
Submitted:
01 August 2024
Posted:
05 August 2024
You are already at the latest version
Abstract
Familial pancreatic cancer (FPC) represents a significant yet underexplored area in pancreatic cancer research. Basic research efforts are notably limited, and when present, they are predominantly centered on BRCA1 and BRCA2 mutations due to the scarcity of other genetic variants associated with FPC, leading to a limited understanding of the broader genetic landscape of FPC. This review examines the current state of FPC research, focusing on the molecular mechanisms driving pancreatic ductal adenocarcinoma (PDAC) progression. It highlights the role of homologous recombination (HR) and its therapeutic exploitation via synthetic lethality with PARP inhibitors in BRCA1/2-deficient tumors. The review discusses various pre-clinical models of FPC, including conventional two-dimensional (2D) cell lines, patient-derived organoids (PDOs), patient-derived xenografts (PDXs), and genetically engineered mouse models (GEMMs) as well as new advancements in FPC research.
Keywords:
familial pancreatic cancer
; BRCA2
; BRCA1
; PARP inhibitor
; Pancreatic Ductal Adenocarcinoma
; Homologous recombination
Introduction
Pancreatic cancer is one of the most aggressive and deadliest cancers. With a late diagnosis due to the lack of early-stage symptoms and a poor prognosis, it has a current 5-year survival rate of 13% and is estimated to become the second-leading cause of cancer-related mortality by 2030 [1,2]. The most common type of pancreatic cancer is termed pancreatic ductal adenocarcinoma (PDAC). It stems from the exocrine pancreas, which is primarily responsible for the secretion of digestive enzymes, ions, and water into the duodenum of the gastrointestinal tract. While surgical resection is the sole curative option, only around 15–20% of patients can undergo resection due to the early metastasis of PDAC, making the early detection of PDAC a necessary but challenging goal [3,4]. The National Comprehensive Cancer Network (NCCN) recommends either chemotherapy combinations, gemcitabine with nab-paclitaxel or FOLFIRINOX (folinic acid, 5-FU, irinotecan, oxaliplatin) as first-line treatment [1]. However, clinical benefits from the standard chemotherapies for PDAC patients remain modest.
The progression of PDAC is prompted by the somatic mutations of key driver genes. Prevalence of the somatic mutation in PDAC includes: activating mutations in KRAS, 90%; inactivating mutations in TP53, 50–74%; inactivating mutations in CDKN2A, 46–60%; inactivating mutations in SMAD4, 31–38% [2]. Oncogenic mutations in KRAS in pancreatic ductal epithelial cells are known to initiate pancreatic intraepithelial neoplasia (PanIN), ultimately resulting in PDAC with subsequent mutations in other tumor suppressor genes. KRAS is a membrane-bound guanosine triphosphate (GTP) binding protein and mainly functions in cell growth and proliferation. CDKN2A plays essential roles in the expression and functionality of cell-cycle regulators including p53 [3]. Furthermore, mutations in TP53 and SMAD4 are frequently detected in PanIN-3 and invasive tumors, which drives the expansion of pancreatic cancers [4]. In addition to these somatic mutations, a portion of patients with a family history of PDAC carry germline mutations, which increase their risk of developing PDAC, known as familial pancreatic cancer (FPC) [5].
Statistical Risk of Familial Pancreatic Cancer
Since its establishment in 1994, the National Familial Pancreas Tumor Registry (NFPTR) has been recruiting cases of FPC, which defines as patients with two or more first-degree relatives diagnosed with pancreatic cancer. Patients without such a family history are classified as having sporadic pancreatic cancer (SPC) [6,7]. These definitions have become the consensus for FPC and SPC. FPC cases comprise approximately 10% of total pancreatic cancer cases [8]. Individuals with first-degree relatives diagnosed with pancreatic cancer are found to have a significantly higher risk of developing pancreatic cancer compared to the general population. The extent of pancreatic cancer risk is directly proportional to the number of affected first-degree relatives [7,8,9]. Having one diagnosed first-degree relative increases the risk by 4.5-fold, having two diagnosed first-degree relatives leads to a 6.4-fold increase, and having three or more diagnosed first-degree relatives increases the risk by 32-fold. However, the elevated risk is not observed in spouses and other genetically unrelated relatives, highlighting the significant role of genetic factors in the etiology of FPC [7].
A meta-analysis of seven case-control and two cohort studies, independently conducted in different geological locations and across a 40-year time span, verified that family history is a risk factor for pancreatic cancer, despite variations in location and methodology. This meta-analysis further supports previous findings, concluding that having more than one affected first or second-degree relative harbors a nearly 2-fold increased risk of pancreatic cancer [10]. In addition, when comparing successive generations within FPC families, each generation showed a lower age of death and a higher risk of pancreatic cancer death than its previous generation [11]. Individuals with family histories of pancreatic cancer also have higher risks of developing other types of cancers including prostate cancer, liver carcinoma, lymphoma, and colon cancer [12]. These meta-analyses statistically demonstrate the familial aggregation of pancreatic cancer and suggest a strong correlation between genetic factors and the development of pancreatic cancers. Investigations on the genetic basis of FPC will provide critical support for evolving areas such as cancer screening, prevention, management, and genetic counseling for high-risk individuals. This has led to various research aimed at pinpointing the genetic variants associated with FPC.
Genes Associated with Familial Pancreatic Cancer
Early studies have identified BRCA2 as one of the most commonly mutated genes in FPC [13,14]. BRCA1 and BRCA2 encode for key proteins activated in the presence of DNA double-strand breaks (DSBs) and subsequently mediate the DNA repair pathway. Failures in repairing DNA DSBs lead to genome instability and the generation of disruptive and harmful mutations that cause severe diseases and cancers [15,16,17,18,19]. Consistent with this, individuals with mutations in BRCA1 and BRCA2 harbor a high risk of developing various types of cancer including breast, ovary, prostate, esophagus, stomach, and uveal cancers [19,20]. By direct sequencing of constitutional DNA, Murphy et al. identified five (17.2%) deleterious BRCA2 mutations in FPC patients [13]. In addition, in 26 European families that met the criteria of FPC, 12% of the families carried germline frameshift mutations in BRCA2 (6672insT, 6819delTG, and 4075delGT) that resulted in a truncated and non-functional BRCA2 protein. Additional two families were identified with sequence variants of BRCA2, resulting in an overall 19% prevalence of BRCA2 mutations in FPC [14]. Among Ashkenazi Jewish breast cancer patients, a higher BRCA1 and BRCA2 mutation rate was found to be associated with a family history of pancreatic cancer. In patients who have first-, second-, and third-degree relatives with pancreatic cancer, the mutation prevalence was 15.4%, 15.3%, and 8.6%, respectively [21].
The development of next-generation sequencing has enabled the identification of a wider range of pathogenic germline mutations that increase the carriers’ risk of being diagnosed with pancreatic cancer, including ATM, BRCA1, BRCA2, MLH1, MSH2, MSH6, TP53, PALB2, PMS2, PRSS1, STK11, and CDKN2A [12,22]. Roberts et al. using whole genome sequencing confirmed the existing FPC susceptibility genes mentioned above and identified novel genes such as BUB1B, CPA1, FANCC, and FANCG. This study reported that 1077 genes were found to have two or more heterozygous premature truncating variants (loss-of-function mutations), demonstrating the high genetic heterogeneity within FPC. Notably, many top-hit candidate genes are involved in the DNA damage repair pathway and genome stability regulation, hereafter referred to as “FPC genes” [23]. Among these genes, BRCA2 remained to be the most commonly mutated gene. Other studies employed nearly or more than 700 patient samples collected from multiple institute sites, without being limited to a single ethnicity, tested and compared the prevalence of BRCA1, BRCA2, PALB2, and CDKN2A mutations, four of the top FPC-associated DNA repair gene mutations, in FPC and SPC patients (Table 1) [12,22,23]. These studies identified multiple novel BRCA2 variants such as 6224insT, confirmed FPC patients carry more mutations in the above four genes than SPC patients, and concluded that BRCA2 and CDKN2A account for the majority of mutations within FPC. In a separate study comparing ATM mutants between FPC and SPC groups, ATM mutants have a higher prevalence in FPC patients compared to the general population [24].
Taken together, these findings suggest that inheritable mutations in multiple genes involved in DNA damage repair and genome stability maintenance lead to a higher risk of developing pancreatic cancer, with BRCA2 being the most commonly mutated gene compared to others in FPC.
Double-Stranded Breaks (DSB) and Homologous Recombination (HR) Pathway
Since defects in FPC genes predispose individuals to the development of pancreatic cancer by disrupting DNA damage response (DDR), a better understanding of how these contribute to the pathogenesis could help in exploiting parts of the pathways therapeutically. DNA can get damaged through exogenous and endogenous damages, leading to DSBs, which pose a serious threat to cell viability and genome stability. DSBs can be generated naturally when replication forks encounter blocking lesions such as those produced as a byproduct of cellular respiration, mainly reactive oxygen species (ROS) leading to fork collapse [25]. DSBs are also produced when cells are exposed to DNA damaging agents such as ionizing radiation, chemical agents, UV light, transposons, and replication of a region that has a nick in the backbone. DSBs can also occur during programmed genome rearrangements induced by nucleases, and physical stress when chromosomes are pulled to opposite poles during mitosis [25,26]. The failure to properly repair DSBs can result in cell death or large-scale chromosome changes, including deletions, translocations, and chromosome fusions that enhance genome instability and are hallmarks of cancer [28].
Two major pathways, non-homologous end joining (NHEJ) and homologous recombination (HR), are used to repair DNA DSBs. NHEJ is an error-prone pathway in which the ends of the DSB are ligated back together nonspecifically, potentially causing insertions and deletions. On the contrary, HR is a high-fidelity pathway in which cells repair DSBs using a DNA template typically from the sister chromatid. When using the sister chromatid strand as a template, HR can result in the loss of heterozygosity with information transferred non-reciprocally from the unbroken donor locus to the broken recipient locus in a process called gene conversion [29]. HR occurs during the S and G2 phases of the mammalian cell cycle when the homologous chromosome or the sister chromatid is available due to CDK-dependent phosphorylation of CtIP, a factor known to stimulate end resection [30]. CtIP also prevents the diploid cell from using the sister chromatid as a template for repair, which can cause a loss of heterozygosity [31].
HR deficiency is particularly relevant to FPC due to BRCA1/2’s involvement in HR, as they are the most commonly mutated genes. HR is carried out by three main steps starting when HR is triggered from a DDR signal cascade that is aided by DDR proteins (Figure 1). The first step of HR pathway, termed ‘pre-synapsis’, involves DNA end-resection to generate a 3′ ssDNA overhang. The DSB signaling is initiated via the binding of the MRN complex (MRE11, RAD50, and NBS1) to the broken DNA ends. The MRN complex facilitates endonucleolytic cleavages near the DSB towards the DNA end in 3′-5′ direction from the nick site to generate a 3′ ssDNA overhang and recruit endonuclease ExoI or helicase BLM to perform bulk 5′-3′ DNA resection. The MRN complex also plays a critical role in recruiting and activating ATM at DSB sites to orchestrate the repair process [32]. The second step, termed ‘synapsis’, is homologous search and DNA strand invasion. Replication protein A (RPA) is then loaded onto the ssDNA by the MRN complex, which preserves the integrity of the resultant ssDNA [33]. In cells, BRCA2 can be recruited to DSBs by BRCA1 and PALB2 [32]. RPA, with the help of BRCA2, can recruit RAD51 recombinase, a DNA-dependent ATPase that serves as the main catalyst involved in DNA strand invasion repair. RAD51, recruited by BRCA2, then competes with RPA to gain access to the ssDNA and initiate HR-directed repair [32,34]. After recruitment, RAD51 forms a nucleoprotein filament with ssDNA, which promotes strand invasion and displacement loop (D-loop) formation [31,34]. The D-loop allows the 3′ end of the invading strand to prime DNA synthesis of the template duplex DNA [35]. The last step is ‘strand extension’. In this stage, DNA polymerase elongates the invading strand using the homologous template strand. After strand synthesis, the intermediate structures are resolved, and DNA ligases seal the nicks in the newly synthesized DNA to the complete the repair, finally resulting in a repaired DNA [34].
How genetic predispositions in FPC genes promote PDAC progression and how we can exploit these genetic defects therapeutically are active research areas. One promising approach is the concept of synthetic lethality between HR defects and Poly(ADP-ribose)-polymerase (PARP) inhibition. In BRCA1/2-deficient pancreatic cancer, defects in HR DNA damage repair create vulnerabilities that can be targeted by chemotherapies or targeted therapies. PARP is an enzyme involved in DNA base excision repair. The concurrent loss of the BRCA1/2-mediated HR pathway and PARP pathway results in an excessive accumulation of DNA damage, thereby leading to synthetic lethality. Indeed, BRCA1/2-deficient tumor cells are more sensitive to PARP inhibition, thus making PARP inhibitor an effective treatment strategy for BRCA1/2-deficient pancreatic cancer [36]. BRCA1/2-deficient tumor cells are also more sensitive to platinum-based chemotherapies and anthracyclines, which are selectively lethal in HR-deficient cells [37]. According to the National Comprehensive Cancer Network (NCCN) guidelines, genetic testing is currently recommended for all PDAC patients, partly because HR defects can benefit from PARP inhibition [1]. However, other HR-related gene defects require more epidemiological and molecular evidence to provide more informed and comprehensive genetic counseling and guidelines for both FPC patients and those at elevated risk of pancreatic cancer.
FPC and PARP Inhibitors
Previous research illuminated the pivotal role of BRCA genes in maintaining genomic stability, with mutations leading to defective DNA repair mechanisms. The defect in DNA repair process conferred increased sensitivity to DNA damaging agents like platinum-based chemotherapy and, more importantly, PARP inhibitors. PARP inhibitors emerged as a novel class of drugs exploiting the concept of synthetic lethality, wherein BRCA-mutated cancer cells deficient in DNA repair are particularly susceptible to PARP inhibition [38]. This strategy has been effective in treating BRCA-mutant cancers, including breast and ovarian cancers, as well as pancreatic cancer. In particular, the Pancreas Cancer Olaparib Ongoing (POLO) clinical trial demonstrated that olaparib significantly extended progression-free survival (PFS) compared to placebo in patients with BRCA-mutant pancreatic cancer following platinum-based chemotherapy [39]. Other PARP inhibitors like rucaparib [40], talazoparib [41,42,43], and the novel AZD 5305 have shown promise in clinical trials, indicating the potential of PARP inhibition in targeted cancer therapy, particularly for BRCA-mutated tumors, by exploiting their reliance on specific DNA repair pathways [44].
Subsequent studies expanded the utility of PARP inhibitors, exploring various combinations and settings, with other PARP inhibitors like rucaparib and veliparib also being investigated for their efficacy in BRCA-mutated cancers. In 2020, a study by O’Reilly and colleagues explored the combination of gemcitabine, cisplatin, and veliparib for BRCA-mutated PDAC, finding only a marginal improvement in response rates without statistical significance, with veliparib reducing PFS and increasing adverse events [45]. Concurrently, PARP inhibitors have been recognized for enhancing PD-L1 expression and stimulating neoantigen formation, prompting investigations such as the SWOG S2001 trial, which examines the combination of olaparib with pembrolizumab [46]. Early trials, such as those combining novel BET and WEE1 kinase inhibitors with PARP inhibitors, showed potential synergies and explore safety and efficacy in human studies [47], highlighting the ongoing search for effective combinations and novel therapeutic strategies. Currently, the NCCN guidelines recommend considering Olaparib as maintenance treatment for patients who have a deleterious germline BRCA1/2 mutation with platinum-based chemotherapy.
PARP inhibitors are a crucial treatment strategy for patients with BRCA- or PALB2-mutated pancreatic cancer, offering a valuable maintenance therapy option. These inhibitors exploit the cancer cells’ compromised DNA repair mechanism, leading to selective cancer cell death. However, the effectiveness of PARP inhibitors can be undermined by the development of resistance, a significant hurdle in the long-term management of the disease. One notable mechanism of PARP inhibitor resistance is the emergence of BRCA reversion mutations [48]. These mutations restore the function of the BRCA gene, enabling cancer cells to regain their DNA repair capabilities and resist PARP inhibitor treatment. The presence of these reversion mutations is associated with a faster progression of the disease post-PARP inhibitor therapy and a decreased overall survival rate [49,50,51]. Brown et al. examined advanced, platinum-sensitive pancreatic cancer patients treated with rucaparib. They found that acquired reversion mutations in BRCA or PALB2 were relatively rare but profoundly impacted treatment outcomes [52]. While BRCA reversion mutations upon PARP inhibitors or platinum therapies appear to be common in breast and ovarian cancer patients, these are not commonly observed in pancreatic cancer [53,54]. In the patient cohort under study by Brown et al., only a minority exhibited these reversion mutations, indicating that while they are influential, they do not account for all cases of PARP inhibitor resistance. This rarity underscores the complexity of resistance mechanisms and the need for ongoing research to understand and overcome these challenges.
In summary, while PARP inhibitors offer a promising therapeutic avenue for BRCA-mutated or HR-deficient pancreatic cancer, resistance remains a critical issue, with BRCA reversion mutations playing a significant but uncommon role. Identifying these mutations can provide valuable insights into the patient’s prognosis and guide subsequent therapeutic strategies, emphasizing the need for personalized approaches in treating pancreatic cancer.
Pre-clinical Models of FPC
The limited efficacy of PARP inhibitors and the emergence of resistance mechanisms underscore the importance of understanding precise molecular mechanisms driving FPC development and progression. Current research understandably focuses on BRCA1/2 mutations, as these alterations are the most prevalent among FPC patients. However, mutations in FPC susceptibility genes, although relatively rare, also warrant attention. Therefore, this section reviews available pre-clinical models of FPC, examining their strengths and limitations in advancing our understanding of this complex disease. A comprehensive review of pre-clinical models of PDAC is beyond the scope of this review and has been discussed elsewhere [55]. Here, we will review preclinical models specifically relevant to FPC.
In vitro models play an important role in this endeavor, with conventional cell lines offering a controlled experimental setting for dissecting the intricate genetic and cellular aberrations characteristics of the disease. Among these, the CAPAN-1 cell line commonly serves as a representative model for studying BRCA2-deficient PDAC. The cell line possesses a naturally occurring frameshift mutation in BRCA2 (c.6147delT), which generates a premature stop codon. This genetic alteration causes a pathogenic frameshift mutation (p.S1982fs*22), leading to truncating critical C-terminal amino acids of the BRCA2 protein and compromising HR repair [56]. Despite their utility, conventional two-dimensional cell line models have significant limitations. These models often lack isogenic controls, complicating comparisons of drug responses across different cell lines due to additional genetic mutations and varying cellular contexts. Moreover, these models fail to accurately recapitulate the three-dimensional cellular architecture found in tumors, potentially skewing signaling and cellular behaviors [57]. In response to these challenges, patient-derived organoids (PDO) have emerged as a novel pre-clinical model. PDOs better mimic the pathophysiology of the originating tumors. For instance, a study by Tiriac et al. utilized PDOs as a primary experimental platform to explore the interplay between genomic alterations and drug sensitivity, although the authors did not observe a significant association between sensitivity to PARP inhibitors and mutations in HR-related genes [58]. Importantly, the PDO library used in the study did not exhibit deleterious mutations in these genes, nor did it show biallelic mutations in BRCA1/2. Therefore, this underscores the necessity for a larger and more diverse PDO library, specifically including samples from FPC cases, to comprehensively address these research questions.
In addition to PDOs, patient-derived xenograft (PDX) and patient-derived organoid xenograft (PDOX) models also preserve the genetic heterogeneity and histological features of the patient’s tumor, making them a powerful tool for investigating therapeutic responses in vivo. Studies have shown that these models, harboring mutations in FPC genes, can precisely predict patient responses to various cancer treatments, including those targeting DNA damage repair pathways [59,60]. However, the predicted treatment response does not seem to be based solely on FPC gene mutations. Golan et al. observed diverse responses among BRCA1/2-mutant PDXs to DNA damaging agents, which reflect the wide spectrum of clinical responses seen in patients [61]. This variability likely stems from factors such as the PDX collection sites, BRCA status in PDX (heterozygous vs. loss of heterozygosity) and prior exposure to platinum-based or PARP inhibitor treatments. More importantly, the absence of a functional immune system in these models could further complicate predictions since PDX and PDOX lack immune cells, a critical component of the tumor microenvironment, which might affect drug responses. Given the growing significance of immunotherapy in cancer treatment, it is crucial to evaluate drug responses and immune-targeting strategies in models with an intact immune system, such as genetically engineered mouse models (GEMMs). These models can provide a more comprehensive understanding of tumor-immune interactions and therapeutic efficacy, bridging the gap left by PDX and PDOX models.
GEMMs, with their intact immune system, provide a sophisticated means to study the genetic complexity and disease progression of FPC from early precursor lesions to metastasis. These models are particularly valuable as they incorporate the critical mutations driving PDAC, including the gain-of-function mutation in Kras together with the loss-of-function mutation in Trp53 [62]. In PDAC GEMMs, these mutations are introduced as germline alterations and only activated within pancreatic epithelial cell lineage using a pancreas-specific Cre recombinase (Pdx1-Cre). Prominent examples of such models include KC (Kras+/LSL-G12D; Pdx1-Cre) and KPC (Kras+/LSL-G12D; Trp53+/LSL-R172H; Pdx1-Cre or Kras+/LSL-G12D; Trp53+/LSL-R270H; Pdx1-Cre) mouse lines, which resemble the development of PanIN and its progression to PDAC [63]. GEMMs’ robust representation of disease progression, coupled with their ability to model DDR deficiencies seen in FPC gene contexts, makes them indispensable for researching FPC biology. These models typically employ conditional knock-out alleles of FPC susceptibility genes, such as Brca1, Brca2, Atm, and Palb2, in the KC or KPC background. Studies have shown that homozygous knock-out of these FPC genes significantly accelerates PDAC progression [64,65,66,67,68,69]. These studies confirm that DNA repair deficiency contributes to PDAC progression and provides a mechanistic insight into how these mutations predispose PDAC development in FPC patients. The heterozygous knock-out model of these genes also displayed an intermediate level of PDAC progression, likely due to the dose-dependent impact of these genetic mutations on cancer development [65,66,67,68,69]. Interestingly, the tumors from FPC heterozygous knock-out models retain the wild-type allele of the respective FPC genes, indicating loss-of-heterozygosity (LOH) of FPC genes is not an obligate step in GEMMs [69]. However, this appears to be inconsistent with clinical observations, where a significant proportion of FPC tumors exhibit LOH of these genes [70]. The bi-allelic loss of FPC genes, as seen in clinical cases, appears to be critical for responses to PARP inhibitors or platinum-based drugs due to the compromised DNA repair capabilities of the tumor cells [71,72,73]. While it has clearly been shown in FPC GEMMs that FPC gene mutations promote PDAC progression in Kras or Kras/Trp53 mutant background, it remains to be addressed whether these FPC gene mutations directly co-operate with key driver mutations in PDAC patients, or these mutations facilitate the arrival of key driver somatic mutations due to impaired DNA repair. The latter can be supported by the observation that FPC patients exhibit somatic mutation profiles very similar to those seen in SPC [74,75,76,77]. It is possible both ideas may contribute to PDAC progression to varying extents. Therefore, the detailed roles of genetic variants associated with FPC need to be further dissected using GEMMs, particularly in PDAC progression and response to therapy. Additionally, exploring how to exploit these genetic vulnerabilities therapeutically and prevent drug resistance using these preclinical models will be crucial for developing targeted therapies and improving survival outcomes for FPC patients.
Current Status of Basic Research on FPC
FPC research has primarily focused on clinical and association-based studies, with little effort for mechanistic, basic research on the disease. Not surprisingly, most advancements have been made on BRCA1/2 in the context of FPC. Although there has been progress in identifying genetic drivers of FPC, translating this knowledge into effective clinical practice remains a critical challenge. Such challenge is compounded by the rarity of certain genetic variants and the pleiotropic roles of FPC genes, such as the HR-independent functions of BRCA1/2, which we are still in the process of understanding. Additionally, the significant influence of the tumor microenvironment (TME) on disease progression and treatment response adds to the complexity. Here, we discuss recent advancements in FPC research, highlighting key developments in understanding the genetic landscape, the creation of novel therapeutic strategies, and the novel roles of BRCA genes beyond HR, particularly their impact on the tumor microenvironment (TME) and potential for therapeutic exploitation.
Recent studies have focused on exploiting the unique vulnerabilities of the stroma in PDAC, aiming to develop novel therapies that target the dense and complex TME. PDAC is well-known for its highly dense desmoplastic stroma, which acts as a physical barrier, hindering immune cell penetration and contributing to its classification as a ‘cold tumor’ [78]. Shaashua et al. explored the unique stromal landscape in BRCA-mutated PDAC and found an elevated activation of HSF1 in the stroma of BRCA-mutated PDAC. This activation drives the transcriptional regulation of clusterin (CLU), resulting in the up-regulation of immune-regulatory CLU-positive cancer-associated fibroblasts. This study suggests that this distinct stromal composition, characterized by HSF1-mediated CLU expression, could be a potential therapeutic target in BRCA-mutated PDAC. [79]. Exciting advancements in cancer immunotherapy are focused on converting ‘cold’ tumors into ‘hot’ tumors, allowing for immune cell infiltration and improved treatment response. Oh et al. identified POLQ, a key mediator in the microhomology-mediated end joining (MMEJ) pathway, as a crucial pathway for DSB repair in BRCA2-deficient PDAC. POLQ inhibition represents a synthetic lethal approach to blocking tumor growth while concurrently activating the cGAS-STING signaling pathway, enhancing tumor immune infiltration and offering a novel therapeutic strategy [80]. Thus, these findings raise the possibility of targeting the unique aspects of the tumor microenvironment in BRCA-mutated PDAC to improve therapeutic outcomes.
As part of efforts to identify novel vulnerabilities of BRCA2-mutant PDAC, our group performed a high-throughput drug screening and discovered that Bromodomain and Extra-Terminal domain protein (BET) inhibitors were particularly effective [81]. This heightened sensitivity in BRCA-deficient cells is linked to enhanced autophagic flux, a catabolic process of self-degradation and recycling to maintain cellular homeostasis. The increased autophagic flux is further elevated by BET inhibition, resulting in autophagy-dependent cell death. BET inhibitors have also been shown to be preferentially cytotoxic in the mutant BRCA2 context in both breast cancer and pan-cancer settings in a publicly available database for Genomics of Drug Sensitivity in Cancer [82,83]. Consistent with these findings, Arun et al. also observed that knocking down BRCA1/2 in triple-negative breast cancer cells induced autophagy, as evidenced by increased LC3-II expression – a key autophagy marker [84]. Together, this suggests that BRCA2 may play a role as a negative regulator of autophagy. Counterintuitively, autophagy inhibition has been used to sensitize cells to drug responses. For instance, the sensitivity to cisplatin in BRCA2-depleted ovarian cancer cells was further sensitized by blocking autophagy with chloroquine [85]. These findings suggest a nuanced therapeutic potential in combining BET inhibition with strategies to modulate autophagy in BRCA2-deficient cancers.
Recent advances in cancer research have shed light on the HR-independent functions of BRCA2, such as epigenetic regulation and transcription control, albeit not in PDAC context. Gruber et al. revealed that BRCA2 loss triggers a cascade of events leading to NF-kB signaling activation and increased acetylation of histone 4 (H4), affecting gene expression and cellular phenotype [86]. In addition, BRCA2 has been shown to resolve R-loops to prevent genome instability. R-loop is a specific DNA-RNA hybrids with a displaced single-stranded DNA caused by nascent RNA re-annealing to its DNA template during transcription [87]. Studies have shown that BRCA2 associates with the TREX2- mRNA export factor PCIID2, preventing R-loop accumulation, and facilitates the transition from promoter-proximal pausing to productive elongation of transcription by recruiting PAF1 to PolII [88]. Additionally, BRCA2 recruits DDX5 helicase to the DNA damage site, further aiding R-loop resolution [89]. These functions of BRCA2 appear to be critical for maintaining genomic stability, preventing transcription-replication conflicts, and modulating cellular responses. Elevated R-loop formation and higher autophagic flux in BRCA2-deficient cells can activate the cGAS-STING pathway, potentially leading to chronic cellular stress and influencing tumor development and immune surveillance. Whether or not these findings in non-PDAC contexts can be applied to PDAC remains to be addressed, but they offer promising directions for future research in FPC. Understanding these multifaceted roles of BRCA2 offers new avenues for the personalized medicine approaches for FPC. As BRCA2’s non-HR functions can contribute to FPC pathogenesis, it is highly likely that other FPC genes also have multiple functions in various cellular processes, adding to the complexity of FPC biology. Further research is needed to dissect these detailed roles and exploit these genetic vulnerabilities.
Conclusion
FPC represents a significant yet underexplored area within pancreatic cancer research. Overall, there is a lack of basic research on FPC. Among these, most studies have predominantly centered on BRCA1 and BRCA2 mutations due to the scarcity of other FPC gene defects. This narrow focus has led to a limited understanding of the broader genetic landscape of FPC, thereby hindering the development of comprehensive therapeutic strategies. Advancing our understanding of FPC gene defects holds immense therapeutic potential. Exploiting the unique genetic makeup of FPC can lead to the development of targeted treatments, such as PARP inhibitors for BRCA-mutated tumors. However, the emergence of resistance mechanisms, including BRCA reversion mutations, underscores the necessity for ongoing research to identify and overcome these barriers. Furthermore, exploring the roles of other DDR-related genes and their pathways in FPC could unveil novel therapeutic targets. Investigating the HR-independent functions of BRCA2, such as R-loop resolution, transcription regulation and replication fork protection, may provide additional avenues for intervention. The concept of “BRCAness,” where non-BRCA-deficient tumors exhibit similar vulnerabilities, also opens new possibilities for therapeutic exploitation through epigenetic modulation. Therefore, addressing the gaps in FPC research requires a multi-faceted approach. Expanding the focus beyond BRCA1/2 mutations, developing diverse preclinical models, and leveraging the genetic intricacies of FPC gene defects will be crucial. A better understanding of FPC will pave the way for personalized medicine strategies that improve outcomes for FPC patients (Figure 2).
Acknowledgment
We would like to thank all members of the C.-I.H. laboratory for helpful discussions and suggestions for this review. C.-I.H. is supported by the National Cancer Institute (NCI) R37CA249007, and the University of California (UC) Davis Comprehensive Cancer Center Pilot Grant (NCI P30CA093373). S.A is supported by the Anandamahidol Foundation. F.A. is supported by the UC Davis Provost Undergraduate Fellowship (PUF). P.L. is supported by the Charles and Nanci Cooper Undergraduate Research
References
- Tempero, M.A. NCCN Guidelines Updates: Pancreatic Cancer. J. Natl. Compr. Canc. Netw. 2019, 17, 603–605. [Google Scholar] [CrossRef]
- Wang, S.; Zheng, Y.; Yang, F.; Zhu, L.; Zhu, X.Q.; Wang, Z.F.; Wu, X.L.; Zhou, C.H.; Yan, J.Y.; Hu, B.Y.; et al. The Molecular Biology of Pancreatic Adenocarcinoma: Translational Challenges and Clinical Perspectives. Signal Transduct. Target. Ther. 2021, 6. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.C.; Rajbhandari, N.; Liu, C.; Sakamoto, K.; Zhang, Q.; Triplett, A.A.; Batra, S.K.; Opavsky, R.; Felsher, D.W.; DiMaio, D.J.; et al. Dormant Cancer Cells Contribute to Residual Disease in a Model of Reversible Pancreatic Cancer. Cancer Res. 2013, 73, 1821–1830. [Google Scholar] [CrossRef] [PubMed]
- Brosens, L.A.A.; Hackeng, W.M.; Offerhaus, J.; Hruban, R.H.; Wood, L.D. Pancreatic Adenocarcinoma Pathology: Changing “Landscape. ” J. Gastrointest. Oncol. 2015, 6, 358–374. [Google Scholar] [CrossRef]
- Pilarski, R. The Role of BRCA Testing in Hereditary Pancreatic and Prostate Cancer Families. Am. Soc. Clin. Oncol. Educ. B. 2019, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Petersen, G.M. Familial Pancreatic Cancer. Semin. Oncol. 2016, 43, 548–553. [Google Scholar] [CrossRef] [PubMed]
- Klein, A.P.; Brune, K.A.; Petersen, G.M.; Goggins, M.; Tersmette, A.C.; Offerhaus, G.J.A.; Griffin, C.; Cameron, J.L.; Yeo, C.J.; Kern, S.; et al. Prospective Risk of Pancreatic Cancer in Familial Pancreatic Cancer Kindreds. Cancer Res. 2004, 64, 2634–2638. [Google Scholar] [CrossRef]
- McWilliams, R.R.; Rabe, K.G.; Olswold, C.; De Andrade, M.; Petersen, G.M. Risk of Malignancy in First-Degree Relatives of Patients with Pancreatic Carcinoma. Cancer 2005, 104, 388–394. [Google Scholar] [CrossRef]
- Tersmette, A.C.; Petersen, G.M.; Offerhaus, G.J.A.; Falatko, F.C.; Brune, K.A.; Goggins, M.; Rozenblum, E.; Wilentz, R.E.; Yeo, C.J.; Cameron, J.L.; et al. Increased Risk of Incident Pancreatic Cancer among First-Degree Relatives of Patients with Familial Pancreatic Cancer. Clin. Cancer Res. 2001, 7, 738–744. [Google Scholar]
- Permuth-Wey, J.; Egan, K.M. Family History Is a Significant Risk Factor for Pancreatic Cancer: Results from a Systematic Review and Meta-Analysis. Fam. Cancer 2009, 8, 109–117. [Google Scholar] [CrossRef]
- McFaul, C.D.; Greenhalf, W.; Earl, J.; Howes, N.; Neoptolemos, J.P.; Kress, R.; Sina-Frey, M.; Rieder, H.; Hahn, S.; Bartsch, D.K. Anticipation in Familial Pancreatic Cancer. Gut 2006, 55, 252–258. [Google Scholar] [CrossRef] [PubMed]
- Klein, A.P. Genetic Susceptibility to Pancreatic Cancer. Mol. Carcinog. 2012, 51, 14–24. [Google Scholar] [CrossRef] [PubMed]
- Murphy, K.M.; Brune, K.A.; Griffin, C.; Sollenberger, J.E.; Petersen, G.M.; Bansal, R.; Hruban, R.H.; Kern, S.E. Evaluation of Candidate Genes MAP2K4, MADH4, ACVR1B, and BRCA2 in Familial Pancreatic Cancer: Deleterious BRCA2 Mutations in 17%. Cancer Res. 2002, 62, 3789–3793. [Google Scholar] [PubMed]
- Hahn, S.A.; Greenhalf, B.; Ellis, I.; Sina-Frey, M.; Rieder, H.; Korte, B.; Gerdes, B.; Kress, R.; Ziegler, A.; Raeburn, J.A.; et al. BRCA2 Germline Mutations in Familial Pancreatic Carcinoma. JNCI J. Natl. Cancer Inst. 2003, 95, 214–221. [Google Scholar] [CrossRef] [PubMed]
- Moynahan, M.E.; Pierce, A.J.; Jasin, M. BRCA2 Is Required for Homology-Directed Repair of Chromosomal Breaks. Mol. Cell 2001, 7, 263–272. [Google Scholar] [CrossRef]
- Yoshida, K.; Miki, Y. Role of BRCA1 and BRCA2 as Regulators of DNA Repair, Transcription, and Cell Cycle in Response to DNA Damage. Cancer Sci. 2004, 95, 866–871. [Google Scholar] [CrossRef]
- Stark, J.M.; Pierce, A.J.; Oh, J.; Pastink, A.; Jasin, M. Genetic Steps of Mammalian Homologous Repair with Distinct Mutagenic Consequences. Mol. Cell. Biol. 2004, 24, 9305–9316. [Google Scholar] [CrossRef] [PubMed]
- Hakem, R. DNA-Damage Repair; the Good, the Bad, and the Ugly. EMBO J. 2008, 27, 589–605. [Google Scholar] [CrossRef] [PubMed]
- Narod, S.A.; Foulkes, W.D. BRCA1 and BRCA2: 1994 and Beyond. Nat. Rev. Cancer 2004, 4, 665–676. [Google Scholar] [CrossRef]
- Moran, A.; O’Hara, C.; Khan, S.; Shack, L.; Woodward, E.; Maher, E.R.; Lalloo, F.; Evans, D.G.R. Risk of Cancer Other than Breast or Ovarian in Individuals with BRCA1 and BRCA2 Mutations. Fam. Cancer 2012, 11, 235–242. [Google Scholar] [CrossRef]
- Stadler, Z.K.; Salo-Mullen, E.; Patil, S.M.; Pietanza, M.C.; Vijai, J.; Saloustros, E.; Hansen, N.A.L.; Kauff, N.D.; Kurtz, R.C.; Kelsen, D.P.; et al. Prevalence of BRCA1 and BRCA2 Mutations in Ashkenazi Jewish Families with Breast and Pancreatic Cancer. Cancer 2012, 118, 493–499. [Google Scholar] [CrossRef] [PubMed]
- Zhen, D.B.; Rabe, K.G.; Gallinger, S.; Syngal, S.; Schwartz, A.G.; Goggins, M.G.; Hruban, R.H.; Cote, M.L.; McWilliams, R.R.; Roberts, N.J.; et al. BRCA1, BRCA2, PALB2, and CDKN2A Mutations in Familial Pancreatic Cancer: A PACGENE Study. Genet. Med. 2015, 17, 569–577. [Google Scholar] [CrossRef] [PubMed]
- Roberts, N.J.; Norris, A.L.; Petersen, G.M.; Bondy, M.L.; Brand, R.; Gallinger, S.; Kurtz, R.C.; Olson, S.H.; Rustgi, A.K.; Schwartz, A.G.; et al. Whole Genome Sequencing Defines the Genetic Heterogeneity of Familial Pancreatic Cancer. Cancer Discov. 2016, 6, 166. [Google Scholar] [CrossRef] [PubMed]
- Roberts, N.J.; Jiao, Y.; Yu, J.; Kopelovich, L.; Petersen, G.M.; Bondy, M.L.; Gallinger, S.; Schwartz, A.G.; Syngal, S.; Cote, M.L.; et al. ATM Mutations in Patients with Hereditary Pancreatic Cancer. Cancer Discov. 2012, 2, 41–46. [Google Scholar] [CrossRef] [PubMed]
- Srinivas, U.S.; Tan, B.W.Q.; Vellayappan, B.A.; Jeyasekharan, A.D. ROS and the DNA Damage Response in Cancer. Redox Biol. 2019, 25, 101084. [Google Scholar] [CrossRef] [PubMed]
- Pâques, F.; Haber, J.E. Multiple Pathways of Recombination Induced by Double-Strand Breaks in Saccharomyces Cerevisiae. Microbiol. Mol. Biol. Rev. 1999, 63, 349–404. [Google Scholar] [CrossRef] [PubMed]
- Murnane, J.P. Telomeres and Chromosome Instability. DNA Repair (Amst). 2006, 5, 1082–1092. [Google Scholar] [CrossRef] [PubMed]
- Shrivastav, M.; De Haro, L.P.; Nickoloff, J.A. Regulation of DNA Double-Strand Break Repair Pathway Choice. Cell Res. 2008, 18, 134–147. [Google Scholar] [CrossRef] [PubMed]
- Nickoloff, J.A. Recombination: Mechanisms and Roles in Tumorigenesis. Encycl. Cancer 2002, 4, 49–59. [Google Scholar] [CrossRef]
- Huertas, P.; Jackason, S.P. Human CtIP Mediates Cell Cycle Control of DNA End Resection and Double Strand Break Repair. J. Biol. Chem. 2009, 284, 9558–9565. [Google Scholar] [CrossRef]
- Groelly, F.J.; Fawkes, M.; Dagg, R.A.; Blackford, A.N.; Tarsounas, M. Targeting DNA Damage Response Pathways in Cancer. Nat. Rev. Cancer 2023, 23, 78–94. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; McCorvie, T.J.; Yates, L.A.; Zhang, X. Structural Basis of Homologous Recombination. Cell. Mol. Life Sci. 2020, 77, 3–18. [Google Scholar] [CrossRef]
- Kim, C.; Paulus, B.F.; Wold, M.S. Interactions of Human Replication Protein A with Oligonucleotides. Biochemistry 1994, 33, 14197–14206. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Heyer, W.D. Homologous Recombination in DNA Repair and DNA Damage Tolerance. Cell Res. 2008, 18, 99–113. [Google Scholar] [CrossRef]
- Jasin, M.; Rothstein, R. Repair of Strand Breaks by Homologous Recombination. Cold Spring Harb. Perspect. Biol. 2013, 5. [Google Scholar] [CrossRef]
- Farmer, H.; McCabe, H.; Lord, C.J.; Tutt, A.H.J.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA Repair Defect in BRCA Mutant Cells as a Therapeutic Strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef]
- Golan, T.; Kanji, Z.S.; Epelbaum, R.; Devaud, N.; Dagan, E.; Holter, S.; Aderka, D.; Paluch-Shimon, S.; Kaufman, B.; Gershoni-Baruch, R.; et al. Overall Survival and Clinical Characteristics of Pancreatic Cancer in BRCA Mutation Carriers. 2014. [Google Scholar] [CrossRef]
- Underhill, C.; Toulmonde, M.; Bonnefoi, H. A Review of PARP Inhibitors: From Bench to Bedside. Ann. Oncol. 2011, 22, 268–279. [Google Scholar] [CrossRef]
- Golan, T.; Hammel, P.; Reni, M.; Van Cutsem, E.; Macarulla, T.; Hall, M.J.; Park, J.-O.; Hochhauser, D.; Arnold, D.; Oh, D.-Y.; et al. Maintenance Olaparib for Germline BRCA -Mutated Metastatic Pancreatic Cancer. N. Engl. J. Med. 2019, 381, 317–327. [Google Scholar] [CrossRef] [PubMed]
- Shroff, R.T.; Hendifar, A.; McWilliams, R.R.; Geva, R.; Epelbaum, R.; Rolfe, L.; Goble, S.; Lin, K.K.; Biankin, A. V.; Giordano, H.; et al. Rucaparib Monotherapy in Patients With Pancreatic Cancer and a Known Deleterious BRCA Mutation. JCO Precis. Oncol. 2018, 1–15. [Google Scholar] [CrossRef]
- Murai, J.; Huang, S.Y.N.; Renaud, A.; Zhang, Y.; Ji, J.; Takeda, S.; Morris, J.; Teicher, B.; Doroshow, J.H.; Pommier, Y. Stereospecific PARP Trapping by BMN 673 and Comparison with Olaparib and Rucaparib. Mol. Cancer Ther. 2014, 13, 433–443. [Google Scholar] [CrossRef]
- Shen, Y.; Rehman, F.L.; Feng, Y.; Boshuizen, J.; Bajrami, I.; Elliott, R.; Wang, B.; Lord, C.J.; Post, L.E.; Ashworth, A. BMN673, a Novel and Highly Potent PARP1/2 Inhibitor for the Treatment of Human Cancers with DNA Repair Deficiency. Clin. Cancer Res. 2013, 19, 5003–5015. [Google Scholar] [CrossRef] [PubMed]
- de Bono, J.; Ramanathan, R.K.; Mina, L.; Chugh, R.; Glaspy, J.; Rafii, S.; Kaye, S.; Sachdev, J.; Heymach, J.; Smith, D.C.; et al. Phase I, Dose-Escalation, Two-Part Trial of the PARP Inhibitor Talazoparib in Patients with Advanced Germline BRCA1/2 Mutations and Selected Sporadic Cancers. Cancer Discov. 2017, 7, 620–629. [Google Scholar] [CrossRef]
- Yap, T.A.; Im, S.-A.; Schram, A.M.; Sharp, A.; Balmana, J.; Baird, R.D.; Brown, J.S.; Schwaederle, M.; Pilling, E.A.; Moorthy, G.; et al. Abstract CT007: PETRA: First in Class, First in Human Trial of the next Generation PARP1-Selective Inhibitor AZD5305 in Patients (Pts) with BRCA1/2, PALB2 or RAD51C/D Mutations. Cancer Res. 2022, 82, CT007–CT007. [Google Scholar] [CrossRef]
- O’Reilly, E.M.; Lee, J.W.; Zalupski, M.; Capanu, M.; Park, J.; Golan, T.; Tahover, E.; Lowery, M.A.; Chou, J.F.; Sahai, V.; et al. Randomized, Multicenter, Phase II Trial of Gemcitabine and Cisplatin with or without Veliparib in Patients with Pancreas Adenocarcinoma and a Germline BRCA/ PALB2 Mutation. J. Clin. Oncol. 2020, 38, 1378–1388. [Google Scholar] [CrossRef]
- Ullman, N.A.; Burchard, P.R.; Dunne, R.F.; Linehan, D.C. Immunologic Strategies in Pancreatic Cancer: Making Cold Tumors Hot. J. Clin. Oncol. 2022, 40, 2789–2805. [Google Scholar] [CrossRef]
- Bukhari, A.B.; Chan, G.K.; Gamper, A.M. Targeting the DNA Damage Response for Cancer Therapy by Inhibiting the Kinase Wee1. Front. Oncol. 2022, 12, 1–13. [Google Scholar] [CrossRef]
- Dias, M.P.; Moser, S.C.; Ganesan, S.; Jonkers, J. Understanding and Overcoming Resistance to PARP Inhibitors in Cancer Therapy. Nat. Rev. Clin. Oncol. 2021, 18, 773–791. [Google Scholar] [CrossRef]
- Edwards, S.L.; Brough, R.; Lord, C.J.; Natrajan, R.; Vatcheva, R.; Levine, D.A.; Boyd, J.; Reis-Filho, J.S.; Ashworth, A. Resistance to Therapy Caused by Intragenic Deletion in BRCA2. Nature 2008, 451, 1111–1115. [Google Scholar] [CrossRef] [PubMed]
- Sakai, W.; Swisher, E.M.; Karlan, B.Y.; Agarwal, M.K.; Higgins, J.; Friedman, C.; Villegas, E.; Jacquemont, C.; Farrugia, D.J.; Couch, F.J.; et al. Secondary Mutations as a Mechanism of Cisplatin Resistance in BRCA2-Mutated Cancers. Nature 2008, 451, 1116–1120. [Google Scholar] [CrossRef]
- Dhillon, K.K.; Swisher, E.M.; Taniguchi, T. Secondary Mutations of BRCA1/2 and Drug Resistance. Cancer Sci. 2011, 102, 663–669. [Google Scholar] [CrossRef]
- Brown, T.J.; Yablonovitch, A.; Till, J.E.; Yen, J.; Kiedrowski, L.A.; Hood, R.; O’Hara, M.H.; Teitelbaum, U.; Karasic, T.B.; Schneider, C.; et al. The Clinical Implications of Reversions in Patients with Advanced Pancreatic Cancer and Pathogenic Variants in BRCA1, BRCA2, or PALB2 after Progression on Rucaparib. Clin. Cancer Res. 2023, 29, 5207–5216. [Google Scholar] [CrossRef] [PubMed]
- Weigelt, B.; Comino-Méndez, I.; De Bruijn, I.; Tian, L.; Meisel, J.L.; García-Murillas, I.; Fribbens, C.; Cutts, R.; Martelotto, L.G.; Ng, C.K.Y.; et al. Diverse BRCA1 and BRCA2 Reversion Mutations in Circulating Cell-Free DNA of Therapy-Resistant Breast or Ovarian Cancer. Clin. Cancer Res. 2017, 23, 6708–6720. [Google Scholar] [CrossRef]
- Kondrashova, O.; Topp, M.; Nesic, K.; Lieschke, E.; Ho, G.Y.; Harrell, M.I.; Zapparoli, G. V.; Hadley, A.; Holian, R.; Boehm, E.; et al. Methylation of All BRCA1 Copies Predicts Response to the PARP Inhibitor Rucaparib in Ovarian Carcinoma. Nat. Commun. 2018, 9. [Google Scholar] [CrossRef]
- Hwang, C. Il; Boj, S.F.; Clevers, H.; Tuveson, D.A. Preclinical Models of Pancreatic Ductal Adenocarcinoma. J. Pathol. 2016, 238, 197–204. [Google Scholar] [CrossRef]
- Stoof, J.; Harrold, E.; Mariottino, S.; Lowery, M.A.; Walsh, N. DNA Damage Repair Deficiency in Pancreatic Ductal Adenocarcinoma: Preclinical Models and Clinical Perspectives. Front. Cell Dev. Biol. 2021, 9, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Casolino, R.; Corbo, V.; Beer, P.; Hwang, C. Il; Paiella, S.; Silvestri, V.; Ottini, L.; Biankin, A. V. Germline Aberrations in Pancreatic Cancer: Implications for Clinical Care. Cancers (Basel). 2022, 14. [Google Scholar] [CrossRef]
- Tiriac, H.; Belleau, P.; Engle, D.D.; Plenker, D.; Deschênes, A.; Somerville, T.D.D.; Froeling, F.E.M.; Burkhart, R.A.; Denroche, R.E.; Jang, G.H.; et al. Organoid Profiling Identifies Common Responders to Chemotherapy in Pancreatic Cancer. Cancer Discov. 2018, 8, 1112–1129. [Google Scholar] [CrossRef] [PubMed]
- Lohse, I.; Borgida, A.; Cao, P.; Cheung, M.; Pintilie, M.; Bianco, T.; Holter, S.; Ibrahimov, E.; Kumareswaran, R.; Bristow, R.G.; et al. BRCA1 and BRCA2 Mutations Sensitize to Chemotherapy in Patient-Derived Pancreatic Cancer Xenografts. Br. J. Cancer 2015, 113, 425–432. [Google Scholar] [CrossRef]
- Hirt, C.K.; Booij, T.H.; Grob, L.; Simmler, P.; Toussaint, N.C.; Keller, D.; Taube, D.; Ludwig, V.; Goryachkin, A.; Pauli, C.; et al. Drug Screening and Genome Editing in Human Pancreatic Cancer Organoids Identifies Drug-Gene Interactions and Candidates for off-Label Therapy. Cell Genomics 2022, 2. [Google Scholar] [CrossRef]
- Golan, T.; Stossel, C.; Atias, D.; Buzhor, E.; Halperin, S.; Cohen, K.; Raitses-Gurevich, M.; Glick, Y.; Raskin, S.; Yehuda, D.; et al. Recapitulating the Clinical Scenario of BRCA-Associated Pancreatic Cancer in Pre-Clinical Models. Int. J. Cancer 2018, 143, 179–183. [Google Scholar] [CrossRef]
- Lee, J.W.; Komar, C.A.; Bengsch, F.; Graham, K.; Beatty, G.L. Genetically Engineered Mouse Models of Pancreatic Cancer: The KPC Model (LSL-KrasG12D/+;LSL-Trp53R172H/+;Pdx-1-Cre), Its Variants, and Their Application in Immuno-Oncology Drug Discovery. Curr. Protoc. Pharmacol. 2016, 2016, 14–39. [Google Scholar] [CrossRef] [PubMed]
- Ariston Gabriel, A.N.; Jiao, Q.; Yvette, U.; Yang, X.; Al-Ameri, S.A.; Du, L.; Wang, Y. shan; Wang, C. Differences between KC and KPC Pancreatic Ductal Adenocarcinoma Mice Models, in Terms of Their Modeling Biology and Their Clinical Relevance. Pancreatology 2020, 20, 79–88. [Google Scholar] [CrossRef]
- Feldmann, G.; Karikari, C.; Dal Molin, M.; Duringer, S.; Volkmann, P.; Bartsch, D.K.; Bisht, S.; Koorstra, J.B.; Brossart, P.; Maitra, A.; et al. Inactivation of Brca2 Cooperates with Trp53R172H to Induce Invasive Pancreatic Ductal Adenocarcinomas in Mice: A Mouse Model of Familial Pancreatic Cancer. Cancer Biol. Ther. 2011, 11, 959–968. [Google Scholar] [CrossRef]
- Skoulidis, F.; Cassidy, L.D.; Pisupati, V.; Jonasson, J.G.; Bjarnason, H.; Eyfjord, J.E.; Karreth, F.A.; Lim, M.; Barber, L.M.; Clatworthy, S.A.; et al. Germline Brca2 Heterozygosity Promotes KrasG12D -Driven Carcinogenesis in a Murine Model of Familial Pancreatic Cancer. Cancer Cell 2010, 18, 499–509. [Google Scholar] [CrossRef]
- Rowley, M.; Ohashi, A.; Mondal, G.; Mills, L.; Yang, L.; Zhang, L.; Sundsbak, R.; Shapiro, V.; Muders, M.H.; Smyrk, T.; et al. Inactivation of Brca2 Promotes Trp53-Associated but Inhibits KrasG12D-Dependent Pancreatic Cancer Development in Mice. Gastroenterology 2011, 140, 1303–1313. [Google Scholar] [CrossRef]
- Russell, R.; Perkhofer, L.; Liebau, S.; Lin, Q.; Lechel, A.; Feld, F.M.; Hessmann, E.; Gaedcke, J.; Güthle, M.; Zenke, M.; et al. Loss of ATM Accelerates Pancreatic Cancer Formation and Epithelial-Mesenchymal Transition. Nat. Commun. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Drosos, Y.; Escobar, D.; Chiang, M.Y.; Roys, K.; Valentine, V.; Valentine, M.B.; Rehg, J.E.; Sahai, V.; Begley, L.A.; Ye, J.; et al. ATM-Deficiency Increases Genomic Instability and Metastatic Potential in a Mouse Model of Pancreatic Cancer. Sci. Rep. 2017, 7, 1–14. [Google Scholar] [CrossRef]
- Park, D.; Shakya, R.; Koivisto, C.; Pitarresi, J.R.; Szabolcs, M.; Kladney, R.; Hadjis, A.; MacE, T.A.; Ludwig, T. Murine Models for Familial Pancreatic Cancer: Histopathology, Latency and Drug Sensitivity among Cancers of Palb2, Brca1 and Brca2 Mutant Mouse Strains. PLoS One 2019, 14, 1–22. [Google Scholar] [CrossRef]
- Lucas, A.L.; Shakya, R.; Lipsyc, M.D.; Mitchel, E.B.; Kumar, S.; Hwang, C.; Deng, L.; Devoe, C.; Chabot, J.A.; Szabolcs, M.; et al. High Prevalence of BRCA1 and BRCA2 Germline Mutations with Loss of Heterozygosity in a Series of Resected Pancreatic Adenocarcinoma and Other Neoplastic Lesions. Clin. Cancer Res. 2013, 19, 3396–3403. [Google Scholar] [CrossRef]
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific Killing of BRCA2-Deficient Tumours with Inhibitors of Poly(ADP-Ribose) Polymerase; 2005.
- Van Der Wijngaart, H.; Hoes, L.R.; Van Berge Henegouwen, J.M.; Van Der Velden, D.L.; Zeverijn, L.J.; Roepman, P.; Van Werkhoven, E.; De Leng, W.W.J.; Jansen, A.M.L.; Mehra, N.; et al. Patients with Biallelic BRCA1/2 Inactivation Respond to Olaparib Treatment across Histologic Tumor Types. Clin. Cancer Res. 2021, 27, 6106–6114. [Google Scholar] [CrossRef]
- Stossel, C.; Raitses-Gurevich, M.; Atias, D.; Beller, T.; Gorman, Y.G.; Halperin, S.; Peer, E.; Denroche, R.E.; Zhang, A.; Notta, F.; et al. Spectrum of Response to Platinum and PARP Inhibitors in Germline BRCA–Associated Pancreatic Cancer in the Clinical and Preclinical Setting. Cancer Discov. 2023, 13, 1826–1843. [Google Scholar] [CrossRef] [PubMed]
- Waddell, N.; Pajic, M.; Patch, A.M.; Chang, D.K.; Kassahn, K.S.; Bailey, P.; Johns, A.L.; Miller, D.; Nones, K.; Quek, K.; et al. Whole Genomes Redefine the Mutational Landscape of Pancreatic Cancer. Nature 2015, 518. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.; Zhang, X.; Parsons, D.W.; Lin, J.C.H.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Kamiyama, H.; Jimeno, A.; et al. Core Signaling Pathways in Human Pancreatic Cancers Revealed by Global Genomic Analyses. Science (80-. ). 2008, 321, 1801–1806. [Google Scholar] [CrossRef] [PubMed]
- Biankin, A. V.; Waddell, N.; Kassahn, K.S.; Gingras, M.C.; Muthuswamy, L.B.; Johns, A.L.; Miller, D.K.; Wilson, P.J.; Patch, A.M.; Wu, J.; et al. Pancreatic Cancer Genomes Reveal Aberrations in Axon Guidance Pathway Genes. Nature 2012, 491, 399–405. [Google Scholar] [CrossRef] [PubMed]
- Norris, A.L.; Roberts, N.J.; Jones, S.; Wheelan, S.J.; Papadopoulos, N.; Vogelstein, B.; Kinzler, K.W.; Hruban, R.H.; Klein, A.P.; Eshleman, J.R. Familial and Sporadic Pancreatic Cancer Share the Same Molecular Pathogenesis. Fam. Cancer 2015, 14, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Hartupee, C.; Nagalo, B.M.; Chabu, C.Y.; Tesfay, M.Z.; Coleman-Barnett, J.; West, J.T.; Moaven, O. Pancreatic Cancer Tumor Microenvironment Is a Major Therapeutic Barrier and Target. Front. Immunol. 2024, 15, 1–17. [Google Scholar] [CrossRef]
- Shaashua, L.; Ben-Shmuel, A.; Pevsner-Fischer, M.; Friedman, G.; Levi-Galibov, O.; Nandakumar, S.; Barki, D.; Nevo, R.; Brown, L.E.; Zhang, W.; et al. BRCA Mutational Status Shapes the Stromal Microenvironment of Pancreatic Cancer Linking Clusterin Expression in Cancer Associated Fibroblasts with HSF1 Signaling. Nat. Commun. 2022, 13, 1–21. [Google Scholar] [CrossRef]
- Oh, G.; Wang, A.; Wang, L.; Li, J.; Werba, G.; Weissinger, D.; Zhao, E.; Dhara, S.; Hernandez, R.E.; Ackermann, A.; et al. POLQ Inhibition Elicits an Immune Response in Homologous Recombination-Deficient Pancreatic Adenocarcinoma via CGAS/STING Signaling. J. Clin. Invest. 2023, 133. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.J.; Archasappawat, S.; Ji, K.; Pena, J.; Fernandez-Vega, V.; Gangaraju, R.; Beesabathuni, N.S.; Kim, M.J.; Tian, Q.; Shah, P.S.; et al. A New Vulnerability to BET Inhibition Due to Enhanced Autophagy in BRCA2 Deficient Pancreatic Cancer. Cell Death Dis. 2023, 14. [Google Scholar] [CrossRef]
- Garnett, M.J.; Edelman, E.J.; Heidorn, S.J.; Greenman, C.D.; Dastur, A.; Lau, K.W.; Greninger, P.; Thompson, I.R.; Luo, X.; Soares, J.; et al. Systematic Identification of Genomic Markers of Drug Sensitivity in Cancer Cells. 2012. [Google Scholar] [CrossRef]
- Liu, C.; Zhao, J.; Lu, W.; Dai, Y.; Hockings, J.; Zhou, Y.; Nussinov, R.; Eng, C.; Cheng, F. Individualized Genetic Network Analysis Reveals New Therapeutic Vulnerabilities in 6,700 Cancer Genomes. PLoS Comput. Biol. 2020, 16. [Google Scholar] [CrossRef]
- Arun, B.; Akar, U.; Gutierrez-Barrera, A.M.; Hortobagyi, G.N.; Ozpolat, B. The PARP Inhibitor AZD2281 (Olaparib) Induces Autophagy/Mitophagy in BRCA1 and BRCA2 Mutant Breast Cancer Cells. Int. J. Oncol. 2015, 47, 262–268. [Google Scholar] [CrossRef] [PubMed]
- Wan, B.; Dai, L.; Wang, L.; Zhang, Y.; Huang, H.; Qian, G.; Yu, T. Knockdown of BRCA2 Enhances Cisplatin and Cisplatin-Induced Autophagy in Ovarian Cancer Cells. Endocr. Relat. Cancer 2018, 25, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Gruber, J.J.; Chen, J.; Geller, B.; Jäger, N.; Lipchik, A.M.; Wang, G.; Kurian, A.W.; Ford, J.M.; Snyder, M.P. Chromatin Remodeling in Response to BRCA2-Crisis. Cell Rep. 2019, 28, 2182–2193. [Google Scholar] [CrossRef] [PubMed]
- MacKay, R.P.; Xu, Q.; Weinberger, P.M. R-Loop Physiology and Pathology: A Brief Review. DNA Cell Biol. 2020, 39, 1914–1925. [Google Scholar] [CrossRef] [PubMed]
- Shivji, M.K.K.; Renaudin, X.; Williams, Ç.H.; Venkitaraman, A.R. BRCA2 Regulates Transcription Elongation by RNA Polymerase II to Prevent R-Loop Accumulation. Cell Rep. 2018, 22, 1031–1039. [Google Scholar] [CrossRef]
- Sessa, G.; Gómez-González, B.; Silva, S.; Pérez-Calero, C.; Beaurepere, R.; Barroso, S.; Martineau, S.; Martin, C.; Ehlén, Å.; Martínez, J.S.; et al. BRCA2 Promotes DNA-RNA Hybrid Resolution by DDX5 Helicase at DNA Breaks to Facilitate Their Repair‡. EMBO J. 2021, 40, 1–25. [Google Scholar] [CrossRef]
Figure 1.
Steps of homologous recombination. Homologous recombination (HR) repairs double-stranded breaks (DSB) in three key steps. First, during pre-synapsis, the MRN complex processes the DNA ends to generate 3′ single-stranded DNA (ssDNA) overhang and recruits ATM kinase. During synapsis, RPA then binds the overhang, and BRCA1 is recruited to the site. BRCA1 then facilitates the recruitment of BRCA2, and PALB2, with RAD51 mediates the invasion of the homologous DNA duplex to form a D-loop. Finally, DNA polymerases extend the invading strand using the homologous template, completing the repair process.
Figure 1.
Steps of homologous recombination. Homologous recombination (HR) repairs double-stranded breaks (DSB) in three key steps. First, during pre-synapsis, the MRN complex processes the DNA ends to generate 3′ single-stranded DNA (ssDNA) overhang and recruits ATM kinase. During synapsis, RPA then binds the overhang, and BRCA1 is recruited to the site. BRCA1 then facilitates the recruitment of BRCA2, and PALB2, with RAD51 mediates the invasion of the homologous DNA duplex to form a D-loop. Finally, DNA polymerases extend the invading strand using the homologous template, completing the repair process.
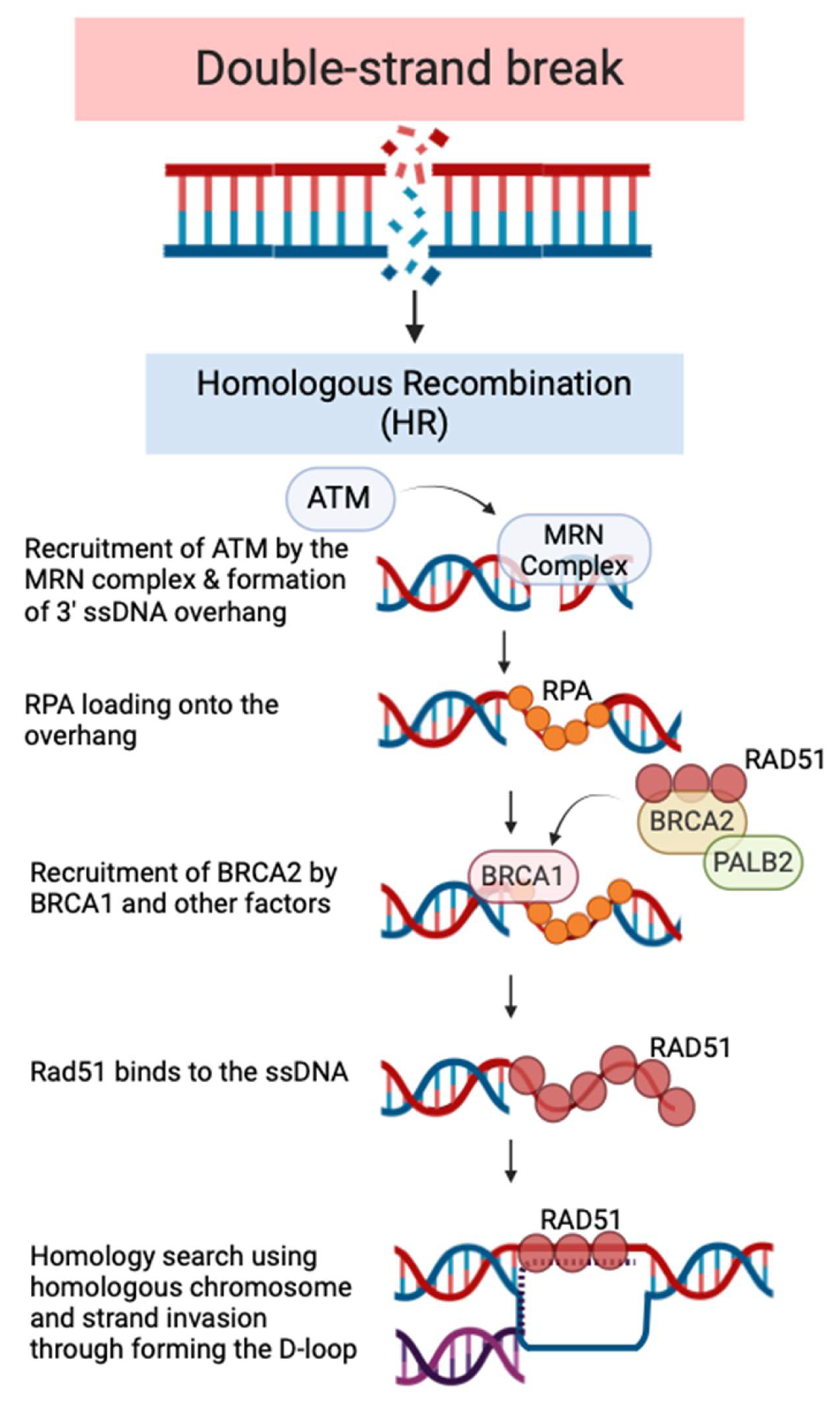
Figure 2.
Future directions for familial pancreatic cancer (FPC). This figure presents a roadmap for future advancements in FPC, highlighting key areas of focus to improve patient outcomes. It emphasizes a multi-pronged approach encompassing the establishment of robust FPC models, identification of new vulnerabilities, personalized medicine strategies, and the application of translational research.
Figure 2.
Future directions for familial pancreatic cancer (FPC). This figure presents a roadmap for future advancements in FPC, highlighting key areas of focus to improve patient outcomes. It emphasizes a multi-pronged approach encompassing the establishment of robust FPC models, identification of new vulnerabilities, personalized medicine strategies, and the application of translational research.
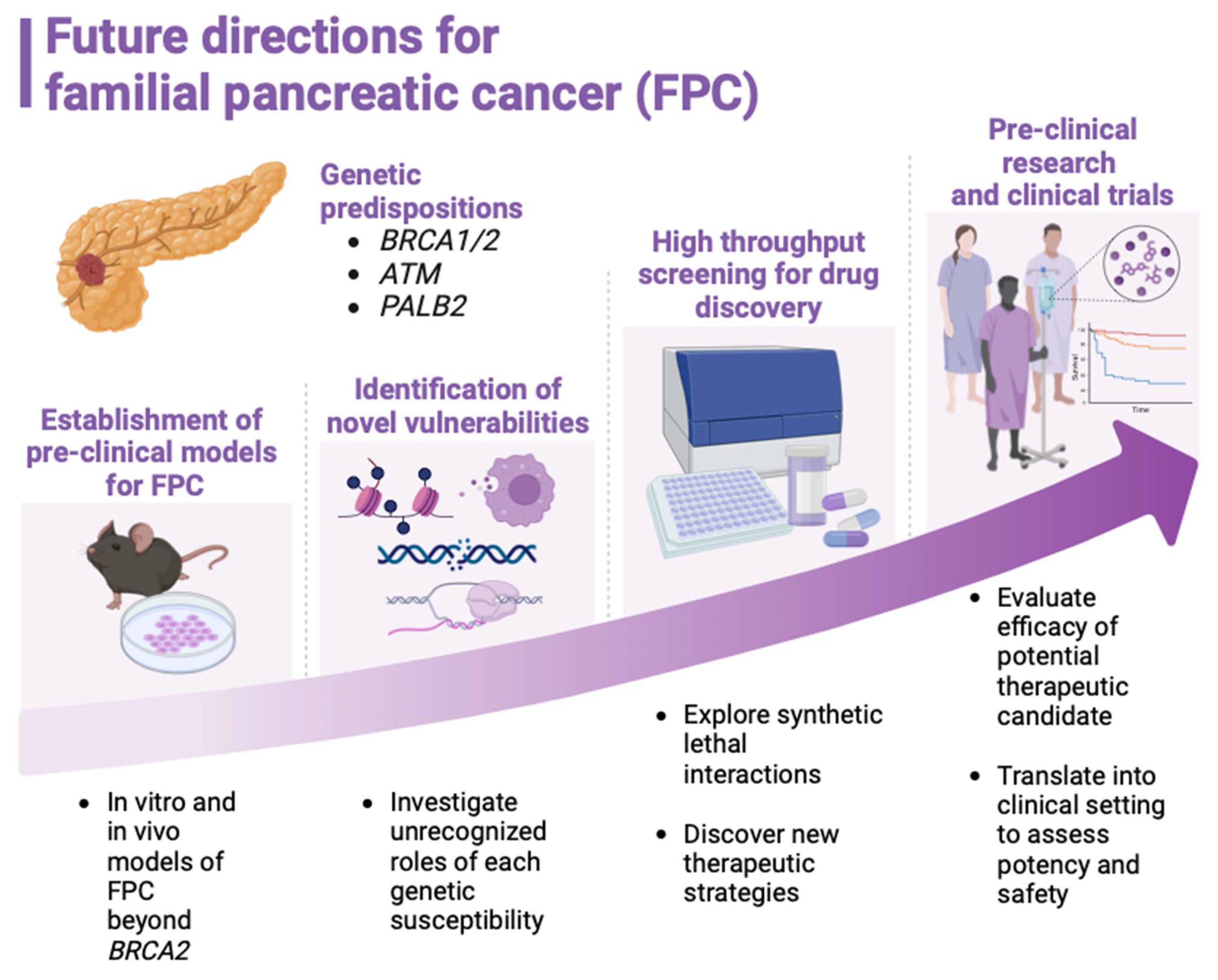

Table 1.
Prevalence of deleterious mutations in the genes BRCA1, BRCA2, PALB2, CDKN2A in both FPC and SPC patients, as well as malignancies/disorders associated with the patients.
Table 1.
Prevalence of deleterious mutations in the genes BRCA1, BRCA2, PALB2, CDKN2A in both FPC and SPC patients, as well as malignancies/disorders associated with the patients.
| Prevalence of deleterious mutations | Associated malignancies and disorders | ||
|---|---|---|---|
| Gene | FPC patients | SPC patients | |
| BRCA1 | 1.2% | 0.0% | Breast, ovary, prostate, esophagus, stomach, uveal |
| BRCA2 | 3.7% | 3.0% | Breast, ovary, prostate, esophagus, stomach, uveal, melanoma |
| PALB2 | 0.6% | 0.5% | Fanconi anemia, breast, prostate, stomach, esophagus |
| CDKN2A | 2.5% | 0.0% | Melanoma |
| Total | 8.0% | 3.5% | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



