
Submitted:
13 August 2024
Posted:
14 August 2024
You are already at the latest version
Abstract
The abstract should be a total of about 250 words and structured to contain the following headings: Background/Objectives, Methods, Results, Conclusions. Background/Objectives: Place the question addressed in a broad context and highlight the purpose of the study; Methods: Describe briefly the main methods or treatments applied. Include any relevant preregistration numbers, and species and strains of any animals used; Results: Summarize the article’s main findings; Conclusions: Indicate the main conclusions or interpretations. The abstract should be an objective representation of the article: it must not contain results which are not presented and substantiated in the main text and should not exaggerate the main conclusions. Clinical trial abstracts should include items that the CONSORT group has identified as essential.
Keywords:
tumor heterogeneity
; transcriptome profile
; cancer classification
; multiomics
; cancer imaging
1. Introduction
Colorectal cancer (CRC) and gastric cancer (STAD) are the foremost gastrointestinal malignancies, and gastrointestinal cancer has the highest incidence rate among all cancers[1]. Gastrointestinal cancers, including esophageal, stomach, colonic, and rectal malignancies, account for over a million deaths annually[2,3]. In China, CRC and STAD exhibit heightened incidence and mortality rates[4]. There is a pronounced reciprocal relationship has developed between CRC and STAD, by CRC frequently emerges as a subsequent primary malignancy in STAD patients, and STAD is the most common initial cancer in CRC patients[5,6,7]. Moreover, CRC and STAD share numerous similarities including pathogenesis, pathological features, treatment approaches, and cellular profiles[8,9]. Hence, a comprehensive analysis of samples from CRC and STAD patients would not only facilitates the discovery of common features, but would also provide a basis for improving the relevance and efficacy of therapeutic strategies to treat cancer.
In 2013, Singaporean researchers were the first to classify gastric cancer based on genomic expression, identifying three primary molecular subtypes: proliferative, metabolic, and mesenchymal[10]. In the following year, The Cancer Genome Atlas (TCGA) research consortium expanded this classification by employing six distinct molecular biology techniques to categorize gastric cancer into four additional molecular subtypes: chromosomal instability (CIN), microsatellite instability (MSI), genomic stability (GS), and Epstein-Barr Virus positive (EBV+)[11]. Budinska et al.[12]reported five distinct subtypes of CRC by analyzing the expression profiles of 1113 colorectal cancer (CRC)-related genes. This expanded classification system presented significant variations in biological traits, clinical outcomes, pathological features, and survival data. CRC was further stratified into four Consensus Molecular Subtypes (CMS) at the molecular level, each distinguished by its oncogenic and oncostatic pathways, mutation profiles, microsatellite instability status, and clinical outcome expression patterns[13,14]. Utilizing miRNA data from colorectal cancer, Paz-Cabezas et al.[15] identified three distinct miRNA-driven tumor subtypes via hierarchical cluster analysis, which showed a strong correlation with mRNA-based tumor classifications (p < 0.001). These findings underscore the pivotal role of transcriptomic data in the identification of tumor subtype biomarkers.
Tumor imaging data are instrumental in revealing the spatial architecture, tissue composition, morphology, and internal organization of tumors, offering vital insights for cancer diagnosis. In the analysis of medical image, edge feature extraction stands out as an essential technique that aids in the identification and characterization of diseases. Several edge detection algorithms are extensively utilized, such as the Sobel[16], Roberts[17], Prewitt[18], and Canny[19] operators. Histopathological images are pivotal in cancer classification and subtyping, enabling a more nuanced understanding of cancer heterogeneity[20]. For instance, N. K. Pratiwi and colleagues utilized the Canny operator to extract edge features from colon cancer images and subsequently applied these features to a classification study of colon cancer[21], thereby validating the efficacy of edge feature extraction in cancer diagnostics.
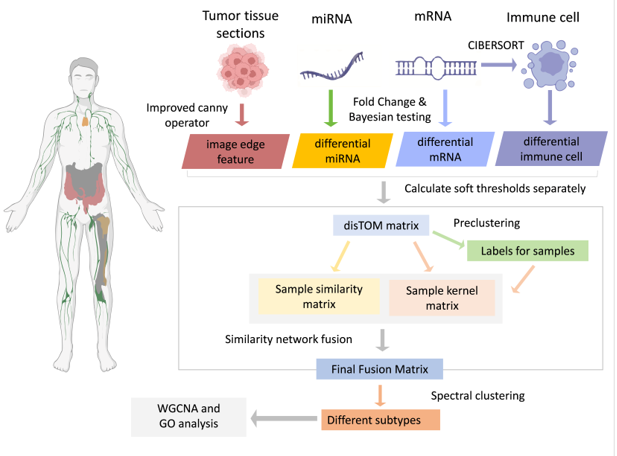
Multimodal data fusion effectively complements and integrates insights from different fields, including pathology, clinical radiology, genetics, and molecular biology[22], resulting in a more advanced and comprehensive analysis of heterogeneity in gastrointestinal cancers. However, integration of multi-omics datasets is challenging. In the present work, we addressed this complexity with an optimized Canny operator to increase the accuracy of extracting image information from tumor tissue sections. Subsequently, we integrated the extracted differential transcriptomes associated with gastrointestinal cancers, immune cell data, and corresponding tumor tissue images. This de novo integrated dataset significantly advanced our cancer classification efforts, delineating three distinct subtypes, each with a distinct set of gene modules. This integrative approach advances our understanding of the intrinsic heterogeneity within gastrointestinal (GI) tumors.
2. Materials and Methods
2.1. Datasets
In the present study, we used The Cancer Genome Atlas (TCGA) [23] to amass mRNA and miRNA expression profiling data taken from primary tumor tissues of gastric and colorectal cancer patients. The mRNA dataset encompassed 937 samples, comprising 74 samples serving as normal controls, whereas the miRNA dataset comprised 964 samples with 53 normal controls. These samples corresponded to 58,387 mRNA transcripts and 2,652 mature miRNAs. Furthermore, leveraging the LM22 gene set from the CIBERSORT algorithm, we estimated the relative abundance of 22 distinct immune cell types within the samples based on their mRNA expression profiles.
Imaging data were sourced from the research conducted by Kather et al.[24] and were retrieved from the publicly accessible Zenodo database at https://zenodo.org/records/2530789.
In further studies, our analysis of differentially expressed mRNAs, miRNAs, and immune cells used data from all samples. We specifically focused on gastrointestinal cancer patients, and we retained the sample at the intersection of the four data types for a total of 515 data samples for in-depth analysis.
2.2. Data Feature Extraction
2.2.1. Edge Feature Extraction of Tumor Images
- Smooth images using bilateral filters
In 1986, John F. Canny introduced an algorithm, which is known as the Canny edge detection operator[25], for image edge detection. In the present work, we optimized this operator by replacing the conventional Gaussian filter, which was originally employed for preliminary filtering, with a more refined bilateral filter.
Bilateral filtering incorporates spatial information about pixel distribution, thus extending beyond the capability of the original Gaussian filter. This sophisticated method optimally refines edge and grayscale details, effectively reduces texture noise, and maintains crucial representative information, thereby efficiently suppressing noise[26]. The formulation of bilateral filtering is as follows:
In Equation (1), represents the output point, where denotes the neighborhood range of size centered at point , and corresponds to the grayscale value of the pixel located at the matrix coordinates.
Utilizing the ‘opencv’ library, we loaded each sample image into a 224x224 pixel matrix, configuring the parameters to , and . The origin of the pixel coordinate system was set at the lower-left corner and denoted as (0,0). Following this, we applied weighted averaging to process the pixel matrices of the images.
- Calculation of gradient change and direction of grayscale values
We applied the Sobel operator to compute the variations and orientations of gray-level values. The operator was utilized to determine the gradients across both positive and negative vertical axes on the horizontal plane. This process enabled us to ascertain the direction angle for each pixel, as detailed in Equation (5):
We defined and , where denotes a 3x3 matrix of gray values centered on the pixel of interest. The Sobel kernels for capturing horizontal and vertical features are constructed as follows:
Upon calculating the image gradients, is is possible that multiple directions can satisfy the threshold conditions. However, we selectively retain the gradient direction with the highest magnitude, while suppressing the others. The magnitude of the gradient at the current pixel is compared with the magnitudes in the four principal directions: 0 (vertical), (one of the diagonals), (horizontal), and (the other diagonal). This comparison is based on the common edge orientations found in images. A pixel is deemed significant and retained if its gradient intensity is greater than the gradient intensities of the adjacent pixels in all four cardinal directions; if not, the pixel is discarded.
- Setting dual thresholds for edge detection
To refine the noise reduction process, we introduce dual thresholds: a high threshold (TH) and a low threshold (TL). Edge pixels with gradient magnitudes exceeding TH are designated as strong edges, while those with magnitudes between TH and TL are categorized as weak edges. Pixels not meeting these criteria are effectively suppressed. The determination of TH and TL is outlined below, where denotes the pixel matrix post nonmaximum suppression:
Differentiating weak edge pixels that belong to true edges from those caused by noise is essential for accurate edge detection. To this end, we set a criterion specifying that a weak edge pixel should be retained and considered part of the image’s edge structure, but only if it is connected to at least one pixel previously identified as a strong edge pixel.
Upon completing the aforementioned image processing steps, we derived the final matrix representing the edge features of the image. We then computed the average value across each column of this matrix to achieve dimensionality reduction for the data corresponding to each sample. This process led to a dimensionally reduced size of 1×224 for the data of each sample.
2.2.2. Transcriptome Feature Extraction
Given the multitude and complexity of mRNA and miRNA data, the presence of redundant or irrelevant features can potentially distort analytic outcomes. To address this, we opted for a feature dimensionality reduction strategy aimed at boosting the precision and efficiency of our research designed to explore the heterogeneity of GI cancer. Furthermore, we prioritized features that are significantly differentially expressed between cancer patients and healthy individuals since we hypothesized that these features might play a crucial role in disease diagnosis and the development of treatment strategies.
To discern variations in gene expression between cancerous and normal samples, we first conducted a differential analysis for mRNAs and miRNAs. This analysis was performed utilizing the “limma” software package within the R programming environment, employing both fold change (FC) and Bayesian statistical testing approaches[27,28]. More specifically, a gene was flagged as differentially expressed if it exhibited a foldchange greater than 1 and yielded a p-value below 0.01 from the Bayesian test.
Through our analysis, we have identified 3,360 down-regulated and 2,484 up-regulated genes, totalling 5,844 differentially expressed mRNAs. Additionally, we found 91 down-regulated and 72 up-regulated miRNAs, totalling 163 differentially expressed miRNAs. As presented in Figure S1, the heat maps showcase the differential expression profiles based on samples from cancer patients and normal subjects, and, as such, validate the reliability of our differential characterization approach.
2.3. Multimodal Data Clustering
2.3.1. Soft Threshold Distance Calculation
Soft threshold distance calculations were conducted autonomously for mRNA, miRNA, immune cell, and image datasets. Let us denote n samples and m features, such as mRNA gene expressions, by matrix , which serves as the sample-feature matrix. We then compute the Pearson correlation coefficient matrices for each of the omics datasets independently.
Within the matrix, elements undergo a nonlinear mapping process as defined by Equation 8, which facilitates the incorporation of a soft threshold.
To ascertain the soft threshold, we utilized an approach akin to a grid search, delineating a spectrum of values ranging from 2 to 20. The criteria for selecting the soft threshold were based on the condition that the coefficient of determination () for the linear regression model should exceed 0.8. In the absence of a satisfactory soft threshold, one was selected that corresponded to an average connectivity of fewer than 100 samples.
Soft thresholding was applied across four distinct data categories, yielding the following outcomes: a threshold of 8 for the mRNA data correlation coefficient matrix, 14 for the miRNA data correlation coefficient matrix, 3 for the image data similarity matrix, and 9 for the immune cell data.
2.3.2. Calculation of DissTOM Distance for the Soft Threshold Matrix
We employed the Topological Overlap Matrix (TOM)[29,30,31] to delineate correlations among samples within the sample network. Thereafter, in the relational equation, we converted the adjacency matrix into the TOM to more precisely capture the complex intersample relationships (Equation 9).
In this context, the association between samples i and j is represented by , and the association of sample i with the remaining samples is given by , which is a direct relationship indicative of exclusive connectivity or related pathways that exist between samples i and j when . Conversely, signifies the nonexistence of a relationship. Subsequently, we defined the dissimilarity measure to construct the dissimilarity matrix, dissTOM.
We employed the k-medoids algorithm to cluster disTOM and utilized the elbow method to identify the optimal number of clusters for each class. To prevent an excessively high number of classes, we limited the range of k to between 2 and 10. The chosen value of k represents the point of maximum deviation in the sum of squared errors (SSE) for each data type. The clustering analysis concluded with k set to 5 for mRNA data, 4 for miRNA data, 4 for image data, and 3 for immune cell data.
2.3.3. Construction of Similarity and Kernel Matrices
Using the dissTOM, we performed data transformation following the procedure specified in Equations (10) and (11):
The value represents the mean distance of sample i from all other samples, excluding itself. Incorporating this step was influenced by the approach pioneered by Bo Wang et al.[32] in 2014, who utilized the Similarity Network Fusion (SNF) model. Distances between samples were transformed using a scaled exponential similarity kernel function, an adaptation of the Gaussian kernel. This transformation adeptly remaps the data onto a specific distribution within a smooth convex space, resulting in a more concentrated depiction of the information[33].
A similarity matrix among samples was established, as detailed in equation (12):
Utilizing the preclustered data, we formulated the kernel matrix as presented in Equation (13):
where denotes the cluster assignment of sample i within the preclustered data categories, such as mRNA.
We iteratively refined for each data modality using Equation (14) and quantified the iterative changes in the data in terms of Frobenius norms, as described by Equation (15):
The final consistent similarity matrix was derived by computing the mean value where the summation is over the four data modalities.
3. Results
3.1. Identification of the Three Subtypes Based on Sample Omics Data
The violin plots depicting the top ten most significantly differentially expressed mRNAs are presented in Figure 1(A). It is of particular interest that elevated expression levels of CLDN3 in gastric cancer influence tumor cell permeability, facilitating their traversal across the basement membrane and extracellular matrix, thus potentially contributing to oncogenesis[34,35]. CDH3, a gene predominantly overexpressed in gastric cancer, is associated with cancer invasion and metastasis. The protein it encodes facilitates the proliferation and mobility of cancer cells[36].
Figure 1.
Significantly different characteristics between cancer and normal samples: (A) top ten most significant mRNAs; (B) most significant differential miRNAs; (C) differential boxplots of 22 immune cells.
Figure 1.
Significantly different characteristics between cancer and normal samples: (A) top ten most significant mRNAs; (B) most significant differential miRNAs; (C) differential boxplots of 22 immune cells.
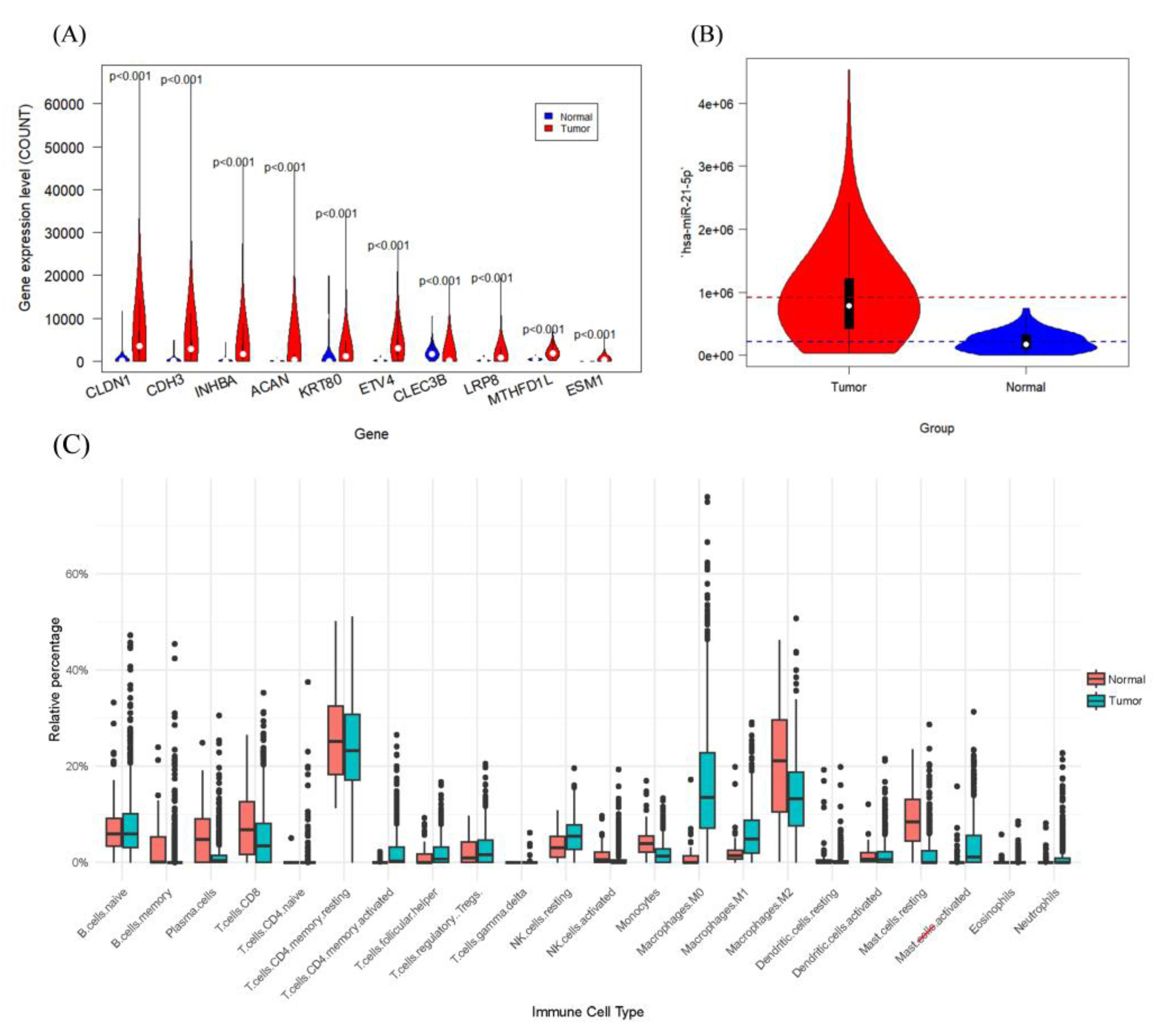
Figure 1(B) illustrates the general up-regulation of hsa-miR-21-5p, a microRNA (miRNA) that expressed in a variety of cancers, especially in colorectal cancer, in which hsa-miR-21-5p contributes to tumor development and progression of tumors through the modulation of multiple biological processes, such as apoptosis and inflammatory responses[37].
Box plots of immune cell percentages for the two samples showed considerable variation in the proportion of certain immune cells, leading to the selection of data from 12 specific immune cells for further analysis.
Following four fusion iterations of the four data models, convergence was successfully attained. The iterative process, as depicted in Figure S2, illustrates the progressive convergence of the four similarity matrices across iterations. Subsequently, spectral clustering was employed to confine the class count (k) within the range of [2,5], utilizing the elbow method to ascertain optimal k. In total, the study classified 515 samples into three subtypes, comprising 212, 132, and 171 samples, respectively. To more vividly exhibit the characteristic differences among the subtypes, the top twenty differential features of mRNA and RNA, along with the comprehensive data of immune cells, were filtered using the Kruskal-Wallis test.
Figures 2(A) and (C) illustrate the expression profiles of samples across different subtypes under the top twenty DEGS, with (C) presenting the mean expression values for samples within each subtype. In these mRNA expressions, samples from subtype I exhibit relatively high levels, while samples from subtype II show comparatively lower levels, and samples from subtype III have the lowest expression. Figures 2(B) and (D) depict the expression of the first twenty differential miRNAs among samples of various subtypes, with (D) also presenting the average expression values for samples of each subtype. For the initial nine miRNA features, the expression levels across the three subtypes are presented in descending order, in alignment with the mRNA results. However, for the last eleven miRNA features, the expression outcomes are nearly inversely related. Corresponding box plots for the first six features are provided in Figure 3, revealing significant differences among subtypes, thereby preliminarily confirming the validity of the cancer typing methodology followed in this study.
Figure 2.
Heatmaps of the top 20 significantly differentially expressed genes(DEGs): (A) mRNA expression heatmap; (B) miRNA expression heatmap; (C) mRNA expression heatmap after taking the mean for the corresponding feature of the same subtype sample; (D) miRNA expression heatmap after taking the mean for the corresponding feature of the same subtype sample.
Figure 2.
Heatmaps of the top 20 significantly differentially expressed genes(DEGs): (A) mRNA expression heatmap; (B) miRNA expression heatmap; (C) mRNA expression heatmap after taking the mean for the corresponding feature of the same subtype sample; (D) miRNA expression heatmap after taking the mean for the corresponding feature of the same subtype sample.
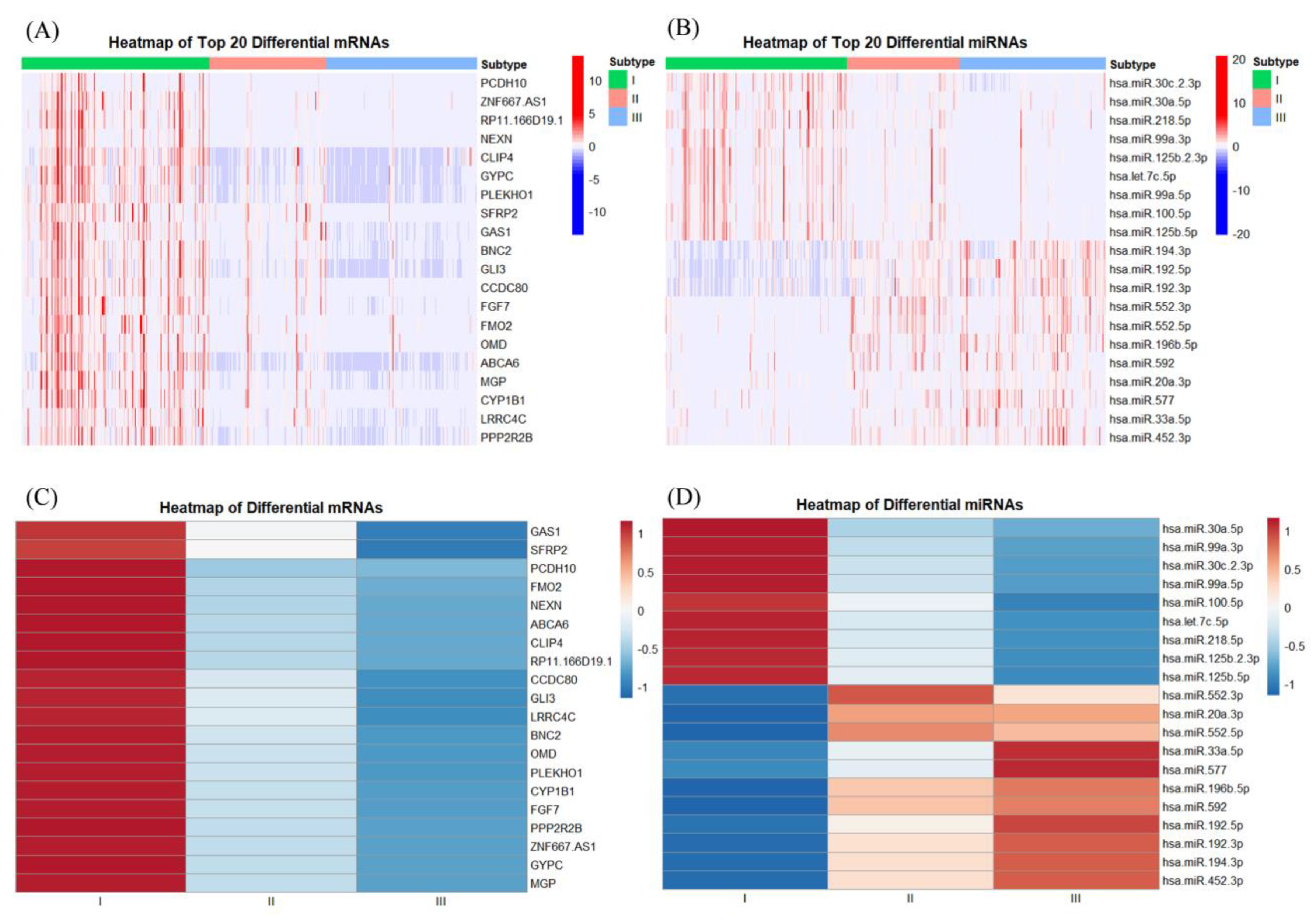
Figure 3.
Six features with the most significant differences among the three subtypes: (A) top six most significantly differentially expressed mRNAs; (B) top six most significantly differentially expressed miRNAs.
Figure 3.
Six features with the most significant differences among the three subtypes: (A) top six most significantly differentially expressed mRNAs; (B) top six most significantly differentially expressed miRNAs.
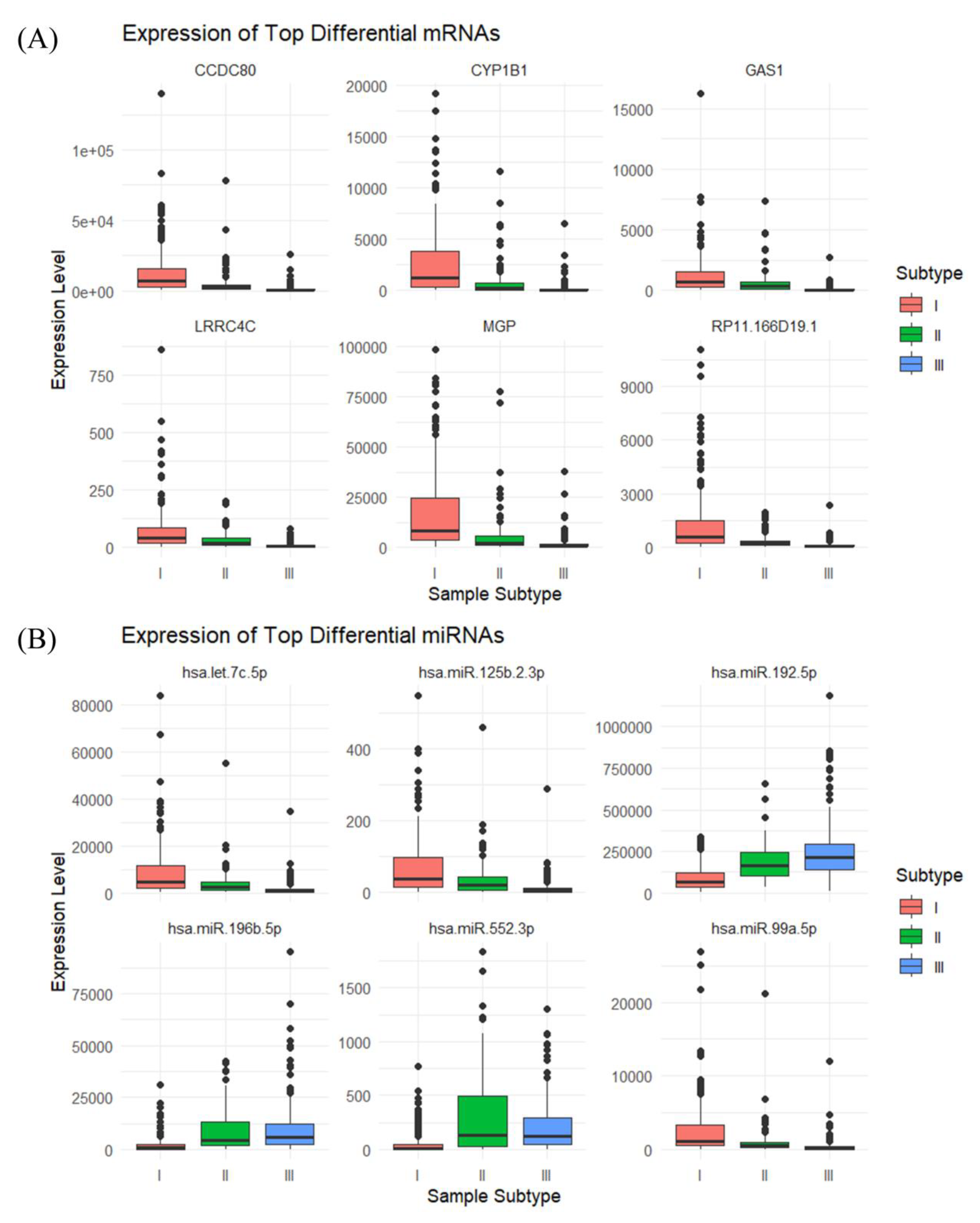
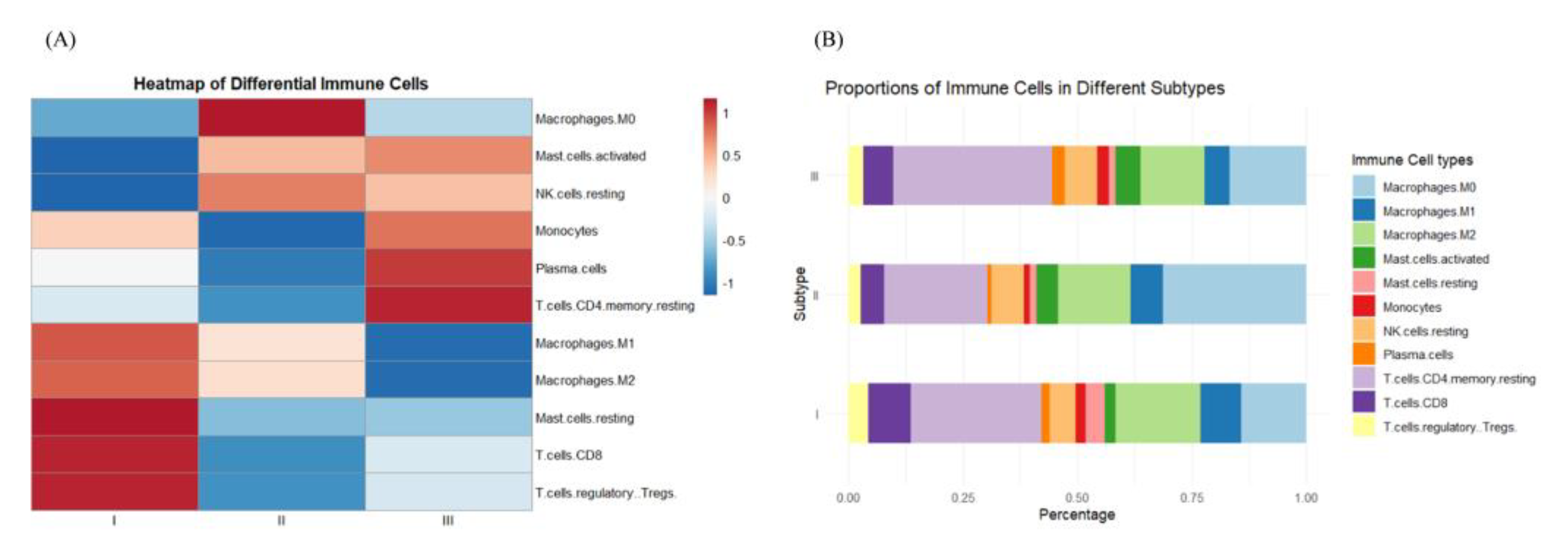
Figure 4 illustrates the expression patterns of various subtypes of immune cells, focusing on the first 11 classes identified through p-value testing. In subtype I, specific immune cells such as resting NK cells and plasma cells exhibit reduced activity (blue), whereas M2 macrophages and regulatory T cells (Tregs) display heightened activity (red). Subtype II presents a pattern distinct from that of subtype I, and subtype III diverges from subtype I in the activity of most cell types, with M1 and M2 macrophages demonstrating moderate to high activity, and resting NK cells and Tregs showing lower activity. As depicted in Figure 4(B), subtype I is characterized by a higher proportion of M2 macrophages and Tregs, which may indicate a more potent anti-inflammatory or immunomodulatory role. Subtype II is marked by an increased presence of CD8 T cells and plasma cells, potentially linked to a more robust immune response or antibody production. In contrast, subtype III exhibits a higher proportion of M1 and M2 macrophages, which could be associated with tissue repair and modulation of the tumor microenvironment.
Figure 4.
Immune cell characteristics of different subtypes: (A) Heat map of immune cell characteristics of samples of the same subtype after taking the mean value; (B) Difference in the percentage of immune cells in samples of different subtypes.
Figure 4.
Immune cell characteristics of different subtypes: (A) Heat map of immune cell characteristics of samples of the same subtype after taking the mean value; (B) Difference in the percentage of immune cells in samples of different subtypes.
3.2. Identification of Hub Genes of Different Subtypes by WGCNA
Weighted Gene Co-expression Network Analysis (WGCNA) was conducted on mRNA data based on the samples from three subtypes, encompassing a total of 515 samples and 5844 genes. Initially, during the analysis, 5000 genes with the highest variability were selected. Using the histogram algorithm, the soft-thresholding power β=3 was identified, thereby achieving an R² value of 0.88 and an average connectivity below 100 to meet our criteria (Figure S3). It can be observed in Figure 5(A) that 1) most mRNAs exhibit low connectivity, 2) only a minority demonstrates high connectivity, and 3) the constructed network exhibits scale-free properties. The dissTOM matrix was constructed by leveraging the topological overlap matrix (TOM) similarities to quantify gene expression dissimilarities. This matrix forms the foundation for clustering and subsequent module identification, and the “cutreeDynamic” algorithm from the WGCNA package was employed for dynamic pruning to discern 16 modules encompassing all genes. Module sizes ranged from a minimum of 33 genes to a maximum of 1747 genes with only 25 genes included in the gray module. The gray module was excluded from subsequent analyses, and the number of genes in each module is detailed in Table S1.
Figure 5.
Results of weighted gene co-expression network analysis (WGCNA):(A) Scale-Free Topology Analysis, frequency distribution of the number of connections (i.e., node degree, k) in the network (left), and a test of the scale-independent nature of the network (right);(B) Clustering of Module Eigengenes;(C) Gene Dendrogram and Module Colors and (D) Module Eigengene Correlation Heatmap.
Figure 5.
Results of weighted gene co-expression network analysis (WGCNA):(A) Scale-Free Topology Analysis, frequency distribution of the number of connections (i.e., node degree, k) in the network (left), and a test of the scale-independent nature of the network (right);(B) Clustering of Module Eigengenes;(C) Gene Dendrogram and Module Colors and (D) Module Eigengene Correlation Heatmap.
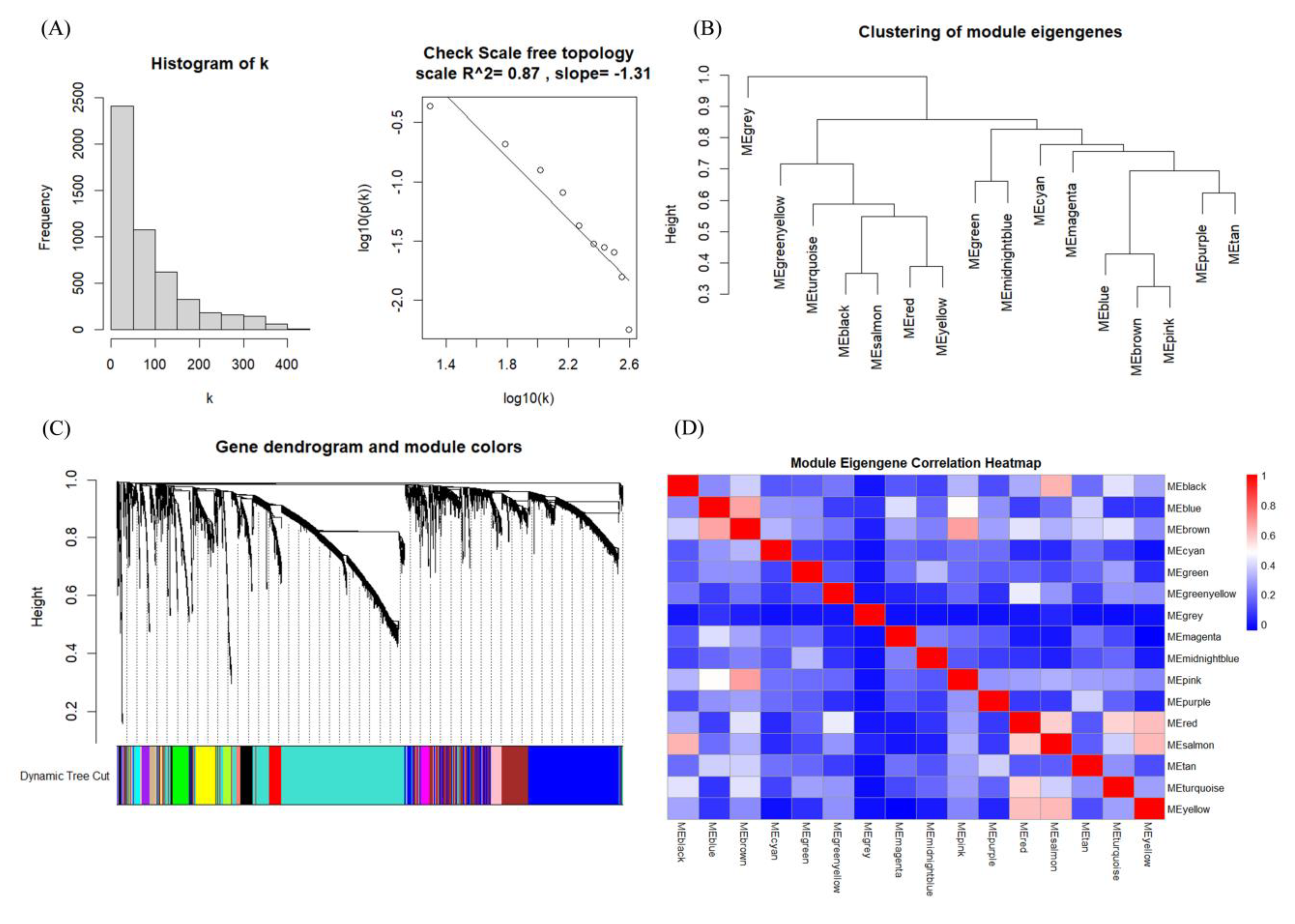
Utilizing the characteristics of Module Eigengenes (MEs), the correlation between individual genes and their corresponding modules can be precisely quantified. This correlation coefficient serves as a pivotal metric for assessing whether a gene functions as a hub gene within its module.
Figure 5(B) presents the correlation clustering tree dendrograms for the 15 identified modules, indicating a similarity threshold below 0.7 (with merge heights exceeding 0.3), thereby avoiding dynamic pruning. To pinpoint the key modules with the most robust correlations to sample traits, notably tumor subtypes, we assessed the gene module-sample subtype correlations (Figure 6). In this evaluation, categorical labels were assigned a value of 1, with non-relevant categories receiving 0. Employing a distinctive heat coding strategy for labels, we conducted three separate analyses to determine the most pertinent central genes for each category. Pearson correlation coefficients were employed to gauge the relationship between feature genes, i.e., those linked to sample characteristics and the categorical variables denoting tumor subtypes. This methodology resulted in quantifying the correlation between tumor subtypes and feature genes across modules. The “Turquoise,” “Brown,” and “Black” modules demonstrated the most pronounced correlation with the three tumor subtypes. Within these pivotal modules, we initially determined the intramodular connectivity (kWithin) for each gene, reflecting the strength of its interaction with other genes within the same module, as calculated using the following formula:
where, is the Pearson correlation coefficient within the module for the two genes.
Figure 7.
Correlation of different modules with different subtypes.
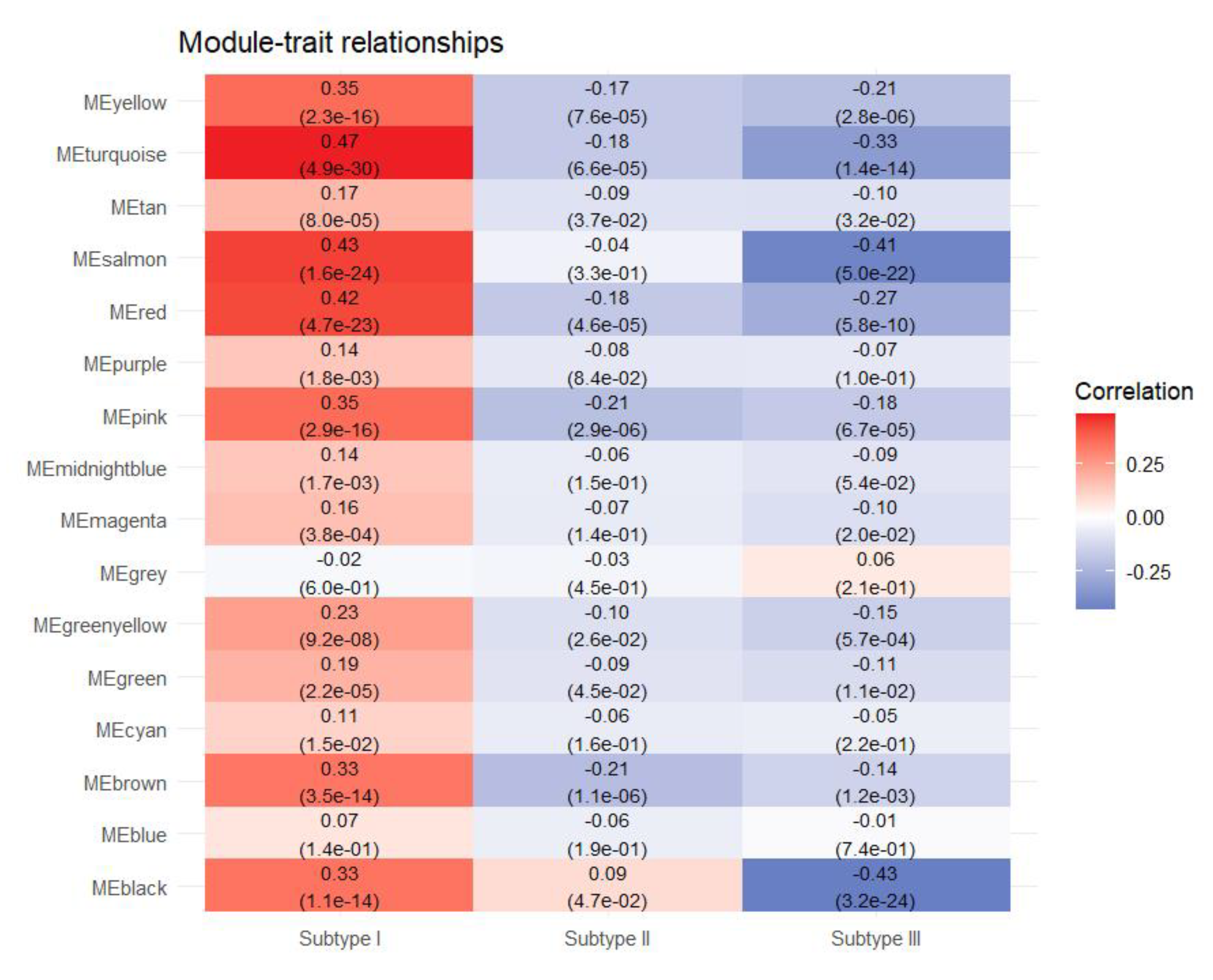
The greater a gene’s intramodular connectivity, the more pivotal its role within the module and the more closely it interacts with other genes. We assessed each gene’s correlation with tumor subtypes (GS) and its agreement with the module’s signature genes (MM). Hub genes are characterized by high GS, high MM, and high within. Accordingly, for subtype I samples, genes were selected with GS>0.45 and |MM|>0.8; for subtype II samples, with GS>0.2 and |MM|>0.7; and for subtype III samples, with GS>0.4 and |MM|>0.8. Thereafter, by ranking genes based on kWithin in descending order, the study identified 16 hub genes for Subtype I, 9 for Subtype II, and 8 for Subtype III.
Table 1 presents an exhaustive compilation of hub genes for the three sample subtypes. Among the hub genes of subtype I, MAGI2-AS3 facilitates the progression of gastric cancer by sequestering miR-141/200a, thereby sustaining the overexpression of ZEB1[38], an epitranscription factor that plays a role in the regulation of epithelial-mesenchymal transition (EMT), a pivotal process in cancer metastasis and invasion. MAGI2-AS3, through its interaction with miRNAs, is implicated in the modulation of the tumor microenvironment, impacting tumor cell proliferation, migration, and invasion. Concurrently, MAGI2-AS3 advances the progression of colorectal cancer by manipulating the miR-3163/TMEM106B axis. It functions as a molecular sponge for miR-3163, inhibiting the suppressive effect of miR-3163 on TMEM106B, which results in the upregulation of TMEM106B expression and consequently fuels tumor cell proliferation and migration[39].

Table 1.
Hub genes for the three subtypes (sorted according to Kwithin).
| Subtype I | kWithin | Subtype II | kWithin | Subtype III | kWithin |
| MAGI2-AS3 | 371.7129 | RP11-416A17.6 | 55.3853 | SPARC | 34.4698 |
| TTC28 | 345.3234 | RP11-166B2.3 | 52.2037 | FAP | 33.0935 |
| RBMS3 | 345.2273 | RP11-192H23.7 | 50.8855 | BGN | 29.6813 |
| CNRIP1 | 338.6266 | MALAT1 | 46.6874 | SULF1 | 29.6049 |
| PLEKHO1 | 323.7333 | RP11-49O14.2 | 46.2525 | CDH11 | 28.0017 |
| GYPC | 315.0139 | CTD-2014D20.1 | 46.0766 | PRRX1 | 26.9047 |
| C20orf194 | 313.7970 | LA16c-431H6.6 | 45.3105 | THY1 | 26.4728 |
| CLIP4 | 312.4037 | NPIPB5 | 40.0239 | NOX4 | 25.9135 |
| FOXN3 | 309.4977 | RYKP1 | 39.8201 | ||
| ATP8B2 | 300.8144 | ||||
| RP11-875O11.1 | 286.9392 | ||||
| PDE1A | 254.0221 | ||||
| NR3C1 | 249.1351 | ||||
| SLC9A9 | 248.7150 | ||||
| NR2F2-AS1 | 245.9516 | ||||
| RP11-730A19.9 | 226.7302 |
Among the hub genes of subtype II, MALAT1 has been identified as intimately linked to the development, progression, and metastasis of various human cancers. It exhibits elevated expression in colorectal cancer tissues, contributing to the enhanced growth of SW480 and HCT116 colorectal cancer cells[40,41]. Additionally, MALAT1 is deeply implicated in gastric carcinogenesis through diverse molecular pathways. For instance, it augments the proliferation of gastric cancer cells by downregulating the expression of miRNAs such as miR-122, miR-1297, miR-22-3p, and miR-202, and by repressing the activity of the oncogene PCDH10, thereby promoting the growth and invasiveness of gastric cancer[42].
Within the hub genes of subtype III, SPARC was shown to amplify the chemosensitivity of 5-FU by facilitating apoptosis. Our findings indicate that both cleaved PARP and cleaved caspase-3 levels were increased after overexpression of SPARC protein. Additionally, Bax, a pivotal protein in the apoptotic process, was significantly upregulated in SGC-7901 and BGC-823 cells with heightened SPARC expression. These outcomes implicate that SPARC may induce apoptosis in gastric cancer through the activation of the PARP/caspase-3 pathway[43]. Expression levels of the SPARC gene are notably correlated with clinical attributes of colorectal cancer, such as tumor stage, suggesting its potential as a biomarker for colorectal cancer[44].
3.3. Impact of Hub Genes on the Development of Gastrointestinal Tumors
Gene Ontology (GO), established by the Gene Ontology Consortium, serves as a comprehensive database that catalogs the functional roles of genes and their transcriptional and translational products within biological processes. Our analysis of the pathways involving hub genes in gastrointestinal cancer subtypes aims to delineate their biological functions and the pathways in which they participate.
Figure 7(A) illustrates the outcomes of GO analysis for subtype I with a primary focus on the Molecular Function (MF) category of GO. In this representation, dots correspond to distinct biological processes, and the magnitude of each dot is proportional to the gene count associated with the process. In addition, the color gradient reflects the adjusted p-value (p.adjust), denoting the statistical significance of enrichment. Cannabinoid receptors contribute to various intestinal physiological processes, including peristalsis, secretion, and epithelial barrier function. Research indicates that the deletion of cannabinoid receptor 1 can result in intestinal inflammation and cancer[45]. Activated cannabinoid receptors, notably CB1 and CB2, are recognized for their role in modulating inflammatory responses and tumor cell proliferation[46]. Glucocorticoid receptors are pivotal in regulating immune responses, inflammation, and cell survival. In the context of gastrointestinal cancers, glucocorticoids may modulate the tumor microenvironment via their receptors, potentially impacting tumor growth and metastasis by suppressing inflammation and regulating immune cell activity. Specifically, in colorectal cancer, glucocorticoids might facilitate cancer cell proliferation and invasion through the GR-CDK1 signaling pathway[47].
Figure 7.
GO analysis results: (A) subtype I (B) subtype III.
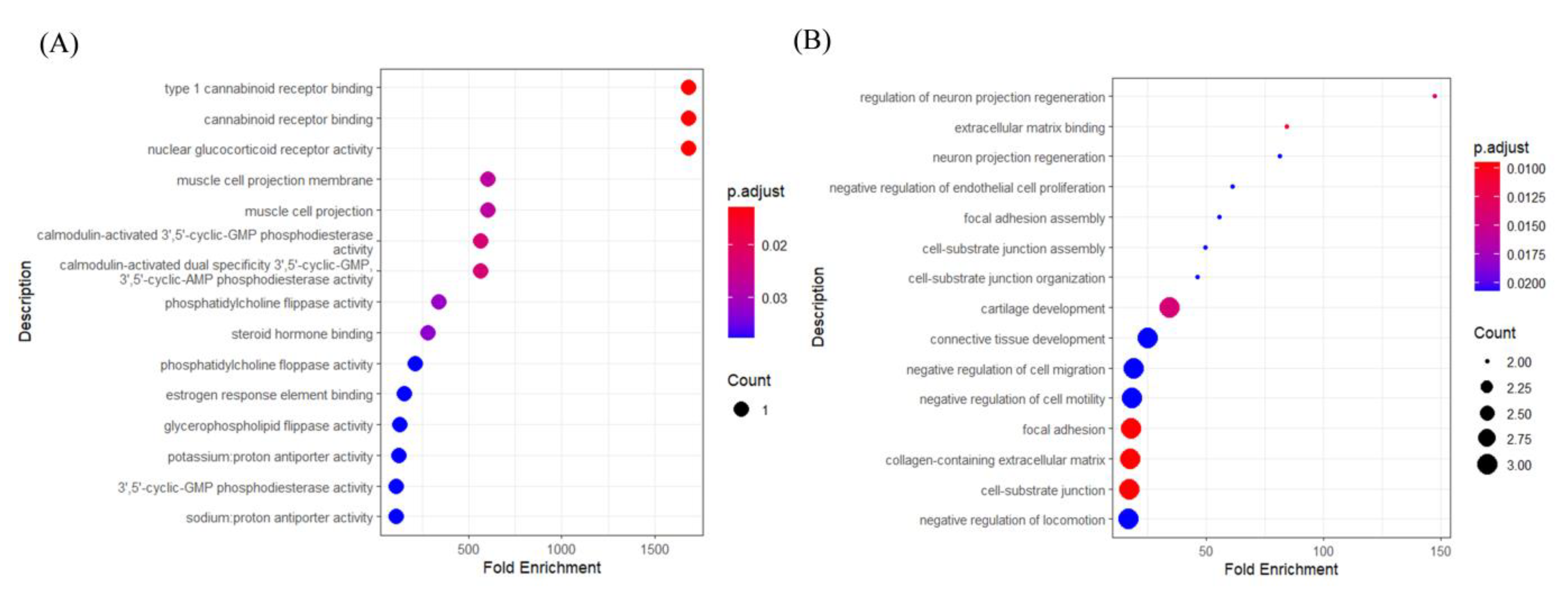
Figure 8(B) shows the results of GO analysis for subtype III, focusing mainly on the GO category ‘Biological Process (BP)’. Most of these functions are associated with extracellular matrix (ECM) interactions, cell migration, and tissue development. Engagement with the extracellular matrix is an essential component of the tumor microenvironment in gastrointestinal cancers, influencing the invasive and migratory capacities of tumor cells. The interaction between tumor cells and the ECM has the potential to either advance or retard tumor progression[48].
Genes like MAGI2 that encodes long non-coding RNA MAGI2-AS3 and RBMS3 that encodes the RNA Binding Motif Single Stranded Interacting protein3 participate in a spectrum of regulatory processes, encompassing signal transduction, gene expression modulation, and intercellular communication. This participation may indicate that subtype I is particularly dynamic in the realms of cellular signaling and gene expression regulation. Genes within subtype II might be more engaged in specialized regulatory roles, such as the involvement of non-coding RNA in transcriptional regulation, and could be pivotal in specific physiological or pathological contexts, including the modulation of gene expression in response to environmental stresses or disease conditions. Genes implicated in extracellular matrix interactions and tissue remodeling, such as SPARC and FAP, frequently contribute to tissue development, repair, and the cancer microenvironment. The roles of these genes suggest that subtype III may be instrumental in governing extracellular matrix dynamics and adaptations under pathological conditions.
This section may be divided by subheadings. It should provide a concise and precise description of the experimental results, their interpretation, as well as the experimental conclusions that can be drawn.
4. Discussion
The prevalence of gastrointestinal cancers is escalating, most notably among younger demographics, constituting a substantial segment of malignant neoplasms within the digestive tract. The emergence of multiomics has unveiled the profuse heterogeneity of these tumors, a key determinant in their evolution, therapeutic response, and metastatic propensity. Our study harnesses multimodal data analysis, integrating diverse technical approaches and data modalities, to deepen our understanding of the intrinsic tumor architecture, molecular makeup, and biological activities.
In our methodology, we integrate edge features derived from the optimized Canny operator in detecting a wide range of edge in images, along with transcriptomic and immunological data. To forge a sample similarity network, we conduct preliminary clustering on disparate data modalities to amplify the significance of localized similarities. This involves an iterative optimization of the similarity and kernel matrices, culminating in convergence. We then deploy spectral clustering on this integrated network to delineate distinct tumor subtypes.
The fusion of diverse data offers a comprehensive view of gene expression profiles, while also yielding insights into cellular and tissue architectures. This dual methodology highlights the tumor’s genotypic and phenotypic traits, leads to a comprehensive elaboration of distinct tumor subtypes and, hence, establishes a foundation for the development of therapeutic modalities.
We conducted an analysis of mRNA gene expression profiles utilizing Weighted Gene Co-expression Network Analysis (WGCNA), identifying distinct gene modules. Synthesizing these findings with the outcomes of sample subtyping, we pinpointed the associated hub genes. These genes constitute critical regulatory pathways, the dysregulation or aberrant expression of which can significantly advance disease progression. Moreover, they represent promising therapeutic targets with the potential to modulate diverse network and pathway activities.
While our optimized Canny operator has demonstrated remarkable efficacy in delineating tumor margin features, it might not encapsulate the full complexity of the tumor microenvironment. Therefore, while not within the scope of the present paper, we plan to pursue more sophisticated image processing methodologies, including deep learning algorithms, with the aim of strengthening the detection and profiling of biomarkers within histopathological assessments.
5. Conclusions
Our study underscores the profound utility of multimodal data analysis in the study of gastrointestinal cancers, and demonstrates that the integration of omics data can be achieved by seamlessly merging the edge features of tumor images with differential transcriptomic and immune cell data. The discovery of hub genes across various tumor subtypes paves the way for innovative diagnostic methods and tailored therapeutic strategies. Moreover, the integration multimodal data deepens insights into the intrinsic heterogeneity of gastrointestinal tumors. Overall, our results lay a robust groundwork for further investigating the complexities of GI cancers with the promise of advancing personalized medicine to achieve superior patient outcomes.
Supplementary Materials
Figure S1: Differences between cancer and normal samples; Figure S2: Figure S2. Reference chart for selecting soft thresholds for WGCNA analysis. Figure S3: Iterative process for four types of data; Table S1: Modules obtained from WGCNA analysis and the number of genes they contain.
Author Contributions
Conceptualization, D.A., Y.D. and Y.W.; methodology, D.A., Y.D. and H.D.; software, Y.D. and Y.W.; validation, H.D. and J.Q.; formal analysis, D.A.; investigation, Y.D.; resources, Y.W.; data curation, D.A.; writing—original draft preparation, D.A., Y.D., H.D. and Y.W.; writing—review and editing, D.A., Y.D., and J.Q.; visualization, H.D.; supervision, D.A.; project administration, D.A.; funding acquisition, D.A. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the National Natural Science Foundation of China, grant number 61873027.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The datasets ANALYZED for this study can be found in the following GitHub repository: https://github.com/gutmicrobes/GI-multimodal.git.
Acknowledgments
Not applicable.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Sung H., Ferlay J., Siegel R.L., Laversanne M., Soerjomataram I., Jemal A., et al. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin, 2021, 71(3): 209-49. [CrossRef]
- Arnold M., Soerjomataram I., Ferlay J., Forman D. Global incidence of oesophageal cancer by histological subtype in 2012. Gut, 2015, 64(3): 381-7. [CrossRef]
- Torre L.A., Siegel R.L., Ward E.M., Jemal A. Global Cancer Incidence and Mortality Rates and Trends--An Update. Cancer Epidemiol Biomarkers Prev, 2016, 25(1): 16-27. [CrossRef]
- Xie Y., Shi L., He X., Luo Y. Gastrointestinal cancers in China, the USA, and Europe. Gastroenterol Rep (Oxf), 2021, 9(2): 91-104. [CrossRef]
- Lee J.H., Bae J.S., Ryu K.W., Lee J.S., Park S.R., Kim C.G., et al. Gastric cancer patients at high-risk of having synchronous cancer. World J Gastroenterol, 2006, 12(16): 2588-92. [CrossRef]
- Lim S.B., Jeong S.Y., Choi H.S., Sohn D.K., Hong C.W., Jung K.H., et al. Synchronous gastric cancer in primary sporadic colorectal cancer patients in Korea. Int J Colorectal Dis, 2008, 23(1): 61-5. [CrossRef]
- Yoon S.N., Oh S.T., Lim S.B., Kim T.W., Kim J.H., Yu C.S., et al. Clinicopathologic characteristics of colorectal cancer patients with synchronous and metachronous gastric cancer. World J Surg, 2010, 34(9): 2168-76. [CrossRef]
- Haffner M.C., Kronberger I.E., Ross J.S., Sheehan C.E., Zitt M., Mühlmann G., et al. Prostate-specific membrane antigen expression in the neovasculature of gastric and colorectal cancers. Human Pathology, 2009, 40(12): 1754-61. [CrossRef]
- Park J.G., Gazdar A.F. Biology of colorectal and gastric cancer cell lines. J Cell Biochem Suppl, 1996, 24: 131-41. [CrossRef]
- Lei Z., Tan I.B., Das K., Deng N., Zouridis H., Pattison S., et al. Identification of molecular subtypes of gastric cancer with different responses to PI3-kinase inhibitors and 5-fluorouracil. Gastroenterology, 2013, 145(3): 554-65. [CrossRef]
- Comprehensive molecular characterization of gastric adenocarcinoma. Nature, 2014, 513(7517): 202-9. [CrossRef]
- Budinska E., Popovici V., Tejpar S., D’Ario G., Lapique N., Sikora K.O., et al. Gene expression patterns unveil a new level of molecular heterogeneity in colorectal cancer. J Pathol, 2013, 231(1): 63-76. [CrossRef]
- Guinney J., Dienstmann R., Wang X., de Reyniès A., Schlicker A., Soneson C., et al. The consensus molecular subtypes of colorectal cancer. Nat Med, 2015, 21(11): 1350-6. [CrossRef]
- Li Q.L., Lin X., Yu Y.L., Chen L., Hu Q.X., Chen M., et al. Genome-wide profiling in colorectal cancer identifies PHF19 and TBC1D16 as oncogenic super enhancers. Nat Commun, 2021, 12(1): 6407. [CrossRef]
- Paz-Cabezas M., Calvo-López T., Romera-Lopez A., Tabas-Madrid D., Ogando J., Fernández-Aceñero M.J., et al. Molecular Classification of Colorectal Cancer by microRNA Profiling: Correlation with the Consensus Molecular Subtypes (CMS) and Validation of miR-30b Targets. Cancers (Basel), 2022, 14(21). [CrossRef]
- Nausheen N., Seal A., Khanna P., Halder S. A FPGA based implementation of Sobel edge detection. Microprocessors and Microsystems, 2018, 56: 84-91. [CrossRef]
- Hoang N.-D., Nguyen Q.-L. Metaheuristic Optimized Edge Detection for Recognition of Concrete Wall Cracks: A Comparative Study on the Performances of Roberts, Prewitt, Canny, and Sobel Algorithms. Advances in Civil Engineering, 2018, 2018: 7163580. [CrossRef]
- Topno P., Murmu G. An Improved Edge Detection Method based on Median Filter; proceedings of the 2019 Devices for Integrated Circuit (DevIC), F 23-24 March 2019, 2019 [C].
- Er-sen L., Shu-long Z., Bao-shan Z., Yong Z., Chao-gui X., Li-hua S. An Adaptive Edge-Detection Method Based on the Canny Operator; proceedings of the 2009 International Conference on Environmental Science and Information Application Technology, F 4-5 July 2009, 2009 [C].
- Luo S., Yingwei F., Chang W., Liao H., Kang H.X., Huo L. Classification of human stomach cancer using morphological feature analysis from optical coherence tomography images. Laser Physics Letters, 2019. [CrossRef]
- Pratiwi N., Magdalena R., Fuadah N., Saidah S. K-Nearest Neighbor for colon cancer identification. Journal of Physics: Conference Series, 2019, 1367: 012023. [CrossRef]
- Toomey P.G., Vohra N.A., Ghansah T., Sarnaik A.A., Pilon-Thomas S.A. Immunotherapy for gastrointestinal malignancies. Cancer Control, 2013, 20(1): 32-42. [CrossRef]
- Tomczak K., Czerwińska P., Wiznerowicz M. The Cancer Genome Atlas (TCGA): an immeasurable source of knowledge. Contemp Oncol (Pozn), 2015, 19(1a): A68-77. [CrossRef]
- Kather J.N., Pearson A.T., Halama N., Jäger D., Krause J., Loosen S.H., et al. Deep learning can predict microsatellite instability directly from histology in gastrointestinal cancer. Nature Medicine, 2019, 25(7): 1054-6. [CrossRef]
- Song R., Zhang Z., Liu H. Edge connection based Canny edge detection algorithm. Pattern Recognit Image Anal, 2017, 27(4): 740–7. [CrossRef]
- Yang Q., Tan K.H., Ahuja N. Shadow Removal Using Bilateral Filtering. IEEE Transactions on Image Processing, 2012, 21(10): 4361-8. [CrossRef]
- Ling Y.Z., Ma W., Yu L., Zhang Y., Liang Q.S. Decreased PSD95 expression in medial prefrontal cortex (mPFC) was associated with cognitive impairment induced by sevoflurane anesthesia. J Zhejiang Univ Sci B, 2015, 16(9): 763-71. [CrossRef]
- Yang D., Parrish R.S., Brock G.N. Empirical evaluation of consistency and accuracy of methods to detect differentially expressed genes based on microarray data. Comput Biol Med, 2014, 46: 1-10. [CrossRef]
- Li A., Horvath S. Network neighborhood analysis with the multi-node topological overlap measure. Bioinformatics, 2007, 23(2): 222-31. [CrossRef]
- Voigt A., Almaas E. Assessment of weighted topological overlap (wTO) to improve fidelity of gene co-expression networks. BMC Bioinformatics, 2019, 20(1): 58. [CrossRef]
- Zhang B., Horvath S. A general framework for weighted gene co-expression network analysis. Stat Appl Genet Mol Biol, 2005, 4: Article17. [CrossRef]
- Wang B., Mezlini A.M., Demir F., Fiume M., Tu Z., Brudno M., et al. Similarity network fusion for aggregating data types on a genomic scale. Nature Methods, 2014, 11(3): 333-7. [CrossRef]
- Wang B., Mezlini A.M., Demir F., Fiume M., Tu Z., Brudno M., et al. Similarity network fusion for aggregating data types on a genomic scale. Nat Methods, 2014, 11(3): 333-7. [CrossRef]
- Hashimoto I., Oshima T. Claudins and Gastric Cancer: An Overview. Cancers, 2022, 14(2): 290.
- Vonniessen B., Tabariès S., Siegel P.M. Antibody-mediated targeting of Claudins in cancer. Frontiers in Oncology, 2024, 14. [CrossRef]
- Shan M.-j., Meng L.-b., Guo P., Zhang Y.-m., Kong D., Liu Y.-b. Screening and Identification of Key Biomarkers of Gastric Cancer: Three Genes Jointly Predict Gastric Cancer. Frontiers in Oncology, 2021, 11. [CrossRef]
- Jiang R., Chen X., Ge S., Wang Q., Liu Y., Chen H., et al. MiR-21-5p Induces Pyroptosis in Colorectal Cancer via TGFBI. Frontiers in Oncology, 2021, 10. [CrossRef]
- Chen Y., Gao X., Dai X., Xia Y., Zhang X., Sun L., et al. Integrated Bioinformatics and Experimental Analysis of Long Noncoding RNA Associated-ceRNA as Prognostic Biomarkers in Advanced Stomach Adenocarcinoma. Journal of Cancer, 2024, 15(6): 1536-50. [CrossRef]
- Ren H., Li Z., Tang Z., Li J., Lang X. Long noncoding MAGI2-AS3 promotes colorectal cancer progression through regulating miR-3163/TMEM106B axis. Journal of Cellular Physiology, 2019, 235: 4824 - 33.
- Wu Q., Meng W.Y., Jie Y., Zhao H. LncRNA MALAT1 induces colon cancer development by regulating miR-129-5p/HMGB1 axis. J Cell Physiol, 2018, 233(9): 6750-7. [CrossRef]
- Chen S., Shen X. Long noncoding RNAs: functions and mechanisms in colon cancer. Mol Cancer, 2020, 19(1): 167. [CrossRef]
- Xu W., Ding M., Wang B., Cai Y., Guo C., Yuan C. Molecular Mechanism of the Canonical Oncogenic lncRNA MALAT1 in Gastric Cancer. Current Medicinal Chemistry, 2021, 28(42): 8800-9. [CrossRef]
- Ma J., Ma Y., Chen S., Guo S., Hu J., Yue T., et al. SPARC enhances 5-FU chemosensitivity in gastric cancer by modulating epithelial-mesenchymal transition and apoptosis. Biochemical and Biophysical Research Communications, 2021, 558: 134-40. [CrossRef]
- Mortezapour M., Tapak L., Bahreini F., Najafi R., Afshar S. Identification of key genes in colorectal cancer diagnosis by weighted gene co-expression network analysis. Computers in Biology and Medicine, 2023, 157: 106779. [CrossRef]
- Taschler U., Hasenoehrl C., Storr M., Schicho R. Cannabinoid Receptors in Regulating the GI Tract: Experimental Evidence and Therapeutic Relevance. Handb Exp Pharmacol, 2017, 239: 343-62. [CrossRef]
- Pagano E., Borrelli F. Targeting cannabinoid receptors in gastrointestinal cancers for therapeutic uses: current status and future perspectives. Expert Rev Gastroenterol Hepatol, 2017, 11(10): 871-3. [CrossRef]
- Tian D., Tian M., Han G., Li J.-L. Increased glucocorticoid receptor activity and proliferation in metastatic colon cancer. Scientific Reports, 2019, 9(1): 11257. [CrossRef]
- Brassart-Pasco S., Brézillon S., Brassart B., Ramont L., Oudart J.-B., Monboisse J.C. Tumor Microenvironment: Extracellular Matrix Alterations Influence Tumor Progression. Frontiers in Oncology, 2020, 10. [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



