
Submitted:
09 August 2024
Posted:
12 August 2024
You are already at the latest version
Abstract
Amphiphilic block copolymers of N-vinyl pyrrolidone (NVP) and various vinyl esters (VEs), PNVP-b-PVEs, namely vinyl butyrate (VBu), vinyl decanoate (VDc) and vinyl stearate (VSt) were synthesized by RAFT polymerization techniques. The sequential addition of monomers methodology was employed starting from the polymerization of NVP followed by the polymerization of the VEs monomer. The reaction was conducted at 80oC in dioxane solutions using AIBN as initiator and O-ethyl S-(phthalimidylmethyl) xanthate as the CTA. The block copolymers were characterized by Size Exclusion Chromatography (SEC) and NMR spectroscopy. The thermal transitions of the copolymers were studied by Differential Scanning Calorimetry (DSC), whereas their thermal stability via Thermogravimetric Analysis (TGA) and Differential Thermogravimetry (DTG).
Keywords:
poly(N-vinyl pyrrolidone) (NVP)
; poly(vinyl esters) (VEs)
; block copolymers
; RAFT polymerization
; thermal analysis
; thermal stability
1. Introduction
Poly(vinyl esters) (PVEs) consist a valuable class of polymeric materials [1], mainly for two reasons. The first one has to do with the numerous applications of these materials in the industrial sector, involving their employment as elastomers, plastics, fibers, coatings, paints, additives, adhesives, textiles, cosmetics, etc. [2,3,4,5,6,7] This broad range of applications is associated with the correlation between the structure and the properties of the PVEs. Depending on the nature of the side ester-group (aliphatic with various carbon atoms, aromatic, alicyclic, olefinic etc.) the PVEs can be either hydrophilic or hydrophobic, either amorphous or semi-crystalline, either liquid-like, or waxy or solid materials etc. [8,9,10,11,12,13,14,15,16,17,18] The second reason has to do with the fact that PVEs can be precursors for the production of poly(vinyl alcohol) (PVA), which is a benchmark water-soluble polymer demonstrating non-toxicity, non-carcinogenic properties and possibilities for bio-conjugation for therapeutical applications. [19,20,21] Therefore, it can be applied as a valuable biomaterial in pharmacological applications. [22,23,24] The transformation of PVEs to PVA is conducted through hydrolysis of the ester group. The nature of the side group affects the kinetics of hydrolysis ranging from a few minutes to several hours. Under these conditions, intermediate degrees of hydrolysis can be reached leading to products with complex behaviour in aqueous solutions. [25]
The most well-known member of the PVEs polymer family is poly(vinyl acetate) (PVAc). It is the most widely used precursor for the synthesis of PVA and numerous studies have been devoted to the synthesis of this material and its copolymers with other VEs or other monomers. [26,27] Much less work has been devoted to the synthesis and the study of the solution and solid-state properties of other members of the PVEs family of polymers. [28,29,30,31,32]
The polymerization of VEs is feasible only through radical polymerization. The conventional radical polymerization, although straightforward in its applications, suffers various disadvantages. These include the lack of control over the molecular weights of the produced materials, the high polydispersity, Ð, values, the inability to produce pure end-functionalized polymers and the very limited possibilities to synthesize complex macromolecular architectures, since the technique is accompanied by several termination and transfer reactions. [33] Especially, for VAc extended studies have been performed to reveal the presence of chain transfer reactions to monomer and polymer leading to the formation of branched structures and the appearance of regio-irregularities with increased head-to-head linkages. [34,35,36,37] For this reason, the polymerization of VEs was not a hot topic in Polymer Chemistry for many years. However, the developments in controlled radical polymerization methodologies opened new horizons for the controlled polymerization of VEs and offered the possibility to manipulate macromolecular engineering towards the synthesis of tailor-made polymers. Among these approaches, nitroxide-mediated, atom transfer radical, iodine transfer, organostibine-mediated, vanadium-mediated and cobalt-mediated polymerization techniques have been employed. [38,39,40,41,42,43,44,45] Although relative success has been reported in several cases these methods present several drawbacks rendering their universal application problematic. By far the most successful polymerization technique for VEs is the Reversible Addition Fragmentation Chain Transfer (RAFT) methodology. It was proven to be the suitable tool towards the synthesis of controlled structures in terms of both the molecular characteristics and the copolymer topology or macromolecular architecture in combination with other polymerization techniques and a variety of monomers. [46,47]
In this work, the synthesis of amphiphilic block copolymers of N-vinyl pyrrolidone (NVP) and various vinyl esters, PNVP-b-PVEs, including vinyl butyrate (VBu), vinyl decanoate (VDc) and vinyl stearate (VSt) is described through RAFT approaches. These materials are expected to provide very interesting properties. On the one hand, PNVP is an amorphous polymer with a high Tg value (equal to 187oC, depending on the humidity), soluble in both aqueous solutions and in organic solvents, without showing lower critical solution temperature (LCST) [48,49,50,51,52] and on the other hand PVBu and PVDc are also amorphous polymers with low Tg values (-5 oC for PVBu and -45 oC for PVDc), whereas PVSt is a semicrystalline polymer with Tm=52-57oC. [54,55,56]
All monomers, NVP and VEs, are considered to belong to the category of less activated monomers (LAMs) in RAFT polymerization. The monomers are classified in two families in RAFT polymerization. They belong either to the more activated (MAMs) or to the less activated monomers (LAMs). The key parameter differentiating these two groups of monomers is their ability to stabilize radicals [47,57]. LAMs have a double bond, which is connected to a saturated carbon atom or conjugated to a lone pair on oxygen or nitrogen. Therefore, the polymerization of these monomers leads to the formation of poorly stabilized radicals. On the other hand, the group of MAMs includes monomers, where the double bond is conjugated to an unsaturated system, such as nitrile, aromatic ring or carbonyl groups. Consequently, these monomers form highly stabilized radicals, due to extended resonance effects. In the present work, the difference in reactivity between NVP and VEs is considered to be low thus, making it easier to combine these monomers in a single polymerization system, thus providing better control over the copolymerization procedure. However, difficulties exist in the specific system of monomers. VAc is considered to be the least active monomer among the common LAMS in RAFT polymerization because it provides highly reactive and thus unstable propagating radicals. Therefore, it is not very easy to control the polymerization reaction. The situation becomes even more problematic upon increasing the size of the n-alkyl side group of the VEs leading to extended retardation of the polymerization reaction and the possibility to have pronounced termination reactions rendering the control of the macromolecular architecture even more difficult. [58,59,60]
This work is focused on the synthesis and characterization of the desired PNVP-b-PVEs block copolymers and the study of their thermal properties. The self-assembly behavior of these copolymers in selective solvents will be presented in a forthcoming publication. In the literature several efforts have been presented for the synthesis of statistical and block copolymers of different VEs, including VAc in all cases and other monomers from the same family, such as vinyl pivalate, vinyl benzoate, vinyl octanoate etc. In the past the corresponding block copolymers of NVP with VAc have been frequently reported in the literature. However, very limited work has been mentioned with the synthesis of block copolymers of NVP with other VEs.
2. Materials and Methods
2.1. Materials
N-Vinyl pyrrolidone (≥97% FLUCA) containing sodium hydroxide as inhibitor was dried overnight over calcium hydride and was distilled prior to use. The vinyl esters (TCI Chemicals) stabilized with monomethylether hydroquinone, were also dried over calcium hydride overnight and then were distilled under vacuum prior the polymerization. VBu has a bp at 115-117οC , whereas VDc has a bp of 119-1200C. On the other hand VSt is a solid monomer and was purified after dissolution in THF and passing through an inhibitor remover column. Azobisisobutyronitrile AIBN (98% ACROS) was purified by recrystallization twice from methanol and was then dried under vacuum. The Chain Transfer Agent, O-ethyl S-(phthalimidylmethyl) xanthate, was synthesized according to the literature protocols [61,62] employing O-Ethyl xanthic acid potassium salt and N-(bromomethyl)phthalimide. Dioxane and benzene were also purified over CaH2 overnight and were distilled just prior to use.
All other reagents and solvents were of commercial grade and were used as received.
2.2. Synthesis of PNVP-b-PVEs Block Copolymers via RAFT Polymerization
The synthesis of the PNVP-b-PVEs block copolymers was accomplished by sequential addition of monomers starting from the polymerization of NVP. O–ethyl S–(phthalimidymethyl) xanthate was employed as the CTA and AIBN as the initiator. The polymerization of NVP was conducted in glass reactors using high vacuum techniques [63,64] at 60oC in benzene solutions for 12 hours.
A typical polymerization procedure for NVP with final Mn=8.9x103 (Table 1, samples PNVP-b-PVBu #3 and #4) is accomplished using a molar ratio of [NVP]0/[CTA]0/[AIBN]0 = 100/1/0.2 and is described as follows: 5g of NVP were polymerized in the presence of 0.1284g CTA and 0.0148g AIBN in 5 ml of benzene. The polymerization solution was subjected to three freeze-thaw pump cycles in order to eliminate the oxygen from the polymerization apparatus. The reactor was flame-sealed and placed in a preheated oil-bath at 60oC for 12h.
The reaction was terminated by removing the reaction flask from the oil-bath and by immediate cooling of the polymerization mixture under the flow of cold water. The apparatus was then opened exposing the mixture to air. The polymer was then precipitated in an excess of diethyl ether. This procedure was repeated at least three times in order to ensure the removal of any unreacted monomer residues. The polymers were finally dried overnight in a vacuum oven at 50oC to remove any residual solvent. The conversions for all homopolymers were near quantitative.
The PNVP homopolymers, served as the macro-CTAs to promote the polymerization of the VEs in a subsequent step. The block copolymerization reactions were performed in dioxane solutions at 80oC for 96 hours in glass reactors under high vacuum conditions, as previously reported for the synthesis of the PNVP macro-CTAs. The quantities of the VEs monomers, the AIBN radical initiator and the dioxane solvent employed for the synthesis of the block copolymers are reported in Table 1. The polymerization mixture was subjected to three freeze-thaw pump cycles in order to eliminate the oxygen from the polymerization apparatus The polymerization was terminated by removing the reactor from the oil-bath and cooling the mixture under a flow of cold water. The reactor was then opened and the copolymerization mixture was exposed to air. The copolymers with the PNBu blocks were precipitated in an excess of hexanes, whereas the copolymers with PVDc and PVSt blocks in methanol. The crude product was dissolved in THF and reprecipitated in the appropriate non-solvent. This procedure was repeated three times in order to ensure the removal of any unreacted monomer residues. Finally, the copolymers were dried overnight in a vacuum oven at 50oC to remove any residual solvent. No further effort was needed to purify the samples.
2.3. Characterization Techniques
The molecular weight (Mw) as well as the molecular weight distribution, Ð= Mw/Mn, were determined by size exclusion chromatography, SEC, employing a modular instrument consisting of a Waters model 510 pump, U6K sample injector, 401 differential refractometer and a set of 5μ-Styragel columns with a continuous porosity range from 500 to 106 Å. The carrier solvent was CHCl3 and the flow rate 1 ml/min. The system was calibrated using nine Polystyrene standards with molecular weights in the range of 970–600,000.
The composition of the copolymers was determined from their 1H NMR spectra, which were recorded in chloroform-d at 30°C with a 400 MHz Bruker Avance Neo spectrometer (Billerica, MA, USA).
The Tg values of the copolymers were determined by a 2910 Modulated DSC Model from TA Instruments. The samples were heated under nitrogen atmosphere at a rate of 10 °C/min from -50 °C up to 220 °C. The second heating results were obtained in all cases.
The thermal stability of the copolymers was studied by thermogravimetric analysis (TGA) employing a Q50 TGA model from TA Instruments. The samples were placed in a platinum pan and heated from ambient temperatures to 600 °C in a 60 mL/min flow of nitrogen at heating rates of 10 °C/min.
3. Results and Discussion
3.1. Synthesis of the Block Copolymers of NVP and VEs
The synthesis of the desired block copolymers was accomplished via RAFT polymerization and sequential addition of monomers. Since both types of monomers have low reactivities the same CTA can be employed for the synthesis of well-defined block copolymers [65,66,67,68]. Several families of CTAs have been applied for the polymerization of LAMs in RAFT polymerization including xanthates and dithiocarbamates. In the present study O-ethyl S-(phthalimidylmethyl) xanthate was used as the CTA, since it is well-known that it promotes very good control over the RAFT polymerization of LAMs. In particular, it has been efficiently employed for the homopolymerization of both NVP and VEs in the past. Specifically, VAc was polymerized with this CTA leading to controlled molecular weights covering the range from values as low as 3000 up to 121000 with relatively low polydispersities, Ð, in the range 1.2 to 1.4. An induction period of about 1 hour was revealed in kinetic experiments performing the polymerization reaction at 60oC. [69,70,71]
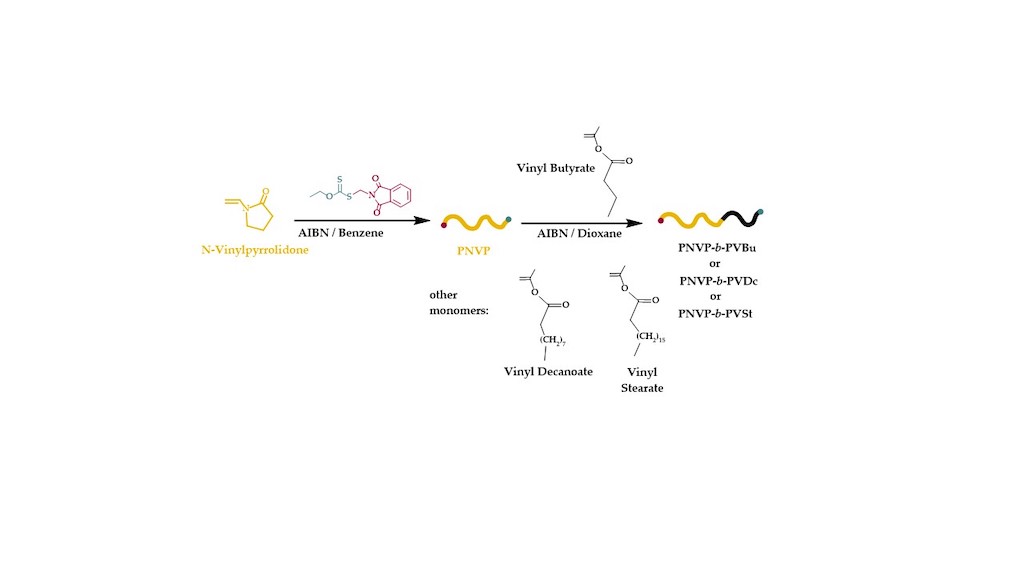
The procedure for the synthesis of the PNVP-b-PVEs block copolymers is depicted in Scheme 1.
NVP was polymerized first providing well-defined macromolecular CTAs (macro-CTAs) having controlled molecular characteristics, ie. relatively narrow molecular weight distribution and molecular weight, which was close to the stoichiometric values. In order to better control the dispersity values and to achieve quantitative incorporation of the CTA’s moieties as the polymer’s end-groups the polymerization was not allowed to proceed to very high yields. In most cases the reaction was terminated by cooling the reaction flask when the conversion was less than 80% in all cases. At higher yields the solution becomes very viscous and the system becomes more or less heterogeneous leading to increased dispersity values. In addition, upon increasing the polymerization yield the possibility to lose the control over the end-groups is increased. The PNVP macro-CTAs were precipitated, re-dissolved and re-precipitated to remove unreacted monomer. The samples were finally dried in a vacuum oven.
The purified and dried macro-CTAs were further employed for the polymerization of various VEs, namely VBu, VDc and VSt. In order to minimize the viscosity problems, the termination and other side reactions involved in RAFT radical polymerization, thus achieving the best degree of control during the copolymerization reaction, the conversion of the polymerization of VEs was not allowed to reach high values, i.e. less than 50% in most cases. The efficiency of this approach was monitored by SEC and 1H NMR measurements. Characteristic SEC traces from the synthesis of the block copolymers are provided in Figure 1, Figure 2 and Figure 3, whereas typical 1H NMR spectra are shown in Figure 4, Figure 5 and Figure 6. Table 2 incorporates the molecular characteristics of all the synthesized block copolymers.
The SEC traces indicate for all block copolymers that the polymerization of the second monomer was efficiently promoted by the macro-CTA. Single peaks with only minor tailing effects in a few cases were obtained, showing that it is impossible to completely avoid termination phenomena, something which is already established in RAFT polymerization.[46] Consequently, no effort was given to further purify the block copolymers. The reverse mode of monomer addition was not tested, since it is known that the chain extension employing PVEs macro-RAFT agents presents difficulties, due to the instability of the PVEs macroradicals. The safer way to minimize chemical heterogeneity in block copolymer synthesis and achieve lower polydispersity values involves the synthesis of the PNVP macro-RAFT agent initially and then the polymerization of the less reactive VEs.
The efficiency of the synthetic procedure was further manifested by the 1H NMR analysis. The characteristic signals of both monomer units are obvious in the spectra of the copolymers. The signal at 3.2 ppm, which is attributed to the methylene protons of the pyrrolidone ring that are adjacent to the nitrogen atom was employed for the calculation of the composition of the PNVP block. On the other hand, for the PVEs blocks the signals at 0.9 ppm, attributed to the methyl end-groups of the side groups of the PVEs were used for the calculation of the content in PVEs. The results, given in Table 2 reveal that it is possible to manipulate the composition of the block copolymers from the synthetic route.
3.2. Thermal Properties
3.2.1. DSC Analysis
The thermal properties of the block copolymers were studied by DSC measurements. The results for the PNVP-b-PVBu copolymers are given in Table 3, whereas the DSC thermograms are provided in Figure 7.
Both components of the blocks are amorphous. Their Tg values are located at 187.1oC [52,53] and -5oC [72] for the PNVP and PVBu homopolymers respectively. In most cases, three different Tg values are obvious from the thermograms. The high Tg value is relatively close to the Tg value of the PNVP homopolymer, although considerably lower. The low Tg value is close to that of the PVBu homopolymers. It has been reported that the Tg value for PVBu is -5oC, which is slightly higher than the one reported in this work for the corresponding homopolymer. This small difference is attributed to the lower molecular weight sample employed in the present study. Finally, the third Tg value is located at intermediate temperatures between those of the two homopolymers. These results clearly indicate that the block copolymers are microphase separated. However, due to the relatively low molecular weight of the copolymers the samples are not located at the strong segregation limit. The third intermediate transition confirms a partial mixing of the two phases. This mixing of the two phases is so substantial that acts as a third phase in the system leading to the appearance of a third Tg from the created mesophase. Only the sample PNVP-b-PVBu #2 shows the absence of a pure PVBu phase, since this sample has the lower content in PVBu (16% mol by NMR analysis). The results confirm that there is an almost pure PNVP microphase (indicated by the highest Tg value, which is relatively close to the corresponding homopolymer) accompanied by a second microphase from the mixing of the PVBu and PNVP components. The sample #1 with the lowest PNVP content (22% mol in PNVP by NMR analysis) has the lowest Tg value for the PNVP microphase, since this phase is “contaminated” by the highest amount of PVBu. Similar microphase separation data by DSC measurements have appeared in the literature for block copolymers of different PVEs. [54,55]
It is obvious that the Tg value of PVDc [54] is much lower than that of PVBu, since the Tg value drastically decreases as the side chain of the main polymer increases. [73,74,75,76] This result is reasonable and is attributed to an internal plasticization effect, which is well-documented in polymer chains carrying aliphatic side chains. The thermal behavior is similar to that observed in the case of the copolymers PNVP-b-PVBu. For all the samples three transitions are observed indicating the existence of microphase separation in the copolymers. However, due to the rather low molecular weights of the copolymers the system is at the low segregation limit with partial mixing of the phases. The difference between the copolymers with PVDc bocks compared to those with the PVBu blocks is that even with very low PVDc content (as low as 7% mol in sample PNVP-b-PVDc #4) the low Tg phase of the PVDc component is clearly observed (even with a much higher Tg value, due to contamination with PNVP chains). This is direct evidence that the PNVP-b-PVDc copolymers have a higher tendency to microphase separation than the PNVP-b-PVBu copolymers, or in other words that the Flory-Huggins χ parameter is much higher between the PNVP and PVDc chains rather that the PNVP and PVBu chains. Consequently, the increase of the size of the side-chain in PVEs facilitates the microphase separation procedure.
The thermal transitions of the PNVP-b-PVSt block copolymers are summarized in Table 5, whereas the DSC traces are shown in Figure 9.
In this case the situation is more complex, since the homopolymer PVSt is a semicrystalline polymer showing side-chain crystallization, due to the extended side-chains of 18 carbon atoms. [55,72] In similar families of polymers bearing long linear alkyl side-groups, such as methacrylates, acrylates and α-olefins it has been shown that when the number of carbom atoms of the linear side-group is higher than 12, then side-chain crystallization occurs. [73,74,75,76] The experimental data, shown in Figure 9, confirm the presence of the melting endotherm during heating for all samples. The Tm value for the PVSt homopolymer has been reported in the range of 52 up to 57oC. [77] The lower Tm value in this study has to do with the relatively low molecular weight of the homopolymer used in this work. However, the enthalpy of melting agrees very well with the literature value and is equal to 87.9 J/g. Compared to the melting point, Tm of the PVSt homopolymer, the Tm values of the copolymers are located at lower temperatures, due to the fact that the crystallization procedure is restricted from the presence of the amorphous block of the PNVP chains. Similar results have been reported in numerous other cases of block copolymers bearing an amorphous and a semicrystalline block. It is characteristic that the lower Tm value was obtained in the sample PNVP-b-PVSt #1 having the lower molecular weight of the PVSt block.
The case of diblock copolymers consisting of a crystallizable block and another amorphous block has been examined theoretically by Flory. [78] Using a lattice model he analyzed the thermodynamics of crystallization of homopolymers and copolymers. For the copolymers he predicted a decrease of the melting point of the crystallizable block compared to the pure homopolymer. This melting point depression, according to Flory, is influenced by the length of the crystallizable block and the thermodynamic interactions between the two blocks.
The Tg of the PVSt, which is expected to be very low (as low as -100oC), was not observed experimentally, since it is a weak transition, due to the crystallinity of the polymer. From the results shown in Table 6 it is obvious that the samples are microphase separated, due to the observation of both the PVSt melting points and the PNVP Tg values, which are very close to the expected ones considering the molecular weight dependence of the Tg of PNVP. This result is further supported by the data obtained by Mayes, Russell et al. [79] concerning the phase behavior of a similar system of block copolymers consisting of polystyrene and poly(n-alkyl methacrylates) having up to 12 carbon atoms. It was found that the interaction parameter, χ, increases by increasing the length of the alkyl side group of the polymethacrylate block.
The degrees of crystallinity were calculated measuring the normalized enthalpy of melting of the PVSt blocks (ΔH, j/PVStg) in the copolymers compared to the same value for the 100% crystalline homopolymer (ΔHo=220 j/PVStg) [55], as follows:
Xc= ΔH (j/PVStg)/ ΔHo (j/PVStg)
The results, shown in Table 5, reveal that the copolymers have relatively low degrees of crystallization (less than 20% in all cases), showing that the presence of the PNVP blocks restricts the organization of the crystalline phases and that the more significant part of the PVSt block remains amorphous.
Taking these results into account it is reasonable to expect that the amorphous phase of the PVSt will be responsible for the appearance of a second Tg value, except that attributed to the amorphous PNVP phases. This hypothesis was proven true from the data presented in Table 5 and the thermograms given in Figure 9. The relative values of the two transitions confirm that the two amorphous phases are partially mixed, due to the low molecular weights of the constituent blocks. Finally, in the case of the PNVP-b-PVSt block copolymers a crystalline phase of the PVSt block and two mixed phases of the amorphous components are present.
3.2.2. TGA Analysis
The thermal stability of the PNVP-b-PVEs block copolymers was studied by TGA and DTG analysis. The results for the PNVP-b-PVBu copolymers are given in Table 6 and the TGA and DTG plots in Figure 10 and Figure 11 respectively.
The PNVP homopolymer is a relatively thermally stable polymer showing a single decomposition peak from the DTG analysis. [80] The maximum temperature of the thermal decomposition peak is located at 437oC. These results indicate a rather simple decomposition mechanism. On the other hand, the PVBu homopolymer reveals a two-step thermal decomposition mechanism having two maxima located at 319 and 416oC respectively. PVBu is a much less thermally stable polymer compared to PNVP, since the main decomposition procedure is the one that takes place at the lower decomposition temperature. [81,82]
All the PNVP-b-PVBu block copolymers revealed two steps of thermal decomposition, indicating similar complex decomposition mechanism as in the case of the PVBu homopolymer. The first thermal decomposition peak was traced at temperatures close to the low temperature decomposition peak of the PVBu homopolymer and obviously is correlated to the decomposition of the PVBu block. The second thermal decomposition is located at higher temperatures and is associated with the decomposition of the PNVP block. This conclusion was further confirmed by comparing the mass loss with the composition of the block copolymers. The first decomposition peak of sample #1, having the highest PBVu content, is the most pronounced among the other samples. On the contrary, in the case of sample #2 with the highest PNVP content the high temperature decomposition peak is very pronounced. The other two samples with similar compositions have also a similar decomposition pattern.
The TGA results for the PNVP-b-PVDc copolymers are given in Table 7, whereas the TGA and DTG plots in Figure 12 and Figure 13 respectively.
The thermal decomposition of PVDc is similar to that of PVBu. It takes place at two different distinct steps having maxima at 323oC and 418oC respectively. As in the case of PVBu the first decomposition step is the most important corresponding to more than 80% mass loss of the initial sample. The increase of the size of the n-alkyl side group does not appreciably increase the thermal stability of the polymer chain. Two thermal degradation steps were also observed for the PNVP-b-PVDc blocks. The low temperature peak corresponds to the decomposition of the PVDc component, whereas the high temperature step to the PNVP component. As in the case of the PNVP-b-PVBu copolymers the composition of the PNVP-b-PVDc is reflected in the relative size of the two decomposition peaks in the DTG analysis.
The TGA results for the PNVP-b-PVSt copolymers are given in Table 8, whereas the TGA and DTG plots in Figure 14 and Figure 15 respectively. The thermal decomposition of PVSt is substantially different compared to that reported for PVBu and PVDc. The main decomposition event is observed at 320oC, as in the other polymers. However, a considerably important degradation step, corresponding to about 40% mass loss, is obvious at much lower temperatures, around 195oC. In addition, a shoulder at temperatures higher than 400oC is visible indicating a complex mechanism of thermal degradation.
Taking into account that the degradation step at 320oC is more or less common to all PVEs this should be attributed to the decomposition of the main poly(vinyl ester) backbone, whereas the lower temperature peak can be attributed to the thermal decomposition of the side n-alkyl group of PVSt.
All the PNVP-b-PVSt copolymers showed three-step degradation patterns. The high temperature step is attributed to the thermally more stable PNVP block, whereas the other peaks from DTG analysis correspond to the less thermally PVSt block. As in the previous cases the composition of the copolymers is reflected in the relative size of the various peaks.
The thermal decomposition of several members of the PVEs family has been thoroughly studied in the literature. [81,82] An autocatalytic degradation mechanism takes place for the first members of the PVEs, specially PVAc. The higher members of this family of polymers degrade through first order kinetics. A combination of autocatalytic and first order kinetics approaches seems to exist for the intermediate members of PVEs, therefore both mechanisms have been examined in these cases. A two-step decomposition process was revealed for most PVEs. The first one involves the removal of the ester group as an acid and then the decomposition of the remaining chain. These conclusions were more or less verified in the present study.
4. Conclusions
Well-defined block copolymers of N-vinyl pyrrolidone (NVP) and various Vinyl Esters (VEs) with aliphatic side groups, namely, vinyl butyrate (VBu), vinyl decanoate (VDc) and vinyl stearate (VSt) were synthesized by RAFT polymerization techniques. The sequential addition of monomers was employed starting from the polymerization of NVP in benzene solutions at 60oC using AIBN as radical initiator and O-ethyl S-(phthalimidylmethyl) xanthate as the CTA. The polymerization of the VEs was conducted in dioxane solutions at 80oC using the PNVP block as the macro-CTA and AIBN as the initiator. Block copolymers of various compositions were obtained. Size exclusion chromatography (SEC) analysis revealed that products of relatively low polydispersity were obtained and that there was no reason for purification of the block copolymers. Differential scanning calorimetry (DSC) analysis showed that the PNVP-b-PVBu and PNVP-b-PVDc block copolymers are microphase separated. However, due to the rather low molecular weight of the samples partial mixing of the two phases was observed. On the other hand, the PNVP-b-PVSt block copolymers are semicrystalline. The samples were again microphase separated, since both the crystalline phase of the PVSt blocks and the amorphous phase of the PNVP blocks were traced in the thermograms. A third amorphous phase, due to the partial mixing of the two constituent blocks was also obtained. The thermal stability of the copolymers was studied via Thermogravimetric Analysis (TGA) and Differential Thermogravimetry (DTG). PNVP is a considerably more thermally stable polymer that the PVEs. A rather simple mechanism of thermal decomposition is applied for PNVP, whereas at least two steps of thermal degradation were traced for the various PVEs homopolymers. The block copolymers combined the thermal decomposition behavior of their constituent components showing a complex pattern of thermal degradation.
Funding
This research is co-financed by Greece and the European Union (European Social Fund- ESF) through the Operational Programme «Human Resources Development, Education and Lifelong Learning» in the context of the project “Strengthening Human Resources Research Potential via Doctorate Research” (MIS-5000432), implemented by the State Scholarships Foundation (ΙΚΥ).
Acknowledgments
This research is co-financed by Greece and the European Union (European Social Fund- ESF) through the Operational Programme «Human Resources Development, Education and Lifelong Learning» in the context of the project “Strengthening Human Resources Research Potential via Doctorate Research” (MIS-5000432), implemented by the State Scholarships Foundation (ΙΚΥ).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Rinno, H. Poly(vinyl esters) In Ullmann’s Encyclopedia of Industrial Chemistry; Wiley-VCH Verlag GmbH&Co. KGaA: Weinheim, Germany, 2000; Volume 28, pp. 469–479. [Google Scholar]
- Ljungberg, N,; Wesslén, B. Preparation and properties of plasticized poly(lactic acid) films. Biomacromolecules, 2005, 6, 1789–1796. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, N.; Kawasumi, M.; Kato, M.; Usuki, A.; Okada, A. Preparation and mechanical properties of polypropylene=clay hybrids using a maleic anhydride-modified polypropylene oligomers. J. Appl. Polym. Sci., 1998, 67, 87–92. [Google Scholar] [CrossRef]
- Mark, H.F.; Bikales, N.; Overberger, C.G.; Menges, G. ; KroschwitzJ.I. Encyclopedia of Polymer Science and Engineering, 2nd Ed. ed; Wiley: New York, 1989; Vol. 17, p. 420. [Google Scholar]
- Ahmad, H.; Hasan, M.K.; Miah, M.A.J.; Ali, A.M.I.; Tauer, K. Solvent effect on the emulsion copolymerization of MMA and LMA in aqueous media. Polymer, 2011, 52, 3925–3932. [Google Scholar] [CrossRef]
- Yang, D.J.; Fu, X.K.; Gong, Y.F. Study on the preparation and performances of P(VAc-MMA) polymer electrolytes for lithium ion battery. J. Polym. Sci., 2008, 26, 375–380. [Google Scholar] [CrossRef]
- Baily, N.; Pound-Lana. ; Klumperman, B. Synthesis, characterization and self-assembly of poly(N-vinylpyrrolidone)-block-poly(vinyl acetate). Aust. J. Chem., 2012, 65, 1124–1131. [Google Scholar] [CrossRef]
- Braun, D.; Leiss, J.; Bergmann, M.J.; Hellmann, G.P. Miscibility behavior of various polymers made of alkyl and carboxyl groups. Eur. Polym. J. 1993, 29, 225–230. [Google Scholar] [CrossRef]
- Saikia, P.J.; Baruah, S.D. Structural and thermal behavior of comb-like polymer having n-octadecyl side chains. J. Appl. Polym. Sci., 2007, 104, 1226–1231. [Google Scholar] [CrossRef]
- Reimschuessel, H.K. Glass-transition temperature of comblike polymers-effects of side chain length and backbone chain structure. J. Polym. Sci. Polym. Chem. Ed., 1979, 17, 2447–2457. [Google Scholar] [CrossRef]
- Inomata, K, Sakamaki, Y. ; Nose, T.; Sasaki, S. Solid-state structure of comb-like polymers having n-octadecyl side chains. 2. Crystalline-amorphous layered structure. Polym. J., 1996, 28, 992–999. [Google Scholar] [CrossRef]
- Morossof, N.; Morawetz, H.; Post, B. Polymerization in crystalline state. 7. A crystallographic study of radiation-initiated polymerization in single crystals of vinyl stearate. J. Am. Chem. Soc. 1965, 87, 3035–3036. [Google Scholar] [CrossRef]
- Platé, N.A.; Shibaev, V.P. Comb-like polymers. Structure and properties. J. Polym. Sci. Macromol. Rev., 1974, 8, 117–253. [Google Scholar] [CrossRef]
- Jordan, E.F.; Feldeisen, D.W.; Wrigsley, A.N. Side-chain crystallinity. 1. Heats of fusion and melting transitions on selected homopolymers having long side chains. J. Polym. Sci. Part A-1, 1971, 9, 1835–1836. [Google Scholar] [CrossRef]
- Shibasaki, Y.; Fukuda, K. Thermal behavior of vinyl esters of long-chain fatty-acids and their comblike polymers. Thermochimica Acta, 1985, 88, 211–216. [Google Scholar] [CrossRef]
- Hsieh, H.W.S.; Post, B.; Morawetz, H. Crystallographic study of polymers exhibiting side-chain crystallization. J. Polym. Sci. Polym. Phys. Ed. 1976, 14, 1241–1255. [Google Scholar] [CrossRef]
- Greenberg, S.A.; Alfrey, T. Side chain crystallization of normal-alkyl polymethacrylates and polyacrylates. J. Am. Chem. Soc., 1954, 76, 6280–6285. [Google Scholar] [CrossRef]
- Port, W.S.; Hansen, J.E.; Jordan Jr., E. F.; Dietz, T.J.; Swern, D. Polymerizable derivatives of long-chain fatty acids. 4. Vinyl esters., J. Polym. Sci. 1951, 7, 207–220. [Google Scholar]
- De Merlis, C.C.; Schoneker, D.R. Review of the oral toxicity of polyvinyl alcohol (PVA). Food Chem. Toxicol. 2003, 41, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Tadavarthy, S.M.; Moller, J.H.; Amplatz, K. Polyvinyl alcohol (Ivalon)--a new embolic material. Am. J. Roentgenol., Radium Ther. Nucl. Med., 1975, 125, 609–616. [Google Scholar] [CrossRef] [PubMed]
- Davidson, G.S.; Terbrugge, K.G. Histologic long-term follow-up after embolization with polyvinyl alcohol particles. Am. J. Neuroradiol., 1995, 16, 843–846. [Google Scholar] [PubMed]
- Kojima, Y.; Maeda, H. Evaluation of poly(vinyl alcohol) for protein tailoring: Improvements in pharmacokinetic properties of superoxide dismutase. J. Bioact. Compat. Polym. 1993, 8, 115–131. [Google Scholar] [CrossRef]
- Hassan, C.M.; Peppas, N.A. Structure and applications of poly(vinyl alcohol) hydrogels produced by conventional crosslinking or by freezing/thawing methods. Adv. Polym. Sci. 2000, 153, 37–65. [Google Scholar]
- Bunck, D.N.; Sorenson, G.P.; Mahanthapp, M.K. Cobalt-mediated radical polymerization routes to poly (vinyl ester) block copolymers J. Polym. Sci. Polym. Part A: Chem. Ed., 2011, 49, 242–249. [Google Scholar] [CrossRef]
- Yamada, K.; Nakano, T.; Okamoto, Y. Synthesis of syndiotactic poly(vinyl alcohol) from fluorine-containing vinyl esters. Polym. J., 1998, 42, 9679–9686. [Google Scholar] [CrossRef]
- Murray, R.E.; Lincoln, D.M. Catalytic route to vinyl esters. Catal. Today, 1992, 13, 93–102. [Google Scholar] [CrossRef]
- Charmot, D.; Corpart, P.; Adam, H.; Zard, S.Z.; Biadatti, T.; Bouhadir, G. Controlled radical polymerization in dispersed media. Macromol. Symp. 2000, 150, 23–32. [Google Scholar] [CrossRef]
- Matioszek, D.; Brusylovets, O.; Wilson, D.J.; Mazières, S.; Destarac, M. Reversible addition-fragmentation chain-transfer polymerization of vinyl monomers with N,N-dimethylsdiselenocarbamates. J. Polym. Sci. Part A: Polym, Chem., 2013, 51, 4361–4368. [Google Scholar] [CrossRef]
- Benard, J.; Favier, A.; Zhang, L.; Nilararoya, A.; Davis, T.P.; Barner-Kowollik, C.; Stenzel, M.H. Poly(vinyl ester) star polymers via xanthate-mediated living radical polymerization: From poly(vinyl alcohol) to glycopolymer stars. Macromolecules 2005, 38, 5475–5484. [Google Scholar] [CrossRef]
- Gu, Y.; He, J.; Li, C.; Zhou, C.; Song, S.; Yang, Y. Block copolymerization of vinyl acetate and vinyl neo-decanoate mediated by dithionosulfide. Macromolecules, 2010, 43, 4500–4510. [Google Scholar] [CrossRef]
- Lipscomb, C.E.; Mahanthappa, M.K. Poly(vinyl ester) block copolymers synthesized by reversible addition-fragmentation chain transfer polymerizations. Macromolecules, 2009, 42, 4571–4579. [Google Scholar] [CrossRef]
- Lipscomb, C.E.; Mahanthappa, M.K. Microphase separation mode-dependent mechanical response in poly(vinyl ester)/PEO triblock copolymers. Macromolecules, 2011, 44, 4401–4409. [Google Scholar] [CrossRef]
- Odian, G. Principles of polymerization, Wiley Interscience, 2004.
- Britton, D.; Heatley, F.; Lovell, P.A. Chain transfer to polymer in free-radical bulk and emulsion polymerization of vinyl acetate studied by NMR spectroscopy. Macromolecules, 1998, 31, 2828–2837. [Google Scholar] [CrossRef]
- Lindemann, M.K. The mechanism of vinyl acetate in vinyl polymerization. Ham, G.E., Ed.; Dekker: New York, NY, USA, 1967; pp. 252–255. [Google Scholar]
- Kwak, Y.; Goto, A.; Fukuda, T.; Kobayashi, Y.; Yamago, S. A systematic study on activation process in organotellurium-mediated living radical polymerization of styrene, methyl methacrylate, methyl acrylate and vinyl acetate. Macromolecules, 2006, 39, 4671–4679. [Google Scholar] [CrossRef]
- Flory, P.J.; Leutner, F.S. Occurrence of head-to-head arrangements of structural units in polyvinyl alcohol. J. Polym. Sci., 1948, 3, 880–890. [Google Scholar] [CrossRef]
- Iovu, M.C.; Matyjaszewski, K. Controlled/living radical polymerization of vinyl acetate by degenerative transfer with alkyl iodides. Macromolecules, 2003, 36, 9346–9354. [Google Scholar] [CrossRef]
- Borkar, S.; Sen, A. Controlled copolymerization of vinyl acetate with 1-alkenes and their fluoro derivatives by degenerative transfer. J. Polym. Sci. Polym. Part A: Chem. Ed., 2005, 43, 3728–3736. [Google Scholar] [CrossRef]
- Koumura, K.; Satoh, K.; Kamigaito, M.; Okamoto, Y. Iodine transfer radical polymerization of vinyl acetate in fluoroalcohols for simultaneous control of molecular weight, stereospecificity, and regiospecificity. Macromolecules, 2006, 39, 4054–4061. [Google Scholar] [CrossRef]
- Yamago, S. Development of organotellurium-mediated and organostibine-mediated living radical polymerization reactions. J. Polym. Sci. Polym. Part A: Chem. Ed., 2006, 44, 1–12. [Google Scholar] [CrossRef]
- Shaver, M.P.; Hanhan, M.F.; Jones, M.R. Controlled radical polymerization of vinyl acetate mediated by a vanadium complex. Chem. Commun., 2010, 46, 2127–2128. [Google Scholar] [CrossRef] [PubMed]
- Debuigne, A.; Caillie, J.-R.; Jérôme, R. Highly Efficient cobalt-mediated radical polymerization of vinyl acetate Angew. Chem., 2005, 44, 1101–1104. [Google Scholar]
- Debuigne, A.; Poli, R.; Jérôme, C.; Jérôme, R.; Detrembleur, C. Overview of cobalt-mediated radical polymerization: Roots, state of the art and future prospects. Progr. Polym. Sci., 2009, 34, 211–239. [Google Scholar] [CrossRef]
- Bryaskova, R.; Detrembleur, C.; Debuigne, A.; Jérôme, R. Cobalt-mediated radical polymerization (CMRP) of vinyl acetate initiated by redox systems: Toward the scale-up of CMRP. Macromolecules, 2006, 39, 8263–8268. [Google Scholar] [CrossRef]
- Roka, N.; Kokkorogianni, O.; Kontoes-Georgoudakis, P.; Choinopoulos, I.; Pitsikalis, M. Recent advances in the synthesis of complex macromolecular architectures based on poly(N-vinyl pyrrolidone) and the RAFT polymerization technique. Polymers 2022, 14, 701. [Google Scholar] [CrossRef] [PubMed]
- Tilottama, B.; Manojkumar, K.; Haribabu, P.M. , Vijayakrishna, K. A short review on RAFT polymerization of less activated monomers. J. Macromol. Sci. Part A, Pure and Appl. Chem., 2022, 59, 180–201. [Google Scholar] [CrossRef]
- Teodorescu, M.; Bercea, M. Poly(vinylpyrrolidone) – A Versatile Polymer for Biomedical and Beyond Medical Applications. Polymer-Plastics Technology and Engineering 2015, 54, 923–943. [Google Scholar] [CrossRef]
- Franco, F.; De Marco, I. The Use of Poly(N-vinyl pyrrolidone) in the Delivery of Drugs: A Review. Polymers 2020, 12, 1114. [Google Scholar] [CrossRef] [PubMed]
- Moulay, S. Molecular iodine/polymer complexes. J. Polym. Eng. 2013, 33(5), 389–443. [Google Scholar] [CrossRef]
- Kurakula, M.; Koteswara Rao, G.S.N. Pharmaceutical assessment of polyvinylpyrrolidone (PVP): As excipient from conventional to controlled delivery systems with a spotlight on COVID-19 inhibition. J. Drug Deliv. Sci. Technol. 2020, 60, 102046. [Google Scholar] [CrossRef] [PubMed]
- Turner, D.T.; Schwartz, A. The glass transition temperature of poly(N-vinyl pyrrolidone) by differential scanning calorimetry. Polymer, 1985, 26, 757–762. [Google Scholar] [CrossRef]
- Del Pilar Buera, M.; Levi, G.; Karel, M. Glass transition in poly(vinylpyrrolidone): Effect of molecular weight and diluents. Biotechnol. Progr. 1992, 8, 144–148. [Google Scholar] [CrossRef]
- Wang, H.; Kolodka, E.; Tande, B.M. Thermomechanical and rheological studies of copolymers of methyl methacrylate with a series of vinyl esters. I&EC Research, 2013, 52, 5111–5119. [Google Scholar]
- Nzé, R.-P.; Colombani, O.; Nicol, E. Synthesis of poly(vinyl laurate)-b-poly(vinyl stearate) deblock copolymers by cobalt-mediated radical polymerization in solution. J. Polym. Sci. Part A: Polym. Chem., 2012, 50, 4046–4054. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, C.; Tian, J.; Xie, Y.; Zhang, K. A comparative study of self-assembled superstructures from cellulose stearoyl ester and poly(vinyl stearate). Macromol. Chem. Phys., 2018, 219, 1800229. [Google Scholar] [CrossRef]
- Perrier, S. 50th Anniversary Perspective: RAFT Polymerization - A User Guide. Macromolecules 2017, 50, 7433–7447. [Google Scholar] [CrossRef]
- Stenzel, M.H.; Cummins, L.; Roberts, G.E.; Davis, T.R.; Vana, P. ; Barner-Kowollik Xanthate mediated living polymerization of vinyl acetate: A systematic variation in MADIX/RAFT agent structure. Macromol. Chem. Phys., 2003, 204, 1160–1168. [Google Scholar] [CrossRef]
- Favier, A.; Barner-Kowollik, C.; Davis, T.P.; Stenzel, M.H. A Detailed On-line FT/NIR and 1 H NMR spectroscopic investigation into factors causing inhibition in xanthate-mediated vinyl acetate polymerization. Macromol. Chem. Phys., 2004, 205, 925–936. [Google Scholar] [CrossRef]
- Tong, Y.-Y.; Dong, Y.-Q.; Du, F.-S.; Li, Z.-C. Synthesis of well-defined poly(vinyl acetate)-b-polystyrene by combination of ATRP and RAFT polymerization. Macromolecules, 2008, 41, 7339–7346. [Google Scholar] [CrossRef]
- Wan, D. , Satoh, K., Kamigaito, M., Okamoto, Y. Xanthate-mediated radical polymerization of N-vinylpyrrolidone in fluoroalcohols for simultaneous control of molecular weight and tacticity. Macromolecules 2005, 38, 10397–10405. [Google Scholar] [CrossRef]
- Postma, A. , Davis, T. P., Li, G., Moad, G., O’Shea, M. S. RAFT polymerization with phthalimidomethyl trithiocarbonates or xanthates. On the origin of bimodal molecular weight distributions in living radical polymerization. Macromolecules 2006, 39, 5307–5318. [Google Scholar] [CrossRef]
- Hadjichristidis, N.; Iatrou, H.; Pispas, S.; Pitsikalis, M. Anionic polymerization: High vacuum techniques. J. Polym. Sci. Part A Polym. Chem. 2000, 38, 3211–3234. [Google Scholar] [CrossRef]
- Uhrig, D.; Mays, J.W. Experimental techniques in high-vacuum anionic polymerization. J. Polym. Sci. Part A Polym. Chem. 2005, 43, 6179–6222. [Google Scholar] [CrossRef]
- Nakabayashi, K.; Mori, H. Recent progress in controlled radical polymerization of N-vinyl monomers. Eur. Polym. J. 2013, 49, 2808–2838. [Google Scholar] [CrossRef]
- Daniel, J.K. A guide to the synthesis of block copolymers using reversible-addition fragmentation chain transfer (RAFT) polymerization. Chem. Soc. Rev. 2014, 43, 495–505. [Google Scholar]
- Renjith, D.; Raveendra, L.B.; Redouane, B.; Nathalie, M.; Yves, G. Controlled Radical Polymerization of N-Vinylpyrrolidone by Reversible Addition-Fragmentation Chain Tranfer Process. Macromol. Symp. 2005, 229, 8–17. [Google Scholar]
- Zard, S.Z. Discovery of the RAFT/MADIX Process: Mechanistic Insights and Polymer Chemistry Implications. Macromolecules. 2020, 53, 8144–8159. [Google Scholar] [CrossRef]
- Smith, A.A.A.; Hussmann, T.; Elich, J.; Postma, A.; Alves, M.-H. Macromolecular design of poly(vinyl alcohol) by RAFT polymerization). Polym. Chem., 2012, 3, 85–88. [Google Scholar] [CrossRef]
- Roka, N.; Pitsikalis, M. Statistical Copolymers of N-Vinylpyrrolidone and Benzyl Methacrylate via RAFT: Monomer Reactivity Ratios, Thermal Properties and Kinetics of Thermal Decomposition. J. Macromol. Sci., Part A Pure Appl. Chem., 2018, 55, 222–230. [Google Scholar] [CrossRef]
- Roka, N.; Pitsikalis, M. Synthesis and micellization behavior of amphiphilic block copolymers of poly(N-Vinylpyrrolidone) and poly(Benzyl Methacrylate): Block vs statistical copolymers. Polymers 2023, 15, 2225. [Google Scholar] [CrossRef] [PubMed]
- Barrales-Rienda, J.M.; Sanchez Chaves, M.; Mazòn-Arechederra, J.M. Polymer precursors of polyacetylene. Thermal degradation of poly(vinyl esters). Part II-Effect of the n-acyl chain length on the autocatalytic thermal degradation of a homologous series of poly(vinyl n-alkyl esters) (PV-n-AEs). Polym. Degrad. Stab. 1989, 23, 279–298. [Google Scholar] [CrossRef]
- Rogers, S.S.; Mandelkern, I. Glass formation in polymers. 1. The glass transitions of the poly(n-alkyl methacrylates). J. Phys. Chem. 1957, 61, 985–990. [Google Scholar] [CrossRef]
- Rehberg, C.E.; Fischer, C.H. Properties of monomeric and polymeric alkyl acrylates and methacrylates. Ind. Eng. Chem. 1948, 40, 1429–1433. [Google Scholar] [CrossRef]
- Wiley, R.H.; Brauer, G.M. Refractometric determination of 2nd-order transition temperatures in polymers. 3. Acrylates and methacrylates. J. Polym. Sci. 1948, 3, 647–651. [Google Scholar] [CrossRef]
- Rehberg, C.E.; Fischer, C.H. Preparation and properties of the n-alkyl acrylates. J. Am. Chem. Soc. 1944, 66, 1203–1207. [Google Scholar] [CrossRef]
- Jordan Jr., E. F.; Feldeisen, D.W.; Wrigley, A.N. Side-chain crystallinity. I. Heat of fusion and melting transitions on selected homopolymers having long side chains. J. Polym. Sci.: Part A-1, 1971, 9, 1835–1852. [Google Scholar] [CrossRef]
- Flory, P.J. Thermodynamics of crystallization in high polymers. IV. A theory of crystalline states and fusion in polymers, copolymers, and their mixtures with diluents. J. Chem. Phys. 1949, 17, 223–240. [Google Scholar] [CrossRef]
- Ruzette, A.-V.G.; Banerjee, P.; Mayes, A.M.; Pollard, M.; Russell, T.P.; Jerome, R.; Slawecki, T.; Hjelm, R.; Thiyagarajan, P. Phase behavior of diblock copolymers between styrene and n-alkyl methacrylates. Macromolecules 1998, 31, 8509–8516. [Google Scholar] [CrossRef]
- Roka, N.; Kokkorogianni, O.; Pitsikalis, M. Statistical copolymers of N-vinylpyrrolidone and 2-(dimethylamino)ethyl methacrylate via RAFT: Monomer reactivity ratios, thermal properties, and kinetics of thermal decomposition. J. Polym. Sci. Polym. Chem. Ed., 2017, 15, 3776–3787. [Google Scholar] [CrossRef]
- Šimon, P.; Rybár, M. Kinetics of polymer degradation involving the splitting off of small molecules: Part 8. Thermal degradation of polyvinyl esters. Polym. Degrad. Stab. 1992, 38, 255–259. [Google Scholar] [CrossRef]
- Gilbert, J.B.; Kipling, J.J.; McEnaney, B.; Sherwood, J.N. Carbonization of polymers. I-Thermogravimetric analysis. Polymer, 1962, 3, 1–10. [Google Scholar]
Scheme 1.
Synthesis of the PNVP-b-PVEs block copolymers via RAFT polymerization.
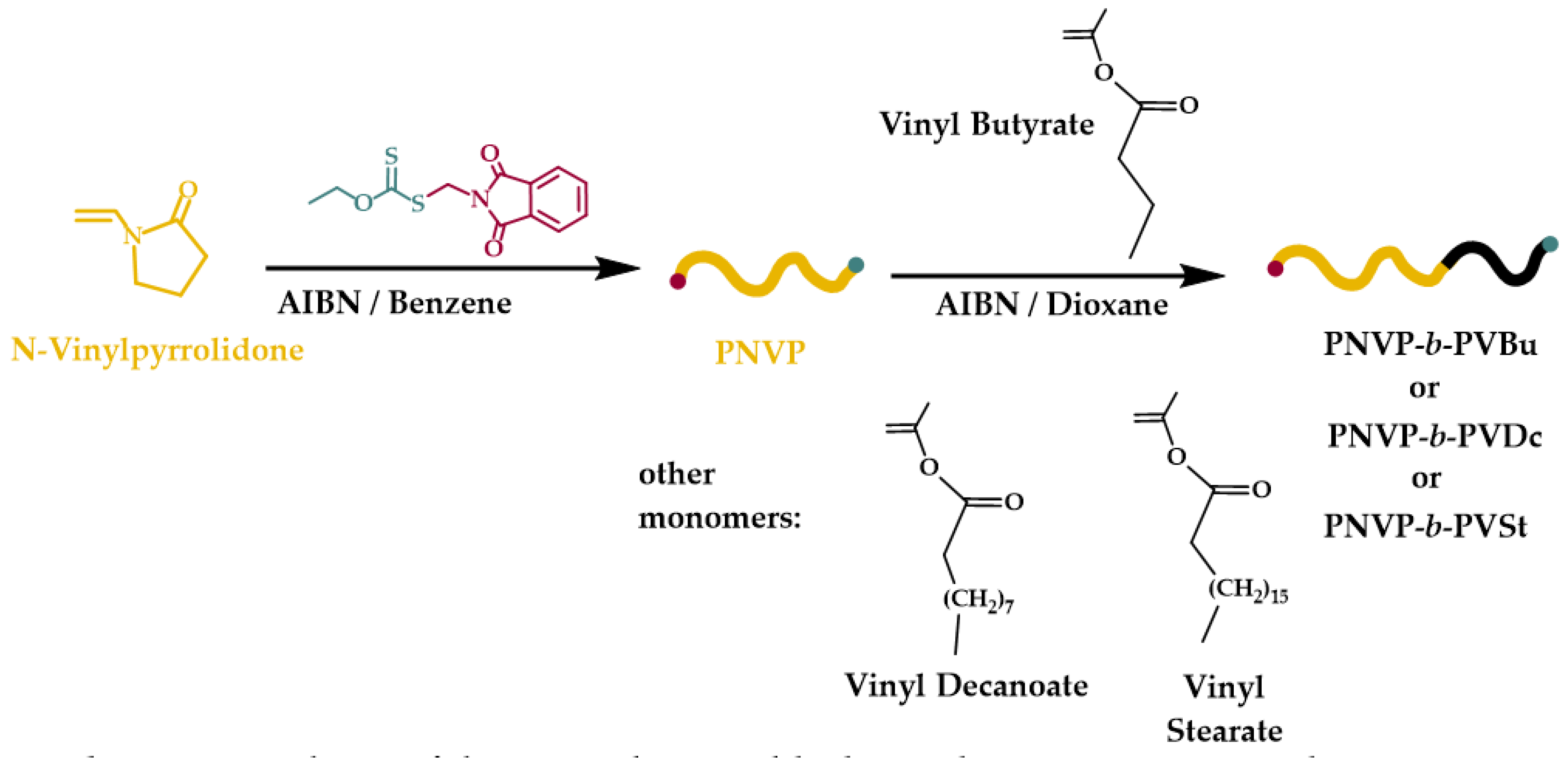
Figure 1.
SEC traces from the synthesis of the block copolymer PNVP-b-PVBu #1 and #2.
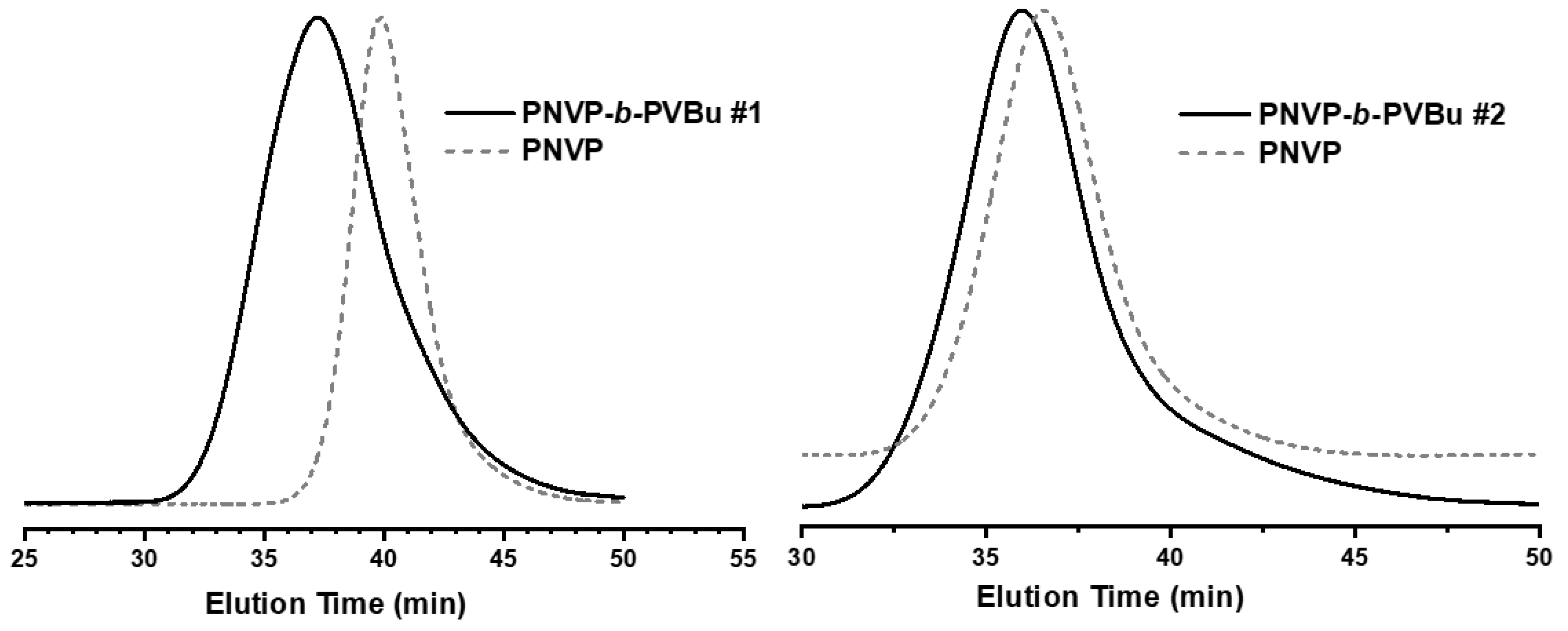
Figure 2.
SEC traces from the synthesis of the block copolymer PNVP-b-PVDc #3 and #4.
Figure 3.
SEC traces from the synthesis of the block copolymer PNVP-b-PVSt #3 and #4.
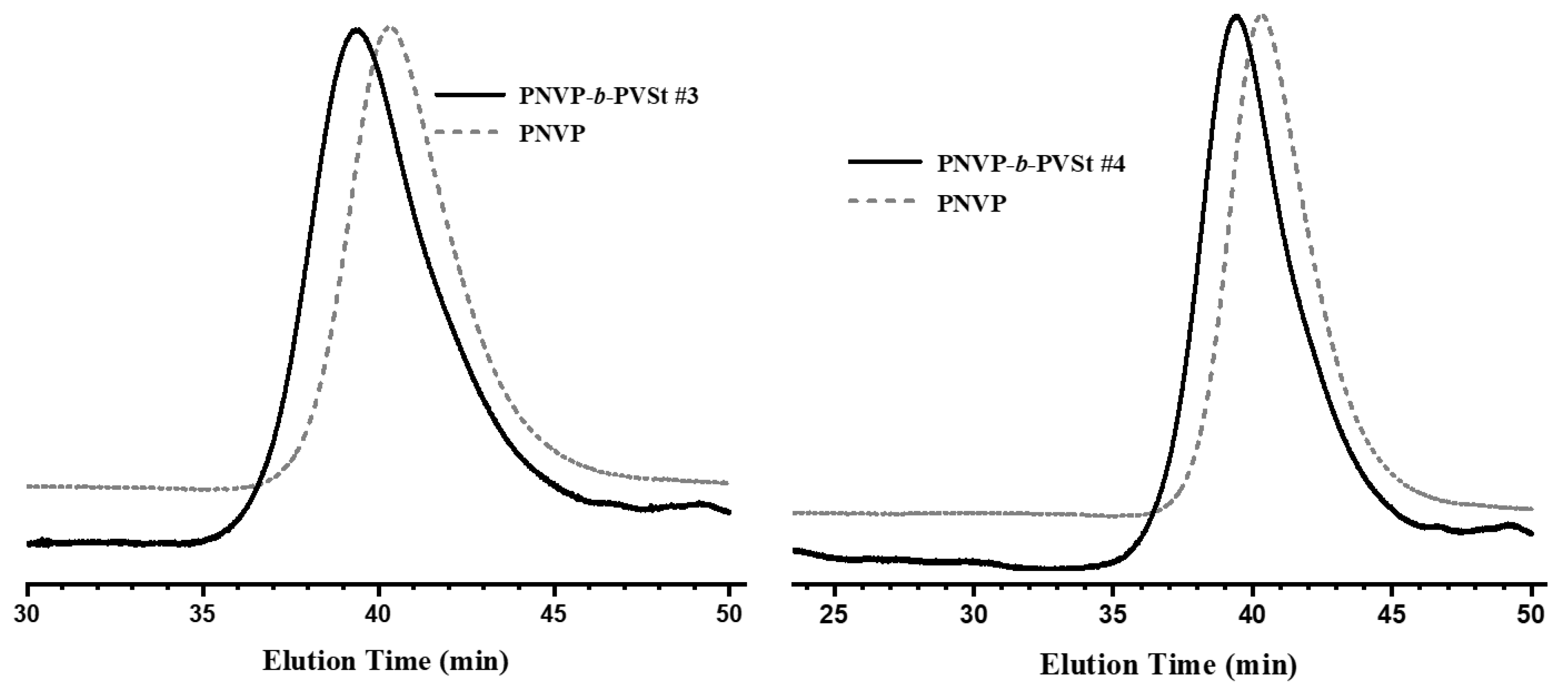
Figure 4.
1H NMR spectrum of the block copolymer PNVP-b-PVBu #2.
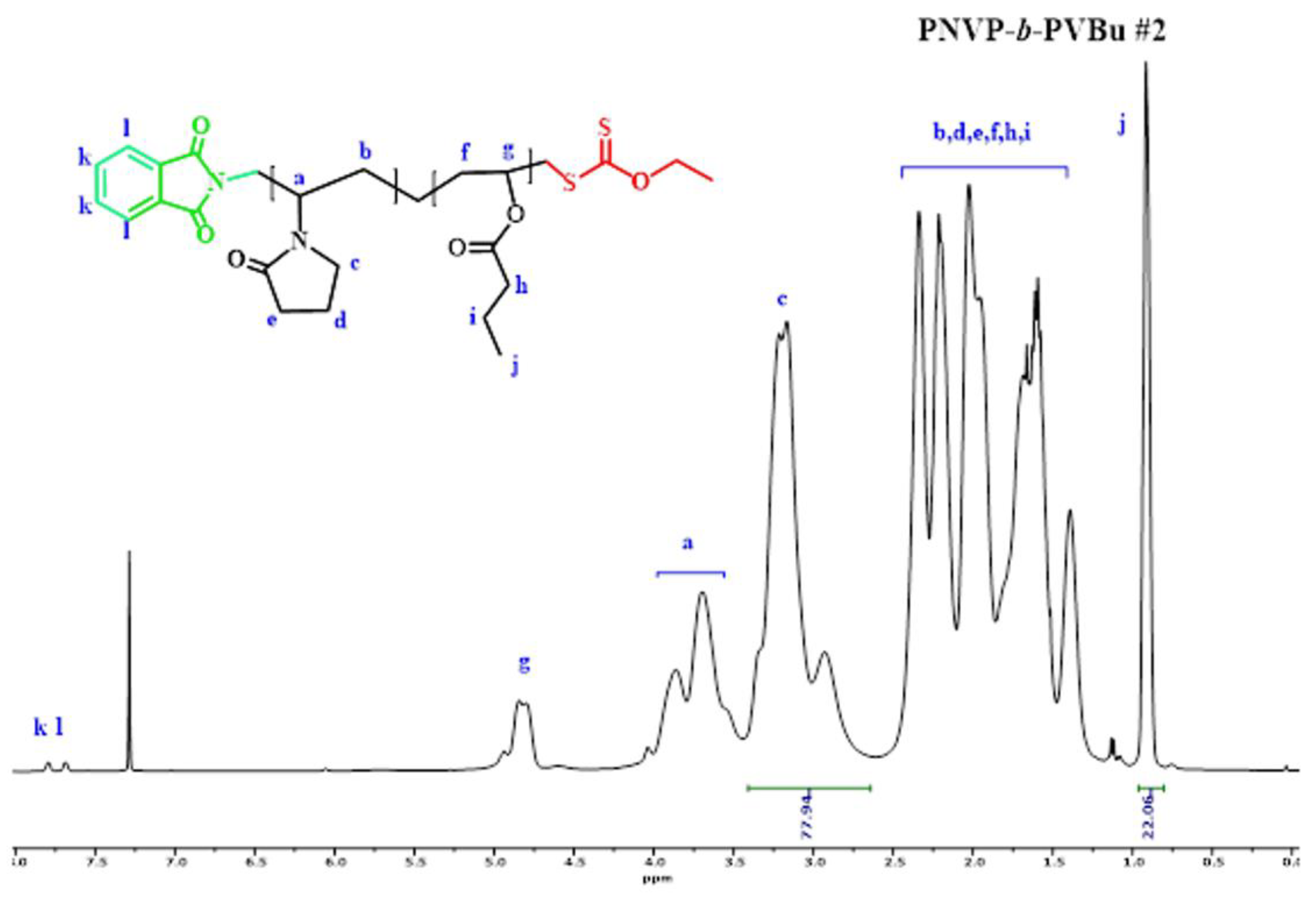
Figure 5.
1H NMR spectrum of the block copolymer PNVP-b-PVDc #1.
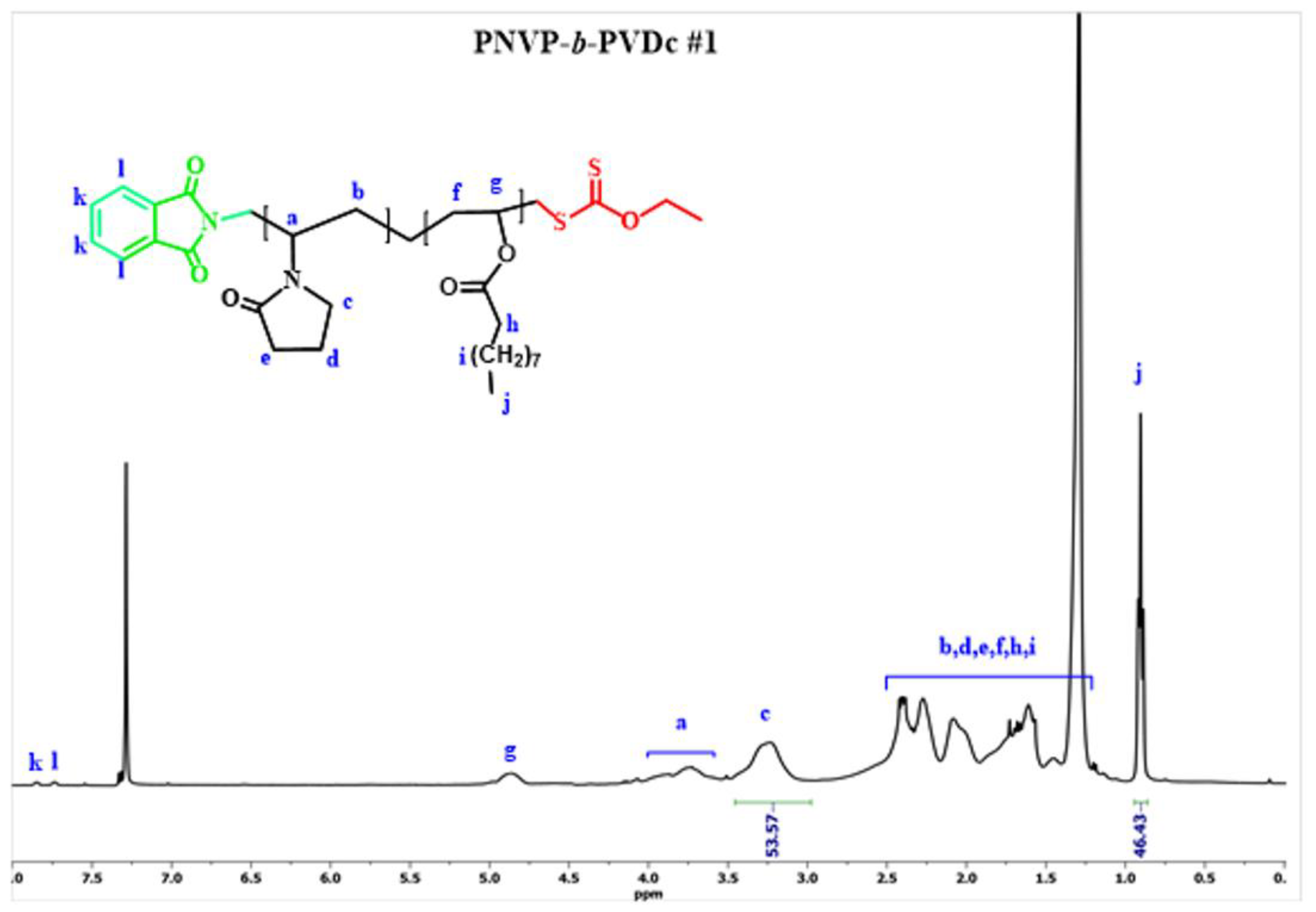
Figure 6.
1H NMR spectrum of the block copolymer PNVP-b-PVSt #2.
Figure 7.
DSC thermograms of the PNVP-b-PVBu copolymers at 10o C/min.
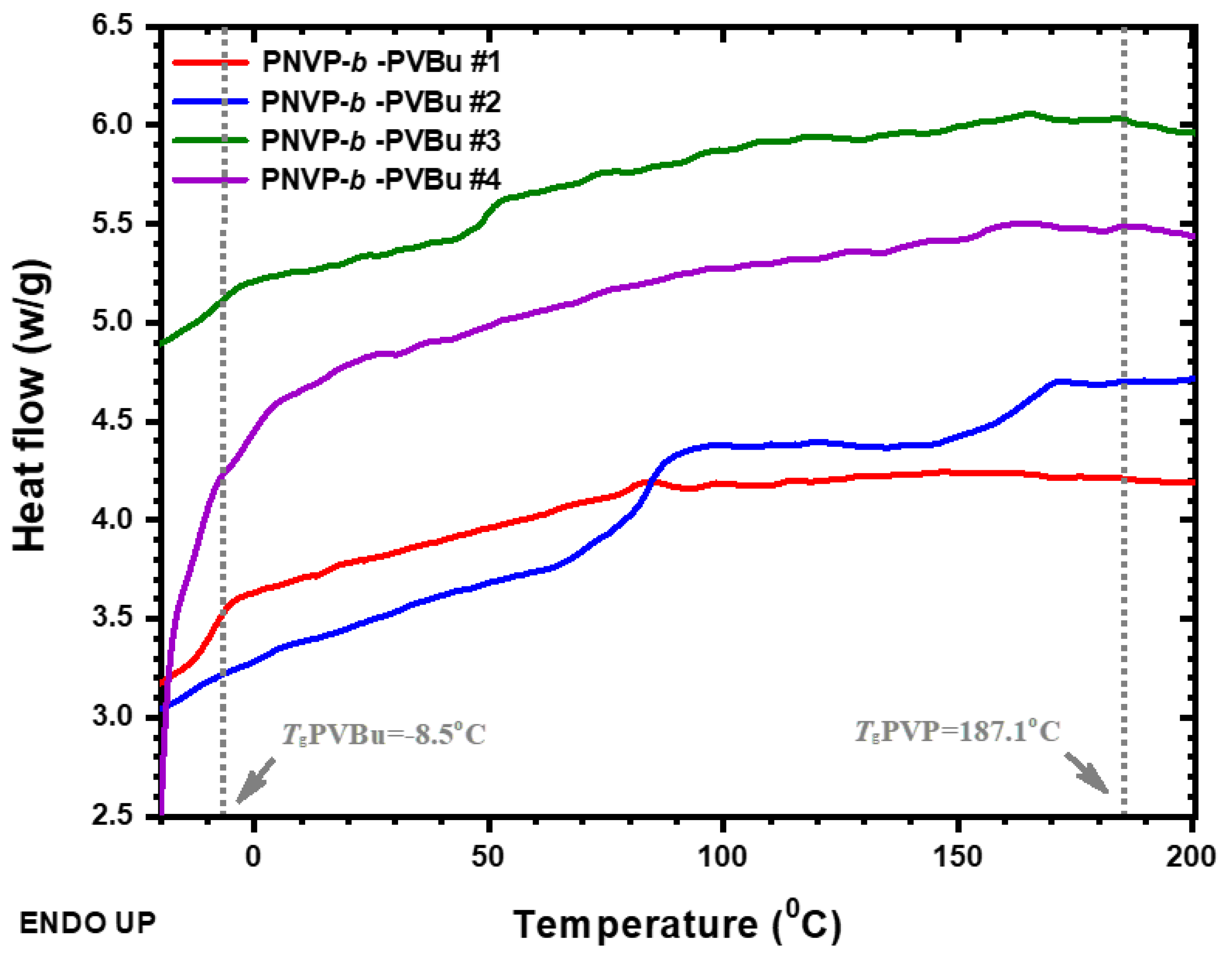
Figure 8.
DSC thermograms of the PNVP-b-PVDc copolymers at 10o C/min.
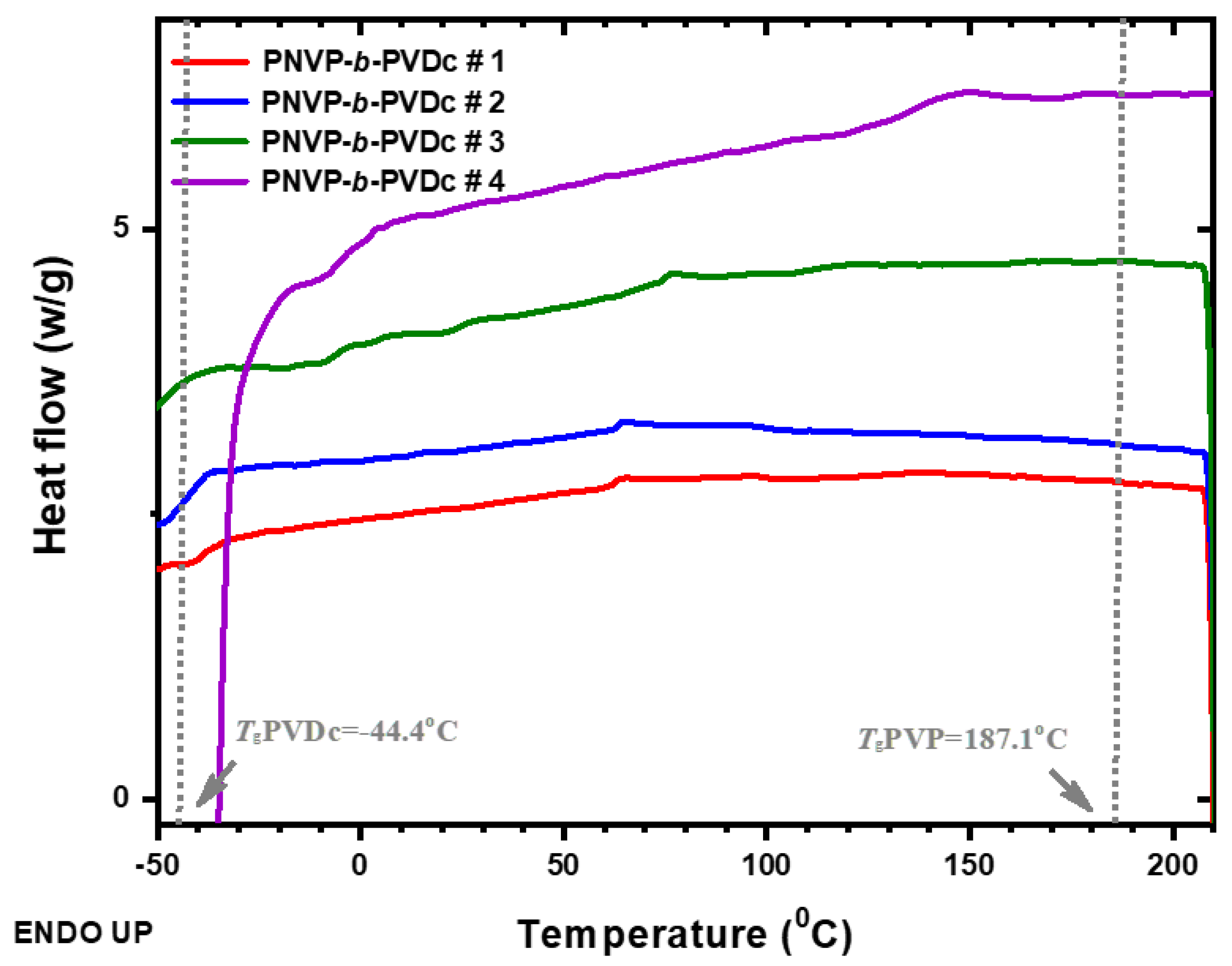
Figure 9.
DSC thermograms of the PNVP-b-PVSt copolymers at 10o C/min.
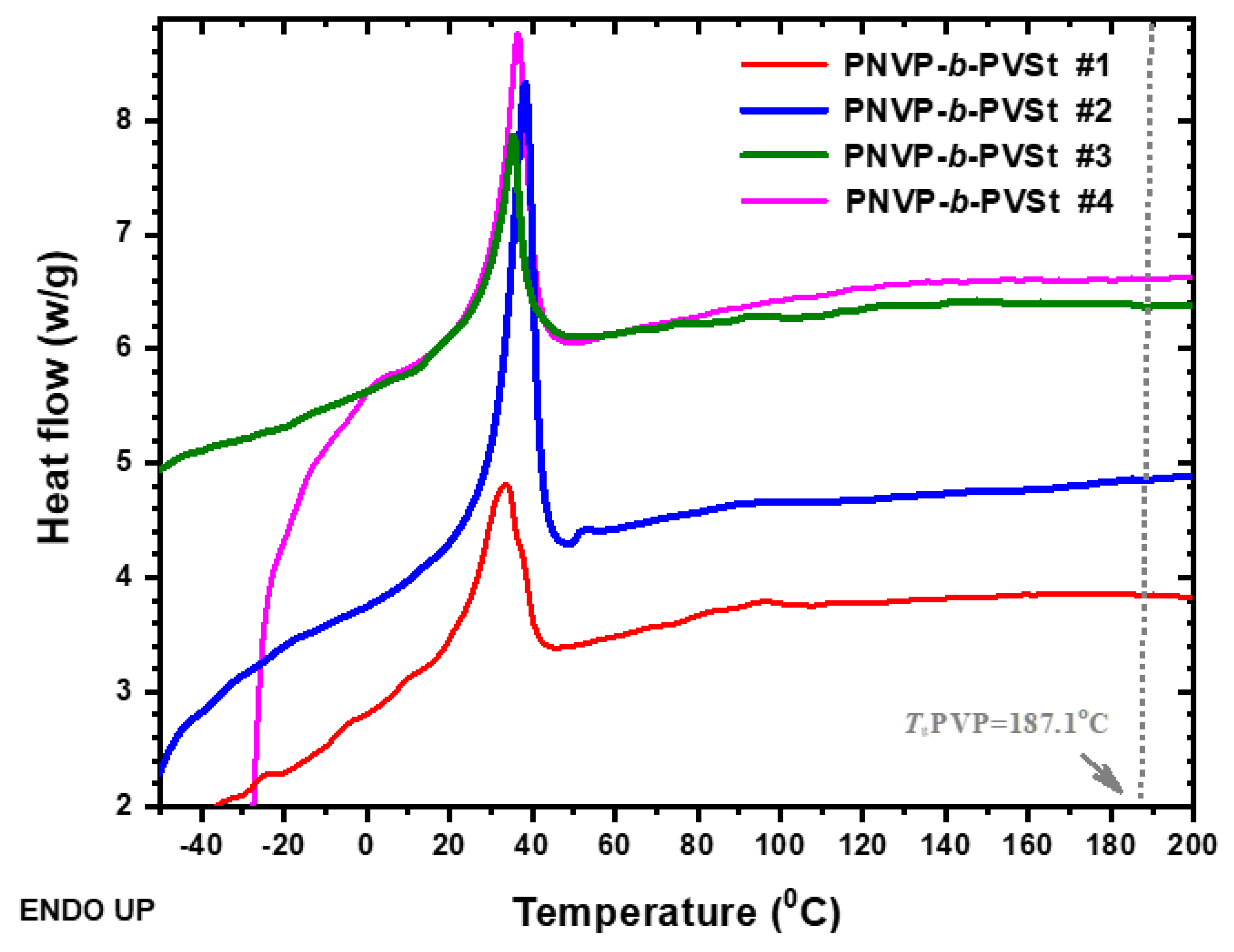
Figure 10.
TGA plots of the PNVP-b-PVBu copolymers at 10o C/min.
Figure 11.
DTG plots of the PNVP-b-PVBu copolymers at 10o C/min.
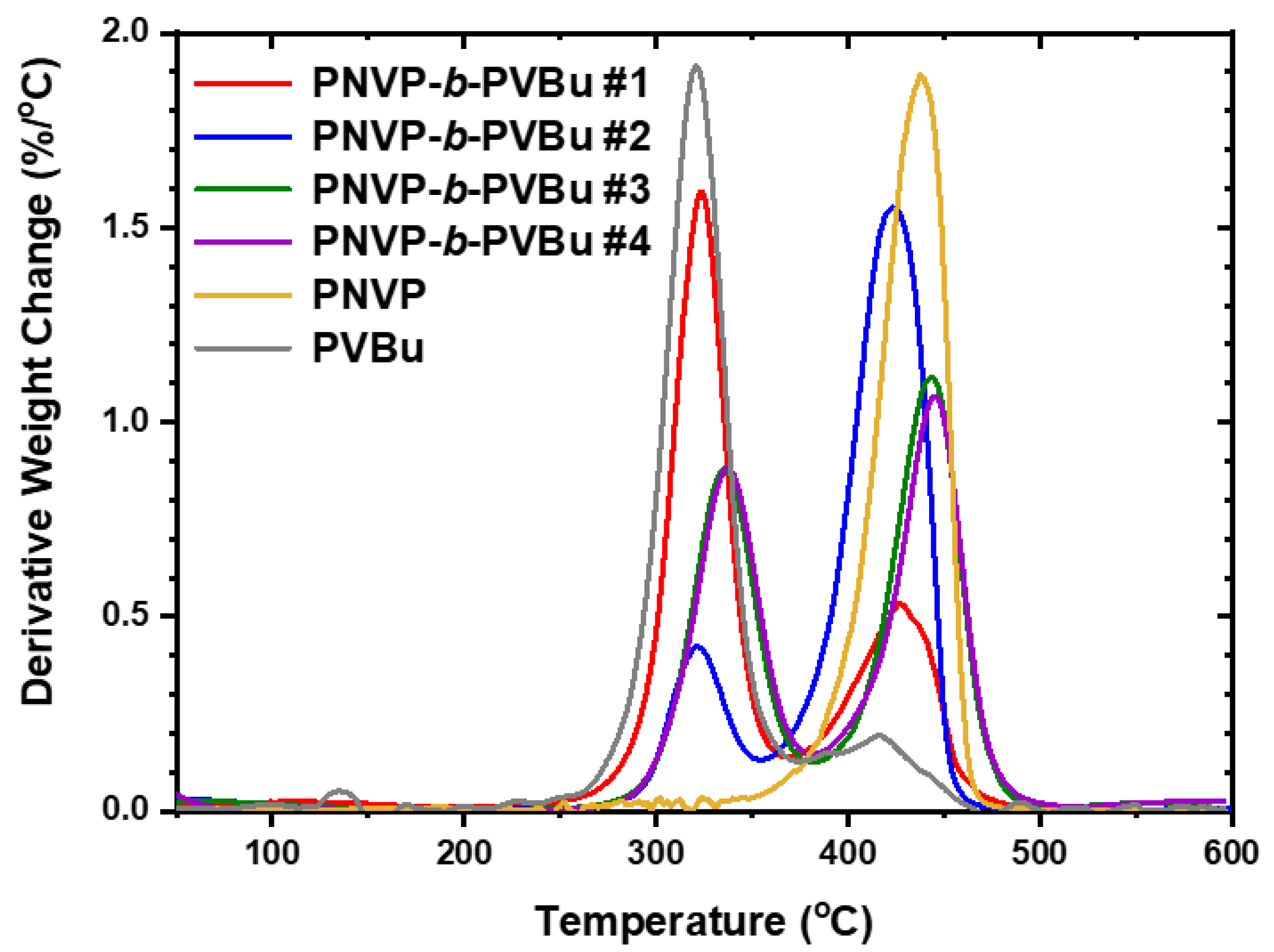
Figure 12.
TGA plots of the PNVP-b-PVDc copolymers at 10o C/min.
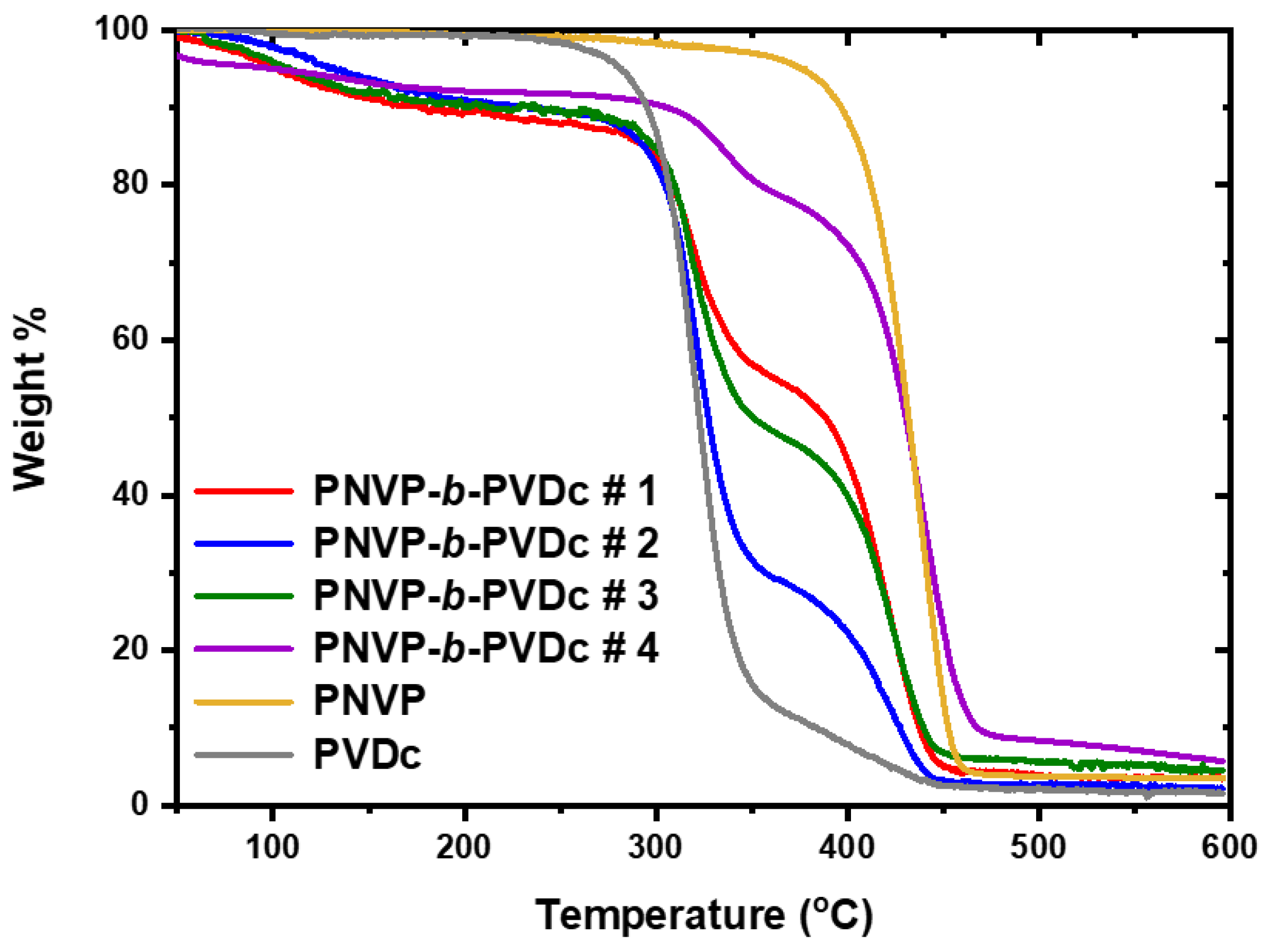
Figure 13.
DTG plots of the PNVP-b-PVDc copolymers at 10o C/min.
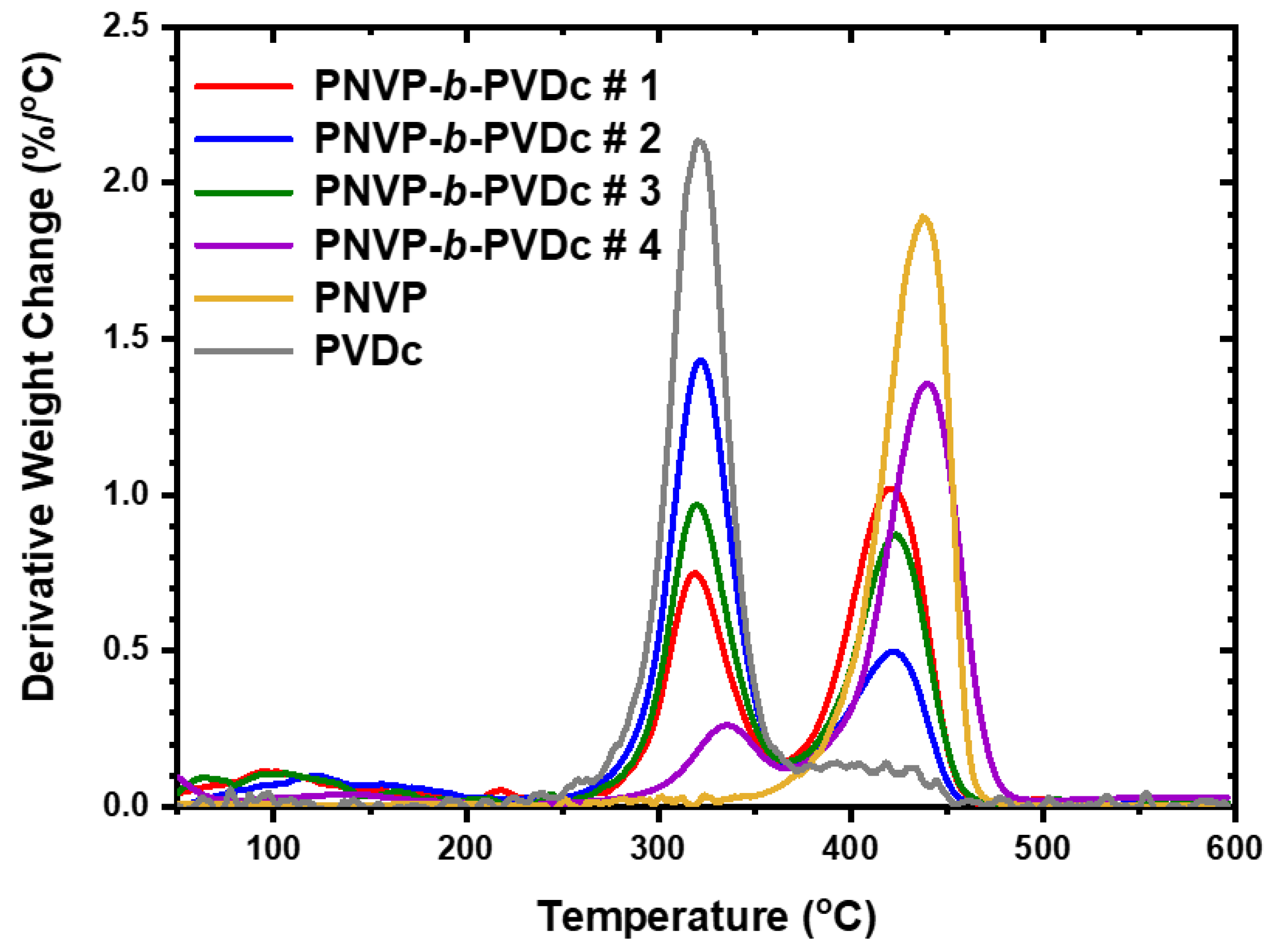
Figure 14.
TGA plots of the PNVP-b-PVSt copolymers at 10o C/min.
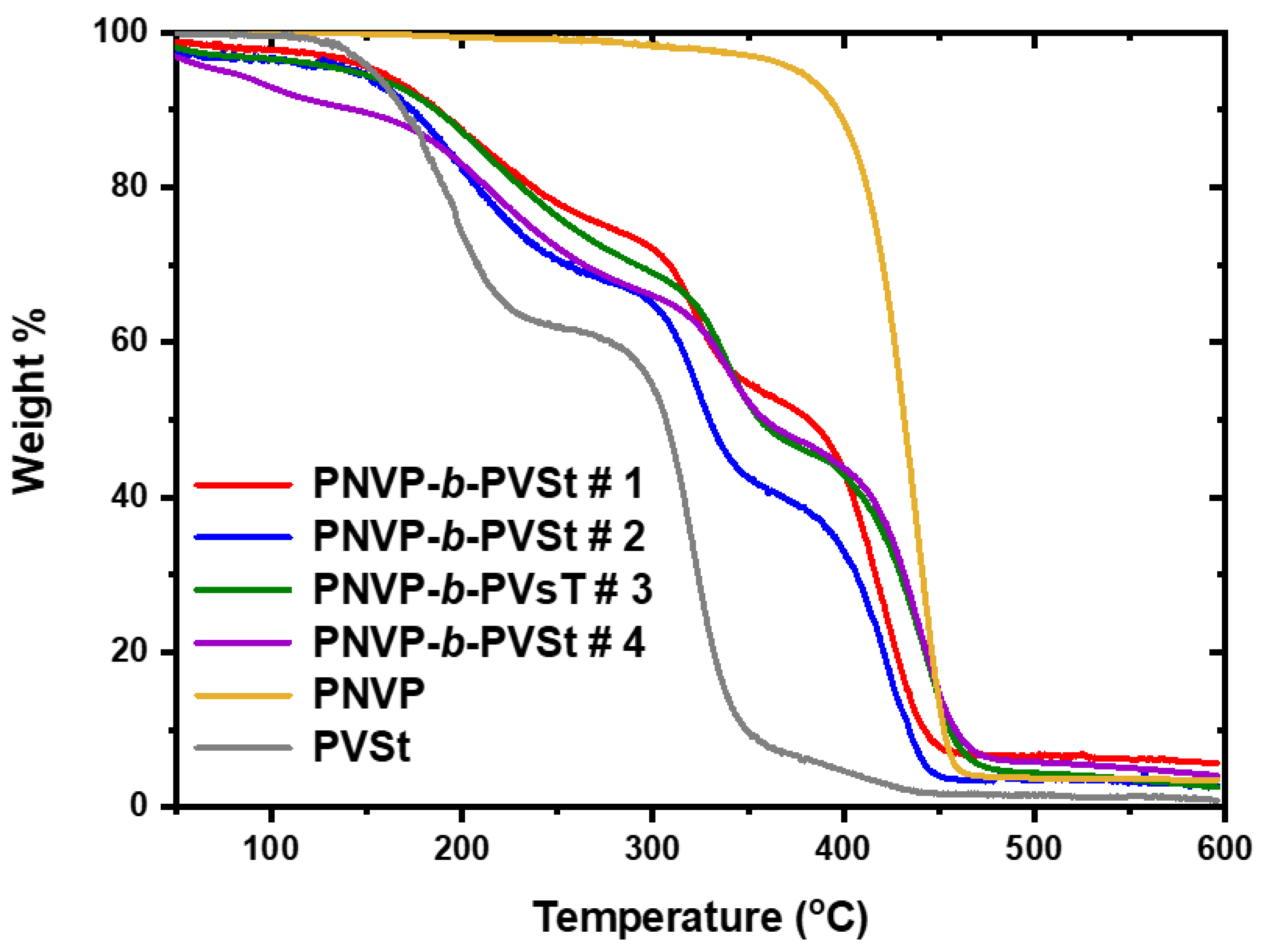
Figure 15.
DTG plots of the PNVP-b-PVSt copolymers at 10o C/min.
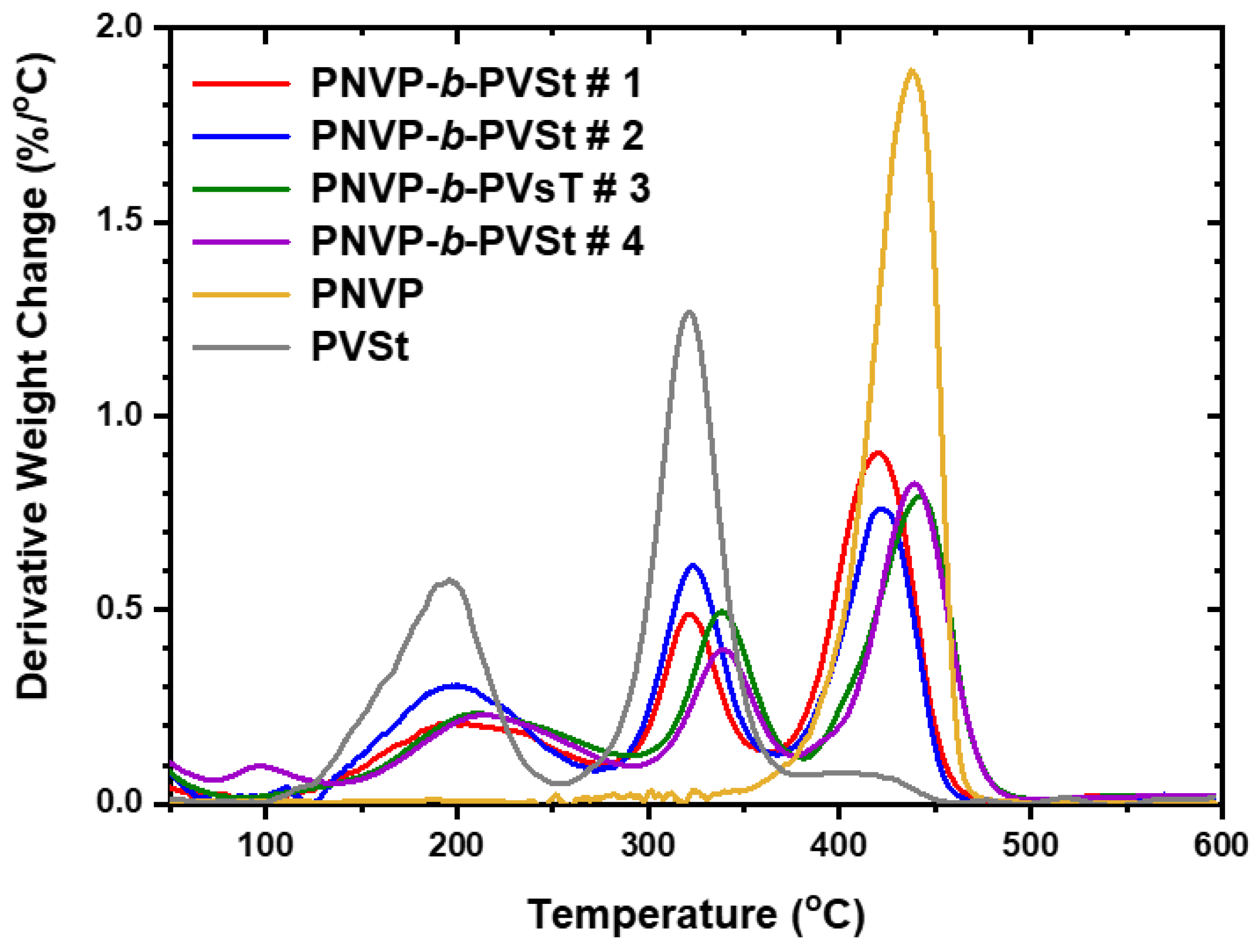

Table 1.
Quantities for the synthesis of the bock copolymers.
| Sample | PNVP (g) | AIBN (g) | Vinyl ester monomer (mL) | Dioxane (mL) | Yield % |
|---|---|---|---|---|---|
| PNVP-b-PVBu #1 | 2 | 0.0410 | 6 | 4 | 40 |
| PNVP-b-PVBu #2 | 2 | 0.0410 | 1 | 4 | 33 |
| PNVP-b-PVBu #3 | 2 | 0.0410 | 7 | 4 | 32 |
| PNVP-b-PVBu #4 | 2 | 0.0410 | 3 | 4 | 38 |
| PNVP-b-PVDc #1 | 2 | 0.0410 | 1.5 | 4 | 70 |
| PNVP-b-PVDc #2 | 2 | 0.0410 | 7 | 4 | 68 |
| PNVP-b-PVDc #3 | 2 | 0.0410 | 2.5 | 4 | 48 |
| PNVP-b-PVDc #4 | 3 | 0.0615 | 1 | 4 | 73 |
| PNVP-b-PVSt #1 | 3 | 0.0615 | 2.25 | 4 | 75 |
| PNVP-b-PVSt #2 | 3 | 0.0615 | 3.75 | 4 | 52 |
| PNVP-b-PVSt #3 | 3 | 0.0615 | 4.30 | 7 | 46 |
| PNVP-b-PVSt #4 | 3 | 0.0615 | 6.00 | 9 | 43 |
Table 2.
Molecular characteristics of the block copolymers.
| macro CTA (PNVP) a |
block copolymers a |
NVP |
Vinyl ester |
|||
|---|---|---|---|---|---|---|
| Sample | Mn 103 (Daltons) |
Ð | Mn 103 (Daltons) | Ð | % molb | % molb |
| PNVP-b-PVBu #1 | 8.5 | 1.30 | 16.0 | 1.90 | 22 | 78 |
| PNVP-b-PVBu #2 | 28.0 | 1.27 | 32.0 | 1.32 | 84 | 16 |
| PNVP-b-PVBu #3 | 8.9 | 1.35 | 17.5 | 1.40 | 57 | 43 |
| PNVP-b-PVBu #4 | 8.9 | 1.35 | 15.5 | 1.54 | 48 | 52 |
| PNVP-b-PVDc #1 | 8.5 | 1.30 | 12.5 | 1.31 | 63 | 37 |
| PNVP-b-PVDc #2 | 5.5 | 1.47 | 12.5 | 1.60 | 38 | 62 |
| PNVP-b-PVDc #3 | 8.5 | 1.30 | 11.0 | 1.45 | 56 | 44 |
| PNVP-b-PVDc #4 | 9.5 | 1.36 | 10.5 | 1.36 | 93 | 7 |
| PNVP-b-PVSt #1 | 8.5 | 1.30 | 10.5 | 1.44 | 78 | 22 |
| PNVP-b-PVSt #2 | 7.5 | 1.30 | 10.4 | 1.51 | 61 | 39 |
| PNVP-b-PVSt #3 | 8.1 | 1.30 | 10.9 | 1.37 | 85 | 15 |
| PNVP-b-PVSt #4 | 8.1 | 1.30 | 12.5 | 1.22 | 83 | 17 |
By SEC in CHCl3. b. By 1H-NMR.
Table 3.
DSC results of the PNVP-b-PVBu block copolymers.
| Sample | Tg Experimental (OC) | ||
|---|---|---|---|
| PNVP-b-PVBu #1 | -9.1 | 80.3 | 126.6 |
| PNVP-b-PVBu #2 | - | 84.0 | 168.7 |
| PNVP-b-PVBu #3 | -8.9 | 93.6 | 153.3 |
| PNVP-b-PVBu #4 | -2.0 | 88.7 | 155.2 |
| PNVP | - | - | 187.1 |
| PVBu | -8.5 | - | - |
Table 4.
DSC results of the PNVP-b-PVDc block copolymers.
| Sample | Tg Experimental (OC) | ||
|---|---|---|---|
| PNVP-b-PVDc #1 | -39.3 | 62.8 | 113.3 |
| PNVP-b-PVDc #2 | -39.7 | 63.3 | 100.8 |
| PNVP-b-PVDc #3 | -52.5 | 66.0 | 112.2 |
| PNVP-b-PVDc #4 | 2.9 | 56.3 | 137.8 |
| PNVP | - | - | 187.1 |
| PVDc | -45.0 | - | - |
Table 5.
DSC results of the PNVP-b-PVSt block copolymers.
| Sample | WVSt % | Tm(°C) | ΔH(j/g VSt) | Xc % | Tg1(°C) | Tg2(°C) |
|---|---|---|---|---|---|---|
| PNVP-b-PVSt #1 | 43.6 | 33.6 | 24.7 | 11.2 | 77.4 | 148.6 |
| PNVP-b-PVSt #2 | 64.2 | 38.2 | 23.0 | 10.4 | 70.2 | 133.3 |
| PNVP-b-PVSt #3 | 33.0 | 36.4 | 36.9 | 16.8 | 64.2 | 116.1 |
| PNVP-b-PVSt #4 | 36.4 | 35.4 | 35.9 | 16.3 | 62.9 | 112.9 |
| PNVP | - | - | - | - | - | 187.1 |
| PVSt | - | 43.9 | 87.7 | 39.9 | - | - |
Table 6.
TGA results of the PNVP-b-PVBu block copolymers.
| Sample | Start | End | Max1 (OC) | Max2 (OC) |
|---|---|---|---|---|
| PNVP-b-PVBu #1 | 266.7 | 475.7 | 321.5 | 429.4 (broad) |
| PNVP-b-PVBu #2 | 284.3 | 459.1 | 321.7 | 423.9 |
| PNVP-b-PVBu #3 | 280.2 | 492.7 | 335.0 | 442.4 |
| PNVP-b-PVBu #4 | 283.6 | 499.7 | 335.8 | 445.7 |
| PNVP | 347.6 | 484.1 | 437.5 | |
| PVBu | 256.2 | 457.6 | 319.5 | 416.1 |
Table 7.
TGA results of the PNVP-b-PVDc block copolymers.
| Sample | Start1 | End1 | Max1 (OC) | Max2 (OC) |
|---|---|---|---|---|
| PNVP-b-PVDc #1 | 266.7 | 463.6 | 320.7 | 421.6 |
| PNVP-b-PVDc #2 | 247.6 | 453.9 | 319.5 | 420.1 |
| PNVP-b-PVDc #3 | 257.7 | 467.9 | 322.2 | 425.3 |
| PNVP-b-PVDc #4 | 286.5 | 483.3 | 335.2 | 439.7 |
| PNVP | 347.63 | 484.06 | - | 437.5 |
| PVDc | 238.6 | 450.8 | 322.7 | 417.8 |
Table 8.
TGA results of the PNVP-b-PVSt block copolymers.
| Sample | Start | End | Max1 (OC) | Max2 (OC) | Max3 (OC) |
|---|---|---|---|---|---|
| PNVP-b-PVSt #1 | 116.2 | 479.6 | 200.8 | 319.1 | 422.9 |
| PNVP-b-PVSt #2 | 128.9 | 465.9 | 196.4 | 322.1 | 419.4 |
| PNVP-b-PVSt #3 | 100.9 | 502.3 | 206.1 | 334.6 | 441.2 |
| PNVP-b-PVSt #4 | 135.0 | 494.7 | 212.5 | 338.8 | 439.8 |
| PNVP | 347.6 | 484.0 | - | 437.5 | |
| PVSt | 113.5 | 412.2 | 194.2 | 320.9 | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



