
Submitted:
04 August 2024
Posted:
05 August 2024
You are already at the latest version
Abstract
Common variable immunodeficiency (CVID) is the most common symptomatic antibody deficiency, characterized by heterogeneous genetic, immunological, and clinical phenotypes. It has no longer been conceived as a sole disease but as an umbrella diagnosis comprising a spectrum of clinical conditions with defects in antibody biosynthesis as their common denominator and complex pathways determining B and T cell developmental impairments due to genetic defects of many receptors and ligands, activating and co-stimulatory molecules, and intracellular signaling molecules. Consequently, these genetic variants may affect crucial immunological processes of antigen presentation, antibody class switch recombination, antibody affinity maturation, and somatic hypermutation. While infections are the most common features of pediatric CVID, variants in genes linked to antibody production defects play a role in pathomechanisms of immune dysregulation with autoimmunity, allergy, and lymphoproliferation reflecting the diversity of the immunogenetic underpinnings of CVID. Herein, we have reviewed the aspects of genetics in CVID, including the monogenic, digenic, and polygenic models of inheritance exemplified by a scope of genes relevant to CVID pathophysiology. We have also briefly discussed the epigenetic mechanisms associated with micro RNA, DNA methylation, chromatin reorganization, and histone protein modification processes as background for CVID development.
Keywords:
antibody deficiency
; common variable immunodeficiency
; epigenetics
; immune dysregulation
; inborn error of immunity
; whole exome sequencing
Introduction
Primary antibody deficiencies (PADs) represent the most common category of inborn errors of immunity (IEI), encompassing a collection of phenotypically and genetically heterogeneous clinical and immunological conditions [1]. PADs are characterized by a broad spectrum of phenotypic expressions, including susceptibility to infections, autoimmunity, autoinflammation, allergy, organ-specific pathology, and malignancy. In young children, significant immaturity of the immune system and developmental delay of antibody production may impede an accurate definitive diagnosis. The greatest and the most dynamic maturational changes among the B cell subsets occur in children below the age of four years. Therefore, the reliable diagnosis of primary antibody deficiency in children aged less than four years cannot yet be established despite their clinical and immunological phenotypes that share common features with specific disease entities. It also needs to be highlighted that the classifications used in adults are not helpful and their application to children with antibody production defects is misleading as they require separate diagnostic and prognostic criteria [2,3].
Common variable immunodeficiency (CVID) is the most prevalent primary symptomatic hypogammaglobulinemia, with the estimated frequency between 1:10000 to 1:50000 in Caucasians, with important morbidity and individual and societal burden. It is a notorious disease with unfortunate and severe associated outcomes, such as recurrent infections and immune dysregulation in the form of autoimmunity, autoinflammation, allergy, lymphoproliferation, and malignancy [4]. According to the international consensus statement and the current revised European Society for Immunodeficiencies (ESID) diagnostic criteria [5,6], CVID is characterized by low serum IgG levels, accompanied by decreased IgM and/or IgA, impaired specific antibody response to vaccines and exclusion of other specific causes of hypogammaglobulinemia [7]. The ESID definition of CVID include clinical criteria, such as increased susceptibility to infection, autoimmune manifestations, granulomatous disease, polyclonal lymphoproliferation, and a positive family history to antibody deficiency. Immunodiagnostic criteria for CVID in children include antibody deficiency interpreted in relation to age-matched reference values, and low switched memory B cell numbers, below 70% of age-related normal values. Importantly, evidence of profound T cell deficiency, low CD4 T helper cell counts, low relative numbers of CD4 T cells in relation to the child’s age, and absent T cell proliferation are exclusion criteria for CVID [6]. In patients with CVID, a spectrum of phenotypic and functional abnormalities in the adaptive and innate immunity reflect impaired immune homeostasis, including defective B-cell differentiation and maturation, isotype-switched memory B-cell development, T-cell dependent costimulation, regulatory T cells, and disturbances in various components of the innate immunity. The data on complex immunophenotype in CVID have led to further stratification of the disease and developing Freiburg [8] and EUROclass [9] classifications. Nonetheless, the application of the criteria based on terminal differentiation of B-cells and formation of isotype-switched memory B-cells used in these classifications in pediatric patients is a matter of discussion because of active maturational processes and dynamic shifts within B and T-cell subsets in childhood [9]. Due to this marked heterogeneity of B and T-cell abnormalities in CVID, it has become clear that CVID is an umbrella diagnosis encompassing a spectrum of disorders with a defective biosynthesis of antibodies as a common denominator.
These complex processes of B cell antigen signaling, activation, survival, migration, and maturation to generate terminal stages of switched memory B cells and plasma cells mirror a composite genetic etiology of CVID [10]. The B cell developmental impairment and hypogammaglobulinemia may result from genetic defects of many receptors and ligands, activating co-stimulatory molecules, and intracellular signaling molecules. However, despite a relatively high prevalence of CVID among IEI in children, the rate of molecular genetic diagnosis remains low, with pathogenic gene variants identifiable in a limited proportion of patients, ranging from merely 2-10% [11] up to 54% in populations with a high rate of consanguinity [12]. Therefore, it strongly implies that beyond the monogenic model of inheritance, another explanation of CVID origin is multifactorial, digenic, or polygenic, and alternatively, that accumulation of rare functional variants, somatic variants, or epigenetic phenomena [13,14,15,16] may show a causal relationship with the regulation of B cell development and functions.
This review aimed to resume and conclude the immunological and genetic underpinnings of pediatric common variable immunodeficiency. It was also conducted to provide data facilitating a better understanding of heterogeneous cellular and genetic immunophenotypes in the context of mechanisms determining infectious and non-infectious manifestations of the disease.
Monogenetic model of CVID in children
The molecular genetic background associated with monogenic causes has been hitherto identified in overall less than 20% of patients affected with CVID, usually in its familial forms that constitute only a small fraction of cases. Furthermore, some of the mutations are characterized by incomplete penetrance, and also sporadic cases remain genetically unexplained, suggesting a complex, non-Mendelian pattern of inheritance [13,14,15,16]. Better understanding a disease immunopathogenesis and a genotype-phenotype relationship could help stratify patients into clinical disease entities to predict complications and possibly individualize treatment. While clinical presentation, disease severity, and immunophenotype in pediatric CVID are highly variable with infectious and non-infectious complications resulting from immune dysregulation [17,18,19,20,21], CVID is currently perceived as a group of disorders with antibody deficiency as their common denominator and a cardinal feature covering a spectrum of genetic subtypes. Although the main tool for diagnosis of CVID remains clinical, it is highly recommended to obtain a genetic signature and molecular analysis in all affected subjects with unclear and severe clinical phenotypes [22].
The genetic etiology of CVID underpins complex processes of B cell antigen signaling, activation, survival, migration, and maturation to generate terminal stages of switched memory B cells and plasma cells. The B cell developmental impairment and antibody deficiency may result from genetic defects of many receptors and ligands, activating and co-stimulatory molecules, and intracellular signaling molecules. Consequently, these genetic variants may affect crucial immunological processes of antigen presentation, antibody class switch recombination (CSR), antibody affinity maturation, and somatic hypermutation (SHM). Furthermore, variants in genes linked to antibody production defects and immune dysregulation with autoimmunity, lymphoproliferation, enteropathy, splenomegaly, and granulomatosis have been identified thus far in a proportion of affected patients [11,19,23]. Genes that have been identified in monogenic CVID on the European background include ICOS (inducible T cell co-stimulator), TNFRSF13B (transmembrane activator and calcium modulator and cyclophilin ligand interactor, TACI), TNFRSF13C (B cell-activating factor belonging to the tumor necrosis factor (TNF) family, BAFF-receptor, BAFF-R), TNFSF12 (TNF-like weak inducer of apoptosis, TWEAK), CD19, CD81, CR2 (CD21), MS4A1 (membrane-spanning 4A1, CD20), TNFRSF7 (CD27), IL21, IL21R, LRBA (lipopolysaccharide (LPS)-responsive beige-like anchor protein), CTLA4 (cytotoxic T lymphocyte-associated antigen 4), PRKCD (Protein kinase C delta), PLCG2 (phospholipase C gamma 2), NFKB1 (nuclear factor kappa B1), NFKB2 (nuclear factor kappa B2), PIK3CD (Phosphoinositide 3-kinase (PI3K) catalytic subunit delta), PIK3R1 (Phosphoinositide 3-kinase (PI3K) regulatory subunit 1), VAV1 (Vav guanine nucleotide exchange factor 1), RAC2 (Ras-related C3 botulinum toxin substrate 2), BLK (B-lymphoid tyrosine kinase), IKZF1 (IKAROS), and IRF2BP2 (Interferon regulatory factor 2 binding protein 2) [11]. An in-depth retrospective review of genes reported in monogenic CVID patients categorized variants involved in multiple molecular pathways and immune compartments such as, but not limited to, B-cell receptor (BCR) costimulatory B-cell surface proteins, tumor necrosis factor superfamily receptors and ligands, lipid signaling molecules, actin cytoskeleton regulators, transcription factors mediating differentiation and crosstalk, metabolic processes of glycosylation and mitochondrial pathways [24]. The spectrum of most frequently reported genetic variants underpinning monogenic CVID stratified according to diverse pathomechanisms are displayed in Table 1.
Beyond infections, pediatric CVID is characterized by impaired immune homeostasis resulting in a high rate of autoimmune complications and inflammatory disorders associated with above mentioned genetic variants, such as PRKCD and LRBA deficiencies, CTLA-4 haploinsufficiency, as well as the activated PI3Kδ syndrome [25]. These monogenic forms of CVID are connected with two predominating immunopathological pathways, disrupted peripheral tolerance due to the impaired T regulatory cell functions in the first ones and hyperactivation of T and B cells in the latter [26,27,28].
Several novel genetic defects, such as variants in BACH2 (BTB and CNC homolog 2), STAT3 (Signal transducer and activator of transcription 3), STAT1, DOCK8 (Dedicator of cytokinesis 8), AIRE (Autoimmune regulator), FOXP3 (Forkhead box protein 3), or CXCR4 (CXC chemokine receptor 4) causing immune dysregulation syndromes presenting as CVID have also been identified [12,19,29,30,31].
Noticeably, variants in DNMT3B (DNA methyltransferase 3 beta), known as the disease-causing gene for Immunodeficiency-Centromeric Instability-Facial anomalies (ICF) syndrome, have also been demonstrated in CVID patients [12,19], thereby linking monogenic underpinnings with epigenetic mechanisms in the pathophysiology of CVID. Several monogenic defects have also been hypothesized to interact with epigenome and impact epigenetic enzymes and their interactors. Therefore, epigenetic alterations and pathogenic variants in CVID-associated genes do not exclude each other as disease-causing immunopathology but provide a novel insight into the mutual relationships between genome and epigenome in CVID [32]. These phenomena of an interplay between genetic variants in transcription factors relevant to the pathogenesis of CVID in conjunction with epigenetic deregulation are well illustrated by NF-κB and JAK (Janus kinase) – STAT signaling pathways. For example, variants in the NFKB1 gene in the form of haploinsufficiency have been reported as the most prominent disease-causing monogenic background for CVID among Europeans [33]. Noteworthy, variants in NFKB1 can impact epigenetic mechanisms such as chromatin remodeling of target genes and histone modifications by interactions with epigenetic enzymes eg. histone deacetylases and methyltransferases thereby resulting in modulation of lymphocyte activity [32,34]. Variants in the JAK-STAT signaling pathway, in particular STAT1 gain-of-function (GOF), and STAT3 GOF that impair binding of STST molecules to their target genes are connected to epigenetic deregulation of histone methylation [32].
The multiplicity of genetic variants found in patients with CVID phenotypes, associated with a constellation of clinical symptoms, from infections to lymphoproliferation, point to the complexity of numerous pathways involved in B lymph cell development and antibody-dependent immune response. The ever-increasing advances in genomics and the consequent growing rate of monogenic causes of CVID indicate the need to reevaluate patients and their reassignment to specific immunogenetic categories of IEI.
Digenic and polygenic CVID
For genetically heterogeneous diseases, such as CVID, for which many disease-causing variants in many genes are known, digenic combinations are likely and they may display different modes of inheritance. Noticeably, in sporadic CVID cases, for which the genetic underpinnings remain unknown, the digenic etiology should be considered [35].
Beyond the genotype-age-related immunophenotype in pediatric CVID, identifying variants not following the Mendelian mode of inheritance in familial cases with variable degrees of penetrance and expressivity determining remarkable diagnostic challenges, the complexity of CVID is even more conveyed by a predisposition to the disease. Noticeably, variants in TACI, a member of the TNF superfamily, may not be causative for CVID but may coexist and interact synergistically with other variants showing deleterious effects. Thereby, the epistatic interactions, synergistic interplay of two genetic loci that substantially modify the disease severity or result in entirely new phenotypes as well as gene additivity effect may determine the disease symptomatology [28,36,37]. The epistasis phenomena may be exemplified by digenic variants in genes in which products are playing roles in the same physiological pathways, eg. variants in TACI, stimulating a T-cell independent class switch recombination, and TCF3 (Transcription Factor 3) aka E2A, a central point of T-cell independent and T-cell dependent immunoglobulin class switching and secretion with a clinical phenotype of immunodeficiency and autoimmunity showing a digenic nature of CVID [38]. The digenic etiologies associated with CVID phenotypes with antibody production deficiencies and autoimmunity relating to clinical epistasis and including variants in genes such as NFKB1 and NOD2 (Nucleotide-binding oligomerization domain containing 2) [39], LRBA and NEIL3 (Nei-Like DNA Glycosylase 3) [40], and CTLA4 and JAK3 (Janus Kinase 3) [41] have been summarized in Table 2.
Whereas the multiple variants found in the monogenic CVID are limited to familial cases with autosomal dominant inheritance and different expression and penetrance, a complex polygenic model of inheritance is likely to be the underlying mechanism for the majority of sporadic cases [42,43]. The data of genetic analysis may also be hampered by a marked intersubject and intrafamilial clinical phenotypic heterogeneity pointing to the modifying effects of additional genetic variants to the broad symptomatology in CVID. Furthermore, a spectrum of environmental contributors, such as dysbiosis of gut microbiota, shifts of microbial composition, and reduced diversity leading to gut-leakage syndrome, the use of drugs reducing the B-cell development, such as rituximab or antiepileptic drugs, valproic acid, as well as viral infection inducing dysregulation of immune cells are risk factors for severe course of CVID [44]. A polygenic burden of CVID has been supported by clinical follow-up and genetic analysis in monozygotic twins concordant for CVID in whom pathogenic variants in genes reported in the monogenic model of the disease have not been found. Nevertheless, as many as seven non-synonymous coding variants in genes involved in relevant immunological pathways such as JUN (Jun Proto-Oncogene, AP-1 Transcription Factor Subunit), PTPRC (Protein Tyrosine Phosphatase Receptor Type C), TLR1 (Toll-like Receptor 1), ICAM1 (Intercellular Adhesion Molecule 1, CD54), and JAK3, with predicted deleterious effect and possible clinical impact have been demonstrated [45].
At this point, referring to the global distribution and the worldwide country-wise prevalence of CVID, a correlation between the rate of CVID diagnosis and medical progress and socio-economic developmental status expressed as the Human Development Index (HDI), has been observed [46]. This conclusion is drawn from assuming that the CVID prevalence does not truly differ between countries and that documentation in registries and referring to databases linked to the country’s developmental status is the major factor contributing to the obstacles in the estimation of the CVID epidemiology. However, an alternative explanation relies on admitting that CVID is presumably a polygenic disease and regionally distinct prevalence of the disease may be explained by influences from genetically diverse founders [47].
Somatic variants in CVID immunogenetics
In contrast to prezygotic germline variants present in the genome of all the body cells, including germ cells, somatic variants arise in the postzygotic DNA from embryonic development through postnatal life to adulthood. The postzygotic variants are associated with the phenomenon of gene mosaicism and consequently, tissues represent a landscape of cells with different genomes. Postzygotic variants are differently distributed in tissues resulting from somatic, gonadal, or gonosomal mosaicism, with the complex and heterogeneous phenotypes and the two latter disorders posing a moderate to high risk of transmission of the variant to the offspring and various implications for disease phenotypes [48]. Somatic variants are often neutral or disadvantageous to cell growth and survival. They may result in clonal expansion conferring a fitness advantage to the cells or clonal regression when expanding clones shrink in size as a response to environmental changes. Whereas age is a key accelerator of somatic mosaicism, it may therefore be assumed, that children show lower risks of disadvantageous consequences of somatic variants, such as clonal hematopoiesis. It has also been hypothesized that gene mosaicism is a relevant and hitherto underrecognized mechanism underlying inborn errors of immunity. Indeed, postzygotic variants in several genes involved in CVID, such as PIK3CD, STAT3, or BTK, have been detected in affected families, suggesting somatic or gonosomal mosaicism and thereby explaining an intrafamilial phenotypic diversity as well as occurrence of IEI symptomatology in sporadic cases without family history [49,50,51]. Somatic mosaicism was first proposed as a cause of autoimmune lymphoproliferative syndrome (ALPS), a disease characterized by lymphocyte dysregulation and autoreactivity, showing common autoimmune and lymphoproliferative features with CVID. Somatic mosaicism has also been demonstrated in patients with autoinflammatory disorders and variants in NOD2 aka CARD15 (Caspase-recruitment domain family, member 2), JAK1 (Janus kinase 1), NLRC4 (NOD-like receptor C), and IL6ST (Interleukin 6 signal transducer) [51,52] as well as KRAS, the gene involved in the RAF/MAPK pathway [53]. Whereas in CVID, T cell abnormalities, such as deficiencies in naïve T CD4+ or regulatory T cells, and expansion of T CD8+ cells have been observed, somatic variants in STAT5B and TET2 might also be linked to the disease pathophysiology and increased susceptibility to autoimmune diseases, lymphoproliferative disorders, and malignant tranformation [53,54,55]. In a patient with a history of opportunistic infections with B and NK cell deficiency, a somatic pathogenic variant in the GATA2 gene has been detected [54].
Epigenetic etiology of CVID
Despite the substantial clinical and genetic heterogeneity of CVID and ever-expanding progress in genomics, a monogenic molecular diagnosis has been found in a small proportion of affected individuals. Furthermore, as most patients with a diagnosis of CVID do not follow a classical Mendelian pattern of inheritance, often representing single sporadic cases, it could suggest that behind the monogenic or polygenic background, epigenetic phenomena may show a causal relationship with the regulation of B cell development and functions [56,57,58]. Epigenetic mechanisms are integrated, dynamic, and potentially reversible changes in gene expression without altering the germline DNA gene sequences, thereby accounting for the alterations in cellular development and differentiation. They comprise the regulation of DNA methylation, histone protein modifications, chromatin remodeling, and changes in transcription [59]. These observations could guide further investigations and epigenetics may, therefore, contribute to explaining the immunopathogenesis of CVID in patients who lack a molecular genetic diagnosis.
Non-coding RNA molecules (ncRNAs) are transcribed from DNA and not translated into proteins, but exert regulatory effects on gene expression and protein translation by influencing DNA transcription and mRNA post-transcriptional changes. In particular, micro RNAs (miRNAs) have critical regulatory functions in cell proliferation, programmed cell death, organ development, and differentiation. MiRNAs are important elements of the molecular pathways in hematopoietic cells and dynamic regulation of their expression suggests their roles in the early cell differentiation and lineage definition [60]. Among miRNAs that showed expression in the immature B cell phase, miRNA-181 and miRNA-150 contributed to the preferential expansion of the B lymph cell compartment [61,62]. It has also been hypothesized that the CD40 B-cell receptor signaling in germinal centers for the differentiation of naïve to memory B-cells modulates levels of several miRNAs, such as miR-150-5p, miR17-5p, miR146a-5p, miR26a-5p or miR-292-5p, and their cognate targets. Consequently, the CD40-miRNA axis controls prospective cell fate during B-cell development [63,64]. A negative modulatory effect on B lymph cell differentiation might, in turn, arise from the expression of miRNA-34a [65], potentially exerting lymphopenia and B-cell developmental disturbances characteristic of CVID. Moreover, miRNA-mediated epigenetic regulation of T cell development and function might play a role in the pathogenesis of CVID. The candidate could be the miRNA-17-92 cluster which has been involved in the development and function of follicular T helper cells, supporting B cells in germinal centers and facilitating class-switch recombination, somatic hypermutation, and antibody affinity maturation [66]. Overexpression of miRNA-210 in T cells has also been postulated in pediatric-onset CVID patients with unsolved PID genetic defects, thereby supporting the hypothesis of the underlying epigenetic pathogenesis [67]. In these patients, the upregulation of miRNA-210 correlated with a reduced count of T CD4+ helper cells and T regulatory cells and an increased count of T CD8+ cytotoxic cells.
B cell-specific miRNAs, such as miRNA-15a-5p, miRNA-199a-3p, and miRNA-103a-3p have been shown to target genes modulating antigen-specific antibody titers following vaccination [68]. This impact on vaccine-induced antibody generation is exerted by different Fc receptors, the phosphatidylinositol-mediated signaling pathways, growth factors signaling pathways, and other processes with relevance to adaptive immunity [69]. The inability to mount a sufficient antigen-specific humoral immune response to vaccines is a fundamental feature and an essential diagnostic criterion of CVID.
In CVID, impaired immunosurveillance also contributes to the autoinflammatory phenotype, which may be determined by a regulatory activity of miRNAs on pro- and anti-inflammatory cytokine profile, production of inflammasome components, effector T lymph cells differentiation, and ultimately, organ-specific inflammatory disorders. Several miRNAs are potential contributors to the inflammatory events, eg. upregulated miRNA-21 and downregulated miRNA-23b may exert their inflammatory effect by influencing the activity of NFκB [70]. Likewise, dynamic regulations of miRNA-155 targetome result in the overrepresentation of target genes involved in the immune response, including, among others, a complex of proinflammatory cytokines IL-6, IL-8, ICAM (intercellular adhesion molecule), and STAT3 [71]. It may, therefore, be assumed that in chronic structural and interstitial lung disease, exacerbated by respiratory viral infections, epigenetic regulation may play a pivotal role. This hypothesis may be supported by the findings showing differently modulated expression of miRNA-6742, miRNA-1825, miRNA-4769, miRNA-1228, miRNA-1972, which are involved in adaptive immune response, by the first immunoglobulin transfusion [72].
Further studies are required to better define mutual relationships between epigenetic alterations and expression of miRNAs and the spectrum of clinical infectious, autoimmune, autoinflammatory, and lymphoproliferative phenotypes in pediatric CVID [19,20,73,74,75,76]. Much light needs to be shed on the role of epigenetics in immune dysregulation in CVID, manifesting as granulomatous lymphocytic interstitial lung disease (GLILD), bronchiectasis, lymphadenopathy, hepatosplenomegaly, arthritis, or inflammatory bowel disease (IBD). Whereas miRNAs are involved in the regulatory activity of a spectrum of B-cell and T-cell intrinsic genes, interrelations between the different miRNAs expression and alterations in the immunophenotype of the B and T lymph cell compartment need to be better understood [77].
With the ever-increasing progress of advanced methods in immunogenetics, analysis of DNA methylation and its role in the control of gene expression thereby shaping the epigenome and related biological processes may become a potential diagnostic and prognostic marker in CVID. DNA methylation is an epigenetic mechanism in which methyl groups are covalently bound to cytosine producing 5-methylcytosine in the CpG dinucleotide domains. The importance of DNA methylation is multidimensional and more complex than repression or activation of gene expression as it also determines interactions between methylated DNA and proteins, including transcription factors, readers of DNA methylation, and effectors involved in methylation signal translation to biological processes [78]. In CVID, aberrant DNA methylation has been demonstrated in 30% of CpG domains during the B lymph cell differentiation process, at the transition from naïve to memory stages. In the early phase of B-cell differentiation, activation of key transcription factors was associated with demethylation of transcription enhancers of genes crucial in the biology of B-cells. In the late phase of differentiation, demethylation of heterochromatin with methylation of repression of genes functionally essential for the biology of B-cells [79,80,81]. In CVID, switched memory B-cells show marked heterogeneity in DNA methylation. Skewed DNA methylation is associated with active demethylation of transcription factors engaged in differentiation and activation of B-cells, such as NF-κB, bZIP family (BATF, JUNB, Fosl2, Fra2), CTCF, IRF, and PU.1, consequently leading to disturbed binding to DNA. Furthermore, abnormally increased or decreased regulation of genes CD70, CD40, NFKB2, ICAM1, and CCL17 has been identified in activated memory B-cells, close to hypermethylated DNA regions [82,83].
Consequently, the transcription program in activated B-cells in CVID is limited, and epigenetic alterations contribute to the impaired transcription during the B-cell immune response. The epigenetic imprinting in switched memory B-cell population occurs in genes and regulatory sequences activated in B cells germinal centers and in plasma cells. Furthermore, immunophenotypic dysregulation within the B-cell compartment may contribute to abnormal interactions between B-cells and other immunological cells in CVID.
Epigenetic mechanisms have been schematically displayed in Figure 1.
Expected advantages of genetic and epigenetic studies in CVID
In children with CVID phenotype, the expected advantages of identifying the causal pathogenic variants primarily include establishing the definitive diagnosis of CVID in its monogenic form. It might be helpful in distinguishing between transient hypogammaglobulinemia of infancy (THI) and CVID in young children with overlapping clinical and immunological phenotypes. In children showing variable serum IgG levels over time, thereby inconsistent with the ESID diagnostic criteria, and in whom timely monitoring of IgG production is not helpful for a clinical diagnosis, demonstration of the genetic underpinning could guide further immunodiagnostics and therapy. Furthermore, in these children, in whom the identified variant is likely to cause a specific disease, such as NFKB2 or CTLA-4 [84,85,86,87], establishing the diagnosis of a CVID-like disorder may lead to excluding them from the umbrella diagnosis of CVID. In monogenic CVID, the identified disease-causing variant may assist in defining the prognosis regarding the clinical course of the disease, possible specific complications, such as an increased risk of autoimmunity, autoinflammation, granulomatous disease, lymphoproliferation, and malignancy. Optimizing the directed, individualized treatment, including an immunoglobulin replacement therapy, antimicrobial therapy, immunosuppression, and eventually a hematopoietic stem cell transplantation (HSCT) is foremostly a variant-specific therapeutic approach. HSCT should be considered as a therapeutic option in children presenting with immunodysregulatory disorders due to genetic variants in PRKCD, DOCK8, and FOXP3 [88]. The presence of a known variant has implications for family members as it is of paramount importance for family counseling and may also facilitate early diagnosis and a prompt starting of replacement Ig therapy in affected family members when they develop symptoms. Finally, the therapeutic aspect means identifying patients with monogenic CVID who may benefit from potential gene therapy in the future.
For researchers, the advantages of identifying the causative variant are also expected, offering a prototypic model of the monogenic CVID and the relationship between genotype and phenotype. It is promising in expanding the knowledge about the function of the immune system and the immunopathogenesis of CVID, CVID-like disorders, and other hypogammaglobulinemias in children that also implicate correlations between the genotype and immunophenotype which may be guiding the classifications of primary immunodeficiency diseases. For clinicians, identifying a disease-causing variant in the monogenic form of CVID could assist with the implementation of specific therapeutic options, such as hematopoietic stem cell transplantation or monoclonal antibodies in some CVID-like disorders as well as implicating the need to elaborate innovative treatment modalities, such as gene therapy in the future (22,23,37).
Elucidating the role of epigenetic alterations in the immunopathogenesis of CVID would be important not only for scientific purposes, but also for the future developments of patient-centered diagnostic, prophylactic, and therapeutic measures. In these children affected with CVID, in whom conventional genetic testing does not provide informative results, miRNAs could serve as a diagnostic biomarker-oriented approach. To address the miRNA investigations in the clinic, determining the existence of potential epigenetic susceptibility to given exposures might also contribute to a novel prophylactic approach. It could facilitate avoiding exposures to environmental factors that might ultimately trigger the development of B cell disorders resulting in a failure in antibody production and development of CVID. Early-life prophylactic interventions in the context of environmental, nutritional, or immunization measures could be implemented in children in high-risk families when other close family members are affected by CVID. The prophylactic measures also apply to these children, in whom the diagnosis of CVID has already been established to minimize the risk of exacerbation of the clinical course of CVID with infectious, autoinflammatory, autoimmune, or lymphoproliferative disorders.
The future perspective of the miRNA approach is pharmacoepigenetics, the novel therapeutic modality serving to improve the health and quality of life of children affected with CVID. The miRNA-based therapeutic strategies might include alterations of miRNA expression specific to CVID pathogenesis and implicate developing targeted treatment with miRNA modifications that could potentially alleviate the disease severity or have a curative effect. The epigenetic approach might pave the way to posttranscriptional and posttranslational modifications of pharmacokinetics and pharmacodynamics of various drugs used in the treatment of CVID to improve effectiveness. The assessment of differential miRNA expression in response to immunoglobulin replacement therapy (Ig-RT) might open a new perspective and warrant the need to assess the role of miRNAs as biomarkers of immunopathology in CVID and precision medicine interventions [67]. Moreover, the administration of drugs used in the treatment of CVID could serve as epigenetic modifiers of miRNA effect on specific gene expression [89].
Unraveling the molecular genetic and epigenetic background of CVID might be helpful in developing personalized approaches to treatment, monitoring, and long-term care of affected patients [90].
Conclusions and Future Direction
The genetics in pediatric inborn errors of immunity has a multidimensional aspect as it not only plays a crucial role in the progress of understanding the immunological pathways involved in the human immune response but also paves the way to the development of personalized precision medicine and implementation of modern curative therapies such as gene therapy or the use of biologic molecules. Moreover, immunogenetics assists decisions on the use of immunoglobulin replacement therapy and hematopoietic cell transplantation [91,92].
Pediatric CVID is a common immunodeficiency among IEI but it is also a phenotypically and genetically variable disease in children, thereby challenging for pediatricians [93]. The increased awareness of CVID is the first successful step in initiating the diagnostic process. For pediatric immunologists, the major task is to evaluate the parameters of the immune system against the maturation of the adaptive immune responses and to perform further advanced diagnostics including the flow cytometric lymphocyte analysis and the genomic approach [94].
Hereupon, several important issues concerning the genetic studies in pediatric IEI need to be highlighted. As it has been emphasized in this review, in a vast majority of children affected with CVID and their relatives, Mendelian patterns of inheritance can not be applied, reflecting the clinical phenotypic heterogeneity and complex phenotype-genotype correlations. Beyond the monogenic etiology of CVID, other genetic causes include digenic or polygenic background, somatic variants as well as structural variations. Furthermore, the identification of novel variants poses the need to demonstrate their pathogenicity to prove the role of newly discovered variants as disease-causing alterations [95,96,97]. In children with complex phenotypes, expanding the genetic testing with whole exome sequencing (WES) together with chromosomal microarray analysis could provide a more precise diagnosis for unexplained IEI cases [98]. The key initiative for the future is improving the accessibility to immunogenetic studies further implicating challenges and opportunities for therapies and improving IEI pediatric patients’ quality of life [99].
Scientific Support:
The manuscript is a part of the scientific National Center of Science grant project „Genetic and epigenetic background of antibody production defects in children: in search of a pathophysiological model of common variable immunodeficiency” (ID 571776, No 2022/47/B/NZ6-0048). The project is led by Aleksandra Szczawińska-Popłonyk and Aleksandra Szczepankiewicz, without honorarium.
Funding Support:
No funds, grants, or other funding support were received during preparation of this manuscript
Conflict of Interest/Competing Interest:
All authors declare no existing financial and non-financial conflict of interest regarding this manuscript
Availability of Data and Material/Data Transparency:
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request
Author Contributions
All authors contributed to the study conception and design. Aleksandra Szczawińska-Popłonyk was responsible for the principal design of the review and its intellectual content, coordinated and supervised data collection, and drafted the final manuscript. The first draft of the manuscript was written by Wiktoria Ciesielska, Marta Konarczak, Jakub Opanowski, Aleksandra Orska, and Julia Wróblewska. These five authors equally contributed to this work. Aleksandra Szczepankiewicz critically revised the manuscript. All authors read and approved the final manuscript.
References
- Driessen G, van der Burg M. Educational paper: primary antibody deficiencies. Eur J Pediatr 2011,170,693-702. [CrossRef]
- Eroglu FK, Kaya FA, Cagdas D, Ozgur TT, Yilmaz T, Tezcan I, et al. B lymphocyte subsets and outcomes in patients with an initial diagnosis of transient hypogammaglobulinemia of infancy. Scand J Immunol 2018,88,12709. [CrossRef] [PubMed]
- Szczawinska-Poplonyk A, Tapolska-Jozwiak K, Samara H. The B-cell compartment in antibody-deficient infants and young children – developing common variable immunodeficiency or transient immune maturation? Ital J Pediatr 2016, 42, 71. [Google Scholar] [CrossRef] [PubMed]
- Odnoletkova I, Kindle G, Quinti I, Grimbacher B, Knerr V, Gathmann B, et al. The burden of common variable immunodeficiency disorders: a retrospective analysis of the European Society for Immunodeficiency (ESID) registry data. Orphanet J Rare Dis 2018,13,201. [CrossRef]
- Bonilla FA, Barlan I, Chapel H, Costa-Carvalho BT, Cunningham-Rundles C, de la Morena MT, et al. International Consensus Document (ICON): common variable immunodeficiency disorders. J Allergy Clin Immunol Pract 2016,4,38-59. [CrossRef]
- Seidel M, Kindle G, Gathmann B, Quinti I, Buckland M, van Monfrans J, et al. The European Society for Immunodeficiencies (ESID) Registry working definitions for the clinical diagnosis of inborn errors of immunity. J Allergy Clin Immunol Pract 2019,7,1763-1770. [CrossRef]
- Patel SY, Carbone J, Jolles S. The expanding field of secondary antibody deficiency: causes, diagnosis, and management. Front Immunol 2019,10,33. [CrossRef]
- Warnatz K, Denz A, Drager R, Braun M, Groth C, Wolff-Vorbeck G, et al. Severe deficiency of switched memory B cell (CD27(+)IgM(-)IgD(-)) in subgroups of patients with common variable immunodeficiency: a new approach to classify a heterogeneous disease. Blood 2002, 99,1544-1551. [CrossRef]
- Wehr C, Kivioja T, Schmitt C, Ferry B, Witte T, Eren E, et al. The EUROclass trial: defining subgroups in common variable immunodeficiency. Blood 2008,111,77-85. [CrossRef]
- Ramirez NJ, Posadas-Cantera S, Caballero-Oteyza A, Camacho-Ordonez N, Grimbacher B. There is no gene for CVID – novel monogenetic causes for primary antibody deficiency. Curr Opin Immunol 2021,72,176-185. [CrossRef]
- Bogaert DJA, Dullaers M, Lambrecht BN, Vermaelen KY, De Baere E, Haerynck F. Genes associated with common variable immunodeficiency: one diagnosis to rule them all? J Med Genet 2016, 53, 575-–590. [CrossRef] [PubMed]
- Abolhassani H, Hammarstrom L, Cunningham-Rundles C. Current genetic landscape in common variable immunodeficiency. Blood 2020,135,:656-667. [CrossRef]
- Aggarval V, Banday AZ, Jindal AK, Das J, Rawat A. Recent advances in elucidating the genetics of common variable immunodeficiency. Genes Dis 2020, 7,26–37. [CrossRef] [PubMed]
- De Valles-Ibanez G, Esteve-Sole A, Piquer M, Gonzalez-Navarro EA, Hernandez-Rodriguez J, Laayouni H, et al. Evaluating the genetics of common variable immunodeficiency: monogenetic model and beyond. Front Immunol 2018,9,636. [CrossRef]
- Edwards ESJ, Bosco JJ, Ojaimi S, O’Heir RE, van Zelm MC. Beyond monogenic rare variants: tackling the low rate of genetic diagnoses in predominantly antibody deficiency. Cell Mol Immunol 2021,18,588-603. [CrossRef]
- Kienzler AK, Hargreaves CE, Patel SY. The role of genomics in common variable immunodeficiency disorders. J Clin Immunol 2017, 188, 326–332. [Google Scholar] [CrossRef] [PubMed]
- Sanchez LA, Maggadottir SM, Pantell MS, Lugar P, Rundles CC, Sullivan KE, et al. Two sides of the same coin: pediatric-onset and adult-onset common variable immunodeficiency. J Clin Immunol 2017,37,592-602. [CrossRef]
- Carrabba M, Salvi M, Baselli LA, Serafino S, Zarantonello M, Trombetta E, et al. Long-term follow-up in common variable immunodeficiency: the pediatric onset and adult-onset landscape. Front Pediatr 2023,11,112994. [CrossRef]
- Esmailzadeh H, Jokar-Derisi A, Hassani AH, Yazdani R, Delavari S, Abolhassani H, et al. Assessment of the first presentations of common variable immunodeficiency in a large cohort of patients. BMC Immunol 2023,24,9. [CrossRef] [PubMed]
- Szczawińska-Popłonyk A, Tąpolska-Jóźwiak K, Schwartzmann E, Popłonyk N. Immune dysregulation in pediatric common variable immunodeficiency: implications for the diagnostic approach. Front Pediatr 2022,10,855200. [CrossRef]
- Costagliola G, Peroni DG, Consolini R. Beyond infections: new warning signs for inborn errors of immunity in children. Front Pediatr 2022,10,855445. [CrossRef]
- Ameratunga R, Lehnert K, Woon ST. All patients with common variable immunodeficiency disorders (CVID) should be routinely offered diagnostic genetic testing. Front Immunol 2019,10,2678. [CrossRef]
- Christiansen M, Offersen R, Jensen JMB, Petersen MS, Larsen CS, Mogensen TH. Identification of novel genetic variants in CVID patients with autoimmunity, autoinflammation, or malignancy. Front Immunol 2020,10,3022. [CrossRef]
- Peng XP, Caballero-Oteyza A, Grimbacher B. Common variable immunodeficiency: more pathways than roads to Rome. Annu Rev Pathol Mech Dis 2023,18,283-310. [CrossRef]
- Giardino G, Gallo V, Prencipe R, Gaudino G, Romano R, De Cataldis M, et al. Unbalanced immune system: immunodeficiencies and autoimmunity. Front Pediatr 2016,4,107. [CrossRef]
- Tessarin G, Baronio M, Lougaris V. Monogenic forms of common variable immunodeficiency and implications on target therapeutic approaches. Curr Opin Allergy Clin Immunol 2023, 23, 461–466. [Google Scholar] [CrossRef] [PubMed]
- Vanselow S, Wahn V, Schuetz C. Activated PI3Kδ syndrome – reviewing challenges in diagnosis and treatment. Front Immunol 2023,14,1208567. [CrossRef]
- Fischer A, Provot J, Jais JP, Alcais A, Mahlaoui N, and CEREDITH PID study group. Autoimmune and inflammatory manifestations occur frequently in patients with primary immunodeficiencies. J Allergy Clin Immunol 2017,140,1388-1393. [CrossRef]
- Grimbacher B, Warnatz K, Yong PFK, Korganow AS, Peter HH. The crossroads of autoimmunity and immunodeficiency: lessons from polygenic traits and monogenic defects. Clin Rev Allergy Immunol 2016,137, 3-17. [CrossRef]
- Ochiai K, Igarashi K. Exploring novel functions of BACH2 in the acquisition of antigen-specific antibodies. Int Immunol 2023,35,257-265. [CrossRef]
- Vogel TP, Leiding JW., Cooper MA, Forbes Satter LR. STAT3 gain-of-function syndrome. Front Pediatr 2022,10,770077. [CrossRef]
- Rodriguez-Ubreva J, Calvillo CL, Forbes Satter LR, Ballestar E. Interplay between epigenetic and genetic alterations in inborn errors of immunity. Trends Immunol 2023,44,902-916. [CrossRef] [PubMed]
- Tuijnenburg P, Lango Allen H, Burns SO, Green D, Jansen MH, Staples E, et al. Loss-of-function nuclear factor κB subunit 1 (NFKB1) variants are the most common monogenic cause of common variable immunodeficiency in Europeans. J Allergy Clin Immunol 2018,142,1285-1296. [CrossRef]
- Zhang L, Xiao X, Arnold PR, Li XC. Transcriptional and epigenetic regulation of immune tolerance: roles of the NF-κB family members. Cell Mol Immunol 2019,16,315-323. [CrossRef]
- Kerner G, Bouaziz M, Cobat A, Bigio B, Timberlake AT, Bustamante J, Lifton RP, et al. A genome-wide case-only test for the detection of digenic inheritance in human genome. Proc Natl Acad Sci USA 2020,117,19367-19375. [CrossRef]
- Ameratunga R, Woon ST, Bryant VL, Steele R, Slade C, Leung EY,; et al Clinical implications of digenic inheritance and epistasis in primary immunodeficiency disorders. Front Immunol 2018,8,1965. [CrossRef] [PubMed]
- Ameratunga R, Edwards ESJ, Lehnert K, Leung E, Woon ST, Lea E, et al. The rapidly expanding genetic spectrum of common variable immunodeficiency-like disorders. J Allergy Clin Immunol Pract 2023,11,1646-1664. [CrossRef]
- Ameratunga R, Koopmans W, Woon ST, Leung E, Lehnert K, Slade CA, et al. Epistatic interactions between mutations in TACI (TNFRSF13B) and TCF3 result in a severe primary immunodeficiency disorder and systemic lupus erythematosus. Clin Transl Immunol 2017,6,159. [CrossRef]
- Dieli-Crimi R, Martinez-Gallo M, Franco-Jarava C, Antolin M, Blasco L, Paramonov I, et al. Th1-skewed profile and excessive production of proinflammatory cytokines in a NFKB1-deficient patient with CVID and severe gastrointestinal manifestations. Clin Immunol 2018,195,49-58. [CrossRef]
- Massaad MJ, Zhou J, Tsuchimoto D, Chou J, Jabara H, Janssen E,; et al Deficiency of base excision repair enzyme NEIL3 drives increased predisposition to autoimmunity. J Clin Invest 2016,126, 4219–4236. [CrossRef] [PubMed]
- Sic H, Speletas M, Cornacchione V, Seidel M, Beibel M, Linghu B, et al. An activating Janus Kinase 3 mutation is associated with Cytotoxic T Lymphocyte Antigen-4-dependent immune dysregulation syndrome. Front Immunol 2017,8,1824. [CrossRef]
- Li J, Wei Z, Li RY, Magadottir SM, Chang X, Desai A, et al. Understanding the genetic and epigenetic basis of common variable immunodeficiency disorder through omics approaches. Biochim Biophys Acta 2016;1860(11):2656-2663. [CrossRef]
- Fang M, Abolhassani H, Lim CK, Zhang J, Hammarstrom L. Next generation sequencing data analysis in primary immunodeficiency disorders – future directions. J Clin Immunol 2016,36,68-75. [CrossRef]
- Jorgensen SF, Fevang B, Aukrust P. Autoimmunity and inflammation in CVID: a possible crosstalk between immune activation, gut microbiota, and epigenetic modification. J Clin Immunol 2019,39,30-36. [CrossRef]
- Silva SL, Fonseca M, Pereira MLM, Silva SP, Barbosa RR, Serra-Caetano A, et al. Monozygotic twins concordant for common variable immunodeficiency: strikingly similar clinical and immune profile associated with a polygenic burden. Front Immunol 2019,10,2503. [CrossRef]
- Weifenbach N, Schneckenburger AAC, Lotters S. Global distribution of common variable immunodeficiency (CVID) in the light of the UNDP Human Development Index (HDI): a preliminary perspective of a rare disese. J Immunol Res 2020,2020,8416124. [CrossRef]
- Selenius JS, Martelius T, Pikkarainen S, Siitonen S, Mattila E, Pietikainen R,; et al. Unexpectedly high prevalence of common variable immunodeficiency in Finland. Front Immunol 2017, 8, 1190. [CrossRef] [PubMed]
- Ogawa H, Horitani K, Izumiya Y, Sano S. Somatic mosaicism in biology and disease. Annu Rev Physol 2022, 84, 113-–133. [CrossRef] [PubMed]
- Mensa-Vilaro A, Garcia-Morato MB, de la Calle-Martin O, Franco-Jarava C, Martinez-Saavedra MT, Gonzalez-Granado LI,; et al. Unexpected relevant role of gene mosaicism in patients with primary immunodeficiency diseases. J Allergy Clin Immunol 2019, 143, 359-–368. [CrossRef] [PubMed]
- Aluri J, Cooper MA. Genetic mosaicism as a cause of inborn errors of immunity. J Clin Immunol 2021, 41, 718–728. [Google Scholar] [CrossRef] [PubMed]
- Aluri J, Cooper MA. Somatic mosaicism in inborn errors of immunity: current knowledge, challenges, and future perspectives. Semin Immunol 2023,67,101761. [CrossRef]
- Materna-Kiryluk A, Pollak A, Gawalski K, Szczawińska-Popłonyk A, Rydzyńska Z, Sosnowska A,; et al. Mosaic IL6ST variant inducing constitutive GP130 cytokine receptor signaling as a cause of neonatal onset immunodeficiency with autoinflammation and dysmorphy. Hum Mol Genet 2021,30, 226–233. [CrossRef] [PubMed]
- Savola P, Martelius T, Kankainen M, Huuhtanen J, Lundgren S, Koski Y, et al. Somatic mutations and T cell clonality in patients with immunodeficiency. Haematologica 2020, 105, 2557–2768. [CrossRef] [PubMed]
- Kwon SS, Cho YK, Hahn S, Oh J, Won D, Shin D,; et al. Genetic diagnosis of inborn errors of immunity using clinical exome sequencing. Front Immunol 2023, 14, 1178582. [CrossRef] [PubMed]
- Guevara-Hoyer K, Fuentes-Antras J, de la Fuente-Munoz E, Fernandez-Arquero M, Solano F, Perez-Segura P, et al. Genomic crossroads between non-Hodgkin lymphoma and common variable immunodeficiency. Front Immunol 2022,13,937872. [CrossRef]
- Rae, W. Indications to epigenetic dysfunction in the pathogenesis of common variable immunodeficiency. Arch Immunol Ther Exp 2017, 65, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Cano J, Campos-Sanchez E, Cobaleda C. Epigenetic priming in immunodeficiencies. Front Cell Dev Biol 2019, 7, 125. [Google Scholar] [CrossRef] [PubMed]
- Campos-Sanchez E, Martinez-Cano J, del Pino Molina L, Lopez-Granados E, Cobaleda C. Epigenetic deregulation in human primary immunodeficiencies. Trends Immunol 2019, 40, 49–65. [CrossRef]
- Camacho-Ordonez N, Ballestar E, Timmers HTM, Grimbacher B. What can clinical immunology learn from inborn errors of epigenetic regulators? J Allergy Clin Immunol 2021, 147, 1602–1618. [CrossRef] [PubMed]
- Ghafouri-Fard S, Niazi V, Taher M. Role of miRNAs and lncRNAs in hematopoietic stem cell differentiation. Noncoding RNA Res 2021, 6, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Chen CZ, Li L, Lodish HF, Bartel DP. Micro RNAs modulate hematopoietic lineage differentiation. Science 2004, 303, 83–86. [CrossRef] [PubMed]
- Xiao C, Calado DP, Galler G, Thai TH, Patterson HC, Wang J, et al. MiR-150 controls B cell differentiation by targeting the transcription factor c-Myb. Cell 2007,131,145-159. [CrossRef]
- Salunkhe S, Vaidya T. CD40-miRNA axis controls prospective cell fate determinants during B cell differentiation. Mol Immunol 2020,126,46-55.
- Bao Y, Cao X. Epigenetic control of B cell development and B cell related immune disorders. Clin Rev Allergy Immunol 2016, 51, 301–311. [Google Scholar] [CrossRef] [PubMed]
- Rao DS, O’Connell, Chaudhuri AA, Garcia-Flores Y, Geiger TI, Baltimore D. MicroRNA-34a perturbs B lymphocyte development by repressing the forkhead box transcription factor Foxp1. Immunity 2010,33,48-59. [CrossRef]
- O’Connell RM, Rao DS, Chaudhuri AA, Baltimore D. Physiological and pathological roles of micro RNAs in the immune system. Nat Rev Immunol 2010, 10, 111–122. [CrossRef] [PubMed]
- Babaha F, Yazdani R, Shahkarami S, Hamidi Esfahani Z, Abolhassani H, Sadr M, et al. Evaluation of miR-210 expression in common variable immunodeficiency: patients with unsolved genetic defect. Allergol Immunopathol 2021,49,84-93. [CrossRef]
- Haralambieva IH, Kennedy RB, Simon WL, Goergen KM, Grill DE, Ovsyannikova IG, et al. Differential miRNA expression in B cells is associated with inter-individual differences in humoral immune response to measles vaccination. PloS ONE 2018,13,e0191812. [CrossRef]
- Rosales, C. Fc gamma receptor heterogeneity in leukocyte functional responses. Front Immunol 2017, 8, 280. [Google Scholar] [CrossRef] [PubMed]
- Surace AEA, Hedrich CM. The role of epigenetics in autoimmune/inflammatory disease. Front Immunol 2019,10,1525. [CrossRef]
- Gutierrez M, Gomez JL, Perez GF, Pancham K, Val S, Pillai DK, et al. Airway secretory microRNAome changes during rhinovirus infection in early childhood. PLoS ONE 2016,11,:e0162244. [CrossRef]
- De Felice B, Nigro E, Polito R, Rossi FW, Pecoraro A, Spadaro G, et al. Differently expressed microRNA in response to the first Ig therapy in common variable immunodeficiency patients. Sci Rep 2020, 10, 21482. [CrossRef] [PubMed]
- Baloh C, Reddy A, Henson M, Prince K, Buckley R, Lugar P. 30-year review of pediatric- and adult-onset CVID: clinical correlates and prognostic factors. J Clin Immunol 2019,39,678-687. [CrossRef]
- Sanchez LA, Magadottir MS, Pantell MS, Lugar P, Cunningham Rundles C, Sullivan K. Two sides of the same coin: pediatric-onset and adult-onset common variable immune deficiency. J Clin Immunol 2017,37,592-602. [CrossRef]
- Szczawinska-Poplonyk A, Schwartzmann E, Bukowska-Olech E, Biernat M, Gattner S, Korobacz T, et al. The pediatric common variable immunodeficiency – from genetics to therapy: a review. Eur J Pediatr 2022,181,1371-1383. [CrossRef]
- Janssen LMA, van der Flier M, de Vries E. Lessons learned from the clinical presentation of common variable immunodeficiency disorders: a systematic review and meta-analysis. Front Immunol 2021:620709. [CrossRef]
- Ammato G, Vita F, Quattrocchi P, Minciullo PL, Pioggia G, Gangemi S. Involvement of miR-142 and miR-155 in non-infectious complications in CVID. Molecules 2020,25,4760. [CrossRef]
- Zhu H, Wang G, Qian J. Transcription factors as readers and effectors of DNA methylation. Nat Rev Genet 2016, 17, 551–565. [Google Scholar] [CrossRef] [PubMed]
- Kulis M, Merkel A, Heath S, Queiros AC, Schuyler RP, Castellano G, et al. Whole-genome fingerprint of the DNA methylome during human B-cell differentiation. Nat Genet 2017,47,746-756. [CrossRef]
- Alvarez-Errico D, Vento-Tormo R, Siewke M, Ballestar E. Epigenetic control of myeloid cell differentiation, identity and function. Nat Rev Immunol 2015,15,7-17. [CrossRef]
- Rodriguez-Cortez VC, de Pino-Molina L, Rodriguez-Ubreva J, Ciudad L, Gomez-Cabrero D, Company C, et al. Monozygotic twins discordant for common variable immunodeficiency reveal impaired DNA demethylation during naiv-to-memory B-cell transition. Nat Commun 2015,6,7335. [CrossRef]
- Rodriguez-Ubreva J, Arutyunyan A, Bonder JM, Del Pino-Molina L, Clark SJ, de la Calle-Fabregat C, et al. Single-cell atlas of common variable immunodeficiency shows germinal center-associated epigenetic dysregulation in B-cell responses. Nat Commun 2022,13,1779. [CrossRef]
- Del Pino-Molina L, Rodriguez-Ubreva J, Torres Canizales J, Coronel-Diaz M, Kulis M, Martin-Subero JI, et al. Impaired CpG demethylation in common variable immunodeficiency associates with B cell phenotype and proliferation rate. Front Immunol 2019, 10, 879. [CrossRef] [PubMed]
- Aird A, Lagos M, Vargas-Hernandez A, Posey JE, Coban-Akdemir Z, Jhangiani S, et al. Novel heterozygous mutation in NFKB2 is associated with early onset CVID and a functional defect in NK cells complicated by disseminated CMV infection and severe nephrotic syndrome. Front Pediatr 2019,7,303. [CrossRef]
- Klemann C, Camacho-Ordonez N, Yang L, Eskandarian Z, Rojas-Restrepo JL, Frede N,; et al Clinical and immunological phenotype of patients with primary immunodeficiency due to damaging mutations in NFKB2. Front Immunol 2019, 10, 297. [CrossRef] [PubMed]
- Lougaris V, Baronio M, Gazzurelli L, Lorenzini T, Fuoti M, Moratto D, et al. A de novo monoallelic CTLA-4 deletion causing pediatric onset CVID with recurrent autoimmune cytopenias and severe enteropathy. Clin Immunol 2018,197,186-188. [CrossRef]
- Rush-Kittle J, Gamez-Diaz L, Grimbacher B. Inborn errors of immunity associated with defects of self-tolerance checkpoints: the CD28 family. Pediatr Allergy Immunol 2022,33,13886. [CrossRef]
- Ballow M, Leiding JW. Precision medicine in the treatment of primary immune deficiency patients with disorders of immune dysregulation. Clin Rev Allergy Immunol 2022,63,1-8. [CrossRef]
- Majchrzak-Celińska A, Baer-Dubowska W. Pharmacoepigenetics: an element of personalized pharmacotherapy? Expert Opin Drug Metab Toxicol 2017, 13, 387–398. [Google Scholar] [CrossRef] [PubMed]
- Lee TK, Gereige JD, Maglione PJ. State-of-the-art diagnostic evaluation of common variable immunodeficiency. Ann Allergy Asthma Immunol 2021,127,19-27. [CrossRef]
- Leiding, JW., Forbes LR. Mechanism-based precision therapy for the treatment of primary immunodeficiency and primary immunodysregulatory diseases. J Clin Immunol Pract, 2019; 7, 761–773. [Google Scholar] [CrossRef]
- Pinto MV, Neves JF. Precision medicine: the use of tailored therapy in primary immunodeficiencies. Front Immunol 2022,13,1029560. [CrossRef]
- Boz V, Zanchi C, Levantino L, Riccio G, Tommasini A. Druggable monogenic immune defects hidden in diverse medical specialties: focus on overlap syndromes. World J Clin Pediatr 2022,11,136-150.
- Fekrvand S, Khanmohammadi S, Abolhassani H, Yazdani R. B- and T-cell subset abnormalities in monogenic common variable immunodeficiency. Front Immunol 2022,13,912826. [CrossRef]
- Heimall JR, Hagin D, Hajjar J, Henrickson SF, Hernandez-Trujillo HS, Itan Y, et al. Use of genetic testing for primary immunodeficiency patients. J Clin Immunol 2018, 38, 320-–329. [CrossRef] [PubMed]
- Schmitt EG, Cooper MA. Genetics of pediatric immune-mediated diseases and human immunity. Annu Rev Immunol 2021,39,227-249. [CrossRef]
- Caballero-Oteyza A, Crisponi L, Peng XP, Yauy K, Volpi S, Giardino S, et al. GenIA, the Genetic Immunology Advisor database for inborn errors of immunity. J Allergy Clin Immunol 2024,153,831-843. [CrossRef]
- Beers BJ, Similuk MN, Ghosh R, Seifert BA, Jamal L, Kamen M, et al. Chromosomal microarray analysis supplements exome sequencing to diagnose children with suspected inborn errors of immunity. Front Immunol 2023, 14, 1172004. [CrossRef] [PubMed]
- Tadros S, Prevot J, Meyts I, Sanchez-Ramon S, Erwa NH, Fischer A, et al. The PID Odyssey 2030: outlooks, unmet needs, hurdles, and opportunities – proceedings from the IPOPI global multi-stakeholdrers’ summit. Front Immunol 2023,14,1245718.
Figure 1.
Epigenetic mechanisms regulating gene expression relevant for CVID.
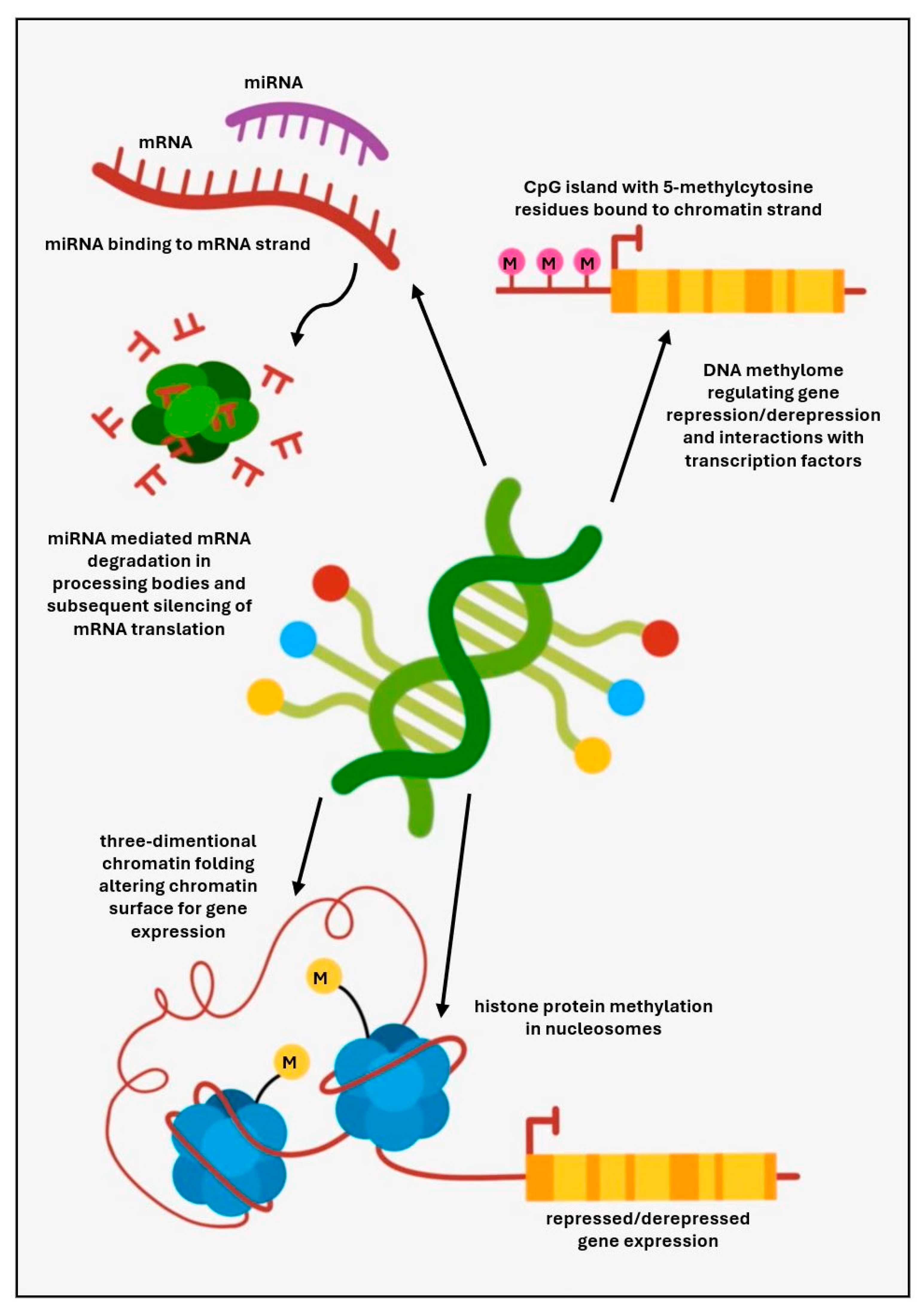

Table 1.
Categories of immune pathways and a spectrum of variants in genes associated with monogenic CVID according to Peng et al. [24].
Table 1.
Categories of immune pathways and a spectrum of variants in genes associated with monogenic CVID according to Peng et al. [24].
| Predominantly antibody deficiencies associated with CVID phenotypes | |||||
|---|---|---|---|---|---|
| BCR costimulatory B cell surface proteins | TNF superfamily receptors and ligands | Lipid signaling molecules | Actin cytoskeleton regulators | Transcription factors mediating differentiation and crosstalk | Metabolic mitochondrial and glycosylation pathways |
|
CD19 MS4A1/ CD20 CR2/ CD21 CD81 |
TNFSF13B/ BAFF/ BLYS/ TALL1 TNFSF13/ APRIL TNFSF12/ TWEAK TNFRSF13C/ BAFF-R TNFRSF13B/ TACI TNFRSF17/ BCMA |
PIK3CD PIK3R1 PTEN PIK3CG TTC7A |
CXCR4 RAC2 ARHGEF1 TTC7A PSTPIP1 DOCK8 WAS |
NFKB1 NFKB2 IKZF1/ IKAROS |
MAGT1 ATP6AP1 PGM3 TNRT1 FNIP1 SBDS TAFAZZIN |
| Hypomorphic variants in other genes associated with predominantly antibody deficiencies | |||||
|
BTK TCF3 FNIP1 ICOS CTNNBL1 ZRSR2 | |||||
| Genes associated with immune dysregulation disorders | |||||
| Transcriptional regulators of central and peripheral tolerance | Membrane- bond organelle dynamics | Genes related to lymphoproliferative conditions | |||
|
AIRE FOXP3 STAT3 SOCS1 BACH2 |
CTLA4 LRBA SEC61A1 SH3KBP1 DEF6 SAMD9 |
CD27 CD70 MAGT1 SH2D1A PRKCD STXBP2 UNC13D FASLG |
|||
| Genes in combined cellular and humoral immunodeficiencies | |||||
| Genes associated with T cell signaling regulators | Genes associated with epigenetic regulation | ||||
|
ICOS CTLA4 FOXP3 GATA2 RFXANK LCK IL21R |
DNMT3B ZBTB24 IGH KMT2D/ MLL2 KDM6A/ UTX |
||||
| Genetic underpinnings of autoinflammatory disorders | |||||
|
DCLRE1C/ Artemis ADA2 RNF31 TNFA1P3 PLCG2 NLRC3, NLRC4, NLRP2, NLRP3, NLRP12 | |||||
Table 2.
Digenic CVID and epistatic effect.
| Digenic variants | Product functions | Clinical phenotype | Immunodeficiency | Authors (references) | ||
|---|---|---|---|---|---|---|
| Genes | Variants | Digenic proband | Monogenic variant | |||
|
TNFSFR13B /TACI (17p11.2) |
rs34557412 C104R |
T-cell independent CSR, MyD88 pathway | CVID Systemic lupus erythematosus |
Mild cytopenia Antibody deficiency | Defective T cell dependent and T cell independent B cell differentiation and activation | Ameratunga et al. [38] |
|
TCF3 (19p13.3) |
T168fsX191 | T-cell dependent and independent CSR, AID pathway | Antibody deficiency Arthritis Diabetes mellitus |
|||
|
NFKB1 (4q24) |
(c.1149delT/ p.Gly384Glu*48) | Key cellular driver of inflammation and immunity | CVID Inflammatory bowel disease Thrombocytopenia |
Asymptomatic | Th1-polarized T cell population B cell lymphopenia and B cell naïvete |
Dieli-Crimi et al. [39] |
|
NOD2 (16q12.1) |
rs 5743272 (p.His352Arg) |
Inflammatory response to pathogens NF-κB pathway |
Crohn disease | |||
|
LRBA (4q31.3) |
C7885delA (p.R2629fs) |
Peripheral B cell tolerance, stimulation of T regulatory cell development and functions, CTLA 4 pathway | Antibody deficiency Recurrent airway infections Sepsis Inflammatory bowel disease Throbocytopenia Autoimmune hemolytic anemia |
Immunodeficiency with autoimmunity | Decreased ability of T regulatory cells to control T effector cells Defective peripheral B cell tolerance Immunodeficiency |
Massaad et al. [40] |
|
NEIL3 (4q34.3) |
rs200055050 (p.D132V) |
Regulation of lymphoid cell proliferation, peripheral B cell tolerance | Asymptomatic High levels of autoantibodies |
|||
|
CTLA4 (2q33.2) |
rs1581573923 (p.Y139C) |
Negative regulator of T cell responses, expressed in activated T cells and T regulatory cells | CVID Recurrent airway infections Lymphoid hyperplasia Gastroenteritis Hypothyroidism |
Asymptomatic | Impaired memory B cell and plasma cell development T cell hyperactivity |
Sic et al. [41] |
|
JAK3 (19p13.11) |
rs200077579 (p.R840C) |
Signal transduction from JAK3-associated cytokine receptor common γ chain | Hashimoto thyroiditis | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



