
Submitted:
07 August 2024
Posted:
08 August 2024
You are already at the latest version
Abstract
Cellular senescence is the irreversible growth arrest subsequent to oncogenic mutations, DNA damage or metabolic insult. Senescence is associated with aging and chronic age associated diseases such as cardiovascular disease and diabetes. The involvement of cellular senescence in Acute Kidney Injury (AKI) and Chronic Kidney Disease (CKD) is not fully understood however, recent studies suggest that such patients have a higher-than-normal level of cellular senescence and accelerated aging. This study aimed to discover key biomarkers of senescence in AKI and CKD patients compared to other chronic ageing diseases as controls. We show that senescence proteins CKAP4 and PTX3 are upregulated in AKI and CKD patients compared with controls with chronic diseases suggesting they may play in a role in overall kidney disease development. Additionally, CKAP4 was found to be differentially expressed in both AKI and CKD when compared to unhealthy controls hence this biomarker could be prognostic senescence biomarker of both AKI and CKD.
Keywords:
Senescence
; chronic kidney disease
; acute kidney injury
; biomarker
; machine learning
1. Introduction
Acute kidney injury (AKI) is the abrupt cessation of kidney function due to heterogenous causes adding complication to critical illness [1]. Chronic kidney disease (CKD) is a global health burden causing a long-term reduction of kidney function resulting in CKD patients being at higher risk of morbidity, specifically type 2 diabetes (T2DM), and cardiovascular disease (CVD) [2,3]. AKI and CKD are now believed to be intertwined conditions acting on similar pathways causing inflammation and fibrosis that lead to worse patient outcomes [4]. AKI-to-CKD progression can be characterised by a long-term reduction in kidney function measured using serum creatinine (sCr) and estimated glomerular filtration rate (eGFR) that does not return to baseline 3-months post-AKI [5]. Current estimates report that 25% of patients who experience AKI progress to CKD, with AKI increasing the likelihood of CKD diagnosis by a hazard ratio of 8.8 [6,7]. Furthermore, for those AKI patients who have CKD at the time of AKI (AKI-on-CKD), AKI leads to more rapid progression of CKD [8]. Progression of CKD is also common and can result in the need for renal replacement therapy (RRT) or transplant, yet progression rate differs for each patient, with some experiencing rapid progression of ≥5mL/min/1.73m2 per year [9,10,11].
Traditional clinical biomarkers such as sCr and urea lack the ability to highlight patients at increased risk of AKI progression or rapid CKD progression [12,13,14]. Previous research has aimed to identify proteins that stratify AKI and CKD patients’ progression risk, however these studies often focused on specific types of CKD, used long follow-up periods, or used proteins already associated with AKI or CKD diagnosis and involved in inflammation and fibrosis pathways [15,16,17,18]. Cellular senescence is the irreversible growth arrest that occurs in response to metabolic insults, oncogenic mutations, and DNA damage, whereby cells cease dividing and undergo distinctive phenotypic alterations [19]. Cellular senescence can also occur in terminally differentiated cells such as cardiomyocytes, neurons, and nephrons, characterised by the accumulation of senescent cells that is hallmark of aging and aging pathologies [20,21]. Acute senescence is known to be beneficial, allowing for regeneration and repair in wound healing and immune clearance [22,23]. By contrast, chronic senescence, with the gradual accumulation of senescent cells over time, promotes deleterious effects accelerating deterioration and hyperplasia in aging [24]. Furthermore, a wide array of intra and extracellular insults including pro-inflammatory mediators, proteotoxic stress, DNA damage and mutation accelerate senescence related pathology [25].
Cells that undergo senescence secrete a variety of inflammatory and stromal regulators collectively known as senescence associated secretory phenotype (SASP) [26,27,28]. SASP is suggested to help immune clearance and elimination of senescent cells [29]. However, SASP can also adversely impact neighboring cells, extracellular matrix, and other structural components, resulting in chronic inflammation, leading to the initiation of senescence in healthy cells and vulnerable tissue [30]. The mechanisms involved in kidney disease are still largely unknown, however there is growing evidence that the reduced regenerative capacity of the kidneys is associated with cellular senescence [31]. Pro-inflammatory and pro-fibrotic senescence associated secretory phenotype (SASP) including: TNFα, IL-6, IL-1B and MCP-1 have been shown to be measurable in mice 24-hours post-AKI [27,32]. Abnormal kidney repair, a maladaptive response following AKI, can lead to tubular epithelial cells assuming a senescence-like phenotype and with downregulation of Klotho expression, increased expression of cyclin kinase inhibitors and telomere shortening can all occur [33]. This results in sustained secretion of profibrotic cytokines that can lead to fibrosis post-AKI and drive the kidney toward CKD [34].
Furthermore, in comparison to the general population, CKD patients experience accelerated aging characterised by increased systemic inflammation, progressive vascular disease, muscle wasting, osteoporosis, and frailty, even before the onset of kidney failure [35]. Early vascular aging (EVA) is characterised by vascular calcification, a cell-mediated process driven by alterations in vascular smooth muscle cells, that has been shown to be a measure of biological age and a predictor of CKD and mortality [36]. This EVA causes the arterial wall of CKD patients to appear older than that of chronological age-matched general population individuals. This is due to the impact of chronic inflammation, reflecting the premature adaptive changes because of repeated cellular insults, allostatic load, and an imbalance of anti-aging and pro-aging systems [37]. Although the mechanisms involved in EVA in CKD patients have not been fully elucidated, SASP causing chronic inflammation appears to play a fundamental role in both initiation and progression [38]. In this study we integrated proteomic targets associated with cardiovascular dysfunction, immune response, inflammation, and neurological impairment. The aim of this study was to investigate SASP [39] from measured plasma proteins or their involvement in AKI and CKD development and progression.
2. Materials and Methods
2.1. Participant Recruitment
Forty-three AKI patients (mean age= 61 years; 47% male) and 155 CKD patients (mean age= 59 years; 63% male) were recruited from Altnagelvin and Letterkenny University hospitals (Northwest of Ireland) as part of Acute Kidney Injury (AKI) outcome study (approved by Greater Manchester West Research Ethics Committee and then NHS GRAMPIAN, North of Scotland Research Ethics Service; REC ref 18/NW/0348 and 18/NS/0067). AKI patients were recruited within 7-days of AKI on ward, and CKD patients were recruited at outpatient clinics with disease stage ranging from 2 – 4 as per American Society of Nephrology. AKI patients were followed up at 3-months post-recruitment using serum creatinine and eGFR data from the hospital electronic care record to establish progression status. One hundred multi-morbid patients who were null of kidney disease, were used as controls (mean age 59.5 years; 47% male), selected to allow for age (p= 0.78), sex (p= 0.024) and BMI (p= 0.30) matching with AKI and CKD patients. The unhealthy control cohort included CVD patients (n= 49), diabetic patients (n= 43) and RA patients (n= 8). AKI progression was determined; AKI patient previously null of CKD, remaining below the cut-off of 60 ml/min/1.73 m2 at time of follow-up or, for AKI patients who had established CKD at the time of AKI, progression was determined by CKD stage progressing to a more severe stage e.g. from stage 2 CKD to stage 3a CKD.
2.2. Proteomic Analysis and Profiling Using Proximity Extension Assay
Levels of 535 unique proteins were measured using 5 µl of plasma per patient using the Proseek Multiplex proximity extension assay (PEA) (Olink Bioscience, Uppsala, Sweden) on the Olink Multiplex Plates: Cardiovascular panel II and Cardiovascular panel III, Immune response, Inflammation, Neuro exploratory, and Neurology. Following quality control (where over 80% of patients had a valid protein value) 476 unique proteins remained and were used for analysis. Using Olink data collected for controls with chronic diseases allowed for 273 proteins to be compared across all three cohorts.
2.3. Statistical Analysis
Statistical analysis was conducted in RStudio comparing base demographics of age, BMI and gender across AKI, CKD and unhealthy control cohorts. AKI and CKD cohorts were tested independently with unhealthy controls in a two-tail analysis. We expected biological differences between cohorts analyzed and because of this we employed welches two-tailed t-test to account of unequal variances between AKI/CKD and unhealthy controls. Code created in RStudio for statistical analysis within these cohorts will be publically available on Github.
2.4. Machine Learning Predictions
Machine learning to determine protein predictive capabilities was carried out in R, primarily using the caret package with additional supporting packages. Support vector machine (SVM) models were selected as the ideal model as they possess the ability to select a linear kernel, allowing for accurate identification of single protein biomarkers. Tenfold cross validation was used when building the model with hyperparameters such as cost being optimized through an iterative grid search function. Once models were generated, they were then fitted onto unseen data and set to predict, with the predictions being used to generate receiver operator characteristic (ROC) curves and area under curve (AUC) scores, which are used to determine predictive accuracy.
3. Results
3.1. Demographics for AKI Patients, CKD Patients, and Controls
Demographic and clinical data was measured for AKI, CKD and controls with chronic disease (Table 1). All cohorts were age and BMI matched. Age and BMI were not found to be significant across AKI, CKD and unhealthy controls, confirming the cohort matching was a success and prevented confounding of the results. Gender was identified as significant within CKD as 62.6% were male and 37.4% were female, however this could be attributed towards the damaging effects testosterone plays versus the protective effects estrogen plays, which would agree with current literature as males tend to progress to end stage renal failure sooner than females.
3.2. MMP3, IL16, TNFRSF10C, CCL23, GDF15, TNFR1 and UPAR that Are Significantly Different between AKI Patients and Controls on Hierarchical Clustered Heatmaps
A heatmap was created using unsupervised clustering of all proteins that were significantly different between AKI patients and controls (n= 143) (Figure 1A). Additionally, clinical classification and age were used to order patients and controls into over 65 years and under 65 years category. The rationale behind accounting for age this way is to ensure that the differential senescence expression that we witness is not attributed to age specifically. For example when we compare the cluster differences between AKI patients and unhealthy control patients, we can clearly see that chronological age plays no part in the direction of expression of these proteins, rather these proteins are specific signatures of AKI (Figure 1A). This suggests the SASP upregulation we see in AKI patients is not a consequence of ageing. Two major clusters of proteins were identified that showed upregulated and downregulated lists of proteins. Comparing the unhealthy controls above the 65 years of age versus AKI patients above 65 years there is a sub-group of upregulated SASP proteins in AKI such as MMP3, IL16, TNFRSF10C, CCL23, GDF15, TNFR1 and 2 and UPAR (p<0.01). This suggests age of patients is not confounding our senescence results.
3.3. TRAILR2, TNFRSF10A, LTBR, EPHB4, TNFR1, UPAR, CKAP4 and IGFBP2 Show Directly Proportional Correlation of Expression between AKI and Unhealthy Control Cohorts
A correlation matrix was constructed using most significant proteins that were different (n= 25) between AKI patients and controls (Figure 1B). Proteins that were most highly correlated are represented by a darker shade of colour with red representing directly proportional protein correlation and blue representing inversely proportional protein correlation. An area of interest was decided upon by selecting the area which had a clustering of the most correlated proteins (Pearson’s corelation coefficient = 0.8). There were 8 proteins in this area of interest which included: TRAILR2, TNFRSF10A, LTBR, EPHB4, TNFR1, UPAR, CKAP4 and IGFBP2. This suggests these SASP protiens may share similar pathways specific to AKI when compared to unhealthy controls.
3.4. Senescence Markers CKAP4, PTX3, UPAR, and TNFRSF10A Are Upregulated in AKI Patients Compared to Unhealthy Cohorts
All proteins were analysed using differential expression analysis and displayed in volcano plot (Figure 1C). The volcano plot displays fold change (log2 fold change) versus significance (-log10 p-value) for AKI patients compared to unhealthy controls. Senescence specific proteins are coloured green. CKAP4, PTX3, UPAR, and TNFRSF10A were the most significantly upregulated proteins in AKI patients vs unhealthy controls (p<1.50E-15). EGFR, TRANCE, DNER and ITGA11 were the most significantly downregulated proteins in AKI patients vs unhealthy controls (p<1.16E-10). SASP biomarkers comprise more than 50% (27/50) of all significantly different proteins.
3.5. Principle Component Analysis (PCA) of AKI Patients Compared to Controls
Principle component analysis (PCA) was conducted on all proteomic signatures between AKI patients and unhealthy controls to identify if there was clinical separation (Figure 1D). Each patients proteomic profile on the PCA is colour coded to match their cohort criteria, red for AKI and blue for controls with chronic disease. Using PC1 and PC2 allowed for approximately 40% of variance to be explained and illustrates that there are two distinct groups formed between AKI patients and controls. We can also see that the majority of class variance between AKI and chronic diseased controls on the PCA is found on PC1, which suggests that the majority of variance that we see is proteomic differences between AKI and controls.
3.5. CKAP4, PTX3, OPN and IGFBP2 Are the Most Differentially Expressed Senescent Proteins between AKI and Unhealthy Control Cohorts
Violin boxplots were generated to visualize individual proteomic differences between AKI patients and unhealthy controls for the top four statistically significant proteins: CKAP4, PTX3, OPN and IGFBP2 (p<5.76E-17): Protein expression was measured using NPX (Normalized Protein Expression) which is a log2 transformed metric quantified by OLINK proteomics (Figure 1E). We can see from the proteomic violin boxplots that CKAP4, PTX3, OPN and IGFBP2 are all upregulated in AKI patients compared to unhealthy controls, suggesting their over-expression could be attributed to initial kidney injury in AKI patients.
3.6. Receiver Operator Characteristic Curves for Individual Proteins for AKI vs. Controls
ROC curves for each protein were generated from univariate SVM models to measure predictive capabilities of each protein. We used the AUC score as a metric to assess predictive accuracy with: CKAP4 ( AUC: 0.98), PTX3 (AUC: 0.90), IGFBP2 (AUC: 0.92) and OPN (AUC: 0.92). (Figure 1F). With the predictions from our SVM models on the test data, we generated ROC curves and calculated AUC scores by using the predicted probability values as decision thresholds, then computed the corresponding sensitivities and specificities. With CKAP4, PTX3, IGFBP2 and OPN all being able to accurately differentiate between AKI and chronic disease controls, it suggests that there is distinct proteomic regulation within AKI patients, which these proteins are able to accurately capture allowing for clear differentiation.
3.7. Senescent Proteins TM, CKAP4 and MMP7 Are Significantly Different between CKD Patients and Controls on Hierarchical Clustered Heatmaps
A heatmap was created using unsupervised clustering of all proteins that were significantly different between CKD patients and controls (n= 255) (Figure 2A). Additionally, clinical classification and age was used to order patients and controls into over 65 years and under 65 years category. The rationale behind accounting for age this way is to ensure that the differential senescence expression that we witness is not attributed to age specifically. For example when we compare the cluster differences between CKD patients over the age of 65 and unhealthy control patients over the age of 65, we can clearly see that chronological age plays no part in the direction of expression of these proteins, rather these proteins are specific signatures of CKD (Figure 2A). Two major clusters of proteins were identified that showed upregulated and downregulated lists of proteins. Comparing the unhealthy controls above the 65 years of age versus CKD patients above 65 years there is a sub-group of upregulated SASP proteins in CKD such as TM, CKAP4 and MMP7 (p<0.01). This once again suggests that age of patients is not confounding our senescence results.
3.8. TNFR1, LTB3, EPHB4 and IL2RA Show Directly Proportional Correlation of Expression between CKD and Unhealthy Control Cohorts
A correlation matrix was constructed using most significant proteins that were different (n= 25) between CKD patients and controls (Figure 2B). Proteins that were most highly correlated are represented by a darker shade of colour with red representing directly proportional protein correlation and blue representing inversely proportional protein correlation. An area of interest was decided upon by selecting the area which had a clustering of the most correlated proteins (Pearson’s corelation coefficient = 0.8). There were 4 proteins in this area of interest which included: TNFR1, LTBR, EPHB4 and IL2RA. This suggests these SASP protiens may share similar pathways specific to CKD when compared to unhealthy controls.
3.9. Senescent Proteins TM, IL2, CKAP4 and MMP7 Are Significantly Differentially Expressed Senescent Proteins between CKD and Unhealthy Control Cohorts
All proteins were analysed using differential expression analysis and displayed in volcano plot (Figure 2C). The volcano plot displays fold change (log2 fold change) versus significance (-log10 p-value) for CKD patients compared to unhealthy controls. Senescence specific proteins are coloured green. TM, IL2, CKAP4 and MMP7 were the most significantly upregulated proteins in CKD patients vs unhealthy controls (p<1.12E-20). DNER, DCBLD2, GLB1 and CD6 were the most significantly downregulated proteins in CKD patients vs unhealthy controls (p<1.37E-07). SASP biomarkers comprise 50% (25/50) of all significantly different proteins.
3.10. Principle Component Analysis (PCA) of CKD Patients Compared to Controls
Principle component analysis (PCA) was conducted on all proteomic signatures between CKD patients and unhealthy controls to identify if there was clinical separation (Figure 2D). Using PC1 and PC2 allowed for approximately 37% of variance to be explained and illustrates that there are two distinct groups formed between CKD patients and controls. We can also see that the majority of class variance between AKI and chronic diseased controls on the PCA is found on PC1, which again suggests that the majority of variance that we see is proteomic differences between CKD and controls and is being captured effectively on that principle component.
3.11. TM, CKAP4, IL2 and MMP7 Are the Most Differentially Expressed Senescent Proteins between CKD and Unhealthy Control Cohorts
Violin boxplots were generated to visualize individual proteomic differences between CKD patients and unhealthy controls for the top four statistically significant proteins: TM, CKAP4, NT3 and MMP7 (p<1.12E-20). Protein expression was measured using NPX (Normalized Protein Expression) which is a log2 transformed metric quantified by OLINK proteomics (Figure 2E). We can see from the proteomic violin boxplots that TM, CKAP4, IL2 and MMP7 are all upregulated in CKD patients compared to unhealthy controls. The magnitude of expression isn’t as large as AKI compared to controls, which is what we expect as we are comparing a chronic disease to a chronic disease as compared to acute injury in AKI. This also suggests persistent over-expression of senescence biomakers could be linked to the pathophysiology of CKD.
3.12. Receiver Operator Characteristic Curves for Individual Proteins for CKD vs. Controls
ROC curves for each protein were retrieved from univariate SVM models to measure predictive capabilities of each protein. We used the area under curve (AUC) as a metric to assess predictive accuracy with: TM (AUC: 0.84), MMP7 (AUC: 0.74), IL2 (AUC: 0.89) and CKAP4 (AUC: 0.83) (Figure 2F). With the predictions from our SVM models on the test data, we generated ROC curves and calculated AUC scores by using the predicted probability values as decision thresholds, then computed the corresponding sensitivities and specificities. With TM, CKAP4, IL2 and MMP7 all being able to accurately differentiate between CKD and chronic disease controls, it suggests that there is distinct senescence specific proteomic regulation within CKD patients, which accurately capture this relationship allowing for clear differentiation.
3.13. Network Analysis of Differentially Expressed Proteins in AKI Compared to Controls
The top 50 statistically significant proteins between AKI and controls were queried in stringDB for network analysis with 48 of these proteins returning a hit search (Figure 3A). Various enriched signalling pathways were identified as statistically significant according to KEGG and Wiki pathways such as NF-kappa B, cytokine-cytokine receptor interactions, Inflammatory response pathways and CKAP4 signalling pathway (p<0.001) (Figure 3B and C). These pathways identified as enriched between AKI patients and chronic disease controls is reassuring, as it suggests that the proteomic signatures that were queried are distinct regulators in AKI pathophysiology and could play an integral role during initial kidney injury.
3.14. Network Analysis of Differentially Expressed Proteins in CKD Compared to Controls
The top 50 statistically significant proteins between AKI and controls were queried in stringDB for network analysis with 48 of these proteins returning a hit search (Figure 4A). Various enriched signalling pathways were identified as statistically significant according to KEGG and Wiki pathways such as NF-kappa B, Cytokine-cytokine receptor interactions, Inflammatory response pathways and CKAP4 signalling pathway (p<0.05) (Figure 4B and C). These pathways identified as enriched between CKD patients and chronic disease, although not as significant as within AKI are still important. These pathways are enriched in CKD patients compared to patients with other chronic diseases, suggesting that these signatures are integral to kidney damage and disease. We can attribute the difference in significance to the immunological response differences between AKI and CKD, as we would expect AKI to have a more pronounced proteomic-inflammatory expression pattern due to initial kidney injury. Whereas CKD would have a lessened but persisting proteomic-inflammatory expression pattern.
4. Discussion
The aim of this study was to investigate the role of cellular senescence in AKI and CKD. We found that levels of SASP cytoskeleton-associated protein 4 (CKAP4) and pentraxin-related protein (PTX3) were increased in AKI and CKD patients when compared to age, sex and BMI matched unhealthy controls, suggesting that these senescence proteins plays a role in AKI and CKD development. CKAP4 is involved in maintenance endoplasmic reticulum sheets and is already known to be associated with development of CKD and is a potential target for drug development for the treatment of Kidney fibrosis [40]. Interestingly, network analysis identified CKAP4 signalling pathway to be significantly enriched in both CKD and AKI cohorts (p<0.05). Analysis in this study using principal component analysis (PCA) and heatmaps highlighted the marked difference between AKI patients and unhealthy controls. This analysis included all measured biomarkers, with PCA showing that there was clear separation between AKI patients and unhealthy controls yet lesser separation between CKD patients and unhealthy controls. This shows that overall, when all biomarkers are included, CKD patients and unhealthy controls may share similar dysregulated pathways. Similarly, heatmap analysis showed that AKI patients and controls were distinctly separated while CKD patients were less separated from controls reinforcing the findings of PCA. This analysis is logical, as AKI patients are acutely seriously ill, often needing lifesaving clinical care as the body is responding to a severe attack. CKD patients, who are chronically ill, have a more similar biomarker response with unhealhty controls who have differing chronic conditions and inflammation, indicating that many chronic conditions react similarly in terms of the magnitude of biomarker upregulation/ downregulation. However, two SASP proteins, cytoskeleton-associated-protein 4 (CKAP4) and pentraxin-related protein (PTX3), were shown to be highly capable of highlighting both AKI and CKD patients for controls and to be of interest for further investigation.
CKAP4 is a type II transmembrane protein consisting of an N-terminal intracellular domain, a single transmembrane domain, and a C-terminal extracellular domain commonly situated in the endoplasmic reticulum and involved in many biological activities within the cell [41]. CKAP4 is well established as a biomarker of differing conditions, including playing a role in lung disease, and in tumor formation, and being upregulated in certain types of cancers [42]. Additionally, it is shown to be upregulated following ischemic injury playing an important role in ventricular fibroblast activation [43] and in recent studies shown to be upregulated in both in vitro and in vivo mice CKD models in vascular smooth muscle cells leading to vascular calcification by modulating YAP phosphorylation and MMP2 [40]. At present however, no other clinical studies have identified an association between CKAP4 and AKI or CKD making this study the first to do so. However, it is possible to hypothesize that with fibrosis and vascular calcification commonly present in both AKI and CKD, CKAP4 may play a role in driving these conditions leading to disease development. Furthermore, CKAP4 is shown to be involved in cell migration where cells with knockdown CKAP4 have a decreased level of cell migration[44]. As AKI is known to cause a reduction in cell migration CKAP4 may also be causing this to occur [45]. Therefore, with CKAP4 possibly affecting numerous aspects in AKI and CKD pathology this SASP protein may be high interest for future investigation.
PTX3, a member of the pentraxins superfamily, PTX3 is shown to play a role in innate immunity, inflammation and tissue remodeling, being mediated by tool-like receptors (TLR) [46,47,48,49]. High levels of PTX3 have been shown to be associated with worse outcomes in other organs including the brain following a stroke [50], and with low levels reported to increase tumor development risk [51]. PTX3 is a well-established senescence protein, is a multifunctional protein that plays complex regulatory roles in extracellular organisation and shown to increase dramatically in inflammatory conditions [52]. In cell models, PTX3 has been shown to stabilise the mitochondrial membrane potential and to suppress apoptosis, with levels shown to increase following ischemic reperfusion injury (IRI) and to be positively correlated with injury severity [53,54].
Mice deficient of PTX3 were shown to have worse recovery post-IRI compared to wild-type controls, and when mice were injected with PTX3 post-IRI, they had improved recovery compared to mice who were not, suggesting that PTX3 increases recovery in renal tubular cells post-injury [55,56]. Clinical studies have shown PTX3 levels to be significantly higher in AKI patients post-AKI than in non-AKI patients and higher in CKD patients compared to controls [57,58,59]. This may suggest that the high PTX3 levels seen in AKI and CKD patients are a protective mechanism aimed at protecting the kidneys post-injury. Furthermore, other clinical studies have shown PTX3 levels to be upregulated in CVD and diabetic patients [60,61]. In our study however, where the control cohort included CVD and diabetic patients, PTX3 levels remained to be significantly upregulated in AKI and CKD when compared to controls. This highlights the extent to which PTX3 is upregulated in AKI and CKD and possibly highlights it as a biomarker of interest for further investigation for disease risk stratification.
If promptly treated, AKI can resolve and the kidneys can recover to baseline function, however, the risk of progression to CKD is greatly increased following AKI [62,63]. The progression to CKD, following AKI, regardless of underlying cause, involves multiple mechanisms including immune cells, proximal tubular cells, and fibroblasts, where inflammation, hypoxia and nephron loss occur [4]. AKI survivors are said to have progressed to CKD when their kidney function has remained permanently below their baseline function for 90-days [64]. A large meta-analysis reported 26% of patients who experienced AKI went on to develop CKD, and 9% progressed to end stage renal disease where renal replacement therapy was required [6]. A longitudinal study involving ICU patients who were followed up after 90-days, reported that 25% of AKI patients progressed to CKD, with another reporting that 43.5% of hospitalized AKI patients progressed to CKD at 36-month follow-up [65]. Therefore, in addition to its ability to potentially aid in the diagnosis of AKI and CKD, CKAP4 and PTX3 may be able to help stratify AKI patients at higher risk of progression to CKD.
5. Conclusions
Senescence protein CKAP4 and PTX3 were shown to be capable of distinguishing AKI and CKD patients from a co-morbid control cohort including patients with diabetes, CVD and RA. This suggests that senescence in playing a role in both AKI and CKD pathology and that these senescence proteins may be useful for disease diagnosis. Furthermore, CKAP4 and PTX3 may be useful for stratification of AKI/CKD patient progression risk.
Supplementary Materials
None
Author Contributions
All the authors have seen and approved the final version of the scientific paper being submitted. All the authors warrant that this article is the authors’ original work, hasn’t received prior publication and isn’t under consideration for publication elsewhere. A.J.B., and T.S.R. conceived the idea, designed the research and secured the funding. SM, TM, and TSR wrote the manuscript. SM did all the experimental and scientific work under supervision of TSR, AJB and EM. TM and SM did majority of data analysis with help from EC, supervised by TSR and SW. MEC, CM, AE, TA, PG, AP, GW, ARE, DM, MOK, AP, DSG, PLM, CK, VM provided unhealthy control datasets. FM and YK lead clinical characterisation of the patients. All authors have read and agreed to the published version of the manuscript.
Funding
This research was mostly funded by a grant to A.J.B. under the EU Regional Development Fund EU Sustainable Competitiveness Programme for NI & the NI Public Health Agency. TSR also acknowledges funding from PHA/HSC R&D Division (COM/5618/20) and the Western Health & Social Care Trust.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Lankadeva, Y.R.; Kosaka, J.; Evans, R.G.; Bailey, S.R.; Bellomo, R.; May, C.N. Intrarenal and urinary oxygenation during norepinephrine resuscitation in ovine septic acute kidney injury. Kidney Int. 2016, 90, 100–108. [CrossRef]
- Levey, A.S. and J. Coresh, Chronic kidney disease. The Lancet, 2012. 379(9811): p. 165-180.
- Luyckx, V.A.; Cherney, D.Z.; Bello, A.K. Preventing CKD in Developed Countries. Kidney Int. Rep. 2019, 5, 263–277. [CrossRef]
- Sato, Y.; Takahashi, M.; Yanagita, M. Pathophysiology of AKI to CKD progression. Semin. Nephrol. 2020, 40, 206–215. [CrossRef]
- Webster, A.C., et al., Chronic Kidney Disease. The Lancet, 2017. 389(10075): p. 1238-1252.
- Coca, S.G.; Singanamala, S.; Parikh, C.R. Chronic kidney disease after acute kidney injury: A systematic review and meta-analysis. Kidney Int. 2012, 81, 442–448. [CrossRef]
- Noble, R.; Taal, M.W. Epidemiology and causes of chronic kidney disease. Medicine 2019, 47, 562–566. [CrossRef]
- Zhang, X.; Agborbesong, E.; Li, X. The Role of Mitochondria in Acute Kidney Injury and Chronic Kidney Disease and Its Therapeutic Potential. Int. J. Mol. Sci. 2021, 22, 11253. [CrossRef]
- Schanstra, J.P.; Zürbig, P.; Alkhalaf, A.; Argiles, A.; Bakker, S.J.; Beige, J.; Bilo, H.J.; Chatzikyrkou, C.; Dakna, M.; Dawson, J.; et al. Diagnosis and Prediction of CKD Progression by Assessment of Urinary Peptides. J. Am. Soc. Nephrol. 2015, 26, 1999–2010. [CrossRef]
- Semaan, V.; Noureddine, S.; Farhood, L. Prevalence of depression and anxiety in end-stage renal disease: A survey of patients undergoing hemodialysis. Appl. Nurs. Res. 2018, 43, 80–85. [CrossRef]
- Zhong, J.; Yang, H.-C.; Fogo, A.B. A perspective on chronic kidney disease progression. Am. J. Physiol. Physiol. 2017, 312, F375–F384. [CrossRef]
- Griffin, B.R.; Gist, K.M.; Faubel, S. Current Status of Novel Biomarkers for the Diagnosis of Acute Kidney Injury: A Historical Perspective. J. Intensiv. Care Med. 2020, 35, 415–424. [CrossRef]
- Levey, A.S.; Cattran, D.; Friedman, A.; Miller, W.G.; Sedor, J.; Tuttle, K.; Kasiske, B.; Hostetter, T. Proteinuria as a Surrogate Outcome in CKD: Report of a Scientific Workshop Sponsored by the National Kidney Foundation and the US Food and Drug Administration. Am. J. Kidney Dis. 2009, 54, 205–226. [CrossRef]
- Takahashi, S.; Nakasatomi, M.; Takei, Y.; Ikeuchi, H.; Sakairi, T.; Kaneko, Y.; Hiromura, K.; Nojima, Y.; Maeshima, A. Identification of Urinary Activin A as a Novel Biomarker Reflecting the Severity of Acute Kidney Injury. Sci. Rep. 2018, 8, 1–10. [CrossRef]
- Colombo, M., et al., Serum kidney injury molecule 1 and beta(2)-microglobulin perform as well as larger biomarker panels for prediction of rapid decline in renal function in type 2 diabetes. Diabetologia, 2019. 62(1): p. 156-168.
- Looker, H.C.; Colombo, M.; Hess, S.; Brosnan, M.J.; Farran, B.; Dalton, R.N.; Wong, M.C.; Turner, C.; Palmer, C.N.; Nogoceke, E.; et al. Biomarkers of rapid chronic kidney disease progression in type 2 diabetes. Kidney Int. 2015, 88, 888–896. [CrossRef]
- Schrauben, S.J.; Shou, H.; Zhang, X.; Anderson, A.H.; Bonventre, J.V.; Chen, J.; Coca, S.; Furth, S.L.; Greenberg, J.H.; Gutierrez, O.M.; et al. Association of Multiple Plasma Biomarker Concentrations with Progression of Prevalent Diabetic Kidney Disease: Findings from the Chronic Renal Insufficiency Cohort (CRIC) Study. J. Am. Soc. Nephrol. 2020, 32, 115–126. [CrossRef]
- Skupien, J.; Warram, J.H.; Niewczas, M.A.; Gohda, T.; Malecki, M.; Mychaleckyj, J.C.; Galecki, A.T.; Krolewski, A.S. Synergism Between Circulating Tumor Necrosis Factor Receptor 2 and HbA1c in Determining Renal Decline During 5–18 Years of Follow-up in Patients With Type 1 Diabetes and Proteinuria. Diabetes Care 2014, 37, 2601–2608. [CrossRef]
- Avelar, R.A.; Ortega, J.G.; Tacutu, R.; Tyler, E.J.; Bennett, D.; Binetti, P.; Budovsky, A.; Chatsirisupachai, K.; Johnson, E.; Murray, A.; et al. A multidimensional systems biology analysis of cellular senescence in aging and disease. Genome Biol. 2020, 21, 1–22. [CrossRef]
- Anderson, R.; Lagnado, A.; Maggiorani, D.; Walaszczyk, A.; Dookun, E.; Chapman, J.; Birch, J.; Salmonowicz, H.; Ogrodnik, M.; Jurk, D.; et al. Length-independent telomere damage drives post-mitotic cardiomyocyte senescence. EMBO J. 2019, 38. [CrossRef]
- Piechota, M., et al., Is senescence-associated β-galactosidase a marker of neuronal senescence? Oncotarget, 2016. 7(49): p. 81099-81099.
- Burton, D.G.; Stolzing, A. Cellular senescence: Immunosurveillance and future immunotherapy. Ageing Res. Rev. 2018, 43, 17–25. [CrossRef]
- Demaria, M.; Ohtani, N.; Youssef, S.A.; Rodier, F.; Toussaint, W.; Mitchell, J.R.; Laberge, R.-M.; Vijg, J.; Van Steeg, H.; Dollé, M.E.; et al. An Essential Role for Senescent Cells in Optimal Wound Healing through Secretion of PDGF-AA. Dev. Cell 2014, 31, 722–733. [CrossRef]
- Faget, D.V.; Ren, Q.; Stewart, S.A. Unmasking senescence: context-dependent effects of SASP in cancer. Nat. Rev. Cancer 2019, 19, 439–453. [CrossRef]
- Childs, B.G.; Baker, D.J.; Wijshake, T.; Conover, C.A.; Campisi, J.; Van Deursen, J.M. Senescent intimal foam cells are deleterious at all stages of atherosclerosis. Science 2016, 354, 472–477. [CrossRef]
- Campisi, J. The biology of replicative senescence. Eur. J. Cancer 1997, 33, 703–709. [CrossRef]
- En, A.; Takauji, Y.; Ayusawa, D.; Fujii, M. The role of lamin B receptor in the regulation of senescence-associated secretory phenotype (SASP). Exp. Cell Res. 2020, 390, 111927. [CrossRef]
- Van Deursen, J.M., The role of senescent cells in ageing. Nature 2014 509:7501, 2014. 509(7501): p. 439-446.
- Xue, W.; Zender, L.; Miething, C.; Dickins, R.A.; Hernando, E.; Krizhanovsky, V.; Cordon-Cardo, C.; Lowe, S.W. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 2007, 445, 656–660. [CrossRef]
- Takasugi, M.; Yoshida, Y.; Hara, E.; Ohtani, N. The role of cellular senescence and SASP in tumour microenvironment. FEBS J. 2022, 290, 1348–1361. [CrossRef]
- Wang, Y., et al., Implication of cellular senescence in the progression of chronic kidney disease and the treatment potencies. Biomedicine & Pharmacotherapy, 2021. 135: p. 111191-111191.
- Jin, H.; Zhang, Y.; Ding, Q.; Wang, S.S.; Rastogi, P.; Dai, D.-F.; Lu, D.; Purvis, M.; Cao, C.; Wang, A.; et al. Epithelial innate immunity mediates tubular cell senescence after kidney injury. J. Clin. Investig. 2019, 4. [CrossRef]
- Andrade, L.; Rodrigues, C.E.; Gomes, S.A.; Noronha, I.L. Acute Kidney Injury as a Condition of Renal Senescence. Cell Transplant. 2018, 27, 739–753. [CrossRef]
- Basile, D.P., et al., Progression after AKI: Understanding Maladaptive Repair Processes to Predict and Identify Therapeutic Treatments. J Am Soc Nephrol, 2016. 27(3): p. 687-97.
- Dai, L.; Qureshi, A.R.; Witasp, A.; Lindholm, B.; Stenvinkel, P. Early Vascular Ageing and Cellular Senescence in Chronic Kidney Disease. Comput. Struct. Biotechnol. J. 2019, 17, 721–729. [CrossRef]
- Benz, K.; Hilgers, K.-F.; Daniel, C.; Amann, K. Vascular Calcification in Chronic Kidney Disease: The Role of Inflammation. Int. J. Nephrol. 2018, 2018, 1–7. [CrossRef]
- Figuer, A.; Bodega, G.; Tato, P.; Valera, G.; Serroukh, N.; Ceprian, N.; de Sequera, P.; Morales, E.; Carracedo, J.; Ramírez, R.; et al. Premature Aging in Chronic Kidney Disease: The Outcome of Persistent Inflammation beyond the Bounds. Int. J. Environ. Res. Public Heal. 2021, 18, 8044. [CrossRef]
- Wang, W.J., G.Y. Cai, and X.M. Chen, Cellular senescence, senescence-associated secretory phenotype, and chronic kidney disease. Oncotarget, 2017. 8(38): p. 64520-64520.
- Rai, T.S.; Cole, J.J.; Nelson, D.M.; Dikovskaya, D.; Faller, W.J.; Vizioli, M.G.; Hewitt, R.N.; Anannya, O.; McBryan, T.; Manoharan, I.; et al. HIRA orchestrates a dynamic chromatin landscape in senescence and is required for suppression of neoplasia. Genes Dev. 2014, 28, 2712–2725. [CrossRef]
- Shi Shanghai Jiao Tong, Y., et al., CKAP4 regulates the progression of vascular calcication in chronic kidney disease by modulating YAP phosphorylation and MMP2 expression. 2020.
- Tan, H., et al., CKAP4 participates in tryptase-induced phenotypic conversion in atrial fibroblasts through PAR2/p38/JNK pathway. American Journal of Translational Research, 2021. 13(4): p. 2270-2270.
- Summers, M.E.; Richmond, B.W.; Kropski, J.A.; Majka, S.A.; Bastarache, J.A.; Hatzopoulos, A.K.; Bylund, J.; Ghosh, M.; Petrache, I.; Foronjy, R.F.; et al. Balanced Wnt/Dickkopf1 signaling by mesenchymal vascular progenitor cells in the microvascular niche maintains distal lung structure and function. Am. J. Physiol. Physiol. 2020, 320, C119–C131. [CrossRef]
- Gladka, M.M.; Molenaar, B.; de Ruiter, H.; van der Elst, S.; Tsui, H.; Versteeg, D.; Lacraz, G.P.; Huibers, M.M.; van Oudenaarden, A.; van Rooij, E. Single-Cell Sequencing of the Healthy and Diseased Heart Reveals Cytoskeleton-Associated Protein 4 as a New Modulator of Fibroblasts Activation. Circulation 2018, 138, 166–180. [CrossRef]
- Osugi, Y.; Fumoto, K.; Kikuchi, A. CKAP4 Regulates Cell Migration via the Interaction with and Recycling of Integrin. Mol. Cell. Biol. 2019, 39. [CrossRef]
- Singbartl, K.; Miller, L.; Ruiz-Velasco, V.; Kellum, J.A. Reversal of Acute Kidney Injury–Induced Neutrophil Dysfunction: A Critical Role for Resistin. Crit. Care Med. 2016, 44, e492–e501. [CrossRef]
- Bottazzi, B.; Inforzato, A.; Messa, M.; Barbagallo, M.; Magrini, E.; Garlanda, C.; Mantovani, A. The pentraxins PTX3 and SAP in innate immunity, regulation of inflammation and tissue remodelling. J. Hepatol. 2016, 64, 1416–1427. [CrossRef]
- Cappuzzello, C.; Doni, A.; Dander, E.; Pasqualini, F.; Nebuloni, M.; Bottazzi, B.; Mantovani, A.; Biondi, A.; Garlanda, C.; D’amico, G. Mesenchymal Stromal Cell-Derived PTX3 Promotes Wound Healing via Fibrin Remodeling. J. Investig. Dermatol. 2016, 136, 293–300. [CrossRef]
- Jaillon, S.; Moalli, F.; Ragnarsdottir, B.; Bonavita, E.; Puthia, M.; Riva, F.; Barbati, E.; Nebuloni, M.; Krajinovic, L.C.; Markotic, A.; et al. The Humoral Pattern Recognition Molecule PTX3 Is a Key Component of Innate Immunity against Urinary Tract Infection. Immunity 2014, 40, 621–632. [CrossRef]
- Wu, X.-Y.; Li, K.-T.; Yang, H.-X.; Yang, B.; Lu, X.; Zhao, L.-D.; Fei, Y.-Y.; Chen, H.; Wang, L.; Li, J.; et al. Complement C1q synergizes with PTX3 in promoting NLRP3 inflammasome over-activation and pyroptosis in rheumatoid arthritis. J. Autoimmun. 2019, 106, 102336. [CrossRef]
- Rodriguez-Grande, B.; Swana, M.; Nguyen, L.; Englezou, P.; Maysami, S.; Allan, S.M.; Rothwell, N.J.; Garlanda, C.; Denes, A.; Pinteaux, E. The Acute-Phase Protein PTX3 is an Essential Mediator of Glial Scar Formation and Resolution of Brain Edema after Ischemic Injury. J. Cereb. Blood Flow Metab. 2013, 34, 480–488. [CrossRef]
- Bonavita, E.; Gentile, S.; Rubino, M.; Maina, V.; Papait, R.; Kunderfranco, P.; Greco, C.; Feruglio, F.; Molgora, M.; Laface, I.; et al. PTX3 Is an Extrinsic Oncosuppressor Regulating Complement-Dependent Inflammation in Cancer. Cell 2015, 160, 700–714. [CrossRef]
- Ristagno, G.; Fumagalli, F.; Bottazzi, B.; Mantovani, A.; Olivari, D.; Novelli, D.; Latini, R. Pentraxin 3 in Cardiovascular Disease. Front. Immunol. 2019, 10, 823. [CrossRef]
- Chen, J.; Matzuk, M.M.; Zhou, X.J.; Lu, C.Y. Endothelial pentraxin 3 contributes to murine ischemic acute kidney injury. Kidney Int. 2012, 82, 1195–1207. [CrossRef]
- de Oliveira, T.H.C., et al., Tissue Dependent Role of PTX3 During Ischemia-Reperfusion Injury. Front Immunol, 2019. 10: p. 1461.
- Lech, M.; Römmele, C.; Gröbmayr, R.; Susanti, H.E.; Kulkarni, O.P.; Wang, S.; Gröne, H.-J.; Uhl, B.; Reichel, C.; Krombach, F.; et al. Endogenous and exogenous pentraxin-3 limits postischemic acute and chronic kidney injury. Kidney Int. 2013, 83, 647–661. [CrossRef]
- Lee, H.H.; Kim, S.Y.; Na, J.C.; Yoon, Y.E.; Han, W.K. Exogenous pentraxin-3 inhibits the reactive oxygen species-mitochondrial and apoptosis pathway in acute kidney injury. PLOS ONE 2018, 13, e0195758. [CrossRef]
- Chu-bing, R., Serum and Urinary PTX-3 for Prediction of the Current Occurrence of Acute Kidney Injury in Critical Patients. JOURNAL OF CLINICAL TRANSFUSION AND LABORATORY MEDICINE, 2020. 22(1): p. 104-108.
- Sjöberg, B.; Qureshi, A.R.; Heimbürger, O.; Stenvinkel, P.; Lind, L.; Larsson, A.; Bárány, P.; Ärnlöv, J. Association between levels of pentraxin 3 and incidence of chronic kidney disease in the elderly. J. Intern. Med. 2015, 279, 173–179. [CrossRef]
- Yilmaz, M.I., et al., Soluble TWEAK and PTX3 in Nondialysis CKD Patients: Impact on Endothelial Dysfunction and Cardiovascular Outcomes. Clinical Journal of the American Society of Nephrology, 2011. 6(4): p. 785-792.
- El-Badawy, A.; Omar, R.; El-Sayed, M.; Maksoud, A.A.; El-Sayed, A. Study of the Relation between Plasma PTX3 Levels and Preclinical Atherosclerotic Cardiovascular Complications in Type 2 Diabetic Patients. Benha J. Appl. Sci. 2019, 4, 9–15. [CrossRef]
- Inoue, K.; Kodama, T.; Daida, H. Pentraxin 3: A Novel Biomarker for Inflammatory Cardiovascular Disease. Int. J. Vasc. Med. 2012, 2012, 1–6. [CrossRef]
- Hannan, M., et al., Risk Factors for CKD Progression: Overview of Findings from the CRIC Study. Clin J Am Soc Nephrol, 2021. 16(4): p. 648-659.
- Jiang, M.; Bai, M.; Lei, J.; Xie, Y.; Xu, S.; Jia, Z.; Zhang, A. Mitochondrial dysfunction and the AKI-to-CKD transition. Am. J. Physiol. Physiol. 2020, 319, F1105–F1116. [CrossRef]
- Gameiro, J.; Fonseca, J.A.; Outerelo, C.; Lopes, J.A. Acute Kidney Injury: From Diagnosis to Prevention and Treatment Strategies. J. Clin. Med. 2020, 9, 1704. [CrossRef]
- Rimes-Stigare, C.; Ravn, B.; Awad, A.; Torlén, K.; Martling, C.-R.; Bottai, M.; Mårtensson, J.; Bell, M. Creatinine- and Cystatin C-Based Incidence of Chronic Kidney Disease and Acute Kidney Disease in AKI Survivors. Crit. Care Res. Pr. 2018, 2018, 1–8. [CrossRef]
Figure 1.
Senescence Proteins with Significant Differences in their Plasma Concentrations in AKI Patients vs Unhealthy controls. A. Heatmap of proteins that were significantly altered between AKI patients and unhealthy controls using unsupervised clustering. Heatmap showing unsupervised clustering of the proteins that were significantly altered between AKI patients and unhealthy controls. Clustering was done for gender, age, BMI and cohort on the x axis with protein clusters shown on the y axis. Protein change is represented by coloured bars which use the UHC as the reference group. Red represents an upregulation in AKI patients and blue represents a down regulation in the AKI patients. Grey represents missing data. The darker the colour represents a greater change. Significance threshold was set at a log2FC of >0.25 & <-0.25, with a p-value < 0.01. B. Correlation matrix of significantly altered protein between AKI patients and unhealthy controls. A correlation matrix of the most significantly different proteins (n= 25) between AKI patients (n= 43) and unhealthy controls (n= 100) was produced to highlight proteins that were strongly correlated. Colours on the graph represent the level of correlation, where red shows positive correlation and white shows no correlation. The darker the shade the stronger the correlation is as can be seen in the colour shade bar below the correlation matrix. The protein quadrant reported on in the analysis contains R2 values all greater than 0.8. C. Volcano plot presenting differential expression analysis of altered proteins in AKI patients compared to unhealthy controls. Volcano plot showing upregulated proteins are represented in red and proteins that were down regulated are represented in blue, with green labelling indicating whether said protein is differentially expressed in senescence according to transcriptomic analysis of cell senescence. The x-axis on the volcano plot uses log2(fold change) with the y-axis using -log10(p-values), with a significance threshold being a log2FC of >0.5 & <-0.5, with a p-value < 0.05. D. PCA analysis of AKI patients and unhealthy controls. PCA conducted on all proteomic signatures between AKI patients and unhealthy controls to identify if there was clinical separation (Figure 2D). Each patients proteomic profile on the PCA is colour coded to match their cohort criteria, red for AKI and blue for controls with chronic disease. Using PC1 and PC2 allowed for approximately 40% of variance to be explained and illustrates that there are two distinct groups formed between AKI patients and controls. E. Violin plots of most significantly altered proteins between AKI patients and unhealthy controls. Violin boxplots visualize individual proteomic differences between AKI patients and unhealthy controls for the top four statistically significant proteins: CKAP4, PTX3, OPN and IGFBP2 (p<5.76E-17): Protein expression was measured using NPX (Normalized Protein Expression) which is a log2 transformed metric quantified by OLINK proteomics. F. Receiver operator curve analysis of most significantly different proteins; TM, IL-2 and CKAP4, to identify AKI patients from UHC. ROC curves for each protein were generated from univariate SVM models to measure predictive capabilities of each protein. We used the AUC score as a metric to assess predictive accuracy with: CKAP4 ( AUC: 0.98), PTX3 (AUC: 0.90), IGFBP2 (AUC: 0.92) and OPN (AUC: 0.92).
Figure 1.
Senescence Proteins with Significant Differences in their Plasma Concentrations in AKI Patients vs Unhealthy controls. A. Heatmap of proteins that were significantly altered between AKI patients and unhealthy controls using unsupervised clustering. Heatmap showing unsupervised clustering of the proteins that were significantly altered between AKI patients and unhealthy controls. Clustering was done for gender, age, BMI and cohort on the x axis with protein clusters shown on the y axis. Protein change is represented by coloured bars which use the UHC as the reference group. Red represents an upregulation in AKI patients and blue represents a down regulation in the AKI patients. Grey represents missing data. The darker the colour represents a greater change. Significance threshold was set at a log2FC of >0.25 & <-0.25, with a p-value < 0.01. B. Correlation matrix of significantly altered protein between AKI patients and unhealthy controls. A correlation matrix of the most significantly different proteins (n= 25) between AKI patients (n= 43) and unhealthy controls (n= 100) was produced to highlight proteins that were strongly correlated. Colours on the graph represent the level of correlation, where red shows positive correlation and white shows no correlation. The darker the shade the stronger the correlation is as can be seen in the colour shade bar below the correlation matrix. The protein quadrant reported on in the analysis contains R2 values all greater than 0.8. C. Volcano plot presenting differential expression analysis of altered proteins in AKI patients compared to unhealthy controls. Volcano plot showing upregulated proteins are represented in red and proteins that were down regulated are represented in blue, with green labelling indicating whether said protein is differentially expressed in senescence according to transcriptomic analysis of cell senescence. The x-axis on the volcano plot uses log2(fold change) with the y-axis using -log10(p-values), with a significance threshold being a log2FC of >0.5 & <-0.5, with a p-value < 0.05. D. PCA analysis of AKI patients and unhealthy controls. PCA conducted on all proteomic signatures between AKI patients and unhealthy controls to identify if there was clinical separation (Figure 2D). Each patients proteomic profile on the PCA is colour coded to match their cohort criteria, red for AKI and blue for controls with chronic disease. Using PC1 and PC2 allowed for approximately 40% of variance to be explained and illustrates that there are two distinct groups formed between AKI patients and controls. E. Violin plots of most significantly altered proteins between AKI patients and unhealthy controls. Violin boxplots visualize individual proteomic differences between AKI patients and unhealthy controls for the top four statistically significant proteins: CKAP4, PTX3, OPN and IGFBP2 (p<5.76E-17): Protein expression was measured using NPX (Normalized Protein Expression) which is a log2 transformed metric quantified by OLINK proteomics. F. Receiver operator curve analysis of most significantly different proteins; TM, IL-2 and CKAP4, to identify AKI patients from UHC. ROC curves for each protein were generated from univariate SVM models to measure predictive capabilities of each protein. We used the AUC score as a metric to assess predictive accuracy with: CKAP4 ( AUC: 0.98), PTX3 (AUC: 0.90), IGFBP2 (AUC: 0.92) and OPN (AUC: 0.92).
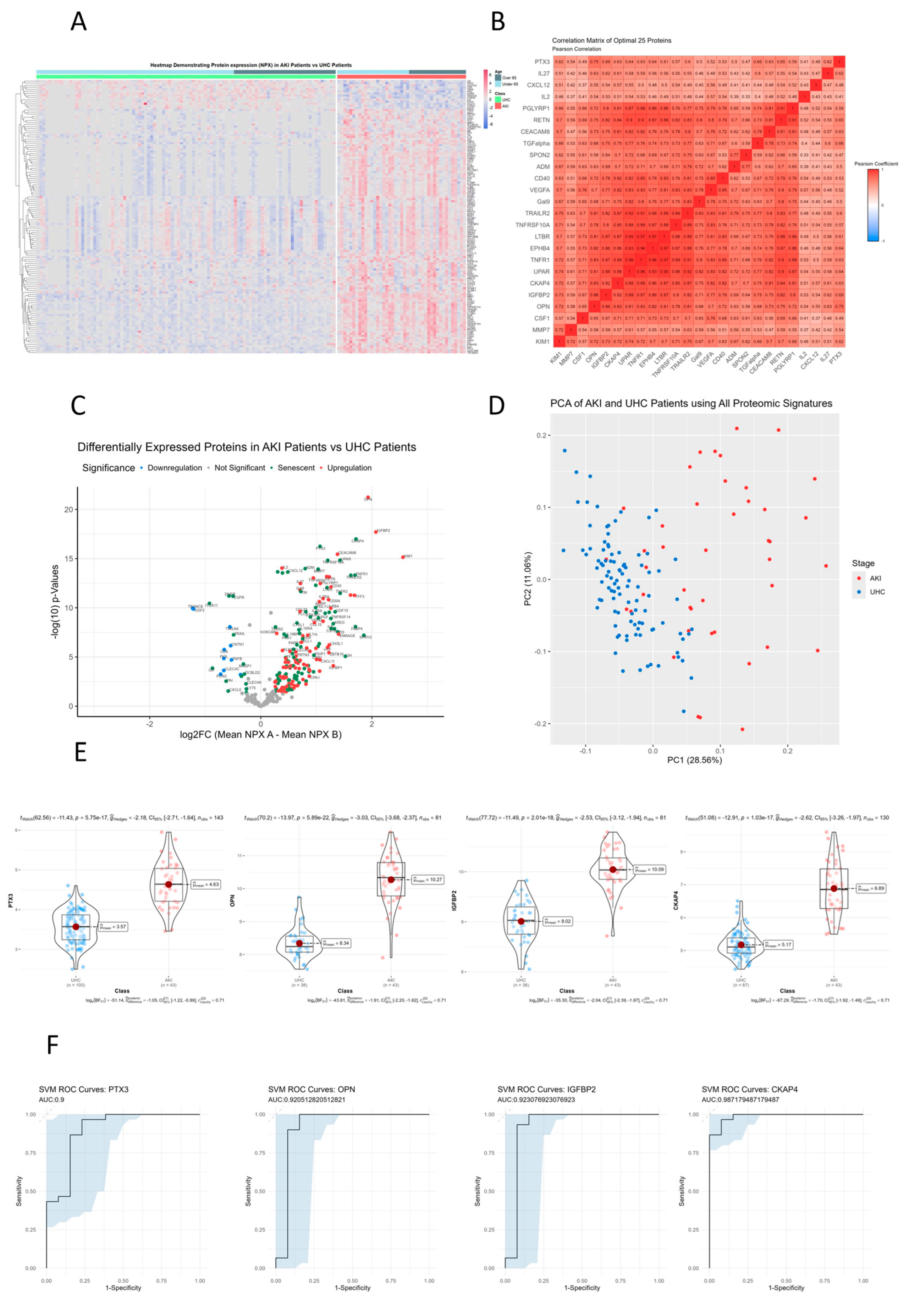
Figure 2.
Senescence Proteins with Significant Differences in their Plasma Concentrations in CKD Patients vs Unhealthy controls. A. Heatmap of proteins that were significantly altered between CKD patients and unhealthy controls using unsupervised clustering. Heatmap showing unsupervised clustering of the proteins that were significantly altered between CKD patients and unhealthy controls. Clustering was done for gender, age, BMI and cohort on the x axis with protein clusters shown on the y axis. Protein change is represented by coloured bars which use the UHC as the reference group. Red represents an upregulation in CKD patients and blue represents a down regulation in the CKD patients. Grey represents missing data. The darker the colour represents a greater change. Significance threshold was set at a log2FC of >0.25 & <-0.25, with a p-value < 0.01. B. Correlation matrix of significantly altered protein between CKD patients and unhealthy controls. A correlation matrix of significantly different proteins (n= 162) between CKD patients (n= 155) and unhealthy controls (n= 100) was produced to highlight proteins that were strongly correlated. Colours on the graph represent the level of correlation were blue represents positive correlation and red represents negative correlation. The darker the shade the stronger the correlation is as can be seen in the colour shade bar below the correlation matrix. The protein quadrant reported on in the analysis contains R2 values all greater than 0.7. C. Volcano plot presenting differential expression analysis of altered proteins in CKD patients compared to unhealthy controls. Volcano plot showing upregulated proteins are represented in red and proteins that were down regulated are represented in blue, with green labelling indicating whether said protein is differentially expressed in senescence according to transcriptomic analysis of cell senescence. The x-axis on the volcano plot uses log2(fold change) with the y-axis using -log10(p-values), with a significance threshold being a log2FC of >0.5 & <-0.5, with a p-value < 0.05. D. PCA analysis of CKD patients and unhealthy controls. Principle component analysis conducted on all proteomic signatures between CKD patients and unhealthy controls to identify if there was clinical separation. Using PC1 and PC2 allowed for approximately 37% of variance to be explained and illustrates that there are two distinct groups formed between CKD patients and controls. E. Violin plots of most significantly altered proteins between CKD patients and unhealthy controls. Violin boxplots visualize individual proteomic differences between CKD patients and unhealthy controls for the top four statistically significant proteins: TM, CKAP4, NT3 and MMP7 (p<1.12E-20). Protein expression was measured using NPX (Normalized Protein Expression) which is a log2 transformed metric quantified by OLINK proteomics. F. Receiver operator curve analysis of most significantly different proteins; TM, IL-2 and CKAP4, to identify CKD patients from UHC. ROC curves for each protein were retrieved from univariate SVM models to measure predictive capabilities of each protein. We used the area under curve (AUC) as a metric to assess predictive accuracy with: TM (AUC: 0.84), MMP7 (AUC: 0.74), IL2 (AUC: 0.89) and CKAP4 (AUC: 0.83).
Figure 2.
Senescence Proteins with Significant Differences in their Plasma Concentrations in CKD Patients vs Unhealthy controls. A. Heatmap of proteins that were significantly altered between CKD patients and unhealthy controls using unsupervised clustering. Heatmap showing unsupervised clustering of the proteins that were significantly altered between CKD patients and unhealthy controls. Clustering was done for gender, age, BMI and cohort on the x axis with protein clusters shown on the y axis. Protein change is represented by coloured bars which use the UHC as the reference group. Red represents an upregulation in CKD patients and blue represents a down regulation in the CKD patients. Grey represents missing data. The darker the colour represents a greater change. Significance threshold was set at a log2FC of >0.25 & <-0.25, with a p-value < 0.01. B. Correlation matrix of significantly altered protein between CKD patients and unhealthy controls. A correlation matrix of significantly different proteins (n= 162) between CKD patients (n= 155) and unhealthy controls (n= 100) was produced to highlight proteins that were strongly correlated. Colours on the graph represent the level of correlation were blue represents positive correlation and red represents negative correlation. The darker the shade the stronger the correlation is as can be seen in the colour shade bar below the correlation matrix. The protein quadrant reported on in the analysis contains R2 values all greater than 0.7. C. Volcano plot presenting differential expression analysis of altered proteins in CKD patients compared to unhealthy controls. Volcano plot showing upregulated proteins are represented in red and proteins that were down regulated are represented in blue, with green labelling indicating whether said protein is differentially expressed in senescence according to transcriptomic analysis of cell senescence. The x-axis on the volcano plot uses log2(fold change) with the y-axis using -log10(p-values), with a significance threshold being a log2FC of >0.5 & <-0.5, with a p-value < 0.05. D. PCA analysis of CKD patients and unhealthy controls. Principle component analysis conducted on all proteomic signatures between CKD patients and unhealthy controls to identify if there was clinical separation. Using PC1 and PC2 allowed for approximately 37% of variance to be explained and illustrates that there are two distinct groups formed between CKD patients and controls. E. Violin plots of most significantly altered proteins between CKD patients and unhealthy controls. Violin boxplots visualize individual proteomic differences between CKD patients and unhealthy controls for the top four statistically significant proteins: TM, CKAP4, NT3 and MMP7 (p<1.12E-20). Protein expression was measured using NPX (Normalized Protein Expression) which is a log2 transformed metric quantified by OLINK proteomics. F. Receiver operator curve analysis of most significantly different proteins; TM, IL-2 and CKAP4, to identify CKD patients from UHC. ROC curves for each protein were retrieved from univariate SVM models to measure predictive capabilities of each protein. We used the area under curve (AUC) as a metric to assess predictive accuracy with: TM (AUC: 0.84), MMP7 (AUC: 0.74), IL2 (AUC: 0.89) and CKAP4 (AUC: 0.83).
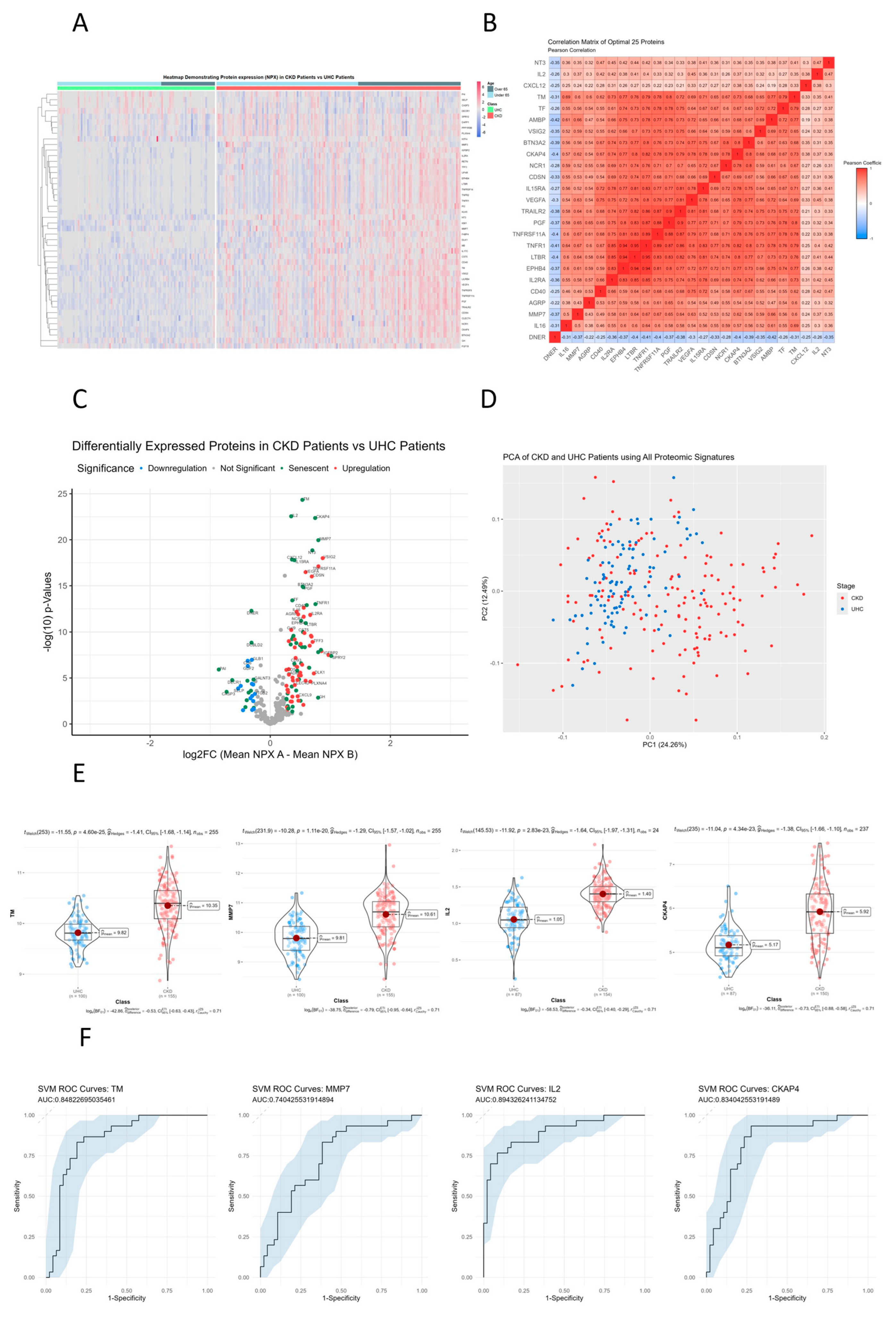
Figure 3.
Pathway Analysis of Proteins with Significant Differences in their Plasma Concentrations in AKI Patients vs Unhealthy controls. A. Protein-Protein interaction network of significant proteins between AKI patients and unhealthy controls. Protein-protein interaction networks generated using stringDB with the input being the corresponding gene symbol for each protein. The network is comprised of multiple nodes, each being a protein, which are interconnected to other proteins which visualize the interaction. Interactions have been found with literature research, scientific experiments or data mining. Networks consisted of the top 50 proteins, with the top ten being labelled as green for senescence or red for non-senescence. B. Enriched KEGG terms between AKI patients and unhealthy controls. Biological enrichment statistics generated on stringDB which provide the enriched KEGG terms based on specific proteins entered in the network. Count in network provides the number of genes present in the PPI map over the number of genes present in the signaling pathway. Strength provides a numerical value indicative of how strong the relationship between the proteins is, with the higher the number the more impactful the relationship, lastly p-value of the network provides how statistically significant each KEGG pathway is enriched. C. Enriched WikiPathway terms between AKI patients and unhealthy controls. Biological enrichment statistics generated on stringDB which provide the enriched WikiPathway terms based on specific proteins entered in the network. Count in network provides the number of genes present in the PPI map over the number of genes present in the signaling pathway. Strength provides a numerical value indicative of how strong the relationship between the proteins is, with the higher the number the more impactful the relationship, lastly p-value of the network provides how statistically significant each WikiPathway pathway is enriched.
Figure 3.
Pathway Analysis of Proteins with Significant Differences in their Plasma Concentrations in AKI Patients vs Unhealthy controls. A. Protein-Protein interaction network of significant proteins between AKI patients and unhealthy controls. Protein-protein interaction networks generated using stringDB with the input being the corresponding gene symbol for each protein. The network is comprised of multiple nodes, each being a protein, which are interconnected to other proteins which visualize the interaction. Interactions have been found with literature research, scientific experiments or data mining. Networks consisted of the top 50 proteins, with the top ten being labelled as green for senescence or red for non-senescence. B. Enriched KEGG terms between AKI patients and unhealthy controls. Biological enrichment statistics generated on stringDB which provide the enriched KEGG terms based on specific proteins entered in the network. Count in network provides the number of genes present in the PPI map over the number of genes present in the signaling pathway. Strength provides a numerical value indicative of how strong the relationship between the proteins is, with the higher the number the more impactful the relationship, lastly p-value of the network provides how statistically significant each KEGG pathway is enriched. C. Enriched WikiPathway terms between AKI patients and unhealthy controls. Biological enrichment statistics generated on stringDB which provide the enriched WikiPathway terms based on specific proteins entered in the network. Count in network provides the number of genes present in the PPI map over the number of genes present in the signaling pathway. Strength provides a numerical value indicative of how strong the relationship between the proteins is, with the higher the number the more impactful the relationship, lastly p-value of the network provides how statistically significant each WikiPathway pathway is enriched.

Figure 4.
Pathway Analysis of Proteins with Significant Differences in their Plasma Concentrations in CKD Patients vs Unhealthy controls. A. Protein-Protein interaction network of significant proteins between CKD patients and unhealthy controls. Protein-protein interaction networks generated using stringDB with the input being the corresponding gene symbol for each protein. The network is comprised of multiple nodes, each being a protein, which are interconnected to other proteins which visualize the interaction. Interactions have been found with literature research, scientific experiments or data mining. Networks consisted of the top 50 proteins, with the top ten being labelled as green for senescence or red for non-senescence. B. Enriched KEGG terms between CKD patients and unhealthy controls. Biological enrichment statistics generated on stringDB which provide the enriched KEGG terms based on specific proteins entered in the network. Count in network provides the number of genes present in the PPI map over the number of genes present in the signaling pathway. Strength provides a numerical value indicative of how strong the relationship between the proteins is, with the higher the number the more impactful the relationship, lastly p-value of the network provides how statistically significant each KEGG pathway is enriched. C. Enriched WikiPathway terms between CKD patients and unhealthy controls. Biological enrichment statistics generated on stringDB which provide the enriched WikiPathway terms based on specific proteins entered in the network. Count in network provides the number of genes present in the PPI map over the number of genes present in the signaling pathway. Strength provides a numerical value indicative of how strong the relationship between the proteins is, with the higher the number the more impactful the relationship, lastly p-value of the network provides how statistically significant each WikiPathway pathway is enriched.
Figure 4.
Pathway Analysis of Proteins with Significant Differences in their Plasma Concentrations in CKD Patients vs Unhealthy controls. A. Protein-Protein interaction network of significant proteins between CKD patients and unhealthy controls. Protein-protein interaction networks generated using stringDB with the input being the corresponding gene symbol for each protein. The network is comprised of multiple nodes, each being a protein, which are interconnected to other proteins which visualize the interaction. Interactions have been found with literature research, scientific experiments or data mining. Networks consisted of the top 50 proteins, with the top ten being labelled as green for senescence or red for non-senescence. B. Enriched KEGG terms between CKD patients and unhealthy controls. Biological enrichment statistics generated on stringDB which provide the enriched KEGG terms based on specific proteins entered in the network. Count in network provides the number of genes present in the PPI map over the number of genes present in the signaling pathway. Strength provides a numerical value indicative of how strong the relationship between the proteins is, with the higher the number the more impactful the relationship, lastly p-value of the network provides how statistically significant each KEGG pathway is enriched. C. Enriched WikiPathway terms between CKD patients and unhealthy controls. Biological enrichment statistics generated on stringDB which provide the enriched WikiPathway terms based on specific proteins entered in the network. Count in network provides the number of genes present in the PPI map over the number of genes present in the signaling pathway. Strength provides a numerical value indicative of how strong the relationship between the proteins is, with the higher the number the more impactful the relationship, lastly p-value of the network provides how statistically significant each WikiPathway pathway is enriched.
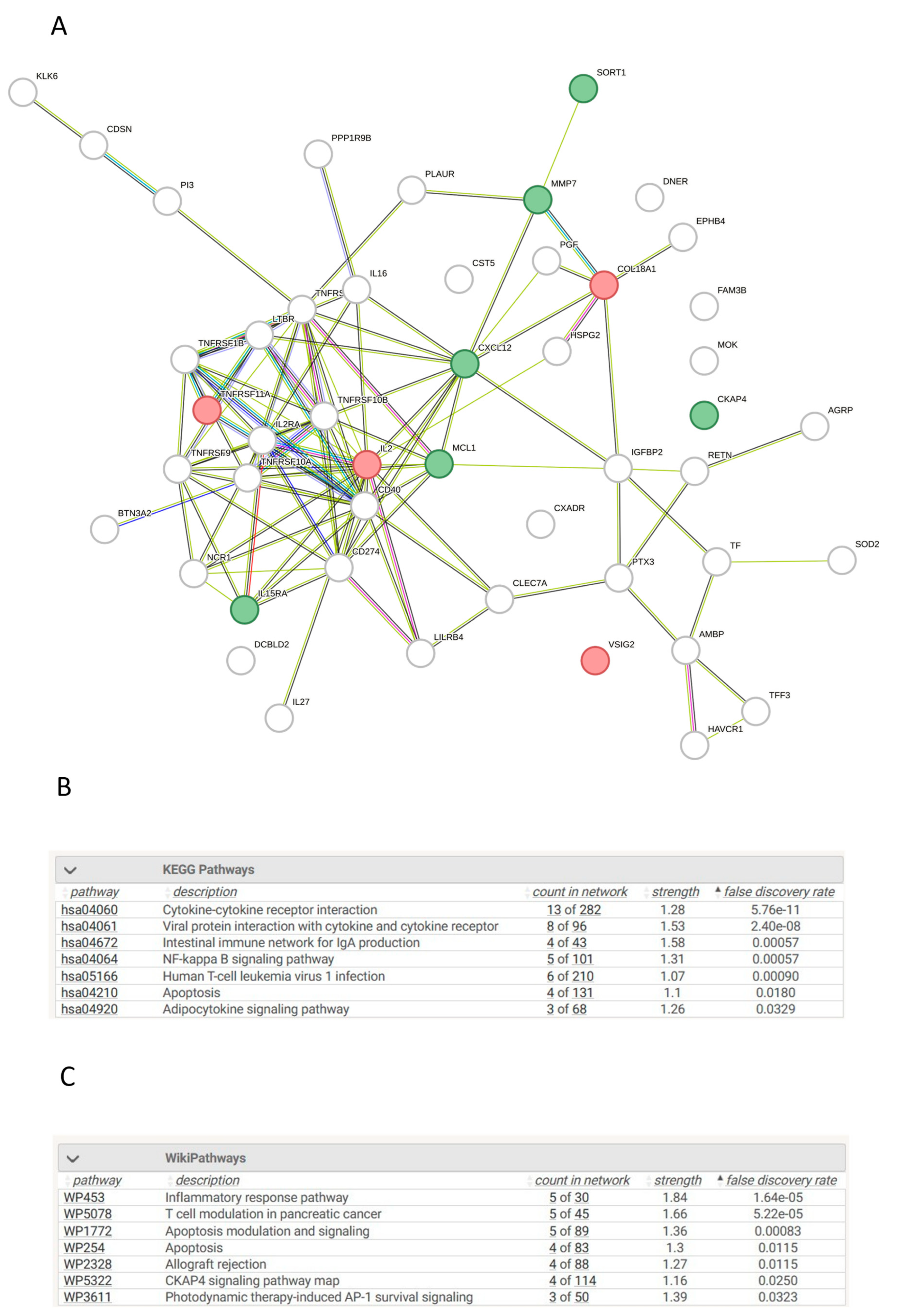

Table 1.
Demographics for acute kidney injury (AKI) patients, chronic kidney disease (CKD) patients and unhealthy controls (UHC).
Table 1.
Demographics for acute kidney injury (AKI) patients, chronic kidney disease (CKD) patients and unhealthy controls (UHC).
| Label | Levels | AKI | CKD | UHC | p-value |
| Gender | Female | 23 | 58 | 53 | |
| (%) | (53.5) | (37.4) | (53.0) | 0.024 | |
| Male | 20 | 97 | 47 | ||
| (%) | (46.5) | (62.6) | (47.0) | ||
| BMI | Mean | 30.1 | 29.3 | 30.7 | |
| (SD) | (10.1) | (5.5) | (7.7) | 0.306 | |
| Age | Mean | 60.5 | 58.7 | 59.5 | |
| (SD) | (14.8) | (17.3) | (12.9) | 0.788 |
This table shows the demographics for AKI patients (n= 43), CKD patients (n= 155) and UHCs (n= 100). All AKI and CKD patients were age, sex, and BMI matched to UHC. Levels of significance are represented with p-values. No urea or CRP data were available for UHCs so no comparison could be completed.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



