Submitted:
06 August 2024
Posted:
08 August 2024
You are already at the latest version
Abstract
Oxidative stress, characterized by an imbalance between the production of reactive oxygen species (ROS) and the body's antioxidant defenses, significantly affects cellular function and viability. It plays a pivotal role in modulating action potentials (APs), essential for properly functioning excitable cells such as neurons, smooth muscles, pancreatic beta cells, and myocytes. The interaction between oxidative stress and AP dynamics is crucial for understanding the pathophysiology of various conditions, including neurodegenerative diseases, cardiac arrhythmias, and ischemia-reperfusion injuries. This review explores how oxidative stress influences APs, focusing on alterations in ion channel functionality, signaling pathways, and membrane properties. By integrating current research, we aim to elucidate how oxidative stress contributes to disease progression and discuss potential therapeutic interventions targeting this interaction.
Keywords:
Oxidative stress
; action potential
; biophysics
; ion channel
; pathophysiology
1. Introduction
Oxidative stress occurs when there is an imbalance between the production of ROS and the body's ability to counteract these harmful molecules with antioxidant defenses. This imbalance results in elevated levels of ROS, which can damage vital cellular components like DNA, proteins, and lipids. Such oxidative damage disrupts normal cellular processes and plays a role in developing various diseases. Oxidative stress is known to play a role in various chronic diseases such as cardiovascular diseases, diabetes, cancer, and neurodegenerative diseases (e.g., Alzheimer's and Parkinson's). A major way oxidative stress contributes to disease is by causing cellular damage. ROS are highly reactive and can alter nucleic acids, leading to mutations and genomic instability [1,2,3,4]. This can initiate or worsen conditions like cancer, particularly in areas where chronic inflammation and oxidative stress are present, such as the gastrointestinal tract in inflammatory bowel disease [5]. Additionally, oxidative modifications to proteins can impair their function, which is linked to several neurodegenerative diseases. For instance, in Alzheimer’s disease, oxidative stress can cause the aggregation of amyloid-beta peptides, a key feature of the disease [6]. Similarly, in Parkinson’s disease, oxidative damage to dopaminergic neurons accelerates neuronal loss and disease progression [7]. Cardiovascular diseases are also closely associated with oxidative stress. Atherosclerosis, characterized by plaque buildup in arterial walls, is partly driven by the oxidation of low-density lipoprotein (LDL) cholesterol [8]. Oxidized LDL is absorbed by macrophages, leading to foam cell formation and atherosclerotic plaques [9]. Additionally, oxidative stress can cause endothelial dysfunction, which is a precursor to hypertension and other vascular conditions [10]. This dysfunction reduces the availability of nitric oxide (NO), a vital molecule for vascular relaxation, thereby increasing vascular resistance and blood pressure [11]. In chronic inflammatory diseases, oxidative stress both results from and contributes to inflammation. For example, rheumatoid arthritis is characterized by ongoing immune cell activation that produces ROS as part of the inflammatory response [12]. These ROS further damage tissues, creating a cycle of inflammation and oxidative stress [13]. In rheumatoid arthritis, oxidative modifications of proteins and lipids exacerbate tissue damage and the autoimmune response [14]. Oxidative stress also plays a role in metabolic disorders such as diabetes mellitus. In diabetes, hyperglycemia leads to excessive ROS production, which in turn causes insulin resistance and beta-cell dysfunction [15]. The oxidative damage to pancreatic beta cells impairs insulin secretion, worsening hyperglycemia and perpetuating metabolic dysregulation [16]. Furthermore, oxidative stress contributes to diabetes complications like nephropathy, retinopathy, and neuropathy by damaging blood vessels and tissues in various organs [17]. Given its central role in these diverse conditions, oxidative stress is a significant target for therapeutic approaches. Figure 1 illustrates the process by which oxidative stress disrupts normal cells through the induction of reactive oxygen species.
Antioxidant therapies aim to restore the balance between ROS production and antioxidant defenses [18,19]. Research into dietary antioxidants, such as vitamins C and E, and pharmacological agents that enhance the body’s antioxidant systems is ongoing. Additionally, lifestyle changes like regular exercise and a diet rich in fruits and vegetables can help improve the body's antioxidant capacity and reduce oxidative stress [20,21].
APs (APs) are rapid, transient electrical signals that travel along the membranes of excitable cells, playing a fundamental role in communication within the nervous system and between different tissues [22,23,24,25]. The generation and propagation of AP are crucial for normal physiological processes, including muscle contraction, sensory perception, and cognitive functions [26,27,28]. It begins with depolarization, where the membrane potential becomes less negative due to the influx of sodium ions (Na+). This change must reach a certain level, known as the threshold potential, to trigger the AP. After reaching this threshold, a rapid rise in membrane potential occurs, followed by repolarization, where potassium ions (K+) exit the cell, restoring the negative membrane potential. Hyperpolarization may occur if the membrane potential temporarily becomes more negative than the resting potential. Some excitable cells can exhibit different firing patterns, such as bursting, where clusters of APs are generated in quick succession, and slow wave firing, characterized by rhythmic, periodic bursts of APs, often seen in certain types of neurons or during specific physiological conditions. Figure 2 is a simulated output that demonstrates the membrane depolarization (black solid line), AP (red solid line), depolarization, repolarization, threshold potential (star mark), and resting membrane potential. The resting membrane potential is maintained at– 52 mV. Table 1 lists the types of excitable cells, their RMP values, and AP/SW generation patterns with references.
However, abnormalities in APs can lead to various pathophysiological conditions, profoundly affecting health. At the core of an AP is the coordinated opening and closing of voltage-gated ion channels, primarily Na⁺ and K⁺ channels [29,30,31,32,33,34]. This orchestrated ion movement generates the rapid depolarization and subsequent repolarization phases characteristic of an AP [35,36,37]. In pathological states, disruptions in these channels' function or expression can alter AP characteristics, impacting cellular communication and function [38,39,40,41,42].
Dysfunctional APs are linked to several neurological conditions [48,49]. In epilepsy, for instance, excessive, synchronous firing of neurons leads to seizures. This hyperexcitability is often due to mutations in ion channels, resulting in prolonged depolarization or insufficient repolarization, thus making neurons more likely to fire inappropriately [50]. Similarly, in multiple sclerosis, demyelination disrupts the efficient propagation of APs, leading to slowed or blocked signal transmission and a range of neurological deficits [51,52]. The heart's rhythm is tightly regulated by APs generated and propagated through cardiac muscle cells. Pathological changes in ion channels or the cellular environment can lead to arrhythmias, where the heart beats too quickly, too slowly, or irregularly [53]. For example, mutations in Na⁺ or K⁺ channels can cause long QT syndrome, a disorder that prolongs the repolarization phase of the cardiac AP, increasing the risk of sudden cardiac death [54]. APs are integral to the perception of pain and other sensory modalities [55]. In chronic pain conditions, such as neuropathic pain, there is often increased excitability of pain pathways due to changes in ion channel function that lower the threshold for AP generation [56]. This heightened sensitivity can lead to pain perception in response to normally non-painful stimuli (allodynia) or exaggerated pain responses to mildly painful stimuli (hyperalgesia) [57]. APs trigger muscle contractions by causing the release of calcium ions (Ca2+) within muscle cells [58,59]. Diseases such as myotonia and periodic paralysis involve mutations in ion channels that disrupt normal AP generation or propagation in muscle fibers [60,61]. These disruptions can lead to symptoms such as muscle stiffness or episodic muscle weakness [62,63]. A range of inherited disorders, collectively termed channelopathies, are caused by mutations in genes encoding ion channels [64]. These disorders illustrate the diverse effects of AP dysregulation, impacting the nervous system, heart, muscles, and other tissues [65]. Examples include cystic fibrosis, where defective chloride (Cl-) channels affect multiple organ systems, and various inherited epilepsies and myopathies linked to specific ion channel mutations [66]. Therefore, understanding the mechanisms underlying AP generation and propagation and their alterations in disease is crucial for developing targeted therapies for these conditions.
Oxidative stress and APs are pivotal in the development of numerous diseases, including neurodegenerative and cardiovascular conditions as well as chronic inflammatory and metabolic disorders. Therefore, understanding how ROS impact AP generation and cause cellular damage is crucial for advancing therapeutic strategies. Recent advances in molecular biology and electrophysiology have provided deeper insights into these processes, offering hope for improved treatments and outcomes for patients with disorders related to oxidative stress and AP dysfunction. Although there is extensive literature on the individual roles of oxidative stress and APs in pathological conditions, comprehensive reviews examining the interplay between these two factors are relatively rare. This gap may be due to the interdisciplinary nature of the subject, requiring knowledge in both biology and physics. However, understanding the interaction between AP biophysics and oxidative stress is essential, highlighting the need for more integrative reviews to bridge basic science and clinical applications. This review aims to deepen our understanding of the interplay between AP biophysics and oxidative stress in cellular and subcellular contexts. It presents experimental evidence illustrating how oxidative stress disrupts ion channel function at both cellular and subcellular levels, thereby influencing the generation of APs. This comprehensive understanding can offer valuable insights into physiological mechanisms and guide the development of therapeutic interventions to address various disorders and promote optimal health.
2. Materials and Methods
We extensively searched the MEDLINE database via PubMed [67,68], focusing on English-language articles published at any time. Our goal was to explore the relationships between oxidative stress, reactive oxygen species, ion channels, biophysics, gap junctions, neurotransmitters, neuromodulators, calcium dynamics, and intracellular electrical activities (such as depolarization, hyperpolarization, APs, and slow waves), including both experimental and computational studies. We meticulously screened all relevant studies, excluding non-English articles and those duplicating information from other sources. Priority was given to the most recent and comprehensive manuscripts in cases of overlap. Our selection criteria included original research articles, randomized and non-randomized clinical trials, experimental studies, prospective observational studies, retrospective cohort studies, case-control studies, and review articles on the impact of oxidative stress on ion channels and AP modulation. Each included article was scrutinized, and supplementary references were consulted to ensure comprehensive coverage. Finally, we designed a model diagram to illustrate the key steps in the relationship between oxidative stress and APs.
3. Ion channel biophysics and oxidative stress
Ion channels are essential membrane proteins that control the movement of ions across cell membranes, playing crucial roles in physiological processes like neuronal signaling, muscle contraction, and hormone release. These channels have diverse structures and functions, with key types including voltage-gated, ligand-gated, and mechanically-gated ion channels [69]. Voltage-gated ion channels, such as those for Na+, K+, Cl-, and Ca2+, respond to changes in membrane potential, facilitating the rapid generation and propagation of APs in excitable cells like neurons and muscle cells [70]. Ligand-gated ion channels, such as nicotinic acetylcholine receptors, are activated by specific neurotransmitters or ligands, resulting in ion flow and cellular responses [71]. Mechanically-gated ion channels, found in sensory neurons, open in response to physical stimuli like pressure or stretch, converting mechanical signals into electrical ones [72]. The activity of ion channels is intricately regulated through mechanisms such as protein phosphorylation, protein-protein interactions, and changes in intracellular ion concentrations [73]. Protein phosphorylation can alter ion channel function by changing their conformation or membrane localization [74]. Additionally, intracellular ion levels, particularly Ca2+, are vital in regulating ion channel activity. Variations in Ca2+ concentrations can directly impact ion channel gating or indirectly affect channel function via Ca2+ -sensitive signaling pathways [75]. Overall, the precise regulation of ion channels is essential for controlling cellular excitability and function, underscoring their importance in maintaining physiological homeostasis. Table 2 illustrates the role of various ion channels in excitable cell AP/SW generation.
Oxidative stress can modify ion channels, which are crucial for AP generation and propagation. ROS can oxidize ion channel proteins, altering their gating properties and functionality. The following section has elaborated on the effects of oxidative stress on various types of ion channels.
Voltage-Dependent Ca2+ Channels
Voltage-dependent Ca2+ channels (VDCCs) are complex structures composed of a central pore-forming α-subunit, which senses voltage changes, along with auxiliary β-, γ-, and α2δ-subunits. VDCCs contribute to APs by allowing Ca2+ ions to enter the cell during depolarization, which is essential for initiating neurotransmitter release and muscle contraction [76]. Their activation prolongs the depolarization phase and enhances the strength and duration of the AP. At least ten different genes encode the α-subunits in mammals, with the Cav1.2 (L-type Ca2+ channel) gene being a prominent example. These channels are subject to redox regulation through the oxidation and nitrosylation of cysteine residues within the α-subunit by ROS and NO. Studies in cardiac myocytes have shown that redox regulation can either enhance or diminish channel function, impacting open time, probability, expression, and trafficking of the channels [77]. This regulation is complex and varies based on cell type, species, and expression systems. In conditions like colonic inflammation, increased oxidative stress results in significant attenuation of Ca2+ influx in smooth muscle cells, leading to decreased muscle contraction. This attenuation is linked to reduced phosphorylation by c-src kinase due to increased tyrosine nitration. While S-nitrosylation can enhance L-type Ca2+ currents, tyrosine nitration during oxidative stress typically inhibits them, indicating the type of posttranslational modification critically influences channel activity [78]. Redox modulation of T-type Ca2+ channels has been studied in rat peripheral nociceptors, where reducing agents like DL-Dithiothreitol (DTT) selectively increased T-type Ca2+ currents, an effect also observed in recombinant channels expressed in HEK cells [79]. Specific cysteines within the extracellular loop of Cav3.1 are potential sites for channel gating affected by redox agents. NO also inhibits Cav3.2 by activating ROS production, indicating that redox regulation has implications for pain therapeutics. Oxidation by external H2O2 enhances the whole-cell Ca2+ currents of P/Q-type Ca2+ channels by accelerating channel opening. Both ROS and reactive nitrogen species (RNS) increase the activity of these channels, which may contribute to neurodegenerative processes [80].
Sodium (Na+) Channels:
Voltage-gated Na+ channels play a crucial role in the depolarization phase of APs in excitable cells. Oxidative modifications can affect Na+ channel inactivation kinetics, leading to prolonged APs and increased cellular excitability, relevant in conditions such as epilepsy and ischemic injury. Agents that oxidize thiol groups, like thimerosal and 4,4-dithiopyridine, can inhibit Na+ currents by shifting the voltage dependence of inactivation toward more hyperpolarized potentials, while leaving activation unaffected [81,82]. RNS impact Na+ channels in baroreceptor neurons through direct nitrosylation of thiol groups [83]. However, they enhance the activity of inactivation-resistant Na+ channels in other neuronal types like hippocampal neurons and posterior pituitary nerve terminals, likely due to differences in channel subtypes [84]. In cardiac cells, the sodium channel SCN5A (Na(V)1.5) is vital for electrical impulse conduction, and oxidative stress is linked to cardiac arrhythmias. Following myocardial infarction, ROS increases, along with lipoxidation products. Oxidants like tert-butylhydroperoxide can shift the availability curve of Na+ currents to hyperpolarized potentials, reducing cardiac Na+ current at the activation threshold. This shift can have significant effects on cardiac function, as even minor changes at the resting membrane potential can impact heart rhythm. Tert-butylhydroperoxide-induced modifications of Na(V)1.5 through lipoxidation, mediated by isoketals from arachidonic acid peroxidation, can be blocked by isoketal scavengers. This indicates that targeting this lipoxidative pathway might protect against oxidant-induced inactivation and could offer novel therapeutic approaches for preventing arrhythmias during myocardial infarction [85]. Additionally, exogenous H2O2 exposure in the heart has been shown to cause early after depolarizations (EADs) and triggered activity by downregulating Na+ current (INa).
Potassium (K+) Channels
Voltage-gated potassium channels (Kv) are crucial for various physiological processes, including muscle contraction, neuronal excitation, regulation of resting membrane potential, and modulation of AP shape, duration, and frequency. The activity of Kv channels can be influenced by ROS and RNS through modifications of both the α-subunits and the auxiliary β-subunits. The β-subunits interact with the α-subunits to modulate the rate of channel inactivation. Kv β-subunits can act as sensors of lipid oxidation due to their catalytic activity, reducing oxidized phospholipids during oxidative stress, which can alter the inactivation properties of Kv channels [86]. Additionally, Kv β1 and Kv β2 function as redox enzymes, converting aldehyde to alcohol with Nicotinamide adenine dinucleotide phosphate (NADPH) as a cofactor, linking the cellular redox state to changes in cellular excitability through the Kv β1 – Kv β2 complex [87]. Ca2+-activated K+ Channels (KCa) channels open in response to intracellular Ca2+ concentrations and/or depolarization, leading to membrane hyperpolarization and a reduced probability of voltage-dependent Ca2+ channel opening. KCa channels are classified into three subtypes based on conductance and toxin sensitivity: large conductance (BKCa), small conductance (SKCa), and intermediate conductance (IKCa). The BK channel α-subunit (hSlo) includes seven transmembrane regions. Oxidation of cysteine residues in hSlo1 reduces channel open probability, while oxidation of methionine by chloramine-T increases it [88,89]. In rabbit colonic muscularis mucosae, monochloramine enhances BK channel activity and shifts voltage dependence to more negative potentials, an effect blocked by the sulfhydryl alkylating agent N-ethylmaleimide. NO donors potentiate BKCa currents in hypophyseal nerve endings and smooth muscle cells by modifying critical thiol groups [88].
ATP-Sensitive K+ Channels (KATP) channels, found in smooth muscles, pancreatic β-cells, myocardium, and neurons, play significant roles in cellular function. They consist of four regulatory sulfonylurea subunits (SUR1, SUR2A, or SUR2B) and four ATP-sensitive pore-forming inwardly rectifying potassium channel subunits (Kir6.1 or Kir6.2). These channels are regulated by various stimuli, including hypoxia, hyperglycemia, ischemia, and oxidative stress, impacting cellular excitability based on metabolic state (Miki and Seino 2005). ROS can modulate KATP activity differently across tissues, reducing activity in cerebral arterioles and coronary arteries, while facilitating opening in cardiac myocytes and pancreatic β-cells [89,90,91,92]. The Cys42 residue in Kir6.2 is crucial for redox regulation [93]. NO activates KATP channels in DRG neurons through S-nitrosylation of the SUR1 subunit, with mutation of Cys717 reducing NO-induced currents (Kawano et al. 2009). S-glutathionylation inhibits Kir6.1/SUR2B channels by targeting Cys176, preventing necessary conformational changes for channel gating [94]. Hydrogen sulfide (H2S) also targets KATP channels through sulfhydration of cysteine residues in Kir6.1 or SUR, enhancing channel activity, particularly during colonic inflammation [95,96]. The human ether-à-go-go-related gene 1 (hERG1, Kv11.1, KCNH2) encodes the pore-forming subunit of the K+ channel responsible for the rapid component of the delayed rectifier current (IKr) in the heart, essential for terminating cardiac APs. Impairment of hERG channel function can lead to long-QT syndrome due to gene mutations, off-target drug effects, or posttranslational modifications [97,98,99]. Oxidative stress reduces hERG function by decreasing protein levels and accelerating deactivation, primarily through thiol modification of Cys723, with contributions from Cys740 and Cys828 [100,101]. The hERG1b isoform, with faster deactivation, is less sensitive to oxidative stress compared to hERG1a.
Transient Receptor Potential (TRP) Channels
TRP channels are proteins with six transmembrane domains and a pore region between the fifth and sixth domains. They assemble into homo- or heterotetramers, forming nonselective and weakly voltage-sensitive channels. TRP channels are vital for maintaining the resting membrane potential by controlling ion flow across the cell membrane [102,103]. During membrane depolarization, these channels permit the entry of cations, initiating the depolarizing phase of APs. Their activation is crucial for generating APs and modulating cellular excitability, thereby supporting both the baseline electrical state of cells and the rapid changes needed for signal transmission. These channels play significant roles in sensory systems, responding to environmental signals such as temperature changes, pH variations, and volatile chemicals. Some TRP channels detect oxidative stress through second messenger production [104], while others are directly influenced by oxidative stress. TRPC3 channels, particularly in the presence of oxidants, contribute to endothelial cell depolarization [105]. These channels likely interact with signaling molecules within caveolae or lipid rafts, with membrane cholesterol oxidation potentially serving as the activating signal during oxidative stress [106]. TRPM2 is a Ca2+-permeable cation channel that also functions as an ADP-ribose hydrolase. It can be activated by H2O2 either through NAD+ [107] or by direct oxidation [108]. The conversion of NAD+ to ADP-ribose, a potent activator, is likely responsible for H2O2-induced TRPM2 activation [109,110]. ADPR generation during oxidative stress involves the PARP-1/PARG pathway, initiated by DNA damage in the nucleus. TRPM2 acts as a cellular oxidant sensor, with Ca2+ influx via ROS-activated TRPM2 inducing chemokine production in monocytes and exacerbating inflammatory neutrophil infiltration [111]. TRPM6, an epithelial Mg2+ channel, is highly expressed in the renal and intestinal cells' luminal membrane and regulates transepithelial Mg2+ transport. TRPM6 and its homologue TRPM7 have ion channel regions combined with serine/threonine kinase domains. RACK1 and REA interact with this domain, inhibiting channel activity in an autophosphorylation-dependent manner [112]. H2O2 decreases TRPM6 activity, but methionine sulfoxide reductase (MsrB1) can restore it by reducing the oxidation of Met1755, suggesting MsrB1 modulates TRPM6 during oxidative stress [113]. TRPV1 channels are activated by noxious heat, capsaicin, and acidic pH. Various signaling pathways modulate TRPV1, affecting nociceptive neuron excitability. Oxidative stress causes potent, long-lasting TRPV1 sensitization via inter-cysteine disulfide bond formation within its cytoplasmic termini. TRPV1 also regulates NADPH oxidase-mediated ROS generation in microglia [114].
Orai Ion Channels
Store-operated calcium entry (SOCE) and Ca2+ release-activated Ca2+ (CRAC) channels play a pivotal role in modulating APs by regulating intracellular Ca2+ levels. When intracellular Ca2+ stores, primarily within the endoplasmic reticulum, are depleted, STIM proteins (STIM1 and STIM2) sense this depletion and activate Orai channels in the plasma membrane, facilitating SOCE. The influx of Ca2+ through CRAC channels replenishes intracellular Ca2+ levels and sustains prolonged Ca2+ signaling, which is critical for various cellular functions, including the modulation of APs. Increased intracellular Ca2+ concentration can activate calcium-dependent potassium channels, leading to membrane hyperpolarization and affecting the firing rate and pattern of APs. These channels consist of Orai family proteins activated by ER calcium-sensing proteins, STIM1 and STIM2 [115]. Both STIM and Orai proteins can be modified by redox agents. Oxidizing agents like thimerosal and H2O2 are known to reduce CRAC currents. ICRAC plays a crucial role in the activation, proliferation, and differentiation of T lymphocytes. During inflammation, lymphocytes encounter highly oxidative environments. Orai1 channels, but not Orai3, are inhibited by H2O2 oxidation. This difference in redox sensitivity is attributed to an extracellular reactive cysteine present in Orai1 but absent in Orai3. Consequently, cellular responses to oxidative stress vary depending on the expression of different Orai isoforms. For instance, naive T helper (TH) cells, which have low Orai3 expression, are more redox-sensitive, whereas effector TH cells, which have increased Orai3 expression, exhibit reduced redox sensitivity [116].
P2X2 Receptors
P2X2 receptors, which are ligand-gated ion channels activated by ATP, play a crucial role in modulating APs by influencing synaptic transmission and neuromuscular communication. When ATP binds to P2X2 receptors, it causes the channel to open, allowing the influx of cations such as Na+ and Ca2+. This influx depolarizes the membrane, thereby influencing the initiation and propagation of APs. Additionally, the activity of P2X2 receptors is modulated by oxidative stress, as evidenced by their potentiation by H2O2 and the enhancement of their function in the presence of mercury. This redox sensitivity can significantly affect neuronal excitability and synaptic plasticity, potentially altering the patterns of AP firing and transmission under conditions of oxidative stress, such as during inflammation or tissue injury. Thus, P2X2 receptors serve as key modulators of AP dynamics, integrating extracellular ATP signals and oxidative cues to regulate neuronal and muscular responses. The P2X2 receptor is a ligand-gated ion channel where ATP binds to mediate synaptic transmission between neurons and neuro-smooth muscle junctions. P2X receptor channels (P2XRs) are allosterically modulated by binding with mercury and copper within the receptor's ectodomain. Mercury, which induces oxidative stress, enhances the activity of P2X2 receptors. The activation of P2X2 receptors is also potentiated by H2O2, and this effect is eliminated by the alkylation of Cys430 [117]. Conversely, research by Mason et al. (2004) demonstrated that hypoxia reduced ATP-induced currents in cells expressing homomeric P2X2 receptors through mitochondrial production of ROS [118].
4. Interstitial cells of Cajal and oxidative stress
Interstitial cells of Cajal (ICCs) are essential pacemakers in the gastrointestinal tract, orchestrating smooth muscle contractions through the generation of rhythmic electrical slow waves [119]. The slow waves propagate through the smooth muscle layers, coordinating peristalsis and other motility patterns by modulating membrane electrical properties through the regulation of various ion channels, including Ca2+ and K+ channels. The depolarization and repolarization phases of these waves, driven by the precise opening and closing of these ion channels, create the electrical signals needed for synchronized muscle contractions. However, oxidative stress negatively affects ICCs by increasing ROS production, which leads to cellular damage and altered ion channel function [120]. This disruption impairs the pacemaker activity of ICCs, resulting in abnormal electrical signaling and contributing to motility disorders such as gastroparesis and chronic intestinal pseudo-obstruction. Understanding these effects is crucial for developing therapeutic strategies to protect ICC function under oxidative stress. In jejunum samples from mice exposed to pro-inflammatory cytokines, Gamma interferon lipopolysaccharide (IFNγ-LPS) mediated inflammation was found to disrupt the pacemaker function of ICCs without causing ultrastructural or apoptotic changes [121]. This inflammatory state induced oxidative stress through NO synthesized by macrophages and smooth muscle cells, leading to phenotypic changes in ICCs. In patients with neurodegenerative diseases such as Alzheimer's and Amyotrophic Lateral Sclerosis, increased serum levels of NADPH oxidase 2 (NOX2) and elevated plasma levels of LPS from intestinal Gram-negative bacteria have been observed [122]. During sepsis, IL-17 activates macrophages in the muscle layer, causing structural damage to ICCs due to NOS-induced oxidative stress [123]. It has been proposed that NO synthesis in ICC enhances intracellular Ca2+, amplifying inhibitory signals; however, abnormal elevation of intracellular Ca2+, can produce cytoplasmic free radicals, increasing cellular damage [124]. Although experimental evidence in ICCs is lacking, NOS overexpression in neurons has been linked to enteropathies such as gastroparesis, achalasia, Hirschsprung’s disease, and diabetic colonic dysfunction [125]. The nitrergic system's involvement in diabetic gastropathy is marked by reduced active neuronal NOS in the antrum, especially in women, and a decrease in myenteric and nitrergic neurons in the jejunum, ileum, and colon of diabetic rats, although this is not observed in the duodenum [126,127]. The cyclic guanidine monophosphate (cGMP)-dependent kinase Prkg1, expressed in ICCs as its β isoform (PKG1β), is essential for ICC survival and gastrointestinal motility by modulating NO neurotransmission [128]. ICCs expressing Prkg1 indicate they are key targets of NO released from enteric neurons and play a role in regulating nitrergic signaling. The absence of ICCs and resulting excess NO at the smooth muscle level may explain reduced intestinal motility, indicating that gastrointestinal motility disorders due to ICC depletion are also linked to defective enteric neurotransmission. Studies in guinea pigs have shown that ICCs are often located near enteric neurons expressing nNOS, especially within the circular and longitudinal muscle layers [129].
5. Gap junction and oxidative stress
Gap junctions are crucial for the electrical properties of excitable cells, such as neurons and cardiac muscle cells, as they facilitate direct intercellular communication [130]. These specialized membrane channels allow the passage of ions and small molecules between adjacent cells, enabling synchronized electrical activity. By permitting the rapid and coordinated spread of APs, gap junctions ensure efficient transmission of electrical signals across cell networks. The permeability and regulatory mechanisms of gap junctions further influence the electrical behavior of excitable cells. Connexins (Cx), the protein subunits that form gap junction channels, can be modified by various factors such as pH, Ca2+ concentration, and phosphorylation [131]. These modifications can alter the conductance and selectivity of gap junctions, thereby impacting the intercellular transfer of ions and signaling molecules. For instance, during cardiac ischemia, changes in intracellular conditions can lead to the closure of gap junctions, contributing to arrhythmias. In neurons, dynamic regulation of gap junctions affects synaptic plasticity and can play a role in the development and modulation of neural circuits. Therefore, gap junctions not only enable direct electrical coupling but also integrate various physiological signals to fine-tune the electrical properties of excitable cells [132]. Figure 3 shows a schematic representation of the gap junction connection between two cells, Cell 1 and Cell 2. In this figure, V1 and V2 represent the membrane potentials of Cell 1 and Cell 2, respectively, while rj denotes the gap junction resistance between them.
Oxidative stress, characterized by the excessive production of ROS, can significantly impact gap junctions, which are crucial for cellular communication and the propagation of APs and membrane potentials. Under oxidative stress, the structure and function of these connexins can be altered, leading to disrupted gap junction communication. Knockdown of Cx43 in cortical astrocytes is reported to increase cell death induced by ROS hydrogen peroxide (H2O2) [133]. It is shown that a localized oxidative insult to endothelial cells (ECs) propagates through gap junction intercellular communication [134]. Consequently, oxidative stress can impair the coordinated contraction of excitable tissues such as cardiac and neural networks, potentially leading to arrhythmias, impaired signal transmission, and various pathophysiological conditions.
6. Calcium dynamics and oxidative stress
Calcium dynamics and calcium signaling pathways play a pivotal role in various cellular processes, serving as a universal signaling mechanism in eukaryotic cells. These pathways regulate diverse functions such as muscle contraction, neurotransmitter release, gene expression, and cell proliferation and differentiation [135,136,137,138,139]. Furthermore, dysregulation of Ca2+ signaling pathways has been implicated in numerous diseases, including cancer, neurodegenerative disorders, and cardiovascular diseases [140]. For instance, aberrant Ca2+ signaling contributes to uncontrolled cell proliferation, apoptosis resistance, and metastasis in cancer [141]. Understanding the intricate mechanisms of Ca2+ signaling and its dysregulation in disease states holds promise for the development of targeted therapies aimed at restoring cellular homeostasis and treating a wide range of pathologies. Calcium dynamics play a crucial role in maintaining the resting membrane potential by regulating ionic balance. During membrane depolarization, the influx of Ca2+ ions contributes to the depolarizing phase of the AP. This Ca2+ influx is essential for triggering AP generation and modulating cellular excitability. Therefore, precise Ca2+ signaling is vital for both establishing the baseline electrical state of cells and driving rapid changes in membrane potential necessary for signal transmission. Intracellular Ca2+ levels are tightly controlled by a delicate balance between Ca2+ influx through various channels and Ca2+ efflux through pumps and exchangers. The dynamic changes in Ca2+ concentration serve as a molecular switch, triggering downstream signaling events that orchestrate cellular responses to internal and external stimuli. Oxidative stress triggers an influx of Ca2+ into the cytoplasm from the extracellular space and from the endoplasmic or sarcoplasmic reticulum (ER/SR) through their respective channels [142,143]. This increase in cytoplasmic Ca2+ leads to further influx into mitochondria and nuclei. In mitochondria, elevated Ca2+ disrupts normal metabolism, potentially resulting in cell death. However, under oxidative stress conditions, mitochondrial Ca2+ accumulation can shift from being a beneficial physiological process to signaling cell death [144]. Within the nuclei, Ca2+ influences gene transcription and nucleases that govern cell apoptosis. Additionally, in both the nuclei and cytoplasm, Ca2+ regulates the phosphorylation and dephosphorylation of proteins, thereby modulating signal transduction pathways, and altering the intracellular electrical properties. Oxidants like superoxide and hydrogen peroxide can significantly inhibit Ca2+ transport by SERCA pumps in smooth muscle cells [145]. This inhibition appears to occur through the oxidation of SERCA's sulfhydryl groups and direct attacks on its ATP binding site. Since oxidative stress is linked to numerous diseases and the aging process, understanding the impact of oxidants on Ca2+ signaling is crucial. This knowledge could provide insights into aging and disease mechanisms and pave the way for new preventive strategies. Mitochondria play a crucial role in cellular energy production, primarily through the generation of ATP via oxidative phosphorylation [146]. This process not only fuels various cellular functions but also produces ROS as by-products, contributing to oxidative stress when not adequately managed. Moreover, mitochondria are central to Ca2+ homeostasis; they buffer intracellular Ca2+ levels by uptaking and releasing Ca²⁺ ions.
7. The model of oxidative stress impact on AP
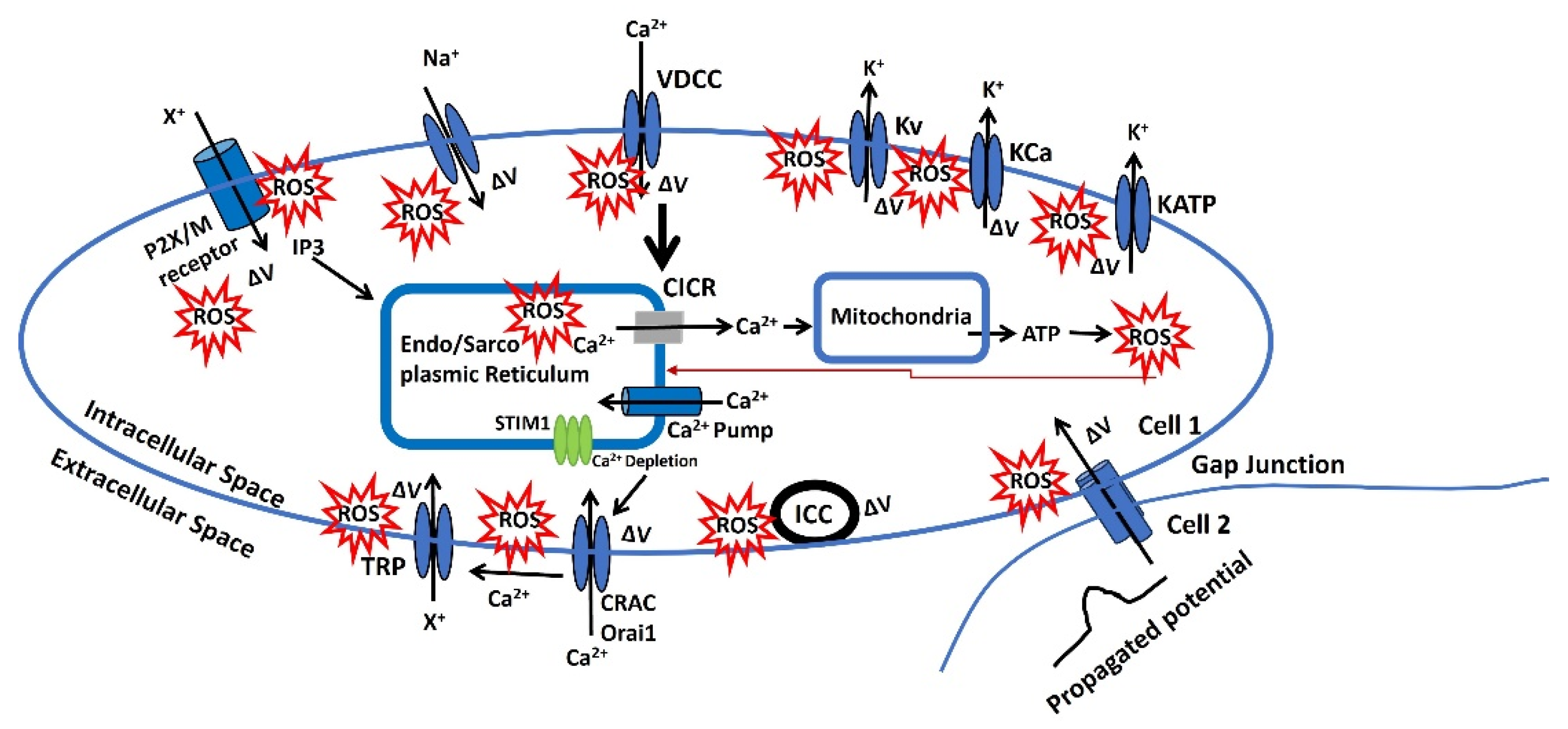
Figure 3 illustrates a representation of the redox modulation on membrane potential via several pathways, which can modulate the AP parameter and cellular excitability. Nevertheless, the pathophysiology of each step for oxidative stress has not been thoroughly investigated. Instead, these findings are based on the analysis previously mentioned that has been experimentally validated. The ROS is in red star symbol and the ΔV is known as a change in membrane potential.
- Endo/Sarcoplasmic Ca²⁺ is sourced from the endoplasmic/sarcoplasmic reticulum (ER/SR), an intracellular reservoir. Ca²⁺ ions are transported from this storage site to the sarcoplasm via Ca²⁺ channels, which are regulated by intracellular agents. Ca²⁺ is replenished in the ER/SR by a pump powered by ATP. An increase in the Ca2+ concentration near the ER/SR triggers further release of Ca2+ which is called the calcium-induced calcium release (CICR). ROS can influence various factors affecting the filling or release of Ca²⁺ in/from the ER/SR. Additionally, Ca²⁺ modulates the release of ATP and ROS from mitochondria, and the ROS released can negatively impact the Endo/Sarcoplasmic Ca²⁺ dynamics. The red arrow indicates this negative feedback loop from mitochondria to the Endo/Sarcoplasmic reticulum.
- There is a potential increase in the concentration of a diffusible second messenger, which links the surface membrane to the release of intracellular Ca2+. This process primarily involves the activation of purinergic receptors (P2X) or M3 muscarinic receptors. Upon activation, these receptors initiate a series of membrane-bound processes that lead to the production of inositol trisphosphate (IP3). IP3, in turn, can influence Ca2+ dynamics as previously described. Changes in the sensitivity or effectiveness of this mechanism can significantly impact the release of intracellular Ca2+. ATP may bind to the purinergic receptor (P2X/M), opening a non-specific cation channel that allows the influx of positive ions (X+), leading to an increase in membrane potential. This depolarization, modulated by ROS, can open L-type Ca2+ channels, facilitate Ca2+ influx, and trigger APs.
- The membrane potential can be transmitted from cell 2 to cell 1 through gap junctions, as some excitable cells function as a syncytium. Moreover, the activation of pacemaking interstitial cells of Cajal (ICC) can also induce an increase in membrane potential. ROS can modulate both gap junction and ICC internal mechanisms, and the resulting depolarization can trigger APs.
Figure 3.
A schematic diagram of the representation of the redox modulation on membrane potential via several pathways, which can modulate the AP parameter and cellular excitability.
Figure 3.
A schematic diagram of the representation of the redox modulation on membrane potential via several pathways, which can modulate the AP parameter and cellular excitability.
- 4.
- The voltage-gated and Ca2+-activated K+ ion channels (Kv, KCa, and KATP) shown in Figure 3 facilitate the flow of K+ from the intracellular to the extracellular space, leading to hyperpolarization. However, the modulating effects of ROS compromise these ion channel mechanisms, resulting in abnormal AP generation. Conversely, VDCC (L-type, T-type, and P/Q type) and voltage-gated Na+ channels allow the influx of Ca2+ and Na+ ions, depolarizing the membrane. ROS also affects these ion channels, contributing to abnormal AP generation.
- 5.
- CRAC channels are activated by intracellular depletion mediated by STIM1 and STIM2, allowing an influx of Ca2+ that depolarizes the membrane. Ca2+, along with other stimuli, can also activate various TRP ion channels, permitting the influx of cations (X+) and further depolarizing the membrane to generate APs. Additionally, ROS influences these ion channels, leading to abnormal AP generation.
- 8.
- Techniques for studying oxidative stress effects on membrane potentials
- a.
- Experimental techniques
- ∙
- Patch-Clamp Electrophysiology: The patch-clamp technique is a powerful method to study ion channel activity and membrane potential in real time [147]. By isolating a small patch of membrane, researchers can measure the ionic currents that flow through individual ion channels or across the entire cell membrane. This technique allows the investigation of how oxidative stress, often induced by reactive oxygen species (ROS), affects ion channel function and AP generation. For example, researchers can compare the ion channel activity in cells treated with ROS to those in untreated cells to determine the impact of oxidative stress.
- ∙
- Fluorescence Imaging and Voltage-Sensitive Dyes: Fluorescent dyes that are sensitive to changes in membrane potential can be used to visualize and measure membrane potential dynamics in live cells [148]. These dyes, such as Di-8-ANEPPS, emit fluorescence in response to voltage changes across the membrane, allowing researchers to monitor how oxidative stress affects membrane potential. Additionally, fluorescent indicators like Fura-2 can be used to measure intracellular calcium levels, providing insights into calcium-dependent processes affected by oxidative stress.
- ∙
- Redox-Selective Probes: To specifically measure oxidative stress levels, redox-sensitive fluorescent probes like roGFP (reduction-oxidation sensitive green fluorescent protein) can be employed [149]. These probes allow for the real-time monitoring of the cellular redox state and ROS levels. When combined with electrophysiological measurements, these probes help elucidate the correlation between oxidative stress and changes in membrane potential or ion channel activity.
- ∙
- Western Blot and Immunoprecipitation: Protein expression and post-translational modifications, such as phosphorylation, of ion channels can be assessed using western blotting [150]. Immunoprecipitation techniques can help identify protein-protein interactions that may be altered under oxidative stress. These biochemical techniques provide information on how oxidative stress may lead to modifications of ion channel proteins, thus affecting their function.
- b.
- Computational Techniques
- ∙
- Molecular Dynamics (MD) Simulations: MD simulations are computational methods used to study the behavior of biomolecules at the atomic level [151]. By simulating ion channels in different redox states, researchers can observe how oxidative stress affects the structure, dynamics, and function of these channels. MD simulations help in understanding the conformational changes that occur in ion channels under oxidative conditions and predict how these changes impact ion flow and membrane potential.
- ∙
- Computational Electrophysiology: This approach involves using mathematical models to simulate the electrical behavior of cells and tissues [152]. By incorporating data on oxidative stress, such as altered ion channel conductance or gating properties, computational models can predict the impact on membrane potential and AP generation. These simulations can help identify potential therapeutic targets for mitigating the effects of oxidative stress on neuronal and cardiac function.
- ∙
- Quantitative Structure-Activity Relationship (QSAR) Models: QSAR models use statistical methods to relate the chemical structure of molecules to their biological activity [153]. By analyzing a series of ion channel modulators or antioxidants, QSAR models can predict which compounds are likely to protect against oxidative stress-induced alterations in ion channel function. This technique is useful for drug discovery and development.
- ∙
- Bioinformatics and Network Analysis: High-throughput data from omics studies (genomics, proteomics, transcriptomics) can be analyzed using bioinformatics tools to identify pathways and networks affected by oxidative stress [154]. Network analysis can reveal key regulatory nodes and interactions between ion channels and other cellular components, providing a holistic view of how oxidative stress impacts cellular electrophysiology.
- c.
- Integrative Approaches
Combining experimental and computational techniques offers a comprehensive understanding of the relationship between oxidative stress and ion channel biophysics. For instance, experimental data from patch-clamp studies can validate computational models, while simulations can predict new experimental conditions to test. Integrative approaches enhance the ability to develop targeted therapies for diseases where oxidative stress plays a pivotal role in disrupting cellular electrophysiology.
- 9.
- Clinical Implications and Future Directions
Understanding the effects of oxidative stress on ion channels, membrane potential, and AP generation has profound clinical implications. Oxidative stress is implicated in numerous pathophysiological conditions, including neurodegenerative diseases (such as Alzheimer's and Parkinson's), cardiovascular diseases, diabetes, and certain types of cancer. Ion channels are crucial for the proper functioning of neurons, muscles, and other excitable cells, so oxidative stress-induced dysfunction can lead to significant clinical consequences:
- Neurodegenerative Diseases: Oxidative stress contributes to neuronal damage and death in diseases like Alzheimer's and Parkinson's. Aberrant ion channel activity due to oxidative modifications can disrupt neuronal signaling, leading to cognitive decline and motor dysfunction. Understanding these mechanisms can guide the development of targeted antioxidants or ion channel modulators to preserve neuronal function.
- Cardiovascular Diseases: In conditions such as ischemia-reperfusion injury and heart failure, oxidative stress alters ion channel function, affecting cardiac excitability and contractility. Therapeutic strategies that protect ion channels from oxidative damage or restore their normal function could improve outcomes in patients with heart disease.
- Diabetes: Oxidative stress plays a role in diabetic complications by affecting ion channels in various tissues, including the pancreas, nerves, and blood vessels. Interventions aimed at reducing oxidative stress or correcting ion channel dysfunction could mitigate these complications.
- Cancer: Some cancer cells exploit oxidative stress to drive proliferation and survival. Ion channels are involved in cancer cell migration, invasion, and metastasis. Targeting ion channel modifications induced by oxidative stress could provide new avenues for cancer therapy.
To fully leverage the insights gained from studying oxidative stress and its impact on ion channel biophysics, several future directions should be pursued:
- Advanced Imaging Techniques: Development of more sensitive and specific fluorescent probes and imaging techniques to measure real-time changes in membrane potential, ROS levels, and ion channel activity in live cells and tissues. This will enhance our understanding of the spatial and temporal dynamics of oxidative stress.
- High-Throughput Screening: Implementing high-throughput screening methods to identify compounds that can protect against oxidative stress-induced ion channel dysfunction. This approach can accelerate the discovery of new therapeutic agents.
- Integrative Multi-Omics Approaches: Combining genomics, proteomics, and metabolomics with electrophysiological data to construct comprehensive models of how oxidative stress impacts ion channel function and cellular excitability. This holistic view can uncover new regulatory mechanisms and potential drug targets.
- Personalized Medicine: Investigating individual variability in oxidative stress responses and ion channel function to develop personalized therapeutic strategies. Genetic and epigenetic factors that influence susceptibility to oxidative stress and ion channel modifications should be identified.
- Animal Models and Clinical Trials: Utilizing animal models to study the in vivo relevance of findings from cellular and molecular studies. Translating these findings into clinical trials to evaluate the efficacy of targeted therapies in mitigating the effects of oxidative stress in human diseases.
- Novel Therapeutics: Developing novel antioxidants, ion channel modulators, and gene therapies to specifically address the ion channel dysfunctions caused by oxidative stress. Combination therapies that target multiple pathways involved in oxidative stress responses could prove particularly effective.
Oxidative stress significantly influences APs by altering ion channel function, activating signaling pathways, and affecting membrane properties. These changes are pivotal in the pathophysiology of diseases such as neurodegenerative disorders, cardiac arrhythmias, and ischemia-reperfusion injury. Understanding the mechanisms behind these alterations is crucial for developing targeted therapeutic strategies. Future research should focus on elucidating the detailed interactions between oxidative stress and ion channels to identify novel interventions that mitigate the detrimental effects on APs. By deepening our understanding of the relationship between oxidative stress and ion channel biophysics and translating these insights into clinical applications, we can enhance the diagnosis, treatment, and prevention of various diseases associated with oxidative stress. Our review offers a fresh perspective on this topic by integrating the latest experimental and computational techniques to study oxidative stress and its impact on ion channels and membrane potentials. This comprehensive approach will be invaluable for future research, providing a robust framework for exploring new therapeutic targets and strategies. By highlighting emerging technologies and methodologies, our review paves the way for innovative research directions and clinical applications, ultimately contributing to better health outcomes in diseases linked to oxidative stress.
Author Contributions
C.M. conceived the idea of the draft, and wrote the manuscript. R.T. and R.K. also helped in writing, editing, and formatting the final draft. C.M. also responded to reviewers’ comments. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
Chitaranjan Mahapatra is thankful to UCSF, San Francisco for providing space and other required facilities for the completion of this manuscript. RK is thankful to UTHSC, Memphis for providing facilities and space for the completion of the manuscript.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Veschetti, L.; Treccani, M.; De Tomi, E.; Malerba, G. Genomic Instability Evolutionary Footprints on Human Health: Driving Forces or Side Effects? Int. J. Mol. Sci. 2023, 24, 11437. [Google Scholar] [CrossRef] [PubMed]
- Salmaninejad, A.; Ilkhani, K.; Marzban, H.; Navashenaq, J.G.; Rahimirad, S.; Radnia, F.; Yousefi, M.; Bahmanpour, Z.; Azhdari, S.; Sahebkar, A. Genomic Instability in Cancer: Molecular Mechanisms and Therapeutic Potentials. Curr. Pharm. Des. 2021, 27, 3161–3169. [Google Scholar] [CrossRef] [PubMed]
- Petrov, D.; Daura, X.; Zagrovic, B. Effect of Oxidative Damage on the Stability and Dimerization of Superoxide Dismutase 1. Biophys. J. 2016, 110, 1499–1509. [Google Scholar] [CrossRef] [PubMed]
- Merlo, D.; Mollinari, C.; Racaniello, M.; Garaci, E.; Cardinale, A. DNA Double Strand Breaks: A Common Theme in Neurodegenerative Diseases. Curr. Alzheimer Res. 2016, 13, 1208–1218. [Google Scholar] [CrossRef] [PubMed]
- Dziąbowska-Grabias, K.; Sztanke, M.; Zając, P.; Celejewski, M.; Kurek, K.; Szkutnicki, S.; Korga, P.; Bulikowski, W.; Sztanke, K. Antioxidant Therapy in Inflammatory Bowel Diseases. Antioxidants 2021, 10, 412. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-G.; Zhu, X.; Nunomura, A.; Perry, G.; Smith, M.A. Amyloid Beta: The Alternate Hypothesis. Curr. Alzheimer Res. 2006, 3, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Lee, Seung-Jae. "α-Synuclein aggregation: a link between mitochondrial defects and Parkinson's disease?." Antioxidants and Redox Signaling 5, no. 3 (2003): 337-348.
- Lorey, M.B.; Öörni, K.; Kovanen, P.T. Modified Lipoproteins Induce Arterial Wall Inflammation During Atherogenesis. Front. Cardiovasc. Med. 2022, 9, 841545. [Google Scholar] [CrossRef] [PubMed]
- Holvoet, P.; Collen, D. Oxidized lipoproteins in atherosclerosis and thrombosis. FASEB J. 1994, 8, 1279–1284. [Google Scholar] [CrossRef] [PubMed]
- Watson, Timothy, Patrick KY Goon, and Gregory YH Lip. "Endothelial progenitor cells, endothelial dysfunction, inflammation, and oxidative stress in hypertension." Antioxidants & redox signaling 10, no. 6 (2008): 1079-1088.
- De Meyer, Guido RY, and Arnold G. Herman. "Nitric oxide and vascular endothelial dysfunction." In Nitric oxide, pp. 547-567. Academic press, 2000.
- Gelderman, K.A.; Hultqvist, M.; Olsson, L.M.; Bauer, K.; Pizzolla, A.; Olofsson, P.; Holmdahl, R. Rheumatoid Arthritis: The Role of Reactive Oxygen Species in Disease Development and Therapeutic Strategies. Antioxidants Redox Signal. 2007, 9, 1541–1568. [Google Scholar] [CrossRef]
- Joshi-Barr, S.; Lux, C.d.G.; Mahmoud, E.; Almutairi, A. Exploiting Oxidative Microenvironments in the Body as Triggers for Drug Delivery Systems. Antioxidants Redox Signal. 2014, 21, 730–754. [Google Scholar] [CrossRef]
- Smallwood, Miranda J., Ahuva Nissim, Annie R. Knight, Matthew Whiteman, Richard Haigh, and Paul G. Winyard. "Oxidative stress in autoimmune rheumatic diseases." Free Radical Biology and Medicine 125 (2018): 3-14.
- Cernea, S.; Dobreanu, M. Diabetes and beta cell function: from mechanisms to evaluation and clinical implications. Biochem. Medica 2013, 23, 266–280. [Google Scholar] [CrossRef] [PubMed]
- González, Ileana, Cristian Lindner, Ivan Schneider, Erik Diaz, MIguel A. Morales, and Armando Rojas. "The Multifaceted Actions of Polyphenols in the Management of Type-2 Diabetes Mellitus." (2023).
- Giacco, F.; Brownlee, M. Oxidative Stress and Diabetic Complications. Circ. Res. 2010, 107, 1058–1070. [Google Scholar] [CrossRef] [PubMed]
- Firuzi, O.; Miri, R.; Tavakkoli, M.; Saso, L. Antioxidant Therapy: Current Status and Future Prospects. Curr. Med. Chem. 2011, 18, 3871–3888. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Marquez, M.E.; Siller-Lopez, F. Current Antioxidant Molecular Therapies for Oxidative Stress-Related Ailments. Curr. Gene Ther. 2008, 8, 256–263. [Google Scholar] [CrossRef] [PubMed]
- Preedy, Victor R., and Vinood Patel, eds. Aging: oxidative stress and dietary antioxidants. Academic Press, 2020.
- Hernández-Ledesma, B.; Martínez-Villaluenga, C. Current Advances for Development of Functional Foods Modulating Inflammation and Oxidative Stress; Elsevier: Amsterdam, NX, Netherlands, 2022. [Google Scholar]
- Król, Elżbieta, H. Dziubinska, and K. Trebacz. "What do plants need action potentials for." Action Potential: Biophysical and Cellular Context, Initiation, Phases and Propagation. DuBois ML.(ed) (2010): 1-26.
- Varró, A.; Tomek, J.; Nagy, N.; Virág, L.; Passini, E.; Rodriguez, B.; Baczkó, I. Cardiac transmembrane ion channels and action potentials: cellular physiology and arrhythmogenic behavior. Physiol. Rev. 2021, 101, 1083–1176. [Google Scholar] [CrossRef] [PubMed]
- Mahapatra, C. , and K. Shanmugam. "Computational Modeling of Sodium Ion Channel-Based Glucose Sensing Biophysics to Study Cardiac Atrial Cell Electrophysiology." (2024).
- MAHAPATRA, CHITARANJAN, and Kirubanandan Shanmugam. "Computational Modeling of Sodium Ion Channel-Based Glucose Sensing Biophysics to Study Abnormal Electrical Activities in Cardiac Atrial Cell." (2024).
- Rybak, Ilya A., and Jessica Ausborn. "Vertebrate pattern generation: overview." Encyclopedia of Computational Neuroscience (2022): 130-140.
- Iaizzo, Paul A. "Introduction to neurophysiology." Neural engineering (2020): 1-64.
- Mahapatra, C.; Kumar, R. Biophysical Mechanisms of Vaginal Smooth Muscle Contraction: The Role of the Membrane Potential and Ion Channels. Pathophysiology 2024, 31, 225–243. [Google Scholar] [CrossRef] [PubMed]
- Bezanilla, Francisco. "Voltage-gated ion channels." Biological Membrane Ion Channels: Dynamics, Structure, and Applications (2007): 81-118.
- Catterall, William A. "Voltage gated sodium and calcium channels: Discovery, structure, function, and Pharmacology." Channels 17, no. 1 (2023): 2281714.
- Pongs, O. Molecular biology of voltage-dependent potassium channels. Physiol. Rev. 1992, 72, S69–S88. [Google Scholar] [CrossRef] [PubMed]
- Mahapatra, C. , and A. Pradhan. "Nifedipine is identified as a potential pharmacological modulator in Parkinson's disease by an in silico electrophysiological study." Parkinsonism & Related Disorders 122 (2024).
- Mahapatra, C.; Samuilik, I. A Mathematical Model of Spontaneous Action Potential Based on Stochastics Synaptic Noise Dynamics in Non-Neural Cells. Mathematics 2024, 12, 1149. [Google Scholar] [CrossRef]
- Mahapatra, Chitaranjan, Keith Brain, and Rohit Manchanda. "Biophysically Realistic Modles of Detrusor Ion Channels: role in shaping spike and excitavility." In Urinary Bladder Physiology: Computational Insights. Publ Narosa Publishing House, 2024.
- Mahapatra, Chitaranjan. "Computational Study of Action Potential Generation in Urethral Smooth Muscle Cell." In Computational Advances in Bio and Medical Sciences: 10th International Conference, ICCABS 2020, Virtual Event, -12, 2020, Revised Selected Papers 10, pp. 26-32. Springer International Publishing, 2021. 10 December.
- Mahapatra, Chitaranjan, and Rohit Manchanda. "Computational studies on ureter smooth muscle: Modeling ion channels and their role in generating electrical activity." In Proceedings of the 2019 Summer Simulation Conference, pp. 1-6. 2019.
- Mahapatra, Chitaranjan, and Rohit Manchanda. "Modeling Vas Deferens Smooth Muscle Electrophysiology: Role of Ion Channels in Generating Electrical Activity." In Soft Computing for Problem Solving: SocProS 2017, Volume 2, pp. 655-663. Springer Singapore, 2019.
- Rajagopal, S.; Ponnusamy, M. Calcium Signaling: From Physiology to Diseases; Springer Nature: Dordrecht, GX, Netherlands, 2017. [Google Scholar]
- Dulla, C.G.; Coulter, D.A.; Ziburkus, J. From Molecular Circuit Dysfunction to Disease. Neurosci. 2015, 22, 295–312. [Google Scholar] [CrossRef]
- Mahapatra, Chitaranjan, and Rohit Manchanda. "Modulating Properties of Hyperpolarization-Activated Cation Current in Urinary Bladder Smooth Muscle Excitability: A Simulation Study." In Recent Findings in Intelligent Computing Techniques: Proceedings of the 5th ICACNI 2017, Volume 1, pp. 261-266. Springer Singapore, 2019.
- Mahapatra, C.; Brain, K.L.; Manchanda, R. Computational Study of Hodgkin-Huxley Type Calcium-Dependent Potassium Current in Urinary Bladder Over Activity. 2018 IEEE 8th International Conference on Computational Advances in Bio and Medical Sciences (ICCABS). LOCATION OF CONFERENCE, United StatesDATE OF CONFERENCE; pp. 1–4.
- Mahapatra, C. , and R. Manchanda. "Contribution of ATP-sensitive potassium channels in the subthalamic nucleus neurons towards Parkinson's disease." In MOVEMENT DISORDERS, vol. 33, pp. S146-S146. 111 RIVER ST, HOBOKEN 07030-5774, NJ USA: WILEY, 2018.
- Mahapatra, C.; Brain, K.L.; Manchanda, R. A biophysically constrained computational model of the action potential of mouse urinary bladder smooth muscle. PLOS ONE 2018, 13, e0200712. [Google Scholar] [CrossRef]
- Bers, Donald M. "Cardiac excitation–contraction coupling." Nature 415, no. 6868 (2002): 198-205.
- Forsberg, A.M.; Bergström, J.; Lindholm, B.; Hultman, E. Resting Membrane Potential of Skeletal Muscle Calculated from Plasma and Muscle Electrolyte and Water Contents. Clin. Sci. 1997, 92, 391–396. [Google Scholar] [CrossRef] [PubMed]
- Hille, Bertil. "Ion channels of excitable membranes (Sinauer, Sunderland, MA)." (2001).
- Ashcroft, Frances M., and Patrik Rorsman. "Electrophysiology of the pancreatic β-cell." Progress in biophysics and molecular biology 54, no. 2 (1989): 87-143.
- DiFrancesco, J.C.; DiFrancesco, D. Dysfunctional HCN ion channels in neurological diseases. Front. Cell. Neurosci. 2015, 6, 174–174. [Google Scholar] [CrossRef] [PubMed]
- Lepeta, K.; Lourenco, M.; Schweitzer, B.C.; Adami, P.V.; Banerjee, P.; Catuara-Solarz, S.; Revenga, M.D.L.F.; Guillem, A.M.; Haidar, M.; Ijomone, O.; et al. Synaptopathies: synaptic dysfunction in neurological disorders - A review from students to students. J. Neurochem. 2016, 138, 785–805. [Google Scholar] [CrossRef] [PubMed]
- A McCormick, D.; Contreras, D. On The Cellular and Network Bases of Epileptic Seizures. Annu. Rev. Physiol. 2001, 63, 815–846. [Google Scholar] [CrossRef] [PubMed]
- Lubetzki, Catherine, and Bruno Stankoff. "Demyelination in multiple sclerosis." Handbook of clinical neurology 122 (2014): 89-99.
- Friese, M.A.; Schattling, B.; Fugger, L. Mechanisms of neurodegeneration and axonal dysfunction in multiple sclerosis. Nat. Rev. Neurol. 2014, 10, 225–238. [Google Scholar] [CrossRef]
- Varró, A.; Tomek, J.; Nagy, N.; Virág, L.; Passini, E.; Rodriguez, B.; Baczkó, I. Cardiac transmembrane ion channels and action potentials: cellular physiology and arrhythmogenic behavior. Physiol. Rev. 2021, 101, 1083–1176. [Google Scholar] [CrossRef] [PubMed]
- Giudicessi, J.R.; Ackerman, M.J. Potassium-channel mutations and cardiac arrhythmias—diagnosis and therapy. Nat. Rev. Cardiol. 2012, 9, 319–332. [Google Scholar] [CrossRef] [PubMed]
- Bourne, S.; Machado, A.G.; Nagel, S.J. Basic Anatomy and Physiology of Pain Pathways. Neurosurg. Clin. North Am. 2014, 25, 629–638. [Google Scholar] [CrossRef] [PubMed]
- Dubin, A.E.; Patapoutian, A. Nociceptors: the sensors of the pain pathway. J. Clin. Investig. 2010, 120, 3760–3772. [Google Scholar] [CrossRef]
- Vinayak, Manjula, and Ajeet Kumar Singh. "Signaling of Nociceptors and Pain Perception: Impact of Age." Models, Molecules and Mechanisms in Biogerontology: Physiological Abnormalities, Diseases and Interventions (2019): 91-107.
- Berridge, M.J. Smooth muscle cell calcium activation mechanisms. J. Physiol. 2008, 586, 5047–5061. [Google Scholar] [CrossRef]
- Endo, M. Calcium-Induced Calcium Release in Skeletal Muscle. Physiol. Rev. 2009, 89, 1153–1176. [Google Scholar] [CrossRef] [PubMed]
- Kuo, Ivana Y., and Barbara E. Ehrlich. "Signaling in muscle contraction." Cold Spring Harbor perspectives in biology 7, no. 2 (2015): a006023.
- Amior, N. "Developing models to study the mechanisms of weakness and myotonia in Periodic Paralysis." PhD diss., UCL (University College London), 2018.
- Lehmann-Horn, Frank, Reinhardt Rüdel, and Karin Jurkat-Rott. "Nondystrophic myotonias and periodic paralyses." Myology 3 (2004): 1257-1300.
- Cannon, S.C. PATHOMECHANISMS IN CHANNELOPATHIES OF SKELETAL MUSCLE AND BRAIN. Annu. Rev. Neurosci. 2006, 29, 387–415. [Google Scholar] [CrossRef] [PubMed]
- Fodstad, Heidi. "Genetic and Functional Studies of Severe Ventricular Arrhythmias." PhD diss., Helsingin yliopisto, 2005.
- Celesia, G.G. Disorders of membrane channels or channelopathies. Clin. Neurophysiol. 2000, 112, 2–18. [Google Scholar] [CrossRef] [PubMed]
- Rose, Michael R. "Neurological channelopathies: Dysfunctional ion channels may cause many neurological diseases." BMJ 316, no. 7138 (1998): 1104-1105.
- Motschall, E.; Falck-Ytter, Y. Searching the MEDLINE Literature Database through PubMed: A Short Guide. Oncol. Res. Treat. 2005, 28, 517–522. [Google Scholar] [CrossRef]
- Schmucker, C.M.; Blümle, A.; Schell, L.K.; Schwarzer, G.; Oeller, P.; Cabrera, L.; von Elm, E.; Briel, M.; Meerpohl, J.J. ; on behalf of the OPEN consortium Systematic review finds that study data not published in full text articles have unclear impact on meta-analyses results in medical research. PLOS ONE 2017, 12, e0176210. [Google Scholar] [CrossRef] [PubMed]
- Alexander, Stephen PH, Jörg Striessnig, Eamonn Kelly, Neil V. Marrion, John A. Peters, Elena Faccenda, Simon D. Harding et al. "The Concise Guide to PHARMACOLOGY 2017/18: Voltage-gated ion channels." British journal of pharmacology 174 (2017): S160-S194.
- Petkov, Georgi V. "Ion channels." In Pharmacology, pp. 387-427. Academic Press, 2009.
- Hucho, Ferdinand, and Christoph Weise. "Ligand-gated ion channels." Angewandte Chemie International Edition 40, no. 17 (2001): 3100-3116.
- Delmas, P.; Coste, B. Mechano-Gated Ion Channels in Sensory Systems. Cell 2013, 155, 278–284. [Google Scholar] [CrossRef] [PubMed]
- Heijman, Jordi, and Dobromir Dobrev. "Ion channels as part of macromolecular multiprotein complexes: Clinical significance." Herzschrittmachertherapie & Elektrophysiologie 29, no. 1 (2018): 30.
- Davis, Michael J., Xin Wu, Timothy R. Nurkiewicz, Junya Kawasaki, Peichun Gui, Michael A. Hill, and Emily Wilson. "Regulation of ion channels by protein tyrosine phosphorylation." American Journal of Physiology-Heart and Circulatory Physiology 281, no. 5 (2001): H1835-H1862.
- Bootman, Martin D., Katja Rietdorf, Holly Hardy, Yana Dautova, Elaine Corps, Cristina Pierro, Eloise Stapleton, Esther Kang, and Diane Proudfoot. "Calcium signalling and regulation of cell function." eLS (2006).
- Huston, Elaine. Involvement of voltage activated calcium channels in neurotransmitter release and its modulation in cultured rat cerebellar granule neurones. University of London, University College London (United Kingdom), 1995.
- Aggarwal, N.T.; Makielski, J.C. Redox Control of Cardiac Excitability. Antioxidants Redox Signal. 2013, 18, 432–468. [Google Scholar] [CrossRef] [PubMed]
- Muralidharan, P.; Ingley, E.; Hool, L. Identifying the Site/S of Modification on Human L-type Calcium Channel Protein Isoforms During Oxidative Stress. Hear. Lung Circ. 2013, 22, S56. [Google Scholar] [CrossRef]
- Todorovic, S.M.; Meyenburg, A.; Jevtovic-Todorovic, V. Redox modulation of peripheral T-type Ca2+ channels in vivo: alteration of nerve injury-induced thermal hyperalgesia. Pain 2004, 109, 328–339. [Google Scholar] [CrossRef]
- Annunziato, L.; Pannaccione, A.; Cataldi, M.; Secondo, A.; Castaldo, P.; Direnzo, G.; Taglialatela, M. Modulation of ion channels by reactive oxygen and nitrogen species: a pathophysiological role in brain aging? Neurobiol. Aging 2002, 23, 819–834. [Google Scholar] [CrossRef]
- Evans, J.; Bielefeldt, K. Regulation of sodium currents through oxidation and reduction of thiol residues. Neuroscience 2000, 101, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Chapleau, M.W.; Bates, J.N.; Bielefeldt, K.; Lee, H.-C.; Abboud, F.M. Nitric Oxide as an Autocrine Regulator of Sodium Currents in Baroreceptor Neurons. Neuron 1998, 20, 1039–1049. [Google Scholar] [CrossRef]
- Nakajima, T.; Hishikari, K.; Ogawa, M.; Watanabe, R.; Suzuki, J.-I.; Nagashima, A.; Masumura, M.; Takayama, K.; Hirata, Y.; Nagai, R.; et al. Clarithromycin attenuates myocardial ischemia–reperfusion injury. Expert Opin. Ther. Targets 2010, 14, 881–893. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.; Bondarenko, V.E.; Morales, M.J.; Strauss, H.C. Closed-state inactivation in Kv4.3 isoforms is differentially modulated by protein kinase C. Am. J. Physiol. Physiol. 2009, 297, C1236–C1248. [Google Scholar] [CrossRef] [PubMed]
- Pan, Yaping, Jun Weng, Yu Cao, Rahul C. Bhosle, and Ming Zhou. "Functional Coupling between the Kv1. 1 Channel and Aldoketoreductase Kvβ1*♦." Journal of Biological Chemistry 283, no. 13 (2008): 8634-8642.
- DiChiara, Timothy J., and Peter H. Reinhart. "Redox modulation of hslo Ca2+-activated K+ channels." Journal of Neuroscience 17, no. 13 (1997): 4942-4955.
- Tang, X.D.; Daggett, H.; Hanner, M.; Garcia, M.L.; McManus, O.B.; Brot, N.; Weissbach, H.; Heinemann, S.H.; Hoshi, T. Oxidative Regulation of Large Conductance Calcium-Activated Potassium Channels. J. Gen. Physiol. 2001, 117, 253–274. [Google Scholar] [CrossRef] [PubMed]
- Mistry, D. K. , and C. J. Garland. "Nitric oxide (NO)-induced activation of large conductance Ca2+-dependent K+ channels (BKCa) in smooth muscle cells isolated from the rat mesenteric artery." British journal of pharmacology 124, no. 6 (1998): 1131-1140.
- Erdos, Benedek. "Cerebrovascular dysfunction in insulin-resistance." PhD diss., Semmelweis Egyetem (Hungary), 2004.
- Miura, T.; Liu, Y.; Goto, M.; Tsuchida, A.; Miki, T.; Nakano, A.; Nishino, Y.; Ohnuma, Y.; Shimamoto, K. Mitochondrial ATP-sensitive K+channels play a role in cardioprotection by Na+-H+exchange inhibition against ischemia/reperfusion injury. Circ. 2001, 37, 957–963. [Google Scholar] [CrossRef] [PubMed]
- Tokube, K.; Kiyosue, T.; Arita, M. Effects of hydroxyl radicals on K ATP channels in guinea-pig ventricular myocytes. Pfl?gers Arch. Eur. J. Physiol. 1998, 437, 155–157. [Google Scholar] [CrossRef]
- Krippeit-Drews, Peter, Claudia Krämer, Susanne Welker, Florian Lang, Hermann PT Ammon, and Gisela Drews. "Interference of H2O2 with stimulus-secretion coupling in mouse pancreatic β-cells." The Journal of physiology 514, no. Pt 2 (1999): 471.
- Trapp, S.; Proks, P.; Tucker, S.J.; Ashcroft, F.M. Molecular Analysis of ATP-sensitive K Channel Gating and Implications for Channel Inhibition by ATP. J. Gen. Physiol. 1998, 112, 333–349. [Google Scholar] [CrossRef]
- Yang, Y.; Shi, W.; Chen, X.; Cui, N.; Konduru, A.S.; Shi, Y.; Trower, T.C.; Zhang, S.; Jiang, C. Molecular Basis and Structural Insight of Vascular KATP Channel Gating by S-Glutathionylation. J. Biol. Chem. 2011, 286, 9298–9307. [Google Scholar] [CrossRef]
- Jiang, G.-L.; Nieves, A.; Bin Im, W.; Old, D.W.; Dinh, D.T.; Wheeler, L. The Prevention of Colitis by E Prostanoid Receptor 4 Agonist through Enhancement of Epithelium Survival and Regeneration. J. Pharmacol. Exp. Ther. 2006, 320, 22–28. [Google Scholar] [CrossRef]
- Gade, A.R.; Kang, M.; Akbarali, H.I. Hydrogen Sulfide as an Allosteric Modulator of ATP-Sensitive Potassium Channels in Colonic Inflammation. Mol. Pharmacol. 2012, 83, 294–306. [Google Scholar] [CrossRef] [PubMed]
- Sanguinetti, M.C.; Tristani-Firouzi, M. hERG potassium channels and cardiac arrhythmia. Nature 2006, 440, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Trudeau, M.C.; Warmke, J.W.; Ganetzky, B.; Robertson, G.A. HERG, a Human Inward Rectifier in the Voltage-Gated Potassium Channel Family. Science 1995, 269, 92–95. [Google Scholar] [CrossRef] [PubMed]
- Fermini, B.; Fossa, A.A. The impact of drug-induced QT interval prolongation on drug discovery and development. Nat. Rev. Drug Discov. 2003, 2, 439–447. [Google Scholar] [CrossRef] [PubMed]
- Vandenberg, Jamie I. "Oxidative stress fine-tunes the dance of hERG K+ channels." The Journal of Physiology 588, no. Pt 16 (2010): 2975.
- Zhang, Y.; Xiao, J.; Wang, H.; Luo, X.; Wang, J.; Villeneuve, L.R.; Zhang, H.; Bai, Y.; Yang, B.; Wang, Z. Restoring depressed HERG K+ channel function as a mechanism for insulin treatment of abnormal QT prolongation and associated arrhythmias in diabetic rabbits. Am. J. Physiol. Circ. Physiol. 2006, 291, H1446–H1455. [Google Scholar] [CrossRef] [PubMed]
- Mahapatra, C.; Thakkar, R. In Silico Electrophysiological Investigation of Transient Receptor Potential Melastatin-4 Ion Channel Biophysics to Study Detrusor Overactivity. Int. J. Mol. Sci. 2024, 25, 6875. [Google Scholar] [CrossRef] [PubMed]
- Mahapatra, Chitaranjan. "Simulation study of transient receptor potential current in urinary bladder over activity: student research abstract." In Proceedings of the 33rd Annual ACM Symposium on Applied Computing, pp. 74-75. 2018.
- Yamamoto, S.; Takahashi, N.; Mori, Y. Chemical physiology of oxidative stress-activated TRPM2 and TRPC5 channels. Prog. Biophys. Mol. Biol. 2010, 103, 18–27. [Google Scholar] [CrossRef] [PubMed]
- Balzer, M.; Lintschinger, B.; Groschner, K. Evidence for a role of Trp proteins in the oxidative stress-induced membrane conductances of porcine aortic endothelial cells. Cardiovasc. Res. 1999, 42, 543–549. [Google Scholar] [CrossRef] [PubMed]
- Groschner, Klaus, Christian Rosker, and Michael Lukas. "Role of TRP channels in oxidative stress." In Mammalian TRP Channels as Molecular Targets: Novartis Foundation Symposium 258, vol. 258, pp. 222-235. Chichester, UK: John Wiley & Sons, Ltd, 2004.
- Hara, Y.; Wakamori, M.; Ishii, M.; Maeno, E.; Nishida, M.; Yoshida, T.; Yamada, H.; Shimizu, S.; Mori, E.; Kudoh, J.; et al. LTRPC2 Ca2+-Permeable Channel Activated by Changes in Redox Status Confers Susceptibility to Cell Death. Mol. Cell 2002, 9, 163–173. [Google Scholar] [CrossRef]
- Wehage, Edith, Jörg Eisfeld, Inka Heiner, Eberhard Jüngling, Christof Zitt, and Andreas Lückhoff. "Activation of the cation channel long transient receptor potential channel 2 (LTRPC2) by hydrogen peroxide: a splice variant reveals a mode of activation independent of ADP-ribose." Journal of Biological Chemistry 277, no. 26 (2002): 23150-23156.
- Kolisek, M.; Beck, A.; Fleig, A.; Penner, R. Cyclic ADP-Ribose and Hydrogen Peroxide Synergize with ADP-Ribose in the Activation of TRPM2 Channels. Mol. Cell 2005, 18, 61–69. [Google Scholar] [CrossRef]
- Perraud, A.-L.; Takanishi, C.L.; Shen, B.; Kang, S.; Smith, M.K.; Schmitz, C.; Knowles, H.M.; Ferraris, D.; Li, W.; Zhang, J.; et al. Accumulation of Free ADP-ribose from Mitochondria Mediates Oxidative Stress-induced Gating of TRPM2 Cation Channels. J. Biol. Chem. 2005, 280, 6138–6148. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, S.; Takahashi, N.; Mori, Y. Chemical physiology of oxidative stress-activated TRPM2 and TRPC5 channels. Prog. Biophys. Mol. Biol. 2010, 103, 18–27. [Google Scholar] [CrossRef] [PubMed]
- Cao, G.; Thébault, S.; van der Wijst, J.; van der Kemp, A.; Lasonder, E.; Bindels, R.J.; Hoenderop, J.G. RACK1 Inhibits TRPM6 Activity via Phosphorylation of the Fused α-Kinase Domain. Curr. Biol. 2008, 18, 168–176. [Google Scholar] [CrossRef] [PubMed]
- Stadtman, E.R.; Levine, R.L. Free radical-mediated oxidation of free amino acids and amino acid residues in proteins. Amino Acids 2003, 25, 207–218. [Google Scholar] [CrossRef] [PubMed]
- Schilling, T.; Eder, C. Importance of the non-selective cation channel TRPV1 for microglial reactive oxygen species generation. J. Neuroimmunol. 2009, 216, 118–121. [Google Scholar] [CrossRef] [PubMed]
- Feng, M.; Grice, D.M.; Faddy, H.M.; Nguyen, N.; Leitch, S.; Wang, Y.; Muend, S.; Kenny, P.A.; Sukumar, S.; Roberts-Thomson, S.J.; et al. Store-Independent Activation of Orai1 by SPCA2 in Mammary Tumors. Cell 2010, 143, 84–98. [Google Scholar] [CrossRef] [PubMed]
- Bogeski, I.; Kummerow, C.; Al-Ansary, D.; Schwarz, E.C.; Koehler, R.; Kozai, D.; Takahashi, N.; Peinelt, C.; Griesemer, D.; Bozem, M.; et al. Differential Redox Regulation of ORAI Ion Channels: A Mechanism to Tune Cellular Calcium Signaling. Sci. Signal. 2010, 3, ra24. [Google Scholar] [CrossRef] [PubMed]
- Coddou, Claudio, Juan F. Codocedo, Shuo Li, Juan G. Lillo, Claudio Acuña-Castillo, Paulina Bull, Stanko S. Stojilkovic, and J. Pablo Huidobro-Toro. "Reactive oxygen species potentiate the P2X2 receptor activity through intracellular Cys430." Journal of Neuroscience 29, no. 39 (2009): 12284-12291.
- Mason, H.S.; Bourke, S.; Kemp, P.J. Selective Modulation of Ligand-Gated P2X Purinoceptor Channels by Acute Hypoxia Is Mediated by Reactive Oxygen Species. Mol. Pharmacol. 2004, 66, 1525–1535. [Google Scholar] [CrossRef] [PubMed]
- Huizinga, J.D.; Wei, R.; Chen, J.-H.; Wright, G.; Bardakjian, B. Generating bowel movements that facilitate nutrient absorption. Can. Young- Sci. J. 2014, 2014, 4–13. [Google Scholar] [CrossRef]
- Morgan, M.J.; Kim, Y.-S.; Liu, Z. Lipid Rafts and Oxidative Stress–Induced Cell Death. Antioxidants Redox Signal. 2007, 9, 1471–1484. [Google Scholar] [CrossRef]
- Kaji, N.; Horiguchi, K.; Iino, S.; Nakayama, S.; Ohwada, T.; Otani, Y.; Firman; Murata, T. ; Sanders, K.M.; Ozaki, H.; et al. Nitric oxide-induced oxidative stress impairs pacemaker function of murine interstitial cells of Cajal during inflammation. Pharmacol. Res. 2016, 111, 838–848. [Google Scholar] [CrossRef] [PubMed]
- Loffredo, L.; Ettorre, E.; Zicari, A.M.; Inghilleri, M.; Nocella, C.; Perri, L.; Spalice, A.; Fossati, C.; De Lucia, M.C.; Pigozzi, F.; et al. Oxidative Stress and Gut-Derived Lipopolysaccharides in Neurodegenerative Disease: Role of NOX2. Oxidative Med. Cell. Longev. 2020, 2020, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Kong, P.; Chen, C.; Tang, J.; Jin, X.; Yan, J.; Wang, Y. Targeting IL-17A Improves the Dysmotility of the Small Intestine and Alleviates the Injury of the Interstitial Cells of Cajal during Sepsis. Oxidative Med. Cell. Longev. 2019, 2019, 1–15. [Google Scholar] [CrossRef]
- Xue, C.; Pollock, J.; Schmidt, H.H.; Ward, S.M.; Sanders, K.M. Expression of nitric oxide synthase immunoreactivity by interstitial cells of the canine proximal colon. J. Auton. Nerv. Syst. 1994, 49, 1–14. [Google Scholar] [CrossRef]
- Rivera, L.R.; Poole, D.P.; Thacker, M.; Furness, J.B. The involvement of nitric oxide synthase neurons in enteric neuropathies. Neurogastroenterol. Motil. 2011, 23, 980–988. [Google Scholar] [CrossRef]
- Bódi, N.; Szalai, Z.; Bagyánszki, M. Nitrergic Enteric Neurons in Health and Disease—Focus on Animal Models. Int. J. Mol. Sci. 2019, 20, 2003. [Google Scholar] [CrossRef]
- Gangula, P.R.R.; Maner, W.L.; Micci, M.-A.; Garfield, R.E.; Pasricha, P.J.; Zhou, H.; Zhou, S.; Gao, J.; Zhang, G.; Lu, Y.; et al. Diabetes induces sex-dependent changes in neuronal nitric oxide synthase dimerization and function in the rat gastric antrum. Am. J. Physiol. Liver Physiol. 2007, 292, G725–G733. [Google Scholar] [CrossRef]
- Klein, S.; Seidler, B.; Kettenberger, A.; Sibaev, A.; Rohn, M.; Feil, R.; Allescher, H.-D.; Vanderwinden, J.-M.; Hofmann, F.; Schemann, M.; et al. Interstitial cells of Cajal integrate excitatory and inhibitory neurotransmission with intestinal slow-wave activity. Nat. Commun. 2013, 4, 1630. [Google Scholar] [CrossRef] [PubMed]
- Iino, S.; Horiguchi, K.; Nojyo, Y. Interstitial cells of Cajal are innervated by nitrergic nerves and express nitric oxide–sensitive guanylate cyclase in the guinea-pig gastrointestinal tract. Neuroscience 2008, 152, 437–448. [Google Scholar] [CrossRef]
- Dhein, S.; Salameh, A. Remodeling of Cardiac Gap Junctional Cell–Cell Coupling. Cells 2021, 10, 2422. [Google Scholar] [CrossRef]
- Moreno, A.P.; Lau, A.F. Gap junction channel gating modulated through protein phosphorylation. Prog. Biophys. Mol. Biol. 2007, 94, 107–119. [Google Scholar] [CrossRef]
- Zong, Yan-Jun, Xiao-Zhou Liu, Lei Tu, and Yu Sun. "Cytomembrane trafficking pathways of Connexin 26, 30, and 43." International Journal of Molecular Sciences 24, no. 12 (2023): 10349.
- Le, H.T.; Sin, W.C.; Lozinsky, S.; Bechberger, J.; Vega, J.L.; Guo, X.Q.; Sáez, J.C.; Naus, C.C. Gap Junction Intercellular Communication Mediated by Connexin43 in Astrocytes Is Essential for Their Resistance to Oxidative Stress. J. Biol. Chem. 2014, 289, 1345–1354. [Google Scholar] [CrossRef] [PubMed]
- Feine, I.; Pinkas, I.; Salomon, Y.; Scherz, A. Local Oxidative Stress Expansion through Endothelial Cells – A Key Role for Gap Junction Intercellular Communication. PLOS ONE 2012, 7, e41633. [Google Scholar] [CrossRef] [PubMed]
- Mahapatra, C. , and R. Manchanda. "Computational assessment of calcium channel effects on subthalamic nucleus neuronal cells: Study of abnormal bursting patterns in Parkinson's disease." In MOVEMENT DISORDERS, vol. 31, pp. S620-S621. 111 RIVER ST, HOBOKEN 07030-5774, NJ USA: WILEY-BLACKWELL, 2016.
- Dave, Vijay, Chitaranjan Mahapatra, and Rohit Manchanda. "A mathematical model of the calcium transient in urinary bladder smooth muscle cells." In 2015 37th Annual International Conference of the IEEE Engineering in Medicine and Biology Society (EMBC), pp. 5359-5362. IEEE, 2015.
- Mahapatra, C.; Brain, K.L.; Manchanda, R. Computational studies on urinary bladder smooth muscle: Modeling ion channels and their role in generating electrical activity. 2015 7th International IEEE/EMBS Conference on Neural Engineering (NER). LOCATION OF CONFERENCE, FranceDATE OF CONFERENCE; pp. 832–835.
- Mahapatra, C.; Manchanda, R. Computational Studies on Bladder Smooth Muscle: Modeling Ion Channels and their Role in Generating Electrical Activity. Biophys. J. 2015, 108, 588a. [Google Scholar] [CrossRef]
- Mahapatra, C. , and R. Manchanda. "Effects of aging in Parkinson’s disease: Role of Ca channel in dopamine neuron computational model." In Front. Neurosci. Conference Abstract: Neuroinformatics. 2015.
- Ureshino, R.P.; Erustes, A.G.; Bassani, T.B.; Wachilewski, P.; Guarache, G.C.; Nascimento, A.C.; Costa, A.J.; Smaili, S.S.; Pereira, G.J.d.S. The Interplay between Ca2+ Signaling Pathways and Neurodegeneration. Int. J. Mol. Sci. 2019, 20, 6004. [Google Scholar] [CrossRef]
- Patergnani, Simone, Alberto Danese, Esmaa Bouhamida, Gianluca Aguiari, Maurizio Previati, Paolo Pinton, and Carlotta Giorgi. "Various aspects of calcium signaling in the regulation of apoptosis, autophagy, cell proliferation, and cancer." International journal of molecular sciences 21, no. 21 (2020): 8323.
- Ermak, G.; Davies, K.J. Calcium and oxidative stress: from cell signaling to cell death. Mol. Immunol. 2002, 38, 713–721. [Google Scholar] [CrossRef] [PubMed]
- Davidson, S.M.; Duchen, M.R. Calcium microdomains and oxidative stress. Cell Calcium 2006, 40, 561–574. [Google Scholar] [CrossRef]
- Ott, Martin, Vladimir Gogvadze, Sten Orrenius, and Boris Zhivotovsky. "Mitochondria, oxidative stress and cell death." Apoptosis 12 (2007): 913-922.
- Tabet, F.; Savoia, C.; Schiffrin, E.L.; Touyz, R.M. Differential Calcium Regulation by Hydrogen Peroxide and Superoxide in Vascular Smooth Muscle Cells from Spontaneously Hypertensive Rats. J. Cardiovasc. Pharmacol. 2004, 44, 200–208. [Google Scholar] [CrossRef]
- Hüttemann, M.; Lee, I.; Samavati, L.; Yu, H.; Doan, J.W. Regulation of mitochondrial oxidative phosphorylation through cell signaling. Biochim. et Biophys. Acta (BBA) - Mol. Cell Res. 2007, 1773, 1701–1720. [Google Scholar] [CrossRef]
- Cahalan, Michael, and Erwin Neher. "[1] Patch clamp techniques: an overview." Methods in enzymology 207 (1992): 3-14.
- Liu, P.; Miller, E.W. Electrophysiology, Unplugged: Imaging Membrane Potential with Fluorescent Indicators. Accounts Chem. Res. 2019, 53, 11–19. [Google Scholar] [CrossRef]
- Cannon, Mark B., and S. James Remington. "Redox-sensitive green fluorescent protein: probes for dynamic intracellular redox responses. A review." Redox-Mediated Signal Transduction: Methods and Protocols (2009): 50-64.
- Moutal, A.; White, K.A.; Chefdeville, A.; Laufmann, R.N.; Vitiello, P.F.; Feinstein, D.; Weimer, J.M.; Khanna, R. Dysregulation of CRMP2 Post-Translational Modifications Drive Its Pathological Functions. Mol. Neurobiol. 2019, 56, 6736–6755. [Google Scholar] [CrossRef] [PubMed]
- Badar, Mohammad Sufian, Shazmeen Shamsi, Jawed Ahmed, and Md Afshar Alam. "Molecular dynamics simulations: concept, methods, and applications." In Transdisciplinarity, pp. 131-151. Cham: Springer International Publishing, 2022.
- Kutzner, C.; Köpfer, D.A.; Machtens, J.-P.; de Groot, B.L.; Song, C.; Zachariae, U. Insights into the function of ion channels by computational electrophysiology simulations. Biochim. et Biophys. Acta (BBA) - Biomembr. 2016, 1858, 1741–1752. [Google Scholar] [CrossRef] [PubMed]
- Winkler, D.A. The role of quantitative structure - activity relationships (QSAR) in biomolecular discovery. Briefings Bioinform. 2002, 3, 73–86. [Google Scholar] [CrossRef] [PubMed]
- Poswar, F.d.O.; Farias, L.C.; Fraga, C.A.d.C.; Bambirra, W.; Brito-Júnior, M.; Sousa-Neto, M.D.; Santos, S.H.S.; de Paula, A.M.B.; D'Angelo, M.F.S.V.; Guimarães, A.L.S. Bioinformatics, Interaction Network Analysis, and Neural Networks to Characterize Gene Expression of Radicular Cyst and Periapical Granuloma. J. Endod. 2015, 41, 877–883. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Illustration of the processes by which oxidative stress disrupts normal cells through the induction of reactive oxygen species. The red positive signs indicate ROS enhancers, while the red negative signs indicate ROS inhibitors. Oxidative stress radicals that modulate the shape of the cell are depicted by red stars and circles.
Figure 1.
Illustration of the processes by which oxidative stress disrupts normal cells through the induction of reactive oxygen species. The red positive signs indicate ROS enhancers, while the red negative signs indicate ROS inhibitors. Oxidative stress radicals that modulate the shape of the cell are depicted by red stars and circles.
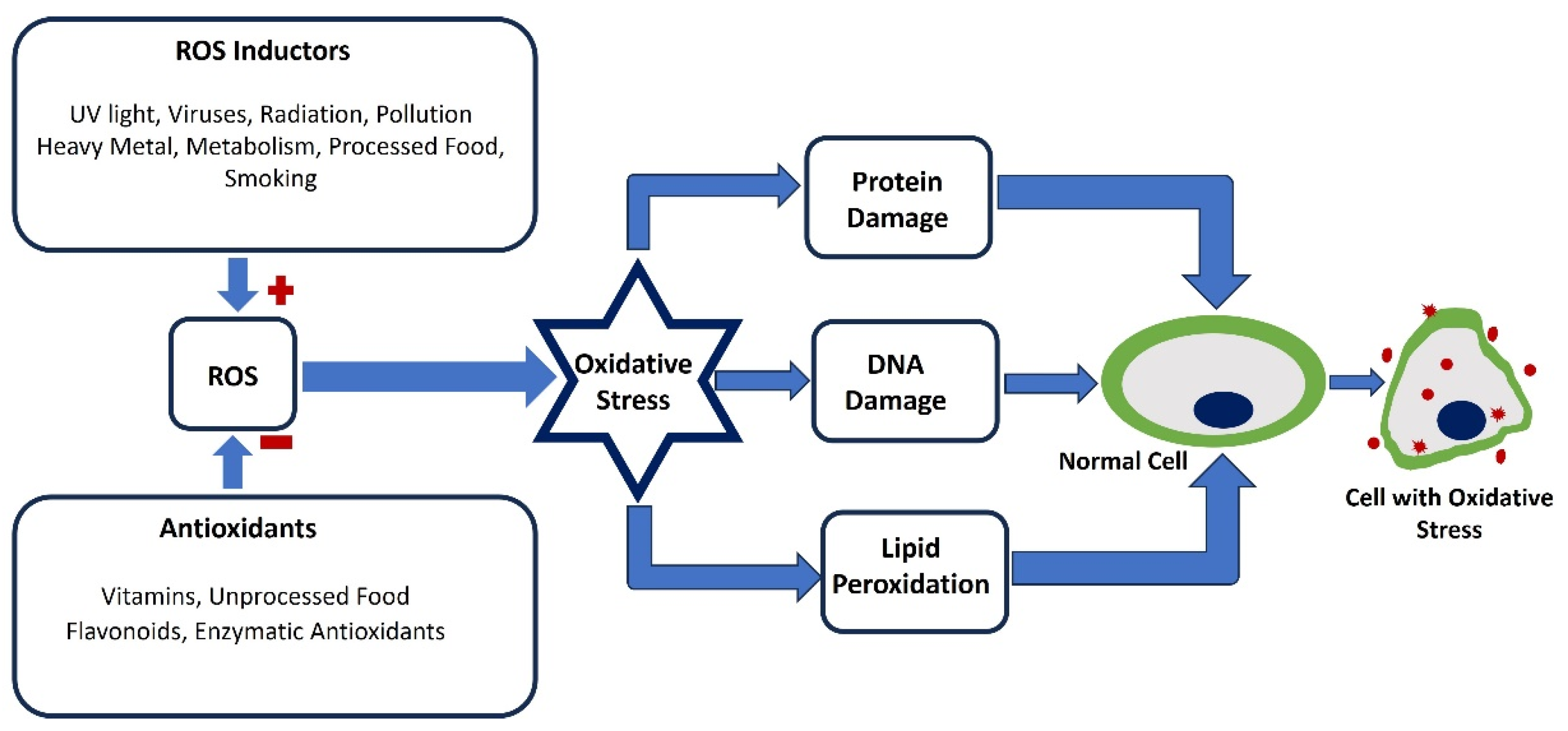
Figure 2.
Illustration of the membrane depolarization (black solid line), AP (red solid line), depolarization, repolarization, threshold potential (star mark), and resting membrane potential, which is maintained at -52 mV.
Figure 2.
Illustration of the membrane depolarization (black solid line), AP (red solid line), depolarization, repolarization, threshold potential (star mark), and resting membrane potential, which is maintained at -52 mV.
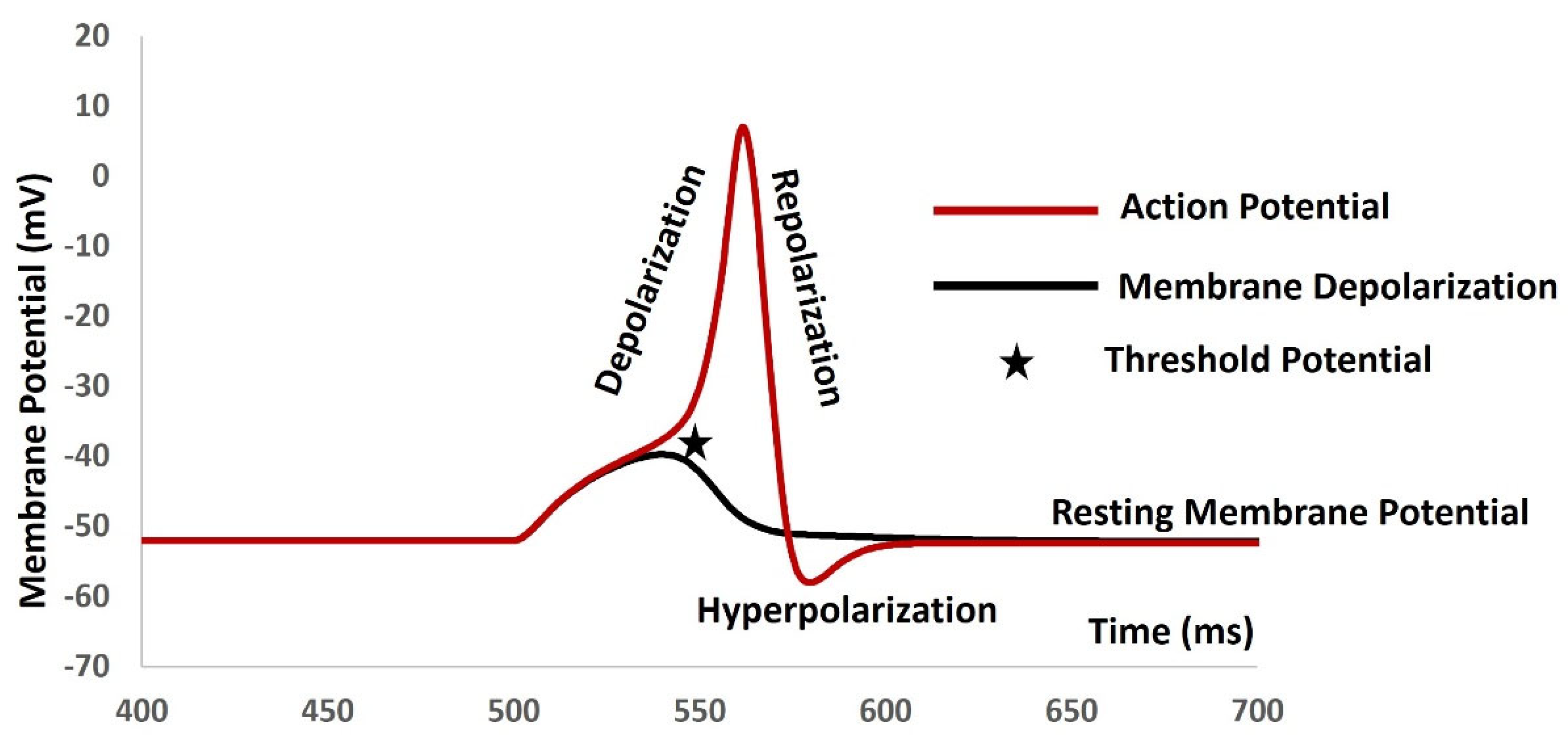
Figure 3.
Illustration of Gap junction connection between two cells Cell 1 and Cell 2. V1 and V2 are membrane potentials of cell 1 and cell 2, where rj value is the gap junction resistance between two cells.
Figure 3.
Illustration of Gap junction connection between two cells Cell 1 and Cell 2. V1 and V2 are membrane potentials of cell 1 and cell 2, where rj value is the gap junction resistance between two cells.
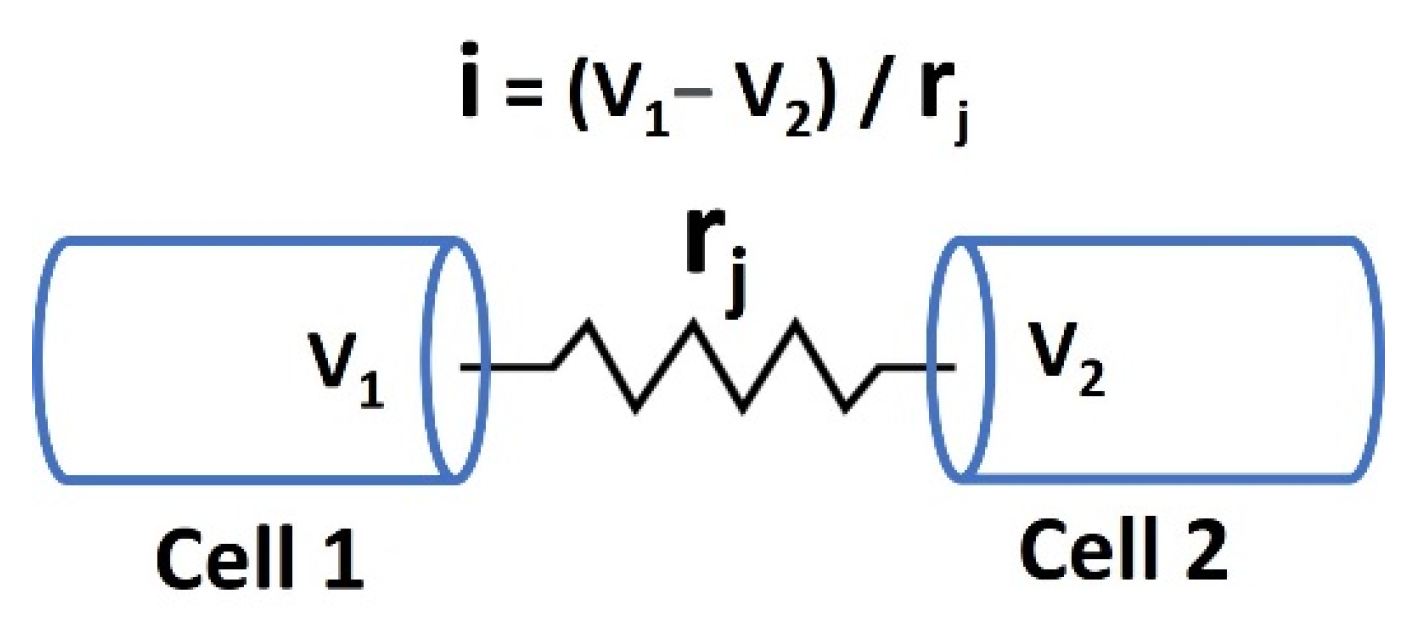

Table 1.
Values of RMP and types of electrical properties generated in major excitable cells.
| Cell type | RMP (mV) | AP/SW | Reference | |
|---|---|---|---|---|
| Smooth Muscle | -45 to -65 | AP/SW | [43] | |
| Cardiac Muscle | -80 to -90 | AP | [44] | |
| Skeletal Muscle | -65 to - 91 | AP | [45] | |
| Neuronal Cell | -60 to -70 | AP | [46] | |
| Pancreatic beta cells | -60 to -70 | SW | [47] |
Table 2.
Role of ion channels in AP/SW generations.
| Ion channel type | Role in AP/SW | |
|---|---|---|
| Ca2+ channels | RMP, AP/SW firing, Depolarization, | |
| Na+ channels | AP/SW firing, Depolarization, | |
| K+ channels | RMP, Repolarization, Hyperpolarization | |
| Cl- channels | RMP, Repolarization | |
| TRP channels | RMP, AP/SW firing, Depolarization, | |
| Leak channels | RMP |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



