
Submitted:
02 August 2024
Posted:
06 August 2024
You are already at the latest version
Abstract
In this work, non-ordered and ordered CeO2-based catalysts are proposed for the CO2 conversion to dimethyl carbonate (DMC). Particularly, a non-ordered mesoporous CeO2, consisting of small nanoparticles of about 8 nm, is compared with two highly porous (635-722 m2/g) ordered CeO2@SBA-15 nanocomposites obtained by two different impregnation strategies (a two-solvent impregnation (TS) and a self-combustion (SC) method), with a final CeO2 loading of 10 wt%. The Rietveld analyses on XRD data combined with TEM imaging evidence the influence of the impregnation strategy on the dispersion of the active phase: nanoparticles of 8 nm for the TS composite vs. 3 nm for the SC composite. The catalytic results show comparable activity for the mesoporous ceria and the CeO2@SBA-15_SC nanocomposite, while a lower DMC yield was found for the CeO2@SBA-15_TS nanocomposite. This finding is presumably ascribed to a partial obstruction of the pores by the CeO2 nanoparticles in the case of the TS composite, which leads to a reduced accessibility of the active phase. On the other hand, in the case of the SC composite, where the CeO2 particle size is much lower than the pore size, there is an improved accessibility of the active phase to the molecules of the reactants.
Keywords:
CO2 utilization
; dimethyl carbonate
; ceria
; mesoporous
; nanocomposites
; catalysis
1. Introduction
Due to the critical consequences of global warming and climate change, caused by the drastic increase in the emissions of anthropogenic greenhouse gases in the atmosphere, one of the main current challenges is the reduction of CO2, the most problematic greenhouse gas. In particular, one of the ways to decrease CO2 concentration is its capture [1,2,3] and its valorization to obtain value-added products (Carbon Capture and Utilization technologies, CCU) like chemicals (e.g. formic acid [4] and dimethyl carbonate [5,6]) and fuels (e.g., methane [7,8], methanol [4,9], and dimethyl ether [4,10,11,12,13]). In this context, the direct synthesis of dimethyl carbonate (DMC) represents a good opportunity to chemically convert CO2 into a valuable solvent applied in different fields, e.g., energy storage (Li-ion batteries) and industrial chemistry (polycarbonates production) [5,6,14]. DMC is traditionally synthesized by many different routes, like the phosgene method, the oxidative carbonylation of methanol, the gas-phase carbonylation of methyl nitrite, the transesterification method, and the alcoholysis of urea. All these methods, however, involve the use of toxic reagents and catalysts or the production of undesired byproducts [5,6,15]. On the other hand, the production of DMC from CO2 and methanol is not only an environmentally friendly route, since it involves the valorization of CO2 as a raw material, but also does not require the use of highly toxic gas-phase reagents or harmful catalysts. Furthermore, this reaction only produces water as a byproduct and requires low amounts of energy and a relatively low-cost equipment [5,6,16]. However, due to the chemical inertness of CO2, the reaction needs to be catalyzed; particularly, ceria-based systems are among the most proposed metal oxide catalysts, due to their chemical stability, redox properties, and high catalytic activity [6,16,17,18,19,20,21,22]. However, cerium is an expensive and critical raw material, making it essential to reduce its use in catalysts to lower both the environmental impact and the cost of the process. For these reasons porous CeO2 and, in particular, mesoporous ceria or nanoceria incorporated into mesostructures could be promising systems to maximize the surface area and favor the diffusion of reactants and products [23]. Mesoporous CeO2 has been synthesized in different ways, such as hard template and soft template processes, sol-gel route, hydrothermal/solvothermal approaches, and precipitation [23]. Hard templating consists in the use of an ordered mesoporous structure (usually silica or carbon) as a template; this template is impregnated with the precursor species of CeO2 and, after CeO2 is formed, the template is removed either by etching (silica) or by combustion (carbon). This approach allows to obtain ordered crystalline mesoporous ceria, with a uniform and adjustable pore size and high thermal stability, but the synthesis processes are often complex, expensive (due to the use of a mesostructured template as a sacrificial material), and the template (particularly silica) is often not easy to remove [23,24,25]. On the other hand, the soft template method involves the use of organic “soft” templating species, like block copolymers, polymers, or smaller organic molecules; these templates are cheaper and easier to remove than the hard templates, however, the obtained mesoporous CeO2 has a disordered pore structure, a broader pore size distribution, and a lower thermal and mechanical stability [23,26,27,28]. The sol-gel methods also often involve the use of organic soft templates, and their use is combined with a sol-gel approach consisting of a hydrolysis reaction, a subsequent condensation, and a final annealing of the obtained material. These syntheses are cheap and sustainable, but also lead to the formation of non-ordered porous structures, often associated with low surface areas [23,29,30,31]. Hydrothermal/solvothermal methods rely on the use of a sealed vessel to perform high-temperature treatments; the use of different temperatures allows to modify the shape of the nanoparticles and their surface area, but an ordered mesoporous structure is often not obtainable [23,32,33,34]. Finally, precipitation methods involve a precipitation reaction between a precursor and a precipitating agent; these approaches, often assisted by the use of soft templating agents, are easy and straightforward, allowing to reach high surface areas but with a non-ordered porous structure [23,35].
Due to its high exposed surface area, several applications have been reported for mesoporous ceria, like catalysis, photocatalysis, water remediation, air purification, degradation of organic pollutants, drug delivery, fuel cells, and sensors. Regarding catalysis, however, only a small number of papers reported the use of mesoporous CeO2 as catalyst for the synthesis of DMC from CO2 and methanol [17,36,37].
Another strategy to design easily accessible CeO2-based systems is to develop mesostructured composites incorporating CeO2 into the channels of inert mesostructures as mesostructured silica (SBA-15, MCM-41). In this framework two-step [38,39,40,41,42,43] and one-step [38,44,45] procedures have been proposed. In the two-step methods CeO2 nanoparticles are incorporated inside the pores of a pre-synthesized support by post-synthesis methods; on the other hand, in the one-step approach the support is synthesized, usually from alkoxide precursors, together with the CeO2 particles, by introducing both the Si and the Ce precursors in the same reaction batch. Regarding the two-step methods, different impregnation strategies have been used to functionalize silica walls, mainly based on the use of aqueous solutions [40,42,43]. Indeed, the silanol groups at the surface render silica hydrophilic and accessible to aqueous solutions containing the cerium precursors. Beside the impregnation approaches involving the use of aqueous solutions, another method, called the molten nitrate method, based on an apolar solvent [39], has been proposed; it consists in the dispersion of the support and cerium nitrate in an apolar solvent (toluene) which is then heated beyond the melting point of cerium nitrate, allowing its introduction into the pores of the support. Furthermore, solid-state approaches, based on the grinding of cerium nitrate with the support, followed by a thermal treatment to induce the decomposition of nitrates [38,41], have been proposed.
Among the cited papers, the only one dealing with the development of CeO2@SBA-15 composites for the synthesis of DMC from CO2 and methanol used a slurry impregnation approach, based on the insertion of an aqueous solution of cerium nitrate into the mesopores of the support, followed by the evaporation of water and a final thermal decomposition of the cerium nitrate to CeO2 [40].
Among the various impregnation methods reported in the literature, a promising strategy is based on the use of two solvents: a polar solvent, like water, and an apolar solvent, to favor the loading of metal precursor solutions into the hydrophilic pores. This method was efficiently used to develop regenerable and efficient sorbents for H2S removal based on ZnO and Fe2O3 in supports like SBA-15 [46,47], MCM-41 [48] and MCM-48 [48] and demonstrated to be more efficient than the conventional water-based incipient impregnation. To allow a complete incorporation of the metal oxide precursors inside the pores of the support, this method relies on using an amount of aqueous solution corresponding to the pore volume of the support.
Another rarely used strategy involves the combination of an impregnation route with a self-combustion reaction. This approach consists in the impregnation of the support with an aqueous solution of the metal nitrates and a reducing agent; after the evaporation of the water the self-combustion reaction (i.e., a redox reaction between the oxidant nitrates and the reducing agent) is ignited with a thermal treatment, leading to the formation of the metal oxides. The self-combustion method has been widely reported and is of particular interest since it allows to obtain various supported and unsupported nano-sized metal oxides [49,50,51,52,53], due to the presence of a reducing agent together with the nitrate (oxidizer) in the reaction environment. Furthermore, this method usually relies on the use of only water as solvent and on cheap and environmentally friendly reducing agents (citric acid, glycine), resulting to be a cheap and green approach. However, regarding its use in combination with an impregnation approach, only a few instances have been reported, focused on the obtainment of Cu-based nanocomposites on SBA-15 or mesostructured γ-Al2O3 [54,55].
In this work we present a non-ordered mesoporous CeO2 catalyst; for its synthesis, a precipitation approach assisted by soft-templating has been chosen, due to its simplicity, quickness, and cheapness. With the aim of investigating both the possibility of reducing the amount of active phase and the effect of an ordered mesoporous structure, the mesoporous CeO2 catalyst is put in comparison with two different CeO2@SBA-15 composites. In this context, SBA-15 has been chosen as support due to its large pore size, which allows it to easily host an active phase in form of nanoparticles, compared to other mesostructures with smaller pores (MCM-41). Since the impregnation strategy has been demonstrated to strongly influence the dispersion of the active phase into the porous support, the particle size and the crystallinity, all critical features for the catalytic activity, we focused on two impregnation routes, namely the two-solvent and the self-combustion impregnation. These strategies, indeed, as mentioned above, proved to be able to efficiently disperse several metal oxides onto different mesostructured siliceous supports (i.e., SBA-15, MCM-41, MCM-48) [46,47,48,54,55]. Furthermore, to the best of our knowledge, these impregnation methods, rarely reported in the literature, have never been used to synthesize CeO2-based composites.
2. Materials and Methods
Chemicals. Cerium(III) nitrate hexahydrate (99.5%, Acros organics) were used in all CeO2 synthesis. NaOH (pellets, Sigma Aldrich) and cetyltrimethyl ammonium bromide (CTAB) (98%, Sigma-Aldrich) were used in soft-template surfactant-assisted precipitation synthesis. Pluronic P123 (Average number average molecular weight ≈ 5800, Sigma-Aldrich), HCl (37%, VWR Chemicals), and TEOS (98%, Acros Organics) were used in the synthesis of the SBA-15 support. Hexane (97%, VWR Chemicals) and bi-distilled water were used in Two-solvent impregnation synthesis. Citric acid (99.5%, Aldrich) was used in the impregnation combined with self-combustion synthesis. All reagents were used as received without further purification.
Synthesis of mesoporous CeO2 (CeO2_Meso). Mesoporous ceria was synthesized using a soft-template surfactant-assisted precipitation method with cetyltrimethyl ammonium bromide (CTAB) as the templating agent, following the procedure reported in [35]. Typically, 1 g of CTAB was added to a cerium nitrate solution (2.17 g of Ce(NO3)3·6H2O in 200 mL) in a 500 mL round-bottom flask at room temperature and stirred gently. Then, a NaOH solution (1 g in 150 mL of distilled water) was added dropwise with continuous stirring at 150 rpm. After the addition was complete, the flask was sealed, and the mixture was maintained under constant stirring for 24 hours. Following thermal aging at 90 °C for 3 hours, the pale-yellow precipitate was filtered and washed twice with 200 mL of hot distilled water (80 °C). The sample was dried in a static oven at 100 °C for 6 hours and then calcined at 450 °C for 4 hours (heating rate 5 °C per minute).
Synthesis of the SBA-15 support (SBA-15). The synthesis of the SBA-15 support was carried out adapting the procedure reported by Zhao et al. [56,57]. Typically, 4 g of Pluronic P123 were dissolved in 120 g of HCl 2M and 30 g of bi-distilled water into an Erlenmeyer flask by stirring at 600 RPM and at 35 °C by a water-bath for 24 hours. Then, the stirring was decrease to 100 RPM and kept overnight. 9 g of TEOS were added dropwise and the stirring was maintained for other 24 h at 35 °C. The resulting suspension was then put into a sealed Teflon-lined autoclave and heated at 100 °C in static conditions for other 24 h. The product was subsequently filtered, washed with warm distilled water (75 °C - 80 °C), and dried at 35 °C for 24 h. The obtained powder was finally calcined at 550 °C for 6 h with a ramp of 5 °C min-1.
Synthesis of CeO2@SBA-15_TS. For the synthesis of the CeO2@SBA-15_TS composite, SBA-15 was impregnated with 10% in weight of CeO2 adapting the two-solvent approach reported in [46,47,48]. In a typical synthesis, the support was firstly dried at 120 °C overnight to remove the adsorbed water; 0.5 g of support was then submerged in 10 mL of hexane into a beaker that was then covered with a watch glass and kept under stirring at 300 RPM for 2 h. The stirring was increased to 400 RPM and 0.57 mL of a 0.56 mM Ce(NO3)3 6H2O aqueous solution was added dropwise. After 2 h the watch glass was removed from the beaker and the temperature was set to 80 °C, to let the hexane evaporate; when the evaporation was almost complete, the beaker was put into an oven at 80 °C overnight. Eventually, the obtained powder was calcined at 500 °C for 2 h with a 2°C min-1 ramp.
Synthesis of CeO2@SBA-15_SC. For the synthesis of the CeO2@SBA-15_SC composite, SBA-15 was impregnated with 10% in weight of CeO2 adapting the self-combustion approach reported in [54,55]. Typically, a 0.56 mM aqueous solution of Ce(NO3)3 6H2O was prepared and mixed with another solution of citric acid, with a citric acid/Ce molar ratio of 1:1. 0.5 g of support, after a previous drying at 120 °C, were then dispersed into 5.7 mL of the mixed solution (i.e., containing the cerium precursor and the citric acid) into a beaker under vigorous stirring until a viscous paste was obtained, then sonicated for 5 min and submitted to a 300 °C treatment for 1 h by putting it into a pre-heated oven to induce the self-combustion reaction between the nitrates (oxidizing agents) and citric acid (reducing agent).
Characterization techniques. Wide-angle X-ray diffraction (WA-XRD) patterns were acquired in the 2θ range 10–100° using a PANalytical X’pert Pro (Malvern PANalytical, Malvern, UK) equipped with Cu Kα source (1.5418 Å). Small-angle X-ray diffraction (SA-XRD) patterns were acquired in the 2θ range 0.7–3° using a Seifert X3000 instrument (Seifert, Radevormwald, Germany) equipped with Cu Kα source. The hexagonal lattice parameter of mesostructured samples was calculated using the equation . Rietveld analysis was performed with the software MAUD. LaB6 from NIST was used as a reference material to determine the instrumental parameters. The CIF structure used for the refinement was 1562989. The simulation of aluminum silica glass was carried out by means of Le Bail model [58,59].
Nitrogen physisorption isotherms were acquired at −196 °C, using a 3Flex physisorption/chemisorption analyzer provided by Micromeritics. All samples were treated under vacuum at 250 °C (heating ramp, 1 °C/min) for 12 h before the analysis. The Brunauer–Emmett–Teller (BET) specific surface area (SA) was calculated from the adsorption branch in the 0.04–0.3 P/P0 interval. The total pore volume (Vp) was determined at P/P0 = 0.99, and the mean pore diameter (Dp) was extrapolated by applying the Barrett–Joyner–Halenda (BJH) model to the desorption data for all samples. The pore wall thickness (Tw) was calculated using the formula.
Transmission Electron Microscopy (TEM) images and Energy Dispersive X-ray (EDX) characterization were carried out using a JEOL JEM 1400-PLUS microscope (JEOL, Akishima, Tokyo, Japan) operating at an accelerating voltage of 120 kV. The samples were first finely ground and dispersed in ethanol by an ultrasound treatment. The obtained suspensions were deposited onto 200 mesh carbon-coated copper grids.
Catalytic tests. The catalytic tests for DMC synthesis were performed in batch conditions under magnetic stirring, using a 100 mL high-pressure reactor manufactured by Berghof (BR-100). For each test, 0.250 g of the catalyst were put into the reactor together with 10 mL of liquid methanol (≥ 99.8%); the reactor was then purged for three times with CO2 in order to remove air and subsequently pressurized at 5.0 MPa with CO2 (99.9%) and heated to 150 °C with a heating rate of 2 °C min-1. After 3h of reaction, the reactor was cooled down to room temperature and the catalyst was recovered by centrifugation. The reaction products were analyzed using a gas-chromatograph equipped with a flame ion detector (Agilent Technologies 6890N GC-FID) and with a capillary column (Zebron ZB-WAX, 30m x 0.25 mm x 0.25 μm), using helium as carrier gas with a flow rate of 1 mL min-1. 1-Propanol (≥99.8%) was added to dimethyl carbonate in methanol as an internal standard to quantitatively analyze the products using the calibration curve method. The DMC yield (mmol gcat-1) was estimated using Eq.1, as follows
3. Results and discussion
The wide-angle XRD patterns (WA-XRD) of the investigated systems are showed in Figure 1a. The sample CeO2_Meso shows the diffraction peaks attributable to cubic CeO2 (PDF card 00-034-0394), no other phases were detected. The Rietveld analysis (Figure S1) points out a mean crystallite size of 7.9 ± 0.1 nm. The same signals from CeO2 are observed in the XRD pattern of the CeO2@SBA-15_TS together with a broad band with a maximum located at a 2θ value of about 23°, ascribed to the amorphous silica of the SBA-15 support, the pattern of which has been reported for reference. Interestingly, also in this case the Rietveld analysis (Figure S2) gives a mean crystallite size of 7.9 ± 0.1 nm, allowing a direct comparison with the unsupported CeO2_Meso system. Differently from what have been observed on the TS composite, the CeO2@SBA-15_SC sample shows very broad signals at 2θ values of about 28.5°, 47.5°, and 56.4°, corresponding to the three most intense crystalline reflections of cubic CeO2, respectively (111), (220), and (311); also in this case, the amorphous band attributed to the support is visible. The Rietveld analysis (Figure S3) points out a mean crystallite size of 2.6 ± 0.1 nm, rather smaller than the pore size of the support (Table 1). This finding suggests that the CeO2 active phase has been dispersed by the impregnation in the form of ultra-small nanoparticles, presumably inside the mesopores of the support. This result is likely to be attributed to the rapid propagation of self-combustion reactions, which leads to a very fast conversion of metal nitrates in metal oxides. On the other hand, the thermal decomposition of a nitrate to an oxide, which takes place during the functionalization with TS impregnation, is presumably slower, being driven merely by the temperature rise. It can be thus inferred that, during the fast self-combustion reaction, the metal oxide particles do not have the time to grow as much as they can during a nitrate decomposition, leading to the formation of smaller nanoparticles.
The small-angle XRD patterns (SA-XRD, Figure 1b) of CeO2_Meso only show an extremely broad band centered at a 2θ value of about 1.8°, suggesting a possible disordered mesoporosity. On the other hand, the patterns of the two composites and their support show a main signal (100) located at a 2θ value of 0.98° and two lower signals at higher 2θ values. These signals are attributable to a hexagonal mesoporous arrangement (p6mm), typical of the SBA-15 structure, indicating that the ordered mesoporous arrangement was maintained after the functionalization process. It can be noticed that the position of the main mesostructure peak (100) is the same (Table 1) for both the support and the two composites, presumably indicating that the lattice parameter of the mesopore arrangement did not change significantly with the impregnation.
All nitrogen physisorption isotherms (Figure 2a) can be described as type IV, typical of mesoporous samples, since all of them feature a capillary condensation branch. The physisorption isotherms of the CeO2_Meso sample, despite being attributable to a mesoporous material (type IV), show a significantly wide hysteresis cycle with not very steep capillary condensation branches, indicating the presence of a disordered mesoporosity, as already suggested by the SA-XRD analysis. On the other hand, the isotherms of the two composites show a very steep capillary condensation adsorption branch, comparable to that of the support (H1 hysteresis cycle), reported for reference, indicating that the mesoporous order was maintained after the incorporation of CeO2. Furthermore, the adsorption branch of the support falls at about the same value of relative pressure of the two composites (about 0.75), indicating that the functionalization did not cause a significant narrowing of the pores. In the TS composite, it can be noticed that the capillary condensation desorption branch is much less steep and present a double concavity, presumably indicating a partial obstruction of the mesopores by the relatively large CeO2 nanoparticles, which causes the formation of inkbottle mesopores (mesopores with a narrow opening) and, consequently, a slower emptying of the pores during the desorption [46,60,61]. As expected, a decrease in terms of surface area and pore volume is observed for both the composite, compared to the support (Table 1), ascribed to the functionalization with the active phase.
The BJH plot (Figure 2b) of CeO2_Meso indicates an extremely wide pore size distribution with a maximum located at 5.8 nm, typical of a non-ordered mesoporous sample. Conversely, the BJH plot of the SC composite and the SBA-15 support show narrow pore size distributions with a similar width and mean pore size (6.1 nm for CeO2@SBA-15_SC and 6.3 nm for SBA-15), indicating, as already suggested by the SA-XRD analysis and the physisorption isotherms, that the impregnation process did not cause neither a significant decrease of the mean pore diameter, nor a loss of mesoporous order in terms of pore size distribution; also the wall thickness does not show a significant change (Table 1). The BJH plot of the TS composite, on the other hand, shows a wide bimodal distribution, with a maximum at 6.1 nm, close to the original value of pore size of the support, and another maximum at about 4 nm, reinforcing the hypothesis of a partial pore obstruction inferred by the observation of the desorption capillary condensation branch. Particularly, the maximum located at 6.1 nm is attributable to unoccupied pores, the one at 4 nm is ascribed to the inkbottle mesopores formed by the incorporation of the CeO2 nanoparticles [46,60,61]. The maximum at 6.1 nm was used to calculate the wall thickness that, as observed for the SC composite, does not show significant differences with that of the support (Table 1).
The TEM micrographs of the samples CeO2_Meso (Figure 3) show a material consisting of large aggregates mainly comprised of nanoparticles of spheroidal shape (with a size of 5-8 nm, in agreement both with the mean crystallite size pointed out by the Rietveld analysis and with the data reported in the literature for the same synthesis process [35]), along with a minor contribution of elongated ones, some of which consisting of chains of spheroidal nanoparticles (indicated by arrows). This finding justifies the mesoporous nature of this system, which is ascribed to a disordered worm-like interparticle porosity.
TEM imaging of the bare SBA-15 support (Figure 4) clearly shows the presence of an ordered mesoporous structure consisting of hexagonally-arranged parallel channels, in agreement with what has been observed in the literature for SBA-15 materials [56].

From the TEM micrographs of the CeO2@SBA-15_TS nanocomposite (Figure 5a-c) it can be observed that the impregnation process led to the formation of elongated CeO2 nanoparticles (the darker spots visible in the micrographs) with a width of about that of the mesopore size (6-7 nm) and a variable length. The dark-field TEM imaging (Figure 5d-f) confirmed the crystalline nature of these particles, as they appear as bright spots. These results are in agreement with the Rietveld analysis, which points out a mean crystallite size of 7.9 nm; this size, indeed, despite being larger than the mean pore diameter of the support (6.3 nm), is attributable to the elongated form of the particles. Therefore, considering their peculiar, elongated shape (always oriented in the same direction of the mesochannels), their size (comparable with the mean pore diameter) and the fact that, at high magnification, the walls of the mesochannels are still visible despite the presence of these particles, it can be assumed that CeO2 is incorporated inside the mesopores. This finding is in agreement with the nitrogen physisorption data, which show a capillary condensation desorption branch less steep that that of the support, presumably due to a partial obstruction of the pores by the CeO2 nanoparticles.
On the other hand, the CeO2@SBA-15_SC nanocomposite did not show, in the bright-field TEM micrographs (Figure 5g-i), any dark spot ascribable to nanoparticles of the active phase. In some zones, however, the mesochannels present a darker color for their whole length (see arrows in Figure 5g), presumably indicating an incorporation of CeO2 inside the pores in a more highly dispersed form (smaller nanoparticles). This assumption is confirmed by the dark-field TEM imaging (Figure 5j-l), which clearly points out the fine functionalization of the mesopores, indicated by the fact that the mesochannels appear bright, due to the presence of finely distributed ultra-small CeO2 crystalline nanoparticles inside them. From these observations, it can be assumed that the active phase has been dispersed in form of very small nanoparticles, but some zones/mesochannels of the support present a higher loading of active phase than others, indicated by a higher contrast (darker zones) in bright-field micrographs (Figure 5g) as well as by brighter zones in the dark-field micrographs, evidenced by arrows in (Figure 5k,l).
The line profile EDX analysis on CeO2@SBA-15_TS shows an overall homogeneity in the distribution of the atomic species attributable to the support and to the active phase (Si and Ce, respectively), indicating that all the CeO2 have presumably been properly dispersed inside the pores, with no segregation of CeO2 particles outside the pores. Furthermore, five EDX spectra were acquired in five different regions (Table S1) and all of them point out a CeO2 weight percentage comprised between 9.5wt% and 12.1wt%, with a mean value of 11(±1) wt%, in very good agreement with the theoretical value of 10wt% and with a low standard deviation.
The low homogeneity of functionalization with active phase of the CeO2@SBA-15_SC composite, suggested by TEM imaging, is also supported by the line-profile EDX analysis which, for CeO2@SBA-15_SC points out a less homogeneous distribution of Ce and Si throughout the material, compared to CeO2@SBA-15_TS. EDX spectra, acquired in thirteen different regions (Table S2), indicate the same average CeO2 wt% loading shown by the TS composite (11%), but with a significantly higher standard deviation (6%), indicating that the total CeO2 loading is the same for the two composites, but CeO2@SBA-15_SC has higher local inhomogeneities. It can be presumed that these inhomogeneities in the functionalization are a consequence of the rapid metal nitrate-metal oxide conversion typical of self-combustion reactions mentioned before. It can be thus inferred that this rapid transition leads to an almost instantaneous formation of the oxide species, which do not have the time to equally distribute inside all the mesochannels of the support.
The catalytic tests evidence that, as expected, the CeO2_Meso sample shows the better performance in terms of DMC yield (0.941 mmol/gcat); this result can be ascribed to the fact that this catalyst is composed of pure CeO2, which is the active phase of the reaction. On the other hand, the two composites show lower values of DMC yield (0.097 mmol/gcat for CeO2@SBA-15_SC and 0.066 mmol/gcat for CeO2@SBA-15_TS) due to the significantly lower amount of active phase contained in these systems (10% in weight), compared to the CeO2_Meso sample. However, it is important to point out that, considering the DMC yield expressed as a function of the amount of active phase, the CeO2@SBA-15_SC sample shows similar performances (0.971 mmol/gact.ph.) than the CeO2_Meso sample (0.941 mmol/gact.ph.). On the other hand, the CeO2@SBA-15_TS composite shows lower performances (0.662 mmol/gact.ph.); this fact can be probably attributed to the lower degree of dispersion (larger nanoparticles) of the active phase into the mesostructure in the case of this sample, compared to the CeO2@SBA-15_SC composite. XRD and TEM analysis, indeed, pointed out how the active phase formed significantly larger nanoparticles for the CeO2@SBA-15_TS composite, compared to the SC one, consequently leading to a less fine dispersion and to a lower area of contact with the reactants, as well as to a reduced accessibility of the active phase and to a lower diffusion of reactants and products due to the partial pore obstruction. The acquired results are in agreement with the data reported in [62] for the theoretical value of yield (mol%) of DMC for this reaction under the investigated conditions of temperature, pressure, and feed ratio. However, Pu et. al. [40] reported much better results for similar composite catalysts (CeO2@SBA-15), probably due significantly different experimental conditions, like a higher CeO2 weight loading (12.3%), a lower temperature (130 °C), a higher reaction time (10 h), a smaller reactor volume (50 mL), and a higher amount of catalyst (500 mg).

Table 2.
Results of the catalytic tests. Conditions: 0.250 g of catalyst; 10 mL of liquid methanol; P = 5.0 MPa; T = 150 °C; reaction time = 3h.
Table 2.
Results of the catalytic tests. Conditions: 0.250 g of catalyst; 10 mL of liquid methanol; P = 5.0 MPa; T = 150 °C; reaction time = 3h.
| Catalyst | Yield (mmol/gcat) | Yield (mmol/gact.ph.) | Yield (mol%) |
| CeO2_Meso | 0.941 | 0.941 | 2x10-3 |
| CeO2@SBA-15_SC | 0.097 | 0.971 | 2x10-4 |
| CeO2@SBA-15_TS | 0.066 | 0.662 | 1x10-4 |
4. Conclusions
In this work a study on different non-ordered and ordered mesoporous CeO2-based catalysts for the CO2 conversion to DMC is presented. Particularly, a non-ordered mesoporous catalyst, consisting of pure CeO2 (CeO2_Meso), has been compared with two composites obtained by dispersing CeO2, with a 10 wt% loading, on an ordered mesoporous siliceous support (SBA-15), with the aim of maximizing the catalytic performance with a low amount of active phase. The two composites have been obtained by functionalizing the support with two different impregnation strategies: a two-solvent impregnation (TS) and an impregnation combined with a self-combustion reaction (SC). The combination of XRD, nitrogen physisorption, and TEM characterization points out that for the composite obtained with the TS strategy (CeO2@SBA-15_TS) the impregnation led to the formation of CeO2 nanoparticles of about 8 nm located inside the mesopores. On the other hand, the composite obtained with the SC method (CeO2@SBA-15_SC) features significantly smaller CeO2 nanoparticles (about 3 nm), also in this case incorporated inside the pores of the mesostructured support. The study of the catalytic performances shows how CeO2_Meso, consisting of pure CeO2, features the best performances; the composites, on the other hand, show lower performances, due to their lower amount of active phase (10%). Normalizing the catalytic activity as a function of the active phase of the catalyst, however, it can be noticed how the CeO2@SBA-15_SC composite shows similar performance (DMC yield = 0.971 mmol/gact.ph.) to CeO2_Meso (DMC yield = 0.941 mmol/gact.ph.), presumably due to the fine dispersion of the active phase throughout the mesostructured matrix which leads to a high exposed area of active phase. This assumption is also supported by the fact that the other composite, obtained by the TS approach and featuring larger CeO2 nanoparticles, with a possible pore obstruction, shows a lower catalytic activity (0.662 mmol/gact.ph.). The combination of an impregnation strategy with a self-combustion reaction, thus, resulted to be the most promising method to obtain supported catalysts with a highly dispersed active phase, leading to an improvement in catalytic performances over the composite obtained by the two-solvent approach. Future studies will focus on the optimization of this approach, by studying different reducing agents, different pH values, and different nitrate/reducing agent ratios, with the aim of improve the dispersion and further reduce the size of nanoparticles. Different loadings of CeO2 will also be studied, in order to maximize the performance while maintaining the lowest possible amount of active phase, due to the fact that cerium is considered a critical raw material, with the consequent necessity of drastically reduce its use. Furthermore, the effect of different parameters (temperature, pressure, MeOH/CO2 ratio) on the catalytic performance will be investigated.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
Conceptualization, C.C.; methodology, C.C., N.R.; validation, C.C., N.R.; formal analysis, N.R., F.S.; investigation, N.R.; resources, C.C.; data curation, N.R., F.S.; writing—original draft preparation, F.S.; writing—review and editing, C.C., N.R., V.M., F.S.; visualization, F.S., N.R.; supervision, C.C.; project administration, C.C.; funding acquisition, C.C. All authors have read and agreed to the published version of the manuscript.”
Funding
Alkeemia S.p.A. is acknowledged for financing the Ph.D. grant and the research project of Nicoletta Rusta. The financial support of the European Union NextGenerationEU under the National Recovery and Resilience Plan (NRRP) of Ministero dell’Università e della Ricerca (MUR) (Project code PE0000021, Network 4 Energy Sustainable Transition, NEST) is acknowledged. University of Cagliari and Fondazione di Sardegna are acknowledged for the financial support – project: “Sorbents for environmental applications: a synergetic computational modelling and experimental approach” CUP F73C22001190007(2021).
Data Availability Statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author/s.
Acknowledgments
Thanks are due to Dr. Andrea Ardu and to the “Centro Servizi di Ateneo per la Ricerca (CeSAR)” for the use of the TEM measurements performed with JEOL JEM 1400 PLUS.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Mikulčić, H.; Skov, I.R.; Dominković, D.F.; Alwi, S.R.W.; Manan, Z.A.; Tan, R.; Duić, N.; Mohamad, S.N.H.; Wang, X. Flexible Carbon Capture and Utilization technologies in future energy systems and the utilization pathways of captured CO2. Renew. Sustain. Energy Rev. 2019, 114, 109338. [Google Scholar] [CrossRef]
- Fu, L.; Ren, Z.; Si, W.; Ma, Q.; Huang, W.; Liao, K.; Huang, Z.; Wang, Y.; Li, J.; Xu, P. Research progress on CO2 capture and utilization technology. J. CO2 Util. 2022, 66. [Google Scholar] [CrossRef]
- Ghiat, I.; Al-Ansari, T. A review of carbon capture and utilisation as a CO2 abatement opportunity within the EWF nexus. J. CO2 Util. 2021, 45. [Google Scholar] [CrossRef]
- Álvarez, A.; Bansode, A.; Urakawa, A.; Bavykina, A.V.; Wezendonk, T.A.; Makkee, M.; Gascon, J.; Kapteijn, F. Challenges in the Greener Production of Formates/Formic Acid, Methanol, and DME by Heterogeneously Catalyzed CO2Hydrogenation Processes. Chem. Rev. 2017, 117, 9804–9838. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Shi, F.; Wang, L. A Review of Catalysts for Synthesis of Dimethyl Carbonate. Catalysts 2024, 14, 259. [Google Scholar] [CrossRef]
- Zhang, M.; Xu, Y.; Williams, B.L.; Xiao, M.; Wang, S.; Han, D.; Sun, L.; Meng, Y. Catalytic materials for direct synthesis of dimethyl carbonate (DMC) from CO2. J. Clean. Prod. 2020, 279, 123344. [Google Scholar] [CrossRef]
- Ashok, J.; Pati, S.; Hongmanorom, P.; Tianxi, Z.; Junmei, C.; Kawi, S. A review of recent catalyst advances in CO2 methanation processes. Catal. Today 2020, 356, 471–489. [Google Scholar] [CrossRef]
- Atzori, L.; Cutrufello, M.G.; Meloni, D.; Secci, F.; Cannas, C.; Rombi, E. Soft-templated NiO–CeO2 mixed oxides for biogas upgrading by direct CO2 methanation. Int. J. Hydrogen Energy 2023, 48, 25031–25043. [Google Scholar] [CrossRef]
- Guil-López, R.; Mota, N.; Llorente, J.; Millán, E.; Pawelec, B.; Fierro, J.L.G.; Navarro, R.M. Methanol Synthesis from CO2: A Review of the Latest Developments in Heterogeneous Catalysis. Materials 2019, 12, 3902. [Google Scholar] [CrossRef] [PubMed]
- Catizzone, E.; Migliori, M.; Purita, A.; Giordano, G. Ferrierite vs. γ-Al2O3: The superiority of zeolites in terms of water-resistance in vapour-phase dehydration of methanol to dimethyl ether. J. Energy Chem. 2019, 30, 162–169. [Google Scholar] [CrossRef]
- Catizzone, E.; Freda, C.; Braccio, G.; Frusteri, F.; Bonura, G. Dimethyl ether as circular hydrogen carrier: Catalytic aspects of hydrogenation/dehydrogenation steps. J. Energy Chem. 2020, 58, 55–77. [Google Scholar] [CrossRef]
- Secci, F.; Mameli, V.; Rombi, E.; Lai, S.; Angotzi, M.S.; Russo, P.A.; Pinna, N.; Mureddu, M.; Cannas, C. On the role of the nature and density of acid sites on mesostructured aluminosilicates dehydration catalysts for dimethyl ether production from CO2. J. Environ. Chem. Eng. 2023, 11. [Google Scholar] [CrossRef]
- Cara, C.; Secci, F.; Lai, S.; Mameli, V.; Skrodczky, K.; Russo, P.A.; Ferrara, F.; Rombi, E.; Pinna, N.; Mureddu, M.; et al. On the design of mesostructured acidic catalysts for the one-pot dimethyl ether production from CO2. J. CO2 Util. 2022, 62. [Google Scholar] [CrossRef]
- Pyo, S.-H.; Park, J.H.; Chang, T.-S.; Hatti-Kaul, R. Dimethyl carbonate as a green chemical. Curr. Opin. Green Sustain. Chem. 2017, 5, 61–66. [Google Scholar] [CrossRef]
- Tan, H.-Z.; Wang, Z.-Q.; Xu, Z.-N.; Sun, J.; Xu, Y.-P.; Chen, Q.-S.; Chen, Y.; Guo, G.-C. Review on the synthesis of dimethyl carbonate. Catal. Today 2018, 316, 2–12. [Google Scholar] [CrossRef]
- Raza, A.; Ikram, M.; Guo, S.; Baiker, A.; Li, G. Green Synthesis of Dimethyl Carbonate from CO2 and Methanol: New Strategies and Industrial Perspective. Adv. Sustain. Syst. 2022, 6. [Google Scholar] [CrossRef]
- Yang, Z.; Zheng, J.T.; Lu, X.; Lin, M.M.; Cai, D.; Wang, Y.; Yu, W.-Y.; Zhu, Y.; Xia, Y. Porous ceria materials for efficient direct conversion of carbon dioxide and methanol to dimethyl carbonate. Mater. Adv. 2024. [Google Scholar] [CrossRef]
- Hou, G.; Wang, Q.; Xu, D.; Fan, H.; Liu, K.; Li, Y.; Gu, X.; Ding, M. Dimethyl Carbonate Synthesis from CO2 over CeO2 with Electron-Enriched Lattice Oxygen Species. Angew. Chem. Int. Ed. 2024, 63, e202402053. [Google Scholar] [CrossRef] [PubMed]
- Marciniak, A.A.; Alves, O.C.; Appel, L.G.; Mota, C.J. Synthesis of dimethyl carbonate from CO2 and methanol over CeO2: Role of copper as dopant and the use of methyl trichloroacetate as dehydrating agent. J. Catal. 2019, 371, 88–95. [Google Scholar] [CrossRef]
- Marciniak, A.A.; Santos, E.C.S.; Caraballo-Vivas, R.J.; Alves, O.C.; da Costa, M.E.H.M.; Garcia, F.; Mota, C.J.A. CeO2-Decorated α-Fe2O3 Nanorings for the Direct Synthesis of Dimethyl Carbonate from CO2 and Methanol. Energy Fuels 2023, 38, 628–636. [Google Scholar] [CrossRef]
- Seeharaj, P.; Saenman, T.; Phiwhom, T.; Muangsuwan, C.; Srinives, S.; Kim-Lohsoontorn, P. Improvement of surface properties of metal doped-CeO2 nanospindle catalysts for direct synthesis of dimethyl carbonate from CO2 and methanol. J. Environ. Chem. Eng. 2023, 11. [Google Scholar] [CrossRef]
- Kulthananat, T.; Kim-Lohsoontorn, P.; Seeharaj, P. Ultrasonically assisted surface modified CeO2 nanospindle catalysts for conversion of CO2 and methanol to DMC. Ultrason. Sonochemistry 2022, 90, 106164. [Google Scholar] [CrossRef] [PubMed]
- Dubey, M.; Wadhwa, S.; Mathur, A.; Kumar, R. Progress in mesoporous ceria: A review on synthesis strategies and catalytic applications. Appl. Surf. Sci. Adv. 2022, 12. [Google Scholar] [CrossRef]
- Sakina, F.; Muñoz-Ocaña, J.M.; Bouziane, A.; Lopez-Haro, M.; Baker, R.T. Synthesis of mesoporous ceria using metal- and halogen-free ordered mesoporous carbon as a hard template. Nanoscale Adv. 2019, 1, 4772–4782. [Google Scholar] [CrossRef] [PubMed]
- Roggenbuck, J.; Schäfer, H.; Tsoncheva, T.; Minchev, C.; Hanss, J.; Tiemann, M. Mesoporous CeO2: Synthesis by nanocasting, characterisation and catalytic properties. Microporous Mesoporous Mater. 2007, 101, 335–341. [Google Scholar] [CrossRef]
- Liang, X.; Xiao, J.; Chen, B.; Li, Y. Catalytically Stable and Active CeO2 Mesoporous Spheres. Inorg. Chem. 2010, 49, 8188–8190. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Sel, O.; Djerdj, I.; Smarsly, B. Preparation of a large Mesoporous CeO2 with crystalline walls using PMMA colloidal crystal templates. Colloid Polym. Sci. 2006, 285, 1–9. [Google Scholar] [CrossRef]
- Junais, P.M.; Athika, M.; Govindaraj, G.; Elumalai, P. Supercapattery performances of nanostructured cerium oxide synthesized using polymer soft-template. J. Energy Storage 2020, 28, 101241. [Google Scholar] [CrossRef]
- Strunk, J.; Vining, W.C.; Bell, A.T. Synthesis of Different CeO2 Structures on Mesoporous Silica and Characterization of Their Reduction Properties. J. Phys. Chem. C 2011, 115, 4114–4126. [Google Scholar] [CrossRef]
- Dhall, A.; Self, W. Cerium Oxide Nanoparticles: A Brief Review of Their Synthesis Methods and Biomedical Applications. Antioxidants 2018, 7, 97. [Google Scholar] [CrossRef]
- Zagaynov, I.V.; Kutsev, S.V. Formation of mesoporous nanocrystalline ceria from cerium nitrate, acetate or acetylacetonate. Appl. Nanosci. 2013, 4, 339–345. [Google Scholar] [CrossRef]
- Borjas-García, S.E.; Medina-Flores, A.; Béjar, L.; Martínez-Torres, P.; Dasgupta-Schubert, N.; Bernal, J.L. Synthesis of Mesoporous Ceria by Using CTAB as Template. Microsc. Microanal. 2016, 22, 1918–1919. [Google Scholar] [CrossRef]
- Kurajica, S.; Minga, I.; Guliš, M.; Mandić, V.; Simčić, I. High Surface Area Ceria Nanoparticles via Hydrothermal Synthesis Experiment Design. J. Nanomater. 2016, 2016, 1–8. [Google Scholar] [CrossRef]
- Zhang, G.; Shen, Z.; Liu, M.; Guo, C.; Sun, P.; Yuan, Z.; Li, B.; Ding, D.; Chen, T. Synthesis and Characterization of Mesoporous Ceria with Hierarchical Nanoarchitecture Controlled by Amino Acids. J. Phys. Chem. B 2006, 110, 25782–25790. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.-M.; He, L.; Liu, Y.-M.; Cao, Y.; He, H.-Y.; Fan, K.-N. Gold supported on mesostructured ceria as an efficient catalyst for the chemoselective hydrogenation of carbonyl compounds in neat water. Green Chem. 2011, 13, 602–607. [Google Scholar] [CrossRef]
- Liu, H.; Zhu, D.; Jia, B.; Huang, Y.; Cheng, Y.; Luo, X.; Liang, Z. Study on catalytic performance and kinetics of high efficiency CeO2 catalyst prepared by freeze drying for the synthesis of dimethyl carbonate from CO2 and methanol. Chem. Eng. Sci. 2022, 254. [Google Scholar] [CrossRef]
- Hu, L.; Hu, K.; Xu, Z.; Yao, W.; Wang, A.; Wu, G.; Xu, W. Preparation and Characterization of Hollow CeO2 Nanoparticles for the Efficient Conversion of CO2 into Dimethyl Carbonate. ChemCatChem 2023, 15. [Google Scholar] [CrossRef]
- Pouretedal, H.R.; Basati, S. Synthesis, Characterization and Photocatalytic Activity of CeO 2-SBA-15; 2012; Vol. 2;
- Mitran, R.-A.; Culita, D.C.; Atkinson, I. Thermal stability enhancement of mesoporous SBA-15 silica through nanoconfinement of ceria nanoparticles. Microporous Mesoporous Mater. 2020, 306, 110484. [Google Scholar] [CrossRef]
- Pu, Y.; Xuan, K.; Wang, F.; Li, A.; Zhao, N.; Xiao, F. Synthesis of dimethyl carbonate from CO2 and methanol over a hydrophobic Ce/SBA-15 catalyst. RSC Adv. 2018, 8, 27216–27226. [Google Scholar] [CrossRef]
- Shen, J.; Hess, C. Controlling the dispersion of ceria using nanoconfinement: application to CeO2/SBA-15 catalysts for NH3-SCR. Mater. Adv. 2021, 2, 7400–7412. [Google Scholar] [CrossRef]
- Yang, J.; Jia, Y.; Huang, B.; Li, X.; Guo, L.; Zheng, A.; Luque, R.; Sun, Y. Functionalized CeO2/SBA-15 Materials as Efficient Catalysts for Aqueous Room Temperature Mono-dehydration of Sugar Alcohols. ACS Sustain. Chem. Eng. 2020, 8, 6371–6380. [Google Scholar] [CrossRef]
- Saadati-Moshtaghin, H.R.; Zonoz, F.M. In situ preparation of CeO2 nanoparticles on the MCM-41 with magnetic core as a novel and efficient catalyst for the synthesis of substituted pyran derivatives. Inorg. Chem. Commun. 2018, 99, 44–51. [Google Scholar] [CrossRef]
- Ngomade, S.B.L.; Fotsop, C.G.; Nguena, K.L.T.; Tchummegne, I.K.; Ngueteu, M.L.T.; Tamo, A.K.; Nche, G.N.-A.; Anagho, S.G. Catalytic performances of CeO2@SBA-15 as nanostructured material for biodiesel production from Podocarpus falcatus oil. Chem. Eng. Res. Des. 2023, 194, 789–800. [Google Scholar] [CrossRef]
- Ma, K.; Zhang, H.; Tang, C.; Dong, L. One-Pot Synthesis of CeO2 Modified SBA-15 With No Pore Clogging for NO Reduction by CO. Front. Environ. Chem. 2021, 2. [Google Scholar] [CrossRef]
- Mureddu, M.; Ferino, I.; Musinu, A.; Ardu, A.; Rombi, E.; Cutrufello, M.G.; Deiana, P.; Fantauzzi, M.; Cannas, C. MeOx/SBA-15 (Me = Zn, Fe): highly efficient nanosorbents for mid-temperature H2S removal. J. Mater. Chem. A 2014, 2, 19396–19406. [Google Scholar] [CrossRef]
- Mureddu, M.; Ferino, I.; Rombi, E.; Cutrufello, M.; Deiana, P.; Ardu, A.; Musinu, A.; Piccaluga, G.; Cannas, C. ZnO/SBA-15 composites for mid-temperature removal of H2S: Synthesis, performance and regeneration studies. Fuel 2012, 102, 691–700. [Google Scholar] [CrossRef]
- Cara, C.; Rombi, E.; Mameli, V.; Ardu, A.; Angotzi, M.S.; Niznansky, D.; Musinu, A.; Cannas, C. γ-Fe2O3-M41S Sorbents for H2S Removal: Effect of Different Porous Structures and Silica Wall Thickness. J. Phys. Chem. C 2018, 122, 12231–12242. [Google Scholar] [CrossRef]
- Varma, A.; Mukasyan, A.S.; Rogachev, A.S.; Manukyan, K.V. Solution Combustion Synthesis of Nanoscale Materials. Chem. Rev. 2016, 116, 14493–14586. [Google Scholar] [CrossRef]
- Cannas, C.; Musinu, A.; Peddis, D.; Piccaluga, G. Synthesis and Characterization of CoFe2O4 Nanoparticles Dispersed in a Silica Matrix by a Sol−Gel Autocombustion Method. Chem. Mater. 2006, 18, 3835–3842. [Google Scholar] [CrossRef]
- Cannas, C.; Ardu, A.; Niznansky, D.; Peddis, D.; Piccaluga, G.; Musinu, A. Simple and fast preparation of pure maghemite nanopowders through sol–gel self-combustion. J. Sol-Gel Sci. Technol. 2011, 60, 266–274. [Google Scholar] [CrossRef]
- Cannas, C.; Musinu, A.; Peddis, D.; Piccaluga, G. New Synthesis of Ferrite–Silica Nanocomposites by a Sol–Gel Auto-Combustion. J. Nanoparticle Res. 2004, 6, 223–232. [Google Scholar] [CrossRef]
- Cannas, C.; Falqui, A.; Musinu, A.; Peddis, D.; Piccaluga, G. CoFe2O4 nanocrystalline powders prepared by citrate-gel methods: Synthesis, structure and magnetic properties. J. Nanoparticle Res. 2006, 8, 255–267. [Google Scholar] [CrossRef]
- Secci, F.; Angotzi, M.S.; Mameli, V.; Lai, S.; Russo, P.A.; Pinna, N.; Mureddu, M.; Rombi, E.; Cannas, C. Mesostructured γ-Al2O3-Based Bifunctional Catalysts for Direct Synthesis of Dimethyl Ether from CO2. Catalysts 2023, 13, 505. [Google Scholar] [CrossRef]
- Mureddu, M.; Ferrara, F.; Pettinau, A. Highly efficient CuO/ZnO/ZrO2@SBA-15 nanocatalysts for methanol synthesis from the catalytic hydrogenation of CO2. Appl. Catal. B: Environ. 2019, 258. [Google Scholar] [CrossRef]
- Zhao, D.; Wan, Y.; Zhou, W. Ordered Mesoporous Materials; Wiley-VCH, 2013; ISBN 9783527326358.
- Zhao, D.; Feng, J.; Huo, Q.; Melosh, N.; Fredrickson, G.H.; Chmelka, B.F.; Stucky, G.D. Triblock Copolymer Syntheses of Mesoporous Silica with Periodic 50 to 300 Angstrom Pores. Science 1998, 279, 548–552. [Google Scholar] [CrossRef] [PubMed]
- Le Bail, A. Modelling the silica glass structure by the Rietveld method. J. Non-Crystalline Solids 1995, 183, 39–42. [Google Scholar] [CrossRef]
- Ennas, G.; Marongiu, G.; Marras, S.; Piccaluga, G. Mechanochemical Route for the Synthesis of Cobalt Ferrite–Silica and Iron–Cobalt Alloy–Silica Nanocomposites. J. Nanoparticle Res. 2004, 6, 99–105. [Google Scholar] [CrossRef]
- Janssen, A.H.; Yang, C.-M.; Wang, Y.; Schüth, F.; Koster, A.J.; de Jong, K.P. Localization of Small Metal (Oxide) Particles in SBA-15 Using Bright-Field Electron Tomography. J. Phys. Chem. B 2003, 107, 10552–10556. [Google Scholar] [CrossRef]
- Delahaye, E.; Escax, V.; El Hassan, N.; Davidson, A.; Aquino, R.; Dupuis, V.; Perzynski, R.; Raikher, Y.L. “Nanocasting”: Using SBA-15 Silicas as Hard Templates to Obtain Ultrasmall Monodispersed γ-Fe2O3 Nanoparticles. J. Phys. Chem. B 2006, 110, 26001–26011. [Google Scholar] [CrossRef]
- Kabra, S.K.; Turpeinen, E.; Keiski, R.L.; Yadav, G.D. Direct synthesis of dimethyl carbonate from methanol and carbon dioxide: A thermodynamic and experimental study. J. Supercrit. Fluids 2016, 117, 98–107. [Google Scholar] [CrossRef]
Figure 1.
WA-XRD (a) and SA-XRD (b) patterns of all the samples.
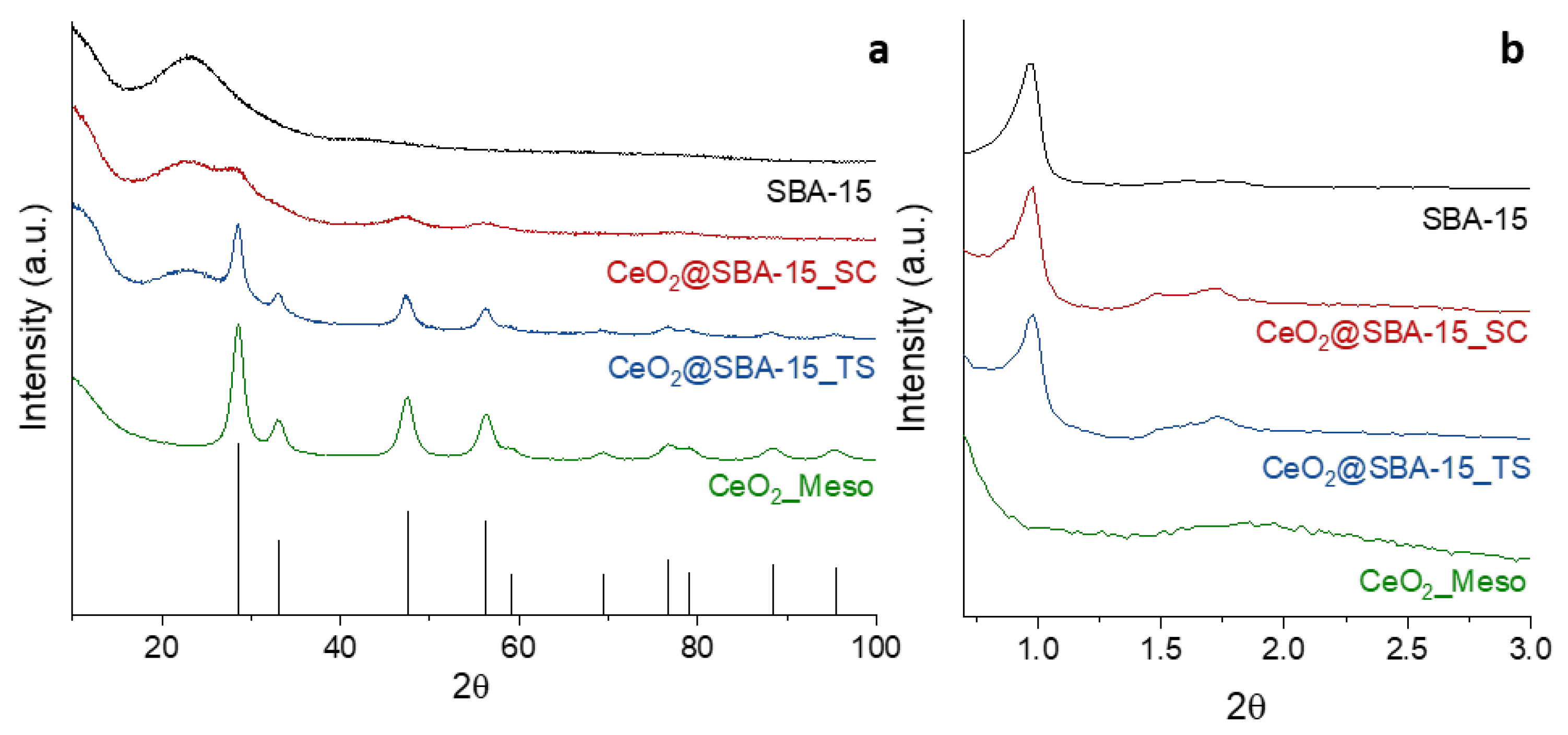
Figure 2.
Nitrogen physisorption isotherms (a) and BJH pore size distributions (b) of all the samples.
Figure 2.
Nitrogen physisorption isotherms (a) and BJH pore size distributions (b) of all the samples.
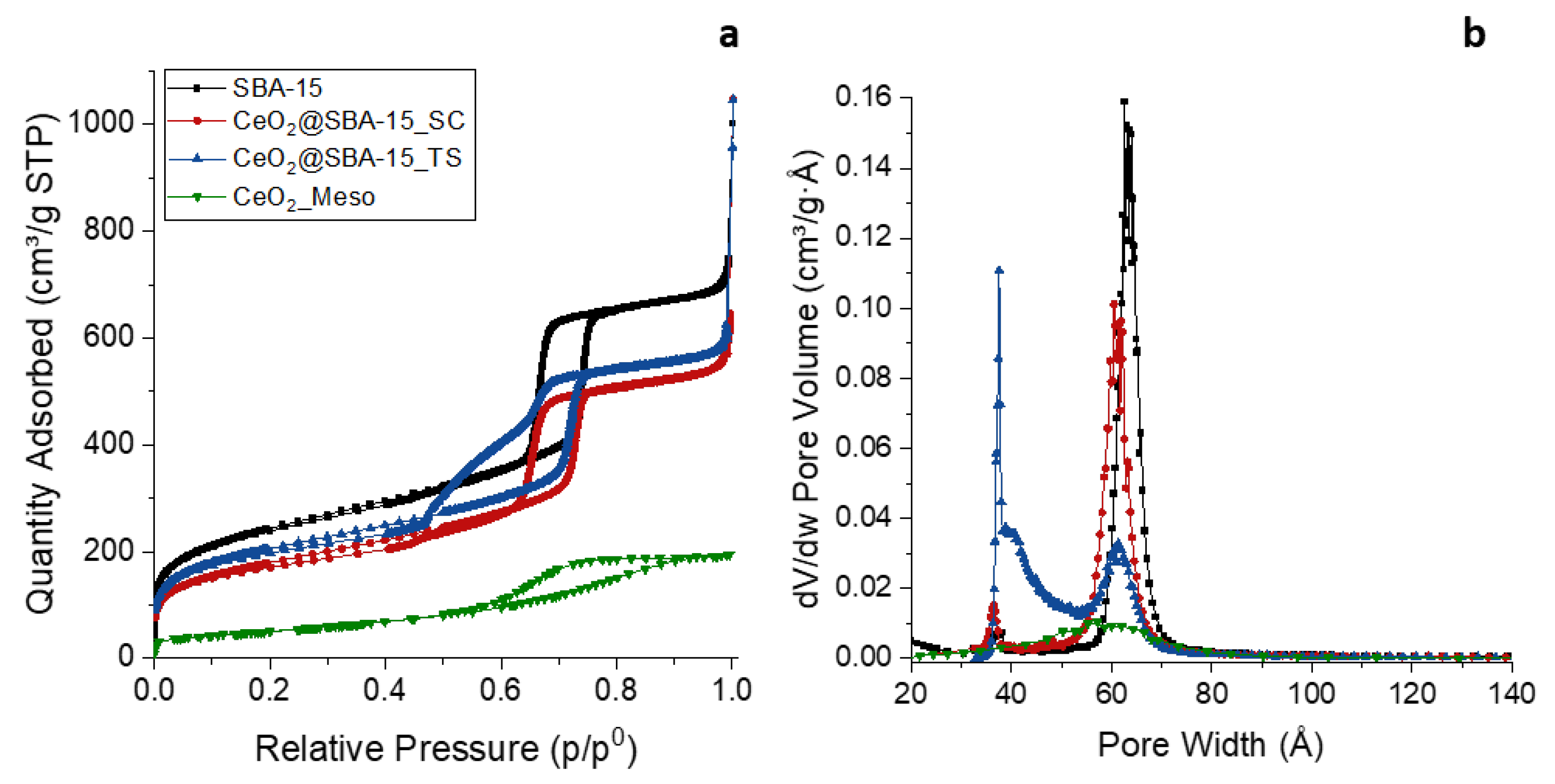
Figure 3.
TEM micrographs of CeO2_Meso.

Figure 5.
Bright-field (a-c; g-i) and dark-field (d-f; j-l) micrographs of CeO2@SBA-15_TS (a-f) and CeO2@SBA-15_SC (g-l).
Figure 5.
Bright-field (a-c; g-i) and dark-field (d-f; j-l) micrographs of CeO2@SBA-15_TS (a-f) and CeO2@SBA-15_SC (g-l).

Figure 6.
Line profile EDX analyses on CeO2@SBA-15_TS (a) and CeO2@SBA-15_SC (b).
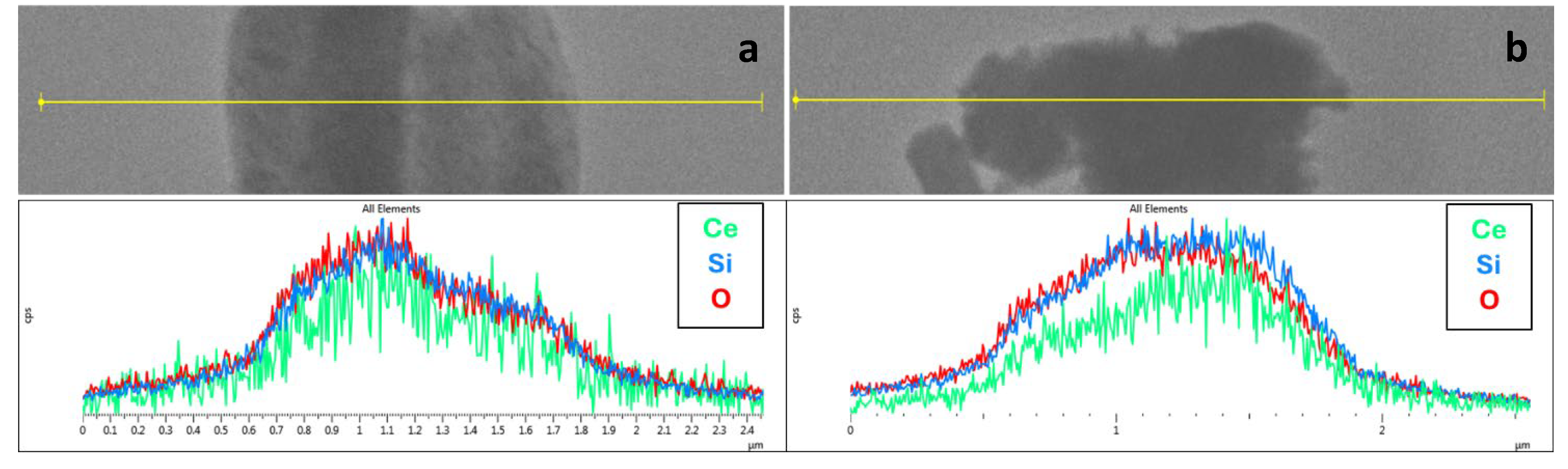
Table 1.
Crystallite size (DXRD), BET surface area (S.A.), pore volume (Vp), mean pore diameter (Dp), mesostructure cell parameter (a0), and wall thickness (Tw) of all the samples.
Table 1.
Crystallite size (DXRD), BET surface area (S.A.), pore volume (Vp), mean pore diameter (Dp), mesostructure cell parameter (a0), and wall thickness (Tw) of all the samples.
| Sample | DXRD (nm) | a0 (nm) | S.A. (m2/g) | Vp (cm3/g) | DP (nm) | Tw (nm) |
| CeO2_Meso | 7.9(1) | - | 182 | 0.27 | 5.8 | - |
| CeO2@SBA-15_TS | 7.9(1) | 10.4 | 722 | 0.93 | 6.1 | 4.3 |
| CeO2@SBA-15_SC | 2.6(1) | 10.4 | 635 | 0.88 | 6.1 | 4.3 |
| SBA-15 | - | 10.5 | 853 | 0.99 | 6.3 | 4.2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



