Submitted:
07 August 2024
Posted:
08 August 2024
You are already at the latest version
Abstract
The prototype misfit layer compound (SnS)1.17NbS2 consists of alternatively a metallic triatomic NbS2 layer in which Nb atoms are sandwiched by S atoms and an insulating SnS double-layer of NaCl-type structure. Here we investigate the effect of lattice misfit on the stability and chemical bonding in the misfit layer compound using a first-principles density-functional theory approach. The calculations show that for the (SnS)1+xNbS2 approximants, the most stable one has x = 0.167, close to the experimental observations. Charge analysis finds a moderate charge transfer from the SnS to NbS2. Sn or S vacancies in the SnS part affect the electronic properties and interlayer interactions. The obtained information here helps understand the mechanism of formation and stability of the misfit layer compounds and ferecrystals, and further to design of novel multilayer compounds and emerging van der Waals heterostructures.
Keywords:
Misfit layer compound
; Commensurate approximation
; Misfit and stability
; Interlayer interactions
; First-principles
; Density-functional theory
1. Introduction
Since the discovery in late 1980s, misfit layer compounds have been a topic of intensive investigation [1,2]. The misfit layer compounds have chemical formula (MX)1+xTX2 (M = Sn, Pb, Bi, Sb, rare earth elements; X = S, Se, Te; T = Ti, V, Cr, Nb and Ta) with planar intergrowth structures [1,2,3,4]. Both MX and TX2 parts have subnano-sized thicknesses. The MX part typically consists of a bi-atom layer of a distorted rock-salt-type constituent and the TX2 a tri-atomic layer with a transition metal atom (T) sandwiched by two X atoms. The intra-interactions in both MX and TX2 parts are strong, whereas the interlayer interactions between MX and TX2 are weak, being van der Waals-type. Structurally the two parts are incommensurate in at least one orientation. Crystallographically these compounds lack three-dimensional periodicity. The misfit layer compounds exhibit unusual physical properties [1,4,5,6,7,8] and potential applications, particularly as two-dimensional electronic devices [4,5,6,9,10,11]. Moreover, lattice misfits provide a platform on which to explore the energetics and local bonding constraints of heterostructures and to engineer novel quantum fabrics and electronic properties [5,6,7].
Both experimental and theoretical efforts have been made to understand the stability of and chemical bonding in the misfit layer compounds. Experimentalists have focused on synthesis for new forms, e.g. nanotubes [1,4,5,9,10,11,12,13,14], structural determination using super-space group theory [1,15,16,17], measurements of physical properties including electrical and thermal transport and electronic and magnetic properties [4,5,6,8,18,19,20] and spectroscopic characteristics [4,21,22,23,24,25,26,27]. The experiments give an improved understanding of the electronic properties and chemical bonding in these systems. Recent progress includes new synthesis approaches and produces new members including the ferecrystals which have thicker MX or TX2 layers [28,29].
Theoretical efforts have been made to understand the chemical bonding in the misfit layer compounds [22,25,29,30,31,32,33,34,35,36,37]. Bond analysis has been applied to investigate the interlayer interactions including charge transfer [22,30,31]. First-principles band structure calculation method have applied to various misfit layer compounds to get insight into the interlayer interactions [22,25,32,33,34,35,36,37]. Meanwhile, the dependence of lattice misfit on stability and chemical bonding is still largely unknown.
(SnS)1.17NbS2 has been regarded as a prototype of the misfit layer compounds [1,4,5,38]. The structure of this compound was determined using the single-crystal X-ray diffraction approach [38,39]. It consists alternatively of a metallic NbS2 triatomic layer with Nb atoms sandwiched by S atoms and an insulating SnS bi-atomic layer of a distorted NaCl-type atomic arrangement. Both NbS2 and SnS subsystems have C-centred orthorhombic unit cells which do not match along the in-plane <100 >orientation. Electrical transport properties measurements revealed this misfit compound is p-type metallic with about 0.87e/Nb holes according to the Hall effect measurements [38]. Photo-emission spectroscopy revealed almost fully occupation of the Nb 4d states indicating charge transfer from the (SnS) part to (NbS2) part [22,23,40]. (SnS)1.17NbS2 was the first misfit layer compound which band structure was investigated by first-principles’ density-functional theory within the local-density approximation (LDA) for an approximant (SnS)1.20NbS2 [22]. Charge transfer from the SnS layer to the NbS2 layer was revealed. However, in the early calculation van der Waals forces and the effect of the misfit parameter, x in the formula (SnS)1+xNbS2 were not included. Recently, Gotoh [31] determined the atomic modulation in two-dimensional (2D) misfit layer (SnS)1.17NbS2 by means of accurate modelling of incommensurate composite crystal structure. The study suggested sulphur vacancy in the SnS part. Based on the first-principles band structure calculations, La vacancy in LaS part was proposed in the misfit layer compound (LaS)1.20CrS2 [36,37]. Kabliman et al performed ab initio investigation on the stability of (PbS)1.14TaS2 using different ratios of PbS and TaS2 and obtained the composition range of high stability with respect to the parental binary compounds [33].
Recent progress in computational materials sciences enables studying larger systems with high reliability and accuracy. The stability of the misfit layer compounds (SnS)1.17NbS2 is here investigated using the approximants (SnS)1+xNbS2 with x = 0.200, 0.167 and 0.143 using first-principles density-functional theory with van der Waals corrections created by Klimes et al. [41,42]. Furthermore, the effects of Sn or S vacancies in the SnS part on the electronic properties and interlayer bonding are examined. The obtained information here is not only helpful to get insight into the stability, chemical compositions, crystal structure and chemical bonding in the misfit layer compound, (SnS)1.17NbS2 and other misfit layer compounds [1,2,3,4,22], but also to that of the ferecrystals and modification via doping [10,24,28], and of other unusual materials, such as FeCl3/graphene of misfit nature [43].
2. Details of Computational Methods
2.1. Supercells for Misfit Layer Compound (SnS)1+xNbS2 Approximants
A plane wave approach employed in the present study utilizes the periodic boundary conditions and thus, supercells were built [44]. According to the experimental observations [1,4,38,39] and previous work [22], the designed supercells have different misfit parameters and symmetry. The created configurations include shifting the SnS part along the misfit direction to contain different types of interlayer bonding. Sn/S vacancies were also included within the (SnS)1.167NbS2. In the formula (SnS)1+xNbS2 seven input configurations were produced: Case a) has x = 0.20 based on the previous work [22], Case b) x = 0.167 and Case c) x = 0.167 with atoms in SnS part shifted along the misfit orientation; Case d) x = 0.143. Two configurations of (i) S vacancy with symmetry Case e) and (ii) without symmetry Case f) were included in the configurations with x = 0.167. Case g) has the configuration with x = 0.167 and Sn vacancies.
Formation energy of the misfit layer compound (SnS)1+xNbS2 with respect to the binaries, SnS and NbS2 is defined as
ΔE((SnS)1+xNbS2) = E((SnS)1+xNbS2) – [(1+x) E(SnS)+E(NbS2)]
Here E((SnS)1+xNbS2), E(SnS) and E(NbS2) represent the calculated total-valence-electron energy of (SnS)1+xNbS2, the parental binaries α-SnS and 3R-NbS2, respectively. The unit of the formation energy ΔE is eV per NbS2. A negative sign of ΔE means an exothermal relation from the binaries to the ternary compound. At 0 K and 0 Pa, the formation energy is equal to the formation enthalpy, ΔE = ΔH if the zero-point vibration is ignored.
2.2. First-Principles Code and Settings
The first-principles code VASP [44] used in the present study employs the projector augmented wave method [45] within the generalized gradient approximation [46]. The van der Waals density functional by Klimes, et al. [41,42] was utilized since it works well for various systems [41,47]. The atomic electronic configurations are Nb ({[Ar] 3d10 4s2} 4p6 4d4 5s2 5p1), S ([Ne] 3s2 3p4) and Sn ({[Kr] 4d10} 5s2 5p2). The symbol {} represents frozen core electrons and the rest are the valence electrons which include the semicore Nb 4p6 electrons.
High cut-off energies of 400.0eV for the wave functions and 550.0eV for the augmentation functions were used. Those values are notably higher than the corresponding default values (EMAX/EAUG = 293.2/455.7eV for Nb, 258.7/335.1eV for Sn and 103.2/263.6eV for Sn, respectively). Dense k-meshes were used for sampling the electronic wave functions, e.g. a 24×24×8 (449 k-points), and 8×24×24 (845 k-points) in the Brillouin zones (BZ) of 3R-NbS2 and α-SnS, respectively, based on the Monkhorst-Pack scheme [48]. Test calculations using different cut-off energies and k-meshes demonstrated that the settings are reliable to produce results with deviations within 1meV/atom.
3. Results
3.1. The Parental SnS and NbS2 Phases
The parental binary compounds, SnS and NbS2 are first investigated using the code and settings in Section 2. There are two phases for SnS reported in the literature [22,49,50,51]. A noval phase of NaCl-type structure was also included. Experiments revealed that both 2H- and 3R-NbS2 exist at ambient conditions [52,53]. There are two types of atomic arrangement for the 2H phase. Table 1 shows the obtained results.
The calculations show that α-SnS is more stable than β-phase while the structure of the NaCl-type is the least stable one. The results agree with the experimental observation [49,50]. 3R-NbS2 is more stable than the 2H-phases (Table 1), agreeing with the experiments [52,53]. Table 1 also shows that the calculated lattice parameters of the SnS and NbS2 phases close to the corresponding experimental values, indicating availability of the DFT-GGA approaches with van der Waals corrections [41,42,54] and the settings. Structurally, α- and β-SnS phases are very similar: both are composed of zigzagged bi-atomic SnS layers along the α-SnS[1 0 0] and β-SnS[0 1 0] axis as shown in Figure 1a and 1b, respectively. The notable difference is the interlayer bonds. In α-SnS there is weak interlayer bonding with Sn-S bond length of 3.43Å, whereas there is no direct bonding in β-SnS. The calculated density of states for α- and β-SnS and 3R-NbS2 are shown in Figure 2.
The calculations showed that the DOS curves of the SnS phases have similar frames and are composed of three parts. The lower part at -14 to -12 eV from the Fermi level (zero eV) is dominated by S 3s states. The second part (valence band) starts from about -8.0eV to the Fermi level. This part is dominated by Sn 5s, 5p and S 3p states. Note that the Sn 5s states have high density at the lower energy part, from -8.0 to -6.0 eV and from -1.0 to the Fermi level. Such large splitting of the lone-pair electrons indicating strong interaction with the S 3p states. However, in the SnS phases both bonding (the lower part) and antibonding (top part) orbitals are fully occupied, indicating not contribution to the chemical bonding in the SnS phases. The conduction band are dominated by Sn 5p 4d states. The calculated band gap for α-SnS is smaller than the experimental value, which is also not unusual for the standard density functionals even with the van der Waals corrections [41,42,54]. Table 2 also shows that i) α-SnS is more stable than β-SnS, and ii) the calculated band gap for α-SnS is larger than those for β-SnS. Such differences from the previous first-principles calculations using the local-density functional (LDA) and spherical wave approach [22,51] correspond to the advantage of the GGA over the LDA [54].
The DOS curve of 3R-NbS2 consists of three parts: the S 3s band, the valence band and the conduction band. The S 3s states form a peak from -13.6 eV to -11.5eV below the Fermi level. The valence band is composed of two parts. The lower part of conduction band starts from -5.5eV to -0.4eV, dominated by Nb 4d and S 3p states. There is a valley at -0.4eV. The upper part of the valence band in the range between -0.4 to 1.6eV is dominated by Nb 4d states. The peak of the Nb 4d states is at -0.1eV. The Fermi level dominated by Nb 4d states hybridized with some S 3p states. Table 2c shows a fraction of unoccupied Nb 4d states. There is a gap between the valence band and the conduction band. The conduction band starts from 2.1eV, consisting of Nb 4d and S 3p states. The electronic structure shows strong covalence nature.
3.2. Stability and Crystal Structure of (SnS)1+xNbS2 Approximants
First-principles calculations were performed for the designed misfit systems as mentioned in Section 2. The obtained results, including lattice parameters and formation energies according to Equation 1 are shown in Table 2. The optimized structures are schematically shown in Figure 3. The atomic coordinates and chemical bonds of the selected systems are included in the Supplementary Materials (Table S2 for case a), S3 for case b), S4 for case d) and S5 for case e), respectively).
Figure 3b and Figure 3c show that the SnS shifts induce slight rearrangements of the S(Nb) and Nb atoms and the calculated lattice parameters differ slightly from the pristine one as (Table 2). However, there is not notable change of the interlayer bonding and the formation energy difference between the two configurations is marginal with the calculation accuracy.
The calculations revealed that all the structures (Cases a-d) have negative formation energies with respect to the stable binary compounds, α-SnS and 3R-NbS2. The structure with x = 0.167 (b and c in Table 2) have the lowest energies. The energy difference between case b and case c is minor, indicating flexibility the misfit layer compound. This composition is close to the experimental value (x = 0.171). Therefore, the previous commensurate approximant with x = 1.20 deviates from the experimental observations.
As shown in the Tables the Nb-S bonding in all the compositions and structure in the NbS2 part have the Nb-S bond-lengths ranging from 2.47 to 2.50 Å, which are close that of the bulk 3R-NbS2 (2.49Å). In the SnS part, each Sn has five S neighbours with Sn-S bond-lengths ranging from 2.59Å to 3.0Å, which are close to those in β-SnS (Table 1). Each Sn has generally one long interlayer Sn-S(Nb) bond with bond-lengths about 3.2 Å, which are in the range of van der Waals bonding.
3.3. Electronic Structure of (SnS)1.167NbS2
First-principles electronic band-structure calculations were performed for the most stable configuration. The obtained density of states (DOS) and band dispersion curves along the high symmetry lines are shown in Figure 4 and 5, respectively.
As shown in Figure 4 and Figure 5, the electronic band structure of (SnS)1.167NbS2 consists of three separated parts, which frame is in line with the previous calculations for (SnS)1.20NbS2 approximant using the DFT-LDA) approximation [22]. Meanwhile, careful analysis revealed subtle differences.
The lowest part starting from -13.8 eV to -11.6 eV consists of S 3s states. The S 3s states in the NbS2 part (S(Nb) 3s in short) are positioned lower in energy than those in the SnS part (S(Sn) 3s in short). The peak of S(Nb) 3s is at -12.0 eV which is about 0.2 eV lower in energy than that of S(Sn). This result is in line with the DOS curves of the binary components (Figure 2). There is an energy gap of about 4.2 eV separate the S 3s states from the valence band.
The valence band starts at -7.4 eV and ends at 1.0 eV. The Sn 5s states dominate lower part in the energy range between -7.4 eV to -5.5 eV. Interestingly, there are Sn 5s states around the Fermi level and there are unoccupied Sn 5s states (Figure 4d), indicating that the inert Sn 5 s2 pair-electrons join the chemical bonding. This phenomenon agrees with the previous study on the transition metal dichalcogenides intercalated by post-transition metals, M1/3TmS2 (M = Pb, Sn; Tm = Nb, Ta) [55]. The unoccupied Sn 5s, 5p and S(Sn) 3s states in the SnS part, which is in contrast with those in pure SnS phases (Figure 2) indicates charge transfer from the SnS part to the NbS2 part in this misfit layer compound. Figure 4 also shows that the valence band is composed of Sn 5s, 5p, Nb 4d and S 3p states. Nb 4d states dominate the states around the Fermi level. There are unoccupied Nb 4d states. Compared with the corresponding pDOS of the Nb 4d peak at -0.1eV of 3R-NbS2, the unoccupied part of the Nb 4d states in the misfit compound is smaller, indicating charge transfer from SnS part. This result agrees with the experimental observations that the Nb 4d states are largely occupied due to the charge transfer. There is a smaller hole in the misfit compound as compared with that in the parental NbS2 [1,4,22,27,38]. Such charge transfer is similar to that in the intercalation compounds [55]. Between the valence band and the conduction band there is a gap of about 0.5eV. The conduction band is composed dominantly of Nb 5d, Sn 5p and S 3p states. The latter indicates covalence between the metals and S.
To assess the interlayer chemical interaction and charge trasnfer, the Bader charge model [56,57] is used. The obtained results for the (SnS)1+xNbS2 approximants are listed in Tables S2-S5. Table 3 shows the averaged values for the (SnS)1.167NbS2 approximant.
As shown in Table 3 each Sn atoms loses 0.89e in the misfit layer compound, which is higher than that in α-SnS but slightly smaller than that in β-SnS. Meanwhile, the value of the lost electrons per Sn is larger than the gain by the S in SnS. Consequently, the (NbS2) part gains net electrons. Thus, there are charge transfer from the SnS part transfer to the NbS2 part. Analysis showed that the averaged value of the charge transfer from the SnS to the NbS2 part in (SnS)1.167NbS2 is 0.18e/NbS2 for both cases b) and c) in Table 1. This corresponds to the long S(Nb)-Sn bonds in the misfit compounds.
The electrons in the atomic spheres in the misfit layer compounds and the components in the misfit layer compound, (SnS)1.167NbS2 (see Table S6). It is revealed no significant differences for each elemental species in the misfit layer compound and in the reference binaries. This means that the charge transfer occurs mainly in the free space outside of the atomic spheres. This corresponds to the long S(Nb)-Sn interlayer bonding (Table 3).
3.4. Effects of Sn or S Vacancy on the Local Bonding in (SnS)1.67NbS2
The optimized structures of the Sn- and S-vacant (SnS)1.167NbS2 are shown in Figure 6. The interlayer Sn-S(Nb) bonds within 3.1Å are included, which helps get more insight into the influences of the S or Sn deficiency on the interlayer interactions. The interatomic bond-lengths and related Bader’s charges at the atomic sites are summarized in Table 3. Moreover, the obtained partial density of states for selected atoms and the total density of states for the S vacant (SnS0.929)1.167NbS2 and Sn vacant (Sn0.929S)1.167NbS2 are displayed in Figure 7a and 7b, respectively.
The influence of the S deficiencies, (SnS0.929)1.167NbS2 approximant revealed the short Sn-S(Nb) bonds (Figure 6b and Table 3). The Sn atoms nearby the S vacancies may have one or two S(Nb) neighbours with the bond lengths varying between 2.8Å to 3.2Å. In comparison, the Sn-S(Nb) bond lengths range from 3.06Å to 3.25Å in the approximant (SnS)1.167NbS2 (Figure 6a). Meanwhile, analysis showed that for the Sn deficiency configuration, (Sn0.929S)1.167NbS2 shortest interlayer Sn-S(Nb) bond length is over 3.1 Å as shown in Figure 6c and Table 3, indicating weaker interlayer interactions.
Bader’s charge analysis revealed more electron loss for the Sn atoms (0.90e/Sn) in the Sn-deficient approximant, (Sn0.929S)1.167NbS2 than those in the S deficient compound (0.80e/Sn). This is reasonable considering the Sn/S(Sn) ratio in the ionic model. Meanwhile, the electrons in the atomic spheres (Table S6) show moderate variation for each species from the corresponding ones in the parental and the vacancy-free misfit layer compound, indicating charge transfers outside of the spheres. Figure 7 showed that the calculated partial of selected atoms in and total density of states of the (SnS0.929)1.167NbS2 (a) and (SnS) part of (Sn0.929S)1.167NbS2 (b) misfit layer compounds.
The frame of the DOS curves for both the S (Figure 7a) and Sn deficient (Figure 7b) systems are similar to that of the stoichiometric system (Figure 4): The S 3s states in the ranges between -13.8eV to -11.5eV; the valence bands between -7.5 to 1.0 eV. The Fermi level is dominated by Nb 4d states. The lone Sn 5s2 pair splits into two parts.
One is at the lower part (-7.5eV to -5.5eV), related to Sn 5s bonding. The other one Sn 5s states are positioned at the upper parts of the valence bands (-1.5eV to 0.2eV), belonging to antibonding. There is a gap between valence band and the conduction band. Meanwhile, there are also some subtle differences between the electronic properties of the S or Sn deficient compounds and that of the stoichiometric compound.
As shown in Figures 7a, in the S deficient (SnS0.929)1.167NbS2 approximant, there are some unoccupied antibonding 5s orbitals of the Sn1 with five S(Sn) neighbours, meanwhile Sn2 atoms which have four S(Sn) and one short Sn-S(Nb) bond (bond length being 2.82Å) and Sn3 with four S(Sn) neighbours are almost fully occupied. It is also notable there are a sharp Sn 5d bonding peak at around -7.0eV for the Sn3 5s states. The S(Sn) 3p states in the valence bands in the S deficient configurations are below the Fermi level. These results indicate that S deficiency in SnS induces electron filling of the S 3p states and enhances interlayer bonding.
In Figure 7b, in the Sn deficient (Sn0.929S)1.167NbS2 approximant, both Sn1 and Sn2 with different numbers of S neighbours have some unoccupied antibonding Sn 5s states. The Fermi level is also in the middles of the S(Sn) 3p states. Meanwhile, the S(Nb) 3s states in the valence band are below the Fermi level, indicating weaker interlayer interactions.
4. Conclusions
The first-principles calculations revealed dependence of the formation energy on the misfit parameter x for the (SnS)1+xNbS2 approximants. The most stable approximant has x = 0.167, close to the experimental observed (SnS)1.171NbS2. Bader charge analysis found a moderate charge transfer (0.18e/NbS2) from the SnS layer to the NbS2 tri-layer part via the interlayer van der Waals interactions. S- or Sn-deficiencies in the SnS part have influences on the local bonding and interlayer interactions. There are short Sn-S(Nb) interlayer bonds formed in the S-deficient system, which enhances the interlayer interactions. Meanwhile for the Sn-deficient (Sn0.929S)1.17NbS2, the interlayer Sn-S(Nb) bonding becomes weaker.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Table S1: Designed approximants and related symmetry for the (SnS)1+xNbS2 (x=0.20, 0.167, 0.143) misfit compounds. *Pseudo-orthorhombic unit cells. The experimental values are included for comparison; Table S2: Calculated results (coordinates of atoms, interatomic distances and Bader charges at the atomic sites) using the DFT + vdW approach for the (SnS)1.20NbS2 misfit layer compound, case a). Table S3: Calculated results (coordinates of atoms, interatomic distances and Bader charges at the atomic sites) using the DFT + vdW correction for the (SnS)1.167NbS2 misfit layer compound, case b); Table S4: Calculated results (coordinates of atoms, interatomic distances and Bader charges at the atomic sites) using the DFT + vdW correction for the (SnS)1.143NbS2 misfit layer compound, case d; Table S5: Calculated results (coordinates of atoms, interatomic distances and Bader charges at the atomic sites) using the DFT + vdW correction for the (SnS1-x)1.167NbS2 (x=0.071) misfit layer compound, case e); Table S6: Calculated electronic configurations in the atomic spheres in the parent binary compounds and the misfit layer compounds, (Sn1-xS)1.167NbS2 (x = 0.00), case b) and (x = 0.0714), case e).
Author Contributions
C. F. is the author who completed the Conceptualization, methodology, validation, formal analysis, writing, visualization, etc.
Funding
Financial support from EPSRC (UK) under grant number EP/S005102/1 is gratefully acknowledged.
Data Availability Statement:
Conflicts of Interest
The author declares no conflicts of interest.
References
- Wiegers, G.A.; Meetsma, A.; van Smaalen, S.; Hannge, R. J.; Wulff, J.; Zeinstra, T.; de Boer, J. L.; Kuypers, S.; van Tendeloo, G.; van Landuyt, J.; Amelinckx, S.; Meerschaut, A.; Rabu, P.; Rouxel, J. Misfit layer compounds (MS)nTS2 (M = Sn, Pb, Bi, rare earth elements; T = Nb, Ta; n = 1.08 – 1.19), a new class of layer compounds, Solid State Commun. 1989, 70, 409-413. [CrossRef]
- Meerschaut, A. Misfit layer compounds, Curr. Op. Solid State Mater. Sci. 1996, 1, 250-259. [CrossRef]
- Rouxel, J.; Meerschaut, A.; Wiegers, G. A. Chalcogenid misfit layer compounds, J. Alloys Compds. 1995, 229, 144-157. [CrossRef]
- Wiegers, G. A. Misfit layer compounds: structures and physical properties, Prog. Solid State Chem. 1996, 24, 1-139. [CrossRef]
- Ng, N.; McQueen, T. M. Misfit layered compounds: Unique, tunable heterostructured materials with untapped properties, APL Mater. 2022, 10, 100901. [CrossRef]
- Leriche, R. T.; Palacio-Morales, A.; Campetella, M.; Tresca, C.; Sasaki, S.; Brun, C.; Debontridder, F.; David, P.; Arfaoui, I.; Šofranko, O.; Samuely, T.; Kremer, G.; Monney, C.; Jouen, T.; Cario, L.; Calandra, M.; Cren, T. Misfit layer compounds: A platform for heavily doped 2D transition metal dichalcogenides, Adv. Func. Mater. 2020, 31, 2007706. [CrossRef]
- Zullo, L.; Marini, G.; Cren, T.; Calandra, M. Misfit layer compounds as ultratunable field effects transistors: From charge transfer control to emergent superconductivity, Nano Lett. 2023, 23, 6658-6663. [CrossRef]
- Nader, A.; Briggs, A.; Gotoh, Y. Superconductivity in the misfit layer compounds (BiSe)1.10(NbSe2) and (BiS)1.11(NbS2), Solid state Commun. 1997, 101, 149-153. [CrossRef]
- Huang, S. T.; Bai, J. H.; Long, H. Y.; Yang S. C.; Chen, W. W.; Wang, Q. Y.; Sa, B. S.; Guo, Z. Y.; Zheng, J. Y.; Pei, J. J.; Du, K.-Z.; Zhan, H. B. Thermally activated photoluminescence induced by tunable interlayer interaction in naturally occurring van der Waals superlattice SnS/TiS2, Nano Lett. 2024, 24, 6061-6068. [CrossRef]
- Merrill, D. R.; Moore, D. B.; Bauers, S. R.; Falmbigl, M.; Johnson, D. C. Misfit layer compounds and ferecrystals: Model systems for thermoelectric nanocomposites, Materials, 2015, 8, 2000-2029. [CrossRef]
- Lorenz, T.; Joswig, J.-O.; Seifert, G. Two-dimensional and tubular structures of misfit compounds: structural and electronic properties, Beilstein J. Nanotechnol. 2014, 5, 2171-2178. [CrossRef]
- Serra, M.; Tenne, R. Nanotubes from misfit layered compounds, J. Coord. Chem. 2018, 71, 1669-1678. [CrossRef]
- Sreedhara, M. B.; Bukvišová, K.; Khadiev, A.; Citterberg, D.; Cohen, H.; Balema, V.; Pathak, A. K.; Novikov, D.; Leitus, G.; Kaplan-Ashiri, I.; Kolíbal, M.; Enyashin, A. N.; Houben, L.; Tenne, R. Nanotubes from the misfit layered compound (SmS)1.19TaS2: Atomic structure, charge transfer, and electric properties, Chem. Mater. 2022, 34, 1838-1853. [CrossRef]
- Lemon, M.; Harvel, F. G.; Gannon, R. N.; Lu, P.; Rudin, S. P.; Johnson, D. C. Targeted synthesis of predicted metastable compounds using modulated elemental reactants, J. Vac. Sci. Technol. A 2023, 41, 022203. [CrossRef]
- van Smaalen, S. A superspace group description of the misfit layer structure of (SnS)1.17(NbS2), J. Phys.: Condens. Matter 1989, 1, 2791-2800. [CrossRef]
- Jobst, A.; van Smaalen, S. Intersubsystem chemical bonds in the misfit layer compounds (LaS)1.13TaS2 and (LaS)1.14NbS2, Acta Cryst. B 2002, 58, 179-190. [CrossRef]
- Fan, H. F.; van Smaalen, S.; Lam, E. J. W.; Beurskens, P. T. Direct methods for incommensurate intergrowth compounds. I. determination of modulation, Acta Cryst. A 1993, 49, 704-708. [CrossRef]
- Shen, X.; Chen, D.; Zhao, H. F.; Yao, Y.; Liu, X. Y.; Yu, R. C. Misfit-layered compound PbTiS3 with incommensurate modulation: transmission electron microscopy analysis and transport properties, Chin. Phys. B 2013, 22, 116102. [CrossRef]
- Meerschaut, A.; Guemas, L.; Auriel, C.; Rouxel, J. Preparation, structure determination and transport properties of a new misfit layer compound: (PbS)114(NbS2)2. Eur. J. Solid State Inorg. Chem. 1990, 27, 557-570.
- Wiegers, G. A.; Haange, R. J. Electric transport and magnestic properties of the misfit layer compound (LaS)1.14NbS2, J. Phys.: Condens. Matter, 1990, 2, 455-463. [CrossRef]
- Sourisseau, C.; Cavagnat, R.; Tirodo, J. L. A Raman study of the misfit layer compounds, (SnS)1.17NbS2 and (PbS)1.18TiS2, J. Ramman Spec. 1992, 23, 647-651. [CrossRef]
- Fang, C. M.; Ettema, A. R. H.; Haas, C.; Wiegers, G. A.; van Leuken, H.; de Groot, R. A. Electronic structure of the misfit -layer compound (SnS)1.17NbS2 deduced from band-structure calculations and photoelectron spectra, Phys. Rev. B 1995, 52, 2336-2347.
- Krasovskki, E.E.; Tiedje, O.; Schottke, W.; Brandt, J.; Kanzow, J.; Rossnagel, K.; Kipp, L.; Skibowski, M.; Hytha, M.; Winkler, B. Electronic structure and UPS of the misfit chalcogenide (SnS)NbS2 and related compounds, J. Elec. Spec. related phenomena, 2001, 114-116, 1133-1138. [CrossRef]
- Göhler, F.; Mitechson, G.; Alemayehu, M. B.; Speck, F.; Wanke, M.; Johnson, D. C.; Seyller, T. Charge transfer in (PbSe)1+δ(NbSe2)2 and (SnSe)1+δ(NbSe2)2 ferecrystals investigated by photoelectron spectroscopy, J. Phys.: Condens. Matter 2018, 30, 055001.
- Fang, C. M.; van Smaalen, S.; Wiegers, G. A.; Haas, C.; de Groot, R. A. Electronic structure of the misfit layer compound (LaS)1.14NbS2: Band-structure calculations and photoelectron spectra, J. Phys.: Condens. Matter 1996, 8, 5367-5382.
- Chikina, A.; Bhattacharyya, G.; Curcio, D.; Sanders, C. E.; Bianchi, M.; Lanatà, N.; Watson, M.; Cacho, C.; Breholm, M.; Hofmann, P. One-dimensional electronic states in a natural misfit structure, Phys. Rev. Mater. 2002, 6, L092001. [CrossRef]
- Fang, C. M.; Wiegers, G. A.; Haas, C. Charge transfer and photoemission spectra of some later rare earth misfit layer compounds, (LnS)1+xTS2 (Ln = Tb, Dy, Ho; T = Nb, Ta), Physica: Condens. Matter 1997, 233, 134-138.
- Beekman, M.; Heideman, C. L.; Johnson, D. C. Ferecrystals: non-epitaxial layered intergrowths, Semicond. Sci. Technol. 2014, 29, 064012. [CrossRef]
- Reisinger, G. R.; Richter, K. W. Review of vanadium-based layered compounds, J. Alloys Compds. 2021, 891, 161976. [CrossRef]
- Deudon, C.; Lafond, A.; Leynaud, O.; Moëlo, Y.; Meerschaut, A. Quantification of the interlayer charge transfer, via bond valence calculation, in a 2D misfit compounds: The case of (Pb(Mn,Nb)0.5S1.5)1.15NbS2, J. Solid State Chem. 2000, 155, 1-18. [CrossRef]
- Gotoh, Y. Structural stability and charge transfer of (SnS0.93)1.17NbS2 by accurate modelling of incommensurate composite crystal structure using information criterion, J. Phys. Soc. Jpn. 2021, 90, 094601.
- Brandt, J.; Kanzow, J.; Roßbagl, K.; Kipp, L.; Skibowski, M.; Krasovskii, E.; Schattke, W.; Traving, M.; Stettner, J.; Press, W.; Dieker, C.; Jäger, W. Band structure of the misfit compound (PbS)NbS2 compared to NbSe2: experiment and theory, J. Electron Spec. Related Phenomena, 2001, 114-116 555-561. [CrossRef]
- Kabliman, E.; Blaha, P.; Schwarz, K. Ab initio study of stabilization of the misfit layer compound (PbS)1.14TaS2, Phys. Rev. B 2010, 82, 125308.
- Abramov, S. P. Varieties of charge transfer and bonding between layers in misfit layer compounds (MX)xTX2, J. Alloys Compds. 1997, 259, 212-218.
- Fang, C. M.; de Groot, R. A.; Wiegers, G. A.; Haas, C. Band structure calculations and photoemission spectra of (SnS)1.20TiS2, J. Phys.: Condens. Matter 1996, 8, 1663-1667.
- Fang, C. M.; de Groot, R. A.; Wiegers, G. A.; Haas, C. Electronic structure and evidence for La vacancies in the misfit layer compound (LaS)1.20CrS2, J. Phys. Chem. Solids, 1997, 58, 1103-1109. [CrossRef]
- Cario, L.; Johrendt, D.; Lafond, A.; Felser, C.; Meerschaut, A.; Rouxel, J. Stability and charge transfer in the misfit layer compound, (LaS)(SrS)0.2CrS2: Ab initio band-structure calculations, Phys. Rev. B 1997, 55, 9409-9414.
- Wiegers, G. A.; Meetsma, A.; Haange, R. J.; de Boer, J. Structure and physical properties of (SnS)1.18NbS2, ‘‘SnNbS3’’, a compound with misfit layer structure, Mat. Res. Bull. 1988, 23, 1551-1559. [CrossRef]
- Meetsma, A.; Wiegers, G. A.; Haange, R. J.; de Boer, J. L. The incommensurate misfit layer structure of (SnS)1.17NbS2, ‘SnNbS3’: I. A study by means of X-ray diffraction, Acta Cryst. A 1989, 45, 285-291. [CrossRef]
- Ohno, Y. Electronic structure of the misfit layer compounds PbTiS3 and SnSbS3, Phys. Rev. B 1991, 44, 1281-1291.
- Klimes, J.; Bowler, D. R.; Michelides, A. Chemical accuracy for the van der Waals density functional, J. Phys.: Cond. Matt. 2010, 22, 022201. [CrossRef]
- Klimes, J.; Bowler, D. R.; Michelides, A. Van der Waals density functionals applied to solids, Phys. Rev. B 2011, 83, 195131. [CrossRef]
- Cowley, J. M.; Ibers, J. A. The structures of some ferric chloride-graphite compounds, Acta Cryst. 1956, 9, 421-431. [CrossRef]
- Kresse, G.; Hafner, J. Ab initio molecular-dynamics simulation of the liquid-metal-amorphous-semiconductor transition in germanium, Phys. Rev. B 1994, 49; 14251–14269.
- Blöchl, P. E. Projector augmented-wave method, Phys. Rev. B 1994, 50, 17953–17978. [CrossRef]
- Perdew, J. P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple, Phys. Rev. Lett. 1996, 77, 3865–3868. [CrossRef]
- Fang, C. M.; Mohammadi, V.; Nihtianov, S.; Sluiter, M. F. H. Stability, geometry and electronic properties of BHn (n= 0 to 3) radicals on the Si(001)3×1:H surface from first-principles, J. Phys. Cond. Matter 2020, 32, 235201.
- Monkhorst, H. J.; Pack, J. P. Special points for Brillouin-zone integrations, Phys. Rev. B 1976, 13, 5188–5192. [CrossRef]
- Wiedemeier, H.; von Schnering, H. G. Refinement of the structure of GeS, GeSe, SnS and SnSe, Z. Kristollogr. 1978, 148, 295-303.
- von Schnering, H. G.; Wiedemeier, H. The high temperature structure of β-SnS and β-SnSe and the B16 to B33 type λ-transition path, Z. Kristollogr. 1981, 156, 143-150.
- Ettema, A. R. H. F.; Groot, R. A.; Haas, C.; Turner, T. S. Electronic structure of SnS deduced from photoelectron spectra and band-structure calculations. Phys. Rev. B, 1992, 46, 7363-7373. [CrossRef]
- Fisher, W. G.;. Sienko, M. J. Stoichiometry, structure, and physical properties of niobium disulfide, Inorg. Chem. 1980, 19, 39-43.
- Morosin, B. Structure refinement on NbS2, Acta Cryst. B 1974, 30, 551-552.
- Jones, R. O. Density functional theory: Its origins, rise to prominence, and future, Rev. Mod. Phys. 2015, 87, 897-923. [CrossRef]
- Fang, C. M.; Wiegers, G. A.; Meetsma, A.; de Groot, R. A.; Haas, C. Crystal structure and band structure calculations of Pb1/3TaS2 and Sn1/3NbS2, Physica B 1996, 226, 259-267.
- Bader, R. F. W. A quantum-theory of molecular-structure and its applications. Chem. Rev. 1991, 91, 893–928. [CrossRef]
- Bader, R. F. W. A bonded path: A universal indicator of bonded interactions. J. Phys. Chem. A 1998, 102, 7314–7323. [CrossRef]
Figure 1.
Schematic structures of orthorhombic α-SnS along the [0 0 1] orientation (a), β-SnS along the [0 0 1] orientation (b) and 3R-NbS2 along the [1 0 0 0] orientation. Sn-S and Nb-S bonds within 3.0 Å are included.
Figure 1.
Schematic structures of orthorhombic α-SnS along the [0 0 1] orientation (a), β-SnS along the [0 0 1] orientation (b) and 3R-NbS2 along the [1 0 0 0] orientation. Sn-S and Nb-S bonds within 3.0 Å are included.
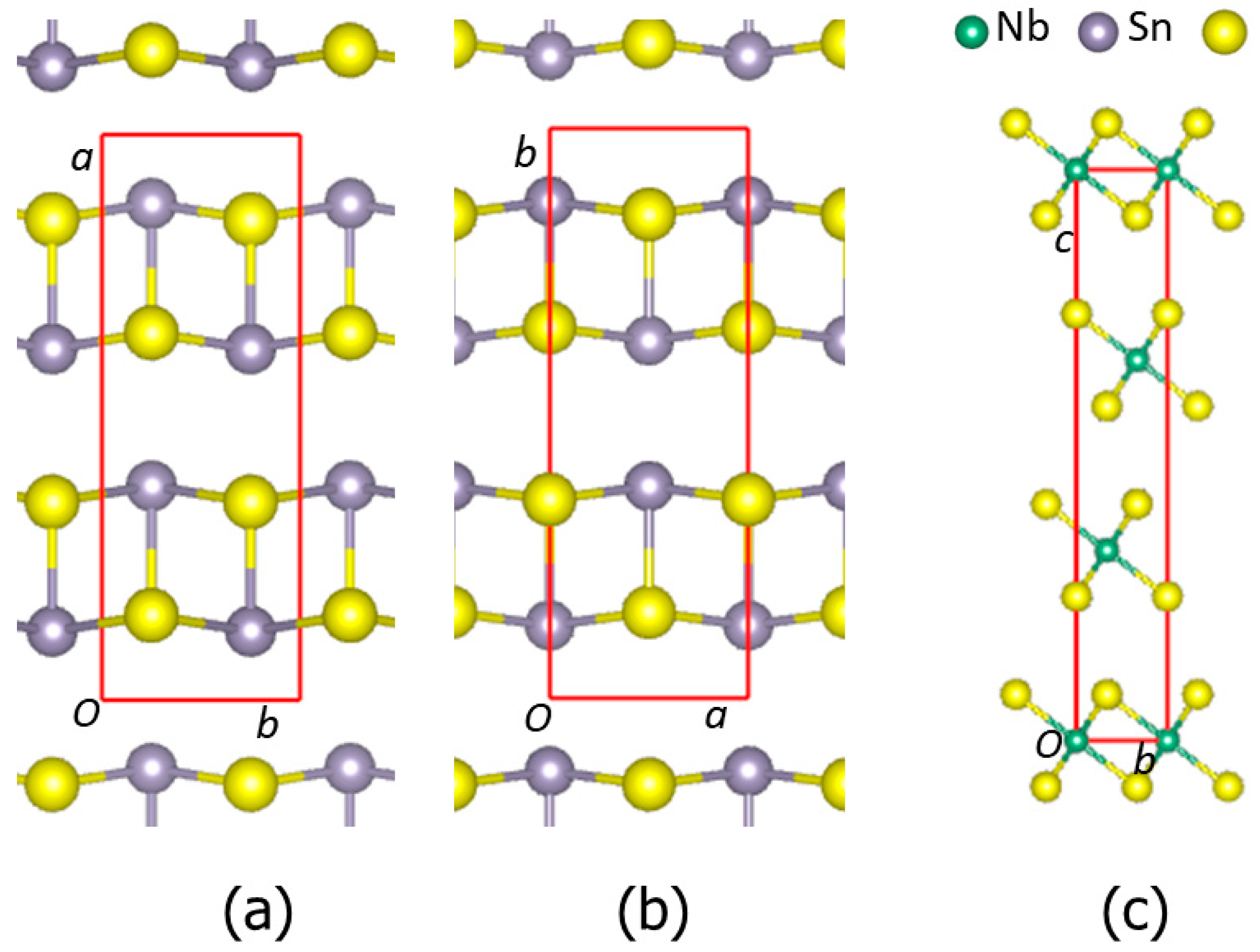
Figure 2.
Calculated partial and total densities of states (PDOS and TDOS) for the reference binary compounds, (a) α-SnS, (b) β-SnS and (c) 3R-NbS2. The perpendicular dotted line represents the Fermi level (zero ev). .
Figure 2.
Calculated partial and total densities of states (PDOS and TDOS) for the reference binary compounds, (a) α-SnS, (b) β-SnS and (c) 3R-NbS2. The perpendicular dotted line represents the Fermi level (zero ev). .
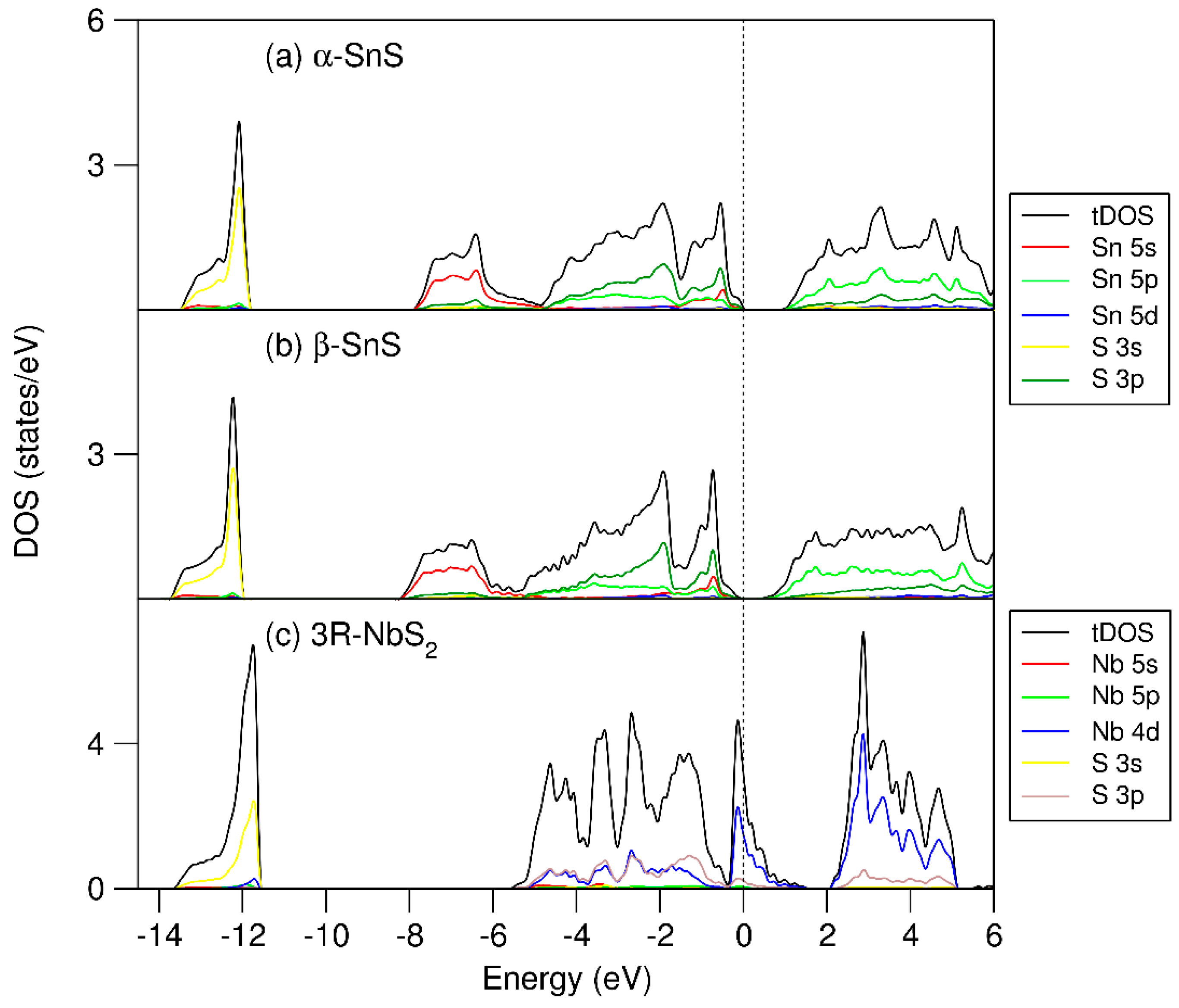
Figure 3.
Schematic structures of the misfit layer compound (SnS)1+xNbS2 with the [0 1 0] projections with the commensurate approximants, x = 0.200 (a), x = 0.143 (d), x = 0.167 (b) and (c) with SnS-shifts. The schematic structure of (SnS)1+xNbS2 with the [1 0 0] projection is shown in (e). The meaning of the spheres is included: The small green spheres represent Nb, dark Sn and golden S. Sn-S and Nb-S bonds with lengths less than 3.5 Å are shown. .
Figure 3.
Schematic structures of the misfit layer compound (SnS)1+xNbS2 with the [0 1 0] projections with the commensurate approximants, x = 0.200 (a), x = 0.143 (d), x = 0.167 (b) and (c) with SnS-shifts. The schematic structure of (SnS)1+xNbS2 with the [1 0 0] projection is shown in (e). The meaning of the spheres is included: The small green spheres represent Nb, dark Sn and golden S. Sn-S and Nb-S bonds with lengths less than 3.5 Å are shown. .
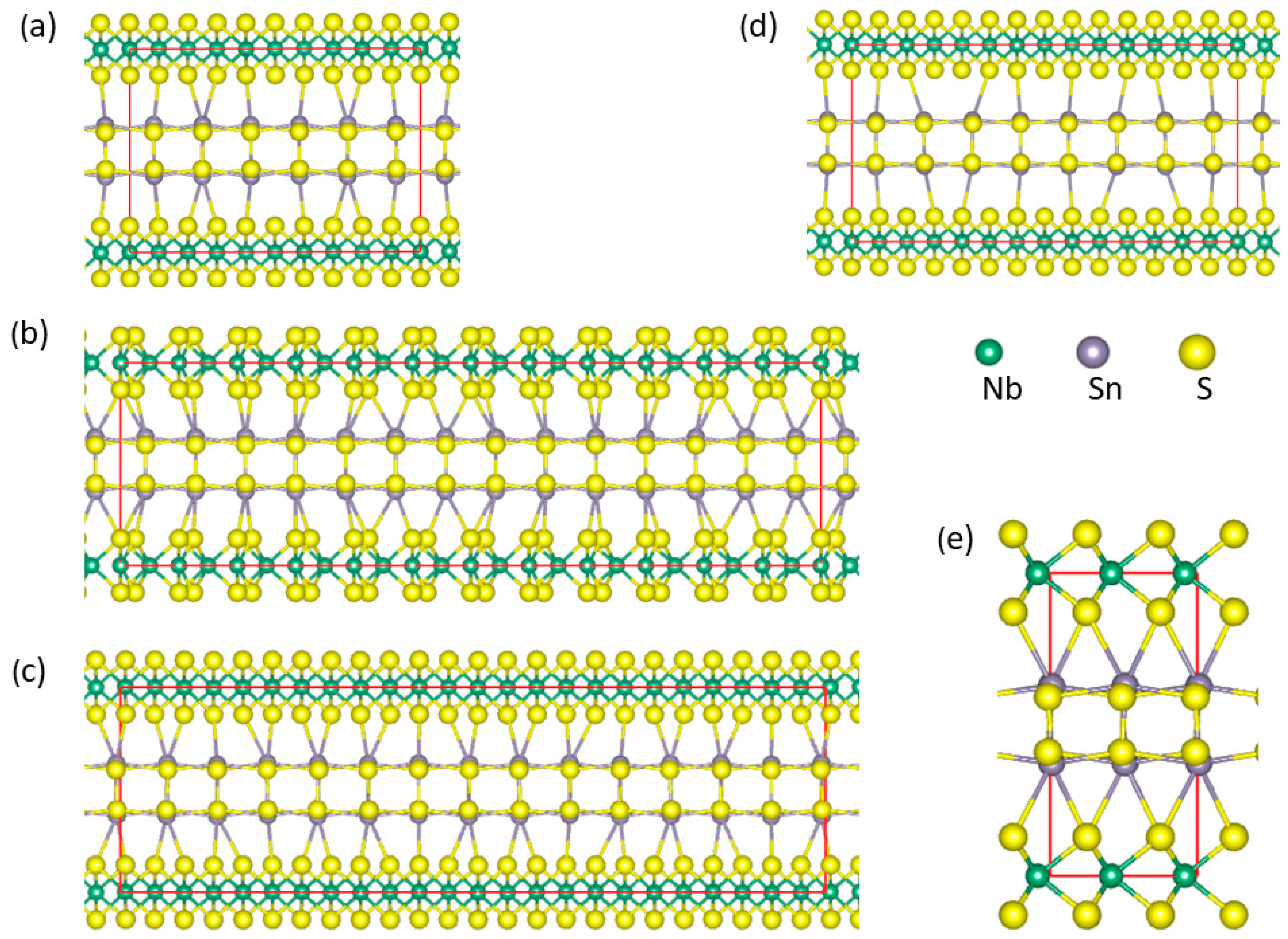
Figure 4.
Partial density of states for selected atoms (a) for Nb, (b) for S in the NbS2 part, (c) for S in the SnS part, (d) for Sn in SnS part, and total density of states (e) for the most stable (SnS)1.167NbS2 structure. The perpendicular dotted line represents the Fermi level at zero eV.
Figure 4.
Partial density of states for selected atoms (a) for Nb, (b) for S in the NbS2 part, (c) for S in the SnS part, (d) for Sn in SnS part, and total density of states (e) for the most stable (SnS)1.167NbS2 structure. The perpendicular dotted line represents the Fermi level at zero eV.
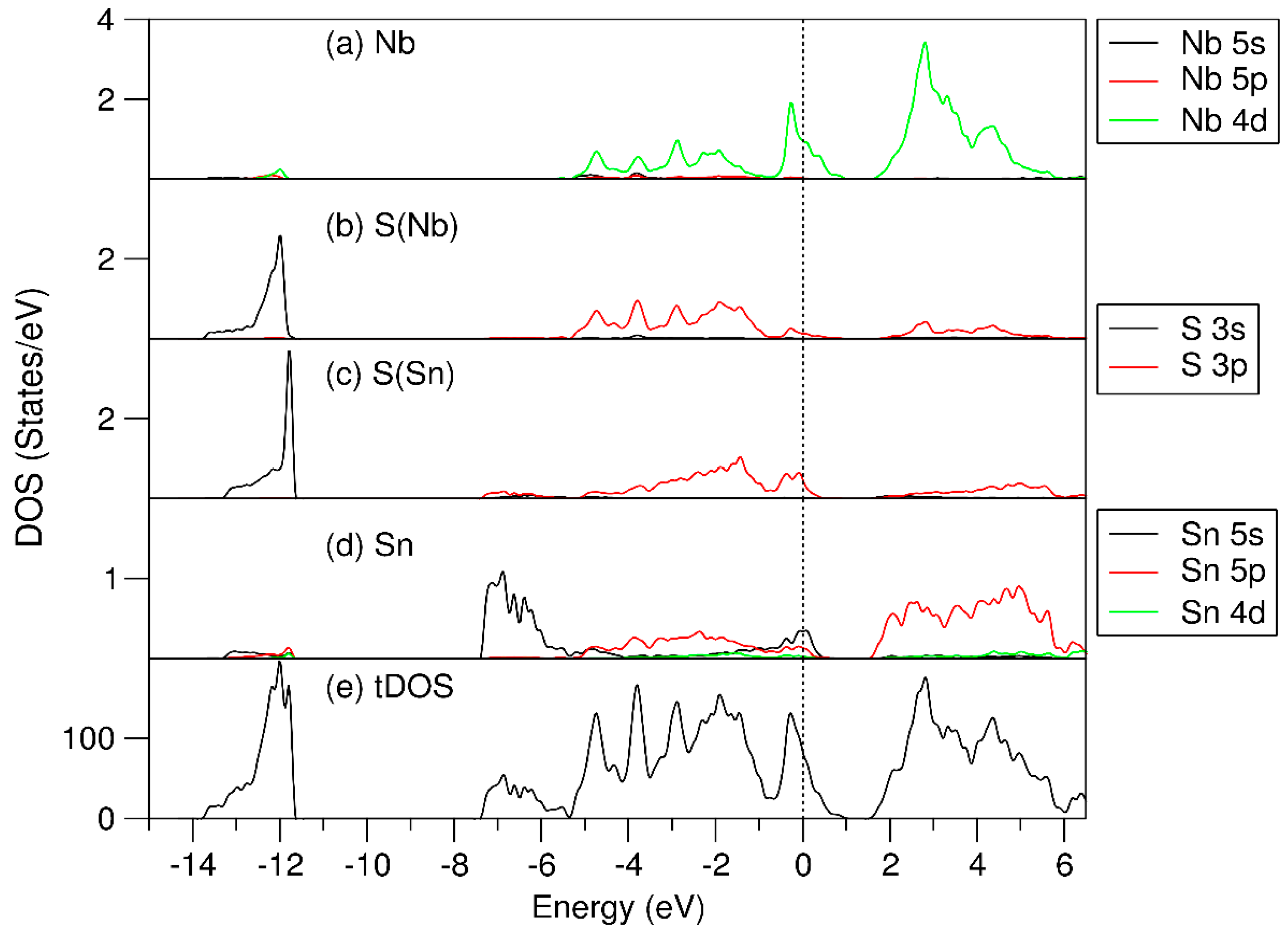
Figure 5.
(a) Dispersion curves along the high-symmetry lines around the Fermi level in (b) the Brillouin zone for the most stable (SnS)1.167NbS2 structure. The filled red horizontal line represents the Fermi level at zero eV.
Figure 5.
(a) Dispersion curves along the high-symmetry lines around the Fermi level in (b) the Brillouin zone for the most stable (SnS)1.167NbS2 structure. The filled red horizontal line represents the Fermi level at zero eV.
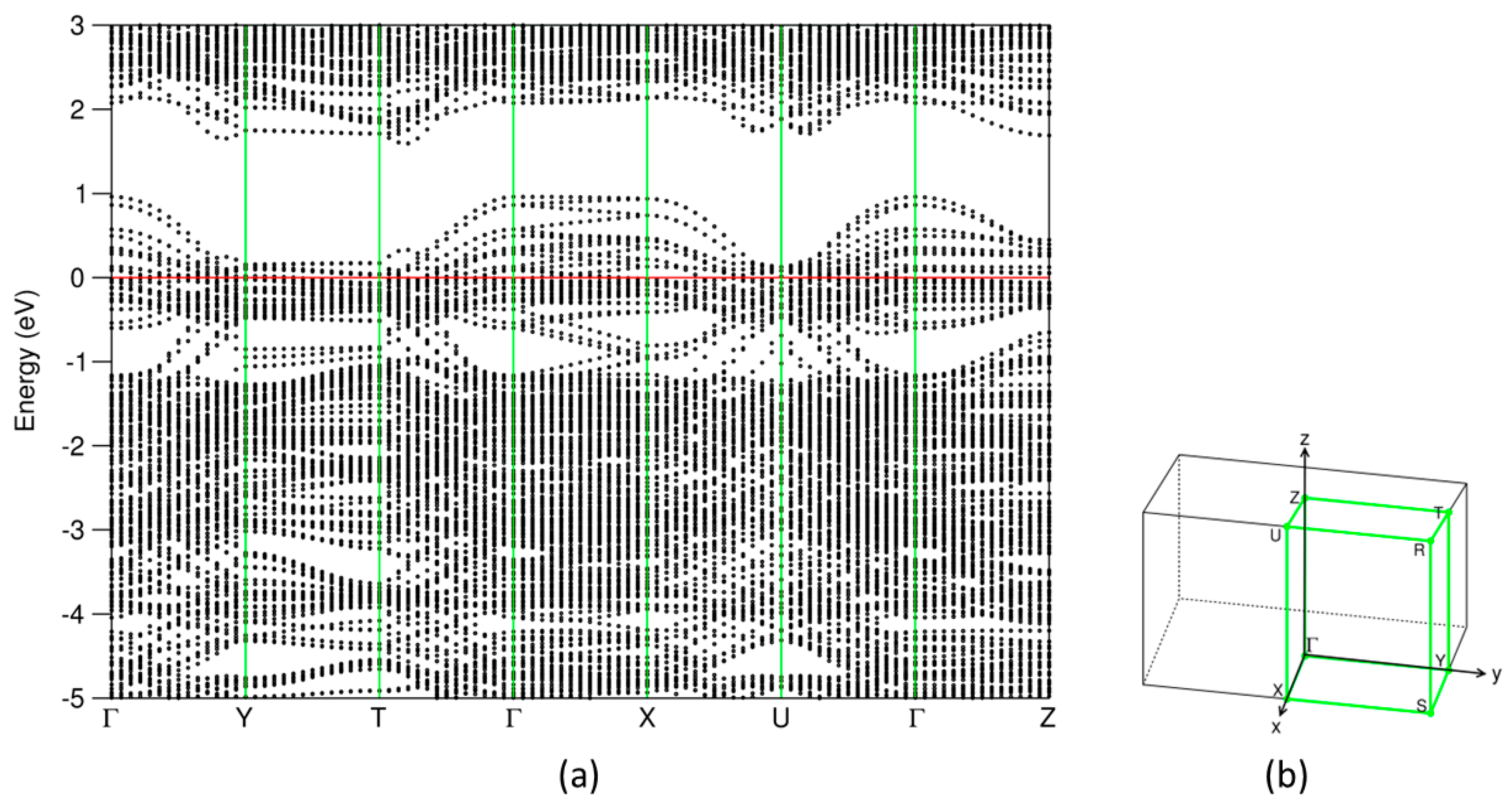
Figure 6.
Schematic structure of optimized atomic arrangements for (SnS)1.167NbS2(a) for comparison, (SnS0.929)1.167NbS2 (b), (Sn0.929S)1.167NbS2 (c). The interlayer bonds Sn-S(Nb) with lengths within 3.1Å are shown.
Figure 6.
Schematic structure of optimized atomic arrangements for (SnS)1.167NbS2(a) for comparison, (SnS0.929)1.167NbS2 (b), (Sn0.929S)1.167NbS2 (c). The interlayer bonds Sn-S(Nb) with lengths within 3.1Å are shown.
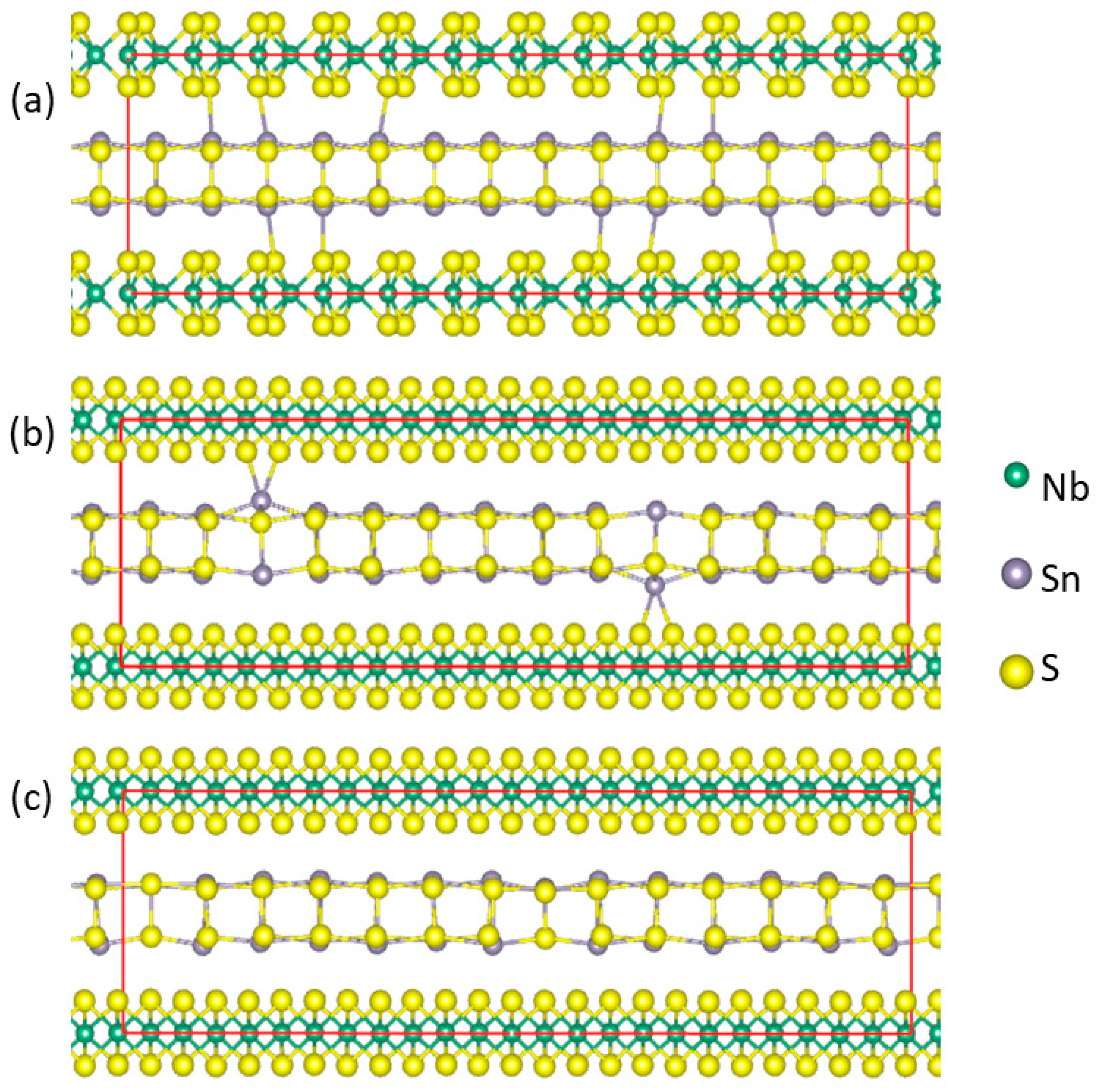
Figure 7.
Partial and total density of states curves for the atoms and cell of the S vacant in (SnS) part of (SnS0.929)1.167NbS2(a) and Sn vacant in (SnS) part of (Sn0.929S)1.167NbS2(b). The dotted lines represent the Fermi level at zero eV. In Figure 7a, Sn1 has five S(Sn) neighbours, Sn2 has four Sn-S(Sn) bonds and two Sn-S(Nb) neighbors and Sn3 has four Sn-S(Sn) bonds and a short Sn-S(Nb) bond (2.82Å). In Figure 7b, Sn1 has five Sn-S(Sn) bonds, Sn2 has five Sn-S(Sn) and two Sn-S(Nb) bonds. S1(Sn) has four Sn neighbours and S2(Sn) has five Sn neighbours.
Figure 7.
Partial and total density of states curves for the atoms and cell of the S vacant in (SnS) part of (SnS0.929)1.167NbS2(a) and Sn vacant in (SnS) part of (Sn0.929S)1.167NbS2(b). The dotted lines represent the Fermi level at zero eV. In Figure 7a, Sn1 has five S(Sn) neighbours, Sn2 has four Sn-S(Sn) bonds and two Sn-S(Nb) neighbors and Sn3 has four Sn-S(Sn) bonds and a short Sn-S(Nb) bond (2.82Å). In Figure 7b, Sn1 has five Sn-S(Sn) bonds, Sn2 has five Sn-S(Sn) and two Sn-S(Nb) bonds. S1(Sn) has four Sn neighbours and S2(Sn) has five Sn neighbours.
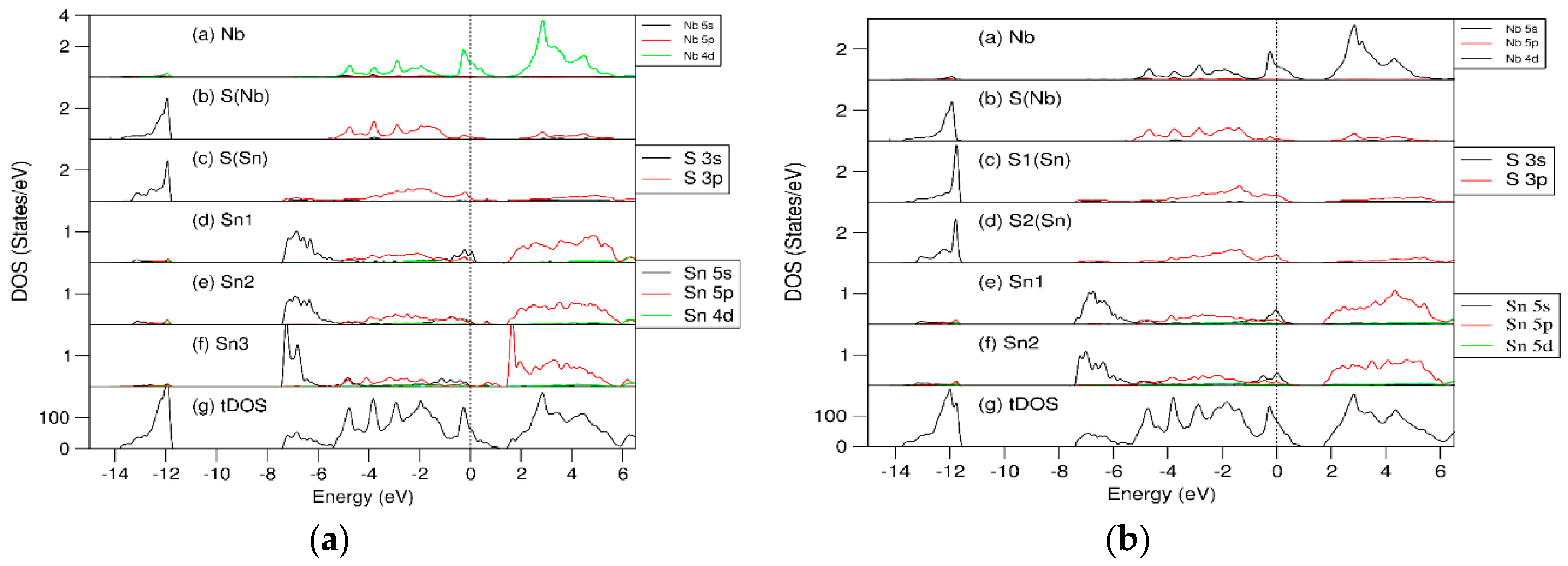

Table 1.
Calculated results (Lattice parameters, volumes and band gaps) for the binary compounds, (α- and β-)SnS and (2H-, 3R-)NbS2 using the DFT-GGA with van der Waals interaction correction.
Table 1.
Calculated results (Lattice parameters, volumes and band gaps) for the binary compounds, (α- and β-)SnS and (2H-, 3R-)NbS2 using the DFT-GGA with van der Waals interaction correction.
| Compound | Latt., spacegroup |
Latt. Parameters (Å) a, b, c |
Energy (eV/f.u.) |
Gap (eV) |
Charge(e) | Remarks |
| α-SnS GGA+vdW Exp. |
orth. Pnma(no.62) |
11.404, 4.040, 4.363 11.200, 3.987, 4.334[49] |
-4.829 - |
0.88 1.11[51] |
Sn+0.45S-0.45 |
Each Sn has three S neighbors and two Sn neighbors |
| β-SnS GGA+vdW Exp. |
orth. Cmcm(no.63) |
4.108, 11.643, 4.124 4.148, 11.480, 4.177[50] |
-4.802 - |
0.32 - |
Sn+0.94S-0.94 |
Each Sn has five S neighbors and two Sn neighbors |
| NaCl-type GGA+vdW Exp. |
Cubic Fm-3m(no.225) |
5.845, 5.845, 5.845 - |
-4.760 - |
0.07 - |
Sn+1.10S-1.10 |
Each Sn has six S neighbors |
| 2Ha-NbS2 GGA+vdW 2Hb-NbS2 GGA+vdW Exp. 3R-NbS2 GGA+vdW Exp. |
Hex. P63/mmc(no.194) Hex. P63/mmc(no.194) Romh. R-3m(no.166) |
3.372, 3.372,12.877 3.374, 3.374, 12.024 3.324, 3.324, 11.95 [52] 3.345, 3.345, 18.258 3.330,3.330,17.918 [53] |
-15.195 - -15.627 - -15.711 - |
- - - - - - |
Nb+1.28(S-0.64)2 Nb+1.14(S-0.57)2 Nb+1.16(S-0.58)2 |
Nb in prismatic coordination Nb in antiprismatic coordination Nb in antiprismatic coordination |
Table 2.
Calculated lattice parameters and formation energies of the approximates of the (SnS)1+xNbS2 compound with respect to α-SnS and 3R-NbS2 from the GGA-vdW calculations (Equation 1). The deviation of the lengths of the axis in the parenthesis is defined as Δ = (dcalc.- dexp)/dexp. ×100 %. *Pseudo-orthorhombic unit cells. The formation energies, ΔE are obtained according to Equation 1.
Table 2.
Calculated lattice parameters and formation energies of the approximates of the (SnS)1+xNbS2 compound with respect to α-SnS and 3R-NbS2 from the GGA-vdW calculations (Equation 1). The deviation of the lengths of the axis in the parenthesis is defined as Δ = (dcalc.- dexp)/dexp. ×100 %. *Pseudo-orthorhombic unit cells. The formation energies, ΔE are obtained according to Equation 1.
| Structure | Symmetry | Latt.paras.(Å) and deviation (Δ) a, b, c Z |
ΔE (eV/NbS2) |
| (a) (SnS)1.200NbS2 | C2 (no. 5) | 16.871(1.6%), 5.770(0.5%), 12.112(3.0%) 10 | -0.16 |
| (b) (SnS)1.167NbS2_a | Pa (no.7) | 40.077(0.6%), 5.781(0.5%), 11.849(0.7%) 24 | -0.40 |
| (c) (SnS)1.167NbS2_b | Pa (no.7) | 40.081(0.6%), 5.778(0.5%), 11.874(1.0%) 24 | -0.40 |
| (d) (SnS)1.143NbS2 | C2 (no. 5) | 23.202(-0.2%),5.791(0.7%), 11.856(0.8%) 14 | -0.22 |
| (e) (SnS0.929)1.167NbS2_a | C2 (no. 5) | 40.001, 5.804, 12.081 24 | - |
| (f) (SnS0.929)1.167NbS2_b | P1 (no. 1) | 39.947, 5.811, 11.901 24 | - |
| (g) (Sn0.929S)1.167NbS2 | C2(no. 5) | 39.888, 5.773, 11.813 24 | - |
| Experimental (SnS)1.171NbS2 Composite [38,39] |
Cm2a C2m2 |
5.673, 5.751, 11.761 4 (SnS) 3.321, 5.751, 11.761 2(NbS2) |
- |
Table 3.
The calculated values (chemical bonds, charges at atomic sites) for the (SnS)1.167NbS2 approximant, case b).
Table 3.
The calculated values (chemical bonds, charges at atomic sites) for the (SnS)1.167NbS2 approximant, case b).
| Element | Bonds(Å) | Charge (e/atom) |
| Nb | Nb-S(Nb): 2.48 to 2.50 (×6) | +0.99 to 1.51 Av.: +1.24 |
| S(Nb) | S-Nb: 2.48 to 2.50 (×6) -Sn: 3.21 to 3.25 |
-0.50 to -0.90 Av.: -0.70 |
| S(Sn) | S-Sn: 2.64 to 3.00 (×5) | -0.55 to -0.93 Av: -0.74 |
| Sn | Sn-S(Sn): 2.64 to 3.00 (×5) -S(Nb): 3.21 to 3.25 |
+0.70 to +1.23 Av. +0.89 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



