
Submitted:
05 August 2024
Posted:
07 August 2024
You are already at the latest version
Abstract
Hidradenitis suppurativa (HS) is a chronic skin disease characterized by painful, recurrent abscesses, nodules, and scarring, primarily in skin folds. The exact causes of HS are multifactorial, involving genetic, hormonal, and environmental factors. It is associated with systemic diseases like metabolic syndrome and inflammatory bowel disease. Genetic studies have identified mutations in the γ-secretase complex, which affects Notch signaling pathways critical for skin cell regulation. Despite its high heritability, most HS reported cases do not follow a simple genetic pattern. In this article, we performed a whole-exome sequencing (WES) on a cohort of 100 individuals with HS and we provide a comprehensive review of the variants known to be described or associated with HS, referencing 91 of them in the γ-secretase and 78 in other genes involved in the Notch pathway, keratinization, or immune response. From this new genetic analysis, we add ten new variants to these catalogs.
Keywords:
hidradenitis suppurativa
; whole-exome sequencing
; γ-secretase
; nicastrin
; notch pathway
; inflammation
1. Introduction
Hidradenitis suppurativa (HS) is a debilitating dermatological disorder that affects approximately 1% of the global population. It is characterized by the formation of large suppurative abscesses, sinuses, nodules, and scars in intertriginous areas, including the axillae, groin, and/or anogenital regions [1]. Although the pathogenesis of HS is multifactorial and poorly understood, its pathomechanism is very likely linked to aberrant keratinization and autoinflammation. Consequently, HS is recognized as an autoinflammatory keratinization disease [2]. It remains uncertain whether autoinflammatory events precede or follow the hyperkeratotic changes in the hair follicle epithelia, although follicular occlusion is typically considered the primary event [3]. Several factors, including genetics, microbiota, and environmental factors such as obesity and smoking, may act as triggers or risk factors for HS. Many patients also suffer from syndromic forms of HS or have comorbidities [4,5,6,7,8], such as inflammatory bowel disease (IBD) like ulcerative colitis or Crohn’s disease (Supp. Figure S1).
Only a small proportion of patients with familial or syndromic HS reveal a monogenic etiology (5%), despite the estimated high heritability of HS (77-80%) [9]. Mutations in genes encoding γ-secretase, an intramembrane multisubunit protease complex, have been identified in several members of Chinese families with severe HS [10]. The γ-secretase complex comprises four protein subunits: the anterior pharynx-defective protein APH-1A or APH-1B (APH1A/B genes), the nicastrin protein NCSTN (NCSTN gene), the presenilin protein PS-1 or PS-2 (PSEN1/2 genes), and the presenilin enhancer PEN-2 (PSENEN gene). It has been established that γ-secretase proteolyzes the transmembrane domain of more than 100 substrates, including those derived from the amyloid precursor protein (APP) and the Notch family of cell surface receptors [11]. To date, 91 mutations have been described in the γ-secretase complex, with more than half located in the nicastrin subunit. Despite several articles implicating γ-secretase variants in HS, evidence for direct causal mechanisms is lacking [12]. Given the pivotal functions of Notch in maintaining epidermal and follicular homeostasis as well as regulating inflammatory processes, Notch deregulation has been proposed to underlie the molecular basis of HS observed in patients with pathogenic variants in γ-secretase protein-coding genes [13].
Furthermore, many other genes have been identified as potentially implicated in HS: AIM2 [14], DCD [15], DEFB126 [16], FGFR2 [17,18], GJB2 [19,20,21,22,23,24], IL1RN [25], IRF2BP2 [26], KDF1 [27,28], KRT6A [29], KRT17 [30,31], MEFV [17,24,25,32,33,34], NBPF12 [16], NF1P6 [16], NLRC4 [24], NLRP3 [25], NOD2 [17,24,25,33], NOTCH1 [35], NOTCH3 [16,36], NOTCH2NLA [16], OCRL [37], OTULIN [24], POFUT1 [38,39], POGLUT1 [40], PSTPIP1 [17,24,25,32,36,41,42,43,44,45,46,47,48], PSMB8 [25], RORC [16], SLC46A2 [16], TCIRG1 [16], and WDR1 [24]. Variants in these genes are often found in individuals with syndromic forms of HS. However, the precise contribution of each gene in HS development is difficult to determine.
Although no fully penetrant variants causing multigenerational disease have been identified, the high heritability rates suggest that sporadic forms of HS have a significant genetic component contributing to their causation [49]. However, the precise nature of the genetic variants causing common non-syndromic forms of HS is still unclear. Here, we performed a whole-exome sequencing (WES) on a cohort of 100 individuals with HS; as controls, we used a cohort of 100 individuals without HS from the French Exome consortium [50,51], examining genetic variants in genes described in the literature as being associated with the disease. We identified seven variants in the NCSTN gene, three of which were not described in dbSNP but were found only in articles. We pinpointed eight other variants (including two new ones) in the other γ-secretase complex genes. Finally, we found 21 variants in our HS individuals, described in the literature affecting other genes than γ-secretase genes, of which only three are of real interest (absent in our controls and with a high deleteriousness score), to which we added two noteworthy new variants in the SLC46A2 and NOTCH3 genes.
2. Results
2.1. Description of the Cohort
We collect data from 100 patients with HS and gathered detailed clinical characteristics (Table 1). Age at enrollment was 34.0 ± 11.5 years; 64 were female (sex ratio 2:1). Twenty-four were obese (BMI ≥ 30), and more than 50% were smokers. Four families were included, and 28 patients reported having an affected first-degree relative. Hurley phenotypes, as described by van der Zee and Jemec [52], were categorized at enrollment, with the majority exhibiting the regular phenotype.
2.2. Variants Identified in Nicastrin (NCSTN) Gene
As most known variants identified in HS patients are located in NCSTN (~37%), we start analyzing SNVs in the NCSTN gene. We identified seven variants, three of which have not been described in dbSNP or gnomAD, and whose bioinformatic predictions indicate that they might have a major impact on the protein structure.
The first variant, p.Leu17SerfsTer30 (c.47dupG; GRCh38:1: 160343443), is a frameshift variant that truncates the protein very prematurely (Supp. Figure S2), and is expected to induce a nonsense-mediated decay (NMD) reaction to eliminate the aberrant transcript. This heterozygous variant is present in a 43-year-old woman with a family history of HS.
The second variant, p.Ala300TrpfsTer20 (c.896_897dupTG; GRCh38:1:160352104), is another frameshift variant that truncates the protein at position 320 (Figure 1b), simply called W300. This would result in a protein without its active site (in orange) and its transmembrane domain (in red), which prevents it from integrating into the cell membrane. It would therefore produce a non-functional protein or a γ-secretase haploinsufficiency, even though the mutation is heterozygous. Although PhyloP shows a score of -7.97, demonstrating rapid evolution of this site in mammals, this specific variant has a CADD Phred-like score of 34 (i.e., in the 0.04% most deleterious variants). It affects three HS individuals in the same family: a father (I:1) and his two daughters (II:1 & II:2).
The third and fourth variants are nonsense variants p.Arg429* (Figure 1c) (rs771414318 already described in a Japanese individual [53]) and p.Trp648* (Figure 1d) (not known in dbSNP or gnomAD), truncating the protein beyond the small lobe of the nicastrin. The transmembrane domain, like the previous two variants, would be lost. These variants are found in sporadic cases of HS affecting a 50-year-old man and a 39-year-old woman, respectively. As expected, their CADD Phred-like scores are very high (39 and 51, respectively), and these variants are in fairly conserved regions (PhyloP=7.04 & 4.42). It is important to note that the p.Trp648* variant has a SpliceAI score that is very close to 0.5, suggesting a potential gain of a splice acceptor site.
With a lower impact, two missense variants p.Glu77Asp (c.231G>C) and p.Asn417Tyr (c.1249A>T), and an intronic variant c.1180-5C>G, as already described in the literature, were also found [17,36,54]. The p.Glu77Asp variant (rs35603924) (Supp. Figure S2) is found in a 17-year-old woman with a Hurley stage III HS. Although very rare in the European population, this variant is not so rare in the African population (AF~6%) and does not seem, according to prediction tools, to have a strong impact on the protein. Unfortunately, we do not have information about the phototype of this young woman. The second missense variant p.Asn417Tyr (rs143039637), affects a 33-year-old woman who also has a Hurley stage III HS. This is a non-familial case. This variant is very rare and, according to SIFT and PolyPhen-2, appears to be “deleterious” and “possibly damaging”. Finally, the intronic variant c.1180-5C>G (rs7528638), although described in the literature [54], is common in the population at over 5%. Similarly, in our cohort, 7.07% of patients and 12% of controls carry this variant. The SpliceAI and Pangolin prediction tools have scores below 0.5, which does not indicate a high probability of alternative splicing induced by this variant.
2.3. Variants Identified in Others Genes Involved in γ-Secretase Complex
Approximately 20% of variants associated with HS and described in the literature affect other γ-secretase genes than the nicastrin one. Yet, we report four variants for PS-1, one for PS-2, and three for APH-1B (Supp. Figure S2), of which three, one and two variants respectively are known from dbSNP and gnomAD.
The most interesting variant is a c.554del(A) frameshift deletion (p.Lys185SerfsTer10, rs745918508) which causes a truncation of the last third of the APH-1B protein. It is observed in a 36-year-old woman with no family history. Although it remains very rare in the population (0.007%), it was also found in one of our control individuals. The CADD Phred-like score of 27.8 is very close to the Ensembl deleteriousness threshold of 30.
The two other variants in the APH1B gene are missense SNVs. The p.Thr27Ala (rs77834210) variant affects two unrelated individuals, one of whom is a phototype 6 male, which is consistent with the greater presence of this variant in the African population (1.05% vs. 0.33% in non-Finnish Europeans). The other p.Leu71Val variant affects only one woman. This second variant has a very high probability of being deleterious (SIFT=0.01; PolyPhen-2=0.998).
Of the four variants detected in the PSEN1 gene, we report a new one: p.Gln325Glu in a single individual, which probably has no particular effect (only the REVEL score exceeds its threshold of 0.5). The other three variants have already been described in the literature [36,54,55] but do not have a strong impact on the protein. These are the variants c.868+16G>T (rs165932), p.Glu318Gly (rs17125721) and c.1248+8T>C (rs362382). The first two are shared by more controls than patients in our cohort (82 vs. 80% and 5 vs. 4%), which is perfectly consistent with what is known from gnomAD: 58% and 1% of the world population. The last variant showed scores of 0 for SpliceAI and Pangolin, demonstrating a null probability of splice site alteration.
Finally, we detected a new variant in PSEN2:p.Gly34Ser which is not listed in the literature or databases and is only carried by an individual with no family history. According to the prediction tools, this variant would not be considered deleterious.
2.4. Comprehensive Catalog of γ-Secretase Variants
To provide a useful resource for the medical and scientific communities, we have listed all the variants affecting γ-secretase described in the literature in patients with HS with or without comorbidities. They are listed in Table 2 and Supplementary Table S1. To ensure the resource is of high quality and value, we have provided the positions of the variants on different reference genomes in relation to the reference transcript defined by the MANE project, the aim of which is to harmonize the annotations of genes and transcripts. We have identified numerous errors in recent reviews (e.g., Supp. Table S2) and have corrected them in this article. The systematic verification of all variants requires a certain level of expertise and is very time-consuming when the variants are poorly annotated in articles. To avoid future issues, we also provide the left-normalized .vcf file of all variants discussed in this article as supplementary data to aid future studies and reviews.
2.5. Variants Identified in Others Genes Described as Being Associated with HS
To present an exhaustive review of all known variants described in the literature with a potential impact or link to HS, we also analyzed the 78 known SNVs that are not related to γ-secretase. These genes include AIM2 [14], DCD [15], DEFB126 [16], FGFR2 [17,18], GJB2 [19,20,21,22,23,24], IL1RN [25], IRF2BP2 [26], KDF1 [27,28], KRT6A [29], KRT17 [30,31], MEFV [17,24,25,32,33,34], NBPF12 [16], NF1P6 [16], NLRC4 [24], NLRP3 [25], NOD2 [17,24,25,33], NOTCH1 [35], NOTCH3 [16,36], NOTCH2NLA [16], OCRL [37], OTULIN [24], POFUT1 [38,39], POGLUT1 [40], PSTPIP1 [17,24,25,32,36,41,42,43,44,45,46,47,48], PSMB8 [25], RORC [16], SLC46A2 [16], TCIRG1 [16], and WDR1 [24]. They are primarily involved in the Notch signaling pathway, immune response pathway (principally the inflammasome) and keratinization, as described by Jfri et al. (Supp. Figure S3). Note that we cannot compare the proportions of variants between those in the γ-secretase and those in other genes since many studies have only performed targeted exome sequencing and have not examined all of these genes.
We found 21 variants in our cohort in common with published studies (Supp. Table S1). Of these, eight variants had an allelic frequency > 2% and five variants had a frequency between 1 and 2%. Among the remaining eight variants, five missense variants had no scores indicating deleteriousness, while the last three variants were of particular interest: RORC:p.Arg10* (rs17582155) [16], GJB2:p.Glu114Gly (rs2274083) [19] and NOD2:p.Ala891Asp (rs104895452) [17]. Their allelic frequencies are 0.279, 0.044, and 0.545% in the general population, respectively.
The RORC p.Arg10* variant is a stop codong gain that truncates the protein from the 10th amino acid (CADD Phred-like score=36). This variant affects two unrelated HS individuals in our cohort, including the youngest daughter (II:2) in the family with the NCSTN W300 mutation. The father (I:1) and the other daughter (II:1) (Hurley II) do not carry this RORC variant, whereas the youngest daughter (II:2) has a more severe form of the disease (Hurley III). The GJB2 variant is a missense mutation with a CADD Phred-like score of 20.8, affecting a sporadic case of HS. The NOD2 variant is a missense mutation with a CADD Phred-like score of 25.5 and the most deleterious SIFT and PolyPhen-2 scores (0 and 1, respectively). Complementary analyses using missense3D and AlphaMissense describe this mutation as “neutral” and “likely benign” effect. This variant affects two unrelated individuals.
Additionally, we highlight two other high-impact variants among the 29 genes reported in the literature and do not affect the individuals in our control cohort. These are two frameshift variants: one affecting SLC46A2 (rs1841700210 - p.Ala326GlyfsTer133) and the other affecting NOTCH3 (rs749829137 - p.Cys43LeufsTer32). As expected, their CADD Phred-like scores are close to 30.
3. Discussion
We analyzed WES data from 100 HS patients, predominantly non-syndromic cases, with a focus on genes already reported to be mutated in HS. We identified 15 variants in the γ-secretase complex, including 12 with rare frequencies (<1%) in the general population. These 12 variants were found in 15 patients, including a family of three individuals affected by the W300 variant. Thus, 15% of the sporadic cases in our HS cohort have a variant in the γ-secretase complex with a predicted moderate to strong effect.
A detailed literature review allowed us to compile a catalogue of potentially HS-causing variants in the γ-secretase complex. Table 2 lists 91 and eight new variants, over two-third of which are predicted to have a significant impact on γ-secretase function (e.g., nonsense mutations, frameshifts leading to premature truncation, alternative splicing or elongation). Approximately 40% of these variants are documented in gnomAD (v. 4.1-exome), an additional 11% are known in dbSNP, and the remaining 49% of variants are described only in articles. Among these, only four variants have an alternative allele frequency greater than 1% in the general population and non-Finnish Europeans: c.436+129A>G (NCSTN), c.1180-5C>G (NCSTN), c.868+16G>T (PSEN1), and c.953A>G (PSEN1; p.E318G), with frequencies of 3.6, 5.1, 57.6, and 1.8%, respectively. These variants have been previously reported [42,54,55] with noted frequencies in the general population or in intrafamilial non-HS controls. Functional studies of some of these variants, such as p.Val75Ile [29], p.Asp185Asn [30], p.Pro211Arg [31] and p.Gln216Pro [29], have shown no impact on γ-secretase activity, suggesting that they are unlikely to be implicated in HS. These findings underscore the necessity for functional studies to elucidate the functional impact of these variants.
Despite its relatively small size of 101 amino acids, the PEN-2 protein has 20 identified variants in the PSENEN gene, with 95% of these having a strong effect. The literature describes that the four proteins of the γ-secretase complex are crucial for its proper function, and deregulation of any single protein can destabilize the entire complex [124]. For instance, PEN-2 is necessary for PS1 endoproteolysis [125], and PS1 mediates NCSTN maturation and intracellular trafficking [124]. Therefore, a strong-effect variant in one of the γ-secretase subunits is likely to have functional consequences. It has been demonstrated that knockdown of NCSTN in HaCaT cells disrupts the interaction between PEN-2 and PS1, impairing γ-secretase activity and leading to abnormal keratinocyte differentiation [64].
In the article by de Oliveira et al. [97], the authors demonstrate that the heterozygous nonsense mutation c.131T>A (p.L44*) in NCSTN gene activates the NMD mechanism, which degrades the aberrant transcript. This degradation can be reversed by the addition of gentamicin, restoring the expression of the truncated NCSTN protein. The resulting haploinsufficiency can affect the expression of genes related to the type I interferon response pathway [126]. Moreover, based on the pLI, pRec and pNull scores (respectively the probabilities of intolerance to loss-of-function, being a recessive gene or being an unconstrained gene) associated with the genes described in this article, NCSTN is among the genes most prone to haploinsufficiency (Supp. Figure S4). However, Pink et al. [87] still assert that haploinsufficiency alone is not sufficient to cause HS.
Among the γ-secretase complex variants reported in this article, there are five heterozygous nonsense or frameshift variants that could potentially lead to the degradation of APH1B and NCSTN RNA. Therefore, these variants warrant further functional analysis to assess their potential role in HS.
Finally, variants affecting proteins other than those of the γ-secretase complex primarily concern syndromic cases of HS or cases with co-morbidities (as noted in Supp. Figure S1). Variants in GJB2 are more commonly associated with the Follicular Occlusion Triad (FO3) and Keratitis-Ichthyosis-Deafness syndrome (KID), KRT17 variants with the Follicular Occlusion Tetrad (FO4), KRT6A and KRT17 variants with Pachyonychia Congenita (PC), MEFV variants with PASH and PAPASH syndromes, OCRL variants with Dent Disease 2 (DD2), POFUT1 and POGLUT1 variants with Dowling Degos Disease (DDD), and PSTPIP1 variants with PASH and PAPASH syndromes. In these cases, it is even more challenging to establish the causative role of the variants in HS. For example, the p.D50N mutation is associated with HS patients who also have KID syndrome [19]. This variant is also found in KID patients without HS, underscoring the importance of careful interpretation of variant data. The two variants that we include in this catalog concern the NOTCH3 and SLC46A2 genes. The first gene is directly involved in the Notch signaling pathway, the second gene is involved in the activation of NOD (nucleotide oligomerization domain) receptors in epithelial cells and initiate an anti-inflammatory response [127]. Both genes are highly deregulated in the lesional tissues of HS patients [128,129]
In conclusion, our study has identified and cataloged 179 new and previously known variants in the γ-secretase complex and other genes potentially associated with HS. These findings underscore the complex genetic landscape of HS, proposing both common and rare variants that may contribute to its pathogenesis. Our analysis emphasizes the potential role of rare variants in the disease’s development and suggests that both haploinsufficiency and more complex genetic interactions are maybe involved. Notably, the presence of multiple low-impact common variants may create a genetic predisposition that, in combination with environmental factors and lifestyle conditions (e.g., smoking behavior or obesity), facilitates the onset of the disease. Further functional studies are necessary to elucidate the precise mechanisms by which these genetic alterations influence HS. Understanding these mechanisms may provide new insights into targeted therapeutic strategies, offering hope for more effective treatments for individuals affected by this debilitating condition.
4. Materials and Methods
4.1. Sample Collection
This article combines data from three independent cohorts: the HS1 cohort, which includes 75 PBMC samples from HS patients; the HS2 cohort, comprising 25 diverse samples (PBMC, keratinocytes, ORS cells in culture, as well as total dermal cells); and the CTL cohort, consisting of 100 non-HS control individuals from the public French Exome consortium [50,51] (http://lysine.univ-brest.fr/FrExAC/). We maintained exactly the same male-to-female ratio between the HS and CTL cohorts. The characteristics of the 100 HS patients are summarized in Table 1. The ethics committee of Henri Mondor Hospital (CPP n°10-026) in agreement with the Declaration of Helsinki approved this study, and written informed consent was received from participants before inclusion in the study.
4.2. Whole Exome Sequencing
Whole Exome sequencing was performed using the Illumina HiSeq2500 sequencer for the HS1 cohort, the HiSeq4000 for the HS2 cohort, and the HiSeq2000 for the CTL cohort according to the following specifications:

| Cohort | Agilent WES Kit | Protocol | Library size |
| HS1 | SureSelectXT Human All Exon V5+UTR 2 | 2 x 100 bp | |
| HS2 | SureSelectXT Human All Exon V6 | 2 x 150 bp | ~70M |
| CTL | SureSelectXT Human All Exon V4+UTR | 2 x 100 bp | ~60M |
4.3. Variant Calling and Annotation
The individual .g.vcf files were obtained through an in-house pipeline using the following tools: bwa-mem (v. 2.2.1) [130,131], PicardTools (v. 2.26.9) [132], Samtools (v. 1.16) [133], Sambamba (v. 0.8.1) [134], GATK (v. 4.2.3.0) [135,136], and bedtools (v. 2.30.0) [137]. The alignment was done on the reference genome GRCh38. The .g.vcf files were aggregated and then transformed into .vcf files using the GATK HaplotypeCaller and GenotypeGVCFs modules. The variants were then limited to the regions captured by the kits (extended from 10 to 100bp) and filtered according to the commonly used criteria [138]: (1) for SNVs: QD < 2, QUAL < 30, SOR > 3, FS > 60, MQ < 40, MQRankSum < -12.5, and ReadPosRankSum < -8; (2) for Indels: QD < 2, QUAL < 30, FS > 200, and ReadPosRankSum < -20. We annotated our variants with the gnomAD (v. 4.1-exome) and dbSNP 2.0 (v. 152) [139] databases using the SnpEff tool [140] and its SnpSift module (v. 4.5) [141]. We complemented missing data with the online tool CADD (v. 1.7) [142]. Variant allele frequencies and deleteriousness scores (using various tools) are provided by gnomAD (v. 4.1). The thresholds used to assess the deleteriousness of a variant according to the tools are as follows:
- CADD Phred-like score ≥ 30: among the 0.1% most deleterious variants
- REVEL ≥ 0.5: deleterious missense
- SIFT < 0.05: deleterious missense
- AlphaMissense > 0.564: possibly deleterious
- PolyPhen-2 ≥ 0.85: probably damaging missense
- SpliceAI ≥ 0.8: variant with the probability of altering splicing
- Pangolin ≥ 0.8: variant with the probability of altering splicing
- PhyloP (mammals) < 1.5: fast evolution of the locus in mammals
4.4. Variant Validation
Each variant of the literature was validated manually. The data retrieved from the articles (electrophoregrams, sequences, genome used, HGVS, dbSNP identifier, transcript concerned, gene position, etc.) were compared with the Ensembl and/or NCBI databases and MANE reference transcripts to harmonize the data. Where data was missing, it was complemented (e.g., if position or exon information was absent, these details were incorporated into the final tables). The tools and databases used were mainly LitVar² (v. 2.0) [143], MutationTaster (v. 2021) [144], gnomAD (v. 2.1.1, v. 3.1.2 & v. 4.1.0-exome) [145], CADD (v. 1.7) [142], dbSNP (v. 156 and previous versions) [139], AlphaMissense (v.04-2024) [146,147], and LiftOver [148].
4.5. Protein Folding
Protein models of NCSTN, PS1 (PSEN1 gene), PEN-2 (PSENEN gene) and APH-1B (APH1B gene) were generated from the canonical Ensembl translated transcripts (i.e., ENST00000294785.10, ENST00000324501.10, ENST00000587708.7 and ENST00000261879.10, respectively) using the AlphaFold tool (v. 2.1.2) [149], which is freely available on the Galaxy France server (https://usegalaxy.fr/ - v. 22.01). The mutated models were processed in the same way, although we aknowledge the limitations of this tool for predicting the consequences of missense mutations [150,151]; it remains the best tool for predicting three-dimensional structures. The visualization and manipulation of the .pdb files was done via the RasTop tool (v.2.2) [152], and their annotation was a mix between InterPro (v.90.0) [153] predictions and cryo-EM structures of γ-secretase (models 2KR6 [154], 6IYC [155] & 5FN [2,3,4,5,154]) available on RCSB Protein Data Bank [156].
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Figure S1: Comorbidities of HS and its involvement in various autoinflammatory syndromes; Figure S2: All γ-secretase exonic variants identified in our HS1 & HS2 cohorts; Figure S3: Protein network of all genes discussed in this article, according to STRING; Figure S4: Distribution of pLI (probability of intolerance to loss of function), pRec (probability of being recessive) and pNull (probability of being unconstrained) gene scores; Table S1: Complete table of all 179 variants found in the literature, including our new variants; Table S2: Example of a mutation presented in 9 distinct articles; Table S3: Additional polymorphisms in other genes associated with HS (78) in the literature, including the two new variants identified in our cohort; Data S1: Complete catolog of variants in a .vcf file left-normalized.
Author Contributions
Conceptualization, S.H., Y.L.; methodology, K.M., C.D.R., V.L.G.; software, K.M., V.L.G., F.S., L.M.; validation, K.M.; formal analysis, C.D.R., K.M., V.L.G., E.L.F.; resources, S.H., J.F.D., J.L.C., B.B., P.W., C.H., F.J.L., D.D., R.O.; investigation, K.M., V.L.G., L.M.; data curation, S.H., K.M., F.J.L.; writing – original draft preparation, K.M.; writing – review and editing, K.M., S.H., J.F.D., E.B.; visualization, K.M.; supervision, S.H., J.F.D., E.B., V.M.; project administration, S.H., J.F.D., E.B., V.M., K.M., Y.L.; funding acquisition, S.H., J.F.D. All authors have read and agreed to the published version of the manuscript.
Funding
This research was partly supported by the French “Agence Nationale de la Recherche” (ANR) under project ANR-20-CE17-0019.
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki, and approved by the Ethics Committee of Henri Mondor Hospital (protocol code 10-026 – 13 septembre 2010).
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Acknowledgments
We would like to acknowledge the French Exome Consortium for granting us access to their cohort as the control group for this study.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Vinkel, C.; Thomsen, S.F. Hidradenitis Suppurativa: Causes, Features, and Current Treatments. J Clin Aesthet Dermatol 2018, 11, 17–23. [Google Scholar]
- Frew, J.W. Hidradenitis Suppurativa Is an Autoinflammatory Keratinization Disease: A Review of the Clinical, Histologic, and Molecular Evidence. JAAD International 2020, 1, 62–72. [Google Scholar] [CrossRef]
- Akiyama, M.; De Vita, V.; Sugiura, K. Editorial: Autoinflammatory Keratinization Disease (AiKD). Front Immunol 2020, 11, 1753. [Google Scholar] [CrossRef] [PubMed]
- Nikolakis, G.; Kaleta, K.P.; Vaiopoulos, A.G.; Wolter, K.; Baroud, S.; Wojas-Pelc, A.; Zouboulis, C.C. Phenotypes and Pathophysiology of Syndromic Hidradenitis Suppurativa: Different Faces of the Same Disease? A Systematic Review. DRM 2021, 237, 673–697. [Google Scholar] [CrossRef] [PubMed]
- Duchatelet, S.; Miskinyte, S.; Delage, M.; Join-Lambert, O.; Coignard, H.; Lortholary, O.; Nassif, X.; Hovnanian, A.; Nassif, A. Coexistence d’une Acné Fulminante et d’une Hidrosadénite Suppurée : Syndrome ASH, Une Nouvelle Entité ? Annales de Dermatologie et de Vénéréologie 2014, 141, S381–S382. [Google Scholar] [CrossRef]
- Phan, K.; Tatian, A.; Woods, J.; Cains, G.; Frew, J.W. Prevalence of Inflammatory Bowel Disease (IBD) in Hidradenitis Suppurativa (HS): Systematic Review and Adjusted Meta-Analysis. Int J Dermatol 2020, 59, 221–228. [Google Scholar] [CrossRef]
- Moltrasio, C.; Tricarico, P.M.; Romagnuolo, M.; Marzano, A.V.; Crovella, S. Hidradenitis Suppurativa: A Perspective on Genetic Factors Involved in the Disease. Biomedicines 2022, 10, 2039. [Google Scholar] [CrossRef]
- Gasparic, J.; Theut Riis, P.; Jemec, G.B. Recognizing Syndromic Hidradenitis Suppurativa: A Review of the Literature. J Eur Acad Dermatol Venereol 2017, 31, 1809–1816. [Google Scholar] [CrossRef]
- Kjaersgaard Andersen, R.; Clemmensen, S.B.; Larsen, L.A.; Hjelmborg, J.V.B.; Ødum, N.; Jemec, G.B.E.; Christensen, K. Evidence of Gene-Gene Interaction in Hidradenitis Suppurativa: A Nationwide Registry Study of Danish Twins. Br J Dermatol 2022, 186, 78–85. [Google Scholar] [CrossRef]
- Sabat, R.; Jemec, G.B.E.; Matusiak, Ł.; Kimball, A.B.; Prens, E.; Wolk, K. Hidradenitis Suppurativa. Nat Rev Dis Primers 2020, 6, 1–20. [Google Scholar] [CrossRef]
- Wolfe, M.S.; Miao, Y. Structure and Mechanism of the γ-Secretase Intramembrane Protease Complex. Curr Opin Struct Biol 2022, 74, 102373. [Google Scholar] [CrossRef] [PubMed]
- Mintoff, D.; Pace, N.P.; Borg, I. Interpreting the Spectrum of Gamma-Secretase Complex Missense Variation in the Context of Hidradenitis Suppurativa—An in-Silico Study. Front Genet 2022, 13, 962449. [Google Scholar] [CrossRef] [PubMed]
- Pace, N.P.; Mintoff, D.; Borg, I. The Genomic Architecture of Hidradenitis Suppurativa-A Systematic Review. Front Genet 2022, 13, 861241. [Google Scholar] [CrossRef] [PubMed]
- Moltrasio, C.; Cagliani, R.; Sironi, M.; Clerici, M.; Pontremoli, C.; Maronese, C.A.; Tricarico, P.M.; Crovella, S.; Marzano, A.V. Autoinflammation in Syndromic Hidradenitis Suppurativa: The Role of AIM2. Vaccines 2023, 11. [Google Scholar] [CrossRef] [PubMed]
- Tricarico, P.M.; Gratton, R.; Dos Santos-Silva, C.A.; de Moura, R.R.; Ura, B.; Sommella, E.; Campiglia, P.; Del Vecchio, C.; Moltrasio, C.; Berti, I.; et al. A Rare Loss-of-Function Genetic Mutation Suggest a Role of Dermcidin Deficiency in Hidradenitis Suppurativa Pathogenesis. Front Immunol 2022, 13, 1060547. [Google Scholar] [CrossRef] [PubMed]
- Theut Riis, P.; Loft, I.C.; Yazdanyar, S.; Kjaersgaard Andersen, R.; Pedersen, O.B.; Ring, H.C.; Huber, R.; Sultan, M.; Loesche, C.; Saunte, D.M.L.; et al. Full Exome Sequencing of 11 Families with Hidradenitis Suppurativa. J Eur Acad Dermatol Venereol 2021, 35, 1203–1211. [Google Scholar] [CrossRef] [PubMed]
- de Oliveira, A.S.L.E.; Bloise, G.; Moltrasio, C.; Coelho, A.; Agrelli, A.; Moura, R.; Tricarico, P.M.; Jamain, S.; Marzano, A.V.; Crovella, S.; et al. Transcriptome Meta-Analysis Confirms the Hidradenitis Suppurativa Pathogenic Triad: Upregulated Inflammation, Altered Epithelial Organization, and Dysregulated Metabolic Signaling. Biomolecules 2022, 12, 1371. [Google Scholar] [CrossRef] [PubMed]
- Higgins, R.; Pink, A.; Hunger, R.; Yawalkar, N.; Navarini, A.A. Generalized Comedones, Acne, and Hidradenitis Suppurativa in a Patient with an FGFR2 Missense Mutation. Front Med (Lausanne) 2017, 4, 16. [Google Scholar] [CrossRef]
- Nyquist, G.G.; Mumm, C.; Grau, R.; Crowson, A.N.; Shurman, D.L.; Benedetto, P.; Allen, P.; Lovelace, K.; Smith, D.W.; Frieden, I.; et al. Malignant Proliferating Pilar Tumors Arising in KID Syndrome: A Report of Two Patients. Am. J. Med. Genet. 2007, 143A, 734–741. [Google Scholar] [CrossRef]
- Lazic, T.; Li, Q.; Frank, M.; Uitto, J.; Zhou, L.H. Extending the Phenotypic Spectrum of Keratitis-Ichthyosis-Deafness Syndrome: Report of a Patient with GJB2 (G12R) Connexin 26 Mutation and Unusual Clinical Findings. Pediatr Dermatol 2012, 29, 349–357. [Google Scholar] [CrossRef]
- Bettoli, V.; Forconi, R.; Pezzini, I.; Martinello, R.; Scuderi, V.; Zedde, P.; Schettini, N.; Pacetti, L.; Corazza, M. KID Syndrome and Hidradenitis Suppurativa: A Rare Association Responding to Surgical Treatment. Skin Appendage Disord 2021, 7, 21–24. [Google Scholar] [CrossRef] [PubMed]
- Oliveira Mendonca, L.; Caroli, F. Hidradenitis Suppurativa in KID’s Syndrome: Genetic Characterisation, Clinical and Surgical Intervention; 2019;
- Montgomery, J.R.; White, T.W.; Martin, B.L.; Turner, M.L.; Holland, S.M. A Novel Connexin 26 Gene Mutation Associated with Features of the Keratitis-Ichthyosis-Deafness Syndrome and the Follicular Occlusion Triad. Journal of the American Academy of Dermatology 2004, 51, 377–382. [Google Scholar] [CrossRef] [PubMed]
- Marzano, A.V.; Genovese, G.; Moltrasio, C.; Tricarico, P.M.; Gratton, R.; Piaserico, S.; Garcovich, S.; Boniotto, M.; Brandão, L.; Moura, R.; et al. Whole-Exome Sequencing in 10 Unrelated Patients with Syndromic Hidradenitis Suppurativa: A Preliminary Step for a Genotype-Phenotype Correlation. Dermatology 2022, 238, 860–869. [Google Scholar] [CrossRef] [PubMed]
- Marzano, A.V.; Ceccherini, I.; Gattorno, M.; Fanoni, D.; Caroli, F.; Rusmini, M.; Grossi, A.; De Simone, C.; Borghi, O.M.; Meroni, P.L.; et al. Association of Pyoderma Gangrenosum, Acne, and Suppurative Hidradenitis (PASH) Shares Genetic and Cytokine Profiles With Other Autoinflammatory Diseases. Medicine (Baltimore) 2014, 93, e187. [Google Scholar] [CrossRef] [PubMed]
- Palmroth, M.; Viskari, H.; Seppänen, M.R.J.; Keskitalo, S.; Virtanen, A.; Varjosalo, M.; Silvennoinen, O.; Isomäki, P. IRF2BP2 Mutation Is Associated with Increased STAT1 and STAT5 Activation in Two Family Members with Inflammatory Conditions and Lymphopenia. Pharmaceuticals (Basel) 2021, 14, 797. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, A.; Ebstein, F.; Shamseldin, H.; Prouteau, C.; Krüger, E.; Binamer, Y.M.; Bonneau, D.; Alkuraya, F.S.; Martin, L. Gain-of-Function Variants in the KDF1 Gene Cause Hidradenitis Suppurativa Associated with Ectodermal Dysplasia by Stabilizing IκB Kinase α. Br J Dermatol 2023, 189, 131–132. [Google Scholar] [CrossRef] [PubMed]
- Shamseldin, H.E.; Khalifa, O.; Binamer, Y.M.; Almutawa, A.; Arold, S.T.; Zaidan, H.; Alkuraya, F.S. KDF1, Encoding Keratinocyte Differentiation Factor 1, Is Mutated in a Multigenerational Family with Ectodermal Dysplasia. Hum Genet 2017, 136, 99–105. [Google Scholar] [CrossRef] [PubMed]
- Pedraz, J.; Peñas, P.; Garcia-Diez, A. Pachyonychia Congenita and Hidradenitis Suppurativa: No Response to Infliximab Therapy. Journal of the European Academy of Dermatology and Venereology 2008, 22, 1500–1501. [Google Scholar] [CrossRef]
- Yan, J.; Zhang, G.; Liao, C.; Wang, X.; Shi, L. ALA-iPDT for Follicular Occlusion Tetrad Concomitant with Pachyonychia Congenital Type Ⅱ and Ankylosing Spondylitis. Photodiagnosis and Photodynamic Therapy 2022, 39, 102891. [Google Scholar] [CrossRef]
- Musumeci, M.L.; Fiorentini, F.; Bianchi, L.; Cascella, R.; Giardina, E.; Caputo, V.; Micali, G. Follicular Occlusion Tetrad in a Male Patient with Pachyonychia Congenita: Clinical and Genetic Analysis. J Eur Acad Dermatol Venereol 2019, 33, 36–39. [Google Scholar] [CrossRef]
- Vural, S.; Gündoğdu, M.; Gökpınar İli, E.; Durmaz, C. d.; Vural, A.; Steinmüller-Magin, L.; Kleinhempel, A.; Holdt, L. m.; Ruzicka, T.; Giehl, K. a.; et al. Association of Pyrin Mutations and Autoinflammation with Complex Phenotype Hidradenitis Suppurativa: A Case–Control Study. British Journal of Dermatology 2019, 180, 1459–1467. [Google Scholar] [CrossRef]
- Jfri, A.; Litvinov, I.V.; Netchiporouk, E.; O’Brien, E. Novel Variants of MEFV and NOD2 Genes in Familial Hidradenitis Suppurativa: A Case Report. SAGE Open Med Case Rep 2020, 8, 2050313X20953113. [Google Scholar] [CrossRef]
- Bueno-Molina, R.C.; Hernández-Rodríguez, J.-C.; Zulueta-Dorado, T.; Pereyra-Rodriguez, J.-J. Pyrin-Associated Autoinflammation with Neutrophilic Dermatosis: A Case Report. The Journal of Dermatology 2024, n/a. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.Y.; Keiser, E.; LeBoit, P.; Yeh, I.; Wei, M.L. A Rare Case of Axillary Keratoacanthoma Arising in Hidradenitis Suppurativa. JAAD Case Rep 2022, 21, 49–51. [Google Scholar] [CrossRef] [PubMed]
- Morales-Heil, D.J.; Cao, L.; Sweeney, C.; Malara, A.; Brown, F.; Milam, P.; Anadkat, M.; Kaffenberger, J.; Kaffenberger, B.; Nagele, P.; et al. Rare Missense Variants in the SH3 Domain of PSTPIP1 Are Associated with Hidradenitis Suppurativa. HGG Adv 2023, 4, 100187. [Google Scholar] [CrossRef] [PubMed]
- Marzuillo, P.; Piccolo, V.; Mascolo, M.; Apicella, A.; Argenziano, G.; Della Vecchia, N.; Guarino, S.; Miraglia del Giudice, E.; La Manna, A. Patients Affected by Dent Disease 2 Could Be Predisposed to Hidradenitis Suppurativa. Journal of the European Academy of Dermatology and Venereology 2018, 32, e309–e311. [Google Scholar] [CrossRef] [PubMed]
- García-Gil, M.F.; Monte Serrano, J.; Ramirez-Lluch, M.; Valero Torres, A.; López-Giménez, M.T.; Lezcano Biosca, V. A Novel Mutation in POFUT1 Gene Associated with Dowling-Degos Disease and Hidradenitis Suppurativa. Int J Dermatol 2021, 60, e25–e27. [Google Scholar] [CrossRef] [PubMed]
- González-Villanueva, I.; Gutiérrez, M.; Hispán, P.; Betlloch, I.; Pascual, J. c. Novel POFUT1 Mutation Associated with Hidradenitis Suppurativa–Dowling–Degos Disease Firm up a Role for Notch Signalling in the Pathogenesis of This Disorder. British Journal of Dermatology 2018, 178, 984–986. [Google Scholar] [CrossRef] [PubMed]
- Duchatelet, S.; Clerc, H.; Machet, L.; Gaboriaud, P.; Miskinyte, S.; Kervarrec, T.; Hovnanian, A. A New Nonsense Mutation in the POGLUT1 Gene in Two Sisters with Dowling–Degos Disease. Journal of the European Academy of Dermatology and Venereology 2018, 32, e440–e442. [Google Scholar] [CrossRef]
- Duchatelet, S.; Miskinyte, S.; Join-Lambert, O.; Ungeheuer, M.-N.; Francès, C.; Nassif, A.; Hovnanian, A. First Nicastrin Mutation in PASH (Pyoderma Gangrenosum, Acne and Suppurative Hidradenitis) Syndrome. Br J Dermatol 2015, 173, 610–612. [Google Scholar] [CrossRef]
- Shen, M.; Yeoh, X.L.A.; Wang, D.Y.; Tey, H.L.; Ren, E.C.; Oon, H.H. Genetic Variations in Gamma-Secretase and PSTPIP1 in Hidradenitis Suppurativa in Singaporean Chinese. J Eur Acad Dermatol Venereol 2021, 35, e348–e350. [Google Scholar] [CrossRef] [PubMed]
- Braun-Falco, M.; Kovnerystyy, O.; Lohse, P.; Ruzicka, T. Pyoderma Gangrenosum, Acne, and Suppurative Hidradenitis (PASH)–a New Autoinflammatory Syndrome Distinct from PAPA Syndrome. Journal of the American Academy of Dermatology 2012, 66, 409–415. [Google Scholar] [CrossRef] [PubMed]
- Kotzerke, M.; Mitri, F.; Marbach, F.; Enk, A.; Haenssle, H. A Case of PAPASH Syndrome in a Young Man Carrying a Novel Heterozygote Missense Variant in PSTPIP1. J Eur Acad Dermatol Venereol 2021, 35, e439–e440. [Google Scholar] [CrossRef]
- Hieta, N.; Nuutinen, H.; Roivas, J.; Salminen, K.; Kujari, H.; Talve, L.; Toivonen, M.; Haanpää, M.K. Severe Ulcerative Proctitis, Pyoderma Gangrenosum, Hidradenitis Suppurativa and Fever in a Patient with a Rare Variant of the PSTPIP1 Gene. Clin Exp Dermatol 2021, 46, 1387–1390. [Google Scholar] [CrossRef] [PubMed]
- Marzano, A.V.; Trevisan, V.; Gattorno, M.; Ceccherini, I.; De Simone, C.; Crosti, C. Pyogenic Arthritis, Pyoderma Gangrenosum, Acne, and Hidradenitis Suppurativa (PAPASH): A New Autoinflammatory Syndrome Associated With a Novel Mutation of the PSTPIP1 Gene. JAMA Dermatology 2013, 149, 762–764. [Google Scholar] [CrossRef] [PubMed]
- Saito, N.; Minami-Hori, M.; Nagahata, H.; Nozaki, H.; Iinuma, S.; Igawa, S.; Kanno, K.; Kishibe, M.; Kanazawa, N.; Ishida-Yamamoto, A. Novel PSTPIP1 Gene Mutation in Pyoderma Gangrenosum, Acne and Suppurative Hidradenitis Syndrome. J Dermatol 2018, 45, e213–e214. [Google Scholar] [CrossRef] [PubMed]
- Calderón-Castrat, X.; Bancalari-Díaz, D.; Román-Curto, C.; Romo-Melgar, A.; Amorós-Cerdán, D.; Alcaraz-Mas, L.A.; Fernández-López, E.; Cañueto, J. PSTPIP1 Gene Mutation in a Pyoderma Gangrenosum, Acne and Suppurative Hidradenitis (PASH) Syndrome. Br J Dermatol 2016, 175, 194–198. [Google Scholar] [CrossRef]
- van Straalen, K.R.; Prens, E.P.; Willemsen, G.; Boomsma, D.I.; van der Zee, H.H. Contribution of Genetics to the Susceptibility to Hidradenitis Suppurativa in a Large, Cross-Sectional Dutch Twin Cohort. JAMA Dermatol 2020, 156, 1359–1362. [Google Scholar] [CrossRef]
- Genin, E.; Redon, R.; Deleuze, J.-F.; Campion, D.; Lambert, J.-C.; Dartigues, J.-F.; Consortium, F. The French Exome (FREX) Project: A Population-Based Panel of Exomes to Help Filter Out Common Local Variants. Genetic Epidemiology 2017, 41, 691. [Google Scholar]
- Herzig, A.F.; Velo-Suárez, L.; Dina, C.; Redon, R.; Deleuze, J.-F.; Génin, E. How Local Reference Panels Improve Imputation in French Populations. Sci Rep 2024, 14, 370. [Google Scholar] [CrossRef]
- Van der Zee, H.H.; Jemec, G.B.E. New Insights into the Diagnosis of Hidradenitis Suppurativa: Clinical Presentations and Phenotypes. J Am Acad Dermatol 2015, 73, S23–26. [Google Scholar] [CrossRef] [PubMed]
- Nishimori, N.; Hayama, K.; Kimura, K.; Fujita, H.; Fujiwara, K.; Terui, T. A Novel NCSTN Gene Mutation in a Japanese Family with Hidradenitis Suppurativa. Acta Derm Venereol 2020, 100, adv00283. [Google Scholar] [CrossRef] [PubMed]
- Ingram, J. r.; Wood, M.; John, B.; Butler, R.; Anstey, A. v. Absence of Pathogenic γ-Secretase Mutations in a South Wales Cohort of Familial and Sporadic Hidradenitis Suppurativa (Acne Inversa). British Journal of Dermatology 2013, 168, 874–876. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Yu, Y.; Yu, G.; Zhang, F. Identification of One Novel Mutation of the NCSTN Gene in One Chinese Acne Inversa Family. Dermatologica Sinica 2014, 32, 126–128. [Google Scholar] [CrossRef]
- Pink, A.E.; Simpson, M.A.; Brice, G.W.; Smith, C.H.; Desai, N.; Mortimer, P.S.; Barker, J.N.W.N.; Trembath, R.C. PSENEN and NCSTN Mutations in Familial Hidradenitis Suppurativa (Acne Inversa). Journal of Investigative Dermatology 2011, 131, 1568–1570. [Google Scholar] [CrossRef] [PubMed]
- Miskinyte, S.; Nassif, A.; Merabtene, F.; Ungeheuer, M.-N.; Join-Lambert, O.; Jais, J.-P.; Hovnanian, A. Nicastrin Mutations in French Families with Hidradenitis Suppurativa. Journal of Investigative Dermatology 2012, 132, 1728–1730. [Google Scholar] [CrossRef] [PubMed]
- Shi, T.-W.; Bai, N.; Zhang, J.-A.; Lu, F.; Chen, X.-B.; Kong, X.-D.; Yu, J.-B. Mutations in the γ-Secretase Genes PSEN1, PSENEN, and NCSTN in a Family with Acne Inversa. Eur J Dermatol 2018, 28, 374–376. [Google Scholar] [CrossRef] [PubMed]
- Duchatelet, S.; Miskinyte, S.; Delage, M.; Ungeheuer, M.-N.; Lam, T.; Benhadou, F.; Del Marmol, V.; Vossen, A.R.J.V.; Prens, E.P.; Cogrel, O.; et al. Low Prevalence of GSC Gene Mutations in a Large Cohort of Predominantly Caucasian Patients with Hidradenitis Suppurativa. Journal of Investigative Dermatology 2020, 140, 2085–2088.e14. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Wang, L.; Chen, L.; Ren, W.; Mei, A.; Chen, X.; Deng, Y. Two Novel Mutations of the NCSTN Gene in Chinese Familial Acne Inverse. J Eur Acad Dermatol Venereol 2013, 27, 1571–1574. [Google Scholar] [CrossRef]
- Pink, A.E.; Simpson, M.A.; Desai, N.; Dafou, D.; Hills, A.; Mortimer, P.; Smith, C.H.; Trembath, R.C.; Barker, J.N.W. Mutations in the γ-Secretase Genes NCSTN, PSENEN, and PSEN1 Underlie Rare Forms of Hidradenitis Suppurativa (Acne Inversa). Journal of Investigative Dermatology 2012, 132, 2459–2461. [Google Scholar] [CrossRef]
- Li, C.-R.; Jiang, M.-J.; Shen, D.-B.; Xu, H.-X.; Wang, H.-S.; Yao, X.; Zhang, Y.; Zhou, W.-Q.; Wang, B. Two Novel Mutations of the Nicastrin Gene in Chinese Patients with Acne Inversa. Br J Dermatol 2011, 165, 415–418. [Google Scholar] [CrossRef] [PubMed]
- Nomura, Y.; Nomura, T.; Sakai, K.; Sasaki, K.; Ohguchi, Y.; Mizuno, O.; Hata, H.; Aoyagi, S.; Abe, R.; Itaya, Y.; et al. A Novel Splice Site Mutation in NCSTN Underlies a Japanese Family with Hidradenitis Suppurativa. British Journal of Dermatology 2013, 168, 206–209. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.; He, Y.; Li, C.; Zhang, X.; Xu, H.; Wang, B. Nicastrin Mutations in Familial Acne Inversa Impact Keratinocyte Proliferation and Differentiation through the Notch and Phosphoinositide 3-Kinase/AKT Signalling Pathways. British Journal of Dermatology 2016, 174, 522–532. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Xu, H.; Li, C.; Zhang, X.; Zhou, P.; Xiao, X.; Zhang, W.; Wu, Y.; Zeng, R.; Wang, B. Nicastrin/miR-30a-3p/RAB31 Axis Regulates Keratinocyte Differentiation by Impairing EGFR Signaling in Familial Acne Inversa. Journal of Investigative Dermatology 2019, 139, 124–134. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Davis, J. w.; Idler, K. b.; Mostafa, N. m.; Okun, M. m.; Waring, J. f. Genetic Analysis of NCSTN for Potential Association with Hidradenitis Suppurativa in Familial and Nonfamilial Patients. British Journal of Dermatology 2016, 175, 414–416. [Google Scholar] [CrossRef] [PubMed]
- Yang, J. -q.; Wu, X. -j.; Dou, T. -t.; Jiao, T.; Chen, X. -b.; Min, M.; Cai, S. -q.; Zheng, M. Haploinsufficiency Caused by a Nonsense Mutation in NCSTN Underlying Hidradenitis Suppurativa in a Chinese Family. Clinical and Experimental Dermatology 2015, 40, 916–919. [Google Scholar] [CrossRef] [PubMed]
- Vural, S.; Baumgartner, M.; Lichtner, P.; Eckstein, G.; Hariry, H.; Chen, W.C.; Ruzicka, T.; Melnik, B.; Plewig, G.; Wagner, M.; et al. Investigation of Gamma Secretase Gene Complex Mutations in German Population with Hidradenitis Suppurativa Designate a Complex Polygenic Heritage. J Eur Acad Dermatol Venereol 2021, 35, 1386–1392. [Google Scholar] [CrossRef] [PubMed]
- Takeichi, T.; Matsumoto, T.; Nomura, T.; Takeda, M.; Niwa, H.; Kono, M.; Shimizu, H.; Ogi, T.; Akiyama, M. A Novel NCSTN Missense Mutation in the Signal Peptide Domain Causes Hidradenitis Suppurativa, Which Has Features Characteristic of an Autoinflammatory Keratinization Disease. British Journal of Dermatology 2020, 182, 491–493. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Gao, M.; Lv, Y.; Yang, X.; Ren, Y.; Jiang, T.; Zhang, X.; Guo, B.; Li, M.; Zhang, Q.; et al. Confirmation by Exome Sequencing of the Pathogenic Role of NCSTN Mutations in Acne Inversa (Hidradenitis Suppurativa). J Invest Dermatol 2011, 131, 1570–1572. [Google Scholar] [CrossRef]
- Li, C.; Xu, H.; Wang, B. Is SAPHO Syndrome Linked to PASH Syndrome and Hidradenitis Suppurativa by Nicastrin Mutation? A Case Report. J Rheumatol 2018, 45, 1605–1607. [Google Scholar] [CrossRef]
- Wu, C.; Yang, J.; Zhang, S.; Li, J.; Jin, H.; Zhang, X. A Novel NCSTN Gene Mutation in a Chinese Family with Acne Inversa. Mol Genet Genomics 2018, 293, 1469–1475. [Google Scholar] [CrossRef]
- Wang, B.; Yang, W.; Wen, W.; Sun, J.; Su, B.; Liu, B.; Ma, D.; Lv, D.; Wen, Y.; Qu, T.; et al. γ-Secretase Gene Mutations in Familial Acne Inversa. Science 2010, 330, 1065–1065. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Xiao, X.; Hui, Y.; Zhang, X.; He, Y.; Li, C.; Wang, B. Phenotype of 53 Chinese Individuals with Nicastrin Gene Mutations in Association with Familial Hidradenitis Suppurativa (Acne Inversa). Br J Dermatol 2016, 174, 927–929. [Google Scholar] [CrossRef]
- Xu, H.; He, Y.; Hui, Y.; Xiao, X.; Zhang, X.; Li, C.; Wang, B. NCSTN Mutations in Hidradenitis Suppurativa/Acne Inversa Do Not Influence Cytokine Production by Peripheral Blood Mononuclear Cells. British Journal of Dermatology 2017, 176, 277–279. [Google Scholar] [CrossRef]
- Chen, S.; Mattei, P.; You, J.; Sobreira, N.L.; Hinds, G.A. γ-Secretase Mutation in an African American Family With Hidradenitis Suppurativa. JAMA Dermatology 2015, 151, 668–670. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Liu, X.; Wang, R.; Chen, L.; Zhao, H.; Zhou, Y. A Novel NCSTN Mutation in a Three-Generation Chinese Family with Hidradenitis Suppurative. J Healthc Eng 2022, 2022, 1540774. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Ge, H.; Fan, Y.; Zhen, Q.; Tang, L.; Sun, L. Novel Mutation of the NCSTN Gene Identified in a Chinese Acne Inversa Family. Ann Dermatol 2020, 32, 237–242. [Google Scholar] [CrossRef]
- Savva, A.; Kanni, T.; Damoraki, G.; Kotsaki, A.; Giatrakou, S.; Grech, I.; Katoulis, A.; Papadavid, E.; Giamarellos-Bourboulis, E. j. Impact of Toll-like Receptor-4 and Tumour Necrosis Factor Gene Polymorphisms in Patients with Hidradenitis Suppurativa. British Journal of Dermatology 2013, 168, 311–317. [Google Scholar] [CrossRef]
- Mintoff, D.; Pace, N.P.; Bauer, P.; Borg, I. A Novel c.671_682del NCSTN Variant in a Family with Hidradenitis Suppurativa: A Pilot Study. Clin Exp Dermatol 2021, 46, 1306–1308. [Google Scholar] [CrossRef]
- Ratnamala, U.; Jhala, D.; Jain, N.K.; Saiyed, N.M.; Raveendrababu, M.; Rao, M.V.; Mehta, T.Y.; Al-Ali, F.M.; Raval, K.; Nair, S.; et al. Expanding the Spectrum of γ-Secretase Gene Mutation-Associated Phenotypes: Two Novel Mutations Segregating with Familial Hidradenitis Suppurativa (Acne Inversa) and Acne Conglobata. Exp Dermatol 2016, 25, 314–316. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, B.; Guo, Y.; Chen, J.; Zheng, S.; Gao, X.; Chen, H.; Xu, X. A Novel Mutation of the NCSTN Gene in a Chinese Hidradenitis Suppurativa Family with Familial Comedones as the Main Clinical Manifestation. Int J Dermatol 2022, 61, e293–e294. [Google Scholar] [CrossRef]
- Xiao, X.-M.; Yang, W.-Z.; Lin, L.-H.; Li, C.-R. Two Novel Nicastrin Mutations in Chinese Families with Acne Inversa. J Dermatol 2020, 47, e449–e451. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Meng, J.; Jiang, M.; Zhao, J. Characterization of a Novel Mutation in the NCSTN Gene in a Large Chinese Family with Acne Inversa. Acta Derm Venereol 2016, 96, 408–409. [Google Scholar] [CrossRef] [PubMed]
- Haines, R.; Common, J.; Teo, D.; Tang, M.; Lane, E. Sequencing of the Gamma-Secretase Complex in Singaporean Patients with Acne Inversa Reveals a Novel Mutation in Nicastrin, but Suggests Other Mechanisms Must Be Present.; April 1 2012; pp. e33–e33.
- Pink, A.E.; Simpson, M.A.; Desai, N.; Trembath, R.C.; Barker, J.N.W. γ-Secretase Mutations in Hidradenitis Suppurativa: New Insights into Disease Pathogenesis. J Invest Dermatol 2013, 133, 601–607. [Google Scholar] [CrossRef] [PubMed]
- Pink, A.E.; Dafou, D.; Desai, N.; Holmes, O.; Hobbs, C.; Smith, C.H.; Mortimer, P.; Simpson, M.A.; Trembath, R.C.; Barker, J.N. Hidradenitis Suppurativa: Haploinsufficiency of Gamma-Secretase Components Does Not Affect Gamma-Secretase Enzyme Activity in Vitro. Br J Dermatol 2016, 175, 632–635. [Google Scholar] [CrossRef] [PubMed]
- Jiao, T.; Dong, H.; Jin, L.; Wang, S.; Wang, J. A Novel Nicastrin Mutation in a Large Chinese Family with Hidradenitis Suppurativa. British Journal of Dermatology 2013, 168, 1141–1143. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.-Y.; Lin, L.-H.; Kang, J.; Li, C.-R.; Xiao, X.-M. Hidradenitis Suppurativa Associated with a Novel NCSTN Mutation and Concomitant Klippel-Trenaunay Syndrome. European Journal of Dermatology 2021, 31, 246–248. [Google Scholar] [CrossRef] [PubMed]
- González-Villanueva, I.; Poveda-Montoyo, I.; Álvarez-Chinchilla, P.; Gutiérrez, M.; Pascual-Ramírez, J.C. Hidradenitis Suppurativa/Dowling-Degos Disease (DDD) Phenotype Associated with Mutations in NCSTN Gene (Poster - Plenary Lectures). Experimental Dermatology 2019, 28, 5–55. [Google Scholar] [CrossRef]
- Qian, Y.-T.; Xiao, M.; Liu, K.; Ma, D.-L.; Zhang, X. Two Novel Mutations of the γ-Secretase Genes in Chinese Acne Inversa (Hidradenitis Suppurativa). Experimental Dermatology 2022, 31, 643–644. [Google Scholar] [CrossRef]
- Faraji Zonooz, M.; Sabbagh-Kermani, F.; Fattahi, Z.; Fadaee, M.; Akbari, M.R.; Amiri, R.; Vahidnezhad, H.; Uitto, J.; Najmabadi, H.; Kariminejad, A. Whole Genome Linkage Analysis Followed by Whole Exome Sequencing Identifies Nicastrin (NCSTN) as a Causative Gene in a Multiplex Family with γ-Secretase Spectrum of Autoinflammatory Skin Phenotypes. J Invest Dermatol 2016, 136, 1283–1286. [Google Scholar] [CrossRef]
- Nomura, Y.; Nomura, T.; Suzuki, S.; Takeda, M.; Mizuno, O.; Ohguchi, Y.; Abe, R.; Murata, Y.; Shimizu, H. A Novel NCSTN Mutation Alone May Be Insufficient for the Development of Familial Hidradenitis Suppurativa. J Dermatol Sci 2014, 74, 180–182. [Google Scholar] [CrossRef]
- Garcovich, S.; Tricarico, P.M.; Nait-Meddour, C.; Giovanardi, G.; Peris, K.; Crovella, S.; Boniotto, M. Novel Nicastrin Mutation in Hidradenitis Suppurativa–Dowling–Degos Disease Clinical Phenotype: More than Just Clinical Overlap? Br J Dermatol 2020, 183, 758–759. [Google Scholar] [CrossRef] [PubMed]
- Vossen, A.R.J.V.; van Straalen, K.R.; Swagemakers, S.M.A.; de Klein, J.E.M.M.; Stubbs, A.P.; Venter, D.J.; van der Zee, H.H.; van der Spek, P.J.; Prens, E.P. A Novel Nicastrin Mutation in a Three-Generation Dutch Family with Hidradenitis Suppurativa: A Search for Functional Significance. J Eur Acad Dermatol Venereol 2020, 34, 2353–2361. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.-J.; Yang, Y.; Liang, Y.-H. Hsa-miR-155 Targeted NCSTN 3′UTR Mutation Promotes the Pathogenesis and Development of Acne Inversa. Int J Clin Exp Pathol 2018, 11, 1878–1889. [Google Scholar]
- de Oliveira, A.S.L.E.; de Siqueira, R.C.; Nait-Meddour, C.; Tricarico, P.M.; Moura, R.; Agrelli, A.; d’Adamo, A.P.; Jamain, S.; Crovella, S.; de Fátima Medeiros Brito, M.; et al. A Loss-of-Function NCSTN Mutation Associated with Familial Dowling Degos Disease and Hidradenitis Suppurativa. Experimental Dermatology 2023, 32, 1935–1945. [Google Scholar] [CrossRef]
- Le Gall, C.; Puca, L.; Madrange, M.; Bal, E.; Choukair, Z.; Hamel, Y.; Fraitag, S.; Brou, C.; Bachelez, H.; Smahi, A. Impairment of Notch 1 Signaling Is a Common Defect in Lesions from Patients with Hidradenitis Suppurativa. J Invest Dermatol 2017, 137, S226. [Google Scholar] [CrossRef]
- Hermasch, M.A.; Schön, M.P.; Frank, J. Comorbid Acne Inversa and Dowling–Degos Disease Due to a Single NCSTN Mutation: Is There Enough Evidence? Br J Dermatol 2021, 184, 374–374. [Google Scholar] [CrossRef]
- Ratnamala, U.; Jain, N.K.; Jhala, D.D.; Prasad, P.V.S.; Saiyed, N.; Nair, S.; Radhakrishna, U. An Updated Mutation Spectrum of the γ-Secretase Complex: Novel NCSTN Gene Mutation in an Indian Family with Hidradenitis Suppurativa and Acne Conglobata. Indian J Dermatol 2023, 68, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Mintoff, D.; Pace, N.P.; Borg, I. NCSTN In-Frame Deletion in Maltese Patients With Hidradenitis Suppurativa. JAMA Dermatol 2023, 159, 939–944. [Google Scholar] [CrossRef]
- Li, A.; Peng, Y.; Taiclet, L.M.; Tanzi, R.E. Analysis of Hidradenitis Suppurativa–Linked Mutations in Four Genes and the Effects of PSEN1-P242LfsX11 on Cytokine and Chemokine Expression in Macrophages. Hum Mol Genet 2019, 28, 1173–1182. [Google Scholar] [CrossRef]
- Ralser, D.J.; Basmanav, F.B.Ü.; Tafazzoli, A.; Wititsuwannakul, J.; Delker, S.; Danda, S.; Thiele, H.; Wolf, S.; Busch, M.; Pulimood, S.A.; et al. Mutations in γ-Secretase Subunit–Encoding PSENEN Underlie Dowling-Degos Disease Associated with Acne Inversa. J Clin Invest 2017, 127, 1485–1490. [Google Scholar] [CrossRef] [PubMed]
- Kan, T.; Takahagi, S.; Shindo, H.; Tanaka, A.; Kawai, M.; Hide, M. A Unique Clinical Phenotype of a Patient Bearing a Newly Identified Deletion Mutation in the PSENEN Gene along with the Pathogenic Serum Desmoglein-1 Antibody. Clin Exp Dermatol 2018, 43, 329–332. [Google Scholar] [CrossRef] [PubMed]
- Peter, D.C.V.; Smith, F.J.D.; Wilson, N.J.; Danda, S. PSENEN Mutation in Coexistent Hidradenitis Suppurativa and Dowling-Degos Disease. Indian Dermatol Online J 2020, 12, 147–149. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.-M.; Zhou, P.-J.; Zhu, C.-H.; Lin, L.-H.; Liu, J.-J.; Han, Y. Coexistence of Acne Inversa with Psoriasis and Dowling-Degos Disease Harboring Impaired PSENEN-Notch Signaling. Chin Med J (Engl) 2020, 133, 2383–2385. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Wen, G.-D.; Soe, L.M.; Xu, H.-J.; Du, J.; Zhang, J.-Z. Novel Mutations in PSENEN Gene in Two Chinese Acne Inversa Families Manifested as Familial Multiple Comedones and Dowling-Degos Disease. Chinese Medical Journal 2016, 129, 2834. [Google Scholar] [CrossRef] [PubMed]
- Pavlovsky, M.; Sarig, O.; Eskin-Schwartz, M.; Malchin, N.; Bochner, R.; Mohamad, J.; Gat, A.; Peled, A.; Hafner, A.; Sprecher, E. A Phenotype Combining Hidradenitis Suppurativa with Dowling-Degos Disease Caused by a Founder Mutation in PSENEN. Br J Dermatol 2018, 178, 502–508. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Li, W.; Xu, H.; Zhang, X.; Su, B.; Zhang, W.; Zhang, X.; Wang, B. PSENEN Mutation Carriers with Co-Manifestation of Acne Inversa (AI) and Dowling-Degos Disease (DDD): Is AI or DDD the Subphenotype? J Invest Dermatol 2017, 137, 2234–2236. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; He, Y.; Xu, H.; Wang, B. First PSENEN Mutation in PASH Syndrome. The Journal of Dermatology 2020, 47, 1335–1337. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Liu, J.; Xu, T.; Guo, Y.; Han, Y.; He, Y.; Lin, L.; Xiao, X. Mutations in γ-Secretase Subunit-Encoding PSENEN Gene Alone May Not Be Sufficient for the Development of Acne Inversa. J Dermatol Sci 2021, 103, 73–81. [Google Scholar] [CrossRef]
- Liu, Y.; Miao, T.; Ma, J.; Shao, L.; Luo, S.; Li, Y.; Liu, Q. PSENEN c.66delG in Sporadic Acne Inversa. Eur J Dermatol 2016, 26, 298–299. [Google Scholar] [CrossRef]
- Chen, A.-W.; Chen, Z.; Bai, X.-M.; Luo, X.-Y.; Wang, H. Successful Treatment of Early-Onset Hidradenitis Suppurativa with Acitretin in an Infant with a Novel Mutation in PSENEN Gene. Indian J Dermatol Venereol Leprol 2022, 88, 445–445. [Google Scholar] [CrossRef]
- Vellaichamy, G.; Dimitrion, P.; Zhou, L.; Ozog, D.; Lim, H.W.; Liao, W.; Hamzavi, I.H.; Mi, Q.-S. Insights from γ-Secretase: Functional Genetics of Hidradenitis Suppurativa. Journal of Investigative Dermatology 2021, 141, 1888–1896. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Yan, Y.; Wang, B. γ-Secretase Genetics of Hidradenitis Suppurativa: A Systematic Literature Review. DRM 2021, 237, 698–704. [Google Scholar] [CrossRef] [PubMed]
- Frew, J.W.; Hawkes, J.E.; Sullivan-Whalen, M.; Gilleaudeau, P.; Krueger, J.G. Inter-Relater Reliability of Phenotypes, and Exploratory Genotype- Phenotype Analysis in Inherited Hidradenitis Suppurativa. Br J Dermatol 2019, 181, 566–571. [Google Scholar] [CrossRef] [PubMed]
- Frew, J.W.; Vekic, D.A.; Woods, J.; Cains, G.D. A Systematic Review and Critical Evaluation of Reported Pathogenic Sequence Variants in Hidradenitis Suppurativa. Br J Dermatol 2017, 177, 987–998. [Google Scholar] [CrossRef] [PubMed]
- Jfri, A.H.; O’Brien, E.A.; Litvinov, I.V.; Alavi, A.; Netchiporouk, E. Hidradenitis Suppurativa: Comprehensive Review of Predisposing Genetic Mutations and Changes. J Cutan Med Surg 2019, 23, 519–527. [Google Scholar] [CrossRef] [PubMed]
- Eble, S.M.; Wisco, O.J.; Boccuto, L.; Laffin, B.; Parker, V.G.; Davis, N.J.; Temples, H.S. Genetic Factors Associated with Hidradenitis Suppurativa, a Literature Review. Int J Womens Dermatol 2024, 10, e158. [Google Scholar] [CrossRef] [PubMed]
- Yudin, A.I.; Generao, S.E.; Tollner, T.L.; Treece, C.A.; Overstreet, J.W.; Cherr, G.N. Beta-Defensin 126 on the Cell Surface Protects Sperm from Immunorecognition and Binding of Anti-Sperm Antibodies1. Biology of Reproduction 2005, 73, 1243–1252. [Google Scholar] [CrossRef]
- Li, Y.; Tang, L.; Yue, J.; Gou, X.; Lin, A.; Weatherbee, S.D.; Wu, X. Regulation of Epidermal Differentiation through KDF1-mediated Deubiquitination of IKKα. EMBO Rep 2020, 21, e48566. [Google Scholar] [CrossRef]
- Körholz, J.; Gabrielyan, A.; Sczakiel, H.L.; Schulze, L.; Rejzek, M.; Laass, M.W.; Leuchten, N.; Tiebel, O.; Aust, D.; Conrad, K.; et al. Novel Mutation and Expanding Phenotype in IRF2BP2 Deficiency. Rheumatology (Oxford) 2023, 62, 1699–1705. [Google Scholar] [CrossRef]
- Sun, Q.; Broadaway, K.A.; Edmiston, S.N.; Fajgenbaum, K.; Miller-Fleming, T.; Westerkam, L.L.; Melendez-Gonzalez, M.; Bui, H.; Blum, F.R.; Levitt, B.; et al. Genetic Variants Associated With Hidradenitis Suppurativa. JAMA Dermatology 2023, 159, 930–938. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Li, Y.; Xu, H.; Zhang, Y. The γ-Secretase Complex: From Structure to Function. Front Cell Neurosci 2014, 8, 427. [Google Scholar] [CrossRef] [PubMed]
- Luo, W.; Wang, H.; Li, H.; Kim, B.S.; Shah, S.; Lee, H.-J.; Thinakaran, G.; Kim, T.-W.; Yu, G.; Xu, H. PEN-2 and APH-1 Coordinately Regulate Proteolytic Processing of Presenilin 1. J Biol Chem 2003, 278, 7850–7854. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Morales-Heil, D.J.; Roberson, E.D.O. Nicastrin Haploinsufficiency Alters Expression of Type I Interferon-Stimulated Genes: The Relationship to Familial Hidradenitis Suppurativa. Clin Exp Dermatol 2019, 44, e118–e125. [Google Scholar] [CrossRef] [PubMed]
- Paik, D.; Monahan, A.; Caffrey, D.R.; Elling, R.; Goldman, W.E.; Silverman, N. SLC46 Family Transporters Facilitate Cytosolic Innate Immune Recognition of Monomeric Peptidoglycans. J Immunol 2017, 199, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Hessam, S.; Gambichler, T.; Skrygan, M.; Scholl, L.; Sand, M.; Meyer, T.; Stockfleth, E.; Bechara, F.G. Increased Expression Profile of NCSTN, Notch and PI3K/AKT3 in Hidradenitis Suppurativa. J Eur Acad Dermatol Venereol 2021, 35, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Gambichler, T.; Hessam, S.; Skrygan, M.; Bakirtzi, M.; Kasakovski, D.; Bechara, F.G. NOD2 Signalling in Hidradenitis Suppurativa. Clin Exp Dermatol 2021, 46, 1488–1494. [Google Scholar] [CrossRef] [PubMed]
- Li, H. Aligning Sequence Reads, Clone Sequences and Assembly Contigs with BWA-MEM 2013.
- Li, H. Toward Better Understanding of Artifacts in Variant Calling from High-Coverage Samples. Bioinformatics 2014, 30, 2843–2851. [Google Scholar] [CrossRef]
- Picard Tools - By Broad Institute . Available online: http://broadinstitute.github.io/picard/ (accessed on 14 October 2022).
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve Years of SAMtools and BCFtools. GigaScience 2021, 10, giab008. [Google Scholar] [CrossRef]
- Tarasov, A.; Vilella, A.J.; Cuppen, E.; Nijman, I.J.; Prins, P. Sambamba: Fast Processing of NGS Alignment Formats. Bioinformatics 2015, 31, 2032–2034. [Google Scholar] [CrossRef]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce Framework for Analyzing next-Generation DNA Sequencing Data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- Poplin, R.; Ruano-Rubio, V.; DePristo, M.A.; Fennell, T.J.; Carneiro, M.O.; Van der Auwera, G.A.; Kling, D.E.; Gauthier, L.D.; Levy-Moonshine, A.; Roazen, D.; et al. Scaling Accurate Genetic Variant Discovery to Tens of Thousands of Samples; Genomics, 2017.
- Quinlan, A.R.; Hall, I.M. BEDTools: A Flexible Suite of Utilities for Comparing Genomic Features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [PubMed]
- Van der Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From FastQ Data to High-Confidence Variant Calls: The Genome Analysis Toolkit Best Practices Pipeline. Current Protocols in Bioinformatics 2013, 43, 11.10.1–11.10.33. [Google Scholar] [CrossRef] [PubMed]
- Sherry, S.T.; Ward, M.-H.; Kholodov, M.; Baker, J.; Phan, L.; Smigielski, E.M.; Sirotkin, K. dbSNP: The NCBI Database of Genetic Variation. Nucleic Acids Res 2001, 29, 308–311. [Google Scholar] [CrossRef] [PubMed]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A Program for Annotating and Predicting the Effects of Single Nucleotide Polymorphisms, SnpEff: SNPs in the Genome of Drosophila Melanogaster Strain w 1118 ; Iso-2; Iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef] [PubMed]
- Cingolani, P.; Patel, V.M.; Coon, M.; Nguyen, T.; Land, S.J.; Ruden, D.M.; Lu, X. Using Drosophila Melanogaster as a Model for Genotoxic Chemical Mutational Studies with a New Program, SnpSift. Front. Gene. 2012, 3. [Google Scholar] [CrossRef] [PubMed]
- Schubach, M.; Maass, T.; Nazaretyan, L.; Röner, S.; Kircher, M. CADD v1.7: Using Protein Language Models, Regulatory CNNs and Other Nucleotide-Level Scores to Improve Genome-Wide Variant Predictions. Nucleic Acids Res 2024, 52, D1143–D1154. [Google Scholar] [CrossRef] [PubMed]
- Allot, A.; Wei, C.-H.; Phan, L.; Hefferon, T.; Landrum, M.; Rehm, H.L.; Lu, Z. Tracking Genetic Variants in the Biomedical Literature Using LitVar 2.0. Nat Genet 2023, 55, 901–903. [Google Scholar] [CrossRef]
- Steinhaus, R.; Proft, S.; Schuelke, M.; Cooper, D.N.; Schwarz, J.M.; Seelow, D. MutationTaster2021. Nucleic Acids Research 2021, 49, W446–W451. [Google Scholar] [CrossRef]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The Mutational Constraint Spectrum Quantified from Variation in 141,456 Humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef]
- Tordai, H.; Torres, O.; Csepi, M.; Padányi, R.; Lukács, G.L.; Hegedűs, T. Analysis of AlphaMissense Data in Different Protein Groups and Structural Context. Sci Data 2024, 11, 495. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Novati, G.; Pan, J.; Bycroft, C.; Žemgulytė, A.; Applebaum, T.; Pritzel, A.; Wong, L.H.; Zielinski, M.; Sargeant, T.; et al. Accurate Proteome-Wide Missense Variant Effect Prediction with AlphaMissense. Science 2023, 381, eadg7492. [Google Scholar] [CrossRef] [PubMed]
- Hinrichs, A.S.; Karolchik, D.; Baertsch, R.; Barber, G.P.; Bejerano, G.; Clawson, H.; Diekhans, M.; Furey, T.S.; Harte, R.A.; Hsu, F.; et al. The UCSC Genome Browser Database: Update 2006. Nucleic Acids Res 2006, 34, D590–598. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly Accurate Protein Structure Prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Buel, G.R.; Walters, K.J. Can AlphaFold2 Predict the Impact of Missense Mutations on Structure? Nat Struct Mol Biol 2022, 29, 1–2. [Google Scholar] [CrossRef]
- Chakravarty, D.; Porter, L.L. AlphaFold2 Fails to Predict Protein Fold Switching. Protein Sci 2022, 31, e4353. [Google Scholar] [CrossRef]
- Valadon, P. RasTop 2007.
- Blum, M.; Chang, H.-Y.; Chuguransky, S.; Grego, T.; Kandasaamy, S.; Mitchell, A.; Nuka, G.; Paysan-Lafosse, T.; Qureshi, M.; Raj, S.; et al. The InterPro Protein Families and Domains Database: 20 Years On. Nucleic Acids Research 2021, 49, D344–D354. [Google Scholar] [CrossRef]
- Bai, X.; Rajendra, E.; Yang, G.; Shi, Y.; Scheres, S.H.W. Sampling the Conformational Space of the Catalytic Subunit of Human γ-Secretase. Elife 2015, 4, e11182. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Yang, G.; Guo, X.; Zhou, Q.; Lei, J.; Shi, Y. Recognition of the Amyloid Precursor Protein by Human γ-Secretase. Science 2019, 363, eaaw0930. [Google Scholar] [CrossRef]
- Burley, S.K.; Bhikadiya, C.; Bi, C.; Bittrich, S.; Chen, L.; Crichlow, G.V.; Christie, C.H.; Dalenberg, K.; Di Costanzo, L.; Duarte, J.M.; et al. RCSB Protein Data Bank: Powerful New Tools for Exploring 3D Structures of Biological Macromolecules for Basic and Applied Research and Education in Fundamental Biology, Biomedicine, Biotechnology, Bioengineering and Energy Sciences. Nucleic Acids Research 2021, 49, D437–D451. [Google Scholar] [CrossRef]
Figure 1.
Computational models of the NCSTN protein. In cyan, red, and grey: the cytosolic, transmembrane (TM), and extracellular regions or domains. In pink: the signal peptide responsible for adressing the protein to the membrane. In green: the small lobe that would interact with the substrate. In dark blue: the large lobe. (a) Reference (WT), (b) p.Ala300TrpfsTer20 (W300), (c) p.Arg429*, and (d) p.Trp648* variant models.
Figure 1.
Computational models of the NCSTN protein. In cyan, red, and grey: the cytosolic, transmembrane (TM), and extracellular regions or domains. In pink: the signal peptide responsible for adressing the protein to the membrane. In green: the small lobe that would interact with the substrate. In dark blue: the large lobe. (a) Reference (WT), (b) p.Ala300TrpfsTer20 (W300), (c) p.Arg429*, and (d) p.Trp648* variant models.
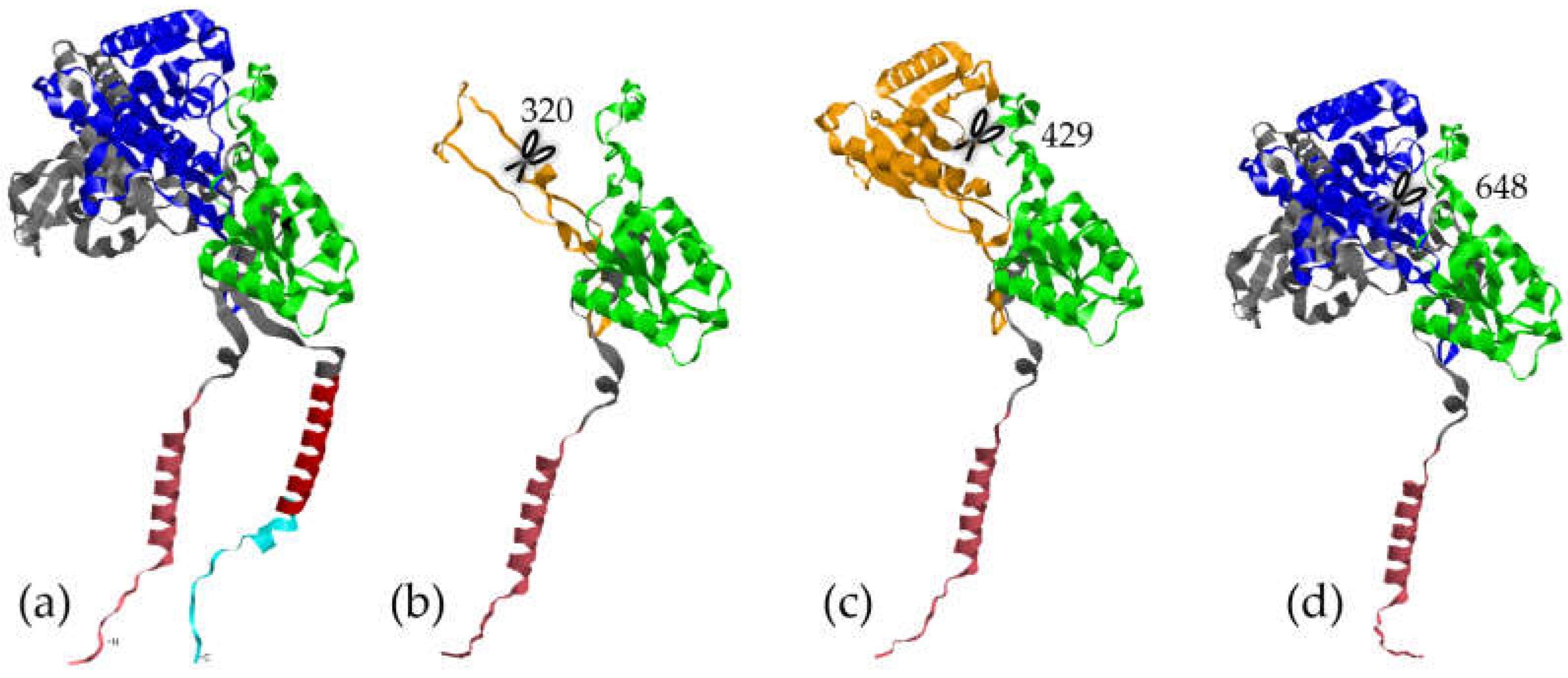
Table 1.
Characteristics of the 100 HS patients.
| Individual characteristics | Male | Female | All | ND |
|---|---|---|---|---|
| N | 36 | 64 | 100 | - |
| Age (mean ± sd) | 33.8 ± 12.2 | 34.6 ± 11.1 | 34.0 ± 11.5 | 2 |
| BMI (mean ± sd) | 26.1 ± 4.5 | 27.1 ± 5.1 | 26.8 ± 4.8 | 4 |
| Smoking status | 21 (67.7%) | 44 (72.1%) | 65 (70.6%) | 8 |
| Familial case | 7 (36.8%) | 21 (37.5%) | 28 (37.3%) | 25 |
| IBD history | 0 | 1 (1.8%) | 1 (1.3%) | 25 |
| Rhumatological history | 0 | 2 (3.6%) | 2 (2.7%) | 25 |
| Hurley | ||||
| I (mild) | 4 (12.5%) | 11 (17.2%) | 15 (15.6%) | 4 |
| II (moderate) | 12 (37.5%) | 30 (46.9%) | 42 (43.7%) | |
| III (severe) | 16 (50.0%) | 23 (35.9%) | 39 (40.6%) | |
Table 2.
All 91 γ-secretase variants found in the literature, including the eight new variants identified in our cohort.
Table 2.
All 91 γ-secretase variants found in the literature, including the eight new variants identified in our cohort.
| Gene | ID | Position (GRCh38) | Ex. | c/p.HGVS | Eff. | rsID | R. | Or. | F/S | Association |
|---|---|---|---|---|---|---|---|---|---|---|
| APH1A (ENST00000369109) | 1 | 1:150267780 | 3 | p.D98E | mis|spl | rs996158631 | 0 | div. | – | – |
| APH1B (ENST00000261879) | 2°N | 15:63277702 | 1 | p.T27A | mis | rs77834210 | 0 | FR* | S | – |
| 3°N | 15:63279258 | 2 | p.L71V | mis | – | 0 | FR* | S | – | |
| 4 | 15:63302375 | 5 | p.H170R | mis | rs139355584 | 2 | div. | F | – | |
| 5°N | 15:63302416 | 5 | p.K185Sfs*10 | fs | rs745918508 | 0 | FR* | S | – | |
| 6 | 15:63305770 | 6 | p.R255C | mis | rs142676640 | 0 | div. | – | – | |
| NCSTN (ENST00000294785) | 7 | 1:160343408 | 1 | p.G6Vfs*22 | fs | rs1266104510 | 0 | div. | – | – |
| 8 | 1:160343434 | 1 | p.G13Efs*15 | fs | – | 3 | DE | S | AC | |
| 9°N | 1:160343443 | 1 | p.L17Sfs*30 | fs | – | 0 | FR* | S | – | |
| 10 | 1:160344733 | 2 | p.G33R | mis|spl | – | 6 | JP | F | – | |
| 11 | 1:160344767 | 2 | p.L44* | non | – | 0 | BR | F | DDD | |
| 12 | 1:160344818 | 2 | p.G61V | mis | – | 3 | FR*|MT* | F | – | |
| 13 | 1:160349018 | 3 | p.V72Yfs*16 | fs | rs1243425689 | 10 | CN | F | – | |
| 14 | 1:160349031 | 3 | p.V75I | mis | rs12045198 | 10 | CN | F | – | |
| 15° | 1:160349039 | 3 | p.E77D | mis | rs35603924 | 0 | div.|FR* | S | – | |
| 16 | 1:160349086 | 3 | p.P93Lfs*15 | fs | – | 8 | CN | FS | SAPHO | |
| 17 | 1:160349578 | 4 | p.T115Nfs*20 | fs | – | 9 | FR* | – | PASH | |
| 18 | 1:160349583 | 4 | p.R117* | non | rs387906896 | 10 | CN|US*|AfUS | F | – | |
| 19 | 1:160349799 | 4-5 | c.436+129A>G | int | rs2274184 | 0 | SG | FS | – | |
| 20 | 1:160350115 | 5 | p.N150Ifs*52 | fs | – | 2 | CN | F | – | |
| 21 | 1:160350118 | 5 | p.S151Qfs*48 | fs | – | 4 | CN | F | – | |
| 22 | 1:160350145 | 5 | p.C159* | non | – | 9 | CN|GR* | F | – | |
| 23 | 1:160350150 | 5 | p.I162Yfs*40 | fs | – | 1 | IT* | S | PASH/SAPHO | |
| 24 | 1:160350155 | 5 | p.Q163Sfs*39 | fs | – | 10 | FR | F | – | |
| 25 | 1:160350165 | 5 | p.S166* | non | – | 6 | CN | F | – | |
| 26 | 1:160350221 | 5 | p.D185N | mis | rs201293070 | 9 | GB* | S | Diab | |
| 27 | 1:160350251 | 5-6 | c.582+1del(G) | spl* | – | 10 | JP | F | – | |
| 28 | 1:160350251 | 5-6 | c.582+1G>A | spl | rs1373027391 | 0 | SG | FS | – | |
| 29 | 1:160351256 | 6 | p.S206* | non | – | 7 | CN* | F | – | |
| 30 | 1:160351271 | 6 | p.P211R | mis | – | 10 | CN | S | – | |
| 31 | 1:160351286 | 6 | p.Q216P | mis | – | 9 | CN|MT* | F | – | |
| 32 | 1:160351310 | 6 | p.V224_T227del | del | – | 3 | MT | F | – | |
| 33 | 1:160351325 | 6 | p.C230Pfs*32 | fs | – | 8 | CN|IN | F | AC | |
| 34 | 1:160351713 | 7 | p.L251Vfs*2 | fs | – | 3 | CN | F | Com | |
| 35 | 1:160351755 | 7 | p.T265Nfs*8 | fs | – | 2 | CN | F | – | |
| 36 | 1:160352097 | 8 | p.E296G | mis | rs758910156 | 7 | CN|MT* | F | – | |
| 37°N | 1:160352104 | 8 | p.A300Wfs*20 | fs | – | 0 | FR* | F | – | |
| 38 | 1:160352154 | 8 | p.A315V | mis | rs1553210405 | 9 | CN | F | – | |
| 39 | 1:160352188 | 8 | p.M326Ifs*31 | fs | – | 5 | SG | – | – | |
| 40 | 1:160352207 | 8-9 | p.E333Q367del(x9) | spl* | – | 5 | GB | F | – | |
| 41 | 1:160352213 | 8-9 | c.996+7G>A | int | rs202046846 | 9 | GB*|FR* | S | AC|Diab | |
| 42 | 1:160352992 | 9-10 | c.1101+1G>A | spl | rs1347055289 | 10 | GB | F | – | |
| 43 | 1:160353001 | 9-10 | c.1101+10A>G | int | rs1048828525 | 9 | GB* | S | – | |
| 44 | 1:160353198 | 10 | p.D381Sfs*7 | fs | – | 1 | IT* | S | PAPASH | |
| 45° | 1:160354113 | 10-11 | c.1180-5C>G | int | rs7528638 | 5 | GB|FR* | F | – | |
| 46 | 1:160354167 | 11 | p.A410V | mis | rs147225198 | 6 | US* | S | – | |
| 47° | 1:160354187 | 11 | p.N417Y | mis | rs143039637 | 0 | div.|FR* | S | – | |
| 48 | 1:160354190 | 11 | p.Q418* | non | – | 0 | div. | – | – | |
| 49 | 1:160354196 | 11 | p.Q420* | non | – | 10 | CN|SG|div. | F | – | |
| 50° | 1:160354223 | 11 | p.R429* | non | rs771414318 | 5 | JP|FR* | F | – | |
| 51 | 1:160354232 | 11 | p.R432* | non | – | 3 | CN* | FS | KTS | |
| 52 | 1:160354238 | 11 | p.R434* | non | rs1085307081 | 10 | CN|ES*|FR | F | DDD | |
| 53 | 1:160354263 | 11 | p.D443Lfs*6 | fs | – | 3 | ES* | – | DDD | |
| 54 | 1:160354291 | 11-12 | c.1352+1G>A | spl | – | 9 | CN | F | – | |
| 55 | 1:160354194 | 11 | c.1381del(C) | fs | – | 2 | FR* | F | – | |
| 56 | 1:160355941 | 13 | p.Q512* | non | – | 3 | CN* | F | – | |
| 57 | 1:160355959 | 13-14 | p.A486_T517del(x13) | spl* | rs1553210984 | 9 | CN | F | – | |
| 58 | 1:160356263 | 14 | p.T519Nfs*10 | fs | – | 3 | CN | S | – | |
| 59 | 1:160356343 | 14 | p.Y545* | non | – | 8 | IR | F | PASH | |
| 60 | 1:160356655 | 15 | p.Y565* | non | – | 10 | CN | F | – | |
| 61 | 1:160356662 | 15 | p.Q568* | non | – | 9 | JP | F | – | |
| 62 | 1:160356687 | 15 | p.G576V | mis | – | 3 | FR*|MT* | F | – | |
| 63 | 1:160356707 | 15 | p.R583* | non | – | 3 | IT* | F | DDD | |
| 64 | 1:160356712 | 15 | p.E584Dfs*44 | fs | rs1553211087 | 10 | CN | F | – | |
| 65 | 1:160356728 | 15 | p.S590G/p.S590Afs*3 | mis|spl | – | 10 | FR | F | – | |
| 66 | 1:160357046 | 16 | p.Y600* | fs | – | 6 | CN|IN | FS | AC | |
| 67 | 1:160357122 | 16 | p.R626* | non | – | 3 | ES*|IN | F | DDD|AC | |
| 68 | 1:160357161 | 16 | p.Q639Gfs*31 | fs | – | 5 | NL | F | – | |
| 69°N | 1:160357189 | 16 | p.W648* | non | – | 0 | FR* | S | – | |
| 70 | 1:160358788 | 3’ | c.*517_*518del(CA) | utr | rs141849450 | 4 | CN | S | – | |
| PSEN1 (ENST00000324501) | 71 | 14:73186897 | 6 | p.S178Ffs*10 | fs | rs1174374799 | 0 | div. | – | – |
| 72 | 14:73192820 | 7 | p.P242Lfs*11 | fs | rs1595035030 | 10 | CN | F | – | |
| 73° | 14:73198145 | 8-9 | c.868+16G>T | int | rs165932 | 4 | CN|div.|FR* | S | Ps | |
| 74° | 14:73206470 | 9 | p.E318G | mis|spl | rs17125721 | 8 | GB|div.|FR* | F | – | |
| 75°N | 14:73211786 | 10 | p.Q325E | mis | – | 0 | FR* | S | – | |
| 76 | 14:73217163 | 11 | p.S390Efs*20 | fs | – | 3 | FR* | F | Crohn | |
| 77° | 14:73217252 | 11-12 | c.1248+8T>C | int | rs362382 | 0 | div.|FR* | S | – | |
| PSEN2 (ENST00000366783) | 78°N | 1:226882007 | 4 | p.G34S | mis | rs200636353 | 0 | FR* | S | – |
| 79 | 1:226894073 | 12 | p.T380K | mis | rs143912759 | 0 | div. | – | – | |
| PSENEN (ENST00000587708) | 80 | 19:35745943 | 2 | p.R5* | non | – | 3 | CN | F | – |
| 81 | 19:35745965 | 2 | p.L12* | non | rs1555738763 | 5 | DE | – | DDD | |
| 82 | 19:35745973 | 2 | p.C15Pfs*101 | fs | – | 5 | JP | F | AC | |
| 83 | 19:35746418 | 2-3 | p.G21_Y56del(x3) | spl* | rs1555738836 | 4 | IN | F | DDD | |
| 84 | 19:35746418 | 2-3 | c.62-1G>T | spl | – | 5 | IN* | F | DDD | |
| 85 | 19:35746423 | 3 | p.F23Vfs*98 | fs | rs1555738837 | 10 | GB*|FR* | FS | DDD | |
| 86 | 19:35746423 | 3 | p.F23Lfs*46 | fs | rs1555738837 | 10 | CN | FS | DDD|Ps | |
| 87 | 19:35746441 | 3 | p.L29Sfs*92 | fs | – | 3 | TH | F | DDD | |
| 88 | 19:35746472 | 3 | p.R39* | non | – | 5 | DE | – | DDD | |
| 89 | 19:35746525 | 3-4 | p.G21_Y56del(x3) | spl* | rs1970575677 | 5 | FR | F | DDD | |
| 90 | 19:35746706 | 3-4 | c.167-2A>G | spl | rs1555738903 | 7 | CN | F | DDD | |
| 91 | 19:35746709 | 4 | p.Y56* | non | rs751542345 | 8 | Jash|FR* | F | DDD|IBD|PS | |
| 92 | 19:35746735 | 4 | p.L65R | mis | rs1555738906 | 7 | CN | F | Com|DDD | |
| 93 | 19:35746756 | 4 | p.S73Pfs*72 | fs | – | 1 | DE | – | DDD | |
| 94 | 19:35746769 | 4 | p.I77Hfs*45 | fs | – | 1 | CN | F | PASH | |
| 95 | 19:35746770 | 4 | p.I77Tfs*45 | fs | – | 4 | CN* | F | DDD | |
| 96 | 19:35746812 | 4 | p.Y91Tfs*54 | fs | – | 2 | div. | F | – | |
| 97 | 19:35746819 | 4 | p.F94Sfs*51 | fs | rs1555738943 | 9 | CN | F | – | |
| 98 | 19:35746833 | 4 | p.L98Wfs*47 | fs | – | 2 | div. | F | – | |
| 99 | 19:35746845 | 4 | p.*102Rxt*50 | elg | – | 3 | FR* | – | DDD|Diab |
This table concerning γ-secretase genes: APH1A [17], APH1B [16,17,36], NCSTN [12,17,24,36,41,42,53,54,55,56,57,58,59,60,61,62,63,64,65,66,67,68,69,70,71,72,73,74,75,76,77,78,79,80,81,82,83,84,85,86,87,88,89,90,91,92,93,94,95,96,97,98,99,100,101], PSEN1 [17,36,54,55,59,73,102], PSEN2 [17], PSENEN [16,17,56,59,73,91,102,103,104,105,106,107,108,109,110,111,112,113]. The lines with an (°) and in bold correspond to the above-mentioned variants (15) found in our HS cohorts. Those with an (N) are the new ones (8), not mentionned in the literature. Ex.: exons; Eff.: effect (del, elg, fs, int, mis, non, spl, spl*, and utr meaning respectively deletion with no frameshift, elongation, frameshift, intronic variant, missense, nonsense, splice site, known alternative splicing event, and untranslated region variant); R.: number of studied reviews [13,59,78,88,100,114,115,116,117,118,119] (out of 11) citing this mutation; Or.: origin. The two-letter country code was used for the various studies (BR:Brazil; CN:China; DE:Germany; ES:Spain; FR:France; GB:United Kingdom; IN:India; IR:Iran; IT:Italy; JP:Japan; MT:Malta; NL:Netherlands; SG:Singapour; TH:Thailand; US:United States and AfUS and Jash for African-American and Jewish Ashkenazi populations). When the code is followed by an asterisk (*), it indicates that the population is not explicitly mentioned in the article, and the country is inferred based on the authors’ affiliations ; F/S: familial and/or sporadic case. The last column lists the disease and/or syndrome associations mentioned in the articles — all acronyms are defined in Supplementary Figure S1.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



