
Submitted:
14 August 2024
Posted:
14 August 2024
You are already at the latest version
Abstract
We report the case of a 45-year-old Caucasian man who was treated upfront with brentuximab vedotin (A) and doxorubicin, vinblastine, and dacarbazine (AVD; A+AVD) for advanced-stage classical Hodgkin's lymphoma (cHL). Right after completing the 6th cycle of induction therapy, he developed serotine fever, progressive clinical deterioration, symptomatic splenomegaly and se-vere anemia. Extensive infectious disease evaluations and serial PET scans were conducted to rule out lymphoma progression. Finally, a diagnosis of visceral leishmaniasis (VL) was performed. The patient was treated with parenteral amphotericin B, with rapid and complete resolution of clinical and laboratory abnormalities. We described this rare case of infectious complication after A+AVD delivered for cHL, and we performed a comprehensive review of the literature on the occurrence of VL in the context of non-HL and cHL, since this infection is becoming increasingly prevalent in non-tropical countries.
Keywords:
Hodgkin’s Lymphoma
; Brentuximab-AVD
; (A+AVD)
; Visceral leishmaniasis
; splenomegaly
1. Introduction
Classical Hodgkin’s lymphoma (cHL) has a global annual incidence of 100,000 cases. It affects approximately 8540 new patients in the United States each year [1]. The reported incidence rate in the European Union is 2.3 per 100.000 habitants [2].
cHL neoplastic cells are large dysplastic mononuclear and multinucleate cells, commonly known as Reed-Sternberg cells. These abnormal cells are surrounded by a combination of mature non-neoplastic inflammatory cells. This tumor microenvironment (TME) has the capacity to expand regulatory T cells, thus inhibiting CD8-positive cells and repolarizing tumor-associated macrophages. Consequently, the interaction between lymphoma cells and TME leads to a state of immune deficiency of the affected subjects [3]. With the increased understanding of the biology and microenvironment of cHL, novel strategies with notable efficacy, including targeted therapies, immunotherapy and cell therapy have been adopted [4]. For example, brentuximab vedotin (A), an anti-CD30 antibody–drug conjugate, that was initially approved for relapsed and refractory cHL, has been recently incorporated into the AVD regimen (A+AVD), with reported survival advantage as compared to the traditional AVD plus bleomycin scheme (ABVD) in advanced stage cHL [5]. Indeed, the ECHELON-1 study has established the superior efficacy in terms of modified progression-free survival of A+AVD over ABVD in the treatment of patients with advanced-stage cHL. All secondary efficacy endpoints were in favor of A+AVD, including the combined risk of progression, death, or noncomplete response, as well as the use of subsequent anticancer therapy at 2 years [5]. The most recent update of the trial has reported a 6-years overall survival advantage of A+AVD (93.9%) as compared to ABVD (89.4%) [1]. However, the A+AVD regimen has also been linked to an increased risk of infection compared to the ABVD backbone, indicating that innovative treatments should always require careful consideration of potential new and rare adverse events.
Leishmaniasis are the most important illnesses transmitted by phlebotomine sand flies [6]. Presenting symptoms vary from ulcerative skin lesions to mucosal deforming lesions (tegumentary leishmaniasis) or liver and spleen hypertrophy, which is the case of visceral leishmaniasis (VL). L. donovani and L. infantum, are the two species that can cause VL. Globally about 500,000 new cases of VL occur worldwide every year. If not appropriately treated, over 95% of patients affected by VL cases will die [7]. In Italy, between 2009 and 2016, a total of 1126 cases of VL were identified, resulting in an incidence rate (IR) of 2.43 cases per 106 person-per-year [8]. This parasite infection can manifest as an opportunistic disease due to a recent infection or as reactivation of a latent condition. The clinical picture is characterized by prolonged fever, weight loss, anemia, and hepatosplenomegaly [7].
Leishmaniasis is an intracellular infection that adversely impacts the activation and functionality of macrophages and dendritic cells, allowing malignant cells to avoid immune destruction. Additionally, chronic leishmaniasis infections result in CD4 lymphopenia and a reduced CD4/CD8 ratio. The noticeable immunosuppression observed during VL is believed to be influenced by elevated levels of regulatory T-cells leading to a compromise in adaptive immune responses [9].
Leishmaniasis have been rarely described in patients with cHL. The case presented here is a rare association of VL and cHL in the setting of post A+AVD chemo-immunotherapy.
2. Case Description
A 43-year-old gentleman was diagnosed with nodular sclerosis stage IV cHL with focal involvement of spleen and bone marrow in February 2023 (Figure 1A). In his medical history, he underwent a total thyroidectomy 12 years before, due to a papillary tumor. Additionally, he was known to be a frequent traveler. He had visited Argentina (2006) and Brazil (2010), with his most recent trip being to Australia in 2022.
The patient underwent six cycles of A+AVD treatment, achieving an early treatment response, as documented by positron emission tomography (PET-TC) after two cycles (Figure 1B). Treatment was completed in August 2023. One month later, a PET-CT scan conducted post-treatment revealed new onset of splenomegaly, which measured 18 cm and had increased uptake of 18FDG, without associated focal lesions or other abnormalities (Figure 1C). This finding contrasted with the imaging conducted at the time of diagnosis, which revealed spleen lesions without splenomegaly. Simultaneously, our patient reported fever and self-limited diffuse erythema for several days. At the same time, his two-year-old child was diagnosed with erythema infectiosum caused by Parvovirus B19 (fifth disease), and for this reason the same disease was initially suspected. A physical examination revealed splenomegaly, confirmed by abdominal ultrasound (splenic span of 18 cm). Laboratory tests indicated mild anemia (hemoglobin Hb: 11 g/dl), with a leukocyte count of 2,400 cells/mm3, neutrophils: 1,200 cell/mm3, and a C-reactive protein level of approximately 95 mg/l. Extensive microbiological tests, including urine analysis, chest x-rays, and blood cultures, were conducted. Subsequently, IgM antibody turned positive for Parvovirus B19, Toxoplasma, as well as Enterovirus. Parvovirus B19 DNA was detected by reverse transcriptase-polymerase chain reaction (RT-PCR), with a count of 658,439 copies/ml. As part of the initial management, we administered granulocyte colony-stimulating factor (G-CSF) and intravenous immunoglobulin therapy with mild clinical benefit.
Despite parvovirus DNA declined to undetectable levels, fever persisted, associated to weight loss of 8 kg in two months, and the blood counts severely impaired in terms of anemia and leukopenia (Hb 8.1 g/dL, WBC: 1,650 cells/mm3). Considering the potential for cHL relapse, another PET-CT scan was performed, confirming splenomegaly (SUV max 6.7) with no other apparent abnormalities (Figure 1D). This was supported by ultrasound, which revealed an increase in splenomegaly to 20 cm. In agreement with our infectious disease specialists, given the clinical and laboratory deterioration of the patient, RT-PCR for Leishmaniasis was performed on peripheral blood. The test turned positive, finally establishing the diagnosis of VL.
The patient was started with amphotericin B at 4 mg/kg for 5 days, followed by an additional 5 weekly doses of 4 mg/kg as part of maintenance therapy. This treatment resulted in a sudden resolution of all symptoms. The abdominal ultrasound showed a significant reduction in splenomegaly with normalization of spleen diameter 1 month after completion of the therapy. Three months later, the patient was asymptomatic, with normal blood counts. Now, approximately one year later, he is doing good and he is in active follow up for his cHL.
3. Discussion and Review of the Literature
Leishmaniasis is recognized as an opportunistic infection. Leishmaniasis is transmitted by sandfly bites in parts of Asia (primarily India), Africa (primarily Sudan) and South America (primarily Brazil) where all together there are an estimated half million cases per year. However, following the global trend of immigrations, the number of populations susceptible to infections due to immunosuppressive factors, co-morbidities and ageing, the disease has demonstrated significant increase in its incidence in non-tropical countries. Therefore, the risk of re-emergence of leishmaniasis associated with increased clinical susceptibility to primary infections or reactivation of latent infections is not negligible [6]. The elderly population (probably because of immunosenescence), adults medically immunosuppressed by chemical or biological drugs, adults with autoimmune diseases or cancer or presenting with immunodeficiencies, who live in or travel to endemic countries are categories particularly at risk [6]. Apart from human immunodeficiency virus (HIV) status, hematological malignancies are the most frequent underlying cause of immunodeficiency in patients who develop VL.
The pathogens Leishmania donovani, L. infantum, and L. chagasi are linked to VL [10]. L. Infantum is the most common species in the Mediterranean basin. In humans, parasites in the amastigote form are typically found intracellularly within the reticuloendothelial system, a flagellate 2–4 μm in diameter (Leishman-Donovan body). Symptoms normally develop after an incubation period of weeks/months. Left untreated, progressive illness can result in death. Asymptomatic phases and relapses suggest that parasites can exist in the tissues for a long time before the clinical onset of the disease[10].
VL is also known as “kala-azar”, which by medical definition is a chronic and potentially fatal parasitic disease of the viscera (the internal organs, particularly the liver, spleen, bone marrow and lymph nodes) due to infection by the parasite, Leishmania donovani. Kala-azar can cause no or few symptoms but typically it is associated with fever, loss of appetite (anorexia), fatigue, enlargement of the liver, spleen and nodes and suppression of the bone marrow. The term "kala-azar" comes from India where it is the Hindi for black fever. The disease is also known as Indian leishmaniasis, visceral leishmaniasis, leishmania infection, dumdum fever, black sickness, and black fever [11].
Non–HIV related VL is progressively growing in non-tropical countries due not only to the rising number of patients with chronic diseases, but also to the rapid development of immune-modulating drugs for treating neoplastic diseases. Various cases of VL in patients with lymphoproliferative disorders have been documented since 1988 [9,12,13,14,15,16,17,18,19,20,21,22,23,24]. These reports describe VL occurring in several lymphoproliferative disorders, such as cHL, splenic marginal zone lymphoma (SMZL), follicular cell lymphoma, lymphoplasmacytic lymphoma, chronic lymphocytic leukemia, angioimmunoblastic T-cell lymphoma, and T-cell prolymphocytic leukemia. In the majority of them (at least 8 of 12), VL was misdiagnosed as a progression of the underlying hematological malignancy. Although it is difficult to prove this with certainty, VL arisen in non-endemic areas may represent the result of parasite reactivation during episodes of immune suppression, or the consequence of a recent acute infection after travel to endemic countries. In the paper by Galith et al for example, both patients had recently traveled to endemic areas from a non-endemic area (central France) [12]. Clinicians should thus be aware of the symptoms of VL and consider it in the differential diagnosis in patients with lymphomas. The clinical symptoms (splenomegaly, fever, weight loss) and laboratory abnormalities (pancytopenia) are often nonspecific.
Serological diagnosis of VL may be delayed or missed in patients treated with drugs that interfere with antibody production (i.e. rituximab or other monoclonal antibodies). Thus, blood or medullary polymerase chain reaction (PCR) may be the preferable tool in the diagnosis of VL in patients with lymphomas.
It has been suggested that leishmaniasis may be implicated into the pathogenesis of some patients who developed SMZL. Indeed, [19,24] some microbiological agents, either bacterial or viral (i.e. hepatitis C virus, helicobacter pylori) have been widely implicated into the pathogenesis of several lymphoid malignancies, including SMZL. There is of course a possible correlation between the two conditions because of chronic antigenic stimulation by microbial agents, but leishmaniasis is still far away to be implicated in such scenario. In fact, the parasites do enter the spleen and activate macrophages of the marginal zone, thus sustaining antigenic stimulation, and possibly triggering polyclonal B-cell proliferation.[24]. We believe that these important aspects should be investigated deeper in future research.
Finally, a case of VL in a 66-year-old female with a history of MALT lymphoma in the gastrointestinal tract has been reported [25]. The patient presented with major hemorrhage and perforation of the small intestine. Due to simultaneous unexplained decreasing platelet count, a bone marrow examination was performed to rule out lymphoma involvement, and, surprisingly, Leishmania donovani bodies were detected. Similarly, to our report, treatment was initiated with lipid formulation of amphotericin B, 4 mg/kg per day for 5 days. After suffering of acute renal dysfunction, which is a well-known complication associated with these antifungal agents, the patient recovered and could proceed to systemic chemotherapy with good response.
The association between cHL and VL appears to be a rare combination. To the best of our knowledge, only four cases (excluding our one) have described in the literature so far in non-HIV cHL. One additional case from an endemic area of Brazil was reported in lymphocyte predominant (LP-) HL (case number 5). In all cases the final outcome appeared favorable, provided that an accurate diagnosis was made as early as possible, in order to prevent severe complications related to the parasite proliferation into the host, favored by the immunosuppressed environment. The main characteristics of patients with cHL reported in the literature are listed in Table 1. Coexistence of lymphoma and leishmaniasis in the same node was described in two patients with HL [9,12], but the majority of diagnosis was performed on marrow smears or by serology. All patients had undergone cytotoxic chemotherapy, except for one patient. VL developed at the time of lymphoma diagnosis in three patients, prior to diagnosis in one, and during the course of the disease in the remaining cases (LP-HL). The latter study emphasizes the importance of considering VL as a potential masquerader and opportunistic infection in HL, even at lymphoma relapse, where symptoms and signs of VL can be easily confused to tumor recurrence.
Our case was apparently the first case of VL in a patient treated upfront with a combination therapy different from ABVD for cHL (A+AVD). The range of side effects mediated by cHL traditional chemotherapy (ABVD), as compared to novel immunotherapy, including the addition of the antibody–drug conjugate A, poses diagnostic and therapeutic challenges, mainly because several infections were not recorded during the studies that enabled these drugs approval. In our particular case, we used the combination A+AVD for the frontline treatment of our patient with advanced stage HL, since the combination was associated with a 23% reduction in the risk of progression compared to ABVD [5], and in a survival advantage in the randomized phase 3 study ECHELON-1. While A may induce an immune imbalance that can facilitate infections, and many opportunistic infections are increasingly described after this combination, no cases of leishmaniasis were included in the safety registry of the ECHELON-1 study. Nevertheless, the combination A+AVD was associated to increased risk of febrile neutropenia, and primary prophylaxis with G-CSF was mandatory along the study. Indeed, in the A+AVD arm, 55% of patients experienced an infectious event and nine died suggesting that this combination may be associated with higher risk of infection than ABVD alone [1].
Interestingly, a retrospective study evaluated the incidence and characteristics of infections in patients undergoing treatment with A (with or without chemotherapy) for cHL, non-Hodgkin T-cell lymphoma, and anaplastic large-cell lymphoma across 14 italian hematology centers [12]. The study included 191 patients treated with A. Overall, 12% of these patients experienced one or more infections during treatment, predominantly pulmonary infections (20%) and CMV/EBV reactivations (20%). The mortality rate was 0.5%. Bacterial infections accounted for 55% of cases, viral infections for 30%, and fungal infections for 15%. No parasites infection was documented during the 335 days covered by this study [12]. No case of VL was reported.
It is well known that VL may present with clinical and computed-tomography (CT) examination mimicking lymphoma. CT imaging may not always provide a straightforward differential diagnosis, due to the involvement of the lymphoid compartment in both diseases [30]. Since the diagnosis of VL is complicated and often unsuspected, limited knowledge exists regarding the efficacy of nuclear imaging techniques. An important study has been conducted in the attempt to discern the utility of fluorodeoxyglucose positron emission tomography/computed tomography (FDG-PET/CT) in cases of VL [31]. Authors retrospectively reviewed VL cases diagnosed at Vall d’Hebron University Hospital between 2012 and 2018 and selected those that had an FDG-PET/CT performed. Four of 43 patients diagnosed with VL had an FDG-PET/CT performed. All four patients presented diffuse splenic uptake of FDG-PET/CT (as was the case of the patient described in the present report, see Figure 1). Adenopathy was not always present, and bone marrow uptake was found in two patients. A posttreatment FDG-PET/CT in one patient revealed normalization of initial findings (again as for our patient). The pattern of spleen involvement described in the literature demonstrated disparate presentations, ranging from “diffuse increased metabolism,” to “nodular pattern “or “patchy and granular”. Other frequent findings were bone marrow uptake and adenopathies. Authors concluded that FDG-PET/CT could become a useful tool for the diagnosis and follow-up of VL and that VL should be taken into account in patients with fever of unknown origin with enhanced splenic uptake in FDG-PET/CT. Of course, differential diagnosis is always mandatory due to the low specificity of this diagnostic technique.
5. Conclusions
To the best of our knowledge, we describe the first case of VL in a patient treated upfront with A+AVD for cHL. Infectious complications when treating these patients should be always considered, though they can resemble tumor progression or resistance, as in our case. A multidisciplinary approach is warranted in such cases. We believe that VL should be ruled out when persistent fever, unexpected cytopenias, or splenomegaly occur in lymphoma patients, regardless of geographical area or epidemiological risk factors. A blood Leishmania PCR should be systematically performed in suspicious cases.
Author Contributions
Conceptualization, D.B., A.B., F.M.Q and C.V.; methodology, A.B.; F.M.Q. and C.V.; investigation, all author D.B., A.M., E.S., E.T., A.B., F.M.Q. and C.V., writing—original draft preparation, D.B., A.B., F.M.Q. and C.V.; writing—review and editing, C.V., A.B. and D.B.; visualization, D.B. and A.B.; supervision, C.V. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Informed consent was obtained from our patient.
Data Availability Statement
Not applicable.
Conflicts of Interest
All the authors declare no conflict of interest.
References
- Ansell, S.M.; Radford, J.; Connors, J.M.; Długosz-Danecka, M.; Kim, W.-S.; Gallamini, A.; Ramchandren, R.; Friedberg, J.W.; Advani, R.; Hutchings, M.; et al. Overall Survival with Brentuximab Vedotin in Stage III or IV Hodgkin’s Lymphoma. New Engl. J. Med. 2022, 387, 310–320. [CrossRef]
- Eichenauer, D.A.; Aleman, B.M.P.; André, M.; Federico, M.; Hutchings, M.; Illidge, T.; Engert, A.; Ladetto, M.; ESMO Guidelines Committee. Hodgkin lymphoma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2018, 29, iv19–iv29. [CrossRef]
- Kennedy-Nasser, A.A.; Hanley, P.; Bollard, C.M. Hodgkin Disease and the Role of the Immune System. Pediatr. Hematol. Oncol. 2011, 28, 176–186. [CrossRef]
- Che, Y.; Ding, X.; Xu, L.; Zhao, J.; Zhang, X.; Li, N.; Sun, X. Advances in the treatment of Hodgkin’s lymphoma (Review). Int. J. Oncol. 2023, 62, 1–14. [CrossRef]
- Connors, J.M.; Jurczak, W.; Straus, D.J.; Ansell, S.M.; Kim, W.S.; Gallamini, A.; Younes, A.; Alekseev, S.; Illés, Á.; Picardi, M.; et al. Brentuximab Vedotin with Chemotherapy for Stage III or IV Hodgkin’s Lymphoma. N. Engl. J. Med. 2018, 378, 331–344. [CrossRef]
- Maia, C. Sand fly-borne diseases in Europe: epidemiological overview and potential triggers for their emergence and re-emergence. J. Comp. Pathol. 2024, 209, 6–12. [CrossRef]
- Telleria EL, Martins-da-Silva A, Tempone AJ, Traub-Csekö YM. Leishmania , microbiota and sand fly immunity. Parasitology [Internet]. 2018 Sep [cited 2024 Apr 21];145(10):1336–53. Available from: https://www.cambridge.org/core/product/identifier/S0031182018001014/type/journal_article.
- Moirano, G.; Ellena, M.; Mercogliano, P.; Richiardi, L.; Maule, M. Spatio-Temporal Pattern and Meteo-Climatic Determinants of Visceral Leishmaniasis in Italy. Trop. Med. Infect. Dis. 2022, 7, 337. [CrossRef]
- Domingues, M.; Menezes, Y.; Ostronoff, F.; Calixto, R.; Florencio, R.; Sucupira, A.; Souto-Maior, A.P.; Ostronoff, M. Coexistence of Leishmaniasis and Hodgkin’s Lymphoma in a Lymph Node. J. Clin. Oncol. 2009, 27, e184–e185. [CrossRef]
- Wilson, M.E.; Jeronimo, S.M.; Pearson, R.D. Immunopathogenesis of infection with the visceralizing Leishmania species. Microb. Pathog. 2005, 38, 147–160. [CrossRef]
- World Health Organization. Leishmaniasis. 2023 Jan 12; Available from: https://www.who.int/news-room/fact-sheets/detail/leishmaniasis.
- Kalmi, G.; Vignon-Pennamen, M.-D.; Ram-Wolff, C.; Battistella, M.; Lafaurie, M.; Bouaziz, J.-D.; Hamane, S.; Bernard, S.; Bretagne, S.; Thieblemont, C.; et al. Visceral leishmaniasis in patients with lymphoma Case reports and review of the literature. Medicine 2020, 99, e22787. [CrossRef]
- Osakwe, N.M.; Paulus, A.; Haggerty, P.F.; Wood, R.A.; Becker, S.J.; Weina, P.J.; Dolan, M.J.; Prakash, V. Visceral Leishmaniasis With Associated Immune Dysregulation Leading to Lymphoma. 2013, 178, e386–e389. [CrossRef]
- van Griensven, J.; Carrillo, E.; López-Vélez, R.; Lynen, L.; Moreno, J. Leishmaniasis in immunosuppressed individuals. Clin. Microbiol. Infect. 2014, 20, 286–299. [CrossRef]
- Chair, Regional Leishmaniasis Control Center (RLCC), Yemen, M Ahmed AK. Cutaneous T Cell Lymphoma (CTCL) Superimposed on Disseminated Cutaneous Leishmaniasis (DCL) in an Immunocompromised Female from Yemen. Int J Clin Dermatol Res [Internet]. 2017 Jul 18 [cited 2024 Aug 9];4–8. Available from: https://scidoc.org/specialissues/IJCDR/S3/IJCDR-2332-2977-S3-002.pdf.
- Nicodemo, A.C.; Duailibi, D.F.; Feriani, D.; Duarte, M.I.S.; Amato, V.S. Mucosal leishmaniasis mimicking T-cell lymphoma in a patient receiving monoclonal antibody against TNFα. PLOS Neglected Trop. Dis. 2017, 11, e0005807. [CrossRef]
- Foulet, F.; Botterel, F.; Buffet, P.; Morizot, G.; Rivollet, D.; Deniau, M.; Pratlong, F.; Costa, J.-M.; Bretagne, S. Detection and Identification ofLeishmaniaSpecies from Clinical Specimens by Using a Real-Time PCR Assay and Sequencing of the CytochromebGene. J. Clin. Microbiol. 2007, 45, 2110–2115. [CrossRef]
- Kawakami, A.; Fukunaga, T.; Usui, M.; Asaoka, H.; Noda, M.; Nakajima, T.; Hashimoto, Y.; Tanaka, A.; Kishi, Y.; Numano, F. Visceral Leishmaniasis Misdiagnosed as Malignant Lymphoma.. Intern. Med. 1996, 35, 502–506. [CrossRef]
- Vase, M..; Hellberg, Y.K.; Larsen, C.S.; Petersen, J.E.; Schaumburg, H.; Bendix, K.; Ravel, C.; Bastien, P.; Christensen, M.; Nyvold, C.G.; et al. Development of a Splenic Marginal Zone Lymphoma Associated with Active Chronic Visceral Leishmaniasis. Blood 2011, 118, 5202–5202. [CrossRef]
- Evers, G.; Pohlen, M.; Berdel, W.E.; Thoennissen, N.H.; Titze, U.; Köhler, G.; Weckesser, M.; Anthoni, C.; Mesters, R.M. Visceral leishmaniasis clinically mimicking lymphoma. Ann. Hematol. 2013, 93, 885–887. [CrossRef]
- Casabianca, A.; Marchetti, M.; Zallio, F.; Feyles, E.; Concialdi, E.; Ferroglio, E.; Biglino, A. Seronegative visceral leishmaniasis with relapsing and fatal course following rituximab treatment. Infection 2011, 39, 375–378. [CrossRef]
- Cencini, E.; Lazzi, S.; Fabbri, A. Atypical clinical presentation of visceral leishmaniasis in a patient with non-Hodgkin lymphoma. Eur. J. Haematol. 2014, 94, 186–186. [CrossRef]
- Liao, H.; Jin, Y.; Yu, J.; Jiang, N. Concomitant T-cell prolymphocytic leukemia and visceral leishmaniasis A case report. Medicine 2018, 97, e12410. [CrossRef]
- Kopterides, P.; Mourtzoukou, E.G.; Skopelitis, E.; Tsavaris, N.; Falagas, M.E. Aspects of the association between leishmaniasis and malignant disorders. Trans. R. Soc. Trop. Med. Hyg. 2007, 101, 1181–1189. [CrossRef]
- Kaae, J.; Nørgaard, P.; Himmelstrup, B. Visceral leishmaniasis diagnosed in a patient with MALT lymphoma. Eur. J. Intern. Med. 2007, 18, 235–237. [CrossRef]
- Kumar, R.; Daga, M.K.; Kamble, N.L.; Sothwal, A.; Singh, T.; Nayak, H.K.; Raizada, N. Rare association of Visceral leishmaniasis with Hodgkin’s disease: A case report. Infect. Agents Cancer 2011, 6, 17–17. [CrossRef]
- Porto, V.B.G.; Carvalho, L.B.; Buzo, B.F.; Litvoc, M.N.; Santos, A.C.S.; Rocci, R.A.; Soares, S.R.C.; Zampieri, R.A.; Duarte, M.I.S.; Lindoso, J.A.L. Visceral leishmaniasis caused by Leishmania (Leishmania) amazonensis associated with Hodgkin’s lymphoma. Rev. do Inst. de Med. Trop. de Sao Paulo 2022, 64, e51. [CrossRef]
- Dereure J. Visceral leishmaniasis. Persistence of parasites in lymph nodes after clinical cure. J Infect [Internet]. 2003 Jul [cited 2024 Aug 9];47(1):77–81. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0163445303000021.
- Marchesini, G.; Nadali, G.; Facchinelli, D.; Candoni, A.; Cattaneo, C.; Cuccaro, A.; Fanci, R.; Farina, F.; Lessi, F.; Visentin, A.; et al. Infections in patients with lymphoproliferative diseases treated with brentuximab vedotin: SEIFEM multicentric retrospective study. Leuk. Lymphoma 2020, 61, 3002–3005. [CrossRef]
- Cohen, D.; Fields, S. CT findings in visceral leishmaniasis mimicking lymphoma. Comput. Med Imaging Graph. 1988, 12, 325–327. [CrossRef]
- Pinnegar, H.P.; Sánchez-Montalvá, A.; Profitos, M.B.; Bosch-Nicolau, P.; Salvador, F.; Molina, I. Utility of Fluorine-18 Fluorodeoxyglucose Positron Emission Tomography/Computed Tomography in Patients with Visceral Leishmaniasis: Case Report and Literature Review. Am. J. Trop. Med. Hyg. 2021, 104, 934–944. [CrossRef]
Figure 1.
PET-TC at cHL diagnosis (A): Increased glycolytic metabolism observed in multiple lymph nodes, specifically above the diaphragm, at the scapular level and a notable lesion in the spleen without splenomegaly. After completing two cycles of immuno-chemotherapy (B), a complete response was achieved, and no evident lesions were observed at the spleen level. PET-CT performed one-month post-treatment (C) revealed homogeneous splenomegaly of 18 cm with no focal lesions (an abdominal ultrasound confirmed the absence of spleen lesions). Two months later, imaging confirmed an increase in splenomegaly measuring 20 cm, without any other findings (D), when the diagnosis of VL was performed.
Figure 1.
PET-TC at cHL diagnosis (A): Increased glycolytic metabolism observed in multiple lymph nodes, specifically above the diaphragm, at the scapular level and a notable lesion in the spleen without splenomegaly. After completing two cycles of immuno-chemotherapy (B), a complete response was achieved, and no evident lesions were observed at the spleen level. PET-CT performed one-month post-treatment (C) revealed homogeneous splenomegaly of 18 cm with no focal lesions (an abdominal ultrasound confirmed the absence of spleen lesions). Two months later, imaging confirmed an increase in splenomegaly measuring 20 cm, without any other findings (D), when the diagnosis of VL was performed.
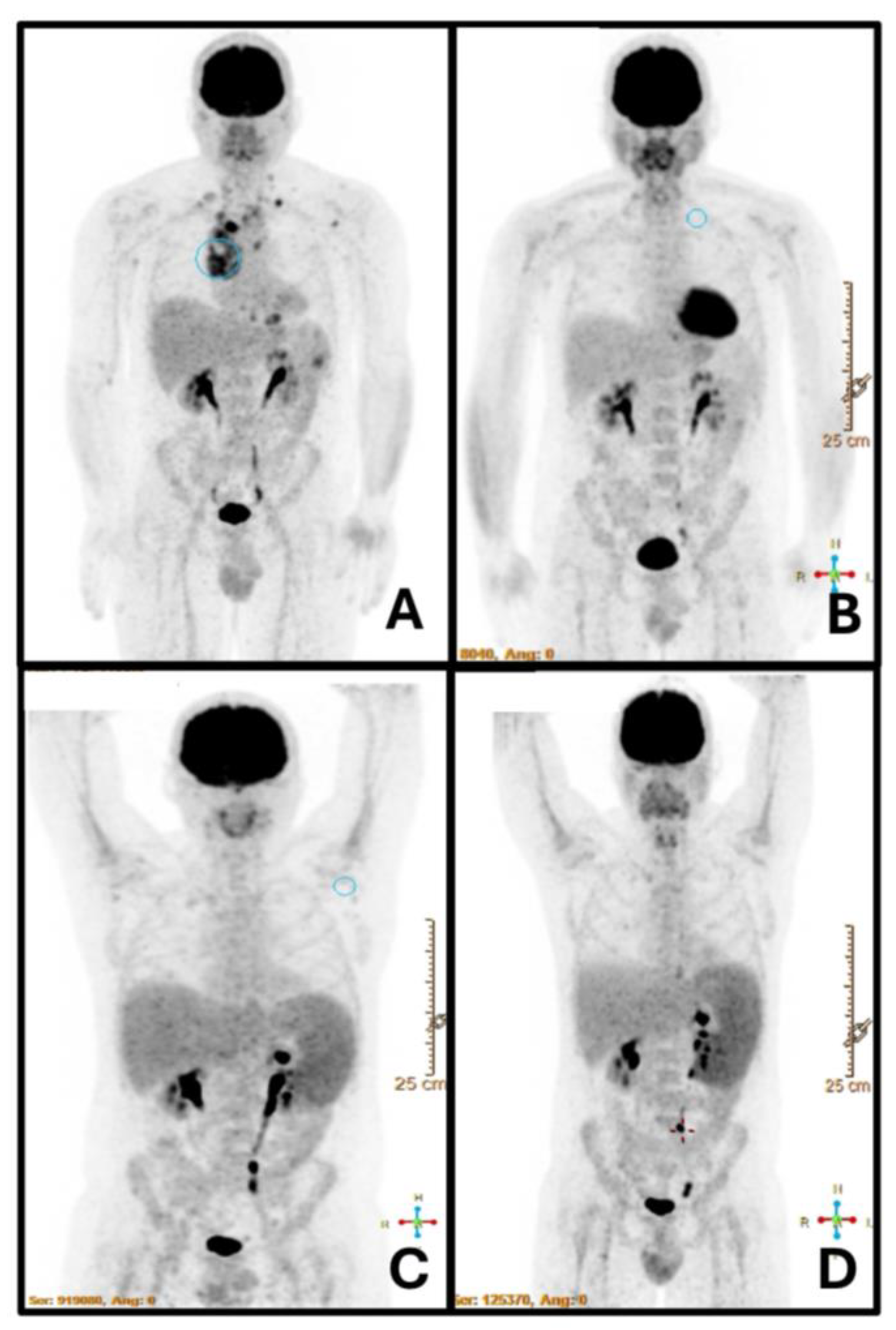

Table 1.
Characteristics and evolution of patients with cHL and VL from the literature.
| Studies Related, Year and Country |
Time Point of VL Diagnosis |
Samples for the Diagnosis of Leishmaniasis | Outcome | cHL First Line Treatment |
|---|---|---|---|---|
| Kumar R, 2011. India [26] | At diagnosis of cHL, pre-chemotherapy | Bone marrow aspirate and serology test | Alive | Chemotherapy not described |
| Gomes Porto VB, 2022. Brazil [27] | At diagnosis of cHL, pre-chemotherapy | Bone marrow aspirate and serology test | Alive | ABVD |
| Magnan A, 1991. France [12] | At diagnosis of cHL, prechemotherapy |
Bone marrow aspirate | Unknown | Not reported |
| Dereure J, 2003. France [28] |
In a patient with history of cHL | Bone marrow aspirate; immunofluorescence antibody test | Unknown | Splenectomy and radiotherapy |
| Domingues M, 2009. Brazil [9] |
During treatment for lymphocyte-predominant HL | Lymphnode | Alive | ABVD |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



