
Submitted:
12 August 2024
Posted:
12 August 2024
You are already at the latest version
Abstract
Neurologic disorders such as traumatic brain injury, multiple sclerosis, Alzheimer’s disease, and drug-resistant epilepsy have a high socioeconomic impact around the world. Current therapies for these disorders are often not effective. This creates a demand for the development of new therapeutic approaches to treat these disorders. Recent data suggest that autoreactive naturally occurring immunoglobulins produced by subsets of B cells called B1 B cells combined with complement are actively involved in the processes of restoration of neuronal functions during pathological conditions and remyelination. The focus of this review is to discuss the possibility of creating specific therapeutic antibodies that can activate and fix complement to enhance neuronal survival and promote central nervous system repair after injuries associated with many types of neurodegenerative diseases.
Keywords:
central nervous system (CNS)
; neurodegeneration
; CNS repair
; complement
; immediate early genes (IEGs)
; immunoglobulins
; natural autoantibodies (NAAs)
; B1 B cells
1. Introduction
An important role of natural autoantibodies (NAAs) as potential therapeutic agent historically arises from applications of intravenous immunoglobulin (IVIG) fraction isolated from serum of human healthy adult subjects. IVIG is a sterile product used for non-specific therapy in neurology, hematology, immunology, nephrology, and dermatology [1]. In dermatology, IVIG is used to treat Kawasaki disease (vasculitis), a disorder recently found to be associated with COVID-19 in children [2]. The most notable results for the usage of IVIG therapy were achieved for treatment of several neurologic disorders including traumatic brain injury [3] and multiple sclerosis [4]. Substantial decrease in plasma level of pathogenic Aβ42 peptides were also observed in IVIG-treated patients with Alzheimer’s disease [5]. The current understanding of the mechanism of action of IVIG is not well understood and mechanistically connected with the deactivation of antigen-presenting cells such as macrophages and dendritic cells via Fc receptors (FcR) in an antigen-independent manner [1]. It is also suggested the therapeutic action is mostly mediated by IgG antibodies that represent ~90-95% of all immunoglobulins in the serum of healthy donors. However, concentrations of other isotypes of immunoglobulins such as IgA and IgM significantly vary from donor to donor [1]. In healthy human subjects IgM and IgA represent on average less than 5-10% of all reaching the peak by the age of 30-40, following a sharp decline during aging [6]. Most of IgM and IgA isotypes in the serum of normal subjects represent NAAs, which have broad spectrum specificity and are often self-reactive binding to carbohydrates and lipids on the surface of damaged or apoptotic cells. It is suggested that these IgM or IgA immunoglobulins recognize known or unknown self-antigens and mediate therapeutic effect via of Fc portion of antibodies that bind recently acknowledged FcRs for IgM and low affinity for IgA (known as FcµR) and both IgM and IgA (known as FcαµR) [7] and by binding of Fab portion of antibodies to self-antigen on the surface of damaged or normal cells [2]. Particularly, it was shown that self-reactive IgM could bind to the surface of oligodendrocytes and their progenitors (cells that comprise myelin sheath in the central nervous system (CNS)) and promote re-myelination after CNS injury acting via FcμR and FcαμR [8]. The antigen specificity of these antibodies is still not well characterized, and they are usually present in the serum of healthy donors in low concentrations, which explains the limited therapeutic efficacy of IVIG. To understand the specificity and properties of therapeutic antibodies in IVIG, it is important to understand the basic biology of specific B cell clones that produce these therapeutic self-reactive IgM or IgA within the CNS. During development, most NAAs are produced by a specific type of B cells called B1 B cells [9]. After birth, the percentage of B1 B cells in the peripheral blood gradually declines over time reaching a minimum level during the aging process similar to the level of NAAs [6,10]. During most of adulthood, B1 B cells remain the main source of NAAs that have therapeutic potential in humans [11]. In this review, we will discuss possible antigen specificity and mechanism of action of NAAs that can activate and fix complement on the surface of neuronal cells to influence the function of neurons including their synaptic activity, response to neurologic insult, survival, and repair.
2. B1 B Cells
2.1. Mouse B1 B Cells
B1 B cells are a subset of innate-like B lymphocytes that are more ancient when compared to conventional B cells (referred to as B2 B cells). B1 cells are phenotypically distinguished from B2 cells by the expression of a panel of specific markers, early appearance during development, and homing in specific locations in the body cavities: peritoneal, pleural, and CNS ventricles [12]. For example, in the peritoneum, more than 70% of all B cells are B1 B cells. Mouse peritoneal B1 B cells express marker CD11b which makes these cells similar to innate immune cells such as macrophages and myeloid dendritic cells that also express this marker. In contrast to body cavities, B1 B cells are found in the secondary lymphoid organs such as the spleen and lymph nodes at very low frequencies (1% and 0.2% of all B cells, respectively) [13]. B1 B cells mostly produce IgM and rarely switch to other classes of antibodies (e.g. IgA or IgG3) compared to the conventional B2 B cells that often switch to IgG1. Finally, B1a B cells are the only type of B lymphocytes that could co-express macrophage CD11b and pan-T-cell marker CD5. In the mouse, B1a cells are conventionally referred to as CD45RlowCD23lowCD19+IgM+CD5+ cells. In the peritoneal cavity, B1 B cells express mentioned earlier marker CD11b, which is absent in B1 B cells in the spleen, lymph nodes, or peripheral blood. Thus, B1 B cells can be CD11b-positive and CD11b-negative. At the same time, B2 B cells are CD45RhiCD23+CD19+IgD+IgMvariableCD5-. Although B1 and B2 cells are not always easy to distinguish in tissues at the site of inflammation, B1a B cells could be distinguished from B2 cells as IgMhiCD5+ [13,14].
2.2. Human B1 B Cells
B cell subsets isolated from immunodeficient patients have CD19hiCD21lowCD23−CD86hiIgMhi phenotype and have many phenotypic and functional similarities with mouse B1 B cells [15]. More recent studies suggest the identification of human B1 B cells as CD19+CD20+CD27+CD38low/intCD43+ [10]. Human B1 B cells are the most attractive targets for the therapy of neurodegenerative diseases as they could produce regulatory NAAs that promote tissue repair in humans [11].
2.3. Function of B1 B Cells and Their Antibodies
Despite intensive investigations for many decades, the main function of B1 B cells remains unclear. It is known that these cells are responsible for the production of naturally occurring germline-encoded antibodies (referred to as NAAs in this review), most of which are IgM and on rare occasions IgA, and even more rare IgG3 [9]; however, as innate-like cells, B1 B cells can also play an important role in debris or apoptotic cell clearance, antigen presentation, and production of immunoregulatory cytokines such as IL-10 [16,17]. Yet, so far, the main function of B1 B cells is mostly connected with the function of B1-derived NAAs. The exact specificity of these antibodies is not well known for most of B1 B cell clones, and their autoimmune nature is related to pathological conditions, but it was shown that these antibodies often recognize carbohydrate and lipid self-antigens [14,18]. It was initially assumed that B1 cell-derived autoimmune IgMs that bind to gangliosides contribute to autoimmune diseases affecting peripheral nerves known as Guillain-Barre syndrome (GBS) [19]. However, more recent studies suggested that GBS is mostly connected with IgG1, but not IgM, IgA, or IgG3 autoantibodies, indicating that this disease is most likely mediated by conventional B2 rather than B1 B cells [20,21]. Recent data also indicate that IgM promotes CNS repair by binding to oligodendrocytes promoting their expansion and differentiation and guiding neurite outgrowth [8,22], confirming the therapeutic potential of B1 B cells and their NAAs. The specificity of most common NAAs that recognize brain-specific antigens is summarized in Table 1.
It is quite interesting that many antigens for NAAs in the CNS are located in neuronal lipid rafts (NLR), which are located in post-synaptic membranes that are enriched in neurotransmitter receptors and brain-specific gangliosides at the area of the synaptic cleft [23] and related to many types of pathologies including traumatic brain injury, Alzheimer’s disease, multiple sclerosis, and epilepsy [24,25,26,27]. How NLR could induce the production of NAAs is currently not clear, but we have previously shown that during blood-brain barrier (BBB) disruption, platelets can recognize brans-specific gangliosides and initiate neuroinflammation, which possibly leads to stimulation activation of B1 B cells among other immune cells such as microglia, macrophages, and T cells [23,24,28]. Currently, the role of B1 B cells and their NAAs in the regulation of neuroinflammation is not well-known. B1-derived IgMs could constitute an active component of IVIG, which requires further investigation. Even though IgM is present in low concentrations in IVIG [1], identification of therapeutic B1 B cell clones that mediate downregulation neuroinflammation and promote CNS repair would allow to create monoclonal antibodies with a high level of efficiency to treat acute neurologic disorders. The role of these antibodies in particular disorders is discussed below.

Table 1.
Antigen specificity of common NAAs that bind to CNS-specific antigens.
| Antigen | Location in the CNS | Isotype (Clone) |
Species (Source of Ab1) |
Relation to pathology and/or repair |
References | |
|---|---|---|---|---|---|---|
| AQP43 | Astrocytes (limitans) | IgG | Human | Neuromyelitis Optica | [29] | |
| NMDAR3 | Neurons (NLR4) | IgM, IgA | Human | Encephalitis, psychosis, seizures | [30] | |
| AMPAR5 | Neurons (NLR) IgG, IgM | Human | Encephalitis, seizures | [30] | ||
| GABAAR6 | Neurons (NLR) IgG1, IgG3, IgM | Human | Encephalitis, psychosis, seizures | [31] | ||
| D2R7 | Neurons (NLR) IgG, IgM | Human | Parkinson's disease, psychosis | [32] | ||
| MOG8 | Oligodendrocytes IgM | Mice | Multiple sclerosis | [33] | ||
| Sulfatide | Oligodendrocytes IgM (hIgM22) | Human | Remyelination | [34] | ||
| Gangliosides | Neurons (NLR) IgM (sHIgM12) | Human | Axonal outgrowth | [35] | ||
| Amyloid | Neurons (NLR) IgM | Human | Alzheimer's disease, neuroprotection | [36] | ||
| MLD9 | Neurons, astrocytes IgM | Human | Schizophrenia | [37] | ||
| PC10 | CNS (including NLR) IgM | Human | Stroke, Alzheimer’s disease | [38,39] | ||
1Ab, antibodies; 2AQP4, aquaporin-4; 3NMDR, N-methyl-D-aspartate receptor; 4NLR, neuronal lipid rafts; 5AMPAR, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor; 6GABAAR, γ-aminobutyric acid type A receptor; 7D2R, dopamine type 2 receptor; 8MOG, myelin oligodendrocyte glycoprotein; 9MLD, malondialdehyde; 10PC, phosphorylcholine.
3. Traumatic Brain Injury
Traumatic brain injury (TBI) is one of the leading causes of death and disability among young adults worldwide with a high socioeconomic impact [40]. TBI could also predispose to the development of other neuropathological conditions such as Alzheimer’s disease, amnesia, and epilepsy [41]. The role of B1 B cells in TBI is not clear and has not been specifically investigated so far. It was reported that certain TBI patients could develop antibodies to gangliosides, but the role of these antibodies in TBI pathology is unclear [42]. We found that st3gal5-deficient mice that lack major brain gangliosides GM1, GM3, GT1b, and GQ1b have less inflammation in the CNS after TBI, but more extensive neuronal damage and cognitive decline [43]. This indicated an important role of brain gangliosides in the development of CNS inflammation and neuronal repair as a potential target for B1 cells. However, the functions of B1 B cells as potential source of anti-ganglioside antibodies have not been extensively investigated in the TBI models. Thus, the role of B1 B cells and B1-derived IgM or IgA in TBI is not clear; however, activation of B cells and production of self-reactive antibodies was reported as a hallmark of TBI or spinal cord injury [44,45], indicating the possibility of stimulating CNS repair after TBI using self-reactive antibodies such as sHIgM12 that bind to brain-specific gangliosides or other CNS autoantigens and promote CNS repair (Table 1).
4. Multiple Sclerosis
Multiple sclerosis (MS) is a chronic autoimmune disease of the CNS that affects predominantly young adults leading to substantial neurological disability that includes upper and lower motor syndrome. MS and experimental autoimmune encephalitis (EAE; an animal model for MS) involve autoimmune T and that recognize myelin self-antigens such as myelin oligodendrocytes glycoprotein (MOG) [46]. MS onset usually occurs with the relapsing-remitting type, which is characterized by multiple relapses followed by spontaneous remission. Recently the role of B cells has been recognized for MS pathology. B cells could play a pathological role in MS by presenting self-antigen (e.g. MOG, see Table 1) stimulating pathogenic T cells, and producing autoantibodies that bind to myelin self-antigens on the surface of oligodendrocytes and activate complement contributing to the destruction of oligodendrocytes. However, it was also proposed that B cells could be beneficial for MS serving as regulatory B cells (Bregs) via the production of IL-10 (and possibly other regulatory cytokines IL-35 and TGFβ) that suppress CNS inflammation [47,48]. At the same time, the role of B1 B cells and B1-derived self-reactive NAA in MS is not well understood; however, it was demonstrated that the level of B1 B cells is decreased during MS progression. We have previously found that antibodies to ganglioside GQ1b (abundant in NLR) dramatically decreased EAE severity, indicating the important role of anti-ganglioside autoantibodies in controlling neuroinflammation and/or preserving neuronal functions [28]. Moreover, certain human autoantibodies such as hIgM22 clone promote remyelination and very perspective for future MS therapy (Table 1).
5. Alzheimer’s Disease
Alzheimer’s disease (AD) is a type of dementia associated with cognitive decline and, commonly, associated with aging. Pathologically AD is characterized by accumulation of aggregated amyloid and tau proteins, synaptic disfunction, and brain atrophy [49]. The role of B cells in the pathology of AD remains controversial. A recent study suggests that regulatory B cells ameliorate AD-associated neuroinflammation by producing regulatory cytokine IL-35 [50] The other study suggests that depletion of B cells using anti-CD20 antibodies or B cell-deficient mice resulted in amelioration of AD in three different mouse models of AD that include 3xTgAD, APP/PS1 and 5xFAD mice [51]. It is interesting that in the study mentioned above, the numbers of B1a and B1b cells were increased in the CNS of the 3xTgAD transgenic mouse model of AD. However, in the 5xFAD model, the numbers of B1a and B1b cells were not significantly different from the control non-transgenic control group of mice, indicating model-specific differences in mouse models of AD [51] Thus, in this study, the therapeutic effect was likely to be due to the depletion or knockout of conventional B2 rather than B1 cells. Indeed, studies in mice and humans indicate a decrease in the number of B1 B cells during aging [52], when AD becomes the prevailing disease. In contrast to the pathological role of B cells, there is more evidence that B1-derived NAAs play a protective role in AD [5,36,39] Our study indicated that deficiency in brain-specific gangliosides that are predominantly located within NLRs significantly ameliorated AD pathology in the 5xFAD mouse model of AD. Moreover, the treatment of mice with sialic-acid binding lectin resulted in substantial improvement of AD pathology by lowering amyloid burden, brain atrophy, and cognitive decline [25]. Since sialic acid is an external part of many types of brain-specific gangliosides and often serves as an external epitope for anti-ganglioside antibodies, this could indicate that NAAs with specificity to gangliosides can be used as future therapeutic agents for AD. In addition, it was also found that NAAs with specificity to phosphorylcholine (also located in NLR) and amyloid significantly ameliorated AD pathology (Table 1).
6. Epilepsy
Epilepsy is a disease characterized by uncontrolled seizures and a disbalance of activating vs. inhibitory neurotransmitters and/or their receptors. Although treatment of epilepsy is available, about 30% of patients do not respond to available drugs that modulate the function of activating or inhibitory neurotransmitter receptors [53]. Epilepsy is associated with an inflammatory response in the CNS and possible involvement of B cells, their antibodies, and complement [54]. The detection of NAAs that recognize activating and inhibitory neurotransmitter receptors (NAMDR and GABAAR, respectively) was associated with the development of epileptic seizures as shown in Table 1. Although the level of Bregs was decreased in patients with epilepsy [55], this could indicate systemic changes of balance towards the development of neuroinflammation [56]. Binging of NAAs to NLR could change signaling downstream of neurotransmitter receptors changing the level of neuronal electric activity and activation, which may have beneficial or detrimental effects on the development of seizures.
7. Emerging Role of B1 B Cells and NAAs in Neurological Diseases
For a very long time, the role of B1 B cells in the development of neurological diseases was not known. Recently it was demonstrated that B1a cells were accumulated in normal CNS during development. These cells were CD45+CD19+CD45R+IgM+CD5+ and were found in the CNS at high frequencies in the brains of developing P1-P21 (post-natal day 1 to day 21) mice but not in adult 8-week-old animals [57]. The authors of this study demonstrated that these cells were located in brain verticals and their IgM played an important role in binding to oligodendrocyte progenitors via the specific receptor for IgM (FcμR) and stimulating the proliferation of oligodendrocyte progenitors and their differentiation into mature oligodendrocytes [57]. FcμR, also known as CD351, is an Fc receptor that binds IgM with a very high affinity and intermediate affinity for IgA, both of which are produced by B1 B cells. Thus, B1a cells play an important role in myelination via producing self-reactive IgM, which may be also important for CNS repair after injury and blood-brain barrier (BBB) disruption, which is associated with MS or TBI. However, the specificity of most self-reactive IgM is not known. Understanding of specificity of B1-derived self-reactive IgM is very important to create highly effective and specific therapeutic antibodies, which would replace current IVIG therapy that uses whole immunoglobulin fraction from healthy donors.
8. New Role of Complement in Neurologic Diseases
The complement system consists of ~30 proteins that comprise an enzymatic self-amplification cascade initiated by carbohydrates or lipids of pathogens, cellular debris, and blood coagulation cascade. There are three paths of complement activation: the classical, lectin, or alternative pathway [58,59]. The classical pathway is triggered by the formation of antigen-antibody complexes that recruit C1q, leading to the proteolytic cleavage of C2 into C2a and C2b and C4 into C4a and C4b. Subsequentially C4b and C2b form the C4bC2b complex (C3-convertase), which cleaves C3 into C3a and C3b. The lectin pathway is activated by carbohydrate antigens that serve as ligands for mannose-binding lectin. This pathway initiates the same cascade as the classical pathway, leading to the formation of C3a and C3b. The alternative pathway is not activated by antigen-antibody complexes. This pathway is constitutively active at a low level and is initiated by the spontaneous hydrolysis of C3 into C3a and C3b. C3b then binds to factor B fragment (fBb) in the presence of co-factor fD to form the alternative C3 convertase C3bfBb [58]. During blood coagulation, activated factor XII (FXIIa) activates the classical complement pathway by cleaving C1q, leading to the cleavage of C2 and C4 and the formation of C4bC2b, which further cleave C3 to generate C3b. Moreover, thrombin, kallikrein, and plasmin directly cleave C3, leading to the formation of C3a and C3b [60]. Active C3b downstream of all pathways leads to the cleavage of C5 (into C5a and C5b), formation of the membrane attack complex (MAC), and lysis of pathogens or target cells. The fragments C3a and C5a activate mast cells and macrophages and thus play a pro-inflammatory role. The active enzymes C3b and C4b are later degraded by factor I (FI) in the presence of other co-factors such as complement receptor (CR)-1 via a two-step process wherein FI and co-factors cleave C3b to make an iC3b fragment, which is then cleaved into C3c and C3d. Similarly, C4b is cleaved by FI into C4c and C4d [58,61]. C3c and C4c have similar sizes and structures, and their functions are not well known. The liver is the dominant source of most complement proteins in the plasma, except C1q which is synthesized in leukocytes [62].
In the CNS, most complement components, including C1q, C4, and C3, are synthesized by astrocytes, oligodendrocytes, neurons, and microglia [63]. Platelets can also secrete C3 [28], and these cells play a key role in the initiation of neuroinflammation after TBI [16]. Microglia express activating complement receptors CR1, CR3, and CR4 [64], while neurons express inhibitory receptors and newly discovered activating CSMD receptors [65]. Recent studies demonstrated the involvement of the complement system in the pathogenesis of TBI, AD, amyotrophic lateral sclerosis (ALS), and MS [61,66,67,68]. An association was also observed between C4 gene polymorphism and schizophrenia [69] similarly as with anti-malondialdehyde IgM (Table 1). In the normal CNS, microglia produce C1q, which recruits C4 and C3 to axons and thus marks them for elimination by microglia via CR3 [70,71]. However, it is currently not clear whether antibodies bind to damaged neurons and/or dysfunctional synapses to fix C1q and activate the C1q-C4-C3 pathway to eliminate damaged or dysfunctional synapses. In our study, we found that C4-deficient mice were susceptible to pentylenetetrazol(GABAAR antagonist)-induced seizures leading to a very high mortality rate [72]. Interestingly in our study, C3-deficient mice had slightly milder seizure scores [72], which was consistent with other studies indicating the pathogenic role of C3 in epilepsy [73,74]. We found that during seizures C4-, but not C3-, deficient mice were unable to upregulate immediate early response genes (IEGs) in the CNS including neuronal Egr1 and Npas4 and their downstream neuronal survival/repair factors such as BDNF, which lead to high mortality rate and cognitive problems [72]. Thus, our data indicate that C4 plays other functions besides activating C3 and promoting synapse pruning. We found that C4 contributed to neuronal activation via induction of IEGs.
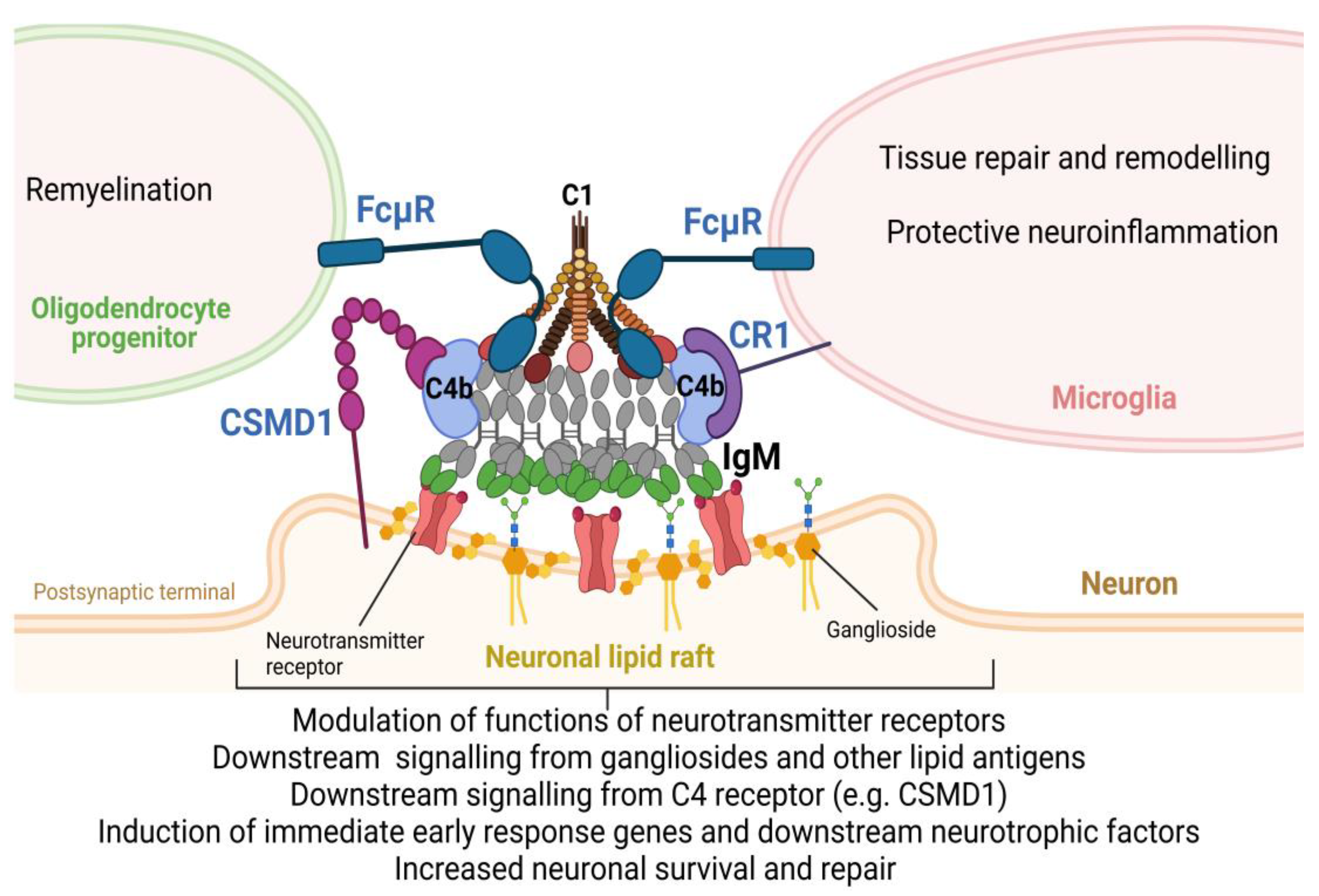
As the new mechanism of action of C4 in CNS, we propose that NAAs produced by B1 B cells bind to self-antigen in the area of damaged synapses that are enriched with NLR. B1-derived IgM or IgA autoantibodies could enter normal CNS via transcytosis from CNS ventricles when B1 B cells reside or could enter CNS from blood vessels during pathological conditions such as TBI, MS, AD, or epilepsy when the BBB becomes compromised. Moreover, B1 B cells accumulate in the CNS during neuroinflammation associated with neurological disorders [75]. Intrathecal synthesis of immunoglobulins is now recognized as the hallmark of many neurological disorders [76]. After binding of IgM to neuronal lipid rafts on the post-synaptic membrane, IgM recruits C1 and C4 to form unified IgM-C1-C4b complexes as was demonstrated [77]. The formation of these complexes plays an important function for neurons and glia. This includes 1) modulation of signaling downstream of neurotransmitter receptors; 2) engaging signaling downstream of gangliosides or other carbohydrate/lipid antigens; 3) engaging complement receptors on neuronal cells (e.g. CSMD family of activating complement receptors) or microglia (e.g. CR1 complement receptor); 4) engaging FcμR receptors on oligodendrocyte progenitors and microglia to stimulate remyelination and tissue repair and remodeling, and 5) induction of IEGs (e.g. Egr1 and Npas4) and its downstream neurotrophic factors (e.g. BDNF) leading to enhanced neuronal survival during CNS insult and its subsequent repair (Figure 1).
9. Conclusion and Future Directions
Traditionally, autoantibodies with complement were considered contributors to neurologic disorders by enhancing inflammation and damaging membranes of target cells by MAC. However, recent data indicates that specific clones of B cells (e.g. IgM-producing B1 B cells) and certain complement subunits (e.g. C4) also play neuroprotective roles and could be used in the future therapy of many types of neurologic disorders including TBI, MS, AD, and epilepsy. Future direction will be focused on the investigation of antigen specificity of particular B1 B cell clones and the role of IgM-C1-C4b complexes in direct modulation of neuronal functions.
Author Contributions
T.V. and E.D.P.: writing—original draft preparation, T.V., N. B., I.V., and E.D.P.: writing—review and editing, E.D.P.: visualization, E.D.P.: supervision, E.D.P.: project administration, E.D.P.: funding acquisition, T.V., N. B., I. V., and E.D.P.: All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Faculty Development Competitive Research Grant Program (FDCRGP), ref. no 201223FD8829, and by the Social Policy Grant from Nazarbayev University (Nazarbayev Fund, Kazakhstan).
Acknowledgments
The image was prepared using BioRender software.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Arumugham, V.B.; Rayi, A. Intravenous Immunoglobulin (IVIG). StatPearls 2023. [Google Scholar]
- Jolles, S.; Sewell, W.A.C.; Misbah, S.A. Clinical Uses of Intravenous Immunoglobulin. Clin Exp Immunol 2005, 142, 1–11. [Google Scholar] [CrossRef]
- Thom, V.; Arumugam, T. V.; Magnus, T.; Gelderblom, M. Therapeutic Potential of Intravenous Immunoglobulin in Acute Brain Injury. Front Immunol 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Soelberg Sorensen, P. Intravenous Polyclonal Human Immunoglobulins in Multiple Sclerosis. Neurodegener Dis 2007, 5, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Relkin, N.R.; Thomas, R.G.; Rissman, R.A.; Brewer, J.B.; Rafii, M.S.; Van Dyck, C.H.; Jack, C.R.; Sano, M.; Knopman, D.S.; Raman, R.; et al. A Phase 3 Trial of IV Immunoglobulin for Alzheimer Disease. Neurology 2017, 88, 1768–1775. [Google Scholar] [CrossRef] [PubMed]
- Jazayeri, M.H.; Pourfathollah, A.A.; Rasaee, M.J.; Porpak, Z.; Jafari, M.E. The Concentration of Total Serum IgG and IgM in Sera of Healthy Individuals Varies at Different Age Intervals. Biomedicine & Aging Pathology 2013, 3, 241–245. [Google Scholar] [CrossRef]
- Li, Y.; Shen, H.; Zhang, R.; Ji, C.; Wang, Y.; Su, C.; Xiao, J. Immunoglobulin M Perception by FcμR. Nature 2023 615:7954 2023, 615, 907–912. [Google Scholar] [CrossRef]
- Trebst, C.; Stangel, M. Promotion of Remyelination by Immunoglobulins: Implications for the Treatment of Multiple Sclerosis. Curr Pharm Des 2005, 12, 241–249. [Google Scholar] [CrossRef]
- Palma, J.; Tokarz-Deptuła, B.; Deptuła, J.; Deptuła, W. Natural Antibodies – Facts Known and Unknown. Cent Eur J Immunol 2018, 43, 466. [Google Scholar] [CrossRef]
- Rodriguez-Zhurbenko, N.; Quach, T.D.; Hopkins, T.J.; Rothstein, T.L.; Hernandez, A.M. Human B-1 Cells and B-1 Cell Antibodies Change with Advancing Age. Front Immunol 2019, 10, 430461. [Google Scholar] [CrossRef]
- Xu, X.; Ng, S.M.; Hassouna, E.; Warrington, A.; Oh, S.H.; Rodriguez, M. Human-Derived Natural Antibodies: Biomarkers and Potential Therapeutics. 2015; 10, 25–39. [Google Scholar] [CrossRef]
- Halperin, S.T.; ’T Hart, B.A.; Luchicchi, A.; Schenk, G.J. The Forgotten Brother: The Innate-like B1 Cell in Multiple Sclerosis. Biomedicines 2022, 10. [Google Scholar] [CrossRef]
- Prieto, J.M.B.; Felippe, M.J.B. Development, Phenotype, and Function of Non-Conventional B Cells. Comp Immunol Microbiol Infect Dis 2017, 54, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Rothstein, T.L. Natural Antibodies as Rheostats for Susceptibility to Chronic Diseases in the Aged. Front Immunol 2016, 7, 127. [Google Scholar] [CrossRef] [PubMed]
- Rakhmanov, M.; Keller, B.; Gutenberger, S.; Foerster, C.; Hoenig, M.; Driessen, G.; Van Der Burg, M.; Van Dongen, J.J.; Wiech, E.; Visentini, M.; et al. Circulating CD21low B Cells in Common Variable Immunodeficiency Resemble Tissue Homing, Innate-like B Cells. Proc Natl Acad Sci U S A 2009, 106, 13451–13456. [Google Scholar] [CrossRef]
- Popi, A.F.; Longo-Maugéri, I.M.; Mariano, M. An Overview of B-1 Cells as Antigen-Presenting Cells. Front Immunol 2016, 7, 177941. [Google Scholar] [CrossRef]
- Valeff, N.J.; Ventimiglia, M.S.; Dibo, M.; Markert, U.R.; Jensen, F. Splenic B1 B Cells Acquire a Proliferative and Anti-Inflamatory Profile During Pregnancy in Mice. Front Immunol 2022, 13, 873493. [Google Scholar] [CrossRef]
- Baumgarth, N. B-1 Cell Heterogeneity and the Regulation of Natural and Antigen-Induced IgM Production. Front Immunol 2016, 7, 324. [Google Scholar] [CrossRef]
- Benboubetra, M.; Nicholl, D.; Wagner, E.R.; Bowes, T.; Willison, H.J.; Cochrane, L.; Conner, J.; Furukawa, K.; Boffey, J. Tolerance to Self Gangliosides Is the Major Factor Restricting the Antibody Response to Lipopolysaccharide Core Oligosaccharides in Campylobacter Jejuni Strains Associated with Guillain-Barre Syndrome. Infect Immun 2002, 70, 5008–5018. [Google Scholar] [CrossRef]
- Fokkink, W.J.R.; Selman, M.H.J.; Dortland, J.R.; Durmuş, B.; Kuitwaard, K.; Huizinga, R.; Van Rijs, W.; Tio-Gillen, A.P.; Van Doorn, P.A.; Deelder, A.M.; et al. IgG Fc N-Glycosylation in Guillain-Barré Syndrome Treated with Immunoglobulins. J Proteome Res 2014, 13, 1722–1730. [Google Scholar] [CrossRef] [PubMed]
- Van Den Berg, B.; Walgaard, C.; Drenthen, J.; Fokke, C.; Jacobs, B.C.; Van Doorn, P.A. Guillain–Barré Syndrome: Pathogenesis, Diagnosis, Treatment and Prognosis. Nature Reviews Neurology 2014 10:8 2014, 10, 469–482. [Google Scholar] [CrossRef]
- Kumar, S.; Rodriguez, M.; Watzlawik, J.O.; Jordan, L.R.; Wittenberg, N.J.; Warrington, A.E.; Xu, X.; Oh, S.-H. A Patterned Recombinant Human IgM Guides Neurite Outgrowth of CNS Neurons. Sci Rep 2013, 3, 2267. [Google Scholar] [CrossRef]
- Ponomarev, E.D. Fresh Evidence for Platelets as Neuronal and Innate Immune Cells: Their Role in the Activation, Differentiation, and Deactivation of Th1, Th17, and Tregs during Tissue Inflammation. Front Immunol 2018, 9. [Google Scholar] [CrossRef]
- Dukhinova, M.; Kuznetsova, I.; Kopeikina, E.; Veniaminova, E.; Yung, A.W.Y.; Veremeyko, T.; Levchuk, K.; Barteneva, N.S.; Wing-Ho, K.K.; Yung, W.H.; et al. Platelets Mediate Protective Neuroinflammation and Promote Neuronal Plasticity at the Site of Neuronal Injury. Brain Behav Immun 2018, 74, 7–27. [Google Scholar] [CrossRef] [PubMed]
- Dukhinova, M.; Veremeyko, T.; Yung, A.W.Y.; Kuznetsova, I.S.; Lau, T.Y.B.; Kopeikina, E.; Chan, A.M.L.; Ponomarev, E.D. Fresh Evidence for Major Brain Gangliosides as a Target for the Treatment of Alzheimer’s Disease. Neurobiol Aging 2019, 77, 128–143. [Google Scholar] [CrossRef] [PubMed]
- Starossom, S.C.; Veremeyko, T.; Yung, A.W.Y.; Dukhinova, M.; Au, C.; Lau, A.Y.; Weiner, H.L.; Ponomarev, E.D. Platelets Play Differential Role during the Initiation and Progression of Autoimmune Neuroinflammation. Circ Res 2015, 117, 779–792. [Google Scholar] [CrossRef] [PubMed]
- Kopeikina, E.; Dukhinova, M.; Yung, A.W.Y.; Veremeyko, T.; Kuznetsova, I.S.; Lau, T.Y.B.; Levchuk, K.; Ponomarev, E.D. Platelets Promote Epileptic Seizures by Modulating Brain Serotonin Level, Enhancing Neuronal Electric Activity, and Contributing to Neuroinflammation and Oxidative Stress. Prog Neurobiol 2020, 188. [Google Scholar] [CrossRef] [PubMed]
- Sotnikov, I.; Veremeyko, T.; Starossom, S.C.; Barteneva, N.; Weiner, H.L.; Ponomarev, E.D. Platelets Recognize Brain-Specific Glycolipid Structures, Respond to Neurovascular Damage and Promote Neuroinflammation. PLoS One 2013, 8, e58979. [Google Scholar] [CrossRef] [PubMed]
- Jarius, S.; Franciotta, D.; Bergamaschi, R.; Wildemann, B.; Wandinger, K.P. Immunoglobulin M Antibodies to Aquaporin-4 in Neuromyelitis Optica and Related Disorders. Clin Chem Lab Med 2010, 48, 659–663. [Google Scholar] [CrossRef] [PubMed]
- Olivero, G.; Roggeri, A.; Pittaluga, A. Anti-NMDA and Anti-AMPA Receptor Antibodies in Central Disorders: Preclinical Approaches to Assess Their Pathological Role and Translatability to Clinic. International Journal of Molecular Sciences 2023, Vol. 24, Page 14905 2023, 24, 14905. [Google Scholar] [CrossRef]
- Pettingill, P.; Kramer, H.B.; Coebergh, J.A.; Pettingill, R.; Maxwell, S.; Nibber, A.; Malaspina, A.; Jacob, A.; Irani, S.R.; Buckley, C.; et al. Antibodies to GABAA Receptor A1 and Γ2 Subunits: Clinical and Serologic Characterization. Neurology 2015, 84, 1233–1241. [Google Scholar] [CrossRef]
- Dale, R.C.; Merheb, V.; Pillai, S.; Wang, D.; Cantrill, L.; Murphy, T.K.; Ben-Pazi, H.; Varadkar, S.; Aumann, T.D.; Horne, M.K.; et al. Antibodies to Surface Dopamine-2 Receptor in Autoimmune Movement and Psychiatric Disorders. Brain 2012, 135, 3453–3468. [Google Scholar] [CrossRef] [PubMed]
- Shurin, M.R.; Wheeler, S.E. Clinical Significance of Uncommon, Non-Clinical, and Novel Autoantibodies. Immunotargets Ther 2024, 13, 215. [Google Scholar] [CrossRef]
- Warrington, A.E.; Rodriguez, M. Method of Identifying Natural Antibodies for Remyelination. J Clin Immunol, 2010; 30 Suppl. [Google Scholar] [CrossRef]
- Denic, A.; Macura, S.I.; Warrington, A.E.; Pirko, I.; Grossardt, B.R.; Pease, L.R.; Rodriguez, M. A Single Dose of Neuron-Binding Human Monoclonal Antibody Improves Spontaneous Activity in a Murine Model of Demyelination. PLoS One 2011, 6, e26001. [Google Scholar] [CrossRef] [PubMed]
- Britschgi, M.; Olin, C.E.; Johns, H.T.; Takeda-Uchimura, Y.; Lemieux, M.C.; Rufibach, K.; Rajadas, J.; Zhang, H.; Tomooka, B.; Robinson, W.H.; et al. Neuroprotective Natural Antibodies to Assemblies of Amyloidogenic Peptides Decrease with Normal Aging and Advancing Alzheimer’s Disease. Proc Natl Acad Sci U S A 2009, 106, 12145–12150. [Google Scholar] [CrossRef] [PubMed]
- Maes, M.; Kanchanatawan, B.; Sirivichayakul, S.; Carvalho, A.F. In Schizophrenia, Deficits in Natural IgM Isotype Antibodies Including Those Directed to Malondialdehyde and Azelaic Acid Strongly Predict Negative Symptoms, Neurocognitive Impairments, and the Deficit Syndrome. Mol Neurobiol 2019, 56, 5122–5135. [Google Scholar] [CrossRef]
- Fiskesund, R.; Stegmayr, B.; Hallmans, G.; Vikström, M.; Weinehall, L.; De Faire, U.; Frostegård, J. Low Levels of Antibodies Against Phosphorylcholine Predict Development of Stroke in a Population-Based Study From Northern Sweden. Stroke 2010, 41, 607–612. [Google Scholar] [CrossRef]
- Eriksson, U.K.; Sjöberg, B.G.; Bennet, A.M.; De Faire, U.; Pedersen, N.L.; Frostegrd, J. Low Levels of Antibodies against Phosphorylcholine in Alzheimer’s Disease. J Alzheimers Dis 2010, 21, 577–584. [Google Scholar] [CrossRef] [PubMed]
- Mckee, A.C.; Daneshvar, D.H. The Neuropathology of Traumatic Brain Injury. In Handbook of Clinical Neurology; 2015; Vol. 127, pp. 45–66 ISBN 1857364570.
- Pearn, M.L.; Niesman, I.R.; Egawa, J.; Sawada, A.; Almenar-Queralt, A.; Shah, S.B.; Duckworth, J.L.; Head, B.P. Pathophysiology Associated with Traumatic Brain Injury: Current Treatments and Potential Novel Therapeutics. Cell Mol Neurobiol 2017, 37, 571–585. [Google Scholar] [CrossRef]
- Shah, M.; Garvin, R.; Shakir, A.; Jackson, C.; Carr, K.R. Post-Traumatic Brain Injury (TBI) Presenting with Guillain-Barre Syndrome and Elevated Anti-Ganglioside Antibodies: A Case Report and Review of the Literature. International Journal of Neuroscience 2014, 125, 486–492. [Google Scholar] [CrossRef]
- Dukhinova, M.; Kuznetsova, I.; Kopeikina, E.; Veniaminova, E.; Yung, A.W.Y.Y.; Veremeyko, T.; Levchuk, K.; Barteneva, N.S.; Wing-Ho, K.K.; Yung, W.-H.H.; et al. Platelets Mediate Protective Neuroinflammation and Promote Neuronal Plasticity at the Site of Neuronal Injury. Brain Behav Immun 2018. [Google Scholar] [CrossRef] [PubMed]
- Kelso, M.L.; Gendelman, H.E. Bridge between Neuroimmunity and Traumatic Brain Injury. Curr Pharm Des 2014, 20, 4284–4298. [Google Scholar] [CrossRef] [PubMed]
- Ankeny, D.P.; Popovich, P.G. B Cells and Autoantibodies: Complex Roles in CNS Injury. Trends Immunol 2010, 31, 332–338. [Google Scholar] [CrossRef] [PubMed]
- Veremeyko, T.; Yung, A.W.Y.; Dukhinova, M.; Kuznetsova, I.S.; Pomytkin, I.; Lyundup, A.; Strekalova, T.; Barteneva, N.S.; Ponomarev, E.D. Cyclic AMP Pathway Suppress Autoimmune Neuroinflammation by Inhibiting Functions of Encephalitogenic CD4 T Cells and Enhancing M2 Macrophage Polarization at the Site of Inflammation. Front Immunol 2018, 9, 50. [Google Scholar] [CrossRef]
- Rangachari, M.; Kerfoot, S.M.; Arbour, N.; Alvarez, J.I. Editorial: Lymphocytes in MS and EAE: More Than Just a CD4+ World. Front Immunol 2017, 8, 133. [Google Scholar] [CrossRef]
- van de Veen, W.; Stanic, B.; Wirz, O.F.; Jansen, K.; Globinska, A.; Akdis, M. Role of Regulatory B Cells in Immune Tolerance to Allergens and Beyond. Journal of Allergy and Clinical Immunology 2016, 138, 654–665. [Google Scholar] [CrossRef] [PubMed]
- Jorfi, M.; Maaser-Hecker, A.; Tanzi, R.E. The Neuroimmune Axis of Alzheimer’s Disease. Genome Med 2023, 15. [Google Scholar] [CrossRef] [PubMed]
- Feng, W.; Zhang, Y.; Ding, S.; Chen, S.; Wang, T.; Wang, Z.; Zou, Y.; Sheng, C.; Chen, Y.; Pang, Y.; et al. B Lymphocytes Ameliorate Alzheimer’s Disease-like Neuropathology via Interleukin-35. Brain Behav Immun 2023, 108, 16–31. [Google Scholar] [CrossRef]
- Kim, K.; Wang, X.; Ragonnaud, E.; Bodogai, M.; Illouz, T.; DeLuca, M.; McDevitt, R.A.; Gusev, F.; Okun, E.; Rogaev, E.; et al. Therapeutic B-Cell Depletion Reverses Progression of Alzheimer’s Disease. Nature Communications 2021 12:1 2021, 12, 1–11. [Google Scholar] [CrossRef]
- de Mol, J.; Kuiper, J.; Tsiantoulas, D.; Foks, A.C. The Dynamics of B Cell Aging in Health and Disease. Front Immunol 2021, 12, 733566. [Google Scholar] [CrossRef]
- Löscher, W.; Potschka, H.; Sisodiya, S.M.; Vezzani, A. Drug Resistance in Epilepsy: Clinical Impact, Potential Mechanisms, and New Innovative Treatment Options. Pharmacol Rev 2020, 72, 606. [Google Scholar] [CrossRef] [PubMed]
- Vezzani, A.; Lang, B.; Aronica, E. Immunity and Inflammation in Epilepsy. Cold Spring Harb Perspect Med 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Sanli, E.; Sirin, N.G.; Kucukali, C.I.; Baykan, B.; Ulusoy, C.A.; Bebek, N.; Yilmaz, V.; Tuzun, E. Peripheral Blood Regulatory B and T Cells Are Decreased in Patients with Focal Epilepsy. J Neuroimmunol 2024, 387, 578287. [Google Scholar] [CrossRef] [PubMed]
- Yamanaka, G.; Morichi, S.; Takamatsu, T.; Watanabe, Y.; Suzuki, S.; Ishida, Y.; Oana, S.; Yamazaki, T.; Takata, F.; Kawashima, H. Links between Immune Cells from the Periphery and the Brain in the Pathogenesis of Epilepsy: A Narrative Review. International Journal of Molecular Sciences 2021, Vol. 22, Page 4395 2021, 22, 4395. [Google Scholar] [CrossRef] [PubMed]
- Tanabe, S.; Yamashita, T. B-1a Lymphocytes Promote Oligodendrogenesis during Brain Development. Nat Neurosci 2018, 21, 506–516. [Google Scholar] [CrossRef] [PubMed]
- Merle, N.S.; Church, S.E.; Fremeaux-Bacchi, V.; Roumenina, L.T. Complement System Part I €“ Molecular Mechanisms of Activation and Regulation. Front Immunol 2015, 6, 262. [Google Scholar] [CrossRef]
- Foley, J.H.; Conway, E.M. Cross Talk Pathways Between Coagulation and Inflammation. Circ Res 2016, 118, 1392–1408. [Google Scholar] [CrossRef]
- Amara, U.; Flierl, M.A.; Rittirsch, D.; Klos, A.; Chen, H.; Acker, B.; Brückner, U.B.; Nilsson, B.; Gebhard, F.; Lambris, J.D.; et al. Molecular Intercommunication between the Complement and Coagulation Systems. The Journal of Immunology 2010, 185, 5628–5636. [Google Scholar] [CrossRef]
- Hammad, A.; Westacott, L.; Zaben, M. The Role of the Complement System in Traumatic Brain Injury: A Review. J Neuroinflammation 2018, 15, 24. [Google Scholar] [CrossRef]
- Lubbers, R.; van Essen, M.F.; van Kooten, C.; Trouw, L.A. Production of Complement Components by Cells of the Immune System. Clin Exp Immunol 2017, 188, 183–194. [Google Scholar] [CrossRef]
- Veerhuis, R.; Nielsen, H.M.; Tenner, A.J. Complement in the Brain. Mol Immunol 2011, 48, 1592–1603. [Google Scholar] [CrossRef]
- Chen, Y.; Chu, J.M.T.; Chang, R.C.C.; Wong, G.T.C. The Complement System in the Central Nervous System: From Neurodevelopment to Neurodegeneration. Biomolecules 2022, Vol. 12, Page 337 2022, 12, 337. [Google Scholar] [CrossRef] [PubMed]
- Kraus, D.M.; Elliott, G.S.; Chute, H.; Horan, T.; Pfenninger, K.H.; Sanford, S.D.; Foster, S.; Scully, S.; Welcher, A.A.; Holers, V.M. CSMD1 Is a Novel Multiple Domain Complement-Regulatory Protein Highly Expressed in the Central Nervous System and Epithelial Tissues. The Journal of Immunology 2006, 176, 4419–4430. [Google Scholar] [CrossRef]
- Wu, T.; Dejanovic, B.; Gandham, V.D.; Gogineni, A.; Edmonds, R.; Schauer, S.; Srinivasan, K.; Huntley, M.A.; Wang, Y.; Wang, T.-M.; et al. Complement C3 Is Activated in Human AD Brain and Is Required for Neurodegeneration in Mouse Models of Amyloidosis and Tauopathy. Cell Rep 2019, 28, 2111–2123.e6. [Google Scholar] [CrossRef]
- Parker, S.E.; Hanton, A.M.; Stefanou, S.N.; Noakes, P.G.; Woodruff, T.M.; Lee, J.D. Revisiting the Role of the Innate Immune Complement System in ALS. Neurobiol Dis 2019, 127, 223–232. [Google Scholar] [CrossRef] [PubMed]
- Ingram, G.; Hakobyan, S.; Robertson, N.P.; Morgan, B.P. Complement in Multiple Sclerosis: Its Role in Disease and Potential as a Biomarker. Clin Exp Immunol 2009, 155, 128–139. [Google Scholar] [CrossRef]
- Sekar, A.; Bialas, A.R.; de Rivera, H.; Davis, A.; Hammond, T.R.; Kamitaki, N.; Tooley, K.; Presumey, J.; Baum, M.; Van Doren, V.; et al. Schizophrenia Risk from Complex Variation of Complement Component 4. Nature 2016, 530, 177–183. [Google Scholar] [CrossRef]
- Chu, Y.; Jin, X.; Parada, I.; Pesic, A.; Stevens, B.; Barres, B.; Prince, D.A. Enhanced Synaptic Connectivity and Epilepsy in C1q Knockout Mice. Proceedings of the National Academy of Sciences 2010, 107, 7975–7980. [Google Scholar] [CrossRef]
- Li, Q.; Barres, B.A. Microglia and Macrophages in Brain Homeostasis and Disease. Nat Rev Immunol 2018, 18, 225–242. [Google Scholar] [CrossRef] [PubMed]
- Veremeyko, T.; Jiang, R.; He, M.; Ponomarev, E.D. Complement C4-Deficient Mice Have a High Mortality Rate during PTZ-Induced Epileptic Seizures, Which Correlates with Cognitive Problems and the Deficiency in the Expression of Egr1 and Other Immediate Early Genes. Front Cell Neurosci 2023, 17. [Google Scholar] [CrossRef]
- Schartz, N.D.; Aroor, A.; Li, Y.; Pinzón-Hoyos, N.; Brewster, A.L. Mice Deficient in Complement C3 Are Protected against Recognition Memory Deficits and Astrogliosis Induced by Status Epilepticus. Front Mol Neurosci 2023, 16, 1265944. [Google Scholar] [CrossRef]
- Wei, Y.; Chen, T.; Bosco, D.B.; Xie, M.; Zheng, J.; Dheer, A.; Ying, Y.; Wu, Q.; Lennon, V.A.; Wu, L.J. The Complement C3-C3aR Pathway Mediates Microglia-Astrocyte Interaction Following Status Epilepticus. Glia 2021, 69, 1155. [Google Scholar] [CrossRef] [PubMed]
- Aspden, J.W.; Murphy, M.A.; Kashlan, R.D.; Xiong, Y.; Poznansky, M.C.; Sîrbulescu, R.F. Intruders or Protectors – the Multifaceted Role of B Cells in CNS Disorders. Front Cell Neurosci 2023, 17, 1329823. [Google Scholar] [CrossRef] [PubMed]
- Negi, N.; Das, B.K. Decoding Intrathecal Immunoglobulins and B Cells in the CNS: Their Synthesis, Function, and Regulation. Int Rev Immunol 2020, 39, 67–79. [Google Scholar] [CrossRef] [PubMed]
- Sharp, T.H.; Boyle, A.L.; Diebolder, C.A.; Kros, A.; Koster, A.J.; Gros, P. Insights into IgM-Mediated Complement Activation Based on in Situ Structures of IgM-C1-C4b. Proc Natl Acad Sci U S A 2019, 116, 11900–11905. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
The proposed model of regulation of the CNS repair process by B1-derived NAAs (IgM) and complement subunits C1 and C4 by forming IgM-C1-C4b complexes that affect neuronal cells, microglia, and oligodendrocyte progenitors.
Figure 1.
The proposed model of regulation of the CNS repair process by B1-derived NAAs (IgM) and complement subunits C1 and C4 by forming IgM-C1-C4b complexes that affect neuronal cells, microglia, and oligodendrocyte progenitors.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



