
Submitted:
01 August 2024
Posted:
06 August 2024
You are already at the latest version
Abstract
Protein dynamics play a crucial role in biological function, encompassing motions ranging from atomic vibrations to large-scale conformational changes. Recent advancements in experimental techniques, computational methods, and artificial intelligence have revolutionized our understanding of protein dynamics. Nuclear magnetic resonance spectroscopy provides atomic-resolution insights, while molecular dynamics simulations offer detailed trajectories of protein motions. The integration of machine learning, exemplified by AlphaFold2, has accelerated structure prediction and dynamics analysis. These approaches have revealed the importance of protein dynamics in allosteric regulation, enzyme catalysis, and intrinsically disordered proteins. The shift towards ensemble representations of protein structures and the application of single-molecule techniques have further enhanced our ability to capture the dynamic nature of proteins. Understanding protein dynamics is essential for elucidating biological mechanisms, designing drugs, and developing novel biocatalysts, marking a significant paradigm shift in structural biology and drug discovery.
Keywords:
Protein Dynamics
; Structural Biology
; Molecular Dynamics
; Allosteric Regulation
; Enzyme Catalysis
; Artificial Intelligence
; Protein Folding
; Conformational Changes
1. Introduction
Proteins are the fundamental workhorses of cellular processes, playing critical roles in signal transduction, cell division, metabolism, and countless other biological functions [1]. While early structural biology studies portrayed proteins as static entities, it has become increasingly clear that proteins are dynamic molecules undergoing complex motions across multiple timescales [2]. These motions range from femtosecond vibrations of individual atoms to large-scale domain movements occurring over microseconds to milliseconds [3]. Understanding protein dynamics is crucial for elucidating how proteins carry out their diverse functions and how these motions relate to disease states and drug interactions [4].
The field of protein dynamics has seen remarkable progress in recent years, driven by advances in biophysical techniques, computational methods, and the integration of artificial intelligence (AI) approaches [5]. Experimental methods such as nuclear magnetic resonance (NMR) spectroscopy, X-ray crystallography, and cryo-electron microscopy (cryo-EM) have provided unprecedented insights into protein structure and dynamics at atomic resolution [6]. NMR relaxation experiments, in particular, have enabled the characterization of protein motions across a wide range of timescales, from picoseconds to seconds [7].
Complementing these experimental approaches, molecular dynamics (MD) simulations have emerged as a powerful tool for studying protein dynamics at atomistic detail [8]. MD simulations can provide a continuous trajectory of protein motions, allowing researchers to observe conformational changes and transient states that may be difficult to capture experimentally [9]. Recent advances in computing power and specialized hardware have enabled simulations to reach biologically relevant timescales of microseconds to milliseconds [8].
The integration of machine learning and AI techniques with traditional biophysical methods has opened up new avenues for studying protein dynamics [6]. Deep learning models, such as AlphaFold2, have revolutionized protein structure prediction and are now being applied to predict protein dynamics and conformational ensembles [10]. These AI-powered approaches can rapidly generate hypotheses about protein motions and interactions, guiding experimental design and accelerating the discovery process.
One area where the study of protein dynamics has had a significant impact is in understanding allosteric regulation [11]. Allostery, the process by which binding at one site affects protein function at a distant site, often involves subtle conformational changes and dynamic processes [12]. Advanced NMR techniques and MD simulations have revealed how allosteric signals propagate through protein structures, providing insights into the design of allosteric drugs and the evolution of protein function [13]. Protein dynamics also play a crucial role in enzyme catalysis [14]. While the static lock-and-key model of enzyme-substrate interactions has long been abandoned, recent studies have shown that enzyme dynamics can contribute to catalysis in various ways [15]. These include promoting the formation of reactive conformations, facilitating the sampling of transition states, and modulating the free energy landscape of the reaction [16,17]. Understanding these dynamic contributions is essential for rational enzyme design and the development of novel biocatalysts [18].
In the field of structural biology, the recognition of protein dynamics has led to a shift from static structural models to ensemble representations [19]. Methods such as ensemble refinement in X-ray crystallography and integrative structural biology approaches combine data from multiple experimental sources to generate more accurate and dynamic models of protein structure and function [20,21]. These ensemble models provide a more realistic picture of protein behavior in solution and cellular environments [22].
The study of intrinsically disordered proteins (IDPs) and intrinsically disordered regions (IDRs) has further highlighted the importance of protein dynamics [23]. These proteins and regions lack a stable three-dimensional structure under physiological conditions but play critical roles in cellular signaling and regulation [24]. Advanced NMR techniques, single-molecule fluorescence, and computational methods have revealed how IDPs/IDRs exploit their conformational flexibility to mediate diverse interactions and functions [25]. Recent developments in single-molecule techniques have provided unprecedented insights into protein dynamics at the individual molecule level [26]. Methods such as single-molecule FRET (smFRET) and high-speed atomic force microscopy (HS-AFM) allow researchers to observe protein motions in real-time, revealing rare conformations and heterogeneous behaviors that may be masked in ensemble measurements [27,28].
The integration of protein dynamics into drug discovery and design has led to new strategies for developing more effective and selective therapeutics [29]. Computational approaches that account for protein flexibility, such as ensemble docking and molecular dynamics-based virtual screening, have improved the ability to identify novel drug candidates and predict their binding modes [30]. Additionally, targeting specific dynamic states or allosteric sites of proteins has emerged as a promising approach for modulating protein function and developing drugs for previously "undruggable" targets [31].
2. Experimental Techniques for Studying Protein Dynamics
Figure 1.
Advanced Experimental Techniques and Applications for Studying Protein Dynamics. (A) Schematic diagram of advanced Cryo-Electron Microscopy (Cryo-EM) techniques, including Time-resolved cryo-EM, Cryo-electron tomography, and Micro-crystal electron diffraction (MicroED). (B) Application fields of protein dynamics research using Nuclear Magnetic Resonance (NMR) Spectroscopy techniques, including Relaxation dispersion experiments, Paramagnetic relaxation enhancement (PRE), and Residual dipolar couplings (RDCs). (C) Application fields of protein dynamics research using Fluorescence-Based Techniques, including Single-molecule FRET, Fluorescence correlation spectroscopy (FCS), and Fluorescence lifetime imaging microscopy (FLIM).
Figure 1.
Advanced Experimental Techniques and Applications for Studying Protein Dynamics. (A) Schematic diagram of advanced Cryo-Electron Microscopy (Cryo-EM) techniques, including Time-resolved cryo-EM, Cryo-electron tomography, and Micro-crystal electron diffraction (MicroED). (B) Application fields of protein dynamics research using Nuclear Magnetic Resonance (NMR) Spectroscopy techniques, including Relaxation dispersion experiments, Paramagnetic relaxation enhancement (PRE), and Residual dipolar couplings (RDCs). (C) Application fields of protein dynamics research using Fluorescence-Based Techniques, including Single-molecule FRET, Fluorescence correlation spectroscopy (FCS), and Fluorescence lifetime imaging microscopy (FLIM).
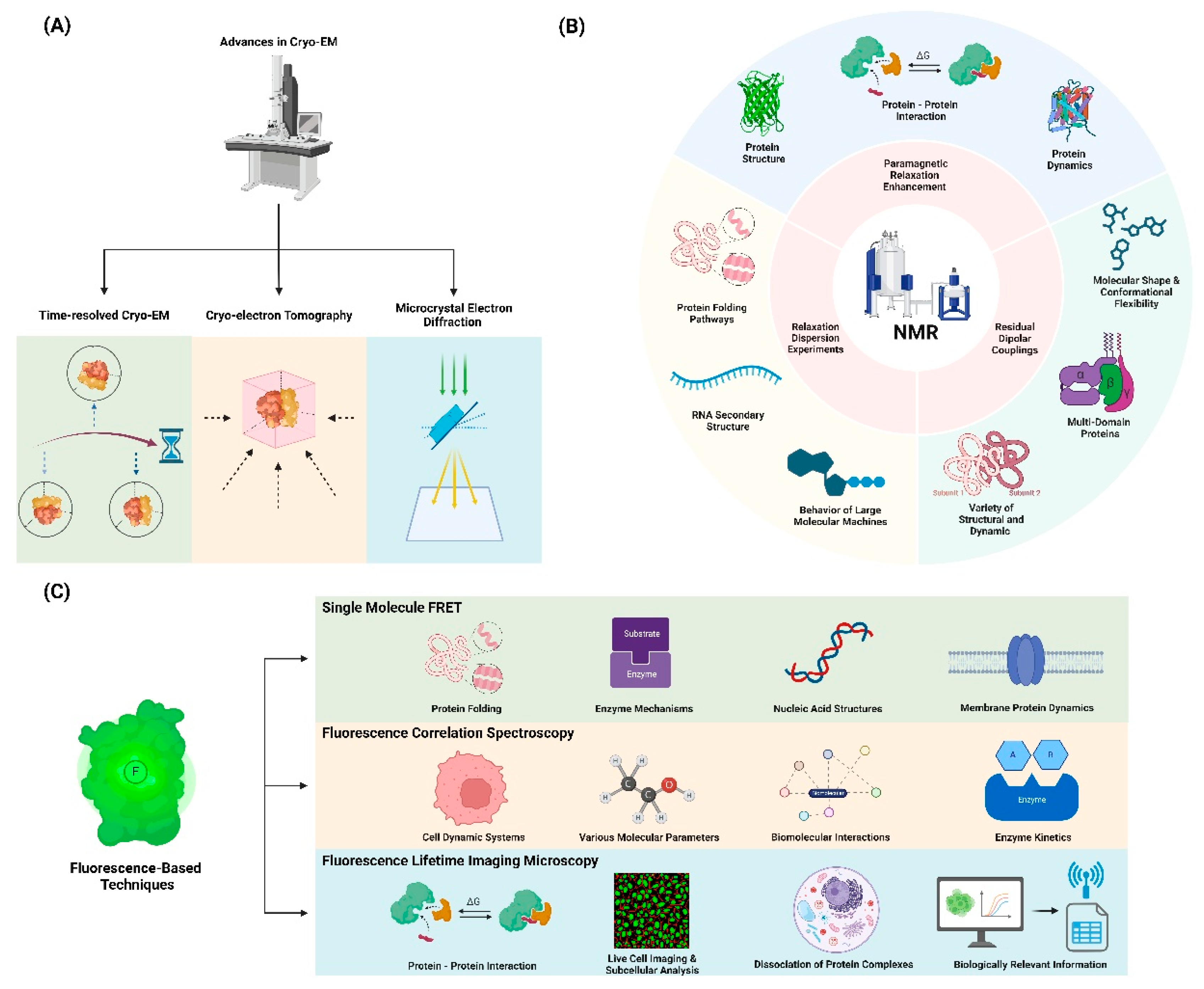
2.1. Advances in Cryo-EM for Dynamics Studies:
Cryogenic Electron Microscopy (Cryo-EM) has revolutionized structural biology by enabling the visualization of proteins in near-native states without the need for crystallization. Recent developments in cryo-EM have provided unprecedented insights into protein dynamics, particularly for large, flexible complexes.
2.1.1. Time-resolved cryo-EM: Capturing protein motions at different time points
Time-resolved cryogenic electron microscopy (cryo-EM) is an emerging technique in structural biology that allows researchers to capture structural states that are too transient for standard methods [32]. This technique has revolutionized the field by enabling the visualization of proteins in near-native states without the need for crystallization [33]. Recent developments in cryo-EM have provided unprecedented insights into protein dynamics, particularly for large, flexible complexes [34]. By freezing samples at different time points, time-resolved cryo-EM can trap non-equilibrium states and determine conformations present after defined periods, typically in the millisecond time frame [35]. This approach has been instrumental in elucidating the mechanics of molecular machines such as ribosomes and polymerases, which undergo complex, multistep processes during their functional cycles [34]. Methods such as microsecond time-resolved cryo-EM have enabled observations of fast protein dynamics, revealing detailed pictures of conformational changes that occur on very short timescales [33]. These advancements highlight the potential of time-resolved cryo-EM to fundamentally advance our understanding of protein function and dynamics [32,35].
2.1.2. Cryo-electron tomography: Visualizing proteins in their cellular context
Cryo-electron tomography (Cryo-ET) is a cutting-edge technique that enables the visualization of proteins within their native cellular environments, offering unprecedented insights into their structural organization and interactions within the complex milieu of living cells [36]. This method combines the principles of electron tomography with cryogenic preservation, allowing researchers to capture three-dimensional images of biological samples without the need for crystallization or chemical fixation, thus preserving their native states [37]. The ability to visualize intact cells and tissues while maintaining the spatial relationships between proteins and other cellular components represents a significant advancement over traditional imaging methods [38]. Recent technological advancements in Cryo-ET have significantly improved resolution and data acquisition speeds, facilitating the observation of dynamic processes at molecular scales [39]. Furthermore, tools such as subtomogram averaging have enhanced the high-resolution analysis of large molecular complexes, providing crucial information about protein localization and conformational states within their cellular contexts [40]. As research progresses, Cryo-ET holds immense potential for advancing our understanding of biological systems by enabling the direct observation of proteins functioning in situ [41].
2.1.3. Microcrystal electron diffraction (MicroED): Studying small molecule dynamics
Microcrystal electron diffraction (MicroED) is an innovative cryo-electron microscopy technique that enables high-resolution structural analysis of small crystals and molecules, providing crucial insights for studying small molecule dynamics [42]. This method, first developed in 2013, allows researchers to obtain structural data from nanocrystals that are one-billionth the size of those required for X-ray crystallography, expanding its applicability to a wide range of small molecules including natural products and drug candidates [43]. Since its inception, MicroED has undergone significant advancements in data collection and analysis protocols, with recent breakthroughs in phasing strategies enhancing its capabilities [44]. The technique has demonstrated remarkable versatility, successfully determining structures for over 40 different proteins, oligopeptides, and organic molecules [42]. MicroED offers unique advantages such as reduced radiation damage and the ability to capture dynamic processes that may be challenging to observe with traditional crystallography methods. By enabling structure determination from seemingly amorphous powders or very fine needle-like crystals, MicroED has opened new avenues for investigating previously inaccessible targets, particularly in the realm of natural products and small molecule research. As the field continues to evolve, MicroED holds immense potential for advancing our understanding of molecular interactions and dynamics, with significant implications for drug discovery and development [45].
2.2. Nuclear Magnetic Resonance (NMR) Spectroscopy
NMR spectroscopy remains a powerful tool for studying protein dynamics, offering atomic-level resolution and the ability to probe motions across a wide range of timescales. Recent advances include:
2.2.1. Relaxation dispersion experiments: Detecting and characterizing excited states
Relaxation dispersion NMR spectroscopy is a powerful technique used to detect and characterize low-populated, transient excited states of biomolecules by quantifying the broadening of NMR resonance lines due to chemical exchange between ground and excited states [46,47]. This method relies on the exchange between highly populated, NMR-visible ground states and sparsely populated, NMR-invisible excited states, transferring information about magnetic resonance properties such as relaxation parameters, chemical shifts, and residual dipolar couplings from the invisible state to the observable species [47]. The technique provides detailed kinetic and thermodynamic data, enabling the study of structural and dynamic properties of these excited states on the millisecond timescale [48]. Various experiments, including Carr–Purcell–Meiboom–Gill (CPMG) and rotating frame relaxation dispersion (R1ρ) methods, are employed to probe these exchange processes, often requiring isotopic labeling of the macromolecules under study [48,49]. Applications of relaxation dispersion NMR have revealed critical insights into protein folding pathways, RNA secondary structure dynamics, and the behavior of large molecular machines, offering a high-resolution view of intermediate states that are otherwise challenging to study [50,51]. This approach is complementary to other biophysical techniques and can be performed in the absence of denaturants, making it a versatile tool for studying biomolecular dynamics in native-like conditions [52].
2.2.2. Paramagnetic relaxation enhancement (PRE): Probing long-range interactions
Paramagnetic relaxation enhancement (PRE) is a powerful technique in nuclear magnetic resonance (NMR) spectroscopy that utilizes the magnetic dipolar interactions between unpaired electrons in a paramagnetic center and nearby nuclei to increase nuclear relaxation rates [53,54]. This effect is measurable at long distances, making it valuable for probing transient, lowly populated states and long-range interactions in macromolecules. PRE can be applied using intrinsic paramagnetic centers in metalloproteins or by introducing paramagnetic labels through chemical modification [54]. The technique is particularly useful for studying protein dynamics, structure, and interactions, as it can provide information on sparsely populated states and conformational changes that are difficult to detect using other methods [55,56]. PRE measurements typically involve comparing nuclear relaxation rates between paramagnetic and diamagnetic samples, with transverse (Γ2) PRE rates generally providing the most reliable and accurate data [54]. Recent advancements have expanded the application of PRE to include solvent accessibility studies, nanostructure determination in materials, and enhancing temporal resolution in NMR experiments [57,58]. The versatility of PRE has made it an invaluable tool in structural biology, materials science, and other fields where understanding molecular interactions and dynamics is crucial [59].
2.2.3. Residual dipolar couplings (RDCs): Characterizing domain orientations and flexibility
Residual dipolar couplings (RDCs) are a valuable tool in nuclear magnetic resonance (NMR) spectroscopy for characterizing the relative orientations and flexibility of molecular domains. RDCs arise when molecules in solution exhibit partial alignment, causing an incomplete averaging of spatially anisotropic dipolar couplings, which provides orientation-dependent restraints that are crucial for structural determination [60]. This partial alignment can be achieved using alignment media such as liquid crystalline phases or stretched polymer gels, which create an anisotropic environment necessary for RDC measurements [61]. RDCs are particularly useful for studying multi-domain proteins, as they provide information on the relative orientation of domains and their dynamic behavior [62]. By measuring the dipolar couplings between NMR-active nuclei, RDCs deliver insights into the global molecular shape and conformational flexibility, which are essential for understanding the functional motions and interactions of biomolecules. The technique has been successf[63]ully applied to a variety of structural and dynamic studies, including the analysis of protein-substrate interactions and the determination of quaternary structures of oligomers in equilibrium with monomers [64]. Recent advancements in RDC measurement and analysis have further expanded its applications, making it an indispensable tool in structural biology and related fields [63].
2.3. Fluorescence-Based Techniques
Fluorescence methods offer high sensitivity and the ability to study proteins in solution or in living cells:
2.3.1. Single-molecule FRET: Probing conformational changes in individual molecules
Single-molecule Förster resonance energy transfer (smFRET) is a powerful technique for studying conformational dynamics and interactions of individual biomolecules with high spatial and temporal resolution [27,65,66]. This method relies on measuring the energy transfer efficiency between donor and acceptor fluorophores attached to specific sites on a molecule of interest [65,67]. smFRET can reveal heterogeneous populations, transient intermediates, and dynamic fluctuations that are often masked in ensemble measurements [66,68]. Recent advances have improved the precision and accuracy of smFRET measurements, with studies reporting distance uncertainties of ±2-5 Å [27,69]. The technique has been successfully applied to investigate protein folding, enzyme mechanisms, nucleic acid structures, and membrane protein dynamics [65-67]. Developments in multicolor FRET schemes allow probing of more complex biomolecular systems [66]. Additionally, progress in data analysis methods, including hidden Markov modeling and photon-by-photon approaches, enables extraction of kinetic information on microsecond to millisecond timescales [68]. While challenges remain in achieving higher temporal resolution and applying smFRET in cellular environments, ongoing innovations continue to expand its capabilities for elucidating biomolecular structure and function at the single-molecule level [27,69].
2.3.2. Fluorescence correlation spectroscopy (FCS): Analyzing diffusion and binding kinetics
Fluorescence correlation spectroscopy (FCS) is a powerful technique for quantifying molecular dynamics and has been widely applied in diverse fields such as biomedicine, biophysics, and chemistry [70]. By analyzing the time-correlation of fluorescence fluctuations induced by molecules diffusing through a focused light, FCS can quantitatively evaluate the concentration, diffusion coefficient, and interactions of molecules both in vitro and in vivo [70,71]. The technique measures the spatial and temporal correlation of individual molecules, providing a bridge between classical ensemble and contemporary single-molecule measurements [71]. Typically implemented on a fluorescence microscope, FCS samples femtoliter volumes, making it especially useful for characterizing small dynamic systems such as biological cells. FCS can investigate various molecular parameters, including diffusion coefficients, chemical rate constants, molecular concentrations, and fluorescence brightness. The method's sensitivity allows for the analysis of extremely low-concentration biomolecules, with applications ranging from studying diffusion and chemical dynamics to monitoring biomolecular interactions and enzyme kinetics. Recent advancements in FCS include dual-color cross-correlation, multi-focus FCS, and scanning FCS, which enhance its capability to probe complex biological environments and interactions [70,72]. Despite its requirement for high signal-to-noise ratios and long time traces, FCS remains a versatile tool, with ongoing developments aimed at improving its temporal resolution and reducing phototoxic effects on living samples [73].
2.3.3. Fluorescence lifetime imaging microscopy (FLIM): Mapping protein interactions in cells
Fluorescence lifetime imaging microscopy (FLIM) is a powerful technique that measures the time-resolved fluorescence decay of fluorophores to generate contrast in microscopy images, providing information beyond traditional intensity-based imaging [74]. FLIM exploits the characteristic excited-state lifetime of fluorophores, which is sensitive to the local molecular environment, enabling the technique to probe various cellular parameters such as pH, viscosity, and protein interactions. The method typically employs time-correlated single-photon counting (TCSPC) to measure fluorescence lifetimes with picosecond resolution, allowing for precise quantification of molecular dynamics. FLIM is particularly valuable for studying protein-protein interactions through Förster resonance energy transfer (FRET), where the fluorescence lifetime of a donor fluorophore decreases in the presence of a nearby acceptor, indicating molecular proximity within 1-10 nm [75]. Recent advances in FLIM instrumentation, including the development of fast FLIM techniques and improved spatial resolution, have expanded its capabilities for live-cell imaging and subcellular analysis. Computational developments in FLIM data analysis, such as phasor approaches and machine learning algorithms, have enhanced the extraction of biologically relevant information from complex lifetime datasets [76]. FLIM has found widespread applications in cell biology, including the study of protein conformational changes, ligand-receptor interactions, and the spatiotemporal dynamics of signaling complexes in living cells [75]. The technique's ability to discriminate between free and bound states of fluorescently labeled proteins makes it particularly suited for mapping the formation and dissociation of protein complexes in various cellular compartments. As FLIM continues to evolve, its integration with other advanced microscopy techniques and the development of novel fluorescent probes promise to further expand its utility in unraveling the intricate molecular interactions that underlie cellular function [76].
3. Computational Approaches to Protein Dynamics
Figure 2.
Advances and Applications of Molecular Dynamics Simulations in Structural Proteomics. (A) Proteins are dynamic molecules that exhibit motions and functions over multiple time scales. The development of time-scaling techniques is crucial for understanding various protein dynamics. Recently, the integration of rapidly advancing computing resources and artificial intelligence models with advanced quantum mechanics (QM)/molecular mechanics (MM) methods has led to significant advancements in Molecular Dynamics Simulations technology. These advancements overcome existing limitations in studying biological processes occurring on time scales from milliseconds to seconds and can be applied to protein engineering and drug discovery, including the exploration of large-scale protein structural changes.
Figure 2.
Advances and Applications of Molecular Dynamics Simulations in Structural Proteomics. (A) Proteins are dynamic molecules that exhibit motions and functions over multiple time scales. The development of time-scaling techniques is crucial for understanding various protein dynamics. Recently, the integration of rapidly advancing computing resources and artificial intelligence models with advanced quantum mechanics (QM)/molecular mechanics (MM) methods has led to significant advancements in Molecular Dynamics Simulations technology. These advancements overcome existing limitations in studying biological processes occurring on time scales from milliseconds to seconds and can be applied to protein engineering and drug discovery, including the exploration of large-scale protein structural changes.
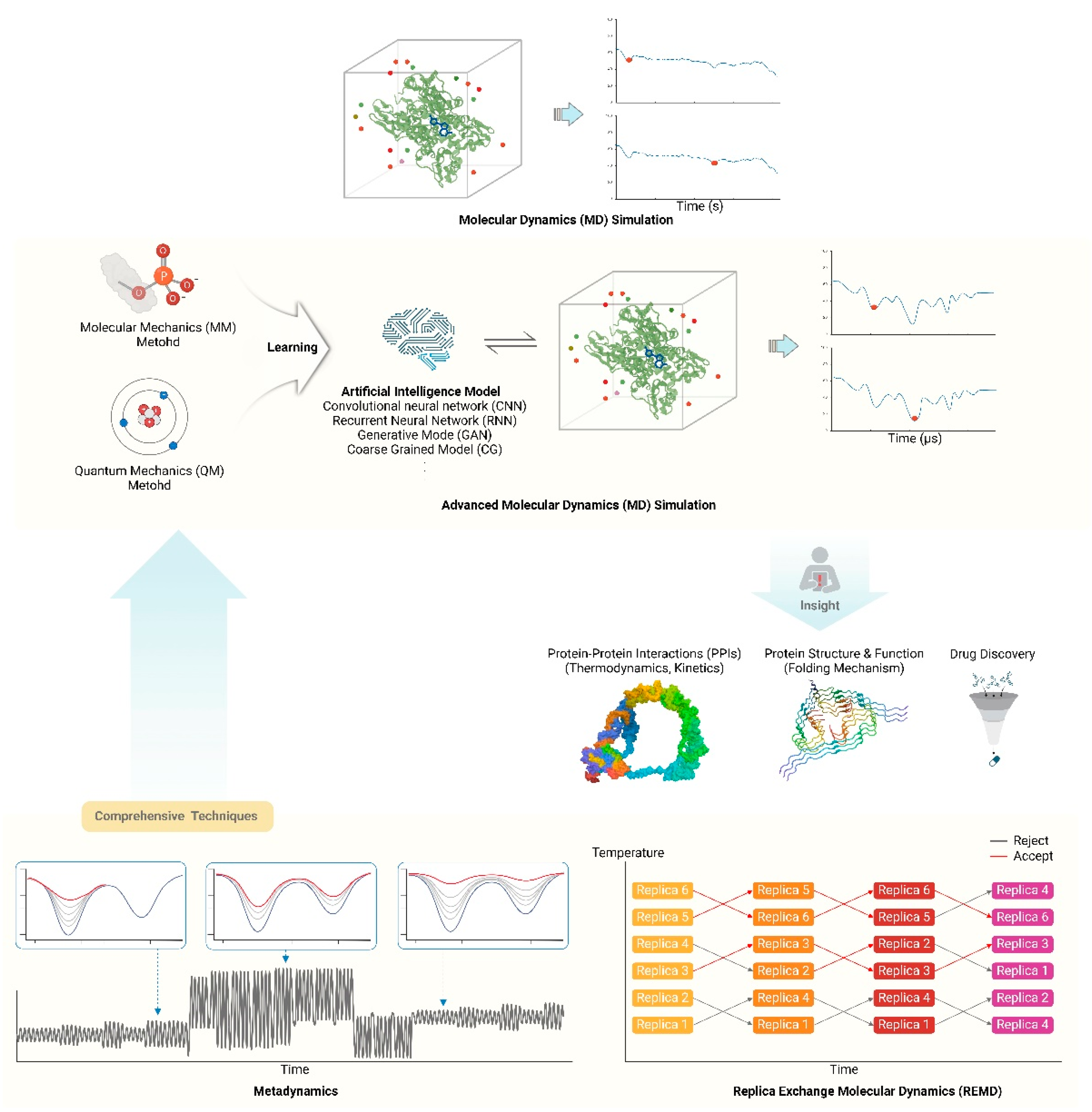
3.1. Molecular Dynamics Simulations
Molecular dynamics (MD) simulations have become an indispensable tool for studying protein dynamics at atomic resolution. Recent advances include:
3.1.1. Long-timescale simulations: Accessing biologically relevant timescales (ms-s)
Long-timescale molecular dynamics simulations have emerged as a powerful tool for studying biological processes occurring on millisecond to second timescales, overcoming traditional limitations of atomistic simulations [77]. Advances in hardware, including specialized supercomputers and graphics processing units (GPUs), have enabled microsecond to millisecond simulations of protein folding, conformational changes, and ligand binding [77,78]. Enhanced sampling techniques like accelerated molecular dynamics, replica exchange, and Markov state models allow more efficient exploration of conformational space [77,79]. Coarse-graining approaches sacrifice atomic detail but permit much longer effective timescales [80]. The development of polarizable force fields and quantum mechanics/molecular mechanics (QM/MM) methods has improved accuracy for modeling electronic effects [78,80]. Machine learning potentials trained on quantum mechanical data show promise for combining accuracy and speed [81]. Distributed computing projects like Folding@home have aggregated enormous computational resources for long timescales. Despite these advances, challenges remain in force field accuracy, sampling of rare events, and bridging timescales from femtoseconds to seconds [77,79]. Careful validation against experimental data and assessment of convergence are critical for reliable results [82]. As the field progresses, long-timescale simulations are providing unprecedented insight into the dynamics and mechanisms of biomolecular systems [77,78,81].
3.1.2. Enhanced sampling techniques: Exploring rare events and conformational transitions
Enhanced sampling techniques have revolutionized molecular dynamics (MD) simulations by enabling the efficient exploration of rare events and conformational transitions that are otherwise computationally prohibitive. These methods address the challenge of rough energy landscapes with many local minima separated by high-energy barriers, which hinder the sampling of relevant conformational states [83,84]. Techniques such as metadynamics and replica exchange molecular dynamics (REMD) have become widely adopted for their ability to enhance sampling by introducing memory effects or simulating multiple replicas of the system at different temperatures [85,86]. Metadynamics, for instance, discourages the re-sampling of previously visited states by adding a history-dependent bias potential, effectively "filling" energy wells with computational sand to facilitate transitions over barriers [78,85]. REMD, on the other hand, allows for the exchange of configurations between replicas at different temperatures, thereby accelerating the sampling of high-energy states that are critical for observing rare events. These methods have been successfully applied to study processes such as protein folding, ligand binding, and phase transitions, providing insights into the underlying mechanisms of these complex phenomena [84]. Recent advancements include hybrid approaches that combine collective variable-based and collective variable-free methods to optimize sampling efficiency further [83,86]. Despite their successes, enhanced sampling techniques continue to evolve, with ongoing developments aimed at improving their robustness and applicability to increasingly complex biological systems [85].
3.1.3. Coarse-grained models: Simulating large systems and complex assemblies
Coarse-grained (CG) models have become indispensable for simulating large systems and complex assemblies by reducing the number of degrees of freedom, thereby enabling the exploration of larger spatial and temporal scales than all-atom simulations [87,88]. These models achieve computational efficiency by averaging out fast local motions and focusing on slow collective dynamics, which is particularly useful for studying biological processes such as protein folding, membrane dynamics, and large biomolecular complexes [89]. The development of CG models involves defining pseudoatoms that represent groups of atoms and parameterizing the interactions between them, often guided by all-atom simulations or experimental data [90]. Recent advancements in machine learning have further enhanced CG models by optimizing the mapping from all-atom to CG representations and improving the accuracy of CG force fields [87,91]. Techniques such as variational auto-encoders and neural network potentials have been employed to create more accurate and generalizable CG models, capable of capturing essential thermodynamic and kinetic properties of the system. These models have been successfully applied to simulate processes that occur on timescales ranging from microseconds to milliseconds, providing insights into the dynamics of large protein complexes, lipid membranes, and other biomolecular assemblies [88]. Despite their advantages, CG models must be carefully validated against experimental data and all-atom simulations to ensure their reliability and accuracy [89,92]. As the field progresses, the integration of CG models with multiscale simulation approaches promises to bridge the gap between molecular and macroscopic descriptions of biological systems, enhancing our understanding of complex biological phenomena [92,93].
3.2. Machine Learning and AI in Protein Dynamics
The integration of machine learning and AI approaches has opened new avenues for understanding protein dynamics:
3.2.1. Deep learning for feature extraction: Identifying relevant collective variables
The integration of machine learning (ML) and AI approaches has opened new avenues for understanding protein dynamics, particularly through deep learning for feature extraction and identifying relevant collective variables (CVs). Deep learning techniques, such as convolutional neural networks (CNNs) and recurrent neural networks (RNNs), have proven effective in extracting meaningful features from large-scale protein data, enabling the prediction of protein structures and dynamics with improved accuracy [94]. These methods leverage vast protein databases to identify patterns and correlations that are not apparent through traditional approaches, thus enhancing our ability to model protein behavior [95]. One notable application is the use of variational autoencoders and neural network potentials to discover CVs that capture the essential dynamics of protein systems, facilitating the exploration of conformational changes and rare events in molecular simulations [96]. For instance, machine learning models have been trained on molecular dynamics (MD) simulation data to generate physically realistic conformational ensembles, significantly reducing computational costs and improving the efficiency of sampling techniques [97,98]. This approach has been particularly beneficial for studying intrinsically disordered proteins (IDPs), which exhibit high conformational variability and are challenging to model using conventional methods. By integrating machine learning with MD simulations, researchers can now achieve a more comprehensive understanding of protein folding mechanisms, energy landscapes, and the thermodynamics and kinetics of protein interactions [95]. As these techniques continue to evolve, they promise to further enhance our ability to predict and manipulate protein functions, with broad implications for drug discovery and protein engineering [94,97].
3.2.2. Generative models: Predicting protein conformations and dynamics
The integration of generative models in understanding protein dynamics has significantly advanced the prediction of protein conformations and dynamics [99]. Generative models, such as variational autoencoders and generative adversarial networks, have been employed to learn low-dimensional representations of protein conformational spaces, which can then be used to generate novel protein structures and dynamic ensembles [96]. These models are trained on extensive datasets from molecular dynamics (MD) simulations, allowing them to capture the complex energy landscapes and conformational transitions of proteins [99,100]. By learning the underlying probability distributions of protein conformations, generative models can efficiently sample new conformations, thereby reducing the computational cost associated with traditional MD simulations [96]. This approach has been particularly effective in studying intrinsically disordered proteins (IDPs), which exhibit a high degree of conformational variability and are challenging to model using conventional methods [100]. The application of generative models has also facilitated the exploration of rare events and large-scale conformational changes, providing deeper insights into the thermodynamics and kinetics of protein folding and function [99]. As these techniques continue to evolve, they promise to enhance our ability to predict and manipulate protein dynamics, with broad implications for drug discovery and protein engineering [96,101].
3.2.3. Reinforcement learning: Optimizing sampling strategies in MD simulations
Reinforcement learning (RL) has emerged as a powerful approach for optimizing sampling strategies in molecular dynamics (MD) simulations, enabling more efficient exploration of protein conformational landscapes and rare events [102,103]. RL-based methods, such as REAP (Reinforcement Learning Based Adaptive Sampling) and AdaptiveBandit, address the exploration-exploitation dilemma by balancing the sampling of known metastable states with the discovery of new, potentially important regions of the conformational space . These algorithms typically define reward functions that encourage exploration of undersampled areas while also prioritizing regions of interest, such as those with low free energy or specific structural features. For example, FAST (Fluctuation Amplification of Specific Traits) uses a multi-armed bandit-inspired approach to initialize simulations that optimize a given property while maintaining exploration of poorly sampled states [104,105]. Similarly, AdaptiveBandit employs the UCB1 algorithm to find minimum free energy configurations by balancing expected rewards with uncertainty in sampling. Tree search molecular dynamics (TS-MD) applies the upper confidence bounds for trees (UCT) algorithm to efficiently sample transition pathways between initial and target protein configurations [102]. These RL-based methods have demonstrated success in enhancing the sampling of complex systems, such as protein folding and conformational changes, often achieving better coverage of the conformational space compared to traditional MD approaches. Moreover, the integration of RL with other machine learning techniques, such as deep learning for feature extraction and dimensionality reduction, has further improved the efficiency and accuracy of MD simulations [106]. As the field progresses, RL-optimized sampling strategies promise to play an increasingly important role in unraveling the intricacies of protein dynamics and facilitating drug discovery efforts [102,106].
4. Applications and Insights from Protein Dynamics Studies
Figure 3.
Applications of Protein Dynamics Studies through Advanced Experimental and Computational Approaches. (A) Schematic diagram of the application of folding protein dynamics through experimental and computational approaches. (B) Schematic diagram of the application of enzyme-substrate complexes using protein dynamics approaches. (C) Application of protein dynamics approaches to membrane proteins.
Figure 3.
Applications of Protein Dynamics Studies through Advanced Experimental and Computational Approaches. (A) Schematic diagram of the application of folding protein dynamics through experimental and computational approaches. (B) Schematic diagram of the application of enzyme-substrate complexes using protein dynamics approaches. (C) Application of protein dynamics approaches to membrane proteins.
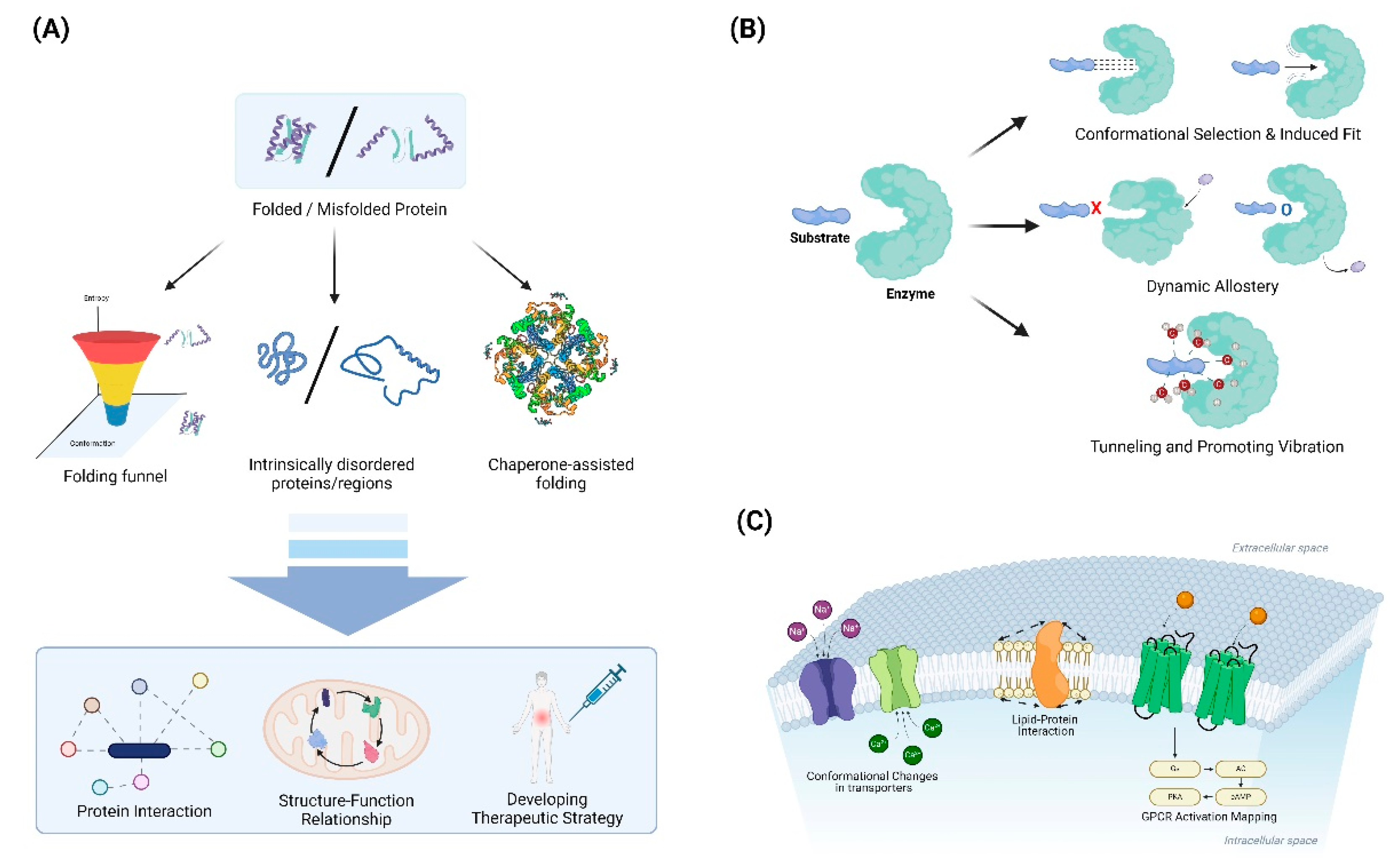
4.1. Protein Folding and Misfolding
Combining experimental and computational approaches has led to significant progress in understanding protein folding mechanisms and the factors contributing to misfolding and aggregation. Key insights include:
4.1.1. Folding funnels and energy landscapes: Characterizing the thermodynamics and kinetics of folding
Folding funnels and energy landscapes provide a powerful framework for understanding the thermodynamics and kinetics of protein folding [107-109]. The funnel-shaped energy landscape concept, introduced in the 1990s, revolutionized our understanding of how proteins navigate the vast conformational space to reach their native states [107]. This model depicts folding as a process of descending a rugged energy landscape, where the vertical axis represents free energy and the horizontal axes represent conformational degrees of freedom [108,110]. The overall funnel shape arises from the principle of minimal frustration, which posits that evolution has selected sequences that minimize conflicting interactions, thereby creating a bias towards the native state [109,111]. Quantitatively, the folding funnel can be characterized by parameters such as the energy gap between the native state and the average of misfolded states, the ruggedness of the landscape, and the configurational entropy [108,112]. These parameters influence the folding rate and mechanism, with smoother funnels generally leading to faster folding [110,113]. The presence of local minima in the energy landscape can give rise to folding intermediates and multiple folding pathways, explaining the complexity observed in folding kinetics [112]. Advanced experimental techniques, such as single-molecule spectroscopy and hydrogen exchange mass spectrometry, have provided empirical support for the funnel model by mapping energy landscapes and detecting transient intermediates [111,113]. Computational methods, including molecular dynamics simulations and statistical mechanical models, have further refined our understanding of folding funnels and their relationship to sequence-structure correlations [109,112]. The folding funnel concept has also been extended to explain protein-protein interactions, allostery, and the evolution of new protein functions, demonstrating its broad applicability in structural biology [107,111].
4.1.2. Intrinsically disordered proteins: Recognizing the functional importance of structural flexibility
Intrinsically disordered proteins (IDPs) and intrinsically disordered regions (IDRs) have emerged as crucial components in cellular signaling and regulation, challenging the traditional structure-function paradigm of proteins [23,114]. These proteins lack a stable three-dimensional structure under physiological conditions, instead existing as dynamic ensembles of interconverting conformations [115,116]. The structural flexibility of IDPs/IDRs enables them to interact with multiple binding partners, facilitating their roles as hubs in protein interaction networks and allowing for fine-tuned regulation through post-translational modifications [23]. This conformational plasticity is encoded in their amino acid sequences, which are typically characterized by a low content of bulky hydrophobic residues and a high proportion of polar and charged amino acids [104,116]. IDPs/IDRs are particularly enriched in eukaryotic proteomes and are involved in various cellular processes, including transcriptional control, cell signaling, and the formation of membrane-less organelles through phase separation [23,114,117]. The functional significance of protein disorder extends to enzymes, where IDRs can play important roles in allosteric regulation and catalysis [115,118]. Recent advances in experimental techniques, such as NMR spectroscopy and single-molecule FRET, combined with computational methods, have greatly enhanced our understanding of the structural ensembles and functional mechanisms of IDPs [116,119]. The recognition of the widespread occurrence and functional importance of intrinsic disorder has led to the establishment of the "disorder-function paradigm," complementing the classical structure-function relationship in protein science [114,120].
4.1.3. Chaperone-assisted folding: Elucidating the role of cellular machinery in protein folding
Chaperone-assisted protein folding is a critical cellular process that ensures the proper folding and functionality of proteins, preventing misfolding and aggregation that can lead to cellular dysfunction [121,122]. Molecular chaperones, such as those from the Hsp70 and chaperonin families, play a pivotal role in this process by stabilizing unfolded or partially folded proteins and facilitating their correct folding through ATP-dependent cycles of binding and release [121,123]. These chaperones recognize exposed hydrophobic regions on nascent or stress-denatured proteins, preventing inappropriate interactions that could lead to aggregation [122,124]. For instance, Hsp70 chaperones not only prevent aggregation but also actively remodel misfolded proteins, promoting their refolding into native conformations [123,125]. Additionally, chaperonins like GroEL/GroES provide a protected environment for protein folding, encapsulating substrate proteins and allowing them to fold without interference from the crowded cellular milieu [126,127]. Recent studies using advanced techniques such as single-molecule fluorescence and cryo-electron microscopy have provided detailed insights into the dynamic interactions between chaperones and their substrate proteins, revealing how these molecular machines guide proteins through their folding landscapes [124,128]. The chaperone system is also integral to the cellular stress response, with heat shock proteins being upregulated to manage increased loads of misfolded proteins under stress conditions [122,129]. Understanding the mechanisms of chaperone-assisted folding not only elucidates fundamental aspects of cellular proteostasis but also has significant implications for developing therapeutic strategies against diseases caused by protein misfolding and aggregation [121,127,129].
4.2. Enzyme Catalysis and Allostery
Studies of protein dynamics have revealed the critical role of motions in enzyme function and regulation:
4.2.1. Conformational selection vs. induced fit: Understanding substrate binding mechanisms
Conformational selection and induced fit are two fundamental mechanisms that explain how proteins interact with ligands, each offering distinct insights into the dynamics of molecular recognition [130,131]. In conformational selection, proteins exist in a pre-existing ensemble of conformations, and the ligand selectively binds to and stabilizes a subset of these conformations, as supported by techniques like NMR and single-molecule FRET [130,132]. Conversely, induced fit posits that the initial protein-ligand interaction triggers a conformational change in the protein to optimize the binding interface, a concept first proposed by Koshland to explain enzymatic specificity [131,133]. These mechanisms are not mutually exclusive and can coexist within the same system, with their relative contributions often dependent on factors such as ligand concentration and specific protein characteristics [130,134]. Advanced computational methods, including molecular dynamics simulations and enhanced sampling techniques, have been instrumental in elucidating the energy landscapes that govern these binding processes [133,134]. Understanding these mechanisms is crucial for drug design and protein engineering, as it provides insights into how proteins achieve specificity and efficiency in their interactions [130,135]. For instance, in enzyme design, considering both conformational selection and induced fit can lead to more effective catalysts by optimizing both the pre-existing conformational ensemble and the induced changes upon substrate binding [133,134]. In the context of drug discovery, this understanding can guide the development of more potent and selective inhibitors by targeting specific conformational states or by designing ligands that can induce favorable conformational changes [131,134]. Recent studies have also highlighted the role of these mechanisms in allosteric regulation and the function of intrinsically disordered proteins, further expanding their relevance in biological processes [130,134].
4.2.2. Dynamic allostery: Recognizing the importance of entropy in allosteric regulation
Dynamic allostery refers to the phenomenon wherein the binding of a ligand leads to conformational changes in a protein that can influence the dynamics of other distant sites within the protein or within associated proteins [136,137]. This process highlights the significance of entropic changes during allosteric regulation, emphasizing that allosteric effects are not merely a consequence of structural changes but also involve shifts in the entropy of the protein states [138]. Entropy plays a crucial role in allosteric regulation as it governs the stability of various protein conformations; thus, changes in entropy upon ligand binding can enhance or inhibit the functional responses of the protein [137]. Additionally, advances in experimental techniques, such as nuclear magnetic resonance (NMR) spectroscopy and molecular dynamics simulations, have demonstrated how conformational flexibility and entropic fluctuations are essential for understanding allosteric mechanisms [138]. This recognition of the importance of entropy in allostery has implications for drug design, providing insights into how small molecules can induce conformational changes that modulate protein activity [139].
4.2.3. Tunneling and promoting vibrations: Exploring quantum effects in enzyme catalysis
Quantum mechanical effects, such as tunneling and promoting vibrations, play a crucial role in enzyme catalysis by influencing the reaction rates and mechanisms involved. Hydrogen tunneling, a process where hydrogen atoms pass through energy barriers rather than over them, has been shown to be significant in enzymatic C-H bond cleavage, linking protein dynamics to catalysis and highlighting the importance of donor-acceptor distance and active-site electrostatics [140]. Promoting vibrations, which are specific localized motions within the enzyme, enhance catalytic efficiency by coupling directly to the reaction coordinate and optimizing the compression along the donor-acceptor distance, thus facilitating hydrogen tunneling [141,142]. These vibrations are believed to be a universal feature of enzymes, contributing to their pre-organized active sites and enabling efficient biochemical transformations. Recent theoretical and experimental studies have demonstrated that these quantum effects are not only significant at cryogenic temperatures but also at physiological conditions, challenging classical transition state theory and emphasizing the dynamic nature of enzyme catalysis [143]. Computational models, particularly quantum mechanical/molecular mechanical (QM/MM) simulations, have provided valuable insights into these mechanisms, underscoring the interplay between electronic structure and protein dynamics in enzyme function [144].
4.3. Membrane Protein Dynamics
Advances in studying membrane protein dynamics have provided insights into transport mechanisms and signal transduction:
4.3.1. Lipid-protein interactions: Characterizing the influence of the membrane environment
Lipid-protein interactions are fundamental in regulating the structure and function of membrane proteins, influencing processes such as protein conformational changes and membrane domain formation [145,146]. These interactions can be specific, involving direct binding of lipids to proteins, or nonspecific, affecting the general properties of the membrane, such as thickness and lateral pressure [147,148]. Experimental techniques, including cryo-electron microscopy and mass spectrometry, have advanced our understanding of these interactions by providing high-resolution structures and identifying lipid compositions in complex membranes [145,146]. Molecular dynamics (MD) simulations have complemented these experimental approaches, offering detailed insights into the dynamics and energetics of lipid-protein interactions at atomic resolution [148,149]. For instance, MD simulations have revealed how lipid composition and membrane properties modulate the conformational equilibrium of ion channels, such as potassium channels, by altering lateral pressure and membrane thickness [149]. Additionally, specific lipid-binding events have been shown to modulate protein-protein interactions and influence the functional states of membrane proteins, further underscoring the complexity and specificity of lipid-protein interactions [145,147]. Overall, the integration of experimental and computational methods continues to enhance our understanding of the intricate roles that lipid-protein interactions play in cellular membranes [146,148].
4.3.2. Conformational changes in transporters: Elucidating alternating access mechanisms
Conformational changes in transporters are essential for elucidating the alternating access mechanisms that facilitate substrate translocation across cell membranes. The alternating access model posits that transporters cycle between outward-facing (OF) and inward-facing (IF) conformations, allowing substrates to bind on one side of the membrane and be released on the other [150,151]. This mechanism has been extensively studied in various transporters, including the Na+/Ca2+ exchanger NCX_Mj, which demonstrates significant structural asymmetry between its topological repeats, with transmembrane helices TM1-TM6 undergoing substantial displacement during the transition [150]. Crystallographic studies of the sodium-hydantoin transporter Mhp1 have captured the transporter in multiple states, providing a detailed mechanism of how it switches from the OF to the IF state through an occluded intermediate [151]. Molecular dynamics (MD) simulations have further elucidated these transitions, revealing that substrate binding induces conformational changes that facilitate the alternating access mechanism, as observed in SWEET-family transporters. Recent advancements in computational approaches, such as AlphaFold2, have enabled the prediction of multiple conformational states of transporters, highlighting the dynamic nature of these proteins and their ability to adopt diverse conformations [152]. The integration of experimental and computational methods continues to enhance our understanding of the alternating access mechanism, underscoring the importance of conformational plasticity in transporter function [153].
4.3.3. GPCR activation: Mapping the energy landscape of receptor activation
The activation of G protein-coupled receptors (GPCRs) involves complex conformational changes that can be conceptualized as transitions across an energy landscape [154,155]. This landscape is characterized by multiple energy wells representing distinct conformational states, including inactive, intermediate, and active configurations. Agonist binding modifies the shape of this energy landscape, lowering energy barriers and stabilizing active conformations [154]. The activation process typically involves outward movement of transmembrane helix 6 (TM6), a hallmark of GPCR activation that creates space for G-protein coupling [156]. Studies have revealed a common activation pathway comprising 34 residue pairs and 35 residues that links the ligand-binding pocket to the G-protein coupling region, unifying previously identified key motifs such as CWxP, DRY, and NPxxY. This pathway allows for the decoupling of ligand binding site and G-protein binding region evolution, potentially facilitating the diversification of GPCRs. Advanced techniques like X-ray crystallography, cryo-electron microscopy, and molecular dynamics simulations have been instrumental in mapping these energy landscapes and identifying key conformational intermediates [157,158]. The energy landscape framework provides insights into mechanisms of constitutive activity, inverse agonism, and allosteric modulation, offering a powerful tool for understanding GPCR function and drug design [154,159]. Recent studies have also explored the vibrational energy landscapes of GPCRs, providing graphical representations of residue-level energy distributions in active and inactive states [160].
5. Future Directions and Challenges
Figure 4.
Future Directions and Challenges in Protein Dynamics Studies through Experimental and Computational Approaches. (A) Integrating atomic simulations, coarse-grained models, and experimental data to connect temporal and spatial scales. (B) Developing methods to study protein motions within their native cellular environments using advanced imaging and spectroscopic techniques. (C) Utilizing machine learning and deep learning to predict functional protein dynamics and design custom proteins. (D) Extending dynamics studies to large macromolecular complexes and cellular machinery. (E) Employing advanced technologies to link protein motions to biological functions and design targeted therapeutics.
Figure 4.
Future Directions and Challenges in Protein Dynamics Studies through Experimental and Computational Approaches. (A) Integrating atomic simulations, coarse-grained models, and experimental data to connect temporal and spatial scales. (B) Developing methods to study protein motions within their native cellular environments using advanced imaging and spectroscopic techniques. (C) Utilizing machine learning and deep learning to predict functional protein dynamics and design custom proteins. (D) Extending dynamics studies to large macromolecular complexes and cellular machinery. (E) Employing advanced technologies to link protein motions to biological functions and design targeted therapeutics.
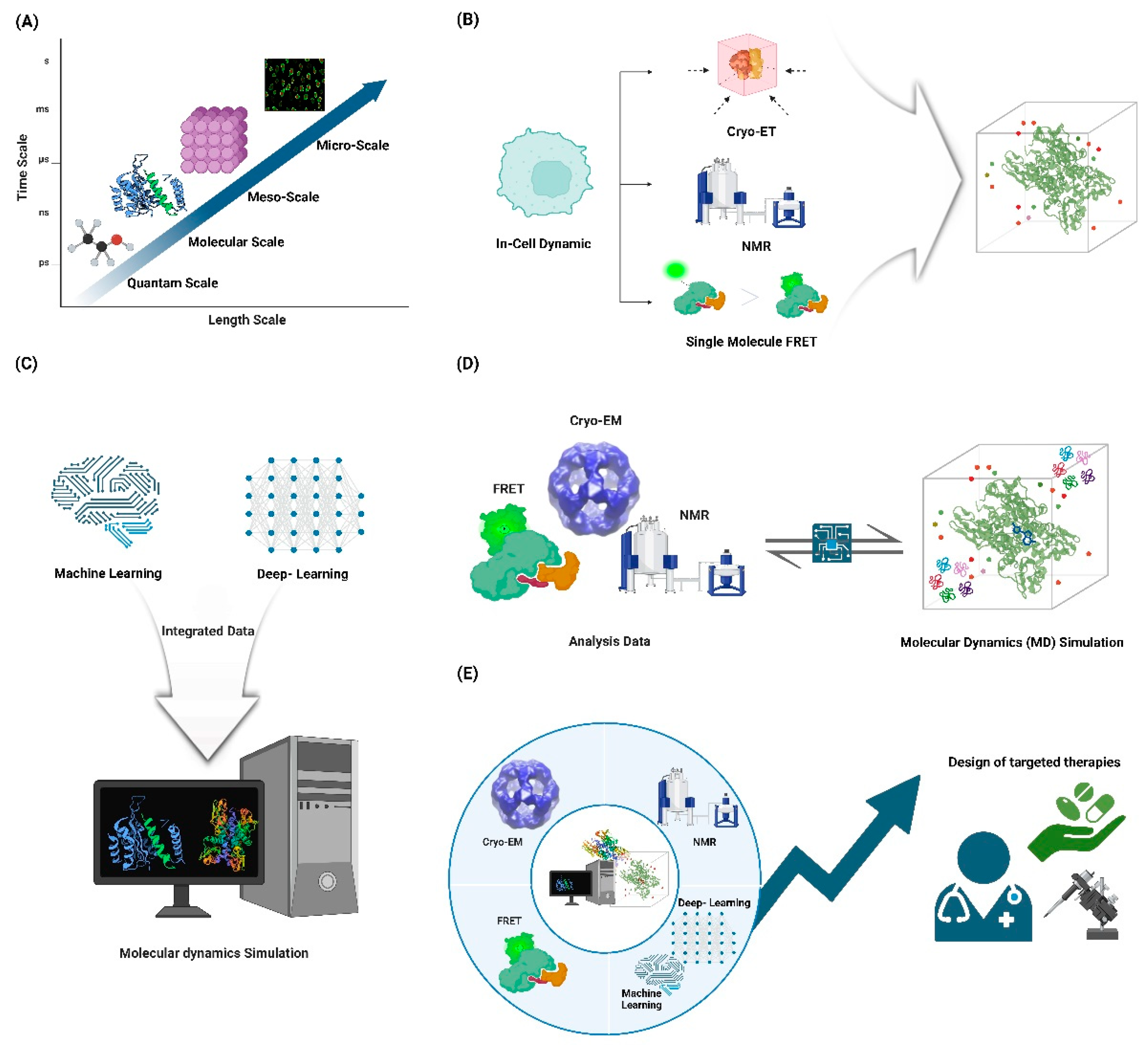
As the field of protein dynamics continues to evolve, several key areas for future research and development emerge:
5.1. Integration of multi-scale approaches: Combining atomistic simulations with coarse-grained models and experimental data to bridge timescales and length scales.
Future directions in computational biology point towards the integration of multi-scale approaches that combine atomistic simulations with coarse-grained models and experimental data to bridge timescales and length scales [161]. Atomistic molecular dynamics simulations provide detailed insights into biomolecular structure and dynamics at the nanoscale, but are limited in the timescales they can access [89]. Coarse-grained models allow for simulations of larger systems over longer timescales by reducing the number of degrees of freedom, but sacrifice atomic-level detail [162]. Integrating these approaches with experimental data can leverage the strengths of each method while overcoming individual limitations [163]. Machine learning techniques are emerging as powerful tools to develop accurate coarse-grained models from atomistic simulation data and to bridge between scales [164]. Multiscale modeling frameworks that dynamically couple different levels of theory show promise for capturing processes that span multiple spatiotemporal scales [93]. Key challenges include developing robust methods to systematically connect different scales, quantifying uncertainties across scales, and efficiently sampling rare events [164]. Overcoming these hurdles will enable more comprehensive and predictive models of complex biological systems across scales.
5.2. In-cell dynamics: Developing methods to study protein motions in their native cellular environment.
Developing methods to study protein motions in their native cellular environment remains a major challenge and frontier in the field of protein dynamics [165]. While significant progress has been made in studying protein dynamics in vitro, the complex and crowded cellular milieu can significantly alter protein behavior, necessitating in-cell approaches [166]. Recent advances in in-cell NMR spectroscopy have enabled the study of protein structure and dynamics directly within living cells, providing insights into how the cellular environment impacts protein function. Fluorescence-based techniques like single-molecule FRET and tracking of transfected biomolecules are also emerging as powerful tools to probe protein conformational dynamics and interactions in cellular with high spatiotemporal resolution [165]. Cryo-electron tomography combined with computational approaches like tomoDRGN show promise for capturing protein structural heterogeneity and dynamics in their native cellular context [167]. However, significant challenges remain, including improving the sensitivity and resolution of in-cell measurements, developing methods to study low-abundance proteins, and integrating data from multiple techniques to build comprehensive models of protein behavior in cells. Overcoming these hurdles will require continued innovation in labeling strategies, instrumentation, and computational analysis methods [157]. As the field progresses, in-cell dynamic studies have the potential to reveal new principles of protein function and regulation within the complex cellular environment.
5.3. AI-driven discovery: Leveraging machine learning to predict functional motions and design proteins with specific dynamic properties.
AI-driven discovery is poised to revolutionize our understanding of protein dynamics and enable the design of proteins with specific dynamic properties. Machine learning approaches have demonstrated remarkable success in predicting protein structures, and are now being extended to capture the dynamic nature of proteins [168]. By integrating sequence, structure, and dynamics information, these methods can provide insights into conformational ensembles and functional motions that are crucial for protein function [98]. Deep learning models, such as those based on geometric deep learning, show promise in predicting protein dynamics from static structures, potentially accelerating the exploration of conformational landscapes [98]. Furthermore, generative models are emerging as powerful tools for designing novel proteins with tailored dynamic properties, opening up new possibilities for protein engineering [169]. The integration of molecular dynamics simulations with machine learning frameworks allows for more accurate predictions of protein flexibility and allosteric communication pathways. Recent advances in physics-informed neural networks offer the potential to incorporate physical constraints and improve the interpretability of AI-driven predictions in protein science [168]. As these methods continue to evolve, they are expected to play an increasingly important role in understanding protein function at a molecular level and designing proteins with specific dynamic properties for biotechnological and therapeutic applications.
5.4. Dynamics in complex assemblies: Extending our understanding to large macromolecular complexes and cellular machines.
Future directions in the study of dynamics in complex assemblies focus on extending our understanding to large macromolecular complexes and cellular machines. The dynamic nature of these assemblies significantly impacts their molecular and cellular functions, necessitating advanced methods to study their conformational changes, interactions, and spatial-temporal dynamics within the cell [170]. Recent progress in coarse-grained (CG) modeling has facilitated the study of large biological systems by bridging the gap between microscopic and macroscopic details, allowing for the simulation of complex macromolecular interactions over extended timescales [171]. Techniques such as cryo-electron microscopy (cryo-EM) have revolutionized structural biology by providing high-resolution models of large complexes, enabling the visualization of their dynamic processes during assembly, remodeling, and disassembly [170]. Single-molecule biophysical approaches have also advanced, offering unique insights into the stochastic behavior of individual molecules within these assemblies, thus revealing the non-linear and highly dynamic nature of cellular machinery [172]. However, challenges remain in improving the sensitivity and resolution of these methods to capture the full spectrum of dynamic behaviors in complex environments. Integrating experimental data with computational models, such as all-atom molecular dynamics (MD) simulations and CG models, is crucial for developing comprehensive and predictive models of macromolecular dynamics [171]. As these methods continue to evolve, they promise to enhance our understanding of the intricate dynamics of large macromolecular complexes and their roles in cellular processes [170-172].
5.5. Linking dynamics to function: Developing quantitative frameworks to relate protein motions to biological function and disease states.
Future directions in linking protein dynamics to function involve developing quantitative frameworks to relate protein motions to biological function and disease states. Proteins are dynamic entities, and their functions are governed by their dynamic personalities, which include fluctuations ranging from atomic vibrations to large domain movements [1]. Understanding these dynamics is crucial, as disease-causing mutations can perturb protein structural networks, leading to altered functions [173]. Integrating experimental data with computational models, such as molecular dynamics (MD) simulations, can reveal how these mutations affect protein stability and function [174]. Techniques like cryo-electron microscopy and NMR spectroscopy have advanced our ability to capture dynamic processes in proteins, providing insights into conformational changes and interactions [1]. Network-based models have also emerged as powerful tools to study the complex spatiotemporal relationships between protein structures and their functions, particularly in the context of disease mutations [175]. These models can help identify allosteric pathways and residues involved in biological activities, linking structural changes to functional outcomes. As we continue to develop and refine these quantitative frameworks, we will enhance our ability to predict how protein dynamics contribute to function and disease, ultimately aiding in the design of targeted therapeutics [176].
6. Conclusion
The study of protein dynamics has advanced significantly in recent years, driven by technological innovations in both experimental and computational approaches. As our understanding of protein motions continues to grow, we gain deeper insights into the fundamental mechanisms of life at the molecular level. The integration of diverse techniques, from cryo-EM to AI-powered simulations, promises to further accelerate progress in this field, with far-reaching implications for drug discovery, protein engineering, and our understanding of cellular processes.
Author Contributions
Conceptualization, investigation, writing, and original draft preparation, A.S. (Ahrum Son); H.K. (Hyunsoo Kim) – Visualization, and proofreading, W.K. (Woojin Kim); J.P. (Jongham Park); W.L. (Wonseok Lee); Y.L. (Yerim Lee) – Supervision, Project Administration, Funding Acquisition, Review and Editing, H.K. (Hyunsoo Kim). All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by BK21 FOUR Program by Chungnam National University Research Grant, 2024. This work was partly supported by Institute of Information & communications Technology Planning & Evaluation (IITP) grant funded by the Korea government (MSIT) (No. RS-2022-00155857, Artificial Intelligence Convergence Innovation Human Resources Development (Chungnam National University). This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (RS-2023-00209456). This work was supported by the Korea Basic Science Institute (National research Facilities and Equipment Center) grant funded by the Korea government (MSIT) (RS-2024-00402298).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were created.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Nam, K.; Wolf-Watz, M. Protein dynamics: The future is bright and complicated! Struct Dyn 2023, 10, 014301. [Google Scholar] [CrossRef] [PubMed]
- Jayaraman, V.; Toledo-Patino, S.; Noda-Garcia, L.; Laurino, P. Mechanisms of protein evolution. Protein Sci 2022, 31, e4362. [Google Scholar] [CrossRef] [PubMed]
- Henzler-Wildman, K.A.; Lei, M.; Thai, V.; Kerns, S.J.; Karplus, M.; Kern, D. A hierarchy of timescales in protein dynamics is linked to enzyme catalysis. Nature 2007, 450, 913–916. [Google Scholar] [CrossRef] [PubMed]
- Klyshko, E.; Kim, J.S.; McGough, L.; Valeeva, V.; Lee, E.; Ranganathan, R.; Rauscher, S. Functional protein dynamics in a crystal. Nat Commun 2024, 15, 3244. [Google Scholar] [CrossRef] [PubMed]
- Roca-Martinez, J.; Lazar, T.; Gavalda-Garcia, J.; Bickel, D.; Pancsa, R.; Dixit, B.; Tzavella, K.; Ramasamy, P.; Sanchez-Fornaris, M.; Grau, I.; et al. Challenges in describing the conformation and dynamics of proteins with ambiguous behavior. Front Mol Biosci 2022, 9, 959956. [Google Scholar] [CrossRef]
- Ghosh, D.; Biswas, A.; Radhakrishna, M. Advanced computational approaches to understand protein aggregation. Biophys Rev (Melville) 2024, 5, 021302. [Google Scholar] [CrossRef]
- Palmer, A.G. , 3rd. NMR characterization of the dynamics of biomacromolecules. Chem Rev 2004, 104, 3623–3640. [Google Scholar] [CrossRef]
- Dror, R.O.; Dirks, R.M.; Grossman, J.P.; Xu, H.; Shaw, D.E. Biomolecular simulation: a computational microscope for molecular biology. Annu Rev Biophys 2012, 41, 429–452. [Google Scholar] [CrossRef]
- Barredo, P.A.; Balanay, M.P. Recent Advances in Molecular Dynamics Simulations of Tau Fibrils and Oligomers. Membranes (Basel) 2023, 13. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Zidek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Grutsch, S.; Bruschweiler, S.; Tollinger, M. NMR Methods to Study Dynamic Allostery. PLoS Comput Biol 2016, 12, e1004620. [Google Scholar] [CrossRef] [PubMed]
- Tzeng, S.R.; Kalodimos, C.G. Protein dynamics and allostery: an NMR view. Curr Opin Struct Biol 2011, 21, 62–67. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Zhou, H.X. Protein Allostery and Conformational Dynamics. Chem Rev 2016, 116, 6503–6515. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, S.D. Protein Dynamics and Enzymatic Catalysis. J Phys Chem B 2023, 127, 2649–2660. [Google Scholar] [CrossRef] [PubMed]
- Kohen, A. Role of dynamics in enzyme catalysis: substantial versus semantic controversies. Acc Chem Res 2015, 48, 466–473. [Google Scholar] [CrossRef] [PubMed]
- McGeagh, J.D.; Ranaghan, K.E.; Mulholland, A.J. Protein dynamics and enzyme catalysis: insights from simulations. Biochim Biophys Acta 2011, 1814, 1077–1092. [Google Scholar] [CrossRef] [PubMed]
- Otten, R.; Liu, L.; Kenner, L.R.; Clarkson, M.W.; Mavor, D.; Tawfik, D.S.; Kern, D.; Fraser, J.S. Rescue of conformational dynamics in enzyme catalysis by directed evolution. Nat Commun 2018, 9, 1314. [Google Scholar] [CrossRef] [PubMed]
- Warshel, A.; Bora, R.P. Perspective: Defining and quantifying the role of dynamics in enzyme catalysis. J Chem Phys 2016, 144, 180901. [Google Scholar] [CrossRef] [PubMed]
- van den Bedem, H.; Fraser, J.S. Integrative, dynamic structural biology at atomic resolution--it's about time. Nat Methods 2015, 12, 307–318. [Google Scholar] [CrossRef]
- Burnley, B.T.; Afonine, P.V.; Adams, P.D.; Gros, P. Modelling dynamics in protein crystal structures by ensemble refinement. Elife 2012, 1, e00311. [Google Scholar] [CrossRef]
- Rout, M.P.; Sali, A. Principles for Integrative Structural Biology Studies. Cell 2019, 177, 1384–1403. [Google Scholar] [CrossRef] [PubMed]
- Boehr, D.D.; Nussinov, R.; Wright, P.E. The role of dynamic conformational ensembles in biomolecular recognition. Nat Chem Biol 2009, 5, 789–796. [Google Scholar] [CrossRef] [PubMed]
- Wright, P.E.; Dyson, H.J. Intrinsically disordered proteins in cellular signalling and regulation. Nat Rev Mol Cell Biol 2015, 16, 18–29. [Google Scholar] [CrossRef] [PubMed]
- Babu, M.M. The contribution of intrinsically disordered regions to protein function, cellular complexity, and human disease. Biochem Soc Trans 2016, 44, 1185–1200. [Google Scholar] [CrossRef] [PubMed]
- Naudi-Fabra, S.; Blackledge, M.; Milles, S. Synergies of Single Molecule Fluorescence and NMR for the Study of Intrinsically Disordered Proteins. Biomolecules 2021, 12. [Google Scholar] [CrossRef] [PubMed]
- Lerner, E.; Cordes, T.; Ingargiola, A.; Alhadid, Y.; Chung, S.; Michalet, X.; Weiss, S. Toward dynamic structural biology: Two decades of single-molecule Forster resonance energy transfer. Science 2018, 359. [Google Scholar] [CrossRef] [PubMed]
- Hellenkamp, B.; Schmid, S.; Doroshenko, O.; Opanasyuk, O.; Kuhnemuth, R.; Rezaei Adariani, S.; Ambrose, B.; Aznauryan, M.; Barth, A.; Birkedal, V.; et al. Precision and accuracy of single-molecule FRET measurements-a multi-laboratory benchmark study. Nat Methods 2018, 15, 669–676. [Google Scholar] [CrossRef] [PubMed]
- Ando, T. High-speed atomic force microscopy and its future prospects. Biophys Rev 2018, 10, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Verkhivker, G.M.; Agajanian, S.; Hu, G.; Tao, P. Allosteric Regulation at the Crossroads of New Technologies: Multiscale Modeling, Networks, and Machine Learning. Front Mol Biosci 2020, 7, 136. [Google Scholar] [CrossRef]
- Govindaraj, R.G.; Thangapandian, S.; Schauperl, M.; Denny, R.A.; Diller, D.J. Recent applications of computational methods to allosteric drug discovery. Front Mol Biosci 2022, 9, 1070328. [Google Scholar] [CrossRef]
- Hu, G.; Doruker, P.; Li, H.; Demet Akten, E. Editorial: Understanding Protein Dynamics, Binding and Allostery for Drug Design. Front Mol Biosci 2021, 8, 681364. [Google Scholar] [CrossRef] [PubMed]
- Maeots, M.E.; Enchev, R.I. Structural dynamics: review of time-resolved cryo-EM. Acta Crystallogr D Struct Biol 2022, 78, 927–935. [Google Scholar] [CrossRef]
- Harder, O.F.; Barrass, S.V.; Drabbels, M.; Lorenz, U.J. Fast viral dynamics revealed by microsecond time-resolved cryo-EM. Nat Commun 2023, 14, 5649. [Google Scholar] [CrossRef]
- Amann, S.J.; Keihsler, D.; Bodrug, T.; Brown, N.G.; Haselbach, D. Frozen in time: analyzing molecular dynamics with time-resolved cryo-EM. Structure 2023, 31, 4–19. [Google Scholar] [CrossRef]
- Klebl, D.P.; Aspinall, L.; Muench, S.P. Time resolved applications for Cryo-EM; approaches, challenges and future directions. Curr Opin Struct Biol 2023, 83, 102696. [Google Scholar] [CrossRef] [PubMed]
- Baumeister, W. Cryo-electron tomography: A long journey to the inner space of cells. Cell 2022, 185, 2649–2652. [Google Scholar] [CrossRef]
- Lucic, V.; Rigort, A.; Baumeister, W. Cryo-electron tomography: the challenge of doing structural biology in situ. J Cell Biol 2013, 202, 407–419. [Google Scholar] [CrossRef]
- Golding, C.G.; Lamboo, L.L.; Beniac, D.R.; Booth, T.F. The scanning electron microscope in microbiology and diagnosis of infectious disease. Sci Rep 2016, 6, 26516. [Google Scholar] [CrossRef]
- Bauerlein, F.J.B.; Baumeister, W. Towards Visual Proteomics at High Resolution. J Mol Biol 2021, 433, 167187. [Google Scholar] [CrossRef] [PubMed]
- Guaita, M.; Watters, S.C.; Loerch, S. Recent advances and current trends in cryo-electron microscopy. Curr Opin Struct Biol 2022, 77, 102484. [Google Scholar] [CrossRef] [PubMed]
- Turk, M.; Baumeister, W. The promise and the challenges of cryo-electron tomography. FEBS Lett 2020, 594, 3243–3261. [Google Scholar] [CrossRef] [PubMed]
- Danelius, E.; Halaby, S.; van der Donk, W.A.; Gonen, T. MicroED in natural product and small molecule research. Nat Prod Rep 2021, 38, 423–431. [Google Scholar] [CrossRef] [PubMed]
- Jones, C.G.; Martynowycz, M.W.; Hattne, J.; Fulton, T.J.; Stoltz, B.M.; Rodriguez, J.A.; Nelson, H.M.; Gonen, T. The CryoEM Method MicroED as a Powerful Tool for Small Molecule Structure Determination. ACS Cent Sci 2018, 4, 1587–1592. [Google Scholar] [CrossRef] [PubMed]
- Danelius, E.; Patel, K.; Gonzalez, B.; Gonen, T. MicroED in drug discovery. Curr Opin Struct Biol 2023, 79, 102549. [Google Scholar] [CrossRef] [PubMed]
- Clark, L.J.; Bu, G.; Nannenga, B.L.; Gonen, T. MicroED for the study of protein-ligand interactions and the potential for drug discovery. Nat Rev Chem 2021, 5, 853–858. [Google Scholar] [CrossRef]
- Walinda, E.; Morimoto, D.; Sugase, K. Overview of Relaxation Dispersion NMR Spectroscopy to Study Protein Dynamics and Protein-Ligand Interactions. Curr Protoc Protein Sci 2018, 92, e57. [Google Scholar] [CrossRef] [PubMed]
- Clore, G.M. NMR spectroscopy, excited states and relevance to problems in cell biology - transient pre-nucleation tetramerization of huntingtin and insights into Huntington's disease. J Cell Sci 2022, 135. [Google Scholar] [CrossRef] [PubMed]
- Neudecker, P.; Lundstrom, P.; Kay, L.E. Relaxation dispersion NMR spectroscopy as a tool for detailed studies of protein folding. Biophys J 2009, 96, 2045–2054. [Google Scholar] [CrossRef] [PubMed]
- Dreydoppel, M.; Lichtenecker, R.J.; Akke, M.; Weininger, U. (1)H R(1rho) relaxation dispersion experiments in aromatic side chains. J Biomol NMR 2021, 75, 383–392. [Google Scholar] [CrossRef]
- Overbeck, J.H.; Kremer, W.; Sprangers, R. A suite of (19)F based relaxation dispersion experiments to assess biomolecular motions. J Biomol NMR 2020, 74, 753–766. [Google Scholar] [CrossRef]
- Xue, Y.; Kellogg, D.; Kimsey, I.J.; Sathyamoorthy, B.; Stein, Z.W.; McBrairty, M.; Al-Hashimi, H.M. Characterizing RNA Excited States Using NMR Relaxation Dispersion. Methods Enzymol 2015, 558, 39–73. [Google Scholar] [CrossRef] [PubMed]
- Vallurupalli, P.; Hansen, D.F.; Kay, L.E. Structures of invisible, excited protein states by relaxation dispersion NMR spectroscopy. Proc Natl Acad Sci U S A 2008, 105, 11766–11771. [Google Scholar] [CrossRef]
- Clore, G.M.; Iwahara, J. Theory, practice, and applications of paramagnetic relaxation enhancement for the characterization of transient low-population states of biological macromolecules and their complexes. Chem Rev 2009, 109, 4108–4139. [Google Scholar] [CrossRef] [PubMed]
- Clore, G.M. Practical Aspects of Paramagnetic Relaxation Enhancement in Biological Macromolecules. Methods Enzymol 2015, 564, 485–497. [Google Scholar] [CrossRef] [PubMed]
- Kocman, V.; Di Mauro, G.M.; Veglia, G.; Ramamoorthy, A. Use of paramagnetic systems to speed-up NMR data acquisition and for structural and dynamic studies. Solid State Nucl Magn Reson 2019, 102, 36–46. [Google Scholar] [CrossRef] [PubMed]
- Lenard, A.J.; Mulder, F.A.A.; Madl, T. Solvent paramagnetic relaxation enhancement as a versatile method for studying structure and dynamics of biomolecular systems. Prog Nucl Magn Reson Spectrosc 2022, 132-133, 113–139. [Google Scholar] [CrossRef] [PubMed]
- Schlagnitweit, J.; Tang, M.; Baias, M.; Richardson, S.; Schantz, S.; Emsley, L. Nanostructure of Materials Determined by Relayed Paramagnetic Relaxation Enhancement. J Am Chem Soc 2015, 137, 12482–12485. [Google Scholar] [CrossRef] [PubMed]
- Bara-Estaun, A.; Harder, M.C.; Lyall, C.L.; Lowe, J.P.; Suturina, E.; Hintermair, U. Paramagnetic Relaxation Agents for Enhancing Temporal Resolution and Sensitivity in Multinuclear FlowNMR Spectroscopy. Chemistry 2023, 29, e202300215. [Google Scholar] [CrossRef] [PubMed]
- Swartjes, A.; White, P.B.; Bruekers, J.P.J.; Elemans, J.; Nolte, R.J.M. Paramagnetic relaxation enhancement NMR as a tool to probe guest binding and exchange in metallohosts. Nat Commun 2022, 13, 1846. [Google Scholar] [CrossRef]
- Chen, K.; Tjandra, N. The use of residual dipolar coupling in studying proteins by NMR. Top Curr Chem 2012, 326, 47–67. [Google Scholar] [CrossRef]
- Born, A.; Henen, M.A.; Nichols, P.J.; Vogeli, B. On the use of residual dipolar couplings in multi-state structure calculation of two-domain proteins. Magn Reson Lett 2022, 2, 61–68. [Google Scholar] [CrossRef]
- Lemak, A.; Wu, B.; Yee, A.; Houliston, S.; Lee, H.W.; Gutmanas, A.; Fang, X.; Garcia, M.; Semesi, A.; Wang, Y.X.; et al. Structural characterization of a flexible two-domain protein in solution using small angle X-ray scattering and NMR data. Structure 2014, 22, 1862–1874. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Traaseth, N.J.; Verardi, R.; Gustavsson, M.; Gao, J.; Veglia, G. Paramagnetic-based NMR restraints lift residual dipolar coupling degeneracy in multidomain detergent-solubilized membrane proteins. J Am Chem Soc 2011, 133, 2232–2241. [Google Scholar] [CrossRef]
- Poveda, A.; Fittolani, G.; Seeberger, P.H.; Delbianco, M.; Jimenez-Barbero, J. The Flexibility of Oligosaccharides Unveiled Through Residual Dipolar Coupling Analysis. Front Mol Biosci 2021, 8, 784318. [Google Scholar] [CrossRef]
- Sasmal, D.K.; Pulido, L.E.; Kasal, S.; Huang, J. Single-molecule fluorescence resonance energy transfer in molecular biology. Nanoscale 2016, 8, 19928–19944. [Google Scholar] [CrossRef]
- Roy, R.; Hohng, S.; Ha, T. A practical guide to single-molecule FRET. Nat Methods 2008, 5, 507–516. [Google Scholar] [CrossRef]
- Mazal, H.; Haran, G. Single-molecule FRET methods to study the dynamics of proteins at work. Curr Opin Biomed Eng 2019, 12, 8–17. [Google Scholar] [CrossRef] [PubMed]
- Meszaros, J.; Geggier, P.; Manning, J.J.; Asher, W.B.; Javitch, J.A. Methods for automating the analysis of live-cell single-molecule FRET data. Front Cell Dev Biol 2023, 11, 1184077. [Google Scholar] [CrossRef]
- Agam, G.; Gebhardt, C.; Popara, M.; Machtel, R.; Folz, J.; Ambrose, B.; Chamachi, N.; Chung, S.Y.; Craggs, T.D.; de Boer, M.; et al. Reliability and accuracy of single-molecule FRET studies for characterization of structural dynamics and distances in proteins. Nat Methods 2023, 20, 523–535. [Google Scholar] [CrossRef]
- Yu, L.; Lei, Y.; Ma, Y.; Liu, M.; Zheng, J.; Dan, D.; Gao, P. A Comprehensive Review of Fluorescence Correlation Spectroscopy. Frontiers in Physics 2021, 9. [Google Scholar] [CrossRef]
- Elson, E.L. Fluorescence correlation spectroscopy: past, present, future. Biophys J 2011, 101, 2855–2870. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zhang, H.; Jian, L.; Ding, B.; Huang, K.; Zhang, W.; Xiao, Q.; Huang, S. Principles of fluorescence correlation spectroscopy applied to studies of biomolecular liquid-liquid phase separation. Biophys Rep 2022, 8, 100–118. [Google Scholar] [CrossRef] [PubMed]
- Jazani, S.; Sgouralis, I.; Shafraz, O.M.; Levitus, M.; Sivasankar, S.; Presse, S. An alternative framework for fluorescence correlation spectroscopy. Nat Commun 2019, 10, 3662. [Google Scholar] [CrossRef] [PubMed]
- Datta, R.; Heaster, T.M.; Sharick, J.T.; Gillette, A.A.; Skala, M.C. Fluorescence lifetime imaging microscopy: fundamentals and advances in instrumentation, analysis, and applications. J Biomed Opt 2020, 25, 1–43. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, T.; Herbert, S.; Hackl, B.; Besold, J.M.; Schramek, C.; Gotzmann, J.; Elsayad, K.; Slade, D. Direct measurement of protein-protein interactions by FLIM-FRET at UV laser-induced DNA damage sites in living cells. Nucleic Acids Res 2020, 48, e122. [Google Scholar] [CrossRef] [PubMed]
- Datta, R.; Gillette, A.; Stefely, M.; Skala, M.C. Recent innovations in fluorescence lifetime imaging microscopy for biology and medicine. J Biomed Opt 2021, 26. [Google Scholar] [CrossRef] [PubMed]
- Durrant, J.D.; McCammon, J.A. Molecular dynamics simulations and drug discovery. BMC Biol 2011, 9, 71. [Google Scholar] [CrossRef] [PubMed]
- Lazim, R.; Suh, D.; Choi, S. Advances in Molecular Dynamics Simulations and Enhanced Sampling Methods for the Study of Protein Systems. Int J Mol Sci 2020, 21. [Google Scholar] [CrossRef]
- Hospital, A.; Goni, J.R.; Orozco, M.; Gelpi, J.L. Molecular dynamics simulations: advances and applications. Adv Appl Bioinform Chem 2015, 8, 37–47. [Google Scholar] [CrossRef]
- Mouvet, F.; Villard, J.; Bolnykh, V.; Rothlisberger, U. Recent Advances in First-Principles Based Molecular Dynamics. Acc Chem Res 2022, 55, 221–230. [Google Scholar] [CrossRef]
- Bhati, A.P.; Hoti, A.; Potterton, A.; Bieniek, M.K.; Coveney, P.V. Long Time Scale Ensemble Methods in Molecular Dynamics: Ligand-Protein Interactions and Allostery in SARS-CoV-2 Targets. J Chem Theory Comput 2023, 19, 3359–3378. [Google Scholar] [CrossRef] [PubMed]
- Henkelman, G.; Jónsson, H.; Lelièvre, T.; Mousseau, N.; Voter, A.F. Long-Timescale Simulations: Challenges, Pitfalls, Best Practices, for Development and Applications. In Handbook of Materials Modeling; 2018; pp. 1–10. [Google Scholar]
- Yang, Y.I.; Shao, Q.; Zhang, J.; Yang, L.; Gao, Y.Q. Enhanced sampling in molecular dynamics. J Chem Phys 2019, 151, 070902. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, C.; Banisch, R.; Sarich, M.; Badowski, T.; Schütte, C. Characterization of Rare Events in Molecular Dynamics. Entropy 2013, 16, 350–376. [Google Scholar] [CrossRef]
- Bernardi, R.C.; Melo, M.C.R.; Schulten, K. Enhanced sampling techniques in molecular dynamics simulations of biological systems. Biochim Biophys Acta 2015, 1850, 872–877. [Google Scholar] [CrossRef] [PubMed]
- Rizzi, V.; Aureli, S.; Ansari, N.; Gervasio, F.L. OneOPES, a Combined Enhanced Sampling Method to Rule Them All. J Chem Theory Comput 2023, 19, 5731–5742. [Google Scholar] [CrossRef] [PubMed]
- Majewski, M.; Perez, A.; Tholke, P.; Doerr, S.; Charron, N.E.; Giorgino, T.; Husic, B.E.; Clementi, C.; Noe, F.; De Fabritiis, G. Machine learning coarse-grained potentials of protein thermodynamics. Nat Commun 2023, 14, 5739. [Google Scholar] [CrossRef]
- Wang, W.; Gómez-Bombarelli, R. Coarse-graining auto-encoders for molecular dynamics. npj Computational Materials 2019, 5. [Google Scholar] [CrossRef]
- Singh, N.; Li, W. Recent Advances in Coarse-Grained Models for Biomolecules and Their Applications. Int J Mol Sci 2019, 20. [Google Scholar] [CrossRef]
- Kmiecik, S.; Gront, D.; Kolinski, M.; Wieteska, L.; Dawid, A.E.; Kolinski, A. Coarse-Grained Protein Models and Their Applications. Chem Rev 2016, 116, 7898–7936. [Google Scholar] [CrossRef]
- Noid, W.G. Perspective: Coarse-grained models for biomolecular systems. J Chem Phys 2013, 139, 090901. [Google Scholar] [CrossRef]
- Liwo, A.; Czaplewski, C.; Sieradzan, A.K.; Lipska, A.G.; Samsonov, S.A.; Murarka, R.K. Theory and Practice of Coarse-Grained Molecular Dynamics of Biologically Important Systems. Biomolecules 2021, 11. [Google Scholar] [CrossRef]
- Saunders, M.G.; Voth, G.A. Coarse-graining methods for computational biology. Annu Rev Biophys 2013, 42, 73–93. [Google Scholar] [CrossRef]
- Lee, M. Recent Advances in Deep Learning for Protein-Protein Interaction Analysis: A Comprehensive Review. Molecules 2023, 28. [Google Scholar] [CrossRef]
- Bhakat, S. Collective variable discovery in the age of machine learning: reality, hype and everything in between. RSC Adv 2022, 12, 25010–25024. [Google Scholar] [CrossRef]
- Janson, G.; Valdes-Garcia, G.; Heo, L.; Feig, M. Direct generation of protein conformational ensembles via machine learning. Nat Commun 2023, 14, 774. [Google Scholar] [CrossRef]
- Saharkhiz, S.; Mostafavi, M.; Birashk, A.; Karimian, S.; Khalilollah, S.; Jaferian, S.; Yazdani, Y.; Alipourfard, I.; Huh, Y.S.; Farani, M.R.; et al. The State-of-the-Art Overview to Application of Deep Learning in Accurate Protein Design and Structure Prediction. Top Curr Chem (Cham) 2024, 382, 23. [Google Scholar] [CrossRef]
- Elia Venanzi, N.A.; Basciu, A.; Vargiu, A.V.; Kiparissides, A.; Dalby, P.A.; Dikicioglu, D. Machine Learning Integrating Protein Structure, Sequence, and Dynamics to Predict the Enzyme Activity of Bovine Enterokinase Variants. J Chem Inf Model 2024, 64, 2681–2694. [Google Scholar] [CrossRef]
- Langmead, C.J. Generative models of conformational dynamics. Adv Exp Med Biol 2014, 805, 87–105. [Google Scholar] [CrossRef]
- Zhu, J.; Li, Z.; Tong, H.; Lu, Z.; Zhang, N.; Wei, T.; Chen, H.F. Phanto-IDP: compact model for precise intrinsically disordered protein backbone generation and enhanced sampling. Brief Bioinform 2023, 25. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Akin, H.; Rao, R.; Hie, B.; Zhu, Z.; Lu, W.; Smetanin, N.; Verkuil, R.; Kabeli, O.; Shmueli, Y.; et al. Evolutionary-scale prediction of atomic-level protein structure with a language model. Science 2023, 379, 1123–1130. [Google Scholar] [CrossRef] [PubMed]
- Shin, K.; Tran, D.P.; Takemura, K.; Kitao, A.; Terayama, K.; Tsuda, K. Enhancing Biomolecular Sampling with Reinforcement Learning: A Tree Search Molecular Dynamics Simulation Method. ACS Omega 2019, 4, 13853–13862. [Google Scholar] [CrossRef] [PubMed]
- Noe, F.; De Fabritiis, G.; Clementi, C. Machine learning for protein folding and dynamics. Curr Opin Struct Biol 2020, 60, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wang, H.; E, W. Reinforced dynamics for enhanced sampling in large atomic and molecular systems. J Chem Phys 2018, 148, 124113. [Google Scholar] [CrossRef] [PubMed]
- Mehdi, S.; Smith, Z.; Herron, L.; Zou, Z.; Tiwary, P. Enhanced Sampling with Machine Learning. Annu Rev Phys Chem 2024, 75, 347–370. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Sidky, H.; Ferguson, A.L. Nonlinear discovery of slow molecular modes using state-free reversible VAMPnets. J Chem Phys 2019, 150, 214114. [Google Scholar] [CrossRef] [PubMed]
- Eaton, W.A. Modern Kinetics and Mechanism of Protein Folding: A Retrospective. J Phys Chem B 2021, 125, 3452–3467. [Google Scholar] [CrossRef] [PubMed]
- Chong, S.H.; Ham, S. Folding Free Energy Landscape of Ordered and Intrinsically Disordered Proteins. Sci Rep 2019, 9, 14927. [Google Scholar] [CrossRef] [PubMed]
- Wolynes, P.G. Evolution, energy landscapes and the paradoxes of protein folding. Biochimie 2015, 119, 218–230. [Google Scholar] [CrossRef] [PubMed]
- Nymeyer, H.; Garcia, A.E.; Onuchic, J.N. Folding funnels and frustration in off-lattice minimalist protein landscapes. Proc Natl Acad Sci U S A 1998, 95, 5921–5928. [Google Scholar] [CrossRef]
- Ma, B.; Kumar, S.; Tsai, C.J.; Nussinov, R. Folding funnels and binding mechanisms. Protein Eng 1999, 12, 713–720. [Google Scholar] [CrossRef]
- Tsai, C.J.; Ma, B.; Nussinov, R. Folding and binding cascades: shifts in energy landscapes. Proc Natl Acad Sci U S A 1999, 96, 9970–9972. [Google Scholar] [CrossRef] [PubMed]
- Bryngelson, J.D.; Onuchic, J.N.; Socci, N.D.; Wolynes, P.G. Funnels, pathways, and the energy landscape of protein folding: a synthesis. Proteins 1995, 21, 167–195. [Google Scholar] [CrossRef] [PubMed]
- Trivedi, R.; Nagarajaram, H.A. Intrinsically Disordered Proteins: An Overview. Int J Mol Sci 2022, 23. [Google Scholar] [CrossRef]
- DeForte, S.; Uversky, V.N. Not an exception to the rule: the functional significance of intrinsically disordered protein regions in enzymes. Mol Biosyst 2017, 13, 463–469. [Google Scholar] [CrossRef]
- Holehouse, A.S.; Kragelund, B.B. The molecular basis for cellular function of intrinsically disordered protein regions. Nat Rev Mol Cell Biol 2024, 25, 187–211. [Google Scholar] [CrossRef] [PubMed]
- Shin, Y.; Brangwynne, C.P. Liquid phase condensation in cell physiology and disease. Science 2017, 357. [Google Scholar] [CrossRef] [PubMed]
- Pancsa, R.; Tompa, P. Structural disorder in eukaryotes. PLoS One 2012, 7, e34687. [Google Scholar] [CrossRef]
- Schuler, B.; Soranno, A.; Hofmann, H.; Nettels, D. Single-Molecule FRET Spectroscopy and the Polymer Physics of Unfolded and Intrinsically Disordered Proteins. Annu Rev Biophys 2016, 45, 207–231. [Google Scholar] [CrossRef]
- Uversky, V.N. Intrinsically Disordered Proteins and Their “Mysterious” (Meta)Physics. Frontiers in Physics 2019, 7. [Google Scholar] [CrossRef]
- Hartl, F.U.; Hayer-Hartl, M. Converging concepts of protein folding in vitro and in vivo. Nat Struct Mol Biol 2009, 16, 574–581. [Google Scholar] [CrossRef]
- Liberek, K.; Lewandowska, A.; Zietkiewicz, S. Chaperones in control of protein disaggregation. EMBO J 2008, 27, 328–335. [Google Scholar] [CrossRef] [PubMed]
- Marzano, N.R.; Paudel, B.P.; van Oijen, A.M.; Ecroyd, H. Real-time single-molecule observation of chaperone-assisted protein folding. Sci Adv 2022, 8, eadd0922. [Google Scholar] [CrossRef] [PubMed]
- Horowitz, S.; Salmon, L.; Koldewey, P.; Ahlstrom, L.S.; Martin, R.; Quan, S.; Afonine, P.V.; van den Bedem, H.; Wang, L.; Xu, Q.; et al. Visualizing chaperone-assisted protein folding. Nat Struct Mol Biol 2016, 23, 691–697. [Google Scholar] [CrossRef]
- Shamsi, Z.; Cheng, K.J.; Shukla, D. Reinforcement Learning Based Adaptive Sampling: REAPing Rewards by Exploring Protein Conformational Landscapes. J Phys Chem B 2018, 122, 8386–8395. [Google Scholar] [CrossRef] [PubMed]
- Balchin, D.; Hayer-Hartl, M.; Hartl, F.U. In vivo aspects of protein folding and quality control. Science 2016, 353, aac4354. [Google Scholar] [CrossRef] [PubMed]
- Bukau, B.; Weissman, J.; Horwich, A. Molecular chaperones and protein quality control. Cell 2006, 125, 443–451. [Google Scholar] [CrossRef] [PubMed]
- Tsutsui, Y.; Wintrode, P.L. Hydrogen/deuterium exchange-mass spectrometry: a powerful tool for probing protein structure, dynamics and interactions. Curr Med Chem 2007, 14, 2344–2358. [Google Scholar] [CrossRef]
- Hartl, F.U. Molecular chaperones in cellular protein folding. Nature 1996, 381, 571–579. [Google Scholar] [CrossRef] [PubMed]
- Nussinov, R.; Ma, B.; Tsai, C.J. Multiple conformational selection and induced fit events take place in allosteric propagation. Biophys Chem 2014, 186, 22–30. [Google Scholar] [CrossRef]
- Paul, F.; Weikl, T.R. How to Distinguish Conformational Selection and Induced Fit Based on Chemical Relaxation Rates. PLoS Comput Biol 2016, 12, e1005067. [Google Scholar] [CrossRef]
- Morando, M.A.; Saladino, G.; D'Amelio, N.; Pucheta-Martinez, E.; Lovera, S.; Lelli, M.; Lopez-Mendez, B.; Marenchino, M.; Campos-Olivas, R.; Gervasio, F.L. Conformational Selection and Induced Fit Mechanisms in the Binding of an Anticancer Drug to the c-Src Kinase. Sci Rep 2016, 6, 24439. [Google Scholar] [CrossRef] [PubMed]
- Nam, K.; Shao, Y.; Major, D.T.; Wolf-Watz, M. Perspectives on Computational Enzyme Modeling: From Mechanisms to Design and Drug Development. ACS Omega 2024, 9, 7393–7412. [Google Scholar] [CrossRef] [PubMed]
- Wlodarski, T.; Zagrovic, B. Conformational selection and induced fit mechanism underlie specificity in noncovalent interactions with ubiquitin. Proc Natl Acad Sci U S A 2009, 106, 19346–19351. [Google Scholar] [CrossRef] [PubMed]
- Henzler-Wildman, K.; Kern, D. Dynamic personalities of proteins. Nature 2007, 450, 964–972. [Google Scholar] [CrossRef] [PubMed]
- Motlagh, H.N.; Wrabl, J.O.; Li, J.; Hilser, V.J. The ensemble nature of allostery. Nature 2014, 508, 331–339. [Google Scholar] [CrossRef] [PubMed]
- Nussinov, R.; Tsai, C.J. Allostery in disease and in drug discovery. Cell 2013, 153, 293–305. [Google Scholar] [CrossRef] [PubMed]
- Kern, D.; Zuiderweg, E.R. The role of dynamics in allosteric regulation. Curr Opin Struct Biol 2003, 13, 748–757. [Google Scholar] [CrossRef]
- Tsai, C.J.; Nussinov, R. A unified view of "how allostery works". PLoS Comput Biol 2014, 10, e1003394. [Google Scholar] [CrossRef]
- Klinman, J.P.; Kohen, A. Hydrogen tunneling links protein dynamics to enzyme catalysis. Annu Rev Biochem 2013, 82, 471–496. [Google Scholar] [CrossRef]
- Schramm, V.L.; Schwartz, S.D. Promoting Vibrations and the Function of Enzymes. Emerging Theoretical and Experimental Convergence. Biochemistry 2018, 57, 3299–3308. [Google Scholar] [CrossRef]
- Chalopin, Y.; Piazza, F.; Mayboroda, S.; Weisbuch, C.; Filoche, M. Universality of fold-encoded localized vibrations in enzymes. Sci Rep 2019, 9, 12835. [Google Scholar] [CrossRef] [PubMed]
- Sutcliffe, M.J.; Scrutton, N.S. Enzymology takes a quantum leap forward. Philos Trans A Math Phys Eng Sci 2000, 358, 367–386. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Mehmood, R.; Wang, M.; Qi, H.W.; Steeves, A.H.; Kulik, H.J. Revealing quantum mechanical effects in enzyme catalysis with large-scale electronic structure simulation. React Chem Eng 2019, 4, 298–315. [Google Scholar] [CrossRef] [PubMed]
- Corradi, V.; Sejdiu, B.I.; Mesa-Galloso, H.; Abdizadeh, H.; Noskov, S.Y.; Marrink, S.J.; Tieleman, D.P. Emerging Diversity in Lipid-Protein Interactions. Chem Rev 2019, 119, 5775–5848. [Google Scholar] [CrossRef] [PubMed]
- Tieleman, D.P.; Sejdiu, B.I.; Cino, E.A.; Smith, P.; Barreto-Ojeda, E.; Khan, H.M.; Corradi, V. Insights into lipid-protein interactions from computer simulations. Biophys Rev 2021, 13, 1019–1027. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Thienpont, B.; Sapuru, V.; Hite, R.K.; Dittman, J.S.; Sturgis, J.N.; Scheuring, S. Membrane-mediated protein interactions drive membrane protein organization. Nat Commun 2022, 13, 7373. [Google Scholar] [CrossRef] [PubMed]
- Muller, M.P.; Jiang, T.; Sun, C.; Lihan, M.; Pant, S.; Mahinthichaichan, P.; Trifan, A.; Tajkhorshid, E. Characterization of Lipid-Protein Interactions and Lipid-Mediated Modulation of Membrane Protein Function through Molecular Simulation. Chem Rev 2019, 119, 6086–6161. [Google Scholar] [CrossRef] [PubMed]
- Gu, R.X.; de Groot, B.L. Lipid-protein interactions modulate the conformational equilibrium of a potassium channel. Nat Commun 2020, 11, 2162. [Google Scholar] [CrossRef] [PubMed]
- Marinelli, F.; Faraldo-Gomez, J.D. Conformational free-energy landscapes of a Na(+)/Ca(2+) exchanger explain its alternating-access mechanism and functional specificity. Proc Natl Acad Sci U S A 2024, 121, e2318009121. [Google Scholar] [CrossRef]
- Weyand, S.; Shimamura, T.; Beckstein, O.; Sansom, M.S.; Iwata, S.; Henderson, P.J.; Cameron, A.D. The alternating access mechanism of transport as observed in the sodium-hydantoin transporter Mhp1. J Synchrotron Radiat 2011, 18, 20–23. [Google Scholar] [CrossRef]
- Del Alamo, D.; Sala, D.; McHaourab, H.S.; Meiler, J. Sampling alternative conformational states of transporters and receptors with AlphaFold2. Elife 2022, 11. [Google Scholar] [CrossRef] [PubMed]
- Badiee, S.A.; Isu, U.H.; Khodadadi, E.; Moradi, M. The Alternating Access Mechanism in Mammalian Multidrug Resistance Transporters and Their Bacterial Homologs. Membranes (Basel) 2023, 13. [Google Scholar] [CrossRef] [PubMed]
- Deupi, X.; Kobilka, B.K. Energy landscapes as a tool to integrate GPCR structure, dynamics, and function. Physiology (Bethesda) 2010, 25, 293–303. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; He, X.; Yang, Z.; Chai, Z.; Zhou, S.; Wang, J.; Rehman, A.U.; Ni, D.; Pu, J.; Sun, J.; et al. Activation pathway of a G protein-coupled receptor uncovers conformational intermediates as targets for allosteric drug design. Nat Commun 2021, 12, 4721. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Yang, D.; Wu, M.; Guo, Y.; Guo, W.; Zhong, L.; Cai, X.; Dai, A.; Jang, W.; Shakhnovich, E.I.; et al. Common activation mechanism of class A GPCRs. Elife 2019, 8. [Google Scholar] [CrossRef]
- Alenghat, F.J.; Golan, D.E. Membrane protein dynamics and functional implications in mammalian cells. Curr Top Membr 2013, 72, 89–120. [Google Scholar] [CrossRef] [PubMed]
- Hauser, A.S.; Kooistra, A.J.; Munk, C.; Heydenreich, F.M.; Veprintsev, D.B.; Bouvier, M.; Babu, M.M.; Gloriam, D.E. GPCR activation mechanisms across classes and macro/microscales. Nat Struct Mol Biol 2021, 28, 879–888. [Google Scholar] [CrossRef]
- Fleetwood, O.; Matricon, P.; Carlsson, J.; Delemotte, L. Energy Landscapes Reveal Agonist Control of G Protein-Coupled Receptor Activation via Microswitches. Biochemistry 2020, 59, 880–891. [Google Scholar] [CrossRef]
- Poudel, H.; Wales, D.J.; Leitner, D.M. Vibrational Energy Landscapes and Energy Flow in GPCRs. J Phys Chem B 2024. [Google Scholar] [CrossRef]
- May, A.; Pool, R.; van Dijk, E.; Bijlard, J.; Abeln, S.; Heringa, J.; Feenstra, K.A. Coarse-grained versus atomistic simulations: realistic interaction free energies for real proteins. Bioinformatics 2014, 30, 326–334. [Google Scholar] [CrossRef]
- van der Kamp, M.W.; Shaw, K.E.; Woods, C.J.; Mulholland, A.J. Biomolecular simulation and modelling: status, progress and prospects. J R Soc Interface 2008, 5 Suppl 3, S173–190. [Google Scholar] [CrossRef]
- Lapatas, V.; Stefanidakis, M.; Jimenez, R.C.; Via, A.; Schneider, M.V. Data integration in biological research: an overview. J Biol Res (Thessalon) 2015, 22, 9. [Google Scholar] [CrossRef] [PubMed]
- Walpole, J.; Papin, J.A.; Peirce, S.M. Multiscale computational models of complex biological systems. Annu Rev Biomed Eng 2013, 15, 137–154. [Google Scholar] [CrossRef] [PubMed]
- Lippincott-Schwartz, J.; Snapp, E.; Kenworthy, A. Studying protein dynamics in living cells. Nat Rev Mol Cell Biol 2001, 2, 444–456. [Google Scholar] [CrossRef] [PubMed]
- Zalejski, J.; Sun, J.; Sharma, A. Unravelling the Mystery inside Cells by Using Single-Molecule Fluorescence Imaging. J Imaging 2023, 9. [Google Scholar] [CrossRef] [PubMed]
- Feng, R.; Gruebele, M.; Davis, C.M. Quantifying protein dynamics and stability in a living organism. Nat Commun 2019, 10, 1179. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.E.; Barethiya, S.; Nordquist, E.; Chen, J. Machine Learning Generation of Dynamic Protein Conformational Ensembles. Molecules 2023, 28. [Google Scholar] [CrossRef] [PubMed]
- Audagnotto, M.; Czechtizky, W.; De Maria, L.; Kack, H.; Papoian, G.; Tornberg, L.; Tyrchan, C.; Ulander, J. Machine learning/molecular dynamic protein structure prediction approach to investigate the protein conformational ensemble. Sci Rep 2022, 12, 10018. [Google Scholar] [CrossRef] [PubMed]
- Ficner, R. Highlight: integrative structural biology of dynamic macromolecular assemblies. Biol Chem 2023, 404, 739. [Google Scholar] [CrossRef]
- Pak, A.J.; Voth, G.A. Advances in coarse-grained modeling of macromolecular complexes. Curr Opin Struct Biol 2018, 52, 119–126. [Google Scholar] [CrossRef]
- Monachino, E.; Spenkelink, L.M.; van Oijen, A.M. Watching cellular machinery in action, one molecule at a time. J Cell Biol 2017, 216, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Prabantu, V.M.; Naveenkumar, N.; Srinivasan, N. Influence of Disease-Causing Mutations on Protein Structural Networks. Front Mol Biosci 2020, 7, 620554. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.Q.; Sang, P.; Tao, Y.; Fu, Y.X.; Zhang, K.Q.; Xie, Y.H.; Liu, S.Q. Protein dynamics and motions in relation to their functions: several case studies and the underlying mechanisms. J Biomol Struct Dyn 2014, 32, 372–393. [Google Scholar] [CrossRef] [PubMed]
- Pacini, L.; Dorantes-Gilardi, R.; Vuillon, L.; Lesieur, C. Mapping Function from Dynamics: Future Challenges for Network-Based Models of Protein Structures. Front Mol Biosci 2021, 8, 744646. [Google Scholar] [CrossRef]
- Ruiz, C.; Zitnik, M.; Leskovec, J. Identification of disease treatment mechanisms through the multiscale interactome. Nat Commun 2021, 12, 1796. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



