
Submitted:
17 August 2024
Posted:
19 August 2024
You are already at the latest version
Abstract
Many definitions of cancer have been proposed over the years to explain cancer’s origins and pathophysiology, but the disease is too variable to allow any single description to completely encompass all of the myriad traits. A true, unifying theory of cancer must not only reveal how all known carcinogens contribute toward malignancy, but must also elucidate why cancers occur predominately in older patients, why they express so many similar behaviors, as well as account for each cancer’s individual variations. A conceptual reorganization of decades of cancer data may reveal the fundamental process that underlies the malignant transformation of cells, which not only provides a new understanding of the etiology of cancers, but also helps to interpret the subsequent events that occur after transformation, which is of significant clinical interest. The revised concepts of initiation, promotion, and progression suggest that cancer is a failed or incomplete attempt at regeneration that occurs when ordinary cellular events unintentionally coincide over time to force the use of a DNA repair pathway that is normally restricted for use in germ cells.
Keywords:
cancer biology
; etiology
; endopolyploidy
; transformation
; senescence
; carcinogen
; cell cycle regulation
; unifying theory
Introduction
What, ultimately, is cancer? What does it take to cause a cell to malfunction to the point of seeming like a new organism with its own agenda and survival skills? Despite the preponderance of data accumulated over more than a century of observations and study, the biology of cancer eludes us [1]. Cancers arise from many different tissues, and often after exposure to a variety of factors including, but not limited to: heavy metals, radiation, smoking, asbestos, toxins, and occasionally viruses. Susceptibility to cancers is influenced by an individual’s own inherited genetic traits, poor diet, chronic inflammation, and more [2,3]. As a consequence of all of these variables, no two cancers are identical [2]. To date, no single feature, be it a specific genetic flaw or any other defect, is shared by all kinds of cancers [2,4,5,6]. It has led many to conclude that cancer is chaos; that cancer is simply the loss of control in a cell.
Despite the heterogeneity, the 200 or so different kinds of cancers share many behaviors and characteristics, such as unrestricted growth and invasion of tissues, migration to distant tissues, evasion of the body’s defenses, angiogenesis, and more, now commonly categorized as “Hallmarks of Cancer” [7]. Each cancer is as individual as the cell from which it originated, yet is connected into a family of diseases by virtue of these similarities. The relationship implies that a common process or sequence of events must be activated by the various carcinogens, which then triggers cancers to follow a similar pathway or genetic program [8]. The tenacity with which cancers adhere to such a pathway or program is suggested when investigators attempt to curb the growth of malignancies through the withdrawal of needed growth signals, inactivation of certain genes, or the blockage of certain receptors, among other targeted therapies. Cancers often find a way around the impediments in order to continue their growth and spread, which may indicate that cancers of all types are not simply the culmination of random defects, or the mere loss of control in a cell [9,10]. There appears to be an underlying direction to their varied activities.
Another shared characteristic, first noted by Otto Warburg nearly a century ago, is that cancers possess altered metabolisms [11]. The altered metabolisms are due to either defects in or suppression of oxidative phosphorylation, which forces an increase in glucose consumption to fulfill cellular energy requirements. Mitochondrial dysfunction and the resulting increase in fermentation is a hallmark of cancer, and was once thought to be the primary cause of cancer [11,12]. Since then, however, the altered metabolisms are considered more of a secondary and essential adaptation to mitochondrial dysfunction, as cancer is not the only possible result from malfunctioning mitochondria [13,14]. Error-prone mitochondria are suspected in a number of ailments from Alzheimer’s and Parkinson’s diseases to late onset diabetes, migraines, infertility and many other disorders [13]. Mitochondria are among the most easily damaged organelles as they are constantly exposed to reactive oxygen species and have poor DNA repair capabilities [15]. As a result, mitochondrial mutations and dysfunction increase with age [16]. Interestingly, mitochondria are responsible for the earliest onset of apoptosis, as cells engineered to lack mitochondria cannot undergo programmed cell death, while enucleated cells with intact mitochondria can [17,18]. Mitochondrial impairment may account for at least one method by which some cancers resist therapies [15,19]. Although increased fermentation is a common idiosyncracy, there are myriad ways to affect respiration and no single mitochondrial defect predominates among cancers [14,15]. Also of note is how glycolysis can be upregulated in cancers without mitochondrial dysfunction, or that oxidative phosphorylation can continue normally in some tumors [20]. If there is a theme to cancer, it is that no two are identical.
Yet another frequent observation is that cancers of all types have demonstrated the errant reactivation of embryonic genes to varying degrees [21,22,23,24,25]. It happens so often that many of the biomarkers used for early cancer detection or monitoring are hormones or proteins that are present only during pregnancy or fetal development [25,26,27]. Many comparisons have been made between malignant tumors and embryos, as both share similar methods of metastasis and invasion, immune avoidance, angiogenesis, toxin resistance, cell surface antigens and receptors, and much more ([22,23,24,28,29] more examples can be provided on request). So many parallels exist between embryonic development and tumorigenesis, that some investigators have attempted to explain cancer’s traits as a pathologic deviation of a developmental process [22,24,28,29,30,31]. Yet, again, although the activation of embryonic genes is commonplace, no single embryonic gene or gene sequence predominates among the different kinds of cancers. No two cancers are alike. They are as individual as the cells from which they originated.
Many definitions of cancer have been proposed over the years to explain its origins and pathophysiology, but the disease has been too variable to allow any single description to completely encompass all of the myriad traits. Flaws in our understanding of this baffling disease have resulted in only incremental advances for metastatic cancer care over the past several decades. There is not even a consensus on how the multitudinous carcinogens initiate or promote cancers, as even they do not have identical mechanisms of action [32]. A true, unifying theory of cancer must not only reveal how all known carcinogens contribute toward malignancy, but must also elucidate why cancers occur predominately in older patients, why they express so many similar behaviors, as well as interpret each cancer’s individual variations.
Most investigators agree that the emergence of cancer is a multistep process, with many presuming that passage through a so-called precancerous stage is a necessary part of the development [32,33]. Biopsies of suspicious lesions often reveal cells that are altered or abnormal, and those anomalous cells, while often still benign, indicate local conditions conducive to carcinogenesis [33]. Most of the altered cells never develop into cancers, and it is unknown what may trigger malignancy, or if indeed cancer must pass through the stage at all.
From the aforementioned observations, let us presume that cancer is not solely the product of random mutations, and that the variety of carcinogenic factors somehow effects a deviation to a pathway or program that is part of, or perhaps intersects with, the embryonic developmental program. This premise would account for many of the observed similarities between tumors and embryos while also affording insight into their differences. Let us also presume that cancer cannot be the result of statistically improbable events or it would occur with far less frequency than currently observed. Indeed, incidence of cancers worldwide is increasing (concurrently with increasing average life expectancies and industrialization) [34,35]. Therefore let us venture to conclude that cancer occurs when ordinary cellular events unintentionally coincide over time to produce the malignancies.
A conceptual reorganization of over a century’s worth of cancer data provides new insights into fundamental processes of cancer initiation and transformation, which not only establishes a complete understanding of the etiology of cancers, but also helps to elucidate the subsequent events that occur after transformation, which is of significant clinical interest. The chain of events which leads to the development of a precancerous cell, followed by the conditions necessary to “prime” that precancerous cell for malignant transformation, will be generically modeled here in an attempt to identify a unifying theory of cancer, and open up new avenues for research.
Consolidation of Carcinogenic Factors
The first step in the sequence of events requires that we establish a relationship among all identified genetic, environmental, dietary, viral, and other factors, and/or their indirect contributors, as to their roles in the creation of precancerous cells. Many disparate factors have been associated with causing, or somehow contributing to, the formation of malignancies. There does not seem to be a universal mechanism of action among them, and, in some cases, the mechanism of action is not known [32,33,36]. Some carcinogens are directly toxic or mutagenic, while others appear to be somewhat indirect contributors, and none cause neoplasia 100% of the time. If there is one common feature among the assorted carcinogens, it may be the condition of long-term or repetitive exposure versus a single exposure.
Many of the identified carcinogens are genotoxic, which has led to the general consensus that cancer is the result of any genetic mutation that causes too much growth or not enough death in a cell (i.e. uncontrolled cell proliferation). This “mutated gene hypothesis of cancer” adequately describes the rampant overgrowth of virtually any tumor, benign or malignant. However, that general description often neglects the most common, shared traits of malignancies, the existence of which makes it statistically improbable that cancer is merely the haphazard loss of control in a cell [7,8,37]. A single gene mutation, or even a dozen of them, does not a cancer make, as it is also interesting to note that transformation to malignancy isn’t immediate, but often follows a latency period of months to decades in humans after carcinogen-mediated mutation occurs, if transformation occurs at all [2,4,37,38,39]. The mutated gene hypothesis has been reworked over time to describe a gradual development through a progressive series of alterations, followed by a Darwinian selection for survival and growth, which could explain the long latency period. However, despite the innumerable faulty genes that have been identified in cancers, no single causative gene, or gene sequence, has been discovered that links all malignancies [1,2,4,5,6]. Complicating matters further is that even the most commonly observed genetic mutations don’t occur in every malignancy [4,37,38]. Other exceptions are the roles of nonmutagenic agents in carcinogenesis [1,32,39,40].
The widely accepted “multiple hit hypothesis” — which assumes a single cell must experience a series of gene-altering events in a stepwise, gradual transformation to full malignancy — applies to a multitude of carcinogenic factors when one broadens the definition. Since many carcinogens lack direct genotoxicity, it might be preferable to rework the concept to mean repeated stress or damage to a single cell over time. The term damage is defined here as any physical injury to a cell, including that which harms genes, proteins, membranes, electron transport system, or other processes within a cell. Using this broad definition, a single factor or a combination may contribute toward the malignant transformation of cells. The most common characteristic among the abundant environmental carcinogens appears to be chronic, intermittent, sublethal cellular stress or injury. Intermittence, which allows cells to recover somewhat between challenges, is an important factor, as too much damage over too short a period of time leads more often to cell death (via necrosis, or apoptosis, or variations thereof), thus decreasing the chances for the development of abnormal cells. The chronic nature of the stresses is necessary as too much time between abuses allows for full cellular repair or degradation, again precluding the development of abnormal cells. Repetitive stresses and limited recovery times seems necessary for the appearance of aberrations [41,42].
Non-damaging events, such as exposures to electromagnetic frequencies (EMF), perhaps should be included in this version of the “multiple hit hypothesis” as well, as intermittent cellular stresses (i.e. anything that triggers checkpoint kinases and cell cycle arrest) can induce greater-than-constitutive levels of protective proteins within a cell [43,44]. Some lines of evidence have shown that increasing levels of heat shock proteins (HSP), also known as molecular chaperones or stress proteins, not only protect, repair, and transport proteins, but also inhibit cell death, therefore, cells repeatedly stressed over time gradually build up both extra protection and the inability to die, further encouraging the development of aberrant cells [44,45]. This will be explored further in the next section.
An individual’s susceptibility to cancers often takes the shape of inherited genetic abnormalities which inhibit a cell’s ability to repair itself after injury and/or its ability to commit organized suicide when damage is too great to repair [3,46,47]. In effect, this decreases the “contact time” necessary for chronic, intermittent, sublethal stresses to promote precancerous development. A diet low in antioxidants (which help defend against cellular damage) contributes indirectly to the formation of anomalous cells via a decrease in protection from both intracellular and extracellular insults. Short term inflammatory responses defend the body against pathogens, often using peroxides and other reactive oxygen or nitrogen species to destroy the foreign bodies. Chronic inflammation exposes cells to long term contact with reactive oxygen or nitrogen species, which may eventually outpace a cell’s ability to protect and repair itself [48]. Chronic inflammation turns short term protection into near constant stress, adding to the “hits” a cell takes over its lifetime.
But this version of the “multiple hit hypothesis” does not at first glance seem to explain the role of virus in carcinogenesis. Rather than inflicting direct physical injuries, viruses insert genes into a cell that usurp control over its normal behaviors. Viral genes can alter apoptotic mechanisms, overexpress mitotic signals, and/or increase a cell’s normal life span (for example, with the addition of genes that code for telomerase), among other effects [49,50]. Indirectly, oncogenic viruses can contribute to chronic inflammation or immunosuppression [50]. Some viruses have been noted for their tendency to influence the development of specific cancers, however, viral carcinogens merely increase the probability of developing certain types of cancers, as infection by a virus alone does not guarantee cancer [12,49]. Viruses may be more of a susceptibility factor in the development of specific malignancies [49,50].
One way that viruses contribute to the formation of cancers, or perhaps add to a future cancer’s individuality, is via the insertion of genes that increase the rate of mitosis of a cell. Although the induction of cell proliferation per se may not be sufficient to induce carcinogenesis, it creates a favorable environment for tumor development as a rapidly dividing cell is at greater risk of acquiring further mutations via the weakening of checkpoints or error correction. Or, through the insertion of genes that inhibit apoptosis, a virus promotes the survival of a cell that might normally eliminate itself. The inability to commit suicide forces a cell to make alterations to exist, encouraging the development of further aberrations, especially if the infected cell’s microenvironment includes other stressors. Viruses may not cause direct physical damage to a cell — the “hits” of this version of the multiple hit hypothesis — but if their genetic alterations are viewed as “hits” that assist in the creation of abnormal cells, then perhaps it does apply.
The basic concept of “repeated damage to a single cell over time” ultimately holds up, but perhaps this version of the multiple hit hypothesis should be reworked to include other, more indirect contributors to cancer emergence. When viewing all of these elements as a whole, it may be posited that any factor, or combination of factors, that contributes to damage, affects a cell’s ability to recover from damage, and/or promotes a cell’s survival after damage, has the ability to promote — directly or indirectly — the formation of a precancerous cell. In this way, we consolidate the huge variety of carcinogenic factors. [see Fig.1] To test this premise, let us develop and examine a model for the development of a precancerous cell from a normal cell undergoing repetitive stress and recovery over time.
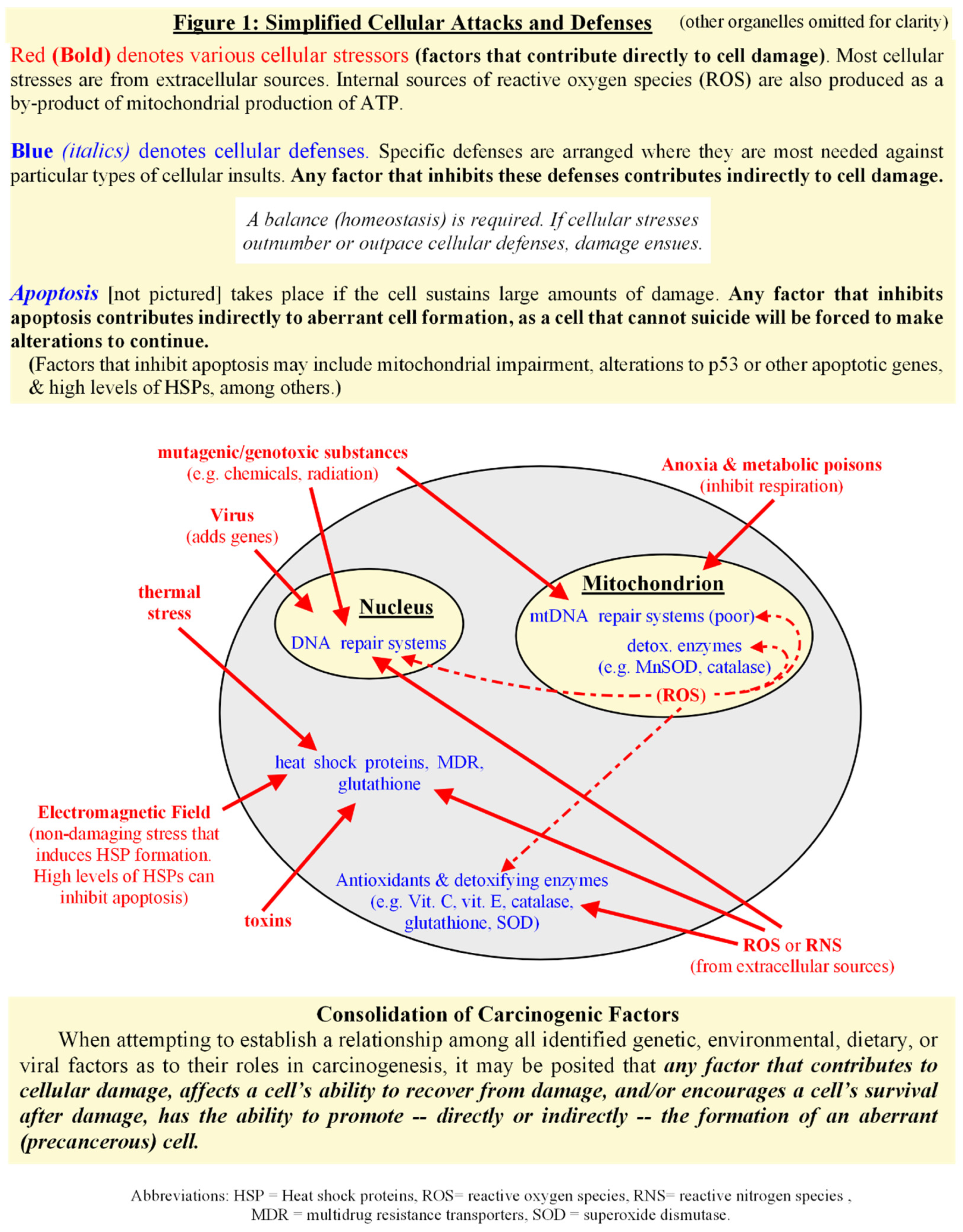
Chaperones as Protectors, Regulators, and Autonomic Decision-Makers.
One point mentioned earlier is the role of protective proteins in cellular responses before, during, and after stresses and injuries, which requires some additional information for the development of the repetitive-stress-and-recovery model. Originally named for the discovery that they protect cells from temperature extremes — so long as a cell receives a sublethal advance notice of future dangers — heat shock proteins, also called molecular chaperones or stress proteins, have since been found to do quite a lot more, which must be superficially addressed here. They are a diverse family that collectively stabilize proteins to prevent denaturing, assist in the folding of newly synthesized or corrupted proteins, or transport flawed proteins for controlled destruction [45]. Chaperone occupancy, and/or overload, governs various aspects of cellular stress responses as well as protein interactions that take place during routine cellular activities [51,52]. Chaperones are constitutively expressed at all times in a healthy cell to maintain proteostasis (known as Heat Shock Cognates, HSC), with more induced as needed (inducible forms are classified as HSP to distinguish them from constitutive versions), and although the holding or folding of defective proteins takes precedence over all other functions, they also sequester or chaperone many other proteins, and therefore help to regulate many important signal transduction pathways [45,51,52]. Molecular chaperones also play a role in shuttling cyclins into and out of the nucleus, which places chaperone occupancy into a primary position for controlling cell proliferation or arrest during periods of stress [53,54]. HSPs themselves repress transcription factor Heat Shock Factor 1 (HSF1) during non-stressful conditions, holding them in the cytoplasm and only releasing the transcription factors during proteotoxic challenge, thus functioning as “stress detectors” for the stress-induced production of supplemental HSPs when overburdened [55,56]. [see Fig. 2A & Fig. 2B]

The ratio of misfolded or aggregated proteins to chaperone availability guides cellular outcomes during and after damaging events. Denatured proteins that greatly outnumber protective chaperones lead to a rapid death via necrosis. The sudden release of cellular contents to the extracellular space during necrosis incites an inflammatory reaction which can stress or damage neighboring cells, so whenever possible a more controlled, less inflammatory, method of removing permanently injured or defective cells is preferred [57]. Increasing levels of intracellular HSPs, in response to recurring environmental challenges, can delay and convert potential necrotic cell death into programmed suicide or senescence, much like deploying a drogue chute behind an incapacitated airplane to change a wild tumble into a more controlled crash [45]. [see Fig.2C] The chaperone occupancy hypothesis can be reviewed more thoroughly in [45,51]
The competition for finite chaperone partners can also dampen the DNA Damage Response (DDR), if chaperone buffers are overburdened [58,59,60,61,62]. As an example, damaged DNA must be sensed by MRE11-RAD50-NBN/NBS1 (MRN complex), and it has been found that HSP90 interacts with NBN, and although its role has not been fully elucidated, it may be involved in either stabilization of NBN protein, or responsible for its nuclear translocation, or both [58,59,60,61,62]. Inhibition, or overloading, of HSP90 leads to defective DNA damage signaling and impaired repair pathways [58,59].
While protective, an overabundance of stress proteins — resulting from a frequently stressful environment — can become something of a double-edged sword, as they can inadvertently hide gene mutations by sequestering, refolding, or transporting flawed gene products for destruction before they cause harm. Only when additional stress and damage occurs, overloading all available chaperones, will the mutated phenotype be revealed [45,51].
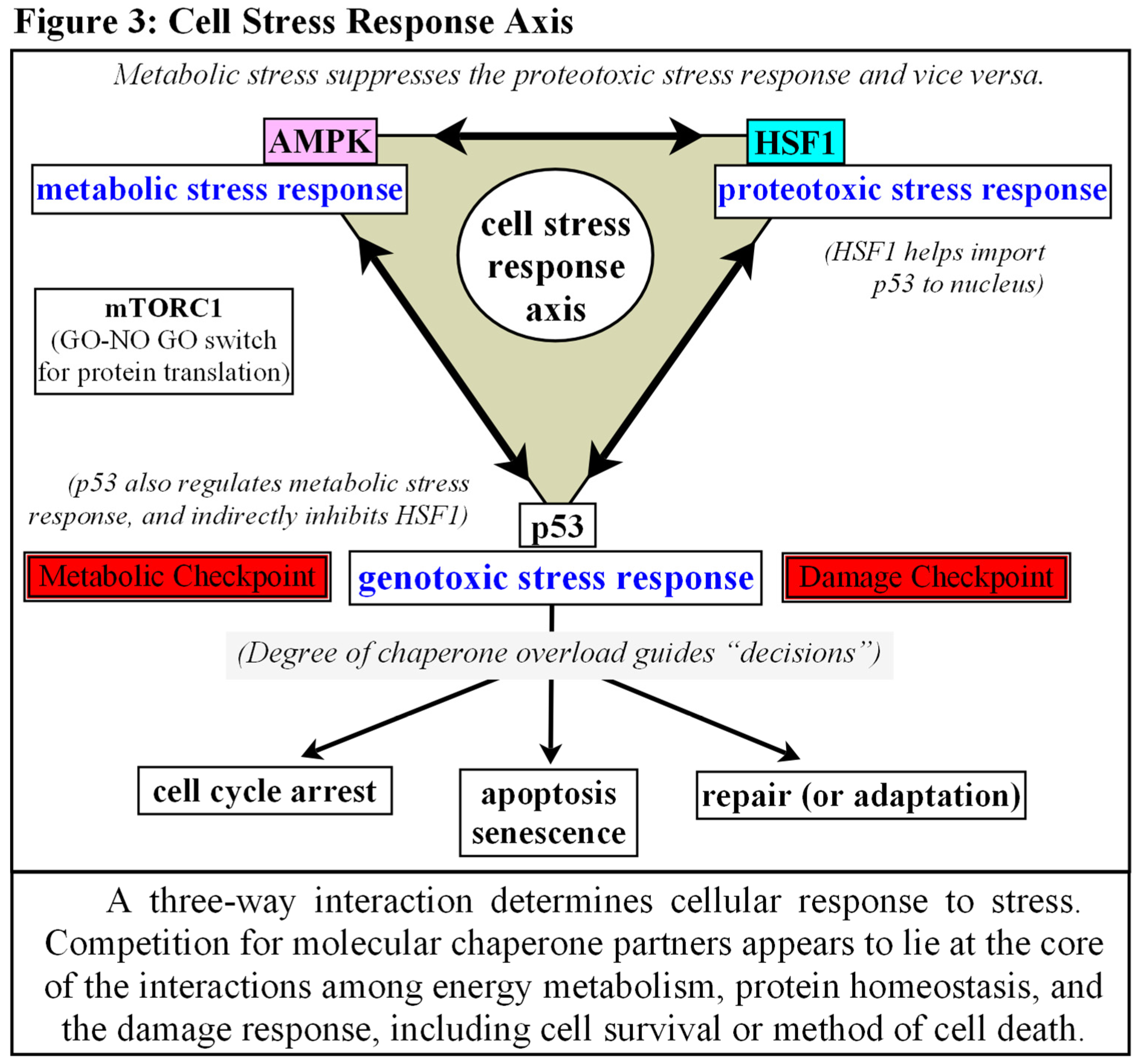
Another activity of note is the discovery of how HSF1 and metabolic sensor AMP-activated protein kinase (AMPK) antagonize each other, both directly and indirectly. Metabolic stress suppresses the proteotoxic stress response and vice versa [63,64,65]. HSF1 induces conformational changes to limit LKB1 binding and activation of AMPK, while AMPK phosphorylates HSF1 to eject it from the nucleus, or to prevent, or possibly alter, transcription once DNA bound [63,64,65]. Perhaps even more interesting is the observation that HSF1 is not only a regulator of the canonical Heat Shock Response, but also coordinates bioenergetics as well. When glucose is limited, HSF1 can bind directly with the Nampt promoter (nicotinamide phosphoribosyltransferase, a rate limiting enzyme for recycling NAD+ from nicotinamide), perhaps due to AMPK phosphorylation [65,66,67]. [see Fig.2D] NAD+ levels affect the sirtuin family of enzymes, and SIRT1/3 play critical roles in mitochondrial “quality control”, among other effects [66,67,68]. Thus a high population of molecular chaperones, induced by recurring environmental challenges, artificially creates low levels of activated HSF1 because they can simultaneously restrain HSF1 and manage aggregated and misfolded proteins [56,66]. Low levels of activated HSF1 during nutrient deprivation affects Nampt levels, which decreases available NAD+, which in turn decreases SIRT1/3 activity, which eventually leads to an accumulation of dysfunctional mitochondria via a reduction in mitochondrial fission and quality control [66,67,68]. In a similar manner, artificially low levels of activated HSF1, induced by an overabundance of stress proteins, can affect NAD+dependent PARP1 (Poly(ADP-ribose) polymerase 1) activity, thus also affecting nuclear DNA break repair [69,70].
Also of note is how both HSF1 and AMPK competitively interact with p53. HSF1 appears to be responsible for the enhanced import of p53 into the nucleus during stress, while p53 may help HSF1 with its binding and transcriptional activation [71,72,73]. Activated AMPK on the other hand, phosphorylates and stabilizes p53, indirectly inhibiting HSF1 via a downregulation of mTORC1 [74,75,76].
Much of the subcellular localization, timing, and activities of these molecules are still being worked out. This subject is too broad to adequately cover in this space and remains to be explored at a later date, but it appears that competition for chaperone partners lies at the core of the interactions among protein homeostasis, energy metabolism, and the initiating sequences of the DDR, as well as determining cellular survival or the method of cell death after damage [45,58,59,60,61]. [See Fig. 3]
The Role of Senescence
Another point that requires mention is the role of senescence in cell biology. The exact details are beyond the scope of this work, but some basic points can be made. It must also be noted that the vast majority of senescence research is conducted with fibroblasts, which are among the most common cells in the human body, while investigation into senescence of epithelial/endothelial cells is limited [69]. The relevance of this must be addressed in future works. For the majority of this discussion, however, we shall group general characteristics under the category of “senescence”, without distinguishing among cell types, unless specifically mentioned.
Senescence was originally thought to be an age-related permanent cessation of the cell cycle. The enzyme telomerase is not active in most adult human cells and as a result, with every cell division, the telomeres shorten. Telomere length was once considered as a sort of “timing device” for the absolute number of cell divisions allowed in a cell’s lifetime. After a finite number of cell divisions, referred to as Hayflick’s limit, the cell stops dividing and enters the permanent state of cell cycle arrest termed replicative senescence [77]. More recently, however, it has been discovered that senescence is more than simply the “retirement” of an older cell. Senescent cells can be found in culture and in vivo without telomere shortening [78,79,80].
Senescence is activated in response to double-stranded DNA breaks (DSB) or other events that activate checkpoint kinases [79,80,81]. Such “premature senescence” has been termed Stress-Induced Premature Senescence, or SIPS, and is virtually indistinguishable from replicative senescence. Unraveling telomeres are perceived by the cell as double stranded DNA breaks, so it appears that replicative senescence actually occurs in response to severe damage to a cell, rather than the advanced age of the cell [79]. It would then be no coincidence that senescence occurs most often in older cells. As a cell ages, the number of injuries it sustains over time accumulates, and the ability to repair itself declines [16]. The most important variable to the induction of senescence appears to be the inhibition of apoptosis [82]. Cellular senescence is controlled by many of the same tumor suppressor genes as apoptosis, and both apoptosis and senescence work to prevent the growth of cells at risk for malignant transformation. Whereas apoptosis eliminates damaged cells, cellular senescence permanently arrests their proliferation [80,83]. Stress-induced senescence is often considered to be a “failsafe” mechanism to prevent neoplastic formation in the event a severely damaged cell cannot undergo programmed cell death [84,85].
Although arrested, senescent cells remain metabolically active, and become secretory cells, releasing chemokines, inflammatory cytokines, proteases, collagenases, and growth factors into the extracellular space — many of which are associated with wound healing [85,86]. The secretory products of senescent cells may, in fact, be a resolution stage of the wound response, where degradation of the extracellular matrix prevents fibrosis, scarring, and the loss of tissue function [87]. As populations of senescent cells accumulate over time in an aging organism, however, benefits become detriments. The secretory products of senescent cells, perhaps meant only as a short-term resolution of wound healing, begin to adversely alter the tissue microenvironment, affecting neighboring cells in myriad ways and contributing to both age-related pathologies and the inadvertent progression of mutant cells [83,85,86]. A growing body of evidence also suggests that precancerous cells must somehow escape senescence in order to progress to malignancy [69,88,89]. However, artificially forcing a cell past senescence (via viral telomerase, or bypassing checkpoint kinases, for instance) causes it to reenter mitosis regardless of damage, yet, without the transformation to cancer, the cell will continue only until crisis [89,90]. Merely bypassing senescence provides a kind of “extended life”, but does not initiate immortalization [88,89]. How a precancerous cell might not only escape senescence but transform into malignancy will be discussed after the next section.
Model for the Development of a Senescent Cell with Malignant Potential
[Please refer to Fig.4 for the following discussion]
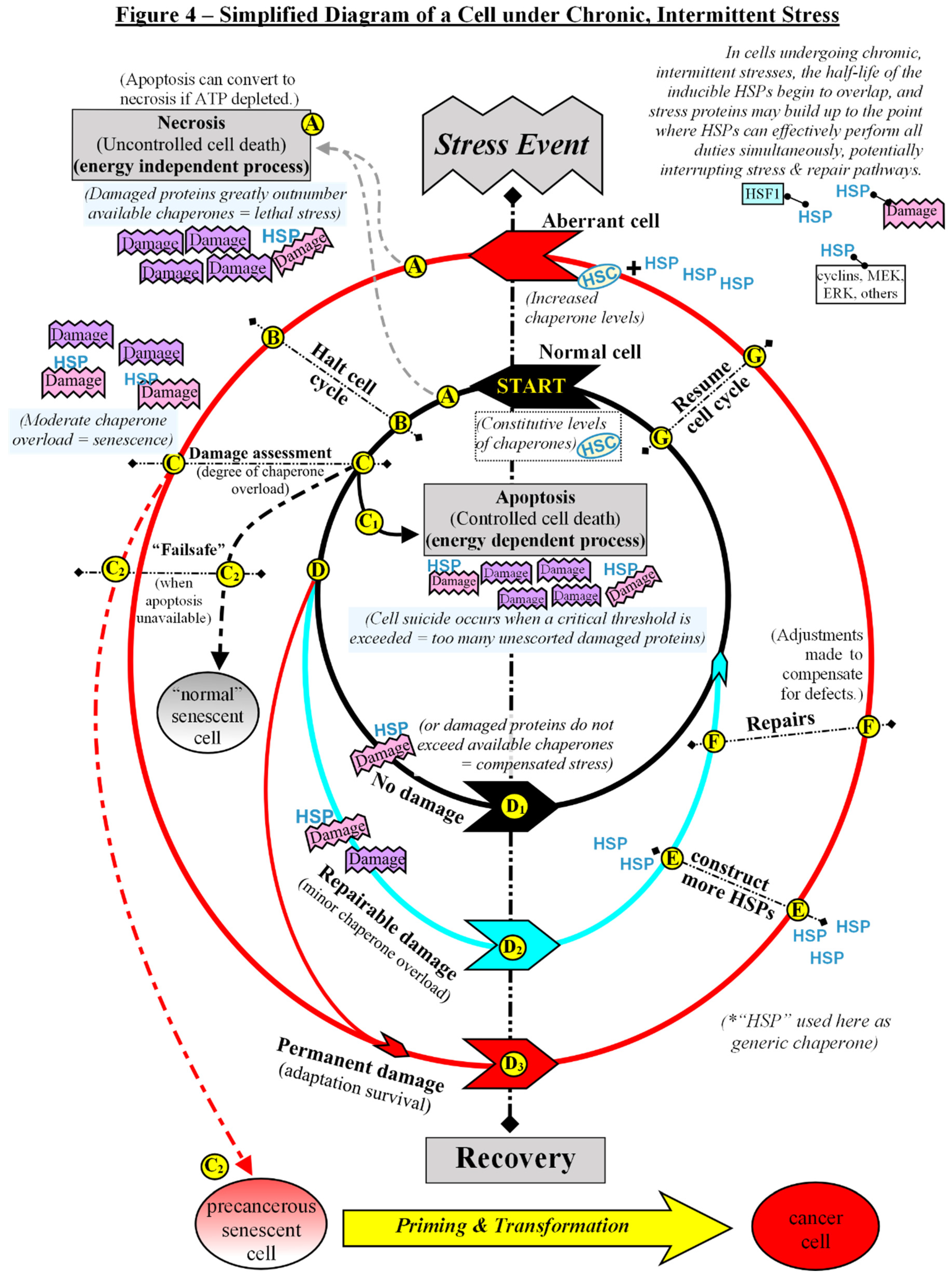
For ease of description, Figure 4 depicts a simplified model for a generalized single cell undergoing stress and then recovery in a repetitive cycle, as chronic, intermittent stresses appear to be significant to the formation of precancerous cells. A stress event could be exposure to toxins, anoxia, heat, radiation, ROS, and more, although detectable cell injury may not always occur (as appears to be the case with EMF) [44]. A stress event is merely anything that triggers a pause to assess and repair or degrade the cell. The term damage will again be defined here as any physical injury to a cell, including that which harms genes, proteins, membranes, electron transport system, or other processes within a cell. The concentric circles represent the various pathways a cell could take following each stress event. The innermost circle (black pathway) represents the “normal” cell, and this discussion begins there. Each successive circle is akin to the expanding electron orbitals of atoms, wherein each jump to a higher orbital indicates less stability in the system. In the case of the cell, each progressively wider circle represents increasing impairment and/or genetic instability. At the starting point of our generic “normal” cell, molecular chaperones, which perform essential roles in the daily cellular housekeeping and repair, are constitutively expressed at a level to accommodate the balance of the competing duties from intrinsic processes.
Immediately following a stress event, there are two possible outcomes: either the cell undergoes a rapid death via necrosis or it doesn’t (A). If the damage greatly exceeds the available stress proteins to mitigate, the cell dies abruptly, releasing its cellular contents to the extracellular space, resulting in an inflammatory reaction which can stress or damage neighboring cells [57]. If a cell possesses enough chaperones to buffer the damage somewhat so that it does not die outright following a stress event, then it temporarily halts its cell cycle (B). Cell cycle arrest allows time for assessment of damage, and then the controlled elimination (C) or recovery (D) of the cell [91].
If our hypothetical simplified cell sustains an injury too great to repair, it will be shunted to an “elimination” pathway (C) where it will undergo programmed suicide or, if restricted from that option, senescence. The dominant method of removing a defective cell from operation is apoptosis (C1), a process by which membrane-bound cell fragments, marked for disposal, are quietly eliminated by macrophages without disturbing nearby cells [57,91]. However, there are multiple ways to inhibit apoptosis, including inherited or acquired genetic mutations, the insertion of viral genes, high levels of molecular chaperones, and/or mitochondrial defects, among others. A cell that has sustained severe damage, but cannot undergo apoptosis, follows the “failsafe” tumor suppressor pathway and enters a state of permanent cell cycle arrest known as senescence (C2) [80,85,92]. Although this model uses a cycling cell to describe events that ensue during recurring stresses, senescence can also occur in post-mitotic cells [93]. The most consistent feature of senescence — aside from the inability to commit suicide — is a persistent, unresolved DNA Damage Response (DDR) [93].
Once the cell has cleared the initial damage appraisal, there are three possible recovery pathways for a cell to take (D), depending on the level of injury sustained:
If no damage occurred during the stress event, or rather, if the available chaperones were capable of handling all tasks of protein transport, degradation, or refolding (compensated stress), then the cell will continue on the black pathway (D1).
If minor, repairable damage occurred during the stress event (a low level of chaperone overload), the cell will begin to follow the blue pathway (D2). Note that the blue pathway is only a temporary detour and once repairs are completed the cell will eventually return to the black (fully restored) pathway. HSF1 is normally held latent in unstressed cells, however, as chaperones become fully occupied with damage control duties, HSF1 is released, allowing it to trimerize and translocate to the nucleus, where it can begin the stress-induced transcription of extra HSPs to compensate for future stresses. (E)[45,55,56] In this way, moderate stress can prepare a cell for more severe stress in the immediate future. Stresses need not cause cellular injury for HSP induction to occur, for example, electromagnetic fields can induce HSPs without obvious cell damage [44,94]. In this simplified model, the generalized term “repair” (F) takes in several methods of restoring order in an injured cell, and so “repairs” will merely be mentioned, rather than going into detail. Once the cell has recovered from its stress, it can resume its activities (G).
If intermittent stresses occur close enough together, the half-life of the inducible HSPs can begin to overlap, and the population of stress proteins increases [43]. This can be something of a double-edged sword, as the extra HSPs simultaneously protect a cell from a repetitive damaging environment, yet also prevent cell death, and even mask gene mutations by sequestering or degrading flawed gene products, as already mentioned previously [45,51]. Some lines of evidence suggest that increasing molecular chaperones above constituent levels may be an important event which steers a cell away from apoptosis and toward adaptation-survival or senescence when it receives irreparable injury [45,92]. If our model injured cell is able to bypass various forms of elimination via its higher HSP ratio, the cell will begin to follow the red pathway (D3). Note that the deviation to the red pathway is a permanent one, as a cell that has irreparable damage can never resume completely normal operation. The permanently injured cell following the red pathway must make whatever repairs it can and adapt to its disabilities, allowing it to continue functioning on some new level. Those adjustments may directly or indirectly cause other changes due to extensive crosstalk among the organelles. For example, one of the most easily damaged organelles of a cell are its mitochondria. As a cell’s metabolism changes, the expression of nuclear genes may be altered to compensate [95,96]. The converse is also true, as there is crosstalk between the nuclear and mitochondrial genomes, and any changes to nuclear gene expression (through mutation or other means) may cause even undamaged mitochondria to alter their functions to accommodate [95,96]. Cells following the red pathway are identified during biopsies of suspicious growths as “altered”, “abnormal”, or “mutant” cells, or benign neoplasias, among other terms. Once repairs or adaptations are completed, the cell may then resume its activities (G).
This model, as depicted in figure 4, predicts two potential subtypes of senescent cells: “normal” -vs- precancerous. “Normal” senescent cells would be those derived from cells which were following the black pathway that, for whatever reason, were unable to undergo programmed cell death after a serious injury. “Precancerous” senescent cells would be those derived from the aberrant cells which were already following the red pathway prior to the stress event, thereby taking their mutations and other alterations into the senescent state. At present, actual evidence for such a distinction is lacking, although the heterogeneity of senescent cell phenotypes in vivo has been noted. It has even been suggested that there exists at least two subtypes of senescence: acute and chronic, depending upon the stressors that created them [84,97]. If, in the future, it is found that the two potential senescent cell subtypes do not exist, it is unlikely to greatly alter this theoretical discourse. Therefore, for the duration of this discussion, let us presume that such variation may exist according to the previous description.
The result of the cell’s journey through repetitive injury and healing over time (in this model) is a senescent cell with malignant potential, awaiting priming and a stimulus to escape senescence barriers and transform to malignancy.
Latency, Microenvironment, and the “Priming” of a Precancerous Senescent Cell
In addition to permanently arresting the proliferation of aberrant cells, senescent cells secrete numerous proinflammatory molecules and mediators, matrix remodeling factors, and growth factors into the extracellular space. That common attribute has been termed “Senescence-Associated Secretory Phenotype” (SASP), “Senescence Messaging Secretome” (SMS), or “Senescence Inflammatory Response” (SIR), by different investigators, and the mix of factors varies slightly from one cell type to another, suggesting that the senescent induction pathway, cell types, and/or microenvironment might promote different outcomes [84,97,98]. The secreted factors have both autocrine and paracrine effects, and their makeup gradually alters over time, suggesting a two-way signal-and-response among cells in the local environment [84,85,97,99].
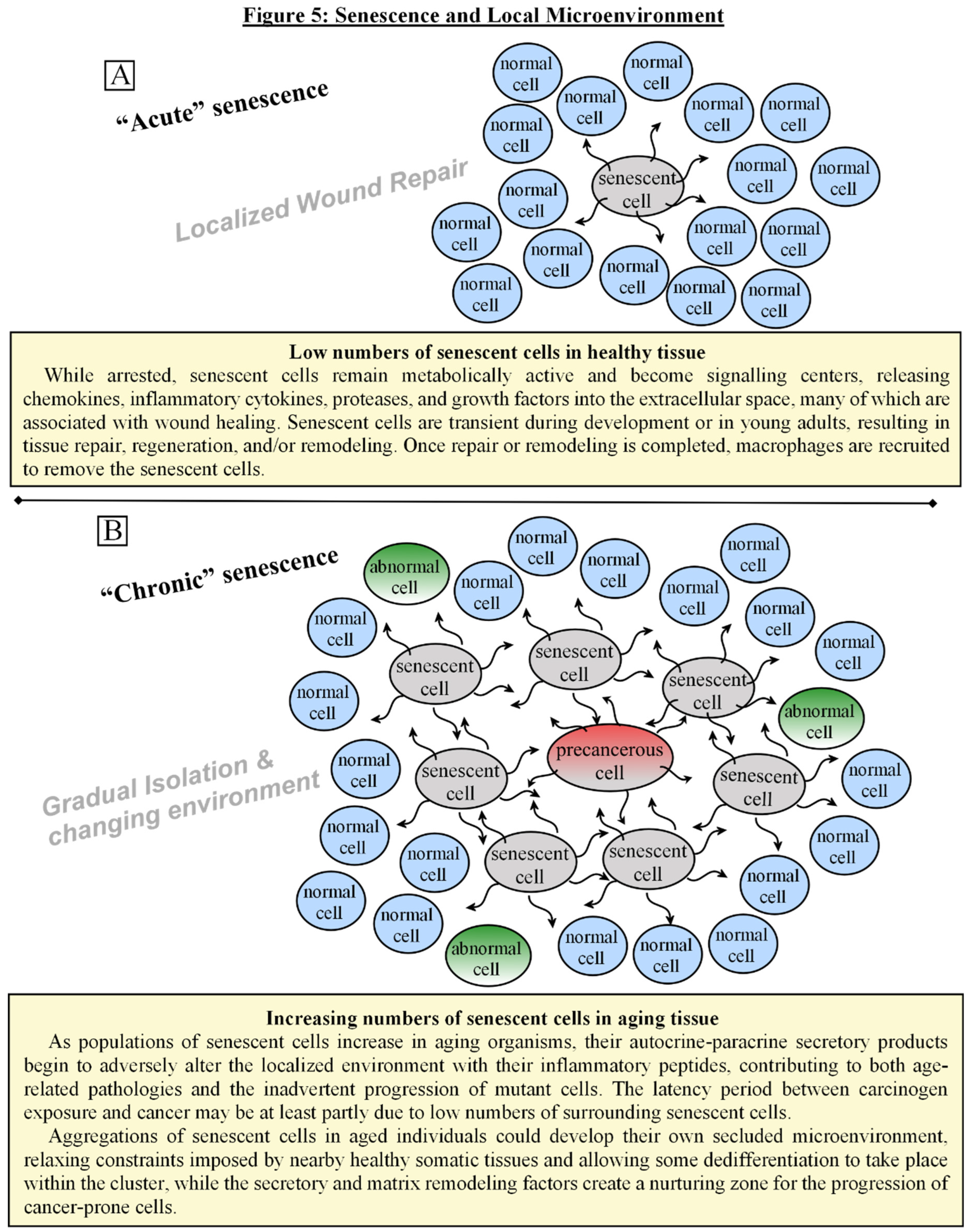
In low numbers, such as in young individuals, senescent cells not only protect against malignancies, but also promote tissue recovery at the site of severe stresses via these growth and inflammatory signals. Evidence shows that senescence is part of the “cleanup and shutdown”, resolution phase of wound healing, where the same cells that initially proliferated, closed wounds, and produced extracellular matrix components, then arrest and begin to degrade the matrix proteins, as a self-limiting mechanism to avoid fibrosis and scarring [86,87,97] [See Fig. 5A]. While undergoing senescence arrest, their secretory products transiently produce stem cell-like regenerative properties in themselves and in nearby cells before complete withdrawal from the cell cycle, as a way to facilitate tissue repair in the local environment [100,101]. Once fully arrested, however, they can induce a DNA damage response in neighboring — and undamaged — normal cells, as a way to counter excessive regeneration. Senescence can be somewhat “contagious”, promoting a so-called bystander effect with the cell to cell signaling [102]. Some investigators have surmised that this bystander effect is the reason for the exponential increase of senescent cells in aging organisms, and that senescence, or more appropriately, the secretory products, is to blame for the tissue degradation associated with aging [103,104]. The idea remains somewhat controversial, yet aggregations of cells with senescent characteristics can be found in vivo in rats, mice, primates, and humans at sites of age-related pathologies [85,104]. Other investigators have found that impaired immune clearance of senescent cells may be more to blame for the higher burden in aging organisms, but the result is similar [85,105]. As senescent cells accumulate, the secretome that initially promoted tissue repair, regeneration, wound resolution, and growth inhibition gradually becomes detrimental, creating an environment of chronic inflammation and permissive conditions for the inadvertent proliferation and progression of malignant and premalignant cells [83,85,99,104,105]. [see Fig. 5B] All of this may help to account for the exponential increase in late life cancers. The latency period often observed between carcinogen exposure and cancer may be at least partly due to low numbers of nearby senescent cells. As the senescent cell population climbs mainly due to stress and damage, not necessarily to age, it also offers an explanation as to why some individuals with inherited genetic disabilities in cellular repair or programmed suicide develop malignancies at an earlier than average age [2,34,42]. Aggregations of senescent cells in aged individuals could develop their own secluded microenvironment, relaxing constraints imposed by nearby healthy somatic tissues and allowing some dedifferentiation to take place within the cluster, while the secretory and matrix remodeling factors create a nurturing zone for the progression of cancer-prone cells [83,85,99,106,107].
Comparisons may also be made to somatic embryogenesis — the process by which embryonic development arises from differentiated somatic, not gonadal, plant cells — which is more effectively induced in plants when the target cell(s) are isolated during or after stresses to eliminate external communications from neighboring cells, and often this occurs naturally deep within a callus of un- and de-differentiated cells that forms after wounding [108,109,110]. The callus itself also demonstrates a gradient of differentiation, depending upon where cells are located within the tissue and how much exposure to hormones and cytokinins they receive. Interestingly, stress, especially intermittent sublethal stresses, and the subsequent expression of members of the heat shock family, are important for the initiation of somatic embryogenesis [108,109,110,111].
Stress is found to be important during mammalian cloning trials as well, where somatic cells are subjected to serum starvation, chilling, or other nonlethal stresses until they fall into a quiescent state before the donor nuclei are harvested [112,113]. It is thought that quiescence modifies the donor chromatin structure to allow reprogramming and subsequent development when fused with an enucleated oocyte [114]. Transient hypoxia, which inhibits mitochondrial respiration and upregulates glycolysis in cultured donor cells, alters gene expressions and appears to facilitate nuclear reprogramming after transfer, improving the survival probability of clones of more difficult species [112]. Serial passage of donor cells before removal of the nucleus also improves reprogramming after transfer, possibly due to some dedifferentiation that occurs via isolation from the original tissues [113,114].
The ethical pressures restricting the study and utilization of human embryonic tissues to treat various diseases encouraged others to explore the factors within oocytes or embryonic stem cells that can reprogram adult somatic cells to toti- or pluripotency, in order to develop alternatives. Induced pluripotent stem cells (iPSC) is the process of reprogramming differentiated somatic cells into pluripotent cells via the ectopic overexpression of four transcription factors: Oct4, Sox2, Klf4, and c-Myc (OSKM) [115,116]. Although the precise mechanism for somatic reprogramming remains unclear, it is interesting to note that OSKM appears to stress some cells into a senescent state prior to reprogramming nearby cells, and that the DNA damage response (DDR) and senescence seems to provide a tissue microenvironment that is required for OSKM reprogramming in vivo [101,107]. A positive correlation was noted between the extent and proximity of senescent cells and the number of reprogrammed cells [107]. While ectopic expression of OSKM can itself induce senescence, the presence of existing senescent populations in aged or injured tissues also increases reprogramming efficiency [107], revealing a pattern reminiscent to the one modeled in Fig#5b, wherein aggregations of senescent cells shield the innermost cell(s) from outside influences while simultaneously bombarding it/them with inflammatory and regenerative signals — producing an exclusive microenvironment conducive for reprogramming cells.
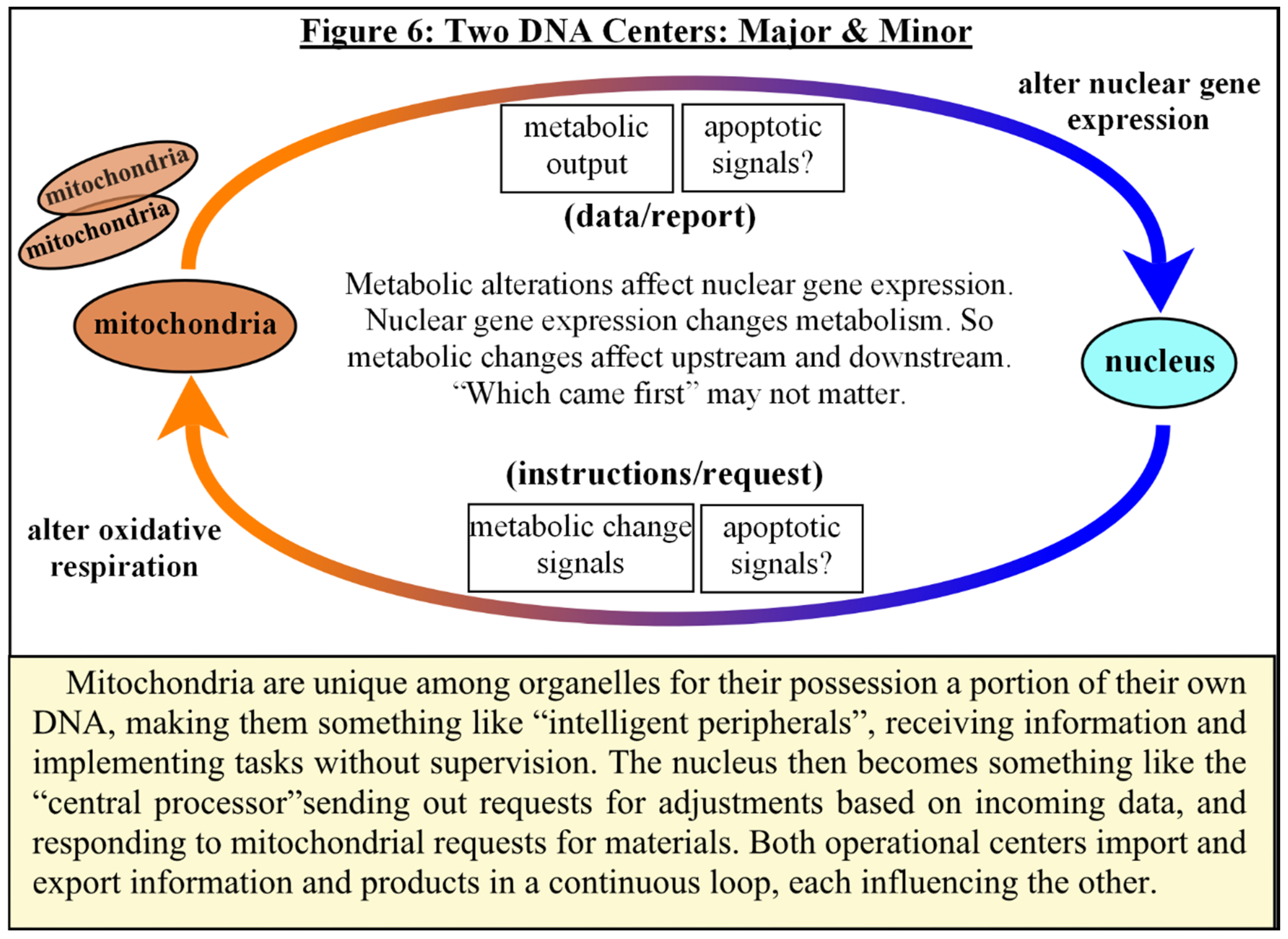
Another interesting observation that occurs during somatic reprogramming is a metabolic switch to glycolysis that is similar to that seen in both cancer and embryonic development [117,118,119], which appears to invert the cause and effect of cloning trials, where it was noted that deliberate inhibition of cellular respiration increases reprogramming efficiencies after nuclear transfer [112]. Mitochondria begin to resemble their embryonic counterparts in both form and function during reprogramming [117,118]. Mitochondria are unique among organelles for their possession of a portion of their own DNA, making them something like “intelligent peripherals”, receiving instructions and implementing tasks without supervision. The nucleus then becomes something like the “central processor” sending out requests for metabolic adjustments based on incoming data, and responding to mitochondrial output and requests for materials. Both operational centers import and export information and products in a continuous loop, each influencing the other. Metabolic alterations affect nuclear gene expression, and nuclear gene expression alters metabolism. [see Fig #6] Due to the extensive nuclear-mitochondrial crosstalk, it is difficult to determine which comes first, nuclear reprogramming, or mitochondrial form and function, or perhaps it does not matter, as the altered metabolism affects gene expressions and operations in both organelles, which may support the induction of pluripotency [95,96,117,118,119].
A similar metabolic shift occurs prior to blastema formation during the regeneration of complex tissues of non-mammalian vertebrates [120]. Blastema are dedifferentiated somatic cells, induced by injury, that exhibit all of the behaviors of the limb bud that forms during embryogenesis, and, like embryonic stem cells, use glycolysis for energy for self-renewal and only switch to oxidative phosphorylation when differentiating and losing pluripotency [120,121]. In both cases, oxygen levels in the localized environment play a role in the metabolic transitions that lead to changes in gene expressions [121]. The limb bud AER of the developing embryo and blastema share characteristics, yet one process is for de novo generation of tissues (organogenesis) while the other is for regeneration of tissues after injury (morphogenesis) [122]. Blastema formation and the process of regeneration has been generally accepted as a crossover between an adult wound response and developmental processes [123].
Senescent cells play a role in both embryogenesis and epimorphic regeneration as well, becoming transient signaling centers that do not participate in proliferation, while their SASP produces a gradient that contributes to tissue patterning and plasticity, becoming something of a temporary waypoint, and once repair or remodeling is completed, macrophages are recruited to remove the senescent cells [101]. In fact, a key role of SASP is the recruitment of immune cells, most notably macrophages, to initiate tissue modeling/remodeling, and the mix of SASP factors exert tight control over macrophage phenotypes and functions [124]. Macrophages, like senescent cells themselves, are only beginning to be appreciated as a highly diverse group with a range of characteristics from inflammatory and destructive to anti-inflammatory and constructive and all variations in between [124,125]. Macrophages are ubiquitous immune cells that acquire tissue-specific phenotypes, and are found at sites of injury or infection as well as throughout the ovarian region, participating in various events such as folliculogenesis, ovulation, placental maintenance, and even parturition [126]. Evidence indicates that ovulation itself is a modified inflammatory process, mediated by macrophages, serving as another example of crossover between adult stress responses and developmental pathways [126,127,128].
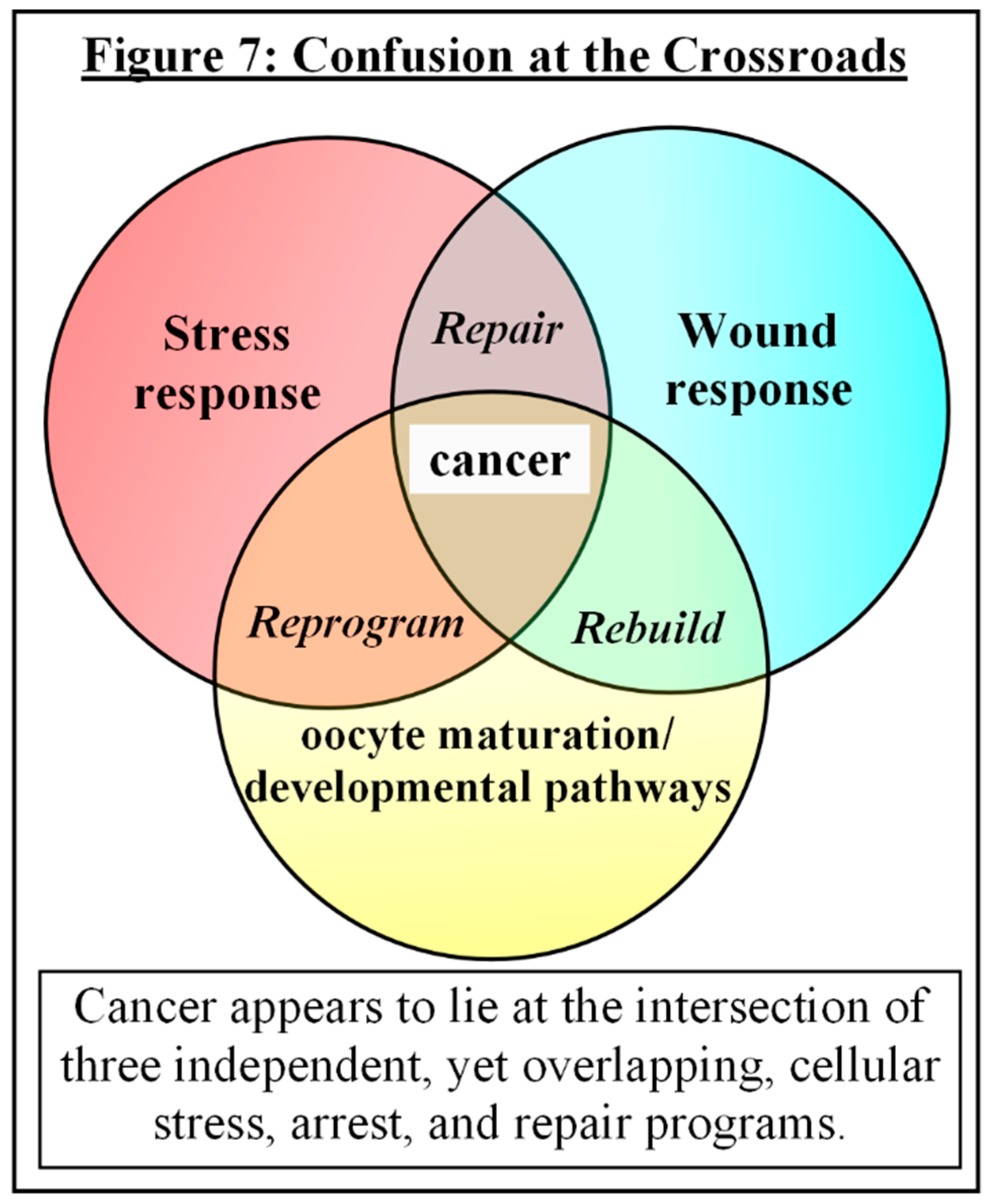
There seems an intriguing pattern of convergence among stress responses, wound responses, and developmental processes that should be explored. Cancer appears to lie at the conjunction of three independent, yet overlapping, cellular programs, suggesting that it is a failed or incomplete attempt at regeneration following chronic cellular trauma. [see Fig. #7] Although some authors have surmised that cancer is the result of stress-triggered atavistic reprogramming (STAR), where dedifferentiation and subsequent redifferentiation occurs, similar to blastema, leading to accidental in vivo reprogramming to pluripotency, followed by Darwinism and adaptation to mutations and environmental hardships — and while the concept of STAR may work to describe the apparent sliding scale of aggressiveness and dedifferentiation that is observed in malignancies — it seems that once again, things may not be as straightforward as that [129]. Many non-germ cell cancers not only display the errant reactivation of embryonic genes and characteristics, but also of pre-embryonic meiotic genes and events, hinting at something much more fundamental [25,130,131,132]. It may not be simply a “back up and drive forward” reversion of attributes, but instead potentially starting over with an entirely new vehicle.
Many of the events that take place during repeated stress responses that ultimately lead to permanent cessation of the cell cycle might, via time and a gradual isolation and localized alteration in the stromal milieu, result in an inadvertent overlap of mitotic pathways onto meiotic pathways in the arrested cells, leading to an accidental resetting of the embryonic developmental program, with the primed pre-malignant senescent cell performing the part of a primed and activated oocyte when surrounded by senescent stroma forming something akin to a cumulus-oocyte complex [83,133,134,135]. Perhaps the world’s most feared disease is demonstrating that somatic embryogenesis is not exclusive to plants — albeit in an incidental and incomplete way, as timing and a specialized organ matters greatly to higher organisms, even without the impediments of mutations and defective cells. Before this conjecture is dismissed, consider both direct and circumstantial evidences.
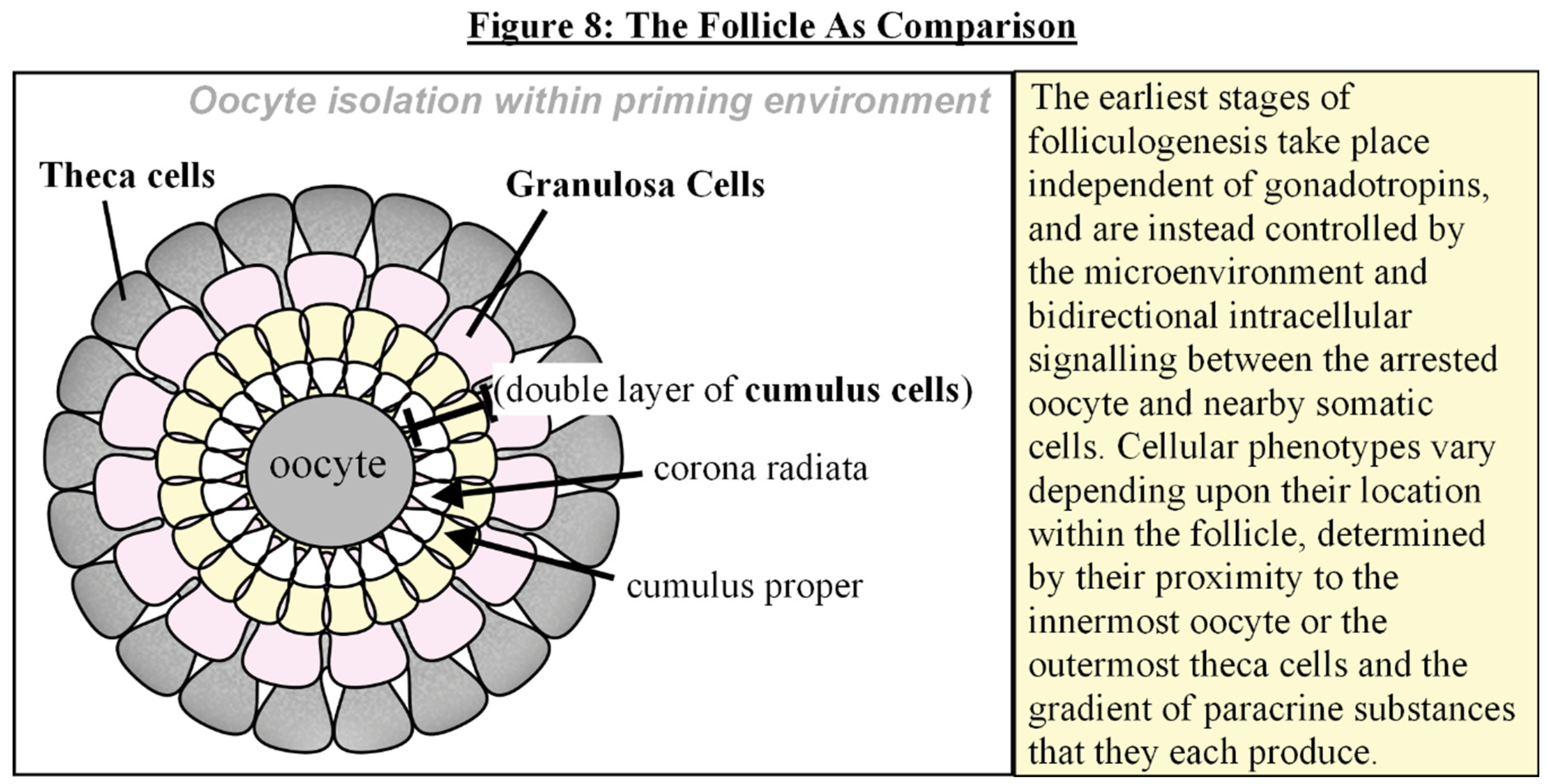
The earliest stages of folliculogenesis take place independent of gonadotropins, and are instead controlled by the microenvironment and intracellular signalling between the arrested oocyte and nearby somatic cells [133,134,135]. [see Fig. 8] Cellular phenotypes vary depending upon their location within the follicle, determined by their proximity to the innermost oocyte or the outermost theca cells and the gradient of paracrine and autocrine substances that they each produce [134,135]. Senescent cells, likewise, display heterogeneous phenotypes depending upon the trigger and cell type of origin, as well as their stage of progression from time of induction, and their interactions with one another [84,85,97,98,99,101].
Meiosis is something of an enigma to evolutionary biologists, as it is highly conserved among eukaryotes, including asexual organisms, yet its overall purpose is subject to debate, since it is slow, costly, and — contrary to classical teachings — recombination doesn’t necessarily increase genetic variability, and at times can even remove beneficial gene arrangements [136,137]. Genes and proteins for meiosis have much in common with bacterial transformation, which uses homologous recombination for repair of DNA DSBs and other physical damage caused by reactive oxygen species, and both bacterial transformation and meiosis are often induced by stress [136,137,138]. Evidence indicates that the primary function of meiosis is restoration of DNA, with gene exchange occurring more as a side effect [137]. Meiosis and reduction division averts Muller’s Ratchet by weeding out deleterious mutations and restoring ploidy levels after endoreduplication [137,139,140]. The ROS repair hypothesis may be reviewed more thoroughly in [136,137,138].
Of the various DNA DSB repair pathways, homologous recombination (HR), which involves a second chromosome, is the most accurate, yet is unavailable to post-mitotic somatic cells [136]. Non-homologous end-joining (NHEJ) and single-strand annealing (SSA) can occur at any phase of the cell cycle without the requirement for replication, yet are more error-prone. Suicide or senescence are the only options available to post-mitotic cells when such repairs become impossible. Yet the process which leads to cessation may also attempt continuation. Senescence, it has been noted, is not quiescence. It is much like meiotic arrest in that the cell is ready to cycle, yet is inhibited [81,141,142]. This manuscript’s hypothesis relies on the assumption that meiosis is an ancient DSB repair pathway, utilized mainly for removing DNA damages in the immortal germ line and normally repressed in somatic cells [131,132,137,143], yet when “regular” mitotic repair pathways are unavailable, a cell defaults to the only remaining repair pathway left, especially if other conditions potentially guide it that way [143,144,145,146,147,148]. In effect, halting mitosis initiates meiosis, given certain conditions [149,150,151].
Due to space constraints, many topics are discussed only superficially here, and must be delved into more deeply at a later date. Among the first generalizations is a mention of how cells impede or promote interactions among proteins and transcription factors and more via subcellular localizations. In order for actions to occur, the various players must be in the same place at the same time, and this realization helps to elucidate the often contradictory reported activities of various proteins. They behave one way in, for example, the cytoplasm, and may have entirely different roles within the nuclear space. [(as examples) [152,153,154,155,156,157,158], more examples can be provided on request] Additionally, other factors compete for, or block, binding sites, and these competing interactions, as well as post-translational modifications, conformational changes, or quick degradation, determine when and how proteins are activated or inactivated or when and how genes are read. Molecular chaperones assist in the trafficking or regulation of various proteins as previously discussed [45,51,52,53,54]. And all of these interactions are enhanced or limited at yet another level by the metabolic status of the cell. Studying the trafficking of proteins is dizzying, especially when attempting to view whole cell operations at once, for as one factor enters the nucleus, for instance, others may be expelled as a direct or indirect consequence, altering potential partnerships and interactions, but the effort leads to yet more revelations.
One interesting — and relatively unstudied — aspect of mitochondria is that they are not static “powerhouses of the cell” as described in most undergraduate biology texts. Mitochondria are dynamic, changing form, function, and location during various cellular events. And of especial interest, mitochondria translocate along cytoskeletal networks to the perinuclear space during stresses, meiosis, embryonic development, and senescence [121,140,159,160,161,162]. Mitochondria have other roles beyond ATP production, including ROS signaling, generating other metabolites, and Ca2+buffering, and their spatial positioning meets the functional needs of the cell [96,163,164,165,166]. Concomitantly, mitochondrial perinuclear repositioning involves several which-comes-first events involving both HSF1 and Hypoxia Inducible Factor-1∀ (HIF-1∀), suggesting an interdependence of those two stress response pathways, as, ultimately, oxidative stress is common factor, and one primary outcome, either directly or indirectly, is a lower respiratory rate and increase in glycolysis [122,159,167]. [see also supplemental poster]
Decreased respiration reduces mitochondrial ROS production, but also reduces energy production, which initiates pathways that, at least transiently, decrease proliferation rate, such as the downregulation, increased degradation of, or competition with, MYC [168]. HIF-1∀ activation is one such pathway that leads to a decrease in MYC-Associated factor X (MAX), which is a DNA-binding factor normally constitutively expressed at stable levels, as it is a necessary partner of the MYC network of proteins. HIF-1∀ activation induces “purposefully defective” isoforms of MAX that, in essence, deplete MAX levels [169]. It is worth mentioning that transient reduction of MAX in embryonic stem cells is one of the triggers that initiates the switch from mitotic self-renewal to meiotic cell division [146,149,170]. While HIF-1∀ antagonizes MYC function, limited data suggests that HIF-2∀ may takeover and enhance MYC activity during prolonged hypoxia, potentially altering a common pathway and promoting a stem cell-like phenotype [171,172]. More study is required to determine HIF-2∀ role(s) during hypoxic stress responses.
The examination of mitochondrial perinuclear translocation provides an important demonstration of reciprocal signaling. For example, hypoxic activation of HIF-1∀ causes mitochondrial perinuclear clustering, or, the converse, perinuclear accumulation of mitochondria activates HIF-1∀ in normoxia [167]. Molecular motors transporting cargoes such as mitochondria cause friction forces along microtubule highways that affect cytoskeletal tension, and the process works both ways: cytoskeletal tension elicits a stress response that modifies mitochondrial functions and position [173]. Cytoskeletal reorganization is both cause and effect of organelle transport [174].
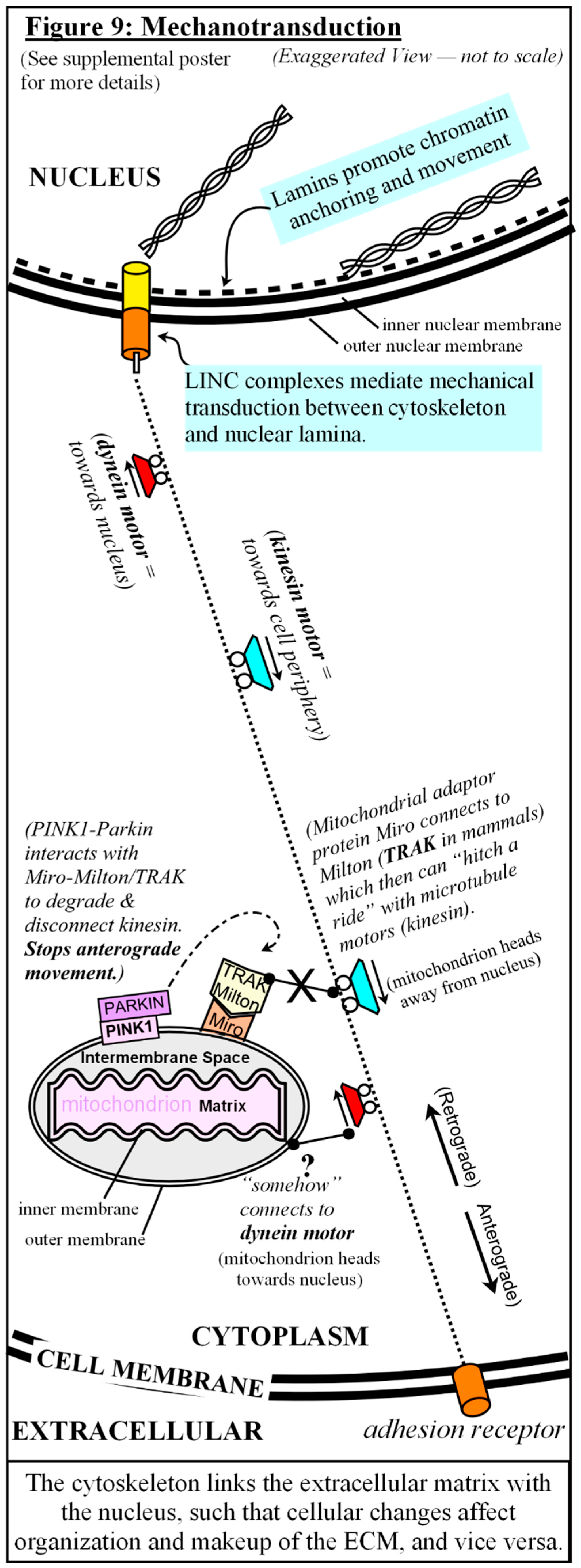
As cytoskeleton filaments connect to the nuclear envelope through Linkers of Nuclear & Cytoskeleton (LINC) complexes, cytoskeletal forces ultimately affect chromatin movements, and the converse, chromatin rearrangements that occur during the cell cycle, or during stresses or senescence, get relayed back to the cytoskeleton, affecting organelle transport and signaling [174,175,176]. [see Fig #9 and supplemental poster] The cytoskeleton also links the extracellular matrix (ECM) with the nucleus, such that cellular changes affect the organization and makeup of the ECM, and vice versa. Nearly 40 years ago, the concept of Dynamic Reciprocity was introduced, describing the ongoing, bidirectional communications between cells and their microenvironment [177,178,179,180]. Extracellular tension is relayed through the cell membrane and cytoskeleton to the nuclear envelope, which ties extracellular microenvironment to cellular processes, and even chromatin remodeling, via mechanotransduction [177,178,179]. Gene expression, then, is not governed merely by conditions within the cell itself, but also by its immediate surroundings, which also elucidates why cells in culture lose their original structure and functions once removed from their tissues [177,178,179,180].
Among the myriad events that occur during its establishment, senescence alters both ECM and nuclear lamina in such a way as to allow dramatic chromatin reorganization, such as association of telomeres toward the nuclear periphery, a shift that has also been observed in meiosis [176,181,182,183,184,185,186]. In meiosis, clustering of telomeres at the nuclear periphery is thought to facilitate the aligning of homologs as a precondition to synaptic pairing [182,186]. Also, a key feature of meiotic homologous recombination repair is a bias toward inter-homolog pairing rather than inter-sister pairing [130,132,145]. A sharp decrease in nuclear PARP1 levels is often observed in aging and senescent cells, that leads to unrepaired single-strand DNA breaks, and a default to homologous recombination after their conversion to DSB, with a bias toward inter-homolog pairing [69,130,144,145]. The mitochondrial perinuclear accumulation that occurs during stresses and senescence, along with the associated cytoskeletal reorganization and metabolic reprogramming, and the resulting transient decrease in MAX all may help to trigger the switch from mitotic to meiotic pathways [121,146,149,169,170].
Additionally, another consequence of mitochondrial movement into the perinuclear space under stressful situations is the role they may play in the nuclear availability of certain proteins as cytoplasmic pools of, for example, p53 or PARP1, are titrated away into oxidatively-stressed mitochondria [154,158,187,188,189]. HIF-1∀ activation can also potentially contribute to senescence escape via induction of PP1 Nuclear Targeting Subunit (PNUTS), which both attenuates the G2 checkpoint and prevents Protein Phosphatase 1 (PP1) from dephosphorylating retinoblastoma protein (Rb), thus leaving Rb in a hyperphosphorylated state and unable to restrain E2F [190,191]. All of this will have to be addressed in more detail in future publications.
Ultimately, senescence, and the stresses that precede it, halts mitosis and appears to set up conditions within a cell that lean it toward meiotic repair, should such a cell bypass the senescent block on proliferation.
The decision to leave the mitotic cell cycle and enter meiotic cell cycle in germ cells occurs prior to the meiotic S-phase [150]. [see Fig #10] Many of the DNA replication regulatory processes are initiated in G1 phase [192]. It has been proposed by others that, similar to the “pre-replication complexes” that bind to replication origin sites during G1, there is also “pre-cohesion” loading of specific cohesin complexes during G1 at Cohesion Attachment Regions (CARs) along the chromosomes and centromeres [193,194]. Then, as the replication fork machinery encounters these complexes in S phase, polymerase switching occurs as the site is replicated, establishing cohesion, and the type of cohesins involved determines subsequent chromosome dynamics [150,193,194,195]. The more complicated meiotic pre-cohesion setup is proposed to account for the slower S-phase replication machinery — as compared to mitotic replication — and embed meiosis-specific cohesins that would repair damages via meiotic homologous recombination, including observed crossover events [145,194]. Meiotic cohesins resist degradation at the end of MI, preventing sister chromatid separation, then with resumption of meiosis and exit from arrest, the cell follows a more mitotic-like division through the rest of MII, and arrests again, awaiting fertilization (an event similar to stress) [196,197]. Gamete fusion at fertilization suppresses cell death pathways to support embryogenesis, and initial replication cycles occur without checkpoints, allowing DNA synthesis and cell division to proceed despite any defects [198,199].
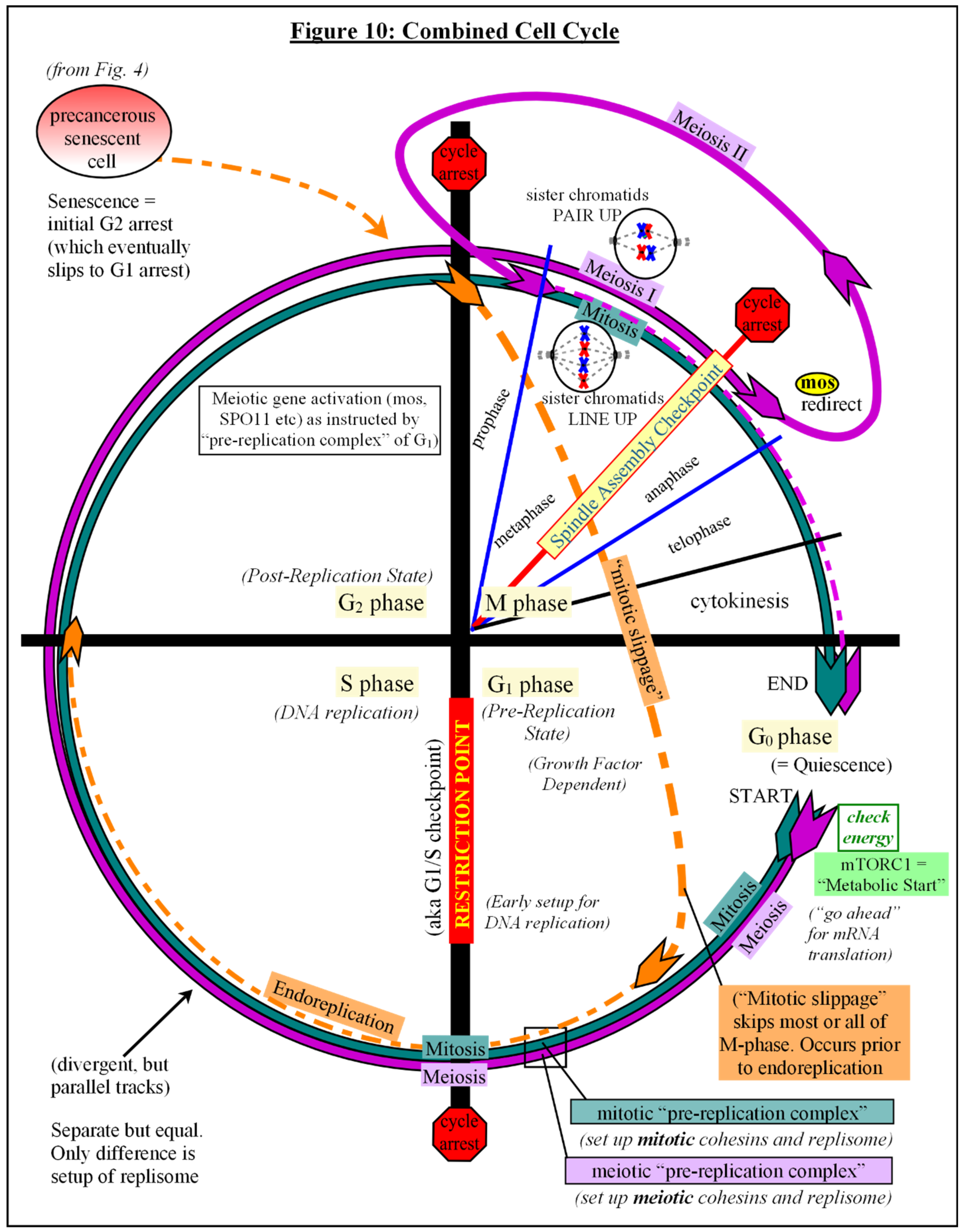
Accumulating evidence suggests that somatic cell polyploidization, followed by depolyploidization, is an important feature of senescence escape and transformation to malignancy [200,201]. Tetraploidy is a common observation during the evolution of cancer, which could potentially result from arrested diploid cells that undergo mitotic slippage and endoreduplication [201,202]. [see Fig #10] Mitotic slippage is most often triggered by stress, especially when p53 (and/or its downstream effectors) is absent or dysregulated. which would allow bypass (“adaptation”) of checkpoints, asynchronous resynthesis of proteins, cell cycle progression and endoreduplication [81,200,202,203,204,205,206]. Unfortunately, terms such as “absence of” p53, or “p53-deficiency”, are commonly understood to mean nuclear p53 levels, without taking into account its presence in any other subcellular compartments [154,187,188]. Its absence from the nucleus may not always be due to gene mutation, but limited studies demonstrate that localized depletion of cytoplasmic supplies via uptake by other organelles create a loss of nuclear availability [154,187,188]. Stressed mitochondria, localized to the perinuclear space, may influence mitotic slippage and endoreduplication by way of competition for supplies, and is a topic for verification.
Endoreduplicated senescent cells that later undergo depolyploidization (genome reduction), have been seen to activate meiotic genes, and it has been noted that daughter cells often have different genotypes from the originating cells [200,204,205,207]. It has been suggested that this form of repair and recovery is an unfortunate side effect of therapy-induced senescent cells that escape to disease relapse and subsequently develop increased resistance to therapy [200,201,205,207,208].
Putting It All Together
The aforementioned observations and interpretation must be regarded as speculation without further confirmation, but to sum up, I propose a model of the sequence of events leading to the malignant transformation of cells as follows: that normal, well-established reactions to multiple, intermittent cellular insults over time trigger senescence in damaged cells where apoptosis is limited. Then cumulative, age-related changes in the stromal milieu, brought about by the secretory products from an increasing population of senescent cells, inadvertently facilitates the progression of mutant cells, contributing to the exponential increase in the probability of late life cancers, by inducing follicle-like conditions within and around the affected cell that mimic those conditions immediately preceding egg fertilization. Additionally, stresses and senescence induce mitochondrial perinuclear accumulations, altering metabolism, reorganizing cytoskeletal networks, and influencing nuclear availability of proteins, among other events. Within a relatively secluded inflammatory environment that promotes tissue repair, regeneration, and wound resolution, yet precluded from using “regular” somatic repair pathways, a precancerous senescent cell is forced onto a repair avenue reserved only for germ cells, via alterations of nuclear lamina, chromosome reorganization, metabolic rewiring, mitochondrial-nuclear associations and crosstalk, and other activities, that lean it toward senescence escape and a switch from mitotic to meiotic cell cycle, and a transition from a soma to germ cell-like state.
A further stress in the primed senescent cell triggers checkpoint adaptation and mitotic bypass to G1 phase, where it assembles pre-replication complexes and pre-loads meiosis-specific cohesin complexes while awaiting Restriction Point transition to begin endoreduplication — reminiscent of pre-meiosis DNA replication — with pairing and meiotic gene activation during G2 phase, resulting in tetraploidy and a subsequent re-arrest similar to Meiosis I. [see Fig #10] The tetraploid cell later undergoes a pseudo “resumption of meiosis” (weakening of SAC) and follows a more mitotic-like division reminiscent of Meiosis II, which either restores a diploid state with concomitant completion of DNA repair and quality control, or mitotic catastrophe and cell death. The surviving primed cell arrests again, awaiting a further stress to activate it.
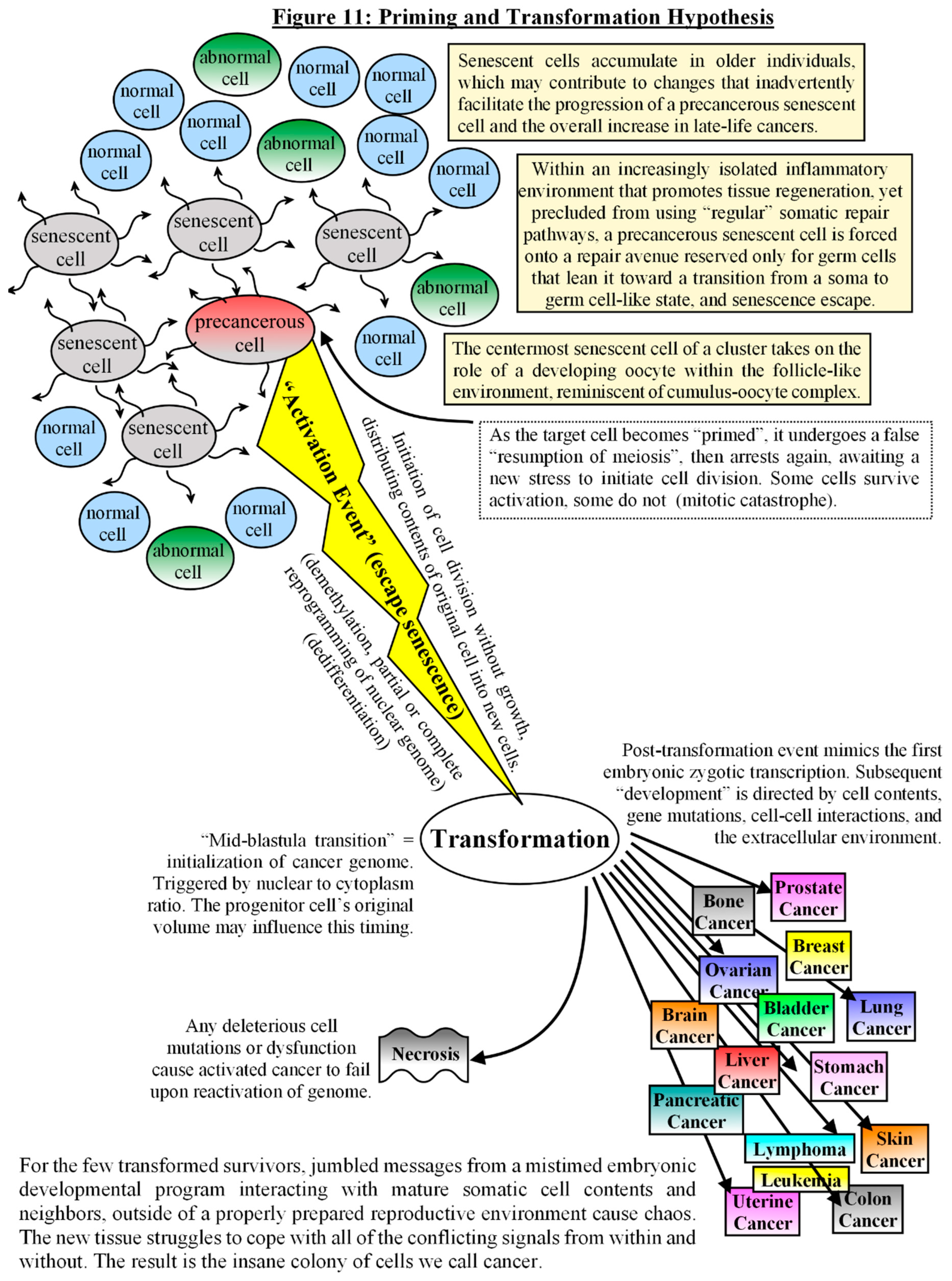
The next stress event would then mimic a false “Fertilization Event”, which could account for much of the aneuploidy that occurs in cancers, either via cell fusion or parthenogenic activation, followed by an automated cell cleavage program that commences cell division without growth, as in an activated egg. Checkpoint kinases are deactivated to allow the initial quick cell divisions. Much of the aberrant methylation patterns and dedifferentiation witnessed in cancers originates during this period of nuclear reprogramming. Nuclear-to-cytoplasm ratios control the timing of the “mid-blastula transition” and the initiation of “zygotic” (cancerous) transcription [209]. [see Fig #11] Post-transformational ontogeny of the dedifferentiated colony of cells is directed by cell contents, gene mutations, cell-cell interactions, and the extracellular environment, much of which predisposes a deviant cell/colony to failure. For the few transformed survivors, jumbled messages from a mistimed embryonic developmental program interacting with mature somatic cell contents and neighbors, and distant from a properly prepared reproductive environment cause chaos. The “development” goes awry as the new tissue struggles to cope as best it can with all of the conflicting signals it receives from within and without. The result is the insane colony of cells we call cancer.
Discussion
Although speculative, this sequence of events, as proposed, goes far to explain the etiology of cancers, as well as the development of the common “hallmarks” and metabolic alterations, and some of the additional changes that occur during and after metastasis. In addition, this model offers insights into the behaviors and development of the supporting tumor microenvironment. If substantiated, this model allows for innovations using currently available drugs, and suggests alternate interpretations for results of diagnostic tests to assist in therapy choices. It also may provoke some measure of alarm at the potential for therapy-induced senescence to produce a reorganized cancer, renewed and repaired via meiosis-like processes to eliminate DNA damage. Current PARP1 inhibition therapies, designed to take advantage of deficiencies in mitotic HR repair (ex: BRCA2) may inadvertently facilitate a push toward an alternate meiotic-like HR repair and subsequent genetic rearrangement and disease relapse in surviving cells, as localized initialization conditions persist to permit an encore performance.
It should also be emphasized that although meiosis-like repair may lead to cancer’s inception and a recapitulation of germ cell through embryonic developmental pathways, including pseudo-blastomere formation[210], cancer is not a true embryo. It originates from a damaged adult somatic cell, and it resides within a differentiated somatic environment, thus reversing the scenario that occurs during cloning trials, where an adult nucleus is placed into an enucleated egg and then into a primed and ready-to-respond uterine environment. The consequence is something akin to a Theory of Relativity in biological tissues, where context means everything [211]. The tissue microenvironment controls, and is controlled by, the crosstalk among neighboring cells. The result is a sliding scale of dedifferentiation and aggressiveness due to the degree that the tissue environment answers the communications from the newly formed “somatic embryonic” tissues and at what point its “development” derails. Misconstrued signal-and-response among cells leads to potential metastasis and a new neighborhood of conflicting signals. Also, rather than the formation of an embryonic mass, it may only form the embryonic tissues of the originating cell’s line via stromal interactions (e.g. mesothelioma from mesothelium from mesoderm, leading primarily to formation of pseudo-extraembryonic membranes). Each cancer is unique, but underneath their complexity is a similar origin story, which can assist us in unraveling each cancer’s idiosyncracies and devising more effective therapies.
Ultimately, cancer may be a flawed attempt at tissue regeneration in a high stress environment, and learning about its etiology reveals unexpected and largely unexplored variations in the cell cycle, cellular mechanotransduction and signaling, microenvironment influences, subcellular control of protein interactions and transcription factors, and more. Rather than closing out a chapter on the origins and pathophysiology of a confounding disease, the information opens up a whole new library of further discoveries for multiple fields of study.
Funding Sources
N/A.
Acknowledgments
Many thanks go to those who encouraged me over the years: Scott Quackenbush, PhD, Suzanne Koptur, PhD, Gregory Bossart, VMD, PhD, Olga Wollinka, MS, Lisa Palmer, M.Ed, Shirley Collar, and Nolan Kirkendoll.
Conflict of Interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
- 1) Weinberg RA. 2014. Coming Full Circle — From Endless Complexity to Simplicity and Back Again. Cell, 157: 267-271. [CrossRef]
- 2) Pérez-Losada J, Castellanos-Martína A, Mao J-H. 2011. Cancer Evolution and Individual Susceptibility. Integr Biol (Camb),3(4): 316–328. [CrossRef]
- 3) Carbone M, Arron ST, Beutler B, Bononi A, Cavenee W, Cleaver JE, et al. 2020. Tumour Predisposition and Cancer Syndromes as Models to Study Gene X Environment Interactions. Nat Rev Cancer, 20(9):533-549. [CrossRef]
- 4) Adjiri A. 2017. DNA Mutations May Not Be the Cause of Cancer. Oncol Ther, 5:85-101. [CrossRef]
- 5) Krzyszczyk P, Acevedo A, Davidoff EJ, Timmins LM, Marrero-Berrios I, et al. 2018. The Growing Role of Precision and Personalized Medicine for Cancer Treatment. Technology (Singap World Sci), 6(3-4):79-100. [CrossRef]
- 6) Hoeben A, Joosten EA, van den Beuken-van Everdingen MHJ. 2021. Personalized Medicine: Recent Progress in Cancer Therapy. Cancers, 13:242. [CrossRef]
- 7) Fouad YA, Aanei C. 2017. Revisiting the Hallmarks of Cancer. Am J Cancer Res, 7(5):1016-1036.
- 8) Edelman EJ, Guinney J, Chi JT, Febbo PG, Mukherjee S. (2008) Modeling Cancer Progression via Pathway Dependencies. PLoS Comput Biol 4(2): e28. [CrossRef]
- 9) Gabriela Jiménez-Valerio G, Casanovas O. 2013. Anti-Angiogenic Therapy for Cancer and the Mechanisms of Tumor Resistance Contributions to Science, 9:67-73. [CrossRef]
- 10) Bergers G, Hanahan D. 2008. Modes of Resistance to Anti-Angiogenic Therapy. Nat Rev Cancer, 8(8): 592–603. [CrossRef]
- 11) Warburg O. 1956. On The Origin of Cancer Cells. Science, 123(3191):309-314. [CrossRef]
- 12) Warburg O. 1966. The Prime Cause and Prevention of Cancer — Part 2. Revised lecture at the meeting of the Nobel-Laureates on June 30, 1966 at Lindau, Lake Constance, Germany.
- 13) Enns GM. 2003. The Contribution of Mitochondria to Common Disorders. Mol. Genet. Metab., 80(1-2):11-26. [CrossRef]
- 14) Bartnik E, Lorenc A, Mroczek K. 2001. Human Mitochondria in Health, Disease, Ageing, and Cancer. J. Appl. Genet., 42(1): 65-71. [PubMed]
- 15) Carew JS, Huang P. 2002. Mitochondrial Defects in Cancer. Mol. Cancer, 1(1):9. [CrossRef]
- 16) Wei Y-H., Ma Y-S, Lee H-C, Lee CF, Lu C-Y. 2001. Mitochondrial Theory of Aging Matures — Roles of mtDNA Mutation and Oxidative Stress in Human Aging. Zhonghua Yi Xue Za Zhi, 64(5):259-270. [PubMed]
- 17) Galluzzi L, Kepp O, Trojel-Hansen C, Kroemer G. 2012. Mitochondrial Control of Cellular Life, Stress, and Death. Circ Res. 00:1198-1207. [CrossRef]
- 18) Zamzami N, Susin SA, Marchetti P, Hirsch T, Gomez-Monterray I, Castedo M, Kroemer G. 1996. Mitochondrial Control of Nuclear Apoptosis. Jour. Exp. Med., 183(4):1533-1544. [CrossRef]
- 19) Singh KK, Russell J, Sigala B, Zhang Y, Williams J, Keshav KF. 1999. Mitochondrial DNA Determines the Cellular Response to Cancer Therapeutic Agents. Oncogene, 18(48): 6641-6646. [CrossRef]
- 20) Vaupel P, Multhoff G. 2021. Revisiting the Warburg effect: Historical Dogma Versus Current Understanding. J Physiol 599.6: 1745–1757. [CrossRef]
- 21) Monk M, Holding C. 2001. Human Embryonic Genes Re-Expressed in Cancer Cells. Oncogene, 20(56):8085-8091. [CrossRef]
- 22) Aiello NM., Stanger BZ. 2016. Echoes of the Embryo: Using the Developmental Biology Toolkit to Study Cancer. Dis. Model Mech, 9(2): 105-114. [CrossRef]
- 23) Dreesen O, Brivanlou AH. 2007. Signaling Pathways in Cancer and Embryonic Stem Cells. Stem Cell Rev. [CrossRef]
- 24) Costanzo V, Bardelli A, Siena S, Abrignani S. 2018. Exploring the Links Between Cancer and Placenta Development. Open Biol., 8:180081. [CrossRef]
- 25) Feichtinger J, Aldeailij I, Anderson R, Almutairi M, Almatrafi A, Alsiwiehri N, et al. 2012. Meta-Analysis of Clinical Data Using Human Meiotic Genes Identifies a Novel Cohort of Highly-Restricted Cancer-Specific Marker Genes. Oncotarget, 3(8): 843-853. [CrossRef]
- 26) Wepsic HT. 1983. Overview of Oncofetal Antigens in Cancer. Ann.Clin.Lab.Sci., 13(4): 261-266. [PubMed]
- 27) Hall C, Clarke L, Pal A, Buchwald P, Eglinton T, Wakeman C, et al. 2019. A Review of the Role of Carcinoembryonic Antigen in Clinical Practice. Ann Coloproctol, 35(6): 294-305. [CrossRef]
- 28) Beard J. 1902. Embryological Aspects and Etiology of Carcinoma. Lancet, 1:1758-1761. [CrossRef]
- 29) Leach SD. 2005. Epithelial Differentiation in Pancreatic Development and Neoplasia. J Clin Gastroenterol, 39(2): S78-S82. [CrossRef]
- 30) Murray MJ, Lessey BA. 1999. Embryo Implantation and Tumor Metastasis: Common Pathways of Invasion and Angiogenesis. Sem Reprod. Endocrin., 17(3): 275-290. [CrossRef]
- 31) Manzo G. 2019. Similarities Between Embryo Development and Cancer Process Suggest New Strategies for Research and Therapy of Tumors: A New Point of View. Front. Cell Dev. Biol., 7:20. [CrossRef]
- 32) Barrett JC. 1993. Mechanisms of Multistep Carcinogenesis and Carcinogen Risk Assessment. Environmental Health Perspectives, 100:9-20. [CrossRef]
- 33) Braakhuis BJM, Brakenhoff RH, Leemans CR. 2005. Second Field Tumors: A New Opportunity for Cancer Prevention? The Oncologist,10:493–500. [CrossRef]
- 34) Ahmad AS, Ormiston-Smith N, Sasieni PD. 2015. Trends in the Lifetime Risk of Developing Cancer in Great Britain: Comparison of Risk for Those Born From 1930 to 1960. BJCancer, 112: 943-947. [CrossRef]
- 35) Huang R-X, Zhou P-K. 2020. DNA Damage Response Signaling Pathways and Targets for Radiotherapy Sensitization in Cancer. Signal Transduction and Targeted Therapy 5:60. [CrossRef]
- 36) Stewart BW. 2019. Mechanisms of Carcinogenesis: from Initiation and Promotion to the Hallmarks. In: Tumour Site Concordance and Mechanisms of Carcinogenesis. Lyon (FR): International Agency for Research on Cancer; 2019. (IARC Scientific Publications, No. 165.) Chapter 11. Baan RA, Stewart BW, Straif K, editors. Available from: https://www.ncbi.nlm.nih.gov/books/NBK570326/.
- 37) Hajri QA, Dash S, Feng W, Garner HR, Anandakrishnan R. 2020. Identifying Multi-Hit Carcinogenic Gene Combinations: Scaling Up a Weighted Set Cover Algorithm Using Compressed Binary Matrix Representation on a GPU. Scientific Reports,10:2022. [CrossRef]
- 38) Li R, Sonik A, Stindl R, Rasnick D, Duesberg P. 2000. Aneuploid -vs- Gene Mutation Hypothesis of Cancer: Recent Study Claims Mutation But is Found to Support Aneuploidy. PNAS, 97(7): 3236-3241. [CrossRef]
- 39) Zhu S, Wang J, Zellmer L, Xu N, Liu M, Hu Y, et al. 2022. Mutation or Not, What Directly Establishes a Neoplastic State, Namely Cellular Immortalilty and Autonomy, Still Remains Unknown and Should Be Prioritized in our Research. J. Cancer, 13(9):2810-2843. [CrossRef]
- 40) Nohmi T. 2018. Thresholds of Genotoxic and Non-Genotoxic Carcinogens. Toxicol Res, 34(4):281-290. [CrossRef]
- 41) Goldblatt H, Cameron G. 1953. Induced Malignancy in Cells From Rat Myocardium Subjected to Intermittent Anaerobiosis during Long Propagation in vitro. J.Exp.Med., 97(4):525-552. [CrossRef]
- 42) Poljsak B, Milisav I. 2012. Clinical Implications of Cellular Stress Responses. Basic Med Sci, 12(2): 122-126. [CrossRef]
- 43) Li D, Duncan RF. 1995. Transient Acquired Thermotolerance in Drosophila, Correlated with Rapid Degradation of Hsp70 During Recovery. Eur J Biochem., 231:454-465. [CrossRef]
- 44) George I, Geddis MS, Lill Z, Lin H, Gomez T, Blank M, et al. 2008. Myocardial Function Improved by Electromagnetic Field Induction of Stress Protein hsp70. J Cell Physiol, 216(3): 816-823. [CrossRef]
- 45) Soti C, Sreedhart AS, Csermely P. 2003. Apoptosis, Necrosis, and Cellular Senescence: Chaperone Occupancy as a Potential Switch. Aging Cell, 2:39-45. [CrossRef]
- 46) Ishikawa T, Zhang SS-M, Qin X, Takahashi Y, Oda H, Nakatsure Y, et al. 2003. DNA Repair and Cancer: Lessons from Mutant Mouse Models. Cancer Sci, 95(2): 112-117. [CrossRef]
- 47) Knudson AG. 1971. Mutation and Cancer: Statistical Study of Retinoblastoma. Proc Nat Acad Sci, 68(4): 820-823. [CrossRef]
- 48) Lamech LT, Haynes CM. 2015. The Unpredictability of Prolonged Activation of Stress Response Pathways. J Cell Biol, 209(6): 781-787. [CrossRef]
- 49) Ameya G, Birri DJ. 2023. The Molecular Mechanisms of Virus-Induced Human Cancers. J Mic. Path, 183:106292. [CrossRef]
- 50) Hatano Y, Ideta T, Hirata A, Hatano K, Tomita H, Okada H, et al. 2021. Virus-Driven Carcinogenesis. Cancers, 13:2625. [CrossRef]
- 51) Sree BR, Joy JM. 2011. A Review on Molecular Chaperones and Chaperone Overload. IJPDT, 1(2): 58-65.
- 52) Jeng W, Lee S, Sung N, Lee J, Tsai FTF. 2015. Molecular Chaperones: Guardians of the Proteome in Normal and Disease States. F1000 Research2015, 4(F1000 Faculty Rev):1448. [CrossRef]
- 53) Moreno DF, Parisi E, Yahya G, Vaggi F, Csikasz-Nagy A, Aldea M. 2019. Competition in the Chaperone-Client Network Subordinates Cell-Cycle Entry to Growth and Stress. Life Science Alliance, 2(2):201800277. [CrossRef]
- 54) Truman AW, Kristjansdottir K, Wolfgeher D, Hasin N, Polier S, Zhang H, et al. 2012. CDK-Dependent Hsp70 Phosphorylation Controls G1 Cyclin Abundance and Cell-Cycle Progression. Cell, 151: 1308-1318. [CrossRef]
- 55) Ali A, Bharadwaj S, O’Carroll R, Ovsenek N. 1998. HSP90 Interacts with and Regulates the Activity of Heat Shock Factor 1 in Xenopus Oocytes. Mol. Cell. Biol., 18(9): 4949-4960. [CrossRef]
- 56) Masser AE, Ciccarelli M, Andreasson C. 2020. Hsf1 on a Leash – Controlling the Heat Shock Response by Chaperone Titration. Exp. Cell Res., 396: 112246. [CrossRef]
- 57) Taylor RC, Cullen SP, Martin SJ. 2008. Apoptosis: Controlled Demolition at the Cellular Level. Nat. Rev. Mol. Cell Biol., 9: 231-241. [CrossRef]
- 58) Stracker TH. 2017. Chaperoning the DNA Damage Response. The FEBS Jour, 284:2375-2377. [CrossRef]
- 59) Knighton LE, Truman AW. 2019. Role of the Molecular Chaperones Hsp70 and Hsp90 in the DNA Damage Response. In: Heat Shock Proteins in Signaling Pathways. Heat Shock Proteins, vol. 17: 345-358. Springer, Cham. [CrossRef]
- 60) Pennisi R, Ascenzi P, di Masi A. 2015. Hsp90: A New Player in DNA Repair? Biomolecules, 5:2589-2618. [CrossRef]
- 61) Dote H, Burgan WE, Camphausen K, Tofilon PJ. 2006. Inhibition of Hsp90 Compromises the DNA Damage Response to Radiation. Cancer Res, 66(18): 9211-9220. [CrossRef]
- 62) Huiting W, Dekker SL, van der Lienden JCJ, Mergener R, Musskopf MK, Furtado GV, et al. 2022. Targeting DNA Topoisomerases or Checkpoint Kinases Results in an Overload of Chaperone Systems, Triggering Aggregation of a Metastable Subproteome. eLife, 11:e70726. [CrossRef]
- 63) Su K-H, Dai S, Tang Z, Xu M, Dai C. 2019. Heat Shock Factor 1 is a Direct Antagonist of AMP-Activated Protein Kinase. Molecular Cell, 76: 546-561. [CrossRef]
- 64) Dai S, Tang Z, Cao J, Zhou W, Li H, Sampson S, et al. 2015. Suppression of the HSF1-Mediated Proteotoxic Stress Response by the Metabolic Stress Sensor AMPK. The EMBO Jour, 34(3):275-293. [CrossRef]
- 65) Li J, Labbadia, J, Morimoto RL. 2017. Rethinking HSF1 in Stress, Development and Organismal Health. Trends Cell Biol, 27(12):895-905. [CrossRef]
- 66) Qiao A, Jin X, Pang J, Moskophidis D, Mivechi NF. 2017. The Transcriptional Regulator of the Chaperone Response HSF1 Controls Hepatic Bioenergetics and Protein Homeostasis. J. Cell. Biol., 216(3): 723-741. [CrossRef]
- 67) Canto C. 2017. The Heat Shock Factor HSF1 Juggles Protein Quality Control and Metabolic Regulation. J. Cell. Biol., 216(3): 551-553. [CrossRef]
- 68) Yang Q, Wang Y, Wang H, Li H, Zhu J, Cong L, et al. 2021. NAD+ Repletion Attenuates Obesity-Induced Oocyte Mitochondrial Dysfunction and Offspring Metabolic Abnormalities via a SIRT3-Dependent Pathway. Clin. Transl. Med., 11:e628. [CrossRef]
- 69) Nassour J, Martien S, Martin N, Deruy E, Tomellini E, Malaquin N, et al. 2016. Defective DNA Single-Strand Break Repair is Responsible for Senescence and Neoplastic Escape of Epithelial Cells. Nat. Comm., 7: 10399. [CrossRef]
- 70) Li J, Bonkowski MS,Moniot S, Zhang D, Hubbard BP, Ling AJY, et al. 2017. A Conserved NAD+ Binding Pocket That Regulates Protein-Protein Interactions During Aging. Science, 355(6331): 1312-1317. [CrossRef]
- 71) Li Q, Feldman RA, Radhakrishnan VM, Carey S, Martinez JD. 2008. Hsf1 is Required for the Nuclear Translocation of p53 Tumor Suppressor. Neoplasia, 10(10):1138-1145. [CrossRef]
- 72) Li Q, Martinez, JD. 2011. P53 is Transported into the Nucleus via an Hsf1-Dependent Nuclear Localization Mechanism. Mol Carcinog., 50(2): 143-152. [CrossRef]
- 73) Toma-Jonik A, Vydra N, Janus P, Widlak W. 2019. Interplay Between HSF1 and p53 Signaling Pathways in Cancer Initiation and Progression: Non-Oncogene and Oncogene Addiction. Cell. Onc., 42:579-589. [CrossRef]
- 74) Hasty P, Sharp ZD, Curiel TJ, Campisi J. 2013. MTORC1 and p53. Clash of the Gods? Cell Cycle, 12(1):20-25. [CrossRef]
- 75) Humpton TJ, Vousden KH. 2016. Regulation of Cellular Metabolism and Hypoxia by p53. Cold Spring Harb Perspect Med, 6(7):a026146. [CrossRef]
- 76) Su K-H, Cao J, Tang Z, Dai S, He Y, Sampson SB, et al. 2016. HSF1 Critically Attunes Proteotoxic Stress Sensing by mTORC1 to Combat Stress and Promote Growth. Nat. Cell. Biol., 18(5):527-539. [CrossRef]
- 77) Hayflick L, Moorhead PS. 1961. The Serial Cultivation of Human Diploid Cell Strains. Exp. Cell Res., 25:585-621. [CrossRef]
- 78) Ohtani N, Hara E. 2013. Roles and Mechanisms of Cellular Senescence in Regulation of Tissue Homeostasis. Cancer Sci., 104(5): 525-530. [CrossRef]
- 79) Ben-Porath I, Weinberg RA. 2004. When Cells Get Stressed: An Integrative View of Cellular Senescence. J. Clin. Invest. 113:8–13. [CrossRef]
- 80) Kuilman T, Michaloglou C, Mooi WJ, Peeper DS. 2010. The Essence of Senescence. Genes & Dev., 24:2463-2479. [CrossRef]
- 81) Gire V, Dulic V. 2015. Senescence From G2 Arrest, Revisited. Cell Cycle, 14(3):297-304. [CrossRef]
- 82) Soto-Gamez A, Quax WJ, Demaria M. 2019. Regulation of Survival Networks in Senescent Cells: From Mechanisms to Interventions. J. Mol. Biol., 431:2629-2643. [CrossRef]
- 83) Krtolica A, Parrinello S, Locket S, Desprez P-Y, Campisi J. 2001. Senescent Fibroblasts Promote Epithelial Cell Growth and Tumorigenesis: A Link Between Cancer and Aging. PNAS, 98(21): 12072-12077. [CrossRef]
- 84) van Deursen JM. 2014. The Role of Senescent Cells in Ageing. Nature, 509(7501): 439-446. [CrossRef]
- 85) Campisi J. 2013. Aging, Cellular Senescence, and Cancer. Annu Rev Physiol.,75:685–705. [CrossRef]
- 86) Demaria M, Ohtani N, Youssef SA, Rodier F, Toussaint W, Mitchell JR, et al. 2014. An Essential Role for Senescent Cells in Optimal Wound Healing through Secretion of PDGF-AA. Develop. Cell, 31:722-733. [CrossRef]
- 87) Jun J-I, Lau LF. 2010. Cellular Senescence Controls Fibrosis in Wound Healing. Aging, 2(9): 627-631. [CrossRef]
- 88) Stewart SA, Weinberg RA. 2000. Telomerase and Human Tumorigenesis. Sem in Canc. Biol., 10(6):399-406. [CrossRef]
- 89) Schwarze SR, DePrimo SE, Grabert LM, Fu VX, Brooks JD, Jarrard DF. 2002. Novel Pathways Associated with Bypassing Cellular Senescence in Human Prostate Epithelial Cells. J. Biol. Chem., 277(17):14877-14883. [CrossRef]
- 90) Shay JW, Wright WE. 2005. Senescence and Immortalization: Role of Telomeres and Telomerase. Carcinogenesis, 26(5): 867-874. [CrossRef]
- 91) Morel Y, Barouki R. 1999. Repression of Gene Expression by Oxidative Stress. Biochem. J., 342:481-496. [CrossRef]
- 92) Childs BG, Baker DJ, Kirkland JL, Campisi J, van Deursen JM. 2014. Senescence and Apoptosis: Dueling or Complementary Cell Fates? EMBO Reports,15(11):1139-1153. [CrossRef]
- 93) von Zglinicki T, Wan T, Miwa S. 2021. Senescence in Post-Mitotic Cells: A Driver of Aging? Antioxidants & Redox Signaling, 34(4):308-323. [CrossRef]
- 94) DiCarlo AL, Farrell JM, Litovitz TA. 1999. Myocardial Protection Conferred by Electromagnetic Fields. Circulation, 99:813-816. [CrossRef]
- 95) Wiese M, Bannister AJ. 2020. Two Genomes, One cell: Mitochondrial-Nuclear Coordination via Epigenetic Pathways. Molec. Metab., 38:100942. [CrossRef]
- 96) Walker BR, Moraes CT. 2022. Nuclear-Mitochondrial Interactions. Biomolecules, 12:427. [CrossRef]
- 97) Herranz N, Gil J. 2018. Mechanisms and Functions of Cellular Senescence. J. Clin. Invest., 128(4): 1238-1246. [CrossRef]
- 98) Maciel-Baron LA, Morales-Rosales SL, Aquino-Cruz AA, Triana-Martínez F, Galván-Arzate S, Luna-López A, et al. 2016. Senescence Associated Secretory Phenotype Profile from Primary Lung Mice Fibroblasts Depends on the Senescence Induction Stimuli. Age,38:26. [CrossRef]
- 99) Young ARJ, Narita M. 2009. SASP Reflects Senescence. EMBO Reports, 10(3):228-230. [CrossRef]
- 100) Ritschka B, Storer M, Mas A, Heinzmann F, Ortells MC, Morton JP, et al. 2017. The Senescence-Associated Secretory Phenotype Induces Cellular Plasticity and Tissue Regeneration. Genes & Develop., 31:172-183. [CrossRef]
- 101) Rhinn M, Ritschka B, Keyes WM. 2019. Cellular Senescence in Development, Regeneration and Disease. Development, 146:dev151837. [CrossRef]
- 102) Nelson G, Wordsworth J, Wang C, Jurk D, Lawless C, Martin-Ruiz C, et al. 2012. A Senescent Cell Bystander Effect: Senescence-Induced Senescence. Aging Cell,11:345-349. [CrossRef]
- 103) Olivieri F, Albertini MC, Orciani M, Ceka A, Cricca M, Procopio AD, et al. 2015. DNA Damage Response (DDR) and Senescence: Shuttled Inflamm-miRNAs on the Stage of Inflamm-aging. Oncotarget, 6(34): 35509-33521. [CrossRef]
- 104) Parrinello S, Coppe J-P, Krtolica A, Campisi J.2004. Stromal-Epithelial Interactions in Aging and Cancer: Senescent Fibroblasts Alter Epithelial Cell Differentiation. Journal of Cell Science, 118: 485-496. [CrossRef]
- 105) Ovadya Y, Landsberger T, Leins H, Vadai E, Gal H, Biran A, et al. 2018. Impaired Immune Surveillance Accelerates Accumulation of Senescent Cells and Aging. Nature Communications, 9:5435. [CrossRef]
- 106) Sprenger CC, Plymate SR, Reed MJ. 2010. Aging-Related Alterations in the Extracellular Matrix Modulate the Microenvironment and Influence Tumor Progression. Int. J. Cancer, 127(12): 2739-2748. [CrossRef]
- 107) Mosteiro L, Pantoja C, Alcazar N, Marión RM, Chondronasiou D, Rovira M, et al. 2016. Tissue Damage and Senescence Provide Critical Signals for Cellular Reprogramming in vivo. Science, 354(6315). [CrossRef]
- 108) Von Aderkas P, Bonga JM. 2000. Influencing Micropropagation and Somatic Embryogenesis in Mature Trees by Manipulation of Phase Change, Stress and Culture Environment. Tree Physiol., 20:921-928. [CrossRef]
- 109) Feher A, Pasternak T, Miskolczi P, Ayaydin F, Dudits D. 2000. Induction of the Embryogenic Pathway in Somatic Plant Cells. Acta Hort., 560:293-298. [CrossRef]
- 110) Dudits D, Bogre L, Gyorgyey J. 1991. Molecular and Cellular Approaches to the Analysis of Plant Embryo Development from Somatic Cells in vitro. J. Cell. Sci., 99:475-484. [CrossRef]
- 111) Ikeda-Iwai M, Umehara M, Satoh S, Kamada H. 2003. Stress-Induced Somatic Embryogenesis in Vegetative Tissues of Arabidopsis thaliana. The Plant Jour., 34:107-114. [CrossRef]
- 112) Mordhorst BR, Benne JA, Cecil RF, Whitworth KM, Samuel MS, Spate LD, et al. 2019. Improvement of in vitro and Early in utero Porcine Clone Development after Somatic Donor Cells are Cultured Under Hypoxia. Mol Reprod Dev., 86:558-565. [CrossRef]
- 113) Liu J-L, Wang M-K, Sun Q-Y, Zhang X-R, Jiang L-K, Chen D-Y. 2001. Refrigeration of Donor Cells in Preparation for Bovine Somatic Nuclear Transfer. Reproduction, 122:801-808. [CrossRef]
- 114) Wilmut I, Beaujean N, de Sousa PA, Dinnyes AA, King TJ, Paterson LA, et al. 2002. Somatic Cell Nuclear Transfer. Nature, 419(6907):583-587. [CrossRef]
- 115) Takahashi K, Yamanaka S. 2006. Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell, 126:663-676. [CrossRef]
- 116) Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, et al. 2007. Induction of Pluripotent Stem Cells from Adult Human Fibroblasts by Defined Factors. Cell, 131:861-872. [CrossRef]
- 117) Folmes CDL, Nelson TJ, Martinez-Fernandez A, Arrell DK, Lindor JZ, Dzeja PP, et al. 2011. Somatic Oxidative Bioenergetics Transitions into Pluripotency-Dependent Glycolysisto Facilitate Nuclear Reprogramming. Cell Metab., 14(2): 264–271. [CrossRef]
- 118) Folmes CDL, Nelson TJ, Terzic A. 2011. Energy Metabolism in Nuclear Reprogramming. Biomark. Med., 5(6):715-729. [CrossRef]
- 119) Ishida T, Nakao S, Ueyama T, Harada Y, Kawamura T. 2020. Metabolic Remodeling During Somatic Cell Reprogramming to Induced Pluripotent Stem Cells: Involvement of Hypoxia-Inducible Factor 1. Inflammation and Regeneration, 40:8. [CrossRef]
- 120) Sinclair JW, Hoying DR, Bresciani E, Nogare DD, Needle CD, Berger A, et al. 2021. The Warburg Effect is Necessary to Promote Glycosylation in the Blastema During Zebrafish Tail Regeneration. Npj Regenerative Medicine, 6:55. [CrossRef]
- 121) Forristal CE, Christensen DR, Chinnery FE, Petruzzelli R, Parry KL, Sanchez-Elsner T, et al. 2013. Environmental Oxygen Tension Regulates the Energy Metabolism and Self-Renewal of Human Embryonic Stem Cells. PLoS ONE 8(5): e62507. [CrossRef]
- 122) McCusker C, Bryant SV, Gardiner DM. 2015. The Axolotl Limb Blastema: Cellular and Molecular Mechanisms Driving Blastema Formation and Limb Regeneration in Tetrapods. Regeneration:54-71. [CrossRef]
- 123) Simkin J, Sammarco MC, Dawson LA, Schanes PP, Yu L, Muneoka K. 2015. The Mammalian Blastema: Regeneration at our Fingertips. Regeneration:93-105. [CrossRef]
- 124) Elder SS, Emmerson E. 2020. Senescent Cells and Macrophages: Key Players for Regeneration? Open Biol., 10:200309. [CrossRef]
- 125) Mosser DM, Edwards JP. 2008. Exploring the Full Spectrum of Macrophage Activation. Nat Rev Immunol., 8(12): 958–969. [CrossRef]
- 126) Wu R, Van der Hoek KH, Ryan NK, Norman RJ, Robker RL. 2004. Macrophage Contributions to Ovarian Function. Human Reproduction Update, 10(2): 119-133. [CrossRef]
- 127) Espey LL. 1980. Ovulation as an Inflammatory Reaction — A Hypothesis. Biol. Reprod., 22:73-106. [CrossRef]
- 128) Duffy DM, Ko C, Jo M, Brannstrom M, Curry, Jr. TE. 2019.Ovulation: Parallels with Inflammatory Process. Endocrine Reviews, 40:369-416. [CrossRef]
- 129) Masuda M, Wakasaki T, Toh S. 2016. Stress-Triggered Atavistic Reprogramming (STAR) Addiction: Driving Force Behind Head and Neck Cancer? Am. J. Cancer Res., 6(6):1149-1166. [PubMed]
- 130) Feichtinger J, McFarlane RJ. 2019. Meiotic Gene Activation in Somatic and Germ Cell Tumours. Andrology, 7:415–427. [CrossRef]
- 131) Gantchev J, Villarreal AM, Gunn S, Zetka M, Ødum N, Litvinov IV. 2020. The Ectopic Expression of meiCT Genes Promotes Meiomitosis and May Facilitate Carcinogenesis. Cell Cycle, 19(8):837–854. [CrossRef]
- 132) Hosoya N, Miyagawa K. 2020. Synaptonemal Complex Proteins Modulate the Level of Genome Integrity in Cancers. Cancer Science, 112:989–996. [CrossRef]
- 133) Uyar A, Torrealday S, Seli E. 2013. Cumulus and Granulosa Cell Markers of Oocyte and Embryo Quality. Fert. Steril., 99(4):979-997. [CrossRef]
- 134) Jones ASK, Shikanov A. 2019. Follicle Development as an Orchestrated Signaling Network in a 3D Organoid. Jour. Biol. Eng., 13(2). [CrossRef]
- 135) Kreeger PK, Deck JW, Woodruff TK, Shea LD. 2006. The in vitro Regulation of Ovarian Follicle Development Using Alginate-Extracellular Matrix Gels. Biomaterials, 27(5):714-723. [CrossRef]
- 136) Horandl E, Hadacek F. 2013. The Oxidative Damage Initiation Hypothesis for Meiosis. Plant Reprod, 26: 351–367. [CrossRef]
- 137) Mirzaghaderi G, Horandl E. 2016. The Evolution of Meiotic Sex and its Alternatives. Proc. R. Soc. B, 283: 20161221. [CrossRef]
- 138) Bernstein H, Bernstein C. 2010. Evolutionary Origin of Recombination During Meiosis. BioScience, 60(7):498-505. [CrossRef]
- 139) Lenormand T, Engelstadter J, Johnston SE, Wijnker E, Haag CR. 2016. Evolutionary Mysteries in Meiosis. Trans. R. Soc. B, 371:20160001. [CrossRef]
- 140) Garg SG, Martin WF. 2016. Mitochondria, the Cell Cycle, and the Origin of Sex via a Syncytial Eukaryote Common Ancestor. Genome Biol. Evol., 8(6):1950–1970. [CrossRef]
- 141) Blagosklonny MV. 2011. Cell Cycle Arrest is Not Senescence. Aging, 3(2):94-101.
- 142) Duckworth BC, Weaver JS, Ruderman JV. 2002. G2 Arrest in Xenopus Oocytes Depends on Phosphorylation of Cdc25 by Protein Kinase A. PNAS, 99(26):16794–16799. [CrossRef]
- 143) Tsang M, Gantchev J, Netchiporouk E, Moreau L, Ghazawi FM, Glassman S, et al. 2018. A Study of Meiomitosis and Novel Pathways of Genomic Instability in Cutaneous T-Cell Lymphomoas (CTCL). Oncotarget, 9(102):37647-37661. [CrossRef]
- 144) Claybon A, Karia B, Bruce C, Bishop AJR. 2010. PARP1 Suppresses Homologous Recombination Events in Mice in vivo. Nucleic Acids Research, 38(21):7538–7545. [CrossRef]
- 145) Lao JP, Hunter N. 2010. Trying to Avoid Your Sister. PLoS Biol., 8(10):e1000519. [CrossRef]
- 146) Okuda A, Suzuki A. 2016. Unexpected Link Between MAX and Meiotic Onset. Cell Cycle, 15(17):2235–2236. [CrossRef]
- 147) Yamashita T, Higashi M, Momose S, Adachi A, Watanabe T, Tanaka Y, et al.2020. Decreased MYC-Associated Factor X (MAX) Expression is a New Potential Biomarker for Adverse Prognosis in Anaplastic Large Cell Lymphoma. Sci Rep., 10:10391. [CrossRef]
- 148) Janisiw E, Raices M, Balmir F, Paulin LF, Baudrimont A, von Haeseler A, et al. 2020. Poly(ADP-ribose) Glycohydrolase Coordinates Meiotic DNA Double-Strand Break Induction and Repair Independent of its Catalytic Activity. Nature Communications, 11:4869. [CrossRef]
- 149) Suzuki, A, Hirasaki M, Okuda A. 2017. Does MAX Open Up a New Avenue for Meiotic Research? Develop. Growth Differ., 59:61–69. [CrossRef]
- 150) Kimble J. 2011. Molecular Regulation of the Mitosis/Meiosis Decision in Multicellular Organisms. Cold Spring Harb Perspect Biol, 3:a002683. [CrossRef]
- 151) Honigberg SM, Purnapatre K. 2003. Signal Pathway Integration in the Switch from the Mitotic Cell Cycle to Meiosis in Yeast. Jour. Cell Sci, 116:2137-2147. [CrossRef]
- 152) Kodiha M, Rassi JG, Brown CM, Stochaj U. 2007. Localization of AMP-Kinase is Regulated by Stress, Cell Density, and Signaling Through the MEK–> ERK1/2 Pathway. Am J Physiol Cell Physiol 293: C1427–C1436. [CrossRef]
- 153) Clements D, Mayer RJ, Johnson SR. 2007. Subcellular Distribution of the TSC2 Gene Product Tuberin in Human Airway Smooth Muscle Cells is Driven by Multiple Localization Sequences and is Cell-Cycle Dependent. Am J Physiol Lung Cell Mol Physiol 292: L258 –L266, 2007. [CrossRef]
- 154) Karni-Schmidt O, Friedler A, Zupnick A, McKinney K, Mattia M, Beckerman R, et al. 2007. Energy-Dependent Nucleolar Localization of p53 in vitro Requires Two Discrete Regions within the p53 Carboxyl Terminus. Oncogene (2007) 26, 3878–3891. [CrossRef]
- 155) Xu-Monette ZY, Medeiros LJ, Li Y, Orlowski RZ, Andreeff M, Bueso-Ramos CE, et al. 2012. Dysfunction of the TP53 Tumor Suppressor Gene in Lymphoid Malignancies. Blood, 119(16):3668-3683. [CrossRef]
- 156) Nilsson K, Landberg G. 2006. Subcellular Localization, Modification and Protein Complex Formation of the Cdk-Inhibitor p16 in Rb-Functional and Rb-Inactivated Tumor Cells. Int. J. Cancer, 118:1120–1125. [CrossRef]
- 157) Apostolova MD, Ivanova IA, Dagnino C, D’Souza SJA, Dagnino L. 2002. Active Nuclear Import and Export Pathways Regulate E2F-5 Subcellular Localization. J Biol Chem, 277(37):34471–34479. [CrossRef]
- 158) Trinh DLN, Elwi AN, Kim S-W. 2010. Direct Interaction Between p53 and Tid1 Proteins Affects p53 Mitochondrial Localization and Apoptosis. Oncotarget, 1(6):396 - 404. [CrossRef]
- 159) Agarwal S, Ganesh S. 2020. Perinuclear Mitochondrial Clustering, Increased ROS Levels, and HIF1are Required for the Activation of HSF1 by Heat Stress. Jour. Cell Sci., 133: jcs245589. [CrossRef]
- 160) Takahashi Y, Hashimoto S, Yamochi T, Goto H, Yamanaka M, Amo A, et al. 2016. Dynamic Changes in Mitochondrial Distribution in Human Oocytes During Meiotic Maturation. J Assist Reprod Genet, 33:929–938. [CrossRef]
- 161) Lees JG, Gardner DK, Harvey AJ. 2017. Pluripotent Stem Cell Metabolism and Mitochondria: Beyond ATP. Stem Cells International, 2017(2874283):17 pages. [CrossRef]
- 162) Moiseeva O, Bourdeau V, Roux A, Deschenes-Simard X, Ferbeyre G. 2009. Mitochondrial Dysfunction Contributes to Oncogene-Induced Senescence. Mol. Cell. Biol, 29(16):4495–4507. [CrossRef]
- 163) Chakrabarty RP, Chandel NS. 2021. Mitochondria as Signaling Organelles Control Mammalian Stem Cell Fate. Cell Stem Cell, 28:394-408. [CrossRef]
- 164) Saxton WM, Hollenbeck PJ. 2012. The Axonal Transport of Mitochondria. J Cell Sci, 125:2095–2104. [CrossRef]
- 165) Voglhuber J, Holzer M, Radulovic S, Thai PN, Djalinac N, Matzer I, et al. 2022. Functional Remodelling of Perinuclear Mitochondria Alters Nucleoplasmic Ca2+ Signalling in Heart Failure. Phil. Trans. R. Soc. B, 377: 20210320. [CrossRef]
- 166) Fenton AR, Jongens TA, Holzbaur ELF. 2021. Mitochondrial Adapter TRAK2 Activates and Functionally Links Opposing Kinesin and Dynein Motors. Nature Comm, 12:4578. [CrossRef]
- 167) Thomas LW, Staples O, Turmaine M, Ashcroft M. 2017. CHCHD4 Regulates Intracellular Oxygenation and Perinuclear Distribution of Mitochondria. Front. Oncol., 7:71. [CrossRef]
- 168) Giardino Torchia ML, Ashwell JD. 2018. Getting MAD at MYC. PNAS,115(40):9821–9823. [CrossRef]
- 169) Kemmerer K, Weigand JE. 2014. Hypoxia Reduces MAX Expression in Endothelial Cells by Unproductive Splicing. FEBS Letters, 588:4784–4790. [CrossRef]
- 170) Suzuki A, Hirasaki M, Hishida T, Wu J, Okamura D, Ueda A, et al. 2016. Loss of MAX Results in Meiotic Entry in Mouse Embryonic and Germline Stem Cells. Nature Comm, 7:11056. [CrossRef]
- 171) Gordon JD, Bertovrt JA, Hu C-J, Diehl JA, Simon MC. 2007. HIF-2∀ Promotes Hypoxic Cell Proliferation by Enhancing c-Myc Transcriptional Activity. Cancer Cell, 11(4):335-347. [CrossRef]
- 172) Yan Y, Liu F, Han L, Zhao L, Chen J, Olopade OI, et al. 2018. HIF-2∀ Promotes Conversion to a Stem Cell Phenotype and Induces Chemoresistance in Breast Cancer Cells by Activating Wnt and Notch Pathways. J Exp. Clin. Canc. Res., 37:256. [CrossRef]
- 173) Tharp KM, Higuchi-Sanabria R, Timblin GA, Ford B, Garzon-Coral C, Schneider C, et al. 2021. Adhesion-Mediated Mechanosignaling Forces Mitohormesis. Cell Metab., 33:1322-1341. [CrossRef]
- 174) Koppers M, Özkan N, Farías GG. 2020. Complex Interactions Between Membrane-Bound Organelles, Biomolecular Condensates and the Cytoskeleton. Front. Cell Dev. Biol., 8:618733. [CrossRef]
- 175) Heffler J, Shah PP, Robison P, Phyo S, Veliz K, Uchida K, et al. 2020. A Balance Between Intermediate Filaments and Microtubules Maintains Nuclear Architecture in the Cardiomyocyte. Circ Res., 126(3): e10–e26. [CrossRef]
- 176) Spichal M, Fabre E. 2017. The Emerging Role of the Cytoskeleton in Chromosome Dynamics. Front. Genet., 8:60. [CrossRef]
- 177) Bissell MJ, Barcellos-Hoff MH. 1987. The Influence of Extracellular Matrix on Gene Expression: Is Structure the Message? J Cell Sci. Suppl., 8: 327-343. [CrossRef]
- 178) Thorne JT, Segal TR, Chang S, Jorge S, Segars JH, Leppert PC. 2015. Dynamic Reciprocity Between Cells and Their Microenvironment in Reproduction. Biol. Reprod., 92(1):25, 1–10. [CrossRef]
- 179) Spencer VA, Xu R, Bissell MJ. 2007. Extracellular Matrix, Nuclear and Chromatin Structure, and Gene Expression in Normal Tissues and Malignant Tumors: A Work in Progress. Adv Cancer Res., 97:275–294. [CrossRef]
- 180) Schultz GS, Davidson JM, Kirsner RS, Bornstein P, Herman IM. 2011. Dynamic Reciprocity in the Wound Microenvironment. Wound Repair Regen., 19(2):134–148. [CrossRef]
- 181) Lenain C, de Graaf CA, Pagie L, Visser NL, de Haas M, de Vries SS, et al. 2017. Massive Reshaping of Genome-Nuclear Lamina Interactions During Oncogene-Induced Senescence. Genome Res, 27:1634–1644. [CrossRef]
- 182) Burla R, La Torre M, Saggio I. 2016. Mammalian Telomeres and their Partnership with Lamins. Nucleus, 7(2):187-202. [CrossRef]
- 183) Meqbel BRM, Gomes M, Omer, A, Gallouzi IE, Horn HF. 2022. LINCing Senescence and Nuclear Envelope Changes. Cells, 11:1787. [CrossRef]
- 184) Link J, Jahn D, Schmitt J, Gob E, Baar J, Ortega S, et al. 2013. The Meiotic Nuclear Lamina Regulates Chromosome Dynamics and Promotes Efficient Homologous Recombination in the Mouse. PloS Genet., 9(1):e1003261. [CrossRef]
- 185) Morimoto, A, Shibuya H, Zhu X, Kim J, Ishiguro K, Han M, et al. 2012. A Conserved KASH Domain Protein Associates with Telomeres, SUN1, and Dynactin During Mammalian Meiosis. J. Cell Biol.,198(2): 165–172. [CrossRef]
- 186) Davis L, Smith GR. 2006. The Meiotic Bouquet Promotes Homolog Interactions and Restricts Ectopic Recombination in Schizosaccharomyces pombe. Genetics, 174:167–177. [CrossRef]
- 187) Zhuang J, Wang P, Huang X, Chen X, Kang J-G, Hwang PM. 2013. Mitochondrial Disulfide Relay Mediates Translocation of p53 and Partitions its Subcellular Activity. PNAS, 110(43):17356–17361. [CrossRef]
- 188) Saleem A, Hood DA. 2013. Acute Exercise Induces Tumor Suppressor p53 Translocation to the Mitochondriaand Promotes a p53-Tfam-Mitochondrial DNA Complex in Skeletal Muscle. J Physiol, 591.14:3625–3636. [CrossRef]
- 189) Brunyanszki A, Szczesny B, Virág L, Szabo C. 2016. Mitochondrial Poly(ADP-Ribose) Polymerase: The Wizard of Oz at Work. Free Radic Biol Med., 100: 257–270. [CrossRef]
- 190) Lee S-J, Lim C-J, Min J-K, Lee J-K, Kim Y-M, Lee J-Y, et al. 2007. Protein Phosphatase 1 Nuclear Targeting Subunit is a Hypoxia Inducible Gene: Its Role in Post-Translational Modification of p53 and MDM2. Cell Death and Differentiation, 14:1106–1116. [CrossRef]
- 191) Landsverk HB, Mora-Bermudez F, Landsverk OJB, Hasvold G, Naderi S, Bakke O, et al. 2010. The Protein Phosphatase 1 Regulator PNUTS is a New Component of the DNA Damage Response. EMBO reports, 11(11): 868–875. [CrossRef]
- 192) Leman AR, Noguchi E. 2013. The Replication Fork: Understanding the Eukaryotic Replication Machinery and the Challenges to Genome Duplication. Genes, 4:1-32. [CrossRef]
- 193) Carson DR, Christman MF. 2001. Evidence That Replication Fork Components Catalyze Establishment of Cohesion Between Sister Chromatids. PNAS, 98(15):8270 – 8275. [CrossRef]
- 194) Forsburg SL. 2002. Only Connect: Linking Meiotic DNA Replication with Chromosome Dynamics. Molecular Cell, 9:703–711. [CrossRef]
- 195) Skibbens RV. 2000. Holding Your Own: Establishing Sister Chromatid Cohesion. Genome Research, 10:1664-1671. [CrossRef]
- 196) Toth A, Rabitsch KP, Galova M, Schleiffer A, Buonomo SBC, Nasmyth K. 2000. Functional Genomics Identifies Monopolin: A Kinetochore Protein Required for Segregation of Homologs During Meiosis I. Cell, 103:1155–1168. [CrossRef]
- 197) Solc P, Schultz RM, Motlik J. 2010. Prophase I Arrest and Progression to Metaphase I in Mouse Oocytes: Comparison of Resumption of Meiosis and Recovery from G2-Arrest in Somatic Cells. Molecular Human Reproduction, 16(9): 654– 664. [CrossRef]
- 198) Sasaki K, Chiba K. 2001. Fertilization Blocks Apoptosis of Starfish Eggs by Inactivation of the MAP Kinase Pathway. Develop Biol, 237:18-28. [CrossRef]
- 199) Harrison RH, Kuo HC, Scriven PN, Handyside AH, Ogilvie CM. 2000. Lack of Cell cycle Checkpoints in Human Cleavage Stage Embryos Revealed by a Clonal Pattern of Chromosomal Mosaicism Analysed by Sequential Multicolour FISH. Zygote, 8(3):217-224. [CrossRef]
- 200) Lee HO, Davidson JM, Duronio RJ. 2009. Endoreplication: Polyploidy with Purpose. Genes & Development, 23:2461–2477. [CrossRef]
- 201) Tan Z, Chu DZV, Chan YJA, Lu YE, Rancati G. 2018. Mammalian Cells Undergo Endoreduplication in Response to Lactic Acidosis. Scientific Reports,8:2890. [CrossRef]
- 202) Walen KH. 2015. Wound Healing is a First Response in a Cancerous Pathway: Hyperplasia Developments to 4n Cell Cycling in Dysplasia Linked to RB-Inactivation. Journal of Cancer Therapy, 2015, 6, 906-916. [CrossRef]
- 203) Salmina K, Huna A, Kalejs M, Pjanova D, Scherthan H, Cragg MS, et al. 2019. The Cancer Aneuploidy Paradox: In the Light of Evolution. Genes, 10: 83. [CrossRef]
- 204) Salmina K, Vainshelbaum NM, Kreishmane M, Inashkina I, Cragg MS, Pjanova D, et al. 2023. The Role of Mitotic Slippage in Creating a “Female Pregnancy-like System” in a Single Polyploid Giant Cancer Cell. Int. J. Mol. Sci., 24:3237. [CrossRef]
- 205) Erenpreisa J, Cragg MS. 2013. Three Steps to the Immortality of Cancer Cells: Senescence, Polyploidy and Self-Renewal. Cancer Cell International, 13:92. [CrossRef]
- 206) Lok TM, Wang Y, Xu WK, Xie S, Ma HT, Poon RYC. 2020. Mitotic Slippage is Determined by p31comet and the Weakening of the Spindle-Assembly Checkpoint. Oncogene, 39:2819–2834. [CrossRef]
- 207) Ianzini F, Kosmacek EA, Nelson ES, Napoli E, Erenpreisa J, Kalejs M, et al. 2009. Activation of Meiosis-Specific Genes is Associated with Depolyploidization of Human Tumor Cells Following Radiation-Induced Mitotic Catastrophe. Cancer Res.,69(6):2296–2304. [CrossRef]
- 208) Mosieniak G, Sliwinska MA, Alster O, Strzeszewska A, Sunderland P, Piechota M, et al. 2015. Polyploidy Formation in Doxorubicin-Treated Cancer Cells Can Favor Escape from Senescence. Neoplasia, 17: 882–893. [CrossRef]
- 209) Amodeo AA, Jukam D, Straight AF, Skotheim JM. 2015. Histone Titration Against the Genome Sets the DNA-to-Cytoplasm Threshold for the Xenopus Midblastula Transition. PNAS:E1086-E1095. [CrossRef]
- 210) Niu N, Mercado-Uribe I, Liu J. 2017. Dedifferentiation into Blastomere-like Cancer Stem Cells via Formation of Polyploid Giant Cancer Cells. Oncogene, 36:4887-4900. [CrossRef]
- 211) Tu, S-M, Zhang, M, Wood, CG, Pisters, LL. 2021. Stem Cell Theory of Cancer: Origin of Tumor Heterogeneity. Cancers, 13:4006. [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



