
Submitted:
02 August 2024
Posted:
02 August 2024
You are already at the latest version
Abstract
This review explores the challenges and emerging trends in pancreatic cancer therapy. In particular, we focus on the tumor microenvironment and the potential of immunotherapy in pancreatic cancer. We discuss novel strategies targeting the desmoplastic barrier and immunosuppressive cells to enhance immune cell infiltration and activation. Recent clinical trials, particularly those involving novel immunotherapeutic agents and tumor vaccines, are examined to understand their efficacy and limitations. This review aims to highlight the ongoing efforts to refine immunotherapy approaches for better patient outcomes.
Keywords:
Pancreatic cancer
; Cancer trials
; Immunotherapy
; Vaccine
Introduction
Pancreatic cancer, currently ranked as the tenth most common cancer in the United States, is predicted to become the second leading cause of cancer death by 2030 [1]. The typical progression of the disease involves a transition from acinar cell alterations to ductal metaplasia, advancing to pancreatic intraepithelial neoplasia and culminating in pancreatic ductal adenocarcinoma (PDAC) [2]. Nearly all pancreatic cancer originates in the exocrine cells rather than the endocrine cells. Currently, the gold standard treatment includes chemotherapy, such as gemcitabine or FOLFIRINOX, a combination of chemotherapy drugs: fluorouracil, irinotecan, and oxaliplatin. However, treatment efficacy is hindered by therapy-associated toxicities and the development of resistance to chemotherapy [3,4]. Moreover, the complex tumor microenvironment characteristic of PDAC makes treatment responses unpredictable and complicates the development of new therapies [5]. Addressing these challenges is crucial for enhancing the efficacy of systemic therapies and improving overall patient outcomes.
As the field of targeted therapy continues to evolve, there is an increased focus on a new generation of treatments, with immunotherapy playing a significant role [6]. Agents such as immune checkpoint inhibitors have shown remarkable efficacy in tumors with a high mutation burden [7]. Yet, the response rate of immunotherapy in PDAC patients remains disappointingly low. This issue is in part due to the dense stromal architecture of the disease, which acts as a physical and functional barrier to immune cell infiltration and activity, and partly because the tumor microenvironment (TME) is enriched with immunosuppressive cells [8,9,10].
These issues necessitate a deeper understanding of the PDAC TME to enhance the delivery and efficacy of immunotherapies. In this review, we examine the emerging body of immunotherapeutic interventions in PDAC, focusing on recent clinical trials. We analyze the shortcomings of recent studies and offer insights and potential directions to refine immunotherapy strategies for PDAC.
Tumor Microenvironment of PDAC
Understanding the tumor microenvironment (TME) and identifying tumor-intrinsic and -extrinsic factors are critical for advancing immunotherapy in PDAC. The TME, consisting of cancer cells, stromal cells, immune cells, and the extracellular matrix, plays a pivotal role in tumor progression and therapeutic response [11,12]. Tumor intrinsic factors, such as genetic mutations and aberrant signaling pathways, drive cancer proliferation and resistance to therapy. Conversely, tumor extrinsic factors, including immune cell infiltration, cytokine milieu, and angiogenesis, influence the efficacy of immunotherapies. In particular, PDAC TME is marked by a dense population of immunosuppressive cells such as regulatory T cells (Tregs), myeloid-derived suppressor cells, and tumor-associated macrophages (TAMs) [13]. These cells release inhibitory cytokines, like IL-10 and TGF-β, that dampen the anti-tumor immune response [14]. Additionally, PDAC often presents fewer tumor-specific mutations compared to cancers like melanoma [15]. This results in fewer neoantigens that the immune system can recognize as "foreign," making it difficult for immune cells to target [16,17]. In addition, tumors often upregulate immune checkpoint proteins, such as PD-L1, on their surfaces [18,19]. When these proteins bind to receptors on immune cells (like PD-1 on T cells), they send an inhibitory signal, preventing the immune cell from attacking the tumor.
PDAC tumors also can alter the metabolic landscape of the TME, creating unfavorable conditions for immune cell function. For instance, they can induce hypoxia or increase the production of immunosuppressive metabolites like adenosine [20,21,22]. PDAC tumors can also reduce the expression of major histocompatibility complex (MHC) class I molecules [23,24]. These molecules are essential for presenting tumor antigens to immune cells. Without them, T cells cannot recognize and target tumor cells effectively [25]. Some PDAC tumors can also release tumor-intrinsic factors that induce apoptosis in effector immune cells, further reducing the body's ability to mount a defense against the tumor [26,27]. Together, these features create a PDAC TME that is mostly immunosuppressive and makes immunotherapy less effective. By understanding these components, we can develop strategies to modulate the TME, overcoming barriers to immune cell infiltration and activation, thereby enhancing the efficacy of immunotherapeutic approaches in PDAC.
Targeting the Desmoplastic Barrier in PDAC
The dense desmoplastic stroma in PDAC fosters an immunosuppressive tumor microenvironment. Therefore, targeting stromal components such as cancer-associated fibroblasts (CAFs) and the signaling pathways facilitating stromal proliferation offers a promising strategy to mitigate this obstruction and enhance immune cell infiltration and activation. Claudins are key constituents of tight junctions, critical structures that regulate the passage of molecules between cells [28,29]. As such, they preserve tissue integrity by establishing barriers between neighboring cells [30]. Two isoforms of CLDN18, CLDN18.2, and CLDN18.1, have distinct localization patterns in specific tissue types [31]. CLDN18.2 is the dominant isoform in normal gastric tissue and is often retained during malignant transformation [32]. This isoform is highly expressed in PDAC, whereas its expression in healthy tissues is limited to the stomach mucosa's differentiated epithelial cells. It is presumed to be a potential biomarker and target for immunotherapy for PDAC [33]. A recent open-label, multi-site, phase I/IIa clinical trial (NCT04683939) investigating the efficacy of targeting claudin 18.2 (CLDN18.2), using BNT141, a lipid nanoparticle-encapsulated RNA-based therapy containing two pseudouridine-modified mRNAs that code for monoclonal IgG antibodies against CLDN18.2 [34,35,36] is currently recruiting. The antibody generated by BNT141 is identical in sequence to zolbetuximab (IMAB362), a CLDN18.2-targeted antibody that demonstrated benefit as an adjunct to chemotherapy in gastric cancers [36].The fundamental concept of the trial is that anti-CLDN18.2 antibodies encoded by the BNT141 mRNA circulate within the body and selectively bind to cancer cells that exhibit high levels of CLDN18.2 expression and induce tumor cell death predominantly through mechanisms such as antibody-dependent cellular cytotoxicity (ADCC) and complement-dependent cytotoxicity (CDC). A challenge inherent in this therapy is that single-stranded RNA entering dendritic cells may bind to toll-like receptor 7 (TLR7), triggering the release of type I interferons [37,38]. These interferons may promote cancer immune subversion by driving the body toward an immunosuppressive state. This occurs by stimulating increased expression of indoleamine 2,3-dioxygenase (IDO) and interleukin-10 (IL-10), among other anti-inflammatory mediators, promoting a tumor microenvironment conducive to cancer survival [39,40]. In addition to fostering an immune subversive TME, the BNT141 antibody can have toxic side effects due to binding to healthy gastric epithelia. The resultant loss of epithelia may lead to gastrointestinal side effects. Indeed, a multicenter clinical trial reported that up to 6% of participants experienced treatment-related mild adverse events (mainly gastrointestinal toxicities) [41].
Neutralizing Immunosuppressive Cells
Targeting immunosuppressive cells like Tregs, myeloid-derived suppressor cells (MDSCs), and TAMs offers a pathway to restore anti-tumor immunity [42,43,44]. Exploiting this approach can potentially unlock significant clinical benefits in PDAC patients [45]. For example, agents that deplete MDSCs or inhibit their function can shift the balance toward an immune-promoting environment [46,47,48]. For example, TAMs are known to express leukemia inhibitory factor (LIF), which promotes tumor progression, inflammation, and therapeutic resistance by creating an immunosuppressive TME by inducing the differentiation of Tregs and enhancing the production of other immunosuppressive cytokines [49,50,51]. AZD0171 (also known as MSC-1), a humanized monoclonal antibody targeting LIF with high affinity, induces phenotypic and functional changes in TAMs [52]. These changes promote anti-tumor inflammation and inhibit the proliferation and metabolism of PDAC stem cells, leading to a reduction in tumor growth. This dual targeting, along with new LIF-targeting therapeutic strategies, is currently being evaluated in a clinical trial [NCT04999969]. This trial aims to explore the safety, pharmacokinetics, and overall efficacy of AZD0171 in combination with durvalumab and chemotherapies (gemcitabine and nab-paclitaxel) in patients with metastatic solid cancers. A potential clinical side effect of AZD0171 treatment could be due to its targeting of LIF, which can suppress hematopoietic stem cells. These stem cells depend on LIF for their proliferation, and interference with this pathway may adversely affect their function [53].
Overcoming Metabolic Barriers
Another strategy to improve immunotherapy response in PDAC patients could be to target metabolic byproducts, as several metabolic byproducts induce immune cell exhaustion and foster a pro-tumor environment [54,55,56]. One such byproduct, CD73, catalyzes the conversion of extracellular ATP/ADP into free adenosine, which then binds to the A2A and A2B receptors, promoting immunosuppression [57,58]. Inhibition of CD73 or targeting A2A/A2B receptors to block adenosine receptors may counteract immunosuppression and mitigate the establishment of an immunologically “cold” TME. Efforts to target these pathways are currently underway in multiple clinical trials. One such approach involves the small molecule inhibitor INCB106385, a potent antagonist of A2A/A2B, currently under investigation as a monotherapy or in combination with retifanlimab (INCMGA00012), a humanized monoclonal antibody targeting (PD-1) [NCT04580485] [59,60]. As an eligibility criterion, the TME of participants in this clinical trial must have CD8+ infiltrating T cells.
While targeting A2A/A2B can prevent adenosine-mediated immunosuppression, CD73 is mainly responsible for the production of free extracellular adenosine [61]. Therefore, clinical trials targeting both A2A/A2B and CD73 [NCT04989387] could be effective. The drug used to target CD73, INCA00186, is a monoclonal antibody that binds CD73 and inhibits the production of free adenosine [62]. A potential challenge in targeting the adenosine production pathway is side effects such as decreased vasodilation, which could impair lymphocyte migration and infiltration into the TME, and tachycardia as adenosine slows the heart rate [63,64]. Retifanlimab may counteract these effects directly by alleviating lymphocyte exhaustion through cell-to-cell interaction or/and indirectly by inhibiting the synthesis of adenosine and blocking the receptors that mediate immunosuppression. The dual targeting of CD73 and PD-1 plus established chemotherapeutic regimens like mFOLFIRINOX is currently being evaluated in a clinical trial [NCT05688215] for PDAC. Though CD73 blockade increases the efficacy of PD-1 blockade and reduces levels of IL-1β in the TME, the challenge remains that antibody-based immunotherapeutic such as retifanlimab must first pass through the desmoplastic stroma to reach their targets [65].
Several cytokines, including the immunosuppressive cytokine TGFβ, can collaborate with adenosine to further enhance the immunosuppression [66]. Therefore, targeting TGFβ with CD73 could significantly improve treatment outcomes. Based on that premise, a phase II trial [NCT05632328] that employs AGEN1423, a humanized bifunctional antibody targeting both CD73 and TGFβ, alongside balstilimab, a PD-1 inhibitor, is currently ongoing [67]. This dual targeting system allows the TME to be transformed from an immunologically “cold” state to a more active immunologically “hot” condition. One potential caveat of this trial is that treatment with AGEN1423 is associated with increased soluble CD73 (sCD73) levels. This may cause a systemic reduction of tissue-resident memory T cells, which are crucial for long-term immune protection against recurrent infections and cancer surveillance.
Blocking Immune Checkpoints
Immune checkpoint proteins depend on cell-to-cell interactions that inhibit effector functions of immune cells [68,69]. The TME often upregulates immune checkpoint ligands like PD-L1, inhibiting T cell functions [18,70]. Checkpoint inhibitors that block PD-1/PD-L1 or CTLA-4 interactions can reinvigorate exhausted T cells and allow tumor recognition, restoring their cytotoxic functions [71,72,73,74]. Utilizing antibodies that block this inhibitory signal has shown encouraging results in various cancers [75,76,77]. For example, PD-1 antibodies have been investigated as a monotherapy or in combination with various regimens in PDAC patients [78,79]. In one phase II clinical trial [NCT05562297], PD-1 blockade through sintilimab, a human IgG monoclonal antibody against PD-1, is being combined with DNA-damaging agent gemcitabine alongside nab-paclitaxel [80]. As blocking PD-1 can prevent immune cell inhibition, combining such therapy with a DNA-damaging agent such as gemcitabine can maximize the efficacy of the immune response activated by the increased neoantigen load from the DNA-damaging agents [81,82].
Similarly, in another phase II clinical trial [NCT04753879], PD-1 blockade (pembrolizumab) is being combined with a PARP inhibitor (olaparib), which blocks DNA repair, alongside a gemcitabine, nab-paclitaxel, capecitabine, cisplatin, and irinotecan (GAX-CI) regimen [83,84]. Combining PD-1 blockade and PARP inhibition alongside DNA-damaging therapeutic agents aims to significantly enhance the immune response against the increased production of neoantigens formed by the PDAC. Currently, this treatment strategy is being evaluated in PDAC patients who carry germline BRCA mutations (9% of all PDAC cases).
PD-1 blockade is also being investigated in a phase II trial [NCT04887805] through combination with the receptor tyrosine kinase (RTK) inhibitor lenvatinib for PDAC patients. Lenvatinib is a well-established multiple kinase inhibitor that has seen success across cancers [85,86,87]. As a multiple kinase inhibitor, lenvatinib inhibits vascular endothelial growth factor receptors (VEGFR1, VEGFR2, and VEGFR3) and fibroblast growth factor receptors (FGFR1-4). By blocking these pathways, lenvatinib reduces the formation of new blood vessels that supply tumors, thereby starving the tumor of nutrients and oxygen necessary for growth and survival. Lenvatinib also targets additional RTKs involved in tumor proliferation, such as platelet-derived growth factor receptor (PDGFR), c-KIT, and RET. By inhibiting these receptors, lenvatinib can directly reduce tumor cell proliferation and induce apoptosis. Combining PD-1 blockade with lenvatinib can have promising therapeutic outcomes by halting tumor progression alongside an enhanced immune response. Only patients with unresectable disease are eligible in this trial, and the treatment strategy is designed primarily as maintenance therapy. In addition, CTLA-4, a critical immune checkpoint protein found in T cells, is being explored in clinical trials for PDAC patients. CTLA-4 interacts with its ligands (CD80/CD86) to induce T cell exhaustion [88,89]. A novel Fc-enhanced CTLA-4 antibody, botensilimab, is currently under investigation alongside nab-paclitaxel and gemcitabine as a potent immunotherapeutic strategy [NCT05630183]. The Fc-enhanced modification of the CTLA-4 antibody increases T cell priming within the tumor microenvironment and elicits a strong response in the peritumoral lymph nodes, which become enriched with Treg CTLA-4+ cells [90]. Although CTLA-4 levels are typically low in PDAC patients, prior trials targeting CTLA-4 have shown some benefit in individuals with impaired mismatch repair (MMR) systems, which are directly responsible for error correction during DNA replication [91]. Impaired MMR increases quantities of neoantigens and thus favors cancer cells' sensitivity to immunotherapy by enhancing the expression of inflammatory cytokines and T cell activation [92].
Currently, CTLA-4 is also under investigation in combination with NLM-001, an inhibitor of the sonic hedgehog (SHH) pathway [NCT04827953] [93]. Tumor-derived SHH activates CAFs and increases extracellular matrix deposition within the desmoplastic stroma [94,95,96]. Inhibiting the SHH pathway in PDAC disrupts tumor growth and modifies the composition of CAFs, increasing inflammatory CAFs and a decrease in myofibroblastic CAFs. However, SHH pathway inhibition can also decrease cytotoxic T lymphocytes within the tumor, alongside an increase in CD4+ T helper cells, resulting in no net change in overall CD3+ T cell infiltration [97].
Expanding on novel approaches in immune checkpoint blockade in treating pancreatic cancers, a phase 2 trial [NCT02866383] introduced a novel therapeutic strategy for treating refractory metastatic PDAC. This strategy combines stereotactic body radiation therapy (SBRT) with checkpoint inhibitors nivolumab and ipilimumab, under the study name Checkpoint Inhibition in Pancreatic Adenocarcinoma (CheckPAC). Nivolumab binds to PD-1 and blocks its interactions with PD-L1 and PD-L2, allowing T cells to remain activated and target cancer cells more effectively [98]. On the other hand, ipilimumab binds to CTLA-4 and prevents its interactions with CD80/CD86 on antigen-presenting cells, allowing for enhanced activation and function of T cells [99]. Because nivolumab and ipilimumab target different immune checkpoints, they are often combined for a synergistic effect, maximizing the immune system's response to cancer [100,101,102]. In the CheckPAC trial, eighty-four patients received at least one dose of study treatment, which led to decreased levels of serum interleukin-6, interleukin-8, and C-reactive protein [103]. These reductions were associated with better overall survival. These findings support the continuation of the Checkpoint Inhibition and Vaccination (CheckVAC) trial, which is discussed later.
Lastly, a recent phase 2 trial [NCT05116917] investigates the potential synergy between influenza vaccination and immune checkpoint inhibitors in PDAC patients. The inspiration for this trial was the finding that influenza vaccination correlated with improved survival outcomes independent of anticancer treatment efficacy [104]. This may be due to the influenza vaccine stimulating increased T- and B-cell activation and promoting an interferon-gamma response. Therefore, it would be a promising candidate with immune checkpoint inhibitors. The overall aim of this trial is to determine the overall response rate (ORR), duration of response (DoR), disease control rate (DCR), progression-free survival (PFS), and overall survival (OS) in patients.
The clinical trials conducted so far have provided valuable insights into both the challenges and potential of immunotherapy in treating PDAC. These studies have also indicated that understanding the complex tumor immune microenvironment of PDAC is critical for devising novel approaches in immunotherapy to overcome the ability of PDAC to adapt. Penetrating the dense stroma of PDAC is also of therapeutic concern, as this fibrotic barrier poses a substantial barrier in the delivery and efficacy of immunotherapeutic agents. Overcoming this stromal barrier is essential for improving immune cell infiltration and function within the tumor microenvironment. In the following sections, we will explore emerging strategies that may further enhance the efficacy of immunotherapy for PDAC patients.
Potential Novel Approaches to PDAC Immunotherapy
Enhancing Antigen Presentation
Enhancing a tumor's antigen-presentation machinery can make the tumor more visible to the immune system. This strategy can be achieved by upregulating MHC molecules or by introducing agents that promote the release of tumor-specific antigens. In PDAC, low MHC I expression helps tumors evade immune surveillance by avoiding recognition by cytotoxic T lymphocytes. This downregulation contributes to the immunologically “cold” tumor microenvironment typical of PDAC [105]. Methods currently being explored to increase the amount of antigen presentation and enhance immune recognition of tumors include FLT3L and CD40 agonists [106,107,108,109]. FLT3L can improve the migration of conventional dendritic cells from the bone marrow, while CD40 promotes cDC activation and increases overall MHC expression [110,111]. These targets are particularly appealing for integrating the innate and adaptive immune systems to combat cancer more effectively. One way in which targeting MHC I has been leveraged is by developing mRNA vaccines focused on MHC antigen presentation [112].
Cancer Vaccines
Cancer vaccines represent a promising frontier in oncology, leveraging the body’s immune system to target and eradicate cancer cells [113]. These vaccines can be designed to elicit an immune response specifically against tumor-associated antigens, providing a personalized approach to cancer treatment that avoids the complications of traditional chemo and radiotherapy. Various strategies have been employed in developing cancer vaccines, primarily focusing on three main approaches: DNA-based, mRNA-based, and peptide-based vaccines [114,115]. DNA vaccines utilize plasmid DNA to encode tumor antigens, inducing an immune response. mRNA vaccines, like those developed for COVID-19, involve the delivery of messenger RNA encoding cancer antigens, prompting the body to produce and present these antigens to the immune system. Peptide vaccines, conversely, directly introduce tumor-specific peptides to stimulate an immune response. Each approach offers unique advantages and challenges, which will be discussed in detail.
mRNA vaccines are emerging as prominent candidates for precision treatment of PDAC [116,117,118,119,120,121]. mRNA vaccines generate robust anti-tumor responses that engage innate and adaptive immune systems. Initially, the innate immune system recognizes foreign mRNA via pattern recognition receptors on antigen-presenting cells like dendritic cells [122]. This detection, in turn, triggers a cascade of pro-inflammatory signaling pathways that enhance innate immune function. mRNA vaccines can also stimulate adaptive immunity by facilitating the processing of non-self mRNA-encoded proteins into peptides, which then present on MHC-I and are transported to the cell surface, where they activate CD8+ T cells.
Additionally, these neo-antigens can be directed through the Golgi bodies to endosomes to engage in the MHC-II presentation pathway to activate CD4+ T cells. mRNA vaccine response is amplified by the upregulation of co-stimulatory molecules (such as CD40 and CD86) on antigen-presenting cells, which enhances antigen presentation and T-cell activation. Activated antigen-presenting cells, including macrophages and dendritic cells, also present antigens to B cells, initiating an antibody response. Recent clinical trials involving an mRNA-based vaccine for pancreatic cancer have shown promising results, particularly in a study at Memorial Sloan Kettering Cancer Center [123]. The vaccine, known as autogene cevumeran (RO7198457), includes an individualized mRNA neoantigen vaccine containing up to 20 neoantigens identified in each patient’s tumor. It led to durable and functional T-cell responses in patients with resectable pancreatic cancer and was associated with a reduced risk of disease recurrence. In this phase I trial [NCT04161755], 16 patients received R07198457 combined with the checkpoint inhibitor atezolizumab and a chemotherapy regimen. Half the participants developed robust immune responses against one or more tumor neoantigens. Importantly, these T cells were long-lasting and maintained their ability to respond to neoantigens for up to three years post-vaccination. Such findings underscore the potential of a vaccine to induce a robust immune response and contribute to delayed disease recurrence in pancreatic cancer.
Safety is paramount in precision therapy, and mRNA vaccines exhibit several key safety advantages over other vaccine platforms. For example, the production and delivery of mRNA vaccines does not involve toxic chemicals, reducing potential harm to manufacturing personnel and patients [124,125]. Additionally, mRNA vaccine production mitigates the risk of contamination with adventitious viruses that can be introduced during the culture of host cells, a concern associated with other vaccine platforms like viral vectors, inactivated viruses, live viruses, and subunit protein vaccines. The rapid manufacture of mRNA vaccines also reduces the window of opportunity for contaminating microorganisms during production. Unlike other therapeutic modalities, mRNA cannot integrate into the host genome. Moreover, the adjustable half-life of mRNA allows for precise control over the duration and intensity of protein expression. This approach enhances safety by allowing modulation of immune responses and potential side effects.
Nonetheless, some challenges need to be addressed with mRNA vaccines. The inherent properties of naked mRNA, such as its size, degradability, and charge, can impede efficient cellular uptake and cytoplasmic entry, except in cases like immature dendritic cells that can efficiently internalize mRNA via the macropinocytosis pathway [126]. To enhance the effective delivery of mRNA into antigen-presenting cells, appropriate mRNA formulations (e.g., liposomes, polyplexes, polysomes, and lipoplexes) and administration routes must be judiciously selected and optimized. Once successful mRNA delivery is achieved, the in vivo half-life of transcribed mRNA requires careful regulation, as various factors influence the pharmacodynamic and pharmacokinetic properties of mRNA-based therapeutics. Structural improvements to mRNA, such as optimizing poly(A) tails, 5' cap structures, and untranslated regions, are vital for enhancing mRNA stability and overall durability. In addition to delivery and stability considerations, immunogenicity must be a focal point in mRNA vaccine design. Emerging evidence suggests a complex interplay between mRNA and its associated immune response. For example, exogenous RNA stimulates the production of type I interferon through innate immunity pathways, but excessive production can promote the degradation of both ribosomal RNA and cellular mRNA [127]. Strategies to mitigate immunogenicity include sequence optimization and post-transcriptional purification, which can reduce innate immune responses while preserving mRNA translation [128].
Furthermore, enhancing the immunostimulatory properties of mRNA by incorporating adjuvants, such as TriMix (mRNA encoding CD70, CD40L, and TLR4), can augment the potency of cancer mRNA vaccines [129]. TriMix, for instance, enhances the immunogenicity of unmodified naked mRNA, facilitating the cytotoxicity of T lymphocytes and the maturation of dendritic cells [130]. These advancements in mRNA vaccine construction are essential for improving their efficacy in treating pancreatic cancer. Furthermore, targeting KRAS mutations, one of the most commonly present mutations in PDAC, through immunotherapies has been especially challenging. In a trial [NCT03948763] utilizing mRNA-5671 (V941), which is a tetravalent vaccine targeting KRAS G12D, G12V, G13D, and G12C (Moderna Inc.) was eventually discontinued in 2022 as it did not meet efficacy endpoints and, consequently, no further progress or updates on V941 have been announced. This issue underscores the necessity for improved vaccine platforms and combinatorial therapies that can levy the immunological response provoked to infiltrate and target antigen-expressing cancer cells.
As mentioned in the CheckPAC trial, some patients experienced improved clinical response rates associated with lowered TGF-β levels, which led to the initiation of the CheckVAC trial [NCT05721846]. CheckVAC is currently exploring the combination of a TGFβ-15 peptide vaccine with nivolumab and ipilimumab treatment. TGFβ-15 is a formulated peptide vaccine containing a TGFβ-derived peptide alongside Montanide ISA-51 as an adjuvant [131]. Upon administration, TGFβ-15 aims to restore and enhance an immunological anti-tumor response by stimulating the host immune system to mount a cytotoxic T-lymphocyte response against TGFβ-expressing immunosuppressive cells in the TME, including TAMs, MDSCs, DCs, Tregs, and CAFs.
Another cancer vaccine that is currently being tested is the combination of TG01 Vaccine / QS-21 Stimulon with or without immune checkpoint inhibitor balstilimab as maintenance therapy following adjuvant chemotherapy in patients with resected pancreatic cancer (TESLA) trial [NCT05638698]. TG01 is an experimental vaccine designed to provoke an immune response against cancer cells by targeting the seven most prevalent codon 12 and 13 oncogenic mutations in KRAS with synthetic RAS peptides. QS-21, derived from the soap bark tree, is a vaccine adjuvant known for stimulating both humoral and cell-mediated immunity, further boosting the immune response induced by TG01.
A new cancer vaccine clinical trial [NCT05964361] focuses on enhancing the body's immune response by targeting the Wilms tumor 1 (WT1) protein. WT1 is a transcription factor that plays a crucial role in both normal development and tumorigenesis [132]. WT1 is highly overexpressed in various malignancies, including pancreatic cancer [133]. WT1 supports tumor progression by promoting cell proliferation, inhibiting apoptosis, and enhancing angiogenesis. This trial investigates the feasibility of developing vaccines specifically targeting the Wilms' Tumor-1 (WT1) antigen alongside a novel IL15-trans presentation mechanism on their cell surface. IL15, known for its role in supporting natural killer cell function, promoting T cell memory formation, and enhancing immune response, is expected to enhance the immunogenicity of dendritic cells towards WT1-expressing cancer cells [134,135].
Another clinical trial [NCT05846516] evaluates the experimental immunotherapy KISIMA-02 for pancreatic cancer patients. The KISIMA platform is a single therapeutic vaccine that is comprised of three components: a cell-penetrating peptide (CPP) for transportation of the vaccine contents across the cell membrane, antigens that can be tailored to each indication, and a TLR peptide agonist that acts as an adjuvant [136]. KISIMA-02 is a combination treatment for patients with KRAS G12D/G12V mutated PDAC. This regimen includes the KISIMA therapeutic protein vaccine (ATP150 or ATP152), an oncolytic viral vector (VSV-GP154), and an immune checkpoint inhibitor. This study is designed to assess the safety and tolerability of the KISIMA-02 regimen before examining its impact on delaying tumor recurrence. Participants will receive a therapeutic protein vaccine (either ATP150 or ATP152), a viral vector VSV-GP154, and an immune checkpoint inhibitor, ezabenlimab, or be in an observational group. The ATP-150 vaccine, specifically developed for KRAS G12D and G12V mutated PDAC, employs a heterologous prime-boost approach, integrating both protein and viral vector components. This vaccine is administered parenterally and is part of the KISIMA immunization platform, designed to enhance immune response through self-adjuvant mechanisms.
The pancreatic GVAX platform is another example of a novel strategy that has seen success in other cancers, with the pancreatic version being a vaccine composed of genetically modified pancreatic cancer cells [137,138]. These engineered pancreatic cells secrete granulocyte-macrophage colony-stimulating factor (GM-CSF) to facilitate a strong dendritic cell-dependent immunological response. The GVAX induces the formation of tertiary lymphoid structures within the tumor, providing a localized site for immune cell activation for an effective immune response. In certain trial arms, GVAX is combined with immune checkpoint inhibitors like nivolumab and ipilimumab. Combining GVAX with these checkpoint inhibitors aims to prime the immune system against the tumor and prevent immune escape. GVAX is also commonly administered with low-dose cyclophosphamide, which selectively depletes Tregs that suppress immune responses, thus enhancing the vaccine's effectiveness [139]. A three-arm, phase 2 clinical trial [NCT02451982] is currently underway, aiming to determine the effect of co-treatment of GVAX with CY on patient outcomes. This trial aims to compare the DFS and OS of patients in three treatment arms: Arm A (GVAX alone), Arm B (GVAX and then low-dose CY), and Arm C (low-dose CY and then GVAX). The study evaluates the effect of the sequencing and combination of GVAX and low-dose CY on patient outcomes. By comparing these three treatment strategies, this trial aims to determine whether the addition of low-dose CY before or after GVAX could enhance or impair the efficacy of the immunotherapy. Another GVAX-related phase 2 clinical trial [NCT03190265] is focused on the investigation of ORR and AEs with combination therapy of CRS-207, nivolumab, and ipilimumab, with or without the GVAX (and CY)). CRS-207 is a live-attenuated, double-deleted Listeria monocytogenes strain engineered to express the pancreatic tumor-associated antigen mesothelin [140,141]. By infecting antigen-presenting cells, CRS-207 helps break immune tolerance by inducing a potent innate and adaptive immune response, including activating T cells specific to mesothelin. Overall, GVAX provides a broad array of tumor antigens in the context of GM-CSF. At the same time, CRS-207 focuses on a specific antigen, mesothelin, enhancing the overall presentation of tumor antigens to the immune system. This combination can lead to a more robust activation and expansion of tumor-specific T cells, as both therapies independently activate and mature dendritic cells. CRS-207’s ability to break immune tolerance complements GVAX’s ability to stimulate an immune response, which may lead to a more effective and sustained anti-tumor immune response. Targeting multiple tumor-associated antigens between GVAX and CRS-207 would also reduce the likelihood of immune escape by tumor cells.
Neoantigen-Based Peptide Vaccines for Patients with Advanced Pancreatic Cancer Refractory to Standard Treatment
In addition to mRNA vaccines, peptide vaccines are another emerging alternative for PDAC patients. Peptide vaccines are designed to induce the host’s immune system to target neoantigens, which are unique to tumor cells due to mutations and are absent in normal cells. Production of neoantigen-based peptide vaccines typically involves several steps. Initially, tumor samples are obtained from the patient, and the genetic material of the tumor cells is sequenced to identify neoantigens produced by the cancer cells. Bioinformatics tools are then employed to predict which neoantigens are most likely to be recognized by the patient's immune system. Once suitable neoantigens are identified, corresponding synthetic peptides are manufactured. These peptides are then formulated into a vaccine, often with an adjuvant to enhance the immune response. The personalized vaccine is subsequently administered to the patient. This process tailors the treatment to the unique genetic profile of each patient's cancer. It also minimizes damage to healthy tissue akin to other vaccination methods previously discussed – a common side effect of broad-based chemotherapeutic and radiotherapeutic treatments.
An example of the successful development of a personalized neoantigen-based peptide vaccine is iNeo-Vac-P01 for patients with advanced PDAC who had exhausted standard treatment options [142]. Notably, the vaccine was well-tolerated, recording no severe adverse events. The median overall survival (OS) and progression-free survival (PFS) for this cohort surpassed those of prior clinical studies, suggesting the vaccine's potential to extend life for patients with this aggressive form of cancer. Some participants also received the neoantigen-based peptide vaccine with chemotherapy or immune checkpoint inhibitors to boost clinical outcomes. Central to the vaccine's success was its ability to stimulate T-cell solid responses against specific tumor antigens. The study identified induced T-cell reaction alterations in peripheral blood T-cell subsets and reported that higher interferon-gamma levels were detected in patients with better OS. The research also signifies strategic timing for vaccine administration, suggesting that minimized tumor burden could optimize immune infiltration. The study proposed combining the neoantigen-based peptide vaccine with anti-CTLA-4 antibodies, presenting a potent therapeutic combination for further evaluation.
Another peptide-based vaccine strategy for pancreatic cancer involves harnessing neoantigens' specificity to stimulate an immune response against cancer cells. One example is SLP vaccines, synthetic peptides that mimic the neoantigens found in cancer cells [143]. They are typically longer than traditional short peptides and can encompass more of the neoantigen sequence. When these SLP vaccines are administered, dendritic cells capture the synthetic peptides, process them, and present them on their cell surface along with MHC molecules. T cells become activated when they encounter the dendritic cells presenting synthetic peptides. The activated T cells undergo clonal expansion, resulting in a population of T cells primed to target the tumor. SLP vaccines have also been demonstrated to stimulate the formation of memory T cells, leading to long-term immune surveillance. A new phase 1 trial [NCT05013216] is set to evaluate the safety and immunogenicity of a pooled mutant-KRAS (mKRAS) SLP peptide vaccine combined with the poly-ICLC adjuvant in 20 patients at high risk of developing PDAC. These high-risk groups were identified as individuals who: 1) have familial pancreatic cancer relatives or 2) are germline mutation carriers with an estimated lifetime risk of pancreatic cancer of ~10% or higher. The vaccine formulation includes synthetic long peptides representing six common mKRAS subtypes: G12D, G12R, G12V, G12A, G12C, and G13D. These peptides are combined with the poly-ICLC adjuvant to enhance the vaccine's effectiveness.
A promising SLP immunotherapy platform has recently been underway involving ELI-002. ELI-002 is composed of a lipid-conjugated immune-stimulatory oligonucleotide (Amph-CpG-7909) and a combination of lipid-conjugated peptide-based antigens (Amph-Peptides) designated by a “NP” to target a broad spectrum of KRAS mutations expressed in various cancers. The first trial [NCT04853017] involving ELI-002 was conducted with ELI-002 2P, which codes for Amph modified KRAS peptides: Amph-G12D and Amph-G12R. The dose-escalation study was designed to assess the safety and efficacy of SLP ELI-002 as immunotherapy that could be an adjuvant treatment for patients with KRAS/NRAS viral oncogene homolog mutated PDAC, among other solid tumors and has since been transitioned into another trial [NCT04853017] utilizing the ELI-002 7P formulation [144]. ELI-002 7P incorporates seven different Amph-Peptides: G12D, G12R, G12V, G12A, G12C, G12S, and G13D. The trial currently assesses 156 subjects across three planned dose levels, with the potential for additional cohorts based on safety reviews and preliminary pharmacodynamic responses. The primary objective is establishing a recommended Phase 2 dose and setting a maximum tolerated dose.
Another SLP immunotherapy trial [NCT04117087] underway is a single-arm, open-label, first-in-human phase I study of a pooled SLP vaccine targeting six mKRAS subtypes: G12D, G12R, G12V, G12A, G12C, and G13D. Treatment is combined with checkpoint inhibitors ipilimumab and nivolumab in patients with resected PDAC. The study aims to evaluate the fold change in interferon-producing mutant-KRAS-specific CD4/CD8+ T cells after vaccination alongside any drug-related adverse events. Secondary measures are determining the overall DFS, ORR, OS, and PFS in these treated patients.
While many trials have been focused on inducing robust CD4 and CD8 T cell responses through peptide and mRNA vaccines, another open-label phase I trial [NCT03122106] aimed to study and evaluate the safety and immunogenicity of a neoantigen DNA vaccine strategy in PDAC patients following surgical resection and adjuvant chemotherapy [145]. The neoantigen DNA vaccine incorporates prioritized neoantigens and personalized mesothelin epitopes and is administered via electroporation. Results indicated that the neoantigen DNA vaccine could stimulate immune responses, evidenced by the presence of these T cells in patients' blood samples after vaccination. However, challenges such as variability in neoantigen identification and vaccine formulation were noted, highlighting the complexity of developing personalized cancer vaccines.
Discussion
There is an urgent need to innovate and improve therapeutic strategies for treating pancreatic cancer, as it is predicted to become the second leading cause of cancer-related death by 2030. Current treatment protocols heavily rely on chemotherapeutic agents such as gemcitabine and FOLFIRINOX; however, their efficacy is significantly compromised by high toxicity and the development of chemoresistance. Despite advancements in cancer therapeutics such as immunotherapy, PDAC remains a dauntingly complex cancer to treat due to its distinctive structural composition. The dense fibrotic stroma characteristic of PDAC creates a physical barrier that blocks immune cell infiltration. In addition, PDAC’s TME poses a unique challenge as it is replete with immunosuppressive elements, including Tregs, MDSCs, and TAMs, which contribute to a robust barrier against effective immune response. Recent efforts are focused on enhancing the delivery and efficacy of immunotherapy by dissecting the complexities of the PDAC TME. Novel strategies aim to target the dense desmoplastic stroma, neutralize the effects of immunosuppressive cytokines, and enhance the presentation of tumor antigens to induce a more robust immune response. For example, personalized neoantigen vaccines and precision targeting of cancer cells have shown considerable promise in enhancing the immune response directly at the tumor site, thus improving the specificity and effectiveness of treatment. Emerging approaches also involve the utility of dual-targeting strategies where immune checkpoint blockade is used with agents that modulate the immune environment. Approaches focused on targeting specific tumor antigens to enhance the immunogenicity of the tumors have also gained attention. Furthermore, developing targeted therapies such as the TG01 vaccine, specifically addressing KRAS mutations common in pancreatic cancer, indicates the shift towards genetic precision in immunotherapy. The direct targeting of the genetic abnormalities driving cancer progression promised a more tailored and potentially effective treatment regimen.
In summary, the promising trajectory of immunotherapy in PDAC underscores a shift towards more personalized and precisely targeted treatment regimens. Continued research and clinical trials will be crucial in moving these innovative strategies from the bench to the clinic. Integrating multidisciplinary research covering molecular biology, pharmacology, and immunology, along with robust patient engagement, will further refine and enhance the therapeutic landscape of PDAC, offering hope for improved survival and quality of life for patients with this challenging disease.

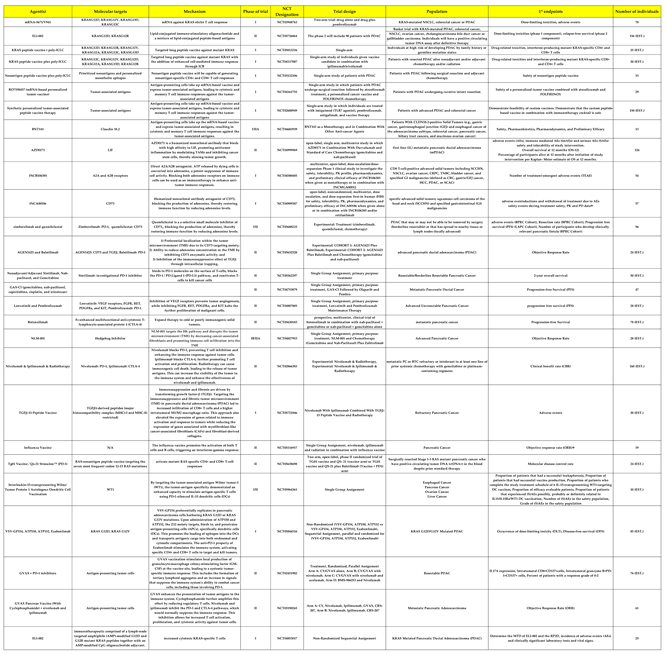
Table 1.
Author Contributions
Chris TP Do and Jack Y. Prochnau contributed equally to this paper. Writing—original draft preparation, CTD, JYP, AD.; writing—review and editing, CTD, JYP, AD; supervision, MKR, PW; funding acquisition, MKR. All authors have read and agreed to the published version of the manuscript.
Funding
Rao MK is supported by NIH (NCI) Grants R01CA179120-01A1 and R01CA239227-A1.
Acknowledgments
We thank Karen P. Klein (Clarus Editorial Services; https://www.claruseditorialservices.com/) for editing.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- “Annual Cancer Facts & Figures | American Cancer Society.” Accessed: Jul. 01, 2024. [Online]. Available: https://www.cancer.org/research/cancer-facts-statistics/all-cancer-facts-figures.html.
- L. Marstrand-Daucé, D. Lorenzo, A. Chassac, P. Nicole, A. Couvelard, and C. Haumaitre, “Acinar-to-Ductal Metaplasia (ADM): On the Road to Pancreatic Intraepithelial Neoplasia (PanIN) and Pancreatic Cancer,” Int. J. Mol. Sci., vol. 24, no. 12, p. 9946, Jun. 2023. [CrossRef]
- V. Tonini and M. Zanni, “Pancreatic cancer in 2021: What you need to know to win,” World J. Gastroenterol., vol. 27, no. 35, pp. 5851–5889, Sep. 2021. [CrossRef]
- D. R. Principe, P. W. Underwood, M. Korc, J. G. Trevino, H. G. Munshi, and A. Rana, “The Current Treatment Paradigm for Pancreatic Ductal Adenocarcinoma and Barriers to Therapeutic Efficacy,” Front. Oncol., vol. 11, p. 688377, Jul. 2021. [CrossRef]
- L.-H. Truong and S. Pauklin, “Pancreatic Cancer Microenvironment and Cellular Composition: Current Understandings and Therapeutic Approaches,” Cancers, vol. 13, no. 19, p. 5028, Oct. 2021. [CrossRef]
- A. N. Hosein, R. A. Brekken, and A. Maitra, “Pancreatic cancer stroma: an update on therapeutic targeting strategies,” Nat. Rev. Gastroenterol. Hepatol., vol. 17, no. 8, pp. 487–505, Aug. 2020. [CrossRef]
- J. S. Bowers, S. R. Bailey, M. P. Rubinstein, C. M. Paulos, and E. R. Camp, “Genomics meets immunity in pancreatic cancer: Current research and future dire4ctions for pancreatic adenocarcinoma immunotherapy,” Oncol. Rev., vol. 13, no. 2, Aug. 2019. [CrossRef]
- S. K. Daniel, K. M. Sullivan, K. P. Labadie, and V. G. Pillarisetty, “Hypoxia as a barrier to immunotherapy in pancreatic adenocarcinoma,” Clin. Transl. Med., vol. 8, no. 1, p. e10, Dec. 2019. [CrossRef]
- S. K. Gautam, S. K. Batra, and M. Jain, “Molecular and metabolic regulation of immunosuppression in metastatic pancreatic ductal adenocarcinoma,” Mol. Cancer, vol. 22, no. 1, p. 118, Jul. 2023. [CrossRef]
- J. Fan, M.-F. Wang, H.-L. Chen, D. Shang, J. K. Das, and J. Song, “Current advances and outlooks in immunotherapy for pancreatic ductal adenocarcinoma,” Mol. Cancer, vol. 19, no. 1, p. 32, Dec. 2020. [CrossRef]
- T. Murakami, Y. Hiroshima, R. Matsuyama, Y. Homma, R. M. Hoffman, and I. Endo, “Role of the tumor microenvironment in pancreatic cancer,” Ann. Gastroenterol. Surg., vol. 3, no. 2, pp. 130–137, Mar. 2019. [CrossRef]
- B. Ren et al., “Tumor microenvironment participates in metastasis of pancreatic cancer,” Mol. Cancer, vol. 17, no. 1, p. 108, Dec. 2018. [CrossRef]
- M. Huber et al., “The Immune Microenvironment in Pancreatic Cancer,” Int. J. Mol. Sci., vol. 21, no. 19, p. 7307, Oct. 2020. [CrossRef]
- M. Pascual-García et al., “LIF regulates CXCL9 in tumor-associated macrophages and prevents CD8+ T cell tumor-infiltration impairing anti-PD1 therapy,” Nat. Commun., vol. 10, no. 1, p. 2416, Jun. 2019. [CrossRef]
- R. Akbani et al., “Genomic Classification of Cutaneous Melanoma,” Cell, vol. 161, no. 7, pp. 1681–1696, Jun. 2015. [CrossRef]
- M. Łuksza et al., “Neoantigen quality predicts immunoediting in survivors of pancreatic cancer,” Nature, vol. 606, no. 7913, pp. 389–395, Jun. 2022. [CrossRef]
- Australian Pancreatic Cancer Genome Initiative et al., “Identification of unique neoantigen qualities in long-term survivors of pancreatic cancer,” Nature, vol. 551, no. 7681, pp. 512–516, Nov. 2017. [CrossRef]
- M. Feng et al., “PD-1/PD-L1 and immunotherapy for pancreatic cancer,” Cancer Lett., vol. 407, pp. 57–65, Oct. 2017. [CrossRef]
- W. Zou and L. Chen, “Inhibitory B7-family molecules in the tumour microenvironment,” Nat. Rev. Immunol., vol. 8, no. 6, pp. 467–477, Jun. 2008. [CrossRef]
- J. Tao et al., “Targeting hypoxic tumor microenvironment in pancreatic cancer,” J. Hematol. Oncol.J Hematol Oncol, vol. 14, no. 1, p. 14, Jan. 2021. [CrossRef]
- Q. Hu et al., “UHRF1 promotes aerobic glycolysis and proliferation via suppression of SIRT4 in pancreatic cancer,” Cancer Lett., vol. 452, pp. 226–236, Jun. 2019. [CrossRef]
- M. Erkan et al., “The role of stroma in pancreatic cancer: diagnostic and therapeutic implications,” Nat. Rev. Gastroenterol. Hepatol., vol. 9, no. 8, pp. 454–467, Aug. 2012. [CrossRef]
- K. Yamamoto et al., “Autophagy promotes immune evasion of pancreatic cancer by degrading MHC-I,” Nature, vol. 581, no. 7806, pp. 100–105, May 2020. [CrossRef]
- K. Yamamoto, A. Venida, R. M. Perera, and A. C. Kimmelman, “Selective autophagy of MHC-I promotes immune evasion of pancreatic cancer,” Autophagy, vol. 16, no. 8, pp. 1524–1525, Aug. 2020. [CrossRef]
- I. A. York and K. L. Rock, “ANTIGEN PROCESSING AND PRESENTATION BY THE CLASS I MAJOR HISTOCOMPATIBILITY COMPLEX,” Annu. Rev. Immunol., vol. 14, no. 1, pp. 369–396, Apr. 1996. [CrossRef]
- N. Pu et al., “Cell-intrinsic PD-1 promotes proliferation in pancreatic cancer by targeting CYR61/CTGF via the hippo pathway,” Cancer Lett., vol. 460, pp. 42–53, Sep. 2019. [CrossRef]
- E. Hessmann et al., “Microenvironmental Determinants of Pancreatic Cancer,” Physiol. Rev., vol. 100, no. 4, pp. 1707–1751, Oct. 2020. [CrossRef]
- S. Tsukita and M. Furuse, “The Structure and Function of Claudins, Cell Adhesion Molecules at Tight Junctions,” Ann. N. Y. Acad. Sci., vol. 915, no. 1, pp. 129–135, Dec. 2000. [CrossRef]
- S. Tsukita, H. Tanaka, and A. Tamura, “The Claudins: From Tight Junctions to Biological Systems,” Trends Biochem. Sci., vol. 44, no. 2, pp. 141–152, Feb. 2019. [CrossRef]
- T. Otani and M. Furuse, “Tight Junction Structure and Function Revisited,” Trends Cell Biol., vol. 30, no. 10, pp. 805–817, Oct. 2020. [CrossRef]
- K. J. Hewitt, R. Agarwal, and P. J. Morin, “The claudin gene family: expression in normal and neoplastic tissues,” BMC Cancer, vol. 6, no. 1, p. 186, Dec. 2006. [CrossRef]
- U. Sahin et al., “Claudin-18 Splice Variant 2 Is a Pan-Cancer Target Suitable for Therapeutic Antibody Development,” Clin. Cancer Res., vol. 14, no. 23, pp. 7624–7634, Dec. 2008. [CrossRef]
- W. Cao et al., “Claudin18.2 is a novel molecular biomarker for tumor-targeted immunotherapy,” Biomark. Res., vol. 10, no. 1, p. 38, Dec. 2022. [CrossRef]
- H. Bähr-Mahmud et al., “Preclinical characterization of an mRNA-encoded anti-Claudin 18.2 antibody,” OncoImmunology, vol. 12, no. 1, p. 2255041, Dec. 2023. [CrossRef]
- K. P. Papadopoulos et al., “A phase I/II dose escalation and expansion trial to evaluate safety and preliminary efficacy of BNT141 in patients with claudin-18.2-positive solid tumors.,” J. Clin. Oncol., vol. 41, no. 16_suppl, pp. TPS2670–TPS2670, Jun. 2023. [CrossRef]
- K. Shitara et al., “Zolbetuximab plus mFOLFOX6 in patients with CLDN18.2-positive, HER2-negative, untreated, locally advanced unresectable or metastatic gastric or gastro-oesophageal junction adenocarcinoma (SPOTLIGHT): a multicentre, randomised, double-blind, phase 3 trial,” The Lancet, vol. 401, no. 10389, pp. 1655–1668, May 2023. [CrossRef]
- S. S. Diebold, T. Kaisho, H. Hemmi, S. Akira, and C. Reis E Sousa, “Innate Antiviral Responses by Means of TLR7-Mediated Recognition of Single-Stranded RNA,” Science, vol. 303, no. 5663, pp. 1529–1531, Mar. 2004. [CrossRef]
- Z. Zhang et al., “Structural Analysis Reveals that Toll-like Receptor 7 Is a Dual Receptor for Guanosine and Single-Stranded RNA,” Immunity, vol. 45, no. 4, pp. 737–748, Oct. 2016. [CrossRef]
- G. Landskron, M. De La Fuente, P. Thuwajit, C. Thuwajit, and M. A. Hermoso, “Chronic Inflammation and Cytokines in the Tumor Microenvironment,” J. Immunol. Res., vol. 2014, pp. 1–19, 2014. [CrossRef]
- G. A. Gastl et al., “Interleukin-10 production by human carcinoma cell lines and its relationship to interleukin-6 expression,” Int. J. Cancer, vol. 55, no. 1, pp. 96–101, Aug. 1993. [CrossRef]
- R. Inamoto, N. Takahashi, and Y. Yamada, “Claudin18.2 in Advanced Gastric Cancer,” Cancers, vol. 15, no. 24, p. 5742, Dec. 2023. [CrossRef]
- W. L. Byrne, K. H. G. Mills, J. A. Lederer, and G. C. O’Sullivan, “Targeting Regulatory T Cells in Cancer,” Cancer Res., vol. 71, no. 22, pp. 6915–6920, Nov. 2011. [CrossRef]
- V. Fleming et al., “Targeting Myeloid-Derived Suppressor Cells to Bypass Tumor-Induced Immunosuppression,” Front. Immunol., vol. 9, p. 398, Mar. 2018. [CrossRef]
- Y. Ohue and H. Nishikawa, “Regulatory T (Treg) cells in cancer: Can Treg cells be a new therapeutic target?,” Cancer Sci., vol. 110, no. 7, pp. 2080–2089, Jul. 2019. [CrossRef]
- J.-E. Jang, C. H. Hajdu, C. Liot, G. Miller, M. L. Dustin, and D. Bar-Sagi, “Crosstalk between Regulatory T Cells and Tumor-Associated Dendritic Cells Negates Anti-tumor Immunity in Pancreatic Cancer,” Cell Rep., vol. 20, no. 3, pp. 558–571, Jul. 2017. [CrossRef]
- I. M. Stromnes et al., “Targeted depletion of an MDSC subset unmasks pancreatic ductal adenocarcinoma to adaptive immunity,” Gut, vol. 63, no. 11, pp. 1769–1781, Nov. 2014. [CrossRef]
- R. Trovato et al., “Immunosuppression by monocytic myeloid-derived suppressor cells in patients with pancreatic ductal carcinoma is orchestrated by STAT3,” J. Immunother. Cancer, vol. 7, no. 1, p. 255, Dec. 2019. [CrossRef]
- E. Eriksson, J. Wenthe, S. Irenaeus, A. Loskog, and G. Ullenhag, “Gemcitabine reduces MDSCs, tregs and TGFβ-1 while restoring the teff/treg ratio in patients with pancreatic cancer,” J. Transl. Med., vol. 14, no. 1, p. 282, Dec. 2016. [CrossRef]
- Y. Shi et al., “Targeting LIF-mediated paracrine interaction for pancreatic cancer therapy and monitoring,” Nature, vol. 569, no. 7754, pp. 131–135, May 2019. [CrossRef]
- F. Peng, J. Zhou, W. Sheng, D. Zhang, and M. Dong, “ [Expression and significance of leukemia inhibitory factor in human pancreatic cancer],” Zhonghua Yi Xue Za Zhi, vol. 94, no. 2, pp. 90–95, Jan. 2014.
- D. Wang et al., “Prognostic value of leukemia inhibitory factor and its receptor in pancreatic adenocarcinoma,” Future Oncol., vol. 16, no. 3, pp. 4461–4473, Jan. 2020. [CrossRef]
- E. Borazanci et al., “Phase I, first-in-human study of MSC-1 (AZD0171), a humanized anti-leukemia inhibitory factor monoclonal antibody, for advanced solid tumors,” ESMO Open, vol. 7, no. 4, p. 100530, Aug. 2022. [CrossRef]
- R. L. Williams et al., “Myeloid leukaemia inhibitory factor maintains the developmental potential of embryonic stem cells,” Nature, vol. 336, no. 6200, pp. 684–687, Dec. 1988. [CrossRef]
- C. J. Halbrook, M. Pasca Di Magliano, and C. A. Lyssiotis, “Tumor cross-talk networks promote growth and support immune evasion in pancreatic cancer,” Am. J. Physiol.-Gastrointest. Liver Physiol., vol. 315, no. 1, pp. G27–G35, Jul. 2018. [CrossRef]
- D. Saka et al., “Mechanisms of T-Cell Exhaustion in Pancreatic Cancer,” Cancers, vol. 12, no. 8, p. 2274, Aug. 2020. [CrossRef]
- F. Marcon et al., “NK cells in pancreatic cancer demonstrate impaired cytotoxicity and a regulatory IL-10 phenotype,” OncoImmunology, vol. 9, no. 1, p. 1845424, Jan. 2020. [CrossRef]
- R. Yang et al., “Conversion of ATP to adenosine by CD39 and CD73 in multiple myeloma can be successfully targeted together with adenosine receptor A2A blockade,” J. Immunother. Cancer, vol. 8, no. 1, p. e000610, May 2020. [CrossRef]
- D. Zahavi and J. Hodge, “Targeting Immunosuppressive Adenosine Signaling: A Review of Potential Immunotherapy Combination Strategies,” Int. J. Mol. Sci., vol. 24, no. 10, p. 8871, May 2023. [CrossRef]
- C. Kang, “Retifanlimab: First Approval,” Drugs, vol. 83, no. 8, pp. 731–737, Jun. 2023. [CrossRef]
- H. Wang et al., “Abstract LB157: Discovery and characterization of INCB106385: a novel A2A/A2B adenosine receptor antagonist, as a cancer immunotherapy,” Cancer Res., vol. 81, no. 13_Supplement, pp. LB157–LB157, Jul. 2021. [CrossRef]
- N. Bach, R. Winzer, E. Tolosa, W. Fiedler, and F. Brauneck, “The Clinical Significance of CD73 in Cancer,” Int. J. Mol. Sci., vol. 24, no. 14, p. 11759, Jul. 2023. [CrossRef]
- S. Stewart et al., “Abstract LB174: Discovery and preclinical characterization of INCA00186, a humanized monoclonal antibody antagonist of CD73, as a cancer immunotherapy,” Cancer Res., vol. 81, no. 13_Supplement, pp. LB174–LB174, Jul. 2021. [CrossRef]
- M. Hori and M. Kitakaze, “Adenosine, the heart, and coronary circulation.,” Hypertension, vol. 18, no. 5, pp. 565–574, Nov. 1991. [CrossRef]
- L. Belardinelli et al., “The A2A adenosine receptor mediates coronary vasodilation,” J. Pharmacol. Exp. Ther., vol. 284, no. 3, pp. 1066–1073, Mar. 1998.
- B. Allard, S. Pommey, M. J. Smyth, and J. Stagg, “Targeting CD73 Enhances the Antitumor Activity of Anti-PD-1 and Anti-CTLA-4 mAbs,” Clin. Cancer Res., vol. 19, no. 20, pp. 5626–5635, Oct. 2013. [CrossRef]
- F. S. Regateiro et al., “Generation of anti-inflammatory adenosine byleukocytes is regulated by TGF-β,” Eur. J. Immunol., vol. 41, no. 10, pp. 2955–2965, Oct. 2011. [CrossRef]
- A. W. Tolcher et al., “Phase 1 first-in-human study of dalutrafusp alfa, an anti–CD73-TGF-β-trap bifunctional antibody, in patients with advanced solid tumors,” J. Immunother. Cancer, vol. 11, no. 2, p. e005267, Feb. 2023. [CrossRef]
- X. He and C. Xu, “Immune checkpoint signaling and cancer immunotherapy,” Cell Res., vol. 30, no. 8, pp. 660–669, Aug. 2020. [CrossRef]
- S. Lim et al., “Interplay between Immune Checkpoint Proteins and Cellular Metabolism,” Cancer Res., vol. 77, no. 6, pp. 1245–1249, Mar. 2017. [CrossRef]
- M. E. Keir, M. J. Butte, G. J. Freeman, and A. H. Sharpe, “PD-1 and Its Ligands in Tolerance and Immunity,” Annu. Rev. Immunol., vol. 26, no. 1, pp. 677–704, Apr. 2008. [CrossRef]
- J. B. A. G. Haanen and C. Robert, “Immune Checkpoint Inhibitors,” in Progress in Tumor Research, vol. 42, O. Michielin and G. Coukos, Eds., S. Karger AG, 2015, pp. 55–66. [CrossRef]
- J. R. Brahmer et al., “Safety and Activity of Anti–PD-L1 Antibody in Patients with Advanced Cancer,” N. Engl. J. Med., vol. 366, no. 26, pp. 2455–2465, Jun. 2012. [CrossRef]
- K. D. McCoy and G. Le Gros, “The role of CTLA-4 in the regulation of T cell immune responses,” Immunol. Cell Biol., vol. 77, no. 1, pp. 1–10, Feb. 1999. [CrossRef]
- F. A. Schildberg, S. R. Klein, G. J. Freeman, and A. H. Sharpe, “Coinhibitory Pathways in the B7-CD28 Ligand-Receptor Family,” Immunity, vol. 44, no. 5, pp. 955–972, May 2016. [CrossRef]
- A. Rotte, “Combination of CTLA-4 and PD-1 blockers for treatment of cancer,” J. Exp. Clin. Cancer Res., vol. 38, no. 1, p. 255, Dec. 2019. [CrossRef]
- M. Nishio et al., “First-line nivolumab plus ipilimumab in metastatic non-small cell lung cancer: 5-year outcomes in Japanese patients from CheckMate 227 Part 1,” Int. J. Clin. Oncol., vol. 28, no. 10, pp. 1354–1368, Oct. 2023. [CrossRef]
- F. S. Hodi et al., “Improved Survival with Ipilimumab in Patients with Metastatic Melanoma,” N. Engl. J. Med., vol. 363, no. 8, pp. 711–723, Aug. 2010. [CrossRef]
- M. Gao et al., “Direct therapeutic targeting of immune checkpoint PD-1 in pancreatic cancer,” Br. J. Cancer, vol. 120, no. 1, pp. 88–96, Jan. 2019. [CrossRef]
- N. Pu, W. Lou, and J. Yu, “PD-1 immunotherapy in pancreatic cancer: current status,” J. Pancreatol., vol. 2, no. 1, pp. 6–10, Mar. 2019. [CrossRef]
- L. Zhang, W. Mai, W. Jiang, and Q. Geng, “Sintilimab: A Promising Anti-Tumor PD-1 Antibody,” Front. Oncol., vol. 10, p. 594558, Nov. 2020. [CrossRef]
- J. S. Brown, R. Sundar, and J. Lopez, “Combining DNA damaging therapeutics with immunotherapy: more haste, less speed,” Br. J. Cancer, vol. 118, no. 3, pp. 312–324, Feb. 2018. [CrossRef]
- C. J. Langer et al., “Carboplatin and pemetrexed with or without pembrolizumab for advanced, non-squamous non-small-cell lung cancer: a randomised, phase 2 cohort of the open-label KEYNOTE-021 study,” Lancet Oncol., vol. 17, no. 11, pp. 1497–1508, Nov. 2016. [CrossRef]
- R. M. Poole, “Pembrolizumab: First Global Approval,” Drugs, vol. 74, no. 16, pp. 1973–1981, Oct. 2014. [CrossRef]
- E. D. Deeks, “Olaparib: First Global Approval,” Drugs, vol. 75, no. 2, pp. 231–240, Feb. 2015. [CrossRef]
- Z. Hao and P. Wang, “Lenvatinib in Management of Solid Tumors,” The Oncologist, vol. 25, no. 2, pp. e302–e310, Feb. 2020. [CrossRef]
- M. Kudo et al., “Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: a randomised phase 3 non-inferiority trial,” The Lancet, vol. 391, no. 10126, pp. 1163–1173, Mar. 2018. [CrossRef]
- M. Schlumberger et al., “Lenvatinib versus Placebo in Radioiodine-Refractory Thyroid Cancer,” N. Engl. J. Med., vol. 372, no. 7, pp. 621–630, Feb. 2015. [CrossRef]
- P. Waterhouse et al., “Lymphoproliferative Disorders with Early Lethality in Mice Deficient in Ctla-4,” Science, vol. 270, no. 5238, pp. 985–988, Nov. 1995. [CrossRef]
- E. A. Tivol, F. Borriello, A. N. Schweitzer, W. P. Lynch, J. A. Bluestone, and A. H. Sharpe, “Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4,” Immunity, vol. 3, no. 5, pp. 541–547, Nov. 1995. [CrossRef]
- A. Bullock et al., “LBA O-9 Botensilimab, a novel innate/adaptive immune activator, plus balstilimab (anti-PD-1) for metastatic heavily pretreated microsatellite stable colorectal cancer,” Ann. Oncol., vol. 33, p. S376, Jun. 2022. [CrossRef]
- R. Balsano, V. Zanuso, A. Pirozzi, L. Rimassa, and S. Bozzarelli, “Pancreatic Ductal Adenocarcinoma and Immune Checkpoint Inhibitors: The Gray Curtain of Immunotherapy and Spikes of Lights,” Curr. Oncol., vol. 30, no. 4, pp. 3871–3885, Mar. 2023. [CrossRef]
- G. Germano et al., “Inactivation of DNA repair triggers neoantigen generation and impairs tumour growth,” Nature, vol. 552, no. 7683, pp. 116–120, Dec. 2017. [CrossRef]
- T. Macarulla Mercade et al., “331P Phase Ib/IIa study to evaluate safety and efficacy of priming treatment with the hedgehog inhibitor NLM-001 prior to gemcitabine and nab-paclitaxel plus zalifrelimab as first-line treatment in patients with advanced pancreatic cancer: NUMANTIA study,” Ann. Oncol., vol. 35, pp. S139–S140, Jun. 2024. [CrossRef]
- S. S. Karhadkar et al., “Hedgehog signalling in prostate regeneration, neoplasia and metastasis,” Nature, vol. 431, no. 7009, pp. 707–712, Oct. 2004. [CrossRef]
- G. Feldmann et al., “Blockade of Hedgehog Signaling Inhibits Pancreatic Cancer Invasion and Metastases: A New Paradigm for Combination Therapy in Solid Cancers,” Cancer Res., vol. 67, no. 5, pp. 2187–2196, Mar. 2007. [CrossRef]
- D. M. Berman et al., “Widespread requirement for Hedgehog ligand stimulation in growth of digestive tract tumours,” Nature, vol. 425, no. 6960, pp. 846–851, Oct. 2003. [CrossRef]
- S. V. Outram, A. Varas, C. V. Pepicelli, and T. Crompton, “Hedgehog Signaling Regulates Differentiation from Double-Negative to Double-Positive Thymocyte,” Immunity, vol. 13, no. 2, pp. 187–197, Aug. 2000. [CrossRef]
- S. L. Topalian et al., “Safety, Activity, and Immune Correlates of Anti–PD-1 Antibody in Cancer,” N. Engl. J. Med., vol. 366, no. 26, pp. 2443–2454, Jun. 2012. [CrossRef]
- A. Hoos et al., “Development of Ipilimumab: Contribution to a New Paradigm for Cancer Immunotherapy,” Semin. Oncol., vol. 37, no. 5, pp. 533–546, Oct. 2010. [CrossRef]
- F. S. Hodi et al., “Nivolumab plus ipilimumab or nivolumab alone versus ipilimumab alone in advanced melanoma (CheckMate 067): 4-year outcomes of a multicentre, randomised, phase 3 trial,” Lancet Oncol., vol. 19, no. 11, pp. 1480–1492, Nov. 2018. [CrossRef]
- H. A. Tawbi et al., “Combined Nivolumab and Ipilimumab in Melanoma Metastatic to the Brain,” N. Engl. J. Med., vol. 379, no. 8, pp. 722–730, Aug. 2018. [CrossRef]
- M. A. Postow et al., “Nivolumab and Ipilimumab versus Ipilimumab in Untreated Melanoma,” N. Engl. J. Med., vol. 372, no. 21, pp. 2006–2017, May 2015. [CrossRef]
- I. M. Chen et al., “Randomized Phase II Study of Nivolumab With or Without Ipilimumab Combined With Stereotactic Body Radiotherapy for Refractory Metastatic Pancreatic Cancer (CheckPAC),” J. Clin. Oncol., vol. 40, no. 27, pp. 3180–3189, Sep. 2022. [CrossRef]
- A. Valachis et al., “Improved survival without increased toxicity with influenza vaccination in cancer patients treated with checkpoint inhibitors,” OncoImmunology, vol. 10, no. 1, p. 1886725, Jan. 2021. [CrossRef]
- X. Wu, T. Li, R. Jiang, X. Yang, H. Guo, and R. Yang, “Targeting MHC-I molecules for cancer: function, mechanism, and therapeutic prospects,” Mol. Cancer, vol. 22, no. 1, p. 194, Dec. 2023. [CrossRef]
- H. Salmon et al., “Expansion and Activation of CD103+ Dendritic Cell Progenitors at the Tumor Site Enhances Tumor Responses to Therapeutic PD-L1 and BRAF Inhibition,” Immunity, vol. 44, no. 4, pp. 924–938, Apr. 2016. [CrossRef]
- A. R. Sánchez-Paulete et al., “Cancer Immunotherapy with Immunomodulatory Anti-CD137 and Anti–PD-1 Monoclonal Antibodies Requires BATF3-Dependent Dendritic Cells,” Cancer Discov., vol. 6, no. 1, pp. 71–79, Jan. 2016. [CrossRef]
- S. Khubchandani, M. S. Czuczman, and F. J. Hernandez-Ilizaliturri, “Dacetuzumab, a humanized mAb against CD40 for the treatment of hematological malignancies,” Curr. Opin. Investig. Drugs Lond. Engl. 2000, vol. 10, no. 6, pp. 579–587, Jun. 2009.
- P. W. Johnson et al., “A Cancer Research UK phase I study evaluating safety, tolerability, and biological effects of chimeric anti-CD40 monoclonal antibody (MAb), Chi Lob 7/4.,” J. Clin. Oncol., vol. 28, no. 15_suppl, pp. 2507–2507, May 2010. [CrossRef]
- H. J. McKenna, “Role of hematopoietic growth factors/flt3 ligand in expansion and regulation of dendritic cells:,” Curr. Opin. Hematol., vol. 8, no. 3, pp. 149–154, May 2001. [CrossRef]
- K. T. Byrne and R. H. Vonderheide, “CD40 Stimulation Obviates Innate Sensors and Drives T Cell Immunity in Cancer,” Cell Rep., vol. 15, no. 12, pp. 2719–2732, Jun. 2016. [CrossRef]
- L. Miao, Y. Zhang, and L. Huang, “mRNA vaccine for cancer immunotherapy,” Mol. Cancer, vol. 20, no. 1, p. 41, Dec. 2021. [CrossRef]
- M. Saxena, S. H. Van Der Burg, C. J. M. Melief, and N. Bhardwaj, “Therapeutic cancer vaccines,” Nat. Rev. Cancer, vol. 21, no. 6, pp. 360–378, Jun. 2021. [CrossRef]
- D. Fioretti, S. Iurescia, V. M. Fazio, and M. Rinaldi, “DNA Vaccines: Developing New Strategies against Cancer,” J. Biomed. Biotechnol., vol. 2010, pp. 1–16, 2010. [CrossRef]
- W. Liu, H. Tang, L. Li, X. Wang, Z. Yu, and J. Li, “Peptide-based therapeutic cancer vaccine: Current trends in clinical application,” Cell Prolif., vol. 54, no. 5, p. e13025, May 2021. [CrossRef]
- Y. Yuan, F. Gao, Y. Chang, Q. Zhao, and X. He, “Advances of mRNA vaccine in tumor: a maze of opportunities and challenges,” Biomark. Res., vol. 11, no. 1, p. 6, Jan. 2023. [CrossRef]
- Z. Wijfjes, F. J. Van Dalen, C. M. Le Gall, and M. Verdoes, “Controlling Antigen Fate in Therapeutic Cancer Vaccines by Targeting Dendritic Cell Receptors,” Mol. Pharm., vol. 20, no. 10, pp. 4826–4847, Oct. 2023. [CrossRef]
- U. Sahin et al., “An RNA vaccine drives immunity in checkpoint-inhibitor-treated melanoma,” Nature, vol. 585, no. 7823, pp. 107–112, Sep. 2020. [CrossRef]
- S. S. Rosa, D. M. F. Prazeres, A. M. Azevedo, and M. P. C. Marques, “mRNA vaccines manufacturing: Challenges and bottlenecks,” Vaccine, vol. 39, no. 16, pp. 2190–2200, Apr. 2021. [CrossRef]
- X. Huang, G. Zhang, T.-Y. Tang, X. Gao, and T.-B. Liang, “Personalized pancreatic cancer therapy: from the perspective of mRNA vaccine,” Mil. Med. Res., vol. 9, no. 1, p. 53, Oct. 2022. [CrossRef]
- Y. L. Vishweshwaraiah and N. V. Dokholyan, “mRNA vaccines for cancer immunotherapy,” Front. Immunol., vol. 13, p. 1029069, Dec. 2022. [CrossRef]
- T. Uehata and O. Takeuchi, “RNA Recognition and Immunity—Innate Immune Sensing and Its Posttranscriptional Regulation Mechanisms,” Cells, vol. 9, no. 7, p. 1701, Jul. 2020. [CrossRef]
- L. A. Rojas et al., “Personalized RNA neoantigen vaccines stimulate T cells in pancreatic cancer,” Nature, vol. 618, no. 7963, pp. 144–150, Jun. 2023. [CrossRef]
- A. Wadhwa, A. Aljabbari, A. Lokras, C. Foged, and A. Thakur, “Opportunities and Challenges in the Delivery of mRNA-Based Vaccines,” Pharmaceutics, vol. 12, no. 2, p. 102, Jan. 2020. [CrossRef]
- U. Sahin, K. Karikó, and Ö. Türeci, “mRNA-based therapeutics — developing a new class of drugs,” Nat. Rev. Drug Discov., vol. 13, no. 10, pp. 759–780, Oct. 2014. [CrossRef]
- M. Diken et al., “Selective uptake of naked vaccine RNA by dendritic cells is driven by macropinocytosis and abrogated upon DC maturation,” Gene Ther., vol. 18, no. 7, pp. 702–708, Jul. 2011. [CrossRef]
- H. Kato and T. Fujita, “RIG-I-like receptors and autoimmune diseases,” Curr. Opin. Immunol., vol. 37, pp. 40–45, Dec. 2015. [CrossRef]
- A.-K. Minnaert et al., “Strategies for controlling the innate immune activity of conventional and self-amplifying mRNA therapeutics: Getting the message across,” Adv. Drug Deliv. Rev., vol. 176, p. 113900, Sep. 2021. [CrossRef]
- H.-G. Hu and Y.-M. Li, “Emerging Adjuvants for Cancer Immunotherapy,” Front. Chem., vol. 8, p. 601, Jul. 2020. [CrossRef]
- S. Van Lint et al., “Preclinical Evaluation of TriMix and Antigen mRNA-Based Antitumor Therapy,” Cancer Res., vol. 72, no. 7, pp. 1661–1671, Apr. 2012. [CrossRef]
- M. H. Andersen, “Novel immune modulatory vaccines targeting TGFβ,” Cell. Mol. Immunol., vol. 20, no. 5, pp. 551–553, Mar. 2023. [CrossRef]
- H. Sugiyama, “Wilms’ Tumor GeneWT1: Its Oncogenic Function and Clinical Application,” Int. J. Hematol., vol. 73, no. 2, pp. 177–187, Feb. 2001. [CrossRef]
- Y. Oji et al., “Overexpression of the Wilms’ tumor gene WT1 in pancreatic ductal adenocarcinoma,” Cancer Sci., vol. 95, no. 7, pp. 583–587, Jul. 2004. [CrossRef]
- M. Shourian, J.-C. Beltra, B. Bourdin, and H. Decaluwe, “Common gamma chain cytokines and CD8 T cells in cancer,” Semin. Immunol., vol. 42, p. 101307, Apr. 2019. [CrossRef]
- J. Leonard, J.-X. Lin, and J. J. O’Shea, “The γc Family of Cytokines: Basic Biology to Therapeutic Ramifications,” Immunity, vol. 50, no. 4, pp. 832–850, Apr. 2019. [CrossRef]
- E. Belnoue et al., “Targeting self- and neoepitopes with a modular self-adjuvanting cancer vaccine,” JCI Insight, vol. 4, no. 11, p. e127305, Jun. 2019. [CrossRef]
- D. T. Le et al., “Evaluation of Ipilimumab in Combination With Allogeneic Pancreatic Tumor Cells Transfected With a GM-CSF Gene in Previously Treated Pancreatic Cancer,” J. Immunother., vol. 36, no. 7, pp. 382–389, Sep. 2013. [CrossRef]
- E. J. Lipson et al., “Safety and immunologic correlates of Melanoma GVAX, a GM-CSF secreting allogeneic melanoma cell vaccine administered in the adjuvant setting,” J. Transl. Med., vol. 13, no. 1, p. 214, Dec. 2015. [CrossRef]
- D. Laheru et al., “Allogeneic Granulocyte Macrophage Colony-Stimulating Factor–Secreting Tumor Immunotherapy Alone or in Sequence with Cyclophosphamide for Metastatic Pancreatic Cancer: A Pilot Study of Safety, Feasibility, and Immune Activation,” Clin. Cancer Res., vol. 14, no. 5, pp. 1455–1463, Mar. 2008. [CrossRef]
- D. T. Le et al., “Safety and Survival With GVAX Pancreas Prime and Listeria Monocytogenes –Expressing Mesothelin (CRS-207) Boost Vaccines for Metastatic Pancreatic Cancer,” J. Clin. Oncol., vol. 33, no. 12, pp. 1325–1333, Apr. 2015. [CrossRef]
- D. T. Le et al., “A Live-Attenuated Listeria Vaccine (ANZ-100) and a Live-Attenuated Listeria Vaccine Expressing Mesothelin (CRS-207) for Advanced Cancers: Phase I Studies of Safety and Immune Induction,” Clin. Cancer Res., vol. 18, no. 3, pp. 858–868, Feb. 2012. [CrossRef]
- Z. Chen et al., “A Neoantigen-Based Peptide Vaccine for Patients With Advanced Pancreatic Cancer Refractory to Standard Treatment,” Front. Immunol., vol. 12, p. 691605, Aug. 2021. [CrossRef]
- C. J. M. Melief and S. H. Van Der Burg, “Immunotherapy of established (pre)malignant disease by synthetic long peptide vaccines,” Nat. Rev. Cancer, vol. 8, no. 5, pp. 351–360, May 2008. [CrossRef]
- S. Pant et al., “First-in-human phase 1 trial of ELI-002 immunotherapy as treatment for subjects with Kirsten rat sarcoma (KRAS)-mutated pancreatic ductal adenocarcinoma and other solid tumors.,” J. Clin. Oncol., vol. 40, no. 16_suppl, pp. TPS2701–TPS2701, Jun. 2022. [CrossRef]
- D. Cullinan et al., “Preliminary Results Of A Phase Ib Clinical Trial Of A Neoantigen Dna Vaccine For Pancreatic Cancer,” HPB, vol. 22, pp. S12–S13, 2020. [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



