
Submitted:
09 August 2024
Posted:
12 August 2024
You are already at the latest version
Abstract
Aluminum (Al) toxicity and low phosphorus availability (LP) are the top two co-existing edaphic constraints limiting agriculture productivity in acid soils. Plants have evolved versatile mechanisms to cope with the two stresses alone or simultaneously. However, the specific and common molecular mechanisms, especially those on flavonoids and carbohydrate metabolism, are still unclear. Laboratory studies were conducted on two wheat genotypes, Fielder (Al-tolerant and P-efficient) and Ardito (Al-sensitive and P-inefficient), exposed to 50 μM Al and 2 μM Pi (LP) in hydroponic solutions. After 4 days of stress, wheat roots were analyzed via transcriptomic and targeted metabolomic techniques. A total of 2296 differentially expressed genes (DEGs, 1535 up and 761 down) under Al and 3029 DEGs (1591 up and 1438 down) under LP were identified in Fielder. Similarly, 4404 DEGs (3191 up and 1213 down) under Al and 1430 DEGs (1176 up and 254 down) under LP were identified in Ardito. The GO annotation analysis results that 4079 DEGs annotated to the metabolic processes term. These DEGs were significantly enriched in the phenylpropanoid, flavonoid, flavone and flavonol biosynthesis, and carbohydrate metabolism pathways by performing the KEGG enrichment analysis. The targeted metabolome analysis detected 19 flavonoids and 15 carbohydrate components in Fielder and Ardito under Al and LP. More responsive genes and metabolites were involved in flavonoid metabolism under LP than Al in the Fielder, while an opposite trend in the Ardito. In the carbohydrate metabolism pathway, the genes and metabolites were higher in Fielder than in Ardito. The combined transcriptome and metabolome analysis revealed that the flavonoid and carbohydrate genes and metabolites differed between Fielder and Ardito under Al and LP, which may be an important reason for Fielder’s high Al and LP resistance. The results of this study lay a foundation for pyramiding genes and breeding multi-resistance varieties.
Keywords:
wheat root
; transcriptomic
; metabolomic
; Al tolerance
; P efficiency
1. Introduction
Over half of the world’s potentially arable lands are comprised of acid soils, posing numerous challenges to plant performance, primarily in the form of aluminum (Al) toxicity and inorganic phosphorus (Pi) deficiency, which are the two largest abiotic stress in acid soil [1]. Al toxicity and Pi deficiency often coexist in acid soil, with phosphorus (P) fixation in Al-P precipitates significantly contributing to low P availability [2]. Al toxicity inhibits root development and directly interferes with plant P metabolism and Pi signaling, diminishing the plant’s ability to acquire Pi from soils [3]. Therefore, crops grown in acid soil are confront with a dilemma: simultaneously increase the uptake of primary macronutrient Pi while reducing the absorption of toxic Al3+. For example, the great challenge for plants in balancing the absorption of Pi and Al is changing the root system morphology and architecture. Al toxicity inhibits root cell expansion and elongation, along with later cell division, causing the root system to become swollen to reduce Al3+ entry into root cells [3]. The strategy of Pi uptake by plants is to enlarge the contact area between the roots and the soil by increasing the lateral roots and root hairs to improve Pi acquisition efficiency [4].
Plants have developed defense mechanisms to simultaneously navigate the challenges Al and LP pose in acid soils encompassing external and internal forms [5]. Plants regulate Al uptake, translocation, and distribution to avoid Al toxicity and efficiently take up Pi from the soil by orchestrating a set of transport mechanisms [6]. A well-documented external tolerance mechanism involves the secretion of organic acid (OAs) from root apices, crucial components in Al detoxification and Pi acquisition [7]. The malate transporter aluminum activated malate transporter 1 (ALMT1) and its positive transcriptional regulator sensitive to proton rhizotoxicity 1 (STOP1) play an essential role in tolerance to Al toxicity [8] and Pi deficiency [9]. In addition, two ABC transporters sensitive to Al rhizotoxicity 1/aluminum sensitive 3 (STAR1/ALS3) [10,11] and wall-associated kinases (WAKs) [12,13] are involved in cell wall-mediated responses to Al toxicity and P deficiency. The internal mechanisms include isolating excess Al3+ into vacuoles and recycling P from vacuoles, increasing the activity of antioxidant enzymes, and common or specific physiological metabolic reactions [7]. In addition, plant hormones and some transcription factors (TFs) such as WRKY, MYB, bHLH, NAC, and ERF/AP2 are the common determinants of Al tolerance and Pi efficiency [14].
Previous studies have investigated physiological metabolites and the changes involved in P starvation and Al tolerance response. Flavonoids are an important class of secondary metabolites widely found in plants, contributing to plant growth and development [15]. Similar to organic acids, secondary metabolites produced from the phenylpropanoid biosynthesis pathway, such as phenylpropanoids and flavonoids, play a crucial role in scavenging reactive oxygen species (ROS), delaying microbial degradation of OAs, and enhancing mobilization of rhizosphere Pi [16,17]. Carbohydrate metabolism plays a vital role in resistance to abiotic stress in plants, such as providing energy and raw materials, regulating cellular osmotic pressure and ion homeostasis, and enhancing stress resistance [18]. Plants could provide glycolytically yielded energy (ATP) by upregulating the glycolysis, starch, and sucrose synthesis genes to crop with Al toxicity [19] and Pi deficiency [20] and maintain basic respiration.
Although Al toxicity and Pi deficiency exist simultaneously, most previous studies were independent, and few studies have focused on the commonalities and specificities in the effects of these two stresses on the regulation of secondary metabolism in plants. This study integrated transcriptome and metabolome analysis to reveal the specificity and commonality in the toxicity and tolerance mechanisms of flavonoids and carbohydrate metabolism in wheat under Al and LP stresses, providing a new perspective on the molecular network of plants responding to multiple stresses in acid soil and further discovering candidate genes for wheat Al and LP tolerance, providing genetic resources.
2. Results
2.1. Wheat Root Transcriptome Profiling in Response to Al and LP Stresses
Whole genome transcriptome sequencing analysis was applied to investigate the molecular responses of wheat roots under Al and LP stress. A total of 18 cDNA libraries were constructed, combining two genotypes, three treatments, and three biological replicates. These constructed libraries generated an average of 51.20 M clean reads. Quality control data indicated a Phred quality score Q30 ranging from 92% to 93.22%, with an average GC content of 54.39%. All samples exhibited total mapping and unique mapping of 91.41% and 84.08% on average, demonstrating high-quality sequencing suitable for the subsequent gene expression analysis. Finally, 118095 genes (91852 known and 26243 new) and 192506 transcripts (113961 known and 78545 new) were assembled. The functions of 91852 genes and 113961 transcripts were annotated using KEGG, Swiss-Prot, Pfam, GO, COG, and NR databases (Table S2).
In total, 67902, 68158, and 67624 expressed genes were detected in the three treatments (CK, Al, and LP) of Fielder and 66758, 68430, and 67864 in Ardito, respectively. The proportion of low-expressing (0 < FPKM ≤ 1), mid-expressing (1 < FPKM ≤ 10), and high-expressing (FPKM > 10) genes across the three treatments in each genotype were similar. Fielder shows a higher proportion of high-expressing genes than Ardito (Figure 1A). Principal component analysis (PCA) was conducted to identify the major sources of variation among the different treatments showing that the samples within each group were accurately reproducible, and noticeable differences existed between the different varieties and treatment samples (Figure 1B). The correlation heatmap between the samples showed that the transcriptome activities within a genotype were more relevant than identical treatment between the genotypes (Figure S1).
2.2. Identified Differentially Expressed Genes Response to Al and LP Stresses
The differentially expressed genes (DEGs) were categorized into nine groups to compare their expression under Al and LP stresses in wheat root (Figure 2A and Table S3). The comparison of the first six groups reflects the response of genes under Al and LP stresses in two wheat genotypes, and the last three groups reflect the changes in expression between genotypes. Based on the degree and importance of the observed stress impacts, six groups were selected to evaluate further. When subjected to Al and LP stresses in Fielder, the number of DEGs under LP (3029 DEGs, 1591 up and 1438 down) is higher than Al (2296 DEGs,1535 up and 761 down), while in Ardito, the number of DEGs under Al (4404 DEGs, 3191 up and 1213 down) is much more than under LP (1430 DEGs, 1176 up and 254 down). A total of 7358 (3217 up and 4141 down) in Al_Fielder vs. Ardito and 6704 DEGs (3328 up and 3376 down) in LP_Fielder vs. Ardito were expressed. The number of DEGs is higher between varieties than between treatments (Figure 2A). The Venn diagram illustrates how the DEGs interact with each other across various treatments and genotypes (Figure 2B and Table S3). Only 145 and 239 common DEGs under Al and LP were discovered in Fielder and Ardito, respectively. Moreover, 112 and 150 common DEGs among the three comparisons (Al vs. CK, LP vs. CK, and Al vs. LP) in Fielder and Ardito were discovered (Figure 2B). These genes were considered valuable candidate genes contributing to common toxicity or tolerant mechanisms.
2.3. GO Annotations and KEGG Enrichment Analysis of DEGs under Al and LP Stresses
GO annotation and KEGG enrichment analysis were employed to examine the function of these DEGs concerning the treatments and genotypes and to compare the similarities and differences in metabolic pathways. DEGs were divided into 50 functional groups within the biological process (BP), cellular component (CC), and molecular function (MF) were the most prominent terms (Table S4). Binding (GO:0005488, 7124 genes) and catalytic activity (GO:0003824, 5595 genes) in BP; cell part (GO:0044464, 4332 genes) and membrane part (GO:0044425, 3742 genes) in CC; and cellular process (GO:0009987, 4260 genes) and metabolic process (GO:0008152, 4079 genes) in MF were the most commonly associated GO terms with the DEGs in all comparisons (Table S4). The responsive genes enriched in these GO terms differed between Fielder and Ardito under Al and LP. For instance, 622 and 766 DEGs under Al and LP compared to CK in Fielder, and 1420 and 447 DEGs in Ardito were annotated to the metabolic process term, respectively. In addition, 2035 and 1893 DEGs were annotated compared to Fielder and Ardito under Al and LP (Table S4).
Consistent with GO annotations, the KEGG enriched analysis revealed the intricate metabolic pathways in wheat under Al and LP stresses (Figure 3). Some DEGs responsive to Al and LP are enriched in common metabolic pathways. DEGs of both Fielder and Ardito were significantly enriched in the diterpenoid biosynthesis (ko00904) and thiamine metabolism (ko00730) pathways under Al and LP. However, some pathways responded explicitly to one genotype or treatment. For instance, several DEGs were significantly enriched in the benzoxazinoid biosynthesis (ko00402) pathway under Al only. Surprisingly, DEGs of all comparisons were significantly enriched in the phenylpropanoid biosynthesis (ko00940) pathway under Al and LP stresses, within 53 and 94 DEGs under Al and LP compared to CK in Fielder, and 94 and 222 DEGs in Ardito were enriched, respectively. A total of 180 and 169 DEGs were enriched compared to Fielder and Ardito under Al and LP. In addition, several DEGs were enriched in the flavonoid biosynthesis (ko00941), flavone and flavonol biosynthesis (ko00943), and starch and sucrose metabolism (ko00500) pathways under Al and LP in two wheat genotypes. These DEGs involved in the phenylpropanoid and flavonoid biosynthesis and carbohydrate metabolism were studied to analyze the potential common and specific toxicity or tolerant mechanisms to Al toxicity and P deficiency.
2.4. DEGs Related to Transcription Factors
When wheat roots were exposed to Al and LP stress, a significant number of transcription factors (TFs) displayed varying levels of expression. A total of 652 TFs were identified, spanning 29 TF families. The top five most abundant TF families were MYB (136), AP2 (84), WRKY (58), bHLH (56), and NAC (47) (Table S5). Among these TFs, 42 were significantly differentially expressed under Al and LP stresses in the two wheat genotypes, encompassing families such as AP2, B3, bHLH, DBB, E2F/DP, FAR1, HB-other, HSF, MYB, NAC, and SRF. The majority of TFs exhibited the consistent expression trend, with 3 AP2, 3 bHLH, 3 MYB, 1 B3, 1 ANC, 1 HSF, and 1 HB-other were upregulated under Al and LP stresses, and 10 MYB, 5 DBB, 3 HB-other, 2 AP2, 2 NAC, 1 SRF, 1 bHLH, and 1 FAR1 were downregulated in two wheat genotypes. Three TFs showed a different expression pattern with One HB-other (TraesCS3B02G000100) downregulated under Al but upregulated under LP. One AP2 (TraesCS2B02G054000) was downregulated in Fielder under Al but upregulated under LP, while one SRF (TraesCS1D02G203300) revealed an opposite trend (Figure 4).
2.5. Confirmation of RNA-Seq Data Using qRT-PCR
To assess the quality of the RNA-Seq data and confirm the DEGs, the expression levels of 10 genes related to the phenylpropanoid and flavonoid biosynthesis pathway were measured using qRT-PCR. Correlation analysis with the transcriptome data showed that the qRT-PCR results supported the RNA-Seq quantification results (R = 0.8885) (Figure S2). The expression levels of genes encoding 4CL, C4H, CHS, and F3H were upregulated, and one gene encoding CHI was downregulated under Al and LP in Fielder. The expression of CHS, CHI, and FNS were upregulated under LP but downregulated under Al in Fielder. The expression levels of genes encoding PAL, C4H, 4CL, CHI, and FNS were significantly upregulated under Al and LP in Ardito. The gene encoding C4H was downregulated under Al but upregulated under LP (Figure 5). These genes can be used to verify the reliability of transcriptional data further.
2.6. Joint Analysis of Genes and Metabolites Involved in Flavonoid Metabolism
The KEGG enrichment analysis showed that several DEGs were significantly enriched in the phenylpropanoid, flavonoid, and flavone and flavonol biosynthesis pathways (Figure 3). The genes involved in flavonoid biosynthesis differed between Al and LP in the two wheat genotypes. Some genes were significantly upregulated under LP in Fielder, including 4CL, C4H, CHS, F3H, and FNSI, but only one C4H was upregulated under Al. Al and LP upregulated the ANR expression and downregulated the FLS expression in Fielder. Similarly, the genes encoding 4CL, C4H, CHS, HIDH, F3H, and FLS were significantly upregulated under Al in Ardito, but only one FG2 was upregulated under LP. Al and LP upregulated the PAL, CHI, and CHS expression and downregulated the F3’5’H expression in Ardito. In addition, four F3’5’H, three ANR, one ANS, and one HIDH were upregulated, and one FLS and one FG2 were downregulated compared to Fielder and Ardito under Al and LP (Figure 6 and Table S7).
Flavonoid-metabolic targeted metabolomics was conducted to further investigate the flavonoid synthesis mechanism in wheat roots within 19 differential flavonoid metabolites that significantly influenced two wheat roots. Like transcriptome results, the flavonoid metabolites showed a different response under Al and LP in two wheat genotypes. The daidzein, formononetin, rhoifolin, and astragalin content were significantly increased under LP, and the (+)-gallocatechin and glycitein were increased under Al in Fielder. Al and LP increased the (−)-epigallocatechin and rutin contents and decreased the prunin, naringenin, and naringin contents. The naringin and isoliquiritigenin contents were increased under Al but decreased under LP. The daidzein and formononetin content were decreased under LP but not significantly under Al, and the apigenin and naringenin contents showed an opposite trend. Al and LP increased the vitexin and isovitexin contents and decreased the glycitein, prunin, dihydrokaempferol, (−)-epigallocatechin, (+)-gallocatechin contents in Ardito. It was found that the content of the 8 and 9 differential flavonoid metabolites was increased in Fielder than Ardito under Al and LP, respectively. The prunin, naringin, and dihydrokaempferol were increased compared to Fielder and Ardito under Al and LP. In addition, the rutin was detected in Fielder, and the vitexin was detected in Ardito only (Figure 6 and Table S8).
The changes in the expression levels of these genes were consistent with the changes in the contents of related metabolites regulated by the enzymes encoded by the genes that can be used to verify the reliability of transcriptional data. For instance, the genes encoding PAL, 4CL, C4H, CHI, CHS, and FNSI related to the general phenylpropanoid and flavonoid biosynthesis pathway were significantly upregulated with the vitexin and isovitexin content increased in Ardito but not significantly in Fielder. The astragalin and rutin content significantly increased in Fielder but not was detected in Ardito, which is consistent with the elevated expression of its upstream genes encoding F3GT, FG2, and F3’5’H, which were upregulated in Fielder but downregulated in Ardito, although they were not significant. One ANR was significantly upregulated in Fielder, while another was downregulated in Ardito, so the (−)-epigallocatechin and (+)-gallocatechin contents showed the same trend in two wheat genotypes (Figure 6).
2.7. Joint Analysis of Carbohydrate Metabolism Genes and Metabolites
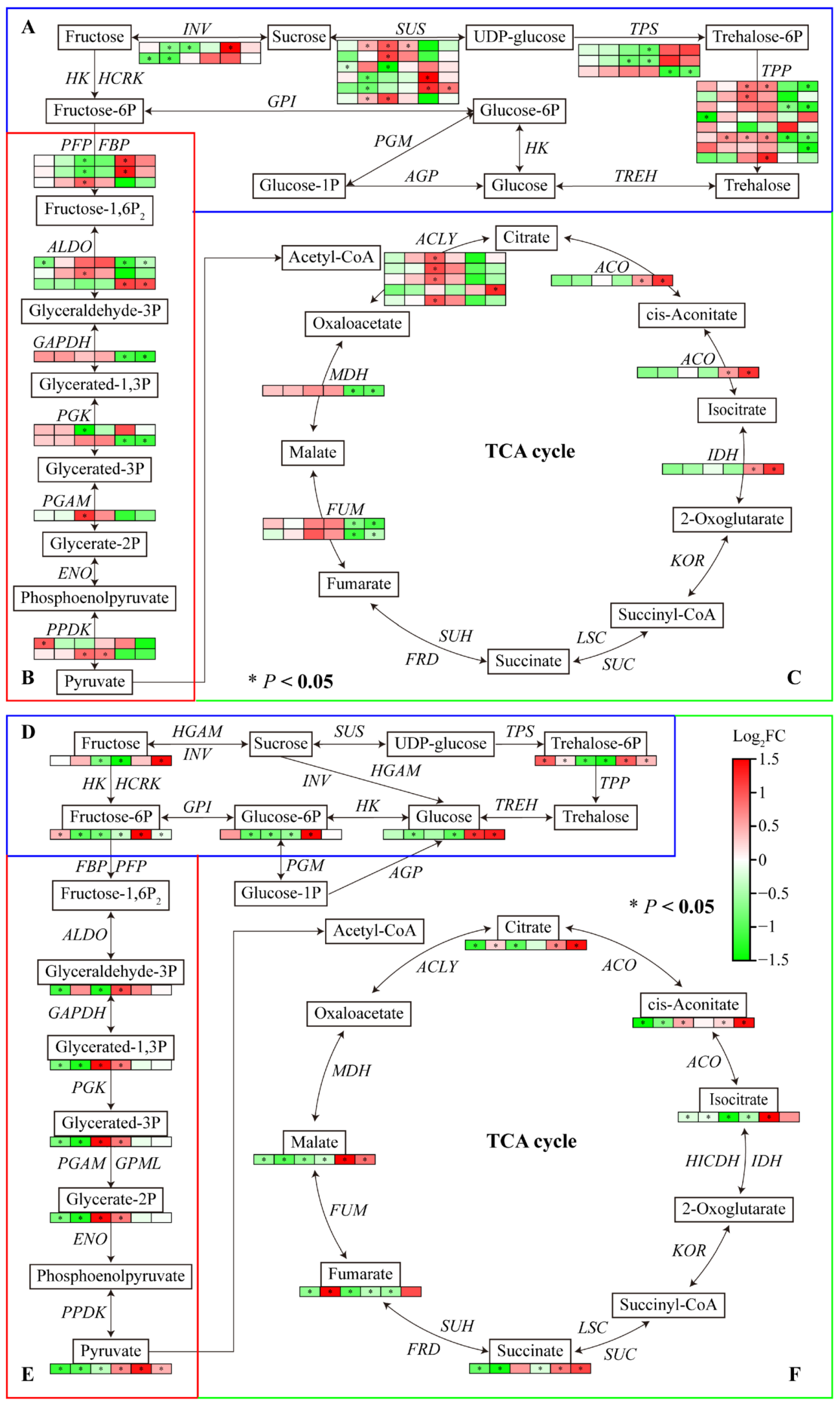
The transcriptome data showed that several DEGs were enriched in starch and sucrose metabolism, glycolysis/gluconeogenesis, and the TCA cycle pathway. Most genes involved in the carbohydrate metabolism were downregulated under Al and LP. One INV under Al and LP, one SUS, TPP, and ALDO under Al, and one INV and two SUS under LP were significantly downregulated in Fielder. One PPDK under Al, two SUS and one TPP under LP were upregulated. One INV, one SUS, two PFP, and one PGK under Al, and one TPS under LP were downregulated. Al and LP upregulated one SUS and two TPP, and downregulated one TPS expression in Ardito. In addition, the genes encoding SUS, ALDO, ACO, and IDH were upregulated and the genes encoding TPP, ALDO, GAPDH, OGK, MDH, and FUM were downregulated compared to Fielder and Ardito.
Carbohydrate-metabolic targeted metabolomics was conducted further to investigate the carbohydrate synthesis mechanism in wheat roots. A total of 15 differential carbohydrate metabolites were identified (Table S7 and Table S9). In consistent with transcriptome results, most carbohydrate metabolites were downregulated under Al and LP compared to CK but showed an upregulated compared to Fielder and Ardito. For instance, the glucose, glucose-6P, fructose, fructose-6P, trehalose-6P, pyruvate, citrate, cis-aconitate, isocitrate, and succinate contents were higher in Fielder than in Ardito. A few metabolites were upregulated within the trehalose-6P content under Al and LP, the fructose-6-P content under Al, and the citrate and fumarate contents under LP in Fielder. The glyceric acid contents under Al and LP, cis-aconitate under Al, and glyceraldehyde under LP were increased in Ardito.
Most of the changes in the expression levels of these genes were in consistent with the changes in the contents of related metabolites regulated by the enzymes encoded by the genes (Figure 7). For instance, the genes encoding INV, SUS, PFP, ALDO, ACLY, ACO, and IDH were significantly upregulated, and the glucose, glucose-6P, fructose, fructose-6P, trehalose-6P, pyruvate, citrate, cis-aconitate, isocitrate, and succinate contents were higher in Fielder than Ardito. Two TPS were downregulated, and the trehalose-6P significantly decreased in Ardito. One ALDO and one PGAM were upregulated in Ardito and one ALDO was downregulated in Fielder which caused the glyceric acid content to decrease in Fielder and increase in Ardito.
3. Discussion
Due to the coexistence of Al toxicity and P deficiency in acid soils, researchers have been motivated to study the interaction between Al toxicity and P deficiency in plant adaptations to acid soils [7]. Previous studies have consistently reported a positive correlation between P efficiency and Al tolerance in various plant species. For example, Al-tolerant buckwheat plants exhibit high P contents or efficient P acquisition and translocation to shoots [21]. Correspondingly, P-efficient genotypes tend to display greater Al tolerance than P-inefficient lines in Stylosanthes [22]. Understanding how plants adapt to multiple simultaneous presence of limiting factors in acid soils is vital for elucidating their survival under such conditions. However, the commonalities and specificities in synthesizing secondary metabolites in roots under Al and LP stress still need to be clarified. This study used two wheat lines, Fielder (Al-tolerant and P-efficient) and Ardito (Al-intolerant and P-inefficient), revealing their flavonoids and carbohydrates profiles and investigating the synthesis mechanisms in two genotypes by integrating transcriptomic and metabolite analyses.
Secondary metabolites, such as phenylpropanoids and flavonoids, play a crucial role in plant growth and development [15]. Flavonoids are involved in plant resistance to Al by forming Al-chelating complexes or scavenging free radicals through their Al-binding affinity [17]. Flavonoids facilitate P solubilization and cooperate with beneficial microorganisms in the rhizosphere, contributing to P acquisition and utilization [16,23]. However, the commonalities and differences in the content of flavonoids and the molecular mechanisms of flavonoid biosynthesis in wheat roots under Al and LP are still unclear. The KEGG enrichment analysis results showed that several DEGs were enriched in the phenylpropanoid, flavonoid, and flavone and flavonol biosynthesis pathways under Al and LP stresses in the two wheat genotypes (Figure 3). However, the response of secondary metabolism-related genes in Al and LP differs. For instance, the genes encoding 4CL, CHS, F3H, and FNSI were upregulated under LP but not significantly under Al in Fielder. The genes encoding 4CL, CHS, F3H, FLS, and HIDH were significantly upregulated, and the genes encoding ANR and F3GT were downregulated under Al but not significantly under LP in Ardito. The flavonoid-targeted metabolomics results showed that 19 differential flavonoid metabolites significantly influenced two wheat roots (Table S8). The (−)-epigallocatechin and rutin contents were increased under Al and LP; the (+)-gallocatechin, astragalin, rhoifolin, daidzein, and formononetin contents were increased under LP but not significantly under Al in Fielder. The vitexin and isovitexin contents were increased under Al and LP; the naringin and isoliquiritigenin contents were increased under Al but not significantly under LP in Ardito. Due to the inability to balance the absorption of Al and P by altering the root structure, plants have developed internal and external mechanisms to resist Al toxicity and P deficiency [7]. Like organic acids, flavonoids participate in internal and external Al detoxification by forming solid complexes with toxic Al ions [17,24]. The accumulation of flavonoids in roots can alter the structure of plant roots (internal mechanism) and release them to dissolve soil P (external mechanism), thereby obtaining more Pi [25,26]. Transcriptome and metabolomics consistently indicate that flavonoid metabolism in roots significantly impacted in both wheat genotypes, but the difference between the Al and LP stress. The flavonoid genes and metabolites respond more to LP than Al in Fielder, suggesting that the increased synthesis of flavonoids in roots mainly contributes to secretion outside of the wheat roots (external mechanism), which facilitates the acquisition of Pi and can also bind to Al outside the roots to prevent it from entering root cells. While, in Ardito, the increased synthesis of flavonoids in the roots may play a more significant role within the roots (internal mechanism), as they respond more actively to Al stress and bind with Al entering the cells, leading to a decrease in P absorption. These results suggest that the difference in response of flavonoids in Al and LP is the reason for the difference in tolerance between the two wheat genotypes. In addition, the content of some flavonoids, including (−)-epigallocatechin, rutin, vitexin, and isovitexin increases under both Al and LP stress in two wheat genotypes, play a joint role in clearing ROS caused by Al and LP stress [7,27].
Carbohydrate metabolism comprises sugar, glycolysis, TCA cycle, and organic acids metabolism. Sucrose is the end product of photosynthesis and is responsible for energy metabolism and the synthesis of complex carbohydrates. ATP in plant cells is mainly generated by the TCA cycle in the mitochondria [18,28]. The transcriptome data showed that several DEGs were enriched in starch and sucrose metabolism, glycolysis/gluconeogenesis, and the TCA cycle pathways (Figure 4). Al and LP reduced the carbohydrate genes and metabolites in two wheat genotypes but showed an upregulation compared to Fielder and Ardito (Figure 7). Al-toxicity reduced energy (ATP) production and accumulation in rice roots, and nonstructural carbohydrates can directly provide energy for plant growth [29]. Pi-deficiency increases carbohydrate translocation via the phloem to roots to favor root growth for better acquisition of Pi from soil [30]. In the previous study, the root and shoot glucose, fructose, sucrose, and starch contents in P-tolerant genotype cotton Jimain169 were increased than P-intolerant genotype one DES926 [28]. The P-efficient genotype had a more remarkable ability to maintain phosphorylated sugars (i.e., glucose-6-P and fructose-6P) by upregulating the genes involved in glycolysis, starch, and sucrose synthesis that are important for glycolysis as well as the biosynthesis of sugars and starch in rice and cotton [20,31]. The Al-tolerant rice cultivar [19] and Citrus species [32,33] could provide more glycolytically yielded energy (ATP) by enhancing the glycolytic pathway in Al-stressed roots. The genes encoding INV, SUS, PFP, ALDO, ACLY, ACO, and IDH were significantly upregulated compared to Fielder and Ardito under Al and LP. Consistent with the transcriptome results, the carbohydrate-targeted metabolomics results showed that the glucose, glucose-6P, fructose, fructose-6P, trehalose-6P, pyruvate, citrate, cis-aconitate, isocitrate, and succinate contents were significantly higher in Fielder than in Ardito (Figure 7). The accumulation of the carbohydrate metabolites in the roots showed that the injury level under Al and LP in Fielder was lower than in Ardito. The transcriptome and metabolome consistently suggested that Al and LP stress significantly impact on the carbohydrate metabolism of roots. The difference between the genotypes suggested that carbohydrate metabolism is critical to improving the tolerance against Al toxicity and P deficiency.
Transcription factors (TFs) proteins play crucial roles in regulatory and signaling networks to respond to Al and LP conditions [7]. AtSTOP1 and OsART1 are two related members of the Cys2His2-type zinc-finger (C2H2) protein family TF that confer Al resistance functions in Arabidopsis and rice [34]. Phosphate starvation response (PHR) transcription factors, such as AtPHR1 in Arabidopsis and OsPHR2 in rice, serve as central regulators of systemic Pi signaling. AtPHR1/OsPHR2 activates the expression of a large set of low-Pi responsive genes by binding to PHR1 binding site (P1BS) elements [3]. TFs may offer the best option for pleiotropic control of multiple abiotic stress genes due to their small and often multiple binding sequences in the genome. TFs such as C2H2, MYB, WRKY, ERFs, NAC, and bHLH may be critical determinants for a plant’s ability to tolerate Al toxicity and P deficiency and withstand drought conditions in acid soil [14]. Promising connections may also exist between plant adaptation to Al toxicity and P deficiency through the regulation of TFs [14]. The transcriptome results showed that 42 TFs, including MYB, bHLH, NAC, and AP2/ERF, were differentially expressed simultaneously under Al and LP stresses. Importantly, their expression changes after Al and LP treatments are similar, indicating that these TFs were associated with response to Al toxicity and P deficiency. Several TFs have been identified as hub genes involved in flavonoid, carbohydrate, and P metabolism [28]. MsMYB741 transcriptionally activates MsPAL1 and MsCHI expression to increase flavonoid accumulation in roots and secretion from root tips, leading to increased resistance of alfalfa to Al stress [17]. MhMYB15 actively responds to Bacillus B2, regulating the accumulation of flavonoids and phosphorus uptake, thereby influencing plant growth and development [35]. MYB-related transcription factor PHR1 is involved in carbohydrate metabolism. The knockout mutant has an altered phosphate (Pi) allocation between root and shoot and accumulates less anthocyanins, sugars, and starch than P-starved WT [36]. Thirteen common MYB TFs were identified, suggesting that MYB TFs may be involved in flavonoid and carbohydrate metabolism; regulating targeting these TFs is a valuable approach for molecular breeding of plants towards regulating tolerance to Al and LP.
4. Materials and Methods
4.1. Plant Materials and Treatment
Based on the morphological performance in preliminary experiments, two wheat genotypes, Fielder (Al-tolerant and P-efficient) and Ardito (Al-sensitive and P-inefficient) were used in this study. Healthy seeds were surface-sterilized in a solution of 0.5% NaClO (v/v) for 30 minutes and then rinsed thoroughly with deionized water. These sterilized seeds were then placed on wet paper in a petri dish for 4 days at 4°C for germinating. Uniform seedlings were selected and transferred to a 10 L barrel with continuously aerated 1/5 Hoagland nutrient solution (pH 4.2) for a 2-day acclimation to low pH. Subsequently, the seedlings were subjected to stress by replacing fresh 1/5 Hoagland nutrient solution with an addition of 50 µM KAl(SO4)2 (for Al stress), a reduced NaH2PO4 (2 µM, for LP stress), and normal Pi levels (200 µM, for control). The growth chamber environments were 14/10 h of light/dark period, 60-80% relative humidity, 23°C, and 400 μmol m−2·s−1 illumination level. Each treatment was replicated three times (3 barrels), and the pH of the solution was maintained at 4.2 by adding 0.5 mM HCl. After 4 d of treatments, about 1 cm in length wheat root apices were harvested separately in triple biological repeats, immersed immediately in liquid nitrogen, and stored at −80°C until transcriptomic and metabolomic analyses were conducted. All chemicals used were of analytical grade and were purchased from Sigma-Aldrich (St. Louis, MO, USA).
4.2. RNA Extraction, Library Preparation, Sequencing, and Read Mapping
The total RNA was separately extracted from the collected 1-cm root apices using TRIzol reagent (Invitrogen Co., Carlsbad, CA, USA) following the manufacturer’s instructions. Subsequently, the Plant RNA Purification Reagent (Invitrogen) was employed to purify the RNA samples. The quality, quantification, and integrity were determined using a 5300 Bioanalyser (Agilent Co., Santa Clara, CA, USA) and ND-2000 (Thermo Fisher NanoDrop, Waltham, MA, USA), respectively. RNA samples meeting the integrity number values above 7.5, the total amount of ≥ 1 ug, and 28S: 18S ≥ 1.0 were utilized to construct the transcriptome libraries for sequencing, following the manufacturer’s protocols (Illumina, San Diego, CA, USA) at Majorbio Bio-pharm Biotechnology Co., Ltd. (Shanghai, China). In brief, mRNA was isolated from the total RNA by the polyA selection method using oligo (dT) beads, followed by fragmentation into short fragments. Subsequently, cDNA was synthesized using a Superscript double-stranded cDNA synthesis kit (Invitrogen) with random hexamer primers. The synthesized cDNA was subjected to decoration according to Illumina’s library construction protocol. The cDNA target fragments of 300 bp were PCR amplified using Phusion DNA polymerase (NEB, Ipswich, MA, USA). After quantification with Qubit 4.0, the paired-end RNA-seq library was sequenced using the NovaSeq 6000 sequencer with a 2 × 150 bp read length.
Finally, 18 RNA-seq transcriptome libraries (2 genotypes × 3 treatments × 3 biological replicates) were constructed and sequenced. The raw paired-end reads were trimmed and quality controlled by FASTP [37] with default parameters, resulting in high-quality sequencing data (clean data). Then, the clean reads were separately aligned to the wheat genome with orientation mode using HISAT2 software Ver. 2.2.1 [38]. The mapped reads of each sample were assembled by StringTie Ver. 1.3.6 [39] in a reference-based approach for subsequent transcript assembly and expression calculation. The assembled genes were annotated against public databases of NCBI non-redundant protein sequences (NR), Protein family (Pfam), Clusters of Orthologous Groups of Proteins (COG), Swiss-Prot, Kyoto Encyclopedia of Genes and Genomes (KEGG) and Gene Ontology (GO) libraries. The RAN-seq data were uploaded to the Comprehensive Gene Expression Database with the accession number PRJNA1033153.
4.3. Differential Expression and Functional Enrichment Analysis
The expression levels of each transcript were calculated using the Fragments Per Kilobase per Million reads (FPKM) method to identify differential expression genes (DEGs) between two different samples. The gene abundances were quantified using RSEM software [40]. DESeq2 was used to identify DEGs based on the criteria of |log2FC| ≥ 1 and FDR ≤ 0.05 [41]. Cluster heat diagrams were generated using Toolkit for Biologists (TB) tools Ver. 2.042 with default settings [42]. The Plant TFDB database was employed for transcription factors (TFs) prediction and analysis, while TFs were identified by BLAST and then subjected to functional annotation and enrichment analysis. The GO annotation information and functional classification of DEGs were performed using Blast2GO based on the GO database [43]. KOBAS 2.0 was utilized to determine the KEGG enrichment pathways at Bonferroni-corrected P-value ≤ 0.05, which were compared with the whole transcriptome background [44].
4.4. Quantitative Real-Time PCR (qRT-PCR) Validation
Ten differentially expressed genes involved in phenylpropanoid and flavonoid biosynthetic pathways were selected from the RNA-seq data to validate the transcriptome results. The identical RNA/cDNAs for RNA-seq were used as templates to determine their transcript levels via the qRT-PCR method. The first-strand cDNA was synthesized using the PrimeScript™ RT Master Mix (Takara Standard Co., Osaka, Japan). The gene-specific primers were designed by Primer3 web v4.1.0 (https://primer3.ut.ee/) and listed in Table S1. A total of 10 μL PCR reaction mixture contained TB Green®Premix Ex Taq™ II (Takara Standard Co., Osaka, Japan) 5 μL, 10 μM primers each 0.4 μL, cDNA template 1 μL, and ddH2O 3.2 μL. The mixture was amplified using the LightCycler 480 machine (Roche, Basel, Switzerland) with a two-step thermal cycling method (95°C for 30 s, followed by 40 cycles of 95°C for 5 s and 60°C for 30 s). TaActin (accession number: XM_044554036.1) served as an internal control for calculating relative gene transcript levels using the 2−ΔΔCT method [45]. Four technical replicates were performed for qRT-PCR, and a representative result from at least three biological replicates was shown for each gene.
4.5. Targeted Metabolite Extraction and Profiling
The frozen wheat root apices were dispatched to Shanghai Majorbio Biopharmaceutical Biotechnology Co., Ltd. (Shanghai, China) for targeted metabolite extraction and analysis. Precision weighing was conducted on 0.1 g root tip samples and metabolite extraction was performed in a low-temperature environment. The supernatant was filtered through a 0.2 μm membrane and then transferred to the injection vial for analysis. Flavonoids and carbohydrates standard solutions were prepared with concentration gradients. LC-ESI-MS/MS (UHPLC-Qtrap) was used to qualitatively and quantitatively detect the target substances in the sample. The chromatographic apparatus was the ExionLCTM CAD system (AB Sciex, Los Angeles, CA, USA). The Mass spectrometry was the SCIEX QTRAP 6500+ (AB Sciex, Los Angeles, CA, USA).
The specific conditions and parameters for flavonoid determination were as follows: Agilent Poroshell 120EC-C18 (3 × 100 mm, 1.9 μm, Agilent Co., Santa Clara, CA, USA) liquid chromatography column; column temperature, 40℃; injection volume, 1 μL; Mobile phase A, 0.1% formic acid water; mobile phase B, 0.1% formic acid methanol; Negative mode detection; Curtain Gas, 35 psi; Collision Gas, Medium; IonSpray Voltage, −4500 V; Temperature, 350°C; Ion Source Gas1, 55 psi; Ion Source Gas2, 55 psi.
The specific conditions and parameters for carbohydrates determination were as follows: Waters HSS T3 (2.1 × 150 mm, 1.8 μm, Waters, Milford, MA, USA) liquid chromatography column; column temperature, 40℃, injection volume, 2 μL. Mobile phase A, 0.03% formic water; mobile phase B, 0.03% formic methanol; Positive/negative mode detection; Curtain Gas, 35 psi; Collision Gas, Medium; IonSpray Voltage, +4500/−4500 V; Temperature, 550°C; Ion Source Gas1, 55 psi; Ion Source Gas2, 55 psi.
4.6. Statistical Analysis
All data were subjected to statistical analysis using SPSS 20.0. Analysis of variance was performed on the datasets. Data are presented as the mean ± standard deviation (SD) of three independent biological experiments. The significant differences between treatments and control were determined with one-way ANOVA followed by the Dunnett post hoc test compared to the control.
5. Conclusions
This study revealed the commonalities and specificities in the expression of genes and metabolites related to flavonoid and carbohydrate metabolism between two wheat genotypes, Fielder (Al-tolerant and P-efficient) and Ardito (Al-intolerant and P-inefficient), by integrating transcriptome and metabolome under Al and LP stress. More responsive genes and metabolites were involved in flavonoid metabolism under LP than Al in the Fielder, while an opposite trend in the Ardito. Al and LP reduced the carbohydrate genes and metabolites in two wheat genotypes while showing an upregulation compared to Fielder and Ardito. The upregulation of these genes and metabolites is one reason for the high Al and LP resistance of Fielder. Numerous genes related to TFs, such as MYB, bHLH, NAC, and AP2/ERF families, were activated to tolerate Al and LP stresses. The present study provides novel insights into elucidating molecular mechanisms of wheat Al and LP tolerance.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
Conceptualization, D.D. and Y.X.; investigation, D.L.; formal analysis, Q.L. and F.P.; validation, W.Z.; writing—original draft preparation, D.L.; writing—review and editing, D.D.; supervision, Y.L.; funding acquisition, D.D. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Guangxi Natural Science Foundation, grant number 2023GXNSFAA026445, and the National Natural Science Foundation of China, grant number 31960633.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The original contributions presented in this study are available in the article or Supplementary Materials. The RNA-seq raw data can be found on the NCBI repository, accession number PRJNA1033153.
Acknowledgments
We are highly grateful to Liu Jiping form the Robert W. Holley Center for providing the wheat seeds.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Kochian, L.V.; Pineros, M.A.; Liu, J.; Magalhaes, J.V. , Plant adaptation to acid soils: The molecular basis for crop aluminum resistance. Annu. Rev. Plant Biol. Merchant, S.S., Ed. 2015; Vol. 66, pp 571-598. [CrossRef]
- Liao, H.; Wan, H.; Shaff, J.; Wang, X.; Yan, X.; Kochian, L.V. , Phosphorus and aluminum interactions in soybean in relation to aluminum tolerance, exudation of specific organic acids from different regions of the intact root system. Plant Physiol. 2006, 141, 674–684. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Ai, S.; Liao, H. , Deciphering interactions between phosphorus status and toxic metal exposure in plants and rhizospheres to improve crops reared on acid soil. Cells 2023, 12. [Google Scholar] [CrossRef] [PubMed]
- Lambers, H.; Finnegan, P.M.; Laliberte, E.; Pearse, S.J.; Ryan, M.H.; Shane, M.W.; Veneklaas, E.J. , Phosphorus nutrition of proteaceae in severely phosphorus-impoverished soils: Are there lessons to be learned for future crops? Plant Physiol. 2011, 156, 1058–1066. [Google Scholar] [CrossRef] [PubMed]
- Kochian, L.V.; Hoekenga, O.A.; Pineros, M.A. , How do crop plants tolerate acid soils? - Mechanisms of aluminum tolerance and phosphorous efficiency. Annu. Rev. Plant Biol. 2004, 55, 459–493. [Google Scholar] [CrossRef] [PubMed]
- Che, J.; Zhao, X.Q.; Shen, R.F. , Molecular mechanisms of plant adaptation to acid soils: A review. Pedosphere 2023, 33, 14–22. [Google Scholar] [CrossRef]
- Chen, W.; Tang, L.; Wang, J.; Zhu, H.; Jin, J.; Yang, J.; Fan, W. , Research advances in the mutual mechanisms regulating response of plant roots to phosphate deficiency and aluminum toxicity. Int. J. Mol. Sci. 2022, 23. [Google Scholar] [CrossRef] [PubMed]
- Iuchi, S.; Koyama, H.; Iuchi, A.; Kobayashi, Y.; Kitabayashi, S.; Kobayashi, Y.; Ikka, T.; Hirayama, T.; Shinozaki, K.; Kobayashi, M. , Zinc finger protein STOP1 is critical for proton tolerance in Arabidopsis and coregulates a key gene in aluminum tolerance. PNAS. 2007, 104, 9900–9905. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Deng, S.; Zhang, C.; Yang, X.; Shi, L.; Xu, F.; Wang, S.; Wang, C. , Brassinosteroid signaling regulates phosphate starvation-induced malate secretion in plants. J. Integr. Plant Biol. 2023, 65, 1099–1112. [Google Scholar] [CrossRef]
- Dong, J.; Pineros, M.A.; Li, X.; Yang, H.; Liu, Y.; Murphy, A.S.; Kochian, L.V.; Liu, D. , An Arabidopsis ABC transporter mediates phosphate deficiency-induced remodeling of root architecture by modulating iron homeostasis in roots. Mol. Plant 2017, 10, 244–259. [Google Scholar] [CrossRef]
- Godon, C.; Mercier, C.; Wang, X.; David, P.; Richaud, P.; Nussaume, L.; Liu, D.; Desnos, T. , Under phosphate starvation conditions, Fe and Al trigger accumulation of the transcription factor STOP1 in the nucleus of Arabidopsis root cells. Plant J. 2019, 99, 937–949. [Google Scholar] [CrossRef]
- Hufnagel, B.; de Sousa, S.M.; Assis, L.; Guimaraes, C.T.; Leiser, W.; Azevedo, G.C.; Negri, B.; Larson, B.G.; Shaff, J.E.; Pastina, M.M.; Barros, B.A.; Weltzien, E.; Frederick, H.; Rattunde, W.; Viana, J.H.; Clark, R.T.; Falcao, A.; Gazaffi, R.; Garcia, A.A.F.; Schaffert, R.E.; Kochian, L.V.; Magalhaes, J.V. , Duplicate and conquer: Multiple homologs of PHOSPHORUS-STARVATION TOLERANCE1 enhance phosphorus acquisition and sorghum performance on low-phosphorus soils. Plant Physiol. 2014, 166, 659–U323. [Google Scholar] [CrossRef] [PubMed]
- Lou, H.Q.; Fan, W.; Jin, J.F.; Xu, J.M.; Chen, W.W.; Yang, J.L.; Zheng, S.J. , A NAC-type transcription factor confers aluminium resistance by regulating cell wall-associated receptor kinase 1 and cell wall pectin. Plant, Cell Environ. 2019, 43, 463–478. [Google Scholar] [CrossRef] [PubMed]
- Barros, V.A.; Chandnani, R.; de Sousa, S.M.; Maciel, L.S.; Tokizawa, M.; Guimaraes, C.T.; Magalhaes, J.V.; Kochian, L.V. , Root adaptation via common genetic factors conditioning tolerance to multiple stresses for crops cultivated on acidic tropical soils. Front. Plant Sci. 3389. [Google Scholar]
- Liu, W.; Feng, Y.; Yu, S.; Fan, Z.; Li, X.; Li, J.; Yin, H. , The flavonoid biosynthesis network in plants. Int. J. Mol. Sci. 2021, 22. [Google Scholar] [CrossRef] [PubMed]
- Tomasi, N.; Weisskopf, L.; Renella, G.; Landi, L.; Pinton, R.; Varanini, Z.; Nannipieri, P.; Torrent, J.; Martinoia, E.; Cesco, S. , Flavonoids of white lupin roots participate in phosphorus mobilization from soil. Soil Biol. Biochem. 2008, 40, 1971–1974. [Google Scholar] [CrossRef]
- Su, L.; Lv, A.; Wen, W.; Fan, N.; Li, J.; Gao, L.; Zhou, P.; An, Y. , MsMYB741 is involved in alfalfa resistance to aluminum stress by regulating flavonoid biosynthesis. Plant J. 2022, 112, 756–771. [Google Scholar] [CrossRef] [PubMed]
- Halford, N.G.; Curtis, T.Y.; Muttucumaru, N.; Postles, J.; Mottram, D.S. , Sugars in crop plants. Ann. Appl. Biol. 2011, 158, 1–25. [Google Scholar] [CrossRef]
- Wang, Z.Q.; Xu, X.Y.; Gong, Q.Q.; Xie, C.; Fan, W.; Yang, J.L.; Lin, Q.S.; Zheng, S.J. , Root proteome of rice studied by iTRAQ provides integrated insight into aluminum stress tolerance mechanisms in plants. J. Proteomics 2014, 98, 189–205. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, V.L.; Palmer, L.; Stangoulis, J. , Higher photochemical quenching and better maintenance of carbon dioxide fixation are key traits for phosphorus use efficiency in the wheat breeding line, RAC875. Front. Plant Sci. 2021, 12, 816211–10. [Google Scholar] [CrossRef]
- Jemo, M.; Abaidoo, C.; Nolte, C.; Horst, W.J. , Aluminum resistance of cowpea as affected by phosphorus-deficiency stress. J. Plant Physiol. 2007, 164, 442–451. [Google Scholar] [CrossRef]
- Sun, C.; Lu, L.; Liu, L.; Liu, W.; Yu, Y.; Liu, X.; Hu, Y.; Jin, C.; Lin, X. , Nitrate reductase-mediated early nitric oxide burst alleviates oxidative damage induced by aluminum through enhancement of antioxidant defenses in roots of wheat (Triticum aestivum). New Phytol. 2014, 201, 1240–1250. [Google Scholar] [CrossRef]
- Luo, J.; Liu, Y.; Zhang, H.; Wang, J.; Chen, Z.; Luo, L.; Liu, G.; Liu, P. , Metabolic alterations provide insights into Stylosanthes roots responding to phosphorus deficiency. BMC Plant Biol. 2020, 20. [Google Scholar] [CrossRef] [PubMed]
- Kidd, P.S.; Llugany, M.; Poschenrieder, C.; Gunsé, B.; Barceló, J. , The role of root exudates in aluminium resistance and silicon-induced amelioration of aluminium toxicity in three varieties of maize (Zea mays L.). J. Exp. Bot. 2001, 52, 1339–1352. [Google Scholar]
- Zhou, Y.; Olt, P.; Neuhauser, B.; Moradtalab, N.; Bautista, W.; Uhde-Stone, C.; Neumann, G.; Ludewig, U. , Loss of LaMATE impairs isoflavonoid release from cluster roots of phosphorus-deficient white lupin. Physiol. Plant. 2021, 173, 1207–1220. [Google Scholar] [CrossRef] [PubMed]
- Grunewald, W.; De Smet, I.; Lewis, D.R.; Loefke, C.; Jansen, L.; Goeminne, G.; Bossche, R.V.; Karimi, M.; De Rybel, B.; Vanholme, B.; Teichmann, T.; Boerjan, W.; Van Montagu, M.C.E.; Gheysen, G.; Muday, G.K.; Friml, J.; Beeckman, T. , Transcription factor WRKY23 assists auxin distribution patterns during Arabidopsis root development through local control on flavonol biosynthesis. PNAS. 2012, 109, 1554–1559. [Google Scholar] [CrossRef] [PubMed]
- You, Y.; Wang, L.; Ju, C.; Wang, X.; Wang, Y. , How does phosphorus influence Cd tolerance strategy in arbuscular mycorrhizal-Phragmites australis symbiotic system? J. Hazard. Mater. 2023, 452. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, A.; Qiang, D.; Wang, X.; Gui, H.; Zhang, H.; Zhang, X.; Song, M. , Integrative physiological, transcriptome and metabolome analysis reveals the involvement of carbon and flavonoid biosynthesis in low phosphorus tolerance in cotton. Plant Physiol. Biochem. 2023, 196, 302–317. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.Q.; Wei, Q.; Hu, W.J.; Li Kong, Y.; Xiang, X.J.; Zhang, H.; Cao, X.C.; Zhu, L.F.; Liu, J.; Tian, W.H.; Jin, Q.Y.; Zhang, J.H. , Unearthing the alleviatory mechanisms of hydrogen sulfide in aluminum toxicity in rice. Plant Physiol. Biochem. 2022, 182, 133–144. [Google Scholar] [CrossRef]
- Iqbal, A.; Qiang, D.; Wang, X.; Gui, H.; Zhang, H.; Zhang, X.; Song, M. , Phosphorus and carbohydrate metabolism contributes to low phosphorus tolerance in cotton. BMC Plant Biol. 2023, 23. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S. ; Pallavi; Chugh, C.; Seem, K.; Kumar, S.; Vinod, K.K.; Mohapatra, T., Characterization of contrasting rice (Oryza sativa L.) genotypes reveals the Pi-efficient schema for phosphate starvation tolerance. BMC Plant Biol. [CrossRef]
- Guo, P.; Qi, Y.P.; Yang, L.T.; Lai, N.W.; Ye, X.; Yang, Y.; Chen, L.S. , Root adaptive responses to aluminum-treatment revealed by RNA-seq in two citrus species with different aluminum-tolerance. Front. Plant Sci. 2017, 8, 330–10. [Google Scholar] [CrossRef]
- Jiang, H.-X.; Yang, L.-T.; Qi, Y.-P.; Lu, Y.-B.; Huang, Z.-R.; Chen, L.-S. , Root iTRAQ protein profile analysis of two Citrus species differing in aluminum-tolerance in response to long-term aluminum-toxicity. BMC Genomics 2015, 16. [Google Scholar] [CrossRef]
- Liu, J.; Pineros, M.A.; Kochian, L.V. , The role of aluminum sensing and signaling in plant aluminum resistance. J. Integr. Plant Biol. 2014, 56, 221–230. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Zhai, L.; Chai, X.; Liu, Y.; Lv, J.; Pi, Y.; Gao, B.; Wang, X.; Wu, T.; Zhang, X.; Han, Z.; Wang, Y. , Bacillus B2 promotes root growth and enhances phosphorus absorption in apple rootstocks by affecting MhMYB15. Plant J. 2024. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, L.; Mueller, R.; Nielsen, T.H. , Increased expression of the MYB-related transcription factor, PHR1, leads to enhanced phosphate uptake in Arabidopsis thaliana. Plant Cell Environ. 2007, 30, 1499–1512. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. , Fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, 884–890. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Langmead, B.; Salzberg, S.L. , HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–U121. [Google Scholar] [CrossRef] [PubMed]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.; Mendell, J.T.; Salzberg, S.L. , StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–+. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Dewey, C.N. , RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 2011, 12. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. , Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15. [Google Scholar] [CrossRef]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. , TBtools: An integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef]
- Ye, J.; Zhang, Y.; Cui, H.; Liu, J.; Wu, Y.; Cheng, Y.; Xu, H.; Huang, X.; Li, S.; Zhou, A.; Zhang, X.; Bolund, L.; Chen, Q.; Wang, J.; Yang, H.; Fang, L.; Shi, C. , WEGO 2.0: a web tool for analyzing and plotting GO annotations, 2018 update. Nucleic Acids Res. 2018, (W1), W71–W75. [Google Scholar] [CrossRef]
- Xie, C.; Mao, X.; Huang, J.; Ding, Y.; Wu, J.; Dong, S.; Kong, L.; Gao, G.; Li, C.; Wei, L. , KOBAS 2.0: a web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011, 39, W316–W322. [Google Scholar] [CrossRef] [PubMed]
- Livak. , K.J.; Schmittgen., T.D., Analysis of relative gene expression data using realtime quantitative PCR and the 2-ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Gene expression profiling in each sample. (A) Numbers of detected genes in each sample. (B) Principal component analysis (PCA) of the RNA-Seq data of each sample.
Figure 1.
Gene expression profiling in each sample. (A) Numbers of detected genes in each sample. (B) Principal component analysis (PCA) of the RNA-Seq data of each sample.
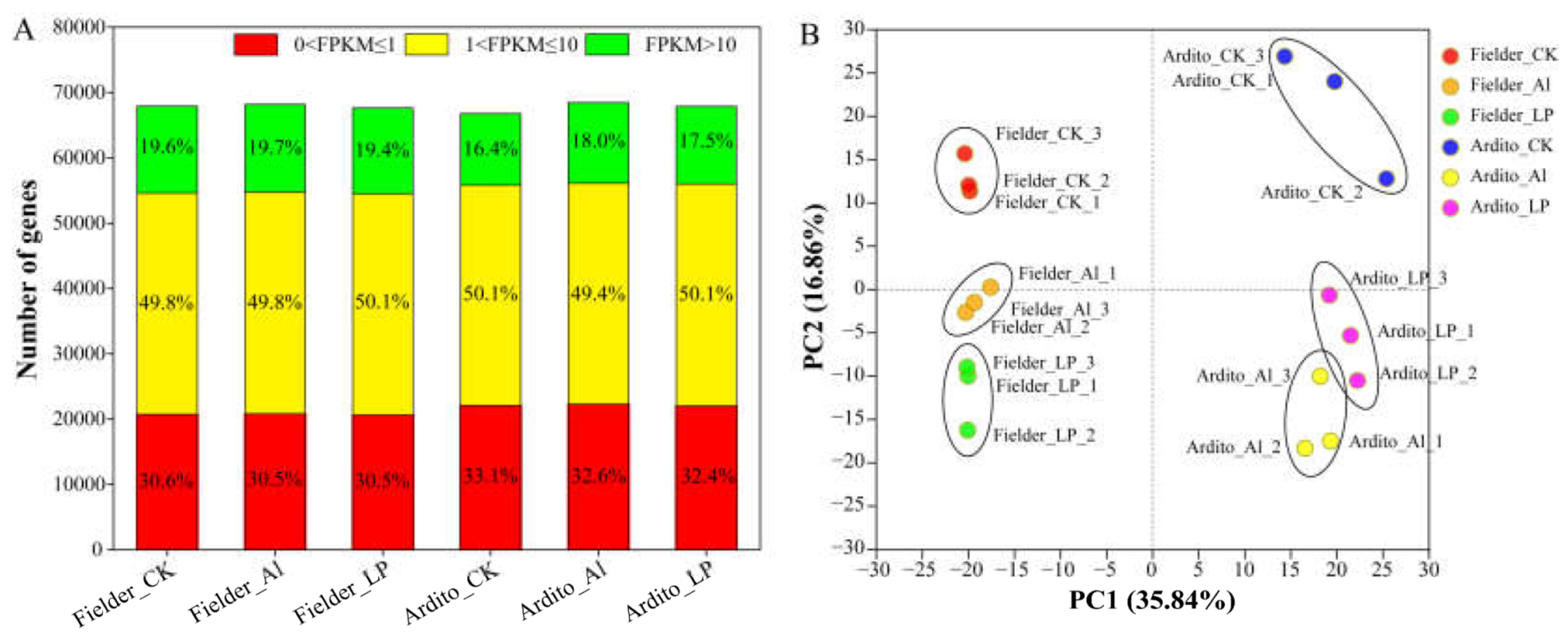
Figure 2.
Al and LP responsive gene expression profiles. (A) The number of differentially expressed genes (DEGs) in each comparison. (B-E) The Venn diagram displays the relationship between differentially expressed genes (DEGs) of various treatments Al (B) and LP (C), and varieties Fielder (D) and Ardito (E).
Figure 2.
Al and LP responsive gene expression profiles. (A) The number of differentially expressed genes (DEGs) in each comparison. (B-E) The Venn diagram displays the relationship between differentially expressed genes (DEGs) of various treatments Al (B) and LP (C), and varieties Fielder (D) and Ardito (E).
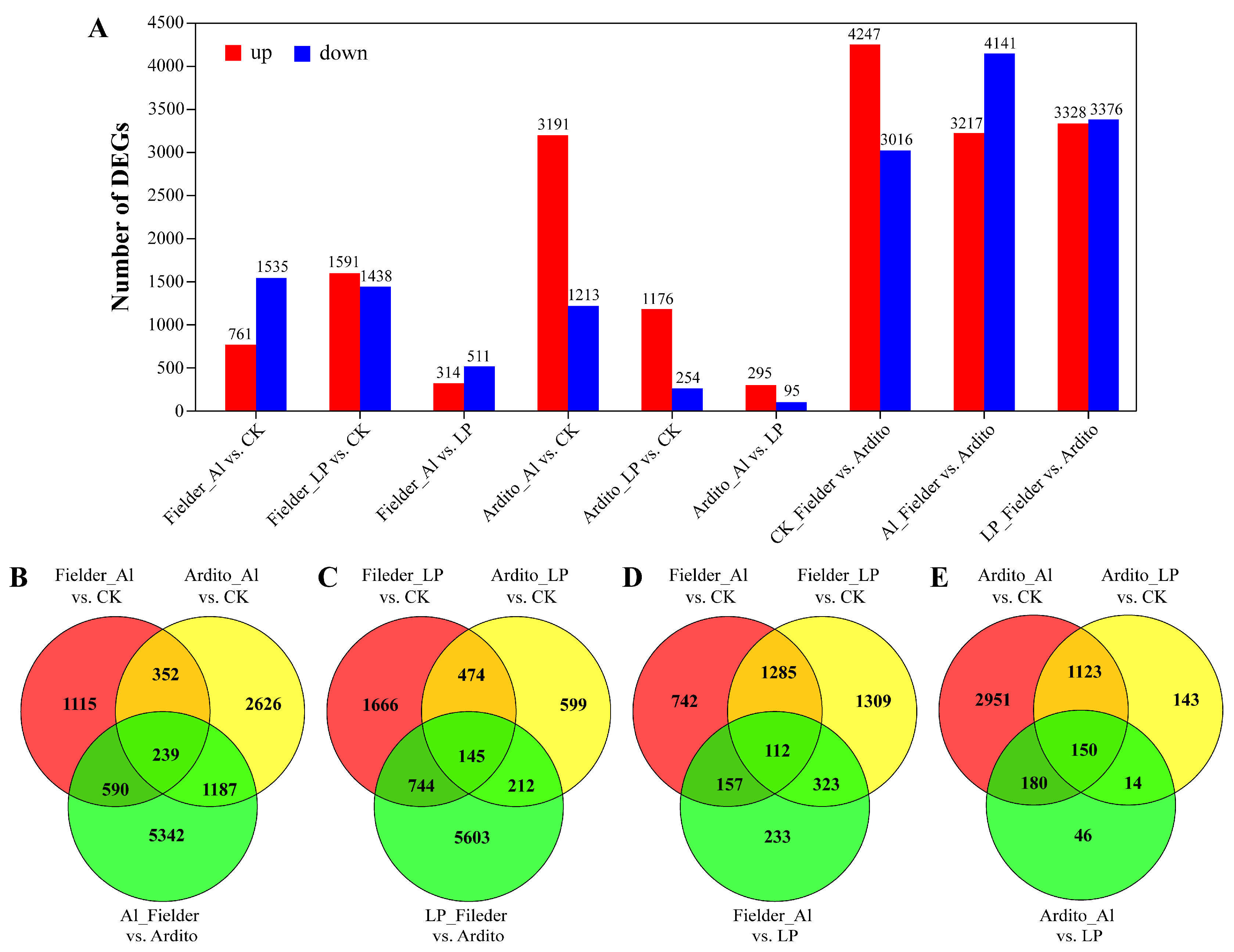
Figure 3.
KEGG enrichment analysis for eight DEGs under Al and LP stresses. The bubble size indicates the number of genes involved in the terms and pathways.
Figure 3.
KEGG enrichment analysis for eight DEGs under Al and LP stresses. The bubble size indicates the number of genes involved in the terms and pathways.
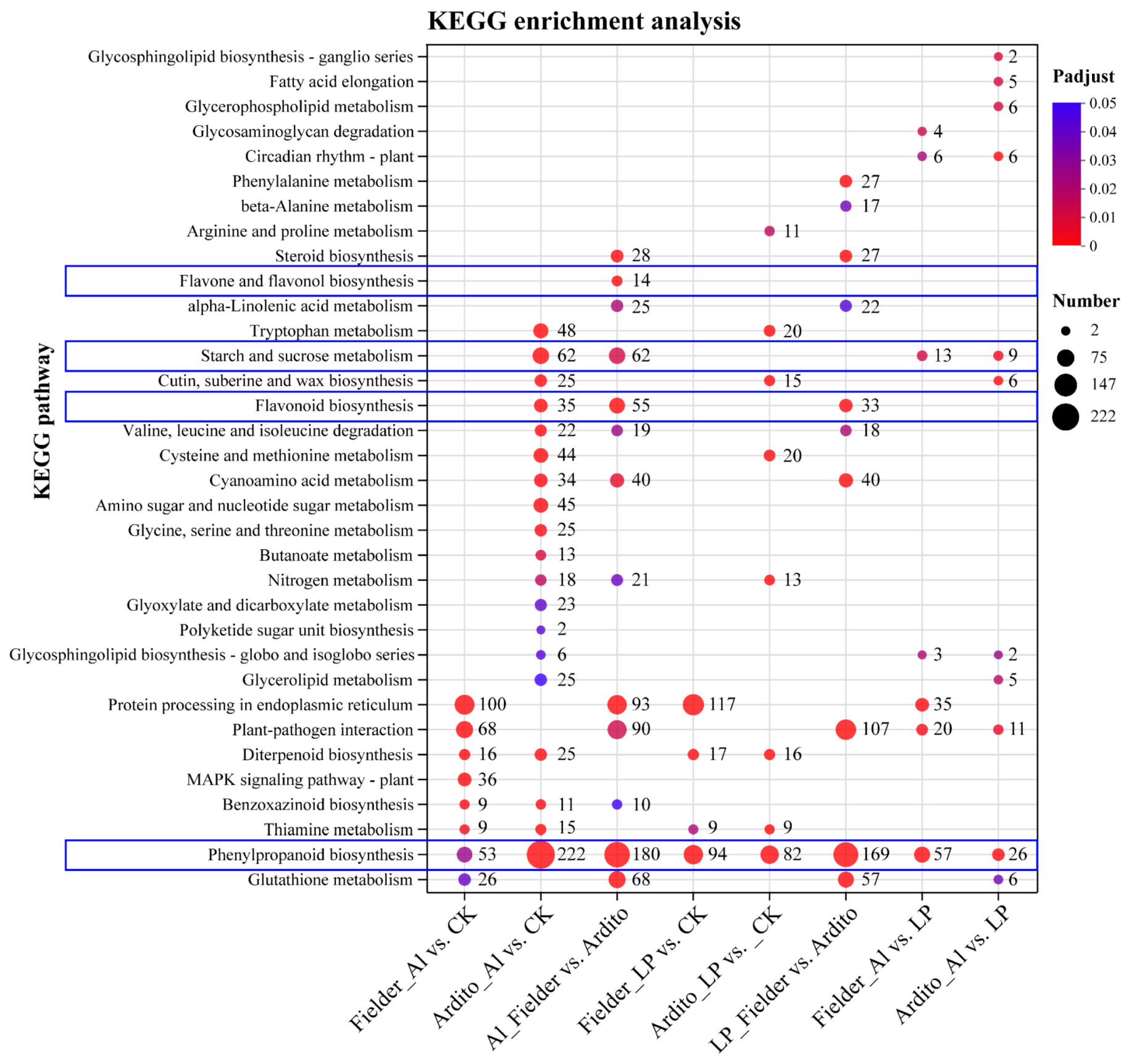
Figure 4.
The changes in expression of common significantly differentially expressed TFs during Al and LP stresses in the two wheat genotypes.
Figure 4.
The changes in expression of common significantly differentially expressed TFs during Al and LP stresses in the two wheat genotypes.
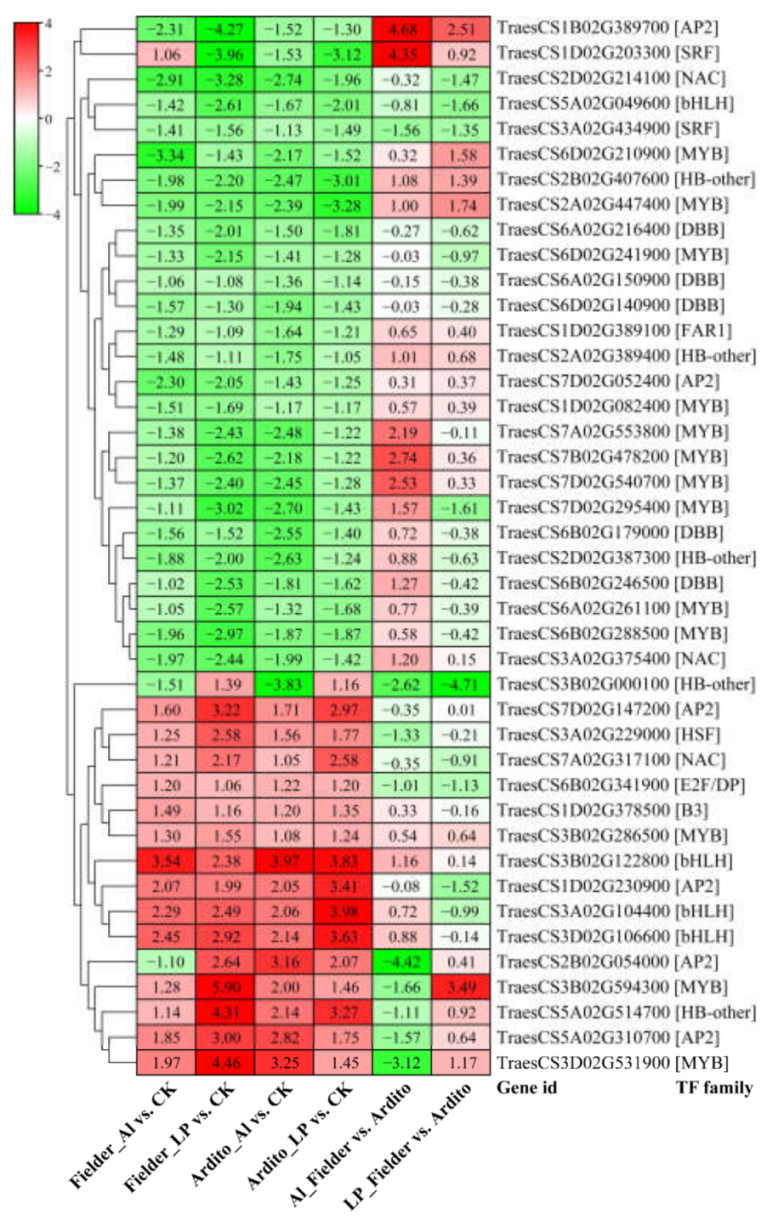
Figure 5.
The relative expression levels detected by qRT-PCR using the 2−ΔΔCT method are shown by the blue bars, while the red line graph represents the FPKM ratio, indicating transcript abundance in the RNA-Seq data. TaActin was used as an internal control. Values are means ± SD; n = 4. PAL: Phenylalanine ammonia-lyase; 4CL: 4-coumarate-CoA ligase; C4H: Cinnamic acid 4-hydroxylase; CHS: Chalcone synthase; CHI: Chalcone isomerase; F3H: Flavanone 3-hydroxylase; FNSI: flavone synthase.
Figure 5.
The relative expression levels detected by qRT-PCR using the 2−ΔΔCT method are shown by the blue bars, while the red line graph represents the FPKM ratio, indicating transcript abundance in the RNA-Seq data. TaActin was used as an internal control. Values are means ± SD; n = 4. PAL: Phenylalanine ammonia-lyase; 4CL: 4-coumarate-CoA ligase; C4H: Cinnamic acid 4-hydroxylase; CHS: Chalcone synthase; CHI: Chalcone isomerase; F3H: Flavanone 3-hydroxylase; FNSI: flavone synthase.
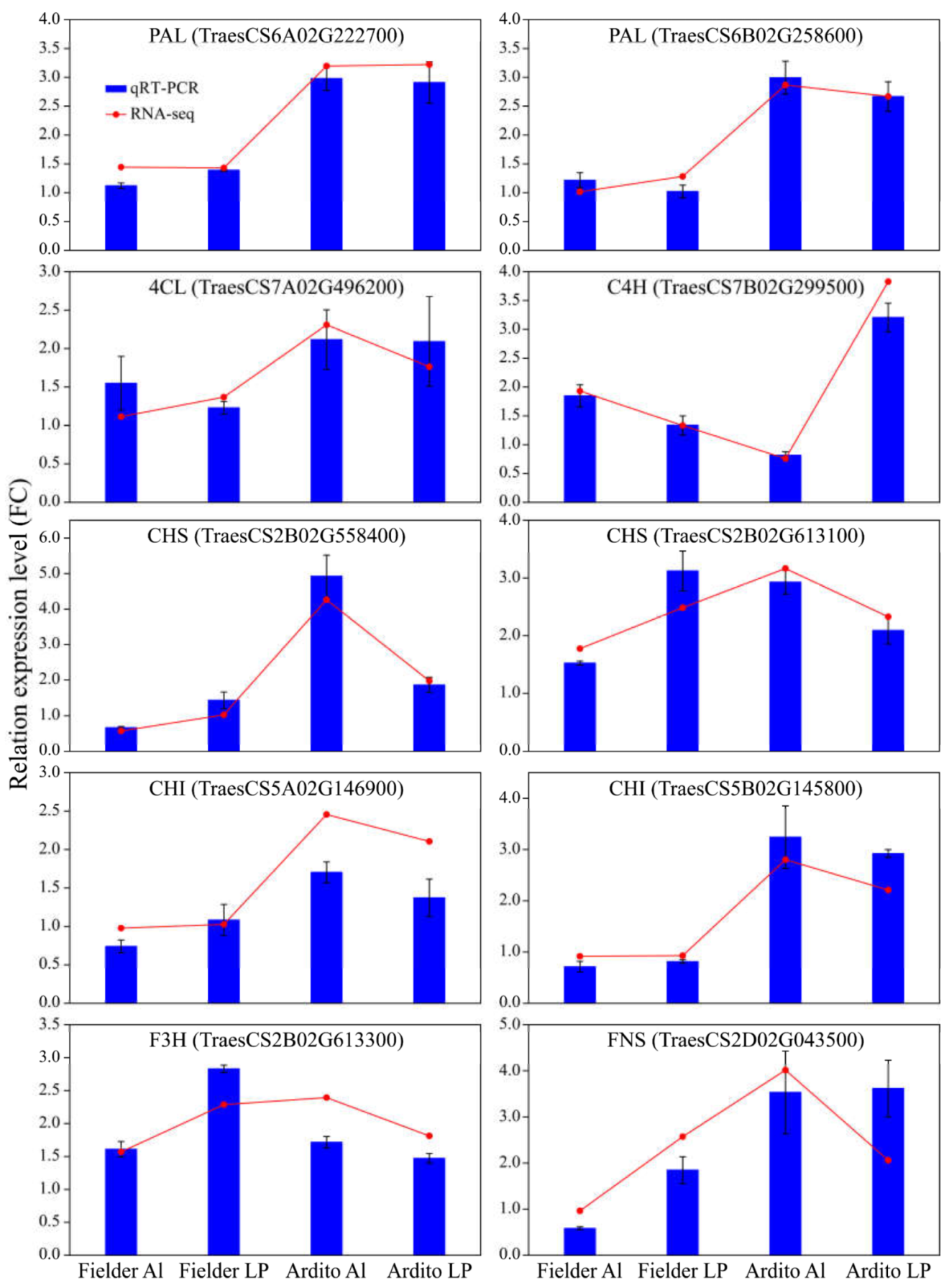
Figure 6.
Joint analysis of flavonoid metabolism genes and metabolites. The changes in expression of DEGs (A-D) and metabolites (E-H) related to the pathways of (A, E) general phenylpropanoid, (B, F) flavonoid, (C, G) isoflavonoid, and (D, H) flavone and flavonol biosynthesis under Al and LP stress in the two wheat genotypes. ND: not detected. ANS: Anthocyanidin synthase, ANR: Anthocyanidin reductase; F3’5’H: Flavonoid 3’,5’-hydroxylase; F3GT: Flavonol 3-O-glucosyltransferase; FG2: Anthocyanidin-3-O-glucoside rhamnosyltransferase; HIDH: 2-hydroxyisoflavanone dehydratase.
Figure 6.
Joint analysis of flavonoid metabolism genes and metabolites. The changes in expression of DEGs (A-D) and metabolites (E-H) related to the pathways of (A, E) general phenylpropanoid, (B, F) flavonoid, (C, G) isoflavonoid, and (D, H) flavone and flavonol biosynthesis under Al and LP stress in the two wheat genotypes. ND: not detected. ANS: Anthocyanidin synthase, ANR: Anthocyanidin reductase; F3’5’H: Flavonoid 3’,5’-hydroxylase; F3GT: Flavonol 3-O-glucosyltransferase; FG2: Anthocyanidin-3-O-glucoside rhamnosyltransferase; HIDH: 2-hydroxyisoflavanone dehydratase.
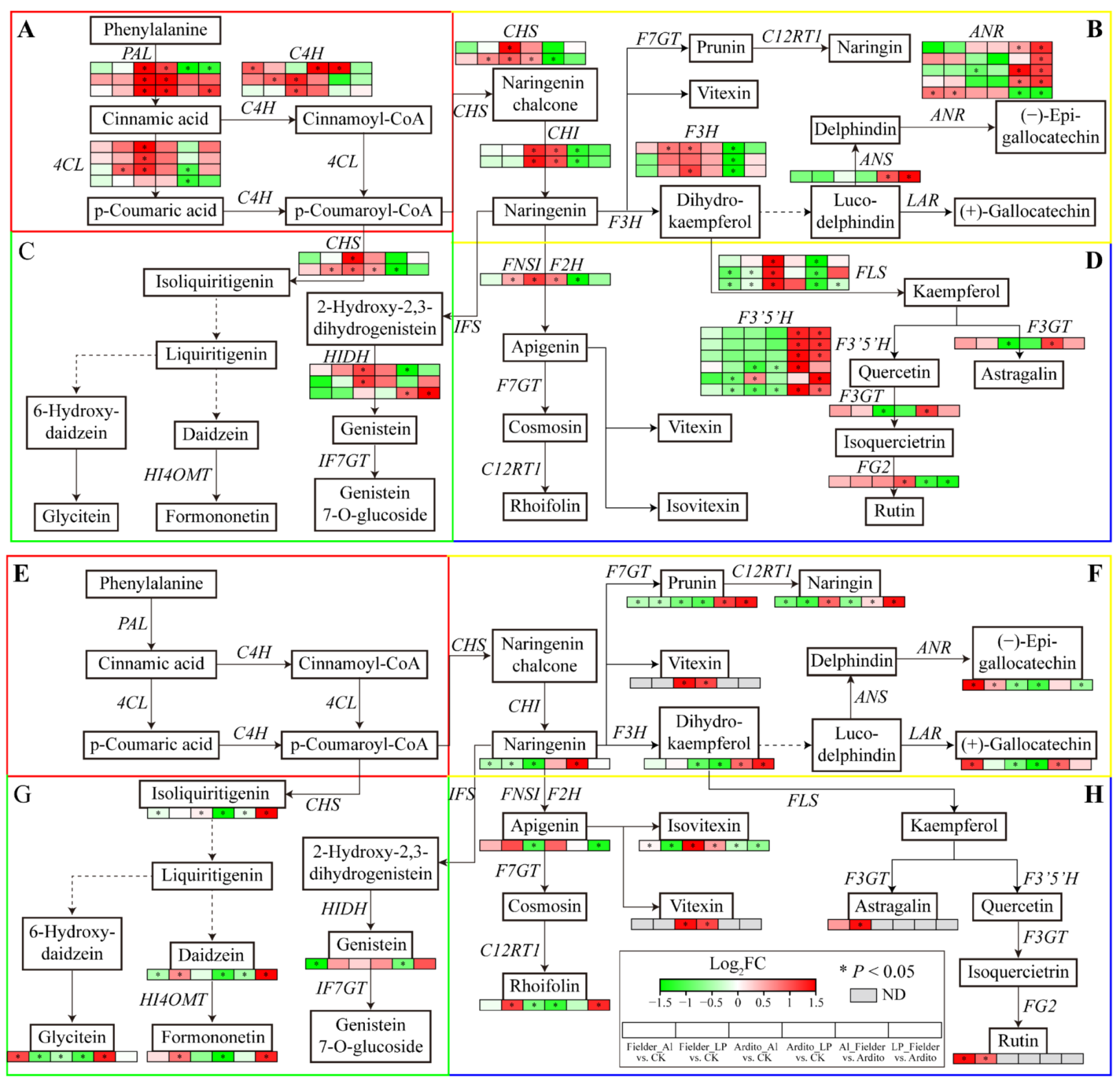
Figure 7.
Joint analysis of carbohydrate metabolism genes and metabolites. The changes in expression of DEGs (A-C) and metabolites (D-F) related to the pathways of (A, D) starch and sucrose metabolism, (B, E) glycolysis/gluconeogenesis, and (C, F) TCA cycle under Al and LP stress in the two wheat genotypes. TPP: Trehalose-phosphatase; TPS: Alpha, alpha-trehalose-phosphate synthase; SUS: Sucrose synthase; INV: β-fructofuranosidase; ALDO: Fructose-bisphosphate aldolase; GAPDH: Glyceraldehyde-3-phosphate dehydrogenase; PGK: Phosphoglycerate kinase; PGAM: Phosphoglycerate mutase, PFP: Phosphofructokinase; FBP: Fructose-1-6-bisphosphatase, PPDK: Pyruvate, phosphate dikinase; ACLY: ATP-citrate synthase; IDH: Isocitrate dehydrogenase; ACO: Aconitate hydratase; MDH: Malate dehydrogenase; SDH: Succinate dehydrogenase; FUM: Fumarate hydratase.
Figure 7.
Joint analysis of carbohydrate metabolism genes and metabolites. The changes in expression of DEGs (A-C) and metabolites (D-F) related to the pathways of (A, D) starch and sucrose metabolism, (B, E) glycolysis/gluconeogenesis, and (C, F) TCA cycle under Al and LP stress in the two wheat genotypes. TPP: Trehalose-phosphatase; TPS: Alpha, alpha-trehalose-phosphate synthase; SUS: Sucrose synthase; INV: β-fructofuranosidase; ALDO: Fructose-bisphosphate aldolase; GAPDH: Glyceraldehyde-3-phosphate dehydrogenase; PGK: Phosphoglycerate kinase; PGAM: Phosphoglycerate mutase, PFP: Phosphofructokinase; FBP: Fructose-1-6-bisphosphatase, PPDK: Pyruvate, phosphate dikinase; ACLY: ATP-citrate synthase; IDH: Isocitrate dehydrogenase; ACO: Aconitate hydratase; MDH: Malate dehydrogenase; SDH: Succinate dehydrogenase; FUM: Fumarate hydratase.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



