Submitted:
08 August 2024
Posted:
12 August 2024
You are already at the latest version
Abstract
The disease of transthyretin (TTR) amyloidosis (ATTR) has been known since the 1960s, and during these past 60 or so years, there has been a sustained peri-od of steady discoveries that have led to the current model of ATTR pathogene-sis. More recent research has achieved major advances in both diagnostics and therapeutics for ATTR, which are having a significant impact on ATTR patients today. Aiding these recent achievements has been the remarkable ability of cryo-electron microscopy (EM) to determine high-resolution structures of amy-loid fibrils obtained from individual patients. Here, we will examine cryo-EM structures of transthyretin amyloid fibrils to explore the structural basis of re-cent monoclonal antibody therapies for ATTR, including ALXN-2220 and Co-ramitug; as well as to point out potential applications of this approach to other systemic amyloid diseases.
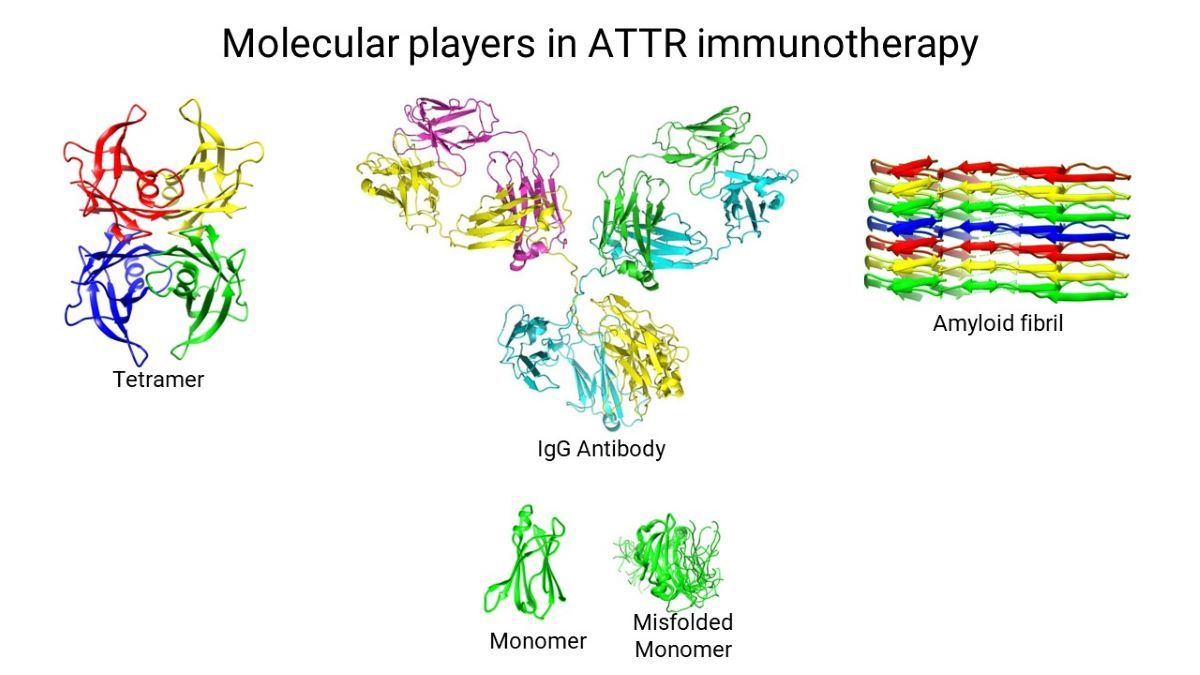
Keywords:
transthyretin
; amyloidosis
; antibody therapy
; protein structure
; cryo-electron microscopy
1. Introduction
Transthyretin amyloidosis is a protein misfolding disease with both sporadic and hereditary forms. In this disease, TTR, the serum transport protein for thyroxine and vitamin A, misfolds and deposits as amyloid in peripheral nerves causing polyneuropathy (ATTR-PN), in the heart causing cardiomyopathy (ATTR-CM), and sometimes in other organs like the kidney, digestive tract, and eyes, causing various organ dysfunctions. Recent advances in the diagnosis and treatment of ATTR, which began in 2011 with the approval of Tafamidis [1], are having a significant impact on patients today. Diagnostic successes include new minimally invasive imaging technologies that precisely locate and measure the amyloid load in the patient [2]. Treatment successes involve the generation of three new types of therapy: silencers, stabilizers, and depleters [3,4]. Silencers reduce the expression of TTR protein through either gene-editing technology or RNA-based therapy, and consequently exhaust the supply of TTR molecules available to form amyloid. Stabilizers are small molecules that bind and stabilize the native tetramers of TTR, thereby diverting TTR molecules away from the misfolding pathway. Depleters are usually antibodies that remove amyloid fibrils, the final product of the misfolding pathway. Antibody depleters of misfolded TTR amyloid will be the major focus of this article.
The TTR misfolding pathway [5], elucidated by a combination of X-ray crystallography, NMR spectroscopy, cryo-EM, and various biochemical techniques, describes a plausible process by which genetic and/or environmental factors destabilize the native tetrameric structure of TTR, causing the molecule to dissociate into monomers. The unstable nature of the isolated TTR monomers induces conformational fluctuations that result in the transient formation of a flattened monomer that stacks with other flattened monomers as they form. This flattened stackable monomer represents the growth unit of the amyloid fibril. Continued growth of the amyloid fibril is believed to be a spontaneous downhill polymerization process [5]. This pathway is illustrated in Figure 1. This pathway is meant to be conceptual and is not a complete depiction of the process; it introduces the players of the misfolding process.
Early experiments, which were crucial for establishing the misfolding pathway, showed that incubation of TTR at pH 3.6 to 4.8 was all that was required to initiate amyloid fibril formation [6,7]. Surprisingly, the mild acidic incubation that produced amyloid fibrils, also caused the appearance of monomers and the disappearance of tetramers. This led to the conclusion that TTR might need to dissociate into monomers before forming amyloid fibrils, thus establishing the monomer as a key misfolding intermediate [6,7].
It was later found that ATTR-causing mutants of TTR dissociated into monomers more readily than the wild type when exposed to mildly acidic conditions. This difference raised the possibility that ATTR mutations cause disease by destabilizing the tetramer relative to the monomer [8,9]. Studies using an engineered monomeric mutant of TTR demonstrated that the engineered monomer formed a well-folded structure at neutral pH that was nearly identical to that of the monomeric subunit in the native tetramer [10] (Figure 1; pdb code 1GKO). At acidic pH, however, the engineered monomer formed amyloid fibrils at a much faster rate than tetrameric TTR [10], implicating the dissociation of the TTR tetramer as the rate limiting step of the TTR misfolding pathway [10].
The engineered monomer still exhibited the same preference for slightly acidic aggregation conditions as wild type tetrameric TTR, suggesting that the monomer converts from a relatively stable conformation at neutral pH to a misfolded monomer at slightly acidic pH [10]. Amide proton exchange experiments have provided evidence that in this acid pH range, specific subdomains comprising several residues in β-strands C, B, E, and F are partially unfolded [11]. The solution structure of the engineered monomer under conditions close to aggregation [12] (Figure 1; pdb code 2NBO) reveals significant partial unfolding. The structure of this partially unfolded engineered monomer is likely to be very similar to the misfolded monomer postulated to be transiently populated during amyloid fibrillogenesis [6,7].
Cryo-EM has provided the highest resolution structures of the conformational states of amyloid fibrils, the final product of the TTR misfolding pathway (Figure 1; pdb code 8ADE). Currently, there are over a dozen cryo-EM structures of TTR amyloid fibrils in the Protein Data Bank. The structures of these amyloid fibrils will be discussed in detail in a later section.
2. Silencers, Stabilizers, Depleters, and Mass Action
Operating through the laws of mass action and Le Chatelier's principle, the newly emerging/available therapies all target the misfolding pathway of TTR. The silencers include CRISPR/Cas9 editing of the TTR gene [4,13], and targeting TTR mRNA with small interfering RNAs [14] and antisense oligonucleotides [15]. Silencers work by drastically reducing the concentration of TTR tetramers in the blood. Based on the misfolding pathway, eliminating the tetramers through nucleic acid-based therapeutics prevents further formation of monomers and thus fibrils. According to Le Chatelier's principle, treatment with silencers should also promote reversal of the reaction to re-establish equilibrium, by causing the release of monomers from the amyloid fibril, as well as the reassembly of tetramers from the released monomers. In practice, however, the slow rate at which monomers dissociate from the fibril may hinder the silencers from causing the total dissolution of existing amyloid. Support for this possibility comes from the clinical trial data of the silencer, Patisiran, which shows a clearly observable reduction, but not elimination, in cardiac amyloid over a one to three-year period [16,17].
The development of the stabilizer drug, Tafamidis [3] began with the discovery that the natural ligand of TTR, thyroxine binds to the TTR tetramer and inhibits amyloid fibril formation [18]. The demonstration that the unnatural ligand, flufenamic acid also binds to the thyroxine binding sites of TTR and inhibits amyloid fibril formation [19] initiated targeted high throughput screening efforts to find high-affinity TTR tetramer binders [20]. The drug discovery efforts led to the development of Tafamidis by Kelly and coworkers [21], the first approved treatment for ATTR [1]. The mechanism of action of Tafamidis is to offer a mass action alternative to the misfolding pathway, whereby the tetramer can become more stable by binding a stabilizing ligand like Tafamidis, and thus avoid misfolding. The effectiveness of Tafamidis has been shown in the ATTR-ACT clinical trial that shows Tafamidis treatment was associated with a lower all-cause mortality, a lower rate of cardiovascular-related hospitalizations, and a lower rate of decline in tests of exercise tolerance (6MWT) and in tests to assess health status among heart failure patients (KCCQ-OS) [22]. However, the stabilizers, like the silencers, target the tetramer and are also potentially prone to the hampering slow rate at which monomers dissociate from the fibril. These slow kinetics may impede stabilizers from causing the total dissolution of existing amyloid. While clinical results demonstrate that Tafamidis slows down disease progression, immediate signs of symptom improvement have not been observed. The kinetic barrier of slow dissociation of monomers from the fibril may limit the fibril shrinkage achievable with Tafamidis treatment.
Certain depleter antibodies can also function through mass action. These types of depleter antibodies bind to monomers and fibrils, but they do not bind to native tetramers. In this scenario, the antibody acts as a molecular chaperone that sequesters monomers and thereby prevents fibril formation. However, once released from the antibodies, monomers will reattach to the antibody unless they have formed native tetramers. The only way for the monomers to escape from the antibody is by forming native tetramers. The exceptional stability of the tetramer ensures that its dissociation into monomers is a rare event. This selective binding of the monomer, and rejection of the tetramer, will result in a gradual conversion of monomers to tetramers. However, the most potent effect of the antibody will be the removal of antibody-tagged fibrils and precursors by macrophages and the immune system. Thus, unlike silencers and stabilizers, these antibodies possess the potential for dual action in reducing fibril deposits. First, through chaperone-like mass action, and second, by facilitating the immune clearance of amyloid fibrils and fibril intermediates. This dual mechanism of action distinguishes these depleter antibodies from silencers and stabilizers. The development of depleter antibodies is described in detail below.
3. Antibodies to TTR and Their Binding Specificities
Antibodies to TTR have been commercially available since the mid-1960s when TTR (originally called prealbumin) was discovered [23,24]. These early antibodies were used in immunoelectrophoresis experiments to demonstrate that TTR is a vitamin A transport protein [25]. They were also used in immunoadsorption experiments to demonstrate that TTR is a thyroxine transport protein [26]. In a classic paper in 1978, it was shown that antibodies raised against amyloid fibrils isolated from patients with familial amyloid polyneuropathy (now known as ATTR-PN) cross-reacted with purified TTR protein [27]. Reciprocally, commercial antibodies raised against purified TTR protein cross-reacted with amyloid fibrils from patients, thus establishing that the amyloid deposits from ATTR-PN patients are composed of TTR [27]. The immunohistochemical demonstration that cardiac amyloid deposits observed in ATTR-CM were also composed of TTR was achieved shortly afterward in 1981 [28].
Antibodies have also been used to identify regions of TTR that are exposed in the amyloid fibril versus regions that are exposed in the native tetramer. In 1994, Westermark and coworkers [29] used a collection of polyclonal antibodies to TTR peptide fragments, TTR amyloid fibrils, and TTR tetramers to map amino acid exposure in fibrils and tetramers. They found antibodies directed to residues 24-35, 50-60, and 90-100 bound both TTR tetramers and amyloid fibrils, while antibodies to residues 115-124 bound specifically to TTR amyloid fibrils. Examination of the TTR tetramer structure does show that both residue segments 24-35 and 50-60 are widely exposed in the tetramer and should be available for antibody binding. However, the 90-100 residue segment is buried in the native tetramer and should not be able to bind to the antibody. Cross-reactivity or nonspecific binding with other antigenic components in the sample may explain why antibodies to TTR (90-100) appear to bind native TTR tetramers. The antibody to residues 115-124 does appear to bind specifically to amyloid fibrils but not native tetramers, which is consistent with this epitope being significantly buried in the native tetramer. By implication, this epitope, residues 115-124, should be exposed in the amyloid fibril structure.
An interesting study produced two monoclonal antibodies that specifically bound to amyloid fibrils, but not to native wild type tetramers [30]. That study used an unnatural, highly amyloidogenic mutant of TTR as an antigen [30]. The hybridoma screening test identified two different monoclonals that showed specific binding to fibrils, but not tetramers. Epitope mapping of the two monoclonals indicates that one monoclonal specifically binds residues 39-44, while the other binds residues 56-61. The anti-TTR (39-44) antibody has been shown to be a useful clinical laboratory tool. It produces a positive immunoassay signal in blood plasma samples from ATTR-PN patients and asymptomatic carriers, but no signal from patients who do not have any mutations in TTR or have nonamyloidogenic mutations [31]. The anti-TTR (39-44) antibody immunoassay signal from plasma has high potential to be a biomarker or diagnostic for ATTR. The anti-TTR (39-44) has also proved to be a useful research tool to investigate amyloid fibril formation of TTR variants [32,33]. Other antibodies specific for TTR amyloid fibrils have also been produced using amyloid fibrils generated from recombinant TTR and TTR mutants as antigen [34]. The epitopes of these TTR fibril-specific antibodies have not been determined.
Ando and coworkers have developed RT24 [35], which is a humanized monoclonal form of the polyclonal anti-TTR (115-124) described above [29]. RT24 has been shown to bind to TTR fibrils but not tetramers. More importantly, RT24 has been shown to inhibit fibril formation [35]. Additionally, RT24 has been shown to form immune complexes with fibrillar TTR that can be readily phagocytosed by human macrophages [35]. RT24 could be one of those potential dual-action therapeutic antibodies discussed earlier, which act through both chaperone-like mass action and immune clearance of amyloid.
Humans sometimes produce antibodies against their own TTR. Unique IgM molecules (catabodies), which were isolated from pools of blood serum obtained from healthy individuals with no history of amyloidosis, were found to hydrolyze TTR fibrils but not TTR tetramers [36]. The hypothesis is that a certain level of TTR misfolding occurs naturally in all individuals, and the immune system generates IgM catabodies to break down the misfolded TTR. No stable immune complexes formed between the misfolded TTR and the IgM catabodies. This may be due to spontaneous hydrolysis of the misfolded TTR molecules while bound to the IgM catabody.
The apparent stochastic generation of antibodies that aid in the clearance of amyloid has been observed in three different ATTR-CM patients [37]. These three male patients, aged 68, 76, and 82 years old, all had heart failure caused by ATTR-CM but they had not yet received any disease-modifying treatments. Remarkably, during routine management of these patients, and before any ATTR specific treatments were initiated, a reversion to near-normal cardiac structure and function was observed. High titers of polyclonal IgG antibodies to TTR were detected in the serum of each patient. These antibodies were shown to bind to ATTR amyloid deposits and synthetic TTR fibrils. The clinical significance of these antibodies is being investigated. But most importantly, these findings show that ATTR in these cases is reversible.
4. ATTR Depleter Antibodies in Clinical Trials
ALXN-2220 (NI006, NI301A, anti-TTR (41-45)) Neuroimmune AG, a company from Schlieren/Zurich, Switzerland, has also taken advantage of the fact that some humans produce antibodies against their own TTR [38,39]. They screened a human memory B cell library generated from healthy elderly adults to identify expressed antibodies that bind to TTR fibrils but not TTR tetramers. The antibody identified from this screening, NI006, binds to fibrils made from both wild type and mutant TTR with nanomolar affinity, but does not bind to tetramers made of the same proteins. Epitope mapping of NI006 revealed that this antibody binds to residues 41-45 of TTR, which are partially buried in the TTR tetramer [39]. The epitope of NI006 overlaps with the epitope of anti-TTR (39-44), a previously developed monoclonal antibody mentioned earlier [30]. Both NI006 and anti-TTR (39-44) bind to TTR monomers, dimers, oligomers, and large aggregates; however, there are some differences in the extent of binding to monomers and large aggregates [39]. NI006 stains amyloid deposits in the heart and other tissues of both wild type cases and several hereditary ATTR cases [39]. Immune complexes formed between NI006 and fibrils, composed of the aggregation-prone L55P mutant of TTR, were readily phagocytosed by cultured macrophages. When these immune complexes were added to patient myocardial tissue sections, they were taken up by resident macrophages in the patient tissue sample. NI006 was shown to aid in the clearance of amyloid material in a non-transgenic mouse model of ATTR [39]. In that experiment, amyloid material from an ATTR patient was injected into the thigh of a mouse, which was then injected with varying amounts of fluorescent NI006 antibody. The administered antibody caused an initial large increase in fluorescence at the injection site followed by a steady decrease indicating that the antibody is binding to the injected material and facilitating its removal.
Based on these positive laboratory results, the company sponsored a phase 1 trial of antibody NI006 for the depletion of cardiac transthyretin amyloid [40]. This clinical trial demonstrated that NI006 met all the safety requirements for a therapeutic IgG antibody. But more importantly, cardiac imaging with bisphosphonate scintigraphy clearly shows a visible reduction in amyloid load with NI006 treatment [40]. Cardiac MRI measures of extracellular volume, which is an index of cardiac amyloid load, also showed significant decreases with NI006 treatment. Cardiac biomarker levels did show a reduction with NI006 treatment at the 12-month mark of the study, indicating a degree of healing. With the fortunate circumstance of having shown efficacy at the phase 1 level, we all wait in anticipation for further clinical trial results. ALXN-2220 is the new designation for NI006, and Alexion-AstraZeneca will be sponsoring the next clinical trial.
Coramitug (PRX004, anti-TTR (89-97)) This depleter antibody started out as a polyclonal antibody developed in this author's academic lab at the University of Toronto [41]. The epitope (TTR 89-97) was selected through a structure-based approach where the structures of tetrameric TTR and monomeric TTR were compared segment by segment. Five to ten continuous residue segments of the protein that were exposed in the monomer but buried in the tetramer were identified. Out of those identified segments, TTR 89-97 was one that was highly buried (13% accessible) in the tetramer and freely accessible in the monomer. Rabbit polyclonal antibodies were raised against TTR 89-97 [41]. These antibodies were shown to bind TTR monomers and fibrils but not tetramers. From immunohistochemistry experiments, the antibody was shown to stain amyloid deposits in cardiac and other tissues from various hereditary and wild type cases of ATTR, but not in pathological controls and healthy tissue. Notably, anti-TTR (89-97) was able to inhibit fibril formation at substoichiometric concentrations. Nanomolar amounts of antibody were sufficient to inhibit fibrillation of micromolar amounts of TTR. Furthermore, these experiments utilized TTR protein, buffer, antibody, and no other components of the immune system. Thus, the fibril inhibition in this experiment is a result of chaperone-like mass action activity of the antibody and not the immune system. Other experiments show that this antibody can activate the immune system. Immune complexes formed between the antibody and TTR fibrils were recognized and phagocytosed by human macrophages in culture [41]. Thus, this antibody may be one of those depleter antibodies that possess dual action in reducing fibril deposits, through chaperone-like mass action, and facilitating the immune clearance of amyloid fibrils and fibril intermediates. It was at this point that a collaboration with Prothena, a company from South San Francisco, USA, was initiated to develop and humanize monoclonals that are equivalent to the anti-TTR (89-97) polyclonal. The antigen that was used to generate rabbit polyclonals was also used to generate mouse monoclonals. The hybridoma screen revealed several monoclonal antibodies that possessed very similar properties to the rabbit polyclonal [42]. These antibodies specifically bind monomers and fibrils, but not tetramers. They specifically recognized TTR amyloid in the heart and other affected tissue. They inhibit fibril formation of TTR and they form immune complexes with misfolded TTR that activate the immune system [42]. One of these monoclonal antibodies was humanized and was designated PRX004.
The phase 1 clinical trial for PRX004 was started in 2018 but had to be terminated in 2020 due to the COVID-19 pandemic. Fortunately, the data gathered during that time were sufficient to aid decision-making on whether to progress to a phase 2 clinical trial. PRX004 was shown to be safe and well-tolerated at all doses examined. No serious adverse effects related to PRX004 administration were observed. The neuropathy impairment score (NIS) was used to evaluate muscle weakness, reflex loss, and sensory loss in the trial participants. All evaluable participants receiving treatment had NIS scores that were more favorable than published historical data, indicating that the rate of neuropathy decline is reduced with treatment. Some patients actually showed improvement in muscle strength, reflexes, and sensory perception. The cardiac function of the evaluable participants was assessed with the echocardiographic parameter of global longitudinal strain (GLS). These participants had improved GLS scores indicating improvement in cardiac health. Prothena has made this information available on their website (https://s201.q4cdn.com/351053094/files/doc_presentations/2021/04/1/AAN-PRX004-Ph1_20March21-FINAL.pdf). A phase 2 clinical trial sponsored by Novo Nordisk A/S., Copenhagen, Denmark was approved and is underway [43]. Coramitug is now the new designation for PRX004.
5. Cryo-EM Structure of the TTR Amyloid Fibril
Several techniques such as X-ray diffraction [44] and solid-state NMR [45] have been used to investigate the structures of TTR amyloid fibrils. While these techniques are capable of determining high resolution structures of amyloid fibrils, cryo-EM is currently the only technique capable of producing 3D structures of single-patient-derived TTR amyloid fibrils. Here we will only be discussing cryo-EM structures of patient-derived TTR fibrils.
The cryo-EM structure of amyloid fibrils from a patient with wild type ATTR [46] is shown in Figure 1. For demonstration purposes, the fibrils are displayed as stacks of flattened monomers ranging from one to seven monomers per fibril. The natural lengths of TTR amyloid fibrils are between 60-220 monomers per fibril, as estimated from studies of the Alzheimer amyloid peptide [47]. Thus, the fibrils shown in Figure 1 are minute compared to natural fibrils. Each monomer is approximately 7 nm x 6 nm x 1 nm and its polypeptide chain lies within this flat slab in an irregular coiled conformation, which can be better appreciated in Figure 2, the top-down view of the flattened monomer.
The models start with the first 9 or 10 residues (in red) being either unstructured in the wild type or missing in the I48S mutant (Figure 2). The fibril structure starts at residue 10 (the last red residue shown). Residues 10-35 form a non-hydrogen bonded hairpin-like structure. At residue 36, the polypeptide chain leaves the fibril, forming an unstructured loop spanning residues 36-57 (in blue). The polypeptide chain re-enters the fibril at residue 58 (first residue after the loop in blue ends), where it continues to form its irregular coil conformation up to residue 123. It then leaves the fibril for 4 residues (in purple) and terminates at residue 127. The main difference between the open and closed gate models is in the conformation of residues 58-65, which, in turn, alters the position of the unstructured loop, residues 36-57 (in blue).
The unstructured loop of residues 36-57 is also the site of proteolysis (Figure 2), a process which affects amyloid morphology [49]. Early studies have indicated a preference for proteolysis at residues 46, 49, and 52 [49]. However, recently solved cryo-EM structures of TTR fibrils also indicate a cleavage site at residue 58 [46,48]. The current data indicates considerable variability in the location of the cleavage sites, which may not yet be completely determined. After some consideration by the research community [50], the current opinion is that the proteolysis occurs on-fibril rather than at the tetramer stage [46,48]. The on-fibril proteolysis is likely a defense mechanism. Proteolyzed TTR likely dissociates from the fibril more easily than intact TTR which would promote fibril disassembly. Similarly, proteolyzed TTR fragments are unlikely to reassemble and add on to the fibril, thus they do not aid fibril growth. The role of loop-proteolysis in ATTR is still being investigated.
6. Structural Disposition of Depleter Antibody Epitopes on the Cryo-EM Structures of TTR Amyloid Fibrils
Earlier, we discussed five potential depleter antibodies that have been shown to stain amyloid deposits of TTR, and to bind to TTR fibrils but not bind to TTR tetramers. They are anti-TTR (39-44), anti-TTR (56-61), RT24, ALXN-2220, and Coramitug. In Figure 3, we indicate the locations of these epitopes on the closed and open gate models of TTR amyloid fibrils.
It is significant and satisfying to see that the potential depleter antibody epitopes all occupy solvent-exposed regions around the periphery of the flattened monomer subunit, because it means the epitopes would be accessible for binding by the antibody. It is noteworthy that these epitopes were selected before the cryo-EM structures of these fibrils were known. ALXN-2220, anti-TTR (39-44), and anti-TTR (56-61) epitopes were selected through screening tests, while Coramitug and RT24 were selected through hypothesis-testing. This provides an example of how two different approaches led to the successful development of antibodies sharing similar important properties.
The anti-TTR (56-61) epitope is part of the unstructured loop spanning residues 36-57 and it contains the known proteolysis site at residue 58 [46,48]. In those patients where this proteolysis may happen, this epitope may either have a reduced availability or be completely lost. The ALXN-2220 epitope and anti-TTR (39-44) epitope are also part of the unstructured loop; however, they do not contain any known proteolysis sites. The position of the unstructured loop differs in the closed versus open gate models. Consequently, antibodies that target the loop, like anti-TTR (39-44) and ALXN-2220, may differ in their affinity for the closed versus open gate model structures. Coramitug and RT24 epitopes are part of the fibril structure and the local structure around those epitopes does not differ greatly between the two models. Thus, Coramitug and RT24 may bind their respective epitopes in both the open and closed gate models with similar affinity.
Proteolysis generates short N-terminal fragments, 46-58 residues long, and longer C-terminal fragments, 69-81 residues long. The Coramitug epitope is located on the C-terminal fragment, while the ALXN-2220 epitope is located on the N-terminal fragment.
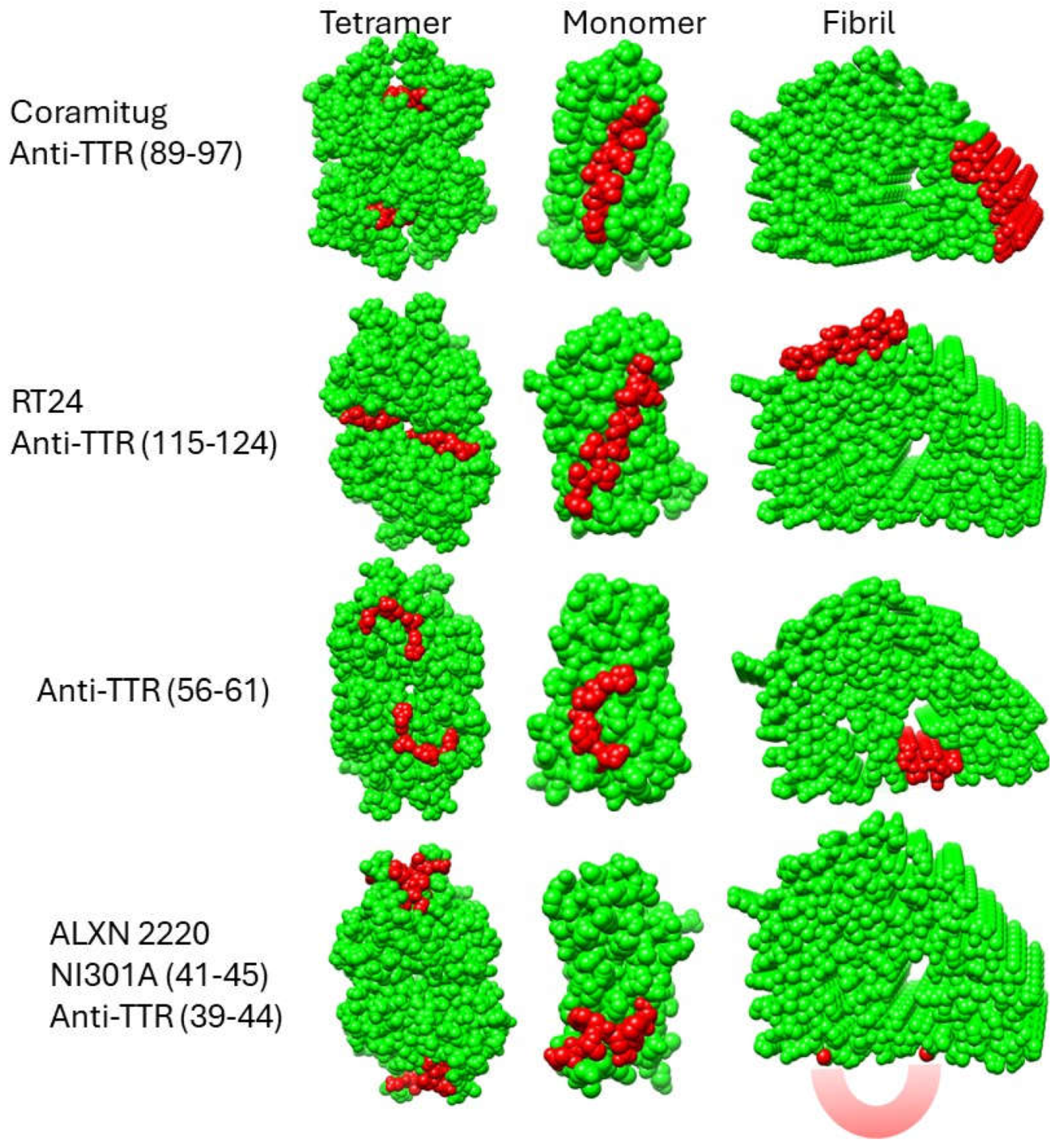
Figure 4 shows a comparison of the accessibility of the epitopes on space filling models in the three structural states of the tetramer, monomer, and fibril.
Based on the fibril structures shown in Figure 4, all five epitopes appear accessible in the fibril state but display contextual differences. The epitopes of Coramitug and RT24 are part of the fibril structure and are likely to show dynamic movements that are similar to surface residues on globular proteins. The ALXN-2220 epitope, on the other hand, is part of the unstructured loop spanning residues 36-57. Consequently, the ALXN-2220 epitope is likely to be very flexible, which would aid antibody binding but might increase susceptibility to proteolysis. Fortunately, no protease sites are known to exist within the ALXN-2220 epitope. The anti-TTR (56-61) epitope is partly located within the central channel of the fibril structure, is less accessible compared to the others, and contains the proteolysis site at residue 58. These properties may potentially result in a reduced ability of this epitope to bind the antibody in the fibril.
In the monomer, all five depleter epitopes appear to be very accessible. Thus, all the antibodies should have similar utility in removing monomers by mass action and immune clearance, if tetramer-binding is sufficiently low. In the tetramer, however, there is significant variation in the accessibility of the epitopes. The Coramitug epitope is buried in the tetramer, the RT24 epitope is slightly exposed, and the ALXN-2220 and anti-TTR (56-61) epitopes are slightly more exposed. Even weak binding to the tetramer can have consequences for monoclonal therapy. Because native tetrameric TTR is present at the relatively high blood concentration of 3.6-7.2 µM (20 to 40 mg/dl) [51], it may sequester antibodies that show micromolar binding to the native TTR tetramer. In such cases, the native TTR tetramer serves to competitively bind the antibody, and consequently reduce the effective dose of antibody for therapy.
In conclusion, by examining the conformation of the depleter epitopes in the cryo-EM structures of the different TTR fibril models, we can better understand the properties of these antibodies. The anti-TTR (56-61) antibody appears to be the weakest of the group. It has the least accessible epitope on the fibril, and it contains a known proteolysis site at residue 58. Only RT24 and Coramitug were shown to inhibit TTR fibril formation, and the epitopes of these two antibodies also displayed the least accessibility in the native tetramer. For therapeutic purposes, the antibody needs to bind the monomer and the fibril, but not the tetramer. The Coramiug and RT24 epitopes are more deeply buried in the tetramer, and thus may result in minimal binding by the antibody. ALXN-2220 targets the loop spanning residues 36-57. The flexible nature of the ALXN-2220 epitope could facilitate antibody binding through induced fit into the antigen binding site. The epitopes of Coramitug and RT24 antibodies are likely rigid and bind with lock and key mechanisms.
The process to develop and understand TTR therapeutic antibodies by analyzing the structures of native and diseased forms of TTR has been described. This process includes the analysis of the structures of the native state, and the misfolding intermediates (like the monomer) and the amyloid fibril. Identification of epitopes differentially exposed between the native and diseased states has also been described. The ability to identify epitopes that are exposed in the fibril and fibril intermediates, but hidden in the native state, can potentially be used to develop future therapeutic antibodies for the many other systemic protein misfolding diseases [52].
Funding
The author has received research funds from Prothena Inc., South San Francisco, CA, USA.
Acknowledgments
The author thanks Bjarke Follin, Douglas V. Laurents, Nancy F. L. Ng and Sumon Chakrabartty for helpful discussions.
Conflicts of Interest
The author has received royalties from Prothena Inc., South San Francisco, CA, USA and consultancy from Novo Nordisk A/S., Copenhagen, Denmark. The author is listed as inventor on several anti-transthyretin antibody patents.
References
- Said, G.; Grippon, S.; Kirkpatrick, P. Tafamidis. Nat Rev Drug Discov. 2012 11, 185–186. [CrossRef]
- Brito, D.; Albrecht, F.C.; de Arenaza, D.P.; Bart, N.; Better, N.; Carvajal-Juarez, I.; Conceição, I.; Damy, T.; Dorbala, S.; Fidalgo, J.-C.; et al. World Heart Federation Consensus on Transthyretin Amyloidosis Cardiomyopathy (ATTR-CM). Glob. Hear. 2023, 18, 59. [Google Scholar] [CrossRef] [PubMed]
- Morfino, P.; Aimo, A.; Vergaro, G.; Sanguinetti, C.; Castiglione, V.; Franzini, M.; Perrone, M.A.; Emdin, M. Transthyretin Stabilizers and Seeding Inhibitors as Therapies for Amyloid Transthyretin Cardiomyopathy. Pharmaceutics 2023, 15, 1129. [Google Scholar] [CrossRef]
- Ioannou, A.; Fontana, M.; Gillmore, J.D. RNA Targeting and Gene Editing Strategies for Transthyretin Amyloidosis. BioDrugs 2023, 37, 127–142. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.M.; Connelly, S.; Fearns, C.; Powers, E.T.; Kelly, J.W. The Transthyretin Amyloidoses: From Delineating the Molecular Mechanism of Aggregation Linked to Pathology to a Regulatory-Agency-Approved Drug. J. Mol. Biol. 2012, 421, 185–203. [Google Scholar] [CrossRef] [PubMed]
- Colon, W.; Kelly, J.W. Partial denaturation of transthyretin is sufficient for amyloid fibril formation in vitro. Biochemistry 1992, 31, 8654–8660. [Google Scholar] [CrossRef] [PubMed]
- Lai, Z.; Colón, W.; Kelly, J.W. The Acid-Mediated Denaturation Pathway of Transthyretin Yields a Conformational Intermediate That Can Self-Assemble into Amyloid. Biochemistry 1996, 35, 6470–6482. [Google Scholar] [CrossRef] [PubMed]
- McCutchen, S.L.; Colon, W.; Kelly, J.W. Transthyretin mutation Leu-55-Pro significantly alters tetramer stability and increases amyloidogenicity. Biochemistry 1993, 32, 12119–12127. [Google Scholar] [CrossRef] [PubMed]
- Colon, W.; Lai, Z.; McCutchen, S.L.; Miroy, G.J.; Strang, C.; Kelly, J.W. FAP mutations destabilize transthyretin facilitating conformational changes required for amyloid formation. Ciba Found Symp. 1996 199, 228-238; discussion 239-242.
- Jiang, X.; Smith, C.S.; Petrassi, H.M.; Hammarström, P.; White, J.T.; Sacchettini, J.C.; Kelly, J.W. An Engineered Transthyretin Monomer that Is Nonamyloidogenic, Unless It Is Partially Denatured. Biochemistry 2001, 40, 11442–11452. [Google Scholar] [CrossRef]
- Liu, K.; Cho, H.S.; Lashuel, H.A.; Kelly, J.W.; Wemmer, D.E. A glimpse of a possible amyloidogenic intermediate of transthyretin. Nat. Struct. Biol. 2000, 7, 754–757. [Google Scholar] [PubMed]
- Oroz, J.; Kim, J.H.; Chang, B.J.; Zweckstetter, M. Mechanistic basis for the recognition of a misfolded protein by the molecular chaperone HspNat. Struct. Mol. Biol. 2017, 24, 407–413. [Google Scholar] [CrossRef]
- Gillmore, J.D.; Gane, E.; Taubel, J.; Kao, J.; Fontana, M.; Maitland, M.L.; Seitzer, J.; O’connell, D.; Walsh, K.R.; Wood, K.; et al. CRISPR-Cas9 In Vivo Gene Editing for Transthyretin Amyloidosis. New Engl. J. Med. 2021, 385, 493–502. [Google Scholar] [CrossRef] [PubMed]
- Adams, D.; Gonzalez-Duarte, A.; O'Riordan, W.D.; et al. Patisiran, an RNAi Therapeutic, for Hereditary Transthyretin Amyloidosis. N Engl J Med. 2018 379, 11–21. [CrossRef]
- Benson, M.D.; Waddington-Cruz, M.; Berk, J.L.; Polydefkis, M.; Dyck, P.J.; Wang, A.K.; Planté-Bordeneuve, V.; Barroso, F.A.; Merlini, G.; Obici, L.; et al. Inotersen Treatment for Patients with Hereditary Transthyretin Amyloidosis. N. Engl. J. Med. 2018, 379, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Fontana, M.; Martinez-Naharro, A.; Chacko, L.; Rowczenio, D.; Gilbertson, J.A.; Whelan, C.J.; Strehina, S.; Lane, T.; Moon, J.; Hutt, D.F.; et al. Reduction in CMR Derived Extracellular Volume With Patisiran Indicates Cardiac Amyloid Regression. JACC: Cardiovasc. Imaging 2020, 14, 189–199. [Google Scholar] [CrossRef] [PubMed]
- Ioannou, A.; Fontana, M.; Gillmore, J.D. RNA Targeting and Gene Editing Strategies for Transthyretin Amyloidosis. BioDrugs 2023, 37, 127–142. [Google Scholar] [CrossRef] [PubMed]
- Miroy, G.J.; Lai, Z.; Lashuel, H.A.; Peterson, S.A.; Strang, C.; Kelly, J.W. Inhibiting transthyretin amyloid fibril formation via protein stabilization. Proc. Natl. Acad. Sci. USA 1996, 93, 15051–15056. [Google Scholar] [CrossRef] [PubMed]
- Peterson, S.A.; Klabunde, T.; Lashuel, H.A.; Purkey, H.; Sacchettini, J.C.; Kelly, J.W. Inhibiting transthyretin conformational changes that lead to amyloid fibril formation. Proc. Natl. Acad. Sci. USA 1998, 95, 12956–12960. [Google Scholar] [CrossRef] [PubMed]
- Baures, P.W.; Peterson, S.A.; Kelly, J.W. Discovering transthyretin amyloid fibril inhibitors by limited screening. Bioorganic Med. Chem. 1998, 6, 1389–1401. [Google Scholar] [CrossRef] [PubMed]
- Bulawa, C.E.; Connelly, S.; DeVit, M.; Wang, L.; Weigel, C.; Fleming, J.A.; Packman, J.; Powers, E.T.; Wiseman, R.L.; Foss, T.R.; et al. Tafamidis, a potent and selective transthyretin kinetic stabilizer that inhibits the amyloid cascade. Proc. Natl. Acad. Sci. USA 2012, 109, 9629–9634. [Google Scholar] [CrossRef] [PubMed]
- Maurer, M.S.; Schwartz, J.H.; Gundapaneni, B.; Elliott, P.M.; Merlini, G.; Waddington-Cruz, M.; Kristen, A.V.; Grogan, M.; Witteles, R.; Damy, T.; et al. Tafamidis Treatment for Patients with Transthyretin Amyloid Cardiomyopathy. N. Engl. J. Med. 2018, 379, 1007–1016. [Google Scholar] [CrossRef] [PubMed]
- Robbins, J.; Rall, J.E.; Petermann, M.L. Thyroxine-Binding by Serum and Urine Proteins in Nephrosis. Qualitative AspectsJ. Clin. Investig. 1957, 36, 1333–1342. [Google Scholar] [CrossRef] [PubMed]
- Ingbar. Endocrinology. 1958 63, 256–259.
- Alvsaker, J.; Haugli, F.; Laland, S. The presence of vitamin A in human tryptophan-rich prealbumin. Biochem. J. 1967, 102, 362–366. [Google Scholar] [CrossRef] [PubMed]
- Woeber, K.A.; Ingbar, S.H. The contribution of thyroxine-binding prealbumin to the binding of thyroxine in human serum, as assessed by immunoadsorption. J. Clin. Investig. 1968, 47, 1710–1721. [Google Scholar] [CrossRef] [PubMed]
- Costa, P.P.; Figueira, A.S.; Bravo, F.R. Amyloid fibril protein related to prealbumin in familial amyloidotic polyneuropathy. Proc. Natl. Acad. Sci. USA 1978, 75, 4499–4503. [Google Scholar] [CrossRef] [PubMed]
- Cornwell, G.G. 3r; Westermark, P.; Natvig, J.B.; Murdoch, W. Senile cardiac amyloid: evidence that fibrils contain a protein immunologically related to prealbumin. Immunology. 1981 44, 447–452.
- Gustavsson, A.; Engström, U.; Westermark, P. Mechanisms of transthyretin amyloidogenesis. Antigenic mapping of transthyretin purified from plasma and amyloid fibrils and within in situ tissue localizations.. 1994, 144, 1301–11. [Google Scholar]
- Goldsteins, G.; Persson, H.; Andersson, K.; Olofsson, A.; Dacklin, I.; Edvinsson. ; Saraiva, M.J.; Lundgren, E. Exposure of cryptic epitopes on transthyretin only in amyloid and in amyloidogenic mutants. Proc. Natl. Acad. Sci. USA 1999, 96, 3108–3113. [Google Scholar] [CrossRef] [PubMed]
- Palha, J.A.; Moreira, P.; Olofsson, A.; Lundgren, E.; Saraiva, M.J. Antibody recognition of amyloidogenic transthyretin variants in serum of patients with familial amyloidotic polyneuropathy. J. Mol. Med. 2000, 78, 703–707. [Google Scholar] [CrossRef] [PubMed]
- Eneqvist, T.; Olofsson, A.; Ando, Y.; Miyakawa, T. ; Katsuragi. S.; Jass, J.; Lundgren, E.; Sauer-Eriksson, A.E. Disulfide-bond formation in the transthyretin mutant Y114C prevents amyloid fibril formation in vivo and in vitro. Biochemistry. 2002 41, 13143-13151.
- Karlsson, A.; Olofsson, A.; Eneqvist, T.; Sauer-Eriksson, A.E. Cys114-Linked Dimers of Transthyretin Are Compatible with Amyloid Formation, Biochemistry 2005, 44, 13063–13070. [Google Scholar] [CrossRef] [PubMed]
- Phay, M.; Blinder, V.; Macy, S.; Greene, M.J.; Wooliver, D.C.; Liu, W.; Planas, A.; Walsh, D.M.; Connors, L.H.; Primmer, S.R.; et al. Transthyretin Aggregate-Specific Antibodies Recognize Cryptic Epitopes on Patient-Derived Amyloid Fibrils. Rejuvenation Res. 2014, 17, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Hosoi, A.; Su, Y.; Torikai, M.; Jono, H.; Ishikawa, D.; Soejima, K.; Higuchi, H.; Guo, J.; Ueda, M.; Suenaga, G.; et al. Novel Antibody for the Treatment of Transthyretin Amyloidosis. J. Biol. Chem. 2016, 291, 25096–25105. [Google Scholar] [CrossRef] [PubMed]
- Planque, S.A.; Nishiyama, Y.; Hara, M.; Sonoda, S.; Murphy, S.K.; Watanabe, K.; Mitsuda, Y.; Brown, E.L.; Massey, R.J.; Primmer, S.R.; et al. Physiological IgM Class Catalytic Antibodies Selective for Transthyretin Amyloid. J. Biol. Chem. 2014, 289, 13243–13258. [Google Scholar] [CrossRef] [PubMed]
- Fontana, M.; Gilbertson, J.; Verona, G.; Riefolo, M.; Slamova, I.; Leone, O.; Rowczenio, D.; Botcher, N.; Ioannou, A.; Patel, R.K.; et al. Antibody-Associated Reversal of ATTR Amyloidosis–Related Cardiomyopathy. New Engl. J. Med. 2023, 388, 2199–2201. [Google Scholar] [CrossRef] [PubMed]
- Michalon, A.; Combaluzier, B.; Varela, E.; Hagenbuch, A.; Suhr, O.; Saraiva, M.; Grimm, J. Characterization of conformation-specific, human-derived monoclonal antibodies against TTR aggregates with potential for diagnostic and therapeutic use. Orphanet J. Rare Dis. 2015, 10, P39–P39. [Google Scholar] [CrossRef]
- Michalon, A.; Hagenbuch, A.; Huy, C.; Varela, E.; Combaluzier, B.; Damy, T.; Suhr, O.B.; Saraiva, M.J.; Hock, C.; Nitsch, R.M.; et al. A human antibody selective for transthyretin amyloid removes cardiac amyloid through phagocytic immune cells. Nat. Commun. 2021, 12, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Pavia, P.; Siepen, F.A.D.; Donal, E.; Lairez, O.; van der Meer, P.; Kristen, A.V.; Mercuri, M.F.; Michalon, A.; Frost, R.J.; Grimm, J.; et al. Phase 1 Trial of Antibody NI006 for Depletion of Cardiac Transthyretin Amyloid. New Engl. J. Med. 2023, 389, 239–250. [Google Scholar] [CrossRef] [PubMed]
- Galant, N.J.; Bugyei-Twum, A.; Rakhit, R.; Walsh, P.; Sharpe, S.; Arslan, P.E.; Westermark, P.; Higaki, J.N.; Torres, R.; Tapia, J.; et al. Substoichiometric inhibition of transthyretin misfolding by immune-targeting sparsely populated misfolding intermediates: a potential diagnostic and therapeutic for TTR amyloidoses. Sci. Rep. 2016, 6, 25080. [Google Scholar] [CrossRef] [PubMed]
- Higaki, J.N.; Chakrabartty, A.; Galant, N.J.; Hadley, K.C.; Hammerson, B.; Nijjar, T.; Torres, R.; Tapia, J.R.; Salmans, J.; Barbour, R.; et al. Novel conformation-specific monoclonal antibodies against amyloidogenic forms of transthyretin. Amyloid 2016, 23, 86–97. [Google Scholar] [CrossRef] [PubMed]
- Fontana, M.; Buchholtz, K.; Engelmann, M.D.M.; Grogan, M.; Hovingh, G.K.; Kristen, A.V.; Poulsen, P.; Shah, S.J.; Maurer, M.S. NNC6019–0001, a humanized monoclonal antibody, in patients with transthyretin amyloid cardiomyopathy (ATTR-CM): rationale and study design of a phase 2, randomized, placebo-controlled trial. Eur. Hear. J. 2022, 43. Issue Supplement_2, October, ehac544.1767. [Google Scholar] [CrossRef]
- Eisenberg, D.S.; Sawaya, M.R. Structural Studies of Amyloid Proteins at the Molecular Level. Annu. Rev. Biochem. 2017, 86, 69–95. [Google Scholar] [CrossRef] [PubMed]
- Tycko, R. Molecular structure of amyloid fibrils: insights from solid-state NMR. Q. Rev. Biophys. 2006, 39, 1–55. [Google Scholar] [CrossRef] [PubMed]
- Steinebrei, M.; Gottwald, J.; Baur, J.; Röcken, C.; Hegenbart, U.; Schönland, S.; Schmidt, M. Cryo-EM structure of an ATTRwt amyloid fibril from systemic non-hereditary transthyretin amyloidosis. Nat. Commun. 2022, 13, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Dubnovitsky, A.; Sandberg, A.; Rahman, M.M.; Benilova, I.; Lendel, C.; Härd, T. Amyloid-β protofibrils: size, morphology and synaptotoxicity of an engineered mimic. PLoS One. 2013 8, e66101. [CrossRef]
- Nguyen, B.A. Singh, V.; Afrin, S.; Yakubovska, A.; Wang, L.; Ahmed, Y.; Pedretti, R.; Fernandez-Ramirez, M.D.C.; Singh, P.; Pękała, M.; Cabrera Hernandez, L.O.; Kumar, S.; Lemoff, A.; Gonzalez-Prieto, R.; Sawaya, M.R.; Eisenberg, D.S.; Benson, M.D.; Saelices, L. Structural polymorphism of amyloid fibrils in ATTR amyloidosis revealed by cryo-electron microscopy. Nat Commun. 2024; 15. [Google Scholar]
- Suhr, O.B.; Lundgren, E.; Westermark, P. One mutation, two distinct disease variants: unravelling the impact of transthyretin amyloid fibril composition. J. Intern. Med. 2017, 281, 337–347. [Google Scholar] [CrossRef] [PubMed]
- Schonhoft J,D.; Monteiro, C.; Plate, L.; Eisele, Y.S.; Kelly, J.M.; Boland, D.; Parker, C.G.; Cravatt, B.F.; Teruya, S.; Helmke, S.; Maurer, M.; Berk, J.; Sekijima, Y.; Novais, M.; Coelho, T. Powers, E.T.; Kelly, J.W. Peptide probes detect misfolded transthyretin oligomers in plasma of hereditary amyloidosis patients. Sci Transl Med. 2017 9, eaam7621.
- Stabilini, R.; Vergani, C.; Agostoni, A.; Agostoni, R.V. Influence of age and sex on prealbumin levels. Clin. Chim. Acta 1968, 20, 358–359. [Google Scholar] [CrossRef] [PubMed]
- Muchtar, E.; Dispenzieri, A.; Magen, H.; Grogan, M.; Mauermann, M.; McPhail, E.D.; Kurtin, P.J.; Leung, N.; Buadi, F.K.; Dingli, D.; et al. Systemic amyloidosis from A (AA) to T (ATTR): a review. J. Intern. Med. 2020, 289, 268–292. [Google Scholar] [CrossRef]
Figure 1.
Based on experimental findings, a proposed pathway for the misfolding of TTR is presented. The protein data bank (PDB) codes are provided for each class of structure shown. The native tetrameric TTR (3TCT), when subjected to mutation and/or exposed to environmental stressors, becomes unstable and dissociates into monomers (1GKO). The overall instability of the isolated monomers causes conformational fluctuations and misfolding (2NBO), resulting in the transient formation of a flattened stackable monomer (8ADE). The spontaneous stacking of these flattened monomers as they are formed is energetically favorable and leads to the growth of amyloid fibrils.
Figure 1.
Based on experimental findings, a proposed pathway for the misfolding of TTR is presented. The protein data bank (PDB) codes are provided for each class of structure shown. The native tetrameric TTR (3TCT), when subjected to mutation and/or exposed to environmental stressors, becomes unstable and dissociates into monomers (1GKO). The overall instability of the isolated monomers causes conformational fluctuations and misfolding (2NBO), resulting in the transient formation of a flattened stackable monomer (8ADE). The spontaneous stacking of these flattened monomers as they are formed is energetically favorable and leads to the growth of amyloid fibrils.
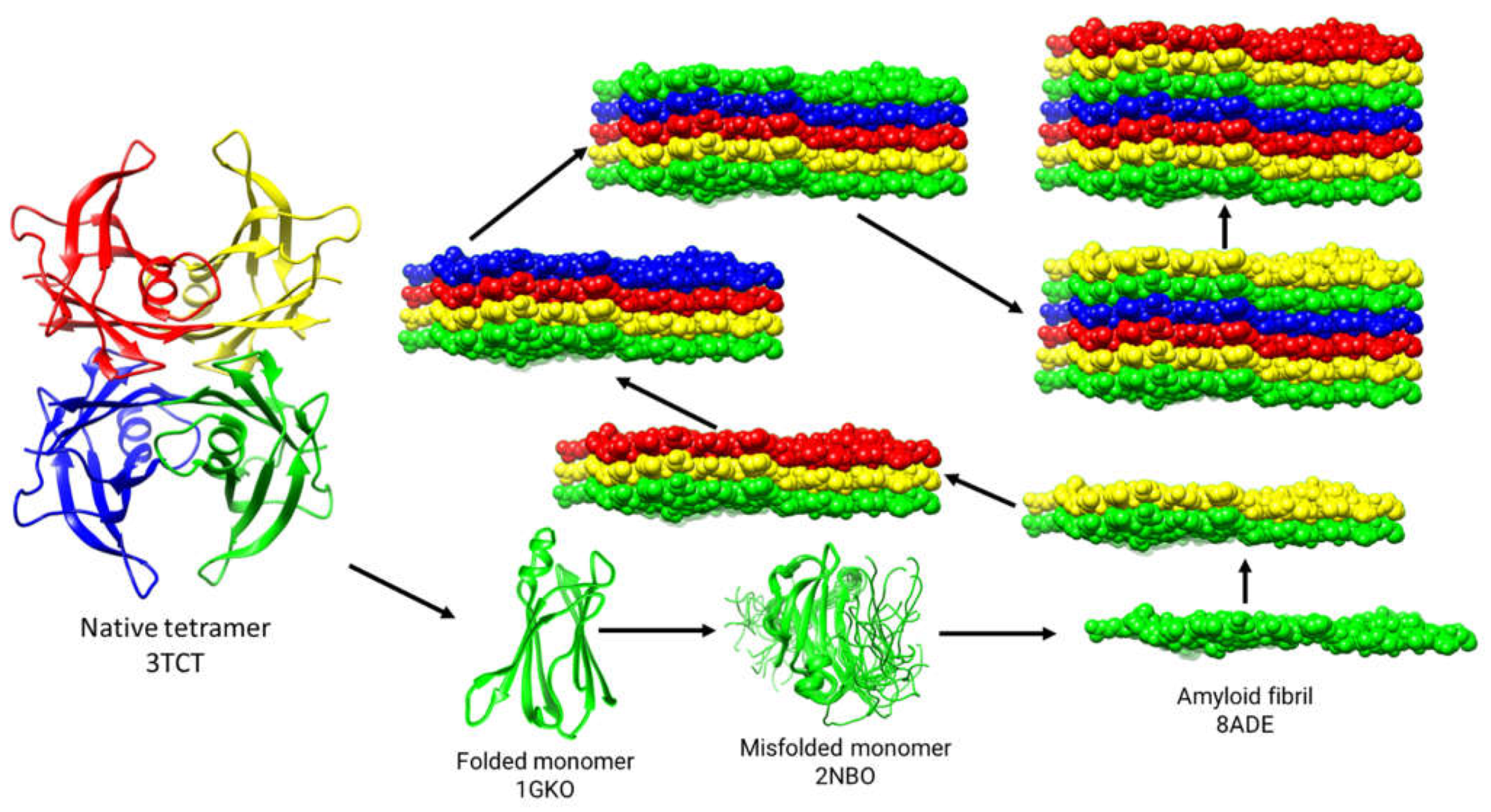
Figure 2.
Top-down view of the closed gate and open gate model structures of TTR fibril subunits. The structures of TTR amyloid fibrils show variation between patients and within a single patient. A wild-type ATTR patient [46] shows a preference for the closed gate model on the left; an I48S patient can adopt either closed or open gate model [48]. In the above structures, amino acid residues represented as encircled one-letter codes are residues that are present in the sequence but not detectable in the cryo-EM structures. The arrows indicate some common protease cleavage sites [49].
Figure 2.
Top-down view of the closed gate and open gate model structures of TTR fibril subunits. The structures of TTR amyloid fibrils show variation between patients and within a single patient. A wild-type ATTR patient [46] shows a preference for the closed gate model on the left; an I48S patient can adopt either closed or open gate model [48]. In the above structures, amino acid residues represented as encircled one-letter codes are residues that are present in the sequence but not detectable in the cryo-EM structures. The arrows indicate some common protease cleavage sites [49].
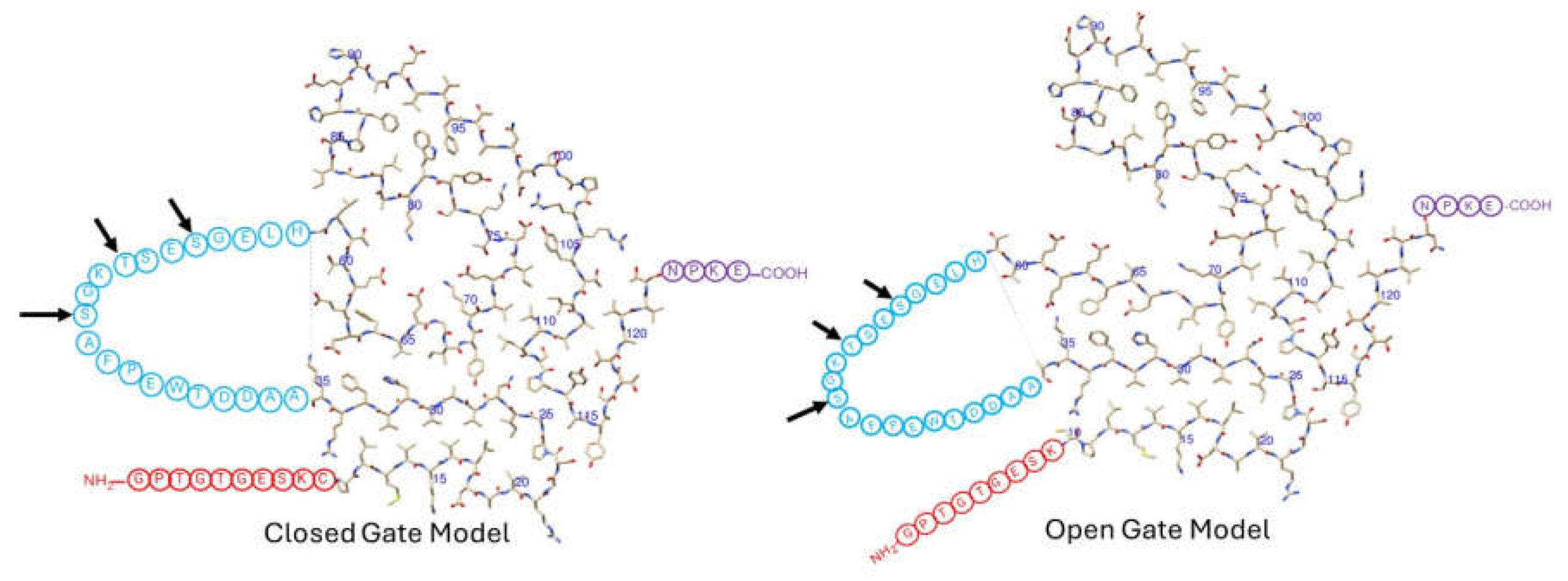
Figure 3.
Top-down view of closed gate and open gate models of TTR fibril subunits, with potential depleter antibody epitopes labeled. The residue numbers specifying the location of the epitope are given in parentheses. The PDB codes for the closed and open gate models are 8ADE (in cyan) and 8TDO (in blue), respectively.
Figure 3.
Top-down view of closed gate and open gate models of TTR fibril subunits, with potential depleter antibody epitopes labeled. The residue numbers specifying the location of the epitope are given in parentheses. The PDB codes for the closed and open gate models are 8ADE (in cyan) and 8TDO (in blue), respectively.
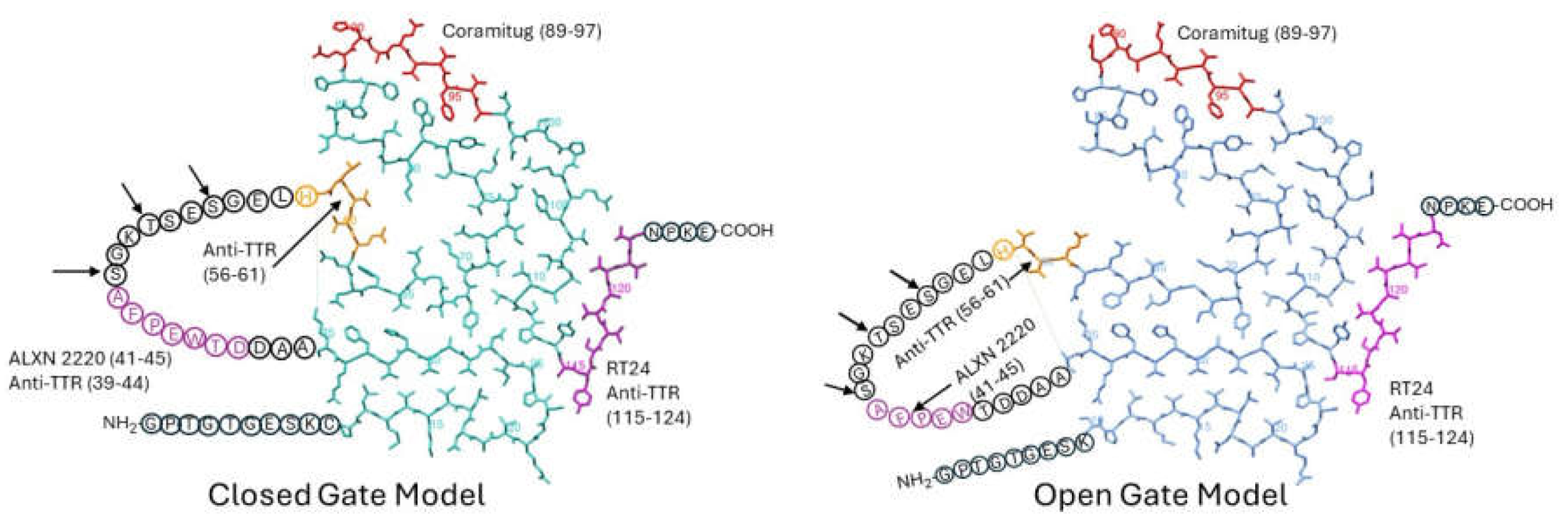
Figure 4.
Structural disposition of potential depleter epitopes on TTR tetramers, monomers, and fibrils. Space-filling models of the TTR tetramer (3TCT), monomer (1GKO), and fibril (8ADE) are shown with the epitope residues in red and all other residues in green. Regarding the fibril model showing the NI006 epitope in the bottom right corner of the figure, because this epitope is part of the unstructured loop spanning residues 36-57, it is not visible in the cryo-EM structure. Instead, it is portrayed as a reddish crescent shape. All the structures are oriented to maximize the view of the epitope. The fibril structures shown are derived from the closed gate model (8ADE). The structural disposition of depleter epitopes on the open gate model (8TDO) is qualitatively similar to those shown in the figure and thus is not shown here.
Figure 4.
Structural disposition of potential depleter epitopes on TTR tetramers, monomers, and fibrils. Space-filling models of the TTR tetramer (3TCT), monomer (1GKO), and fibril (8ADE) are shown with the epitope residues in red and all other residues in green. Regarding the fibril model showing the NI006 epitope in the bottom right corner of the figure, because this epitope is part of the unstructured loop spanning residues 36-57, it is not visible in the cryo-EM structure. Instead, it is portrayed as a reddish crescent shape. All the structures are oriented to maximize the view of the epitope. The fibril structures shown are derived from the closed gate model (8ADE). The structural disposition of depleter epitopes on the open gate model (8TDO) is qualitatively similar to those shown in the figure and thus is not shown here.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



