
Submitted:
02 August 2024
Posted:
06 August 2024
You are already at the latest version
Abstract
ATP is an essential molecule in many biocatalytic processes. The industrial implementation of these processes is hampered by the fact that they require stoichiometric quantities of this expen-sive co-factor. To avoid this problem, recycling systems are needed. This study characterizes a putative polyphosphate kinase enzyme from Burkholderia cenocepacia that can regenerate ATP from AMP. Sequence analysis shows that BcPPK2-III exhibits the characteristic structural motifs of known PPK2 enzymes, including conserved motifs Walker A and B, and the subclass-specific res-idue E137. Molecular docking simulations showed ADP had the highest binding affinity (-7.340 kcal/mol), followed by AMP (-7.284 kcal/mol), with ATP having the lowest affinity (-6.464 kcal/mol). The enzyme was overexpressed in E. coli. Protein expression and purification were monitored by SDS-PAGE and quantified by densitometric analysis. Enzymatic activity assays re-vealed that BcPPK2-III converts 70% ATP with a TTN of 334,000 after 24 hours and remains sta-ble over a pH range of 6-9 and temperatures up to 70°C. BcPPK2-III shows significant potential for industrial ATP regeneration and offers high conversion rates and stability. The enzyme's high conversion rate and stability make BcPPK2-III a promising candidate for the improvement of bio-catalytic processes that rely on ATP at industrial level.
Keywords:
biocatalysis
; ATP regeneration
; polyphosphate
; polyphosphate kinase
; cofactor recycling
; cascade reactions
; Burkholderia cenocepacia
1. Introduction
Adenosine 5′-triphosphate (ATP) is one of the most important molecules in living systems and probably the foremost phosphate compound of all. Normally known as the universal energy “currency” or “store” of the cell [1], it is also used as a building block for nucleic acid synthesis and is involved in the synthesis of key phosphorus-containing intermediates for biosyntheses and cofactors such as flavin adenine dinucleotide (FAD), nicotinamide adenine dinucleotide (NAD+), S-adenosylmethionine (SAM), and 3′-phosphoadenosine 5′-phosphosulfate (PAPS) [2,3,4,5]. ATP is an essential molecule in many biocatalytic processes, but industrial implementation of these processes is hampered by the fact that they require stoichiometric amounts of this expensive cofactor. ATP recycling systems can provide higher yields in these reactions by shifting the reaction equilibrium toward the desired product [6]. Other benefits of ATP recycling systems include reduced product inhibition by decreasing the concentration of the side product ADP, while keeping the ATP concentration high to facilitate phosphorylation reactions, simplified product purification, and cost savings by reducing the required amount of this expensive substrate [7].
The most commonly used ATP regeneration systems from ADP are based on the use of phosphoenolpyruvate as the phosphate donor in a coupled reaction catalyzed by pyruvate kinase (PK) and on the use of acetylphosphate coupled with acetate kinase (AK) [8]. In recent decades, the regeneration system based on the use of polyphosphate kinases (PPK) that catalyze the reversible transfer of γ-phosphate from ATP to inorganic polyphosphate (polyP) has attracted considerable attention [9,10]. Inorganic polyphosphate is a linear polymer consisting of up to hundreds of orthophosphate (Pi) residues linked by high-energy phosphoanhydride bonds [11,12,13]. Nobel laureate Arthur Kornberg theorized that polyP is a “bioenergy fossil” that was the prominent “energy currency” in prebiotic evolution [14].
Regeneration systems based on the use of PPKs have several advantages that make them very attractive in terms of their application in biocatalysis. First and foremost, polyP is a low-cost phosphate group donor. A large amount of polyP is regularly produced as sodium hexametaphosphate (about 13-18 residues) for industrial purposes such as food additives, which also makes polyP inexpensive compared to other phosphoryl group donors. To get an idea of the magnitude of savings from using polyP as a phosphate donor, consider that a commercial form of polyP costing about $9/pound can provide ATP equivalents that would cost more than $2,000/pound, and that other phosphate donors commonly used in ATP regeneration, such as phosphoenolpyruvate (PEP), are even more expensive than ATP [15,16,17]. The other major advantage of this system is that there are PPKs that allow regeneration of ATP from AMP.
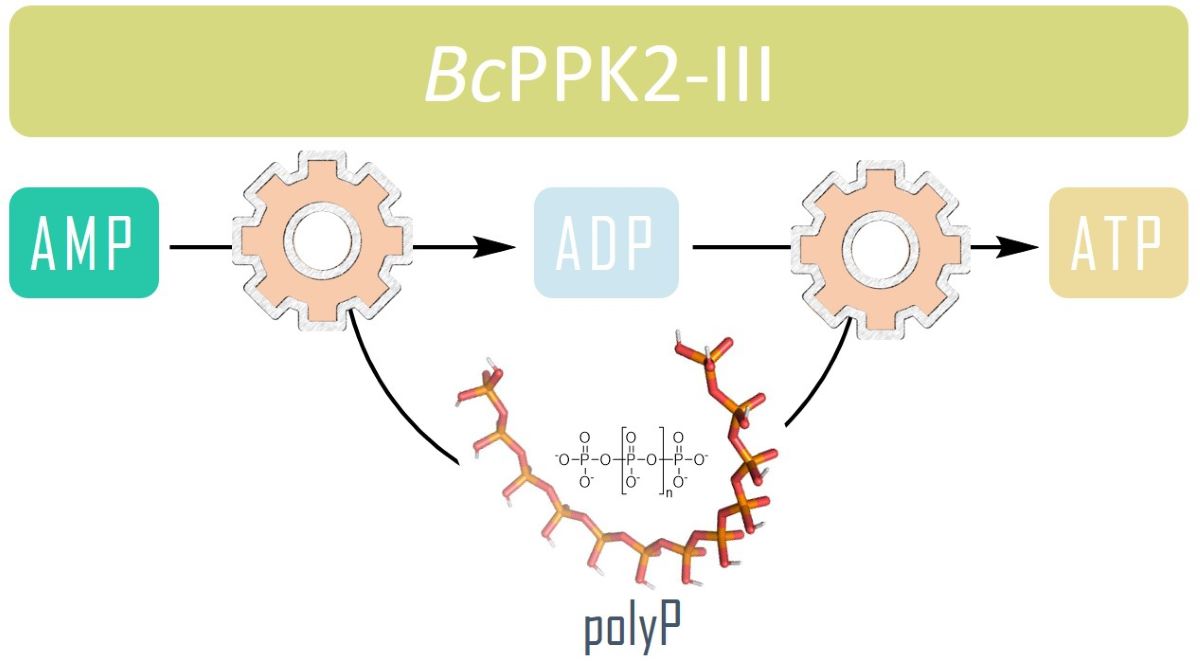
Methods to regenerate ATP from AMP or adenosine are not well established, although they have great potential for biocatalysis [9,18]. There are several cases where the regeneration of ATP from AMP based on low-cost polyphosphate as the sole energy source would be of great value, such as the regeneration of acyl-CoA with integrated ATP regeneration [19] or the in situ synthesis of PAPS in reactions catalyzed by sulfotransferases [20]. PAPS is synthesized from sulfate and two ATP molecules through ATP sulfurylase, forming adenosine 5′-phosphosulfate (APS), and APS kinase, which phosphorylates APS to PAPS, consuming a second ATP molecule [21,22]. Polyphosphate kinase enzymes play an important role in regulating intracellular nucleotide concentrations in vivo by participating in the reversible transfer of phosphate groups between nucleotides and polyP [23]. To date, two major families of highly conserved PPKs have been characterized in bacteria that are neither sequentially nor structurally related. Polyphosphate kinase 1 (PPK1) is responsible for the synthesis of polyP using ATP as a donor of phosphate groups, while polyphosphate kinase 2 (PPK2) enzymes are responsible for its degradation.[23] PPK2 can be divided into three subclasses (I, II, and III) depending on the reaction they catalyze, as shown in Figure 1. They all use polyP as a phosphate group donor to catalyze the phosphorylation of the various nucleotides (AMP, ADP and ATP). The substrate preference of PPK2-II is AMP from which they can synthesize ADP. The substrate preference of PPK2-I and PPK2-III are ADP and AMP, respectively [23,24].
Given the interest that class III of PPK2 represents from a biocatalysis point of view, we consider it necessary to identify and describe new members of this enzyme family in order to increase the tools available for the development of efficient ATP regeneration processes. Although several gene sequences have been found that could encode PPK2-III enzymes, most of them are putatively assigned, i.e., they lack experimental verification. In this regard, Motomura et al. (2014) [25] constructed an unrooted phylogenetic tree of PPK2 using amino acid sequences from 17 characterized and 192 putative PPK2 homologs. This phylogenetic analysis revealed 40 sequences belonging to the class III subfamily. However, only the sequence from the bacterium Meiothermus ruber was amplified and expressed to characterize the biochemical properties of the PPK2-III enzymes.
Burkholderia cenocepacia is a Gram-negative bacterium and belongs to the Burkholderia cepacia complex (Bcc). Although common in the environment, it can also cause disease in plants and act as an opportunistic pathogen that frequently infects patients with cystic fibrosis and chronic granulomatous diseases. Studies have shown that B. cepacia isolates from various sources accumulate higher levels of intracellular polyphosphate (polyP) and increase the uptake of inorganic phosphate when grown in acidic environments [26]. The importance of polyP metabolism in this bacterium suggests that B. cenocepacia could be an interesting source for the overexpression and characterization of a new PPK2-III with interesting properties for its application in biocatalysis.
Herein, in this work, we addressed the characterization of the putative polyphosphate kinase from Burkholderia cenocepacia. A study of its secondary and tertiary structure was performed using theoretical structural analysis and 3D modeling. The enzyme was overexpressed in E. coli, and after its purification, its enzymatic activity, divalent cation dependence and reaction mechanism were studied. Enzyme kinetic parameters and stability were also analyzed and discussed. In addition, we have been concerned with elucidating the mechanism of action of this enzyme, as it is controversial whether it is a stepwise sequential diphosphorylation mechanism or a pyrophosphorylation mechanism and thus occurs in a single step [24].
2. Results and Discussion
2.1. Sequence Analysis of the Putative PPK2-III from Burkholderia cenocepacia
PPK2 belongs to the P-loop kinases. This protein fold is by far the most common among enzymes that bind or hydrolyze nucleoside triphosphates (NTPs) and is characterized by the presence of two conserved sequence motifs (Walker A and Walker B) and a lid module (RxxxxxxPxxxxxxxD). The Walker A (DxxGK) and Walker B (RSxY) motifs are conserved in all PPK2 subclasses, whereas the amino acid following the Walker B motif appears to be subclass-specific. These amino acids are asparagine (subclass I), glycine (subclass II), and glutamic acid (subclass III) [25,27,28].
Using as query the sequence of the PPK2-III enzyme from Meiothermus ruber (Mrub_2488), a sequence alignment of the putative PPK2-III from B. cenocepacia was performed using the BLAST® program (Figure S1) [29,30]. Sequence analysis shows a 51% of identity and the E-value, indicating the probability of alignment occurring by chance, was 10-95. Thus, the alignment between these two enzymes was significant and of high quality. In addition to the high sequence similarity, the Walker A and B motifs, as well as the cap module, were also found in the BcPPK2-III sequence (Figure S1). It also contains glutamine (Q74), which precedes the Walker A motif in the PPK2 subclass III, and glutamic acid (E137), located downstream of the Walker B motif which contributes to the binding of the adenine base [28]. This glutamic acid residue is the class III signature residue of PPK2. However, mutation of this residue led to a very significant reduction in the phosphorylation activity of AMP, ADP and GDP, indicating that this residue alone does not determine the substrate specificity of PPK2 [31].
The sequence of BcPPK2-III was also analyzed for possible transmembrane regions using the protein topology prediction method, TMHMM, based on a hidden Markov model [32]. As no such zone was found, we can assume that the protein is free in the cytosol, which increases the chances that it will be expressed soluble.
2.2. Heterologous Overexpression of BcPPK2-III
Expression of the recombinant protein was induced with 0.5 mM IPTG at an OD600 nm of 0.5-0.6. The expression and purification of the BcPPK2-III enzyme was analyzed by SDS-PAGE (Figure S2). The system yielded a high degree of overexpression of the target enzyme. The CFE (Figure S2, lane 1) showed a majority band that would correspond to the recombinant protein given its theoretical size (31.92 kDa). Densitometric analysis revealed that the recombinant protein accounted for 60% of the total soluble proteins in the CFE. The analysis of the IMAC purification process of the recombinant protein shows that the recombinant protein binds very specifically to the resin, as it was not detected in the combined fractions collected during the CFE loading (Figure S2, lane 2), nor in the subsequent washes performed to remove proteins not specifically bound to the resin (Figure S2, lane 3). On the contrary, in the combined fractions obtained after elution of the recombinant protein with imidazole (Figure S2, lane 4), a single band corresponding to BcPPK2-III was observed with a purity higher than 98%. Interestingly, when the resin was analyzed after elution of the recombinant protein (Figure S2, lane 5), it was found that a considerable amount of the protein remained bound to the resin with a high degree of purity (77.4%).
2.2. Identification and Molecular Modeling of BcPPK2-III
Identity of the recombinant protein was confirmed by the peptide mass fingerprinting (Figure S3). Seventeen different peptides were identified, corresponding to 68% of the protein sequence. This result confirmed that the sequence of the purified protein matched the sequence of BcPPK2-III.
The 3D structure of the protein was modeled using the ColabFold open-source software [33,34] completed by a 3D structure modeling. The high Model Confidence and Sequence coverage (Figure S4 a and b respectively) indicate a good quality in the predicted enzyme structure. The vast majority of the structure has been predicted with a very high (>90%) degree of confidence (Figure S4a). The obtained results were visualized using PyMOL. The three conserved motifs (Walker A motif, Walker B motif, and lid module) are highlighted in red, blue, and green, respectively (Figure 2). The spatial position and proximity of the three motifs supports the hypothesis that the active center is located below the lid module, between motifs A and B [25,28].
2.3. Modelling of the Enzyme-Substrate Complexes
Molecular docking is a helpful tool in predicting the affinity and biding mode of a native molecule in the enzyme active site. In order to harness a more complete understanding BcPPK2-III and the molecular foundation of interaction and conformation of the three main compounds within the binding pose a docking study was performed using Maestro Schrödinger’s Glide module. The results of the molecular docking are summarized in Table 1, which lists the conformer of each substrate that yields the best docking score, as well as the residues of the active site of the enzyme involved in the interaction with the substrates.
The docking scores indicated that ADP had the highest binding affinity with a score of -7.340 kcal/mol, closely followed by AMP with a score of -7.284 kcal/mol, while ATP showed a relatively lower affinity with a score of -6.464 kcal/mol. The Glide Emodel results provided further insights into the stability and quality of the binding poses. ADP exhibited the most favorable Emodel score of -122.995, considering the lower docking score, suggesting a highly stable interaction within the active site. AMP also demonstrated a strong binding pose with an Emodel score of -98.475. Interestingly, ATP had the highest Emodel score of -138.999. However, taking into account the docking score this value could indicate significant internal strain or a less optimal binding conformation.
The biding pose and residue interaction of the three ligands are displayed in Figure 3. ADP interaction with particularly multiple charged and polar residues such as Arg133 (Walker B), Arg193, and Asp204 (Lid module) indicates a stable binding conformation. This is reflected in both its favorable docking score and Emodel score. In the case of AMP, the presence of aromatic Phe102 and multiple polar/charged residues contributes to a strong interaction profile. As stated before, AMP’s docking score and Emodel score suggest that it fits well in the binding site but slightly less effectively than ADP. Finally, considering ATP complex binding mode (interaction with 8 different residues) and different scores might have significant internal strain or suboptimal initial positioning in the active site. These results underscore the nuanced interplay between binding affinity and pose stability, highlighting ADP as the most suitable ligand.
2.4. Biochemical Characterization of BcPPK2-III
Characterization of BcPPK2-III initially focused on the enzyme metal cofactor determination, and four different divalent cations were tested. BcPPK2-III shows activity across all tested cations (Figure 4) and exhibits substantial and similar activity towards the production of ATP when Co2+, Mg2+, and Mn2+ are used as cofactors. Interestingly, when the enzyme used Ca2+ exhibited a distinct behavior wherein the production of ADP surpassed that of ATP. Determining this specificity is of great interest because it has been described that metal cofactor preference plays a crucial role in the efficiency of the enzyme towards ATP production. Generally PPK2 enzymes show dependence on Mn2+ [25] or Mg2+ [28]. In the cases of PPK2-III from Meiothermus ruber and PPK2-III from Ralstonia eutropha accept both divalent cations were used by the enzyme to produce ATP [25,29].
However, to the best of our knowledge, this is the first time that a PPK2-III has been reported to exhibit high and similar activity with three different cations, illustrating the versatility of the enzyme along with its distinct Ca2+dependent activity. Currently, we have no data that allow us to support a hypothesis that explains the different behavior found with Ca2+, apart from attributing it to the smaller ionic radius and solvation shell of the cation.
In addition, it is worth to mention that 24 h reaction using Mn2+ as enzyme cofactor resulted in a precipitate that could indicate the formation of a nanoflower-like structure [36] between the shorter polyP chains and the cation, resulting after enzyme activity (Figure S5). This could translate in lower availability of the phosphate groups, which could have effect in the enzymatic efficiency of the enzyme to produce ATP. In this sense, further characterization of the enzyme was carried out using Mg2+ as a metal cofactor.
PPK2-III family can perform the ATP formation from AMP through two main reaction mechanisms. The most common mechanism is step-by-step phosphorylation, in such a way that, starting from AMP, the enzyme catalyzes the formation of ATP with an intermediate formation of ADP. On the other hand, it has been reported the direct production of ATP from AMP in the so-called pyrophosphorylation mechanism, adding two phosphate groups to AMP, without forming the intermediate ADP [24,28,37]. To assess the enzymatic mechanism of BcPPK2-III, the progress of the reaction over 48h was analyzed (Figure 5a). According with the time-course experiment, in the first 10 minutes of the reaction, the enzyme produces ADP and, in a lower rate, forms ATP reaching a high yield (70%) after 24h. PPK2-III enzymes prefer larger chains of polyPn and the shortening of the phosphate donor during the reaction is a limiting factor in enzyme activity [25,31]. Subsequently, the reaction was supplemented with polyP12 and the analysis after 48h did not show a significant yield in ATP increment, probably due to enzyme stability or inhibition.
To further support the hypothesis that the reaction mechanism proceeds by a stepwise phosphorylation process and that the observed decrease in ADP is not due to an adenylate kinase side reactivity, a reaction was performed with ADP as starting substrate instead of AMP. Figure 5b shows the reaction time-course using ADP as substrate. After only 3.5 h the reaction yield in terms of the formation of ATP is 60%, which indicates a higher enzyme efficiency when ADP is the reaction substrate.
This enzymatic behavior agrees with the performed molecular docking as ADP is clearly a more suitable substrate for BcPPK2-III. In addition, this results experimentally confirms that the enzyme BcPPK2-III belongs to subclass III, as it is able to catalyze the phosphorylation of AMP to ADP and from this to ATP and the step-by-step phosphorylation enzymatic mechanism. On the other hand, the occurrence of the AMP formation must be due to dephosphorylation processes. This dephosphorylation reaction could be related to the adenylate kinase (ADK) activity observed in the three PPK2 subclasses [31]. As a result of this activity, the enzymes are able to convert two ADP molecules into ATP and AMP [24,31,37]. On the other hand, they could also be related to the reversibility of the reaction catalyzed by PPK2-III. Thus, ADP or newly formed ATP could be used as a donor for short-chain polyP phosphorylation.
The efficiency of the enzyme in the production of ATP was assessed by the total turnover number (TTN) after 24h and the Initial Turnover Rates (ITR), calculated in the first 6h of the reaction, when the production of ATP is linear with time (Table 1). TTN is a dimensionless parameter with a high significance in determining the industrial implementation of an enzyme [23]. In this case, considering that the order of BcPPK2-III TTNs is 105, it can be concluded that the enzyme is very efficient in the formation of ATP.

Table 2.
Enzyme activity and kinetic parameters of BcPPK2-III.
| Enzyme activity (U/ml) |
Specific Activity (U/mg) |
ITR (μmol ATP/μmol enzyme x min) |
TTN 24h (μmol ATP/μmol enzyme) |
|---|---|---|---|
| 100.77 | 1660.64 | 513.44 | 3.34 x 105 |
2.4. Enzyme Stability
The T50 (the temperature at which the enzyme retains 50% of its activity after a ten-minute incubation) was determined to be 62.2 °C (Figure 6a). Notably, the enzyme holds 100% activity from 30-58 ºC, which indicates a high degree of thermostability considering other enzymes described belonging to the same family [25]. In addition, the stability of the enzyme has been evidenced by pH profile experiments (Figure 6b) since the enzyme retains more than 97% of activity from pH 6 to 9. The wide range of pH activity exhibited by the novel BcPPK2-III is a distinctive attribute that enhances its versatility to be included in several systems as ATP regeneration module. Enzymes from this family have been described to show activity across a wide pH range, including neutral, acidic and alkaline conditions [38,39].
3. Materials and Methods
3.1. Materials and General Procedures
Synthesis of the gene ppk2-III from Burkholderia cenocepacia, optimization of the nucleotide sequence, and cloning into the plasmid pET-28b (+) were performed by GenScript (Piscataway, NJ). Escherichia coli strain BL21 (DE3) was obtained from Promega Biotech Ibérica S.L. (Madrid, Spain). The GenEluteTM Plasmid Miniprep Kit from Sigma-Aldrich (Darmstadt, Germany) was used to purify the plasmid. Promega 1kb DNA Step Ladder Molecular Weight Marker and restriction enzymes were purchased from ThermoFisher Scientific Inc. (Waltham, MA). Kanamycin, DNase I, and other reagents were purchased from Sigma-Aldrich (Darmstadt, Germany). Isopropyl-β-d-thiogalactopyranoside (IPTG) was purchased from Applichem GmBH (Darmstadt, Germany). The acrylamide/bis-acrylamide 30% (29:1) solution used for protein analysis by SDS-PAGE was purchased from Bio-Rad (Hercules, CA). The Low Molecular Weight Calibration Kit from GE Healthcare was used. SDS-PAGE gels were run on a MiniProtean® Tetracell cuvette from Bio-Rad (Hercules, CA). Agarose gels were run in a RunOne™ Electrophoresis Cell cuvette from EmbiTec (San Diego, CA) using SYBR Safe from ThermoFisher Scientific (Waltham, MA). Analysis of the gels was performed using a Gene Flash Bio Imaging Photodocumenter from Syngene LTD (Bengaluru, Karnataka). Iminodiacetic acid agarose (IDA-agarose) was purchased from Agarose Bead Technologies (Miami, FL). Solvents were of analytical grade. DNA manipulation was performed according to standard procedures [40].
3.2. Cloning of ppk2-III Gene, Expression and Purification of Recombinant BcPPK2-III
The ppk2-III gene from B. cenocepacia was synthesized and optimized for expression in E. coli. This significantly improved the codon adaptation index (CAI) from 0.68 to 0.97. Considering that a CAI value of 1 occurs when the sequence is perfect to be expressed in the specific organism, the performed optimization significantly improves the chances of correct expression of the ppk2-III gene in E. coli. The percentage of guanines (G) and cytosines (C) could be decreased by 8%, which could significantly increase the stability of DNA and the secondary structure of the mRNA. Cloning of the optimized ppk2-III gene was performed in the expression plasmid pET-28b(+), which allows induction of protein overexpression with isopropyl-β-d-1-thiogalactopyranoside (IPTG). The protein is expressed fused to a 6-His tag at the N-terminus, allowing its one-step purification by ion metal affinity chromatography (IMAC) from cell-free extract (CFE). The plasmid, designated pET-28b(+)-ppk2-III, was then transformed into competent E. coli BL21 (DE3) cells. Restriction analysis of the plasmid after its purification revealed two bands (Figure S6): the band corresponding to the plasmid pET-28-b(+), with a size of 5368 bp, and a band of 279 bp corresponding to the expected size of the ppk2-III gene.
For expression of the BcPPK2-III protein, preinocula of recombinant colonies were prepared in 5 ml LB medium containing 5 μl kanamycin (50 μg/ml) and incubated overnight at 37 ºC with shaking (160 rpm). The culture was extended and incubated under the same conditions until the exponential phase (OD600nm = 0.5-0.7) was reached. At this point, protein expression was induced with IPTG at a final concentration of 1 mM. Orbital agitation was kept constant and the temperature was lowered to 30 °C to avoid the formation of inclusion bodies. The culture was then centrifuged at 4000 rpm for 20 min to recover the cells. The pellet was resuspended in Na2HPO4 buffer (50 mM, 300 mM NaCl, pH = 8.0). Cell disruption was performed by sonication (70% amplitude, 10 seconds pulse and 15 seconds pause). The lysed cells were separated by centrifugation of the mixture at 8000 rpm for 20 minutes. The supernatant obtained was treated with DNaseI (10 µg/ml cells) and MgCl2 (0.95 µg/ml cells) for 20 minutes on ice. Streptomycin (1% weight/volume) was then added and allowed to act for an additional 20 minutes. Finally, the cell-free extract (CFE) was obtained by centrifugation, which, after aliquoting, was frozen with liquid NO2 and stored at -80 °C.
Purification by divalent ion affinity chromatography (IMAC) was performed using a Co2+-IDA-agarose column. The resin was washed with milliQ H2O and equilibrated with 10 mM imidazole, 50 mM Na2HPO4 and 300 mM NaCl buffer, pH = 8.0. CFE was added to the resin at a ratio of 1:1 (v/v). To remove proteins non-specifically bound to the resin, the column was washed with 2 volumes of the same buffer. The recombinant protein was then eluted with 1 volume of the same buffer containing 2.0 M imidazole. The eluates were dialyzed to remove the imidazole on 30 kDa cut-off Vivaspin centricones. Finally, the enzyme was kept in Tris-hydroxymethyl-aminomethane (Tris-HCl) buffer (50 mM pH = 8.0 at 4 ºC). The protein concentration in the purification fractions was quantified by Ab280nm using the theoretical size of the protein (31.92 kDa) and the theoretical molar extinction coefficient (ε) determined by the content of tryptophan (W), tyrosine (Y), and cysteine (C) and calculated using Equation (1):
ε = (nW × 5500) + (nY ×1490) + (nC ×125)
Expression and successive purification steps were monitored by SDS-PAGE (Figure S2) at 11% in the resolving gel. Quantification of gels and protein purity was performed by densitometric analysis (GeneTools 3.07).
3.3. Identification, Modeling and Sequence Analysis of BcPPK2-III
Peptide mass fingerprinting was performed at the Proteomics Unit of the Complutense University of Madrid. Secondary structural motifs of the protein were identified by the theoretical structural analysis performed in the PSIPRED Protein Analysis Workbench [41]. 3D structure modeling without template was performed using the ColabFold open-source software [33,34].
3.4. Molecular Docking Modelling of the Enzyme-Substrate Complexes
The docking study was conducted using Maestro (version 13.2.128) for molecular modeling and visualization, and MMshare (version 5.8.128, Release 2022-2) for molecular mechanics. The protein structure was prepared using Maestro. Ligand structures were sketched using Chemdraw and subsequently prepared using Maestro’s ligand preparation tools. This preparation involved generating 3D coordinates, optimizing the ligand geometry, and assigning appropriate force field parameters. The ligand structures were energy minimized using the OPLS3e force field within Maestro to ensure accurate docking results. Docking simulations were performed using the Glide module within Maestro. The protein’s active site was defined by setting a grid box around the binding pocket, and the ligands were docked into this grid to explore potential binding modes. Default Glide settings, including standard precision (SP) mode, were used for the docking simulations. Each ligand was docked in multiple poses, and the results were analyzed to identify binding modes with favorable docking scores. The top-ranked binding poses were selected based on their docking scores and interactions with the active site residues. Binding interactions and the best docking poses were visualized and analyzed using Maestro’s visualization tools to assess the quality of ligand binding.
3.5. HPLC Analysis of the Enzymatic Activity of BcPPK2-III
Analysis of the enzymatic activity of BcPPK2-III in all cases was performed by reverse high-performance liquid chromatography (RHPLC) with an Agilent 1260 Infinity II equipped with an InfinityLab Poroshell 120 EC-18 column (150 mm × 4.6 mm, 4 μm) and photodiode array (PDA). The injections were performed by an autosampler and 3 μL of enzyme reaction were injected. The solvent system used was solvent (A): 0.1% (v/v) trifluoroacetic acid (TFA) in H2O and solvent (B): 0.05% (v/v) TFA in CH3CN, flow rate 1 mL min−1, detection at 260 nm and column temperature at 30 °C. Samples were analyzed using a gradient elution of 100% A over 4 min and to 0 to 10% of B over 4 min. Commercial standards of ATP, ADP, and AMP with retention times of 2.8 min, 3.5 min and 7.3 min, respectively, were used to identify the different nucleotides present in the reaction mixture (Figure S7).
3.5. Enzyme Activity Assay, Metal Ion Preference and Reaction Progress
Unless stated otherwise, enzyme activity assays were performed using 100 mM Tris-HCl buffer pH= 7.0, polyP (10 mM) with a chain length of 12 units as the donor of phosphate groups, AMP or ADP as substrates with a concentration of 2 mM and MgSO4 (10 mM) as the metal ion cofactor. Reactions were initiated by the addition of BcPPK2-III (10µg/ml) and maintained at 30 °C with orbital shaking (700 rpm). BcPPK2-III reaction progress was monitored by RHPLC each 10 minutes for the first 6h and after 24h. Additionally, 10 mM of polyP were supplemented to the same reaction and measured after 48h.
A study of the enzymatic activity of BcPPK2-III was performed using the standard conditions and introducing various metal ions (Ca2+, Co2+, Mg2+ and Mn2+) with a final concentration of 10 mM in the reaction.
3.6. Enzyme Stability
Thermostability and pH profile assays were performed with a final enzyme concentration of 10 µg/ml. The temperature gradient scale ranging from 30 to 70 °C was established as follows: 30.0, 31.6, 34.6, 39.5, 45.3, 49.6, 52.8, 55, 56.1, 57.9, 60.7, 64, 67, 68.9, and 70 °C. This gradient profile was achieved using a thermocycler (Bio-rad T100 Thermal Cycler). After 10 min of incubation, BcPPK2-III samples were removed and chilled out on ice for 10 min and incubated further at room temperature for 10 min. Finally, samples of 20 μl were added to 180 μl volume of reaction mixture containing 100 mM Tris-HCl buffer, pH= 7.0, polyP (10 mM), AMP (2 mM) and MgSO4 (10 mM).
The optimum pH activity was determined using 100mM Britton-Robinson buffer at different pH values (2.0, 3.0, 4.0, 5.0, 6.0, 7.0, 8.0, 9.0, 10.0, 11.0 and 12.0) containing the reaction mixture with the aforementioned concentrations of AMP, polyP and MgSO4.
3.7. Scanning Transmission Electron Microscopy
Transmission electron microscopy images were recorded with a field emission scanning electron microscope (FESEM) Hitachi SU-8000 operated at 30 kV in transmitted electron imaging mode (sTEM) equipped with a charge-coupled device (CCD) camera. Briefly, for sample preparation, one drop of reaction solution in water (∼5–10 μL) was left to dry over ultrathin carbon type-A film supported on a 400 mesh copper grid (3 mm in diameter) (from Ted Pella, Inc.).
4. Conclusions
It was experimentally demonstrated that the enzyme encoded by the ppk2-III gene is a polyphosphate kinase of family 2, subclass III which is capable of produce ATP from AMP. Sequence analysis confirmed the presence of conserved motifs, including the Walker A and B motifs, characteristic of PPK2 enzymes. Structural characterization through 3D modeling, molecular docking and peptide mass fingerprinting verified the identity and integrity of the recombinant protein.
The biochemical characterization of BcPPK2-III revealed that the enzyme is active with several divalent cations, exhibiting substantial activity with Mg2+, Mn2+, and Co2+, and a distinct ADP production preference with Ca2+. The enzyme achieved a 70% conversion of ATP from AMP after 24 hours, with a total turnover number of 334,000. The reaction mechanism was determined to proceed through a stepwise phosphorylation process, forming ADP as an intermediate before generating ATP.
Enzyme stability tests showed that BcPPK2-III retains full activity at temperatures between 30-58 °C, with a T50 of 62.2 °C, indicating high thermostability. The enzyme also maintained more than 97% of its activity across a pH range of 6-9, with optimal activity at pH 9.0. These two properties are very valuable from a biocatalysis point of view, as in many processes —such as ATP regeneration— it is necessary to combine several enzymes with different activity optima or to work under very demanding conditions in order to adapt the process to an industrial scale.
These findings suggest that BcPPK2-III is a robust and versatile enzyme with significant potential for use in ATP regeneration, reducing costs and improving the efficiency of biocatalytic processes that require stoichiometric amounts of ATP. The enzyme’s ability to function with multiple metal cofactors and its stability under various conditions make it a valuable candidate for industrial applications.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Figures S1-S7.
Author Contributions
Conceptualization, D.T.M. and E.G.-J.; methodology, L.A., and D.T.M.; formal analysis, L.A., D.T.M., I.S.-M and E.G.-J.; investigation, L.A. and D.T.M.; writing—original draft preparation, E.G.-J; writing—review and editing, D.T.M., I.S.-M. and J.R.; visualization, L.A., D.T.M. and E.G.-J.; supervision, D.T.M. and E.G.-J.; funding acquisition, J.R. and E.G.-J.; project administration, E.G.-J. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by MICIN/AEI/10.13039/501100011033 and by “ERDF A way of making Europe”, grant number PID2019-105337RB-C21.
Data Availability Statement
The data that support the findings of this study are available from the corresponding authors upon reasonable request.
Acknowledgments
The authors thank Dr. Miguel Alcalde (ICP-CSIC) for the facilities given to complete this work. We gratefully acknowledge Dr. Olga García from ICTP-CSIC for her assistance with obtaining the sTEM images.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Hanson, R.W. The Role of ATP in Metabolism. Biochem. Educ. 1989, 17, 86–92. [CrossRef]
- Richter, M. Functional Diversity of Organic Molecule Enzyme Cofactors. Nat. Prod. Rep. 2013, 30, 1324. [CrossRef]
- Fischer, J.D.; Holliday, G.L.; Rahman, S.A.; Thornton, J.M. The Structures and Physicochemical Properties of Organic Cofactors in Biocatalysis. J. Mol. Biol. 2010, 403, 803–824. [CrossRef]
- Wohlgemuth, R.; Liese, A.; Streit, W. Biocatalytic Phosphorylations of Metabolites: Past, Present, and Future. Trends Biotechnol. 2017, 35, 452–465. [CrossRef]
- Mueller, J.W.; Shafqat, N. Adenosine-5′-Phosphosulfate – a Multifaceted Modulator of Bifunctional 3′-Phospho-Adenosine-5′-Phosphosulfate Synthases and Related Enzymes. FEBS J. 2013, 280, 3050–3057. [CrossRef]
- Abu, R.; Woodley, J.M. Application of Enzyme Coupling Reactions to Shift Thermodynamically Limited Biocatalytic Reactions. ChemCatChem 2015, 7, 3094–3105. [CrossRef]
- Fehlau, M.; Kaspar, F.; Hellendahl, K.F.; Schollmeyer, J.; Neubauer, P.; Wagner, A. Modular Enzymatic Cascade Synthesis of Nucleotides Using a (d)ATP Regeneration System. Front. Bioeng. Biotechnol. 2020, 8, 1–10. [CrossRef]
- Iturrate, L.; García-Junceda, E. DHAP-Dependent Aldolases in the Core of Multistep Processes. In Multi-Step Enzyme Catalysis: Biotransformations and Chemoenzymatic Synthesis; García-Junceda, E., Ed.; Wiley-VCH Verlag GMBH & Co. KGaA: Weinheim, Germany, 2008; pp. 61–81.
- Andexer, J.N.; Richter, M. Emerging Enzymes for ATP Regeneration in Biocatalytic Processes. ChemBioChem 2015, 16, 380–386. [CrossRef]
- Zhao, H.; van der Donk, W.A. Regeneration of Cofactors for Use in Biocatalysis. Curr. Opin. Biotechnol. 2003, 14, 583–589. [CrossRef]
- Achbergerová, L.; Nahálka, J. Polyphosphate - an Ancient Energy Source and Active Metabolic Regulator. Microb. Cell Fact. 2011, 10, 63. [CrossRef]
- Kornberg, A.; Rao, N.N.; Ault-Riché, D. Inorganic Polyphosphate: A Molecule of Many Functions. Annu. Rev. Biochem. 1999, 68, 89–125. [CrossRef]
- Rao, N.N.; Gómez-García, M.R.; Kornberg, A. Inorganic Polyphosphate: Essential for Growth and Survival. Annu. Rev. Biochem. 2009, 78, 605–647. [CrossRef]
- Kornberg, A. Inorganic Polyphosphate: Toward Making a Forgotten Polymer Unforgettable. J. Bacteriol. 1995, 177, 491–496. [CrossRef]
- Bordes, I.; García-Junceda, E.; Sánchez-Moreno, I.; Castillo, R.; Moliner, V. Computational Study of the Phosphoryl Donor Activity of Dihydroxyacetone Kinase from ATP to Inorganic Polyphosphate. Int. J. Quantum Chem. 2018, 118, e25520. [CrossRef]
- Iwamoto, S.; Motomura, K.; Shinoda, Y.; Urata, M.; Kato, J.; Takiguchi, N.; Ohtake, H.; Hirota, R.; Kuroda, A. Use of an Escherichia Coli Recombinant Producing Thermostable Polyphosphate Kinase as an ATP Regenerator To Produce Fructose 1,6-Diphosphate. Appl. Environ. Microbiol. 2007, 73, 5676–5678. [CrossRef]
- Sánchez-Moreno, I.; Bordes, I.; Castillo, R.; Ruiz-Pernía, J.; Moliner, V.; García-Junceda, E. Tuning the Phosphoryl Donor Specificity of Dihydroxyacetone Kinase from ATP to Inorganic Polyphosphate. An Insight from Computational Studies. Int. J. Mol. Sci. 2015, 16, 27835–27849. [CrossRef]
- Tavanti, M.; Hosford, J.; Lloyd, R.C.; Brown, M.J.B. ATP Regeneration by a Single Polyphosphate Kinase Powers Multigram-Scale Aldehyde Synthesis in Vitro. Green Chem. 2021, 23, 828–837. [CrossRef]
- Mordhorst, S.; Maurer, A.; Popadić, D.; Brech, J.; Andexer, J.N. A Flexible Polyphosphate-Driven Regeneration System for Coenzyme A Dependent Catalysis. ChemCatChem 2017, 9, 4164–4168. [CrossRef]
- Xu, R.; Wang, Y.; Huang, H.; Jin, X.; Li, J.; Du, G.; Kang, Z. Closed-Loop System Driven by ADP Phosphorylation from Pyrophosphate Affords Equimolar Transformation of ATP to 3′-Phosphoadenosine-5′-Phosphosulfate. ACS Catal. 2021, 11, 10405–10415. [CrossRef]
- Monterrey, D.T.; Benito-Arenas, R.; Revuelta, J.; García-Junceda, E. Design of a Biocatalytic Cascade for the Enzymatic Sulfation of Unsulfated Chondroitin with in Situ Generation of PAPS. Front. Bioeng. Biotechnol. 2023, 11, 1–10. [CrossRef]
- Datta, P.; Fu, L.; He, W.; Koffas, M.A.G.; Dordick, J.S.; Linhardt, R.J. Expression of Enzymes for 3′-Phosphoadenosine-5′-Phosphosulfate (PAPS) Biosynthesis and Their Preparation for PAPS Synthesis and Regeneration. Appl. Microbiol. Biotechnol. 2020, 104, 7067–7078. [CrossRef]
- Mordhorst, S.; Singh, J.; Mohr, M.K.F.; Hinkelmann, R.; Keppler, M.; Jessen, H.J.; Andexer, J.N. Several Polyphosphate Kinase 2 Enzymes Catalyse the Production of Adenosine 5′-Polyphosphates. ChemBioChem 2019, 20, 1019–1022. [CrossRef]
- Ogawa, M.; Uyeda, A.; Harada, K.; Sato, Y.; Kato, Y.; Watanabe, H.; Honda, K.; Matsuura, T. Class III Polyphosphate Kinase 2 Enzymes Catalyze the Pyrophosphorylation of Adenosine-5′-Monophosphate. ChemBioChem 2019, 20, 2961–2967. [CrossRef]
- Motomura, K.; Hirota, R.; Okada, M.; Ikeda, T.; Ishida, T.; Kuroda, A. A New Subfamily of Polyphosphate Kinase 2 (Class III PPK2) Catalyzes Both Nucleoside Monophosphate Phosphorylation and Nucleoside Diphosphate Phosphorylation. Appl. Environ. Microbiol. 2014, 80, 2602–2608. [CrossRef]
- Moriarty, T.F.; Mullan, A.; McGrath, J.W.; Quinn, J.P.; Elborn, J.S.; Tunney, M.M. Effect of Reduced PH on Inorganic Polyphosphate Accumulation by Burkholderia Cepacia Complex Isolates. Lett. Appl. Microbiol. 2006, 42, 617–623. [CrossRef]
- Leipe, D.D.; Koonin, E. V.; Aravind, L. Evolution and Classification of P-Loop Kinases and Related Proteins. J. Mol. Biol. 2003, 333, 781–815. [CrossRef]
- Nocek, B.; Kochinyan, S.; Proudfoot, M.; Brown, G.; Evdokimova, E.; Osipiuk, J.; Edwards, A.M.; Savchenko, A.; Joachimiak, A.; Yakunin, A.F. Polyphosphate-Dependent Synthesis of ATP and ADP by the Family-2 Polyphosphate Kinases in Bacteria. Proc. Natl. Acad. Sci. 2008, 105, 17730–17735. [CrossRef]
- Altschul, S.F.; Wootton, J.C.; Gertz, E.M.; Agarwala, R.; Morgulis, A.; Schäffer, A.A.; Yu, Y. Protein Database Searches Using Compositionally Adjusted Substitution Matrices. FEBS J. 2005, 272, 5101–5109. [CrossRef]
- Altschul, S.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A New Generation of Protein Database Search Programs. Nucleic Acids Res. 1997, 25, 3389–3402. [CrossRef]
- Nocek, B.P.; Khusnutdinova, A.N.; Ruszkowski, M.; Flick, R.; Burda, M.; Batyrova, K.; Brown, G.; Mucha, A.; Joachimiak, A.; Berlicki, Ł.; et al. Structural Insights into Substrate Selectivity and Activity of Bacterial Polyphosphate Kinases. ACS Catal. 2018, 8, 10746–10760. [CrossRef]
- Krogh, A.; Larsson, B.; von Heijne, G.; Sonnhammer, E.L.. Predicting Transmembrane Protein Topology with a Hidden Markov Model: Application to Complete Genomes11Edited by F. Cohen. J. Mol. Biol. 2001, 305, 567–580. [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly Accurate Protein Structure Prediction with AlphaFold. Nature 2021, 596, 583–589. [CrossRef]
- Mirdita, M.; Schütze, K.; Moriwaki, Y.; Heo, L.; Ovchinnikov, S.; Steinegger, M. ColabFold: Making Protein Folding Accessible to All. Nat. Methods 2022, 19, 679–682. [CrossRef]
- Hildenbrand, J.C.; Teleki, A.; Jendrossek, D. A Universal Polyphosphate Kinase: PPK2c of Ralstonia Eutropha Accepts Purine and Pyrimidine Nucleotides Including Uridine Diphosphate. Appl. Microbiol. Biotechnol. 2020, 6659–6667. [CrossRef]
- Lee, S.J.; Jang, H.; Lee, D.N. Recent Advances in Nanoflowers: Compositional and Structural Diversification for Potential Applications. Nanoscale Adv. 2023, 5, 5165–5213. [CrossRef]
- Mordhorst, S.; Singh, J.; Mohr, M.K.F.; Hinkelmann, R.; Keppler, M.; Jessen, H.J.; Andexer, J.N. Several Polyphosphate Kinase 2 Enzymes Catalyse the Production of Adenosine 5′-Polyphosphates. ChemBioChem 2019, 20, 1019–1022. [CrossRef]
- Batten, L.E.; Parnell, A.E.; Wells, N.J.; Murch, A.L.; Oyston, P.C.F.; Roach, P.L. Biochemical and Structural Characterization of Polyphosphate Kinase 2 from the Intracellular Pathogen Francisella Tularensis. Biosci. Rep. 2016, 36. [CrossRef]
- Zhang, X.; Cui, X.; Li, Z. Characterization of Two Polyphosphate Kinase 2 Enzymes Used for ATP Synthesis. Appl. Biochem. Biotechnol. 2020, 191, 881–892. [CrossRef]
- Sambrook, J.; Fritsch, E.F.; Maniatis, T. Molecular Cloning : A Laboratory Manual; 2nd ed.; Cold Spring Harbour: New York, NY, 1989; ISBN 0-87969-309-6.
- Buchan, D.W.A.; Jones, D.T. The PSIPRED Protein Analysis Workbench: 20 Years On. Nucleic Acids Res. 2019, 47, W402–W407. [CrossRef]
Figure 1.
Reactions catalysed by the different PPK family 2 (PPK2) subclasses.
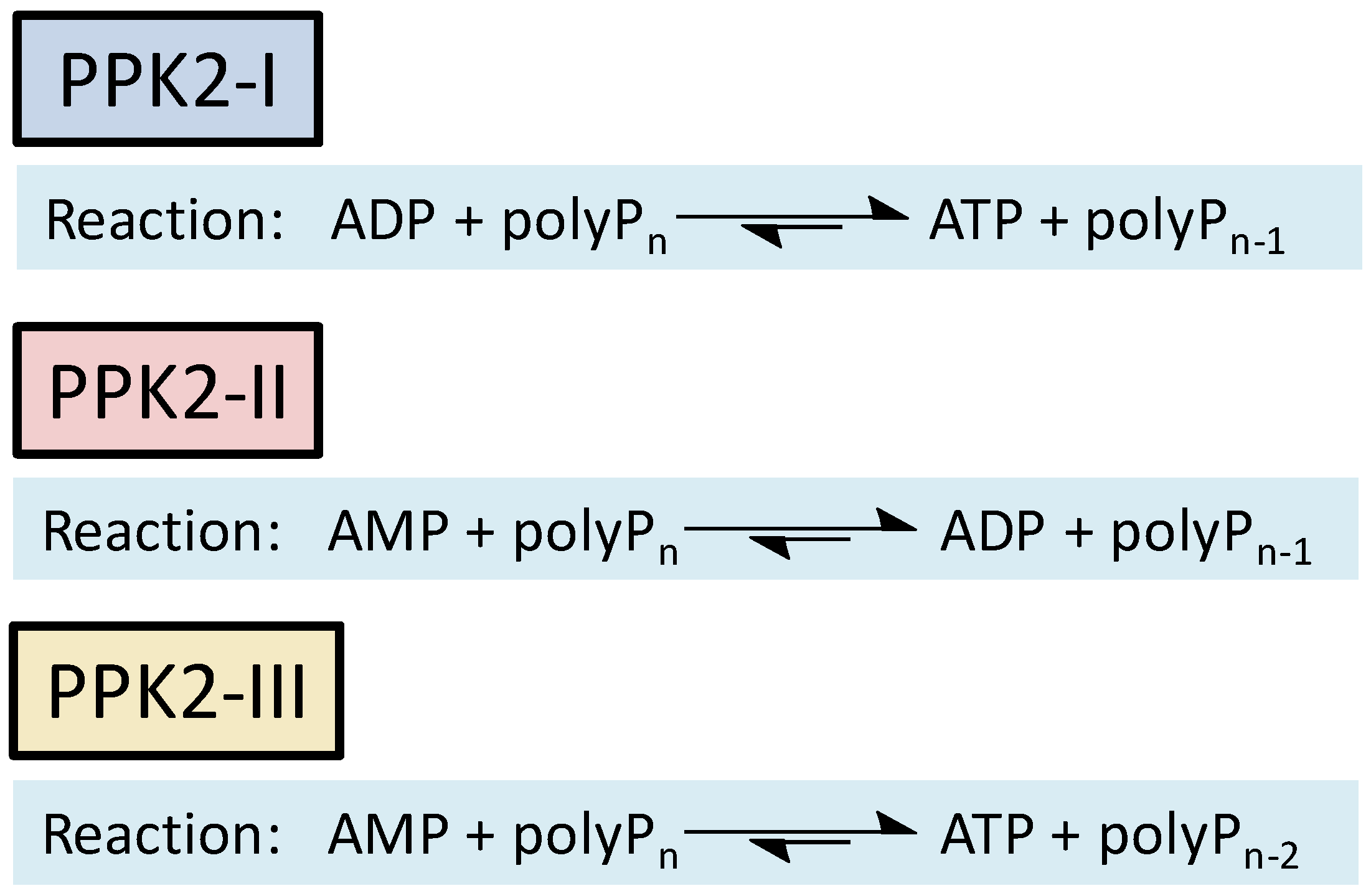
Figure 2.
(a) Model of the 3D structure of the recombinant protein BcPPK2-III. The conserved motifs are shown in colors: Walker A is shown in red, Walker B in blue and the lid module in violet. The conserved amino acids Q74 (yellow) and E137 (green) are displayed in sticks; (b) and (c) detail of the spatial arrangement of the conserved motifs, showing that the active center is located bellow the lid.
Figure 2.
(a) Model of the 3D structure of the recombinant protein BcPPK2-III. The conserved motifs are shown in colors: Walker A is shown in red, Walker B in blue and the lid module in violet. The conserved amino acids Q74 (yellow) and E137 (green) are displayed in sticks; (b) and (c) detail of the spatial arrangement of the conserved motifs, showing that the active center is located bellow the lid.
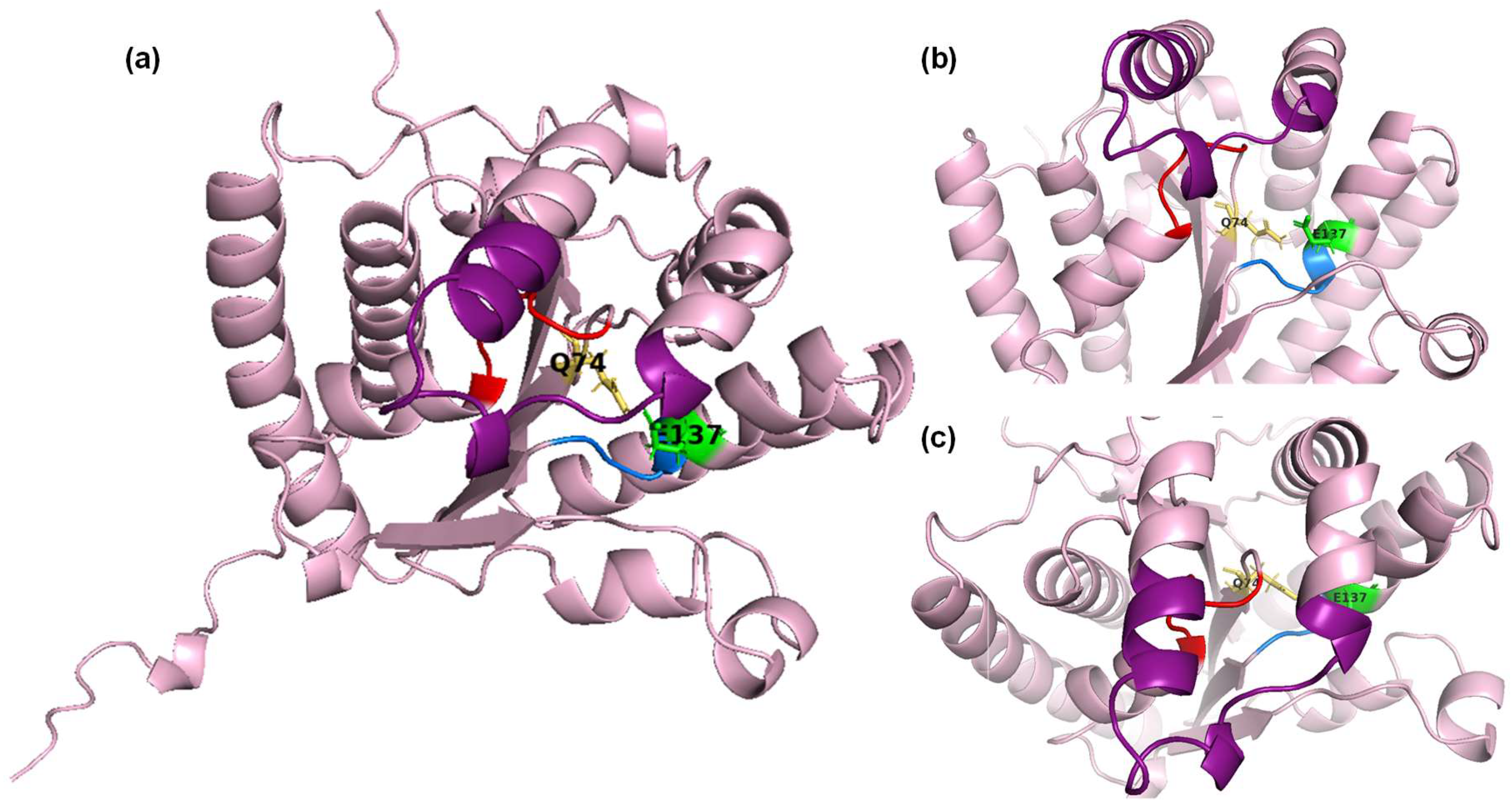
Figure 3.
Structural representation of the enzyme with three different substrates bound in the active site. (a) The enzyme bound with AMP, (b) ADP, and (c) with ATP. Each panel includes a 3D structure of the enzyme-substrate complex and a 2D representation showing the residues interacting with the compounds. In the 3D structures, key residues are highlighted: Walker A motif residues in red, Walker B motif residues in blue, glutamic acid (E) in green, and the lid domain residues in violet.
Figure 3.
Structural representation of the enzyme with three different substrates bound in the active site. (a) The enzyme bound with AMP, (b) ADP, and (c) with ATP. Each panel includes a 3D structure of the enzyme-substrate complex and a 2D representation showing the residues interacting with the compounds. In the 3D structures, key residues are highlighted: Walker A motif residues in red, Walker B motif residues in blue, glutamic acid (E) in green, and the lid domain residues in violet.
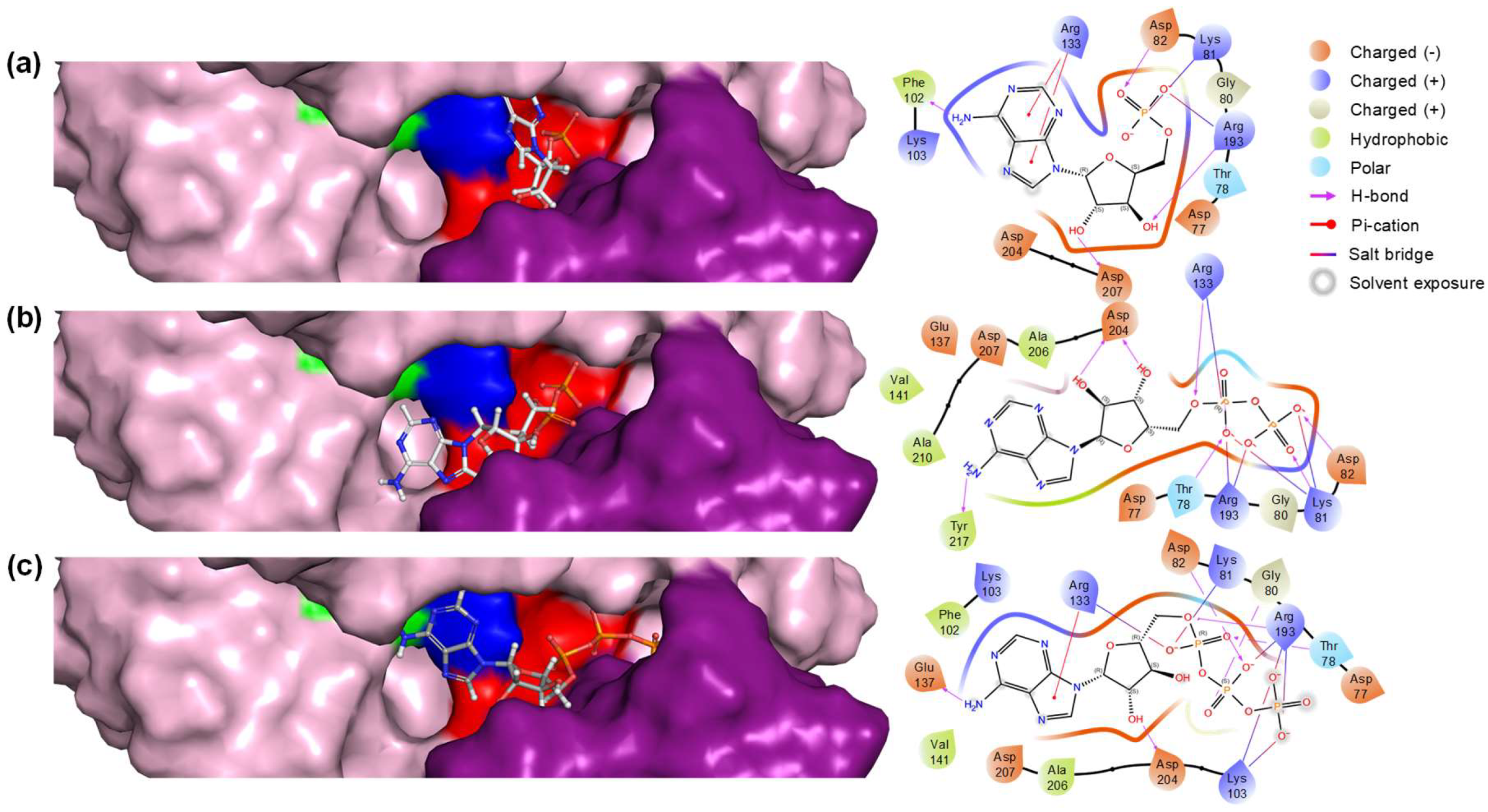
Figure 4.
Effect of metal ions in enzyme activity. Reaction after 24h using the same concentration of polyP12 (10 mM) and same concentration of divalent cation (10 mM), for Ca2+, Co2+, Mg2+, Mn2+. Concentration of produced ATP (blue) and ADP (green) is represented.
Figure 4.
Effect of metal ions in enzyme activity. Reaction after 24h using the same concentration of polyP12 (10 mM) and same concentration of divalent cation (10 mM), for Ca2+, Co2+, Mg2+, Mn2+. Concentration of produced ATP (blue) and ADP (green) is represented.
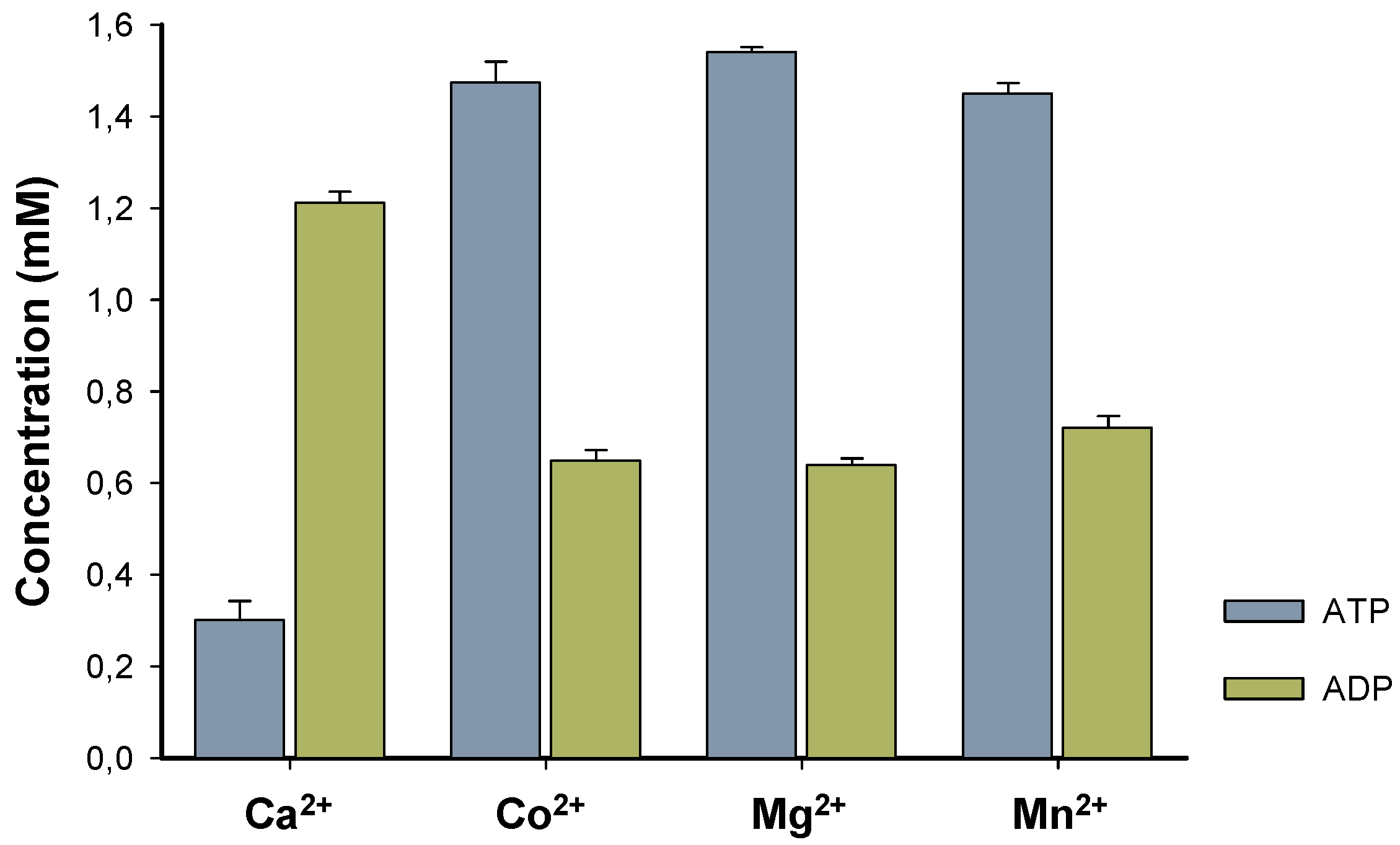
Figure 5.
Reaction time-course, showing the concentration of AMP (yellow triangles), ADP (green circles) and ATP (blue circles). In both reactions the starting material concentration was 2mM, Mg2+ was used as divalent cation and PolyP12 (10 mM) as a phosphate donor and the experiments were perfomed in triplicate. (a) Starting material of he reaction was AMP and red arrow indicates the supplementation of the reaction with PolyP12 (10 mM). (b) Reaction was carried out with ADP as the initial substrate.
Figure 5.
Reaction time-course, showing the concentration of AMP (yellow triangles), ADP (green circles) and ATP (blue circles). In both reactions the starting material concentration was 2mM, Mg2+ was used as divalent cation and PolyP12 (10 mM) as a phosphate donor and the experiments were perfomed in triplicate. (a) Starting material of he reaction was AMP and red arrow indicates the supplementation of the reaction with PolyP12 (10 mM). (b) Reaction was carried out with ADP as the initial substrate.
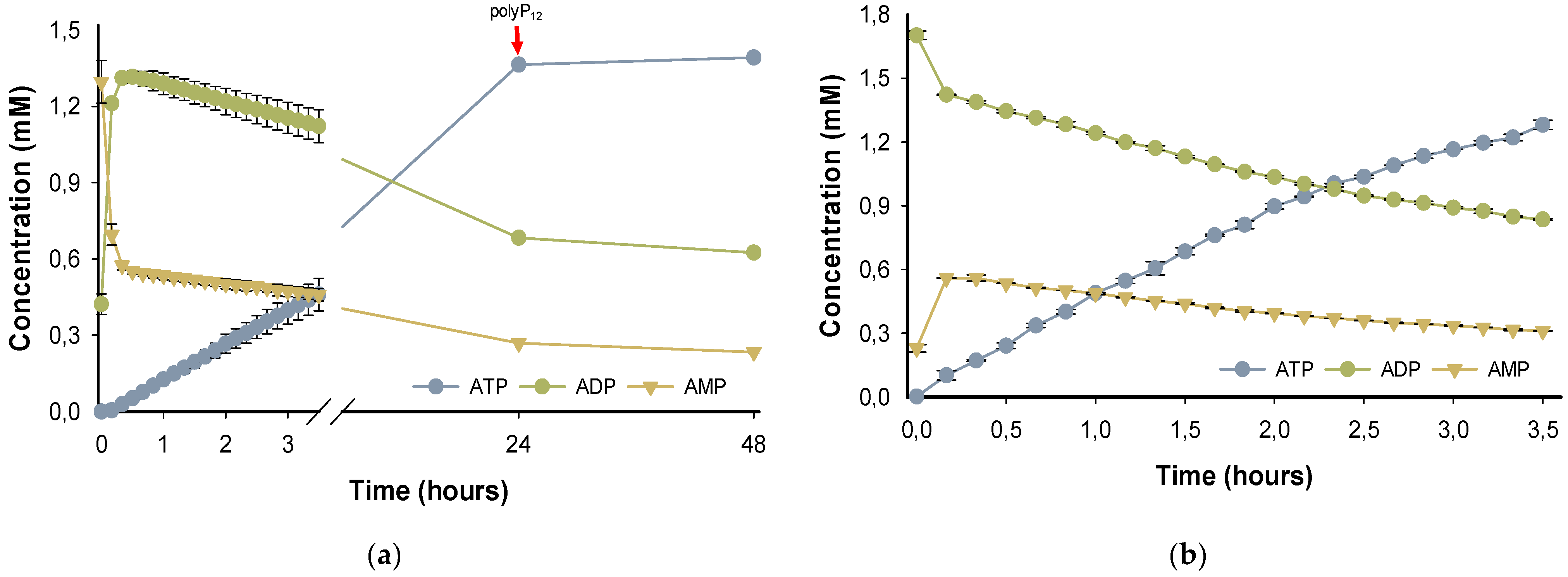
Figure 6.
(a) Kinetic thermostability (T50) of BcPPK2-III (b) pH profile. Values are presented as the mean ± standard deviation of three independent experiments.
Figure 6.
(a) Kinetic thermostability (T50) of BcPPK2-III (b) pH profile. Values are presented as the mean ± standard deviation of three independent experiments.
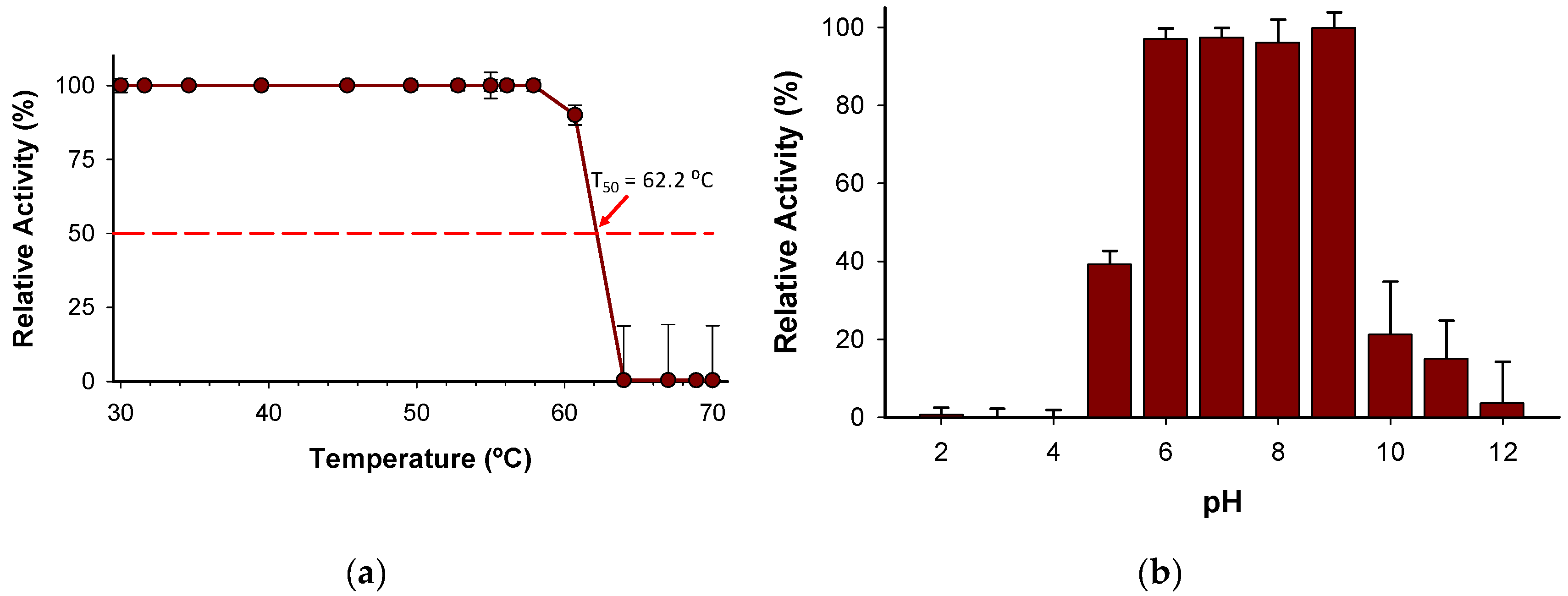
Table 1.
Molecular docking results.
| Compound | Interacting residues | Docking score (kcal/mol) |
Glide Emodel |
|---|---|---|---|
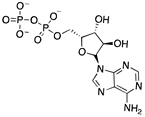
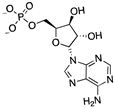 |
Lys81, Asp82, Phe102, Arg133, Arg193, Asp207 | -7.284 | -98.475 |
 |
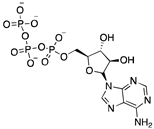
Thr78, Lys81, Asp82, Arg133, Arg193, Asp204, Tyr217 | -7.340 | -122.995 |
 |
Thr78, Gly80, Lys81, Asp82, Arg133, Arg193, Lys202, Asp204 | -6.464 | -138.999 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



