holds the potential to improve healing outcomes, prevent long-term joint degeneration, and restore full functional capacity to the injured knee.
Inflammatory Response
The inflammatory response to an ACL injury is a highly orchestrated molecular process that involves intricate signaling pathways aimed at damage control and tissue repair.
Vascular Response: Immediately following ACL rupture, the disruption of blood vessels within the ligament leads to hemorrhage and the formation of a hematoma, creating a hypoxic microenvironment. Hypoxia triggers a cascade of molecular events, primarily through the activation of hypoxia-inducible factors (HIFs), which are transcription factors sensitive to oxygen levels. In response to low oxygen, HIF-1α and HIF-2α are stabilized and translocate to the nucleus, where they bind to hypoxia-responsive elements (HREs) in the promoters of target genes. These genes include vascular endothelial growth factor (VEGF), a key mediator of angiogenesis. VEGF stimulates the proliferation and migration of endothelial cells to form new blood vessels, thereby restoring oxygen supply to the injured tissue. Moreover, HIFs upregulate glycolytic enzymes, enabling cells to generate energy under anaerobic conditions, which is crucial for cell survival in the hypoxic environment. Other downstream targets of HIFs include erythropoietin (EPO) and nitric oxide synthase (NOS), both of which contribute to tissue perfusion and oxygen delivery.
Cellular Infiltration: The initial vascular injury and tissue damage result in the release of damage-associated molecular patterns (DAMPs), such as high-mobility group box 1 (HMGB1), ATP, and heat shock proteins. These DAMPs are recognized by pattern recognition receptors (PRRs) on immune cells, including Toll-like receptors (TLRs) and NOD-like receptors (NLRs). TLR4, for instance, recognizes HMGB1 and activates downstream signaling pathways involving nuclear factor-kappa B (NF-κB). This leads to the transcription of pro-inflammatory cytokines, such as tumor necrosis factor-alpha (TNF-α), interleukin-1β (IL-1β), and interleukin-6 (IL-6). These cytokines amplify the inflammatory response, recruiting additional immune cells to the site of injury.
Neutrophils are among the first immune cells to arrive at the injury site, where they release a variety of molecules, including reactive oxygen species (ROS) like superoxide and hydrogen peroxide. ROS generation is driven by NADPH oxidase, an enzyme complex that becomes activated in neutrophils. These ROS serve as signaling molecules, enhancing the recruitment of other immune cells and contributing to the breakdown of damaged cellular components. Neutrophils also secrete proteolytic enzymes, including matrix metalloproteinases (MMPs) and elastase, which degrade the extracellular matrix (ECM), clearing the way for subsequent repair processes. In addition, neutrophils can undergo a unique form of cell death known as NETosis, where they release neutrophil extracellular traps (NETs). These NETs consist of DNA and histones coated with antimicrobial proteins like myeloperoxidase (MPO) and neutrophil elastase, which help to trap and neutralize pathogens. However, excessive NET formation can exacerbate tissue damage by promoting thrombosis and enhancing the inflammatory milieu.
Macrophage Activation: Following neutrophil infiltration, macrophages are recruited to the injury site. The phenotype of macrophages is influenced by the local cytokine environment. Initially, macrophages adopt a pro-inflammatory M1 phenotype in response to signals such as interferon-gamma (IFN-γ) and microbial products recognized by TLRs. M1 macrophages produce large amounts of pro-inflammatory cytokines, including TNF-α, IL-1β, and IL-6, which further activate NF-κB signaling and promote inflammation. In addition to cytokines, M1 macrophages release more MMPs, especially MMP-9 and MMP-12, which contribute to ECM degradation and the removal of damaged tissue. MMP activity is tightly regulated by tissue inhibitors of metalloproteinases (TIMPs), balancing matrix breakdown and synthesis.
As inflammation progresses, macrophages undergo a phenotypic switch to an anti-inflammatory M2 phenotype. This transition is driven by cytokines such as interleukin-4 (IL-4) and interleukin-13 (IL-13), which activate the STAT6 signaling pathway. M2 macrophages secrete anti-inflammatory cytokines, including interleukin-10 (IL-10) and transforming growth factor-beta (TGF-β), which suppress NF-κB activity and promote tissue repair. In addition to their anti-inflammatory role, M2 macrophages release growth factors such as insulin-like growth factor 1 (IGF-1) and platelet-derived growth factor (PDGF), which stimulate fibroblast proliferation and collagen synthesis, key processes in tissue remodeling. Furthermore, M2 macrophages play a crucial role in promoting angiogenesis by secreting VEGF and TGF-β, enhancing the formation of new blood vessels and supporting tissue regeneration.
The M1-to-M2 transition is essential for the resolution of inflammation and the initiation of tissue repair. Failure to switch from a pro-inflammatory to an anti-inflammatory state can result in chronic inflammation and impaired healing, as seen in some ACL injuries that develop into chronic conditions. The balance between pro-inflammatory and anti-inflammatory signals, orchestrated by a complex network of cytokines, growth factors, and matrix-remodeling enzymes, is therefore crucial for successful recovery following ACL injury.
Extracellular Matrix (ECM) Degradation and Remodeling
The extracellular matrix (ECM) is fundamental to the structural integrity of the ACL, consisting of proteins like collagen (mainly type I), elastin, and proteoglycans, which contribute to the ligament’s strength and elasticity. Following an ACL injury, the balance between ECM degradation and synthesis becomes a pivotal factor in the ligament’s ability to heal and regain function. At the molecular level, several key players regulate this balance.
Matrix Metalloproteinases (MMPs) are a family of zinc-dependent proteolytic enzymes responsible for breaking down various ECM components. MMPs are tightly regulated at the transcriptional and post-transcriptional levels, and their activity is crucial for both normal tissue remodeling and pathological ECM degradation. MMP-1 (collagenase-1) and MMP-13 (collagenase-3) specifically target type I collagen, the most abundant collagen type in ligaments. Their activity is enhanced in the injured ACL due to the increased expression of pro-inflammatory cytokines like tumor necrosis factor-alpha (TNF-α), interleukin-1β (IL-1β), and interleukin-6 (IL-6), which activate the MMP gene transcription via pathways like nuclear factor-kappa B (NF-κB) and mitogen-activated protein kinase (MAPK) signaling. MMP-3 (stromelysin-1) plays a dual role by degrading non-collagenous ECM components like proteoglycans and fibronectin, while also activating other MMPs in a proteolytic cascade. The increased activity of MMPs leads to excessive ECM breakdown, undermining ligament structure and impeding healing if not controlled.
The activity of MMPs is kept in check by Tissue Inhibitors of Metalloproteinases (TIMPs), a family of proteins that bind to MMPs in a 1:1 ratio, inhibiting their enzymatic activity. TIMPs, particularly TIMP-1 and TIMP-2, regulate ECM turnover by limiting the extent of collagen and other matrix component degradation. However, following an ACL injury, the expression of TIMPs is often insufficient to counterbalance the heightened MMP activity, leading to excessive degradation of the ECM. The transcription of TIMPs is regulated by the same pathways that control MMP expression, including NF-κB, but can also be modulated by anti-inflammatory signals such as transforming growth factor-beta (TGF-β). A fine molecular equilibrium between MMPs and TIMPs dictates the degree of ECM degradation and remodeling. Disruption of this balance, with a tilt toward excessive MMP activity, can lead to detrimental tissue destruction and delayed ligament healing.
Growth factors released in response to ACL injury are critical regulators of ECM synthesis and overall tissue remodeling. Among the most important is Transforming Growth Factor-beta (TGF-β), which plays a central role in promoting collagen synthesis and restoring ECM integrity. TGF-β signals through its receptors (TGF-βRI and TGF-βRII), initiating the phosphorylation of Smad proteins (Smad2 and Smad3). These phosphorylated Smads translocate to the nucleus, where they regulate the transcription of genes involved in ECM production, including those coding for collagen, fibronectin, and proteoglycans. TGF-β also upregulates the production of TIMPs, providing a mechanism to limit excessive MMP activity and protect ECM integrity. Furthermore, TGF-β modulates fibroblast differentiation into myofibroblasts, cells that are highly active in ECM production and tissue contraction, which is essential for ligament healing.
Other growth factors such as Fibroblast Growth Factor (FGF) and Platelet-Derived Growth Factor (PDGF) are also key players in ECM repair post-ACL injury. FGF and PDGF bind to their respective tyrosine kinase receptors on fibroblasts, initiating intracellular signaling through the MAPK and PI3K-Akt pathways. These pathways stimulate fibroblast proliferation, migration, and collagen synthesis. FGF, in particular, enhances the production of collagen types I and III, both of which are crucial for the tensile strength and repair of the ACL. PDGF, on the other hand, not only promotes fibroblast proliferation but also recruits mesenchymal stem cells (MSCs) to the injury site, which can differentiate into fibroblasts and other cell types that contribute to tissue repair. The PI3K-Akt pathway also activates downstream effectors such as mTOR, which enhances protein synthesis and cell survival, facilitating the regeneration of the ECM.
Additional molecular mechanisms involve the regulation of ECM synthesis and degradation by integrins, which are transmembrane receptors that mediate cell-ECM interactions. Integrins interact with ECM components like collagen and fibronectin, transmitting signals that regulate cell adhesion, migration, and survival. These signals are integrated with growth factor signaling to coordinate the repair process. Dysregulation of integrin signaling can impair fibroblast function and ECM assembly, further complicating ligament healing.
Moreover, cytokines like interleukin-10 (IL-10) and interleukin-4 (IL-4) exert anti-inflammatory effects and promote tissue repair by inhibiting MMP expression and enhancing TIMP production. These cytokines activate the JAK-STAT signaling pathway, leading to the transcription of genes involved in anti-inflammatory responses and tissue regeneration. IL-10, for instance, suppresses the activation of NF-κB, thereby reducing the expression of pro-inflammatory cytokines and MMPs, while boosting the anti-inflammatory and pro-repair activities of macrophages and fibroblasts.
The interplay between these molecular regulators—MMPs, TIMPs, growth factors, integrins, and cytokines—determines the extent and rate of ECM remodeling in response to ACL injury. A finely tuned balance between ECM degradation and synthesis is essential for restoring ligament structure, function, and mechanical strength. Disruptions in this balance, such as uncontrolled MMP activity or inadequate growth factor signaling, can lead to poor healing outcomes, including chronic instability or the development of fibrosis.
Fibroblast Activation and Proliferation
Fibroblasts play a pivotal role in the synthesis of the extracellular matrix (ECM) and are essential for ligament repair following an ACL injury. At a molecular level, their activation, proliferation, and ECM production are tightly regulated by a series of growth factors, cytokines, and signaling pathways.
Fibroblast Proliferation: After an ACL injury, fibroblast activation and proliferation are initiated by several key growth factors, particularly transforming growth factor-beta (TGF-β) and platelet-derived growth factor (PDGF). These growth factors bind to their specific cell surface receptors, triggering intracellular signaling cascades that are crucial for fibroblast function. For instance, TGF-β binds to its receptor complex (TGF-βRI and TGF-βRII), leading to the phosphorylation and activation of the Smad pathway. Phosphorylated Smad2 and Smad3 proteins translocate to the nucleus where they act as transcription factors, regulating genes involved in cell proliferation, migration, and ECM synthesis. TGF-β also activates non-Smad pathways like the MAPK/ERK and PI3K-Akt pathways, which further drive fibroblast proliferation and survival by regulating genes involved in cell cycle progression, such as cyclins and cyclin-dependent kinases. PDGF signals through its receptor, PDGFR, primarily activating the PI3K-Akt and MAPK pathways. These pathways not only promote fibroblast proliferation but also enhance their migratory ability, enabling them to move to the injury site and actively participate in tissue repair.
Collagen Synthesis: Fibroblasts are the primary producers of type I collagen, the most abundant collagen in ligaments, which is critical for re-establishing the structural integrity of the ACL. The process of collagen synthesis begins with the transcriptional activation of COL1A1 and COL1A2 genes, which encode the alpha chains of type I collagen. This transcriptional activation is heavily regulated by TGF-β/Smad signaling, where Smad proteins bind to promoter regions of collagen genes, enhancing their expression. The production of type I collagen is followed by post-translational modifications such as hydroxylation of proline and lysine residues, catalyzed by enzymes like prolyl-4-hydroxylase and lysyl hydroxylase. These modifications are critical for the stability and triple-helix formation of collagen molecules. Subsequently, glycosylation of hydroxylysine residues occurs, facilitating the correct folding and assembly of procollagen. Once secreted into the extracellular space, procollagen undergoes cleavage of its propeptides by specific proteases (e.g., procollagen N-proteinase and procollagen C-proteinase), yielding mature collagen fibrils. These fibrils then undergo cross-linking, a process mediated by the enzyme lysyl oxidase, which strengthens the collagen network, giving the ligament its tensile strength. Mechanical stimuli during rehabilitation are important because they regulate the molecular pathways involved in collagen fiber alignment through integrin-mediated signaling. Integrins, transmembrane receptors that connect the ECM to the cytoskeleton, activate focal adhesion kinase (FAK) and downstream pathways like RhoA and p38 MAPK, which promote the reorganization and proper alignment of collagen fibers, improving the biomechanical properties of the healing tissue.
ECM Production: Besides collagen, fibroblasts synthesize other critical ECM components, including elastin and proteoglycans like decorin and biglycan, which play vital roles in maintaining the ligament’s biomechanical properties. Elastin provides elasticity, allowing the ligament to stretch and recoil, while proteoglycans help retain water, contributing to the viscoelastic properties of the tissue. The production of these ECM components is also regulated by multiple signaling pathways. For instance, TGF-β not only stimulates collagen synthesis but also regulates the expression of genes encoding proteoglycans. Through Smad-dependent pathways, TGF-β upregulates the synthesis of proteoglycans like decorin and biglycan, which bind to collagen fibrils, influencing their spacing and organization. Proteoglycans also have glycosaminoglycan (GAG) chains that attract water, ensuring tissue hydration and flexibility. The MAPK and PI3K-Akt pathways further regulate the production of ECM components by controlling fibroblast metabolism and promoting the synthesis of proteins necessary for ECM assembly. Fibroblast growth factor (FGF) and PDGF activate these pathways, driving fibroblast proliferation and ECM production.
In addition to these well-established signaling pathways, mechanosensitive molecules like YAP/TAZ (Yes-associated protein and transcriptional co-activator with PDZ-binding motif) are activated in response to mechanical stress. These molecules sense changes in the physical environment and regulate the expression of genes involved in ECM synthesis and fibroblast activation. When fibroblasts experience mechanical loading, YAP/TAZ translocate to the nucleus and promote the transcription of ECM-related genes, further enhancing tissue repair and regeneration.
Moreover, fibroblast differentiation into myofibroblasts, a specialized cell type that produces a higher amount of ECM, is induced by TGF-β and mechanical stress. Myofibroblasts express alpha-smooth muscle actin (α-SMA), which allows them to contract and contribute to wound closure. The balance between fibroblast proliferation, ECM synthesis, and collagen fiber organization is critical for successful ACL healing. A disruption in these molecular processes, such as overactivation of MMPs leading to excessive ECM degradation or insufficient growth factor signaling, can impair ligament repair and result in compromised mechanical properties of the regenerated tissue.
Mesenchymal Stem Cell (MSC) Recruitment and Differentiation
Mesenchymal stem cells (MSCs) are multipotent progenitor cells with the capacity to differentiate into various cell types, including fibroblasts, chondrocytes, and osteoblasts, making them indispensable for tissue regeneration and repair following ACL injury. At the molecular level, their involvement in the repair process begins with MSC recruitment to the injury site, orchestrated by chemotactic factors. Stromal cell-derived factor-1 (SDF-1), secreted by damaged tissues, binds to its receptor CXCR4 on MSCs, initiating the SDF-1/CXCR4 signaling axis. This axis activates downstream pathways like PI3K-Akt and MAPK, which promote cytoskeletal reorganization, cell polarization, and directed migration toward the injury site. Additionally, vascular endothelial growth factor (VEGF), produced in response to hypoxia at the injury site, binds to VEGFR2 on MSCs, further enhancing their migration through the activation of VEGFR-mediated signaling pathways, including ERK1/2 and Akt, which increase MSC motility and survival during migration.
Upon reaching the injured ACL, MSCs encounter various microenvironmental cues that influence their differentiation into fibroblasts, chondrocytes, or other cell types needed for tissue repair. These cues include mechanical forces, oxygen levels, and the presence of specific growth factors. Transforming growth factor-beta (TGF-β), a key regulator of MSC differentiation, binds to the TGF-β receptor complex (TGF-βRI/II), leading to the phosphorylation of Smad2/3 proteins. These Smad proteins then form complexes with Smad4 and translocate to the nucleus, where they regulate the transcription of genes involved in fibroblast differentiation, such as COL1A1 (collagen type I alpha 1 chain) and FN1 (fibronectin). TGF-β also activates non-Smad pathways, such as p38 MAPK and PI3K-Akt, which further promote fibroblast differentiation and enhance MSC survival and proliferation in the injury site. Fibroblast growth factor (FGF) and platelet-derived growth factor (PDGF) also play significant roles by binding to their respective tyrosine kinase receptors (FGFR and PDGFR), activating MAPK/ERK and PI3K-Akt pathways, which drive MSC differentiation into fibroblasts, chondrocytes, and other cell types involved in ligament healing.
In addition to direct differentiation, MSCs exert powerful paracrine effects that influence the repair process through the secretion of a wide array of cytokines, growth factors, and bioactive molecules. For instance, MSCs secrete interleukin-10 (IL-10) and TGF-β, both of which play anti-inflammatory roles by suppressing the activation of pro-inflammatory pathways such as NF-κB in immune cells like macrophages. This helps shift macrophages from a pro-inflammatory M1 phenotype to an anti-inflammatory M2 phenotype, thereby facilitating tissue repair and reducing excessive inflammation. MSCs also secrete vascular endothelial growth factor (VEGF) and angiopoietin-1 (Ang-1), which stimulate the formation of new blood vessels, a process known as angiogenesis. These factors bind to receptors like VEGFR2 and Tie2 on endothelial cells, activating signaling pathways such as ERK1/2 and PI3K-Akt, which promote endothelial cell proliferation, migration, and tube formation, enhancing blood supply to the injured tissue and supporting regeneration.
One of the emerging molecular mechanisms through which MSCs exert their paracrine effects is via the release of extracellular vesicles (EVs), including exosomes and microvesicles. These vesicles are packed with bioactive molecules, including microRNAs (miRNAs), proteins, and lipids, which can be delivered to target cells, modulating their behavior. For example, MSC-derived exosomes have been shown to contain miR-126, which promotes angiogenesis by targeting inhibitors of the PI3K-Akt pathway, thereby enhancing endothelial cell function. Additionally, exosomes carry anti-inflammatory cytokines and growth factors that can modulate the activity of immune cells and fibroblasts, promoting a pro-regenerative environment.
Furthermore, MSCs themselves are influenced by the mechanical properties of their surroundings, including matrix stiffness and mechanical loading. The mechanical environment is sensed by integrins, which activate intracellular signaling pathways such as focal adhesion kinase (FAK), RhoA, and YAP/TAZ, leading to the nuclear translocation of transcriptional co-activators like YAP/TAZ. Once in the nucleus, YAP/TAZ interact with transcription factors such as TEAD, promoting the expression of genes involved in cell proliferation, ECM production, and differentiation into repair cells like fibroblasts. These mechanical cues are crucial in determining whether MSCs differentiate into fibroblasts, chondrocytes, or other cell types, influencing the overall success of ligament repair.
In summary, MSCs contribute to ACL repair through a combination of differentiation into critical cell types like fibroblasts and their paracrine effects, which modulate the immune response, promote angiogenesis, and stimulate tissue regeneration. The recruitment, differentiation, and paracrine activity of MSCs are regulated by a complex interplay of signaling pathways, including the SDF-1/CXCR4, VEGF/VEGFR, TGF-β/Smad, MAPK, PI3K-Akt, and YAP/TAZ pathways, which ensure that the repair process proceeds efficiently and effectively. This molecular orchestration of MSC activity is essential for restoring the structure and function of the damaged ACL.
Angiogenesis
Angiogenesis, the formation of new blood vessels, is a vital molecular process that significantly influences tissue repair following an ACL injury. This process is intricately regulated at the cellular and molecular levels, primarily orchestrated by vascular endothelial growth factor (VEGF), which acts as a master regulator of angiogenesis. VEGF is upregulated in response to hypoxia, which occurs rapidly after tissue injury due to disrupted blood flow. Hypoxia activates hypoxia-inducible factor-1 alpha (HIF-1α), a transcription factor that binds to hypoxia-responsive elements (HREs) on the VEGF gene promoter, enhancing VEGF expression. VEGF binds to its receptor, VEGFR-2, on endothelial cells, initiating a cascade of intracellular signaling through the MAPK/ERK, PI3K-Akt, and PLCγ pathways. These pathways lead to endothelial cell proliferation, survival, migration, and increased permeability of blood vessels, key processes for angiogenic sprouting. In particular, PI3K-Akt signaling promotes endothelial cell survival by inhibiting apoptotic pathways, while MAPK/ERK enhances proliferation. PLCγ activation increases intracellular calcium, promoting cytoskeletal reorganization, which is necessary for endothelial cell migration and vessel formation.
Additionally, other angiogenic factors, such as fibroblast growth factor (FGF) and platelet-derived growth factor (PDGF), contribute to the angiogenic process. FGF binds to FGFR and activates similar signaling pathways, particularly MAPK and PI3K-Akt, facilitating endothelial cell proliferation and migration. PDGF plays a complementary role, particularly in stabilizing newly formed vessels by recruiting pericytes and smooth muscle cells to the endothelial network. PDGF signaling through its receptor, PDGFR, activates downstream effectors that promote the maturation and stabilization of nascent blood vessels, preventing regression and ensuring long-term vascular integrity. Moreover, angiopoietins (Ang-1 and Ang-2), which bind to the Tie2 receptor on endothelial cells, fine-tune the angiogenic process. Ang-1/Tie2 signaling promotes vessel stability and maturation, while Ang-2 acts as a context-dependent antagonist that facilitates vessel sprouting in the presence of VEGF but destabilizes vessels in its absence, enhancing vascular remodeling.
The role of angiogenesis extends beyond merely providing oxygen and nutrients. The newly formed blood vessels also deliver essential reparative cells, such as mesenchymal stem cells (MSCs) and immune cells, to the injury site. These cells, in turn, secrete a variety of cytokines and growth factors that influence both angiogenesis and tissue regeneration. VEGF, for instance, acts not only as an angiogenic factor but also as a chemoattractant for endothelial progenitor cells (EPCs), which further contribute to vascular repair. Additionally, HIF-1α and VEGF signaling induce the expression of matrix metalloproteinases (MMPs), which degrade the extracellular matrix (ECM), allowing endothelial cells to migrate through the tissue and form new blood vessels. However, MMP activity must be tightly regulated, as excessive degradation can impair tissue repair and destabilize the healing process.
Angiogenesis is also influenced by the mechanical forces acting on the healing tissue, as the biomechanical environment modulates cellular responses. Mechanosensitive pathways, such as YAP/TAZ signaling, respond to changes in mechanical load and directly influence the expression of VEGF and other angiogenic genes. This mechanotransduction ensures that blood vessel formation aligns with the mechanical needs of the regenerating tissue. Moreover, integrins, which mediate cell-ECM interactions, play a critical role in endothelial cell adhesion and migration during angiogenesis. Integrin activation leads to the recruitment of signaling molecules like focal adhesion kinase (FAK) and Src, further integrating mechanical and biochemical signals to promote vessel formation.
As angiogenesis progresses, it facilitates the removal of metabolic byproducts and cellular debris from the injury site, a process that is essential for maintaining tissue homeostasis and preventing inflammation from becoming chronic. The interplay between angiogenic and inflammatory signals is crucial for balancing the repair process. Pro-inflammatory cytokines like interleukin-1 beta (IL-1β) and tumor necrosis factor-alpha (TNF-α) can promote VEGF expression in the early phases of healing, linking inflammation to angiogenesis. However, these inflammatory mediators must be modulated by anti-inflammatory signals such as interleukin-10 (IL-10) to avoid excessive angiogenesis, which could lead to pathological neovascularization or poor tissue quality.
In summary, angiogenesis following ACL injury is a highly regulated process involving a network of growth factors, signaling pathways, and mechanosensitive elements that work together to form functional blood vessels. These newly formed vessels are critical for supplying the regenerating tissue with oxygen, nutrients, and reparative cells, while also ensuring the removal of waste products. The interplay between VEGF, FGF, PDGF, Ang-1, HIF-1α, and integrin/FAK signaling highlights the complexity of angiogenic regulation and underscores the importance of this process in successful ACL repair. Understanding these molecular mechanisms opens avenues for targeted therapies, such as the use of VEGF modulators, FGF-based treatments, or MSC-derived exosomes, to enhance angiogenesis and improve the outcomes of ACL rehabilitation.
Mechanotransduction and Mechanical Loading
Mechanical loading plays a crucial role in ACL repair and remodeling by influencing cellular behavior through a variety of mechanotransduction pathways. At the molecular level, these pathways enable cells within the ligament, such as fibroblasts and mesenchymal stem cells (MSCs), to sense mechanical forces and convert them into biochemical signals that regulate essential processes like cell proliferation, differentiation, and extracellular matrix (ECM) production. One of the key molecular mechanisms involved in this response is integrin signaling. Integrins are transmembrane receptors that connect the intracellular cytoskeleton to the ECM, providing a physical and signaling link between the cell and its surrounding environment. Upon mechanical loading, integrins are activated and form clusters at focal adhesions, sites where the cell adheres to the ECM. This clustering leads to the recruitment and activation of focal adhesion kinase (FAK) and Src family kinases, which serve as key mediators in transducing mechanical signals. FAK activation triggers the phosphorylation of downstream molecules, including paxillin and talin, which link integrins to the actin cytoskeleton. This signaling cascade not only promotes cytoskeletal reorganization—a critical process for maintaining cell shape and integrity under mechanical stress—but also stimulates gene expression and protein synthesis involved in ECM production.
FAK signaling is closely integrated with other key mechanotransduction pathways, such as MAPK/ERK and PI3K-Akt, both of which regulate cellular responses to mechanical stress. The MAPK pathway plays a central role in coordinating the repair process by regulating fibroblast and MSC proliferation, differentiation, and ECM synthesis. Mechanical loading activates ERK1/2 and p38 MAPK, which phosphorylate downstream transcription factors such as c-Fos, c-Jun, and AP-1 that regulate genes involved in cell proliferation, collagen production, and matrix remodeling. ERK1/2 is particularly important in promoting the proliferation of fibroblasts, while p38 modulates stress responses and drives MSC differentiation into fibroblasts, which are essential for producing the ECM components necessary to restore the structural integrity of the ligament. The PI3K-Akt pathway, also activated by integrin signaling, promotes cell survival by inhibiting pro-apoptotic factors like Bad and activating the mTOR pathway, which is essential for protein synthesis and cell growth. This pathway ensures that fibroblasts and MSCs survive and function optimally during mechanical stress, contributing to efficient ligament repair.
In addition to integrin signaling, mechanical loading activates stretch-activated ion channels, particularly calcium channels, which play a significant role in regulating cellular responses to mechanical stimuli. When mechanical forces are applied, these channels open, allowing calcium ions (Ca²⁺) to flow into the cell. The increase in intracellular calcium triggers several downstream signaling pathways that are crucial for the cellular adaptation to mechanical stress. One such pathway is the calcineurin/NFAT (nuclear factor of activated T cells) pathway, in which the phosphatase calcineurin dephosphorylates NFAT, allowing it to translocate into the nucleus and regulate gene expression involved in cell proliferation, differentiation, and ECM remodeling. Similarly, calcium influx activates the calmodulin-dependent kinase (CaMK) pathway, which influences cytoskeletal organization and gene transcription, further supporting the adaptation of cells to mechanical forces. These calcium-dependent pathways also interact with other mechanotransduction signaling networks, creating a highly coordinated cellular response to mechanical loading.
Another critical player in mechanotransduction is the YAP/TAZ pathway, part of the Hippo signaling pathway, which is sensitive to changes in mechanical forces and cellular tension. YAP (Yes-associated protein) and TAZ (transcriptional co-activator with PDZ-binding motif) are transcriptional regulators that shuttle between the cytoplasm and nucleus depending on mechanical stimuli. Under mechanical loading, YAP and TAZ are dephosphorylated, allowing them to enter the nucleus, where they interact with transcription factors such as TEAD to activate genes involved in cell proliferation, ECM synthesis, and tissue regeneration. This pathway is especially important in maintaining tissue homeostasis and ensuring that cellular responses are appropriately matched to the mechanical environment of the ACL during repair. YAP/TAZ activation leads to the upregulation of connective tissue growth factor (CTGF) and other ECM-related genes, promoting collagen production and tissue remodeling.
Mechanical stress also induces the activation of RhoA/ROCK signaling, which regulates cytoskeletal tension and cell contractility. This pathway plays a crucial role in maintaining cell shape, promoting actin filament assembly, and ensuring the proper alignment of collagen fibers during ECM remodeling. By controlling the actomyosin contractility of fibroblasts and MSCs, RhoA/ROCK signaling helps the healing ligament withstand mechanical forces while restoring its mechanical strength and structural integrity.
The coordination of these mechanotransduction pathways—integrin/FAK, MAPK/ERK, PI3K-Akt, calcium signaling, YAP/TAZ, and RhoA/ROCK—ensures that cells within the injured ACL respond appropriately to mechanical stimuli, promoting efficient tissue repair and remodeling. Mechanical loading not only stimulates the production of new ECM components, such as collagen, but also influences the alignment and organization of collagen fibers, critical for restoring the biomechanical properties of the ACL. However, excessive or inappropriate mechanical loading can dysregulate these pathways, leading to maladaptive responses such as excessive ECM degradation through matrix metalloproteinase (MMP) upregulation or fibrosis, ultimately compromising the healing process. Understanding the molecular mechanisms by which mechanical loading influences ACL repair opens up new therapeutic avenues, such as targeted modulation of these signaling pathways through pharmacological agents or optimizing rehabilitation protocols that apply controlled mechanical forces to enhance tissue regeneration while minimizing the risk of re-injury or degeneration.

Table 1.
The table explains how different factors contribute to ACL injury, including high-impact sports, gender-specific risk factors, and improper landing or deceleration movements. It also details the biological cascade that occurs after an ACL tear, including inflammation, cellular recruitment, tissue remodeling, and the critical role of mechanical forces in tissue repair. The final rows discuss therapeutic interventions like platelet-rich plasma (PRP) and mesenchymal stem cell (MSC) therapies, which are designed to enhance healing and restore knee function.
Table 1.
The table explains how different factors contribute to ACL injury, including high-impact sports, gender-specific risk factors, and improper landing or deceleration movements. It also details the biological cascade that occurs after an ACL tear, including inflammation, cellular recruitment, tissue remodeling, and the critical role of mechanical forces in tissue repair. The final rows discuss therapeutic interventions like platelet-rich plasma (PRP) and mesenchymal stem cell (MSC) therapies, which are designed to enhance healing and restore knee function.
| Category | Details | Processes Involved | Key Molecular/Cellular Players |
| ACL Function and Vulnerability | ACL stabilizes the knee by limiting excessive anterior translation of the tibia relative to the femur and controlling rotational movements. | Stabilizes the knee during dynamic activities like pivoting, jumping, and sudden stops, which put high stress on the ACL. Sports with rapid directional changes like soccer, basketball, and skiing are high-risk. | ACL, tibia, femur, dynamic activities (pivoting, jumping, sudden stops), rotational forces, anterior translation |
| Risk Factors for ACL Injuries | Common in sports with sudden directional changes and high-impact landings. Women are at higher risk due to hormonal influences, neuromuscular control differences, and anatomical factors. | Hormonal fluctuations, neuromuscular control deficits, and biomechanics like greater Q-angle in women increase susceptibility. | Hormones (estrogen, relaxin), Q-angle, neuromuscular control, biomechanics, deceleration, valgus collapse |
| Mechanisms of Injury | ACL injuries are often non-contact, accounting for 70% of cases, particularly during rapid deceleration, pivoting, or improper landing from jumps. | High knee stress due to valgus stress, rotational forces, and rapid changes in movement, often without direct contact. | Valgus stress, rotational forces, knee mechanics, pivoting, deceleration, improper landing |
| Consequences of ACL Rupture | Immediate joint instability occurs with ACL rupture, leading to increased risk of secondary injuries to the meniscus and cartilage. Long-term consequences include osteoarthritis. | Altered knee mechanics after ACL injury lead to abnormal wear patterns in the joint, increasing the likelihood of degenerative joint disease like osteoarthritis. | Meniscus, articular cartilage, joint instability, osteoarthritis, abnormal joint kinematics |
| Inflammatory Response to Injury | The body initiates a biochemical cascade involving cytokine release, immune cell recruitment, and ECM degradation. This starts with an acute inflammatory phase. | Neutrophils and macrophages clear debris; cytokines (TNF-α, IL-1β, IL-6) promote inflammation, while MMPs degrade ECM, allowing tissue repair but potentially causing excessive damage. | Cytokines (TNF-α, IL-1β, IL-6), neutrophils, macrophages (M1 to M2), MMPs (MMP-1, MMP-3, MMP-13), ECM, fibroblasts |
| Tissue Repair and Remodeling | After inflammation, the repair phase begins with fibroblast activation and ECM synthesis, particularly collagen production to restore ligament strength. | Growth factors (TGF-β, FGF, PDGF) activate fibroblasts, leading to ECM synthesis. Mechanical forces influence collagen fiber alignment for proper tissue remodeling. | Fibroblasts, growth factors (TGF-β, FGF, PDGF), collagen (type I), mechanical forces, ECM, integrins, YAP/TAZ |
| Extracellular Matrix (ECM) Degradation and Remodeling | ECM, composed of collagen, elastin, and proteoglycans, is crucial for ACL structural integrity. Injury disrupts this balance, leading to both degradation and remodeling. | MMPs degrade ECM components like collagen, while growth factors like TGF-β promote ECM synthesis. TIMPs regulate MMP activity to balance degradation and repair. | MMPs (MMP-1, MMP-3, MMP-13), TIMPs (TIMP-1, TIMP-2), TGF-β, collagen (type I), ECM, fibroblasts, proteoglycans |
| Cellular Recruitment and MSC Involvement | Mesenchymal stem cells (MSCs) are recruited to the injury site, where they differentiate into fibroblasts and other cell types essential for tissue repair. | Chemotactic signals (SDF-1, VEGF) recruit MSCs, which differentiate into fibroblasts to aid in ECM production and remodeling. MSCs also release growth factors that modulate immune responses. | MSCs, chemotactic signals (SDF-1/CXCR4, VEGF), fibroblasts, immune cells, ECM, cytokines, paracrine signaling |
| Mechanical Loading and Tissue Repair | Mechanical forces play a key role in ACL healing, influencing cellular behaviors such as collagen alignment and fibroblast activity. Proper loading can improve tissue repair, while excessive loading can cause further damage. | Mechanical forces are sensed by integrins and mechanoreceptors like YAP/TAZ, which modulate gene expression related to ECM production, collagen fiber alignment, and cell proliferation. | Integrins, mechanoreceptors (Piezo1), YAP/TAZ, focal adhesion kinase (FAK), fibroblasts, ECM, collagen alignment |
| Role of Angiogenesis in ACL Healing | Angiogenesis is crucial for delivering nutrients and oxygen to the injured tissue. Growth factors like VEGF promote new blood vessel formation, which supports tissue regeneration. | Hypoxia after injury triggers HIF-1α, which upregulates VEGF and promotes endothelial cell proliferation and migration, leading to new blood vessel formation. | VEGF, HIF-1α, endothelial cells, angiogenesis, fibroblasts, PDGF, endothelial progenitor cells (EPCs) |
| Chronic Effects of Dysregulated Healing | If inflammation is not properly resolved, or if mechanical loading is inappropriate, ACL repair can be impaired, leading to chronic instability or the development of fibrosis. | Chronic inflammation or excessive ECM degradation due to unchecked MMP activity can impair tissue regeneration, increasing the risk of long-term complications like osteoarthritis. | Chronic inflammation, fibrosis, osteoarthritis, MMPs, TIMPs, cytokines, abnormal joint kinematics |
| Therapeutic Interventions and Potential Strategies | Therapies like platelet-rich plasma (PRP) and MSC-derived exosomes aim to enhance healing by modulating inflammation and promoting tissue regeneration. Rehabilitation strategies emphasize controlled mechanical loading. | Biologics (PRP, MSC exosomes) and targeted therapies modulate the healing environment by reducing inflammation, enhancing ECM production, and promoting proper mechanical loading for optimal healing. | PRP, MSC exosomes, cytokines, growth factors, rehabilitation, mechanical loading, integrins, YAP/TAZ |
Understanding Biological and Physiological Processes
A comprehensive understanding of the biological and physiological processes underlying ACL injury and repair is essential for designing effective strategies aimed at promoting healing, repair, and regeneration not only of the ACL but also of associated muscle and tendon injuries. The intricate interplay of inflammation, extracellular matrix (ECM) remodeling, cellular proliferation, and tissue regeneration governs the repair process and dictates the outcomes of rehabilitation efforts. By understanding how cells such as fibroblasts, chondrocytes, and mesenchymal stem cells (MSCs) respond to mechanical and biochemical signals, clinicians can better tailor rehabilitation exercises and treatments to align with the natural cascade of cellular and molecular responses. These responses involve complex signaling pathways, such as TGF-β/Smad, MAPK/ERK, PI3K/Akt, and YAP/TAZ, that regulate processes like ECM synthesis, cell migration, and tissue remodeling, all of which are crucial for restoring ligament structure and function. Advanced molecular biology techniques, such as gene expression profiling, RNA sequencing, and proteomics, offer deep insights into the specific genes, proteins, and regulatory networks involved in ACL repair. These tools allow researchers and clinicians to identify key molecular targets that could be manipulated to enhance healing—whether by accelerating tissue regeneration, modulating inflammation, or improving ECM organization. For example, by profiling the expression of genes involved in collagen production or ECM degradation, scientists can develop targeted therapies, such as growth factor treatments or gene-editing technologies like CRISPR, that specifically boost beneficial cellular responses while mitigating negative factors like excessive scarring or fibrosis. Furthermore, proteomics can uncover changes in protein levels during various stages of healing, guiding the use of biologic therapies such as platelet-rich plasma (PRP) or MSC-derived exosomes, which deliver bioactive molecules to stimulate repair. This molecular understanding also aids in the design of rehabilitation protocols that optimize mechanical loading, ensuring that exercises are introduced at the right stage of healing to promote tissue regeneration without exacerbating injury. Ultimately, leveraging these molecular insights allows for more precise interventions that minimize complications, improve tissue quality, and enhance long-term outcomes for patients recovering from ACL injuries.
Table 2.
Cellular pathway in the process of ligament regeneration and remodelling bounded with rehabilitation regimes.
Table 2.
Cellular pathway in the process of ligament regeneration and remodelling bounded with rehabilitation regimes.
| Inflammation phase | |
| Injury/ incident | Immediate vasoconstriction of the blood flow. Immobilization to reduce pain and swelling |
| 24–48 h post | Vasodilatation and proliferation of tissue. Inflammation. Icing is not recommended as it slows down healing by decreasing lymphatic flow, proliferation, and cell– cell-interactions. Same rules apply for anti-inflammatory drugs. Ice has numbing effects and should only be used for a few minutes for pain relief |
| Proliferation phase | |
| 5 days | Type III collagen is produced and will be transferred to type I over time. Reconstruction and orientation of the type III fibers depend on stress of movement and weight bearing. That is why exercises in full range of motion allowed and weight bearing are so important to guarantee good healing of tissue and scars |
| Remodeling/ maturation | |
| ~3 weeks | Type III collagen is transferred into type I. regaining range of motion, proprioceptive and contractile information to allow good healing. Regaining biomechanical qualities of the tissue. Formation of cross-links for greater stiffness. This process is supported by load and mobilization into the end of ROM in exercises. 300–500 days until tissue regains its former function |
Molecular Description of Ligament Healing Process.
The molecular process of ligament healing is a highly orchestrated sequence of cellular and biochemical events that unfolds in distinct but overlapping phases: the inflammatory phase, the proliferative phase, and the remodeling phase. Each of these phases is governed by specific molecular signals that regulate cell recruitment, tissue repair, and extracellular matrix (ECM) remodeling, ultimately restoring the structural integrity and mechanical function of the ligament.
Immediately following ligament injury, the inflammatory phase begins, characterized by the activation of immune cells and the release of pro-inflammatory mediators. Damage-associated molecular patterns (DAMPs), such as high mobility group box 1 (HMGB1), are released from damaged cells and extracellular matrix, triggering the activation of pattern recognition receptors (PRRs), including Toll-like receptors (TLRs), on resident cells. This initiates a cascade of pro-inflammatory signaling, including the upregulation of nuclear factor-kappa B (NF-κB), which controls the expression of cytokines such as tumor necrosis factor-alpha (TNF-α), interleukin-1 beta (IL-1β), and interleukin-6 (IL-6). These cytokines promote the recruitment of neutrophils and macrophages to the injury site, where they remove cellular debris and secrete growth factors that promote the next phase of healing.
At the same time, matrix metalloproteinases (MMPs), particularly MMP-1, MMP-3, and MMP-13, are activated to degrade damaged collagen and other ECM components. This ECM breakdown is crucial for clearing space for new tissue formation but must be tightly regulated to avoid excessive tissue destruction. Inhibitors of MMPs, known as tissue inhibitors of metalloproteinases (TIMPs), work in tandem to control MMP activity and prevent excessive degradation.
The proliferative phase is marked by the activation and proliferation of fibroblasts, the main cell type responsible for ECM synthesis in ligaments. This phase is largely driven by growth factors such as transforming growth factor-beta (TGF-β), fibroblast growth factor (FGF), platelet-derived growth factor (PDGF), and insulin-like growth factor 1 (IGF-1), which are released by immune cells, fibroblasts, and platelets. These growth factors activate key intracellular signaling pathways, such as the TGF-β/Smad pathway, MAPK/ERK, and PI3K/Akt, which regulate gene expression and drive fibroblast proliferation and differentiation.
TGF-β, in particular, plays a critical role by promoting the production of collagen types I and III, which form the backbone of the new ligament matrix. Fibroblasts synthesize collagen precursors, procollagen, which undergo post-translational modifications such as hydroxylation and glycosylation before being secreted into the ECM. Once outside the cell, procollagen is cleaved by enzymes like procollagen peptidase to form mature collagen fibrils. These fibrils are then cross-linked by enzymes such as lysyl oxidase, which strengthens the collagen network.
In addition to collagen, fibroblasts produce other ECM components, including proteoglycans and elastin, which contribute to the biomechanical properties of the ligament. Decorin, a small leucine-rich proteoglycan, is particularly important in regulating collagen fibril formation and spacing. As fibroblasts deposit new matrix, angiogenesis is stimulated by growth factors like vascular endothelial growth factor (VEGF), which promotes the formation of new blood vessels to supply the healing tissue with oxygen and nutrients.
The final remodeling phase is critical for the maturation and strengthening of the newly formed ligament. During this phase, collagen fibers are realigned along the lines of mechanical stress, a process that is influenced by mechanical loading and activity. Mechanical forces are sensed by integrins on fibroblasts, which activate mechanotransduction pathways such as focal adhesion kinase (FAK), RhoA/ROCK, and YAP/TAZ, leading to cytoskeletal reorganization and increased ECM synthesis.
Collagen fibers undergo a process of cross-linking and reorganization, with type III collagen (initially deposited during the proliferative phase) being gradually replaced by the stronger type I collagen, which is better suited to bear mechanical loads. The cross-linking of collagen fibers, mediated by lysyl oxidase, is essential for the tensile strength of the ligament. During this phase, matrix metalloproteinases (MMPs) continue to play a role, breaking down disorganized or excess matrix components, while TIMP-1 and TIMP-2 regulate their activity to prevent excessive degradation.
As the ligament matures, there is a gradual reduction in cellularity and vascularity, with the ligament becoming more densely packed with well-aligned collagen fibers. TGF-β, which initially promotes collagen synthesis, later shifts to suppress excessive ECM production, ensuring a balance between tissue regeneration and scar formation. This phase is also marked by a reduction in the expression of inflammatory cytokines, signaling the resolution of the inflammatory response.
Throughout the ligament healing process, various signaling pathways interact and regulate each other to ensure the proper progression of healing. Cross-talk between TGF-β/Smad, Wnt/β-catenin, and PI3K/Akt pathways helps coordinate fibroblast activity and ECM remodeling. Additionally, microRNAs (miRNAs) play a regulatory role by modulating gene expression involved in collagen synthesis, inflammation, and fibroblast proliferation. For example, miR-29 has been shown to regulate collagen production, while miR-146a modulates inflammatory responses by targeting NF-κB signaling.
The role of mechanical loading in this process cannot be understated, as it directly influences molecular signaling through mechanotransduction. The application of controlled mechanical forces during rehabilitation stimulates pathways such as YAP/TAZ, which are activated by changes in cellular tension and matrix stiffness, promoting the alignment and maturation of collagen fibers. Mechanical stimuli also regulate the expression of integrins, which interact with the ECM to transmit signals that drive tissue remodeling.
In conclusion, the molecular description of ligament healing involves a complex interplay of inflammatory, proliferative, and remodeling phases, each regulated by specific signaling pathways, growth factors, and cellular responses. Understanding these molecular mechanisms provides critical insights for developing targeted therapies, optimizing rehabilitation protocols, and improving outcomes for ligament injuries.
- Injury incydent (0h).
At the moment of injury, vasoconstriction is initiated by a highly regulated molecular response that minimizes blood loss and sets the stage for tissue repair. The primary driver of this process is the release of endothelin-1 (ET-1) by endothelial cells, which are activated by mechanical damage, hypoxia, and inflammatory signals such as TNF-α and IL-1β. The production of ET-1 begins with the transcription of the preproendothelin gene, followed by the cleavage of big endothelin into its active form by endothelin-converting enzyme (ECE). Once released, ET-1 binds to ETA and ETB receptors, which are G-protein-coupled receptors (GPCRs) located on vascular smooth muscle cells. This interaction triggers the Gq protein signaling pathway, which activates phospholipase C (PLC).
PLC catalyzes the hydrolysis of phosphatidylinositol 4,5-bisphosphate (PIP2) into two critical second messengers: inositol trisphosphate (IP3) and diacylglycerol (DAG). IP3 binds to its receptors on the sarcoplasmic reticulum (SR), releasing calcium ions (Ca²⁺) into the cytoplasm. The rise in intracellular calcium is a key event that activates calmodulin, a calcium-binding protein. Calmodulin, in turn, activates myosin light-chain kinase (MLCK), an enzyme that phosphorylates myosin light chains, enabling the interaction between myosin and actin filaments, which drives the contraction of smooth muscle cells and reduces the vessel lumen. This smooth muscle contraction is essential for maintaining hemostasis and limiting blood flow at the injury site.
Simultaneously, the sympathetic nervous system is activated almost immediately, releasing norepinephrine from postganglionic sympathetic neurons. Norepinephrine binds to alpha-1 adrenergic receptors on smooth muscle cells, which are also GPCRs linked to the Gq protein pathway. This activation mirrors ET-1 signaling, as PLC is stimulated to produce IP3 and DAG, leading to the same cascade of calcium release from the SR and smooth muscle contraction. DAG, in addition to facilitating calcium release, also activates protein kinase C (PKC), which enhances the contractile response by modulating calcium channels and increasing calcium sensitivity. PKC also inhibits myosin phosphatase, thereby maintaining the phosphorylated state of myosin light chains, prolonging smooth muscle contraction.
In parallel to these vasoconstrictive pathways, the body downregulates nitric oxide (NO) signaling, which normally promotes vasodilation. NO, synthesized by endothelial nitric oxide synthase (eNOS), would typically diffuse into smooth muscle cells and activate soluble guanylate cyclase (sGC), leading to the production of cyclic GMP (cGMP). cGMP activates protein kinase G (PKG), which reduces intracellular calcium levels by enhancing calcium reuptake into the SR and reducing calcium influx through channels, leading to smooth muscle relaxation. During injury, the suppression of NO by damaged endothelial cells, combined with the dominance of ET-1 and adrenergic signaling, ensures that vasoconstriction predominates, allowing for rapid reduction in blood flow and blood loss.
Another key molecular player is the regulation of reactive oxygen species (ROS), which are often produced at the injury site due to oxidative stress. ROS interact with NO, forming peroxynitrite (ONOO⁻), a reactive nitrogen species that further depletes NO levels. This leads to endothelial dysfunction and exacerbates vasoconstriction by tipping the balance even more toward ET-1 and norepinephrine-mediated pathways. The continued suppression of NO not only supports vasoconstriction but also prevents the usual vasodilatory feedback mechanisms, ensuring sustained control over blood flow.
The inflammatory response following injury is also molecularly complex. Pro-inflammatory cytokines such as TNF-α, IL-1β, and IL-6 are released by immune cells like macrophages and neutrophils. These cytokines bind to their respective receptors, including TNF receptors (TNFRs) and IL-1 receptors (IL-1Rs), on surrounding cells. This binding activates intracellular pathways such as NF-κB and MAPK, which drive the expression of genes involved in inflammation and tissue repair. NF-κB, in particular, upregulates the production of adhesion molecules like ICAM-1 and VCAM-1, which are essential for the recruitment of additional immune cells to the site of injury. NF-κB also increases the expression of matrix metalloproteinases (MMPs), such as MMP-1, MMP-9, and MMP-13, which degrade extracellular matrix (ECM) components, facilitating tissue remodeling.
The pain response is modulated through prostaglandins, specifically prostaglandin E2 (PGE2), produced via the cyclooxygenase (COX-2) pathway. PGE2 binds to EP receptors on nociceptors, activating adenylate cyclase and increasing cyclic AMP (cAMP) levels. cAMP activates protein kinase A (PKA), which phosphorylates voltage-gated sodium channels and TRPV1 receptors, enhancing nociceptor sensitivity and amplifying pain signals. This sensitization ensures that the body remains alert to potential further injury by increasing pain perception.
As part of the tissue remodeling process, MMPs are tightly regulated by tissue inhibitors of metalloproteinases (TIMPs). This balance ensures controlled ECM degradation and prevents excessive tissue breakdown. In response to growth factors like TGF-β and PDGF, fibroblasts are recruited to synthesize new ECM components, particularly collagen, while TIMPs inhibit MMP activity to ensure the integrity of the newly forming tissue.
In summary, the molecular mechanisms driving immediate vasoconstriction and tissue response to injury involve the coordinated actions of ET-1, norepinephrine, PKC, and the suppression of NO. These pathways ensure rapid control over blood flow, modulate inflammation via cytokines and MMPs, and regulate pain through prostaglandin signaling. The interaction between these molecular systems provides a robust framework for the body’s initial injury response, balancing vasoconstriction, inflammation, and tissue remodeling to facilitate healing. Understanding these molecular players offers insights into therapeutic targets for enhancing tissue repair and recovery following injury.
- 24–48 h after ACL injury.
Vasodilatation is a critical physiological process that ensures increased blood flow to tissues, especially following injury or inflammation. At the molecular level, this process is tightly regulated and involves a cascade of signaling mechanisms, primarily driven by endothelial cells, nitric oxide (NO), and prostaglandins, which coordinate the relaxation of vascular smooth muscle cells. These mechanisms not only help deliver oxygen and nutrients to injured tissues but also facilitate the removal of metabolic waste and immune cell infiltration to support the healing process.
Endothelial cells, which line the inner walls of blood vessels, serve as central regulators of vascular tone. These cells respond to various stimuli such as mechanical shear stress, inflammatory mediators, and hypoxia by releasing vasoactive substances like nitric oxide (NO) and prostaglandins. Shear stress, caused by increased blood flow, activates mechanosensitive pathways in endothelial cells, leading to the activation of endothelial nitric oxide synthase (eNOS). The enzyme eNOS converts L-arginine into NO, a process modulated by cofactors like tetrahydrobiopterin (BH4) and regulated by intracellular calcium concentrations and the Akt kinase pathway. The phosphorylation of eNOS by Akt, a downstream effector of the PI3K pathway, enhances NO production, ensuring rapid endothelial responses to vascular signals.
NO, once synthesized, rapidly diffuses across the endothelial membrane due to its lipophilic properties, allowing it to easily penetrate the adjacent smooth muscle cells in the vessel wall. NO binds to soluble guanylate cyclase (sGC) in smooth muscle cells, activating this enzyme. sGC converts guanosine triphosphate (GTP) into cyclic guanosine monophosphate (cGMP), a crucial second messenger in vasodilation. The increased cGMP levels trigger a cascade of downstream effects, most notably the activation of protein kinase G (PKG). PKG phosphorylates several proteins involved in smooth muscle relaxation, including myosin light-chain phosphatase, which dephosphorylates myosin light chains, thereby inhibiting the interaction between myosin and actin filaments required for contraction. In parallel, PKG facilitates the opening of potassium channels, leading to hyperpolarization of the smooth muscle cell membrane, and reduces intracellular calcium levels by promoting calcium reuptake into the sarcoplasmic reticulum (SR) and extrusion through calcium ATPases on the plasma membrane. The combined reduction in intracellular calcium and myosin light-chain phosphorylation results in smooth muscle relaxation, causing vasodilatation and the widening of the blood vessel.
Beyond NO, prostaglandins, particularly prostaglandin E2 (PGE2), are also key regulators of vasodilatation. PGE2 is synthesized through the cyclooxygenase (COX) pathway, which converts arachidonic acid into prostaglandins via COX-1 and COX-2 enzymes. While COX-1 is constitutively expressed in many tissues, COX-2 is inducible and upregulated during inflammation. PGE2 binds to specific EP receptors on smooth muscle cells, activating the adenylate cyclase (AC) pathway. This activation increases the levels of cyclic adenosine monophosphate (cAMP), another crucial second messenger in smooth muscle relaxation. Similar to cGMP, elevated cAMP activates protein kinase A (PKA), which reduces intracellular calcium concentrations by enhancing calcium reuptake into the sarcoplasmic reticulum and inhibiting calcium influx through voltage-gated calcium channels. PKA also phosphorylates and inactivates myosin light-chain kinase (MLCK), preventing the phosphorylation of myosin light chains and thus contributing to smooth muscle relaxation and vasodilatation.
Calcium signaling plays a pivotal role in regulating smooth muscle contraction and relaxation, acting as a molecular switch between these two states. The binding of calmodulin to calcium in smooth muscle cells activates MLCK, which phosphorylates myosin and facilitates contraction. In contrast, reducing intracellular calcium levels via NO/cGMP or PGE2/cAMP signaling pathways inhibits MLCK activity, promoting vasodilatation. The fine balance between contraction and relaxation is tightly controlled by these molecular mechanisms, ensuring appropriate vascular responses to physiological needs.
In addition to NO and prostaglandins, endothelin-1 (ET-1), another vasoactive molecule produced by endothelial cells, plays a role in modulating vascular tone. Although primarily known for its vasoconstrictive properties through ETA receptors, ET-1 can also interact with ETB receptors on endothelial cells to promote vasodilatation by stimulating NO and prostacyclin (PGI2) release. This dual role of ET-1 highlights the complexity of molecular interactions in regulating blood vessel diameter, with ET-1 balancing between vasoconstriction and vasodilation based on receptor context and physiological demands.
Another important aspect of vasodilatation is its role in tissue proliferation and repair following injury. The increased blood flow resulting from vasodilatation ensures the delivery of key growth factors, such as vascular endothelial growth factor (VEGF), fibroblast growth factor (FGF), and platelet-derived growth factor (PDGF), which are essential for cell proliferation, angiogenesis, and ECM remodeling. VEGF stimulates the formation of new blood vessels by binding to VEGFR receptors on endothelial cells, activating pathways like MAPK/ERK and PI3K/Akt that promote endothelial cell proliferation and migration. FGF and PDGF act on fibroblasts and other mesenchymal cells to drive cell division, ECM synthesis, and tissue repair. These growth factors also regulate matrix metalloproteinases (MMPs), enzymes that degrade the ECM, allowing for tissue remodeling and the formation of new, functional tissue. MMP activity is tightly regulated by tissue inhibitors of metalloproteinases (TIMPs), ensuring a balance between ECM degradation and synthesis, which is critical for proper tissue repair.
Inflammation plays a central role in coordinating the vasodilatory response and tissue repair. Pro-inflammatory cytokines such as TNF-α, IL-1, and IL-6 not only promote immune cell recruitment but also upregulate COX-2 expression, enhancing prostaglandin production and vasodilatation. This ensures that sufficient blood flow reaches inflamed or injured tissues, bringing oxygen, nutrients, and immune cells necessary for clearing debris and initiating the repair process. As the inflammation subsides, anti-inflammatory cytokines like IL-10 and TGF-β help resolve the inflammatory response, while promoting tissue repair and angiogenesis. Macrophages, particularly those transitioning from an M1 (pro-inflammatory) to M2 (anti-inflammatory) phenotype, play a pivotal role in orchestrating tissue repair by secreting factors that regulate vasodilation, inflammation, and ECM remodeling.
In conclusion, vasodilatation is a highly regulated molecular process driven by a complex interplay of signaling molecules such as NO, prostaglandins, and growth factors that coordinate smooth muscle relaxation, tissue proliferation, and inflammation. These molecular pathways ensure an appropriate physiological response to injury and inflammation, promoting efficient blood flow, nutrient delivery, and tissue repair. Understanding these processes at the molecular level provides insights into therapeutic interventions that can enhance or modulate vasodilatation to improve outcomes in conditions involving tissue damage, inflammation, or impaired vascular function.
- 5 days after ACL injury.
Type III collagen production is a critical step in the wound healing process, particularly in the early stages of tissue repair, where it serves as a provisional matrix that provides structural support to the injured tissue. Type III collagen forms a soft and flexible framework that facilitates cell migration, proliferation, and angiogenesis, laying the foundation for more permanent tissue repair. The molecular regulation of type III collagen production and its subsequent replacement by type I collagen involves a highly coordinated interplay of growth factors, transcriptional regulators, and signaling pathways.
Immediately following injury, fibroblasts are recruited to the wound site, and growth factors such as transforming growth factor-beta (TGF-β) and platelet-derived growth factor (PDGF) play a central role in activating these cells. TGF-β, a major regulator of ECM synthesis, binds to TGF-β receptors on fibroblasts, triggering the activation of the SMAD pathway. Upon ligand binding, SMAD2/3 proteins are phosphorylated by the receptor and form a complex with SMAD4, which translocates to the nucleus. Once in the nucleus, this complex acts as a transcriptional regulator that binds to the promoters of target genes, including COL3A1, the gene encoding type III collagen. PDGF further amplifies this process by activating PI3K/Akt and MAPK pathways, which promote fibroblast proliferation and survival, ensuring a sustained supply of ECM components. MAPK signaling, in particular, is critical for upregulating transcription factors such as AP-1, which further enhance the transcription of collagen genes.
As fibroblasts synthesize and secrete type III collagen, this collagen forms a provisional ECM, providing a scaffold for cell migration and the formation of new capillaries during the angiogenesis phase of healing. The synthesis of type III collagen involves several post-translational modifications, including hydroxylation of proline and lysine residues by prolyl hydroxylase and lysyl hydroxylase, which require vitamin C as a cofactor. These modifications are essential for stabilizing the collagen triple helix and ensuring proper fibril formation. After secretion into the ECM, procollagen is cleaved by procollagen peptidases, generating mature collagen molecules that spontaneously assemble into fibrils.
During the remodeling phase, the initial type III collagen scaffold is gradually replaced by the stronger and more durable type I collagen, which provides the healed tissue with increased tensile strength and resilience. This transition is orchestrated by matrix metalloproteinases (MMPs), a family of enzymes responsible for degrading ECM components. MMP-1 (collagenase-1) specifically targets type III collagen, breaking down the loose, temporary matrix to allow for the deposition of type I collagen. MMP-2 (gelatinase A) further degrades denatured collagen and gelatin, facilitating ECM remodeling. The activity of MMPs is tightly regulated by tissue inhibitors of metalloproteinases (TIMPs), which prevent excessive collagen degradation and ensure balanced ECM turnover.
The synthesis of type I collagen is a key feature of the tissue maturation phase. Similar to type III collagen production, type I collagen synthesis is upregulated by growth factors like TGF-β and PDGF. The COL1A1 and COL1A2 genes, which encode type I collagen, are regulated by transcription factors activated through the MAPK/ERK and PI3K/Akt pathways. As type I collagen replaces type III collagen, the ECM undergoes significant cross-linking to enhance its mechanical properties. Lysyl oxidase, an enzyme activated during the remodeling phase, catalyzes the formation of covalent cross-links between lysine residues in adjacent collagen molecules, increasing the tensile strength of the collagen fibrils. These cross-links are essential for stabilizing the ECM and providing long-term durability to the repaired tissue.
A key factor in the organization and strength of the newly formed collagen matrix is the response to mechanical stress during the healing process. Fibroblasts and other cells involved in tissue repair are mechanosensitive, meaning they respond to changes in mechanical forces in their environment. These cells detect mechanical stimuli through integrins, which are transmembrane receptors that anchor cells to the ECM. Upon binding to ECM components such as collagen, integrins cluster at focal adhesions, where they activate intracellular signaling molecules, including focal adhesion kinase (FAK) and RhoA/ROCK. FAK signaling transmits mechanical signals from the ECM to the nucleus, where it influences gene expression and cellular behavior, promoting collagen realignment along the lines of mechanical tension.
In addition to integrin signaling, mechanically activated ion channels—such as stretch-activated calcium channels—play a role in the cellular response to mechanical stress. These channels allow calcium ions (Ca²⁺) to enter the cell in response to mechanical deformation, raising intracellular calcium levels. Calcium binds to calmodulin, activating downstream effectors like myosin light-chain kinase (MLCK), which facilitates cytoskeletal rearrangement and enhances the mechanical strength of the tissue. Calcium signaling also influences the MAPK pathway, which is essential for the continued synthesis and remodeling of collagen fibers. AP-1, a transcription factor activated by MAPK signaling, upregulates genes involved in collagen production and matrix remodeling, ensuring that the ECM is properly structured and capable of withstanding mechanical forces.
As the ECM matures, collagen fibers undergo a process of realignment, where fibroblasts reorganize the fibers according to the direction of mechanical stress. This realignment is critical for optimizing the structural integrity of the tissue. The mechanical loading of the tissue during rehabilitation, through exercises like range of motion (ROM) and weight-bearing activities, provides the necessary stimuli for collagen fibers to align correctly. Mechanical loading activates integrin signaling and mechanotransduction pathways that drive the synthesis of type I collagen and improve the overall quality of the ECM.
The role of mechanotransduction in collagen fiber orientation and strength is further supported by the YAP/TAZ pathway, which senses mechanical signals and regulates cellular responses to tissue stiffness and tension. YAP/TAZ transcriptional co-activators are regulated by the Hippo pathway and are translocated to the nucleus in response to mechanical cues, where they promote the expression of genes involved in cell proliferation, ECM remodeling, and tissue regeneration.
In conclusion, the molecular processes underlying type III collagen production and its transition to type I collagen involve intricate signaling pathways, growth factors, and mechanotransduction mechanisms. Understanding these molecular events is essential for developing therapeutic strategies that enhance wound healing and tissue repair. By targeting the TGF-β/SMAD, PI3K/Akt, and MAPK/ERK pathways, as well as optimizing mechanical loading during rehabilitation, clinicians can promote efficient ECM remodeling and restore tissue function.
- ~3 weeks post ACL injury.
The transformation from type III collagen to type I collagen during tissue repair involves a highly orchestrated molecular process, integrating growth factor signaling, enzymatic degradation, mechanical stimuli, and cellular responses to ensure proper tissue restoration. In the early phase of injury, fibroblasts are activated by key growth factors such as TGF-β, PDGF, and FGF. These growth factors bind to their specific receptors on fibroblasts, triggering multiple intracellular signaling cascades, particularly the SMAD, MAPK, and PI3K/Akt pathways. In the TGF-β/SMAD pathway, SMAD2/3 proteins are phosphorylated and form a complex with SMAD4, which then translocates to the nucleus, where it regulates the transcription of collagen genes, including COL3A1 (responsible for type III collagen) and later COL1A1 and COL1A2 (responsible for type I collagen). This early synthesis of type III collagen forms a provisional ECM, providing structural support for cell migration and tissue stabilization.
Matrix metalloproteinases (MMPs), particularly MMP-1 and MMP-2, are critical enzymes that mediate the degradation of type III collagen as the tissue transitions to a more mature ECM composed of type I collagen. These MMPs are tightly regulated by inflammatory cytokines like TNF-α, IL-1β, and IL-6, which are produced by immune cells in response to injury. The NF-κB and AP-1 pathways are activated by these cytokines, upregulating MMP gene expression to ensure efficient degradation of the provisional ECM. MMP activity is tightly controlled by tissue inhibitors of metalloproteinases (TIMPs) to prevent excessive breakdown of the ECM, ensuring a balanced remodeling process. This regulated degradation clears the way for the deposition of the stronger type I collagen, which provides the necessary tensile strength and long-term stability to the tissue.
The synthesis of type I collagen is induced by both biochemical and mechanical signals. The PI3K/Akt pathway, activated by growth factors like TGF-β and PDGF, promotes fibroblast survival, proliferation, and collagen production. The MAPK pathway is also crucial for type I collagen synthesis, as it activates transcription factors such as AP-1, which binds to promoter regions of collagen genes, particularly COL1A1 and COL1A2. These pathways ensure the upregulation of type I collagen production, which is required for the transition from a flexible, early-stage matrix to a more rigid and durable ECM.
Mechanical stimuli play a central role in regulating the production and alignment of type I collagen. Fibroblasts sense mechanical forces through integrins, which act as mechanotransducers that link the ECM to the intracellular cytoskeleton. Integrin activation leads to clustering at focal adhesions, where they activate key signaling pathways, including focal adhesion kinase (FAK) and RhoA/ROCK. These pathways are critical for regulating fibroblast adhesion, migration, and the reorganization of the cytoskeleton. The FAK pathway, in particular, promotes the realignment of collagen fibers in response to mechanical stress, a process crucial for the functional organization of the tissue. Mechanical loading during rehabilitation exercises enhances this mechanotransduction process, ensuring that collagen fibers are properly aligned along lines of tension, thereby strengthening the tissue.
Another key molecular process during the transformation of collagen is collagen cross-linking, which is catalyzed by the enzyme lysyl oxidase. Lysyl oxidase facilitates the formation of covalent cross-links between lysine residues in collagen fibers, stabilizing the ECM and significantly increasing the mechanical strength and stiffness of the tissue. TGF-β regulates the expression and activity of lysyl oxidase, ensuring that cross-linking is tightly coupled with collagen synthesis. Cross-linking is essential for providing the ECM with the structural integrity needed to withstand mechanical stresses over time.
Additionally, calcium signaling plays a significant role in fibroblast contractility and collagen alignment. Mechanically activated stretch-activated calcium channels open in response to mechanical deformation, allowing calcium ions (Ca²⁺) to enter the cell. This increase in intracellular calcium activates calmodulin, which then activates myosin light-chain kinase (MLCK). MLCK phosphorylates myosin light chains, promoting actin-myosin interactions and fibroblast contraction. This contractility is critical for the realignment of collagen fibers, as fibroblasts physically manipulate the ECM during the healing process.
The process of reactive oxygen species (ROS) generation during injury also influences collagen synthesis and remodeling. Low levels of ROS act as signaling molecules that can enhance TGF-β signaling and increase the activity of lysyl oxidase, thereby promoting ECM cross-linking. However, elevated levels of ROS can lead to oxidative damage and interfere with collagen maturation, highlighting the need for controlled ROS regulation during tissue repair.
Throughout this process, the Wnt/β-catenin pathway plays a role in modulating fibroblast differentiation and ECM remodeling. Wnt signaling is activated in response to mechanical cues and biochemical signals during the later stages of healing. β-catenin translocates to the nucleus, where it regulates the expression of genes involved in fibroblast activity and collagen production, further ensuring the proper formation of type I collagen.
In the final phases of tissue remodeling, type I collagen fibers undergo further maturation through continuous mechanical loading and ECM remodeling. The prolonged regulation of MMPs, TIMPs, and cross-linking enzymes ensures that the ECM achieves the proper balance between collagen degradation and synthesis, allowing for a well-organized, mechanically stable matrix. Over time, the alignment and strengthening of type I collagen fibers through repeated mechanical stimuli promote the restoration of tissue strength and functionality, allowing the tissue to handle daily mechanical stresses and resist future injury.
In conclusion, the transformation from type III collagen to type I collagen is driven by an intricate network of growth factor signaling, mechanotransduction, enzymatic degradation, collagen cross-linking, and calcium dynamics. These molecular processes work together to ensure that the ECM is remodeled in a way that restores tissue structure, strength, and function, highlighting key therapeutic targets for improving wound healing and tissue repair outcomes.
Meniscal Injuries
The menisci are two crescent-shaped pieces of fibrocartilage located between the femoral condyles and the tibial plateau in the knee joint. They play crucial roles in shock absorption, load distribution, joint stability, and lubrication, reducing the stress on the articular cartilage by spreading compressive forces across a larger area. Structurally, the menisci are composed of a dense network of collagen fibers, primarily type I collagen, which provides tensile strength, along with proteoglycans, such as aggrecan, that contribute to the tissue’s compressive properties. The menisci also contain elastin fibers and a small population of specialized cells, primarily fibrochondrocytes, which maintain the extracellular matrix (ECM) through the synthesis of collagen, proteoglycans, and other ECM components.
Meniscal injuries can result from either acute trauma, such as a sudden twisting motion of the knee, or degenerative changes that weaken the meniscal tissue over time. Acute injuries, commonly seen in athletes, often involve tears in the meniscus, while degenerative injuries occur gradually due to wear and tear, leading to fragmentation or fraying of the fibrocartilage. Meniscal injuries are frequently associated with pain, swelling, and mechanical symptoms such as locking, catching, or instability of the knee joint, which can significantly impair mobility.
At the cellular level, meniscal injury induces a series of responses aimed at repairing the damaged tissue, but these responses are often insufficient due to the limited vascular supply in the meniscus, particularly in the inner, avascular zone. In the vascularized outer region of the meniscus, which is referred to as the red-red zone, there is a greater capacity for healing due to the presence of blood vessels, allowing for the recruitment of immune cells and growth factors that can initiate tissue repair. However, the inner white-white zone is largely avascular, leading to poor healing potential in this region.
Following meniscal injury, the activity of fibrochondrocytes, the primary cells in the meniscus, changes significantly. These cells respond to injury by increasing the synthesis of proteolytic enzymes such as matrix metalloproteinases (MMPs) and aggrecanases, which degrade the ECM, particularly collagen and aggrecan. This degradation is part of the tissue’s initial attempt to remodel the damaged ECM, but if unchecked, excessive MMP activity can lead to further tissue breakdown and degeneration. Inflammatory cytokines such as interleukin-1 beta (IL-1β), tumor necrosis factor-alpha (TNF-α), and interleukin-6 (IL-6) are upregulated in response to injury, driving catabolic processes that accelerate ECM degradation. IL-1β and TNF-α are particularly potent in promoting the expression of MMPs and inhibiting the synthesis of type I collagen and proteoglycans, impairing the meniscus’s ability to regenerate and maintain its structural integrity.
The extracellular matrix (ECM) composition of the meniscus is also altered following injury. In healthy meniscal tissue, the ECM is composed of a balanced network of collagens (primarily type I and type II), proteoglycans, glycosaminoglycans (GAGs), and elastin. Following injury, the ratio of collagen to proteoglycans shifts, with a decrease in the synthesis of structural proteins and an increase in the production of fibrotic tissue. This change in ECM composition reduces the biomechanical properties of the meniscus, leading to a loss of elasticity and shock-absorbing capacity. Additionally, proteoglycan loss reduces the ability of the tissue to retain water, further compromising the meniscus’s ability to handle compressive forces, making it more susceptible to further damage.
The inflammatory response in meniscal injuries is also a key factor in the progression of tissue damage. Injury to the meniscus triggers the release of damage-associated molecular patterns (DAMPs) from necrotic cells and ECM fragments, which activate the innate immune system. Macrophages, neutrophils, and other immune cells infiltrate the injured meniscus, releasing pro-inflammatory mediators that exacerbate the breakdown of the ECM. These inflammatory mediators activate signaling pathways such as NF-κB, which increases the transcription of pro-inflammatory cytokines, further amplifying the inflammatory response. Over time, chronic inflammation can lead to fibrosis, the formation of scar tissue, and an overall reduction in the biomechanical properties of the meniscus.
Chondrocytes, the cells responsible for maintaining cartilage homeostasis, also play a role in meniscal repair. In response to injury, chondrocytes in the adjacent articular cartilage can become hypertrophic and increase their production of collagen type X, MMPs, and vascular endothelial growth factor (VEGF). While these changes are aimed at remodeling the ECM, they can contribute to cartilage degeneration over time, particularly in the context of osteoarthritis (OA), a common long-term consequence of meniscal injury. The interplay between meniscal injury and OA is significant, as meniscal damage can disrupt normal joint mechanics, increasing the load on the articular cartilage and accelerating the degenerative processes associated with OA.
At the molecular level, the TGF-β signaling pathway plays a dual role in meniscal injury and repair. While TGF-β promotes ECM synthesis and fibroblast proliferation, contributing to tissue repair, it can also drive fibrosis if overactivated, leading to the excessive deposition of type I collagen and scar tissue formation. Additionally, hypoxia-inducible factors (HIFs) are activated in response to the avascular nature of the inner meniscus, leading to the upregulation of angiogenic factors such as VEGF, which attempts to promote vascularization, although the success of neovascularization is limited in the white-white zone.
Efforts to enhance meniscal healing focus on biological therapies, such as the use of growth factors, scaffolds, and stem cells to improve tissue regeneration. Platelet-rich plasma (PRP) and mesenchymal stem cells (MSCs) have been explored as potential treatments to stimulate meniscal healing by promoting chondrocyte activity, enhancing ECM synthesis, and reducing inflammation. These therapies aim to overcome the meniscus’s inherent limitations in healing, especially in the avascular zones, by providing the necessary biological cues to support tissue repair and regeneration.
In conclusion, the cellular and molecular responses to meniscal injury involve complex interactions between chondrocytes, fibrochondrocytes, and the ECM, alongside inflammatory processes that drive tissue degradation and repair. Understanding these processes at a molecular level provides insights into potential therapeutic approaches that could improve meniscal healing and prevent long-term joint damage, such as osteoarthritis.
Mechanisms of Injury
Meniscal injuries are typically caused by rotational forces or direct impact on the knee, which place excessive stress on the menisci, leading to tears or other forms of damage. Acute meniscal injuries are particularly common in high-impact sports such as football, soccer, basketball, and skiing, where sudden changes in direction, twisting motions, and rapid deceleration place significant strain on the meniscus. During these activities, the knee is often subjected to high torsional forces, especially when the foot is planted, and the body pivots, causing the meniscus to be pinched between the femur and tibia. This can lead to various types of tears, such as bucket-handle, radial, or horizontal tears, which vary in severity and location depending on the force and mechanics involved.
In contrast, degenerative meniscal tears are more prevalent in older adults, typically due to the cumulative effects of aging and the gradual wear and tear of the meniscus over time. As people age, the meniscus becomes more prone to microtrauma, which leads to structural weakening. The collagen network and proteoglycan content within the meniscus decrease, reducing the tissue’s ability to withstand mechanical stress. This degradation is often exacerbated by osteoarthritic changes in the knee joint, where repetitive loading and altered joint mechanics contribute to the breakdown of the meniscal ECM. In these cases, even minor stresses such as squatting, rising from a seated position, or stepping awkwardly can result in a tear. Degenerative meniscal tears are commonly associated with horizontal cleavage, leading to a frayed or fragmented meniscus, which can cause pain, swelling, and joint stiffness.
The limited blood supply of the meniscus is a critical factor that impairs its ability to heal following an injury. The meniscus is divided into three zones based on vascularity: the outer red-red zone, the middle red-white zone, and the inner white-white zone. The red-red zone, located in the peripheral third of the meniscus, has a relatively rich blood supply derived from the genicular arteries, which penetrate the outer edges of the meniscus. This vascularization provides essential oxygen, nutrients, and immune cells to the damaged tissue, allowing for a more robust healing response. As a result, tears that occur in the red-red zone have a higher likelihood of healing, particularly when treated with conservative approaches like physical therapy or surgical repair techniques such as meniscal suturing.
In contrast, the red-white zone has a mixed blood supply, with partial vascularization extending from the periphery but diminishing as it moves toward the central portion of the meniscus. Tears in this zone may have a moderate healing potential, depending on the size and location of the injury, as well as the treatment strategy employed. Surgical techniques, such as meniscal repair or biological augmentation, which aims to stimulate vascularization, are often necessary to promote healing in this area.
The white-white zone, located in the inner two-thirds of the meniscus, is avascular and relies entirely on diffusion from synovial fluid for nutrient supply. This lack of a blood supply severely limits the intrinsic healing capacity of the meniscus in this zone. Tears in the white-white zone are less likely to heal on their own and are more commonly associated with meniscectomy, a surgical procedure in which the damaged portion of the meniscus is removed. However, meniscectomy can lead to long-term complications, such as increased stress on the articular cartilage, which raises the risk of osteoarthritis due to the loss of the meniscus’s shock-absorbing and load-distributing functions.
At the cellular level, the response to meniscal injury in the different zones varies depending on the availability of vascular supply. In the red-red zone, the presence of blood vessels allows for the recruitment of immune cells, including macrophages and neutrophils, which are critical for clearing debris and initiating tissue repair. These immune cells release cytokines and growth factors, such as platelet-derived growth factor (PDGF) and vascular endothelial growth factor (VEGF), which stimulate fibrochondrocyte proliferation and ECM remodeling. Fibrochondrocytes in this zone produce type I collagen, which is essential for rebuilding the fibrous structure of the meniscus.
In the white-white zone, the lack of vascularization and limited access to immune cells hinder the repair process. Fibrochondrocytes in this region rely on diffusion of nutrients from the synovial fluid, which is a less efficient mechanism for tissue regeneration. Consequently, the synthesis of type I and type II collagen, as well as proteoglycans like aggrecan, is reduced, and the overall ability of the meniscus to regenerate is diminished. Without adequate repair, the injured meniscal tissue in the white-white zone becomes progressively weaker, further compromising the structural integrity of the knee joint.
The molecular response to meniscal injury also involves the activation of matrix metalloproteinases (MMPs), which degrade the ECM as part of the tissue remodeling process. MMP-1, MMP-3, and MMP-13 are upregulated in response to injury, particularly in the avascular regions, contributing to collagen breakdown. While MMP activity is essential for clearing damaged tissue, excessive activity can lead to further degradation and impede healing. Tissue inhibitors of metalloproteinases (TIMPs) play a crucial role in regulating MMP activity and ensuring that ECM remodeling occurs in a controlled manner. In the absence of sufficient TIMP regulation, ECM degradation can outpace synthesis, exacerbating tissue loss and leading to degeneration.
Recent advances in biological therapies aim to address the meniscus’s limited healing capacity, particularly in the avascular regions. Platelet-rich plasma (PRP) injections, for example, have been explored as a treatment option to enhance meniscal repair by delivering growth factors directly to the injured site, promoting cell proliferation, angiogenesis, and ECM synthesis. Similarly, mesenchymal stem cells (MSCs) are being investigated for their potential to differentiate into fibrochondrocytes and facilitate meniscal regeneration. These therapies seek to improve the healing environment within the meniscus, particularly in the white-white zone, by stimulating cellular activity and enhancing the reparative processes.
In summary, meniscal injuries are influenced by the location of the tear within the meniscus, with vascularized zones demonstrating a higher capacity for healing than avascular zones. The limited blood supply in the inner meniscus significantly impairs its ability to heal, necessitating surgical intervention or biological augmentation to restore function. Understanding the molecular and cellular responses to meniscal injury provides valuable insights into potential therapeutic strategies aimed at improving healing outcomes and preventing long-term complications such as osteoarthritis.
Cellular Responses to Meniscal Injury
Meniscal injuries are not only among the most common knee injuries but also pose significant challenges for maintaining joint function and ensuring long-term joint health. The meniscus, a crescent-shaped fibrocartilaginous structure located between the femoral condyles and the tibial plateau, serves as a key component in load distribution, shock absorption, and joint stabilization. By dispersing compressive forces across the knee joint, the meniscus helps prevent excessive stress on the underlying articular cartilage, thus preserving the integrity of the knee joint during weight-bearing and dynamic activities. However, when the meniscus is injured, this delicate balance is disrupted, and the joint becomes vulnerable to further damage, potentially leading to joint instability, pain, and degenerative changes such as osteoarthritis (OA).
The repair capacity of the meniscus is inherently limited by its complex structure and regional variations in vascularity. The meniscus is divided into three zones based on its blood supply: the outer red-red zone, which is well vascularized, the middle red-white zone, with a more limited blood supply, and the inner white-white zone, which is avascular. The red-red zone has a greater potential for healing due to its access to a blood supply that facilitates the delivery of oxygen, nutrients, and immune cells. This vascularity allows for a more efficient inflammatory response and subsequent tissue repair. In contrast, the white-white zone, located in the inner two-thirds of the meniscus, relies on diffusion from synovial fluid for nutrient supply, significantly reducing its healing potential. As a result, tears or injuries in this avascular region often do not heal spontaneously and may require surgical intervention, such as meniscectomy or meniscal transplantation, to restore function.
Following a meniscal injury, a cascade of cellular and molecular responses is initiated to attempt tissue repair. The initial phase of this response is characterized by an acute inflammatory reaction, primarily in the more vascularized zones. Vascular disruption in the red-red zone leads to the formation of a hematoma and increases vascular permeability, allowing immune cells and plasma proteins to infiltrate the injury site. Neutrophils are the first immune cells to arrive, releasing reactive oxygen species (ROS) and proteolytic enzymes to clear damaged tissue and debris. These neutrophils set the stage for the recruitment of macrophages, which are critical for coordinating the later stages of repair. Macrophages initially adopt a pro-inflammatory M1 phenotype, secreting cytokines such as tumor necrosis factor-alpha (TNF-α), interleukin-1 beta (IL-1β), and interleukin-6 (IL-6), which amplify the inflammatory response and promote the recruitment of additional immune cells to the site of injury.
As the inflammatory phase progresses, M1 macrophages transition to the reparative M2 phenotype, which is characterized by the secretion of anti-inflammatory cytokines such as interleukin-10 (IL-10) and transforming growth factor-beta (TGF-β). These cytokines help resolve the inflammation and promote fibrochondrocyte activation. Fibrochondrocytes, the primary cells responsible for maintaining the meniscal extracellular matrix (ECM), respond to the signals from M2 macrophages by increasing the synthesis of type I collagen and proteoglycans, key components of the meniscal ECM that are essential for tissue repair and regeneration. TGF-β, in particular, plays a crucial role in driving the SMAD signaling pathway, which upregulates collagen synthesis and supports the rebuilding of the damaged meniscal tissue.
However, despite these repair mechanisms, the healing process is often compromised in the avascular white-white zone, where the absence of a direct blood supply severely limits the recruitment of immune cells and reparative factors. In this region, fibrochondrocyte activity is reduced, and the production of critical ECM components is diminished. Moreover, the lack of sufficient vascular endothelial growth factor (VEGF) signaling in this avascular zone limits the potential for neovascularization, further hindering the healing process.
In addition to the limitations imposed by vascularity, the repair process is influenced by the activity of matrix metalloproteinases (MMPs), which are enzymes responsible for degrading ECM components such as collagen and aggrecan. MMP-1, MMP-3, and MMP-13 are particularly important in the context of meniscal injuries, as they mediate the breakdown of damaged collagen fibers, clearing the way for new ECM deposition. While MMP activity is essential for tissue remodeling, it must be tightly regulated by tissue inhibitors of metalloproteinases (TIMPs) to prevent excessive ECM degradation. Uncontrolled MMP activity can lead to further breakdown of the meniscal tissue, exacerbating the injury and impairing the repair process.
As the inflammatory response subsides and tissue remodeling begins, the meniscal ECM undergoes a complex process of collagen turnover and reorganization. Type III collagen, which is initially deposited during the early stages of repair, is gradually replaced by the stronger and more organized type I collagen, providing the meniscus with the tensile strength needed to withstand mechanical loads. However, in many cases, the newly formed ECM is not as structurally robust as the original tissue, leading to long-term changes in the mechanical properties of the meniscus. This can result in joint instability, increased stress on the surrounding articular cartilage, and an elevated risk of developing osteoarthritis.
In cases where natural healing is insufficient, surgical repair techniques or biological therapies may be employed to enhance meniscal regeneration. Meniscal suturing and partial meniscectomy are common surgical options, depending on the location and severity of the tear. Additionally, biological augmentations such as platelet-rich plasma (PRP) and mesenchymal stem cells (MSCs) are being investigated for their potential to stimulate meniscal healing by promoting fibrochondrocyte proliferation, enhancing ECM synthesis, and modulating the inflammatory response. These therapies aim to overcome the intrinsic limitations of meniscal healing by delivering growth factors and reparative cells directly to the site of injury, particularly in the avascular zones where natural repair processes are limited.
In conclusion, meniscal injuries initiate a complex series of cellular, molecular, and mechanical responses aimed at repairing the damaged tissue. However, the success of this repair is highly dependent on the location of the injury within the meniscus and the availability of a vascular supply. While the red-red zone has a relatively high capacity for healing due to its vascularity, the white-white zone remains a significant challenge for repair due to its avascular nature. Understanding the molecular pathways involved in meniscal injury and repair provides crucial insights into potential therapeutic strategies that could enhance healing outcomes and preserve long-term joint health, especially in the context of preventing degenerative changes such as osteoarthritis.
Initial Inflammatory Response
The immediate molecular response following a meniscal injury begins with vascular changes in the more vascularized regions. Damage to blood vessels results in hematoma formation and increased vascular permeability, allowing immune cells and plasma proteins to enter the injury site. This increase in permeability facilitates the movement of signaling molecules such as cytokines and growth factors that are critical for initiating the inflammatory and repair processes. The entry of plasma proteins also contributes to the formation of a provisional matrix, which acts as a scaffold for the migration of immune and reparative cells.
Neutrophils are the first immune cells recruited to the site of injury. These cells are activated by signals such as damage-associated molecular patterns (DAMPs) released from injured meniscal cells and extracellular matrix components. Neutrophils release reactive oxygen species (ROS) and proteolytic enzymes, including neutrophil elastase and cathepsins, which break down damaged tissues and clear cellular debris. These early catabolic activities are crucial for preparing the tissue for subsequent repair. Neutrophils also secrete cytokines such as IL-1β and TNF-α, which amplify the inflammatory response and signal for further immune cell recruitment.
Following the neutrophil response, macrophages are recruited to the injury site. These cells play a dual role in inflammation and tissue repair. In the early phase, M1 macrophages dominate and release pro-inflammatory cytokines such as TNF-α, IL-1β, and IL-6, which further enhance the inflammatory response. These cytokines activate intracellular signaling pathways such as NF-κB and JAK-STAT, leading to the upregulation of genes involved in inflammation and tissue remodeling. Macrophages also secrete matrix metalloproteinases (MMPs), such as MMP-1, MMP-3, and MMP-13, which degrade the extracellular matrix, including collagen and proteoglycans, facilitating the breakdown of damaged tissue. This degradation process clears space for new tissue formation and ECM reconstruction.
As the inflammatory response progresses, macrophages undergo a phenotypic shift from the M1 (pro-inflammatory) state to the M2 (reparative) phenotype. M2 macrophages are essential for the resolution of inflammation and the promotion of tissue repair. These cells secrete anti-inflammatory cytokines such as IL-10 and TGF-β, which inhibit further inflammation and stimulate fibrochondrocyte activity. TGF-β, in particular, is critical for promoting the synthesis of type I collagen and proteoglycans by fibrochondrocytes, aiding in the rebuilding of the meniscal extracellular matrix. The activation of TGF-β receptors on fibrochondrocytes leads to the activation of the SMAD signaling pathway, which regulates the transcription of ECM-related genes.
Cytokine release following a meniscal injury is a key driver of both inflammation and repair. IL-1, IL-6, and TNF-α are the primary pro-inflammatory cytokines involved in meniscal injuries. These cytokines not only recruit additional immune cells but also stimulate the production of MMPs and other catabolic enzymes that degrade the ECM. TNF-α activates NF-κB, a transcription factor that upregulates the expression of genes involved in inflammation and ECM breakdown. IL-1β and TNF-α also inhibit the synthesis of collagen and proteoglycans by fibrochondrocytes, leading to a further imbalance in the ECM composition. Additionally, IL-6 promotes the activation of JAK-STAT signaling, which influences cell survival, proliferation, and inflammatory responses, further amplifying the inflammatory cascade.
The matrix metalloproteinases produced during this phase are key regulators of tissue remodeling. MMPs degrade various ECM components, including collagen type I, type II, and aggrecan, to remove damaged tissue and facilitate repair. MMP-13, a collagenase, is particularly effective at degrading type II collagen, which is a major component of the meniscal ECM. While the controlled activity of MMPs is necessary for tissue remodeling, excessive MMP activity can lead to further degradation of the meniscus, resulting in tissue weakness and a higher risk of degenerative changes such as osteoarthritis. To counterbalance MMP activity, tissue inhibitors of metalloproteinases (TIMPs) are secreted to regulate ECM degradation and promote tissue stability.
The extracellular matrix (ECM) itself undergoes significant changes during the repair process. In healthy meniscal tissue, the ECM is rich in collagens (type I and II), proteoglycans, and glycosaminoglycans. Following injury, there is an initial loss of these structural components due to the activity of MMPs, which degrades the collagen network and reduces proteoglycan content. As the repair process progresses, the synthesis of new collagen, particularly type I collagen, increases under the influence of growth factors like TGF-β. However, the newly formed ECM is often less organized and mechanically weaker than the original tissue, which may lead to long-term functional deficits.
Overall, the molecular response to meniscal injury is characterized by a tightly regulated interplay of inflammatory signals, cellular responses, and ECM remodeling. While this process aims to restore meniscal structure and function, the limited vascularity of the meniscus, particularly in the inner zones, often impairs the effectiveness of these repair mechanisms, leading to incomplete healing and a higher risk of joint degeneration over time. Understanding these molecular pathways is critical for developing targeted therapies that can enhance meniscal repair, reduce inflammation, and prevent the progression of degenerative changes in the knee joint.
Extracellular Matrix Degradation and Remodeling
The extracellular matrix (ECM) of the meniscus is a dynamic and highly regulated structure, composed primarily of collagens, proteoglycans, and glycoproteins, which provide both mechanical strength and elasticity to the tissue. The ECM not only supports the structural integrity of the meniscus but also plays a crucial role in regulating cellular behavior through mechanotransduction and signaling pathways. Following injury, the meniscal ECM undergoes substantial molecular remodeling, which is necessary for tissue repair but can lead to tissue degeneration if these processes are dysregulated.
Matrix metalloproteinases (MMPs) are a family of zinc-dependent proteolytic enzymes that are central to ECM remodeling. MMPs, such as MMP-1 (collagenase-1), MMP-3 (stromelysin-1), and MMP-13 (collagenase-3), are upregulated in response to injury, particularly through inflammatory signals like interleukin-1 beta (IL-1β) and tumor necrosis factor-alpha (TNF-α). These cytokines activate the NF-κB and MAPK pathways, which drive the transcription of MMP genes, leading to increased degradation of ECM components, particularly type I and type II collagens and aggrecan. The cleavage of collagen by MMP-1 and MMP-13 is a key step in ECM turnover, as these enzymes break down the fibrillar collagen network, facilitating the removal of damaged tissue and the subsequent formation of a new matrix. MMP-3 not only degrades proteoglycans like aggrecan but also activates other MMPs, amplifying the overall matrix degradation process.
Although MMP-mediated degradation is essential for clearing damaged ECM and allowing for tissue repair, unregulated MMP activity can result in the excessive breakdown of healthy matrix components, leading to further weakening of the meniscus and impaired function. This overactivity can compromise the mechanical properties of the meniscus, such as its ability to absorb shock and distribute loads, making it more susceptible to degeneration. Excessive ECM degradation also disrupts the cell-ECM interactions that are crucial for fibrochondrocyte function, further hindering tissue repair. Integrin-mediated signaling, which is sensitive to the structural integrity of the ECM, is disrupted by excessive matrix breakdown, leading to impaired cellular adhesion, proliferation, and differentiation—all of which are necessary for proper meniscal healing.
To counterbalance MMP activity and prevent excessive matrix degradation, tissue inhibitors of metalloproteinases (TIMPs) play a critical regulatory role. TIMPs bind to active MMPs, inhibiting their proteolytic activity and ensuring that ECM breakdown is controlled. The TIMP-MMP ratio is a key determinant of ECM homeostasis. In the context of meniscal injury, a disruption in the TIMP/MMP balance can lead to pathological tissue remodeling. When TIMP levels are insufficient, unchecked MMP activity can lead to the over-degradation of collagen and other matrix components, resulting in tissue instability and impaired healing. Conversely, an excessive increase in TIMP levels could inhibit necessary ECM turnover, potentially leading to the formation of fibrotic tissue rather than proper tissue regeneration. The regulation of MMP-TIMP interactions is thus essential for maintaining a balance between matrix degradation and synthesis, allowing for effective tissue repair without excessive breakdown or fibrosis.
Growth factors such as transforming growth factor-beta (TGF-β), fibroblast growth factor (FGF), and insulin-like growth factor 1 (IGF-1) play pivotal roles in promoting ECM synthesis and regulating MMP and TIMP activity. TGF-β, in particular, is a potent inducer of collagen synthesis and ECM remodeling. It signals through the TGF-β/SMAD pathway, where activated SMAD2/3 proteins translocate to the nucleus and promote the transcription of COL1A1 and COL1A2, the genes encoding type I collagen. This process ensures the production of new collagen fibers, which are critical for restoring the structural integrity of the meniscus after injury. TGF-β also plays a role in modulating fibrochondrocyte differentiation, driving these cells to adopt a matrix-producing phenotype that enhances ECM regeneration.
FGF and IGF-1 further support the repair process by stimulating cell proliferation and ECM component synthesis. FGF activates the MAPK pathway, promoting cell division and the upregulation of genes involved in ECM production. Meanwhile, IGF-1 enhances cell survival and boosts the production of proteoglycans, essential for maintaining the compressive strength of the meniscus. Both growth factors interact with MMPs and TIMPs, fine-tuning the balance between matrix degradation and synthesis to promote tissue repair while preventing excessive breakdown.
In addition to growth factors, mechanical forces play a significant role in modulating ECM remodeling in the meniscus. Mechanical loading, such as that experienced during rehabilitation exercises, activates mechanotransduction pathways in fibrochondrocytes through integrins and other mechanosensitive proteins. These pathways, including focal adhesion kinase (FAK) and RhoA/ROCK signaling, regulate cytoskeletal dynamics and influence ECM synthesis. Mechanical forces also stimulate the production of collagen cross-links via the enzyme lysyl oxidase, which strengthens collagen fibers and enhances the mechanical stability of the repaired meniscal tissue. Lysyl oxidase-mediated cross-linking ensures that newly synthesized collagen fibers are properly stabilized, providing the tissue with the necessary tensile strength to withstand joint loading.
The extracellular matrix (ECM) remodeling process is further influenced by hypoxia in the avascular regions of the meniscus. Hypoxic conditions activate hypoxia-inducible factor 1-alpha (HIF-1α), which induces the expression of genes involved in angiogenesis and ECM synthesis. HIF-1α upregulates vascular endothelial growth factor (VEGF), which promotes neovascularization in the peripheral regions of the meniscus. However, in the avascular inner regions, the lack of oxygen and nutrient supply remains a significant challenge for tissue repair. Hypoxia can also induce ROS production, which, while necessary for certain signaling processes, can lead to oxidative damage if not properly regulated, further complicating ECM remodeling.
In conclusion, the molecular processes governing ECM remodeling in the meniscus involve a complex interplay between MMPs, TIMPs, growth factors, and mechanical signals. MMP activity is necessary for clearing damaged ECM components, but its activity must be tightly regulated by TIMPs to prevent excessive tissue breakdown. Growth factors like TGF-β, FGF, and IGF-1 play essential roles in promoting ECM synthesis and modulating the balance between matrix degradation and repair. Proper coordination of these molecular pathways is crucial for ensuring effective tissue regeneration and maintaining the structural and functional integrity of the meniscus. Disruptions in these processes, whether due to excessive MMP activity, insufficient TIMP regulation, or imbalances in growth factor signaling, can lead to tissue degeneration, impaired healing, and long-term joint dysfunction. Understanding these molecular mechanisms is key to developing targeted therapies aimed at enhancing meniscal repair and preventing degenerative joint diseases such as osteoarthritis.
Chondrocyte and Fibrochondrocyte Activation
The meniscus is populated by two key cell types: chondrocytes and fibrochondrocytes, which are crucial for maintaining the extracellular matrix (ECM) and orchestrating the repair process following injury. Both cell types engage in intricate molecular processes that involve responding to injury through shifts in their metabolic and biosynthetic activities. Understanding these molecular responses provides insights into how meniscal healing occurs and the challenges posed by the limited vascularity of the meniscus.
Chondrocyte Activation: Chondrocytes, particularly in the inner avascular zone of the meniscus (white-white zone), become highly active following injury due to the release of pro-inflammatory cytokines such as interleukin-1 beta (IL-1β), tumor necrosis factor-alpha (TNF-α), and interleukin-6 (IL-6). These cytokines trigger intracellular signaling pathways like NF-κB, MAPK, and JAK-STAT, leading to the upregulation of catabolic enzymes such as matrix metalloproteinases (MMPs) and aggrecanases (like ADAMTS-4 and ADAMTS-5). MMP-1, MMP-3, and MMP-13 degrade type I and type II collagen, while aggrecanases cleave the proteoglycan aggrecan, which is essential for maintaining the compressive strength of the meniscus. This breakdown of ECM components helps remove damaged tissue, clearing space for new matrix synthesis.
However, the persistent activation of chondrocytes and the prolonged release of MMPs and aggrecanases can result in excessive ECM degradation. If this degradation is not properly regulated, it can lead to further tissue breakdown, reducing the mechanical properties of the meniscus and exacerbating degenerative changes. Chondrocytes also produce reactive oxygen species (ROS) in response to injury, which, while playing a role in signaling, can lead to oxidative damage if not controlled, further contributing to tissue degradation.
In addition to their catabolic role, chondrocytes can switch to an anabolic mode in response to factors such as insulin-like growth factor 1 (IGF-1), fibroblast growth factor (FGF), and bone morphogenetic proteins (BMPs). These growth factors activate pathways such as PI3K/Akt and TGF-β/SMAD, promoting collagen synthesis and ECM repair. IGF-1 enhances chondrocyte survival and proliferation, while TGF-β regulates the production of type II collagen and proteoglycans, helping balance ECM degradation with repair. This switch between catabolic and anabolic activity is crucial for effective tissue regeneration and is tightly regulated by signaling pathways that integrate inflammatory and reparative cues.
Fibrochondrocyte Activation: Fibrochondrocytes, which are more prevalent in the vascularized outer zone (red-red zone) of the meniscus, play a central role in ECM repair following injury. These cells are highly responsive to growth factors such as TGF-β, platelet-derived growth factor (PDGF), and fibroblast growth factor (FGF), which stimulate the production of type I and type II collagen. TGF-β, in particular, activates the SMAD2/3 signaling pathway, which upregulates genes involved in collagen synthesis, including COL1A1 and COL2A1, essential for restoring the meniscal ECM. PDGF promotes fibrochondrocyte proliferation, while FGF enhances their ability to synthesize ECM proteins.
Fibrochondrocytes are also sensitive to mechanical stimuli, such as those encountered during normal joint movement or rehabilitation exercises. Mechanical forces activate mechanotransduction pathways through integrins, which link the ECM to the cell cytoskeleton. The activation of focal adhesion kinase (FAK), RhoA/ROCK, and the MAPK/ERK pathway regulates fibrochondrocyte proliferation and ECM remodeling. Mechanical loading also stimulates the production of lysyl oxidase, an enzyme that promotes the cross-linking of collagen fibers, enhancing the tensile strength and stability of the repaired ECM. This mechanical stimulation is critical for proper collagen alignment and ensuring that the newly synthesized ECM can withstand the mechanical stresses of joint function.
Cellular Hypertrophy: Both chondrocytes and fibrochondrocytes can undergo cellular hypertrophy in response to injury, particularly under the influence of factors such as hypoxia, TGF-β, and mechanical stress. Hypoxic conditions, common in the avascular regions of the meniscus, activate hypoxia-inducible factor-1 alpha (HIF-1α), which drives the expression of genes involved in cellular hypertrophy and ECM production. Hypertrophic chondrocytes increase their production of type X collagen and alkaline phosphatase, both of which are associated with the endochondral ossification process. This hypertrophic response leads to an increase in ECM production, aiding in tissue repair, but can also result in calcification and altered mechanical properties if it persists unchecked.
Hypertrophy is often accompanied by increased metabolic activity and enhanced production of MMPs, further accelerating ECM degradation if not properly regulated by tissue inhibitors of metalloproteinases (TIMPs). TIMP-1 and TIMP-2 are critical for controlling MMP activity and ensuring that ECM degradation does not outpace ECM synthesis. An imbalance between MMPs and TIMPs can lead to tissue stiffening, disrupting the mechanical properties of the meniscus and reducing its ability to distribute loads and absorb shocks.
In hypertrophic cells, mechanosensitive ion channels such as TRPV4 respond to mechanical stress by regulating calcium influx, which in turn modulates calmodulin and myosin light-chain kinase (MLCK), influencing cytoskeletal dynamics and cell contractility. These processes are essential for maintaining the structural integrity of the meniscus, but excessive hypertrophy can compromise tissue flexibility and lead to dysfunctional repair outcomes.
In summary, the molecular response of chondrocytes and fibrochondrocytes to meniscal injury involves a complex interplay between catabolic and anabolic pathways, growth factor signaling, and mechanical stimuli. Chondrocytes in the avascular zone play a dual role in ECM degradation and repair, while fibrochondrocytes in the vascularized outer zone are key to ECM synthesis and remodeling. Cellular hypertrophy represents a critical aspect of the repair process, but if not properly regulated, it can lead to altered tissue mechanics and contribute to long-term dysfunction. Understanding these molecular mechanisms is essential for developing targeted therapies that can enhance meniscal repair, promote balanced ECM remodeling, and prevent degenerative changes.
Mesenchymal Stem Cell (MSC) Recruitment and Differentiation
Mesenchymal stem cells (MSCs) play a pivotal role in meniscal repair due to their ability to differentiate into various cell types, modulate the inflammatory response, and promote tissue regeneration through molecular signaling pathways. Following a meniscal injury, the recruitment, differentiation, and paracrine actions of MSCs are orchestrated by a complex network of growth factors, cytokines, and mechanical signals, all of which contribute to the molecular dynamics of tissue repair.
MSC Recruitment: The migration of MSCs to the site of injury is primarily driven by chemotactic gradients established by signaling molecules such as stromal cell-derived factor 1 (SDF-1) and vascular endothelial growth factor (VEGF). SDF-1 binds to the CXCR4 receptor on MSCs, activating the G-protein-coupled receptor (GPCR) signaling cascade that promotes cell migration. This pathway involves the activation of downstream effectors such as PI3K/Akt and Rho GTPases, which regulate the cytoskeletal reorganization necessary for MSC movement through the extracellular matrix (ECM). VEGF, through its interaction with VEGFR2, not only recruits MSCs but also promotes angiogenesis at the injury site, improving blood supply to enhance nutrient and oxygen delivery. The hypoxic environment often found in meniscal injuries further stimulates VEGF production via hypoxia-inducible factor-1 alpha (HIF-1α), which amplifies MSC recruitment by enhancing the chemotactic gradient. The convergence of these molecular signals enables MSCs to migrate effectively toward the damaged tissue, particularly in avascular zones, where natural healing is limited.
MSC Differentiation: Once MSCs reach the injury site, they are exposed to local biochemical signals that drive their differentiation into chondrocytes and fibrochondrocytes. Transforming growth factor-beta (TGF-β) is a major regulator of MSC differentiation, activating the TGF-β/SMAD signaling pathway. Binding of TGF-β to its receptor activates SMAD2/3, which translocates to the nucleus and upregulates genes essential for chondrogenesis, such as SOX9, which is critical for the synthesis of type II collagen and aggrecan. Additionally, bone morphogenetic proteins (BMPs), especially BMP-2 and BMP-7, promote MSC differentiation through the activation of SMAD-independent pathways, such as MAPK, ERK, and p38, which enhance the production of ECM components necessary for meniscal repair.
Insulin-like growth factor 1 (IGF-1) plays a supportive role by promoting cell survival, proliferation, and anabolic activity in MSCs. Through the PI3K/Akt signaling pathway, IGF-1 enhances the expression of genes involved in ECM production and inhibits apoptosis, ensuring that MSCs contribute to tissue regeneration rather than undergoing programmed cell death. Furthermore, mechanical signals generated by joint movement and loading activate integrins on MSCs, which link the ECM to the cytoskeleton. This mechanotransduction activates pathways such as focal adhesion kinase (FAK), RhoA/ROCK, and YAP/TAZ, which further enhance MSC differentiation into fibrochondrocytes. These cells synthesize type I and type II collagen, proteoglycans, and other ECM components necessary for restoring the structural and functional properties of the meniscus.
Paracrine Effects of MSCs: Beyond their ability to differentiate, MSCs exert significant influence on the healing environment through paracrine signaling. MSCs secrete a wide array of cytokines, growth factors, and extracellular vesicles (including exosomes) that modulate the local microenvironment. These secreted factors include interleukin-10 (IL-10), TGF-β, and prostaglandin E2 (PGE2), which have potent anti-inflammatory effects. IL-10 and TGF-β suppress the activity of M1 macrophages, shifting them toward an M2 reparative phenotype, which promotes tissue repair rather than further inflammation. This transition is crucial for resolving the inflammatory phase of meniscal injury and moving toward tissue regeneration.
MSC-derived exosomes are rich in microRNAs (miRNAs), such as miR-21, miR-29, and miR-146a, which regulate gene expression in target cells by inhibiting pro-inflammatory pathways and promoting collagen synthesis. These exosomes are taken up by resident meniscal cells and immune cells, influencing processes like cell proliferation, ECM remodeling, and angiogenesis. For example, miR-29 inhibits the expression of collagen-degrading enzymes such as MMP-9, thereby reducing excessive matrix degradation and supporting ECM integrity.
In addition to modulating inflammation, MSCs promote angiogenesis through the secretion of VEGF, basic fibroblast growth factor (bFGF), and angiopoietin-1. These factors enhance the formation of new blood vessels in the peripheral zones of the meniscus, improving nutrient and oxygen delivery to the healing tissue. The creation of a vascularized environment is particularly important in the red-red zone, where the presence of blood vessels enhances the regenerative capacity of the meniscus.
MSC paracrine signaling also stimulates the proliferation and activity of resident meniscal cells. Through the release of IGF-1, TGF-β, and FGF, MSCs enhance the anabolic activity of fibrochondrocytes, promoting the production of collagen, proteoglycans, and other key ECM molecules. This paracrine effect ensures that tissue regeneration is not solely reliant on MSC differentiation but also involves the activation of endogenous repair mechanisms within the meniscus.
In summary, MSCs are recruited to the site of meniscal injury through chemotactic signaling, where they contribute to tissue repair both through differentiation into chondrocytes and fibrochondrocytes and through extensive paracrine signaling. The combination of growth factor-mediated differentiation, mechanotransduction, and the secretion of anti-inflammatory cytokines and pro-regenerative growth factors enables MSCs to play a central role in meniscal repair. These molecular mechanisms make MSCs a valuable target for regenerative medicine and tissue engineering strategies, with the potential to enhance meniscal healing and prevent long-term joint degeneration. Understanding and harnessing these molecular pathways is key to developing more effective treatments for meniscal injuries and improving outcomes in tissue repair.
Angiogenesis
Angiogenesis, the process of forming new blood vessels, is an essential molecular mechanism in the repair of the meniscus, particularly in the peripheral, vascularized regions where oxygen and nutrients are required to support tissue regeneration. At the core of this process is vascular endothelial growth factor (VEGF), a potent angiogenic factor that is upregulated in response to hypoxia and inflammatory signals at the site of injury. When meniscal tissue is damaged, local oxygen deprivation leads to the stabilization of hypoxia-inducible factor 1-alpha (HIF-1α), a transcription factor that drives the expression of VEGF and other genes involved in angiogenesis. VEGF, through its primary receptor VEGFR2, activates several intracellular signaling cascades, including the PI3K/Akt, ERK/MAPK, and p38 MAPK pathways, which collectively regulate endothelial cell proliferation, migration, and survival. This molecular signaling leads to the sprouting of new capillaries, which deliver much-needed oxygen and nutrients to the regenerating meniscal tissue, enabling cellular survival, proliferation, and extracellular matrix (ECM) production by meniscal cells, including fibrochondrocytes and mesenchymal stem cells (MSCs).
The process of angiogenesis is further modulated by other growth factors such as fibroblast growth factor (FGF) and platelet-derived growth factor (PDGF). FGF, through FGFR, enhances the proliferation and migration of endothelial cells, working synergistically with VEGF to form stable and functional blood vessels. PDGF, on the other hand, stabilizes the nascent blood vessels by recruiting pericytes and smooth muscle cells, which are necessary for the maturation and stability of the newly formed vasculature. This ensures that the new capillaries are structurally sound and can efficiently deliver blood to the regenerating tissue, facilitating the removal of metabolic waste products such as lactic acid and preventing ischemia-induced cell death in the injured meniscus.
VEGF and the associated angiogenic processes also play a critical role in immune modulation at the injury site. The influx of blood through new capillaries brings immune cells, including macrophages, which are involved in clearing debris and orchestrating the repair process. Macrophages themselves secrete additional VEGF, creating a positive feedback loop that further promotes angiogenesis. Additionally, VEGF enhances the permeability of the endothelial barrier, allowing more immune cells and growth factors to infiltrate the injury site, which is crucial for the initial inflammatory response and for creating an environment conducive to tissue repair. The sustained presence of VEGF and other angiogenic factors ensures that this vascular remodeling process continues through the proliferative and remodeling phases of healing.
Angiogenesis is particularly critical in the red-red zone of the meniscus, the outer third that is vascularized and has a higher regenerative capacity. In this zone, endothelial cells are readily available to respond to angiogenic signals, and the existing vascular network provides the structural framework for new blood vessel formation. This vascularity ensures that reparative cells such as fibrochondrocytes and MSCs receive the necessary signals and resources to proliferate and synthesize ECM components, including collagen types I and II and proteoglycans. The outer meniscal zones have the most favorable environment for repair due to their rich blood supply and robust angiogenic response.
In contrast, the white-white zone of the meniscus, which is avascular, poses a significant challenge for healing because it lacks both the initial blood supply and the capacity for angiogenic remodeling. Growth factors, nutrients, and oxygen must diffuse from the synovial fluid, a much slower and less efficient process. In this region, VEGF expression and activity are limited, resulting in a lower likelihood of spontaneous healing. This highlights the molecular distinction between the vascularized outer zone and the avascular inner zone, and underscores the need for therapeutic interventions that target angiogenesis in meniscal repair. Emerging strategies include VEGF gene therapy and the use of pro-angiogenic scaffolds, which aim to artificially induce or enhance blood vessel formation in the avascular regions of the meniscus to improve healing outcomes.
At the molecular level, the balance between pro-angiogenic and anti-angiogenic factors is critical for ensuring that angiogenesis proceeds in a controlled manner. Excessive angiogenesis can lead to over-vascularization, which may impair tissue function or promote inappropriate remodeling of the meniscus. Thus, tissue inhibitors of metalloproteinases (TIMPs), thrombospondin-1 (TSP-1), and endostatin are often upregulated in parallel with VEGF to prevent uncontrolled blood vessel growth. These anti-angiogenic molecules work by inhibiting VEGF signaling, blocking endothelial cell migration, and promoting endothelial cell apoptosis to fine-tune the angiogenic response. Thrombospondin-1, in particular, binds to CD36 on endothelial cells and suppresses VEGFR2 signaling, reducing angiogenesis when the repair process is sufficiently advanced. This precise regulation of angiogenesis ensures that new blood vessels form only where they are needed and that they regress once their function is no longer necessary.
In conclusion, angiogenesis is a molecularly complex and finely regulated process that is essential for meniscal repair, particularly in the vascularized red-red zone. Through the coordinated actions of VEGF, FGF, and PDGF, along with the inflammatory response and immune cell recruitment, angiogenesis supplies the injured meniscus with oxygen, nutrients, and reparative cells necessary for effective healing. The challenge of limited vascularity in the white-white zone underscores the importance of developing targeted therapies that promote angiogenesis in avascular regions to enhance the overall healing capacity of the meniscus. Molecular interventions that modulate the balance between pro-angiogenic and anti-angiogenic factors hold great promise for improving outcomes in meniscal injuries and tissue engineering approaches.
Mechanotransduction and Mechanical Loading
Mechanical loading significantly influences the repair and remodeling of the meniscus through a series of molecular mechanotransduction pathways, which convert external mechanical forces into biochemical signals that guide the cellular responses of chondrocytes, fibrochondrocytes, and mesenchymal stem cells (MSCs). These cells rely on intricate molecular networks to regulate processes like cell proliferation, ECM production, and tissue adaptation, which are critical for maintaining the structural integrity of the meniscus and ensuring effective healing after injury.
Integrin Signaling: At the core of mechanical sensing in meniscal cells are integrins, which are transmembrane proteins that connect the ECM to the intracellular actin cytoskeleton. Mechanical loading causes integrins to cluster and activate focal adhesion kinase (FAK), a key regulator of focal adhesions, which are multiprotein complexes that mediate cell adhesion to the ECM. FAK activation leads to the recruitment of signaling proteins such as Src kinases, paxillin, and talin, which further amplify the signal by triggering downstream pathways like PI3K/Akt and RhoA/ROCK. These pathways regulate cytoskeletal remodeling, allowing cells to adjust their structure in response to mechanical stimuli. FAK-mediated signaling also activates transcription factors such as YAP/TAZ, which translocate to the nucleus and modulate the expression of genes involved in ECM synthesis, including COL1A1 and COL2A1, which encode for type I and II collagen, respectively. This process is crucial for reinforcing the ECM, promoting cell survival, and ensuring the meniscus can endure mechanical stresses.
In addition, integrins interact with growth factor receptors, such as TGF-β receptors, enhancing TGF-β signaling, which is pivotal for chondrogenic differentiation and ECM production. This interaction between integrins and growth factor receptors ensures that mechanical signals are integrated with biochemical signals, optimizing the meniscus’s response to mechanical loads.
Ion Channels: Stretch-activated ion channels play a vital role in translating mechanical forces into biochemical signals, particularly through the regulation of intracellular calcium levels. Mechanical deformation of the meniscus activates mechanosensitive ion channels, such as Piezo1 and TRPV4, leading to an influx of calcium ions (Ca²⁺). The rise in intracellular calcium triggers the activation of the calcineurin/NFAT and calmodulin-dependent kinase (CaMK) pathways, both of which are critical for regulating gene expression and cellular adaptation to mechanical stress. Calcineurin, a calcium-dependent phosphatase, dephosphorylates NFAT (nuclear factor of activated T cells), allowing it to enter the nucleus and promote the expression of genes involved in ECM remodeling and cell migration. CaMK, in contrast, phosphorylates various targets involved in cytoskeletal dynamics, enhancing cell movement and facilitating the restructuring of the meniscus to accommodate mechanical loads.
In addition to these pathways, calcium signaling also influences matrix metalloproteinase (MMP) activity, which is essential for ECM turnover. For instance, mechanical loading can activate MMP-2 and MMP-13, enzymes responsible for degrading collagen, thereby allowing the removal of damaged ECM and facilitating the deposition of new matrix components. The regulation of MMP activity is tightly controlled by calcium levels to prevent excessive ECM degradation, ensuring a balanced remodeling process that supports tissue repair.
MAPK Pathway: The mitogen-activated protein kinase (MAPK) pathway is a central mechanotransduction pathway activated by mechanical loading. Mechanical forces, such as compression and shear stress, trigger the activation of MAPKs, including ERK1/2, p38, and JNK. ERK1/2 is primarily involved in promoting cell proliferation and differentiation, while p38 plays a crucial role in regulating ECM production and responding to cellular stress. Mechanical activation of the MAPK pathway leads to the phosphorylation of transcription factors such as AP-1 and c-Fos, which regulate the expression of genes involved in collagen synthesis (such as COL2A1), proteoglycan production, and other ECM components that are critical for maintaining meniscal structure and function.
In addition to its role in ECM synthesis, the p38 MAPK pathway also regulates MMP expression, particularly MMP-13, which is involved in collagen degradation during ECM turnover. By coordinating the activities of MMPs and ECM synthesis, the MAPK pathway ensures that the meniscus adapts to mechanical forces while maintaining its structural integrity. The ERK1/2 pathway also interacts with growth factors like IGF-1 and FGF, amplifying the anabolic response and promoting cell proliferation, which is essential for repairing damaged meniscal tissue.
Hippo-YAP/TAZ Pathway: Another critical mechanotransduction pathway activated by mechanical loading is the Hippo-YAP/TAZ pathway. YAP and TAZ are transcriptional coactivators that respond to mechanical cues by shuttling between the cytoplasm and nucleus. Under mechanical stress, cytoskeletal tension mediated by RhoA/ROCK activation suppresses the Hippo pathway, allowing YAP/TAZ to accumulate in the nucleus, where they bind to transcription factors like TEAD and promote the expression of genes involved in cell proliferation and ECM production. This pathway is particularly important for the maintenance of tissue homeostasis and the regeneration of meniscal tissue, as YAP/TAZ activity enhances the production of collagen and other ECM components in response to mechanical forces.
Wnt/β-catenin Signaling: Mechanical loading also influences the Wnt/β-catenin signaling pathway, which plays a critical role in chondrogenesis and tissue regeneration. Activation of this pathway under mechanical stress leads to the stabilization of β-catenin, which accumulates in the nucleus and promotes the transcription of genes involved in ECM production, including collagen and proteoglycans. The Wnt pathway interacts with other mechanotransduction pathways, such as the TGF-β/SMAD pathway, to coordinate cellular responses to mechanical loading, ensuring that the meniscus produces the necessary matrix components to repair and reinforce the tissue.
In conclusion, mechanical loading activates a variety of molecular pathways, including integrin signaling, ion channels, the MAPK pathway, Hippo-YAP/TAZ, and Wnt/β-catenin, which collectively regulate the cellular and molecular processes essential for meniscal repair and remodeling. By understanding these molecular mechanisms, therapeutic strategies and rehabilitation protocols can be designed to enhance tissue regeneration, promote ECM synthesis, and restore meniscal function following injury.
Table 3.
The table also describes the impact of inflammation, ECM degradation, and fibrosis on meniscal healing, and it emphasizes the importance of molecular signaling pathways (e.g., SMAD, NF-κB, HIF) in the repair process. Lastly, it discusses therapeutic approaches such as platelet-rich plasma (PRP), mesenchymal stem cells (MSCs), and surgical interventions like meniscal suturing and meniscectomy. Overall, this table provides a detailed analysis of the mechanisms underlying meniscal injury, the biological challenges to healing, and potential therapeutic strategies to improve outcomes, particularly in avascular regions of the meniscus.
Table 3.
The table also describes the impact of inflammation, ECM degradation, and fibrosis on meniscal healing, and it emphasizes the importance of molecular signaling pathways (e.g., SMAD, NF-κB, HIF) in the repair process. Lastly, it discusses therapeutic approaches such as platelet-rich plasma (PRP), mesenchymal stem cells (MSCs), and surgical interventions like meniscal suturing and meniscectomy. Overall, this table provides a detailed analysis of the mechanisms underlying meniscal injury, the biological challenges to healing, and potential therapeutic strategies to improve outcomes, particularly in avascular regions of the meniscus.
| Category | Details | Processes Involved | Key Molecular/Cellular Players |
| Meniscus Structure and Function | The meniscus is a fibrocartilaginous structure located between the femoral condyles and tibial plateau in the knee joint. It plays a crucial role in load distribution, shock absorption, joint stability, and lubrication. | The meniscus spreads compressive forces, reducing stress on articular cartilage and maintaining knee joint function. | Meniscus, fibrocartilage, type I collagen, proteoglycans (aggrecan), fibrochondrocytes, ECM (extracellular matrix) |
| Meniscal Injury Types | Meniscal injuries can result from acute trauma (e.g., twisting motion) or degenerative changes (wear and tear over time). Acute injuries are common in athletes, while degenerative injuries are more prevalent in older adults. | Acute injuries lead to tears, while degenerative injuries cause fragmentation or fraying. Both result in knee pain, swelling, and mechanical symptoms (e.g., locking, instability). | Meniscal tears, acute trauma, degeneration, knee joint instability, swelling, pain, locking |
| Healing Capacity Based on Vascular Zones | The meniscus has different healing capacities depending on its vascular zones: the red-red (vascularized), red-white (partially vascularized), and white-white (avascular) zones. | The outer (red-red) zone has better healing potential due to vascular supply, while the inner (white-white) zone has limited healing ability due to avascularity. | Red-red zone, white-white zone, blood supply, vascularization, synovial fluid, meniscal healing |
| Cellular Response to Injury | Meniscal injury triggers fibrochondrocytes to produce enzymes (MMPs) that degrade ECM components, aiming to remodel damaged tissue. Inflammatory cytokines (e.g., IL-1β, TNF-α) are upregulated, promoting tissue breakdown. | Increased synthesis of proteolytic enzymes (MMPs) and cytokines drives ECM degradation. However, excessive degradation impairs tissue repair. | MMPs (MMP-1, MMP-3, MMP-13), inflammatory cytokines (IL-1β, TNF-α, IL-6), ECM, fibrochondrocytes |
| Extracellular Matrix (ECM) Changes | ECM composition shifts after injury, with a decrease in collagen and proteoglycan synthesis, leading to reduced biomechanical properties. This loss of elasticity compromises shock absorption and load distribution. | Reduced synthesis of collagen and proteoglycans results in diminished ability to retain water, further weakening the meniscus. | ECM, collagen (type I, type II), proteoglycans, elastin, glycosaminoglycans (GAGs), fibrotic tissue |
| Inflammatory Response and ECM Breakdown | Meniscal injuries trigger the release of DAMPs and the recruitment of immune cells (e.g., macrophages, neutrophils), which exacerbate ECM degradation. Chronic inflammation can lead to fibrosis and joint degeneration. | Immune cell infiltration and cytokine release activate inflammatory pathways (e.g., NF-κB), amplifying the degradation of the ECM. | DAMPs, macrophages, neutrophils, pro-inflammatory cytokines (TNF-α, IL-1β, IL-6), NF-κB, ECM |
| Chondrocyte and Fibrochondrocyte Role in Repair | Chondrocytes in articular cartilage respond to meniscal injury by producing ECM components (e.g., collagen X) but may also contribute to cartilage degeneration, particularly in osteoarthritis. | Hypertrophic chondrocytes produce collagen X and MMPs, while fibrochondrocytes in the meniscus increase collagen and ECM synthesis in response to injury. | Chondrocytes, fibrochondrocytes, collagen (type I, type II, type X), MMPs, vascular endothelial growth factor (VEGF) |
| Molecular Signaling Pathways in Healing | TGF-β signaling plays a dual role, promoting ECM synthesis and fibroblast proliferation but also driving fibrosis if overactivated. Hypoxia-induced factors (HIFs) promote limited neovascularization in the avascular zones. | TGF-β promotes collagen synthesis through SMAD signaling, while hypoxia activates HIFs, inducing VEGF and angiogenesis attempts. | TGF-β, SMAD, hypoxia-inducible factors (HIF-1α), VEGF, fibroblasts, collagen synthesis, fibrosis |
| Therapeutic Strategies for Meniscal Repair | Biological therapies (e.g., PRP, MSCs) aim to stimulate meniscal healing, especially in avascular zones, by enhancing ECM synthesis, reducing inflammation, and promoting chondrocyte activity. | Platelet-rich plasma (PRP) and mesenchymal stem cells (MSCs) support regeneration by providing growth factors that stimulate chondrocyte activity and ECM repair. | PRP, MSCs, growth factors (TGF-β, FGF, IGF-1), chondrocytes, ECM, biological scaffolds |
| Mechanisms of Meniscal Injury | Meniscal injuries are caused by rotational forces, direct impacts, and repetitive microtrauma, common in high-impact sports (e.g., football, basketball). Degenerative tears are more common in older adults. | Acute injuries result from sudden twisting or deceleration, while degenerative tears are due to cumulative microtrauma and aging-related tissue degradation. | Rotational forces, direct impact, meniscal tears, degenerative changes, high-impact sports, aging |
| Meniscal Healing and Surgical Interventions | Tears in the red-red zone are more likely to heal with conservative treatments or suturing. Tears in the white-white zone may require meniscectomy due to poor healing potential. | Surgical options like meniscal suturing and meniscectomy are determined by tear location and the meniscus’s vascular supply. | Meniscal suturing, meniscectomy, vascular supply, red-red zone, white-white zone, conservative treatment |
Articular Cartilage and Its Functionality with Molecular Biology Insights
Articular cartilage is a highly specialized tissue found on the surfaces of bones within synovial joints, including the knee, hip, and shoulder, where it plays a critical role in facilitating frictionless movement and distributing mechanical loads efficiently. Structurally, it is composed of chondrocytes, which are the only cell type found in this tissue, embedded within an extracellular matrix (ECM) rich in type II collagen and proteoglycans like aggrecan. The unique biochemical and biophysical properties of articular cartilage arise from the composition and organization of its ECM. Type II collagen forms a dense, fibrillar network that provides the tissue with tensile strength and prevents over-stretching under mechanical stress. This collagen network is interwoven with proteoglycans, particularly aggrecan, which are large, heavily glycosylated molecules that contain glycosaminoglycans (GAGs) such as chondroitin sulfate and keratan sulfate. These GAG chains are highly hydrophilic, allowing them to bind water molecules, which gives cartilage its compressive strength and elasticity. This water-binding capacity enables cartilage to deform under load and then recover its shape, making it highly resistant to compressive forces encountered during activities such as walking, running, or jumping.
The organization of the ECM in articular cartilage is stratified into different zones—the superficial zone, middle zone, and deep zone—each characterized by distinct collagen fiber orientation and chondrocyte density. In the superficial zone, collagen fibers are aligned parallel to the surface, which enhances the tissue’s resistance to shear forces and reduces friction during joint movement. The middle zone features more randomly oriented collagen fibers and is rich in proteoglycans, allowing for greater resistance to compressive forces. The deep zone has collagen fibers oriented perpendicularly to the subchondral bone, providing strong anchorage to the underlying bone and further enhancing the cartilage’s ability to withstand compressive loads. The chondrocytes within these zones display different morphological and functional characteristics, adapting to the varying mechanical environments within the tissue. In the superficial zone, chondrocytes are flatter and more active in maintaining the collagen network, while in the deeper zones, chondrocytes are more spherical and contribute to ECM remodeling and proteoglycan production.
One of the most significant challenges with articular cartilage is its limited regenerative capacity. The avascular nature of cartilage means that it lacks a direct blood supply, relying instead on diffusion from the synovial fluid to deliver nutrients and remove waste products. This limits the availability of progenitor cells and growth factors, which are essential for tissue repair, making cartilage injuries difficult to heal. Furthermore, cartilage is devoid of lymphatic and nervous systems, which further impedes the tissue’s ability to respond to damage. When cartilage is injured, the chondrocytes have a limited ability to proliferate and migrate to the injury site, and ECM components like collagen and proteoglycans are slowly synthesized, leading to incomplete or ineffective repair. Over time, this can result in progressive degeneration, as seen in conditions like osteoarthritis, where the loss of cartilage leads to increased joint friction, pain, and reduced mobility.
At the molecular level, the repair response of articular cartilage is hampered by the lack of vascularization, which limits the infiltration of immune cells and growth factors typically involved in tissue repair. Injured cartilage produces matrix metalloproteinases (MMPs), which degrade the ECM, particularly collagen and aggrecan, and if not adequately regulated by tissue inhibitors of metalloproteinases (TIMPs), this leads to further tissue breakdown. In addition, the inflammatory cytokines released after injury, such as interleukin-1 beta (IL-1β) and tumor necrosis factor-alpha (TNF-α), can upregulate MMP expression and inhibit the synthesis of new matrix components, exacerbating cartilage degeneration.
Recent advances in cartilage repair strategies have focused on overcoming these limitations. Tissue engineering approaches, such as the use of scaffolds made from natural or synthetic materials, aim to provide a structural framework that supports chondrocyte activity and ECM deposition. Stem cell therapies, particularly those involving mesenchymal stem cells (MSCs), are being explored for their ability to differentiate into chondrocytes and promote cartilage regeneration. Growth factor delivery, especially of factors like transforming growth factor-beta (TGF-β) and insulin-like growth factor 1 (IGF-1), is also being used to enhance the anabolic activity of chondrocytes, encouraging the production of collagen and proteoglycans to repair damaged tissue.
In summary, articular cartilage’s unique structure, with its specialized ECM and zonal organization, allows it to withstand the mechanical forces of joint movement. However, its avascular nature and limited regenerative capacity make cartilage injuries challenging to repair, often leading to progressive degeneration and joint dysfunction. Understanding the molecular mechanisms underlying cartilage repair and developing new therapeutic strategies are critical for improving outcomes in cartilage-related injuries and diseases.
Cartilage Injuries: Causes and Types
Cartilage injuries can be broadly categorized into focal and diffuse types, each with distinct causes, progression, and impact on joint function. Focal injuries are localized to a specific area of the cartilage and typically result from acute trauma, such as a direct impact, twisting motion, or sudden mechanical overload. These injuries are common among athletes, particularly those engaged in high-impact sports like football, basketball, or skiing, where rapid directional changes or collisions occur. Focal injuries can involve small, localized cartilage defects that, if untreated, may progress to osteoarthritis (OA) or more significant cartilage degeneration over time. These injuries often lead to the exposure of the underlying subchondral bone, which can result in increased joint friction, pain, and further cartilage breakdown. The healing response in focal injuries is limited by the avascularity of cartilage, as the lack of a blood supply restricts the migration of reparative cells and the delivery of nutrients necessary for repair. If left untreated, focal injuries can evolve into more diffuse degeneration, affecting the surrounding cartilage tissue.
In contrast, diffuse cartilage injuries involve a broader area of the joint and are more commonly associated with chronic degenerative conditions like osteoarthritis (OA). Osteoarthritis is a progressive joint disease that affects millions of people worldwide, particularly in aging populations. It is characterized by the gradual breakdown of articular cartilage, along with associated changes in the subchondral bone, synovial inflammation, and the formation of osteophytes (bone spurs). The underlying causes of OA are multifactorial, involving a combination of biomechanical stress, genetic predisposition, and biochemical factors such as inflammatory cytokines and matrix-degrading enzymes. Prolonged mechanical loading, particularly in weight-bearing joints like the knee and hip, exacerbates cartilage wear, leading to thinning of the cartilage and the formation of fissures. As the cartilage deteriorates, the ability of the tissue to withstand compressive forces diminishes, resulting in increased joint stress and accelerated cartilage loss.
The biochemical environment in osteoarthritic joints is marked by elevated levels of pro-inflammatory cytokines, such as interleukin-1 beta (IL-1β) and tumor necrosis factor-alpha (TNF-α), which promote matrix degradation by upregulating the production of matrix metalloproteinases (MMPs) and aggrecanases (such as ADAMTS-4 and ADAMTS-5). These enzymes break down key ECM components, including type II collagen and aggrecan, leading to a loss of cartilage’s structural integrity and its water-retaining properties, which are crucial for resisting compressive forces. The imbalance between anabolic and catabolic activities in the cartilage further accelerates tissue degradation. Additionally, oxidative stress and the generation of reactive oxygen species (ROS) contribute to chondrocyte senescence and apoptosis, reducing the ability of chondrocytes to maintain and repair the ECM.
Both focal and diffuse cartilage injuries disrupt the structural integrity and biomechanical function of the joint. In focal injuries, the localized cartilage defect can alter the load distribution within the joint, leading to increased stress on the surrounding cartilage. Over time, this can cause the adjacent cartilage to break down, resulting in a larger, more diffuse injury pattern. In diffuse injuries, such as those seen in osteoarthritis, the widespread loss of cartilage leads to joint space narrowing, bone-on-bone contact, and changes in the subchondral bone, such as sclerosis and the development of subchondral cysts. This further impairs joint function, causing pain, stiffness, and loss of mobility. The synovium also becomes inflamed in osteoarthritis, contributing to the overall disease process through the production of pro-inflammatory mediators and proteolytic enzymes.
The clinical management of focal and diffuse cartilage injuries requires distinct approaches. For focal injuries, surgical interventions such as microfracture, autologous chondrocyte implantation (ACI), and osteochondral autograft transplantation are commonly employed to promote cartilage repair and restore joint function. These techniques aim to stimulate the formation of new cartilage tissue, either through the recruitment of bone marrow-derived stem cells (as in microfracture) or the transplantation of healthy cartilage from non-weight-bearing areas. However, the repair tissue that forms in response to these treatments is often fibrocartilage, which lacks the mechanical properties and durability of hyaline cartilage, limiting long-term success.
For diffuse cartilage injuries, particularly in the context of osteoarthritis, treatment typically focuses on pain management and symptom relief, with options ranging from nonsteroidal anti-inflammatory drugs (NSAIDs) to intra-articular injections of corticosteroids or hyaluronic acid. In advanced cases, joint replacement surgery may be necessary. Emerging therapies, such as biologics (including platelet-rich plasma (PRP) and mesenchymal stem cells (MSCs)), are being investigated for their potential to modulate the inflammatory environment and promote cartilage repair, although their efficacy in diffuse, late-stage osteoarthritis remains an area of active research.
In conclusion, both focal and diffuse cartilage injuries present significant challenges for joint function and long-term health, with distinct etiologies and progression. Focal injuries, often caused by trauma, can lead to localized cartilage defects that may evolve into more widespread degeneration if not treated promptly. Diffuse injuries, typically associated with osteoarthritis, involve a more comprehensive degradation of cartilage due to chronic wear and tear, biomechanical stress, and inflammatory processes. Understanding the molecular mechanisms driving these injuries is critical for developing effective treatments and improving patient outcomes.
Cellular Responses to Cartilage Injury
The cellular responses to cartilage injury involve a highly regulated interplay of molecular processes that significantly affect tissue integrity, including chondrocyte death, ECM degradation, and the triggering of an inflammatory response. Chondrocytes, the specialized cells responsible for synthesizing and maintaining the extracellular matrix (ECM), are central to these processes. The ECM of cartilage is predominantly composed of type II collagen and proteoglycans, such as aggrecan, which confer tensile strength and compressive resistance. Injury to the cartilage disrupts the balance between anabolic and catabolic processes in chondrocytes, leading to complex molecular cascades that further exacerbate tissue damage.
Chondrocyte Death: Chondrocyte death occurs through two major mechanisms—necrosis and apoptosis—both of which are influenced by distinct molecular triggers. Necrosis is primarily associated with acute mechanical trauma, where mechanical overload directly damages the chondrocytes, causing an abrupt loss of membrane integrity. This leads to the release of intracellular contents, including damage-associated molecular patterns (DAMPs), such as high-mobility group box 1 (HMGB1) and ATP, which act as signals for the immune system. These molecules engage pattern recognition receptors (PRRs) like Toll-like receptors (TLRs) on surrounding cells, triggering a robust inflammatory response. In contrast, apoptosis is a more regulated form of cell death that can be induced by various factors, including oxidative stress, inflammatory cytokines like tumor necrosis factor-alpha (TNF-α) and interleukin-1 beta (IL-1β), and ECM degradation. Apoptosis is mediated by the activation of caspases, particularly caspase-3, which orchestrate the cleavage of key cellular proteins, leading to DNA fragmentation and chromatin condensation. One critical pathway involves the mitochondrial (intrinsic) apoptotic pathway, where signals such as reactive oxygen species (ROS) and mitochondrial membrane depolarization cause the release of cytochrome c, leading to the activation of the apoptosome and subsequent caspase activation. Additionally, the Fas receptor/Fas ligand pathway (extrinsic) also activates caspases via Fas-associated death domain (FADD) recruitment, further contributing to chondrocyte apoptosis.
Extracellular Matrix Damage: The ECM is composed primarily of collagen fibers and proteoglycans, both of which are crucial for cartilage’s biomechanical properties. Cartilage injury results in ECM degradation through the upregulation of catabolic enzymes, particularly matrix metalloproteinases (MMPs) and aggrecanases (such as ADAMTS-4 and ADAMTS-5). The activation of MMP-13 is a key factor in the breakdown of type II collagen, while ADAMTS-5 cleaves aggrecan, leading to a loss of cartilage’s ability to retain water, thereby compromising its compressive properties. These enzymes are regulated by inflammatory mediators such as IL-1β, TNF-α, and nitric oxide (NO), which amplify the degradation process by increasing MMP expression through signaling pathways like nuclear factor-kappa B (NF-κB) and mitogen-activated protein kinase (MAPK). NF-κB activation, driven by the binding of cytokines to receptors such as IL-1 receptor (IL-1R) and TNF receptor (TNFR), leads to the transcription of pro-inflammatory genes, further promoting ECM degradation. Additionally, the breakdown of the ECM releases matrix fragments, which act as DAMPs, feeding back into the inflammatory loop by stimulating TLRs and further activating NF-κB signaling.
The interaction between chondrocyte death and ECM degradation creates a feedback loop that perpetuates cartilage damage. Chondrocyte apoptosis results in reduced ECM synthesis, particularly of collagen type II and aggrecan, further weakening the tissue structure. Simultaneously, the release of matrix fragments, such as degraded collagen peptides and hyaluronan, exacerbates inflammation, which continues to drive MMP and ADAMTS activity, creating a cycle of progressive tissue destruction. Moreover, oxidative stress, largely driven by increased levels of ROS, contributes to both chondrocyte apoptosis and ECM breakdown. ROS can directly damage cellular components and activate signaling pathways like p38 MAPK, which promote the expression of catabolic enzymes.
Additionally, the loss of ECM integrity impacts chondrocyte mechanotransduction, the process by which cells sense and respond to mechanical forces. The integrin receptors that link chondrocytes to the ECM play a critical role in transmitting mechanical signals, which regulate cellular activities like proliferation, differentiation, and ECM synthesis. When the ECM is degraded, chondrocytes lose their connection to these mechanical cues, leading to altered mechanotransduction signaling through pathways like FAK/Src and RhoA/ROCK, further impairing their function and contributing to tissue degeneration.
In summary, the molecular mechanisms involved in the cellular response to cartilage injury are highly complex and interconnected. Chondrocyte death, whether by necrosis or apoptosis, triggers an inflammatory cascade that perpetuates ECM degradation. Inflammatory cytokines, ROS, and matrix-degrading enzymes such as MMPs and ADAMTSs form a vicious cycle of tissue breakdown. Understanding these molecular processes is crucial for developing therapeutic strategies aimed at halting cartilage degeneration and promoting effective tissue repair.
Inflammatory Response to Cartilage Injury
The inflammatory response to cartilage injury is a highly regulated molecular process, crucial for initiating tissue repair but often exacerbating damage if it becomes chronic. Cartilage, being avascular, relies on neighboring structures such as the synovium and subchondral bone to trigger the inflammatory response. Chondrocytes, the primary cell type in cartilage, play a central role in this process by releasing inflammatory mediators in response to injury. These mediators activate a cascade of molecular events that can either promote repair or, in chronic conditions, drive cartilage degeneration.
Cytokine Release: The release of pro-inflammatory cytokines such as interleukin-1 beta (IL-1β) and tumor necrosis factor-alpha (TNF-α) is an immediate response to cartilage injury. These cytokines are secreted by chondrocytes, synovial fibroblasts, and macrophages present in the synovium. Upon binding to their respective receptors, IL-1R and TNFR, on chondrocytes and surrounding cells, these cytokines activate key signaling pathways, including the nuclear factor-kappa B (NF-κB) and mitogen-activated protein kinase (MAPK) pathways. NF-κB activation is pivotal in driving the transcription of inflammatory genes, leading to the production of interleukin-6 (IL-6), prostaglandins, and additional TNF-α, creating a feed-forward loop that amplifies inflammation. The MAPK pathway, including ERK1/2, p38, and JNK, is also critical in mediating cellular responses, including the production of matrix metalloproteinases (MMPs) and aggrecanases. IL-1β and TNF-α also induce the production of reactive oxygen species (ROS) via the upregulation of NADPH oxidase and the mitochondrial dysfunction, which further drives chondrocyte apoptosis and exacerbates tissue damage.
Matrix Metalloproteinases (MMPs): Inflammatory cytokines like IL-1β and TNF-α stimulate the expression of MMPs, including MMP-1 (collagenase-1), MMP-3 (stromelysin-1), and MMP-13 (collagenase-3). These enzymes are central to ECM degradation, particularly the breakdown of type II collagen, which provides structural integrity to cartilage. MMP-13, specifically, cleaves collagen into smaller fragments, impairing the tensile strength of cartilage. The activation of MMPs is tightly controlled by signaling pathways such as NF-κB and p38 MAPK, both of which are responsive to inflammatory cytokines and mechanical stress signals. Additionally, the MAPK pathway regulates the phosphorylation of transcription factors like AP-1 (c-Fos and c-Jun), which directly bind to MMP gene promoters, driving their expression. MMPs are normally regulated by tissue inhibitors of metalloproteinases (TIMPs), particularly TIMP-1 and TIMP-2, but in the inflammatory environment following injury, the balance shifts towards increased MMP activity, overwhelming TIMP control and resulting in excessive matrix breakdown. MMPs are also activated through proteolytic cleavage by other enzymes like plasmin and furin, adding another layer of regulation in response to injury.
Aggrecanases: Alongside MMPs, ADAMTS-4 and ADAMTS-5 (aggrecanases) are upregulated following cartilage injury and inflammation. These enzymes specifically degrade aggrecan, a large proteoglycan responsible for retaining water in cartilage, thus providing compressive strength. ADAMTS-5 is particularly important in pathological cartilage degradation, as it cleaves aggrecan at specific sites within the core protein, leading to the release of glycosaminoglycan (GAG) chains. This loss of aggrecan reduces the cartilage’s ability to resist compressive forces, further compromising its functional integrity. Similar to MMPs, the activity of aggrecanases is regulated by cytokines like IL-1β and TNF-α, which drive their expression through the NF-κB and MAPK pathways. The proteolytic activity of ADAMTS enzymes is modulated by TIMP-3, but like TIMP regulation of MMPs, inflammatory signaling reduces the effectiveness of this inhibition, resulting in unchecked ECM degradation.
Chondrocyte Mechanotransduction: Cartilage injury and the subsequent degradation of the ECM disrupt the normal mechanotransduction processes of chondrocytes. Under normal conditions, integrins on the surface of chondrocytes interact with ECM components like collagen, allowing the cells to sense mechanical forces and respond by adjusting ECM synthesis. This interaction is mediated by focal adhesion complexes that activate signaling pathways such as focal adhesion kinase (FAK), RhoA/ROCK, and PI3K/Akt. After injury, the degradation of ECM components impairs integrin-ECM interactions, disrupting mechanotransduction. This leads to altered gene expression and diminished production of anabolic ECM components like type II collagen and aggrecan, further reducing the capacity for cartilage repair. The loss of ECM integrity also triggers abnormal calcium signaling in chondrocytes, as mechanosensitive ion channels such as Piezo1 and TRPV4 are dysregulated, contributing to increased intracellular calcium levels that promote cell death and catabolic activity.
Reactive Oxygen Species (ROS) and Nitric Oxide (NO): In the inflammatory microenvironment, increased levels of ROS and NO play key roles in promoting chondrocyte apoptosis and further ECM degradation. ROS, generated by the activity of enzymes such as NADPH oxidase and mitochondrial dysfunction, can directly damage cellular components, including lipids, proteins, and DNA. ROS also activate signaling pathways like p38 MAPK, leading to the upregulation of catabolic enzymes, including MMPs and aggrecanases. NO, produced by inducible nitric oxide synthase (iNOS) in response to IL-1β and TNF-α, further contributes to cartilage degradation by inhibiting ECM synthesis and promoting chondrocyte apoptosis through mitochondrial dysfunction.
In summary, the inflammatory response following cartilage injury involves a complex interplay of cytokine signaling, MMP and aggrecanase upregulation, oxidative stress, and disrupted mechanotransduction. The molecular events driven by cytokines such as IL-1β and TNF-α activate catabolic pathways that degrade the ECM, particularly through the activity of MMPs and aggrecanases, while also promoting chondrocyte death via oxidative stress. The resulting degradation of type II collagen and aggrecan leads to a loss of cartilage structure and function, perpetuating a cycle of inflammation and degeneration. Understanding these molecular pathways is critical for developing therapeutic strategies that target inflammation, protect the ECM, and promote cartilage regeneration in conditions like osteoarthritis.
Extracellular Matrix Remodeling and Repair
The balance between ECM degradation and synthesis in cartilage repair is tightly controlled by a series of molecular processes involving both catabolic and anabolic signaling pathways. The initial phase of ECM degradation is orchestrated by matrix metalloproteinases (MMPs) and aggrecanases like ADAMTS-4 and ADAMTS-5, which are upregulated in response to inflammatory cytokines such as interleukin-1 beta (IL-1β) and tumor necrosis factor-alpha (TNF-α). These cytokines activate key signaling pathways, including nuclear factor-kappa B (NF-κB) and mitogen-activated protein kinase (MAPK) pathways, which drive the expression of catabolic enzymes. MMP-13 specifically degrades type II collagen, the primary structural protein in cartilage, while ADAMTS-5 cleaves aggrecan, a proteoglycan essential for cartilage’s compressive resistance. The degradation of these ECM components results in the loss of structural integrity and mechanical function, especially as the ECM fragments released during this process act as damage-associated molecular patterns (DAMPs), further activating inflammatory pathways and perpetuating tissue breakdown.
On the anabolic side, chondrocytes initiate ECM synthesis in response to growth factors such as transforming growth factor-beta (TGF-β), insulin-like growth factor-1 (IGF-1), and bone morphogenetic proteins (BMPs). TGF-β is particularly crucial in driving chondrocyte proliferation and matrix synthesis by activating the Smad signaling pathway, which regulates the transcription of genes encoding type II collagen and aggrecan. Once activated, TGF-β receptors phosphorylate Smad2/3, which form complexes with Smad4 and translocate to the nucleus to initiate ECM gene transcription. This anabolic response is further supported by IGF-1, which stimulates the PI3K/Akt pathway, enhancing cell survival and promoting protein synthesis. BMPs, through the MAPK and Smad pathways, also contribute to chondrocyte differentiation and ECM production. This ensures that the cartilage can synthesize new matrix components to replace those degraded during injury.
A critical regulatory mechanism in this process involves the balance between MMPs and their inhibitors, the tissue inhibitors of metalloproteinases (TIMPs). TIMP-1 and TIMP-3 inhibit MMPs and aggrecanases, respectively, preventing excessive ECM breakdown. However, in the presence of chronic inflammation, pro-inflammatory cytokines reduce the expression and activity of TIMPs, tipping the balance toward catabolic activity and uncontrolled matrix degradation. This results in sustained ECM breakdown, impairing the ability of cartilage to recover and leading to progressive damage, as seen in diseases like osteoarthritis.
The interplay between oxidative stress and inflammation further complicates this balance. Inflammatory cytokines such as IL-1β and TNF-α induce the production of reactive oxygen species (ROS) and nitric oxide (NO) via inducible nitric oxide synthase (iNOS) and NADPH oxidase. ROS can activate p38 MAPK and JNK, leading to increased MMP expression and chondrocyte apoptosis. NO, on the other hand, impairs ECM synthesis by inhibiting chondrocyte mitochondrial function and reducing the production of anabolic growth factors. ROS and NO also disrupt mitochondrial homeostasis, promoting chondrocyte apoptosis and further limiting the capacity for matrix regeneration.
Another key element in maintaining ECM homeostasis is the mechanotransduction pathway, which allows chondrocytes to sense mechanical forces and adjust ECM synthesis accordingly. Integrins, transmembrane receptors that mediate cell-ECM interactions, play a crucial role in this process. When integrins engage with ECM components like collagen, they activate focal adhesion kinase (FAK), which in turn stimulates downstream signaling pathways, including the RhoA/ROCK and PI3K/Akt pathways. These pathways regulate cytoskeletal dynamics, chondrocyte survival, and ECM protein production. Disruption of integrin signaling due to ECM degradation impairs mechanotransduction, reducing the chondrocyte’s ability to respond to mechanical stimuli and further compromising matrix synthesis.
In summary, the molecular balance between ECM degradation and synthesis is governed by a complex network of cytokine signaling, protease activity, growth factor pathways, and mechanotransduction mechanisms. The degradation of ECM components by MMPs and aggrecanases, driven by pro-inflammatory signals, is counterbalanced by the anabolic effects of TGF-β, IGF-1, and BMPs, which promote matrix repair. However, chronic inflammation, oxidative stress, and disruptions in mechanotransduction can tip this balance in favor of degradation, leading to progressive cartilage damage. Understanding these molecular pathways is critical for developing therapeutic strategies aimed at enhancing cartilage repair by inhibiting excessive catabolic activity and promoting ECM regeneration.
Chondrocyte Responses to Injury
The molecular responses of chondrocytes to cartilage injury involve intricate signaling pathways that regulate proliferation, phenotypic modulation, and autophagy—all critical for tissue repair, but potentially detrimental if not properly balanced. Following injury, chondrocyte proliferation is driven by growth factors such as insulin-like growth factor-1 (IGF-1), fibroblast growth factor (FGF), and transforming growth factor-beta (TGF-β), which activate key signaling pathways including PI3K/Akt and MAPK. These pathways regulate cell cycle progression through the activation of proteins like cyclin D1 and CDK4/6, leading to enhanced chondrocyte proliferation. IGF-1, through the Akt/mTOR pathway, not only promotes cell growth but also stimulates the synthesis of extracellular matrix (ECM) components such as type II collagen and aggrecan. However, the formation of chondrocyte clusters in response to excessive proliferation can result in altered cartilage mechanics. These clusters produce fibrocartilage instead of the native hyaline cartilage, leading to the deposition of type I collagen—a hallmark of degraded cartilage with inferior biomechanical properties.
The phenotypic modulation of chondrocytes during cartilage injury is largely driven by inflammatory cytokines such as interleukin-1 beta (IL-1β) and tumor necrosis factor-alpha (TNF-α), which activate nuclear factor-kappa B (NF-κB) and p38 MAPK signaling pathways. Under normal conditions, chondrocytes maintain a stable phenotype that supports the production of hyaline cartilage matrix components, including type II collagen and aggrecan. However, in the presence of chronic inflammation, these cells undergo a shift toward a fibroblastic phenotype, characterized by increased expression of type I collagen and MMPs (matrix metalloproteinases). MMP-13, for example, is upregulated via p38 MAPK and contributes to the degradation of type II collagen, further diminishing the structural integrity of the ECM. This phenotypic shift is also regulated by mechanical stress, where the activation of integrins and focal adhesion kinase (FAK) leads to changes in cytoskeletal organization and gene expression, reinforcing the production of fibrocartilage, which lacks the durability and load-bearing capacity of healthy hyaline cartilage.
Autophagy, a key survival mechanism, is upregulated in chondrocytes in response to oxidative and mechanical stress, particularly during cartilage injury. Autophagy is regulated through the AMPK/mTOR axis, where AMPK activation promotes autophagy by inhibiting mTOR, a master regulator of cell growth and autophagy suppression. This process involves the formation of autophagosomes, which encapsulate and degrade damaged organelles and misfolded proteins via lysosomal degradation. Autophagy is particularly crucial in mitigating oxidative damage caused by reactive oxygen species (ROS), which are produced in excess during inflammation. The activation of autophagy-related proteins like LC3, Beclin-1, and ATG5 helps maintain cellular homeostasis by preventing apoptosis and supporting the survival of chondrocytes. Dysregulation of autophagy, however, can accelerate cell death through apoptosis, contributing to cartilage degeneration. Excessive oxidative stress can impair mitochondrial function, promoting chondrocyte apoptosis via the intrinsic pathway, where mitochondrial cytochrome c release activates caspase-9, leading to caspase-3-mediated cell death.
In the context of injury, chondrocytes also experience disrupted mechanotransduction due to changes in the ECM composition. Integrins, which mediate cell-ECM interactions, are key receptors that transmit mechanical signals from the ECM to the cell. These integrins, when bound to degraded matrix components, activate signaling through FAK and RhoA/ROCK, leading to altered gene expression and matrix production. Disrupted mechanotransduction further contributes to the downregulation of anabolic processes and upregulation of catabolic enzymes, exacerbating cartilage breakdown. Additionally, autophagy interacts with mechanotransduction pathways, as mechanical stress can modulate autophagy levels, with mild stress promoting protective autophagy and excessive stress inhibiting it, pushing cells toward apoptosis.
The balance between catabolic and anabolic processes is crucial for chondrocyte survival and ECM maintenance. TGF-β promotes anabolic activity by activating the Smad2/3 pathway, leading to the transcription of ECM components such as type II collagen and aggrecan. In contrast, inflammatory cytokines like IL-1β and TNF-α drive catabolic pathways through NF-κB, inducing the expression of MMPs and ADAMTS (a disintegrin and metalloproteinase with thrombospondin motifs), which degrade the ECM. These catabolic enzymes are responsible for breaking down both collagen and proteoglycans, creating a feedback loop that perpetuates inflammation and tissue destruction. Reactive nitrogen species (RNS), including nitric oxide (NO), also play a role in cartilage catabolism by inhibiting ECM synthesis and promoting chondrocyte apoptosis through mitochondrial dysfunction.
In summary, chondrocyte responses to cartilage injury involve complex molecular interactions between proliferation, phenotypic modulation, and autophagy. Proliferation and clustering are driven by growth factors like IGF-1 and TGF-β, but if unchecked, lead to fibrocartilage formation. Phenotypic modulation, induced by inflammatory cytokines and mechanical stress, results in the production of type I collagen and MMPs, contributing to tissue degeneration. Autophagy, regulated by the AMPK/mTOR pathway, protects chondrocytes from oxidative stress and apoptosis, but its dysregulation exacerbates cell death and cartilage breakdown. These molecular processes are tightly regulated, and imbalances can lead to chronic cartilage degeneration, emphasizing the need for targeted therapeutic interventions to modulate these pathways and enhance cartilage repair.
Mesenchymal Stem Cell (MSC) Recruitment and Differentiation
Mesenchymal stem cells (MSCs) are pivotal in cartilage repair, leveraging their multipotency to differentiate into chondrocytes and influence tissue repair through molecular mechanisms. Upon cartilage injury, MSC recruitment is driven by chemotactic signals such as stromal cell-derived factor-1 (SDF-1), which binds to the CXCR4 receptor on MSCs, and platelet-derived growth factor (PDGF), which activates the PDGFR receptor. These signals, along with others like vascular endothelial growth factor (VEGF) and fibroblast growth factor (FGF), guide MSC migration from surrounding tissues, such as the synovium, subchondral bone, and bone marrow. The recruitment process involves the activation of intracellular signaling cascades, including the PI3K/Akt and MAPK pathways, which regulate MSC motility, adhesion, and survival at the injury site. Once MSCs are localized to the damaged cartilage, they enter the next phase: differentiation.
MSC differentiation into chondrocytes is highly dependent on local cues, particularly the presence of growth factors such as transforming growth factor-beta (TGF-β), bone morphogenetic proteins (BMPs), and insulin-like growth factor-1 (IGF-1). TGF-β plays a central role by binding to the TGF-β receptor, activating the Smad2/3 signaling pathway. This results in the phosphorylation of Smad2/3, which forms a complex with Smad4 and translocates to the nucleus, where it upregulates the expression of chondrogenic markers such as SOX9, type II collagen (COL2A1), and aggrecan (ACAN). BMPs activate similar pathways, specifically through Smad1/5/8, which also promotes chondrocyte differentiation. Additionally, the Wnt/β-catenin signaling pathway, while generally inhibitory in excess, can fine-tune chondrogenesis depending on its regulation, interacting with other pathways like Hedgehog (Hh) and Notch to balance MSC differentiation.
During this differentiation process, MSCs undergo epigenetic modifications, such as changes in histone acetylation and DNA methylation, which regulate the expression of chondrocyte-specific genes. For example, histone acetyltransferases (HATs) increase the accessibility of chromatin to transcription factors such as SOX9, enhancing the transcription of cartilage-specific ECM proteins like type II collagen and aggrecan. Meanwhile, microRNAs (miRNAs), particularly miR-140, are upregulated in differentiating MSCs and play a role in stabilizing chondrocyte-specific gene expression while inhibiting pathways that promote osteogenic differentiation, thus ensuring MSCs commit to a chondrogenic lineage.
Beyond differentiation, MSCs exert profound paracrine effects through the secretion of cytokines, growth factors, and extracellular vesicles (including exosomes). These secreted molecules modulate the injury microenvironment, reducing inflammation, protecting chondrocytes from apoptosis, and enhancing ECM synthesis. For instance, MSC-derived exosomes contain bioactive molecules like microRNAs (miR-21, miR-24) and growth factors (TGF-β, IGF-1) that regulate gene expression in resident chondrocytes and suppress inflammatory pathways by inhibiting NF-κB signaling. Furthermore, IL-10 and TGF-β secreted by MSCs play anti-inflammatory roles by inhibiting the production of pro-inflammatory cytokines such as interleukin-1 beta (IL-1β) and tumor necrosis factor-alpha (TNF-α), which are commonly elevated after cartilage injury. By suppressing MMPs (such as MMP-13) and aggrecanases (like ADAMTS-5), MSC-derived factors help prevent further ECM degradation.
Additionally, MSCs promote angiogenesis by releasing VEGF and basic fibroblast growth factor (bFGF), which enhance the formation of new blood vessels in the surrounding tissues, improving nutrient supply and waste removal at the injury site. This is particularly important in cartilage, where the lack of vascularization limits nutrient diffusion. Angiogenesis indirectly supports the survival and function of transplanted or endogenous MSCs, creating a more favorable microenvironment for tissue regeneration. The combination of these paracrine effects and direct differentiation into chondrocytes forms the backbone of MSC-mediated cartilage repair.
At the molecular level, MSC activity is tightly regulated by various signaling networks, including the Notch, Hedgehog, and Hippo pathways, which balance MSC proliferation, differentiation, and paracrine function. These pathways interact with growth factor signaling to either promote or inhibit chondrogenesis, depending on the context. For instance, Notch signaling tends to inhibit excessive differentiation and is involved in maintaining MSCs in a more undifferentiated state until the proper cues are present. Hedgehog signaling, through Sonic hedgehog (Shh), can enhance MSC proliferation while influencing their differentiation towards either cartilage or bone, depending on the strength and duration of the signal.
In conclusion, MSCs contribute to cartilage repair via chemotactic recruitment, differentiation into chondrocytes, and the secretion of paracrine factors that modulate inflammation, promote ECM synthesis, and enhance tissue regeneration. The orchestration of molecular signals such as TGF-β/Smad, PI3K/Akt, NF-κB, and exosomal miRNAs highlights the sophisticated network that governs MSC behavior during cartilage repair, making them a powerful tool for regenerative therapies. Understanding these molecular pathways is essential for optimizing the therapeutic potential of MSCs in treating cartilage injuries and degenerative diseases like osteoarthritis.
Angiogenesis and Subchondral Bone Response
The interaction between articular cartilage and the subchondral bone is regulated by complex molecular pathways, influencing both tissue repair and degeneration, especially in the context of angiogenesis and subchondral bone remodeling. VEGF (vascular endothelial growth factor) is a key mediator of angiogenesis in the subchondral bone. VEGF, produced by osteoblasts, chondrocytes, and synovial cells, binds to its receptor VEGFR-2 on endothelial cells, triggering a signaling cascade that includes the activation of PI3K/Akt, MAPK, and ERK1/2 pathways. This cascade promotes endothelial cell proliferation, migration, and the formation of new blood vessels. However, excessive angiogenesis leads to increased vascular permeability and allows for the infiltration of inflammatory cells such as macrophages and neutrophils. These cells secrete pro-inflammatory cytokines like IL-1β and TNF-α, which activate catabolic pathways in chondrocytes through the NF-κB signaling pathway, leading to upregulation of MMPs (MMP-13) and ADAMTS-5. These enzymes degrade key components of the cartilage extracellular matrix, such as type II collagen and aggrecan, contributing to the progressive breakdown of cartilage.
In addition to VEGF, other angiogenic factors like fibroblast growth factor (FGF-2) and hypoxia-inducible factor-1 alpha (HIF-1α) play significant roles in promoting vascularization in the subchondral bone. HIF-1α is particularly crucial in hypoxic conditions, which are common in osteoarthritic joints, as it induces the expression of VEGF and enhances the angiogenic response. While this increased vascularization can initially support tissue repair by providing necessary nutrients and oxygen, chronic hypoxia and persistent VEGF signaling contribute to pathological changes, including the formation of abnormal blood vessels, which further exacerbates inflammation and matrix degradation.
Subchondral bone remodeling in response to cartilage injury involves tightly regulated molecular processes, balancing bone resorption and formation. Osteoclasts, responsible for bone resorption, are activated by RANKL (receptor activator of nuclear factor kappa-B ligand), which binds to its receptor RANK on osteoclast precursors, initiating the NF-κB and JNK pathways that lead to osteoclast differentiation and activity. In contrast, osteoblasts, the cells responsible for bone formation, are regulated by Wnt/β-catenin signaling, which promotes osteoblast proliferation and activity. In the context of cartilage injury, this balance is disrupted, often favoring subchondral bone sclerosis, where increased bone formation leads to a stiffer, denser bone matrix. This increased density, driven by increased Wnt signaling and reduced sclerostin (a Wnt pathway inhibitor), results in the formation of osteophytes and altered mechanical loading on the overlying cartilage. The increased mechanical stress transmitted to the cartilage accelerates its degradation, as chondrocytes respond to mechanical overload by further upregulating catabolic enzymes like MMP-13 and ADAMTS-4, compounding the loss of ECM integrity.
Additionally, the TGF-β pathway is critical in both subchondral bone remodeling and cartilage degradation. TGF-β, released from the bone matrix during remodeling, acts through the Smad signaling pathway to regulate osteoblast and chondrocyte activity. In the bone, TGF-β promotes osteoblast differentiation and matrix production, but in cartilage, it can enhance the expression of proteases like MMP-13 and ADAMTS, contributing to ECM breakdown. The bidirectional signaling between cartilage and subchondral bone, mediated by molecules like TGF-β, VEGF, and RANKL, emphasizes the integrated nature of joint tissue health.
Moreover, changes in subchondral bone vasculature and the influx of inflammatory mediators also influence subchondral bone marrow lesions, which are commonly seen in conditions like osteoarthritis. These lesions are associated with increased bone turnover, which further disrupts the mechanical environment and perpetuates the cycle of cartilage degeneration. Bone morphogenetic proteins (BMPs), particularly BMP-2, are involved in the abnormal bone formation seen in osteoarthritic joints, enhancing osteoblast activity and contributing to osteophyte development. BMP signaling can also influence chondrocyte hypertrophy, driving the pathological differentiation of chondrocytes into a hypertrophic phenotype, which is more prone to producing type X collagen and contributing to cartilage calcification.
In summary, the molecular interplay between angiogenesis, inflammation, and subchondral bone remodeling is critical in the pathophysiology of cartilage injuries. VEGF, TGF-β, Wnt/β-catenin, and RANKL signaling pathways regulate the balance between repair and degeneration in both cartilage and subchondral bone, with disruptions leading to chronic joint diseases like osteoarthritis. Understanding these molecular mechanisms provides insights into potential therapeutic targets aimed at modulating both cartilage and subchondral bone to enhance joint repair and prevent further degeneration.
Mechanotransduction and Mechanical Loading
Mechanical loading profoundly influences cartilage homeostasis and repair by engaging a network of mechanotransduction pathways that convert physical stimuli into biochemical signals, directing cellular responses like proliferation, ECM synthesis, and repair. Central to this process is integrin signaling, where integrins, primarily α5β1 and αVβ3, bind to ECM components such as fibronectin and collagen. Upon mechanical stress, these integrins cluster and recruit focal adhesion kinase (FAK), which activates downstream signaling pathways including PI3K/Akt, RhoA/ROCK, and MAPK/ERK. FAK phosphorylation triggers actin cytoskeleton remodeling and promotes cell survival, adhesion, and migration by activating Akt. This pathway also regulates mTOR, which drives protein synthesis and cell growth, ensuring that chondrocytes produce the necessary ECM components like type II collagen and aggrecan. Through this mechanotransduction, integrins help maintain cartilage structure and stimulate anabolic processes that counteract degeneration.
Ion channels, specifically stretch-activated calcium channels (SACs), are key players in mechanotransduction. Mechanical loading induces the opening of SACs, leading to an influx of Ca²⁺ into chondrocytes. This rise in intracellular calcium levels activates calmodulin, which binds to and activates calmodulin-dependent kinase (CaMK) and calcineurin, initiating a cascade that includes the NFAT (nuclear factor of activated T-cells) signaling pathway. NFAT translocates to the nucleus, where it regulates gene expression, including the upregulation of ECM components and survival factors. Calcium signaling also activates the PKC (protein kinase C) pathway, which modulates chondrocyte metabolism, proliferation, and matrix production. The CaMK and calcineurin pathways are particularly important for gene transcription that supports anabolic functions in response to mechanical stress, driving the synthesis of proteoglycans and collagen, which reinforce the cartilage matrix.
The MAPK pathway, activated by mechanical stress, plays a pivotal role in coordinating the cellular response to mechanical loading. ERK1/2 activation promotes chondrocyte proliferation and increases ECM synthesis by upregulating type II collagen and aggrecan production. ERK1/2 phosphorylates transcription factors like Elk-1 and CREB, which bind to the promoters of matrix-related genes, promoting the synthesis of structural proteins essential for cartilage integrity. In parallel, p38 MAPK is activated in response to mechanical stress and contributes to both repair and degradation processes. Under appropriate mechanical loading, p38 MAPK promotes ECM synthesis by regulating the expression of SOX9, a transcription factor critical for chondrogenesis and the production of cartilage-specific ECM proteins. However, in cases of excessive or abnormal loading, p38 MAPK can also upregulate matrix metalloproteinases (MMPs) like MMP-13, which degrade type II collagen and aggrecan, accelerating cartilage breakdown.
Additionally, mechanical stress can activate the YAP/TAZ (Yes-associated protein/transcriptional coactivator with PDZ-binding motif) pathway, a crucial regulator of cellular mechanotransduction. YAP/TAZ responds to changes in cell tension and ECM stiffness by relocating to the nucleus, where it regulates gene expression related to ECM synthesis, cell proliferation, and survival. In cartilage, YAP/TAZ activation promotes anabolic activities, enhancing the production of collagen and proteoglycans necessary for cartilage maintenance and repair. Conversely, dysregulation of this pathway under excessive mechanical loading can lead to chondrocyte hypertrophy and ECM degradation, contributing to conditions such as osteoarthritis.
Mechanical loading also influences ion channel signaling, particularly through TRPV4 and Piezo1/2 channels. TRPV4 is a mechanosensitive ion channel that responds to osmotic and mechanical changes, allowing the influx of Ca²⁺. Activation of TRPV4 stimulates the production of SOX9 and matrix proteins, thereby promoting ECM synthesis. However, prolonged or excessive activation of these channels may lead to oxidative stress and catabolic enzyme release, contributing to cartilage breakdown. Piezo1/2 channels, similarly, respond to mechanical deformation and mediate calcium influx, influencing chondrocyte behavior. The activation of Piezo channels modulates the expression of matrix genes and regulates cellular responses to mechanical compression, helping balance anabolic and catabolic activities in the cartilage.
Mechanical loading also impacts the balance of TGF-β signaling, which is integral to cartilage repair. Under appropriate mechanical stress, TGF-β signals through the Smad2/3 pathway, promoting chondrocyte differentiation and enhancing type II collagen synthesis. This anabolic effect helps maintain cartilage integrity and supports tissue regeneration. However, excessive mechanical loading can activate non-canonical TGF-β pathways, such as the p38 MAPK and RhoA/ROCK pathways, which can lead to chondrocyte hypertrophy and increased expression of type X collagen, contributing to cartilage calcification and degeneration.
In summary, mechanical loading exerts its effects on cartilage through a complex network of mechanotransduction pathways, including integrin/FAK, ion channel-mediated calcium signaling, MAPK, YAP/TAZ, and TGF-β signaling. These pathways regulate key processes such as chondrocyte proliferation, differentiation, and ECM synthesis, which are critical for maintaining cartilage homeostasis and promoting repair. However, excessive or abnormal mechanical stress can shift these pathways towards catabolic activities, accelerating cartilage degeneration. Understanding these molecular mechanisms is crucial for developing therapeutic interventions that harness mechanical cues to enhance cartilage repair and prevent degenerative joint diseases such as osteoarthritis.
Table 4.
This table categorizes the molecular biology and biomechanical functionality of articular cartilage, detailing its structure, cellular responses, and repair mechanisms. It provides insights into cartilage composition, injury types, and the molecular processes involved in cartilage degeneration and regeneration.
Table 4.
This table categorizes the molecular biology and biomechanical functionality of articular cartilage, detailing its structure, cellular responses, and repair mechanisms. It provides insights into cartilage composition, injury types, and the molecular processes involved in cartilage degeneration and regeneration.
| Category | Details | Processes Involved | Key Molecular/Cellular Players |
| Articular Cartilage Structure | Articular cartilage is found on the surfaces of bones in synovial joints, providing frictionless movement and efficient load distribution. It consists of chondrocytes embedded in an extracellular matrix (ECM) composed of type II collagen and proteoglycans like aggrecan. The ECM’s unique properties enable the cartilage to resist mechanical forces such as compression and shear stress. | The ECM consists of type II collagen for tensile strength and proteoglycans, particularly aggrecan, to retain water for compressive resistance. Cartilage is stratified into superficial, middle, and deep zones, each with varying collagen fiber orientation and chondrocyte density, which contribute to the tissue’s mechanical functionality. | Chondrocytes maintain the ECM by synthesizing collagen and proteoglycans. Type II collagen provides tensile strength, while aggrecan retains water. Glycosaminoglycans (GAGs), like chondroitin sulfate, enable water retention. Superficial zone chondrocytes handle shear forces, while deep zone chondrocytes remodel the ECM. |
| Limited Regenerative Capacity | Articular cartilage has limited healing ability due to its avascularity, meaning it lacks a direct blood supply. Nutrient delivery and waste removal rely on diffusion from synovial fluid. Injuries lead to insufficient chondrocyte proliferation and slow synthesis of ECM components, often resulting in incomplete repair or progressive degeneration, such as in osteoarthritis (OA). | Cartilage injuries trigger a slow repair response due to limited availability of growth factors and progenitor cells. Matrix metalloproteinases (MMPs) degrade the ECM, particularly collagen and aggrecan, and if left unchecked, can lead to further breakdown. Inflammatory cytokines like IL-1β and TNF-α exacerbate ECM degradation and inhibit new matrix synthesis. | MMPs degrade ECM components, while tissue inhibitors of metalloproteinases (TIMPs) regulate this activity. IL-1β and TNF-α upregulate MMPs and inhibit matrix synthesis, worsening tissue degeneration. Chondrocytes have limited ability to proliferate and migrate, further limiting repair. Synovial fluid is critical for nutrient delivery in the absence of blood supply. |
| Cartilage Injury Types | Cartilage injuries are either focal (localized, often caused by acute trauma) or diffuse (widespread degeneration due to conditions like osteoarthritis). Focal injuries are common in athletes and can evolve into broader degeneration if untreated. Diffuse injuries, associated with OA, result from chronic wear and tear, leading to progressive loss of cartilage and joint dysfunction. | Focal injuries arise from mechanical trauma and often lead to cartilage defects that expose subchondral bone, causing joint friction and pain. Diffuse injuries like OA involve pro-inflammatory cytokines (IL-1β, TNF-α) that promote the breakdown of ECM by MMPs and aggrecanases, leading to joint degeneration. Reactive oxygen species (ROS) contribute to chondrocyte apoptosis and matrix damage. | Pro-inflammatory cytokines (IL-1β, TNF-α) are key drivers of inflammation and cartilage degradation. MMPs and aggrecanases (ADAMTS-4, ADAMTS-5) degrade type II collagen and aggrecan. Reactive oxygen species (ROS) contribute to oxidative stress and further tissue damage. Chondrocytes experience apoptosis and reduced ability to maintain ECM in injured cartilage. |
| Inflammatory Response to Injury | Cartilage injury triggers an inflammatory response, with the release of cytokines (IL-1β, TNF-α) that initiate tissue repair but can exacerbate damage if prolonged. This inflammation activates catabolic pathways that degrade the ECM, especially type II collagen and aggrecan, leading to tissue degeneration in conditions like osteoarthritis. | Inflammatory cytokines activate signaling pathways such as NF-κB and MAPK, which upregulate the production of MMPs and aggrecanases, driving ECM degradation. ROS and nitric oxide (NO) further damage chondrocytes and impair ECM synthesis. Excessive degradation disrupts chondrocyte mechanotransduction, leading to a loss of mechanical cues that regulate ECM production and survival. | Cytokines (IL-1β, TNF-α) upregulate MMPs and aggrecanases, promoting matrix breakdown. NF-κB and MAPK pathways mediate the inflammatory response and ECM degradation. ROS and NO drive oxidative stress and chondrocyte apoptosis, exacerbating tissue damage. Chondrocytes lose mechanotransduction abilities, further impairing ECM repair. |
| Extracellular Matrix Remodeling | Cartilage repair involves balancing ECM degradation and synthesis. During injury, MMPs and aggrecanases degrade key ECM components, but this is counteracted by anabolic signaling pathways like TGF-β, IGF-1, and BMPs, which promote matrix synthesis. However, prolonged inflammation and oxidative stress tip this balance towards degradation, leading to progressive cartilage breakdown. | MMPs and aggrecanases degrade type II collagen and aggrecan during injury, while growth factors like TGF-β, IGF-1, and BMPs promote ECM synthesis. The Smad signaling pathway, activated by TGF-β, stimulates the production of cartilage ECM components. Oxidative stress and excessive inflammation can impair ECM synthesis, pushing cartilage toward degeneration. | MMPs and ADAMTS enzymes drive ECM degradation. TGF-β, IGF-1, and BMPs promote ECM repair through anabolic pathways like Smad signaling. ROS and NO impair chondrocyte function and matrix synthesis. Chondrocytes produce matrix components in response to growth factors but are limited by chronic inflammation and oxidative damage. |
| Therapeutic Strategies | Recent advances in cartilage repair focus on overcoming its limited regenerative capacity. Tissue engineering with scaffolds, stem cell therapies (particularly mesenchymal stem cells (MSCs)), and growth factor delivery (like TGF-β, IGF-1) are being explored to enhance cartilage regeneration and promote ECM synthesis. | Stem cell therapies aim to differentiate MSCs into chondrocytes that can synthesize ECM components. Scaffolds provide a framework for cells to regenerate tissue. Growth factors like TGF-β and IGF-1 stimulate chondrocyte activity, enhancing collagen and proteoglycan synthesis. MSC-derived exosomes and growth factors have paracrine effects that modulate inflammation and repair. | Mesenchymal stem cells (MSCs) differentiate into chondrocytes and secrete growth factors like TGF-β and IGF-1 to promote repair. Scaffolds provide structural support for tissue regeneration. MSCs also secrete exosomes containing anti-inflammatory molecules, aiding in cartilage healing. Growth factors enhance ECM synthesis and chondrocyte activity. |
| Mechanotransduction in Cartilage | Cartilage cells respond to mechanical loading through mechanotransduction pathways, where external forces are converted into biochemical signals that regulate ECM synthesis, repair, and cell survival. Integrins, ion channels, and mechanosensitive signaling pathways like FAK, MAPK, and YAP/TAZ play critical roles in directing cellular responses to mechanical stimuli. | Integrins activate FAK and PI3K/Akt signaling in response to mechanical stress, promoting cell survival and ECM synthesis. Ion channels like TRPV4 mediate calcium influx, triggering signaling pathways that regulate matrix production. MAPK and YAP/TAZ pathways also respond to mechanical cues, driving anabolic processes that maintain cartilage structure. Excessive mechanical stress can shift these pathways towards catabolic activities, promoting ECM degradation. | Integrins activate mechanotransduction pathways like FAK and PI3K/Akt, which regulate ECM synthesis and survival. TRPV4 and Piezo channels mediate calcium signaling in response to mechanical loading. MAPK and YAP/TAZ pathways influence chondrocyte activity in response to mechanical stress. Excessive loading can activate MMPs and drive cartilage degeneration. |
Cellular Responses to Knee Joint Injuries
Knee joint injuries, whether traumatic or degenerative, involve a wide range of structural damage to key components such as the ligaments, menisci, and articular cartilage, triggering complex cellular responses aimed at repair and recovery. These responses involve interactions among various cell types—including chondrocytes, fibroblasts, synoviocytes, and immune cells—along with intricate signaling pathways and the remodeling of the extracellular matrix (ECM). Understanding these molecular mechanisms is essential for designing targeted therapies that promote tissue regeneration and optimize rehabilitation outcomes.
In the case of ligament injuries such as anterior cruciate ligament (ACL) tears, the inflammatory response is one of the earliest reactions. Damaged tissue releases damage-associated molecular patterns (DAMPs), which activate immune cells such as neutrophils and macrophages. These immune cells release pro-inflammatory cytokines like tumor necrosis factor-alpha (TNF-α), interleukin-1 beta (IL-1β), and interleukin-6 (IL-6), which further recruit immune cells to the injury site and initiate the release of matrix metalloproteinases (MMPs) that degrade damaged ECM components. Macrophages, which shift from an inflammatory M1 phenotype to a reparative M2 phenotype, play a central role in coordinating the transition from inflammation to tissue repair by secreting growth factors such as transforming growth factor-beta (TGF-β) and platelet-derived growth factor (PDGF). These growth factors stimulate the activity of fibroblasts, which synthesize collagen and other ECM proteins necessary for ligament repair. TGF-β, in particular, activates the Smad signaling pathway, promoting the transcription of genes involved in collagen production and ECM remodeling.
For meniscal injuries, particularly tears, the response varies depending on the vascularization of the affected region. The meniscus is divided into three zones: the red-red zone (highly vascularized), the red-white zone (moderately vascularized), and the white-white zone (avascular). Meniscal tears in the vascularized regions tend to heal better due to an enhanced inflammatory response and greater recruitment of mesenchymal stem cells (MSCs), fibroblasts, and chondrocytes. In response to meniscal injury, MSCs are recruited to the site through chemotactic signals such as stromal cell-derived factor-1 (SDF-1) and VEGF. Once at the injury site, MSCs differentiate into chondrocytes or fibrochondrocytes under the influence of growth factors like TGF-β, bone morphogenetic proteins (BMPs), and insulin-like growth factor-1 (IGF-1). These cells contribute to ECM repair by synthesizing type I collagen in the vascularized regions and type II collagen in the avascular regions, restoring some of the meniscus’s biomechanical properties.
Articular cartilage injuries present a unique challenge due to the avascular nature of cartilage, limiting the influx of immune cells and nutrients necessary for repair. Following cartilage damage, chondrocytes—the main cell type in cartilage—undergo changes in phenotype, shifting from a quiescent state to an active state where they attempt to repair the damaged ECM. This involves upregulating the production of type II collagen and aggrecan, as well as proteoglycans that are essential for cartilage resilience. However, injured chondrocytes also produce catabolic enzymes like MMP-13 and ADAMTS-5, which degrade ECM components, exacerbating cartilage damage if not tightly regulated. Additionally, hypoxia in the cartilage microenvironment activates hypoxia-inducible factor-1 alpha (HIF-1α), which modulates both anabolic and catabolic processes, influencing the balance between matrix synthesis and degradation. VEGF, produced in response to hypoxia, promotes angiogenesis in the subchondral bone, which, while improving nutrient supply, can also lead to pathological changes in the bone and further cartilage degeneration.
In all these injuries, mechanical loading plays a critical role in directing the healing process. Mechanotransduction pathways, such as integrin-mediated signaling and ion channel activation, convert mechanical forces into biochemical signals that influence cell behavior. Integrins on the surface of chondrocytes and fibroblasts interact with ECM components, activating focal adhesion kinase (FAK) and downstream pathways like MAPK and PI3K/Akt, which regulate cell proliferation, differentiation, and ECM production. Stretch-activated calcium channels also respond to mechanical stress by increasing intracellular calcium levels, activating the calcineurin/NFAT pathway, which regulates the expression of genes involved in ECM synthesis and tissue repair. Proper mechanical stimulation through rehabilitation exercises can promote ECM synthesis and tissue regeneration, whereas excessive or abnormal loading can exacerbate tissue breakdown.
Extracellular matrix remodeling is a central aspect of the repair process in knee injuries. MMPs and aggrecanases (such as ADAMTS-4 and ADAMTS-5) are upregulated in response to injury and inflammation, degrading damaged ECM components to allow for new matrix synthesis. Tissue inhibitors of metalloproteinases (TIMPs) regulate MMP activity to prevent excessive ECM degradation and ensure a controlled remodeling process. Growth factors like TGF-β, IGF-1, and fibroblast growth factor (FGF) stimulate fibroblasts and chondrocytes to synthesize new ECM, restoring tissue integrity.
In summary, the cellular and molecular responses to knee joint injuries are multifaceted and involve a complex interplay of immune cells, chondrocytes, fibroblasts, MSCs, and signaling molecules that regulate inflammation, tissue repair, and ECM remodeling. These processes are influenced by the vascularity of the injured tissue, the mechanical environment, and the balance between anabolic and catabolic activities. Understanding these mechanisms is crucial for developing targeted therapeutic strategies that enhance tissue repair, minimize degeneration, and improve long-term joint function.
Inflammation and Immune Response
Following knee joint injury, the acute inflammatory response is governed by a tightly regulated cascade of molecular events, involving multiple cell types, cytokines, and signaling pathways. The vascular response is the first key event, where damage to blood vessels causes endothelial cells to upregulate the expression of adhesion molecules such as E-selectin, P-selectin, and ICAM-1 (intercellular adhesion molecule-1). These adhesion molecules facilitate the rolling, adhesion, and extravasation of leukocytes from the bloodstream to the injured site. The release of VEGF from endothelial cells, macrophages, and platelets not only increases vascular permeability but also stimulates the formation of new blood vessels through angiogenesis. VEGF binds to its receptor, VEGFR-2, activating downstream pathways like PI3K/Akt and MAPK, promoting endothelial cell migration and proliferation to restore blood flow to the damaged area.
The infiltration of neutrophils, one of the earliest immune responses, is driven by the release of chemokines such as CXCL8 (IL-8), which bind to CXCR1 and CXCR2 receptors on neutrophils. Upon activation, neutrophils release reactive oxygen species (ROS) via NADPH oxidase and proteolytic enzymes like MMP-8 and neutrophil elastase, which degrade damaged ECM proteins. Neutrophils also secrete TNF-α and IL-1β, which further amplify the inflammatory response by activating the NF-κB signaling pathway in nearby cells. This induces the production of additional cytokines, chemokines, and MMPs, creating a feed-forward loop that sustains inflammation. Neutrophil apoptosis is a critical regulatory mechanism, as their timely clearance by macrophages through efferocytosis prevents the excessive release of harmful enzymes and limits tissue damage.
Macrophage activation follows the neutrophil phase, with M1 macrophages initially driving inflammation. M1 macrophages are activated by interferon-gamma (IFN-γ) and lipopolysaccharides (LPS), and they produce pro-inflammatory cytokines such as IL-6, IL-1β, and TNF-α, which further stimulate the inflammatory cascade by activating JAK/STAT and MAPK pathways. Additionally, M1 macrophages release MMP-9 and ROS, contributing to the breakdown of damaged matrix and the clearance of necrotic tissue. Over time, the local environment shifts, and M2 macrophages emerge in response to signals like IL-4, IL-10, and TGF-β. M2 macrophages secrete anti-inflammatory cytokines and growth factors, including VEGF and TGF-β, which activate Smad-dependent pathways, promoting ECM synthesis, collagen deposition, and tissue remodeling. They also upregulate arginase-1, an enzyme involved in the production of ornithine, a precursor for collagen synthesis.
The recruitment of lymphocytes, particularly T cells, introduces further regulatory complexity. T helper 1 (Th1) cells contribute to maintaining the inflammatory state by releasing IFN-γ, which activates macrophages and perpetuates the M1 phenotype. Conversely, T helper 2 (Th2) cells produce IL-4 and IL-13, which support the transition to the M2 macrophage phenotype, fostering repair and anti-inflammatory processes. Regulatory T cells (Tregs) play a key role in dampening excessive inflammation by releasing IL-10 and TGF-β, which inhibit the activity of Th1 cells and reduce the production of pro-inflammatory cytokines. Tregs suppress NF-κB activation in macrophages and other immune cells, helping to resolve inflammation and promote tissue repair.
At the molecular level, the regulation of matrix metalloproteinases (MMPs) is critical for controlled ECM degradation. MMP-13, upregulated by IL-1β and TNF-α, degrades type II collagen, a major structural component of cartilage. This ECM degradation is balanced by the action of tissue inhibitors of metalloproteinases (TIMPs), which are upregulated during the resolution phase of inflammation. TIMP-1 and TIMP-2 bind to MMPs and inhibit their proteolytic activity, preventing excessive matrix degradation and promoting ECM stability. The balance between MMPs and TIMPs is essential for effective tissue remodeling and repair.
Mechanical signals also play an important role in the injury response. Integrin-mediated mechanotransduction helps cells sense mechanical stress in the injured tissue, triggering downstream signaling cascades that regulate cell survival, proliferation, and ECM synthesis. Integrins such as αvβ3 bind to ECM components like fibronectin and collagen, activating focal adhesion kinase (FAK) and initiating pathways like PI3K/Akt and MAPK/ERK. These pathways promote cell migration, cytoskeletal remodeling, and matrix synthesis, contributing to tissue repair.
In summary, the acute inflammatory response following knee injury involves a highly coordinated interplay of vascular responses, neutrophil and macrophage activity, lymphocyte recruitment, and matrix remodeling. This process is regulated by complex molecular signaling pathways, including NF-κB, MAPK, PI3K/Akt, and Smad-dependent pathways, which balance inflammation, tissue repair, and ECM remodeling. Understanding these molecular mechanisms is critical for developing targeted therapies that modulate the inflammatory response, promote tissue regeneration, and prevent chronic joint damage.
Extracellular Matrix (ECM) Remodeling
The extracellular matrix (ECM) in knee joint tissues is a dynamic structure that provides physical support and regulates cellular processes like adhesion, migration, proliferation, and differentiation. After injury, ECM remodeling is essential for repair, but it must be tightly controlled to prevent tissue degeneration. Matrix metalloproteinases (MMPs), including MMP-1 (collagenase) and MMP-13 (collagenase-3), degrade collagen, while MMP-3 (stromelysin-1) breaks down other ECM components and activates additional MMPs, amplifying their degradative effects. These enzymes are upregulated in response to pro-inflammatory cytokines such as TNF-α and IL-1β, which activate intracellular signaling pathways like NF-κB and MAPK, driving the transcription of MMP genes. In addition to cytokine signaling, mechanical stress from injury further enhances MMP activity through integrin-mediated mechanotransduction, which feeds into these same pathways, leading to further ECM breakdown.
Excessive MMP activity, if unchecked, compromises the ECM’s structural integrity, making tissue repair difficult. The degradation of ECM components also releases damage-associated molecular patterns (DAMPs), such as fragmented collagen and proteoglycans. These DAMPs activate pattern recognition receptors (PRRs), like Toll-like receptors (TLRs), on immune cells and chondrocytes, promoting an inflammatory response that perpetuates the cycle of tissue degradation and inflammation. This feedback loop accelerates cartilage and joint destruction, especially in chronic conditions like osteoarthritis, where MMP overactivity leads to progressive cartilage loss.
Tissue inhibitors of metalloproteinases (TIMPs) play a critical regulatory role by binding to and inhibiting active MMPs. TIMP-1 and TIMP-2, for instance, form complexes with MMPs, preventing excessive ECM degradation. The balance between MMPs and TIMPs is crucial for controlled ECM remodeling. Growth factors and cytokines, such as TGF-β and IL-10, regulate TIMP expression, ensuring that tissue repair processes are not overwhelmed by excessive proteolysis. Disruption of this balance in favor of MMP activity leads to uncontrolled ECM degradation, impaired healing, and tissue instability. However, sufficient TIMP activity allows for proper ECM turnover, facilitating regeneration and the replacement of damaged matrix components with new ones, thereby maintaining tissue integrity.
Growth factors like TGF-β, fibroblast growth factor (FGF), and platelet-derived growth factor (PDGF) are central to stimulating ECM synthesis and promoting tissue repair. TGF-β, via the SMAD signaling pathway, induces the production of type II collagen and aggrecan, essential components for cartilage repair and restoration of the joint’s biomechanical properties. FGF and PDGF, through the PI3K/AKT pathway, stimulate the proliferation of fibroblasts and chondrocytes, further enhancing ECM production. These growth factors also regulate the expression of MMPs and TIMPs, ensuring a balance between ECM breakdown and synthesis. Proper growth factor signaling orchestrates the repair process by promoting cell proliferation, differentiation, and matrix production, critical for functional recovery. Dysregulation of these pathways can result in incomplete or disordered tissue regeneration, leading to long-term joint dysfunction and increased susceptibility to degenerative diseases. Understanding these molecular interactions is crucial for developing targeted therapies to promote effective tissue repair and prevent further degeneration.
Chondrocyte Responses
Chondrocytes, as the primary cells maintaining cartilage integrity, exhibit intricate molecular responses to injury, driven by various signaling pathways aimed at managing tissue repair. In cases of chondrocyte apoptosis and necrosis, injury can activate different death mechanisms. Necrosis typically follows severe mechanical damage, leading to uncontrolled cellular content release, which intensifies inflammation through the activation of damage-associated molecular patterns (DAMPs). These DAMPs, such as HMGB1 and S100 proteins, interact with Toll-like receptors (TLRs) on immune cells, further amplifying the inflammatory response. In contrast, apoptosis involves regulated cell death mediated by signaling cascades such as the intrinsic (mitochondrial) pathway and the extrinsic (death receptor) pathway. The intrinsic pathway is controlled by the Bcl-2 family of proteins, which regulate mitochondrial membrane permeability. This leads to the release of cytochrome c, triggering the formation of the apoptosome and the activation of caspase-9, which then activates downstream caspases, such as caspase-3, ultimately leading to apoptosis. The extrinsic pathway, involving Fas and TNF receptors, activates caspase-8, which directly cleaves and activates effector caspases. The balance between apoptotic triggers and survival pathways, such as the PI3K/AKT pathway, determines chondrocyte fate. PI3K/AKT promotes cell survival by inhibiting pro-apoptotic proteins like Bad and by activating survival-promoting proteins like mTOR and NF-κB.
Chondrocyte proliferation and clustering in response to injury are driven by growth factors such as IGF-1, TGF-β, and FGF, which bind to their respective receptors and activate downstream signaling pathways like MAPK/ERK and PI3K/AKT. These pathways promote the proliferation of chondrocytes to repair the damaged tissue. However, the clustering of chondrocytes can alter the cartilage’s structural and mechanical properties. The transition from a homogeneously distributed cell population to clustered chondrocytes can lead to the formation of fibrocartilage, which lacks the mechanical resilience of hyaline cartilage due to its increased production of type I collagen. This process is modulated by mechanical signals through mechanotransduction, where integrins on chondrocyte surfaces activate focal adhesion kinase (FAK) and YAP/TAZ signaling, linking mechanical stimuli to changes in gene expression and ECM production. Mechanical loading enhances chondrocyte proliferation and matrix synthesis, but unregulated mechanical stress can lead to over-clustering and poor cartilage repair outcomes.
Phenotypic modulation of chondrocytes involves a shift from a matrix-producing phenotype to a more fibroblastic state, driven by inflammatory cytokines like IL-1β and TNF-α. This modulation results in the increased production of type I collagen and other fibrous matrix components, leading to the formation of mechanically inferior fibrocartilage. Signaling pathways like Wnt/β-catenin and Hedgehog play critical roles in controlling chondrocyte differentiation and phenotypic stability. For instance, Wnt signaling, through β-catenin stabilization, promotes chondrocyte hypertrophy and matrix degradation, while Hedgehog signaling regulates the expression of genes involved in cartilage-specific matrix production. Dysregulation of these pathways contributes to the loss of the hyaline cartilage phenotype and impairs tissue function.
Autophagy, a key survival mechanism under stress conditions, is regulated in chondrocytes by the mTOR pathway. Autophagy involves the formation of autophagosomes that engulf damaged organelles and proteins, which are then degraded by lysosomes. Under stress conditions, such as nutrient deprivation or oxidative stress, AMP-activated protein kinase (AMPK) inhibits mTOR, promoting autophagy and allowing chondrocytes to maintain homeostasis and avoid apoptosis. Autophagy is also linked to the secretion of extracellular vesicles, including exosomes, which carry signaling molecules like microRNAs (miRNAs) and cytokines that modulate inflammation and repair in the surrounding tissue. For example, miR-140, secreted in extracellular vesicles, has been shown to suppress MMP expression and support ECM preservation, thus playing a protective role in cartilage repair.
In addition to these pathways, chondrocyte metabolism shifts in response to injury and inflammation. Hypoxia-inducible factor-1 alpha (HIF-1α) is activated in the hypoxic environment of cartilage, particularly following injury, and regulates the expression of glycolytic enzymes to adapt to low oxygen conditions. HIF-1α also promotes the expression of COL2A1 (type II collagen) and ACAN (aggrecan), which are essential for maintaining the cartilage matrix. Additionally, oxidative stress, through the production of reactive oxygen species (ROS), can exacerbate chondrocyte apoptosis and matrix degradation. ROS activate signaling pathways such as JNK and p38 MAPK, which contribute to the upregulation of catabolic enzymes like MMPs and aggrecanases, further damaging the ECM.
Overall, the molecular responses of chondrocytes to injury are governed by a balance of pro-survival and pro-death signals, as well as the regulation of ECM remodeling and phenotypic stability. These complex molecular mechanisms are critical for maintaining cartilage integrity and guiding the repair process following injury. Understanding these pathways provides insight into potential therapeutic targets for promoting cartilage regeneration and preventing degenerative joint diseases.
Synoviocyte Activation
Synoviocytes, particularly type A and type B, play critical roles in the molecular orchestration of inflammation and repair in the knee joint after injury. Type A synoviocytes, which are macrophage-like cells, are activated through pattern recognition receptors (PRRs), such as Toll-like receptors (TLRs), upon encountering damage-associated molecular patterns (DAMPs) from necrotic cells and ECM fragments. This activation leads to the production of pro-inflammatory cytokines like TNF-α, IL-1, and IL-6 via the NF-κB and MAPK signaling pathways. These cytokines further enhance the recruitment of immune cells by upregulating cell adhesion molecules (CAMs) on the endothelial cells lining blood vessels, which facilitate leukocyte extravasation into the synovium. Additionally, type A synoviocytes engage in antigen presentation through major histocompatibility complex (MHC) class II molecules, linking the innate and adaptive immune responses by activating T cells, which contributes to prolonged inflammation if not properly regulated.
Type B synoviocytes, fibroblast-like cells, are crucial for maintaining synovial fluid composition and producing ECM components like collagen and hyaluronan. These cells respond to injury by upregulating growth factors such as TGF-β, FGF, and PDGF, which signal through pathways like SMAD (for TGF-β) and PI3K/AKT (for FGF and PDGF). This signaling promotes the synthesis of key ECM components and lubricating molecules like hyaluronan, which is critical for joint function by providing viscoelastic properties to the synovial fluid. Moreover, type B synoviocytes produce pro-inflammatory cytokines, including IL-1, IL-6, and TNF-α, that not only propagate inflammation but also induce the expression of matrix metalloproteinases (MMPs) such as MMP-1, MMP-3, and MMP-13. These MMPs degrade collagen and other ECM components, facilitating tissue remodeling but also contributing to cartilage breakdown if not properly controlled.
The production of these cytokines and chemokines by synoviocytes is regulated by complex feedback mechanisms. For example, TGF-β and IL-10, which are anti-inflammatory cytokines, work through the SMAD and STAT3 pathways, respectively, to suppress the excessive inflammatory responses driven by NF-κB and AP-1. TGF-β also promotes fibroblast proliferation and ECM synthesis, balancing the destructive effects of MMPs and supporting tissue regeneration. However, dysregulation in these signaling pathways, particularly chronic activation of NF-κB, can lead to the persistent production of pro-inflammatory mediators and MMPs, resulting in joint tissue degradation, a hallmark of conditions like osteoarthritis and rheumatoid arthritis.
In addition to cytokine regulation, synoviocyte autophagy has been observed to play a role in controlling inflammation and tissue damage. Autophagy is mediated by the AMPK/mTOR pathway, where AMP-activated protein kinase (AMPK) inhibits mTOR, promoting autophagy. This process helps clear damaged organelles and proteins, protecting synoviocytes from apoptosis and maintaining cellular homeostasis. The activation of autophagy in synoviocytes can reduce inflammation by limiting the production of ROS and other inflammatory mediators, helping to protect joint tissues from excessive damage.
The dynamic balance between the pro-inflammatory and anti-inflammatory activities of synoviocytes, as well as their role in ECM remodeling, is critical for joint homeostasis and repair. Molecular pathways such as PI3K/AKT, SMAD, NF-κB, and MAPK play crucial roles in regulating synoviocyte function. Targeting these pathways pharmacologically could offer therapeutic potential for modulating synovial inflammation and improving outcomes in joint injury and degenerative diseases. Understanding the molecular mechanisms governing synoviocyte behavior provides insights into new treatments aimed at reducing inflammation, promoting tissue regeneration, and preventing long-term joint damage.
Fibroblast Activation and Proliferation
Fibroblasts play a crucial molecular role in the repair of ligament and tendon tissues post-injury by synthesizing new ECM components, regulating collagen organization, and responding to growth factors and mechanical signals. The proliferation of fibroblasts is triggered by growth factors such as PDGF, FGF, and TGF-β, which bind to their respective receptors on the fibroblast surface, activating downstream signaling pathways like PI3K/AKT, MAPK/ERK, and JAK/STAT. These pathways regulate gene expression to promote fibroblast proliferation and migration to the injury site. TGF-β is particularly important for activating the SMAD signaling pathway, which translocates to the nucleus to upregulate the transcription of collagen and other ECM-related genes. This growth factor signaling ensures the rapid proliferation of fibroblasts and prepares them for ECM synthesis, essential for the early phases of tissue repair.
The synthesis of type I collagen, the predominant collagen in ligaments and tendons, is a tightly regulated process at the molecular level. The TGF-β/SMAD pathway plays a central role by upregulating the expression of COL1A1 and COL1A2 genes, which encode the α1 and α2 chains of type I collagen. These collagen molecules are secreted into the ECM, where they undergo post-translational modifications such as hydroxylation and glycosylation, essential for proper collagen fiber assembly. Lysyl oxidase, an enzyme crucial for collagen cross-linking, oxidizes lysine residues on collagen molecules, enabling the formation of covalent cross-links between collagen fibers. This cross-linking is critical for enhancing the tensile strength and durability of the ECM, allowing the repaired tissue to withstand mechanical forces.
Fibroblasts also respond to mechanical loading through mechanotransduction pathways. Integrins, transmembrane receptors on fibroblast surfaces, sense mechanical stress and activate intracellular signaling cascades such as focal adhesion kinase (FAK), RhoA/ROCK, and YAP/TAZ, which regulate cytoskeletal organization and ECM production. Mechanical loading not only promotes the alignment of collagen fibers in the direction of force but also enhances fibroblast proliferation and matrix synthesis. The activation of YAP/TAZ, in particular, leads to the transcription of genes involved in cell proliferation and ECM remodeling, ensuring that the tissue adapts to mechanical stress during the healing process. Mechanical cues also regulate the balance between collagen synthesis and degradation, ensuring proper ECM remodeling and avoiding fibrosis or scar tissue formation.
In addition to collagen, fibroblasts produce proteoglycans such as decorin and biglycan, which play roles in regulating collagen fibrillogenesis and organizing collagen fibers within the ECM. These proteoglycans also help retain water within the tissue, contributing to its viscoelastic properties, which are crucial for the biomechanical functionality of ligaments and tendons. The synthesis of proteoglycans is regulated by growth factors like FGF and mechanical signals that activate the PI3K/AKT pathway, promoting fibroblast survival and ECM production. Furthermore, fibroblasts produce elastin, which provides elasticity to the ECM, allowing it to return to its original shape after deformation. The proper regulation of elastin and collagen ensures that the tissue has both tensile strength and flexibility.
Fibroblasts also regulate ECM degradation by producing matrix metalloproteinases (MMPs) and their inhibitors, tissue inhibitors of metalloproteinases (TIMPs). MMPs, such as MMP-1 and MMP-9, degrade collagen and other ECM components, facilitating tissue remodeling. However, an imbalance between MMPs and TIMPs can lead to excessive ECM degradation, impairing tissue repair. The NF-κB and MAPK signaling pathways, activated by pro-inflammatory cytokines like IL-1 and TNF-α, upregulate MMP expression, while TIMPs are regulated by growth factors like TGF-β, which ensures controlled ECM remodeling during the healing process.
In summary, the molecular regulation of fibroblasts following knee joint injury involves a complex interplay of growth factors, mechanical signals, and ECM remodeling enzymes. Key signaling pathways, such as TGF-β/SMAD, PI3K/AKT, FAK, and YAP/TAZ, drive fibroblast proliferation, migration, and ECM synthesis, while MMPs and TIMPs ensure controlled matrix degradation. Proper alignment and cross-linking of collagen fibers, regulated by both molecular and mechanical cues, are essential for restoring the biomechanical integrity of the injured tissue, enabling it to regain function and withstand mechanical stress.
Mesenchymal Stem Cell (MSC) Recruitment and Differentiation
Mesenchymal stem cells (MSCs) are central to tissue repair due to their ability to differentiate into multiple cell types, including chondrocytes, fibroblasts, and osteoblasts, and their role in modulating the repair environment via molecular signaling. Following knee joint injury, MSC recruitment is driven by chemotactic signals such as stromal cell-derived factor-1 (SDF-1) and vascular endothelial growth factor (VEGF). These factors bind to receptors like CXCR4 and VEGFR on MSCs, activating intracellular pathways such as PI3K/AKT and Rho/ROCK, which facilitate MSC migration through the extracellular matrix (ECM). This migration is highly regulated by integrins, which interact with ECM components like collagen and fibronectin, mediating adhesion and motility. Focal adhesion kinase (FAK) signaling further promotes cytoskeletal reorganization, ensuring efficient MSC homing to the injury site. Additionally, the interaction of MSCs with the vascular endothelium involves selectins and VCAM-1, enabling MSC transmigration into the damaged tissue, where they can exert their reparative functions.
Upon reaching the injury site, MSCs differentiate into repair-specific cell types. Local growth factors such as TGF-β, IGF-1, and bone morphogenetic proteins (BMPs) drive MSC differentiation. TGF-β activates the SMAD pathway, leading to the nuclear translocation of SMAD proteins, which regulate the transcription of genes involved in chondrogenesis, including SOX9 and COL2A1. Similarly, BMPs activate the Wnt/β-catenin signaling pathway, which promotes osteogenic and chondrogenic differentiation. Mechanical forces also influence MSC fate through integrins and mechanosensitive ion channels like Piezo1, which translate mechanical stimuli into biochemical signals. Compressive forces favor chondrogenesis via YAP/TAZ regulation, while tensile forces drive fibroblastic differentiation by activating pathways like ERK/MAPK. This mechanotransduction ensures MSCs differentiate in accordance with the mechanical environment, optimizing tissue-specific repair.
MSCs also exert significant paracrine effects, secreting a broad range of cytokines, growth factors, and extracellular vesicles that modulate the local tissue environment. IL-10 and TGF-β secreted by MSCs play anti-inflammatory roles by inhibiting the NF-κB signaling pathway, reducing the production of pro-inflammatory cytokines such as TNF-α and IL-1β. This helps to resolve inflammation and creates a microenvironment conducive to tissue repair. VEGF secretion by MSCs promotes angiogenesis, enhancing blood supply to the injured tissue, which is crucial for delivering oxygen, nutrients, and reparative cells. Additionally, MSC-derived exosomes contain microRNAs (miRNAs), mRNAs, and proteins that regulate gene expression in neighboring cells, including resident chondrocytes and fibroblasts, enhancing their proliferation, ECM synthesis, and survival. For instance, MSC-derived miR-21 has been shown to inhibit apoptosis in chondrocytes by downregulating pro-apoptotic genes, while miR-140 promotes cartilage regeneration by enhancing collagen synthesis.
The ability of MSCs to modulate the inflammatory response and support tissue regeneration via paracrine signaling is further enhanced by their interaction with the ECM. The ECM composition influences MSC behavior through integrin-mediated signaling, regulating their secretion of paracrine factors and extracellular vesicles. For instance, hyaluronan and collagen type II in the ECM can bind to specific integrins on MSCs, promoting chondrogenic differentiation and anti-inflammatory signaling through pathways such as PI3K/AKT and MAPK. Additionally, MSCs express matrix metalloproteinases (MMPs), which play a role in ECM remodeling by breaking down damaged matrix components and creating space for new tissue formation. However, MMP activity is tightly regulated by tissue inhibitors of metalloproteinases (TIMPs) to prevent excessive matrix degradation, which could impair tissue repair.
In summary, MSCs contribute to knee joint repair through a combination of differentiation into key cell types and paracrine signaling that modulates inflammation, enhances ECM synthesis, and promotes cell survival. Their recruitment is driven by chemotactic signals like SDF-1 and VEGF, and their differentiation is regulated by TGF-β, BMPs, and mechanical forces. Paracrine effects, including the secretion of cytokines, growth factors, and extracellular vesicles, create a regenerative microenvironment that supports tissue repair. Understanding these molecular mechanisms is critical for harnessing the therapeutic potential of MSCs in regenerative medicine.
Angiogenesis
Angiogenesis, the process of forming new blood vessels, is a highly regulated and intricate molecular process that plays a crucial role in tissue repair following knee joint injury. VEGF (vascular endothelial growth factor) is the primary driver of angiogenesis. Its expression is induced by hypoxia and regulated by hypoxia-inducible factors (HIFs). Under hypoxic conditions, HIFs stabilize and translocate to the nucleus, where they bind to hypoxia-response elements (HREs) in the VEGF gene promoter, initiating transcription. VEGF binds to its receptor VEGFR-2 on endothelial cells, activating several key signaling cascades, including the PI3K/AKT pathway, which promotes endothelial cell survival, and the ERK/MAPK pathway, which drives endothelial cell proliferation and migration. Src kinases and p38 MAPK are also involved in enhancing endothelial cell permeability and cytoskeletal reorganization, allowing the cells to migrate toward the hypoxic site. These pathways collectively initiate the formation of vascular sprouts that expand into the injury zone.
Additionally, VEGF not only stimulates endothelial cell proliferation but also recruits pericytes and smooth muscle cells through signaling pathways involving platelet-derived growth factor (PDGF). PDGF binds to its receptor, PDGFR, on pericytes and smooth muscle cells, promoting their proliferation and migration to stabilize the nascent blood vessels. FGF (fibroblast growth factor) also plays a complementary role by activating its receptor, FGFR, to further enhance endothelial cell migration and proliferation via the RAS/RAF/MEK/ERK pathway. VEGF, FGF, and PDGF work in concert to ensure that the new vasculature forms efficiently and is structurally stable.
In addition to VEGF and FGF, the angiogenic process is tightly controlled by matrix metalloproteinases (MMPs), particularly MMP-2 and MMP-9, which degrade the extracellular matrix (ECM) surrounding blood vessels. This degradation allows endothelial cells to invade the ECM and form new capillary networks. The activity of MMPs is tightly regulated by tissue inhibitors of metalloproteinases (TIMPs), which ensure that matrix degradation does not become excessive and disrupt tissue integrity. MMPs also release ECM-bound VEGF, further enhancing angiogenic signaling in a localized manner.
As the new blood vessels form, angiopoietins (Ang-1 and Ang-2) play critical roles in vessel maturation and stabilization. Ang-1 interacts with the Tie2 receptor on endothelial cells, promoting vessel stabilization through the recruitment of pericytes and the strengthening of endothelial junctions. Conversely, Ang-2 can act as an antagonist to Ang-1, destabilizing vessels during early angiogenesis to allow for remodeling, though its effects are context-dependent and can switch between promoting angiogenesis or vessel regression based on the local microenvironment.
The regulation of angiogenesis also involves balancing pro-angiogenic signals with anti-angiogenic factors. Thrombospondin-1 (TSP-1), an important anti-angiogenic molecule, interacts with cell surface receptors like CD36, which inhibits endothelial cell proliferation and migration. Endostatin and angiostatin, derived from cleavage of collagen XVIII and plasminogen, respectively, also serve as potent inhibitors of angiogenesis by downregulating VEGF signaling and inducing endothelial cell apoptosis. The fine-tuning between these pro-angiogenic and anti-angiogenic signals is crucial for ensuring that angiogenesis is controlled and does not lead to aberrant or excessive vascular growth, which could compromise tissue function and healing.
Furthermore, the role of extracellular vesicles (EVs) in angiogenesis is emerging as a significant area of molecular regulation. Exosomes and microvesicles secreted by MSCs (mesenchymal stem cells) and other repair cells carry pro-angiogenic factors such as VEGF, miRNAs like miR-126 and miR-210, and proteins that modulate endothelial cell activity. These vesicles facilitate intercellular communication and can enhance endothelial cell migration, proliferation, and survival. miR-126, for example, targets negative regulators of the VEGF signaling pathway, thereby promoting angiogenesis.
The newly formed vasculature not only supports the metabolic demands of proliferating cells, such as fibroblasts and chondrocytes, but also plays a role in resolving inflammation. Enhanced vascularization ensures the efficient delivery of immune cells and the clearance of waste products, including reactive oxygen species (ROS) and debris generated during the inflammatory response. The interaction between the endothelial cells and pericytes ensures vessel stability, which is critical for long-term tissue repair and regeneration. Proper angiogenesis also supports the formation of a functional ECM, promoting structural integrity and sustaining tissue regeneration in the long term.
In summary, the molecular regulation of angiogenesis following knee joint injury involves a complex network of signals including VEGF, FGF, PDGF, and their downstream pathways (PI3K/AKT, ERK/MAPK), alongside the controlled activity of MMPs and the balance between pro- and anti-angiogenic factors such as TSP-1 and endostatin. Understanding these molecular mechanisms is crucial for developing targeted therapies to enhance tissue repair and control abnormal vascularization.
Table 5.
This table provides a detailed breakdown of the cellular and molecular responses involved in knee joint injuries, covering different tissue types such as ligaments, menisci, and articular cartilage. It highlights the specific cellular processes, signaling pathways, and extracellular matrix remodeling events that occur during the injury and repair phases, emphasizing the role of immune cells, fibroblasts, chondrocytes, and mesenchymal stem cells in tissue regeneration.
Table 5.
This table provides a detailed breakdown of the cellular and molecular responses involved in knee joint injuries, covering different tissue types such as ligaments, menisci, and articular cartilage. It highlights the specific cellular processes, signaling pathways, and extracellular matrix remodeling events that occur during the injury and repair phases, emphasizing the role of immune cells, fibroblasts, chondrocytes, and mesenchymal stem cells in tissue regeneration.
| Cell Type/Process | Response to Knee Joint Injury | Key Molecular Pathways | Role in Tissue Repair |
| Chondrocytes | Shift from a quiescent to an active state after injury, producing ECM components like type II collagen and aggrecan. Also release catabolic enzymes (MMP-13, ADAMTS-5) that degrade the ECM. Hypoxia in cartilage activates HIF-1α, which influences both anabolic (repair) and catabolic (degradation) processes. | TGF-β/SMAD: Promotes ECM synthesis; HIF-1α: Regulates response to hypoxia; MMP-13, ADAMTS-5: Degrade ECM; VEGF: Promotes angiogenesis, influencing subchondral bone. | Repair ECM through collagen and proteoglycan production, but also contribute to degradation via MMP-13 and ADAMTS-5. Regulate balance between ECM synthesis and degradation. Participate in cartilage degeneration in osteoarthritis and other chronic conditions when not properly regulated. |
| Fibroblasts | Fibroblasts proliferate and migrate to injury sites, synthesizing ECM components like type I collagen and proteoglycans. They also align newly formed collagen fibers in response to mechanical cues. Produce MMPs to degrade damaged ECM and facilitate new tissue formation. | TGF-β/SMAD: Induces ECM production; PI3K/AKT, FAK: Regulate proliferation and migration; MAPK/ERK: Controls gene expression for ECM synthesis. | Produce collagen and other ECM components necessary for ligament and tendon repair. Regulate collagen fiber organization. Contribute to ECM remodeling by balancing matrix degradation and synthesis. Mechanotransduction pathways guide their alignment of collagen based on mechanical stress. |
| Macrophages | Initiate the inflammatory response by releasing pro-inflammatory cytokines (TNF-α, IL-1β) in their M1 phenotype. Transition to the M2 phenotype as healing progresses, secreting growth factors (TGF-β, PDGF) to promote tissue repair. Macrophages also clear debris via phagocytosis and promote fibroblast activity for ECM remodeling. | TGF-β, PDGF: Induce tissue repair; NF-κB, JAK/STAT: Regulate cytokine production; MMP-9: Break down ECM for repair. | Early-stage macrophages (M1) promote inflammation, while later-stage macrophages (M2) support repair and ECM synthesis. Macrophages regulate the inflammatory response, clear necrotic tissue, and stimulate fibroblasts and chondrocytes to produce collagen and other ECM proteins essential for healing. |
| Mesenchymal Stem Cells (MSCs) | MSCs are recruited to injury sites via chemotactic signals like SDF-1 and VEGF. They differentiate into repair-specific cell types such as chondrocytes, fibroblasts, and osteoblasts. MSCs also secrete paracrine factors (IL-10, TGF-β) and exosomes that modulate inflammation, reduce apoptosis, and enhance ECM production by resident cells. | SDF-1/CXCR4, VEGF: Recruit MSCs; TGF-β/SMAD, Wnt/β-catenin: Drive chondrogenic and osteogenic differentiation; PI3K/AKT, ERK/MAPK: Control cell survival and migration. | MSCs contribute to tissue repair by differentiating into key cell types (chondrocytes, fibroblasts). Their paracrine signaling modulates inflammation, enhances tissue regeneration, and promotes ECM synthesis. Their exosomes and microRNAs help regulate surrounding cells’ behavior to optimize tissue recovery. |
| Extracellular Matrix (ECM) Remodeling | Injured tissues undergo ECM degradation to clear damaged components, driven by matrix metalloproteinases (MMPs) like MMP-13 and aggrecanases (ADAMTS-5). Tissue inhibitors of metalloproteinases (TIMPs) regulate MMP activity to prevent excessive ECM breakdown. Growth factors stimulate the synthesis of new ECM components like collagen and proteoglycans. | MMP-13, ADAMTS: Degrade ECM; TIMPs: Inhibit MMPs to regulate ECM turnover; TGF-β, IGF-1, FGF: Promote new ECM synthesis. | Clear damaged ECM, enabling new matrix formation. TIMPs ensure a controlled balance between ECM degradation and synthesis. Growth factors stimulate fibroblasts and chondrocytes to synthesize new ECM, restoring tissue integrity and function after injury. Excessive MMP activity leads to tissue degeneration. |
| Inflammation and Immune Response | The initial inflammatory response involves neutrophils, which release reactive oxygen species (ROS) and cytokines (TNF-α, IL-1β), followed by macrophages that regulate inflammation and repair. Prolonged inflammation, if uncontrolled, leads to chronic joint damage. Lymphocytes, including T cells, regulate the immune response to limit excessive inflammation. | TNF-α, IL-1β: Drive inflammation; NF-κB, MAPK: Regulate inflammatory response; PI3K/AKT: Modulates cell survival; TGF-β, IL-10: Control inflammation and repair. | Orchestrates the body’s response to injury by recruiting immune cells and regulating cytokine production. Balances pro-inflammatory and anti-inflammatory signals to initiate repair while avoiding excessive tissue damage. Proper immune regulation ensures a transition from inflammation to tissue healing. |
| Angiogenesis | New blood vessels form in response to injury, driven by VEGF, FGF, and PDGF signaling. These blood vessels supply oxygen and nutrients to the repair site. Excessive angiogenesis can lead to pathological changes in cartilage and subchondral bone, exacerbating cartilage degeneration in chronic conditions. | VEGF/VEGFR: Induce endothelial cell migration and proliferation; PI3K/AKT, ERK/MAPK: Promote survival and migration; PDGF: Stabilize new blood vessels. | Provides necessary blood supply for healing, enhancing oxygen and nutrient delivery. Supports tissue regeneration, but excessive or poorly regulated angiogenesis can contribute to pathological changes, such as bone sclerosis and cartilage breakdown in conditions like osteoarthritis. |
Mechanical Loading and Cellular Mechanisms
Mechanical loading is critical for the health, maintenance, and repair of musculoskeletal tissues, including the knee joint, as these tissues experience constant mechanical forces such as tension, compression, and shear during activities like walking, running, and jumping. The cellular responses to these forces involve intricate mechanotransduction processes, where mechanical signals are converted into biochemical responses, triggering downstream pathways that regulate cell behavior, gene expression, and extracellular matrix (ECM) remodeling. At the molecular level, integrin signaling plays a pivotal role, as integrins, upon mechanical activation, cluster and recruit focal adhesion kinase (FAK), which phosphorylates and activates downstream effectors such as Src kinases, PI3K/Akt, and the MAPK pathway. These cascades lead to cytoskeletal reorganization, cellular adhesion, migration, and ECM production, especially in tissues like cartilage where type II collagen and aggrecan synthesis are crucial for maintaining structural integrity. In parallel, mechanosensitive ion channels, including stretch-activated calcium channels and TRPV4, mediate calcium influx in response to mechanical stress, activating calcium/calmodulin-dependent kinases (CaMK) and calcineurin, which drive nuclear translocation of transcription factors like NFAT. This further regulates the expression of genes essential for ECM synthesis and cellular proliferation, ensuring tissue adaptation to mechanical loads. The MAPK pathway, activated by mechanical stimuli through integrin and growth factor receptors, involves kinases such as ERK, JNK, and p38 MAPK, which modulate gene transcription associated with cell proliferation, differentiation, and inflammatory responses. While ERK signaling promotes anabolic processes by enhancing the production of ECM proteins, excessive mechanical stress activates JNK and p38, leading to the upregulation of matrix-degrading enzymes like matrix metalloproteinases (MMPs) and aggrecanases, contributing to tissue breakdown in pathological conditions such as osteoarthritis. Crosstalk between these pathways, including the YAP/TAZ mechanotransduction system, fine-tunes cellular responses to the mechanical environment, ensuring that tissue integrity is maintained under normal loading conditions while also adapting to injury or stress. Understanding these molecular processes is crucial for developing therapeutic strategies aimed at enhancing tissue repair, preventing degeneration, and managing conditions resulting from mechanical overloading or injury.
Mechanotransduction Pathways
Mechanotransduction is a molecularly intricate process through which cells convert mechanical stimuli into biochemical signals, leading to a range of cellular responses, including changes in gene expression, protein synthesis, and cytoskeletal reorganization. This process begins at the cell membrane with mechanosensitive proteins such as integrins, cadherins, ion channels (e.g., Piezo1, TRPV4), and G-protein coupled receptors (GPCRs), which respond to mechanical forces like tension, compression, or shear stress. Integrins play a pivotal role in this process by linking the extracellular matrix (ECM) to the intracellular cytoskeleton. When activated by mechanical forces, integrins cluster at focal adhesions, where they recruit proteins such as focal adhesion kinase (FAK) and paxillin, initiating a series of downstream signaling cascades. FAK phosphorylates and activates signaling molecules like Src, leading to the activation of the PI3K/Akt and RhoA/ROCK pathways, which regulate cytoskeletal dynamics and cell motility. Additionally, integrin signaling interfaces with the mitogen-activated protein kinase (MAPK) pathway, specifically the ERK1/2 and p38 MAPK branches, which play key roles in regulating gene expression related to cell survival, proliferation, and ECM remodeling.
Mechanosensitive ion channels, including Piezo1, TRPV4, and ASICs (acid-sensing ion channels), are also crucial in mechanotransduction by modulating intracellular calcium levels. These channels open in response to mechanical stimuli, allowing calcium influx, which triggers the activation of calcium-dependent enzymes such as calmodulin and calcineurin. This calcium signaling cascade leads to the activation of transcription factors like NFAT (nuclear factor of activated T-cells), which translocate to the nucleus to regulate genes involved in cellular adaptation to mechanical stress. Additionally, increased intracellular calcium activates protein kinases like CaMKII, influencing processes such as autophagy, apoptosis, and metabolic regulation.
At the nuclear level, mechanical forces are also transmitted through the cytoskeleton via linker of nucleoskeleton and cytoskeleton (LINC) complexes, directly impacting nuclear architecture and chromatin organization. Mechanical signals can modulate the activity of mechanosensitive transcription factors such as YAP/TAZ, components of the Hippo signaling pathway, which are regulated by ECM stiffness and cell shape. YAP/TAZ translocate to the nucleus in response to mechanical cues, promoting the transcription of genes involved in cell proliferation, ECM production, and tissue regeneration.
In parallel, stretch-activated pathways like the RhoA/ROCK axis influence actin-myosin contractility and cellular tension, coordinating changes in cytoskeletal organization. This mechanical feedback loop is essential for maintaining cell shape, adhesion, and migration. Importantly, mechanical signals can also regulate the activity of matrix metalloproteinases (MMPs), aggrecanases (such as ADAMTS-4 and ADAMTS-5), and tissue inhibitors of metalloproteinases (TIMPs), tightly controlling ECM turnover. Dysregulated mechanotransduction, particularly the excessive activation of MMPs and aggrecanases in response to abnormal mechanical loading, can result in accelerated ECM degradation, contributing to conditions like osteoarthritis, fibrosis, and tendon degeneration.
Thus, mechanotransduction is a highly coordinated molecular process involving cross-talk between integrins, ion channels, and various signaling pathways, all of which converge to regulate cellular responses to mechanical stress. These pathways ensure that cells can adapt to changes in their mechanical environment, maintain tissue homeostasis, and repair damaged tissues. Understanding these molecular mechanisms offers crucial insights into developing therapeutic strategies for mechanical stress-related diseases and enhancing tissue repair and regeneration.
Integrin Signaling
Integrins, functioning as key transmembrane receptors, bridge the extracellular matrix (ECM) and the cytoskeleton, thus playing an essential role in mechanotransduction. Integrins are composed of α and β subunits that combine to form heterodimers, allowing specific recognition of ECM ligands such as fibronectin, laminin, and various types of collagen. Mechanical forces applied to cells induce integrin clustering and conformational changes that transition integrins from a low-affinity to a high-affinity state. This structural rearrangement facilitates the binding to ECM ligands and amplifies the mechanical signal, initiating intracellular signaling cascades at focal adhesion sites. These focal adhesions are dynamic, integrin-rich complexes that anchor the cytoskeleton to the ECM and act as hubs for signal transduction.
Once integrins are activated and clustered, focal adhesion kinase (FAK) undergoes autophosphorylation at specific tyrosine residues, particularly Tyr397, creating docking sites for SH2 domain-containing proteins such as Src family kinases. Src kinases phosphorylate additional tyrosine residues on FAK, further amplifying the signal. This process recruits adaptor proteins like p130Cas and paxillin, which in turn activate downstream signaling pathways. The FAK-Src complex serves as a key integrator of mechanical signals, propagating them through multiple molecular pathways.
The MAPK (mitogen-activated protein kinase) pathway, activated by integrin clustering and FAK signaling, involves the phosphorylation cascade of Ras, Raf, MEK, and ERK proteins. This pathway ultimately regulates gene expression by activating transcription factors such as AP-1 and ELK1, which promote cell proliferation, differentiation, and survival. Additionally, the PI3K/Akt pathway is activated through FAK-Src signaling, leading to the phosphorylation of PI3K and subsequent activation of Akt. Akt plays a critical role in promoting cell survival and metabolic activity by inhibiting pro-apoptotic proteins such as Bad and promoting protein synthesis through the activation of mTOR (mechanistic target of rapamycin). This ensures that cells can thrive and adapt under mechanical stress.
Integrin-mediated mechanotransduction also significantly influences the Rho family of GTPases, including RhoA, Rac1, and Cdc42, which regulate cytoskeletal dynamics. RhoA activation leads to the formation of stress fibers composed of actin filaments, enhancing cellular tension and mechanical stability. The cytoskeleton undergoes continuous remodeling in response to integrin signaling, which allows the cell to adjust its morphology and stiffness in response to changes in the mechanical environment. Additionally, Rac1 and Cdc42 modulate the formation of lamellipodia and filopodia, facilitating cell migration and adhesion turnover. This cytoskeletal reorganization is critical for processes such as wound healing, tissue repair, and maintenance of tissue integrity.
Furthermore, mechanical loading can activate stretch-activated ion channels, such as TRPV4 and Piezo1, which allow the influx of calcium (Ca²⁺) into the cell. Elevated intracellular calcium levels activate downstream effectors, including calmodulin and calcineurin, which further modulate gene expression and protein synthesis. Calcium signaling also cooperates with integrin-mediated pathways to regulate cytoskeletal tension and mechanosensitive transcription factors such as YAP/TAZ. YAP/TAZ translocate to the nucleus in response to increased cytoskeletal tension, where they drive the expression of genes involved in cell proliferation, survival, and matrix remodeling.
In summary, integrins play a central role in mechanotransduction by initiating a cascade of molecular events that translate mechanical forces into biochemical signals. These signals regulate multiple cellular processes, including cytoskeletal remodeling, gene expression, protein synthesis, and cell survival, enabling cells to adapt to mechanical changes in their environment and maintain tissue homeostasis. This complex interplay of molecular pathways, including FAK/Src, MAPK, PI3K/Akt, Rho GTPases, and calcium signaling, is crucial for tissue development, repair, and adaptation to mechanical stress, as well as for understanding pathological conditions like fibrosis, osteoarthritis, and tendinopathies.
Ion Channels and Calcium Signaling
Mechanosensitive ion channels, such as Piezo1, TRPV4, and stretch-activated calcium channels (SACs), play a critical role in mechanotransduction by allowing the influx of ions like calcium (Ca²⁺) in response to mechanical deformation of the cell membrane. These channels are activated by mechanical forces, such as stretching or compression, and their opening triggers an immediate rise in intracellular Ca²⁺ levels. This calcium influx serves as a crucial second messenger that activates several downstream signaling cascades, including those mediated by calmodulin, calcineurin, and calmodulin-dependent kinases (CaMKs). Once inside the cell, calcium binds to calmodulin, a key calcium-sensing protein, inducing a conformational change that allows calmodulin to interact with and activate various target proteins, including CaMKs and calcineurin.
The CaMK family of kinases, specifically CaMKII, plays a pivotal role in translating mechanical signals into biochemical responses. Activated CaMKII phosphorylates a variety of substrates involved in gene expression, cytoskeletal reorganization, and cellular proliferation. For instance, CaMKII phosphorylates transcription factors such as CREB (cAMP response element-binding protein), which regulates the expression of genes associated with cellular adaptation to mechanical stress, including those involved in ECM remodeling, cell survival, and tissue repair. This pathway is essential in processes like muscle hypertrophy, bone adaptation to mechanical load, and cartilage maintenance.
Another critical pathway activated by Ca²⁺ influx is the calcineurin-NFAT (nuclear factor of activated T-cells) pathway. Calcineurin, a Ca²⁺/calmodulin-dependent phosphatase, is activated by the calcium-calmodulin complex and subsequently dephosphorylates NFAT. This dephosphorylation allows NFAT to translocate from the cytoplasm to the nucleus, where it acts as a transcription factor to regulate the expression of genes involved in immune responses, cell proliferation, and tissue repair. NFAT plays a key role in the regulation of gene expression in response to mechanical stimuli, particularly in tissues like bone and muscle, where it governs processes such as osteoblast differentiation and muscle fiber remodeling.
In addition to these pathways, the elevated Ca²⁺ levels also influence cytoskeletal dynamics through the Rho GTPase signaling pathway. RhoA, a small GTPase, is activated in response to mechanical stress and calcium signaling, leading to the assembly of actin stress fibers and the formation of focal adhesions. These structures anchor cells to the ECM and are critical for transmitting mechanical forces into the cell. The actin cytoskeleton, in conjunction with integrin receptors, coordinates with mechanosensitive ion channels to modulate cell shape, migration, and ECM interaction. This feedback mechanism allows cells to continuously sense and respond to their mechanical environment, ensuring proper adaptation to mechanical stress.
Furthermore, Ca²⁺-mediated activation of phospholipases and other calcium-sensitive enzymes leads to the production of secondary messengers like inositol trisphosphate (IP₃) and diacylglycerol (DAG). These molecules further amplify the signaling cascades by promoting the release of Ca²⁺ from intracellular stores, such as the endoplasmic reticulum, and activating protein kinase C (PKC). PKC modulates numerous cellular functions, including gene transcription, apoptosis, and cell proliferation, linking mechanical signals to broader cellular responses.
In summary, mechanosensitive ion channels facilitate the entry of Ca²⁺ in response to mechanical stimuli, triggering a cascade of molecular events involving calmodulin, CaMK, calcineurin, and NFAT. These pathways collectively regulate critical cellular functions, such as gene expression, cytoskeletal reorganization, and ECM remodeling, ensuring that cells can adapt to their mechanical environment. This complex network of calcium-dependent signaling not only governs tissue maintenance and repair but also plays a role in pathological conditions where mechanical forces are dysregulated, such as in osteoarthritis and fibrosis.
MAPK Pathway
The MAPK (Mitogen-Activated Protein Kinase) pathway is a critical molecular signaling cascade that integrates mechanical and biochemical stimuli, orchestrating a wide range of cellular processes such as gene expression, proliferation, apoptosis, and differentiation. Mechanically induced activation of the MAPK pathway is initiated by upstream kinases, such as Raf, ASK1, or TAK1, which are activated through membrane receptors like integrins, receptor tyrosine kinases (RTKs), and stretch-activated ion channels. These receptors sense mechanical changes, such as stress and deformation in the extracellular matrix (ECM), and transmit these signals into intracellular biochemical cascades. Upon activation, Raf phosphorylates MEK1/2 (MAPK/ERK kinase), which then phosphorylates ERK1/2 (extracellular signal-regulated kinase), driving a critical branch of the MAPK pathway.
ERK1/2 is highly sensitive to growth factors and mechanical forces, undergoing dual phosphorylation on its threonine and tyrosine residues. Once activated, ERK1/2 translocates into the nucleus and phosphorylates a variety of transcription factors such as Elk-1, c-Fos, and Ets-1. These transcription factors bind to promoter regions of genes involved in cellular proliferation and differentiation, including cyclin D1 and Myc, essential for the G1/S phase transition in the cell cycle. Furthermore, ERK1/2-mediated phosphorylation of cytoplasmic substrates like ribosomal S6 kinase (RSK) regulates mRNA translation, ensuring that the synthesis of proteins required for growth and survival is upregulated under mechanical stress conditions.
The JNK (c-Jun N-terminal kinase) branch of the MAPK pathway is primarily activated by mechanical and oxidative stress, including shear forces and reactive oxygen species (ROS). JNK is activated by upstream kinases such as MKK4/7, which phosphorylate JNK at specific threonine and tyrosine residues. Activated JNK translocates to the nucleus, where it phosphorylates transcription factors, most notably c-Jun, forming part of the activator protein-1 (AP-1) complex. AP-1 regulates the expression of genes involved in stress response, inflammation, and apoptosis, including Bim, a pro-apoptotic protein. JNK also modulates cytoskeletal dynamics by phosphorylating paxillin and other actin-associated proteins, influencing cell migration and mechanosensitivity.
The p38 MAPK pathway, another important component of the MAPK signaling family, is activated by upstream MAPKKs like MKK3/6 in response to mechanical stress, pro-inflammatory cytokines, and environmental stressors. p38 MAPK is known to phosphorylate a broad range of substrates, including transcription factors such as ATF2 and MEF2, which regulate genes involved in inflammation, cell differentiation, and tissue repair. p38 MAPK also interacts with downstream kinases such as MAPK-activated protein kinases (MK2/3), which modulate the stability of mRNA transcripts for pro-inflammatory cytokines like TNF-α and IL-6, thus playing a crucial role in the cellular response to inflammation and injury. The role of p38 in modulating the cellular cytoskeleton via the phosphorylation of proteins such as Hsp27 and its involvement in regulating actin dynamics is essential for mechanosensation and cellular adaptation to mechanical forces.
Cross-talk between the MAPK pathway and other key signaling pathways, such as the PI3K/Akt, NF-κB, and Hippo pathways, adds an additional layer of molecular regulation. For instance, mechanical loading often simultaneously activates both ERK1/2 and PI3K/Akt pathways. PI3K, activated through integrin engagement, phosphorylates and activates Akt, promoting cell survival by inhibiting pro-apoptotic factors such as Bad and activating mTOR, which enhances protein synthesis. The convergence of ERK1/2 and PI3K/Akt pathways ensures that cells under mechanical stress promote anabolic processes, such as ECM synthesis and cell growth, while counteracting catabolic responses mediated by proteases like matrix metalloproteinases (MMPs).
Moreover, feedback loops within the MAPK signaling cascade further fine-tune cellular responses to mechanical loading. Phosphatases like dual-specificity phosphatases (DUSPs) dephosphorylate MAPKs, thereby providing a negative feedback mechanism that limits excessive MAPK activation and ensures tight regulation of cellular proliferation and stress responses. Similarly, scaffold proteins like KSR (Kinase Suppressor of Ras) facilitate the spatial organization of MAPK signaling components, ensuring precise signal transduction and preventing inappropriate crosstalk between parallel pathways.
In summary, the MAPK pathway is a highly interconnected and dynamic signaling network that plays a pivotal role in translating mechanical signals into precise biochemical responses. Through the activation of ERK1/2, JNK, and p38, this pathway regulates crucial cellular functions, including gene transcription, cytoskeletal remodeling, and protein synthesis, ensuring that cells can adapt to mechanical stresses, promote tissue repair, and maintain homeostasis. Understanding the molecular intricacies of MAPK signaling provides valuable insights into therapeutic strategies aimed at enhancing tissue regeneration and mitigating the effects of mechanical injury.
ECM Synthesis and Remodeling
Mechanical loading induces complex molecular responses that govern the synthesis, remodeling, and degradation of the extracellular matrix (ECM), which are essential for maintaining tissue integrity. The process begins with mechanotransduction pathways, where integrins—transmembrane receptors—sense mechanical forces and activate intracellular signaling cascades. Integrin clustering at focal adhesion sites leads to the recruitment of focal adhesion kinase (FAK) and Src family kinases, initiating downstream pathways such as the mitogen-activated protein kinase (MAPK) and phosphoinositide 3-kinase (PI3K/Akt) pathways. These signaling networks regulate gene expression and protein synthesis, promoting the production of ECM components like collagen and proteoglycans.
One of the key effects of mechanical loading is the upregulation of type I and type III collagen synthesis by fibroblasts, chondrocytes, and other ECM-producing cells. This process is driven by the activation of transforming growth factor-beta (TGF-β) and its downstream Smad signaling pathway, which enhances the transcription of collagen genes. The newly synthesized collagen molecules undergo post-translational modifications, including hydroxylation and glycosylation, which are crucial for collagen fibril formation and cross-linking. Lysyl oxidase, an enzyme activated by mechanical signals, catalyzes the formation of covalent cross-links between collagen fibers, reinforcing the ECM’s tensile strength and structural resilience.
In addition to collagen, mechanical loading enhances the production of proteoglycans, such as aggrecan and decorin, through signaling pathways like PI3K/Akt and Smad. Proteoglycans consist of a protein core linked to glycosaminoglycan (GAG) chains, which are highly hydrophilic, allowing them to retain water and confer compressive strength to the ECM. The interaction between proteoglycans and collagen fibers provides the ECM with its viscoelastic properties, enabling it to absorb and dissipate mechanical forces while protecting cells from excessive stress. Mechanosensitive ion channels, particularly calcium channels, also contribute by regulating calcium influx, which activates calcium/calmodulin-dependent kinases (CaMK) that further influence proteoglycan synthesis.
Matrix degradation is equally important for ECM remodeling, ensuring that damaged or excess ECM components are removed to allow for tissue adaptation. Matrix metalloproteinases (MMPs), such as MMP-2, MMP-9, and MMP-13, are upregulated in response to mechanical loading through pathways like MAPK and NF-κB. These MMPs degrade collagen and proteoglycans, creating space for new matrix deposition. However, their activity is tightly regulated by tissue inhibitors of metalloproteinases (TIMPs), which bind to active MMPs and prevent excessive ECM breakdown. The balance between MMPs and TIMPs is crucial for maintaining ECM homeostasis; any dysregulation can lead to pathological conditions such as fibrosis, where excessive ECM deposition occurs, or tissue degeneration, where excessive matrix degradation prevails.
Mechanical loading also influences the expression of genes involved in ECM turnover via the YAP/TAZ pathway, which responds to changes in mechanical tension within the cell. YAP and TAZ are transcriptional co-activators that translocate to the nucleus upon activation, where they regulate the expression of genes involved in cell proliferation, differentiation, and ECM synthesis. This pathway integrates mechanical cues with biochemical signals to ensure that the ECM adapts dynamically to mechanical stress.
In summary, the molecular mechanisms governing the response to mechanical loading involve a tightly coordinated network of signaling pathways, including integrin-mediated mechanotransduction, TGF-β/Smad signaling, and MMP/TIMP regulation. These pathways control the synthesis and degradation of key ECM components such as collagen and proteoglycans, ensuring that tissues can maintain structural integrity, adapt to mechanical stress, and undergo proper remodeling during injury and repair processes.
Cell Proliferation and Differentiation
Mechanical loading initiates a complex network of molecular pathways that regulate cell proliferation and differentiation, critical for tissue repair and regeneration. At the molecular level, mechanical forces are sensed by integrins, transmembrane receptors that link the extracellular matrix (ECM) to the cytoskeleton. Upon mechanical stimulation, integrins cluster and activate focal adhesion kinase (FAK), which triggers multiple downstream signaling pathways, including the PI3K/Akt and MAPK cascades. FAK phosphorylation leads to the recruitment of Src family kinases and the formation of focal adhesion complexes that integrate mechanical signals into biochemical responses. This results in the activation of transcription factors, such as NF-κB and Smads, which regulate gene expression related to cell proliferation, ECM synthesis, and differentiation.
In fibroblasts, mechanical stress enhances the production of growth factors like transforming growth factor-beta (TGF-β) and platelet-derived growth factor (PDGF), which bind to their respective receptors and activate downstream pathways, including PI3K/Akt and RhoA/ROCK, promoting cytoskeletal reorganization, cell migration, and ECM production. TGF-β, in particular, plays a central role by activating the Smad2/3 pathway, leading to the transcription of genes involved in collagen synthesis and fibroblast proliferation. The activated Smad complexes translocate to the nucleus and regulate the expression of ECM-related genes, such as those encoding collagen type I and fibronectin, essential for tissue repair and structural integrity.
Mesenchymal stem cells (MSCs) respond to mechanical cues through mechanotransduction pathways like YAP/TAZ and Wnt/β-catenin. YAP/TAZ, in response to mechanical tension, translocate to the nucleus, where they promote the transcription of genes involved in cell proliferation and differentiation. Wnt/β-catenin signaling is also activated by mechanical forces, leading to β-catenin stabilization and its nuclear translocation, where it regulates the expression of genes involved in MSC differentiation into osteoblasts, chondrocytes, or fibroblasts. Mechanical stimuli further influence MSC behavior through stretch-activated ion channels, such as TRPV4, which allow calcium influx. Increased intracellular calcium levels activate calcineurin, which dephosphorylates nuclear factor of activated T-cells (NFAT), allowing NFAT to enter the nucleus and regulate gene expression related to cell differentiation and matrix remodeling.
In chondrocytes, mechanical loading activates integrin signaling and calcium influx through mechanosensitive ion channels like Piezo1 and TRPV4. This calcium influx triggers the activation of calcium/calmodulin-dependent kinases (CaMK) and calcineurin, which influence the activity of key transcription factors such as NFAT and c-Fos. These transcription factors regulate the synthesis of cartilage-specific ECM components, including type II collagen and aggrecan. Additionally, the activation of the ERK1/2 pathway in response to mechanical stress promotes chondrocyte proliferation and survival, ensuring the maintenance of cartilage under mechanical load.
Together, these molecular mechanisms coordinate cellular responses to mechanical loading, driving tissue repair, regeneration, and the adaptation of cells to their mechanical environment. The intricate balance between proliferative signals, cytoskeletal remodeling, and ECM synthesis ensures that tissues maintain structural integrity and function in response to varying mechanical forces. Understanding these pathways at a molecular level is essential for developing therapeutic interventions that enhance tissue regeneration and address diseases associated with impaired mechanotransduction, such as osteoarthritis or tendon injuries.
Inflammation and Immune Response
Mechanical loading intricately modulates the inflammatory response at the molecular level by activating mechanotransduction pathways that regulate cytokine production, immune cell recruitment, and tissue regeneration. The mechanosensitive receptors, primarily integrins, sense mechanical stress and initiate intracellular signaling cascades such as the MAPK, NF-κB, and PI3K/Akt pathways. These pathways directly influence the production of pro-inflammatory cytokines like interleukin-1 beta (IL-1β), tumor necrosis factor-alpha (TNF-α), and interleukin-6 (IL-6), while also promoting the expression of anti-inflammatory cytokines like transforming growth factor-beta (TGF-β) and interleukin-10 (IL-10). The balance between these cytokines is critical for controlling the inflammatory response, ensuring that it is robust enough to facilitate tissue repair but not excessive enough to cause chronic inflammation or tissue damage. For example, the activation of NF-κB by mechanical signals can lead to the transcription of pro-inflammatory cytokines, while mechanical modulation of the PI3K/Akt pathway can activate anti-inflammatory signals that promote tissue regeneration.
Additionally, mechanical loading influences the recruitment and function of immune cells, such as macrophages and neutrophils, through the regulation of chemokines and adhesion molecules. Integrin clustering and focal adhesion kinase (FAK) activation lead to the production of chemokines like CCL2, which attracts immune cells to the injury site. Once recruited, mechanical signals can polarize macrophages towards the anti-inflammatory M2 phenotype, which is essential for tissue healing. The M2 macrophages secrete growth factors such as platelet-derived growth factor (PDGF) and vascular endothelial growth factor (VEGF), which promote extracellular matrix (ECM) remodeling and angiogenesis, further supporting tissue regeneration. Meanwhile, the regulation of matrix metalloproteinases (MMPs) by mechanical stress ensures that damaged ECM is degraded in a controlled manner, preventing excessive matrix destruction that could impair healing.
Moreover, mechanical loading also affects ion channel activity, particularly stretch-activated calcium channels, which modulate intracellular calcium levels. This calcium influx activates downstream signaling pathways like the calcineurin/NFAT pathway, which can influence gene transcription involved in inflammation and repair. Calcineurin dephosphorylates NFAT, allowing it to enter the nucleus and regulate the expression of genes that control immune cell activation, cytokine production, and tissue regeneration. Additionally, mechanical stress can influence reactive oxygen species (ROS) production, which can act as secondary messengers in signaling pathways like MAPK and NF-κB, further influencing the inflammatory and reparative processes.
This intricate molecular network of cytokine regulation, immune cell recruitment, ion channel activity, and signaling pathways demonstrates how mechanical forces not only initiate but also finely tune the inflammatory response. By modulating these molecular processes, mechanical loading ensures an optimal environment for tissue repair, preventing excessive inflammation while promoting regeneration and tissue homeostasis. Understanding these molecular mechanisms provides crucial insights into potential therapeutic strategies that harness mechanical signals to control inflammation and enhance tissue repair in conditions such as arthritis, tendon injuries, and other mechanically sensitive disorders.
Angiogenesis
Mechanical loading exerts its effects on angiogenesis through intricate molecular mechanisms, primarily involving the regulation of vascular endothelial growth factor (VEGF) and other signaling molecules that mediate endothelial cell activity and extracellular matrix (ECM) remodeling. When tissues experience mechanical stress, cells such as fibroblasts, chondrocytes, and mesenchymal stem cells (MSCs) respond by increasing the expression of VEGF. This upregulation is driven by mechanosensitive pathways like the phosphoinositide 3-kinase (PI3K)/Akt pathway and the mitogen-activated protein kinase (MAPK) pathway, as well as hypoxia-inducible factor-1 alpha (HIF-1α). HIF-1α is stabilized under conditions of hypoxia or mechanical stress, and it binds to hypoxia response elements (HREs) in the promoter region of the VEGF gene, boosting VEGF transcription. Once secreted, VEGF binds to its receptor, VEGFR-2, on the surface of endothelial cells, initiating a cascade of intracellular events that promote angiogenesis. This interaction activates downstream signaling pathways such as the PI3K/Akt and ERK1/2 MAPK pathways, which lead to endothelial cell proliferation, migration, and survival—critical processes for the formation of new capillary networks.
Mechanically induced VEGF signaling also activates focal adhesion kinase (FAK) in endothelial cells. FAK is recruited to integrin clusters at sites where endothelial cells interact with the ECM. The activation of FAK facilitates cytoskeletal reorganization by regulating actin dynamics, enabling endothelial cells to migrate through the ECM and form new vascular structures. This is further supported by the activation of Rho family GTPases, such as RhoA, which modulate cytoskeletal tension and endothelial cell motility. Additionally, mechanical forces modulate the activity of matrix metalloproteinases (MMPs), particularly MMP-2 and MMP-9, which degrade components of the ECM, creating pathways for endothelial cell invasion and capillary sprouting. The controlled degradation of the ECM by MMPs also releases matrix-bound growth factors, such as VEGF and fibroblast growth factor (FGF), which further enhance angiogenesis.
Simultaneously, angiogenic factors like FGF and platelet-derived growth factor (PDGF) contribute to the stabilization and maturation of new blood vessels by recruiting pericytes and smooth muscle cells. PDGF signaling through its receptor, PDGFR-β, supports pericyte attachment to endothelial cells, strengthening the newly formed blood vessels. This is critical for vessel maturation, as pericytes provide structural support and regulate vascular permeability. Mechanical loading also influences the production of angiopoietins (Ang-1 and Ang-2), which modulate the stability and remodeling of blood vessels. Ang-1, through its receptor Tie2 on endothelial cells, promotes vessel stabilization and prevents vascular leakage by enhancing endothelial cell junction integrity and recruiting pericytes. Conversely, Ang-2 can destabilize blood vessels during early angiogenesis, allowing for the necessary remodeling of the vascular network under mechanical stress.
Moreover, mechanical loading regulates the production of nitric oxide (NO) by endothelial cells via endothelial nitric oxide synthase (eNOS) activation. NO acts as a vasodilator, increasing blood flow and vascular permeability, which are essential for facilitating angiogenesis. Mechanical stress stimulates eNOS through calcium influx and phosphorylation events mediated by Akt, enhancing NO production. The vasodilatory effect of NO ensures adequate blood perfusion in growing tissues, supporting the metabolic demands of regenerating cells.
The coordinated interplay between these molecular pathways—VEGF signaling, integrin-FAK interactions, MMP-mediated ECM remodeling, and NO production—ensures that angiogenesis proceeds efficiently and that tissues undergoing repair receive a sufficient blood supply. By integrating mechanical signals into angiogenic processes, tissues can adapt to environmental changes and promote healing following injury. Understanding the molecular regulation of angiogenesis in response to mechanical loading provides valuable insights for developing therapeutic strategies aimed at enhancing tissue regeneration, particularly in conditions where vascularization is critical for recovery, such as in bone fractures, wound healing, and degenerative diseases.
Table 6.
This table outlines the molecular pathways involved in cellular responses to mechanical loading, highlighting key processes such as integrin signaling, mechanosensitive ion channels, and ECM remodeling. It emphasizes the role of mechanical forces in regulating gene expression, protein synthesis, cell proliferation, and tissue repair, providing a detailed overview of the mechanisms that maintain tissue integrity under stress.
Table 6.
This table outlines the molecular pathways involved in cellular responses to mechanical loading, highlighting key processes such as integrin signaling, mechanosensitive ion channels, and ECM remodeling. It emphasizes the role of mechanical forces in regulating gene expression, protein synthesis, cell proliferation, and tissue repair, providing a detailed overview of the mechanisms that maintain tissue integrity under stress.
| Category | Molecular Components | Mechanism of Action | Cellular/Tissue Response |
| Integrin Signaling | Integrins (α and β subunits), Focal Adhesion Kinase (FAK), Src Kinases, PI3K/Akt, RhoA, MAPK, Paxillin, P130Cas | Mechanical loading induces integrin clustering at focal adhesions, activating FAK. FAK phosphorylates Src and recruits downstream effectors, including paxillin and p130Cas, which activate MAPK and PI3K/Akt pathways. This signaling cascade regulates cytoskeletal dynamics, cell adhesion, and migration, while promoting ECM synthesis. | Increased fibroblast adhesion, migration, and survival. Enhanced ECM production (collagen, fibronectin) and cytoskeletal reorganization contribute to stronger tissue integrity and wound healing. |
| Mechanosensitive Ion Channels | Piezo1, TRPV4, ASICs (Acid-Sensing Ion Channels), Calcium (Ca²⁺), Calmodulin, CaMK (Ca²⁺/calmodulin-dependent protein kinase), Calcineurin/NFAT | Mechanical stress opens ion channels (Piezo1, TRPV4), allowing Ca²⁺ influx. Ca²⁺ binds to calmodulin, activating CaMK, calcineurin, and subsequent dephosphorylation of NFAT. This regulates gene expression related to ECM production, cellular proliferation, and adaptation to mechanical forces. | Increased intracellular Ca²⁺ levels drive NFAT translocation to the nucleus, upregulating genes for ECM remodeling, chondrocyte activity, and mesenchymal stem cell (MSC) differentiation. |
| MAPK Pathway | ERK1/2, JNK, p38 MAPK, Raf, MEK, Elk-1, c-Fos, AP-1, MEF2, ATF2 | Integrin-mediated mechanical stress activates MAPK signaling, including ERK1/2, JNK, and p38 MAPK. ERK1/2 promotes cell proliferation and survival by phosphorylating transcription factors such as Elk-1 and c-Fos. JNK is activated by mechanical and oxidative stress, influencing AP-1 activity, while p38 MAPK phosphorylates transcription factors like ATF2 and MEF2, regulating stress responses and inflammation. | ERK1/2 promotes collagen synthesis, cell proliferation, and differentiation. JNK and p38 MAPK are activated in response to excessive mechanical loading, regulating inflammation, apoptosis, and tissue repair processes. |
| ECM Synthesis and Remodeling | Collagen Types I, III, and II, Proteoglycans (Aggrecan, Decorin), Lysyl Oxidase, Matrix Metalloproteinases (MMP-2, MMP-9, MMP-13), Tissue Inhibitors of Metalloproteinases (TIMPs), Smad2/3, TGF-β | Mechanical forces activate TGF-β/Smad2/3 and PI3K/Akt signaling, increasing collagen and proteoglycan synthesis. Lysyl oxidase catalyzes cross-linking of collagen fibers, enhancing ECM tensile strength. MMPs degrade damaged ECM, while TIMPs control MMP activity to prevent excessive breakdown. | Enhanced production of type I and III collagen strengthens ECM structure, while proteoglycans maintain compressive strength. Balanced MMP and TIMP activity allows controlled ECM remodeling during tissue repair. |
| Cell Proliferation & Differentiation | Fibroblasts, Mesenchymal Stem Cells (MSCs), YAP/TAZ, Wnt/β-catenin, Rho GTPases (RhoA, Rac1, Cdc42), TGF-β, PDGF, Smad2/3, NFAT | Mechanical loading activates YAP/TAZ and Wnt/β-catenin pathways, promoting MSC differentiation into fibroblasts, chondrocytes, and osteoblasts. Growth factors (TGF-β, PDGF) trigger Smad2/3 signaling, enhancing fibroblast proliferation and ECM production. Rho GTPases regulate cytoskeletal dynamics, influencing cell shape and migration. | Increased fibroblast proliferation enhances ECM production. MSC differentiation into chondrocytes and osteoblasts accelerates tissue regeneration, while cytoskeletal remodeling supports cell migration and wound healing. |
| Inflammatory Response | NF-κB, PI3K/Akt, IL-1β, TNF-α, IL-6, IL-10, TGF-β, M1/M2 Macrophages, Chemokines (CCL2, CXCL8), Reactive Oxygen Species (ROS), Calcineurin | Mechanical loading modulates cytokine production, balancing pro-inflammatory (IL-1β, TNF-α) and anti-inflammatory (TGF-β, IL-10) signals. NF-κB regulates pro-inflammatory gene transcription, while PI3K/Akt and calcineurin pathways support anti-inflammatory responses. Chemokines recruit immune cells, and mechanical signals polarize macrophages to the reparative M2 phenotype. | Enhanced immune cell recruitment (macrophages, neutrophils) supports tissue repair and controlled inflammation. M2 macrophages secrete growth factors like PDGF and VEGF, promoting angiogenesis and ECM remodeling. |
| Angiogenesis | VEGF, VEGFR-2, Hypoxia-Inducible Factor-1α (HIF-1α), Nitric Oxide (NO), eNOS, FAK, MMPs (MMP-2, MMP-9), Angiopoietins (Ang-1, Ang-2), PDGF, RhoA | Mechanical loading upregulates VEGF via HIF-1α stabilization and PI3K/Akt signaling. VEGF binds to VEGFR-2 on endothelial cells, promoting proliferation, migration, and capillary formation. FAK and MMP activity facilitate endothelial cell migration through ECM remodeling. Nitric oxide produced by eNOS enhances blood flow and vascular permeability. Angiopoietins regulate vessel stability. | Increased vascularization ensures adequate oxygen and nutrient delivery to regenerating tissues. PDGF recruits pericytes for vessel stabilization, while MMP-mediated ECM degradation allows new capillary formation. |
Rehabilitation Strategies Based on Musculoskeletal Healing Stages: Early Mechanical Loading
Rehabilitation strategies for musculoskeletal injuries must be intricately aligned with the body’s natural healing phases—namely inflammation, proliferation, and remodeling—to ensure optimal tissue repair, restore function, and prevent re-injury. During the inflammatory phase, which typically lasts a few days to a week post-injury, the primary goal is to minimize pain, swelling, and further tissue damage while promoting early cellular repair. Gentle, controlled movement within pain-free ranges is encouraged to maintain joint mobility and stimulate blood flow without exacerbating inflammation. Modalities such as cryotherapy, compression, and elevation are commonly used to reduce swelling, while isometric exercises may be introduced to maintain muscle activation without stressing the injured tissue.
In the proliferation phase, which spans several weeks, tissue regeneration and collagen deposition accelerate. At this stage, a gradual increase in mechanical loading is crucial to stimulate cellular processes involved in tissue repair, particularly in tendons, ligaments, and muscles. Low-intensity, controlled resistance exercises and range-of-motion activities are essential to promote collagen alignment and prevent scar tissue formation. Techniques such as eccentric loading, which has been shown to enhance tendon repair, may be introduced gradually, ensuring that the mechanical stimuli are within the tissue’s tolerance to avoid re-injury.
The remodeling phase, lasting from months to over a year, focuses on tissue maturation and functional recovery. Progressive resistance training with higher intensity and specificity to the patient’s sport or activity is essential to enhance the tensile strength of the tissue and support functional movements. Plyometric exercises, proprioceptive training, and dynamic loading are introduced to improve neuromuscular coordination and ensure that the repaired tissue can withstand the stresses of daily activities and sports. Monitoring the patient’s response to increasing mechanical loads is crucial, and rehabilitation should be adjusted based on the tissue’s adaptation, avoiding overloading that could lead to chronic injury.
Understanding the cellular and molecular processes underpinning each healing phase ensures that rehabilitation strategies are appropriately timed and tailored, facilitating optimal recovery and minimizing the risk of complications.
Inflammation Stage
The inflammation stage is a highly orchestrated molecular response involving numerous chemical mediators, immune cells, and signaling pathways, all aimed at initiating tissue repair after a musculoskeletal injury. Central to this process is vasodilation, driven by molecules such as histamine, bradykinin, and prostaglandin E2 (PGE2), which increase blood flow to the injured area. Histamine, released from mast cells and basophils, binds to H1 receptors on endothelial cells, causing endothelial contraction and the formation of intercellular gaps, allowing immune cells and proteins to enter the damaged tissue. Histamine also stimulates endothelial nitric oxide synthase (eNOS), leading to the production of nitric oxide (NO), a potent vasodilator that relaxes vascular smooth muscle cells and further enhances blood flow.
Bradykinin, formed from high-molecular-weight kininogen by the enzyme kallikrein, binds to B2 receptors on endothelial cells, promoting NO release and the synthesis of prostacyclin (PGI2), both of which facilitate vasodilation and increased vascular permeability. Bradykinin also activates phospholipase C (PLC), leading to the generation of inositol trisphosphate (IP3), which triggers calcium release from intracellular stores, further amplifying vascular smooth muscle relaxation. Additionally, bradykinin sensitizes nociceptors, contributing to pain perception, which acts as a protective mechanism, preventing further tissue damage by limiting movement.
PGE2, synthesized from arachidonic acid via the cyclooxygenase-2 (COX-2) pathway, binds to EP2 and EP4 receptors on smooth muscle cells, increasing intracellular cyclic AMP (cAMP) levels and promoting muscle relaxation, leading to vasodilation. PGE2 also sensitizes sensory neurons by increasing transient receptor potential (TRP) channel activity, amplifying pain signaling and enhancing the inflammatory response. PGE2’s role in increasing vascular permeability facilitates immune cell infiltration, which is crucial for clearing pathogens and debris.
The invasion of platelets at the injury site triggers the release of pro-hemostatic and pro-inflammatory mediators. Upon vascular injury, platelets adhere to the exposed collagen and von Willebrand factor (vWF) through glycoprotein receptors (GPVI and GPIb-IX-V complex), initiating platelet activation. This activation results in the release of adenosine diphosphate (ADP), serotonin, and thromboxane A2 (TXA2), which bind to their respective receptors on platelets, reinforcing platelet aggregation and forming a stable hemostatic plug. Platelets also release alpha granules containing growth factors such as platelet-derived growth factor (PDGF) and transforming growth factor-beta (TGF-β), which recruit fibroblasts, smooth muscle cells, and immune cells to the injury site, promoting the transition to the proliferative phase of healing.
Neutrophils, the first immune cells to arrive at the injury site, are recruited by chemotactic signals such as interleukin-8 (IL-8), leukotriene B4 (LTB4), and complement component C5a. Neutrophils engage in phagocytosis, engulfing pathogens and necrotic tissue, and release proteolytic enzymes like elastase and matrix metalloproteinase-9 (MMP-9), which degrade damaged extracellular matrix (ECM) components, facilitating tissue clearance. Neutrophils also generate reactive oxygen species (ROS) via the NADPH oxidase complex, contributing to microbial killing and further signaling in the inflammatory cascade.
As the inflammatory phase progresses, monocytes are recruited to the site by chemokines like monocyte chemoattractant protein-1 (MCP-1), where they differentiate into macrophages. Macrophages undergo polarization into two main phenotypes: M1 macrophages, which produce pro-inflammatory cytokines such as tumor necrosis factor-alpha (TNF-α), interleukin-1 beta (IL-1β), and interleukin-6 (IL-6), and M2 macrophages, which secrete anti-inflammatory cytokines like interleukin-10 (IL-10) and TGF-β. This switch from M1 to M2 macrophages is essential for resolving inflammation and initiating tissue repair and remodeling. M1 macrophages activate additional inflammatory responses via the NF-κB pathway, while M2 macrophages promote tissue repair by secreting growth factors that stimulate fibroblast proliferation and collagen synthesis.
The interplay of these chemical mediators, such as histamine, bradykinin, PGE2, leukotrienes, and nitric oxide, ensures that immune cells are effectively recruited, the injury site is cleared of debris, and the conditions for tissue regeneration are established. Leukotrienes, derived from arachidonic acid via the 5-lipoxygenase (5-LOX) pathway, particularly LTB4, serve as potent chemotactic agents, attracting neutrophils and monocytes to the injury site, further amplifying the inflammatory response. The production of nitric oxide (NO), particularly by macrophages through inducible nitric oxide synthase (iNOS), not only contributes to vasodilation but also has antimicrobial properties, playing a crucial role in pathogen defense during the early inflammatory response. The precise regulation of these processes is critical for transitioning from inflammation to the proliferative phase of healing, where tissue repair and regeneration can proceed.
Fibroblastic Stage
The fibroblastic stage of tissue repair is heavily influenced by a network of molecular interactions that drive fibroblast activation, proliferation, and ECM synthesis, all of which are critical for stabilizing the damaged tissue. Central to this process is Transforming Growth Factor-beta 1 (TGF-β1), which exerts its effects by binding to TGF-β receptors I and II (TGF-βRI and TGF-βRII) on fibroblasts, initiating a cascade of intracellular signaling events. TGF-β1 binding leads to the phosphorylation of Smad2/3 proteins, which subsequently complex with Smad4 and translocate to the nucleus. There, they regulate the transcription of ECM-related genes, including those encoding type I and type III collagen, fibronectin, and proteoglycans. TGF-β1 also suppresses the expression of matrix metalloproteinases (MMPs) such as MMP-1 and MMP-13, which would otherwise degrade newly formed collagen, thus ensuring the integrity and stability of the ECM.
In addition to TGF-β1, Bone Morphogenetic Proteins (BMPs) activate Smad1/5/8 signaling pathways through BMP receptors (BMPR-I and BMPR-II), which are critical for fibroblast differentiation into myofibroblasts. Myofibroblasts play a pivotal role in wound contraction by producing a contractile apparatus composed of α-smooth muscle actin (α-SMA), enabling these cells to physically reduce wound size while also secreting large quantities of ECM proteins. This contractile activity is regulated by intracellular signaling pathways, such as RhoA/ROCK and mechanotransduction, which further influence cytoskeletal organization and fibroblast-mediated force generation within the wound matrix.
Another significant player, Connective Tissue Growth Factor (CTGF), acts downstream of TGF-β1 and enhances the MAPK/ERK signaling pathway in fibroblasts. CTGF binds to receptors such as integrins and heparan sulfate proteoglycans, which activate ERK1/2 kinases that phosphorylate downstream transcription factors like c-Fos and c-Jun, promoting fibroblast proliferation and migration. These processes are essential for fibroblast recruitment to the injury site, where they actively contribute to ECM deposition and remodeling. CTGF also stimulates the expression of collagen, fibronectin, and proteoglycans, further enhancing ECM stability and providing a scaffold for cellular activities involved in tissue repair.
The production of ECM components, particularly type I and III collagen, undergoes post-translational modifications that are essential for the assembly and cross-linking of collagen fibers. Lysyl oxidase (LOX) catalyzes the oxidative deamination of lysine residues on collagen molecules, promoting the formation of covalent cross-links between collagen fibers. This cross-linking is critical for increasing the tensile strength of the ECM, providing the necessary mechanical stability for the repaired tissue. Additionally, procollagen peptidases cleave procollagen into mature collagen, facilitating fiber assembly and organization within the ECM.
While the initial ECM deposition is disorganized, subsequent ECM remodeling is driven by both biochemical and mechanical cues. Fibroblasts and myofibroblasts respond to mechanotransduction signals, generated by the tension exerted on the wound matrix, through focal adhesions and integrin interactions with the ECM. This mechanical feedback modulates intracellular pathways such as focal adhesion kinase (FAK) and Rho GTPase, which influence cytoskeletal dynamics and guide the alignment of collagen fibers along lines of mechanical stress. Although this reorganization improves the mechanical properties of the tissue, incomplete alignment often results in the formation of scar tissue, which lacks the structural and functional properties of native tissue.
Moreover, other molecular signals, including vascular endothelial growth factor (VEGF) and platelet-derived growth factor (PDGF), contribute to the repair process by promoting angiogenesis and further fibroblast recruitment. VEGF stimulates the formation of new blood vessels through its interaction with VEGF receptor 2 (VEGFR2) on endothelial cells, while PDGF recruits fibroblasts and stimulates their proliferation via PDGF receptors (PDGFR). These growth factors ensure that the healing tissue receives an adequate supply of oxygen and nutrients, critical for sustaining the high metabolic demands of ECM synthesis and tissue repair.
In conclusion, the fibroblastic stage of tissue repair involves a highly regulated interplay of growth factors, signaling pathways, and mechanical cues that coordinate fibroblast proliferation, ECM production, and remodeling. These molecular processes ensure the formation of a stable and functional matrix, though the eventual outcome of scar tissue highlights the importance of regulated mechanical and biochemical inputs in achieving optimal tissue repair.
Remodeling Stage
The remodeling stage of tissue repair involves a highly dynamic and regulated process of extracellular matrix (ECM) reorganization, which is driven by the coordinated activity of cells like fibroblasts and myofibroblasts, enzymes, and intricate signaling pathways. Central to this process is the production and remodeling of collagen, the primary structural component of the ECM. Fibroblasts and myofibroblasts, under the influence of transforming growth factor-beta (TGF-β), produce large amounts of type I and type III collagen, which provide tensile strength and structural support. Lysyl oxidase (LOX) catalyzes the formation of covalent cross-links between collagen fibers, enhancing the mechanical stability of the remodeled tissue. This cross-linking is essential for increasing the tissue’s resistance to mechanical stress.
Matrix metalloproteinases (MMPs), especially MMP-1 (collagenase) and MMP-9 (gelatinase), degrade disorganized collagen fibers, allowing for their replacement with newly synthesized, properly aligned collagen. Tissue inhibitors of metalloproteinases (TIMPs) regulate MMP activity, maintaining a balance between collagen degradation and synthesis, which is essential for ensuring proper ECM remodeling. Dysregulation of this balance can lead to excessive fibrosis or inadequate tissue repair, resulting in compromised tissue function.
At the molecular level, myofibroblasts play a crucial role in tissue contraction and collagen alignment. These cells express alpha-smooth muscle actin (α-SMA), which allows them to generate contractile forces that align collagen fibers along the lines of mechanical stress, thereby improving the mechanical properties of the remodeled tissue. Integrin signaling between fibroblasts and the ECM is critical for transmitting mechanical signals, activating pathways such as focal adhesion kinase (FAK) and MAPK/ERK, which regulate cellular migration, ECM synthesis, and survival.
TGF-β is a key growth factor in ECM remodeling, promoting fibroblast proliferation, myofibroblast differentiation, and collagen production while also regulating the expression of MMPs and TIMPs. Connective tissue growth factor (CTGF) and platelet-derived growth factor (PDGF) further enhance fibroblast adhesion, migration, and ECM deposition, ensuring the sustained activity of cells involved in the remodeling process. The overproduction of ECM components by these cells can lead to excessive fibrosis, characterized by a dense, disorganized collagen network that compromises tissue flexibility and function.
In addition to collagen, other ECM components such as fibronectin, elastin, and proteoglycans contribute to the structural and functional properties of the remodeled tissue. Fibronectin is critical for cell adhesion and migration, while elastin provides resilience, allowing the ECM to stretch and recoil under mechanical stress. Proteoglycans and glycosaminoglycans (GAGs), by retaining water, ensure that the ECM remains hydrated and maintains its viscoelastic properties, supporting ongoing cellular activity and tissue repair.
Chronic activation of fibroblasts and continuous ECM synthesis can lead to fibrosis, resulting in the formation of scar tissue. This tissue is often stiffer and less elastic than normal tissue due to the dense collagen network, impairing its functional properties. Additionally, excessive ECM deposition can lead to tendon adhesions, where tendons become bound to surrounding tissues, restricting movement and impairing joint mobility. The molecular pathways involved in these processes include persistent inflammation, which sustains fibroblast and myofibroblast activation through pro-inflammatory cytokines such as interleukin-1 (IL-1) and tumor necrosis factor-alpha (TNF-α). These cytokines, along with TGF-β, promote fibroblast-to-myofibroblast transition, enhancing collagen production and ECM contraction, which contribute to adhesion formation and tissue stiffness.
Ultimately, the remodeling stage is a delicate balance of ECM synthesis and degradation, governed by a network of growth factors, cytokines, and enzymes. Effective regulation of these molecular mechanisms is critical to ensuring optimal tissue repair, preventing excessive fibrosis, and maintaining the functionality of the remodeled tissue. Understanding these processes at a molecular level can inform therapeutic strategies to modulate ECM remodeling and improve recovery outcomes.
| Healing stage | Cellular phase | Biophysical characteristics | Therapeutic intervention |
| Inflammation Stage | Vasodilation, invasion of platelets, and inflammatory cells (neutrophils, monocytes, and macrophages) are crucial processes in the body’s response to injury. These events are orchestrated by a complex interplay of chemical mediators, including histamine, bradykinin, and PGE2, each playing specific roles at the molecular level to injury, facilitating effective tissue repair and restoration of function. |
Swelling, erythema, warmth, pain | Cryotherapy, preferably with compression NSAIDs (unless contraindicated) Manual therapy |
| The strength of the scar depends on the temporary clot and stitches | Methods: electrical stimulation, laser therapy, ultrasound, PEMF, ESWT, isometric and BFR training. | ||
| Fibroblastic stage. |
Growth factors such as Transforming Growth Factor-beta 1 (TGF-β1), Bone Morphogenetic Proteins (BMP), and Connective Tissue Growth Factor (CTGF) play critical roles in wound healing by activating fibroblastic cells. Upon activation, these fibroblastic cells undergo proliferation and upregulate the synthesis of extracellular matrix (ECM) components including collagen, fibronectin, and proteoglycans. |
Expression of inflammatory markers | Manual therapy: passive range of motion, soft tissue mobilization, joint mobilization |
| The scar begins to gain tensile strength | Methods: electrical stimulation, laser therapy, ultrasound, PEMF, ESWT Therapeutic exercises: prescribed to achieve the goal of full weight bearing on the surgical limb while protecting the tissues (slow eccentric tempo) |
||
| Remodelling stage. |
The remodeling of the scar improves the organization and mechanical properties of the extracellular matrix (ECM) through a dynamic process involving the coordinated activity of various cells, enzymes, and signaling pathways. Fibroblasts and myofibroblasts play key roles in this process by synthesizing and remodeling collagen and other ECM components. |
The inflammation should subside; pain, if present, may be due to osteoarthritis, DOMS, re-damage to healing tissue | Manual therapy depending on needs, based on the patient’s assessment of the operated limb and the rest of the body; passive and active range of motion, soft tissue mobilization, including scar mobilization, joint mobilization |
| Methods: Typically discontinued at this stage unless patient assessment indicates special requirements for the surgical limb or rest of the body Therapeutic exercises: prescribed to increase active ROM and flexibility, build muscle strength and endurance, improve proprioception, motor control, and improve cardiovascular fitness | |||
| Abbreviations: BMP, bone morphogenetic protein; CTGF, connective tissue growth factor; DOMS, delayed onset muscle soreness; ECM, extracellular matrix; ESWT, extracorporeal shock wave therapy; NSAIDs, non-steroidal anti-inflammatory drugs; PEMF, pulsed electromagnetic field therapy; BFR, blood flow restriciton; PGE2, prostaglandin E2; ROM, range of motion; TGF-β1, transforming growth factor-β1. | |||
Early Mechanical Loading: Benefits and Risks
Early mechanical loading involves the controlled application of physical stress to injured tissues during the initial stages of healing, leveraging the mechanotransduction pathways that regulate cellular responses to mechanical stimuli. This process stimulates key molecular pathways such as integrin signaling and mechanosensitive ion channels, which trigger the activation of intracellular cascades like focal adhesion kinase (FAK), PI3K/Akt, and the MAPK/ERK pathways. These signaling pathways play a critical role in promoting cell proliferation, extracellular matrix (ECM) synthesis, and tissue remodeling. For example, the activation of integrins by mechanical forces leads to the clustering of integrins at focal adhesions, enhancing fibroblast activity and facilitating collagen synthesis, which strengthens the ECM and accelerates tissue repair. Additionally, mechanosensitive ion channels like TRPV4 allow calcium influx into cells, activating calcium-dependent signaling pathways, such as the calcineurin/NFAT pathway, which further promotes gene expression related to tissue repair and cellular adaptation.
While early mechanical loading supports tissue regeneration and revascularization by enhancing the production of vascular endothelial growth factor (VEGF), which stimulates angiogenesis, it also poses risks. Overloading or applying stress too early can lead to excessive inflammation, aggravating tissue damage and disrupting the healing process. Mechanical stress can upregulate matrix metalloproteinases (MMPs), such as MMP-1 and MMP-9, leading to excessive ECM degradation, which can impair tissue stability and prolong recovery. Thus, the balance between promoting tissue adaptation and avoiding overloading is crucial, requiring careful modulation of the intensity and duration of mechanical stress to prevent re-injury or chronic inflammation. Properly timed and dosed mechanical loading can optimize the molecular pathways involved in healing, leading to improved functional recovery without compromising tissue integrity.
Benefits of Early Mechanical Loading
Benefits of Early Mechanical Loading are rooted in its ability to enhance the healing process through targeted activation of cellular and molecular pathways that regulate tissue repair and regeneration. One of the primary benefits is the stimulation of mechanotransduction, the process by which cells convert mechanical stimuli into biochemical signals. This activates key signaling pathways such as integrin-mediated FAK activation, which promotes cell proliferation, migration, and the synthesis of extracellular matrix (ECM) components like collagen. Controlled mechanical loading also helps maintain the structural integrity of healing tissues by encouraging the proper alignment and organization of collagen fibers, which enhances the tensile strength and resilience of the tissue.
Another major benefit of early mechanical loading is its role in promoting angiogenesis, the formation of new blood vessels. By upregulating factors such as vascular endothelial growth factor (VEGF), mechanical loading improves vascularization of the injured area, ensuring that newly formed tissues receive adequate oxygen and nutrients. This increased blood flow is critical for supporting the metabolic demands of regenerating tissues and for clearing cellular debris and byproducts of inflammation.
Early mechanical loading also accelerates muscle and tendon repair by promoting the activation of satellite cells and fibroblasts, essential for muscle hypertrophy and tendon regeneration. It stimulates the production of growth factors like platelet-derived growth factor (PDGF) and transforming growth factor-beta (TGF-β), which orchestrate cellular activities that contribute to tissue repair, including cell proliferation and matrix synthesis.
In addition, early mechanical loading helps reduce excessive scar formation and fibrosis by regulating the balance between matrix metalloproteinases (MMPs) and their inhibitors (TIMPs). This balance prevents excessive ECM degradation while promoting ECM remodeling, ensuring that tissues heal with minimal scarring and improved functionality. Furthermore, mechanical loading plays a critical role in preventing muscle atrophy and joint stiffness by maintaining neuromuscular function and promoting the activation of motor units, which is essential for restoring strength and mobility.
Ultimately, early mechanical loading enhances functional recovery, allowing patients to regain strength, mobility, and range of motion more quickly than passive rehabilitation approaches. By properly timing and dosing mechanical stress, clinicians can harness the physiological benefits of early mechanical loading to optimize healing outcomes, reduce recovery time, and minimize the risk of long-term complications such as joint stiffness, muscle weakness, and chronic pain.
- 1. Enhanced Extracellular Matrix (ECM) Synthesis
Mechanical loading initiates a cascade of molecular events that are fundamental to the production and organization of collagen and proteoglycans, essential components of connective tissues. At the molecular level, mechanical stress activates mechanoreceptors on the surface of cells, such as integrins, which play a crucial role in translating mechanical signals into biochemical responses. These signals trigger intracellular pathways, including the activation of mitogen-activated protein kinases (MAPKs) and focal adhesion kinase (FAK), which regulate gene expression related to the synthesis of collagen and other ECM proteins. Fibroblasts, the primary cells responsible for collagen production, respond to mechanical loading by upregulating the transcription of collagen genes, particularly COL1A1 and COL3A1 for type I and type III collagen, respectively. This leads to increased collagen mRNA levels, which are then translated into pro-collagen proteins in the rough endoplasmic reticulum (ER) of fibroblasts.
Once synthesized, these pro-collagen molecules undergo post-translational modifications, including hydroxylation of proline and lysine residues, which is catalyzed by prolyl and lysyl hydroxylases in the ER. These modifications are critical for the stability and cross-linking of collagen molecules. After the pro-collagen molecules are secreted into the extracellular space, specific enzymes called procollagen N- and C-proteinases cleave the terminal propeptides, converting pro-collagen into mature collagen. These mature collagen molecules spontaneously assemble into fibrils, a process facilitated by the enzyme lysyl oxidase, which catalyzes the cross-linking of collagen fibers. This cross-linking is a crucial step in forming a strong and organized matrix capable of withstanding mechanical loads.
Proteoglycan synthesis, particularly the production of aggrecan, is similarly regulated by mechanical signals. Mechanical stress stimulates chondrocytes, the cells responsible for cartilage maintenance, to upregulate the expression of aggrecan core protein genes, as well as enzymes involved in the glycosylation of the core protein, which is essential for the formation of glycosaminoglycan (GAG) chains. These GAG chains, primarily chondroitin sulfate and keratan sulfate, are covalently attached to the aggrecan core protein, forming large macromolecular structures. The interaction between aggrecan and hyaluronic acid, mediated by link proteins, results in the formation of large proteoglycan aggregates, which are essential for the load-bearing properties of cartilage.
The retention of water molecules within the ECM, a property largely dependent on the negatively charged GAG chains, provides the cartilage with its characteristic viscoelasticity, enabling it to compress and rebound under mechanical loads. This water-binding capacity is crucial for maintaining cartilage hydration, nutrient diffusion, and overall joint lubrication. Moreover, mechanical loading influences the activity of matrix metalloproteinases (MMPs) and tissue inhibitors of metalloproteinases (TIMPs), which regulate the remodeling of the ECM by controlling collagen degradation and proteoglycan turnover. A delicate balance between these molecules is necessary to prevent excessive ECM degradation, which can lead to degenerative conditions such as osteoarthritis.
In addition to collagen and proteoglycan synthesis, mechanical loading also affects the production of other ECM components, such as elastin, fibronectin, and decorin. Elastin contributes to the elasticity of tissues, while fibronectin and decorin play roles in collagen fibrillogenesis and matrix organization. Decorin, a small leucine-rich proteoglycan, binds to collagen fibrils and regulates their spacing and diameter, ensuring proper matrix architecture. Together, these molecular processes orchestrate the complex and dynamic remodeling of the ECM in response to mechanical loading, optimizing tissue strength, flexibility, and resilience.
- 2. Promotion of Cell Proliferation and Differentiation
Mechanical loading triggers a complex molecular response that significantly impacts fibroblast proliferation, chondrocyte activity, and mesenchymal stem cell (MSC) differentiation, all of which are critical for tissue repair and regeneration. When fibroblasts are exposed to mechanical stress, they enter a proliferative state, increasing their numbers at the injury site. This proliferation is driven by mechanotransduction pathways that activate key signaling molecules, such as MAPKs and transforming growth factor-beta (TGF-β), promoting fibroblast division and ECM production. Fibroblasts synthesize essential ECM components, including type I and III collagen, elastin, and glycosaminoglycans, which are vital for forming a strong and resilient tissue matrix. As mechanical loading continues, fibroblasts increase their production of these molecules, reinforcing the structural integrity of tendons, ligaments, and other connective tissues. The cross-linking of collagen fibers, facilitated by enzymes like lysyl oxidase, further strengthens the matrix, making the tissue more resistant to mechanical forces. Additionally, mechanical loading enhances the secretion of growth factors, such as fibroblast growth factor (FGF) and platelet-derived growth factor (PDGF), which not only stimulate fibroblast proliferation but also recruit additional fibroblasts to the injury site, ensuring a robust supply of ECM-producing cells.
In cartilage, chondrocytes respond to mechanical loading by increasing their synthesis of type II collagen and aggrecan, two critical components of the cartilage matrix. Mechanical stimulation activates mechanosensitive ion channels and integrins on the surface of chondrocytes, which in turn initiate intracellular signaling cascades like the ERK and p38 MAPK pathways. These pathways lead to the upregulation of genes involved in ECM production, ensuring that the cartilage matrix remains resilient and capable of withstanding compressive forces. The enhanced activity of chondrocytes also promotes the synthesis of anabolic factors like insulin-like growth factor-1 (IGF-1) and bone morphogenetic proteins (BMPs), which further support matrix synthesis and repair. This coordinated response ensures that cartilage maintains its structural integrity and ability to absorb mechanical loads, which is crucial for joint function and mobility. Moreover, mechanical loading helps regulate the balance between anabolic and catabolic processes in chondrocytes, preventing the degradation of the ECM and protecting against degenerative conditions like osteoarthritis.
Mechanical loading also plays a pivotal role in guiding the differentiation of MSCs, multipotent stem cells capable of differentiating into various specialized cell types, including chondrocytes, osteoblasts, and fibroblasts. The type and magnitude of mechanical load influence MSC fate, with tensile loading favoring fibroblastic or osteoblastic differentiation and compressive loading promoting chondrogenic differentiation. This process is mediated by mechanotransduction pathways such as the Wnt/β-catenin and TGF-β/Smad signaling pathways, which regulate gene expression and direct MSCs toward specific lineages. The mechanical environment also influences the secretion of extracellular vesicles and exosomes by MSCs, which carry signaling molecules like microRNAs and proteins that modulate the behavior of neighboring cells and enhance tissue repair. By providing a steady supply of differentiated cells tailored to the specific tissue needs, MSCs play an essential role in the long-term regeneration of damaged tissues. This process of MSC differentiation is crucial not only for the initial repair but also for the ongoing maintenance and remodeling of tissues subjected to mechanical stress, ensuring their continued functionality and resilience.
- 3. Modulation of Inflammatory Responses
Mechanical loading exerts a highly orchestrated molecular influence on cytokine regulation, immune cell activity, and the overall inflammatory environment, which is critical for tissue repair and regeneration. At the core of this process are cytokines, small signaling proteins that regulate not only inflammation but also immune responses, cellular migration, and tissue homeostasis. Pro-inflammatory cytokines, such as interleukin-1 (IL-1), interleukin-6 (IL-6), and tumor necrosis factor-alpha (TNF-α), are typically elevated following injury to initiate the inflammatory phase of tissue repair. These molecules act by attracting immune cells like neutrophils and macrophages to the injury site, which help in the clearance of pathogens, dead cells, and other debris. However, mechanical loading plays a pivotal role in modulating this inflammatory response. Controlled mechanical stress has been shown to downregulate the expression of pro-inflammatory cytokines like TNF-α and IL-1, limiting excessive inflammation that can lead to chronic conditions like fibrosis or scar tissue formation. At the same time, mechanical loading upregulates anti-inflammatory cytokines such as interleukin-10 (IL-10) and transforming growth factor-beta (TGF-β), which are essential for shifting the immune environment from one focused on defense to one geared toward repair.
This cytokine modulation directly influences the behavior of immune cells at a molecular level. For instance, mechanical loading has been shown to affect the phenotypic polarization of macrophages, key immune cells that play a dual role in inflammation and repair. Early in the inflammatory response, macrophages predominantly exhibit the M1 phenotype, characterized by the production of pro-inflammatory cytokines and the promotion of tissue clearance. However, mechanical loading accelerates the transition of macrophages to the M2 phenotype, which is associated with anti-inflammatory cytokine production, tissue remodeling, and ECM synthesis. M2 macrophages secrete IL-10 and TGF-β, cytokines that not only suppress the inflammatory response but also enhance fibroblast activity, leading to increased collagen production and tissue regeneration. This shift in macrophage polarization is driven by mechanical stimuli that activate intracellular pathways, such as the PI3K/Akt pathway, which promotes M2-related gene expression and the release of repair-associated cytokines.
In addition to macrophages, other immune cells such as neutrophils and T-cells are also regulated by mechanical loading. Neutrophils, the first responders to injury, are attracted to the site by cytokines and chemokines, where they act to eliminate pathogens and initiate tissue repair. However, prolonged neutrophil activity can lead to excessive tissue damage due to the release of proteolytic enzymes and reactive oxygen species (ROS). Mechanical loading helps regulate neutrophil infiltration and promotes their timely clearance from the injury site, preventing secondary damage. Meanwhile, mechanical loading also influences T-cells, particularly regulatory T-cells (Tregs), which secrete anti-inflammatory cytokines like IL-10 and TGF-β, contributing to the suppression of inflammation and promoting tissue healing.
On a broader molecular level, mechanical loading activates various signaling pathways that govern cytokine production and immune cell behavior. Mechanotransduction, the process by which cells convert mechanical signals into biochemical responses, is mediated by integrins, stretch-activated ion channels, and cytoskeletal tension, all of which initiate intracellular signaling cascades. For instance, the nuclear factor kappa B (NF-κB) pathway, which is typically activated during inflammation and drives the expression of pro-inflammatory cytokines, can be inhibited by mechanical loading, reducing the levels of cytokines like TNF-α and IL-6. Similarly, the activation of the Smad signaling pathway by mechanical loading promotes TGF-β signaling, enhancing tissue repair processes and fibroblast activation.
Ultimately, the molecular effects of mechanical loading on cytokine regulation not only reduce the harmful effects of excessive inflammation but also create a balanced environment that supports tissue repair, regeneration, and long-term recovery. This modulation ensures that the inflammatory response is efficient and tightly controlled, avoiding the risk of chronic inflammation that could lead to degenerative diseases such as osteoarthritis or tendinopathies. By influencing cytokine profiles and immune cell activities at the molecular level, mechanical loading optimizes the conditions for tissue repair, accelerating healing and enhancing the structural and functional integrity of the injured tissues.
- 4. Enhanced Angiogenesis
Mechanical loading plays a pivotal molecular role in stimulating the production of vascular endothelial growth factor (VEGF), a crucial signaling protein that regulates angiogenesis, the formation of new blood vessels. VEGF is primarily produced by cells such as fibroblasts, endothelial cells, and macrophages in response to mechanical stress, hypoxia, and tissue damage. The activation of intracellular signaling pathways, particularly the PI3K/Akt and MAPK pathways, under mechanical loading leads to the upregulation of VEGF expression. This upregulation triggers a cascade of molecular events that promote endothelial cell proliferation, migration, and differentiation, which are critical steps in the formation of new capillary networks. The newly formed blood vessels enhance oxygen and nutrient delivery to the injury site, supporting the metabolic demands of reparative cells, such as fibroblasts and stem cells, which are actively synthesizing extracellular matrix components like collagen.
This increase in vascularization ensures that the healing tissues receive a steady supply of glucose, amino acids, and oxygen, which are essential for cellular respiration, protein synthesis, and overall tissue repair. Furthermore, VEGF-induced angiogenesis also facilitates the removal of metabolic waste products, carbon dioxide, and excess inflammatory mediators from the injury site, reducing local inflammation and preventing the accumulation of harmful by-products that could impede healing. The improved blood flow provided by new vessel formation supports an optimal healing environment, as it allows the efficient distribution of growth factors, cytokines, and other reparative molecules necessary for tissue regeneration. Additionally, VEGF promotes the recruitment of progenitor cells and other regenerative cells to the injury site, further enhancing tissue repair.
By promoting the development of an enhanced vascular network, mechanical loading not only supports immediate tissue healing but also ensures the long-term health and functionality of the regenerated tissues. The ongoing presence of new blood vessels helps maintain tissue viability, prevents ischemic damage, and supports the overall structural integrity of the repaired area. This process is particularly important in tissues such as tendons, ligaments, and cartilage, which have limited intrinsic vascularity and rely on VEGF-driven angiogenesis to restore function after injury. The molecular regulation of VEGF by mechanical loading underscores its critical role in ensuring that tissues can recover efficiently and sustain their physiological functions over time.
- 5. Improved Functional Recovery
Mechanical loading exerts a comprehensive molecular influence on the structural and functional properties of healing tissues, particularly by modulating key processes involved in extracellular matrix (ECM) remodeling, tissue strength, and flexibility. At the molecular level, mechanical loading activates mechanoreceptors such as integrins, which form focal adhesions between the cell cytoskeleton and the ECM. These mechanosensitive receptors translate mechanical signals into biochemical responses, triggering intracellular pathways like the RhoA/ROCK, MAPK, and PI3K/Akt pathways. These cascades enhance the transcription and translation of genes responsible for synthesizing critical ECM proteins, including type I, II, and III collagen, which are fundamental to tissue structure and tensile strength. Mechanical stress also increases the expression of lysyl oxidase, an enzyme responsible for cross-linking collagen fibrils, further solidifying and strengthening the tissue matrix. This cross-linking is essential for withstanding mechanical forces, contributing to the long-term durability of tendons, ligaments, and other load-bearing tissues.
Mechanical loading also stimulates the production of elastin, another key ECM component that provides tissues with elasticity and the ability to return to their original shape after deformation. Elastin synthesis is regulated by growth factors such as fibroblast growth factor-2 (FGF-2) and transforming growth factor-beta (TGF-β), which are upregulated in response to mechanical cues. Elastin fibers, in conjunction with collagen, create a dynamic matrix that allows tissues to flex and stretch without compromising their structural integrity. This balance between tensile strength and elasticity is crucial for tissues that undergo repetitive motion or mechanical strain, such as muscles, ligaments, and skin.
In parallel, mechanical loading enhances the synthesis of glycosaminoglycans (GAGs), including hyaluronic acid and chondroitin sulfate, which are critical for tissue hydration and viscoelasticity. These large polysaccharides bind significant amounts of water, creating a hydrated gel-like matrix that provides cushioning and lubrication to tissues, particularly in cartilage and joint spaces. GAGs contribute to the viscoelastic properties of tissues, enabling them to absorb compressive forces while maintaining flexibility. This hydration effect is essential for preventing tissue dehydration and brittleness, particularly in cartilage, where water retention is crucial for maintaining the smooth, frictionless surface necessary for joint movement.
On a cellular level, mechanical loading influences the behavior of fibroblasts, the primary cells responsible for ECM production. Fibroblasts respond to mechanical cues by proliferating and increasing their secretion of ECM proteins, including collagen and elastin, thereby accelerating the remodeling of the damaged tissue. This cellular activity is tightly regulated by molecular pathways, such as the mechanosensitive YAP/TAZ signaling pathway, which controls fibroblast differentiation and ECM synthesis. Additionally, mechanical loading induces the expression of matrix metalloproteinases (MMPs) and tissue inhibitors of metalloproteinases (TIMPs), which work together to regulate ECM degradation and remodeling. This balance between ECM synthesis and degradation ensures that the tissue is continuously remodeled and reinforced in response to mechanical stress, preventing scar tissue formation and promoting functional recovery.
Moreover, mechanical loading enhances angiogenesis through the upregulation of vascular endothelial growth factor (VEGF), promoting the formation of new blood vessels to supply the healing tissue with oxygen, nutrients, and growth factors. This improved vascularization supports the metabolic needs of fibroblasts, chondrocytes, and other reparative cells, ensuring efficient ECM synthesis and tissue repair. Enhanced blood flow also aids in the removal of waste products and inflammatory mediators, reducing inflammation and promoting a healthier healing environment.
Ultimately, the molecular response to mechanical loading leads to the alignment of collagen fibers along the lines of stress, enhancing the tissue’s tensile strength and flexibility. This organized collagen matrix is better suited to withstand mechanical loads and prevent re-injury, ensuring that the healing tissue regains its functional capacity. By continuously modulating the production of ECM components and regulating cellular activity, mechanical loading optimizes the biomechanical properties of healing tissues, enabling them to recover their structural integrity, elasticity, and resilience. This process is particularly important in rehabilitation, as it allows tissues to adapt to increasing physical demands, reducing the risk of future injuries and supporting long-term functional recovery.
Risks and Considerations
Although early mechanical loading can provide substantial advantages in promoting tissue repair and enhancing recovery, it also comes with inherent risks that must be thoughtfully managed. Excessive or improperly timed mechanical stress can lead to delayed healing, re-injury, or even permanent damage to the affected area. Overloading healing tissues too soon may result in excessive inflammation, compromised tissue integrity, or impaired collagen formation, which can ultimately slow down the recovery process. Additionally, individual factors such as the type and severity of the injury, patient age, and overall health can influence the body’s tolerance to mechanical loading.
Therefore, careful assessment and monitoring by healthcare professionals are essential to tailor mechanical loading protocols to each patient’s specific needs and recovery stage. This helps ensure that the mechanical stimuli provided during rehabilitation are sufficient to promote healing without causing further harm. Incorporating a gradual increase in load, based on real-time feedback from the patient’s response, can mitigate risks and optimize recovery outcomes. Ultimately, understanding and addressing these risks is crucial for developing rehabilitation programs that maximize the positive effects of mechanical loading while safeguarding the healing process from potential setbacks.
- 1. Risk of Exacerbating Injury
Excessive mechanical loading, or overloading, at the molecular level disrupts the delicate balance of tissue repair, leading to significant setbacks in the healing process and potentially causing chronic issues. When tissues are subjected to mechanical stress that exceeds their regenerative capacity, cellular signaling pathways such as the mechanosensitive integrin and focal adhesion kinase (FAK) pathways are overstimulated, resulting in the dysregulation of key molecules involved in tissue repair. Overloading can increase the production of pro-inflammatory cytokines such as interleukin-1 (IL-1) and tumor necrosis factor-alpha (TNF-α), which exacerbate inflammation and impair the synthesis of extracellular matrix (ECM) components like collagen and elastin. This excessive inflammation hinders fibroblast and chondrocyte activity, delaying collagen fibrillogenesis and disrupting the proper alignment of newly formed collagen fibers, which are essential for restoring tissue tensile strength and flexibility. Overloading also accelerates the activity of matrix metalloproteinases (MMPs), enzymes responsible for ECM degradation, leading to the breakdown of newly formed matrix and further compromising tissue integrity.
At the cellular level, excessive mechanical stress can cause fibroblasts, chondrocytes, and other reparative cells to undergo apoptosis or senescence, reducing their ability to produce the ECM components required for effective tissue repair. The overproduction of reactive oxygen species (ROS) in response to high mechanical stress further contributes to cellular damage and oxidative stress, impairing the overall healing environment. Additionally, overloading can alter the mechanotransduction pathways responsible for sensing and responding to mechanical cues, such as the YAP/TAZ signaling axis, leading to aberrant tissue remodeling and scar formation. This disruption of normal cellular processes increases the vulnerability of the tissue to re-injury, as the damaged matrix cannot properly support mechanical loads, leading to prolonged recovery times.
Improper timing of mechanical loading, especially when introduced too early in the healing process, exacerbates these negative molecular effects. Early loading can disrupt the initial inflammatory phase, characterized by the infiltration of immune cells and the secretion of growth factors necessary for initiating tissue repair. This disruption can prevent the transition from the inflammatory phase to the proliferative phase, where fibroblasts and other reparative cells begin synthesizing new ECM components. Premature loading also increases the risk of reactivating pro-inflammatory signaling pathways, further delaying tissue repair and increasing the likelihood of chronic inflammation. Conversely, delaying mechanical loading for too long can lead to detrimental molecular changes, such as decreased production of ECM components, muscle atrophy, and joint stiffness due to the lack of mechanical stimuli required for maintaining tissue health and function.
Clinicians must carefully modulate the intensity, frequency, and timing of mechanical loading to avoid these adverse outcomes. Monitoring molecular markers of inflammation, tissue repair, and cellular stress can provide valuable insights into the patient’s response to loading. This allows for the adjustment of rehabilitation protocols to ensure that mechanical stress is applied in a manner that supports, rather than impairs, tissue regeneration. By doing so, clinicians can optimize the mechanical environment to facilitate the proper alignment of collagen fibers, enhance ECM synthesis, and promote functional recovery, all while minimizing the risk of overloading and its associated molecular consequences.
- 2. Inflammation and Tissue Damage
Inappropriate mechanical loading can significantly increase the production of pro-inflammatory cytokines, such as interleukin-1 (IL-1), tumor necrosis factor-alpha (TNF-α), and interleukin-6 (IL-6), which play pivotal roles in initiating and perpetuating inflammation. These cytokines are key molecular mediators that recruit and activate immune cells, such as neutrophils and macrophages, to the injury site, where they release additional inflammatory mediators and reactive oxygen species (ROS). While inflammation is necessary for initiating the healing process, excessive or prolonged production of these pro-inflammatory cytokines can lead to a chronic inflammatory state, characterized by continuous immune cell activation and elevated ROS levels, resulting in tissue damage and impaired repair. This chronic inflammation triggers a feed-forward loop that promotes ECM degradation and tissue degeneration, undermining the beneficial effects of controlled mechanical loading.
At the molecular level, inappropriate loading upregulates the activity of nuclear factor kappa B (NF-κB), a transcription factor that drives the expression of pro-inflammatory cytokines. Elevated NF-κB activity maintains high levels of IL-1, TNF-α, and IL-6, which not only exacerbate inflammation but also upregulate matrix metalloproteinases (MMPs), enzymes responsible for ECM breakdown. Increased MMP activity, particularly MMP-1, MMP-3, and MMP-9, leads to the degradation of essential ECM components such as collagen and elastin, weakening the structural integrity of the tissue. Excessive ECM degradation disrupts the mechanical stability of the healing tissue, making it more vulnerable to further injury and delaying the repair process. In addition, heightened MMP activity can interfere with collagen fibrillogenesis, preventing the proper alignment and cross-linking of collagen fibers, which are necessary for restoring tissue strength and flexibility.
The persistence of pro-inflammatory cytokines and the overactivity of MMPs can also inhibit the transition from the inflammatory phase to the proliferative phase of tissue healing, where fibroblasts and other reparative cells synthesize new ECM. This disruption prolongs the inflammatory response, reducing the effectiveness of tissue regeneration and promoting scar tissue formation instead of functional tissue repair. Moreover, prolonged inflammation and MMP-driven ECM degradation can lead to the development of fibrotic tissue, which is less flexible and more prone to further damage under mechanical stress.
To mitigate these molecular consequences, it is essential for clinicians to carefully monitor the patient’s inflammatory status during rehabilitation, using biomarkers such as elevated levels of IL-1, TNF-α, and MMPs to gauge the intensity of the inflammatory response. Adjusting the mechanical loading regimen based on these molecular indicators can help maintain a balanced inflammatory response that supports tissue repair without exacerbating injury. By modulating the loading intensity, duration, and frequency, clinicians can regulate cytokine production and MMP activity, ensuring that mechanical loading promotes ECM synthesis and tissue regeneration while minimizing excessive degradation and inflammation. This careful balancing of molecular signals is crucial for optimizing the healing process and preventing the development of chronic inflammation and tissue degeneration.
- 3. Individual Variability
The molecular response to mechanical loading is highly influenced by patient-specific factors, which necessitate personalized rehabilitation strategies to optimize outcomes. Age, for instance, plays a critical role at the molecular level, as older individuals often experience diminished cellular responses to mechanical stimuli due to reduced fibroblast and chondrocyte activity, decreased collagen synthesis, and slower ECM turnover. This is partly driven by the age-related decline in growth factor signaling, such as reduced levels of insulin-like growth factor (IGF-1) and transforming growth factor-beta (TGF-β), which are essential for tissue repair and regeneration. Older patients also tend to have elevated levels of pro-inflammatory cytokines like IL-6, which can lead to prolonged inflammation and delayed healing. In contrast, younger individuals typically exhibit more robust cellular responses to mechanical loading, with greater fibroblast proliferation, enhanced collagen production, and quicker resolution of inflammation. This variability in molecular response underscores the need for a more conservative, gradual approach to loading in older populations, while younger patients may tolerate more aggressive loading protocols without the same risks of overloading or re-injury.
Sex also contributes to molecular differences in tissue response to loading. For example, estrogen has been shown to modulate collagen synthesis and influence the inflammatory response, with higher estrogen levels promoting faster tissue healing and enhanced collagen organization. Women may therefore have different loading tolerances compared to men, particularly in tissues like ligaments, where hormonal fluctuations can affect ECM remodeling. Genetic background is another critical factor, as genetic variations in key ECM proteins, growth factors, and cytokines can influence individual responses to mechanical stress. For instance, polymorphisms in the COL1A1 gene, which encodes type I collagen, can affect collagen fiber alignment and strength, making certain individuals more susceptible to tendon and ligament injuries under mechanical loading.
Injury severity and overall health status also affect the molecular response to mechanical loading. Patients with chronic conditions like diabetes or cardiovascular disease may have impaired angiogenesis and reduced VEGF production, leading to insufficient blood vessel formation and compromised nutrient delivery to healing tissues. These conditions can also elevate levels of advanced glycation end-products (AGEs), which cross-link collagen fibers inappropriately, reducing tissue elasticity and increasing stiffness, thereby altering the optimal mechanical loading parameters. In such cases, rehabilitation must be carefully tailored to account for these systemic effects on tissue repair mechanisms.
Finally, personalizing the rehabilitation program requires consideration of the patient’s metabolic state, immune function, and inflammatory status, all of which are influenced by factors such as diet, physical activity, and co-existing conditions. For instance, patients with heightened levels of systemic inflammation may exhibit an exaggerated pro-inflammatory cytokine response to mechanical loading, which could lead to excessive tissue degradation via increased matrix metalloproteinase (MMP) activity. Clinicians must, therefore, monitor molecular markers of inflammation and ECM turnover, such as IL-1, TNF-α, and MMPs, to adjust the intensity and duration of loading accordingly. By taking into account these patient-specific molecular factors, clinicians can design personalized rehabilitation programs that minimize the risk of adverse outcomes while promoting optimal tissue repair and functional recovery.
- 4. Monitoring and Adjustment
Biomarker monitoring at the molecular level offers a powerful tool for optimizing rehabilitation by providing detailed feedback on the tissue’s biochemical state in response to mechanical loading. As tissues undergo repair, various molecular signals are activated, which can be tracked through biomarkers to provide real-time insights into inflammation, tissue degradation, ECM remodeling, and cellular regeneration. Key inflammatory biomarkers like interleukin-1 (IL-1), tumor necrosis factor-alpha (TNF-α), and interleukin-6 (IL-6) are critical indicators of early inflammatory responses. These cytokines drive immune cell recruitment and local tissue response, but prolonged elevation can indicate excessive inflammation, suggesting the need for modulation in the loading regimen. Persistent elevation of these pro-inflammatory biomarkers could signal the onset of chronic inflammation, which is detrimental to tissue repair, as it promotes catabolic processes that degrade ECM proteins such as collagen and elastin.
At the same time, biomarkers of ECM turnover such as matrix metalloproteinases (MMPs) and their tissue inhibitors (TIMPs) provide crucial information on the balance between matrix degradation and synthesis. MMPs, particularly MMP-1, MMP-3, and MMP-9, are involved in breaking down damaged ECM components, allowing for new matrix deposition. However, excessive MMP activity, indicated by elevated levels, may result in the over-degradation of newly synthesized ECM, compromising the structural integrity of the healing tissue. TIMPs, on the other hand, regulate MMP activity and ensure controlled ECM remodeling. The balance between MMPs and TIMPs, monitored through their respective biomarker levels, allows clinicians to assess whether the tissue is undergoing proper matrix remodeling or if degradation is outpacing repair, signaling the need to adjust the intensity or duration of mechanical loading.
Additionally, growth factors such as transforming growth factor-beta (TGF-β), insulin-like growth factor-1 (IGF-1), and vascular endothelial growth factor (VEGF) play crucial roles in promoting cellular proliferation, differentiation, and tissue regeneration. TGF-β is particularly important for fibroblast activation and collagen synthesis, critical for ECM formation. Tracking TGF-β levels can provide insights into whether the tissue is progressing from the inflammatory phase into the proliferative phase, where tissue regeneration is dominant. Similarly, IGF-1 supports anabolic processes, stimulating muscle and connective tissue growth, while VEGF promotes angiogenesis, ensuring an adequate blood supply to the healing tissues. Elevated levels of these growth factors typically indicate successful tissue repair and suggest that the loading regimen is appropriate for stimulating healing.
Moreover, collagen synthesis biomarkers such as procollagen type I C-peptide (PICP) and procollagen type III N-terminal propeptide (PIIINP) are direct indicators of collagen production, which is fundamental for restoring tissue strength and flexibility. Monitoring the levels of these biomarkers allows clinicians to determine the effectiveness of mechanical loading in stimulating collagen deposition. If collagen synthesis biomarkers increase in response to loading, it suggests that the tissue is responding favorably, with newly synthesized collagen fibers being deposited and organized in alignment with mechanical stress. This is a key indicator that the tissue is regaining its tensile strength and mechanical properties.
Oxidative stress markers such as reactive oxygen species (ROS) or malondialdehyde (MDA) can also be important in evaluating the tissue’s response to mechanical loading. Excessive ROS generation, often associated with high-intensity mechanical loading, can cause oxidative damage to cells and ECM components, leading to impaired healing. Monitoring oxidative stress biomarkers can help clinicians identify if the mechanical loading intensity is causing more harm than benefit, allowing them to adjust the regimen to reduce tissue stress and promote a healthier healing environment.
In conclusion, by regularly monitoring a comprehensive panel of biomarkers—including inflammatory cytokines, MMPs, growth factors, collagen synthesis indicators, and oxidative stress markers—clinicians can gain a molecular-level understanding of how tissues are responding to mechanical loading. This enables precise adjustments to the rehabilitation protocol, optimizing the intensity, timing, and type of loading to maximize tissue healing while minimizing the risk of overloading or re-injury. Biomarker-driven rehabilitation provides a personalized, data-driven approach that enhances the overall effectiveness of mechanical loading, ensuring that healing is progressing optimally and safely for each individual patient.
- 5. Rehabilitation Protocol Design
Progressive loading is a foundational principle in rehabilitation, promoting tissue adaptation at the molecular level through a carefully controlled increase in mechanical stress. As tissues respond to progressive mechanical loading, a cascade of molecular events is triggered that enhances the structural and functional properties of the healing tissue. Initially, low-intensity loading activates mechanosensitive receptors, such as integrins, on the surface of fibroblasts and chondrocytes. These receptors initiate intracellular signaling pathways, including the mitogen-activated protein kinase (MAPK) and phosphoinositide 3-kinase (PI3K)/Akt pathways, which regulate gene expression related to ECM synthesis. This controlled stimulation promotes the production of essential ECM components like type I and III collagen, which are critical for rebuilding tissue strength and resilience. Gradual increases in mechanical load ensure that these proteins are synthesized in a structured, organized manner, allowing collagen fibers to align along the lines of stress, increasing tensile strength and resistance to future mechanical strain.
Progressive loading also modulates the activity of matrix metalloproteinases (MMPs), enzymes responsible for ECM turnover. During the early phases of loading, a balanced MMP-to-TIMP (tissue inhibitors of metalloproteinases) ratio is maintained, ensuring that damaged ECM components are cleared away, making room for newly synthesized collagen and elastin fibers. However, an excessive increase in load too early could shift this balance, leading to over-activation of MMPs and excessive matrix degradation, which would weaken the tissue structure. This is why progressive loading protocols are crucial—they provide enough mechanical stimulus to promote ECM synthesis while avoiding overload-induced tissue breakdown.
In addition to promoting collagen synthesis, progressive mechanical loading also enhances the production of other critical ECM components, such as elastin and proteoglycans like aggrecan. These molecules are responsible for maintaining tissue elasticity and hydration, particularly in cartilage, tendons, and ligaments. Elastin synthesis, regulated by factors like TGF-β and fibroblast growth factor (FGF), allows tissues to recover from deformation without sustaining damage, while proteoglycans bind water to provide shock absorption and lubrication, especially in joint tissues. By incrementally increasing loading intensity and complexity, progressive loading facilitates the deposition and organization of these molecules within the ECM, enhancing the tissue’s ability to withstand both tensile and compressive forces.
Another key molecular effect of progressive loading is its role in angiogenesis, the formation of new blood vessels. Mechanical stress induces the expression of vascular endothelial growth factor (VEGF), a potent angiogenic factor that promotes the proliferation and migration of endothelial cells, leading to the formation of capillaries that supply oxygen and nutrients to the healing tissue. This increase in blood supply is essential for sustaining the metabolic needs of fibroblasts, chondrocytes, and other reparative cells, ensuring that they have the resources necessary for continued ECM synthesis and tissue remodeling. Progressive loading ensures that angiogenesis is well-timed with tissue repair, supporting the metabolic demands of the healing tissue and preventing ischemic damage.
On a cellular level, progressive loading also influences the behavior of stem cells, particularly mesenchymal stem cells (MSCs), which play a critical role in tissue regeneration. Mechanical loading can direct MSC differentiation into fibroblasts, chondrocytes, or osteoblasts, depending on the type and magnitude of the load applied. Low-intensity loading may promote fibroblastic differentiation, enhancing collagen production in tendons and ligaments, while more targeted loading can encourage chondrogenic differentiation, crucial for cartilage repair. This controlled stem cell differentiation is essential for replenishing the cellular pool necessary for ongoing tissue repair and regeneration. Progressive loading helps ensure that stem cell recruitment and differentiation are properly aligned with the tissue’s needs, optimizing the overall healing process.
Furthermore, progressive loading plays a vital role in the regulation of inflammatory mediators. In the initial phases of injury, pro-inflammatory cytokines such as interleukin-1 (IL-1), IL-6, and tumor necrosis factor-alpha (TNF-α) are elevated to initiate the healing process. However, prolonged or excessive inflammation can impair healing by causing tissue damage and increasing MMP activity. Progressive loading, when properly applied, helps modulate the inflammatory response by reducing the expression of these pro-inflammatory cytokines and promoting the release of anti-inflammatory factors like interleukin-10 (IL-10) and transforming growth factor-beta (TGF-β). This shift toward an anti-inflammatory state creates an environment conducive to tissue repair, minimizing the risk of chronic inflammation and fibrosis.
In conclusion, progressive loading is a finely tuned molecular process that gradually strengthens tissues by promoting balanced ECM synthesis, controlled inflammation, and proper cell differentiation. By progressively increasing mechanical demands, tissues can adapt and reorganize at the molecular level, enhancing their structural integrity, flexibility, and ability to withstand future stresses. This approach minimizes the risks of overloading and re-injury while maximizing functional recovery, making it an essential component of rehabilitation strategies. Coupled with regular biomarker monitoring and functional assessments, progressive loading enables clinicians to design personalized, data-driven rehabilitation protocols that support long-term tissue health and resilience.
Clinical Guidelines and Recommendations
To maximize the benefits and minimize the risks of early mechanical loading, the following clinical guidelines and recommendations should be considered: First, it is essential to ensure that the loading is progressively increased, allowing the tissues to adapt and strengthen over time without risking injury. Careful assessment of the patient’s condition, including the stage of healing and individual tolerance to loading, is crucial. Monitoring for any signs of overloading, such as increased pain or inflammation, should guide adjustments in therapy. Additionally, proper technique and posture during exercises or activities should be emphasized to avoid undue stress on vulnerable areas. Incorporating rest and recovery periods between loading sessions is equally important to support healing and prevent fatigue-related injuries. Clinicians should also stay informed about the latest evidence-based practices to optimize outcomes while safeguarding against potential complications.
1. Early Initiation: Beginning mechanical loading as soon as it is deemed safe based on the patient’s injury and condition plays a crucial role in preventing the negative physiological effects of immobilization, such as muscle atrophy, joint stiffness, and tissue degeneration. On a molecular level, prolonged immobilization leads to the downregulation of anabolic pathways and the activation of catabolic processes. For example, the absence of mechanical stress reduces IGF-1 (Insulin-like Growth Factor-1) signaling, a key factor in promoting protein synthesis and muscle regeneration, while increasing the activity of the ubiquitin-proteasome system, which accelerates muscle breakdown.
Early mechanical loading reverses this process by activating mechanosensitive pathways in tissues. Integrins, which are transmembrane receptors sensitive to mechanical stimuli, are crucial in translating physical forces into biochemical signals. These integrins activate intracellular signaling cascades such as the MAPK (mitogen-activated protein kinase) and PI3K/Akt pathways, both of which promote cell proliferation, tissue repair, and protein synthesis. In muscle, early initiation of loading activates mTORC1 (mechanistic target of rapamycin complex 1), which enhances protein synthesis and muscle hypertrophy, countering muscle wasting.
In connective tissues like tendons and ligaments, early mechanical loading stimulates fibroblasts and tenocytes to upregulate the production of structural proteins like collagen I and collagen III, which are essential for restoring tissue integrity and tensile strength. This process is mediated through the activation of TGF-β (Transforming Growth Factor-beta) signaling, which plays a key role in regulating ECM (extracellular matrix) remodeling and scar tissue formation. Mechanical loading also triggers the YAP/TAZ (Yes-associated protein/transcriptional coactivator with PDZ-binding motif) pathway, which is vital for cellular mechanotransduction, influencing gene expression that supports tissue repair and regeneration.
Furthermore, early loading prevents the detrimental effects of immobilization on cartilage. Mechanical stimulation is crucial for chondrocyte activity and ECM production, especially the synthesis of aggrecan and type II collagen, which are important for cartilage resilience and shock absorption. Prolonged immobilization can result in the degradation of these ECM components, leading to joint stiffness and cartilage degeneration. Early mechanical loading restores chondrocyte function by activating the Wnt/β-catenin and SOX9 pathways, promoting cartilage repair and maintenance.
Clinically, the timing of mechanical loading initiation should be carefully assessed. Early loading, initiated too soon, can risk disrupting the healing process by inducing excessive inflammation or causing re-injury. Therefore, clinicians must rely on both molecular markers of tissue healing, such as reduced levels of pro-inflammatory cytokines like TNF-α and IL-6, as well as functional assessments, to determine when it is safe to introduce loading. Once initiated, early mechanical loading not only accelerates tissue repair by activating key molecular pathways but also prevents the chronic effects of immobilization, ultimately promoting a faster, more efficient recovery.
In summary, early initiation of mechanical loading supports tissue repair at the molecular level by activating anabolic signaling pathways, promoting protein synthesis, and restoring ECM integrity, all while countering the catabolic effects of immobilization. By carefully assessing the patient’s condition and timing the introduction of loading, clinicians can enhance recovery outcomes and prevent long-term functional impairments.
2. Controlled Loading: Controlled mechanical loading is a key component of rehabilitation, where the intensity, duration, and complexity of exercises are carefully regulated to stimulate tissue adaptation without overwhelming the healing structures. From a molecular biology perspective, controlled loading promotes optimal cellular responses and ensures that the body’s tissues strengthen and repair in a way that minimizes the risk of re-injury.
At the cellular level, tissues such as muscles, tendons, ligaments, and bones respond to mechanical loading through mechanotransduction, a process where physical forces are converted into biochemical signals. This is primarily mediated by mechanosensitive receptors, including integrins, stretch-activated ion channels, and focal adhesion complexes. These receptors sense the mechanical environment and trigger intracellular signaling pathways such as the MAPK/ERK, PI3K/Akt, and RhoA/ROCK pathways, which regulate cytoskeletal remodeling, gene expression, and protein synthesis necessary for tissue adaptation and repair.
Low-intensity controlled loading activates these pathways in a gradual manner, allowing cells to respond appropriately to mechanical stress. For example, in muscle tissue, controlled loading stimulates mTORC1 (mechanistic target of rapamycin complex 1) signaling, which promotes protein synthesis and muscle hypertrophy while inhibiting protein degradation. This controlled activation allows for muscle fiber repair and growth without causing excessive cellular damage that could occur with sudden, high-intensity loading.
In tendon and ligament tissues, controlled loading enhances the synthesis and alignment of collagen fibers by promoting fibroblast and tenocyte activity. These cells respond to mechanical stress by upregulating the production of key ECM proteins, such as collagen type I and fibronectin, which are essential for strengthening the tissue’s structure. Overloading the tissue too early, however, can trigger excessive activity of matrix metalloproteinases (MMPs), which degrade ECM components and can weaken the tissue if not properly regulated. Gradually increasing the load helps maintain a balance between ECM synthesis and degradation, ensuring that tissue strength is built up in a controlled and sustainable manner.
In bone tissue, controlled mechanical loading stimulates osteoblasts to promote bone formation and mineralization. Osteocytes, which act as mechanosensors within the bone, respond to controlled mechanical stress by activating the Wnt/β-catenin signaling pathway, which is critical for osteogenesis and bone remodeling. Gradual loading also activates the release of sclerostin, a protein that regulates bone formation by modulating Wnt signaling. Overloading can disrupt this balance and lead to bone resorption, increasing the risk of stress fractures or other injuries. Therefore, controlling the intensity of loading is essential to maintaining the delicate balance between bone formation and resorption.
Controlled loading also modulates the inflammatory response during the healing process. Mechanical stress of the appropriate intensity reduces the expression of pro-inflammatory cytokines like TNF-α, IL-6, and IL-1β, which are associated with tissue damage and prolonged inflammation. At the same time, controlled loading upregulates anti-inflammatory molecules like IL-10 and TGF-β, which promote tissue repair and reduce fibrosis. This fine-tuned modulation of the inflammatory response ensures that tissue repair proceeds efficiently without excessive scar tissue formation, which can impair functionality.
Additionally, controlled loading activates mechanosensitive transcription factors like YAP/TAZ in various tissues, including muscles, tendons, and bones. These transcription factors translocate to the nucleus in response to mechanical stress and regulate the expression of genes involved in cell proliferation, differentiation, and ECM remodeling. This process is crucial for long-term tissue adaptation, as it allows cells to ‘learn’ from mechanical stimuli and adjust their behavior to strengthen and protect the tissue.
By gradually increasing the complexity and intensity of exercises, clinicians can optimize these molecular pathways, promoting tissue resilience and reducing the risk of overloading. For example, progressive loading in rehabilitation protocols enhances angiogenesis and capillary density in healing muscle tissues, improving oxygen and nutrient delivery to the affected area. This controlled vascular response is necessary for supporting tissue regeneration and maintaining metabolic activity during the recovery process.
In conclusion, controlled loading plays a critical role in orchestrating the molecular processes involved in tissue repair and strengthening. By ensuring a gradual and progressive increase in mechanical stress, clinicians can activate beneficial molecular pathways while minimizing the risk of tissue overload and re-injury. This balanced approach to rehabilitation promotes long-term recovery and improved tissue function.
3. Patient Education: Educating patients on the importance of adhering to prescribed mechanical loading protocols is fundamental to ensuring successful rehabilitation and minimizing the risk of complications. Mechanical loading, when applied correctly, stimulates critical molecular pathways responsible for tissue repair, adaptation, and regeneration. However, if patients deviate from the plan—either by overloading or underloading the affected tissues—these molecular processes can be disrupted, potentially leading to delayed healing, re-injury, or chronic dysfunction. Educating patients on these potential outcomes fosters a deeper understanding of the importance of sticking to the plan, which in turn improves adherence and promotes optimal recovery.
Clear communication is key to helping patients grasp why each element of their rehabilitation is crucial. For example, explaining how early, controlled mechanical loading activates pathways like mTORC1 for muscle repair, TGF-β for collagen synthesis, and YAP/TAZ for cell proliferation can reinforce the idea that following the protocol step-by-step enables their body to heal and adapt appropriately. On the flip side, overloading the tissue too soon might trigger excessive MMP activity, leading to ECM degradation rather than repair, or cause inflammation to persist due to increased levels of IL-6 and TNF-α. These explanations help patients understand the scientific rationale behind gradual loading and why patience is crucial to long-term success.
Providing patients with detailed information about the benefits of following mechanical loading protocols also includes helping them understand the consequences of non-compliance. For instance, if a patient skips prescribed exercises or increases loading prematurely, it can lead to imbalanced muscle development, joint instability, or tendon rupture due to insufficient collagen maturation. By breaking down these complex issues into easily understandable terms, clinicians can emphasize that even small deviations from the plan could set back their recovery.
Patients should also be educated on the importance of timing and progression in mechanical loading. Introducing mechanical stress too early or advancing too quickly can overwhelm the body’s capacity for repair, leading to cellular damage and inflammation. Conversely, insufficient loading can result in muscle atrophy, fibrosis, or disuse osteoporosis, where bones weaken due to a lack of stimulation. Teaching patients that mechanical stimuli directly influence gene expression and protein synthesis, such as collagen type I in tendons or aggrecan in cartilage, encourages them to follow a slow and steady approach to healing. This molecular insight can help patients appreciate the need for adherence to a well-structured loading protocol.
Beyond scientific explanations, clinicians should provide continuous support and follow-up to ensure patients feel engaged and motivated throughout their rehabilitation journey. Ongoing communication, regular check-ins, and reminders about the progress they are making—both functionally and at a cellular level—can keep them committed to the prescribed regimen. Patients who understand how their actions influence the body’s natural healing processes will feel more empowered to actively participate in their recovery, reducing the likelihood of skipping sessions or pushing themselves too hard.
It’s also essential to address psychological and emotional factors that can affect adherence. Some patients may experience frustration due to perceived slow progress, which can lead them to ignore prescribed guidelines and attempt more intense exercises prematurely. Others might fear re-injury and hesitate to fully engage in prescribed loading exercises. Addressing these concerns through education and reassurance—explaining, for example, how progressive loading reduces the likelihood of future injury by strengthening tissue at the molecular level—can foster confidence and reduce anxiety.
Clinicians can enhance patient engagement by using visual aids, diagrams, or digital tools that demonstrate how their tissues respond to mechanical loading. Interactive tools that track a patient’s progress, showing how improvements in strength, flexibility, and pain reduction correspond with cellular changes, can further motivate patients to stay on course. Integrating real-time feedback into the process can reinforce their understanding of how compliance directly influences their healing at a fundamental biological level.
Moreover, emphasizing the long-term benefits of adhering to mechanical loading protocols can increase patient commitment. For example, explaining how controlled, progressive loading strengthens musculoskeletal tissues and enhances joint stability, potentially reducing the risk of future injuries, provides a long-term perspective on why sticking to the plan is essential. Highlighting the connection between following protocols and regaining full functionality or returning to a favorite sport or activity can serve as additional motivation for patients.
In conclusion, patient education is a multifaceted approach that involves explaining the biological and molecular effects of mechanical loading, detailing the risks of non-compliance, and offering continuous support throughout the rehabilitation process. By ensuring patients understand the “why” behind their treatment plan, clinicians can foster a collaborative and proactive approach to rehabilitation, significantly improving adherence and ultimately leading to better outcomes.
4. Regular Monitoring: Regular monitoring of patients during rehabilitation is essential for ensuring that mechanical loading protocols are tailored to their individual progress and biological responses. Incorporating biomarkers, functional assessments, and patient-reported outcomes into the monitoring process allows clinicians to make informed decisions and adjust loading regimens dynamically. This molecularly-informed approach optimizes tissue repair, minimizes risks, and enhances long-term recovery by aligning rehabilitation efforts with the patient’s specific healing processes.
Biomarkers play a pivotal role in understanding how tissues respond to mechanical loading at a molecular level. For example, monitoring inflammatory biomarkers such as C-reactive protein (CRP), tumor necrosis factor-alpha (TNF-α), interleukin-6 (IL-6), and interleukin-1 beta (IL-1β) can provide real-time feedback on the patient’s inflammatory status. These cytokines are typically elevated in response to tissue damage or excessive mechanical stress. If these markers rise significantly, it could indicate that the patient is being overloaded, and adjustments to the rehabilitation protocol may be necessary to reduce inflammation and avoid further injury.
Conversely, the presence of anti-inflammatory and tissue-repair-promoting biomarkers, such as IL-10, transforming growth factor-beta (TGF-β), and growth factors like IGF-1 and VEGF, indicates that tissues are actively repairing and regenerating. Monitoring levels of these molecules can guide the clinician to increase the mechanical load progressively, knowing that the tissues are responding favorably to the stress. IGF-1, for instance, is closely associated with muscle hypertrophy and tissue repair, while VEGF supports angiogenesis, which is crucial for restoring blood supply to healing tissues.
In addition to inflammatory biomarkers, monitoring the expression of matrix metalloproteinases (MMPs) and their inhibitors (TIMPs) is valuable for understanding ECM turnover. MMPs degrade damaged collagen and other ECM components, which is necessary for tissue remodeling, but excessive MMP activity may suggest tissue breakdown, requiring the clinician to modify loading protocols. On the other hand, an increase in collagen synthesis markers such as procollagen type I or hydroxyproline levels can indicate healthy collagen deposition and ECM repair, signaling that it may be appropriate to increase the mechanical load.
Functional assessments, such as range of motion (ROM), strength tests, and gait analysis, provide direct feedback on how well tissues are adapting to mechanical stress. These physical measures can be linked to molecular events occurring at the tissue level. For instance, improvements in muscle strength are associated with the activation of anabolic pathways, such as the mTORC1 pathway, which promotes protein synthesis in response to controlled loading. Similarly, enhanced ROM may indicate proper collagen alignment in tendons and ligaments, driven by TGF-β signaling and the gradual deposition of organized ECM fibers.
If functional assessments reveal a plateau in strength gains or ROM, it may signal the need for an increase in mechanical load to stimulate further adaptations. Conversely, if assessments indicate regression, such as decreased strength or abnormal gait patterns, this could reflect an imbalance between load and tissue capacity, possibly due to molecular signs of tissue overload or inflammation. This would prompt a reevaluation of the loading regimen to prevent injury.
Patient-reported outcomes, such as pain levels, fatigue, and perceived functional abilities, are equally important in shaping the loading regimen. From a molecular perspective, pain can be an indicator of ongoing inflammation or tissue damage. Elevated levels of prostaglandins and inflammatory cytokines are often associated with nociception, the body’s process of sensing pain. When patients report increasing or unmanageable pain, it suggests that molecular pathways linked to inflammation and nociception are being overstimulated, indicating a need to reduce or modify mechanical loading to allow tissues to recover.
In contrast, reports of decreased pain and improved functionality suggest that the molecular environment is shifting towards repair and regeneration. Reduced levels of substance P (a neuropeptide involved in pain perception) and lower concentrations of inflammatory cytokines could signify that the patient’s tissues are adapting well to the mechanical stress, enabling clinicians to consider progressively increasing the intensity or complexity of exercises.
By integrating biomarker data, functional outcomes, and patient-reported experiences, clinicians can continuously refine the rehabilitation process. For example, if molecular data indicate increased collagen synthesis and reduced MMP activity (suggesting ECM stability), functional assessments show improved strength and ROM, and patients report less pain, clinicians may safely increase the load, progressing from low-intensity to moderate-intensity exercises.
In contrast, if biomarkers show elevated inflammatory markers or excessive MMP activity, functional assessments show decreased strength or abnormal movement patterns, and patients report worsening pain, it may be necessary to reduce the mechanical load or introduce recovery phases to prevent tissue overload and potential setbacks. This ensures that the tissue’s biological response aligns with the rehabilitation protocol, optimizing healing outcomes while minimizing risks.
Long-term monitoring can also provide insights into the chronic adaptation of tissues to mechanical stress. For instance, monitoring bone turnover markers like osteocalcin and bone-specific alkaline phosphatase (BAP) can indicate whether bones are responding positively to mechanical loading, particularly in patients recovering from fractures. A healthy balance between bone formation and resorption suggests that the loading intensity is appropriate for enhancing bone density through Wnt/β-catenin signaling and osteoblast activity.
In conclusion, regular monitoring of biomarkers, functional assessments, and patient-reported outcomes is essential for ensuring that mechanical loading protocols are optimized at the molecular and functional levels. This approach allows clinicians to make informed, real-time adjustments to rehabilitation protocols, ensuring that tissue repair processes are supported without overwhelming the patient’s biological capacity for healing. By continuously tracking these indicators, clinicians can foster a more adaptive and effective rehabilitation program that enhances recovery outcomes and promotes long-term tissue health.
5. Personalized Rehabilitation: Tailoring rehabilitation protocols to the individual patient’s condition is essential for optimizing recovery and ensuring long-term health. Personalized rehabilitation not only considers visible factors like injury severity, age, and overall health but also accounts for the molecular and cellular responses that vary from patient to patient. Different tissues—whether bone, muscle, tendon, or ligament—respond uniquely to mechanical stress, and each patient’s biological makeup influences how their tissues heal and regenerate. Therefore, creating a customized rehabilitation plan that aligns with the patient’s molecular and physiological characteristics can significantly improve outcomes and enhance patient satisfaction.
The severity of an injury dictates the intensity, frequency, and type of rehabilitation exercises that can be safely introduced. In cases of severe injuries, such as tendon ruptures or bone fractures, tissues may be in an acute inflammatory state, with elevated levels of pro-inflammatory cytokines like TNF-α, IL-6, and IL-1β. During this phase, the focus should be on reducing inflammation and allowing time for the initial healing response. High-intensity exercises should be avoided, as they can exacerbate tissue damage and slow down the repair process by overstimulating the NF-κB pathway, which controls inflammation and cellular stress responses.
In contrast, for less severe injuries where inflammation has subsided, molecular pathways involved in tissue regeneration—such as the TGF-β signaling pathway, responsible for collagen synthesis, and the mTORC1 pathway, which governs muscle protein synthesis—are more active. In such cases, controlled loading can be introduced earlier to stimulate tissue repair and promote adaptive responses. Tailoring rehabilitation to the injury’s molecular stage helps ensure that tissues are not overstressed during vulnerable phases and that appropriate mechanical loading can enhance tissue recovery.
Age is a significant factor in determining how tissues respond to mechanical loading and rehabilitation. Younger patients typically have a higher capacity for tissue regeneration due to more robust cellular activity and more responsive signaling pathways. For example, young patients generally exhibit increased levels of growth factors, such as IGF-1 and VEGF, which promote muscle growth, angiogenesis, and tissue repair. These patients may tolerate higher mechanical loads earlier in the rehabilitation process, enabling more aggressive protocols that promote faster recovery.
In older patients, however, the molecular processes involved in tissue repair are often slower and less efficient. The decline in collagen synthesis and increased expression of matrix metalloproteinases (MMPs) can result in weaker ECM structures and slower recovery times. Additionally, age-related changes in muscle, such as reduced satellite cell activity (which is essential for muscle regeneration), mean that muscle repair and hypertrophy occur at a slower rate. These patients may also have a higher risk of developing fibrosis due to prolonged inflammatory responses, driven by elevated levels of pro-inflammatory cytokines like IL-6. For older individuals, rehabilitation protocols should focus on low-intensity, gradual mechanical loading that avoids overwhelming the weakened molecular systems, allowing sufficient time for tissue repair and adaptation.
Patients with comorbidities such as diabetes, cardiovascular disease, or obesity may experience impaired healing due to systemic issues that affect their molecular responses to injury. For instance, in diabetic patients, hyperglycemia can lead to increased levels of advanced glycation end-products (AGEs), which interfere with collagen crosslinking and weaken tissue integrity. High blood sugar levels can also impair angiogenesis by inhibiting VEGF signaling and reducing blood supply to injured areas, slowing down recovery.
Similarly, cardiovascular disease may reduce oxygen and nutrient delivery to healing tissues, while obesity can contribute to chronic low-grade inflammation, characterized by elevated levels of TNF-α and IL-6. In these cases, rehabilitation protocols should be carefully calibrated to account for reduced healing capacity at the molecular level. Lower-intensity, longer-duration exercises may be more appropriate to avoid exacerbating systemic issues while still promoting tissue repair through controlled mechanical stress.
Every patient has unique goals for rehabilitation, whether it’s returning to high-level athletic performance, achieving pain-free mobility, or regaining functional independence. These goals should shape the molecular focus of rehabilitation. For an athlete aiming to return to sports, rehabilitation may need to focus on enhancing muscle hypertrophy and power through higher-intensity loading that activates mTORC1 and increases protein synthesis. For this, incorporating eccentric exercises, which have been shown to stimulate greater muscle fiber recruitment and promote tendon adaptation through collagen remodeling, may be a key component.
In contrast, a patient focused on functional recovery after a joint replacement might benefit from moderate-intensity exercises that emphasize mobility, balance, and joint stability. This approach would target molecular pathways involved in joint remodeling and cartilage health, such as SOX9 and Wnt/β-catenin signaling, both of which are crucial for maintaining cartilage integrity and promoting joint function.
For patients recovering from fractures, loading protocols may focus on stimulating osteogenesis through mechanosensitive pathways in bone cells. Gradual mechanical loading is known to activate osteocytes and promote bone formation via Wnt/β-catenin signaling, encouraging bone density improvement and minimizing the risk of re-fracture. Tailoring exercises to promote bone healing may involve progressive weight-bearing exercises, depending on the stage of bone consolidation, which can be monitored through bone turnover markers like osteocalcin.
By incorporating molecular insights into rehabilitation, clinicians can better understand how each patient’s tissues respond to mechanical stimuli at various stages of healing. For instance, patients with chronic conditions such as tendinopathies may exhibit dysregulated TGF-β signaling, leading to excessive fibrosis rather than healthy collagen remodeling. In these cases, rehabilitation protocols should prioritize low-intensity, high-frequency loading that stimulates proper ECM remodeling without promoting excessive fibrosis. The goal would be to regulate collagen deposition while minimizing scar tissue formation through controlled loading.
In conclusion, personalized rehabilitation protocols are designed to target each patient’s unique molecular and physiological characteristics, ensuring that mechanical loading aligns with their tissue’s capacity for healing and adaptation. By taking into account injury severity, age, comorbidities, and individual goals, clinicians can craft rehabilitation programs that optimize molecular pathways for tissue repair, leading to more efficient recovery and improved long-term outcomes.
Figure 1.
The figure illustrates how mechanical loading, such as ground reaction forces during running, initiates mechanotransduction through the extracellular matrix (ECM), converting mechanical signals into biochemical responses in cells. This leads to tissue remodeling, resulting in sustained or improved function to better handle future mechanical stresses.
Figure 1.
The figure illustrates how mechanical loading, such as ground reaction forces during running, initiates mechanotransduction through the extracellular matrix (ECM), converting mechanical signals into biochemical responses in cells. This leads to tissue remodeling, resulting in sustained or improved function to better handle future mechanical stresses.
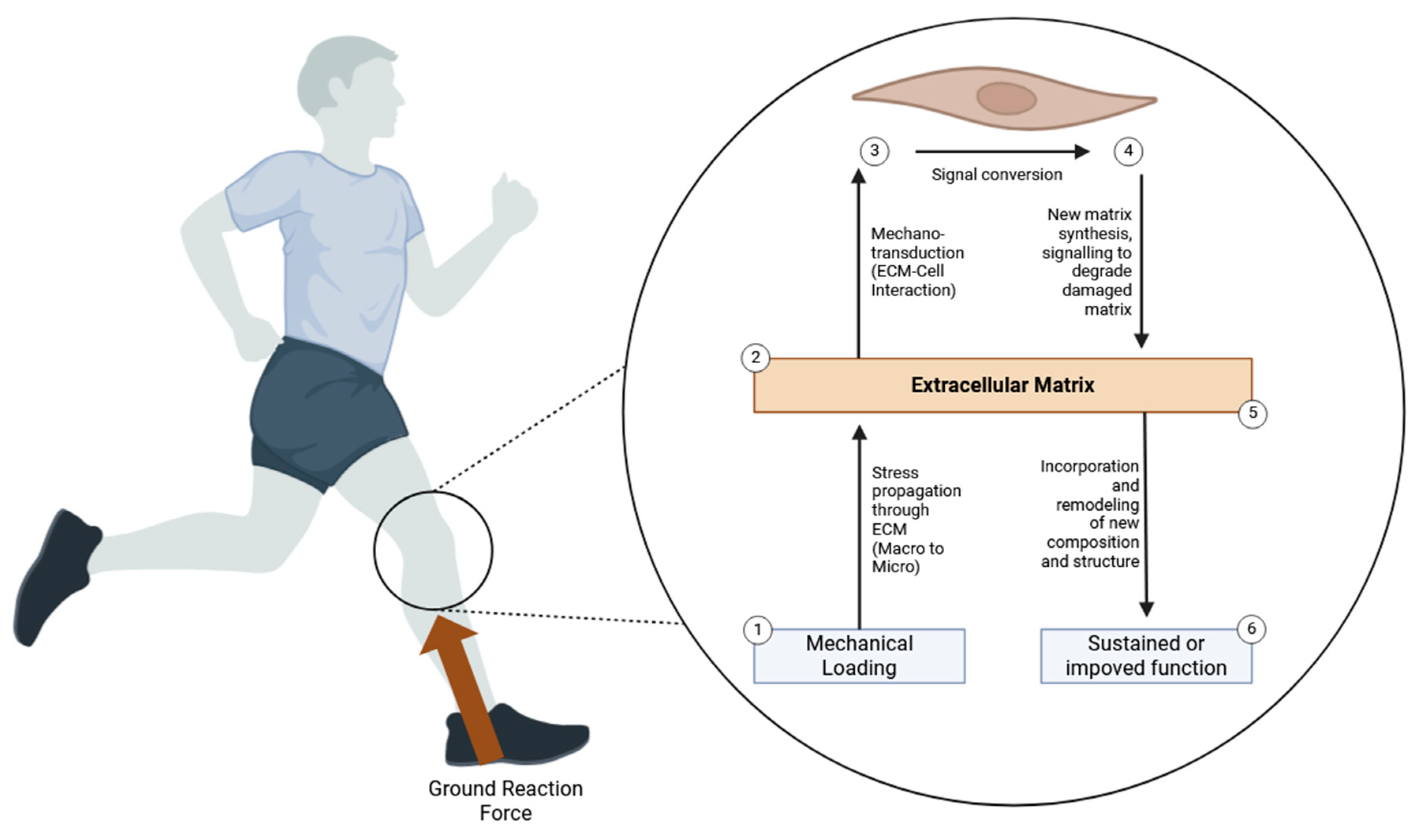
Signaling Pathways Involved in Mechanical Loading
The signaling pathways involved in mechanical loading are critical to how cells sense, respond, and adapt to mechanical stimuli, ultimately driving tissue repair, regeneration, and strengthening. When mechanical stress is applied to tissues, mechanoreceptors on the surface of cells, such as integrins and cadherins, are activated, triggering mechanotransduction. This process converts mechanical signals into biochemical responses through key signaling cascades. One of the primary pathways involved is the focal adhesion kinase (FAK) pathway, which is activated by integrin binding and plays a crucial role in promoting cell adhesion, migration, and survival, as well as stimulating extracellular matrix (ECM) remodeling. Another significant pathway is the mitogen-activated protein kinase (MAPK) pathway, which includes ERK, JNK, and p38 MAPK sub-pathways that regulate gene expression related to cellular proliferation, differentiation, and ECM synthesis. Additionally, the phosphoinositide 3-kinase (PI3K)/Akt pathway is essential for cell survival and metabolism, facilitating tissue repair by promoting anabolic processes and inhibiting apoptosis. The Wnt/β-catenin pathway, often activated by mechanical loading, influences stem cell differentiation, particularly in directing mesenchymal stem cells (MSCs) toward chondrogenic or osteogenic lineages, which is crucial for cartilage and bone repair. YAP/TAZ signaling, part of the Hippo pathway, is another key player in responding to mechanical cues, regulating cell growth and ECM production. These pathways work synergistically to ensure that tissues adapt to mechanical stress by reinforcing their structural integrity, facilitating ECM synthesis, enhancing cellular resilience, and promoting long-term recovery. Understanding these signaling pathways allows for more targeted rehabilitation strategies, enabling clinicians to optimize tissue repair and prevent overloading or injury.
Signaling Pathways Involved in Mechanical Loading
Mechanical loading initiates a complex cascade of biochemical events within cells, translating physical stimuli into coordinated cellular responses in a process called mechanotransduction. This intricate process enables tissues to adapt to mechanical stress by activating multiple signaling pathways that regulate critical cellular functions, such as proliferation, differentiation, migration, and extracellular matrix (ECM) synthesis. At the core of mechanotransduction is the Integrin signaling pathway, which plays a key role in sensing mechanical stress. Integrins are transmembrane receptors that anchor cells to the ECM and transmit mechanical forces from the ECM to the intracellular cytoskeleton. Upon activation by mechanical loading, integrins cluster at focal adhesion sites, activating focal adhesion kinase (FAK) and triggering downstream signaling events, such as the activation of the RhoA/ROCK pathway. This cascade promotes cytoskeletal remodeling, ECM reinforcement, and cell survival, thereby helping tissues strengthen in response to mechanical stress.
Ion channels and calcium signaling are another crucial component of mechanotransduction. Mechanically gated ion channels, such as Piezo1, are sensitive to mechanical stress and open in response to deformation, allowing the influx of ions like calcium (Ca²⁺) into the cell. The rise in intracellular calcium levels acts as a secondary messenger, triggering various downstream pathways, including the activation of calmodulin and calcineurin, which influence gene expression and cellular responses such as contraction, migration, and ECM production. Calcium signaling is integral to cellular responses in muscle, cartilage, and bone, as it regulates the differentiation of stem cells into specialized cell types such as osteoblasts and chondrocytes.
One of the primary downstream signaling cascades activated by mechanical loading is the Mitogen-Activated Protein Kinase (MAPK) pathway, which includes the extracellular signal-regulated kinase (ERK), c-Jun N-terminal kinase (JNK), and p38 MAPK sub-pathways. The ERK pathway is particularly important for cell proliferation and differentiation, while the JNK and p38 pathways are more closely linked to stress responses, inflammation, and apoptosis. In the context of mechanical loading, the MAPK pathway helps regulate cellular adaptation by controlling the expression of genes involved in ECM remodeling, cellular proliferation, and survival. Activation of this pathway ensures that fibroblasts, chondrocytes, and osteoblasts proliferate and contribute to the repair and regeneration of tissues by synthesizing key ECM components such as collagen, elastin, and proteoglycans.
Another critical pathway influenced by mechanical loading is the Wnt/β-Catenin signaling pathway, which is central to stem cell differentiation and tissue regeneration. Mechanical loading activates Wnt signaling, promoting the stabilization and accumulation of β-catenin in the cytoplasm. This leads to its translocation into the nucleus, where it regulates the transcription of target genes associated with cellular proliferation and differentiation. In tissues such as bone and cartilage, Wnt signaling directs mesenchymal stem cells (MSCs) to differentiate into osteoblasts and chondrocytes, respectively, facilitating bone formation and cartilage repair. This pathway is especially important in the context of bone density and strength, as mechanical loading increases Wnt signaling, promoting osteogenesis and bone remodeling.
The YAP/TAZ signaling pathway, part of the Hippo pathway, is another key regulator of mechanotransduction. YAP (Yes-associated protein) and TAZ (transcriptional co-activator with PDZ-binding motif) are mechanosensitive proteins that are activated in response to mechanical cues, such as increased stiffness of the ECM. Under mechanical loading, YAP/TAZ translocate to the nucleus, where they interact with transcription factors to regulate genes involved in cell proliferation, ECM production, and tissue growth. This pathway plays a vital role in regulating organ size, tissue repair, and stem cell behavior. In tissues experiencing mechanical stress, such as tendons, ligaments, and skin, YAP/TAZ signaling promotes cell proliferation and ECM deposition, ensuring the tissue adapts and strengthens in response to loading.
In addition to these primary pathways, mechanical loading can also influence the PI3K/Akt signaling pathway, which is critical for promoting cell survival and growth. The activation of PI3K leads to the phosphorylation of Akt, a serine/threonine kinase that regulates cell metabolism, survival, and protein synthesis. In response to mechanical loading, this pathway enhances anabolic processes, stimulating the synthesis of ECM proteins and preventing apoptosis in stressed cells. PI3K/Akt signaling is particularly important in tissues like muscle and bone, where it promotes hypertrophy and increases tissue strength in response to loading.
Collectively, these signaling pathways act in concert to ensure that mechanical loading promotes tissue repair, adaptation, and resilience. By coordinating the cellular responses to mechanical stimuli, they regulate crucial processes such as stem cell differentiation, ECM synthesis, and tissue regeneration, ensuring that tissues can withstand mechanical stress and recover effectively from injury. Understanding the molecular mechanisms underlying these signaling pathways allows clinicians to develop more targeted rehabilitation protocols, optimizing mechanical loading to promote efficient tissue repair while minimizing the risk of injury or overloading.
Integrin Signaling Pathway
Integrins are transmembrane receptors that play a pivotal role in mechanotransduction, bridging the extracellular matrix (ECM) and the cell’s internal structure. Upon mechanical loading, integrins aggregate at focal adhesion sites, which serve as critical hubs for signal transduction. These focal adhesions link the ECM to the actin cytoskeleton within the cell, a vital component for transmitting mechanical forces into biochemical signals. Integrin clustering at these sites enhances the strength of the integrin-ECM bond, facilitating the mechanical communication between the external environment and the cell’s interior. This clustering is not a passive event; it activates a cascade of intracellular signaling pathways that are essential for cellular adaptation to mechanical stress. One of the earliest responses to integrin clustering is the activation of Focal Adhesion Kinase (FAK). FAK undergoes autophosphorylation at key tyrosine residues, creating docking sites for other signaling molecules like Src family kinases. This initiates the formation of a multi-protein complex at focal adhesions, amplifying the signal and coordinating cellular responses such as migration, growth, and survival.
Activated FAK triggers several downstream pathways, including the MAPK (Mitogen-Activated Protein Kinase), PI3K/Akt, and Rho family GTPases pathways. The MAPK pathway regulates gene expression and cell cycle progression, ensuring that cells proliferate and differentiate in response to mechanical loading. The PI3K/Akt pathway is crucial for cell survival, as it promotes anabolic processes while inhibiting apoptosis, enabling cells to thrive under mechanical stress. Meanwhile, Rho family GTPases control the dynamics of the cytoskeleton, governing cell shape and motility. These pathways are highly integrated, ensuring that cells can coordinate a range of responses—from ECM remodeling to migration and proliferation—essential for tissue repair and adaptation to mechanical loading.
Integrin signaling also drives cytoskeletal remodeling, which is critical for maintaining cell shape, enabling movement, and reinforcing structural integrity. Mechanical loading stimulates the formation of stress fibers, which are composed of actin filaments that provide tensile strength and mechanical stability. These stress fibers, coupled with focal adhesions, help cells anchor to the ECM and withstand further mechanical stress. The dynamic reorganization of the cytoskeleton is crucial for various cellular processes, including migration, adhesion, and division, all of which are necessary for tissue regeneration. Integrin-mediated cytoskeletal remodeling enables cells to adapt structurally to their mechanical environment, ensuring that they can respond effectively to continued mechanical stress. This integrin-driven signaling and cytoskeletal reorganization provide the molecular foundation for tissue resilience, repair, and adaptation in response to mechanical loading.
Ion Channels and Calcium Signaling
Ion channels, particularly those sensitive to mechanical stimuli, play a pivotal role in mechanotransduction by facilitating the rapid influx of ions such as calcium (Ca²⁺), sodium (Na⁺), and potassium (K⁺) into cells in response to mechanical loading. Stretch-activated ion channels are specialized proteins embedded in the cell membrane that respond to mechanical deformation of the cell’s structure. When external mechanical forces, such as stretching or pressure, are applied to the cell, these channels open, allowing ions to flow into the cell, which alters the electrochemical balance inside the cell. This change in ion concentration acts as a trigger for various intracellular signaling cascades. Calcium influx, in particular, is crucial, as it serves as a secondary messenger that activates numerous downstream pathways, leading to adaptations in cellular behavior necessary for responding to mechanical stress. Sodium and potassium also contribute to cellular homeostasis and are vital for maintaining the electrochemical gradients necessary for cell function, but calcium’s role is especially important in initiating mechanotransduction.
The calcium influx through these stretch-activated ion channels is one of the most critical early events in mechanotransduction. When calcium enters the cell, it rapidly binds to a variety of proteins, such as calmodulin, which serves as a central mediator in calcium signaling. This increase in intracellular calcium concentration initiates several cellular processes, including gene expression, cytoskeletal remodeling, and differentiation. Calcium’s ability to act as a versatile secondary messenger allows it to regulate a wide array of cellular functions, ranging from immediate responses like cell migration to long-term adaptations such as changes in gene expression that support tissue repair and remodeling. By facilitating this calcium entry, stretch-activated ion channels serve as the primary gateway for translating mechanical signals into biochemical responses.
One key calcium-dependent pathway is the Calcineurin/NFAT pathway, where the influx of Ca²⁺ activates calcineurin, a calcium/calmodulin-dependent phosphatase. Activated calcineurin dephosphorylates the transcription factor nuclear factor of activated T-cells (NFAT), allowing NFAT to translocate to the nucleus. Once inside the nucleus, NFAT regulates the transcription of genes involved in cell proliferation, differentiation, and survival, which are critical for tissue repair and adaptation to mechanical stress. This pathway exemplifies how mechanical loading, through calcium signaling, can influence gene expression and guide cellular responses essential for tissue regeneration. The Calcineurin/NFAT pathway ensures that mechanical cues lead to specific genetic responses, facilitating long-term adaptations such as enhanced tissue strength and resilience.
Another important calcium-mediated pathway is the Calmodulin-Dependent Kinase (CaMK) pathway, which plays a crucial role in cellular responses to mechanical loading. Upon binding to calcium, calmodulin forms a complex that activates CaMK, a family of protein kinases. Activated CaMK phosphorylates a variety of target proteins, including transcription factors and regulatory proteins that influence gene expression. This phosphorylation alters the activity of these proteins, leading to changes in cellular behavior such as migration, growth, and differentiation. The CaMK pathway ensures that cells can adjust their functions to meet the mechanical demands placed upon them, facilitating processes like ECM remodeling and tissue repair. The activation of CaMK underscores calcium’s central role as a mediator in mechanotransduction, linking external mechanical stimuli to intracellular biochemical changes that drive tissue adaptation and resilience.
Together, these ion channels and calcium-dependent pathways illustrate how mechanical stimuli are translated into precise cellular responses at the molecular level. By initiating signaling cascades such as the Calcineurin/NFAT and CaMK pathways, stretch-activated ion channels enable cells to respond appropriately to their mechanical environment, promoting tissue repair, adaptation, and regeneration. These processes are essential for maintaining the structural integrity and function of tissues subjected to mechanical stress, such as muscles, tendons, and bones, ensuring that they can adapt to increased mechanical demands and prevent injury.
Mitogen-Activated Protein Kinase (MAPK) Pathway
The MAPK (Mitogen-Activated Protein Kinase) pathway is a central signaling cascade that regulates cellular responses to a variety of stimuli, including mechanical loading. Mechanical stress activates the MAPK pathway through integrin signaling and other mechanotransduction mechanisms, allowing cells to adapt to their mechanical environment. The MAPK pathway is divided into three major branches: ERK1/2 (extracellular signal-regulated kinases 1 and 2), JNK (c-Jun N-terminal kinases), and p38 MAPK, each of which plays a distinct role in regulating gene expression, cell proliferation, differentiation, survival, and apoptosis. These kinases respond to mechanical stimuli by phosphorylating a series of downstream targets, which ultimately control transcriptional programs essential for tissue repair and adaptation.
Activation by Mechanical Loading: Mechanical stress activates MAPKs primarily through integrin signaling and other mechanosensitive receptors. When cells experience mechanical loading, integrins cluster at focal adhesions and initiate the activation of upstream kinases like Raf and MEKK1, leading to the activation of MAPKs. ERK1/2 is primarily activated by growth factors and mechanical stress, promoting cell growth and differentiation. JNK and p38 MAPK, on the other hand, are stress-activated protein kinases that respond to more severe mechanical or inflammatory stresses. The activation of these kinases ensures that cells can initiate coordinated responses to mechanical stress, such as the synthesis of extracellular matrix (ECM) components, cytoskeletal reorganization, and tissue regeneration, all of which are crucial for maintaining tissue integrity.
ERK1/2 Pathway: The ERK1/2 pathway begins with the activation of upstream kinases, specifically Raf, which phosphorylates MEK1/2. MEK1/2 then phosphorylates ERK1/2, leading to its activation. Once phosphorylated, ERK1/2 translocates to the nucleus, where it phosphorylates transcription factors such as Elk-1 and c-Fos, both of which regulate the expression of genes involved in cell proliferation, differentiation, and survival. In the context of mechanical loading, the ERK1/2 pathway is crucial for driving cellular responses that promote tissue repair, such as collagen synthesis, fibroblast proliferation, and ECM remodeling. The precise regulation of gene expression by ERK1/2 ensures that cells respond appropriately to mechanical cues, facilitating tissue growth and adaptation without over-proliferation or excessive ECM degradation.
JNK Pathway: The JNK pathway is primarily activated in response to stress signals, including mechanical stress, oxidative stress, and inflammatory stimuli. Mechanical loading, particularly in conditions of high stress, activates JNK, which then translocates to the nucleus. In the nucleus, JNK phosphorylates transcription factors such as c-Jun, a component of the AP-1 (Activator Protein-1) transcription factor complex. c-Jun regulates genes involved in cell proliferation, apoptosis, and differentiation, allowing cells to adapt to mechanical stress by either promoting survival or initiating programmed cell death if damage is too severe. This pathway is especially important in tissues exposed to high mechanical loads, such as tendons and cartilage, where it helps balance cellular repair with the need to remove damaged cells.
p38 MAPK Pathway: The p38 MAPK pathway is activated by various stress signals, including mechanical loading, oxidative stress, and pro-inflammatory cytokines. p38 MAPK activation leads to the phosphorylation of transcription factors like ATF-2 and MEF2, as well as other target proteins involved in cellular differentiation, inflammation, and apoptosis. In the context of mechanical loading, p38 MAPK plays a key role in regulating the inflammatory response, cell differentiation, and the production of matrix metalloproteinases (MMPs), which are involved in ECM remodeling. This pathway is critical for maintaining tissue homeostasis under stress by modulating inflammatory signals and facilitating tissue repair. For instance, in response to mechanical stress, p38 MAPK regulates the production of cytokines like IL-6 and TNF-α, which help coordinate the repair process while preventing excessive inflammation.
Together, these MAPK pathways—ERK1/2, JNK, and p38 MAPK—create a complex, integrated network of signaling events that allow cells to sense and respond to mechanical forces. They regulate diverse processes such as gene expression, cytoskeletal organization, ECM remodeling, and cell survival, ensuring that tissues can adapt to changing mechanical environments. By coordinating these responses, the MAPK pathway helps cells maintain structural integrity and promotes the efficient repair of tissues subjected to mechanical loading. Through the precise modulation of these pathways, cells can fine-tune their responses to ensure that mechanical stress promotes growth, differentiation, and repair while minimizing the risk of damage or dysfunction. Understanding how these pathways interact is crucial for developing targeted therapies that harness mechanical stimuli to enhance tissue regeneration and recovery in various clinical contexts, such as musculoskeletal injuries, wound healing, and tissue engineering.
Wnt/β-Catenin Signaling Pathway
The Wnt/β-catenin signaling pathway plays a pivotal molecular role in regulating cell proliferation, differentiation, and extracellular matrix (ECM) remodeling, especially in response to mechanical loading. This pathway is crucial for translating mechanical stimuli into cellular responses that drive tissue repair and adaptation. When mechanical stress is applied, it enhances the expression of Wnt ligands, which are secreted signaling proteins that bind to Frizzled receptors and their co-receptors on the cell surface. This interaction initiates the Wnt signaling cascade, leading to the stabilization of the key effector molecule β-catenin. Mechanical loading-induced upregulation of Wnt ligands ensures that cells can efficiently detect and respond to changes in their mechanical environment, promoting tissue regeneration and remodeling. By enhancing the expression of these ligands, mechanical loading facilitates the activation of the Wnt pathway, allowing cells to respond with appropriate growth, differentiation, and ECM production to support tissue integrity.
One of the critical steps in the Wnt/β-catenin pathway is the stabilization of β-catenin. In the absence of Wnt signaling, β-catenin is continuously targeted for degradation by a destruction complex that includes proteins such as GSK-3β, APC, and Axin. However, when Wnt ligands bind to Frizzled receptors, the activity of this destruction complex is inhibited, preventing β-catenin degradation. As a result, β-catenin accumulates in the cytoplasm. This accumulation is a critical molecular event in response to mechanical loading, as it enables β-catenin to translocate into the nucleus, where it functions as a transcriptional co-activator. The inhibition of β-catenin degradation is a key mechanism by which mechanical signals stabilize β-catenin, ensuring that cellular adaptations to mechanical stress are initiated at the molecular level.
In the nucleus, β-catenin interacts with transcription factors, particularly TCF/LEF (T-cell factor/lymphoid enhancer factor), to regulate the expression of target genes that are essential for cell proliferation, differentiation, and ECM synthesis. These genes include those encoding components of the ECM, such as collagen and fibronectin, as well as growth factors that promote cellular proliferation and tissue remodeling. The activation of these genetic programs is crucial for maintaining tissue homeostasis and ensuring proper tissue repair in response to mechanical loading. By modulating the expression of Wnt target genes, β-catenin ensures that cells can effectively respond to mechanical stimuli, driving processes like fibroblast proliferation, osteoblast differentiation, and the synthesis of ECM components necessary for tissue strength and resilience.
The Wnt/β-catenin signaling pathway underscores the importance of extracellular signals in regulating cellular responses to mechanical loading. By activating Wnt ligands, stabilizing β-catenin, and driving gene expression, this pathway plays a central role in ensuring that tissues can adapt to mechanical stress and regenerate effectively. Whether in bone, cartilage, or connective tissue, the Wnt/β-catenin pathway integrates mechanical cues into the cellular machinery, enabling cells to coordinate tissue repair, maintain structural integrity, and promote long-term functionality. Understanding how this pathway operates at the molecular level provides valuable insights into tissue engineering, regenerative medicine, and therapeutic strategies aimed at enhancing tissue repair and preventing degeneration.
YAP/TAZ Signaling Pathway
Yes-associated protein (YAP) and transcriptional co-activator with PDZ-binding motif (TAZ) are central regulators of mechanotransduction, translating mechanical signals into cellular responses that govern cell proliferation, differentiation, and survival. These proteins are part of the Hippo pathway, which regulates organ size, tissue growth, and cell fate. In the absence of mechanical loading, the Hippo pathway is active and suppresses YAP/TAZ by phosphorylating them, leading to their retention in the cytoplasm where they are marked for degradation. However, mechanical loading inhibits the Hippo pathway, allowing YAP and TAZ to become dephosphorylated and activated. This inhibition occurs through mechanotransduction mechanisms such as changes in cell shape, cytoskeletal tension, and extracellular matrix (ECM) stiffness, which signal that the tissue is experiencing mechanical stress. By deactivating the Hippo pathway, cells allow YAP and TAZ to translocate to the nucleus, where they can perform their transcriptional regulatory functions. This inhibition of the Hippo pathway is a crucial molecular event, enabling the cell to respond to its mechanical environment by promoting cellular growth and survival, essential for tissue repair and regeneration.
Once YAP and TAZ are activated, they undergo nuclear translocation, moving from the cytoplasm into the nucleus. In the nucleus, YAP/TAZ act as transcriptional co-activators by binding to TEAD (TEA domain transcription factors) and other transcription factors. This interaction is essential for activating the transcription of genes involved in cell proliferation, differentiation, and ECM synthesis. The nuclear translocation of YAP/TAZ is a key step in mechanotransduction, as it links mechanical signals to the genetic programs necessary for cellular adaptation. These programs include genes involved in promoting cell cycle progression, preventing apoptosis, and enhancing ECM production, all of which are critical for tissue repair and growth. The regulation of these genes by YAP/TAZ ensures that cells can adjust their behavior in response to mechanical loading, making them more resilient to stress and better equipped to maintain tissue integrity.
In addition to nuclear translocation, YAP/TAZ regulate cell behavior by modulating the expression of a wide range of genes involved in essential cellular processes. By promoting the expression of genes that drive cell proliferation, YAP/TAZ support the expansion of cell populations needed for tissue repair and regeneration. Their role in inhibiting apoptosis (programmed cell death) ensures that cells survive in environments of mechanical stress, which is particularly important during tissue remodeling. Furthermore, YAP/TAZ also play a role in cell differentiation, helping progenitor and stem cells commit to specific lineages based on mechanical cues. For example, YAP/TAZ activation can promote osteogenic differentiation in bone-forming cells in response to increased ECM stiffness, facilitating bone repair. Similarly, YAP/TAZ can influence the differentiation of other cell types, such as fibroblasts and myoblasts, depending on the mechanical properties of the surrounding tissue. This ability to fine-tune cell fate based on mechanical signals is critical for the regeneration of functional tissues that are structurally and functionally adapted to their environment.
Overall, the YAP/TAZ pathway is a sophisticated molecular mechanism that integrates mechanical signals into gene regulatory networks, ensuring that cells can proliferate, differentiate, and survive in response to mechanical stress. This pathway plays a central role in tissue homeostasis, enabling effective tissue repair and adaptation by translating physical forces into biochemical responses. By coordinating these processes, YAP/TAZ contribute to the maintenance of long-term tissue health and prevent degeneration. The inhibition of the Hippo pathway, nuclear translocation of YAP/TAZ, and subsequent regulation of gene expression highlight the complexity of mechanotransduction and underscore the importance of YAP/TAZ in linking mechanical stimuli to cellular outcomes. Understanding these molecular mechanisms provides critical insights into therapeutic approaches for enhancing tissue regeneration and preventing disorders associated with impaired mechanotransduction, such as fibrosis or tissue atrophy.
Interactions among Signaling Pathways
The signaling pathways involved in mechanotransduction do not function independently but rather engage in extensive crosstalk to integrate mechanical signals and coordinate comprehensive cellular responses. This interconnectedness allows cells to process and respond to mechanical stimuli in a finely tuned manner, ensuring that tissue repair, growth, and adaptation occur optimally. One key interaction occurs between the Integrin and MAPK pathways, where integrin activation through Focal Adhesion Kinase (FAK) leads to the activation of MAPKs such as ERK1/2, JNK, and p38 MAPK. When integrins cluster at focal adhesions in response to mechanical stress, FAK triggers downstream signaling that activates MAPKs. This crosstalk enhances the cell’s ability to regulate gene expression, cellular behavior, and cytoskeletal remodeling in response to mechanical stimuli. By combining integrin and MAPK signaling, cells can orchestrate processes like cell proliferation, migration, and ECM remodeling, critical for maintaining tissue homeostasis and promoting regeneration in response to stress.
Calcium signaling also plays a pivotal role in interacting with the MAPK pathway. The influx of calcium through stretch-activated ion channels in response to mechanical loading activates calcium-sensitive kinases, such as Calmodulin-dependent protein kinase (CaMK). CaMK can, in turn, modulate the activity of MAPKs, fine-tuning their response to mechanical stress. This crosstalk allows for precise control over cellular processes like gene expression, protein synthesis, and cytoskeletal organization. The integration of calcium signaling with the MAPK pathway ensures that cellular responses are not only reactive to mechanical forces but also sensitive to changes in the intracellular calcium environment, providing an additional layer of regulation. This coordination is crucial for promoting balanced tissue repair, ensuring that mechanical stimuli are translated into effective cellular responses without overwhelming the cell.
Another significant interaction occurs between the Wnt/β-catenin and YAP/TAZ signaling pathways, both of which are activated by mechanical loading and influence gene expression, cell proliferation, and differentiation. These pathways can work synergistically to regulate cellular responses to mechanical stress, promoting tissue repair and regeneration. For instance, β-catenin and YAP/TAZ both regulate genes involved in stem cell renewal, ECM production, and cell fate determination, making their combined activity particularly important for tissues under mechanical strain, such as bone, cartilage, and tendons. The interaction between Wnt and YAP/TAZ signaling allows cells to coordinate the regulation of critical genes involved in mechanotransduction, enhancing the ability of tissues to adapt to mechanical forces and heal from injuries. Additionally, both pathways may converge on common transcriptional targets or signaling intermediates, amplifying the overall cellular response to mechanical stimuli and ensuring that tissue regeneration occurs efficiently.
In summary, the crosstalk between signaling pathways such as Integrin-MAPK, Calcium-MAPK, and Wnt/YAP-TAZ exemplifies the complexity of mechanotransduction. These pathways integrate mechanical signals and biochemical cues, ensuring that cellular responses are both specific and coordinated. Through these interactions, cells can regulate diverse processes like gene expression, cell survival, and ECM remodeling, enabling tissues to maintain structural integrity and function under mechanical stress. This molecular integration of mechanotransduction pathways ensures that cells can adapt appropriately to changes in their environment, promoting long-term tissue health and functionality. Understanding these intricate signaling networks highlights the sophisticated nature of cellular responses to mechanical cues and provides valuable insights into developing therapies for enhancing tissue regeneration and treating degenerative diseases.
Table 7.
The table outlines key signaling pathways involved in mechanotransduction, including the Integrin, Ion Channels and Calcium, MAPK, Wnt/β-Catenin, and YAP/TAZ pathways. It details each pathway’s mechanism of activation, downstream effects, and cellular responses, highlighting their roles in tissue repair, regeneration, and adaptation to mechanical stress.
Table 7.
The table outlines key signaling pathways involved in mechanotransduction, including the Integrin, Ion Channels and Calcium, MAPK, Wnt/β-Catenin, and YAP/TAZ pathways. It details each pathway’s mechanism of activation, downstream effects, and cellular responses, highlighting their roles in tissue repair, regeneration, and adaptation to mechanical stress.
| Signaling Pathway | Mechanism of Activation | Downstream Effects | Cellular Response |
| Integrin Signaling Pathway | Mechanical stress leads to integrin clustering at focal adhesion sites, initiating signaling via FAK and RhoA/ROCK pathways, directly connecting the ECM to intracellular structures. | FAK activation phosphorylates downstream kinases like Src, activating pathways such as PI3K/Akt, leading to enhanced cell survival, migration, and ECM production through cytoskeletal remodeling. | Promotes cell adhesion, migration, proliferation, and ECM remodeling, facilitating tissue repair and regeneration by enhancing cytoskeletal strength and structural integrity. |
| Ion Channels and Calcium Signaling | Mechanically gated ion channels, such as Piezo1, open in response to membrane deformation, allowing calcium influx, which serves as a rapid signal to trigger further cellular pathways. | Calcium influx through ion channels activates calcium-binding proteins like calmodulin, triggering the calcineurin/NFAT and CaMK pathways that regulate gene expression and cytoskeletal organization. | Calcium serves as a key secondary messenger, driving processes like gene expression, cytoskeletal reorganization, and ECM production, ensuring that cells can respond dynamically to mechanical stimuli. |
| Mitogen-Activated Protein Kinase (MAPK) Pathway | Mechanical stress activates MAPK pathways through integrin signaling and mechanosensitive receptors, such as FAK, leading to phosphorylation events that activate ERK1/2, JNK, and p38 MAPK. | ERK1/2 supports proliferation and differentiation, JNK governs stress responses including apoptosis, and p38 MAPK modulates inflammation, ECM remodeling, and cellular survival in response to mechanical signals. | Coordinates cellular responses such as proliferation, apoptosis, and ECM synthesis to help tissues adapt to mechanical stress and maintain tissue integrity, preventing overloading and damage. |
| Wnt/β-Catenin Signaling Pathway | Wnt ligands bind to Frizzled and LRP5/6 receptors, leading to the inhibition of the β-catenin destruction complex, stabilizing β-catenin, allowing it to accumulate in the cytoplasm and translocate to the nucleus. | Stabilized β-catenin interacts with TCF/LEF transcription factors in the nucleus to regulate genes critical for stem cell proliferation, differentiation, and ECM synthesis, crucial for tissue repair. | Directs mesenchymal stem cells toward osteogenic and chondrogenic lineages, ensuring efficient bone and cartilage formation, promoting tissue resilience and functional recovery. |
| YAP/TAZ Signaling Pathway | Mechanical loading inhibits the Hippo pathway, allowing dephosphorylated YAP/TAZ to enter the nucleus, where they regulate transcription in response to mechanical cues such as ECM stiffness. | YAP/TAZ interact with TEAD transcription factors to promote the expression of genes controlling cell cycle progression, apoptosis inhibition, and ECM production, adapting tissues to mechanical loading. | YAP/TAZ modulate the expression of genes involved in cell growth, differentiation, and survival, contributing to tissue regeneration, homeostasis, and adaptation to mechanical changes. |
Molecular and Cellular Biology Aspects of Differences Between Meniscus, Cartilage, Ligament, and Subchondral Bone in Rehabilitation and Injury
Understanding the molecular and cellular differences between the meniscus, cartilage, ligament, and subchondral bone is crucial for developing targeted rehabilitation strategies that address the unique properties and healing capacities of each tissue. These tissues vary not only in their structural composition but also in their response to injury, mechanical loading, and regenerative potential. For example, the meniscus is composed largely of fibrocartilage, which provides both shock absorption and joint stability, while articular cartilage consists of chondrocytes embedded in a dense extracellular matrix (ECM) that facilitates smooth joint movement. In contrast, ligaments are made of dense, fibrous connective tissue that provides tensile strength, and subchondral bone, located beneath cartilage, supports the joint’s structural integrity.
Due to these differences, each tissue type has distinct responses to mechanical stimuli, inflammation, and healing processes. Cartilage, for instance, is avascular and lacks a direct blood supply, which significantly limits its healing capacity, whereas ligaments and subchondral bone have relatively better vascularization, allowing for a more robust regenerative response. Furthermore, mechanical loading plays a critical role in tissue repair; while moderate loading can stimulate collagen synthesis and ECM remodeling in ligaments and bone, excessive loading can cause further damage, particularly in cartilage, where overloading can exacerbate degeneration.
Tailored rehabilitation approaches are, therefore, necessary to optimize healing and restore function. This requires not only an understanding of how each tissue responds to specific mechanical loads but also careful modulation of rehabilitation protocols to prevent re-injury. For example, progressive loading strategies may be beneficial for ligaments and bone, encouraging tissue strengthening, whereas cartilage may require more conservative loading to avoid further wear and tear. By integrating the molecular and cellular characteristics of each tissue type, clinicians can develop more effective rehabilitation protocols that promote tissue-specific repair, enhance functional recovery, and reduce the risk of long-term complications.
Meniscus
The meniscus, a fibrocartilaginous structure in the knee joint, plays a critical role in load distribution, shock absorption, and joint stability. It is composed of type I and type II collagen, arranged in a specific pattern that influences its ability to handle compressive and tensile forces. At the molecular and cellular level, fibrochondrocytes are the primary cell type in the meniscus, with the ability to synthesize both fibrous (type I collagen) and cartilaginous (type II collagen and proteoglycans) components. These cells maintain the extracellular matrix (ECM) by responding to mechanical stress and adjusting the production of matrix components. The ECM itself varies regionally, with type I collagen dominating in the outer meniscus for tensile strength, while type II collagen and proteoglycans like aggrecan in the inner region provide compressive resilience and shock absorption. This sophisticated ECM organization, along with glycosaminoglycans (GAGs) that retain water, helps the meniscus maintain its structural integrity under mechanical stress.
Upon injury, the meniscus responds with an inflammatory cascade characterized by the release of cytokines such as IL-1β, TNF-α, and PGE2. These inflammatory mediators increase matrix metalloproteinase (MMP) activity, particularly MMP-13, which degrades collagen and compromises the meniscus’s structural integrity. This catabolic environment hinders the meniscus’s healing potential by degrading ECM components faster than they can be repaired, especially in the avascular regions of the meniscus, such as the inner two-thirds, where nutrient diffusion from synovial fluid is limited. The outer third, known as the red-red zone, benefits from better vascularization, enabling more efficient healing through cellular infiltration and ECM production, while the avascular regions often require surgical intervention for effective repair.
Rehabilitation strategies for meniscal injuries must consider these molecular characteristics. Progressive biomechanical loading, such as controlled weight-bearing and proprioceptive exercises, stimulates fibrochondrocyte activity and ECM synthesis, helping to strengthen the meniscus without causing further damage. Additionally, biologic therapies involving growth factors like TGF-β and PDGF can enhance fibrochondrocyte proliferation and matrix production, improving healing outcomes. Growth factors also modulate the inflammatory response, reducing ECM degradation and promoting tissue regeneration, offering an advanced therapeutic approach for enhancing meniscal repair and recovery. By integrating these molecular insights, rehabilitation protocols can be tailored to the specific needs of the meniscus, optimizing healing and restoring knee function.
Cartilage
Articular cartilage is a smooth, avascular tissue that covers the ends of bones in joints, facilitating frictionless movement and efficient load distribution. Its molecular composition, dominated by type II collagen and proteoglycans like aggrecan, plays a crucial role in its ability to withstand compressive forces and provide structural resilience. Chondrocytes, the sole cell type in cartilage, are embedded in lacunae within the extracellular matrix (ECM) and are responsible for synthesizing and maintaining this matrix. These cells produce type II collagen, which forms the structural framework, and aggrecan, a proteoglycan that binds water, giving the tissue its ability to absorb impact and distribute loads across the joint. However, chondrocytes have a limited capacity for proliferation and migration, contributing to the poor healing potential of cartilage. The avascular nature of cartilage also restricts nutrient supply to chondrocytes, further limiting their ability to respond to injury.
The extracellular matrix (ECM) of articular cartilage is highly specialized, composed predominantly of type II collagen and proteoglycans. The unique interaction between these components provides cartilage with both strength and flexibility. Aggrecan, which is rich in glycosaminoglycans (GAGs), attracts and retains water, allowing the tissue to resist compressive forces and maintain its shock-absorbing properties. Additionally, other molecules, such as hyaluronan and link proteins, contribute to the stabilization of the ECM structure, ensuring that cartilage can function effectively under mechanical stress. The ECM is integral to the cartilage’s role in maintaining smooth joint movement and distributing mechanical loads, but its complex composition also makes it vulnerable to degradation.
In response to injury, articular cartilage experiences an inflammatory reaction that further limits its repair capacity. Inflammatory mediators, including cytokines like IL-1 and TNF-α, are released, which activate catabolic enzymes such as ADAMTS-4 and ADAMTS-5. These enzymes degrade key ECM components, including aggrecan and type II collagen, leading to a loss of structural integrity and mechanical function. This degradation process creates a cycle of damage, as the breakdown of ECM components weakens the cartilage and impairs its ability to absorb shocks and distribute loads, leaving the joint more vulnerable to further injury. The chronic inflammatory environment also inhibits the anabolic activity of chondrocytes, exacerbating tissue breakdown and contributing to progressive cartilage degeneration.
The limited repair capacity of cartilage is further compounded by its avascularity, which prevents the direct delivery of reparative cells and nutrients to the site of injury. Chondrocytes have a low capacity for cell division and migration, which makes it difficult for the tissue to regenerate naturally. As a result, cartilage relies on the slow diffusion of nutrients from synovial fluid, which is often insufficient for effective repair. This lack of intrinsic healing potential frequently leads to chronic joint problems, such as osteoarthritis, where the gradual degradation of cartilage results in pain, stiffness, and loss of joint function.
Rehabilitation strategies for cartilage injuries focus on providing mechanical stimulation and introducing biologic therapies to enhance chondrocyte activity and ECM production. Controlled mechanical loading, such as hydrotherapy and continuous passive motion (CPM) machines, can stimulate chondrocytes to produce more ECM, maintaining tissue integrity and promoting repair. These methods help enhance joint flexibility and reduce stiffness, supporting cartilage health without overloading the tissue. In addition, biologic therapies such as autologous chondrocyte implantation (ACI) and mesenchymal stem cell (MSC) therapy aim to introduce new cells capable of regenerating damaged ECM. ACI involves harvesting the patient’s chondrocytes, expanding them in vitro, and re-implanting them into the damaged area, while MSC therapy utilizes stem cells that can differentiate into chondrocytes to facilitate cartilage regeneration. These therapies are often combined with scaffolds that support cell growth and integration, offering promising solutions to overcome the natural limitations of cartilage repair.
Ligament
Ligaments are dense connective tissues composed primarily of type I collagen fibers, which are arranged in a hierarchical structure to provide high tensile strength and flexibility, allowing ligaments to stabilize joints and guide joint movement. The molecular and cellular composition of ligaments is specialized to resist stretching and mechanical loads. Fibroblasts are the primary cell type within ligaments, responsible for synthesizing and organizing collagen and other extracellular matrix (ECM) components. These cells play a vital role in the ligament’s response to mechanical stress and injury, adjusting their activity to reinforce the ECM by producing new collagen fibers. Fibroblasts are mechanosensitive, responding to mechanical signals to promote ECM remodeling and repair following injury. They also secrete growth factors and cytokines, such as TGF-β and IL-6, which regulate the healing process and coordinate the actions of other cells involved in tissue repair. The ECM of ligaments is predominantly composed of type I collagen, which provides the strength needed to resist tensile forces, while elastin and proteoglycans contribute to the viscoelastic properties, allowing the ligament to stretch and return to its original shape. This intricate organization of collagen fibers, arranged in a crimped structure, ensures that ligaments can withstand the mechanical demands of joint movement, providing both stability and flexibility.
Upon injury, the inflammatory phase is the first stage of the healing process, where fibroblasts and immune cells release cytokines such as IL-1β, TNF-α, and IL-6, triggering inflammation. This phase is characterized by increased vascular permeability, leukocyte infiltration, and the release of inflammatory mediators that prepare the tissue for repair. The inflammatory response is essential for clearing cellular debris and initiating the healing process but can also cause pain and swelling. Following this phase, the proliferative phase begins, where fibroblasts proliferate and synthesize new ECM components, primarily type I collagen, which replaces the damaged tissue. During this phase, collagen fibers are initially laid down in a disorganized manner. The subsequent remodeling phase involves the realignment and maturation of collagen fibers to restore the ligament’s structural integrity and mechanical properties. This phase can extend for months, with the collagen fibers gradually reorganizing into a more aligned structure that closely mimics the original tissue. The remodeling phase ensures that the ligament regains its ability to resist tensile forces, ultimately restoring its function.
Rehabilitation strategies for ligament injuries emphasize the importance of early mobilization and controlled loading to stimulate fibroblast activity and collagen synthesis without overstressing the tissue. Controlled exercises, such as isometric and isotonic movements, help align newly synthesized collagen fibers, improving the ligament’s strength and elasticity. This gradual loading process encourages fibroblasts to produce robust collagen fibers and reduces the risk of joint stiffness and adhesion formation. Additionally, proprioceptive training is critical for enhancing neuromuscular control and preventing re-injury. This type of training, which includes balance and coordination exercises, helps restore joint stability by improving the body’s ability to sense joint position and respond to movement. Proprioceptive exercises enhance the interaction between the nervous system and musculoskeletal system, ensuring precise and coordinated movements, which is particularly important for athletes and individuals engaging in high-demand activities. These rehabilitation strategies, when combined, support the healing process by promoting the alignment and strengthening of collagen fibers, improving joint stability, and reducing the risk of future ligament injuries.
Subchondral Bone
Subchondral bone, located beneath the articular cartilage, plays a critical role in supporting joint structures and absorbing mechanical loads during movement. It serves as a foundation for the overlying cartilage, ensuring proper load distribution and contributing to joint health. Osteoblasts, one of the key cell types in subchondral bone, are responsible for producing and mineralizing the bone matrix, primarily by secreting type I collagen and other proteins that form the scaffold for bone mineralization. These cells are highly responsive to mechanical stimuli, which enhance their activity, leading to increased bone formation and strengthening. Osteoblasts also produce growth factors, such as TGF-β and BMPs (bone morphogenetic proteins), which promote bone growth and repair.
Osteoclasts are involved in the resorption of bone tissue, breaking down old or damaged bone to allow for the formation of new bone. This process, known as bone remodeling, is tightly regulated by signaling molecules like RANKL (receptor activator of nuclear factor kappa-B ligand) and osteoprotegerin, which control the activity of osteoclasts. Osteoclasts ensure that bone homeostasis is maintained by balancing bone resorption with formation, a process that is essential for the subchondral bone to remain strong and adaptive to mechanical loads. Osteocytes, the mature bone cells embedded within the bone matrix, act as mechanosensors, detecting mechanical stress and coordinating the activity of osteoblasts and osteoclasts to regulate bone remodeling. These cells communicate via an extensive network of canaliculi, allowing them to fine-tune the bone’s response to mechanical forces, ensuring that the bone structure adapts to changing mechanical demands. Osteocytes also produce signaling molecules such as sclerostin, which inhibits bone formation, and RANKL, which stimulates bone resorption, playing a central role in regulating the balance between bone formation and resorption.
When subchondral bone is injured, the release of inflammatory cytokines such as IL-1, IL-6, and TNF-α triggers an inflammatory response that can disrupt the balance between bone resorption and formation. These cytokines stimulate osteoclast activity, leading to increased bone resorption and the weakening of subchondral bone. This can contribute to conditions like subchondral sclerosis (abnormal hardening of the bone) and cyst formation, which can further compromise joint stability and function. The inflammatory environment not only accelerates bone resorption but also inhibits osteoblast function, impairing bone formation and disrupting the bone remodeling process. The imbalance between osteoclast and osteoblast activity can weaken the subchondral bone, which may exacerbate joint degeneration, particularly in conditions like osteoarthritis, where both the bone and cartilage are affected.
Bone remodeling, a dynamic process involving the coordinated actions of osteoclasts and osteoblasts, is essential for maintaining the structural integrity of subchondral bone. In the event of injury, disruptions in this process can lead to suboptimal bone quality, increasing susceptibility to further injury or degenerative changes. Effective remodeling ensures that old or damaged bone is replaced with new bone, adapting the subchondral bone structure to mechanical demands and maintaining joint stability. When remodeling is impaired, subchondral bone becomes more vulnerable to overload, potentially accelerating the progression of joint disorders.
Rehabilitation strategies for subchondral bone injuries focus on load management and pharmacological interventions. Gradual reintroduction of weight-bearing activities, such as progressive resistance training and low-impact exercises like swimming or cycling, stimulates bone remodeling while minimizing the risk of re-injury. Proper load management encourages osteoblasts to form new bone, enhancing the density and strength of the subchondral bone. Pharmacological interventions, including bisphosphonates or anabolic agents, can regulate bone turnover, reduce pain, and support bone health by inhibiting osteoclast activity and promoting bone formation. These medications, combined with physical rehabilitation, help restore the balance between bone formation and resorption, optimizing the recovery of subchondral bone and overall joint health.
Table 8.
The table provides a detailed comparison of the molecular composition, injury response, and rehabilitation strategies for the meniscus, cartilage, ligament, and subchondral bone. It highlights the unique cellular characteristics and healing capacities of each tissue, emphasizing the importance of tailored rehabilitation approaches to optimize recovery and restore joint function.
Table 8.
The table provides a detailed comparison of the molecular composition, injury response, and rehabilitation strategies for the meniscus, cartilage, ligament, and subchondral bone. It highlights the unique cellular characteristics and healing capacities of each tissue, emphasizing the importance of tailored rehabilitation approaches to optimize recovery and restore joint function.
| Tissue Type | Molecular Composition | Response to Injury | Rehabilitation Strategies |
| Meniscus | Primarily Type I and Type II collagen, with proteoglycans like aggrecan. Fibrochondrocytes are responsible for ECM maintenance and adapting to mechanical stress. The ECM also contains glycosaminoglycans (GAGs) that help retain water for viscoelastic properties. | Injury triggers the release of cytokines (IL-1β, TNF-α) and MMPs, degrading collagen. Healing is limited in avascular zones, and surgical intervention may be necessary. Inflammation can exacerbate ECM breakdown and slow repair. | Controlled weight-bearing exercises to stimulate fibrochondrocytes and ECM synthesis. Proprioceptive training enhances joint stability, while biologic therapies (TGF-β, PDGF) promote matrix production. Surgical repair may be required for avascular regions. |
| Cartilage | Rich in Type II collagen and aggrecan, which bind water to resist compression. Chondrocytes are the sole cell type and produce ECM components. The ECM also contains hyaluronan and link proteins to stabilize the matrix. | Cartilage responds with an inflammatory cascade, releasing cytokines (IL-1, TNF-α) that activate enzymes like ADAMTS-4 and -5. This leads to aggrecan and collagen degradation, further weakening the ECM. Avascularity severely limits repair capacity. | Hydrotherapy and continuous passive motion (CPM) stimulate chondrocytes and prevent stiffness. Biologic therapies like autologous chondrocyte implantation (ACI) and mesenchymal stem cell (MSC) therapy introduce new cells for ECM repair. Scaffolds may be used to support cell growth. |
| Ligament | Dense Type I collagen fibers arranged in a crimped pattern, providing high tensile strength. Fibroblasts synthesize collagen and elastin, maintaining ECM structure. Proteoglycans and elastin contribute to the ligament’s viscoelastic properties. | Fibroblasts and immune cells release cytokines like IL-1β and TNF-α, initiating inflammation. Fibroblasts proliferate and synthesize collagen during the proliferative phase. Remodeling realigns collagen fibers to restore mechanical properties. | Early mobilization (isometric and isotonic exercises) promotes fibroblast activity and collagen realignment. Proprioceptive training helps restore neuromuscular control. Gradual loading and strengthening exercises prevent re-injury and promote ligament healing. |
| Subchondral Bone | Subchondral bone matrix is composed of Type I collagen, produced by osteoblasts. Osteocytes within the bone matrix regulate remodeling, while osteoclasts resorb bone. The ECM is mineralized with hydroxyapatite, giving strength to the bone. | Inflammatory cytokines (IL-1, IL-6, TNF-α) activate osteoclasts, increasing bone resorption. Disruption in osteoblast-osteoclast balance leads to weakened bone, subchondral sclerosis, or cyst formation, contributing to joint degeneration. | Gradual reintroduction of weight-bearing activities like resistance training to promote bone remodeling. Pharmacological interventions (bisphosphonates, anabolic agents) regulate bone turnover, while low-impact exercises like swimming maintain joint function without overloading the bone. |
Implications for Treatment and Rehabilitation
Understanding the cellular and molecular mechanisms underlying knee joint injuries, such as those affecting the meniscus, cartilage, ligaments, and subchondral bone, is crucial for developing more effective treatment and rehabilitation strategies. The intricate interplay between mechanical loading, inflammation, and tissue regeneration highlights the need for precise interventions that are tailored to the specific tissue involved. Mechanical loading, when applied appropriately, can stimulate cellular processes like collagen synthesis, extracellular matrix (ECM) remodeling, and overall tissue repair. However, excessive or improperly timed loading can exacerbate damage, particularly in tissues like cartilage that have limited regenerative capacity. Thus, early controlled mechanical loading, implemented in a progressive manner, becomes a critical component of rehabilitation. By carefully modulating mechanical stress, clinicians can encourage beneficial cellular responses while minimizing the risk of further injury.
Tailored rehabilitation protocols are essential, as each tissue type within the knee joint responds differently to mechanical stress and injury. For example, ligaments, with their rich vascularization, can benefit from progressive weight-bearing and proprioceptive exercises that enhance fibroblast activity and collagen realignment. Conversely, the avascular nature of articular cartilage necessitates more conservative loading strategies, such as continuous passive motion (CPM) or hydrotherapy, to stimulate chondrocytes without overwhelming the tissue. Meniscal injuries, especially in the avascular inner regions, may require biologic interventions like growth factors (TGF-β, PDGF) to boost fibrochondrocyte activity and promote ECM production. Subchondral bone injuries call for gradual reintroduction of weight-bearing activities to stimulate bone remodeling, combined with pharmacological support to manage bone turnover and ensure proper healing.
Pharmacological interventions, such as the use of bisphosphonates, nonsteroidal anti-inflammatory drugs (NSAIDs), and anabolic agents, play a supportive role in managing inflammation, promoting bone formation, and inhibiting excessive tissue degradation. For instance, bisphosphonates can help regulate bone resorption in subchondral bone injuries by inhibiting osteoclast activity, while anabolic agents can stimulate osteoblasts to enhance bone repair. Anti-inflammatory medications, though commonly used, must be administered with caution to avoid hindering the natural inflammatory processes necessary for initial tissue healing. These pharmacological strategies, when combined with targeted mechanical loading, can accelerate recovery and improve tissue resilience.
Emerging regenerative medicine approaches, including autologous chondrocyte implantation (ACI), mesenchymal stem cell (MSC) therapy, and gene therapy, offer exciting possibilities for treating knee joint injuries. ACI involves harvesting a patient’s own chondrocytes, expanding them in vitro, and re-implanting them into damaged cartilage, thereby enhancing the tissue’s regenerative capacity. MSC therapy leverages the multipotent nature of stem cells to differentiate into various cell types, including chondrocytes, osteoblasts, and fibroblasts, enabling targeted repair across multiple tissue types. Additionally, gene therapy approaches, which can deliver genes encoding growth factors or other regenerative molecules directly to the injury site, hold promise for enhancing cellular activity and tissue repair. These advanced therapies aim to overcome the limitations of natural healing processes, especially in avascular tissues like cartilage, where traditional approaches are less effective.
By integrating insights into cellular and molecular mechanisms, clinicians can optimize rehabilitation protocols, pharmacological interventions, and regenerative therapies to create a more comprehensive and effective treatment plan. Early controlled loading, tailored exercise regimens, and cutting-edge biologic therapies provide a synergistic approach that not only promotes faster recovery but also enhances long-term joint function and durability. This integrative strategy ensures that the unique healing capacities of different knee joint tissues are maximized, leading to better outcomes for patients with knee injuries.
Rehabilitation Protocols
Early controlled mechanical loading not only enhances tissue repair but also activates key molecular pathways that are fundamental to the healing process. Mechanical stimuli influence cellular behaviors such as proliferation, differentiation, and extracellular matrix (ECM) remodeling by engaging mechanosensitive receptors like integrins, stretch-activated ion channels, and associated intracellular signaling cascades. These mechanotransduction processes are crucial for activating molecular pathways like the Wnt/β-catenin, focal adhesion kinase (FAK), and mitogen-activated protein kinase (MAPK) pathways, all of which regulate gene expression, ECM synthesis, and cell survival. Controlled mechanical loading, therefore, ensures that tissues like ligaments, cartilage, and subchondral bone are biologically stimulated to repair and regenerate optimally.
Progressive loading plays a pivotal role in enhancing the cellular environment by gradually increasing mechanical stimuli that activate fibroblasts, chondrocytes, and osteoblasts. For instance, during the early phase of rehabilitation, low-intensity range-of-motion exercises prevent the formation of fibrotic tissue and joint stiffness, while gently loading the tissues to maintain cellular function. As healing progresses, the introduction of isometric exercises, which engage muscles without joint movement, triggers collagen synthesis in ligaments and strengthens the surrounding musculature without overstressing the healing tissue. From a molecular perspective, this controlled increase in mechanical stress enhances the production of growth factors like transforming growth factor-beta (TGF-β) and insulin-like growth factor (IGF), both of which promote tissue regeneration and reduce inflammation.
When functional exercises are introduced, they simulate real-world activities and progressively challenge the neuromuscular system. These exercises, which may include squats, lunges, and sport-specific drills, enhance cellular communication and adaptation through mechanical cues. For example, functional loading increases the activity of integrins, which interact with the cytoskeleton and activate intracellular signaling networks like the FAK pathway, leading to enhanced ECM remodeling. In cartilage, the stimulation of chondrocytes by functional exercises promotes the synthesis of proteoglycans like aggrecan, which helps maintain the tissue’s ability to resist compressive forces. Similarly, in ligaments, functional loading ensures the alignment of type I collagen fibers, strengthening the tissue’s capacity to withstand tensile loads. These exercises also promote the activation of mechanosensitive ion channels, which facilitate calcium influx and subsequent activation of pathways such as calcineurin-NFAT, driving gene expression related to tissue adaptation.
Joint-specific loading is key to targeting the unique properties of each tissue type within the knee joint. For instance, cartilage benefits from weight-bearing exercises because mechanical compression stimulates chondrocyte activity, increasing the production of type II collagen and proteoglycans necessary for shock absorption and smooth joint articulation. Weight-bearing also triggers the release of lubricin, a glycoprotein that improves joint lubrication and reduces friction during movement. On the other hand, ligament healing is optimized through proprioceptive exercises that restore neuromuscular control and joint stability. These proprioceptive exercises enhance the interaction between sensory neurons and muscle fibers, refining the feedback loop that controls movement. At a molecular level, proprioceptive training activates signaling molecules like brain-derived neurotrophic factor (BDNF), which promotes the repair of neural pathways involved in joint stabilization.
Tailored rehabilitation programs, based on biomechanical assessments, further refine the rehabilitation process by identifying specific weaknesses or compensatory movement patterns that may hinder recovery. Advanced assessments like motion analysis or electromyography (EMG) provide data on joint kinematics and muscle activation, helping clinicians pinpoint areas requiring targeted intervention. For example, if gait analysis reveals abnormal loading patterns, specific exercises can be prescribed to address imbalances, ensuring that the mechanical forces are distributed evenly across the joint. These personalized insights allow for the precise adjustment of rehabilitation protocols, ensuring that mechanical loading is applied in a way that promotes healing without risking further damage.
Patient-specific goals are an essential component of rehabilitation, as they ensure that the rehabilitation process is aligned with the individual’s unique needs and aspirations. Whether the goal is to return to high-performance sports or to regain basic daily function, the rehabilitation program must be flexible enough to adjust to changes in the patient’s condition. Athletes may require intense, sport-specific training that includes plyometrics, agility drills, and cutting movements, all of which simulate the dynamic stresses experienced during competition. These exercises not only strengthen muscles and ligaments but also enhance proprioception and reaction time, which are crucial for injury prevention. Conversely, non-athletic patients may focus on improving joint mobility, balance, and endurance to regain the ability to perform daily activities like walking or climbing stairs.
Adjustable protocols ensure that the rehabilitation process is dynamic and responsive to the patient’s progress. Regular reassessments are essential to track the patient’s recovery and to modify exercise intensity or type based on their response to treatment. For instance, if a patient exhibits signs of joint swelling or pain, the rehabilitation protocol may be adjusted to reduce the mechanical load temporarily. Conversely, if the patient demonstrates improved strength, range of motion, or proprioception, the rehabilitation protocol can be intensified, incorporating more challenging exercises to continue promoting tissue repair and adaptation. This flexibility allows clinicians to maintain an optimal balance between loading and rest, ensuring that the healing tissues are not overstressed.
Monitoring and feedback through biomarkers and functional assessments provide critical insights into the biological and functional status of the patient. Biomarker monitoring allows clinicians to track the levels of inflammatory cytokines like IL-1, TNF-α, and matrix metalloproteinases (MMPs), which are involved in tissue degradation and inflammation. Elevated levels of these biomarkers may indicate excessive tissue stress or ongoing inflammation, signaling the need for adjustments in the rehabilitation protocol. Additionally, monitoring the levels of anabolic factors like growth factors (TGF-β, PDGF) and ECM components provides insight into the tissue’s reparative capacity, allowing clinicians to fine-tune the rehabilitation approach. Functional assessments, including range of motion, strength, and proprioception testing, offer real-time data on the patient’s recovery trajectory. Advanced tools like force plates, wearable sensors, and motion capture systems provide quantitative data on joint loading and movement patterns, allowing clinicians to identify any deficits or imbalances that need to be addressed.
By incorporating early controlled mechanical loading, joint-specific rehabilitation, and ongoing feedback through biomarker monitoring and functional assessments, rehabilitation strategies can be optimized to promote cellular regeneration, tissue repair, and functional recovery. This integrative approach leverages molecular insights into mechanotransduction and tissue-specific responses, ensuring that patients achieve better outcomes and return to their pre-injury levels of activity with minimal risk of re-injury.
Pharmacological Interventions
Pharmacological interventions complement mechanical loading and rehabilitation by targeting specific molecular pathways involved in inflammation, tissue repair, and mechanotransduction, thereby enhancing the effectiveness of treatment. These interventions can modulate cellular behaviors such as inflammation, ECM synthesis, and tissue remodeling, ensuring a more favorable environment for healing.
Anti-inflammatory agents are crucial in the early phases of injury to modulate the inflammatory response and prevent chronic inflammation. For example, Non-Steroidal Anti-Inflammatory Drugs (NSAIDs) work by inhibiting cyclooxygenase enzymes (COX-1 and COX-2), reducing the production of prostaglandins that mediate inflammation and pain. This reduction in prostaglandins alleviates symptoms and promotes early mobilization, allowing patients to participate in mechanical loading exercises without exacerbating pain. However, since prostaglandins also play a role in tissue repair, overuse of NSAIDs may impair healing by slowing the anabolic processes needed for ECM synthesis and tissue regeneration. Therefore, careful management is essential to balance pain relief with tissue repair. Cytokine inhibitors, which block pro-inflammatory cytokines such as IL-1 and TNF-α, target key molecules that drive chronic inflammation and ECM degradation. By reducing the excessive activity of these cytokines, these inhibitors create a more controlled inflammatory environment, preventing tissue damage and promoting healing.
MMP inhibitors are valuable in controlling the excessive breakdown of ECM during injury. Matrix metalloproteinases (MMPs), particularly MMP-13, degrade collagen and other ECM components, impairing tissue integrity. Selective MMP inhibitors can prevent excessive ECM degradation by targeting specific MMPs involved in pathological conditions. By inhibiting MMP activity, these drugs preserve collagen and proteoglycans essential for tissue structure and function, ensuring that tissue remodeling occurs in a balanced manner. This selective inhibition allows necessary ECM turnover for tissue repair while minimizing detrimental degradation, especially in chronic conditions like osteoarthritis, where excessive ECM breakdown is a significant concern.
Growth factor therapy targets tissue regeneration at the cellular level by promoting cell proliferation, differentiation, and ECM synthesis. Growth factors like TGF-β (Transforming Growth Factor-Beta), IGF-1 (Insulin-Like Growth Factor-1), and BMPs (Bone Morphogenetic Proteins) activate intracellular signaling pathways that regulate anabolic processes. For instance, TGF-β promotes the synthesis of type II collagen in cartilage and type I collagen in ligaments, aiding in tissue regeneration. Localized delivery of growth factors directly to the injury site through injections or biomaterial scaffolds enhances their therapeutic effectiveness by ensuring high concentrations at the target tissue while minimizing systemic side effects. Additionally, controlled release systems, such as hydrogels or biodegradable scaffolds, offer sustained delivery of growth factors, allowing them to be released gradually in a manner that aligns with the body’s natural healing timeline. This ensures a steady supply of bioactive molecules at the injury site, supporting long-term tissue repair and preventing the need for repeated interventions.
Modulation of mechanotransduction pathways through pharmacological agents enhances the cellular responses to mechanical loading, thereby improving tissue repair. Integrin modulators can strengthen cell-ECM interactions, which are essential for mechanotransduction. By enhancing integrin signaling, fibroblasts, chondrocytes, and osteoblasts can better respond to mechanical cues, leading to more effective ECM remodeling and tissue regeneration. For example, integrins activate pathways like FAK (Focal Adhesion Kinase), which coordinates cell migration and ECM production. FAK inhibitors help regulate these downstream pathways, preventing excessive cellular proliferation and ensuring controlled tissue repair. By fine-tuning FAK signaling, these inhibitors support a more organized and balanced ECM synthesis, enhancing tissue integrity without promoting fibrosis or abnormal tissue growth.
Lastly, calcium signaling modulators target another critical aspect of mechanotransduction. Calcium ions act as second messengers in a variety of cellular processes, including contraction, secretion, and gene expression. Modulating calcium influx through stretch-activated ion channels or calcium channel blockers can optimize cellular responses to mechanical loading, improving processes like cell proliferation and differentiation. This modulation supports the proper activation of mechanotransduction pathways that are essential for tissue repair, such as the calcineurin-NFAT pathway, which regulates gene expression related to ECM production and cell survival.
In conclusion, pharmacological interventions that modulate inflammation, ECM degradation, growth factor activity, and mechanotransduction pathways work synergistically with mechanical loading and rehabilitation to promote more effective tissue repair and regeneration. These approaches ensure a comprehensive and targeted strategy for enhancing cellular responses to injury, facilitating faster recovery and improved long-term outcomes.
Advanced Regenerative Medicine Approaches
Regenerative medicine presents innovative strategies for repairing and regenerating damaged tissues in knee joint injuries by leveraging tissue engineering, stem cell therapy, and gene therapy. These approaches focus on restoring the structure and function of tissues such as cartilage, ligaments, and bone by targeting specific cellular pathways and enhancing the body’s intrinsic repair mechanisms.
Tissue engineering utilizes biomaterial scaffolds made from materials like collagen, hyaluronic acid, or synthetic polymers to create structures that mimic the mechanical properties of native tissues. These scaffolds provide a supportive environment for cell attachment, proliferation, and differentiation. By mimicking the biomechanical environment, scaffolds guide tissue regeneration, allowing cells like chondrocytes, fibroblasts, and osteoblasts to rebuild the extracellular matrix (ECM). Scaffolds can be functionalized with bioactive molecules, such as growth factors, to further enhance their regenerative capacity, influencing cellular behavior, ECM production, and tissue remodeling. 3D bioprinting offers an advanced approach by creating complex tissue constructs with precise control over scaffold architecture, enabling the integration of multiple cell types and ECM components. This technique replicates native joint structures, supporting more accurate tissue regeneration and functional recovery.
Stem cell therapy is another promising avenue, utilizing mesenchymal stem cells (MSCs) that can differentiate into various cell types necessary for tissue repair, including chondrocytes, fibroblasts, and osteoblasts. Autologous MSCs, harvested from the patient’s own tissue, minimize immune rejection and improve therapeutic outcomes by leveraging the body’s own regenerative potential. In contrast, allogeneic MSCs from donor sources provide an off-the-shelf solution, offering immediate availability and standardized quality. In addition to their differentiation capacity, MSCs exert significant paracrine effects by secreting cytokines and growth factors that modulate inflammation, promote angiogenesis, and enhance the activity of resident cells. These paracrine effects help create a favorable environment for tissue repair, supporting both structural regeneration and cellular activity at the injury site.
Gene therapy focuses on modifying cellular genetic material to promote tissue repair and regeneration through targeted techniques. Gene overexpression strategies aim to enhance the cellular response to mechanical loading by upregulating genes involved in ECM synthesis, mechanotransduction, or anti-inflammatory processes, boosting tissue repair. Conversely, gene knockdown involves silencing genes that inhibit tissue regeneration, such as those involved in chronic inflammation or excessive ECM degradation, creating a more favorable environment for healing. Techniques like CRISPR/Cas9 technology provide precise gene-editing capabilities, allowing for specific modifications that promote tissue repair while minimizing off-target effects. This cutting-edge gene-editing method enables highly targeted interventions to improve the effectiveness of regenerative therapies by directly influencing cellular behavior and tissue outcomes.
These regenerative medicine strategies—whether through tissue engineering, stem cell therapy, or gene therapy—integrate with mechanical loading and rehabilitation protocols to enhance the body’s natural healing processes, offering new avenues for treating knee joint injuries and promoting long-term recovery.
Combination Therapies
Combining mechanical loading with other therapeutic modalities, such as pharmacological interventions and biologics, offers a powerful strategy for enhancing tissue repair and regeneration at the molecular level. Mechanical loading stimulates cellular processes like mechanotransduction, ECM synthesis, and cell differentiation, while pharmacological agents and biologics provide biochemical support that complements these mechanical cues. By integrating these approaches, synergistic effects can be achieved, optimizing the healing process.
Pharmacological and Mechanical Interventions leverage the complementary actions of drugs and mechanical stimuli. For example, anti-inflammatory drugs, such as NSAIDs or cytokine inhibitors, can be used to modulate the inflammatory response during the early stages of rehabilitation. These agents reduce the levels of pro-inflammatory cytokines like IL-1 and TNF-α, which are known to exacerbate tissue damage and inhibit repair. When combined with controlled mechanical loading, which enhances mechanotransduction and promotes cellular activity, the reduced inflammation facilitates more effective tissue repair and accelerates recovery. MMP inhibitors can also be used to limit ECM degradation while mechanical loading stimulates fibroblast or chondrocyte activity, further enhancing ECM synthesis and strengthening the healing tissue. The key to success lies in timing and dosage, ensuring that pharmacological agents are administered at the appropriate stage of healing to maximize their effects while minimizing side effects, such as delayed tissue repair associated with excessive use of anti-inflammatory drugs.
Biologics and Mechanical Loading, particularly the use of stem cell therapy and growth factor administration, can greatly benefit from the mechanical cues provided by loading exercises. Stem cells, such as mesenchymal stem cells (MSCs), rely on mechanical signals to guide their differentiation into specific cell types, such as chondrocytes, osteoblasts, or fibroblasts. When combined with controlled mechanical loading, MSCs can respond more effectively, producing ECM components and promoting tissue regeneration in injured ligaments, cartilage, or bone. Growth factors like TGF-β or IGF-1, which are critical for ECM synthesis and cell proliferation, can be delivered through scaffolds or direct injection, providing biochemical signals that enhance the effects of mechanical loading. The interaction between mechanical stimuli and growth factors stimulates mechanotransduction pathways, such as the integrin-FAK and MAPK pathways, promoting robust tissue regeneration.
Scaffold-based therapies represent a sophisticated approach, where biomaterials are engineered to mimic the mechanical properties of native tissues while also being functionalized with growth factors or bioactive molecules. These scaffolds not only provide structural support for tissue regeneration but also deliver biochemical signals that enhance the healing process. When combined with mechanical loading, the scaffold provides both physical and biochemical cues to cells, promoting ECM synthesis, cell proliferation, and differentiation. By aligning the mechanical properties of the scaffold with the specific requirements of the injured tissue, and applying controlled mechanical loading, this approach creates an optimal environment for tissue repair, accelerating recovery and improving functional outcomes.
In summary, the combination of pharmacological interventions, biologics, and mechanical loading offers a comprehensive, multi-modal approach to tissue regeneration. This synergistic strategy targets multiple molecular pathways involved in inflammation, mechanotransduction, and tissue remodeling, optimizing the healing process and promoting long-term recovery.
Advanced Research and Future Directions
Advances in the molecular and cellular understanding of knee joint injuries have dramatically expanded possibilities for more effective and personalized treatment and rehabilitation strategies. These developments center on optimizing how we address the different phases of injury repair— from inflammation to tissue regeneration—by targeting specific cellular pathways and integrating insights from clinical and preclinical studies, biomarker research, and new therapeutic technologies.
In vivo models are indispensable for capturing the complexity of joint injuries and their repair in a biological context. Rodent models provide a cost-effective and genetically manipulable platform to dissect molecular pathways, such as those involved in inflammation, mechanotransduction, or ECM remodeling, which are crucial to tissue repair. These models allow researchers to study gene knockout effects or manipulate signaling pathways to investigate how specific proteins like integrins, MMPs, or cytokines contribute to the healing process. On the other hand, large animal models, with joint sizes and mechanics more similar to human knees, offer an essential step closer to human clinical conditions. They are particularly valuable for evaluating regenerative medicine approaches like tissue-engineered scaffolds, stem cell therapies, and surgical interventions. Studies using these models can assess the physiological responses to therapies, which helps bridge the gap between laboratory research and clinical application.
Clinical research is essential for translating these findings into human care. Randomized controlled trials (RCTs) offer the most reliable means of determining the efficacy of new interventions, while longitudinal studies allow for the long-term tracking of patient outcomes, providing insights into factors like recurrence rates, joint function preservation, and the durability of mechanical loading protocols. For example, these trials may explore how progressive loading schedules influence outcomes in ligament or cartilage repair or compare the impact of various biologic therapies on the regenerative capacity of damaged tissues over time. Understanding how different patient populations respond to interventions, including those with comorbidities or varying levels of activity, helps refine rehabilitation approaches.
Biomarker discovery offers another frontier in personalized rehabilitation and injury management, with molecular markers providing real-time data on injury status and tissue healing. Inflammatory biomarkers such as IL-6, TNF-α, and IL-1β can help guide the use of pharmacological interventions, such as NSAIDs or cytokine inhibitors, by providing feedback on the level of inflammation within the joint. Likewise, monitoring MMPs and their tissue inhibitors (TIMPs) can reveal the balance between ECM degradation and synthesis during healing, ensuring that treatments promote tissue repair without contributing to excessive breakdown. Biomarkers that reflect mechanotransduction processes, such as integrins or calcium signaling molecules, are particularly important in adjusting mechanical loading protocols to optimize tissue repair, especially in the early stages of rehabilitation.
The use of omics technologies, such as genomics, proteomics, and metabolomics, offers an unprecedented level of detail about the molecular changes occurring after knee joint injuries. These technologies enable the identification of genes, proteins, and metabolic pathways that are activated or suppressed during injury and healing. For instance, genomic analyses might uncover genetic variations that make some individuals more susceptible to ligament tears or cartilage degeneration, providing insights into patient-specific vulnerabilities and guiding personalized rehabilitation programs. Proteomics and metabolomics, by analyzing protein expression and metabolite profiles, respectively, help track the biochemical processes governing inflammation, tissue repair, and cellular metabolism. Integrating multi-omics data— combining genomics, proteomics, and metabolomics— creates a comprehensive view of the molecular landscape of injury and recovery, helping to identify complex biomarker networks that can be leveraged for personalized therapy.
Incorporating these biomarkers into clinical practice allows for real-time monitoring and tailored interventions. For example, point-of-care devices that measure inflammatory cytokines or ECM degradation products could allow clinicians to adjust treatment strategies on the fly, providing more responsive care. Similarly, monitoring markers like growth factors or collagen fragments could indicate the success of biologic therapies, such as stem cell or growth factor injections, and guide the timing of rehabilitation protocols to match the underlying tissue’s regenerative capacity.
Regenerative medicine, including tissue engineering, stem cell therapy, and gene therapy, represents the future of knee joint injury treatment, offering potential solutions for overcoming the limitations of traditional treatments. Tissue engineering approaches, like the use of biomaterial scaffolds that mimic the mechanical properties of native tissues, are being developed to provide structural support and promote cell attachment, proliferation, and differentiation. These scaffolds can be customized to release growth factors over time, creating a biologically active environment that supports long-term tissue repair. Furthermore, 3D bioprinting allows for the creation of complex, patient-specific constructs that replicate the structure and function of native tissues, incorporating multiple cell types and ECM components to enhance regenerative potential.
Stem cell therapies, particularly those using mesenchymal stem cells (MSCs), hold promise for regenerating damaged cartilage, ligaments, and other knee joint tissues. Autologous MSC therapy—harvesting a patient’s own stem cells from tissues like bone marrow or adipose tissue—reduces the risk of immune rejection and offers a personalized regenerative option. In contrast, allogeneic MSCs provide an off-the-shelf solution, enabling immediate intervention with standardized cells. Stem cells not only replace damaged cells but also exert paracrine effects, secreting cytokines and growth factors that modulate inflammation, enhance ECM synthesis, and promote angiogenesis, contributing to a more favorable healing environment.
Gene therapy is another advanced approach that can be used to enhance tissue repair by either upregulating beneficial genes or silencing those that impede healing. For example, gene overexpression could boost the production of key ECM components like collagen or anti-inflammatory proteins, while gene knockdown approaches could silence genes responsible for excessive ECM degradation or inflammation. The precision of CRISPR/Cas9 gene-editing technology allows for targeted manipulation of these genes, potentially transforming the ability to repair and regenerate joint tissues.
Combining these regenerative therapies with mechanical loading could further enhance their effectiveness. For instance, loading protocols tailored to the type and timing of biologic therapy, such as progressive weight-bearing exercises following stem cell injections, may optimize tissue integration and functional recovery. Similarly, scaffold-based therapies that release growth factors in response to mechanical stimuli could create a synergistic environment that maximizes tissue regeneration.
Looking forward, future research will focus on integrating these innovations into clinical practice through personalized rehabilitation protocols, advanced biomarker-guided treatments, and long-term clinical trials that evaluate the efficacy of combined therapies. As point-of-care technologies and wearable biosensors become more sophisticated, they will provide real-time feedback on tissue healing and mechanical load responses, enabling clinicians to continually optimize treatment and rehabilitation plans. The integration of molecular insights, cutting-edge technologies, and personalized medicine holds the potential to revolutionize knee joint injury care, offering patients more effective, long-lasting recovery options.
Novel Therapeutic Targets
Recent advances in the understanding of cellular and molecular mechanisms underlying knee joint injuries have led to the identification of novel therapeutic targets that present exciting opportunities for enhancing tissue repair and regeneration. These advancements are transforming traditional rehabilitation approaches and opening new avenues for integrative therapies that combine molecular interventions with mechanical loading and biologic treatments. At the forefront of this innovation are strategies focused on modulating key signaling pathways involved in mechanotransduction, cell-ECM interactions, and tissue remodeling. By targeting critical molecules like integrins, FAK, ion channels, and components of the MAPK and Wnt/β-catenin pathways, therapies can be tailored to achieve more precise control over cellular responses to injury and rehabilitation.
Integrin signaling modulators are becoming particularly important for their ability to influence how cells interact with the extracellular matrix (ECM). By modulating integrin function, these therapies can either enhance or inhibit cellular activities like adhesion, migration, and differentiation. For instance, integrin activators strengthen cell adhesion to the ECM, which is critical for tissue regeneration, while integrin inhibitors help control pathological processes such as fibrosis by reducing excessive cell-ECM interactions. This dual capability offers flexibility in treating a wide range of conditions, from promoting tissue repair in acute injuries to managing chronic inflammatory conditions where overactive cell adhesion may cause excessive tissue scarring.
FAK (Focal Adhesion Kinase) has emerged as another promising therapeutic target due to its central role in mechanotransduction and its downstream effects on pathways like MAPK and PI3K/Akt. FAK activators can enhance cell proliferation, migration, and ECM synthesis, making them useful for promoting the healing of ligaments, cartilage, and subchondral bone. These activators could be particularly beneficial when combined with stem cell therapies, where FAK signaling enhances the integration and effectiveness of transplanted cells. On the other hand, FAK inhibitors are valuable for conditions where uncontrolled cell migration and proliferation contribute to disease progression, such as in cancer or fibrotic diseases. By inhibiting FAK activity, these therapies can slow down the pathological remodeling of tissues.
Modulating mechanotransduction pathways—the cellular machinery that converts mechanical signals into biochemical responses—offers another powerful approach to enhancing tissue repair. Ion channels and calcium signaling modulators are critical components of these pathways, influencing how cells perceive and respond to mechanical stress. By targeting calcium channels or manipulating calcium-binding proteins like calmodulin, therapies can enhance the cellular responses necessary for regeneration, particularly in response to mechanical loading during rehabilitation. Similarly, MAPK pathway modulators such as ERK1/2 activators or JNK inhibitors can regulate key cellular processes like proliferation and inflammation, either promoting tissue growth or reducing harmful inflammatory responses, depending on the therapeutic goals.
Gene therapy is a cutting-edge tool that allows for the direct manipulation of genetic pathways involved in tissue repair and mechanotransduction. By using gene overexpression techniques, such as viral vector delivery or CRISPR/Cas9 technology, genes that promote tissue repair—like those involved in collagen production or growth factor synthesis—can be amplified, enhancing the healing potential of injured tissues. In contrast, gene knockdown techniques can silence genes that contribute to excessive inflammation or ECM degradation, creating a more favorable environment for tissue regeneration. This precision in controlling gene expression opens the door to highly personalized treatment strategies that can be tailored to individual patients based on their specific injury profiles and genetic predispositions.
The integration of combination therapies represents one of the most promising trends in the field of regenerative medicine and rehabilitation. Combining pharmacological agents, such as anti-inflammatory drugs or growth factor therapies, with mechanical loading can create synergistic effects that accelerate healing. For example, anti-inflammatory drugs can reduce pain and swelling, allowing patients to engage in early mechanical loading, which in turn stimulates the production of collagen and ECM components needed for tissue repair. Similarly, stem cell therapies can be enhanced by mechanical loading, as the physical forces applied during rehabilitation promote the differentiation of stem cells into the specific tissue types required for regeneration, whether cartilage, bone, or ligament.
Scaffold-based therapies provide another layer of innovation by offering a structural framework that supports tissue growth while delivering bioactive molecules like growth factors. These scaffolds can be engineered to mimic the mechanical properties of native tissues, providing both mechanical support and biochemical cues that guide cell behavior. When combined with mechanical loading, these scaffolds enhance the natural healing process by ensuring that cells receive the necessary mechanical stimuli to produce ECM components and integrate properly into the surrounding tissue.
Advanced research continues to explore novel therapeutic targets and optimize these approaches through in vivo models, biomarker discovery, and clinical trials. Animal models, particularly large mammals, provide crucial insights into how these therapies function in a complex, biomechanically active environment that closely mimics human physiology. As clinical studies progress, the translation of these therapies from bench to bedside is becoming increasingly feasible, with a growing body of evidence supporting their efficacy in improving patient outcomes.
In summary, these molecular advances are revolutionizing the way knee joint injuries are treated, moving from purely mechanical rehabilitation strategies to a more integrated, multi-modal approach that leverages pharmacological, genetic, and cellular therapies to achieve superior results. The ability to fine-tune therapeutic interventions based on molecular and cellular mechanisms promises to not only enhance the speed and quality of tissue repair but also reduce the risk of re-injury and chronic complications, setting the stage for more effective and personalized patient care.
Personalized Rehabilitation Strategies
Personalized rehabilitation strategies are gaining increasing recognition as a critical approach to optimizing recovery for patients with knee joint injuries. These strategies go beyond standardized protocols by incorporating the unique biological, physiological, and lifestyle characteristics of each patient, which can significantly impact their response to treatment. Factors such as age, sex, genetic predispositions, injury severity, body composition, and the presence of comorbidities like diabetes or osteoarthritis all influence how well a patient may recover from injury and respond to specific rehabilitation interventions. By developing individualized rehabilitation programs that account for these variables, clinicians can enhance the effectiveness of treatment, reduce recovery times, and minimize the risk of re-injury or long-term complications.
Integration of patient-specific data plays a crucial role in the design of personalized rehabilitation protocols. Genetic background, for example, can influence tissue healing rates, inflammatory responses, and susceptibility to injury. Advances in genomics and the identification of genetic biomarkers allow for the assessment of a patient’s genetic predisposition to factors like collagen synthesis, mechanotransduction, and inflammatory processes. By incorporating this data, clinicians can predict a patient’s healing potential and adjust rehabilitation intensity accordingly. For instance, patients with genetic markers that indicate slower collagen synthesis may require extended periods of lower-intensity mechanical loading to prevent over-stressing the tissues before they are fully healed. This tailored approach helps to ensure that rehabilitation is not only safe but also optimally timed for maximal tissue regeneration.
Age and sex differences are also pivotal in personalizing rehabilitation strategies. Older adults, for instance, typically experience slower tissue healing due to age-related declines in cellular function, ECM turnover, and reduced vascularization. In such cases, rehabilitation protocols may need to incorporate longer recovery periods between sessions or use more conservative loading strategies to prevent excessive strain on the healing tissues. In contrast, younger patients may be able to tolerate more aggressive rehabilitation, with faster progression in mechanical loading and functional exercises. Additionally, sex-based differences in muscle mass, hormonal levels, and joint biomechanics can influence how patients respond to rehabilitation. For example, women may be at higher risk of ligament injuries due to differences in neuromuscular control and joint laxity, necessitating tailored proprioceptive and strengthening exercises aimed at stabilizing the knee joint.
Injury severity and comorbidities further highlight the need for personalized approaches. Patients with more severe injuries, such as complete ligament tears or complex cartilage damage, often require slower, more cautious rehabilitation programs that incorporate both mechanical and biological therapies to promote tissue repair. In contrast, those with milder injuries may benefit from faster progression in strength and mobility exercises. Patients with comorbidities like obesity, diabetes, or chronic inflammatory conditions may experience delayed healing or increased susceptibility to complications like infection or fibrosis. As such, their rehabilitation programs may need to incorporate strategies to manage these conditions concurrently, such as nutritional counseling or pharmacological interventions aimed at controlling inflammation and metabolic imbalances.
The development of predictive models and decision-support systems is another key component of personalized rehabilitation. Using advanced algorithms and artificial intelligence (AI), these models can analyze large datasets that include patient demographics, injury characteristics, genetic information, and previous treatment outcomes. By identifying patterns and correlations within the data, predictive models can provide clinicians with personalized rehabilitation recommendations, optimizing the choice of exercises, loading regimens, and recovery timelines. For instance, AI-driven decision-support systems can help clinicians predict which patients are at higher risk of developing post-injury complications like osteoarthritis or chronic pain and adjust rehabilitation strategies to mitigate these risks. These tools also allow for continuous adjustment of rehabilitation protocols based on real-time data, ensuring that the treatment remains responsive to the patient’s progress and changing needs.
Advanced technologies for monitoring and adjustment further enhance the ability to personalize rehabilitation. Wearable devices, such as smart braces, sensors, and fitness trackers, can provide real-time feedback on joint movements, loading patterns, and muscle activation. This data can be used to monitor the patient’s adherence to prescribed exercises, detect compensatory movement patterns, and adjust the rehabilitation program as needed. For example, if a wearable device detects that a patient is overloading the injured knee during daily activities, the clinician can modify the rehabilitation exercises or recommend assistive devices to reduce strain. Additionally, these technologies allow for remote monitoring, enabling patients to continue rehabilitation at home while maintaining communication with their healthcare providers. This can be especially beneficial for patients with limited access to in-person physical therapy due to geographic or logistical barriers.
Functional assessments such as gait analysis, range of motion testing, and strength measurements are also essential for tracking progress and making real-time adjustments to rehabilitation protocols. These assessments provide valuable insights into the biomechanics of the injured joint and surrounding musculature, helping to identify deficits in joint stability, muscle strength, or flexibility that need to be addressed. For example, gait analysis can reveal compensatory patterns that may lead to secondary injuries if left untreated, while range of motion testing can highlight areas of stiffness or restriction that need targeted intervention. By continuously monitoring these functional outcomes, clinicians can ensure that rehabilitation is progressing as planned and make adjustments to optimize recovery.
Neuroplasticity and motor learning principles are increasingly being integrated into personalized rehabilitation strategies, particularly in patients recovering from knee surgeries or complex joint injuries. Tailored proprioceptive and neuromuscular control exercises are designed to retrain the patient’s sensory-motor pathways, enhancing joint stability and movement coordination. Neuroplasticity, the brain’s ability to adapt and reorganize itself in response to training, is a key factor in restoring functional movement patterns after injury. Personalized rehabilitation exercises that challenge balance, coordination, and fine motor control can help improve the body’s ability to sense joint position (proprioception) and restore more natural movement patterns, reducing the risk of re-injury.
In conclusion, personalized rehabilitation strategies represent a paradigm shift in the treatment of knee joint injuries, moving away from one-size-fits-all protocols to tailored interventions that account for individual variability. By integrating patient-specific data, utilizing predictive models, and incorporating advanced monitoring technologies, these strategies offer the potential to significantly improve clinical outcomes, accelerate recovery, and reduce the risk of long-term complications. Personalized rehabilitation not only enhances the efficacy of mechanical and biological therapies but also empowers patients to take an active role in their recovery, fostering better adherence and long-term success.
Integration of Patient-Specific Data
Integrating comprehensive patient-specific data is essential for developing personalized rehabilitation protocols that optimize recovery and improve outcomes for knee joint injuries. This approach involves collecting and analyzing a wide range of data, including biomechanical assessments, imaging results, biomarker profiles, and patient-reported outcomes, each of which provides critical insights into the patient’s unique condition. Biomechanical assessments, such as gait analysis, joint kinematics, and muscle strength testing, help to identify functional deficits and compensatory movement patterns that may need to be addressed through targeted exercises. These assessments allow clinicians to evaluate how the patient moves post-injury, revealing issues like uneven weight distribution, improper joint alignment, or muscle imbalances that, if left uncorrected, could lead to re-injury or delayed healing. For example, gait analysis can highlight deviations in walking mechanics that may increase the risk of joint degeneration or additional ligament strain, enabling early intervention with corrective strategies.
Imaging results—including MRI, CT scans, and ultrasound—offer a detailed view of the structural integrity of the joint and surrounding tissues, such as cartilage, ligaments, and subchondral bone. These images provide crucial information on the extent of tissue damage, such as cartilage degradation, ligament tears, or bone edema, helping to guide the intensity and type of rehabilitation exercises. For instance, a patient with significant cartilage damage seen on an MRI may require a more conservative loading regimen to avoid further wear, while a patient with mild ligament strain might be able to engage in more aggressive strengthening exercises early on. Imaging also helps monitor the healing process, allowing for adjustments to the rehabilitation protocol as the tissue regenerates or remodels.
Biomarker profiles add a molecular dimension to personalized rehabilitation, offering insights into the biological processes underlying injury and recovery. Biomarkers such as cytokines, growth factors, and enzymes involved in tissue remodeling can indicate the level of inflammation, tissue repair activity, and degradation of extracellular matrix components. For example, elevated levels of pro-inflammatory cytokines like IL-1β or TNF-α could suggest an ongoing inflammatory process that might necessitate anti-inflammatory interventions or a reduction in mechanical loading to prevent further tissue damage. Conversely, biomarkers indicating high levels of tissue repair, such as increased collagen synthesis or growth factor activity (e.g., TGF-β, IGF-1), could signal that the patient is ready for more intensive rehabilitation exercises aimed at tissue strengthening and regeneration. Monitoring these biomarkers throughout the rehabilitation process allows clinicians to dynamically adjust the treatment plan based on real-time biological feedback, ensuring that interventions are timed appropriately to the patient’s healing stages.
Patient-reported outcomes (PROs) provide valuable qualitative data on the patient’s subjective experience of pain, functional limitations, and overall quality of life. This data is crucial for tailoring rehabilitation to meet the patient’s specific goals, whether that involves returning to high-level sports, regaining full mobility, or simply reducing pain during daily activities. PROs also help to identify psychological factors such as fear-avoidance behaviors or anxiety about re-injury, which can significantly impact rehabilitation progress. Addressing these concerns through education, graded exposure to activity, and patient empowerment can improve adherence to the rehabilitation program and enhance recovery outcomes. Moreover, ongoing collection of patient-reported outcomes allows for continuous feedback on the effectiveness of the rehabilitation protocol, highlighting areas that may require modification.
By integrating biomechanical data, imaging results, biomarker profiles, and patient-reported outcomes, clinicians can develop highly individualized rehabilitation protocols that are tailored to the patient’s specific physiological, biomechanical, and psychological needs. This comprehensive approach ensures that rehabilitation is not only based on the structural and functional aspects of the injury but also aligned with the underlying molecular processes and the patient’s personal experience. Furthermore, this integration allows for real-time adjustments to the rehabilitation plan, making it a dynamic and responsive process that evolves with the patient’s progress. The result is a more precise, effective, and patient-centered rehabilitation strategy that promotes optimal healing, functional recovery, and long-term joint health.
- 1. Biomechanical Assessments
Biomechanical assessments are essential for creating effective, personalized rehabilitation strategies, offering detailed, objective data on movement mechanics, muscle performance, and joint function. These assessments not only identify specific functional deficits but also enable the development of interventions tailored to the unique needs of each patient. Gait analysis, for instance, is a powerful tool for examining the dynamic aspects of walking, capturing details such as stride length, cadence, foot positioning, and joint angles at different phases of the gait cycle. This level of analysis helps clinicians detect subtle deviations in movement patterns that may not be immediately apparent but could contribute to long-term knee joint instability, pain, or dysfunction. By addressing these deviations early in the rehabilitation process, clinicians can design interventions that specifically target problematic areas, such as muscle imbalances, joint stiffness, or improper weight distribution, helping prevent re-injury and promoting more efficient movement. Additionally, integrating digital gait analysis systems with feedback mechanisms enables real-time monitoring, giving patients immediate visual or auditory cues to correct their movement patterns, thus accelerating the learning of proper walking mechanics.
Joint kinematics assessments focus on understanding how the knee and surrounding joints move during various activities, providing a three-dimensional view of movement. These analyses are critical for evaluating how different tasks—such as walking, running, jumping, or climbing stairs—impact the joint. Precise tools, including wearable sensors and advanced motion capture systems, can record joint angles, rotations, and accelerations, offering comprehensive data that reveals abnormalities in joint movement that might not be visible to the naked eye. For example, subtle alterations in knee flexion or extension angles during weight-bearing activities can place excessive stress on the cartilage, ligaments, or surrounding muscles, increasing the risk of injury or delaying recovery. Based on joint kinematics data, rehabilitation exercises can be precisely targeted to improve range of motion, stability, and neuromuscular coordination, thus optimizing joint function and reducing mechanical stress on the knee.
Muscle strength testing is vital in determining the strength and endurance of key muscle groups that support the knee joint, such as the quadriceps, hamstrings, and calf muscles. Weakness or imbalance between these muscles can lead to compensatory movements, which increase the risk of strain or further injury. For instance, insufficient quadriceps strength may lead to poor control during knee extension, while hamstring weakness can affect knee flexion stability. Utilizing tools like isokinetic dynamometers provides an accurate, quantitative assessment of muscle force output across different joint movements, enabling the identification of specific muscle deficiencies that may contribute to knee instability or impaired function. Strength testing can also reveal muscle endurance deficits, which are equally important for long-term joint health, particularly in activities that require sustained physical effort. Based on these insights, a personalized strength training program can be designed to progressively improve muscle power, endurance, and balance, focusing on restoring the proper functional relationship between the quadriceps and hamstrings. This tailored approach ensures that rehabilitation exercises not only address immediate muscle weaknesses but also build a foundation for long-term knee joint health, reducing the risk of re-injury.
Moreover, combining these biomechanical assessments with patient-reported outcomes and clinical markers like pain levels, swelling, or range of motion offers a more holistic view of recovery progress. This data-driven approach allows for real-time adjustments to the rehabilitation protocol, ensuring that exercises remain effective and aligned with the patient’s evolving capabilities. For example, if a patient shows improvements in strength but continues to exhibit gait abnormalities, further adjustments can be made to correct movement patterns, enhancing the overall rehabilitation outcome. Continuous monitoring and reassessment ensure that the rehabilitation process remains dynamic and responsive to the patient’s needs, fostering more efficient recovery and reducing the risk of chronic issues. Additionally, the integration of emerging technologies such as wearable motion sensors, artificial intelligence (AI)-driven analytics, and virtual reality (VR)-assisted rehabilitation could further enhance the precision and customization of these assessments, making personalized rehabilitation even more effective.
- 2. Imaging
Expanding on the role of Magnetic Resonance Imaging (MRI), this imaging modality plays a pivotal role in diagnosing and monitoring soft tissue injuries within the knee joint. MRI’s ability to produce high-resolution images of ligaments, menisci, cartilage, and other joint structures makes it essential for assessing the extent of both acute and chronic injuries. For example, in the case of a meniscal tear or ligament rupture, MRI can precisely define the size, location, and type of tear, which directly influences treatment planning. Moreover, MRI allows for the detection of microstructural changes in cartilage, including early-stage degeneration or matrix breakdown, which are critical for diagnosing conditions such as osteoarthritis. Advanced MRI techniques, like T2 mapping and delayed gadolinium-enhanced MRI of cartilage (dGEMRIC), provide quantitative data on cartilage health by assessing water content and glycosaminoglycan (GAG) concentrations. This level of detail helps clinicians identify early degenerative changes and implement interventions that might slow or reverse cartilage deterioration. Additionally, functional MRI (fMRI) can assess neuromuscular changes, providing insights into muscle activation patterns that are essential for designing personalized rehabilitation strategies. Over time, repeated MRI scans help track the healing of injured tissues, offering a non-invasive method to monitor tissue regeneration and adjust treatment protocols accordingly.
Ultrasound, particularly with advancements like elastography and Doppler ultrasound, adds significant value to the dynamic assessment of knee injuries. The real-time nature of ultrasound enables clinicians to observe joint movement and evaluate soft tissue structures during functional activities, such as flexion and extension of the knee. This is particularly important in cases of tendon or ligament injuries where dynamic imaging can reveal abnormalities that static imaging might miss. For instance, an ultrasound can capture the movement of tendons in real time, showing abnormalities in tendon gliding or detecting partial tears that are only visible during motion. Furthermore, Doppler ultrasound is valuable for evaluating vascularity and inflammation in the affected area, helping to detect increased blood flow associated with acute inflammation or chronic overuse injuries. This modality is also instrumental in guiding therapeutic interventions, such as platelet-rich plasma (PRP) injections, ensuring precise delivery of treatments to the injured tissues. Ultrasound’s portability and ease of use make it a practical tool for continuous monitoring throughout the rehabilitation process, and its biofeedback capabilities provide patients with immediate visual feedback, allowing them to actively participate in their rehabilitation by correcting movements in real time.
Computed Tomography (CT) continues to be the gold standard for evaluating bony structures, particularly in trauma cases involving fractures or significant joint deformities. 3D reconstructions from CT scans allow for a detailed view of complex fracture patterns, enabling surgeons to plan the most effective approach for reconstructing the joint. This is especially useful in cases requiring surgical intervention, where precise anatomical details are crucial for ensuring correct alignment and fixation. Low-dose CT techniques have made it possible to reduce radiation exposure, allowing for repeated imaging without significant risk to the patient, which is particularly important in follow-up assessments. CT imaging is also beneficial post-surgery for evaluating the success of fixation techniques, checking for proper alignment, and monitoring for potential complications such as implant loosening or joint misalignment. Dual-energy CT adds another layer of diagnostic capability by differentiating between bone and soft tissue, making it useful for detecting soft tissue abnormalities like gouty tophi or calcifications in addition to fractures.
In summary, the integration of advanced imaging techniques, including MRI, ultrasound, and CT, into the diagnostic and monitoring processes for knee injuries significantly enhances the clinician’s ability to accurately assess and treat these injuries. The detailed, high-resolution images provided by MRI and CT, combined with the real-time dynamic capabilities of ultrasound, offer a comprehensive approach to understanding the structural and functional aspects of knee joint injuries. These technologies not only aid in diagnosing the extent of injury but also play a critical role in tracking healing, guiding interventions, and tailoring personalized rehabilitation protocols, ultimately improving patient outcomes.
- 3. Biomarker Profiles
Expanding on the role of biomarker profiles in personalized rehabilitation, the molecular insights provided by these markers enable clinicians to fine-tune interventions in a highly specific manner, addressing the dynamic nature of tissue injury and healing. Inflammatory markers such as IL-1, IL-6, TNF-α, and CRP are not just indicators of inflammation; they are also predictive tools for identifying patients at risk of chronic inflammation, delayed healing, or secondary injuries. Persistent elevation of these cytokines can contribute to a prolonged inflammatory phase, leading to tissue fibrosis, cartilage degradation, or joint stiffness. By targeting these markers, clinicians can introduce anti-inflammatory agents (e.g., NSAIDs, corticosteroids, or biologics like TNF-α inhibitors) at critical points in the rehabilitation process. This approach minimizes the destructive aspects of inflammation while preserving its beneficial role in clearing cellular debris and initiating the healing process.
Tissue repair biomarkers, including growth factors like TGF-β and IGF-1, play essential roles in promoting cellular proliferation, differentiation, and extracellular matrix (ECM) synthesis. These biomarkers provide direct feedback on the anabolic processes occurring within the injured tissue. Elevated levels of TGF-β, for example, could indicate active ECM synthesis, which is crucial for cartilage repair and ligament regeneration. Clinicians can use this information to increase or decrease mechanical loading based on the tissue’s current repair status, preventing overloading that could disrupt the healing process. Additionally, collagen breakdown products (e.g., CTX-II) and MMP activity offer insights into the balance between tissue degradation and regeneration. High levels of these degradation markers might suggest that the tissue is undergoing excessive breakdown, requiring interventions to slow down degradation processes, such as MMP inhibitors or other ECM-stabilizing therapies.
Mechanotransduction markers, like integrins, focal adhesion kinase (FAK), and MAPK pathway components, are central to understanding how mechanical forces translate into biochemical signals that drive tissue repair. The regulation of mechanotransduction is crucial in tissues like cartilage, ligaments, and tendons, where mechanical loading directly influences cellular activity. Monitoring these markers can help determine the optimal types of mechanical stimuli—whether low-intensity stretching, progressive weight-bearing exercises, or more dynamic movements like plyometric training—that are most beneficial at different stages of healing. For instance, elevated FAK levels may suggest that cells are actively responding to mechanical stimuli, promoting ECM production and tissue regeneration. This could encourage a clinician to gradually increase the intensity of mechanical loading exercises, enhancing tissue adaptation while minimizing injury risk.
Moreover, the integration of biomarker profiles with advanced imaging techniques (such as MRI and ultrasound) provides a comprehensive view of both the structural and biochemical state of the injured tissue. Imaging can visualize structural changes such as tissue thickening, fibrosis, or cartilage wear, while biomarkers offer real-time data on cellular and molecular activities. This combination allows for a multi-dimensional approach to rehabilitation, where structural and biochemical feedback informs personalized exercise regimens, pharmacological interventions, and mechanical loading protocols.
Finally, longitudinal monitoring of biomarkers throughout the rehabilitation process allows for adaptive management of treatment protocols. By tracking changes in biomarkers over time, clinicians can adjust interventions to match the current healing stage. For example, during the early inflammatory phase, anti-inflammatory treatments might be prioritized, while later stages could focus on stimulating tissue repair through growth factor therapies or advanced regenerative medicine approaches such as stem cell therapy. This dynamic approach ensures that patients receive the right treatments at the right time, optimizing the repair process and preventing complications like chronic inflammation, fibrosis, or joint instability.
In summary, biomarker profiles are integral to personalizing rehabilitation strategies, offering a molecular window into the healing processes that are otherwise invisible. By leveraging this biochemical data, clinicians can design more precise, effective, and adaptive rehabilitation protocols, ultimately improving patient outcomes and accelerating recovery.
- 4. Patient-Reported Outcomes
Expanding on the molecular and cellular aspects, patient-reported outcomes (PROs) serve as critical feedback mechanisms that can help clinicians personalize and optimize rehabilitation protocols for knee joint injuries. These outcomes offer valuable insights into how the patient’s body responds to interventions at the molecular and tissue levels, particularly in relation to pain, functional recovery, and overall activity levels. Pain assessment, for instance, when combined with biomarker analysis, provides a nuanced understanding of the patient’s inflammatory state. Self-reported pain scores can be directly correlated with inflammatory markers such as pro-inflammatory cytokines (e.g., IL-1β, IL-6, and TNF-α) or acute phase proteins like C-reactive protein (CRP). Elevated levels of these markers often indicate ongoing tissue damage or excessive inflammation, which may be responsible for increased pain. This correlation allows clinicians to make informed decisions about introducing or adjusting anti-inflammatory agents, such as NSAIDs or biologics that inhibit specific cytokines, to manage the inflammatory response more precisely. Thus, patient-reported pain, when integrated with molecular data, becomes a valuable tool for tracking not only subjective discomfort but also the underlying biochemical environment influencing tissue healing.
In terms of functional assessments, tools such as KOOS or WOMAC provide insights into how well the patient is regaining joint function and mobility, which can be mapped against the progress of tissue repair at the molecular level. For example, as the patient reports improvements in knee function, corresponding molecular markers like collagen synthesis or growth factor activity (e.g., TGF-β, IGF-1) may also show positive trends, indicating successful tissue remodeling. By using PROMs alongside molecular biomarkers, clinicians can track how mechanical stimuli from rehabilitation exercises affect the mechanotransduction pathways. These pathways, particularly those involving integrins, FAK, and MAPK signaling, play crucial roles in translating mechanical loading into cellular responses that drive tissue regeneration and repair. Functional improvements reported by the patient may be a sign of enhanced ECM production or the reorganization of collagen fibers within ligaments, tendons, or cartilage. Clinicians can adjust the rehabilitation protocol based on both the patient’s functional feedback and the molecular indicators of tissue adaptation, ensuring that the loading patterns are optimal for promoting healing while minimizing the risk of re-injury.
Activity levels reported by the patient or tracked through wearables also offer valuable insights into the mechanical stimuli being applied to the healing tissue. Mechanotransduction markers, such as changes in FAK activity, calcium signaling, or MAPK activation, can be influenced by the amount and intensity of mechanical loading during rehabilitation exercises. If a patient reports lower-than-recommended activity levels or if wearable data shows inconsistent adherence to prescribed exercises, clinicians can expect a slower rate of tissue repair, which may be reflected in lower levels of key growth factors or an increase in MMPs, which degrade the ECM. Conversely, overloading during early stages of recovery, as indicated by excessive activity, may lead to elevated levels of inflammation or tissue damage, requiring a reduction in intensity to avoid further injury. The combination of patient-reported activity levels and molecular markers enables clinicians to modulate the rehabilitation intensity, ensuring that it aligns with the biological processes necessary for optimal healing. Additionally, advanced analytics from wearables can monitor specific movements and joint angles, helping to identify potential overloading patterns that could compromise the integrity of the healing tissue.
By continuously integrating patient-reported outcomes with biomechanical assessments and biomarker profiles, rehabilitation protocols can be dynamically tailored to reflect the real-time needs of the patient. This holistic approach ensures that interventions are not only safe but also biologically effective, promoting tissue repair at the molecular level while addressing the patient’s subjective experience of pain, function, and activity. Ultimately, the interplay between molecular insights and patient feedback leads to a more adaptive and personalized rehabilitation process, improving clinical outcomes and reducing the likelihood of long-term complications such as chronic pain or joint instability.
Predictive Models and Decision-Support Systems
Advances in data science and artificial intelligence (AI) are revolutionizing the way personalized rehabilitation strategies are developed by leveraging vast amounts of patient data to create predictive models and decision-support systems. These technologies can process complex datasets, including biomarker profiles, imaging results, biomechanical assessments, and patient-reported outcomes, to identify patterns and correlations that are difficult for humans to detect. By integrating data from multiple sources, AI can provide real-time analysis and insights that enhance the personalization of treatment protocols, ensuring that each patient receives the most effective and timely intervention based on their unique biological and functional status.
For example, machine learning (ML) algorithms can analyze historical patient data to predict how individuals might respond to specific rehabilitation exercises or pharmacological interventions. These algorithms can identify subtle trends in recovery trajectories by correlating molecular data—such as levels of inflammatory markers, growth factors, and collagen synthesis—with patient-reported outcomes like pain levels or functional improvements. Predictive models developed through ML can thus anticipate potential setbacks in recovery, such as delayed tissue healing or excessive inflammation, allowing clinicians to adjust treatment protocols preemptively. This can prevent complications like chronic pain or reinjury by ensuring that rehabilitation is tailored to the patient’s ongoing physiological responses.
Deep learning models, which are a subset of ML, can also be applied to imaging data, such as MRI or CT scans, to detect early signs of tissue damage or poor healing outcomes that may not be immediately visible to the human eye. These models can be trained to recognize patterns associated with early cartilage degradation, ligament damage, or subchondral bone changes, helping clinicians adjust rehabilitation protocols or consider surgical interventions at the right time. By providing an automated, objective analysis of imaging data, deep learning systems enhance decision-making accuracy, improving patient outcomes through timely interventions.
Moreover, natural language processing (NLP) tools can analyze unstructured data such as clinical notes, patient surveys, and feedback collected from wearables or mobile apps. By extracting relevant information from these data sources, NLP can provide insights into the patient’s adherence to rehabilitation protocols, their subjective experience of recovery, and potential psychosocial factors affecting their progress. This comprehensive data analysis helps clinicians understand not only the biological aspects of recovery but also the psychological and behavioral factors that can influence rehabilitation outcomes. AI-driven chatbots or virtual assistants can also engage with patients remotely, providing guidance, monitoring adherence, and collecting feedback, ensuring that the rehabilitation process is continuously aligned with the patient’s needs.
Predictive models can be used to create dynamic rehabilitation plans that evolve based on the patient’s ongoing response to treatment. For instance, if data from wearables indicate that a patient is overloading the knee joint during daily activities, the system can recommend a modification in exercise intensity or frequency. Conversely, if biomarkers show signs of slow tissue healing, the system might suggest extending the duration of a specific rehabilitation phase or incorporating additional therapeutic modalities like growth factor therapy or low-intensity ultrasound. In this way, AI-driven systems provide real-time decision support, helping clinicians deliver more precise, adaptive rehabilitation strategies that respond to the patient’s current physiological state.
AI-driven platforms also hold great potential in personalized pharmacological interventions. By integrating molecular data with clinical outcomes, these platforms can predict how individual patients might respond to specific drugs, such as NSAIDs, cytokine inhibitors, or MMP inhibitors. For example, an AI model trained on biomarker and drug response data could predict which patients are likely to benefit from anti-inflammatory treatments versus those who may experience adverse effects or insufficient responses. This enables precision medicine approaches that minimize the risk of side effects while maximizing therapeutic efficacy. By combining molecular insights with AI-generated treatment recommendations, clinicians can tailor pharmacological interventions to the patient’s unique biological profile, improving outcomes while reducing unnecessary drug use.
In addition to predictive modeling, AI-based decision-support systems can facilitate personalized exercise regimens by analyzing data from biomechanical assessments and motion capture systems. These systems can recommend specific exercises that optimize joint mechanics and muscle activation patterns, ensuring that patients engage in activities that promote healing while avoiding those that could exacerbate their condition. AI systems can also provide virtual coaching through interactive platforms, guiding patients through exercises and offering real-time feedback based on their movement patterns. This can enhance patient engagement, improve adherence to rehabilitation protocols, and reduce the risk of improper form during exercises, further supporting optimal recovery.
In the future, AI and data science may also facilitate the development of digital twins—virtual replicas of the patient’s knee joint that simulate biological processes in real-time. These digital twins can incorporate data from genomic, proteomic, and metabolomic profiles alongside imaging and functional assessments, allowing clinicians to test different rehabilitation or pharmacological interventions in a virtual environment before applying them to the patient. By predicting how specific treatments will affect tissue healing, joint mechanics, and overall function, digital twins can help refine personalized rehabilitation strategies to achieve the best possible outcomes.
In summary, advances in AI and data science are transforming the field of personalized rehabilitation by enabling the development of predictive models and decision-support systems that analyze complex datasets to optimize treatment protocols. These tools provide valuable insights into individual patient responses, enhancing the precision and effectiveness of rehabilitation strategies while improving overall outcomes for patients with knee joint injuries.
- 1. Machine Learning
Expanding further on the application of machine learning in personalized rehabilitation, predictive analytics not only anticipates treatment outcomes but also continuously refines the rehabilitation process by adapting to each patient’s evolving condition. For example, machine learning models can monitor real-time data from wearable devices, such as sensors tracking joint movement, muscle activity, or step counts, alongside biometric data like heart rate or blood pressure. By analyzing this dynamic information, algorithms can detect subtle trends that may indicate the onset of fatigue, overuse, or improper technique during exercises, allowing clinicians to intervene early and adjust the rehabilitation plan before further complications arise. This approach also enables real-time feedback loops, where the algorithm instantly updates the patient’s progress and provides suggestions for modifications in the exercise routine, ensuring that rehabilitation remains effective and aligned with the patient’s recovery trajectory.
In the realm of classification algorithms, these models can integrate diverse datasets, such as genetic profiles, inflammation markers, and previous injury history, to predict how a patient’s body will respond to specific interventions. For instance, a classification algorithm might identify that a particular patient’s genetic predisposition to collagen disorders could influence the healing time for ligament injuries, guiding the clinician to choose therapies that bolster collagen synthesis or reduce tissue degradation. By stratifying patients into distinct categories based on such individualized factors, clinicians can implement precision medicine approaches that customize not only the rehabilitation exercises but also pharmacological treatments, nutritional plans, and mechanical loading regimens. This personalized stratification ensures that patients with different biological or clinical profiles receive the most appropriate care for their specific needs, avoiding the one-size-fits-all pitfalls that may not work for everyone.
Additionally, reinforcement learning models in machine learning can be designed to optimize rehabilitation protocols over time. These algorithms learn from trial and error by simulating various treatment scenarios and receiving feedback based on patient outcomes. Over time, reinforcement learning can fine-tune treatment strategies, learning which combination of exercises, intensities, and therapies yield the best results for each patient cohort. This can result in dynamic treatment pathways, where rehabilitation strategies evolve based on real-time patient responses, ensuring that the interventions are constantly improving and adapting to individual progress.
Furthermore, machine learning-driven natural language processing (NLP) can analyze patient-reported outcomes, such as responses from surveys or unstructured data from medical records, to gain insights into subjective experiences like pain levels, emotional well-being, or treatment satisfaction. This enables a more holistic understanding of patient needs, ensuring that both physical and psychological aspects of recovery are addressed. By incorporating this data into predictive models, the rehabilitation process can become even more personalized, catering to not just the biological aspects of healing but also the emotional and psychological components that significantly influence recovery outcomes.
In essence, the integration of advanced machine learning techniques into rehabilitation offers a comprehensive, adaptable, and highly individualized approach, improving both the effectiveness and efficiency of treatments. It promises a future where patient care is increasingly data-driven, responsive, and tailored to the unique physiological and psychological needs of each individual, ultimately enhancing recovery outcomes and reducing long-term complications.
- 2. Decision-Support Systems
Decision-support systems (DSS) that integrate patient-specific data, predictive models, and clinical guidelines are pushing the boundaries of personalized rehabilitation, especially for complex injuries such as those involving the knee joint. By incorporating a wide range of patient data—such as genomic profiles, muscle activity, gait mechanics, and detailed imaging—these systems can make highly individualized recommendations that go far beyond traditional care protocols. For example, in the case of knee joint rehabilitation, DSS can analyze molecular data like cytokine levels to detect ongoing inflammation and suggest targeted anti-inflammatory interventions, all while aligning this with biomechanical data to adjust load-bearing exercises or movement patterns.
These systems also enable clinicians to combine predictive modeling with real-time monitoring, which is crucial for adjusting rehabilitation plans as the patient’s condition evolves. Machine learning algorithms embedded in DSS can continuously learn from patient responses, refining predictions on how an individual might progress, or identifying early signs of suboptimal recovery, such as delayed tissue healing or recurring pain. This allows for proactive interventions, such as adjusting exercise intensity or introducing pharmacological support, before problems become more severe. The dynamic nature of DSS makes it possible to not only tailor rehabilitation in real-time but also to predict and prevent long-term complications like osteoarthritis or joint instability, improving overall patient outcomes.
The integration of wearable technology into DSS enhances its capabilities even further by allowing for continuous real-time monitoring. Wearable sensors can track parameters like joint angles, muscle activation, step count, and even biochemical markers in sweat or interstitial fluid. This real-time data stream feeds into the decision-support system, which then adjusts the treatment regimen to optimize recovery while preventing injury. For example, if the system detects that a patient is placing abnormal stress on their knee during certain activities, it might automatically suggest changes to the exercise program or alert the clinician to reassess the patient’s biomechanics. Such granular, data-driven insights ensure that rehabilitation protocols are not only personalized but also dynamic, adapting to the real-time physiological changes of the patient.
In addition to optimizing patient care, DSS can facilitate better communication between clinicians, physiotherapists, and other members of the healthcare team. The system can generate comprehensive reports and visualizations that help all stakeholders understand the patient’s progress, any adjustments made to the rehabilitation plan, and the reasoning behind those changes. Automated recommendations generated by the system can be discussed and tailored further in multidisciplinary meetings, ensuring a cohesive approach that aligns with the patient’s evolving needs. Moreover, DSS can bridge the gap between patient-reported outcomes and clinical decision-making. With patients continuously providing feedback through digital platforms on pain levels, mobility, or fatigue, the DSS can process this qualitative data alongside quantitative clinical metrics, creating a holistic view of the patient’s recovery journey.
Future developments in DSS are likely to involve even more advanced algorithms capable of integrating molecular data from biomarkers, genomic information, and even epigenetic markers to predict long-term recovery trends. For example, a system might incorporate genetic predispositions for tissue repair or inflammatory responses, using this information to adjust the rehabilitation timeline or introduce specific therapies like stem cells or gene therapy. The future of personalized rehabilitation will be one where every therapeutic decision is informed by both molecular insights and biomechanical data, with DSS serving as a crucial tool in ensuring that every patient receives the most precise, effective, and individualized care possible.
- 3. Personalized Rehabilitation Protocols
Adaptive rehabilitation protocols are underpinned by an understanding of the molecular processes that govern tissue healing, allowing for precise adjustments as the patient’s recovery progresses. Mechanical loading, a critical factor in rehabilitation, triggers mechanotransduction pathways such as the FAK, YAP/TAZ, and MAPK signaling cascades, which regulate cellular responses to physical stress. These pathways influence cell proliferation, ECM remodeling, and tissue repair, making it essential to modulate the intensity and type of exercise to optimize these responses. For instance, early rehabilitation might focus on light loading to prevent excessive inflammation and promote fibroblast activation, which stimulates collagen deposition and strengthens the extracellular matrix. As healing progresses, mechanical loading can be increased to further enhance mechanosensitive pathways like Wnt/β-catenin signaling, driving tissue regeneration and remodeling.
Moreover, personalized rehabilitation protocols leverage real-time data from biomarker monitoring to adjust treatments based on individual molecular responses. By tracking inflammation markers (e.g., IL-1, TNF-α), clinicians can gauge whether anti-inflammatory strategies are needed, while markers of collagen turnover (e.g., CTX-II) can indicate the pace of tissue repair. Mechanotransduction markers such as integrins and FAK levels provide insights into how well cells are responding to mechanical stimuli, allowing clinicians to optimize exercise intensity. This biomarker-driven approach ensures that rehabilitation remains aligned with the patient’s specific healing trajectory, reducing the risk of overloading tissues and preventing complications like fibrosis or reinjury.
Tailored exercises, such as proprioceptive training for ligament injuries or resistance exercises for muscle weakness, are designed to activate the precise cellular pathways involved in tissue regeneration. Proprioceptive exercises, for instance, stimulate neural pathways and enhance joint stability by modulating calcium signaling through mechanosensitive ion channels. Meanwhile, resistance training can activate the mTOR pathway, promoting muscle hypertrophy and restoring strength. Adjusting these exercises based on continuous feedback ensures that the rehabilitation program remains both effective and safe.
The multidisciplinary nature of adaptive rehabilitation protocols, involving physical therapists, orthopedic specialists, nutritionists, and sometimes even genetic counselors, ensures a holistic approach to recovery. Nutritional strategies that enhance collagen synthesis, such as increased intake of vitamin C or omega-3 fatty acids, support molecular processes at the cellular level, facilitating tissue repair. By incorporating diverse inputs from various medical disciplines, adaptive protocols address the full spectrum of the patient’s recovery needs, from inflammation control and tissue regeneration to metabolic and nutritional support. This comprehensive, patient-specific approach maximizes the likelihood of successful outcomes and long-term recovery.
Advanced Technologies for Monitoring and Adjustment
Advanced technologies play a crucial role in enhancing the precision and responsiveness of personalized rehabilitation protocols by providing real-time, molecular-level insights into the patient’s physiological and biomechanical state. Wearable sensors, for instance, can continuously monitor biomechanical data such as joint angles, muscle activity, and gait patterns, offering a detailed analysis of how mechanical forces are distributed across injured tissues. This data, integrated with mechanotransduction pathways like FAK, MAPK, and YAP/TAZ signaling, can help clinicians understand how cells are responding to mechanical loading at a molecular level. For example, changes in sensor data might indicate suboptimal tissue stress or overloading, which can disrupt key cellular processes such as collagen synthesis or fibroblast activity, leading to delayed healing or excessive inflammation. In response, rehabilitation protocols can be dynamically adjusted to ensure that mechanical stimuli are aligned with the optimal activation of repair mechanisms.
Furthermore, biofeedback devices utilizing electromyography (EMG) or real-time ultrasound imaging can provide immediate insights into muscle activation patterns and tissue integrity. These tools can detect molecular changes, such as variations in muscle fiber recruitment or tissue stiffness, which are often regulated by calcium signaling and integrin-mediated mechanotransduction pathways. Clinicians can use this data to fine-tune exercises, enhancing the activation of specific muscles or tissues and ensuring that cellular responses like ECM remodeling and angiogenesis are progressing as expected. Real-time imaging technologies, such as MRI and ultrasound elastography, further allow the visualization of soft tissue structures, monitoring changes in collagen density or vascularization at a molecular level, and adjusting protocols accordingly.
By incorporating advanced technologies such as wearable sensors, biofeedback devices, and real-time imaging, clinicians can precisely tailor rehabilitation programs to the patient’s molecular and biomechanical needs. This technology-driven approach ensures that rehabilitation protocols are not only customized but continuously optimized to enhance tissue repair, reduce the risk of complications, and improve overall clinical outcomes.
- 1. Wearable Sensors
Wearable sensors not only offer real-time monitoring but also play a critical role in translating mechanical stimuli into molecular and cellular responses that influence tissue healing and recovery. By capturing biomechanical data such as joint angles, force distribution, and muscle activation during movement, wearable sensors can provide insights into how mechanical loading impacts critical pathways like integrin signaling and mechanotransduction. These pathways are responsible for converting mechanical stress into biochemical signals that regulate cell behaviors such as proliferation, differentiation, and ECM synthesis. For example, sensors that detect joint instability or excessive loading during rehabilitation exercises can prompt adjustments that prevent overloading, which might otherwise lead to excessive activation of matrix metalloproteinases (MMPs), accelerating cartilage degradation or slowing tissue repair.
Physiological sensors, like heart rate monitors or EMG sensors, offer molecular insights into how the body’s systems respond to exercise and rehabilitation efforts. Elevated heart rates, for instance, can indicate systemic inflammation, while abnormal muscle activation patterns might suggest an imbalance in muscle recovery, potentially linked to altered signaling in calcium-regulated pathways such as calmodulin or calcineurin, which are critical for muscle adaptation and repair. By tracking these physiological parameters, wearable sensors help clinicians fine-tune the rehabilitation intensity to modulate the body’s healing response at a cellular level, ensuring that exercise regimens promote anabolic processes such as collagen deposition and cell proliferation, without triggering catabolic pathways associated with inflammation and tissue breakdown.
Moreover, the integration of wearable sensor data with advanced algorithms and predictive models can further refine rehabilitation strategies. Machine learning models trained on large datasets of sensor data can predict recovery trajectories and identify early signs of complications, such as suboptimal tissue regeneration or delayed healing, which might be linked to molecular dysregulation. For example, if sensors indicate that a patient’s movement patterns are not improving as expected, predictive models can suggest adjustments to the rehabilitation protocol, such as incorporating more proprioceptive exercises that promote neuromuscular retraining, ultimately enhancing cellular responses involved in tissue remodeling.
This continuous data stream from wearable sensors provides a feedback loop that can optimize rehabilitation by dynamically adjusting to the patient’s evolving molecular and physiological needs, improving long-term outcomes by ensuring that rehabilitation is both responsive and personalized at the cellular level.
- 2. Telemedicine and Remote Monitoring
Telemedicine and remote monitoring technologies represent a major advancement in personalized rehabilitation, offering the ability to seamlessly integrate molecular and biomechanical data into continuous patient care. These platforms provide real-time insights into a patient’s physiological status, allowing for precise, data-driven interventions that go beyond traditional methods. By monitoring molecular markers like inflammatory cytokines (IL-1, IL-6, TNF-α), remote systems can detect early signs of excessive inflammation or tissue stress, which could signal a need to alter the rehabilitation protocol before further damage occurs. Such early detection is crucial, as prolonged inflammation could lead to delayed healing or even chronic conditions like osteoarthritis if left unaddressed.
Remote monitoring also tracks vital physiological responses—such as heart rate variability, muscle tension, and joint movement—offering an in-depth understanding of how the patient’s body is responding to mechanical loading and therapeutic exercises. For example, continuous monitoring of joint kinematics through wearable sensors can identify subtle deviations in movement that may not be apparent during periodic clinical assessments. These deviations can result in abnormal loading patterns, increasing the risk of re-injury or inefficient healing, especially in tissues with limited regenerative capacity like cartilage. By providing immediate feedback, clinicians can quickly adjust exercise intensity or introduce corrective strategies, such as neuromuscular re-education, to restore optimal biomechanics and reduce the risk of further damage.
From a molecular perspective, telemedicine platforms can guide personalized interventions based on the patient’s biomarker profile. For example, if remote data shows inadequate production of anabolic growth factors like IGF-1 or TGF-β, which are critical for tissue regeneration, clinicians can modify the rehabilitation plan to incorporate regenerative therapies like stem cell injections or tissue-engineered scaffolds. These approaches can then be monitored to assess their molecular impact on tissue repair, allowing for a highly responsive, adaptive treatment plan.
The integration of remote monitoring with predictive models powered by machine learning can further enhance patient care by identifying patterns that suggest optimal recovery trajectories. Predictive analytics can analyze massive datasets from previous patients to recommend personalized rehabilitation pathways based on factors like age, genetics, injury type, and molecular markers. This precision approach allows clinicians to anticipate potential complications or bottlenecks in healing, providing a roadmap that continuously adjusts as the patient progresses.
Additionally, wearable sensors and telemedicine platforms help maintain patient engagement, a critical factor in successful rehabilitation. Real-time feedback loops, such as alerts for improper movement patterns or notifications when biomarker levels reach thresholds that require attention, keep patients active participants in their recovery process. This continuous engagement ensures that patients adhere to their rehabilitation programs, reducing the risk of setbacks due to inactivity or incorrect exercise techniques.
Overall, telemedicine and remote monitoring enable a more molecularly informed, patient-centered approach to rehabilitation. The ability to continuously assess both physiological and biochemical data ensures that rehabilitation protocols remain flexible, adaptive, and optimized for each patient’s unique recovery process, ultimately improving clinical outcomes and enhancing the overall quality of care.
- 3. 3D Motion Analysis
Expanding further on the molecular and biomechanical integration provided by 3D motion analysis systems, these advanced tools enable clinicians to dive deeper into the cellular mechanisms affected by improper movement patterns and loading forces, linking movement mechanics directly to tissue health and healing at a molecular level. For example, tissues such as ligaments, tendons, cartilage, and muscles rely on optimal mechanical stimuli to maintain cellular homeostasis. When these tissues are exposed to abnormal movement patterns, such as improper joint alignment or overcompensation from injury, mechanosensitive pathways like the integrin-FAK (focal adhesion kinase) signaling pathway and other mechanotransduction processes become dysregulated. This dysregulation can affect the production and degradation of essential extracellular matrix (ECM) components, such as collagen and proteoglycans, which are crucial for maintaining tissue structure and resilience.
The precise measurements provided by 3D motion analysis systems allow clinicians to fine-tune rehabilitation interventions to target these molecular responses. For example, excessive strain on a joint due to improper movement can lead to increased matrix metalloproteinase (MMP) activity, accelerating the degradation of cartilage ECM. By identifying the faulty movement pattern and implementing corrective exercises, clinicians can reduce the mechanical stress on the tissue, thereby downregulating the activity of MMPs and supporting ECM preservation and regeneration. This approach ensures that the tissues experience the right amount of mechanical load—enough to stimulate beneficial cellular responses, such as collagen synthesis in ligaments or fibrocartilage repair in the meniscus—without causing further damage or triggering excessive catabolism.
In addition to targeting cellular pathways, 3D motion analysis systems can provide valuable feedback during rehabilitation sessions, offering real-time corrections that enhance the precision of exercises. This immediate feedback loop ensures that patients adopt correct movement patterns early in their recovery, minimizing the risk of reinforcing harmful biomechanics. Moreover, this dynamic feedback can positively influence neuroplasticity, helping to retrain the neuromuscular system to adopt proper motor control and coordination. This retraining is essential for restoring proprioception and preventing future injuries, as neuromuscular deficits often persist after an initial injury and can lead to recurrent problems if not addressed.
Furthermore, these systems can be integrated with wearable devices and biomechanical sensors to continuously monitor movement patterns throughout daily activities, providing a more comprehensive view of the patient’s progress. This level of detail allows for the early detection of compensatory movements that may not be noticeable during clinical evaluations but could contribute to long-term joint damage if left uncorrected. For instance, subtle changes in gait or posture may lead to imbalanced loading on the knee joint, exacerbating conditions such as osteoarthritis or delaying ligament healing. By capturing these details and making timely adjustments to the rehabilitation protocol, 3D motion analysis can prevent the cascade of molecular changes that lead to chronic injury or degeneration.
Overall, the use of 3D motion analysis systems not only enhances the precision of biomechanical interventions but also creates a direct link between mechanical forces, cellular responses, and molecular pathways, ensuring that rehabilitation strategies are both effective and responsive to the complex needs of the injured tissue. This integration of real-time movement analysis with an understanding of the underlying molecular biology optimizes tissue repair and improves long-term functional outcomes, making rehabilitation more personalized and data-driven than ever before.
Future Directions in Personalized Rehabilitation
The field of personalized rehabilitation is advancing through breakthroughs in molecular biology, biomechanics, and data science, offering increasingly sophisticated approaches to individualized care. One promising direction for future research lies in the integration of genomics and proteomics to better understand the genetic and molecular underpinnings of tissue repair and recovery. For example, identifying specific genetic polymorphisms related to collagen synthesis, inflammation, or mechanotransduction could enable clinicians to tailor rehabilitation strategies based on an individual’s genetic predisposition to healing or injury. Moreover, future studies are expected to focus on molecular signaling pathways such as the Wnt/β-catenin and TGF-β pathways, which regulate tissue regeneration and scarring, to develop more targeted interventions. By combining these molecular insights with advanced technologies like wearable biosensors and artificial intelligence (AI)-driven predictive models, rehabilitation programs can be dynamically adapted in real time. These AI tools could predict how a patient’s cellular and molecular responses to different mechanical stimuli influence recovery, ensuring that interventions are optimized for maximum efficacy. Additionally, future research may explore personalized biologics, such as growth factor or stem cell therapies, that can be tailored to the molecular profile of the patient, enhancing tissue repair and reducing the risk of complications like fibrosis or delayed healing. Overall, the intersection of molecular biology, advanced diagnostics, and personalized treatment will continue to push the boundaries of rehabilitation science, leading to highly customized, data-driven protocols.
- 1. Integration of Multi-Omics Data
Integrating multi-omics data, such as genomics, proteomics, metabolomics, and transcriptomics, holds immense potential for advancing personalized rehabilitation by providing a more precise understanding of knee joint injuries at the molecular level. This comprehensive approach enables the identification of critical genes, proteins, metabolites, and RNA molecules involved in the injury and repair processes. For instance, genomics can reveal specific mutations or genetic variations that predispose an individual to slower recovery or higher risk of complications, while proteomics uncovers the protein-level changes that govern inflammation, ECM remodeling, and tissue repair. Metabolomics provides insights into the biochemical pathways involved in energy metabolism, oxidative stress, and nutrient utilization during healing, offering clues about how metabolic disturbances could impact rehabilitation progress.
By integrating these datasets, researchers can map out intricate signaling pathways and molecular networks, leading to the identification of key regulatory molecules that control tissue regeneration and inflammation. This integrated analysis allows for a more complete characterization of the patient’s biological state, enabling clinicians to predict which patients might benefit from particular interventions, such as anti-inflammatory drugs, growth factors, or mechanical loading regimens. Additionally, multi-omics data can help identify novel biomarkers that track the effectiveness of rehabilitation and signal when adjustments are needed to prevent setbacks or re-injury.
Systems biology techniques further enhance the utility of multi-omics data by simulating how different molecular pathways interact and respond to therapies. For example, it can model how inflammation interacts with tissue repair processes, helping clinicians anticipate and mitigate potential complications like fibrosis or chronic inflammation. Ultimately, multi-omics data integration leads to highly personalized rehabilitation strategies, optimized for each patient’s unique molecular profile, and offers the possibility of real-time adjustments as the patient’s biological responses evolve during treatment. This advanced approach not only improves outcomes but also minimizes recovery time and reduces the risk of long-term complications.
- 2. Advanced Analytics and Machine Learning
The integration of advanced analytics and machine learning into personalized rehabilitation offers unprecedented capabilities for refining treatment plans, improving outcomes, and enhancing patient engagement. Predictive Modeling goes beyond traditional approaches by analyzing multiple layers of patient data—such as biomarker profiles, genetic predispositions, imaging results, and real-time physiological feedback—to forecast individual responses to rehabilitation. This allows clinicians to predict with greater accuracy which patients are likely to experience complications or delayed healing, enabling them to intervene early. Machine learning algorithms can also highlight the most significant factors contributing to successful rehabilitation, such as specific biomarkers linked to better tissue repair or optimal mechanical loading thresholds. These insights can help optimize the timing and intensity of interventions, allowing for personalized protocols that evolve based on continuous data input rather than static treatment plans.
Moreover, Dynamic Adjustment powered by machine learning algorithms creates a continuously evolving rehabilitation process, where real-time data informs ongoing modifications to the treatment plan. For instance, wearable sensors can track gait patterns, muscle activity, and joint movement, while also capturing physiological responses like heart rate and muscle fatigue. By feeding this data into machine learning models, clinicians can receive alerts if the patient’s recovery deviates from the expected trajectory or if their activity patterns suggest they are not performing exercises correctly. The system can then recommend modifications to exercise intensity, duration, or type, ensuring that the rehabilitation remains both safe and effective. This continuous feedback loop between patient performance and data-driven adjustment ensures that treatment is not only personalized but dynamically responsive to each patient’s progress.
In addition, Enhanced Resource Allocation is another benefit of using machine learning in rehabilitation. By categorizing patients based on their predicted rehabilitation needs and responses, healthcare systems can more efficiently allocate resources, directing more intensive therapies to patients who are likely to benefit most, while using less resource-intensive approaches for those with straightforward recovery paths. Predictive models can also help schedule follow-up visits, ensuring that patients who need more frequent monitoring are seen promptly, while others can manage with less frequent check-ins. This not only improves patient outcomes but also reduces unnecessary healthcare costs.
Patient Engagement and Motivation are also enhanced through the use of real-time feedback and predictive insights. When patients receive personalized, data-driven updates about their progress, they are more likely to stay engaged with their rehabilitation program. Interactive apps or interfaces connected to wearable devices can provide daily feedback, showing patients how their efforts are contributing to their recovery. Moreover, gamification elements can be integrated, rewarding patients for reaching certain milestones or adhering to their rehabilitation plan. This increased engagement can lead to better adherence, faster recovery times, and improved patient satisfaction.
Finally, Continuous Learning and Improvement are intrinsic benefits of applying machine learning to rehabilitation. As more patient data is gathered and analyzed, the models become more accurate and effective over time. This leads to progressively better treatment recommendations and more precise predictive models, enabling clinicians to stay at the cutting edge of personalized care. This iterative process ensures that personalized rehabilitation protocols are constantly updated with the latest evidence, improving the overall quality of care provided. As machine learning algorithms process more data from diverse patient populations, they can also identify patterns that may lead to the discovery of new rehabilitation techniques or interventions, further advancing the field of rehabilitation science.
- 3. Patient Engagement and Empowerment
Enhancing patient engagement and empowerment through personalized rehabilitation protocols rooted in molecular insights can revolutionize patient adherence and recovery outcomes. By providing patients with an understanding of how their unique molecular profile, including genetic predispositions and biomarker fluctuations, influences their recovery, they can make more informed decisions and feel more connected to their rehabilitation process. For example, explaining how specific inflammatory markers like IL-1 or TNF-α are contributing to pain and how certain exercises can modulate their levels can make the treatment plan feel more relevant and actionable. This molecularly informed education can be delivered through digital platforms that offer personalized learning experiences, such as tailored content on genetic factors influencing cartilage repair or metabolic insights that impact exercise performance.
Gamification, combined with molecular feedback, offers a powerful strategy to maintain patient motivation. Patients could receive real-time rewards or unlock achievements when they reach molecular milestones, such as a reduction in inflammatory biomarkers or improvements in tissue regeneration, which are tracked via wearables or lab tests. Personalized games could be designed based on the patient’s biological data, where achieving certain biomarker targets or completing specific movements correlates with virtual rewards or progress within the game. For instance, if a patient’s muscle activity or collagen production improves, it could be reflected in the game, offering tangible evidence of biological progress and creating a sense of accomplishment. This makes the rehabilitation process more engaging and enjoyable while fostering a strong connection between physical activities and molecular healing.
Moreover, advanced analytics can further optimize this experience by dynamically adjusting rehabilitation challenges and gamified content based on the patient’s evolving molecular profile. For example, if metabolomic data indicates that a patient is experiencing oxidative stress, the system can suggest modifications in exercise intensity or nutrition to improve recovery. Similarly, personalized virtual coaching could offer encouragement based on real-time feedback from wearable sensors, ensuring that patients are receiving tailored support throughout their rehabilitation journey. This integration of molecular data, education, and gamification empowers patients not only to adhere more consistently to their rehabilitation protocols but also to take ownership of their healing process, fostering long-term improvements in both their physical health and overall well-being.
By integrating these components into personalized rehabilitation strategies, healthcare providers can optimize treatment outcomes for patients with knee joint injuries, ensuring that each patient receives the most appropriate and effective care based on their unique needs and conditions. The future of personalized rehabilitation lies in the continued development and integration of advanced technologies, data-driven approaches, and patient-centered care models that prioritize the individual needs and experiences of each patient. By embracing these advancements, healthcare providers can enhance the effectiveness of rehabilitation programs, improve patient outcomes, and ensure that each patient receives the highest quality of care possible.
Conclusion
Knee joint injuries, such as meniscal tears, ligament damage, and cartilage degradation, are particularly challenging due to the intricate balance required between mechanical forces and biological healing processes. Early mechanical loading plays a pivotal role in influencing cellular behavior through mechanotransduction—the conversion of mechanical signals into biochemical responses within cells. This process involves the activation of mechanosensitive receptors like integrins, which connect the extracellular matrix (ECM) to the cell cytoskeleton, and key signaling molecules such as focal adhesion kinase (FAK) and the MAPK/ERK pathways. These signaling cascades regulate cellular activities like proliferation, migration, and ECM synthesis, which are vital for tissue repair. For example, chondrocytes and fibroblasts respond to controlled mechanical stimuli by increasing the production of type II collagen and proteoglycans, which are crucial for cartilage and ligament regeneration, respectively.
Additionally, mechanical loading can influence the expression of inflammatory mediators, promoting an anti-inflammatory environment that supports healing rather than prolonged inflammation, which can lead to tissue degradation. Growth factors such as transforming growth factor-beta (TGF-β) and insulin-like growth factor-1 (IGF-1) are also upregulated in response to mechanical stimuli, enhancing cell proliferation, differentiation, and matrix production. Moreover, the modulation of matrix metalloproteinases (MMPs), enzymes responsible for ECM breakdown, is tightly regulated by mechanical forces to ensure balanced tissue remodeling without excessive degradation. These molecular responses to mechanical loading are highly dependent on the timing, intensity, and type of load applied, making it crucial to personalize rehabilitation protocols to each patient’s specific injury and biological profile.
Future research aims to further elucidate how mechanical loading can be optimized to maximize tissue repair, incorporating advanced techniques like multi-omics profiling and machine learning to develop predictive models. These insights will enable clinicians to create tailored rehabilitation strategies that not only accelerate recovery but also reduce the risk of chronic conditions, such as osteoarthritis, which often follow knee joint injuries.
| Aspect | Cartilage | Ligaments | Tendons | Meniscus |
| Biophysical Stimulation | - Compression - Hydrostatic pressure - Shear stress - Dynamic loading to mimic joint movements |
- Tensile loading - Cyclic stretching - Controlled dynamic loading to prevent overstretching |
- Tensile loading - Cyclic stretching - Gradual progressive loading |
- Compression - Shear stress - Tensile loading - Cyclic and static loading for comprehensive stimulation |
| Mechanotransduction | - Integrin signaling (e.g., α5β1 integrin) - Ion channels (e.g., stretch-activated Ca²⁺ channels) - MAPK pathway (ERK1/2, JNK, p38) - Wnt/β-Catenin signaling - YAP/TAZ activation |
- Integrin signaling (e.g., αvβ3 integrin) - Focal Adhesion Kinase (FAK) activation - Rho family GTPases (RhoA, Rac1) - MAPK pathway (ERK1/2, JNK, p38) |
- Integrin signaling (e.g., α5β1 integrin) - Ion channels (e.g., stretch-activated Ca²⁺ channels) - FAK activation - MAPK pathway (ERK1/2, JNK, p38) |
- Integrin signaling (e.g., α5β1 and α6β1 integrins) - Ion channels (e.g., stretch-activated Ca²⁺ channels) - MAPK pathway (ERK1/2, JNK, p38) - Wnt/β-Catenin signaling - YAP/TAZ activation |
| Stress/Strain | - Moderate compressive stress (optimal to stimulate chondrocytes) - Cyclic loading to promote ECM production - Avoid excessive stress to prevent chondrocyte apoptosis |
- Tensile stress aligned with ligament fibers - Gradual increase in load to stimulate fibroblasts - Cyclic loading to enhance collagen synthesis |
- Tensile stress aligned with tendon fibers - Gradual increase in load to stimulate tenocytes - Cyclic loading to enhance collagen synthesis |
- Combination of compressive and tensile stress - Cyclic loading to stimulate chondrocytes and fibrochondrocytes - Avoid excessive stress to prevent further tearing |
| Stress-Relaxation | - Gradual application and release of load - Allows time for ECM adaptation - Prevents cell damage and apoptosis |
- Gradual relaxation phases - Reduces risk of re-injury - Enhances ligament compliance and function |
- Gradual relaxation phases - Reduces risk of tendinopathy - Enhances tendon compliance and function |
- Gradual relaxation phases - Allows time for ECM adaptation - Prevents further damage and promotes healing |
| Hysteresis | - Minimizes energy loss during loading/unloading - Maintains cartilage resilience and function - Promotes efficient load-bearing capacity |
- Reduces energy loss during cyclic loading - Enhances ligament elasticity and function - Promotes efficient load transfer and shock absorption |
- Reduces energy loss during cyclic loading - Enhances tendon elasticity and function - Promotes efficient force transmission and load-bearing capacity |
- Minimizes energy loss during loading/unloading - Maintains meniscal resilience and function - Promotes efficient load distribution and shock absorption |
| Cell Biology of Early Mechanical Loading | - Chondrocyte proliferation and differentiation - ECM synthesis (collagen II, aggrecan) - Autophagy activation for cell survival - Modulation of inflammatory response (decreased IL-1, TNF-α) - Enhanced synthesis of proteoglycans and glycosaminoglycans |
- Fibroblast proliferation and migration - Collagen synthesis (type I and III) - ECM remodeling and organization - Modulation of inflammatory response (decreased IL-6, MMPs) - Enhanced ligament strength and flexibility |
- Tenocyte proliferation and alignment - Collagen synthesis (type I) - ECM remodeling and organization - Modulation of inflammatory response (decreased MMPs, increased TIMPs) - Enhanced tendon strength and flexibility |
- Chondrocyte and fibrochondrocyte activity - ECM synthesis (collagen I and II, proteoglycans) - MSC recruitment and differentiation - Modulation of inflammatory response (decreased pro-inflammatory cytokines) - Enhanced meniscal function and integration |
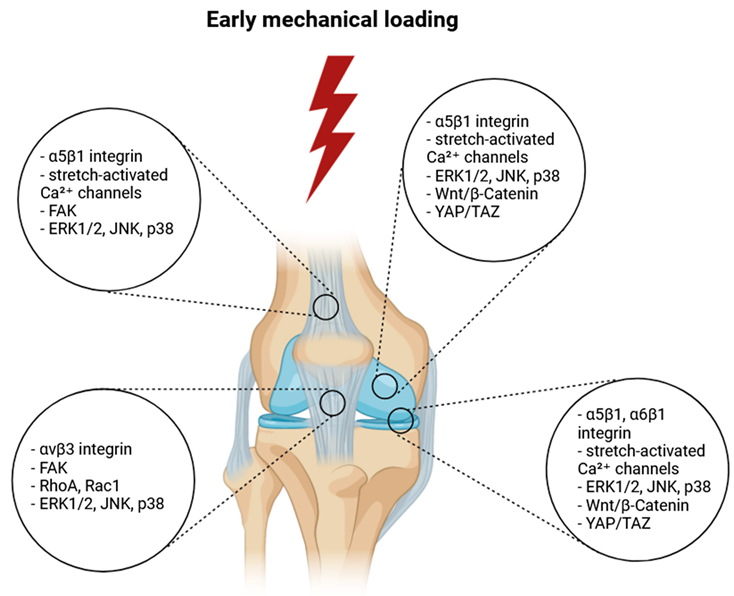 |
References
- Achenbach, L.; Bloch, H.; Klein, C.; Damm, T.; Obinger, M.; Rudert, M.; Krutsch, W.; Szymski, D. Four distinct patterns of anterior cruciate ligament injury in women’s professional football (soccer): A systematic video analysis of 37 match injuries. Br J Sports Med. 2024, 58, 709–716. [Google Scholar] [CrossRef] [PubMed]
- Gholipour Aghdam, G.M.; Alizadeh, M.H.; Minoonejad, H.; Shirzad, E.; Wilke, J. Knee Biomechanics During Neurocognitively Challenged Drop Landings in Male Elite Soccer Players with Anterior Cruciate Ligament Reconstruction. Sports Med Open. 2024, 10, 19. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Aizawa, J.; Hirohata, K.; Ohji, S.; Ohmi, T.; Mitomo, S.; Koga, H.; Yagishita, K. Cross-sectional study on relationships between physical function and psychological readiness to return to sport after anterior cruciate ligament reconstruction. BMC Sports Sci Med Rehabil. 2022, 14, 97. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ananías, J.; Vidal, C.; Ortiz-Muñoz, L.; Irarrázaval, S.; Besa, P. Use of electromyographic biofeedback in rehabilitation following anterior cruciate ligament reconstruction: A systematic review and meta-analysis. Physiotherapy. 2024, 123, 19–29. [Google Scholar] [CrossRef] [PubMed]
- Andrushko, J.W.; Carr, J.C.; Farthing, J.P.; Lepley, L.K.; DeFreitas, J.M.; Goodall, S.; Hendy, A.M.; Howatson, G.; Grooms, D.R.; Zult, T.; Hortobagyi, T.; Harput, G.; Papandreou, M.; Nosaka, K.; Carson, R.G.; Manca, A.; Deriu, F.; Behm, D.G.; Kidgell, D.J.; Clark, N.C.; Boyd, L.A. Potential role of cross-education in early-stage rehabilitation after anterior cruciate ligament reconstruction. Br J Sports Med. 2023, 57, 1474–1475. [Google Scholar] [CrossRef] [PubMed]
- Armento, A.; Keeter, C.; Gagliardi, A.; Rossing, H.; Giachino, C.; VandenBerg, C.; Howell, D.; Albright, J. Association of Grit With Postoperative Knee Outcomes and Physical Function After ACL Reconstruction in Adolescent Athletes. Am J Sports Med. 2023, 51, 2900–2907. [Google Scholar] [CrossRef] [PubMed]
- Baez, S.; Harkey, M.; Birchmeier, T.; Triplett, A.; Collins, K.; Kuenze, C. Psychological Readiness, Injury-Related Fear, and Persistent Knee Symptoms After Anterior Cruciate Ligament Reconstruction. J Athl Train. 2023, 58, 998–1003. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Bahr, R. Why screening tests to predict injury do not work-and probably never will…: A critical review. Br J Sports Med. 2016, 50, 776–780. [Google Scholar] [CrossRef] [PubMed]
- Bakhsh, H.R.; Metikala, S.; Billy, G.G.; Vairo, G.L. Association Between Self-Reported Kinesiophobia and Single-Leg Hop for Distance in Patients With ACL Reconstruction: A Systematic Review. Sports Health. 2022, 14, 674–680. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Balendra, G.; Jones, M.; Borque, K.A.; Willinger, L.; Pinheiro, V.H.; Williams, A. Factors affecting return to play and graft re-rupture after primary ACL reconstruction in professional footballers. Knee Surg Sports Traumatol Arthrosc. 2022, 30, 2200–2208. [Google Scholar] [CrossRef] [PubMed]
- Banios, K.; Raoulis, V.; Fyllos, A.; Chytas, D.; Mitrousias, V.; Zibis, A. Anterior and Posterior Cruciate Ligaments Mechanoreceptors: A Review of Basic Science. Diagnostics 2022, 12, 331. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Belcher, S.; Whatman, C.; Brughelli, M. A systematic video analysis of 21 anterior cruciate ligament injuries in elite netball players during games. Sports Biomech. 2022, 7, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Bencke, J.; Aagaard, P.; Zebis, M.K. Muscle Activation During ACL Injury Risk Movements in Young Female Athletes: A Narrative Review. Front Physiol. 2018, 9, 445. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Benjaminse, A.; Otten, B.; Gokeler, A.; Diercks, R.L.; Lemmink, K.A.P.M. Motor learning strategies in basketball players and its implications for ACL injury prevention: A randomized controlled trial. Knee Surg Sports Traumatol Arthrosc. 2017, 25, 2365–2376. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Bertozzi, F.; Fischer, P.D.; Hutchison, K.A.; Zago, M.; Sforza, C.; Monfort, S.M. Associations Between Cognitive Function and ACL Injury-Related Biomechanics: A Systematic Review. Sports Health. 2023, 15, 855–866. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Betsch, M.; Darwich, A.; Chang, J.; Whelan, D.; Ogilvie-Harris, D.; Chahal, J.; Theodoropoulos, J. Wide Variability in Return-to-Sport Criteria used by Team Physicians After Anterior Cruciate Ligament Reconstruction in Elite Athletes-A Qualitative Study. Arthrosc Sports Med Rehabil. 2022, 4, e1759–e1766. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Beynnon, B.D.; Tourville, T.W.; Hollenbach, H.C.; Shultz, S.; Vacek, P. Intrinsic Risk Factors for First-Time Noncontact ACL Injury: A Prospective Study of College and High School Athletes. Sports Health. 2023, 15, 433–442. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Boden, B.P.; Sheehan, F.T. Mechanism of non-contact ACL injury: OREF Clinical Research Award 2021. J Orthop Res. 2022, 40, 531–540. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Bodkin, S.G. Time to Reflect on Return to Sport Timing Following ACL Reconstruction. Sports Med. 2024. [Google Scholar] [CrossRef] [PubMed]
- Bolt, R.; Heuvelmans, P.; Benjaminse, A.; Robinson, M.A.; Gokeler, A. An ecological dynamics approach to ACL injury risk research: A current opinion. Sports Biomech. 2021, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Brophy, R.H.; Wojtys, E.M.; Mack, C.D.; Hawaldar, K.; Herzog, M.M.; Owens, B.D. Factors Associated With the Mechanism of ACL Tears in the National Football League: A Video-Based Analysis. Orthop J Sports Med. 2021, 9, 23259671211053301. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Buckthorpe, M.; Gokeler, A.; Herrington, L.; Hughes, M.; Grassi, A.; Wadey, R.; Patterson, S.; Compagnin, A.; La Rosa, G.; Della Villa, F. Optimising the Early-Stage Rehabilitation Process Post-ACL Reconstruction. Sports Med. 2024, 54, 49–72. [Google Scholar] [CrossRef] [PubMed]
- Byrne, S.; Lay, B.; Staynor, J.; Alderson, J.; Donnelly, C.J. The effect of planning time on penultimate and ultimate step kinematics and subsequent knee moments during sidestepping. Scand J Med Sci Sports. 2022, 32, 1366–1376. [Google Scholar] [CrossRef] [PubMed]
- Cacolice, P.A.; Starkey, B.E.; Carcia, C.R.; Higgins, P.E. Research Dominance Definitions May Not Identify Higher Risk Limb for Anterior Cruciate Ligament Injury in NCAA D3 Student-Athletes. Int J Sports Phys Ther. 2022, 17, 622–627. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Carlson, V.R.; Sheehan, F.T.; Boden, B.P. Video Analysis of Anterior Cruciate Ligament (ACL) Injuries: A Systematic Review. JBJS Rev. 2016, 4, e5. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Chaaban, C.R.; Hearn, D.; Goerger, B.; Padua, D.A. Are Elite Collegiate Female Athletes PRIME for a Safe Return to Sport after ACLR? An Investigation of Physical Readiness and Integrated Movement Efficiency (PRIME). Int J Sports Phys Ther. 2022, 17, 445–455. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Chaaban, C.R.; Turner, J.A.; Padua, D.A. Think outside the box: Incorporating secondary cognitive tasks into return to sport testing after ACL reconstruction. Front Sports Act Living. 2023, 4, 1089882. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Chaput, M.; Onate, J.A.; Simon, J.E.; Criss, C.R.; Jamison, S.; McNally, M.; Grooms, D.R. Visual cognition associated with knee proprioception, time to stability, and sensory integration neural activity after ACL reconstruction. J Orthop Res. 2022, 40, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Larson, D.; Vu, V.; Ness, B.M.; Wellsandt, E.; Morrison, S. A Multi-Systems Approach to Human Movement after ACL Reconstruction: The Musculoskeletal System. Int J Sports Phys Ther. 2021, 17, 27–46. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Chen, Z.; Li, Y.; Zhang, Y.; Zhang, Z.; Wang, J.; Deng, X.; Liu, C.; Chen, N.; Jiang, C.; Li, W.; Song, B. Analysis of Visual Risk Factors of Anterior Cruciate Ligament Injury of Knee Joint. J Clin Med. 2022, 11, 5602. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ciceklidag, M.; Kaya, I.; Ayanoglu, T.; Ayas, I.H.; Ozer, M.; Ataoglu, M.B.; Kanatli, U. Proprioception After Primary Repair of the Anterior Cruciate Ligament. Am J Sports Med. 2024, 52, 1199–1208, Erratum in Am J Sports Med. 2024, 3635465241253575. https://doi.org/10.1177/03635465241253575. [Google Scholar] [CrossRef] [PubMed]
- Colapietro, M.; Portnoff, B.; Miller, S.J.; Sebastianelli, W.; Vairo, G.L. Effects of Blood Flow Restriction Training on Clinical Outcomes for Patients With ACL Reconstruction: A Systematic Review. Sports Health. 2023, 15, 260–273. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Collings, T.J.; Diamond, L.E.; Barrett, R.S.; Timmins, R.G.; Hickey, J.T.; DUMoulin, W.S.; Williams, M.D.; Beerworth, K.A.; Bourne, M.N. Strength and Biomechanical Risk Factors for Noncontact ACL Injury in Elite Female Footballers: A Prospective Study. Med Sci Sports Exerc. 2022, 54, 1242–1251. [Google Scholar] [CrossRef] [PubMed]
- Crotti, M.; Heering, T.; Lander, N.; Fox, A.; Barnett, L.M.; Duncan, M.J. Extrinsic Risk Factors for Primary Noncontact Anterior Cruciate Ligament Injury in Adolescents Aged between 14 and 18 years: A Systematic Review. Sports Med. 2024, 54, 875–894. [Google Scholar] [CrossRef] [PubMed]
- Crotty, N.M.N.; Daniels, K.A.J.; McFadden, C.; Cafferkey, N.; King, E. Relationship Between Isokinetic Knee Strength and Single-Leg Drop Jump Performance 9 Months After ACL Reconstruction. Orthop J Sports Med. 2022, 10, 23259671211063800. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Culvenor, A.G.; Girdwood, M.A.; Juhl, C.B.; Patterson, B.E.; Haberfield, M.J.; Holm, P.M.; Bricca, A.; Whittaker, J.L.; Roos, E.M.; Crossley, K.M. Rehabilitation after anterior cruciate ligament and meniscal injuries: A best-evidence synthesis of systematic reviews for the OPTIKNEE consensus. Br J Sports Med. 2022, 56, 1445–1453. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Cunha, J.; Solomon, D.J. ACL Prehabilitation Improves Postoperative Strength and Motion and Return to Sport in Athletes. Arthrosc Sports Med Rehabil. 2022, 4, e65–e69. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Cuyul-Vásquez, I.; Álvarez, E.; Riquelme, A.; Zimmermann, R.; Araya-Quintanilla, F. Effectiveness of Unilateral Training of the Uninjured Limb on Muscle Strength and Knee Function of Patients With Anterior Cruciate Ligament Reconstruction: A Systematic Review and Meta-Analysis of Cross-Education. J Sport Rehabil. 2022, 31, 605–616. [Google Scholar] [CrossRef] [PubMed]
- D’Hooghe, P.; Grassi, A.; Villa, F.D.; Alkhelaifi, K.; Papakostas, E.; Rekik, R.; Marin, T.; Tosarelli, F.; Zaffagnini, S. The injury mechanism correlation between MRI and video-analysis in professional football players with an acute ACL knee injury reveals consistent bone bruise patterns. Knee Surg Sports Traumatol Arthrosc. 2023, 31, 121–132. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Daggett, M.C.; Witte, K.A.; Cabarkapa, D.; Cabarkapa, D.V.; Fry, A.C. Evidence-Based Data Models for Return-to-Play Criteria after Anterior Cruciate Ligament Reconstruction. Healthcare 2022, 10, 929. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Dalvandpour, N.; Zareei, M.; Abbasi, H.; Abdoli, B.; Mohammadian, M.A.; Rommers, N.; Rössler, R. Focus of Attention During ACL Injury Prevention Exercises Affects Improvements in Jump-Landing Kinematics in Soccer Players: A Randomized Controlled Trial. J Strength Cond Res. 2023, 37, 337–342. [Google Scholar] [CrossRef] [PubMed]
- D’Argenio, E.M.; Eckard, T.G.; Frank, B.S.; Prentice, W.E.; Padua, D.A. Examining the Dynamic Nature of Anterior Cruciate Ligament Injury Risk Factors in Women’s Collegiate Soccer. J Sport Rehabil. 2022, 31, 286–293. [Google Scholar] [CrossRef] [PubMed]
- Das, L.; Johri, A.S.; Abdusamad, V.; Schuh, A.; Goyal, T. Joint awareness and return to pre-injury level of activities after ACL reconstruction in athletes vs non-athletes. Eur J Orthop Surg Traumatol. 2023, 33, 819–827. [Google Scholar] [CrossRef] [PubMed]
- Dauty, M.; Crenn, V.; Louguet, B.; Grondin, J.; Menu, P.; Fouasson-Chailloux, A. Anatomical and Neuromuscular Factors Associated to Non-Contact Anterior Cruciate Ligament Injury. J Clin Med. 2022, 11, 1402. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Davies, W.T.; Ryu, J.H.; Graham-Smith, P.; Goodwin, J.E.; Cleather, D.J. Stronger Subjects Select a Movement Pattern That May Reduce Anterior Cruciate Ligament Loading During Cutting. J Strength Cond Res. 2022, 36, 1853–1859. [Google Scholar] [CrossRef] [PubMed]
- Della Villa, F.; Buckthorpe, M.; Grassi, A.; Nabiuzzi, A.; Tosarelli, F.; Zaffagnini, S.; Della Villa, S. Systematic video analysis of ACL injuries in professional male football (soccer): Injury mechanisms, situational patterns and biomechanics study on 134 consecutive cases. Br J Sports Med. 2020, 54, 1423–1432. [Google Scholar] [CrossRef] [PubMed]
- Detherage, J.P.; Divine, J.G.; Donaworth, M.A.; Palmer, T.G.; Hagen, J.A.; Hasselfeld, K.A.; Eifert-Mangine, M.; Mangine, R.E.; Clark, J.F.; Grawe, B.M. Physiological Monitoring Detected Changes During Women’s Soccer Anterior Cruciate Ligament Injury. Cureus. 2021, 13, e14838. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Devana, S.K.; Solorzano, C.A.; Vail, J.; Jackson, N.; Pham, D.; Jones, K.J. Outcomes of Blood Flow Restriction Training After ACL Reconstruction in NCAA Division I Athletes. Orthop J Sports Med. 2024, 12, 23259671241248589. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Alejandra Díaz, M.; Smeets, A.; Hagen, M.; Sankey, S.P.; Verschueren, S.; Vanrenterghem, J. Postural balance strategies during landing at the moment of return-to-sports after anterior cruciate ligament reconstruction. J Biomech. 2022, 145, 111381. [Google Scholar] [CrossRef] [PubMed]
- Di Paolo, S.; Bragonzoni, L.; Della Villa, F.; Grassi, A.; Zaffagnini, S. Do healthy athletes exhibit at-risk biomechanics for anterior cruciate ligament injury during pivoting movements? Sports Biomech. 2022, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Dischiavi, S.L.; Wright, A.A.; Heller, R.A.; Love, C.E.; Salzman, A.J.; Harris, C.A.; Bleakley, C.M. Do ACL Injury Risk Reduction Exercises Reflect Common Injury Mechanisms? A Scoping Review of Injury Prevention Programs. Sports Health. 2022, 14, 592–600. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Drole, K.; Paravlic, A.H. Interventions for increasing return to sport rates after an anterior cruciate ligament reconstruction surgery: A systematic review. Front Psychol. 2022, 13, 939209. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Dos’Santos, T.; McBurnie, A.; Donelon, T.; Thomas, C.; Comfort, P.; Jones, P.A. A qualitative screening tool to identify athletes with ‘high-risk’ movement mechanics during cutting: The cutting movement assessment score (CMAS). Phys Ther Sport. 2019, 38, 152–161. [Google Scholar] [CrossRef] [PubMed]
- Duthon, V.B.; Barea, C.; Abrassart, S.; Fasel, J.H.; Fritschy, D.; Ménétrey, J. Anatomy of the anterior cruciate ligament. Knee Surg Sports Traumatol Arthrosc. 2006, 14, 204–213. [Google Scholar] [CrossRef] [PubMed]
- Emami, F.; Negahban, H.; Sinaei, E.; Mostafaee, N.; Shahtahmassebi, B.; Ebrahimzadeh, M.H.; Mehravar, M. The Effects of Various Cognitive Tasks Including Working Memory, Visuospatial, and Executive Function on Postural Control in Patients With Anterior Cruciate Ligament Injury. Motor Control. 2024, 28, 193–209. [Google Scholar] [CrossRef] [PubMed]
- Fältström, A.; Hägglund, M.; Hedevik, H.; Kvist, J. Poor Validity of Functional Performance Tests to Predict Knee Injury in Female Soccer Players With or Without Anterior Cruciate Ligament Reconstruction. Am J Sports Med. 2021, 49, 1441–1450. [Google Scholar] [CrossRef] [PubMed]
- Fausett, W.A.; Reid, D.A.; Larmer, P.J. Current perspectives of New Zealand physiotherapists on rehabilitation and return to sport following anterior cruciate ligament reconstruction: A survey. Phys Ther Sport. 2022, 53, 166–172. [Google Scholar] [CrossRef] [PubMed]
- Figueroa, D.; Arce, G.; Espregueira-Mendes, J.; Maestu, R.; Mosquera, M.; Williams, A.; Parker, D.; Cohen, M.; Karahan, M.; Ochoa Perea, G.A.; Zaffagnini, S.; Neyret, P.; Karlsson, J.; Musahl, V.; Radice, F.; van der Merwe, W.M.; Landreau, P.; Imhoff, A.; Menetrey, J.; Ayeni, O.R.; Arliani, G.G.; Sherman, S.L.; Monllau, J.C.; D’Hooghe, P.; Pinczewski, L.; Feller, J.; Patnaik, S. Return to sport soccer after anterior cruciate ligament reconstruction: ISAKOS consensus. J ISAKOS. 2022, 7, 150–161. [Google Scholar] [CrossRef] [PubMed]
- Fleming, B.C. Fifty Years of ACL Biomechanics: What’s Next? Am J Sports Med. 2022, 50, 3745–3748. [Google Scholar] [CrossRef] [PubMed]
- García-Rodríguez, P.; Pecci, J.; Vázquez-González, S.; Pareja-Galeano, H. Acute and Chronic Effects of Blood Flow Restriction Training in Physically Active Patients With Anterior Cruciate Ligament Reconstruction: A Systematic Review. Sports Health. 2023, 19417381231208636. [Google Scholar] [CrossRef] [PubMed]
- Gholami, F.; Letafatkar, A.; Moghadas Tabrizi, Y.; Gokeler, A.; Rossettini, G.; Ghanati, H.A.; Schöllhorn, W.I. Comparing the Effects of Differential and Visuo-Motor Training on Functional Performance, Biomechanical, and Psychological Factors in Athletes after ACL Reconstruction: A Randomized Controlled Trial. J Clin Med. 2023, 12, 2845. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Giesche, F.; Vieluf, S.; Wilke, J.; Engeroff, T.; Niederer, D.; Banzer, W. Cortical Motor Planning and Biomechanical Stability During Unplanned Jump Landings in Men With Anterior Cruciate Ligament Reconstruction. J Athl Train. 2022, 57, 547–556. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Gokeler, A.; Seil, R.; Kerkhoffs, G.; Verhagen, E. A novel approach to enhance ACL injury prevention programs. J Exp Orthop. 2018, 5, 22. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Gokeler, A.; Neuhaus, D.; Benjaminse, A.; Grooms, D.R.; Baumeister, J. Principles of Motor Learning to Support Neuroplasticity After ACL Injury: Implications for Optimizing Performance and Reducing Risk of Second ACL Injury. Sports Med. 2019, 49, 853–865, Erratum in Sports Med. 2019, 49, 979. https://doi.org/10.1007/s40279-019-01078-w. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Gokeler, A.; Dingenen, B.; Hewett, T.E. Rehabilitation and Return to Sport Testing After Anterior Cruciate Ligament Reconstruction: Where Are We in 2022? Arthrosc Sports Med Rehabil. 2022, 4, e77–e82. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Gokeler, A.; Grassi, A.; Hoogeslag, R.; van Houten, A.; Lehman, T.; Bolling, C.; Buckthorpe, M.; Norte, G.; Benjaminse, A.; Heuvelmans, P.; Di Paolo, S.; Tak, I.; Villa, F.D. Return to sports after ACL injury 5 years from now: 10 things we must do. J Exp Orthop. 2022, 9, 73, Erratum in J Exp Orthop. 2022, 9, 111. https://doi.org/10.1186/s40634-022-00548-x. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Gokeler, A.; Tosarelli, F.; Buckthorpe, M.; Della Villa, F. Neurocognitive Errors and Noncontact Anterior Cruciate Ligament Injuries in Professional Male Soccer Players. J Athl Train. 2024, 59, 262–269. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Golberg, E.; Sommerfeldt, M.; Pinkoski, A.; Dennett, L.; Beaupre, L. Anterior Cruciate Ligament Reconstruction Return-to-Sport Decision-Making: A Scoping Review. Sports Health. 2024, 16, 115–123. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Gopinatth, V.; Smith, M.V.; Matava, M.J.; Brophy, R.H.; Knapik, D.M. Most Anterior Cruciate Ligament Injuries in Professional Athletes Occur Without Contact to the Injured Knee: A Systematic Review of Video Analysis Studies. Arthroscopy 2024. [Google Scholar] [CrossRef] [PubMed]
- Grooms, D.R.; Page, S.J.; Nichols-Larsen, D.S.; Chaudhari, A.M.; White, S.E.; Onate, J.A. Neuroplasticity Associated With Anterior Cruciate Ligament Reconstruction. J Orthop Sports Phys Ther. 2017, 47, 180–189. [Google Scholar] [CrossRef] [PubMed]
- Grooms, D.; Appelbaum, G.; Onate, J. Neuroplasticity following anterior cruciate ligament injury: A framework for visual-motor training approaches in rehabilitation. J Orthop Sports Phys Ther. 2015, 45, 381–393. [Google Scholar] [CrossRef] [PubMed]
- Grooms, D.R.; Chaput, M.; Simon, J.E.; Criss, C.R.; Myer, G.D.; Diekfuss, J.A. Combining Neurocognitive and Functional Tests to Improve Return-to-Sport Decisions Following ACL Reconstruction. J Orthop Sports Phys Ther. 2023, 53, 415–419. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Gureck, A.E.; Crockett, Z.; Barsky, B.W.; Samuels, S.; Frank, J.S.; Storer, S.K.; Fazekas, M.L. Do Differences Exist in Impact Test Domains between Youth Athletes with and without an Anterior Cruciate Ligament Injury? Healthcare 2023, 11, 2764. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Hamdan, M.; Haddad, B.; Alshrouf, M.A.; Azzam, M.I.; Isleem, U.; Hamasha, R.; Albtoush, O.M.; Alhusban, M.T.; Mubarak, N.; Alryalat, S.A. Can MRI knee joint measurements predict the population at risk of ACL injury? BMC Sports Sci Med Rehabil. 2022, 14, 98. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Hamoongard, M.; Hadadnezhad, M.; Abbasi, A. Effect of combining eight weeks of neuromuscular training with dual cognitive tasks on landing mechanics in futsal players with knee ligament dominance defect: A randomized controlled trial. BMC Sports Sci Med Rehabil. 2022, 14, 196. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Harput, G.; Demirci, S.; Soylu, A.R.; Bayrakci Tunay, V. Association between quadriceps muscle thickness and knee function in anterior cruciate ligament reconstructed athletes: A cross-sectional study. Physiother Theory Pract. 2023, 39, 2171–2179. [Google Scholar] [CrossRef] [PubMed]
- Henderson, F.J.; Konishi, Y.; Shima, N.; Shimokochi, Y. Effects of 8-Week Exhausting Deep Knee Flexion Flywheel Training on Persistent Quadriceps Weakness in Well-Trained Athletes Following Anterior Cruciate Ligament Reconstruction. Int J Environ Res Public Health. 2022, 19, 13209. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Hewett, T.E.; Myer, G.D.; Ford, K.R.; Paterno, M.V.; Quatman, C.E. Mechanisms, prediction, and prevention of ACL injuries: Cut risk with three sharpened and validated tools. J Orthop Res. 2016, 34, 1843–1855. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Homan, M.D.; Braaten, J.A.; Banovetz, M.T.; Monson, J.K.; Kennedy, N.I.; LaPrade, R.F. Principles for optimizing anterior cruciate ligament reconstruction outcomes in elite athletes: A review of current techniques. Ann Jt. 2024, 9, 19. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Hoogeslag, R.A.G.; Huis In ‘t Veld, R.; Brouwer, R.W.; de Graaff, F.; Verdonschot, N. Acute Anterior Cruciate Ligament Rupture: Repair or Reconstruction? Five-Year Results of a Randomized Controlled Clinical Trial. Am J Sports Med. 2022, 50, 1779–1787. [Google Scholar] [CrossRef] [PubMed]
- Hurley, E.T.; Mojica, E.S.; Haskel, J.D.; Mannino, B.J.; Alaia, M.; Strauss, E.J.; Jazrawi, L.M.; Gonzlaez-Lomas, G. Return to play testing following anterior cruciate reconstruction—A systematic review & meta-analysis. Knee. 2022, 34, 134–140. [Google Scholar] [CrossRef] [PubMed]
- Imai, S.; Harato, K.; Morishige, Y.; Kobayashi, S.; Niki, Y.; Sato, K.; Nagura, T. Effects of Dual Task Interference on Biomechanics of The Entire Lower Extremity During the Drop Vertical Jump. J Hum Kinet. 2022, 81, 5–14. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Jack, R.A., 2nd; Lambert, B.S.; Hedt, C.A.; Delgado, D.; Goble, H.; McCulloch, P.C. Blood Flow Restriction Therapy Preserves Lower Extremity Bone and Muscle Mass After ACL Reconstruction. Sports Health. 2023, 15, 361–371. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Jenkins, S.M.; Guzman, A.; Gardner, B.B.; Bryant, S.A.; Del Sol, S.R.; McGahan, P.; Chen, J. Rehabilitation After Anterior Cruciate Ligament Injury: Review of Current Literature and Recommendations. Curr Rev Musculoskelet Med. 2022, 15, 170–179. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Jiang, L.; Zhang, L.; Huang, W.; Zeng, Q.; Huang, G. The effect of proprioception training on knee kinematics after anterior cruciate ligament reconstruction: A randomized control trial. J Back Musculoskelet Rehabil. 2022, 35, 1085–1095. [Google Scholar] [CrossRef] [PubMed]
- Johnston, J.T.; Mandelbaum, B.R.; Schub, D.; Rodeo, S.A.; Matava, M.J.; Silvers-Granelli, H.J.; Cole, B.J.; ElAttrache, N.S.; McAdams, T.R.; Brophy, R.H. Video Analysis of Anterior Cruciate Ligament Tears in Professional American Football Athletes. Am J Sports Med. 2018, 46, 862–868. [Google Scholar] [CrossRef] [PubMed]
- Jones, H.S.R.; Moore, I.S.; King, E.; Stiles, V.H.; Laudani, L.; McCarthy-Ryan, M.; McFadden, C.; Daniels, K.A.J. Movement strategy correspondence across jumping and cutting tasks after anterior cruciate ligament reconstruction. Scand J Med Sci Sports. 2022, 32, 612–621. [Google Scholar] [CrossRef] [PubMed]
- Josse, C.M. Gym-Based Training Interventions for Anterior Cruciate Ligament Injury Reduction in American Football Players. HSS J. 2023, 19, 285–291. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kadlec, D.; Miller-Dicks, M.; Nimphius, S. Training for “Worst-Case” Scenarios in Sidestepping: Unifying Strength and Conditioning and Perception-Action Approaches. Sports Med Open. 2023, 9, 22. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kakavas, G.; Malliaropoulos, N.; Pruna, R.; Traster, D.; Bikos, G.; Maffulli, N. Neuroplasticity and Anterior Cruciate Ligament Injury. Indian J Orthop. 2020, 54, 275–280. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kakavas, G.; Forelli, F.; Malliaropoulos, N.; Hewett, T.E.; Tsaklis, P. Periodization in Anterior Cruciate Ligament Rehabilitation: New Framework Versus Old Model? A Clinical Commentary. Int J Sports Phys Ther. 2023, 18, 541–546. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kellis, E.; Sahinis, C.; Baltzopoulos, V. Is hamstrings-to-quadriceps torque ratio useful for predicting anterior cruciate ligament and hamstring injuries? A systematic and critical review. J Sport Health Sci. 2023, 12, 343–358. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- King, E.; Richter, C.; Daniels, K.A.J.; Franklyn-Miller, A.; Falvey, E.; Myer, G.D.; Jackson, M.; Moran, R.; Strike, S. Can Biomechanical Testing After Anterior Cruciate Ligament Reconstruction Identify Athletes at Risk for Subsequent ACL Injury to the Contralateral Uninjured Limb? Am J Sports Med. 2021, 49, 609–619. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Koc, B.B.; Truyens, A.; Heymans, M.J.L.F.; Jansen, E.J.P.; Schotanus, M.G.M. Effect of Low-Load Blood Flow Restriction Training After Anterior Cruciate Ligament Reconstruction: A Systematic Review. Int J Sports Phys Ther. 2022, 17, 334–346. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kotsifaki, R.; Korakakis, V.; King, E.; Barbosa, O.; Maree, D.; Pantouveris, M.; Bjerregaard, A.; Luomajoki, J.; Wilhelmsen, J.; Whiteley, R. Aspetar clinical practice guideline on rehabilitation after anterior cruciate ligament reconstruction. Br J Sports Med. 2023, 57, 500–514. [Google Scholar] [CrossRef] [PubMed]
- Kiani Haft Lang, M.; Mofateh, R.; Orakifar, N.; Goharpey, S. Differences in Neurocognitive Functions Between Healthy Controls and Anterior Cruciate Ligament-Reconstructed Male Athletes Who Passed or Failed Return to Sport Criteria: A Preliminary Study. J Sport Rehabil. 2023, 32, 645–654. [Google Scholar] [CrossRef] [PubMed]
- Larson, D.; Vu, V.; Ness, B.M.; Wellsandt, E.; Morrison, S. A Multi-Systems Approach to Human Movement after ACL Reconstruction: The Musculoskeletal System. Int J Sports Phys Ther. 2021, 17, 27–46. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Liew, B.X.W.; Feller, J.A.; Webster, K.E. Understanding the psychological mechanisms of return to sports readiness after anterior cruciate ligament reconstruction. PLoS ONE. 2022, 17, e0266029. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Louw, Q.; Gillion, N.; van Niekerk, S.M.; Morris, L.; Baumeister, J. The effect of vision on knee biomechanics during functional activities—A systematic review. J Sci Med Sport. 2015, 18, 469–474. [Google Scholar] [CrossRef] [PubMed]
- Lucarno, S.; Zago, M.; Buckthorpe, M.; Grassi, A.; Tosarelli, F.; Smith, R.; Della Villa, F. Systematic Video Analysis of Anterior Cruciate Ligament Injuries in Professional Female Soccer Players. Am J Sports Med. 2021, 49, 1794–1802. [Google Scholar] [CrossRef] [PubMed]
- Maestroni, L.; Turner, A.; Papadopoulos, K.; Sideris, V.; Read, P. Total Score of Athleticism: Profiling Strength and Power Characteristics in Professional Soccer Players After Anterior Cruciate Ligament Reconstruction to Assess Readiness to Return to Sport. Am J Sports Med. 2023, 51, 3121–3130. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Mangine, R.; Tersak, J.; Palmer, T.; Hill-Lindsay, A.; Patton, B.; Eifert-Mangine, M.; Jacobs, B.; Colosimo, A.J. The Longitudinal Neurophysiological Adaptation of a Division I Female Lacrosse Player Following Anterior Cruciate Rupture and Repair: A Case Report. Int J Sports Phys Ther. 2023, 18, 467–476. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Maniar, N.; Cole, M.H.; Bryant, A.L.; Opar, D.A. Muscle Force Contributions to Anterior Cruciate Ligament Loading. Sports Med. 2022, 52, 1737–1750. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Meierbachtol, A.; Obermeier, M.; Yungtum, W.; Bottoms, J.; Paur, E.; Nelson, B.J.; Tompkins, M.; Russell, H.C.; Chmielewski, T.L. Injury-Related Fears During the Return-to-Sport Phase of ACL Reconstruction Rehabilitation. Orthop J Sports Med. 2020, 8, 2325967120909385. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Mok, A.C.; Fancher, A.J.; Vopat, M.L.; Baker, J.; Tarakemeh, A.; Mullen, S.; Schroeppel, J.P.; Templeton, K.; Mulcahey, M.K.; Vopat, B.G. Sex-Specific Outcomes After Anterior Cruciate Ligament Reconstruction: A Systematic Review and Meta-analysis. Orthop J Sports Med. 2022, 10, 23259671221076883. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Momaya, A.M.; Wood, A.S.; Benson, E.M.; Kwapisz, A.L. The Influence of Psychosocial Factors on Patients Undergoing Anterior Cruciate Ligament Reconstruction. Sports Health. 2024, 16, 230–238. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Mørtvedt, A.I.; Krosshaug, T.; Bahr, R.; Petushek, E. I spy with my little eye … a knee about to go ‘pop’? Can coaches and sports medicine professionals predict who is at greater risk of ACL rupture? Br J Sports Med. 2020, 54, 154–158. [Google Scholar] [CrossRef] [PubMed]
- Nae, J.Ä.; Cronström, A. Association between sensorimotor function and visual assessment of postural orientation in patients with ACL injury. Phys Ther Sport. 2022, 55, 160–167. [Google Scholar] [CrossRef] [PubMed]
- Nessler, T.; Denney, L.; Sampley, J. ACL Injury Prevention: What Does Research Tell Us? Curr Rev Musculoskelet Med. 2017, 10, 281–288. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ngatuvai, M.S.; Yang, J.; Kistamgari, S.; Collins, C.L.; Smith, G.A. Epidemiological Comparison of ACL Injuries on Different Playing Surfaces in High School Football and Soccer. Orthop J Sports Med. 2022, 10, 23259671221092321. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Nilstad, A.; Petushek, E.; Mok, K.M.; Bahr, R.; Krosshaug, T. Kiss goodbye to the ‘kissing knees’: No association between frontal plane inward knee motion and risk of future non-contact ACL injury in elite female athletes. Sports Biomech. 2023, 22, 65–79. [Google Scholar] [CrossRef] [PubMed]
- Oladeji, L.; Reynolds, G.; Gonzales, H.; DeFroda, S. Anterior Cruciate Ligament Return to Play: Where Are We Now? J Knee Surg. 2024, 37, 586–592. [Google Scholar] [CrossRef] [PubMed]
- Mohammadi Orangi, B.; Dehghani, M.; Jones, P.A. Manipulation of task constraints is the most effective motor learning method for reducing risk factors for ACL injury during side-step cutting in both male and female athletes. Res Sports Med. 2024, 32, 631–647. [Google Scholar] [CrossRef] [PubMed]
- Parsons, J.L.; Coen, S.E.; Bekker, S. Anterior cruciate ligament injury: Towards a gendered environmental approach. Br J Sports Med. 2021, 55, 984–990. [Google Scholar] [CrossRef] [PubMed]
- Piskin, D.; Benjaminse, A.; Dimitrakis, P.; Gokeler, A. Neurocognitive and Neurophysiological Functions Related to ACL Injury: A Framework for Neurocognitive Approaches in Rehabilitation and Return-to-Sports Tests. Sports Health. 2022, 14, 549–555. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Rambaud, A.J.; Neri, T.; Dingenen, B.; Parker, D.; Servien, E.; Gokeler, A.; Edouard, P. The modifying factors that help improve anterior cruciate ligament reconstruction rehabilitation: A narrative review. Ann Phys Rehabil Med. 2022, 65, 101601. [Google Scholar] [CrossRef] [PubMed]
- Read, P.J.; Pedley, J.S.; Eirug, I.; Sideris, V.; Oliver, J.L. Impaired Stretch-Shortening Cycle Function Persists Despite Improvements in Reactive Strength After Anterior Cruciate Ligament Reconstruction. J Strength Cond Res. 2022, 36, 1238–1244. [Google Scholar] [CrossRef] [PubMed]
- Read, P.J.; Davies, W.T.; Bishop, C.; McAuliffe, S.; Wilson, M.G.; Turner, A.N. Residual Deficits in Reactive Strength After Anterior Cruciate Ligament Reconstruction in Soccer Players. J Athl Train. 2023, 58, 423–429. [Google Scholar] [CrossRef] [PubMed]
- Reiche, E.; Collins, K.; Genoese, F.; Walaszek, M.; Triplett, A.; Kuenze, C.; Harkey, M.; Baez, S. Lower Extremity Reaction Time in Individuals With Contact Versus Noncontact Anterior Cruciate Ligament Injuries After Reconstruction. J Athl Train. 2024, 59, 66–72. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Reyes, M.A.; Probasco, M.O.; Worby, T.N.; Loertscher, D.E.; Soderbeck, L.K.; Huddleston, W.E. Lower Kinetic Chain, Meet the Thinking Brain: A Scoping Review of Cognitive Function and Lower Extremity Injury Risk. Int J Sports Phys Ther. 2022, 17, 787–815. [Google Scholar] [PubMed] [PubMed Central]
- Robey, N.J.; Buchholz, K.O.; Murphy, S.P.; Smith, J.D.; Heise, G.D. Do Athletes With a Reconstructed Anterior Cruciate Ligament Respond Differently Than Controls to Visual Challenges When Dynamic Postural Stability is Assessed? J Appl Biomech. 2021, 37, 611–618. [Google Scholar] [CrossRef] [PubMed]
- Rolley, T.; Gill, S.D.; Keast, M.; Reade, T.; Page, R.; Bonacci, J.; Stella, J.; Johnson, B.; Fox, A. Anticipatory effects on side-step cutting biomechanics in Women’s Australian Football League players. BMJ Open Sport Exerc Med. 2023, 9, e001587. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Romanchuk, N.J.; Livock, H.; Lukas, K.J.; Del Bel, M.J.; Benoit, D.L.; Carsen, S. Criteria Used to Determine Unrestricted Return to Activity After ACL Reconstruction in Pediatric and Adolescent Patients: A Systematic Review. Orthop J Sports Med. 2023, 11, 23259671231154540. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Saadat, S.; Stephenson, M.L.; Gillette, J.C. Entry angle during jump landing changes biomechanical risk factors for ACL injury. Sports Biomech. 2022, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Saadat, S.; Bricarell, K.M.; Gillette, J.C. Dual tasking increases kinematic and kinetic risk factors of ACL injury. Sports Biomech. 2023, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Schilaty, N.D.; McPherson, A.L.; Nagai, T.; Bates, N.A. Differences in psychological readiness for return to sport after anterior cruciate ligament injury is evident in thigh musculature motor unit characteristics. BMJ Open Sport Exerc Med. 2023, 9, e001609. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Schweizer, N.; Strutzenberger, G.; Franchi, M.V.; Farshad, M.; Scherr, J.; Spörri, J. Screening Tests for Assessing Athletes at Risk of ACL Injury or Reinjury-A Scoping Review. Int J Environ Res Public Health. 2022, 19, 2864. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Schwery, N.A.; Kiely, M.T.; Larson, C.M.; Wulf, C.A.; Heikes, C.S.; Hess, R.W.; Giveans, M.R.; Solie, B.S.; Doney, C.P. Quadriceps Strength following Anterior Cruciate Ligament Reconstruction: Normative Values based on Sex, Graft Type and Meniscal Status at 3, 6 & 9 Months. Int J Sports Phys Ther. 2022, 17, 434–444. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Sheean, A.J.; DeFoor, M.T.; Spindler, K.P.; Arner, J.W.; Athiviraham, A.; Bedi, A.; DeFroda, S.; Ernat, J.J.; Frangiamore, S.J.; Nuelle, C.W.; Sheean, A.J.; Spindler, K.P.; Bedi, A. The Psychology of ACL Injury, Treatment, and Recovery: Current Concepts and Future Directions. Sports Health. 2024, 19417381241226896. [Google Scholar] [CrossRef] [PubMed]
- Shekhar, A.; Pilar, A.; Ponnanna, K.M.; Tapasvi, S. ACL repair for athletes? J Orthop. 2022, 31, 61–66. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Sherman, D.A.; Baumeister, J.; Stock, M.S.; Murray, A.M.; Bazett-Jones, D.M.; Norte, G.E. Inhibition of Motor Planning and Response Selection after Anterior Cruciate Ligament Reconstruction. Med Sci Sports Exerc. 2023, 55, 440–449. [Google Scholar] [CrossRef] [PubMed]
- Shibata, S.; Takemura, M.; Miyakawa, S. Kinematics, Kinetics and Muscle Activity Analysis during Single-leg Drop-jump Landing Followed by an Unanticipated Task: Focusing on Differences in Neurocognitive Function. Int J Sports Phys Ther. 2023, 18, 1085–1093. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Sim, K.; Rahardja, R.; Zhu, M.; Young, S.W. Optimal Graft Choice in Athletic Patients with Anterior Cruciate Ligament Injuries: Review and Clinical Insights. Open Access J Sports Med. 2022, 13, 55–67. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Slater, L.V.; Hart, J.M. Quantifying the relationship between quadriceps strength and aerobic fitness following anterior cruciate ligament reconstruction. Phys Ther Sport. 2022, 55, 106–110. [Google Scholar] [CrossRef] [PubMed]
- Smeets, A.; Willems, M.; Gilson, L.; Verschueren, S.; Staes, F.; Vandenneucker, H.; Claes, S.; Vanrenterghem, J. Neuromuscular and biomechanical landing alterations persist in athletes returning to sport after anterior cruciate ligament reconstruction. Knee. 2021, 33, 305–317. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Li, L.; Hughes, G.; Dai, B. Trunk motion and anterior cruciate ligament injuries: A narrative review of injury videos and controlled jump-landing and cutting tasks. Sports Biomech. 2023, 22, 46–64. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Li, L.; Layer, J.; Fairbanks, R.; Jenkins, M.; Hughes, G.; Smith, D.; Wilson, M.; Zhu, Q.; Dai, B. Indirect contact matters: Mid-flight external trunk perturbation increased unilateral anterior cruciate ligament loading variables during jump-landings. J Sport Health Sci. 2023, 12, 534–543. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Stearns, K.M.; Powers, C.M. Improvements in hip muscle performance result in increased use of the hip extensors and abductors during a landing task. Am J Sports Med. 2014, 42, 602–609. [Google Scholar] [CrossRef] [PubMed]
- Stojanović, E.; Terrence Scanlan, A.; Radovanović, D.; Jakovljević, V.; Faude, O. A multicomponent neuromuscular warm-up program reduces lower-extremity injuries in trained basketball players: A cluster randomized controlled trial. Phys Sportsmed. 2023, 51, 463–471. [Google Scholar] [CrossRef] [PubMed]
- Straub, R.K.; Powers, C.M. Is muscular strength a predictor for primary or secondary ACL injury? A scoping review of prospective studies. Phys Ther Sport. 2023, 61, 91–101. [Google Scholar] [CrossRef] [PubMed]
- Sugarman, B.S.; Sullivan, Z.B.; Le, D.; Killelea, C.; Faherty, M.S.; Diehl, L.H.; Wittstein, J.R.; Riboh, J.C.; Toth, A.P.; Amendola, A.; Taylor, D.C.; Sell, T.C. Isometric Knee Strength is Greater in Individuals Who Score Higher on Psychological Readiness to Return to Sport After Primary Anterior Cruciate Ligament Reconstruction. Int J Sports Phys Ther. 2022, 17, 1330–1339. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Taberner, M.; van Dyk, N.; Allen, T.; Jain, N.; Richter, C.; Drust, B.; Betancur, E.; Cohen, D.D. Physical preparation and return to performance of an elite female football player following ACL reconstruction: A journey to the FIFA Women’s World Cup. BMJ Open Sport Exerc Med. 2020, 6, e000843. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Romain, T.; Francois, F. The Role of Foot-Ankle Complex in Rehabilitation After ACLR—Make Miracles Happen. 2023, 12, 310–316.
- Van Cant, J.; Pairot de Fontenay, B.; Douaihy, C.; Rambaud, A. Characteristics of return to running programs following an anterior cruciate ligament reconstruction: A scoping review of 64 studies with clinical perspectives. Phys Ther Sport. 2022, 57, 61–70. [Google Scholar] [CrossRef] [PubMed]
- van Melick, N.; Pronk, Y.; Nijhuis-van der Sanden, M.; Rutten, S.; van Tienen, T.; Hoogeboom, T. Meeting movement quantity or quality return to sport criteria is associated with reduced second ACL injury rate. J Orthop Res. 2022, 40, 117–128. [Google Scholar] [CrossRef] [PubMed]
- Vitharana, T.N.; King, E.; Moran, K. Sensorimotor Dysfunction Following Anterior Cruciate Ligament Reconstruction- an Afferent Perspective: A Scoping Review. Int J Sports Phys Ther. 2024, 19, 1410–1437. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Weingart, A.; Rynecki, N.; Pereira, D. A Review of Neuromuscular Training and Biomechanical Risk Factor Screening for ACL Injury Prevention Among Female Soccer Players. Bull Hosp Jt Dis (2013). 2022, 80, 253–259. [Google Scholar] [PubMed]
- Weir, G. Anterior cruciate ligament injury prevention in sport: Biomechanically informed approaches. Sports Biomech. 2021, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Welling, W.; Frik, L. On-Field Tests for Patients After Anterior Cruciate Ligament Reconstruction: A Scoping Review. Orthop J Sports Med. 2022, 10, 23259671211055481. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Welling, W. Return to sports after an ACL reconstruction in 2024—A glass half full? A narrative review. Phys Ther Sport. 2024, 67, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Wilk, K.; Thomas, Z.M.; Arrigo, C.A.; Davies, G.J. The Need To Change Return to Play Testing in Athletes Following ACL Injury: A Theoretical Model. Int J Sports Phys Ther. 2023, 18, 272–281. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wohl, T.R.; Criss, C.R.; Grooms, D.R. Visual Perturbation to Enhance Return to Sport Rehabilitation after Anterior Cruciate Ligament Injury: A Clinical Commentary. Int J Sports Phys Ther. 2021, 16, 552–564. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- An, Y.W.; Kim, K.M.; DiTrani Lobacz, A.; Baumeister, J.; Higginson, J.S.; Rosen, J.; Swanik, C.B. Cognitive Training Improves Joint Stiffness Regulation and Function in ACLR Patients Compared to Healthy Controls. Healthcare 2023, 11, 1875. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zago, M.; Esposito, F.; Stillavato, S.; Zaffagnini, S.; Frigo, C.A.; Della Villa, F. 3-Dimensional Biomechanics of Noncontact Anterior Cruciate Ligament Injuries in Male Professional Soccer Players. Am J Sports Med. 2024, 52, 1794–1803. [Google Scholar] [CrossRef] [PubMed]
- Zebis, M.K.; Aagaard, P.; Andersen, L.L.; Hölmich, P.; Clausen, M.B.; Brandt, M.; Husted, R.S.; Lauridsen, H.B.; Curtis, D.J.; Bencke, J. First-time anterior cruciate ligament injury in adolescent female elite athletes: A prospective cohort study to identify modifiable risk factors. Knee Surg Sports Traumatol Arthrosc. 2022, 30, 1341–1351. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zhang, L.; Hacke, J.D.; Garrett, W.E.; Liu, H.; Yu, B. Bone Bruises Associated with Anterior Cruciate Ligament Injury as Indicators of Injury Mechanism: A Systematic Review. Sports Med. 2019, 49, 453–462. [Google Scholar] [CrossRef] [PubMed]
- Kacprzak, B.; Rosińska, K.; Siuba-Jarosz, N. Hyalofast Cartilage Repair Surgery with a Full Load-Bearing Rehabilitation Program One Day after Operation Reduces the Time for Professional Athletes to Return to Play. Medicina. 2023, 59, 804. [Google Scholar] [CrossRef]
- Loeser, R.F.; Goldring, S.R.; Scanzello, C.R.; Gldring, M.B. Osteoarthritis: A Disease of the Joint as an Organ. Arthritis Rheum 2012, 64, 1697–1707. [Google Scholar] [CrossRef]
- Osteoarthritis Society International—OA Definition. Available online: https://oarsi.org/ (accessed on 4 February 2023).
- Zheng, L.; Zhang, Z.; Sheng, P.; Mobasheri, A. The role of metabolism in chondrocyte dysfunction and the progression of osteoarthritis. Ageing Res. Rev. 2021, 66, 101249. [Google Scholar] [CrossRef]
- Fenwick, S.A.; Gregg, P.J.; Rooney, P. Osteoarthritic cartilage loses its ability to remain avascular. Osteoarthr. Cart. 1999, 7, 441–452. [Google Scholar] [CrossRef]
- Fahy, N.; de Vries-van, M.M.I.; Lehmann, W.; Grotenhuis, N.; Farrel, E.; van der Kraan, P.M.; Murphy, J.M.; Bastiaansen-Jenniskens, Y.M.; van Osch, G.J.V.M. Human osteoarthritic synovium impacts chondrogenic differentiation of mesenchymal stem cells via macrophage polarization state. Osteoarthr. Cart. 2014, 22, 1167–1175. [Google Scholar] [CrossRef]
- Gómez-Aristizábal, A.; Gadhi, R.; Mahomed, N.N.; Marshall, K.W.; Viswanathan, S. Synovial fluid monocyte/macrophage subsets and their correlation to patient-reported outcomes in osteoarthritic patients: A cohort study. Arthritis Res. Ther. 2019, 21, 26. [Google Scholar] [CrossRef] [PubMed]
- Kemble, S.; Croft, A.P. Critical Role of Synovial Tissue–Resident Macrophage and Fibroblast Subsets in the Persistence of Joint Inflammation. Front. Immunol. 2021, 12, 715894. [Google Scholar] [CrossRef] [PubMed]
- Kawanishi, M.; Kami, K.; Nishimura, Y.; Minami, K.; Senba, E.; Umemoto, Y.; Kinoshita, T.; Tajima, F. Exercise-induced increase in M2 macrophages accelerates wound healing in young mice. Physiol. Rep. 2022, 10, e15447. [Google Scholar] [CrossRef] [PubMed]
- Jang, J.S.; Lee, K.; Ju, J.H. Recent Updates of Diagnosis, Pathophysiology, and Treatment on Osteoarthritis of the Knee. Int. J. Mol. Sci. 2021, 22, 2619. [Google Scholar] [CrossRef]
- Katsuola, G.; Kreitmaier, P.; Zeggini, E. Insights into the molecular landscape of osteoarthritis in human tissues. Curr. Opin. Rheumatol. 2022, 34, 79–90. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.; Jeong, S.; Kim, H.; Kang, D.; Lee, J.; Kang, S.-J.; Kim, J.-H. Disease-modifying therapeutic strategies in osteoarthritis: Current status and future directions. Exp. Mol. Med. 2021, 53, 1689–1696. [Google Scholar] [CrossRef] [PubMed]
- Yu, P.; Zhang, X.; Liu, N.; Tang, L.; Peng, C.; Chen, X. Pyroptosis: Mechanisms and diseases. Signal Transduct. Target. Ther. 2021, 6, 128. [Google Scholar] [CrossRef]
- Jiang, W.; Wang, X.; Su, S.; Du, S.S.H. Identifying the shared genes and KEGG pathways of Resolvin D1-targeted network and osteoarthritis using bioinformatics. Bioengineered 2022, 13, 9839–9854. [Google Scholar] [CrossRef]
- Franceschi, C.; Bonafè, M.; Valensin, S.; Olivieri, F.; De Luca, M.; Ottaviani, E.; De Benedictis, G. Inflamm-aging. An evolutionary perspective on immunosenescence. Ann. N.Y. Acad. Sci. 2000, 908, 244–254. [Google Scholar] [CrossRef]
- Rausch Osthoff, A.-K.; Niedermann, K.; Braun, J.; Adams, J.; Brodin, N.; Dagfinrud, H.; Duruoz, T.; Esbensen, B.A.; Günther, K.-P.; Hurkmans, E.; et al. 2018 EULAR recommendations for physical activity in people with inflammatory arthritis and osteoarthritis. Ann. Rheum. Dis. 2018, 77, 1251–1260. [Google Scholar] [CrossRef]
- Dhillon, R.J.-S.; Hasni, S. Pathogenesis and Management of Sarcopenia. Clin. Geriatr. Med. 2017, 33, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Elagizi, A.; Kachur, S.; Carbone, S.; Lavie, C.J.; Blair, S.N. A Review of Obesity, Physical Activity and Cardiovascular Disease. Curr. Obes. Rep. 2020, 9, 571–581. [Google Scholar] [CrossRef] [PubMed]
- Boutron, I.; Tubach, F.; Giradeau, B.; Ravaud, P. Blinding was judged more difficult to achieve and maintain in nonpharmacologic than pharmacologic trials. J. Clin. Epidemiol. 2004, 57, 543–550. [Google Scholar] [CrossRef] [PubMed]
- Fregni, F.; Imamura, M.; Chien, H.F.; Lew, H.L.; Boggio, P.; Kaptchuk, T.J.; Riberto, M.; Hsing, W.T.; Battistella, L.R.; Furlan, A. Challenges and Recommendations for Placebo Controls in Randomized Trials in Physical and Rehabilitation Medicine: A Report of the International Placebo Symposium Working Group. Am. J. Phys. Med. Rehabil. 2010, 89, 160–172. [Google Scholar] [CrossRef]
- Villamar, M.F.; Contreras, V.S.; Kuntz, R.E.; Fregni, F. The Reporting of Blinding in Physical Medicine and Rehabilitation Randomized Controlled Trials: A Systematic Review. J. Rehabil. Med. 2013, 45, 6–13. [Google Scholar] [CrossRef]
- Lv, Z.; Yang, Y.X.; Li, J.; Fei, Y.; Guo, H.; Sun, Z.; Lu, J.; Xu, X.; Jiang, Q.; Ikegawa, S.; et al. Molecular Classification of Knee Osteoarthritis Front. Cell Dev. Biol. 2021, 9, 725568. [Google Scholar] [CrossRef]
- Vassao, P.G.; de Souza, A.C.F.; da Silveira Campos, R.M.; Garcia, L.A.; Tucci, H.T.; Renno, A.C.M. Effects of photobiomodulation and a physical exercise program on the expression of inflammatory and cartilage degradation biomarkers and functional capacity in women with knee osteoarthritis: A randomized blinded study. Adv. Rheumatol. 2021, 61. [Google Scholar] [CrossRef]
- Kim, J.-R.; Yoo, J.J.; Kim, H.A. Therapeutics in Osteoarthritis Based on an Understanding of Its Molecular Pathogenesis. Int. J. Mol. Sci. 2018, 19, 674. [Google Scholar] [CrossRef]
- American Kinesiotherapy Association. Available online: https://akta.org/about/history (accessed on 4 February 2023).
- Alves, M.D.d.J.; Silva, D.d.S.; Pereira, E.V.M.; Pereira, D.D.; de Sousa Fernandes, M.S.; Santos, D.F.C.; Oliveira, D.P.M.; Vieira-Souza, L.M.; Aidar, F.J.; de Souza, R.F. Changes in Cytokines Concentration Following Long-Distance Running: A Systematic Review and Meta-Analysis. Front. Physiol. 2022, 13, 838069. [Google Scholar] [CrossRef]
- Balan, E.; De Groote, E.; Bouillon, M.; Vieconte, N.; Mahieu, M.N.; Aslain, D.; Nielens, H.; Decottignies, A.; Deldicque, L. No effect of the endurance training status on senescence despite reduced inflammation in skeletal muscle of older individuals. Am. J. Physiol. Endocrinol. Metab. 2020, 319, E447–E454. [Google Scholar] [CrossRef]
- Lehner, C.; et al. Tenophages: A novel macrophage-like tendon cell population expressing CX3CL1 and CX3CR1. DMM Dis Model Mech. 2019, 12. [Google Scholar] [CrossRef] [PubMed]
- Bi, Y.; Ehirchiou, D.; Kilts, T.M.; Inkson, C.A.; Embree, M.C.; Sonoyama, W.; Li, L.; Leet, A.I.; Seo, B.-M.; Zhang, L.; Shi, S.; Young, M.F. Identification of tendon stem/progenitor cells and the role of the extracellular matrix in their niche. Nat Med. 2007, 13, 1219–1227. [Google Scholar] [CrossRef] [PubMed]
- Rui, Y.F.; Lui, P.P.Y.; Ni, M.; Chan, L.S.; Lee, Y.W.; Chan, K.M. Mechanical loading increased BMP-2 expression which promoted osteogenic differentiation of tendon-derived stem cells. J Orthop Res. 2011, 29, 390–396. [Google Scholar] [CrossRef]
- Spiesz, E.; Thorpe, C.T.; Chaudhry, S.; Riley, G.P.; Birch, H.L.; Clegg, P.D.; Screen, H.R.C. Tendon extracellular matrix damage, degradation and inflammation in response to in vitro overload exercise. J Orthop Res. 2015, 33, 889–897. [Google Scholar] [CrossRef] [PubMed]
- Cadby, J.A.; Buehler, E.; Godbout, C.; Van Weeren, P.R.; Snedeker, J.G.; Lionetti, V. Differences between the cell populations from the peritenon and the tendon core with regard to their potential implication in tendon repair. PLoS ONE. 2014, 9, e92474. [Google Scholar] [CrossRef] [PubMed]
- Dyment, N.A.; Hagiwara, Y.; Matthews, B.G.; Li, Y.; Kalajzic, I.; Rowe, D.W. Lineage tracing of resident tendon progenitor cells during growth and natural healing. PLoS ONE. 2014, 9, e96113. [Google Scholar] [CrossRef]
- Yoshida, R.; Alaee, F.; Dyrna, F.; Kronenberg, M.S.; Maye, P.; Kalajzic, I.; Rowe, D.W.; Mazzocca, A.D.; Dyment, N.A. Murine supraspinatus tendon injury model to identify the cellular origins of rotator cuff healing. Connect Tissue Res. 2016, 57, 507–515. [Google Scholar] [CrossRef] [PubMed]
- Carroll, C.C.; Dickinson, J.M.; Haus, J.M.; Lee, G.A.; Hollon, C.J.; Aagaard, P.; Magnusson, S.P.; Trappe, T.A. Influence of aging on the in vivo properties of human patellar tendon. J Appl Physiol. 2008, 105, 1907–1915. [Google Scholar] [CrossRef]
- Light, N.D.; Bailey, A.J. Changes in crosslinking during aging in bovine tendon collagen. FEBS letters 1979, 97, 183–188. [Google Scholar] [CrossRef]
- Li, Y.; Fessel, G.; Georgiadis, M.; Snedeker, J.G. Advanced glycation end-products diminish tendon collagen fiber sliding. Matrix Biol. 2013, 32, 169–177. [Google Scholar] [CrossRef]
- Thorpe, C.T.; Riley, G.P.; Birch, H.L.; Clegg, P.D.; Screen, H.R.C. Fascicles from energy-storing tendons show an age-specific response to cyclic fatigue loading. J R Soc Interface. 2014, 11, 20131058. [Google Scholar] [CrossRef] [PubMed]
- Zuskov, A.; et al. Tendon biomechanics and crimp properties following fatigue loading are influenced by tendon type and age in mice. J Orthop Res. 2020, 54, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Thorpe, C.T.; McDermott, B.T.; Goodship, A.E.; Clegg, P.D.; Birch, H.L. Ageing does not result in a decline in cell synthetic activity in an injury prone tendon. Scand J Med Sci Sports. 2016, 26, 684–693. [Google Scholar] [CrossRef] [PubMed]
- Connizzo, B.K.; Piet, J.M.; Shefelbine, S.J.; Grodzinsky, A.J. Age-associated changes in the response of tendon explants to stress deprivation is sex-dependent. Connect Tissue Res. 2020, 61, 48–62. [Google Scholar] [CrossRef]
- Zhou, Z.; Akinbiyi, T.; Xu, L.; Ramcharan, M.; Leong, D.J.; Ros, S.J.; Colvin, A.C.; Schaffler, M.B.; Majeska, R.J.; Flatow, E.L.; Sun, H.B. Tendon-derived stem/progenitor cell aging: Defective self-renewal and altered fate. Aging Cell. 2010, 9, 911–915. [Google Scholar] [CrossRef]
- Veres, S.P.; Harrison, J.M.; Lee, J.M. Repeated subrupture overload causes progression of nanoscaled discrete plasticity damage in tendon collagen fibrils. J Orthop Res. 2013, 31, 731–737. [Google Scholar] [CrossRef]
- Egerbacher, M.; Arnoczky, S.P.; Caballero, O.; Lavagnino, M.; Gardner, K.L. Loss of homeostatic tension induces apoptosis in tendon cells: An in vitro study. Clin Orthop Relat Res. 2008, 466, 1562–1568. [Google Scholar] [CrossRef]
- Thorpe, C.T.; et al. Tendon overload results in alterations in cell shape and increased markers of inflammation and matrix degradation. Scand J Med Sci Sports. 2015, 25, e381–e391. [Google Scholar] [CrossRef]
- Lavagnino, M.; Arnoczky, S.P.; Gardner, K.L. In situ deflection of tendon cell-cilia in response to tensile loading: An in vitro study. J Orthop Res. 2011, 29, 925–930. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, E.; Ascenzi, M.-G.; Farnum, C. Primary cilia are highly oriented with respect to collagen direction and long axis of extensor tendon. J Orthop Res. 2009, 28. [Google Scholar] [CrossRef]
- Gardner, K.; Arnoczky, S.P.; Lavagnino, M. Effect of in vitro stress-deprivation and cyclic loading on the length of tendon cell cilia in situ. J Orthop Res. 2011, 29, 582–587. [Google Scholar] [CrossRef] [PubMed]
- Rowson, D.T.; Shelton, J.C.; Screen, H.R.C.; Knight, M.M. Mechanical loading induces primary cilia disassembly in tendon cells via TGFβ and HDAC6. Sci Rep. 2018, 8, 11107. [Google Scholar] [CrossRef] [PubMed]
- Rowson, D.; Knight, M.M.; Screen, H.R.C. Zonal variation in primary cilia elongation correlates with localized biomechanical degradation in stress deprived tendon. J Orthop Res. 2016, 34, 2146–2153. [Google Scholar] [CrossRef] [PubMed]
- Fang, F.; Schwartz, A.G.; Moore, E.R.; Sup, M.E.; Thomopoulos, S. Primary cilia as the nexus of biophysical and hedgehog signaling at the tendon enthesis. Sci Adv. 2020, 6, eabc1799. [Google Scholar] [CrossRef]
- Cohen, M.; Kam, Z.; Addadi, L.; Geiger, B. Dynamic study of the transition from hyaluronan- to integrin-mediated adhesion in chondrocytes. EMBO J. 2006, 25, 302–311. [Google Scholar] [CrossRef]
- Chopra, A.; Murray, M.E.; Byfield, F.J.; Mendez, M.G.; Halleluyan, R.; Restle, D.J.; Raz-Ben Aroush, D.; Galie, P.A.; Pogoda, K.; Bucki, R.; et al. Augmentation of integrin-mediated mechanotransduction by hyaluronic acid. Biomaterials. 2014, 35, 71–82. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.M.; Thomas, C.E.; Birk, D.E. Pericellular proteins of the developing mouse tendon: A proteomic analysis. Connect Tissue Res. 2012, 53, 2–13. [Google Scholar] [CrossRef]
- Docheva, D.; Popov, C.; Alberton, P.; Aszodi, A. Integrin signaling in skeletal development and function. Birth Defects Res Part C Embryo Today Rev. 2014, 102, 13–36. [Google Scholar] [CrossRef]
- Wang, D.; Pun, C.C.M.; Huang, S.; Tang, T.C.M.; Ho, K.K.W.; Rothrauff, B.B.; Yung, P.S.H.; Blocki, A.M.; Ker, E.D.F.; Tuan, R.S. Tendon-derived extracellular matrix induces mesenchymal stem cell tenogenesis via an integrin/transforming growth factor-β crosstalk-mediated mechanism. FASEB J. 2020, 34, 8172–8186. [Google Scholar] [CrossRef]
- Mousavizadeh, R.; Hojabrpour, P.; Eltit, F.; McDonald, P.C.; Dedhar, S.; McCormack, R.G.; Duronio, V.; Jafarnejad, S.M.; Scott, A. β1 integrin, ILK and mTOR regulate collagen synthesis in mechanically loaded tendon cells. Sci Rep. 2020, 10. [Google Scholar] [CrossRef]
- Popov, C.; Burggraf, M.; Kreja, L.; Ignatius, A.; Schieker, M.; Docheva, D. Mechanical stimulation of human tendon stem/progenitor cells results in upregulation of matrix proteins, integrins and MMPs, and activation of p38 and ERK1/2 kinases. BMC Mol Biol. 2015, 16, 6. [Google Scholar] [CrossRef]
- Hannafin, J.A.; Attia, E.A.; Henshaw, R.; Warren, R.F.; Bhargava, M.M. Effect of cyclic strain and plating matrix on cell proliferation and integrin expression by ligament fibroblasts. J Orthop Res. 2006, 24, 149–158. [Google Scholar] [CrossRef]
- Kaneko, D.; Sasazaki, Y.; Kikuchi, T.; Ono, T.; Nemoto, K.; Matsumoto, H.; Toyama, Y. Temporal effects of cyclic stretching on distribution and gene expression of integrin and cytoskeleton by ligament fibroblasts in vitro. Connect Tissue Res. 2009, 50, 263–269. [Google Scholar] [CrossRef]
- Bayer, M.L.; Schjerling, P.; Herchenhan, A.; Zeltz, C.; Heinemeier, K.M.; Christensen, L.; Krogsgaard, M.; Gullberg, D.; Kjaer, M. Release of tensile strain on engineered human tendon tissue disturbs cell adhesions, changes matrix architecture, and induces an inflammatory phenotype. PLoS ONE. 2014, 9, e86078. [Google Scholar] [CrossRef]
- Sun, H.B.; Andarawis-Puri, N.; Li, Y.; Fung, D.T.; Lee, J.Y.; Wang, V.M.; Basta-Pljakic, J.; Leong, D.J.; Sereysky, J.B.; Ros, S.J.; Klug, R.A.; Braman, J.; Schaffler, M.B.; Jepsen, K.J.; Flatow, E.L. Cycle-dependent matrix remodeling gene expression response in fatigue-loaded rat patellar tendons. J Orthop Res. 2010, 28, 1380–1386. [Google Scholar] [CrossRef]
- McNeilly, C.M.; Banes, A.J.; Benjamin, M.; Ralphs, J.R. Tendon cells in vivo form a three-dimensional network of cell processes linked by gap junctions. J Anat. 1996, 189, 593–600. [Google Scholar] [CrossRef]
- Waggett, A.D.; Benjamin, M.; Ralphs, J.R. Connexin 32 and 43 gap junctions differentially modulate tenocyte response to cyclic mechanical load. Eur J Cell Biol. 2006, 85, 1145–1154. [Google Scholar] [CrossRef]
- Shen, H.; Schwartz, A.G.; Civitelli, R.; Thomopoulos, S. Connexin 43 is necessary for murine tendon enthesis formation and response to loading. J Bone Miner Res. 2020, 35, 4018. [Google Scholar] [CrossRef]
- Foolen, J.; Wunderli, S.L.; Loerakker, S.; Snedeker, J.G. Tissue alignment enhances remodeling potential of tendon-derived cells—Lessons from a novel microtissue model of tendon scarring. Matrix Biol. 2018, 65, 14–29. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Tytell, J.D.; Ingber, D.E. Mechanotransduction at a distance: Mechanically coupling the extracellular matrix with the nucleus. Nat Rev Mol Cell Biol. 2009, 10, 75–82. [Google Scholar] [CrossRef]
- Lavagnino, M.; Arnoczky, S.P. In vitro alterations in cytoskeletal tensional homeostasis control gene expression in tendon cells. J Orthop Res. 2005, 23, 1211–1218. [Google Scholar] [CrossRef] [PubMed]
- Lavagnino, M.; Gardner, K.L.; Arnoczky, S.P. High magnitude, in vitro, biaxial, cyclic tensile strain induces actin depolymerization in tendon cells. Muscles Ligaments Tendons J. 2015, 5, 124–128. [Google Scholar] [CrossRef] [PubMed]
- Lavagnino, M.; Brooks, A.E.; Oslapas, A.N.; Gardner, K.L.; Arnoczky, S.P. Crimp length decreases in lax tendons due to cytoskeletal tension, but is restored with tensional homeostasis. J Orthop Res. 2017, 35, 573–579. [Google Scholar] [CrossRef]
- Lavagnino, M.; Gardner, K.; Arnoczky, S.P. Age-related changes in the cellular, mechanical, and contractile properties of rat tail tendons. Connect Tissue Res. 2013, 54, 70–75. [Google Scholar] [CrossRef]
- Lavagnino, M.; Bedi, A.; Walsh, C.P.; Sibilsky Enselman, E.R.; Sheibani-Rad, S.; Arnoczky, S.P. Tendon contraction after cyclic elongation is an age-dependent phenomenon: In vitro and in vivo comparisons. Am J Sports Med. 2014, 42, 1471–1477. [Google Scholar] [CrossRef]
- Arnoczky, S.P.; Lavagnino, M.; Whallon, J.H.; Hoonjan, A. In situ cell nucleus deformation in tendons under tensile load; a morphological analysis using confocal laser microscopy. J Orthop Res. 2002, 20, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Henderson, J.T.; Shannon, G.; Veress, A.I.; Neu, C.P. Direct measurement of intranuclear strain distributions and RNA synthesis in single cells embedded within native tissue. Biophys J. 2013, 105, 2252–2261. [Google Scholar] [CrossRef] [PubMed]
- Long, H.; Liu, Q.; Yin, H.; Wang, K.; Diao, N.; Zhang, Y.; et al. Prevalence trends of site-specific osteoarthritis from 1990 to 2019: Findings from the Global Burden of Disease Study 2019. Arthritis Rheumatol. 2022, 74, 1172–1183. [Google Scholar] [CrossRef] [PubMed]
- Cieza, A.; Causey, K.; Kamenov, K.; Hanson, S.W.; Chatterji, S.; Vos, T. Global estimates of the need for rehabilitation based on the Global Burden of Disease Study 2019: A systematic analysis for the Global Burden of Disease Study 2019. Lancet 2021, 396, 2006–2017. [Google Scholar] [CrossRef]
- Safiri, S.; Kolahi, A.A.; Smith, E.; Hill, C.; Bettampadi, D.; Mansournia, M.A.; et al. Global, regional and national burden of osteoarthritis 1990-2017: A systematic analysis of the Global Burden of Disease Study 2017. Ann. Rheum. Dis. 2020, 79, 819–828. [Google Scholar] [CrossRef]
- Poole, A.R. Osteoarthritis as a whole joint disease. HSS J. 2012, 8, 4–6. [Google Scholar] [CrossRef]
- Blanco, F.J. Osteoarthritis and atherosclerosis in joint disease. Reumatol. Clin. (Engl Ed) 2018, 14, 251–253. [Google Scholar] [CrossRef] [PubMed]
- Uhlen, M.; Fagerberg, L.; Hallstrom, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; et al. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, M.; Zhang, C.; Méar, L.; Zhong, W.; Digre, A.; Katona, B.; et al. A single-cell type transcriptomics map of human tissues. Sci. Adv. 2021, 7, eabh2169. [Google Scholar] [CrossRef] [PubMed]
- Loeser, R.F.; Goldring, S.R.; Scanzello, C.R.; Goldring, M.B. Osteoarthritis: A disease of the joint as an organ. Arthritis Rheum. 2012, 64, 1697–1707. [Google Scholar] [CrossRef] [PubMed]
- Aebersold, R.; Bader, G.D.; Edwards, A.M.; van Eyk, J.E.; Kussmann, M.; Qin, J.; et al. The biology/disease-driven human proteome project (B/D-HPP): Enabling protein research for the life sciences community. J. Proteome Res. 2013, 12, 23–27. [Google Scholar] [CrossRef]
- Han, Y.; Wennersten, S.A.; Lam, M.P.Y. Working the literature harder: What can text mining and bibliometric analysis reveal? Expert Rev. Proteomics 2019, 16, 871–873. [Google Scholar] [CrossRef]
- Ruiz-Romero, C.; Lam, M.P.Y.; Nilsson, P.; Önnerfjord, P.; Utz, P.J.; Van Eyk, J.E.; et al. Mining the proteome associated with rheumatic and autoimmune diseases. J. Proteome Res. 2019, 18, 4231–4239. [Google Scholar] [CrossRef]
- Kraus, V.B.; Blanco, F.J.; Englund, M.; Henrotin, Y.; Lohmander, L.S.; Losina, E.; et al. OARSI clinical trials recommendations: Soluble biomarker assessments in clinical trials in osteoarthritis. Osteoarthritis Cartilage 2015, 23, 686–697. [Google Scholar] [CrossRef]
- Lau, E.; Venkatraman, V.; Thomas, C.T.; Wu, J.C.; Van Eyk, J.E.; Lam, M.P.Y. Identifying high-priority proteins across the human diseasome using semantic similarity. J. Proteome Res. 2018, 17, 4267–4278. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Gable, A.L.; Nastou, K.C.; Lyon, D.; Kirsch, R.; Pyysalo, S.; et al. The STRING database in 2021: Customizable protein-protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 2021, 49, D605–D612. [Google Scholar] [CrossRef] [PubMed]
- Supek, F.; Bošnjak, M.; Škunca, N.; Šmuc, T. REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS ONE 2011, 6, e21800. [Google Scholar] [CrossRef] [PubMed]
- Thysen, S.; Luyten, F.P.; Lories, R.J. Loss of Frzb and Sfrp1 differentially affects joint homeostasis in instability-induced osteoarthritis. Osteoarthritis Cartilage 2015, 23, 275–279. [Google Scholar] [CrossRef]
- Cai, S.; Zou, Y.; Zhao, H.; Lin, H.; Zheng, D.; Xu, L.; et al. Mechanical stress reduces secreted frizzled-related protein expression and promotes temporomandibular joint osteoarthritis via Wnt/β-catenin signaling. Bone 2022, 161, 116445. [Google Scholar] [CrossRef]
- Claudel, M.; Jouzeau, J.Y.; Cailotto, F. Secreted Frizzled-related proteins (sFRPs) in osteo-articular diseases: Much more than simple antagonists of Wnt signaling? FEBS J. 2019, 286, 4832–4851. [Google Scholar] [CrossRef]
- Wang, R.; Zhu, J.; Dong, X.; Shi, M.; Lu, C.; Springer, T.A. GARP regulates the bioavailability and activation of TGFβ. Mol. Biol. Cell 2012, 23, 1129–1139. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.J.; Chang, W.A.; Wu, L.Y.; Hsu, Y.L.; Chen, C.H.; Kuo, P.L. Systematic analysis of transcriptomic profile of chondrocytes in osteoarthritic knee using next-generation sequencing and bioinformatics. J. Clin. Med. 2018, 7, 535. [Google Scholar] [CrossRef]
- Jayaseelan, V.P.; Arumugam, P. A computational data mining strategy to identify the common genetic markers of temporomandibular joint disorders and osteoarthritis. Glob. Med. Genet. 2022, 9, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Lourido, L.; Calamia, V.; Mateos, J.; Fernández-Puente, P.; Fernández-Tajes, J.; Blanco, F.J.; et al. Quantitative proteomic profiling of human articular cartilage degradation in osteoarthritis. J. Proteome Res. 2014, 13, 6096–6106. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Choi, J.; Hwang, N.S. Regulation of lubricin for functional cartilage tissue regeneration: A review. Biomater. Res. 2018, 22, 9. [Google Scholar] [CrossRef]
- Carballo, C.B.; Nakagawa, Y.; Sekiya, I.; Rodeo, S.A. Basic science of articular cartilage. Clin. Sports Med. 2017, 36, 413–425. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Puente, P.; González-Rodríguez, L.; Calamia, V.; Picchi, F.; Lourido, L.; Camacho-Encina, M.; et al. Analysis of endogenous peptides released from osteoarthritic cartilage unravels novel pathogenic markers. Mol. Cell. Proteomics 2019, 18, 2018–2028. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.J.; Cheng, J.H.; Chou, W.Y.; Hsu, S.L.; Chen, J.H.; Huang, C.Y. Changes of articular cartilage and subchondral bone after extracorporeal shockwave therapy in osteoarthritis of the knee. Int. J. Med. Sci. 2017, 14, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Goldring, S.R.; Goldring, M.B. Changes in the osteochondral unit during osteoarthritis: Structure, function and cartilage-bone crosstalk. Nat. Rev. Rheumatol. 2016, 12, 632–644. [Google Scholar] [CrossRef]
- Funck-Brentano, T.; Cohen-Solal, M. Subchondral bone and osteoarthritis. Curr. Opin. Rheumatol. 2015, 27, 420–426. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, C.; Gabay, C.; Salvat, C.; Henrotin, Y.E.; Berenbaum, F. Mechanical loading highly increases IL-6 production and decreases OPG expression by osteoblasts. Osteoarthritis Cartilage 2009, 17, 473–481. [Google Scholar] [CrossRef]
- Sanchez, C.; Deberg, M.A.; Piccardi, N.; Msika, P.; Reginster, J.Y.; Henrotin, Y.E. Subchondral bone osteoblasts induce phenotypic changes in human osteoarthritic chondrocytes. Osteoarthritis Cartilage 2005, 13, 988–997. [Google Scholar] [CrossRef]
- Yu, D.; Xu, J.; Liu, F.; Wang, X.; Mao, Y.; Zhu, Z. Subchondral bone changes and the impacts on joint pain and articular cartilage degeneration in osteoarthritis. Clin. Exp. Rheumatol. 2016, 34, 929–934. [Google Scholar] [PubMed]
- Chan, P.M.B.; Zhu, L.; Wen, C.Y.; Chiu, K.Y. Subchondral bone proteomics in osteoarthritis: Current status and perspectives. J. Orthop. Translat. 2015, 3, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, C.; Pesesse, L.; Gabay, C.; Delcour, J.P.; Msika, P.; Baudouin, C.; et al. Regulation of subchondral bone osteoblast metabolism by cyclic compression. Arthritis Rheum. 2012, 64, 1193–1203. [Google Scholar] [CrossRef]
- Sanchez, C.; Deberg, M.A.; Piccardi, N.; Msika, P.; Reginster, J.Y.; Henrotin, Y.E. Osteoblasts from the sclerotic subchondral bone downregulate aggrecan but upregulate metalloproteinases expression by chondrocytes. This effect is mimicked by interleukin-6, -1beta and oncostatin M pre-treated non-sclerotic osteoblasts. Osteoarthritis Cartilage 2005, 13, 979–987. [Google Scholar] [CrossRef] [PubMed]
- Chim, S.M.; Tickner, J.; Chow, S.T.; Kuek, V.; Guo, B.; Zhang, G.; et al. Angiogenic factors in bone local environment. Cytokine Growth Factor Rev. 2013, 24, 297–310. [Google Scholar] [CrossRef] [PubMed]
- Creecy, J.G.; Damrath, J.M.; Wallace, J.M. Control of bone matrix properties by osteocytes. Front. Endocrinol. 2020, 11, 578477. [Google Scholar] [CrossRef] [PubMed]
- Sakao, K.; Takahashi, K.A.; Arai, Y.; Saito, M.; Honjyo, K.; Hiraoka, N.; et al. Asporin and transforming growth factor-beta gene expression in osteoblasts from subchondral bone and osteophytes in osteoarthritis. J. Orthop. Sci. 2009, 14, 738–747. [Google Scholar] [CrossRef]
- MacDonald, I.J.; Liu, S.C.; Su, C.M.; Wang, Y.H.; Tsai, C.H.; Tang, C.H. Implications of angiogenesis involvement in arthritis. Int. J. Mol. Sci. 2018, 19, 2012. [Google Scholar] [CrossRef] [PubMed]
- Mishra, A.; Awasthi, S.; Raj, S.; Mishra, P.; Srivastava, R.N. Identifying the role of ASPN and COMP genes in knee osteoarthritis development. J. Orthop. Surg. Res. 2019, 14, 337. [Google Scholar] [CrossRef] [PubMed]
- Mathiessen, A.; Conaghan, P.G. Synovitis in osteoarthritis: Current understanding with therapeutic implications. Arthritis Res. Ther. 2017, 19, 18. [Google Scholar] [CrossRef]
- Orr, C.; Vieira-Sousa, E.; Boyle, D.L.; Buch, M.H.; Buckley, C.D.; Cañete, J.D.; et al. Synovial tissue research: A state-of-the-art review. Nat. Rev. Rheumatol. 2017, 13, 463–475. [Google Scholar] [CrossRef]
- Kemble, S.; Croft, A.P. Critical role of synovial tissue-resident macrophage and fibroblast subsets in the persistence of joint inflammation. Front. Immunol. 2021, 12, 715894. [Google Scholar] [CrossRef]
- Wehling, P.; Evans, C.; Wehling, J.; Maixner, W. Effectiveness of intra-articular therapies in osteoarthritis: A literature review. Ther. Adv. Musculoskelet. Dis. 2017, 9, 183–196. [Google Scholar] [CrossRef]
- Lambert, C.; Dubuc, J.E.; Montell, E.; Vergés, J.; Munaut, C.; Noël, A.; et al. Gene expression pattern of cells from inflamed and normal areas of osteoarthritis synovial membrane. Arthritis Rheumatol. 2014, 66, 960–968. [Google Scholar] [CrossRef] [PubMed]
- Plsikova Matejova, J.; Spakova, T.; Harvanova, D.; Lacko, M.; Filip, V.; Sepitka, R.; et al. A preliminary study of combined detection of COMP, TIMP-1, and MMP-3 in synovial fluid: Potential indicators of osteoarthritis progression. Cartilage 2020, 13, 1421S–1430S. [Google Scholar] [CrossRef] [PubMed]
- Ravalli, S.; Szychlinska, M.A.; Lauretta, G.; Di Rosa, M.; Musumeci, G. Investigating lubricin and known cartilage-based biomarkers of osteoarthritis. Expert Rev. Mol. Diagn. 2020, 20, 443–452. [Google Scholar] [CrossRef]
- Qadri, M.; Jay, G.D.; Zhang, L.X.; Schmidt, T.A.; Totonchy, K.A.; Elsaid, K.A. Proteoglycan-4 is an essential regulator of synovial macrophage polarization and inflammatory macrophage joint infiltration. Arthritis Res. Ther. 2021, 23, 241. [Google Scholar] [CrossRef] [PubMed]
- Mucke, J.; Hoyer, A.; Brinks, R.; Bleck, E.; Pauly, T.; Schneider, M.; et al. Inhomogeneity of immune cell composition in the synovial sublining: Linear mixed modelling indicates differences in distribution and spatial decline of CD68+ macrophages in osteoarthritis and rheumatoid arthritis. Arthritis Res. Ther. 2016, 18, 170. [Google Scholar] [CrossRef]
- Tamer, T.M. Hyaluronan and synovial joint: Function, distribution and healing. Interdiscip. Toxicol. 2013, 6, 111–125. [Google Scholar] [CrossRef] [PubMed]
- Posey, K.L.; Coustry, F.; Hecht, J.T. Cartilage oligomeric matrix protein: COMPopathies and beyond. Matrix Biol. 2018, 71–72, 161–173. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, D.J. Serpins in cartilage and osteoarthritis: What do we know? Biochem. Soc. Trans. 2021, 49, 1013–1026. [Google Scholar] [CrossRef]
- Wilkinson, D.J.; Falconer, A.M.D.; Wright, H.L.; Lin, H.; Yamamoto, K.; Cheung, K.; et al. Matrix metalloproteinase-13 is fully activated by neutrophil elastase and inactivates its serpin inhibitor, alpha-1 antitrypsin: Implications for osteoarthritis. FEBS J. 2021, 289, 121–139. [Google Scholar] [CrossRef]
- Heit, C.; Jackson, B.C.; McAndrews, M.; Wright, M.W.; Thompson, D.C.; Silverman, G.A.; et al. Update of the human and mouse SERPIN gene superfamily. Hum. Genomics 2013, 7, 22. [Google Scholar] [CrossRef]
- Fox, A.J.; Bedi, A.; Rodeo, S.A. The basic science of human knee menisci: Structure, composition, and function. Sports Health 2012, 4, 340–351. [Google Scholar] [CrossRef] [PubMed]
- Englund, M.; Roemer, F.W.; Hayashi, D.; Crema, M.D.; Guermazi, A. Meniscus pathology, osteoarthritis and the treatment controversy. Nat. Rev. Rheumatol. 2012, 8, 412–419. [Google Scholar] [CrossRef] [PubMed]
- Folkesson, E.; Turkiewicz, A.; Ali, N.; Rydén, M.; Hughes, H.V.; Tjörnstrand, J.; et al. Proteomic comparison of osteoarthritic and reference human menisci using data-independent acquisition mass spectrometry. Osteoarthritis Cartilage 2020, 28, 1092–1101. [Google Scholar] [CrossRef]
- Sun, Y.; Mauerhan, D.R. Meniscal calcification, pathogenesis and implications. Curr. Opin. Rheumatol. 2012, 24, 152–157. [Google Scholar] [CrossRef]
- Kiraly, A.J.; Roberts, A.; Cox, M.; Mauerhan, D.; Hanley, E.; Sun, Y. Comparison of meniscal cell-mediated and chondrocyte-mediated calcification. Open Orthop. J. 2017, 11, 225–233. [Google Scholar] [CrossRef]
- Schulze-Tanzil, G. Intraarticular ligament degeneration is interrelated with cartilage and bone destruction in osteoarthritis. Cells 2019, 8, 990. [Google Scholar] [CrossRef] [PubMed]
- Harkey, M.S.; Luc, B.A.; Golightly, Y.M.; Thomas, A.C.; Driban, J.B.; Hackney, A.C.; et al. Osteoarthritis-related biomarkers following anterior cruciate ligament injury and reconstruction: A systematic review. Osteoarthritis Cartilage 2015, 23, 1–12. [Google Scholar] [CrossRef]
- Yoshida, H.; Kojima, T.; Kurokouchi, S.; Takahashi, S.; Hanamura, H.; Kojima, M.; et al. Relationship between pre-radiographic cartilage damage following anterior cruciate ligament injury and biomarkers of cartilage turnover in clinical practice: A cross-sectional observational study. Osteoarthritis Cartilage 2013, 21, 831–838. [Google Scholar] [CrossRef]
- Tang, N.; Zhang, W.; George, D.M.; Su, Y.; Huang, T. The Top 100 most cited articles on anterior cruciate ligament reconstruction: A bibliometric analysis. Orthop. J. Sports Med. 2021, 9, 2325967120976372. [Google Scholar] [CrossRef]
- Zhang, D.; Cheriyan, T.; Martin, S.D.; Gomoll, A.H.; Schmid, T.M.; Spector, M. Lubricin distribution in the torn human anterior cruciate ligament and meniscus. J. Orthop. Res. 2011, 29, 1916–1922. [Google Scholar] [CrossRef] [PubMed]
- Jay, G.D.; Fleming, B.C.; Watkins, B.A.; McHugh, K.A.; Anderson, S.C.; Zhang, L.X.; et al. Prevention of cartilage degeneration and restoration of chondroprotection by lubricin tribosupplementation in the rat following anterior cruciate ligament transection. Arthritis Rheum. 2010, 62, 2382–2391. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhang, W. The role of HOPX in normal tissues and tumor progression. Biosci. Rep. 2020, 40, BSR20191953. [Google Scholar] [CrossRef] [PubMed]
- Tarka, M.C.; Davey, A.; Lonza, G.C.; O’Brien, C.M.; Delaney, J.P.; Endres, N.K. Alpine ski racing injuries. Sports Health 2019, 11, 265–271. [Google Scholar] [CrossRef]
- Chin, B.Z.; Wee, I.J.Y.; Syn, N.L.; Krishna, L. Arthroscopic anterior cruciate ligament reconstruction: A meta-analysis comparing semitendinosus alone and semitendinosus with gracilis tendon autografts. J. Knee Surg. 2019, 32, 796–803. [Google Scholar] [CrossRef] [PubMed]
- Lotz, M.; Martel-Pelletier, J.; Christiansen, C.; Brandi, M.L.; Bruyère, O.; Chapurlat, R.; et al. Value of biomarkers in osteoarthritis: Current status and perspectives. Ann. Rheum. Dis. 2013, 72, 1756–1763. [Google Scholar] [CrossRef] [PubMed]
- Styrkarsdottir, U.; Lund, S.H.; Thorleifsson, S.; Saevarsdottir, S.; Gudbjartsson, D.F.; Thorsteinsdottir, U.; et al. The CRTAC1 protein in plasma associates with prevalent osteoarthritis and predicts future risk as well as progression to joint replacements—Results from the UK Biobank resource. Arthritis Rheumatol. 2022, 75, 544–552. [Google Scholar] [CrossRef]
- Valdes, A.M.; Loughlin, J.; Oene, M.V.; Chapman, K.; Surdulescu, G.L.; Doherty, M.; et al. Sex and ethnic differences in the association of ASPN, CALM1, COL2A1, COMP, and FRZB with genetic susceptibility to osteoarthritis of the knee. Arthritis Rheum. 2007, 56, 137–146. [Google Scholar] [CrossRef] [PubMed]
- King, K.B.; Lindsey, C.T.; Dunn, T.C.; Ries, M.D.; Steinbach, L.S.; Majumdar, S. A study of the relationship between molecular biomarkers of joint degeneration and the magnetic resonance-measured characteristics of cartilage in 16 symptomatic knees. Magn. Reson. Imaging 2004, 22, 1117–1123. [Google Scholar] [CrossRef] [PubMed]
- Liem, Y.; Judge, A.; Kirwan, J.; Ourradi, K.; Li, Y.; Sharif, M. Multivariable logistic and linear regression models for identification of clinically useful biomarkers for osteoarthritis. Sci. Rep. 2020, 10, 11328. [Google Scholar] [CrossRef] [PubMed]
- Arden, N.; Richette, P.; Cooper, C.; Bruyère, O.; Abadie, E.; Branco, J.; et al. Can we identify patients with high risk of osteoarthritis progression who will respond to treatment? A focus on biomarkers and frailty. Drugs Aging 2015, 32, 525–535. [Google Scholar] [CrossRef]
- Kraus, V.B.; McDaniel, G.; Huebner, J.L.; Stabler, T.V.; Pieper, C.F.; Shipes, S.W.; et al. Direct in vivo evidence of activated macrophages in human osteoarthritis. Osteoarthritis Cartilage 2016, 24, 1613–1621. [Google Scholar] [CrossRef] [PubMed]
- Yao, Q.; Wu, X.; Tao, C.; Gong, W.; Chen, M.; Qu, M.; et al. Osteoarthritis: Pathogenic signaling pathways and therapeutic targets. Signal Transduct. Target. Ther. 2023, 8, 56. [Google Scholar] [CrossRef] [PubMed]
- Teeple, E.; Elsaid, K.A.; Jay, G.D.; Zhang, L.; Badger, G.J.; Akelman, M.; et al. Effects of supplemental intra-articular lubricin and hyaluronic acid on the progression of posttraumatic arthritis in the anterior cruciate ligament-deficient rat knee. Am. J. Sports Med. 2011, 39, 164–172. [Google Scholar] [CrossRef] [PubMed]
- Watkins, A.R.; Reesink, H.L. Lubricin in experimental and naturally occurring osteoarthritis: A systematic review. Osteoarthritis Cartilage 2020, 28, 1303–1315. [Google Scholar] [CrossRef] [PubMed]
- Teerlink, C.C.; Jurynec, M.J.; Hernandez, R.; Stevens, J.; Hughes, D.C.; Brunker, C.P.; et al. A role for the MEGF6 gene in predisposition to osteoporosis. Ann. Hum. Genet. 2021, 85, 58–72. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Tan, L.J.; Wang, P.; Chen, X.D.; Zhu, L.H.; Zeng, Q.; et al. Functional relevance for associations between osteoporosis and genetic variants. PLoS ONE 2017, 12, e0169926. [Google Scholar] [CrossRef]
- Glyn-Jones, S.; Palmer, A.J.R.; Agricola, R.; Price, A.J.; Vincent, T.L.; Weinans, H.; et al. Osteoarthritis. Lancet 2015, 386, 376–387. [Google Scholar] [CrossRef]
- Abramson, S.B.; Attur, M. Developments in the scientific understanding of osteoarthritis. Osteoarthritis Cartilage 2009, 17, 1610–1615. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



