
Submitted:
15 August 2024
Posted:
20 August 2024
You are already at the latest version
Abstract
The blood-brain barrier (BBB) acts as a structural and functional barrier for brain homeostasis. This review highlights the pathological contribution of BBB dysfunction to neuroimmunological diseases, including multiple sclerosis (MS), neuromyelitis optica spectrum disorder (NMOSD), myelin oligodendrocyte glycoprotein antibody-associated disease (MOGAD), autoimmune encephalitis (AE) and paraneoplastic neurological syndrome (PNS). The transmigration of massive lymphocytes across the BBB caused by the activation of cell adhesion molecules is involved in the early phase of MS, and dysfunction of the cortical BBB is associated with the atrophy of gray matter in the late phase of MS. In the onset of NMOSD, increased permeability of the BBB causes the entry of circulating AQP4 autoantibodies into the central nervous system (CNS). Recent reports have shown the importance of glucose-regulated protein (GRP) autoantibodies as BBB-reactive autoantibodies in NMOSD, inducing antibody-mediated BBB dysfunction. BBB breakdown has also been observed in MOGAD, NPSLE, and AE with anti-NMDAR antibodies. Our recent report demonstrated the presence of GRP78 autoantibodies in patients with MOGAD and the molecular mechanism responsible for GRP78 autoantibody-mediated BBB impairment. Disruption of the BBB may explain the symptoms in the brain and cerebellum in the development of PNS, as it induces the entry of pathogenic autoantibodies or lymphocytes into the CNS by autoimmunity against tumors in the periphery. GRP78 autoantibodies were detected in paraneoplastic cerebellar degeneration and Lambert-Eaton myasthenic syndrome and were associated with cerebellar ataxia with anti-P/Q type voltage-gated calcium channel antibodies. This review reports that therapies affecting the BBB that are currently available for disease-modifying therapies for neuroimmunological diseases have the potential to prevent BBB damage.
Keywords:
blood-brain barrier
; neuroimmunological disease
; multiple sclerosis
; neuromyelitis optica spectrum disorder
; autoimmune encephalitis
; paraneoplastic neurological syndrome
1. Introduction
The blood-brain barrier (BBB) plays an important role in protecting the central nervous system (CNS) from potentially harmful circulating pathogens [1,2]. The BBB consists of brain microvascular endothelial cells (BMECs) surrounded by pericytes and astrocyte endofeet and ensheathed in two basement membranes (vascular basement membrane and glia limitans) [1,2]. These cells in addition to neurons and perivascular microglia constitute the neurovascular unit (NVU) [3,4] (Figure 1A). BMECs form a physical barrier through tight junctions and adhesion junctions to prevent the entry of blood cells and other molecules and maintain brain homeostasis by controlling nutrient, water, and molecule exchanges and removing waste products from the CNS through transporters [5]. Tight junction proteins include claudins (especially claudin-5) and occludin, which have intracellular domains that interact with ZO-1. At the intracellular level, ZO-1, ZO-2, and ZO-3 establish a link between transmembrane proteins and the actin cytoskeleton to maintain cytoskeletal integrity [6,7] (Figure 1B). Adherent junctions are composed of VE-cadherin and catenins [6,7].
The major physiological roles of the intact BBB are (1) restriction of plasma macromolecules into the brain, (2) maintenance of ionic metastasis, (3) the uptake of brain nutrients, (4) regulation of optimal levels of neurotransmitters, (4) protection of the brain against neurotoxins, and (5) elimination of substances from the brain [8,9].
The breakdown of the BBB is associated with several neuroimmunological diseases, including multiple sclerosis (MS), neuromyelitis optica (NMO), myelin oligodendrocyte glycoprotein antibody-associated disease (MOGAD), autoimmune encephalitis (AE) and neuropsychiatric systemic lupus erythematosus (NPSLE) [2,10].
This review discusses the significance and molecular mechanisms of BBB disruption in neuroimmunological diseases.
2. Multiple sclerosis
2.1. The BBB Breakdown in Multiple Sclerosis
Multiple sclerosis is the most common chronic inflammatory demyelinating disease affecting the CNS [11,12]. It affects the brain, spinal cord, and optic nerves. Relapsing-remitting MS (RRMS) is characterized by intermittent neurological disturbance (relapse) followed by complete or incomplete recovery [13]. Throughout the disease course, MS usually begins as RRMS, and 30%-60% of RRMS patients shift to a phase of secondary progressive MS, characterized by gradual clinical worsening without relapse [14,15,16,17]. Multiple genetic polymorphisms with environmental and endogenous triggers are believed to lead to the formation of demyelinating plaques with inflammation and ultimately neurodegeneration [18]. Neurodegenerative processes, including axonal loss and gray matter atrophy, are major causes of neurological disability in SPMS [18,19].
In the early stages of MS, inflammatory BBB malfunction is associated with pathogenic immune cell infiltration, including T cells and B cells, immunoglobulin G, and inflammatory cytokines into the CNS parenchyma, although a normal BBB restricts the entry of immune cells into the CNS [20,21,22]. Clinical findings show that newly formed lesions within the CNS can be detected by gadolinium (Gd) enhancement of the brain on T1-weighted magnetic resonance imaging (MRI) during relapse in MS [23]. This change is considered a feature of BBB disruption [23]. An increase in the cerebrospinal fluid (CSF)/serum albumin ratio in MS patients also reflects the movement of albumin from the blood to the CSF via BBB disruption [24].
In MS, the disruption of the BBB was considered to be transient, as the contrast effect of Gd-enhancement on T1-weighted MRI did not last long. In histopathological findings, acute MS lesions demonstrated disruption of the BBB, supported by post-mortem evidence of focal microvascular leakage of albumin and the accumulation of inflammatory cells around the vessels [23]. Furthermore, abnormalities of the BBB, including vascular leakage and the decreased expression of tight junction proteins, were observed in both active and inactive lesions, as well as in normal-appearing white matter (NAWM) in RRMS and SPMS patients, suggesting that the persistent loss of BBB integrity may be involved in pathogenesis in both disease onset and progression [25].
2.2. Molecular Basis of BBB Disruption in RRMS
After T and B lymphocytes are activated in the periphery as a first step, they infiltrate the CNS and trigger a central autoimmune response, leading to myelin and axonal damage [26]. Both BBB disruption and leukocyte trafficking are the most important pathological processes in the active lesion (“the acute demyelinating brain lesion”) as well as the inactive lesion (“NAWM”) [27]. Leukocyte-derived proinflammatory cytokines activate endothelial cells and upregulate the expression of additional adhesion molecules, mediating the self-sustained CNS infiltration of more immune cells (Figure 2) [26,27]. During tethering, peripheral lymphocytes express the P-selectin glycoprotein ligand-1 (PSGL-1), which interacts with the ligand molecules expressed on endothelial cells (E- and P-selectins) and facilitates the capture of lymphocytes [28].
During the rolling process, endothelial cells express several chemokines, including CCL21 and CCL19, which activate the G protein-coupled receptor (GPCR) on the surface of the lymphocyte and stimulate the expression of integrin α4β1 (VLA-4) and lymphocyte function-associated antigen 1 (LFA-1) [29]. Activated lymphocytes slow their flow speed due to the interaction of VLA-4 and LFA-1 from the surface of lymphocytes with adhesion molecules from the surface of inflamed endothelial cells, including vascular cell adhesion molecule 1 (VCAM-1) and intracellular adhesion molecules (ICAM-1) [30]. In adhesion and transcellular pathways, lymphocytes adhere to endothelial cells and transverse the BBB by coupling VLA-4 and LFA-1 expressed on lymphocytes with endothelial cell receptors (VCAM-1 and ICAM-1) [30]. The interaction between VCAM-1 and ICAM and their ligands on leukocytes induces the arrest of immune cells from the blood in brain endothelial cells [31] (Figure 1C). Importantly, the upregulation of VCAM-1 was observed in the BBB endothelial cells around the active or inactive lesion or NAWM in autopsy cases of MS, suggesting that activation of endothelial cells and upregulation of VCAM-1 precede the formation of demyelination [32].
Natalizumab, a monoclonal antibody against α4β1 integrin, the ligand of VCAM-1, directly interferes with the transmigration of T and B lymphocytes across BBB endothelial cells [33]. In addition, other cell adhesion molecules (CAMs), including melanoma cell adhesion molecules (MCAMs), activated leukocyte cell adhesion molecules (ALCAMs), platelet and endothelial cell adhesion molecules (PECAMs), and dual immunoglobulin domain-containing cell adhesion molecules (DICAMs), play a role in trans-endothelial immune cell infiltration [34,35,36,37]. The upregulation of MCAM in brain endothelial cells recruits pathogenic Th1 and Th17 CD4+ T lymphocytes expressing MCAM from circulation during neuroinflammation in EAE and autopsied brain samples in MS [34]. The upregulation of ALCAMs in brain endothelial cells drives the entry of proinflammatory B lymphocytes expressing ALCAMs into the brain lesion in EAE and MS [35]. An increase in DICAM-expressing Th17 CD4+ T cells and the upregulation of DICAM ligand on the brain endothelial cells upon inflammation and in MS lesions have been observed, and monoclonal antibodies against DICAM have been shown to reduce Th17 cell trafficking across the blood-brain barrier both in vitro and in vivo and to ameliorate both relapsing and progressive EAE [37]. Therefore, CAMs and their interacting ligands are attractive targets for novel therapies for RRMS.
2.3. BBB in Progressive MS
In the late stage of MS, cortical gray matter atrophy is correlated with cognitive decline and gait disturbances [38]. A total of 45% of RRMS patients and 75% of SPMS patients show effects on their daily working memory and verbal fluency tasks [38]. Gd-enhanced lesions on MRI, reflecting disruption of the BBB, were rarely observed in progressive MS, although fibrin deposition and tight junction abnormalities were found in the cortex in progressive MS in both active and inactive lesions and NAWM [25], suggesting persistent dysfunction of cortical BBB integrity in progressive MS [26]. We demonstrated that anti-galectin-3 autoantibodies in SPMS mediate the breakdown of the BBB through degradation of claudin-5 and upregulation of ICAM-1 and reported that anti-galection-3 antibodies were associated with persistent damage to the BBB [39]. Galectin-3 is a β-galactoside-binding lectin expressed both extra- and intracellularly in several cell types, and the activation of intracellular galectin-3 can induce activation of the NFκB pathway [40,41]. Anti-galectin 3 antibodies also may prevent remyelination, causing morphological and functional differentiation of oligodendrocyte progenitor cells [42].
Gray matter atrophy can induce cortical hypoperfusion in progressive MS. Functional MRI reportedly shows that cerebral vascular reactivity, which is the change in cerebral blood flow upon stimulation with vasoactive compounds, is reduced in the gray matter of patients with MS [43]. This change was shown to be correlated with gray matter atrophy and lesion volume in patients with MS [43]. The disturbance of cerebral vascular reactivity reflects the dysfunction of the NVC, which is linked to neurodegeneration. The vascular pathology hypothesis in MS states that vascular changes play a central role in MS pathogenesis [44]. Whether vascular pathology is a cause or consequence of neurodegeneration associated with cognitive impairment in MS remains elusive [45].
2.4. Fluid Biomarkers for BBB Disturbance in MS
Serum molecules associated with CNS cell damage, including neurofilament light chain (neuron), GFAP, and S100B (astrocytes), have been detected in MS and NMO using ultrasensitive single-molecular arrays, reflecting CSF drainage towards the peripheral compartment through a disrupted BBB. Neurofilament light chain concentrations in the blood and CSF are increased in newly diagnosed MS patients and correlate with relapse, new lesions on MRI, disease severity, and the prognosis in MS [46]. An increase in serum neurofilament light chain was shown to be related to elevation of Q Alb and CSF-located CD80+ B cells and the presence of Gd-enhancement lesions on MRI, suggesting increased BBB permeability in MS [47], and treatment with disease-modifying therapies was found to decrease serum neurofilament light chain levels [48,49,50]. The serum concentration of GFAP, reflecting astrocyte damage, was higher in patients with progressive MS during relapse than healthy control [51], but it increased in response to CNS injury, including BBB breakdown after TBI and intracerebral hemorrhaging [52]. Serum S100B is related to permeable BBB and S100B from serum and CSF, is likely secreted from astrocytes or Schwann cells. Increases of S100B is observed in RRMS at the diagnosis, and is related to disease severity and progression in MS [53].
Chemokines play a role in the recruitment of leukocytes to inflamed CNS sites. An increase in some chemokines, including CXCL8 (which mediates the recruitment of neutrophils secreted by macrophages or endothelial cells), CXCL10/interferon gamma-induced protein (IP)-10 (which mediates the recruitment of T cells and macrophages secreted by monocytes and endothelial cells), and CXCL13 (which mediates recruitment of B cells secreted by B cells), can be observed in the CSF of patients with MS compared with non-inflammatory controls [54]. Levels of the soluble form of CAMs secreted by endothelial cells, including VCAM-1 (serum), MCAM (CSF) and PECAM-1, are increased in MS [36,55,56].
2.5. Possible Causes of BBB Disturbance in MS
Genetic and environmental factors associated with MS can contribute directly and indirectly to BBB disturbances. A genome-wide association study showed that more than 230 genetic variants in MS and human leukocyte antigen (HLA)-DRB1 polymorphisms were associated with MS risk [57]. The single-nucleotide polymorphism (SNP) of ALCAM (rs6437585) is associated with the risk and progression of MS, while the SNP of VCAM-1 (rs11581062) is a risk factor for MS [35,58]. VCAM-1 and ALCAM play an important role in immune cell adhesion and transmigration and are linked to BBB disturbance in the development of MS.
A decrease in the serum 25-hydroxyvitamin D level is associated with an increased risk of MS onset and disease progression [59]. In a clinical study, vitamin D3 with interferon-beta (IFN-β) reduced the number of new Gd-enhanced lesions in RRMS compared to placebo with IFN-β, which suggests a role for vitamin D in repairing the BBB function [60]. The active form of vitamin D (1,25(OH)2D3) enhances the barrier function by upregulating claudin-5 and reducing VCAM-1 expression [61,62].
Infection with Epstein-Barr virus (EBV) is an important causal factor for increased risk of subsequent MS The risk of MS was 32-fold higher following EBV infection, and serum concentration of neurofilament light chain was increased after EBV seroconversion in MS [63]. EBV can contribute to the development of MS through molecular mimicry between the chronic presentation of viral antigens as a potential source of autoreactivity and CNS proteins, such as anoctamin-2 and GlialCAM [64]. Autoantibodies against anoctamin-2 (an ion channel expressed in the CNS) or GlialCAM (a component of glial cells in the brain) can recognizes the fragment of EBV nuclear antigen 1 and were increased in MS patients [65,66]. Upregulation of ICAM-1 and CCL5 and increased adherence of leukocytes have been observed in BBB-endothelial cells infected with EBV [67].
Smoking is a risk factor for the onset and progression of MS [68]. Nicotine can enhance BBB permeability by downregulating tight junction proteins [69]. In addition, concussion and brain trauma during adolescence are associated with the onset of MS [70]. Some reports have suggested that brain trauma temporarily increases BBB permeability [71].
2.6. Therapies Modulating the BBB in MS
Methylprednisolone pulse therapy is widely used for the acute treatment of MS relapses and influences the recovery of new Gd-enhanced lesions [72]. Glucocorticoids (GCs) reduce immune cell trafficking and cytokines (IFN-γ, TNF-α and IL-2) from lymphocytes [73,74]. GCs recover BBB dysfunction through an increase in tight junctions (occludin and claudin), a decrease in MMP-1 and MMP-9 expression and downregulation of adhesion molecules, such as VCAM-1, ICAM-1 and E-selectin, in BBB-endothelial cells [75,76,77] (Table 1).
IFN-β therapy is the first approved disease-modifying therapy (DMT) that decreases T cell proliferation. IFN-β prevents trans-endothelial migration of proinflammatory CD4+ Th1 cells and enhances BBB integrity through upregulation of tight junction [78,79,80].
Natalizumab is a monoclonal antibody against α4β1 integrin, which is the cognate ligand of VCAM-1. This drug cannot cross the BBB but blocks the interaction between α4 integrin from the surface of lymphocytes and VCAM-1 from the surface of BBB-endothelial cells, thereby preventing the transendothelial migration of lymphocytes directly [81]. Natalizumab is widely used for RRMS patients and has demonstrated high efficacy by reducing the annualized relapse rate and MS lesion accumulation on MRI and decreasing the sustained progression of disability [82]. Natalizumab dramatically reduces the number of CD4+T cells, CD8+ T cells, Th17 and B cells in the CNS, as lymphocytes cannot adhere to the BBB after treatment with natalizumab [83]. Unfortunately, the use of natalizumab is associated with a potentially fatal complication in progressive multifocal leukoencephalopathy (PML), as it inhibits immune surveillance against viral leukoencephalopathy induced by infection with John Cunningham (JC) virus [84].
Dimethyl fumarate (DMF) is a first-line oral DMT in RRMS patients and has shown efficacy in reducing relapse rates. Activation of the transcription factor pathway nuclear factor (erythroid-derived 2)-like 2 (Nrf2), which maintains intracellular redox homeostasis, is a target of DMF, and activation of the hydroxyl carboxylic acid receptor, independent of the Nrf2 pathway, is another target of DMF [85]. DMF reduces the number of serum lymphocytes. such as CD4+ T cells, CD8+T cells, B cells and type 1 myeloid dendritic cells, through activation of the Nrf2 pathway [86,87]. It also decreases the transendothelial migration of lymphocytes by decreasing α4 integrin on the lymphocyte surface and VCAM-1 on the endothelial cell surface independent of the Nrf2 pathway [88]. Furthermore, DMF can cross the BBB and exert a protective effect on neurons and astrocytes by inducing an antioxidant effect dependent on the Nrf2 pathway and microglia independent of the Nrf2 pathway [88,89].
Fingolimod is a sphingosine 1-phosphatate (S1P) receptor modulator that acts on S1P receptors, such as S1P1, S1P2, S1P3, S1P4 and S1P5. Fingolimod reduces the number of lymphocytes in the periphery by inhibiting the egress of lymphocytes from lymph nodes [90]. It decreases the trans-endothelial migration of lymphocytes by acting on S1P1 and S1P3 on the surface of BBB-endothelial cells [91,92,93,94]. Fingolimod also modifies the barrier function through clausin-5 upregulation and VCAM-1 downregulation [95]. After fingolimod crosses the BBB, it exerts protective effects on neurons by S1P1 and S1P3 modulation and BDNF upregulation [92,96,97], oligodendrocytes by S1P1 and S1P5 modulation [92,96,98], astrocytes by S1P1 modulation and inhibition of proinflammatory cytokines and microglia [99,100,101].
Cladribine is a purine nucleoside analog that reduces activated B and CD4+ T lymphocytes [102]. Cladribine can cross the BBB and act on lymphocyte death that has already entered the CNS [103]. Cladribine plays an effect on inhibits lymphocyte trafficking by interacting with ICAM-1 and E-selectin and reducing MMP-2 and MMP-9 secretion [104].
3. Neuromyelitis Optica Spectrum Disorder (NMOSD)
3.1. Pathophysiological Mechanism Underlying NMOSD
NMOSD is a relapsing neuroinflammatory autoimmune astrocytopathy, and its predominant clinical manifestations are longitudinally extensive transverse myelitis (LETM) and optic neuritis [105]. Most NMOSD patients have autoantibodies against the water channel aquaporin-4 (AQP4) expressed on astrocyte endfeet; thus, anti-AQP4 antibody detection has been used for the clinical diagnosis of NMOSD patients worldwide [106,107]. Most cases of NMOSD show a relapsing disease course and severe disability without any preventative therapies [108]. Satralizumab, an interleukin-6 receptor (IL-6R) inhibitor; inebilizumab, an antibody against CD19+ B cells; and eculizumab/ravulizumab, an antibody that blocks the C5 component of complement, was approved for NMOSD therapies after clinical trials [109].
Regarding the pathophysiological mechanism, AQP4 specific B cells (CD19intCD27highCD38highCD180- B cells) and plasmablasts are selectively expanded in peripheral blood and produce anti-AQP4 antibody following IL-6 stimulation [110,111]. Serum anti-AQP4 antibodies penetrate the BBB and bind to AQP4 in astrocyte endfeet [112]. Anti-AQP4 antibodies bind to orthogonal arrays of particles (OAPs), which are formed by the aggregation of M-23 AQP4 isoforms [113]. Binding to AQP4 autoantibodies results in AQP loss in astrocytes and deposits of IgG and IgM and complements the rosette pattern around the BBB with cellular infiltrate of neutrophils, eosinophils, macrophages/microglia and T cells [114,115]. Binding of anti-AQP4 antibodies to AQP4 activates complement through C1q with anti-AQP4 antibodies, leading to astrocyte death by classical complement cascade activation and membrane attack complex (MAC) formation [114]. C3a and C5a increase vascular permeability and neutrophil [109]. Interactions between pathogenic T cells and B cells in the presence of IL-6, IL-23 and TGF-β differentiate into Th17 cells, which secrete IL-17, promote endothelial activation and increase transendothelial migration of neutrophils and eosinophils [105,116,117]. Regarding the pathological findings of NMOSD, loss of astrocytes, neuronal injury, demyelination, microglial activation and macrophage infiltration were prominent [115].
3.2. Fluid Biomarkers for BBB Disturbance in NMOSD
An increase in Qalb, indicating increased albumin leakage in the CSF, was clinically observed in the acute phase of NMO [118,119]. Well-established fluid biomarkers for predicting the prognosis or treatment response in NMOSD are still insufficient. B cells, neutrophils, and eosinophils infiltrate the CNS across the BBB and contribute to the development of NMOSD lesion [109,120]. Increased levels of B cell activating factor (BAFF), proliferation-inducing ligand (APRIL), IL-6, and CXCL-13, which play a critical role in the survival and homeostasis of B cells, were observed in the CSF of patients with NMOSD, probably playing an important role in AQP4-antibody producing cell recruitment and maintenance [121,122]. The number of neutrophils in the CSF is elevated in approximately 60% of acute and untreated NMOSD patients, and neutrophil chemo-attractants CXCL5 and CXCL8 and neutrophil protease are elevated in the sera of NMOSD patients [123,124]. In addition, an increase in eotaxin-2, eotaxin-3 and eosinophil cationic protein (ECP), which contribute to the recruitment and activation of eosinophils, was observed in the CSF of patients with NMOSD compared to patients with multiple sclerosis and healthy controls, and apparent infiltration of eosinophils around the perivascular and meningeal space was observed in the active NMOSD lesion, suggesting the contribution of eosinophils to the pathogenesis of NMOSD [109,122].
IL-6, produced by astrocytes in NMOSD, is associated with increased BBB permeability [125,126]. IL-6 levels in CSF and serum in NMOSD patients are higher than those in MS patients and healthy controls and are correlated with EDSS and CSF cell counts [127,128]. The concentration of IL-6 in the serum and CSF is elevated in NMOSD during relapse compared with that in remission, and higher serum concentrations of baseline IL-6 levels in remission are correlated with a higher relapse risk [127,128]. Increased levels of IL-6 in the CSF are linked to short relapse-free durations after relapse [126]. GFAP levels are transiently increased in the CSF and serum during NMOSD attacks and correlate with disability in NMOSD [105,129]. Importantly, serum GFAP levels in AQP4-Ab+ NMOSD during remission may be predictive of future attack risk in NMOSD [130]. Some studies suggest that serum NfL may correlate with disability worsening during and after an attack in NMOSD and may serve as an indicator of treatment response in NMOSD [131]. Increased serum GFAP and NfL levels may reflect damage to astrocytes and neurons and the breakdown of the BBB.
3.3. Disruption of BBB in NMOSD
When the BBB is disrupted in NMOSD, massive amounts of AQP4 antibodies enter the CNS. Gd-enhanced lesions on MRI and/or increased Qalb were clinically observed in NMOSD during the acute stage [132]. More anti-AQP4 antibodies are produced from serum plasmablasts than from CSF plasmablasts in NMOSD [133,134]. The anti-AQP4 antibodies cannot induce disease development without pre-existing T cell-mediated CNS inflammation and BBB disruption, as the injection of anti-AQP4 antibodies alone from the periphery is not sufficient to mediate NMO-like histopathology [135,136]. Relatively high titers of anti-AQP4 antibodies are observed in sera from many patients with NMOSD, even during remission, suggesting the importance of BBB disruption in inducing CNS lesions [137,138]. However, the mechanism by which anti-AQP4 antibodies in the sera can bind to AQP4 on astrocyte endfeet behind the BBB in NMOSD has long been unclear [2].
As endothelial cells have weak tight junctions, and the expression of AQP4 is enriched in circumventricular organs (CVOs), including the area postrema, CVOs may be a viable routes for the entry of anti-AQP4 antibodies into the CNS [139]. MRI observations show that NMO lesions are often observed in the hypothalamus, periaqueductal brainstem and area postrema surrounding the CVO [140,141]. After AQP4-IgG enters the CSF space through the CVO, it can affect astrocytes via intrathecal inflammation and induce direct damage to the BBB through astrocyte dysfunction [139]. Another possible route is through direct penetration of the BBB. However, serum anti-AQP4 antibodies cannot affect BBB-endothelial cells because they do not express AQP4 protein [2]. We hypothesized the presence of other specific autoantibodies against BBB-endothelial cells in NMOSD sera that mediate increased penetration of anti-AQP4 antibodies across the BBB [142,143,144,145]. Our data showed that sera from patients with NMOSD during the acute phase decreased the barrier function and claudin-5 protein through VEGF and MMP-2/9 secreted from BBB-endothelial cells in an autocrine manner [142,143,144].
The following results were obtained from our study [145]: (1) IgG from 50 patients with AQP4 antibody-positive NMOSD (NMOSD-IgG) and two monoclonal “not AQP4-specific” antibodies from CSF plasmablasts from NMOSD patients bound to and activated BBB-endothelial cells through the NF-κB signal and increased permeability via the decrease of claudin-5 in vitro; (2) glucose-regulated protein 78 (GRP 78) was identified as the antigen of these 2 monoclonal antibodies; (3) the reduction of GRP78-specific antibodies from NMOSD-IgG decrease the effect on the activation of BBB-endothelial cells; and (4) peripheral injection of GRP78-speficic NMO monoclonal antibody induced increased permeability of the BBB in vivo. Our series of studies demonstrated that GRP78 autoantibodies can directly mediate the increased permeability of BBB endothelial cells, thereby causing the paracellular entry of anti-AQP4 antibodies across the BBB endothelial cells [145] (Figure 3). Our study showed that the positivity rate of anti-GRP78 antibodies differed from the NMOSD phenotype (LETM 71% vs. ON 17%), and positivity of anti-GRP78 antibodies in NMOSD was associated with the LETM phenotype and EDSS severity in each patient [146]. GRP78 autoantibodies have been detected in sera of patients with RA, and these antibodies are produced in response to abundant GRP78 in the synovial fluids of patients with RA [147].
Another important molecule that plays a critical role in BBB breakdown is IL-6. Some in vitro studies have demonstrated that NMOSD-IgG mediates IL-6 release in astrocytes via JAK/STAT or NF-κB signaling [148,149]. Our studies showed that AQP4 Ab-NMOSD IgG-mediated IL-6 production by astrocytes with AQP4 expression and IL-6 signaling to BBB-endothelial cells increases barrier permeability, upregulates the expression of chemokines (CCL2 and CXCL8) and reinforces the transmigration of leukocytes under flow according to in vitro static and flow-based BBB models, including co-culture of human brain microvascular endothelial cells (TY10) and human astrocyte cell lines with or without AQP4 expression [150]. Furthermore, satralizumab, an IL-6R-neutralizing antibody, reversed the increased BBB permeability and infiltration of lymphocytes [151]. Our series of studies showed that secretion of IL-6 from astrocytes on the CNS side after binding of AQP4 antibodies to AQP4 on astrocytes increased permeability of the BBB and enhanced infiltration of inflammatory cells through upregulation of chemokines (CCL2 and CXCL8) from endothelial cells by IL-6 signaling [151].
4. Pathophysiological Mechanism and BBB Breakdown in MOGAD
MOGAD is a recently recognized new entity in the spectrum of CNS inflammatory demyelinating diseases, which differs from both MS and NMOSD [152,153]. The international diagnostic criteria for MOGAD are based on the presence of anti-MOG autoantibodies (MOG-Abs) detected using cell-based assays [152]. The clinical phenotype of MOGAD is broad and includes optic neuritis, transverse myelitis, cerebral cortical encephalitis, brainstem or cerebellar symptoms and acute disseminated encephalomyelitis (ADEM) [152,153]. MOG is a transmembrane protein on the outer surface of the CNS that is expressed in oligodendrocytes [154,155]. Histopathological findings of MOGAD show a distinct pattern of confluent demyelination around small vessels in white matter and deep gray matter structures with abundant myelin-laden macrophages/microglial cells [156,157,158]. The dominant infiltrating lymphocytes are CD4+ T cells, with few CD8+ T cells and B cells [159,160,161]. A study found that early-phase demyelinating lesions of MOGAD showed MOG-dominant myelin loss with relatively preserved oligodendrocytes [159,160,161].
CSF pleocytosis in MOGAD is common during relapses in the spinal cord (85%) and brain/brainstem (60%) [162]. Q Alb is increased in almost one-third of patients with MOGAD [163,164]. The cytokines/chemokines in the CSF showed an increase in proinflammatory cytokines/chemokines, including Th1 (TNF-α, IFN-γ), Th2 (IL-13), Th17 (IL-6, IL8, G-CSF, GM-CSF) and B cells (CXCL12, BAFF, APRIL, CXCL13, and CCL19), in MOGAD patients [165,166].
MOG-specific T cells are activated peripherally [167]. Infections, molecular mimicry and MOG peptide presentation can promote the activation of self-reactive T-cells [168]. The pathogenicity of MOG-IgG purified from patients with MOGAD was observed based on the finding that human MOG-IgG was injected intrathecally in an adoptive transfer EAE model induced by MBP or MOG-specific T cells transferred to Lewis rats [169]. In this study, human MOG-IgG was pathogenic in two different EAE models, suggesting that MOG-Abs have a pathogenic effect coupled with MOG-specific or encephalitogenic T cells when they enter the CNS [169]. MOG-Abs cannot bind to and react with the BBB, as MOG is not expressed in BMECs. MOG-Abs are produced mostly peripherally, and these Abs can penetrate across the impaired BBB (induced by activated T cells, infection, co-existing autoantibodies) [170]. Our data demonstrated that MOG-IgG-purified MOGAD patients had their BBB-endothelial cells activated during the acute phase, resulting in the induction of NF-κB signaling, increased VCAM-1/ICAM-1, increased permeability and decreased Nrf2 [170]. The positivity rate of GRP78 autoantibodies in acute MOGAD was 66%, and removal of GRP78 antibody from MOG-IgG reduced the effect on NF-κB activation, indicating that co-existing anti-GRP78 antibodies with MOG-Ab can facilitate BBB transit of pathogenic MOG-Abs in MOGAD [170].
5. NPSLE and AE
5.1. NPSLE and the Blood-Brain Barrier
About 40%-75% of patients with SLE experience neuropsychiatric symptoms, termed NPSLE [171,172]. The symptoms of NPSLE vary from mild symptoms, such as headache, mood disorder, and cognitive decline, to severe symptoms, such as seizures, cerebrovascular events, an acute confusional state and psychosis [171,174]. The diagnosis of NPSLE is difficult because of the lack of accurate and reliable biomarkers. Several pathological mechanisms, including neuroinflammation and neuronal damage induced by autoantibodies and proinflammatory cytokines (TNF-α, IL-1, IL-8, and IL-17), vascular occlusion and BBB breakdown, have been implicated in NPSLE [172,175]. Elevated CSF Qalb and S100B levels in the CSF have been observed in NPSLE, indicating the involvement of dysfunction of the BBB in the development of the disease [176,177].
Anti-dsDNA, anti-phospholipid (aPL), anti-ribosomal P protein (anti-P), and NMDA receptor antibodies have been associated with NPSLE manifestations [172,173]. Anti-aPL antibodies can induce vascular endothelial cell injury, platelet activation and thrombosis, resulting in focal ischemia and intracranial vascular embolism [178,179]. A subset of anti-dsDNA antibodies (anti-dsDNA/NMDAR-NR2 antibodies) can cross-react with NMDAR-NR2 expressed in neurons [172,180,181]. Anti-dsDNA/NMDAR-NR2 antibodies can damage endothelial cells by secreting inflammatory cytokines from endothelial cells [182,183]. After penetrating the BBB, this antibody was shown to be able to induce neuronal apoptosis and degeneration of surviving neurons in an in vivo model and was associated with behavioral and psychiatric manifestations in NPSLE [184,185,186].
Anti-endothelial cell antibodies against unknown antigens located on the surface of endothelial cells are common in 65% of NPSLE cases and can contribute to the direct cytotoxic effect induced by complement- or antibody-dependent cytotoxicity and mediate coagulation of endothelial cells [187,188,189,190]. In addition, these anti-endothelial cell antibodies from NPSLE patients increased ICAM-1, VCAM-1 and E-selectin expression, as well as the secretion of cytokines, such as IL-1b, Il-8 and MCP-1 [145,188,189]. Anti-GRP78 autoantibodies have been detected in NPSLE, and titers of these antibodies are higher in diffuse NPSLE with acute confusion than in focal NPSLE [191].
5.2. AE and BBB
AE is associated with autoantibodies against synaptic receptors, neuronal cell surface proteins and neuronal intracytoplasmic antigens, including NMDAR encephalitis leucine-rich glioma-inactivated 1 (LGI-1), γ-aminobutyric acid type B receptor (GABAbR) and contactin-associated protein-2 (CASPR2) antibodies [192,193,194]. Anti-NMDAR encephalitis was reported to be the most common AE (81%) [195,196]. Young females are often affected, and some develop ovarian teratomas [196]. Ectopic neural tissue in ovarian teratomas as a source of autoantigen is thought to trigger the production of NMDAR autoantibodies in the sera [196]. The symptoms of the disease start with mood changes and psychosis, followed by consciousness disturbance, seizures, respiratory failure, bizarre involuntary movements and autonomic disturbances [195]. Anti-NMDAR antibodies are detected in the serum and CSF of patients with NMDAR encephalitis, and anti-NMDAR antibody titers are associated with the severity of disease symptoms, outcomes and prognosis [195,196]. Anti-NMDAR antibodies can bind to the NMDAR-NR1 subunit and induce selective internalization of NMDARs, resulting in a decreased glutamate synaptic function [193,197,198,199,200]. Brain-biopsied or autopsied cases of NMDAR encephalitis showed mild perivascular lymphocytic cuffing, microglial activation and a decrease in NMDAR expression in the hippocampus [201,202].
An increase in Q-Alb was observed in anti-NMDAR encephalitis, indicating BBB damage [203], which was shown to be associated with the prognosis and mRS score after two months of follow-up; these findings suggest that BBB damage reflects disease severity [203]. Anti-NMDAR antibodies in the sera may penetrate the damaged BBB and enter the CNS, leading to clinical symptoms. NMDAR is expressed on BBB-endothelial cells, and activation of NMDAR can affect paracellular permeability of the BBB with the altered expression of tight junctions by activation of the PI3K/Akt signaling pathway [204,205]. What mechanism at the molecular level is involved in anti-NMDAR encephalitis remains unclear, as does whether or not BBB dysfunction occurs in other types of AE with anti-LGI-1, anti-CASPR2 or anti-GABAbR autoantibodies. Further analyses are needed to understand the molecular mechanisms responsible for BBB breakdown in AE.
Autoimmune cerebellar ataxia is an emerging disease that affects the cerebellum via autoimmune mechanisms [206,207]. The disease has several etiologies, including gluten ataxia, anti-glutamate decarboxylase (GAD) ataxia, paraneoplastic cerebellar degeneration (PCD), primary autoimmune cerebellar ataxia and postinfectious cerebellar ataxia [206,207]. Breakdown of the BBB could potentially explain the vulnerability of the cerebellum to autoimmune cerebellar ataxia, as it triggers the entry of pathogenic autoantibodies or lymphocytes induced by the autoimmune response in the periphery into the cerebellum.
Whether or not BBB permeability is increased in autoimmune cerebellar ataxia remains unclear.
6. Paraneoplastic Neurological Syndromes (PNSs)
6.1. PNSs and the BBB
PNSs are characterized by acute or subacute neurological manifestations and are mediated by the remote effects of cancer, with an immune-mediated pathogenesis that is not caused by cancer or metastasis [208,209]. Recent diagnostic criteria have demonstrated that high-risk phenotypes of PNS include encephalomyelitis, limbic encephalitis, rapidly progressive cerebellar syndrome, opsoclonus-myoclonus, sensory neuronopathy, gastrointestinal pseudo-obstruction (enteric neuropathy) and Lambert-Eaton myasthenic syndrome (LEMS) [210]. Anti-onconeural antibodies directed to intracellular antigens (Hu, Yo, Ri, MA1/2, CRMP5), intracellular synaptic antigens (GAD65, amphyiphysin) and extracellular/cell membrane antigens (NMDAR, AMPAR, LGI1, CASPR2, GABABR, mGluR1, GlyR, VGCC, mGluR5) have been identified as being associated with PNSs and are thus used for their diagnosis [210,211,212].
Paraneoplastic cerebellar degeneration (PCD) is one of the most common forms of PNSs [208,213]. Approximately half of PCD cases are related to anti-Yo antibodies and other autoantibodies, including anti-Hu, anti-Ri, anti-Tr, anti-Ma2, anti-P/Q-type VGCC and anti-CV2/CRMP5 antibodies [208,214,215]. Dysfunction of the BBB or blood-nerve barrier (BNB) may be responsible for the onset and progression of PNSs, although the precise molecular mechanism underlying the BBB breakdown in PNSs remains elusive.
6.2. PCD-LEMS
LEMS is an autoimmune disease of the neuromuscular junction, characterized by proximal muscle weakness, areflexia and autonomic dysfunction and associated with P/Q type VGCC autoantibodies and small-cell lung carcinoma [212,216,217]. P/Q-type VGCCs are localized at presynaptic motor nerve terminals and play a role in neurotransmitter release [216,217]. Cerebellar symptoms are observed in 10% of LEMS patients diagnosed with PCD with LEMS (PCD-LEMS) [212]. In autopsied brains of PCD-LEMS patients, selective reduction of P/Q-type VGCCs was observed in the molecular layer of the cerebellum. Anti-P/Q-type antibodies can enter the CNS in cases of BBB dysfunction in PCD-LEMS [218].
We recently identified anti-GRP78 antibodies in patients with PCD-LEMS and NMOSD [219]. GRP78 (heat shock protein family A [Hsp70] member 5 HSPA5) plays a role in preventing unfolded protein accumulation and apoptosis as an endoplasmic reticulum (ER) chaperone in all CNS cells [220]. The cell surface GRP78 is abundant in malignant cells and BBB-endothelial cells in vivo and in vitro, leading to the activation of NF-κB signal transduction, which supports the notion that cell-surface GRP78 may be a target for cancer-specific therapy [221,222,223]. GRP78 autoantibodies have been detected in sera from patients with malignant tumors, suggesting that GRP78 autoantibodies may be produced in response to cell-surface overexpression of GRP78 in patients with malignant tumors [224,225,226]. In PCD-LEMS, GRP78 antibodies induced by cross-reactivity with small-cell carcinoma can induce BBB dysfunction and facilitate the penetration of anti-P/Q-type VGCC antibodies into the cerebellum, resulting in cerebellar ataxia [219] (Figure 4).
6.3. Paraneoplastic NMOSD
In addition, cases of paraneoplastic NMOSD have been reported increasingly frequently, and some case reports of patients with paraneoplastic NMOSD have shown the expression of AQP4 in the tumor cells, suggesting that AQP4 autoantibodies may be produced in the autoimmune response to AQP4 protein in the tumor cells of some paraneoplastic NMOSD patients [227,228,229]. Our report describes a case of paraneoplastic NMOSD presenting with LETM with colorectal cancer that was positive for both GRP78 antibodies and AQP4 antibodies [230]. In this case, the tumor cells showed a high expression of GRP78, possibly upregulating the production of GRP78 antibodies because of an autoimmune response mediated by the tumor.
7. Conclusions and Future Direction
In this review, we summarize the pathogenic contribution of the BBB in several neuroimmunological diseases, such as MS, NMOSD, MOGAD, AE and PNSs. The major molecular mechanisms responsible for BBB dysfunction differ among diseases; thus, a detailed understanding of the pathomechanism involved needs to be explored in greater depth to stimulate drug development for the BBB and improve treatment options. Common genetic and environmental factors, including dietary habits, Vitamin D, the gut microbiome, smoking and EBV infection, may contribute to BBB dysfunction in several neuroimmunological diseases, including MS. Although at present several DMTs, such as natalizumab for MS and satralizumab for NMOSD, can modify the BBB function and contribute to relapse prevention, prospective therapeutic approaches, such as monoclonal antibodies targeting another CAM, cytokines or chemokines, may pave the way for new treatments to prevent BBB injury in several neuroimmunological diseases.
Furthermore, investigation of how the BBB is repaired after BBB disruption and how novel medicines against CNS targets penetrate the BBB may lead to the discovery of novel molecular-targeted drugs against the BBB for several neurological diseases. In particular, as dysfunction of the NVU is associated with gray matter atrophy in MS, several platforms, such as organ-on-a-chip models, have been established in the field of the BBB to understand the detailed pathomechanism of neurological disease, drug discovery research, and screening, leading to further novel therapeutic approaches in several neuroimmunological diseases. Thus, the BBB may become a therapeutic target in several neuroimmunological diseases, not only to protect and repair the BBB when damaged but also to reach neuroprotective molecules inside the CNS.
Author Contributions
Fumitaka Shimizu reviewed the reference and wrote the manuscript. Masayuki Nakamori edited the manuscript. Fumitaka Shimizu and Masayuki Nakamori were responsible for planning conception and design of this review.
Funding
Funding had no influence on the design of this research. Grants-in-Aid for Scientific Research (Kakenhi from the Japan Society for the Promotion of Science, Tokyo, Japan, Nos. 24K10621) and Chugai Foundation for Innovative Drug Discovery Science and BRAIN SCIENCE FOUNDATION supported this research.
Provenance and Peer Review
Not commissioned; externally peer reviewed.
Conflicts of Interest
None.
References
- Schreiner, T.G.; Romanescu, C.; Popescu, B.O. The Blood–Brain Barrier—A Key Player in Multiple Sclerosis Disease Mechanisms. Biomolecules 2022, 12, 538. [Google Scholar] [CrossRef]
- Shimizu, F.; Nishihara, H.; Kanda, T. Blood–brain barrier dysfunction in immuno-mediated neurological diseases. Immunol. Med. 2018, 41, 120–128. [Google Scholar] [CrossRef]
- Bell, A.H.; Miller, S.L.; Castillo-Melendez, M.; Malhotra, A. The Neurovascular Unit: Effects of Brain Insults During the Perinatal Period. Front. Neurosci. 2020, 13, 1452. [Google Scholar] [CrossRef] [PubMed]
- Brandl, S.; Reindl, M. Blood–Brain Barrier Breakdown in Neuroinflammation: Current In Vitro Models. Int. J. Mol. Sci. 2023, 24, 12699. [Google Scholar] [CrossRef] [PubMed]
- Abbott, N.J.; Patabendige, A.A.K.; Dolman, D.E.M.; Yusof, S.R.; Begley, D.J. Structure and function of the blood-brain barrier. Neurobiol. Dis. 2010, 37, 13–25. [Google Scholar] [CrossRef]
- Chow, B.W.; Gu, C. The Molecular Constituents of the Blood–Brain Barrier. Trends Neurosci. 2015, 38, 598–608. [Google Scholar] [CrossRef]
- Sweeney, M.D.; Zhao, Z.; Montagne, A.; Nelson, A.R.; Zlokovic, B.V. Blood-Brain Barrier: From Physiology to Disease and Back. Physiol. Rev. 2019, 99, 21–78. [Google Scholar] [CrossRef] [PubMed]
- Zlokovic, B.V. The Blood-Brain Barrier in Health and Chronic Neurodegenerative Disorders. Neuron 2008, 57, 178–201. [Google Scholar] [CrossRef] [PubMed]
- Obermeier, B.; Daneman, R.; Ransohoff, R.M. Development, maintenance and disruption of the blood-brain barrier. Nat. Med. 2013, 19, 1584–1596. [Google Scholar] [CrossRef]
- Brimberg, L.; Mader, S.; Fujieda, Y.; Arinuma, Y.; Kowal, C.; Volpe, B.T.; Diamond, B. Antibodies as Mediators of Brain Pathology. Trends Immunol. 2015, 36, 709–724. [Google Scholar] [CrossRef]
- Chataway, J.; Williams, T.; Li, V.; Marrie, R.A.; Ontaneda, D.; Fox, R.J. Clinical trials for progressive multiple sclerosis: Progress, new lessons learned, and remaining challenges. Lancet Neurol. 2024, 23, 277–301. [Google Scholar] [CrossRef] [PubMed]
- Ortiz, G.G.; Pacheco-Moisés, F.P.; Macías-Islas, M.Á.; Flores-Alvarado, L.J.; Mireles-Ramírez, M.A.; González-Renovato, E.D.; Hernández-Navarro, V.E.; Sánchez-López, A.L.; Alatorre-Jiménez, M.A. Role of the Blood–Brain Barrier in Multiple Sclerosis. Arch. Med. Res. 2014, 45, 687–697. [Google Scholar] [CrossRef]
- Noseworthy, J.H.; Lucchinetti, C.; Rodriguez, M.; Weinshenker, B.G. Multiple sclerosis. N. Engl. J. Med. 2000, 343, 938–952. [Google Scholar] [CrossRef]
- Lublin, F.D.; Reingold, S.C. Defining the clinical course of multiple sclerosis: Results of an international survey. National Multiple Sclerosis Society (USA) Advisory Committee on Clinical Trials of New Agents in Multiple Sclerosis. Neurology 1996, 46, 907–911. [Google Scholar] [CrossRef] [PubMed]
- Mahad, D.H.; Trapp, B.D.; Lassmann, H. Pathological mechanisms in progressive multiple sclerosis. Lancet Neurol. 2015, 14, 183–193. [Google Scholar] [CrossRef]
- Ontaneda, D.; Fox, R.J.; Chataway, J. Clinical trials in progressive multiple sclerosis: Lessons learned and future perspectives. Lancet Neurol. 2015, 14, 208–223. [Google Scholar] [CrossRef]
- Dutta, R.; Trapp, B.D. Relapsing and progressive forms of multiple sclerosis: Insights from pathology. Curr. Opin. Neurol. 2014, 27, 271–278. [Google Scholar] [CrossRef]
- Lassmann, H. Pathogenic Mechanisms Associated With Different Clinical Courses of Multiple Sclerosis. Front. Immunol. 2019, 9, 3116. [Google Scholar] [CrossRef]
- Leray, E.; Yaouanq, J.; Le Page, E.; Coustans, M.; Laplaud, D.; Oger, J.; Edan, G. Evidence for a two-stage disability progression in multiple sclerosis. Brain 2010, 133, 1900–1913. [Google Scholar] [CrossRef]
- Alvarez, J.I.; Cayrol, R.; Prat, A. Disruption of central nervous system barriers in multiple sclerosis. Biochim. et Biophys. Acta (BBA) - Mol. Basis Dis. 2010, 1812, 252–264. [Google Scholar] [CrossRef] [PubMed]
- Larochelle, C.; Alvarez, J.I.; Prat, A. How do immune cells overcome the blood-brain barrier in multiple sclerosis? FEBS Lett. 2011, 585, 3770–3780. [Google Scholar] [CrossRef] [PubMed]
- Kirk, J.; Plumb, J.; Mirakhur, M.; McQuaid, S. Tight junctional abnormality in multiple sclerosis white matter affects all calibres of vessel and is associated with blood–brain barrier leakage and active demyelination. J. Pathol. 2003, 201, 319–327. [Google Scholar] [CrossRef] [PubMed]
- Plumb, J.; McQuaid, S.; Mirakhur, M.; Kirk, J. Abnormal Endothelial Tight Junctions in Active Lesions and Normal-appearing White Matter in Multiple Sclerosis. Brain Pathol. 2002, 12, 154–169. [Google Scholar] [CrossRef]
- Waubant, E. Biomarkers Indicative of Blood-Brain Barrier Disruption in Multiple Sclerosis. Dis. Markers 2006, 22, 235–244. [Google Scholar] [CrossRef]
- Leech, S.; Kirk, J.; Plumb, J.; McQuaid, S. Persistent endothelial abnormalities and blood–brain barrier leak in primary and secondary progressive multiple sclerosis. Neuropathol. Appl. Neurobiol. 2007, 33, 86–98. [Google Scholar] [CrossRef]
- Zierfuss, B.; Larochelle, C.; Prat, A. Blood-brain barrier dysfunction in multiple sclerosis: Causes, consequences, and potential effects of therapies. Lancet Neurol. 2024, 23, 95–109. [Google Scholar] [CrossRef]
- Balasa, R.; Barcutean, L.; Mosora, O.; Manu, D. Reviewing the Significance of Blood–Brain Barrier Disruption in Multiple Sclerosis Pathology and Treatment. Int. J. Mol. Sci. 2021, 22, 8370. [Google Scholar] [CrossRef] [PubMed]
- Ifergan, I.; Kebir, H.; Alvarez, J.I.; Marceau, G.; Bernard, M.; Bourbonnière, L.; Poirier, J.; Duquette, P.; Talbot, P.J.; Arbour, N.; Prat, A. Central nervous system recruitment of effector memory CD8+ T lymphocytes during neuroinflammation is dependent on α4 integrin. Brain 2011, 134, 3560–3577. [Google Scholar] [CrossRef]
- Marchetti, L.; Engelhardt, B. Immune cell trafficking across the blood-brain barrier in the absence and presence of neuroinflammation. Vasc. Biol. 2020, 2, H1–H18. [Google Scholar] [CrossRef]
- Mitroulis, I.; Alexaki, V.I.; Kourtzelis, I.; Ziogas, A.; Hajishengallis, G.; Chavakis, T. Leukocyte integrins: Role in leukocyte recruitment and as therapeutic targets in inflammatory disease. Pharmacol. Ther. 2015, 147, 123–135. [Google Scholar] [CrossRef]
- Holman, D.W.; Klein, R.S.; Ransohoff, R.M. The blood–brain barrier, chemokines and multiple sclerosis. Biochim. et Biophys. Acta (BBA) - Mol. Basis Dis. 2010, 1812, 220–230. [Google Scholar] [CrossRef]
- Alvarez, J.I.; Saint-Laurent, O.; Godschalk, A.; Terouz, S.; Briels, C.; Larouche, S.; Bourbonnière, L.; Larochelle, C.; Prat, A. Focal disturbances in the blood–brain barrier are associated with formation of neuroinflammatory lesions. Neurobiol. Dis. 2015, 74, 14–24. [Google Scholar] [CrossRef]
- Miller, D.H.; Soon, D.; Fernando, K.T.; MacManus, D.G.; Barker, G.J.; Yousry, T.A.; Fisher, E.; O’Connor, P.W.; Phillips, J.T.; Polman, C.H.; et al. MRI outcomes in a placebo-controlled trial of natalizumab in relapsing MS. Neurology 2007, 68, 1390–1401. [Google Scholar] [CrossRef]
- Charabati, M.; Zandee, S.; Fournier, A.P.; Tastet, O.; Thai, K.; Zaminpeyma, R.; Lécuyer, M.-A.; Bourbonnière, L.; Larouche, S.; Klement, W.; et al. MCAM+ brain endothelial cells contribute to neuroinflammation by recruiting pathogenic CD4+ T lymphocytes. Brain 2022, 146, 1483–1495. [Google Scholar] [CrossRef]
- Michel, L.; Grasmuck, C.; Charabati, M.; Lécuyer, M.-A.; Zandee, S.; Dhaeze, T.; Alvarez, J.I.; Li, R.; Larouche, S.; Bourbonnière, L.; et al. Activated leukocyte cell adhesion molecule regulates B lymphocyte migration across central nervous system barriers. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef] [PubMed]
- Wimmer, I.; Tietz, S.; Nishihara, H.; Deutsch, U.; Sallusto, F.; Gosselet, F.; Lyck, R.; Muller, W.A.; Lassmann, H.; Engelhardt, B. PECAM-1 Stabilizes Blood-Brain Barrier Integrity and Favors Paracellular T-Cell Diapedesis Across the Blood-Brain Barrier During Neuroinflammation. Front. Immunol. 2019, 10, 711. [Google Scholar] [CrossRef] [PubMed]
- Charabati, M.; Grasmuck, C.; Ghannam, S.; Bourbonnière, L.; Fournier, A.P.; Lécuyer, M.A.; Tastet, O.; Kebir, H.; Rébillard, R.M.; Hoornaert, C.; et al. DICAM promotes TH17 lymphocyte trafficking across the blood-brain barrier during autoimmune neuroinflammation. Sci. Transl. Med. 2022, 14, eabj0473. [Google Scholar] [CrossRef]
- Benedict, R.H.B.; Amato, M.P.; DeLuca, J.; Geurts, J.J.G. Cognitive impairment in multiple sclerosis: Clinical management, MRI, and therapeutic avenues. Lancet Neurol. 2020, 19, 860–871. [Google Scholar] [CrossRef]
- Nishihara, H.; Shimizu, F.; Kitagawa, T.; Yamanaka, N.; Akada, J.; Kuramitsu, Y.; Sano, Y.; Takeshita, Y.; Maeda, T.; Abe, M.; et al. Identification of galectin-3 as a possible antibody target for secondary progressive multiple sclerosis. Mult. Scler. J. 2016, 23, 382–394. [Google Scholar] [CrossRef]
- Dumic, J.; Dabelic, S.; Flögel, M. Galectin-3: An open-ended story. Biochim. et Biophys. Acta (BBA) - Gen. Subj. 2006, 1760, 616–635. [Google Scholar] [CrossRef] [PubMed]
- Radosavljevic, G.; Volarevic, V.; Jovanovic, I.; Milovanovic, M.; Pejnovic, N.; Arsenijevic, N.; Hsu, D.K.; Lukic, M.L. The roles of Galectin-3 in autoimmunity and tumor progression. Immunol. Res. 2012, 52, 100–110. [Google Scholar] [CrossRef]
- Cristofanilli, M.; Rosenthal, H.; Cymring, B.; Gratch, D.; Pagano, B.; Xie, B.; Sadiq, S.A. Progressive multiple sclerosis cerebrospinal fluid induces inflammatory demyelination, axonal loss, and astrogliosis in mice. Exp. Neurol. 2014, 261, 620–632. [Google Scholar] [CrossRef] [PubMed]
- Granziera, C.; Wuerfel, J.; Barkhof, F.; Calabrese, M.; De Stefano, N.; Enzinger, C.; Evangelou, N.; Filippi, M.; Geurts, J.J.G.; Reich, D.S.; et al. Quantitative magnetic resonance imaging towards clinical application in multiple sclerosis. Brain 2021, 144, 1296–1311. [Google Scholar] [CrossRef] [PubMed]
- Sivakolundu, D.K.; West, K.L.; Maruthy, G.B.; Zuppichini, M.; Turner, M.P.; Abdelkarim, D.; Zhao, Y.; Nguyen, D.; Spence, J.S.; Lu, H.; et al. Reduced arterial compliance along the cerebrovascular tree predicts cognitive slowing in multiple sclerosis: Evidence for a neurovascular uncoupling hypothesis. Mult. Scler. J. 2019, 26, 1486–1496. [Google Scholar] [CrossRef]
- Spencer, J.I.; Bell, J.S.; DeLuca, G.C. Vascular pathology in multiple sclerosis: Reframing pathogenesis around the blood-brain barrier. J. Neurol. Neurosurg. Psychiatry 2018, 89, 42–52. [Google Scholar] [CrossRef] [PubMed]
- Barro, C.; Benkert, P.; Disanto, G.; Tsagkas, C.; Amann, M.; Naegelin, Y.; Leppert, D.; Gobbi, C.; Granziera, C.; Yaldizli. ; et al. Serum neurofilament as a predictor of disease worsening and brain and spinal cord atrophy in multiple sclerosis. Brain 2018, 141, 2382–2391. [Google Scholar] [CrossRef]
- Uher, T.; McComb, M.; Galkin, S.; Srpova, B.; Oechtering, J.; Barro, C.; Tyblova, M.; Bergsland, N.; Krasensky, J.; Dwyer, M.; et al. Neurofilament levels are associated with blood–brain barrier integrity, lymphocyte extravasation, and risk factors following the first demyelinating event in multiple sclerosis. Mult. Scler. J. 2020, 27, 220–231. [Google Scholar] [CrossRef]
- Uher, T.; McComb, M.; Galkin, S.; Srpova, B.; Oechtering, J.; Barro, C.; Tyblova, M.; Bergsland, N.; Krasensky, J.; Dwyer, M.; et al. Neurofilament levels are associated with blood–brain barrier integrity, lymphocyte extravasation, and risk factors following the first demyelinating event in multiple sclerosis. Mult. Scler. J. 2020, 27, 220–231. [Google Scholar] [CrossRef]
- Sejbaek, T.; Nielsen, H.H.; Penner, N.; Plavina, T.; Mendoza, J.P.; Martin, N.A.; Elkjaer, M.L.; Ravnborg, M.H.; Illes, Z. Dimethyl fumarate decreases neurofilament light chain in CSF and blood of treatment naïve relapsing MS patients. J. Neurol. Neurosurg. Psychiatry 2019, 90, 1324–1330. [Google Scholar] [CrossRef]
- Hyun, J.-W.; Kim, Y.; Kim, G.; Kim, S.-H.; Kim, H.J. Longitudinal analysis of serum neurofilament light chain: A potential therapeutic monitoring biomarker for multiple sclerosis. Mult. Scler. J. 2020, 26, 659–667. [Google Scholar] [CrossRef] [PubMed]
- Abdelhak, A.; Antweiler, K.; Kowarik, M.C.; Senel, M.; Havla, J.; Zettl, U.K.; Kleiter, I.; Skripuletz, T.; Haarmann, A.; Stahmann, A.; et al. Serum glial fibrillary acidic protein and disability progression in progressive multiple sclerosis. Ann. Clin. Transl. Neurol. 2023, 11, 477–485. [Google Scholar] [CrossRef] [PubMed]
- Papa, L.; Ladde, J.G.; O’brien, J.F.; Thundiyil, J.G.; Tesar, J.; Leech, S.; Cassidy, D.D.; Roa, J.; Hunter, C.; Miller, S.; et al. Evaluation of Glial and Neuronal Blood Biomarkers Compared With Clinical Decision Rules in Assessing the Need for Computed Tomography in Patients With Mild Traumatic Brain Injury. JAMA Netw. Open 2022, 5, e221302–e221302. [Google Scholar] [CrossRef] [PubMed]
- Uher, T.; McComb, M.; Galkin, S.; Srpova, B.; Oechtering, J.; Barro, C.; Tyblova, M.; Bergsland, N.; Krasensky, J.; Dwyer, M.; et al. Neurofilament levels are associated with blood–brain barrier integrity, lymphocyte extravasation, and risk factors following the first demyelinating event in multiple sclerosis. Mult. Scler. J. 2020, 27, 220–231. [Google Scholar] [CrossRef] [PubMed]
- Brambilla, R. The contribution of astrocytes to the neuroinflammatory response in multiple sclerosis and experimental autoimmune encephalomyelitis. Acta Neuropathol. 2019, 137, 757–783. [Google Scholar] [CrossRef] [PubMed]
- Petersen, E.; Søndergaard, H.; Oturai, A.; Jensen, P.; Sorensen, P.; Sellebjerg, F.; Börnsen, L. Soluble serum VCAM-1, whole blood mRNA expression and treatment response in natalizumab-treated multiple sclerosis. Mult. Scler. Relat. Disord. 2016, 10, 66–72. [Google Scholar] [CrossRef]
- Wang, D.; Duan, H.; Feng, J.; Xiang, J.; Feng, L.; Liu, D.; Chen, X.; Jing, L.; Liu, Z.; Zhang, D.; et al. Soluble CD146, a cerebrospinal fluid marker for neuroinflammation, promotes blood-brain barrier dysfunction. Theranostics 2020, 10, 231–246. [Google Scholar] [CrossRef] [PubMed]
- Cotsapas, C.; Mitrovic, M. Genome-wide association studies of multiple sclerosis. Clin. Transl. Immunol. 2018, 7, e1018. [Google Scholar] [CrossRef] [PubMed]
- International Multiple Sclerosis Genetics Consortium. Multiple sclerosis genomic map implicates peripheral immune cells and microglia in susceptibility. Science 2019, 365, eaav7188. [Google Scholar] [CrossRef]
- Bäcker-Koduah, P.; Bellmann-Strobl, J.; Scheel, M.; Wuerfel, J.; Wernecke, K.-D.; Dörr, J.; Brandt, A.U.; Paul, F. Vitamin D and Disease Severity in Multiple Sclerosis—Baseline Data From the Randomized Controlled Trial (EVIDIMS). Front. Neurol. 2020, 11, 129. [Google Scholar] [CrossRef] [PubMed]
- Hupperts, R.; Smolders, J.; Vieth, R.; Holmøy, T.; Marhardt, K.; Schluep, M.; Killestein, J.; Barkhof, F.; Beelke, M.; Grimaldi, L.M.; et al. Randomized trial of daily high-dose vitamin D 3 in patients with RRMS receiving subcutaneous interferon β-1a. Neurology 2019, 93, e1906–e1916. [Google Scholar] [CrossRef]
- Sangha, A.; Quon, M.; Pfeffer, G.; Orton, S.-M. The Role of Vitamin D in Neuroprotection in Multiple Sclerosis: An Update. Nutrients 2023, 15, 2978. [Google Scholar] [CrossRef]
- Takahashi, S.; Maeda, T.; Sano, Y.; Nishihara, H.; Takeshita, Y.; Shimizu, F.; Kanda, T. Active form of vitamin D directly protects the blood–brain barrier in multiple sclerosis. Clin. Exp. Neuroimmunol. 2017, 8, 244–254. [Google Scholar] [CrossRef]
- Bjornevik, K.; Bjornevik, K.; Cortese, M.; Cortese, M.; Healy, B.C.; Healy, B.C.; Kuhle, J.; Kuhle, J.; Mina, M.J.; Mina, M.J.; et al. Longitudinal analysis reveals high prevalence of Epstein-Barr virus associated with multiple sclerosis. Science 2022, 375, 296–301. [Google Scholar] [CrossRef] [PubMed]
- Lanz, T.V.; Brewer, R.C.; Ho, P.P.; Moon, J.-S.; Jude, K.M.; Fernandez, D.; Fernandes, R.A.; Gomez, A.M.; Nadj, G.-S.; Bartley, C.M.; et al. Clonally expanded B cells in multiple sclerosis bind EBV EBNA1 and GlialCAM. Nature 2022, 603, 321–327. [Google Scholar] [CrossRef] [PubMed]
- Soldan, S.S.; Lieberman, P.M. Epstein–Barr virus and multiple sclerosis. Nat. Rev. Microbiol. 2022, 21, 51–64. [Google Scholar] [CrossRef] [PubMed]
- Tengvall, K.; Huang, J.; Hellström, C.; Kammer, P.; Biström, M.; Ayoglu, B.; Bomfim, I.L.; Stridh, P.; Butt, J.; Brenner, N.; et al. Molecular mimicry between Anoctamin 2 and Epstein-Barr virus nuclear antigen 1 associates with multiple sclerosis risk. Proc. Natl. Acad. Sci. 2019, 116, 16955–16960. [Google Scholar] [CrossRef] [PubMed]
- Casiraghi, C.; Dorovini-Zis, K.; Horwitz, M.S. Epstein-Barr virus infection of human brain microvessel endothelial cells: A novel role in multiple sclerosis. J. Neuroimmunol. 2011, 230, 173–177. [Google Scholar] [CrossRef] [PubMed]
- Degelman, M.L.; Herman, K.M. Smoking and multiple sclerosis: A systematic review and meta-analysis using the Bradford Hill criteria for causation. Mult. Scler. Relat. Disord. 2017, 17, 207–216. [Google Scholar] [CrossRef]
- Hawkins, B.T.; Abbruscato, T.J.; Egleton, R.D.; Brown, R.C.; Huber, J.D.; Campos, C.R.; Davis, T.P. Nicotine increases in vivo blood–brain barrier permeability and alters cerebral microvascular tight junction protein distribution. Brain Res. 2004, 1027, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Johansson, E.; Alfredsson, L.; Strid, P.; Kockum, I.; Olsson, T.; Hedström, A.K. Head trauma results in manyfold increased risk of multiple sclerosis in genetically susceptible individuals. J. Neurol. Neurosurg. Psychiatry 2024, 95, 554–560. [Google Scholar] [CrossRef]
- Cash, A.; Theus, M.H. Mechanisms of Blood–Brain Barrier Dysfunction in Traumatic Brain Injury. Int. J. Mol. Sci. 2020, 21, 3344. [Google Scholar] [CrossRef] [PubMed]
- Milligan, N.M.; Newcombe, R.; A Compston, D. A double-blind controlled trial of high dose methylprednisolone in patients with multiple sclerosis: Clinical effects. J. Neurol. Neurosurg. Psychiatry 1987, 50, 511–516. [Google Scholar] [CrossRef]
- Sloka, J.S.; Stefanelli, M. The mechanism of action of methylprednisolone in the treatment of multiple sclerosis. Mult. Scler. J. 2005, 11, 425–432. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Cáceres, E.M.; A Barrau, M.; Brieva, L.; Espejo, C.; Barberà, N.; Montalban, X. Treatment with methylprednisolone in relapses of multiple sclerosis patients: Immunological evidence of immediate and short-term but not long-lasting effects. Clin. Exp. Immunol. 2002, 127, 165–171. [Google Scholar] [CrossRef] [PubMed]
- Gelati, M.; Corsini, E.; Dufour, A.; Massa, G.; Giombini, S.; Solero, C.L.; Salmaggi, A. High-dose methylprednisolone reduces cytokine-induced adhesion molecules on human brain endothelium. Can. J. Neurol. Sci. 2000, 27, 241–244. [Google Scholar] [CrossRef]
- Xu, J.; Kim, G.-M.; Ahmed, S.H.; Xu, J.; Yan, P.; Xu, X.M.; Hsu, C.Y. Glucocorticoid Receptor-Mediated Suppression of Activator Protein-1 Activation and Matrix Metalloproteinase Expression after Spinal Cord Injury. J. Neurosci. 2001, 21, 92–97. [Google Scholar] [CrossRef]
- Förster, C.; Silwedel, C.; Golenhofen, N.; Burek, M.; Kietz, S.; Mankertz, J.; Drenckhahn, D. Occludin as direct target for glucocorticoid-induced improvement of blood-brain barrier properties in a murine in vitro system. J. Physiol. 2005, 565, 475–486. [Google Scholar] [CrossRef]
- Kraus, J.; Ling, A.K.; Hamm, S.; Voigt, K.; Oschmann, P.; Engelhardt, B. Interferon-β stabilizes barrier characteristics of brain endothelial cells in vitro. Ann. Neurol. 2004, 56, 192–205. [Google Scholar] [CrossRef]
- Kraus, J.; Oschmann, P. The impact of interferon-β treatment on the blood–brain barrier. Drug Discov. Today 2006, 11, 755–762. [Google Scholar] [CrossRef]
- Kuruganti, P.A.; Hinojoza, J.R.; Eaton, M.J.; Ehmann, U.K.; Sobel, R.A. Interferon-β Counteracts Inflammatory Mediator-Induced Effects on Brain Endothelial Cell Tight Junction Molecules—Implications for Multiple Sclerosis. J. Neuropathol. Exp. Neurol. 2002, 61, 710–724. [Google Scholar] [CrossRef]
- McCormack, P.L. Natalizumab: A Review of Its Use in the Management of Relapsing-Remitting Multiple Sclerosis. Drugs 2013, 73, 1463–1481. [Google Scholar] [CrossRef]
- Butzkueven, H.; Kappos, L.; Wiendl, H.; Trojano, M.; Spelman, T.; Chang, I.; Kasliwal, R.; Jaitly, S.; Campbell, N.; Ho, P.-R.; et al. Long-term safety and effectiveness of natalizumab treatment in clinical practice: 10 years of real-world data from the Tysabri Observational Program (TOP). J. Neurol. Neurosurg. Psychiatry 2020, 91, 660–668. [Google Scholar] [CrossRef]
- A Mills, E.; Mao-Draayer, Y. Aging and lymphocyte changes by immunomodulatory therapies impact PML risk in multiple sclerosis patients. Mult. Scler. J. 2018, 24, 1014–1022. [Google Scholar] [CrossRef]
- Cortese, I.; Reich, D.S.; Nath, A. Progressive multifocal leukoencephalopathy and the spectrum of JC virus-related disease. Nat. Rev. Neurol. 2020, 17, 37–51. [Google Scholar] [CrossRef] [PubMed]
- Bresciani, G.; Manai, F.; Davinelli, S.; Tucci, P.; Saso, L.; Amadio, M. Novel potential pharmacological applications of dimethyl fumarate—An overview and update. Front. Pharmacol. 2023, 14, 1264842. [Google Scholar] [CrossRef] [PubMed]
- Mehta, D.; Miller, C.; Arnold, D.L.; Bame, E.; Bar-Or, A.; Gold, R.; Hanna, J.; Kappos, L.; Liu, S.; Matta, A.; et al. Effect of dimethyl fumarate on lymphocytes in RRMS. Neurology 2019, 92, e1724–e1738. [Google Scholar] [CrossRef]
- Hammer, A.; Waschbisch, A.; Kuhbandner, K.; Bayas, A.; Lee, D.; Duscha, A.; Haghikia, A.; Gold, R.; Linker, R.A. The NRF2 pathway as potential biomarker for dimethyl fumarate treatment in multiple sclerosis. Ann. Clin. Transl. Neurol. 2018, 5, 668–676. [Google Scholar] [CrossRef]
- Mills, E.A.; Ogrodnik, M.A.; Plave, A.; Mao-Draayer, Y. Emerging Understanding of the Mechanism of Action for Dimethyl Fumarate in the Treatment of Multiple Sclerosis. Front. Neurol. 2018, 9, 5. [Google Scholar] [CrossRef] [PubMed]
- Galloway, D.A.; Williams, J.B.; Moore, C.S. Effects of fumarates on inflammatory human astrocyte responses and oligodendrocyte differentiation. Ann. Clin. Transl. Neurol. 2017, 4, 381–391. [Google Scholar] [CrossRef]
- Hla, T.; Brinkmann, V. Sphingosine 1-phosphate (S1P). Neurology 2011, 76, S3–S8. [Google Scholar] [CrossRef]
- Van Doorn, R.; Nijland, P.G.; Dekker, N.; Witte, M.E.; Lopes-Pinheiro, M.A.; van het Hof, B.; Kooij, G.; Reijerkerk, A.; Dijkstra, C.; Van Van Der Valk, P.; et al. Fingolimod attenuates ceramide-induced blood–brain barrier dysfunction in multiple sclerosis by targeting reactive astrocytes. Acta Neuropathol. 2012, 124, 397–410. [Google Scholar] [CrossRef] [PubMed]
- Hunter, S.F.; Bowen, J.D.; Reder, A.T. The Direct Effects of Fingolimod in the Central Nervous System: Implications for Relapsing Multiple Sclerosis. CNS Drugs 2015, 30, 135–147. [Google Scholar] [CrossRef] [PubMed]
- Spampinato, S.F.; Obermeier, B.; Cotleur, A.; Love, A.; Takeshita, Y.; Sano, Y.; Kanda, T.; Ransohoff, R.M. Sphingosine 1 Phosphate at the Blood Brain Barrier: Can the Modulation of S1P Receptor 1 Influence the Response of Endothelial Cells and Astrocytes to Inflammatory Stimuli? PLoS ONE 2015, 10, e0133392. [Google Scholar] [CrossRef]
- Prager, B.; Spampinato, S.F.; Ransohoff, R.M. Sphingosine 1-phosphate signaling at the blood–brain barrier. Trends Mol. Med. 2015, 21, 354–363. [Google Scholar] [CrossRef]
- Nishihara, H.; Shimizu, F.; Sano, Y.; Takeshita, Y.; Maeda, T.; Abe, M.; Koga, M.; Kanda, T. Fingolimod Prevents Blood-Brain Barrier Disruption Induced by the Sera from Patients with Multiple Sclerosis. PLoS ONE 2015, 10, e0121488–e0121488. [Google Scholar] [CrossRef] [PubMed]
- Chun, J.; Hartung, H.P. Mechanism of Action of Oral Fingolimod (FTY720) in Multiple Sclerosis. Clin. Neuropharmacol. 2010, 33, 91–101. [Google Scholar] [CrossRef] [PubMed]
- Smith, P.A.; Schmid, C.; Zurbruegg, S.; Jivkov, M.; Doelemeyer, A.; Theil, D.; Dubost, V.; Beckmann, N. Fingolimod inhibits brain atrophy and promotes brain-derived neurotrophic factor in an animal model of multiple sclerosis. J. Neuroimmunol. 2018, 318, 103–113. [Google Scholar] [CrossRef]
- Miron, V.E.; Jung, C.G.; Kim, H.J.; Kennedy, T.E.; Soliven, B.; Antel, J.P. FTY720 modulates human oligodendrocyte progenitor process extension and survival. Ann. Neurol. 2008, 63, 61–71. [Google Scholar] [CrossRef]
- Sorensen, S.D.; Nicole, O.; Peavy, R.D.; Montoya, L.M.; Lee, C.J.; Murphy, T.J.; Traynelis, S.F.; Hepler, J.R. Common Signaling Pathways Link Activation of Murine PAR-1, LPA, and S1P Receptors to Proliferation of Astrocytes. Mol. Pharmacol. 2003, 64, 1199–1209. [Google Scholar] [CrossRef]
- Noda, H.; Takeuchi, H.; Mizuno, T.; Suzumura, A. Fingolimod phosphate promotes the neuroprotective effects of microglia. J. Neuroimmunol. 2013, 256, 13–18. [Google Scholar] [CrossRef]
- Jackson, S.J.; Giovannoni, G.; Baker, D. Fingolimod modulates microglial activation to augment markers of remyelination. J. Neuroinflammation 2011, 8, 76–76. [Google Scholar] [CrossRef] [PubMed]
- Rammohan, K.; Coyle, P.K.; Sylvester, E.; Galazka, A.; Dangond, F.; Grosso, M.; Leist, T.P. The Development of Cladribine Tablets for the Treatment of Multiple Sclerosis: A Comprehensive Review. Drugs 2020, 80, 1901–1928. [Google Scholar] [CrossRef]
- Leist, T.P.; Weissert, R. Cladribine. Clin. Neuropharmacol. 2011, 34, 28–35. [Google Scholar] [CrossRef]
- Mitosek-Szewczyk, K.; Stelmasiak, Z.; Bartosik-Psujek, H.; Belniak, E. Impact of cladribine on soluble adhesion molecules in multiple sclerosis. Acta Neurol. Scand. 2010, 122, 409–413. [Google Scholar] [CrossRef]
- Siriratnam, P.; Huda, S.; Butzkueven, H.; van der Walt, A.; Jokubaitis, V.; Monif, M. A comprehensive review of the advances in neuromyelitis optica spectrum disorder. Autoimmun. Rev. 2023, 22, 103465. [Google Scholar] [CrossRef] [PubMed]
- Wingerchuk, D.M.; Banwell, B.; Bennett, J.L.; Cabre, P.; Carroll, W.; Chitnis, T.; De Seze, J.; Fujihara, K.; Greenberg, B.; Jacob, A.; et al. International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology 2015, 85, 177–189. [Google Scholar] [CrossRef]
- Hamid, S.H.; Elsone, L.; Mutch, K.; Solomon, T.; Jacob, A. The impact of 2015 neuromyelitis optica spectrum disorders criteria on diagnostic rates. Mult. Scler. J. 2016, 23, 228–233. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Kermode, A.G.; Hu, X.; Qiu, W. Risk of relapse in patients with neuromyelitis optica spectrum disorder: Recognition and preventive strategy. Mult. Scler. Relat. Disord. 2020, 46, 102522. [Google Scholar] [CrossRef]
- Contentti, E.C.; Correale, J. Neuromyelitis optica spectrum disorders: From pathophysiology to therapeutic strategies. J. Neuroinflammation 2021, 18, 1–18. [Google Scholar] [CrossRef]
- Chihara, N.; Aranami, T.; Oki, S.; Matsuoka, T.; Nakamura, M.; Kishida, H.; Yokoyama, K.; Kuroiwa, Y.; Hattori, N.; Okamoto, T.; et al. Plasmablasts as Migratory IgG-Producing Cells in the Pathogenesis of Neuromyelitis Optica. PLoS ONE 2013, 8, e83036. [Google Scholar] [CrossRef]
- Chihara, N.; Aranami, T.; Sato, W.; Miyazaki, Y.; Miyake, S.; Okamoto, T.; Ogawa, M.; Toda, T.; Yamamura, T. Interleukin 6 signaling promotes anti-aquaporin 4 autoantibody production from plasmablasts in neuromyelitis optica. Proc. Natl. Acad. Sci. USA 2011, 108, 3701–3706. [Google Scholar] [CrossRef]
- Papadopoulos, M.C.; Verkman, A. Aquaporin 4 and neuromyelitis optica. Lancet Neurol. 2012, 11, 535–544. [Google Scholar] [CrossRef] [PubMed]
- Crane, J.M.; Lam, C.; Rossi, A.; Gupta, T.; Bennett, J.L.; Verkman, A.S. Binding affinity and specificity of neuromyelitis optica autoantibodies to aquaporin-4 M1/M23 isoforms and orthogonal arrays. J. Biol. Chem. 2011, 286, 16516–16524. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulos, M.C.; Bennett, J.L.; Verkman, A.S. Treatment of neuromyelitis optica: State-of-the-art and emerging therapies. Nat. Rev. Neurol. 2014, 10, 493–506. [Google Scholar] [CrossRef]
- Lucchinetti, C.F.; Guo, Y.; Popescu, B.F.G.; Fujihara, K.; Itoyama, Y.; Misu, T. The Pathology of an Autoimmune Astrocytopathy: Lessons Learned from Neuromyelitis Optica. Brain Pathol. 2013, 24, 83–97. [Google Scholar] [CrossRef]
- Linhares, U.C.; Schiavoni, P.B.; Barros, P.O.; Kasahara, T.M.; Teixeira, B.; Ferreira, T.B.; Alvarenga, R.; Hygino, J.; Vieira, M.M.M.; Bittencourt, V.C.B.; et al. The Ex Vivo Production of IL-6 and IL-21 by CD4+ T Cells is Directly Associated with Neurological Disability in Neuromyelitis Optica Patients. J. Clin. Immunol. 2012, 33, 179–189. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Li, X.; Xia, J. Th17 cells in neuromyelitis optica spectrum disorder: A review. Int. J. Neurosci. 2015, 126, 1051–1060. [Google Scholar] [CrossRef]
- Tomizawa, Y.; Yokoyama, K.; Saiki, S.; Takahashi, T.; Matsuoka, J.; Hattori, N. Blood—Brain Barrier Disruption is More Severe in Neuromyelitis Optica than in Multiple Sclerosis and Correlates with Clinical Disability. J. Int. Med Res. 2012, 40, 1483–1491. [Google Scholar] [CrossRef] [PubMed]
- You, X.; Yan, L.; Li, X.; Pang, Y.; Guo, X.; Ye, J.; Hu, H. Disruption of blood-brain barrier integrity associated with brain lesions in Chinese neuromyelitis optica spectrum disorder patients. Mult. Scler. Relat. Disord. 2018, 27, 254–259. [Google Scholar] [CrossRef]
- Rodin, R.E.; Chitnis, T. Soluble biomarkers for Neuromyelitis Optica Spectrum Disorders: A mini review. Front. Neurol. 2024, 15, 1415535. [Google Scholar] [CrossRef]
- Vaknin-Dembinsky, A.; Brill, L.; Orpaz, N.; Abramsky, O.; Karussis, D. Preferential increase of B-cell activating factor in the cerebrospinal fluid of neuromyelitis optica in a white population. Mult. Scler. J. 2010, 16, 1453–1457. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, K.; Sato, D.K.; Nakashima, I.; Ogawa, R.; Akaishi, T.; Takai, Y.; Nishiyama, S.; Takahashi, T.; Misu, T.; Kuroda, H.; et al. CSF cytokine profile in MOG-IgG+ neurological disease is similar to AQP4-IgG+ NMOSD but distinct from MS: A cross-sectional study and potential therapeutic implications. J. Neurol. Neurosurg. Psychiatry 2018, 89, 927–936. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Chen, J.; Wang, Z.; Wang, Y.; Zheng, D.; Wang, H.; Peng, Y. The CSF Levels of Neutrophil-Related Chemokines in Patients with Neuromyelitis Optica. Ann. Clin. Transl. Neurol. 2020, 7, 1245–1251. [Google Scholar] [CrossRef] [PubMed]
- Matsushita, T.; Tateishi, T.; Isobe, N.; Yonekawa, T.; Yamasaki, R.; Matsuse, D.; Murai, H.; Kira, J.-I. Characteristic Cerebrospinal Fluid Cytokine/Chemokine Profiles in Neuromyelitis Optica, Relapsing Remitting or Primary Progressive Multiple Sclerosis. PLoS ONE 2013, 8, e61835. [Google Scholar] [CrossRef] [PubMed]
- Grebenciucova, E.; VanHaerents, S. Interleukin 6: At the interface of human health and disease. Front. Immunol. 2023, 14, 1255533. [Google Scholar] [CrossRef] [PubMed]
- Fujihara, K.; Bennett, J.L.; de Seze, J.; Haramura, M.; Kleiter, I.; Weinshenker, B.G.; Kang, D.; Mughal, T.; Yamamura, T. Interleukin-6 in neuromyelitis optica spectrum disorder pathophysiology. Neurol. - Neuroimmunol. Neuroinflammation 2020, 7. [Google Scholar] [CrossRef] [PubMed]
- Uzawa, A.; Mori, M.; Ito, M.; Uchida, T.; Hayakawa, S.; Masuda, S.; Kuwabara, S. Markedly increased CSF interleukin-6 levels in neuromyelitis optica, but not in multiple sclerosis. J. Neurol. 2009, 256, 2082–2084. [Google Scholar] [CrossRef]
- Uzawa, A.; Mori, M.; Arai, K.; Sato, Y.; Hayakawa, S.; Masuda, S.; Taniguchi, J.; Kuwabara, S. Cytokine and chemokine profiles in neuromyelitis optica: Significance of interleukin-6. Mult. Scler. J. 2010, 16, 1443–1452. [Google Scholar] [CrossRef]
- Misu, T.; Takano, R.; Fujihara, K.; Takahashi, T.; Sato, S.; Itoyama, Y. Marked increase in cerebrospinal fluid glial fibrillar acidic protein in neuromyelitis optica: An astrocytic damage marker. J. Neurol. Neurosurg. Psychiatry 2009, 80, 575–577. [Google Scholar] [CrossRef]
- Aktas, O.; Smith, M.A.; Marignier, R.; Kim, H.J.; Weinshenker, B.G.; Pittock, S.J.; Wingerchuk, D.M.; Cutter, G.R.; Green, A.J.; Mealy, M.A.; et al. Serum Glial Fibrillary Acidic Protein: A Neuromyelitis Optica Spectrum Disorder Biomarker. Ann. Neurol. 2021, 89, 895–910. [Google Scholar] [CrossRef]
- Aktas, O.; Hartung, H.-P.; A Smith, M.; A Rees, W.; Fujihara, K.; Paul, F.; Marignier, R.; Bennett, J.L.; Kim, H.J.; Weinshenker, B.G.; et al. Serum neurofilament light chain levels at attack predict post-attack disability worsening and are mitigated by inebilizumab: Analysis of four potential biomarkers in neuromyelitis optica spectrum disorder. J. Neurol. Neurosurg. Psychiatry 2023, 94, 757–768. [Google Scholar] [CrossRef]
- Kim, S.-M.; Waters, P.; Vincent, A.; Go, M.J.; Park, K.S.; Sung, J.-J.; Lee, K.-W. Cerebrospinal fluid/serum gradient of IgG is associated with disability at acute attacks of neuromyelitis optica. J. Neurol. 2011, 258, 2176–2180. [Google Scholar] [CrossRef] [PubMed]
- Jarius, S.; Franciotta, D.; Paul, F.; Ruprecht, K.; Bergamaschi, R.; Rommer, P.S.; Reuss, R.; Probst, C.; Kristoferitsch, W.; Wandinger, K.P.; et al. Cerebrospinal fluid antibodies to aquaporin-4 in neuromyelitis optica and related disorders: Frequency, origin, and diagnostic relevance. J. Neuroinflamm. 2010, 7, 52. [Google Scholar] [CrossRef] [PubMed]
- Kowarik, M.C.; Dzieciatkowska, M.; Wemlinger, S.; Ritchie, A.M.; Hemmer, B.; Owens, G.P.; Bennett, J.L. The cerebrospinal fluid immunoglobulin transcriptome and proteome in neuromyelitis optica reveals central nervous system-specific B cell populations. J. Neuroinflammation 2015, 12, 1–8. [Google Scholar] [CrossRef]
- Bradl, M.; Misu, T.; Takahashi, T.; Watanabe, M.; Mader, S.; Reindl, M.; Adzemovic, M.; Bauer, J.; Berger, T.; Fujihara, K.; et al. Neuromyelitis optica: Pathogenicity of patient immunoglobulin in vivo. Ann. Neurol. 2009, 66, 630–643. [Google Scholar] [CrossRef] [PubMed]
- Sharma, R.; Fischer, M.-T.; Bauer, J.; Felts, P.A.; Smith, K.J.; Misu, T.; Fujihara, K.; Bradl, M.; Lassmann, H. Inflammation induced by innate immunity in the central nervous system leads to primary astrocyte dysfunction followed by demyelination. Acta Neuropathol. 2010, 120, 223–236. [Google Scholar] [CrossRef] [PubMed]
- Jitprapaikulsan, J.; Fryer, J.P.; Majed, M.; Smith, C.Y.; Jenkins, S.M.; Cabre, P.; Hinson, S.R.; Weinshenker, B.G.; Mandrekar, J.; Chen, J.J.; et al. Clinical utility of AQP4-IgG titers and measures of complement-mediated cell killing in NMOSD. Neurol. - Neuroimmunol. Neuroinflammation 2020, 7. [Google Scholar] [CrossRef]
- Schmetzer, O.; Lakin, E.; Roediger, B.; Duchow, A.; Asseyer, S.; Paul, F.; Siebert, N. Anti-aquaporin 4 IgG Is Not Associated With Any Clinical Disease Characteristics in Neuromyelitis Optica Spectrum Disorder. Front. Neurol. 2021, 12. [Google Scholar] [CrossRef]
- Shimizu, F.; Ransohoff, R.M. GRP78 autoantibodies initiate the breakdown of the blood-brain barrier in neuromyelitis optica. Oncotarget 2017, 8, 106175–106176. [Google Scholar] [CrossRef]
- Kim, H.J.; Paul, F.; Lana-Peixoto, M.A.; Tenembaum, S.; Asgari, N.; Palace, J.; Klawiter, E.C.; Sato, D.K.; de Seze, J.; Wuerfel, J.; et al. MRI characteristics of neuromyelitis optica spectrum disorder: An international update. Neurology 2015, 84, 1165–1173. [Google Scholar] [CrossRef]
- Clarke, L.; Arnett, S.; Bukhari, W.; Khalilidehkordi, E.; Sanchez, S.J.; O’Gorman, C.; Sun, J.; Prain, K.M.; Woodhall, M.; Silvestrini, R.; et al. MRI Patterns Distinguish AQP4 Antibody Positive Neuromyelitis Optica Spectrum Disorder From Multiple Sclerosis. Front. Neurol. 2021, 12. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, F.; Sano, Y.; Takahashi, T.; Haruki, H.; Saito, K.; Koga, M.; Kanda, T. Sera from neuromyelitis optica patients disrupt the blood–brain barrier. J. Neurol. Neurosurg. Psychiatry 2011, 83, 288–297. [Google Scholar] [CrossRef] [PubMed]
- Tasaki, A.; Shimizu, F.; Sano, Y.; Fujisawa, M.; Takahashi, T.; Haruki, H.; Abe, M.; Koga, M.; Kanda, T. Autocrine MMP-2/9 secretion increases the BBB permeability in neuromyelitis optica. J. Neurol. Neurosurg. Psychiatry 2014, 85, 419–430. [Google Scholar] [CrossRef]
- Shimizu, F.; Nishihara, H.; Sano, Y.; Takeshita, Y.; Takahashi, S.; Maeda, T.; Takahashi, T.; Abe, M.; Koga, M.; Kanda, T. Markedly Increased IP-10 Production by Blood-Brain Barrier in Neuromyelitis Optica. PLoS ONE 2015, 10, e0122000. [Google Scholar] [CrossRef]
- Shimizu, F.; Schaller, K.L.; Owens, G.P.; Cotleur, A.C.; Kellner, D.; Takeshita, Y.; Obermeier, B.; Kryzer, T.J.; Sano, Y.; Kanda, T.; et al. Glucose-regulated protein 78 autoantibody associates with blood-brain barrier disruption in neuromyelitis optica. Sci. Transl. Med. 2017, 9, 397. [Google Scholar] [CrossRef]
- Shimizu, F.; Takeshita, Y.; Hamamoto, Y.; Nishihara, H.; Sano, Y.; Honda, M.; Sato, R.; Maeda, T.; Takahashi, T.; Fujikawa, S.; et al. GRP 78 antibodies are associated with clinical phenotype in neuromyelitis optica. Ann. Clin. Transl. Neurol. 2019, 6, 2079–2087. [Google Scholar] [CrossRef]
- Bläss, S.; Union, A.; Raymackers, J.; Schumann, F.; Ungethüm, U.; Müller-Steinbach, S.; De Keyser, F.; Engel, J.M.; Burmester, G.R. The stress protein BiP is overexpressed and is a major B and T cell target in rheumatoid arthritis. Arthritis Rheum 2001, 44, 761–771. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, J.; Chang, H.; Wang, H.; Xu, W.; Cong, H.; Zhang, X.; Liu, J.; Yin, L. NMO-IgG Induce Interleukin-6 Release via Activation of the NF-κB Signaling Pathway in Astrocytes. Neuroscience 2022, 496, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Du, L.; Chang, H.; Xu, W.; Wei, Y.; Wang, Y.; Yin, L.; Zhang, X. Effect of NMO-IgG on the interleukin-6 cascade in astrocytes via activation of the JAK/STAT3 signaling pathway. Life Sci. 2020, 258, 118217. [Google Scholar] [CrossRef]
- Takeshita, Y.; Obermeier, B.; Cotleur, A.C.; Spampinato, S.F.; Shimizu, F.; Yamamoto, E.; Sano, Y.; Kryzer, T.J.; Lennon, V.A.; Kanda, T.; et al. Effects of neuromyelitis optica–IgG at the blood–brain barrier in vitro. Neurol. - Neuroimmunol. Neuroinflammation 2017, 4, e311. [Google Scholar] [CrossRef]
- Takeshita, Y.; Fujikawa, S.; Serizawa, K.; Fujisawa, M.; Matsuo, K.; Nemoto, J.; Shimizu, F.; Sano, Y.; Tomizawa-Shinohara, H.; Miyake, S.; et al. New BBB Model Reveals That IL-6 Blockade Suppressed the BBB Disorder, Preventing Onset of NMOSD. Neurol.-Neuroimmunol. Neuroinflammation 2021, 8. [Google Scholar] [CrossRef]
- Banwell, B.; Bennett, J.L.; Marignier, R.; Kim, H.J.; Brilot, F.; Flanagan, E.P.; Ramanathan, S.; Waters, P.; Tenembaum, S.; Graves, J.S.; et al. Diagnosis of myelin oligodendrocyte glycoprotein antibody-associated disease: International MOGAD Panel proposed criteria. Lancet Neurol. 2023, 22, 268–282. [Google Scholar] [CrossRef] [PubMed]
- Marignier, R.; Hacohen, Y.; Cobo-Calvo, A.; Pröbstel, A.-K.; Aktas, O.; Alexopoulos, H.; Amato, M.-P.; Asgari, N.; Banwell, B.; Bennett, J.; et al. Myelin-oligodendrocyte glycoprotein antibody-associated disease. Lancet Neurol. 2021, 20, 762–772. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Kezuka, T.; Ishikawa, H.; Tanaka, M.; Sakimura, K.; Abe, M.; Kawamura, M. Pathogenesis, Clinical Features, and Treatment of Patients with Myelin Oligodendrocyte Glycoprotein (MOG) Autoantibody-Associated Disorders Focusing on Optic Neuritis with Consideration of Autoantibody-Binding Sites: A Review. Int. J. Mol. Sci. 2023, 24, 13368. [Google Scholar] [CrossRef] [PubMed]
- Ambrosius, W.; Michalak, S.; Kozubski, W.; Kalinowska, A. Myelin Oligodendrocyte Glycoprotein Antibody-Associated Disease: Current Insights into the Disease Pathophysiology, Diagnosis and Management. Int. J. Mol. Sci. 2020, 22, 100. [Google Scholar] [CrossRef]
- Wang, J.J.; Jaunmuktane, Z.; Mummery, C.; Brandner, S.; Leary, S.; Trip, S.A. Inflammatory demyelination without astrocyte loss in MOG antibody–positive NMOSD. Neurology 2016, 87, 229–231. [Google Scholar] [CrossRef]
- Zhou, L.; Huang, Y.; Li, H.; Fan, J.; Zhangbao, J.; Yu, H.; Li, Y.; Lu, J.; Zhao, C.; Lu, C.; et al. MOG-antibody associated demyelinating disease of the CNS: A clinical and pathological study in Chinese Han patients. J. Neuroimmunol. 2017, 305, 19–28. [Google Scholar] [CrossRef]
- Körtvélyessy, P.; Breu, M.; Pawlitzki, M.; Metz, I.; Heinze, H.-J.; Matzke, M.; et al. ADEM-like presentation, anti-MOG antibodies, and MS pathology: Two case reports. Neurol. Neuroimmunol Neuroinflamm 2017, 4, e335. [Google Scholar] [CrossRef]
- Höftberger, R.; Guo, Y.; Flanagan, E.P.; Lopez-Chiriboga, A.S.; Endmayr, V.; Hochmeister, S.; Joldic, D.; Pittock, S.J.; Tillema, J.M.; Gorman, M.; et al. The pathology of central nervous system inflammatory demyelinating disease accompanying myelin oligodendrocyte glycoprotein autoantibody. Acta Neuropathol. 2020, 139, 875–892. [Google Scholar] [CrossRef]
- Machado-Santos, J.; Saji, E.; Tröscher, A.R.; Paunovic, M.; Liblau, R.; Gabriely, G.; Bien, C.G.; Bauer, J.; Lassmann, H. The compartmentalized inflammatory response in the multiple sclerosis brain is composed of tissue-resident CD8+ T lymphocytes and B cells. Brain 2018, 141, 2066–2082. [Google Scholar] [CrossRef]
- Takai, Y.; Misu, T.; Kaneko, K.; Chihara, N.; Narikawa, K.; Tsuchida, S.; Nishida, H.; Komori, T.; Seki, M.; Komatsu, T.; et al. Myelin oligodendrocyte glycoprotein antibody-associated disease: An immunopathological study. Brain 2020, 143, 1431–1446. [Google Scholar] [CrossRef] [PubMed]
- Jarius, S.; Pellkofer, H.; Siebert, N.; Korporal-Kuhnke, M.; Hümmert, M.W.; Ringelstein, M.; Rommer, P.S.; Ayzenberg, I.; Ruprecht, K.; Klotz, L.; et al. Cerebrospinal fluid findings in patients with myelin oligodendrocyte glycoprotein (MOG) antibodies. Part 1: Results from 163 lumbar punctures in 100 adult patients. J. Neuroinflammation 2020, 17, 1–26. [Google Scholar] [CrossRef] [PubMed]
- in cooperation with the Neuromyelitis Optica Study Group (NEMOS); Jarius, S. ; Ruprecht, K.; Kleiter, I.; Borisow, N.; Asgari, N.; Pitarokoili, K.; Pache, F.; Stich, O.; Beume, L.-A.; et al. MOG-IgG in NMO and related disorders: A multicenter study of 50 patients. Part 2: Epidemiology, clinical presentation, radiological and laboratory features, treatment responses, and long-term outcome. J. Neuroinflammation 2016, 13, 1–45. [Google Scholar] [CrossRef]
- Tanaka, S.; Hashimoto, B.; Izaki, S.; Oji, S.; Fukaura, H.; Nomura, K. Clinical and immunological differences between MOG associated disease and anti AQP4 antibody-positive neuromyelitis optica spectrum disorders: Blood-brain barrier breakdown and peripheral plasmablasts. Mult. Scler. Relat. Disord. 2020, 41, 102005. [Google Scholar] [CrossRef]
- Kaneko, K.; Sato, D.K.; Nakashima, I.; Ogawa, R.; Akaishi, T.; Takai, Y.; Nishiyama, S.; Takahashi, T.; Misu, T.; Kuroda, H.; et al. CSF cytokine profile in MOG-IgG+ neurological disease is similar to AQP4-IgG+ NMOSD but distinct from MS: A cross-sectional study and potential therapeutic implications. J. Neurol. Neurosurg. Psychiatry 2018, 89, 927–936. [Google Scholar] [CrossRef]
- Kothur, K.; Wienholt, L.; Tantsis, E.M.; Earl, J.; Bandodkar, S.; Prelog, K.; Tea, F.; Ramanathan, S.; Brilot, F.; Dale, R.C. B Cell, Th17, and Neutrophil Related Cerebrospinal Fluid Cytokine/Chemokines Are Elevated in MOG Antibody Associated Demyelination. PLoS ONE 2016, 11, e0149411. [Google Scholar] [CrossRef] [PubMed]
- Goverman, J. Autoimmune T cell responses in the central nervous system. Nat. Rev. Immunol. 2009, 9, 393–407. [Google Scholar] [CrossRef]
- Corbali, O.; Chitnis, T. Pathophysiology of myelin oligodendrocyte glycoprotein antibody disease. Front. Neurol. 2023, 14, 1137998. [Google Scholar] [CrossRef]
- Spadaro, M.; Winklmeier, S.; Beltrán, E.; Macrini, C.; Höftberger, R.; Schuh, E.; Thaler, F.S.; Gerdes, L.A.; Laurent, S.; Gerhards, R.; et al. Pathogenicity of human antibodies against myelin oligodendrocyte glycoprotein. Ann. Neurol. 2018, 84, 315–328. [Google Scholar] [CrossRef]
- Shimizu, F.; Ogawa, R.; Mizukami, Y.; Watanabe, K.; Hara, K.; Kadono, C.; Takahashi, T.; Misu, T.; Takeshita, Y.; Sano, Y.; et al. GRP78 Antibodies Are Associated With Blood-Brain Barrier Breakdown in Anti–Myelin Oligodendrocyte Glycoprotein Antibody–Associated Disorder. Neurol. - Neuroimmunol. Neuroinflammation 2021, 9. [Google Scholar] [CrossRef]
- Liu, Y.; Tu, Z.; Zhang, X.; Du, K.; Xie, Z.; Lin, Z. Pathogenesis and treatment of neuropsychiatric systemic lupus erythematosus: A review. Front. Cell Dev. Biol. 2022, 10, 998328. [Google Scholar] [CrossRef] [PubMed]
- Stock, A.D.; Gelb, S.; Pasternak, O.; Ben-Zvi, A.; Putterman, C. The blood brain barrier and neuropsychiatric lupus: New perspectives in light of advances in understanding the neuroimmune interface. Autoimmun. Rev. 2017, 16, 612–619. [Google Scholar] [CrossRef] [PubMed]
- Brimberg, L.; Mader, S.; Fujieda, Y.; Arinuma, Y.; Kowal, C.; Volpe, B.T.; Diamond, B. Antibodies as Mediators of Brain Pathology. Trends Immunol. 2015, 36, 709–724. [Google Scholar] [CrossRef] [PubMed]
- Govoni, M.; Bortoluzzi, A.; Padovan, M.; Silvagni, E.; Borrelli, M.; Donelli, F.; Ceruti, S.; Trotta, F. The diagnosis and clinical management of the neuropsychiatric manifestations of lupus. J. Autoimmun. 2016, 74, 41–72. [Google Scholar] [CrossRef] [PubMed]
- Kivity, S.; Agmon-Levin, N.; Zandman-Goddard, G.; Chapman, J.; Shoenfeld, Y. Neuropsychiatric lupus: A mosaic of clinical presentations. BMC Med. 2015, 13, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Stock, A.D.; Wen, J.; Putterman, C. Neuropsychiatric Lupus, the Blood Brain Barrier, and the TWEAK/Fn14 Pathway. Front. Immunol. 2013, 4, 484. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.-Y.; Lin, J.; Lu, X.-Y.; Zhao, X.-Y. Expression of S100B protein levels in serum and cerebrospinal fluid with different forms of neuropsychiatric systemic lupus erythematosus. Clin. Rheumatol. 2007, 27, 353–357. [Google Scholar] [CrossRef]
- Gris, J.-C.; Nobile, B.; Bouvier, S. Neuropsychiatric presentations of antiphospholipid antibodies. Thromb. Res. 2015, 135, S56–S59. [Google Scholar] [CrossRef]
- Fleetwood, T.; Cantello, R.; Comi, C. Antiphospholipid Syndrome and the Neurologist: From Pathogenesis to Therapy. Front. Neurol. 2018, 9, 1001. [Google Scholar] [CrossRef]
- Brimberg, L.; Mader, S.; Fujieda, Y.; Arinuma, Y.; Kowal, C.; Volpe, B.T.; Diamond, B. Antibodies as Mediators of Brain Pathology. Trends Immunol. 2015, 36, 709–724. [Google Scholar] [CrossRef]
- Mader, S.; Brimberg, L.; Diamond, B. The Role of Brain-Reactive Autoantibodies in Brain Pathology and Cognitive Impairment. Front. Immunol. 2017, 8, 1101–1101. [Google Scholar] [CrossRef] [PubMed]
- Yoshio, T.; Okamoto, H.; Hirohata, S.; Minota, S. IgG anti–NR2 glutamate receptor autoantibodies from patients with systemic lupus erythematosus activate endothelial cells. Arthritis Rheum. 2012, 65, 457–463. [Google Scholar] [CrossRef]
- Matus, S.; Burgos, P.V.; Bravo-Zehnder, M.; Kraft, R.; Porras, O.H.; Farías, P.; Barros, L.F.; Torrealba, F.; Massardo, L.; Jacobelli, S.; et al. Antiribosomal-P autoantibodies from psychiatric lupus target a novel neuronal surface protein causing calcium influx and apoptosis. J. Exp. Med. 2007, 204, 3221–3234. [Google Scholar] [CrossRef]
- DeGiorgio, L.A.; Konstantinov, K.N.; Lee, S.C.; Hardin, J.A.; Volpe, B.T.; Diamond, B. A subset of lupus anti-DNA antibodies cross-reacts with the NR2 glutamate receptor in systemic lupus erythematosus. Nat. Med. 2001, 7, 1189–1193. [Google Scholar] [CrossRef] [PubMed]
- Kowal, C.; DeGiorgio, L.A.; Lee, J.Y.; Edgar, M.A.; Huerta, P.T.; Volpe, B.T.; Diamond, B. Human lupus autoantibodies against NMDA receptors mediate cognitive impairment. Proc. Natl. Acad. Sci. 2006, 103, 19854–19859. [Google Scholar] [CrossRef]
- Kowal, C.; DeGiorgio, L.A.; Nakaoka, T.; Hetherington, H.; Huerta, P.T.; et al. Cognition and immunity; antibody impairs memory. Immunity 2004, 21, 179–188. [Google Scholar] [CrossRef]
- Conti, F.; Alessandri, C.; Bompane, D.; Bombardieri, M.; Spinelli, F.R.; Rusconi, A.C.; Valesini, G. Autoantibody profile in systemic lupus erythematosus with psychiatric manifestations: A role for anti-endothelial-cell antibodies. Arthritis Res. Ther. 2004, 6, R366–R372. [Google Scholar] [CrossRef]
- Carvalho, D.; Savage, C.O.S.; Isenberg, D.; Pearson, J.D. IgG anti-endothelial cell autoantibodies from patients with systemic lupus erythematosus or systemic vasculitis stimulate the release of two endothelial cell-derived mediators, which enhance adhesion molecule expression and leukocyte adhesion in an autocrine manner. Arthritis Rheum. 1999, 42, 631–640. [Google Scholar] [CrossRef]
- Cieślik, P.; Semik-Grabarczyk, E.; Hrycek, A.; Holecki, M. The impact of anti-endothelial cell antibodies (AECAs) on the development of blood vessel damage in patients with systemic lupus erythematosus: The preliminary study. Rheumatol. Int. 2022, 42, 791–801. [Google Scholar] [CrossRef]
- Renaudineau, Y.; Dugué, C.; Dueymes, M.; Youinou, P. Antiendothelial cell antibodies in systemic lupus erythematosus. Autoimmun. Rev. 2002, 1, 365–372. [Google Scholar] [CrossRef] [PubMed]
- Matsueda, Y.; Arinuma, Y.; Nagai, T.; Hirohata, S. Elevation of serum anti–glucose-regulated protein 78 antibodies in neuropsychiatric systemic lupus erythematosus. Lupus Sci. Med. 2018, 5, e000281. [Google Scholar] [CrossRef] [PubMed]
- Graus, F.; Titulaer, M.J.; Balu, R.; Benseler, S.; Bien, C.G.; Cellucci, T.; Cortese, I.; Dale, R.C.; Gelfand, J.M.; Geschwind, M.; et al. A clinical approach to diagnosis of autoimmune encephalitis. Lancet Neurol. 2016, 15, 391–404. [Google Scholar] [CrossRef]
- Tanaka, K.; Kawamura, M.; Sakimura, K.; Kato, N. Significance of Autoantibodies in Autoimmune Encephalitis in Relation to Antigen Localization: An Outline of Frequently Reported Autoantibodies with a Non-Systematic Review. Int. J. Mol. Sci. 2020, 21, 4941. [Google Scholar] [CrossRef] [PubMed]
- Shan, W.; Yang, H.; Wang, Q. Neuronal Surface Antibody-Medicated Autoimmune Encephalitis (Limbic Encephalitis) in China: A Multiple-Center, Retrospective Study. Front. Immunol. 2021, 12. [Google Scholar] [CrossRef]
- Dalmau, J.; Armangué, T.; Planagumà, J.; Radosevic, M.; Mannara, F.; Leypoldt, F.; Geis, C.; Lancaster, E.; Titulaer, M.J.; Rosenfeld, M.R.; et al. An update on anti-NMDA receptor encephalitis for neurologists and psychiatrists: Mechanisms and models. Lancet Neurol. 2019, 18, 1045–1057. [Google Scholar] [CrossRef]
- Dalmau, J.; Gleichman, A.J.; Hughes, E.G.; Rossi, J.E.; Peng, X.; Lai, M.; Dessain, S.K.; Rosenfeld, M.R.; Balice-Gordon, R.; Lynch, D.R. Anti-NMDA-receptor encephalitis: Case series and analysis of the effects of antibodies. Lancet Neurol. 2008, 7, 1091–1098. [Google Scholar] [CrossRef] [PubMed]
- Hughes, E.G.; Peng, X.; Gleichman, A.J.; Lai, M.; Zhou, L.; Tsou, R.; Parsons, T.D.; Lynch, D.R.; Dalmau, J.; Balice-Gordon, R.J. Cellular and Synaptic Mechanisms of Anti-NMDA Receptor Encephalitis. J. Neurosci. 2010, 30, 5866–5875. [Google Scholar] [CrossRef]
- Hughes, E.G.; Peng, X.; Gleichman, A.J.; Lai, M.; Zhou, L.; Tsou, R.; Parsons, T.D.; Lynch, D.R.; Dalmau, J.; Balice-Gordon, R.J. Cellular and Synaptic Mechanisms of Anti-NMDA Receptor Encephalitis. J. Neurosci. 2010, 30, 5866–5875. [Google Scholar] [CrossRef]
- Abe, M.; Fukaya, M.; Yagi, T.; Mishina, M.; Watanabe, M.; Sakimura, K. NMDA Receptor GluRϵ/NR2 Subunits Are Essential for Postsynaptic Localization and Protein Stability of GluRζ1/NR1 Subunit. J. Neurosci. 2004, 24, 7292–7304. [Google Scholar] [CrossRef]
- Zhang, Q.; Tanaka, K.; Sun, P.; Nakata, M.; Yamamoto, R.; Sakimura, K.; Matsui, M.; Kato, N. Suppression of synaptic plasticity by cerebrospinal fluid from anti-NMDA receptor encephalitis patients. Neurobiol. Dis. 2011, 45, 610–615. [Google Scholar] [CrossRef] [PubMed]
- Camdessanché, J.-P.; Streichenberger, N.; Cavillon, G.; Rogemond, V.; Jousserand, G.; Honnorat, J.; Convers, P.; Antoine, J.-C. Brain immunohistopathological study in a patient with anti-NMDAR encephalitis. Eur. J. Neurol. 2010, 18, 929–931. [Google Scholar] [CrossRef]
- Dalmau, J.; Tüzün, E.; Wu, H.; Masjuan, J.; Rossi, J.E.; Voloschin, A.; Baehring, J.M.; Shimazaki, H.; Koide, R.; King, D.; et al. Paraneoplastic anti–N-methyl-D-aspartate receptor encephalitis associated with ovarian teratoma. Ann. Neurol. 2007, 61, 25–36. [Google Scholar] [CrossRef]
- Yu, Y.; Wu, Y.; Cao, X.; Li, J.; Liao, X.; Wei, J.; Huang, W. The Clinical Features and Prognosis of Anti-NMDAR Encephalitis Depends on Blood Brain Barrier Integrity. Mult. Scler. Relat. Disord. 2020, 47, 102604. [Google Scholar] [CrossRef] [PubMed]
- Mao, F.; Huang, F.; Nong, W.; Lao, D.; Gong, Z.; Huang, W. N-methyl-D-aspartic acid increases tight junction protein destruction in brain endothelial cell via caveolin-1-associated ERK1/2 signaling. Toxicology 2022, 470, 153139. [Google Scholar] [CrossRef]
- Gong, Z.; Lao, D.; Wu, Y.; Li, T.; Lv, S.; Mo, X.; Huang, W. Inhibiting PI3K/Akt-Signaling Pathway Improves Neurobehavior Changes in Anti-NMDAR Encephalitis Mice by Ameliorating Blood–Brain Barrier Disruption and Neuronal Damage. Cell. Mol. Neurobiol. 2023, 43, 3623–3637. [Google Scholar] [CrossRef]
- Hampe, C.S.; Mitoma, H. A Breakdown of Immune Tolerance in the Cerebellum. Brain Sci. 2022, 12, 328. [Google Scholar] [CrossRef]
- Mitoma, H.; Buffo, A.; Gelfo, F.; Guell, X.; Fucà, E.; Kakei, S.; Schmahmann, J.D. Consensus Paper. Cerebellar Reserve: From Cerebellar Physiology to Cerebellar Disorders. Cerebellum.
- Yshii, L.; Bost, C.; Liblau, R. Immunological Bases of Paraneoplastic Cerebellar Degeneration and Therapeutic Implications. Front. Immunol. 2020, 11, 991. [Google Scholar] [CrossRef] [PubMed]
- Furneaux, H.M.; Rosenblum, M.K.; Dalmau, J.; Wong, E.; Woodruff, P.; Graus, F.; Posner, J.B. Selective Expression of Purkinje-Cell Antigens in Tumor Tissue from Patients with Paraneoplastic Cerebellar Degeneration. New Engl. J. Med. 1990, 322, 1844–1851. [Google Scholar] [CrossRef] [PubMed]
- Graus, F.; Vogrig, A.; Muñiz-Castrillo, S.; Antoine, J.-C.G.; Desestret, V.; Dubey, D.; Giometto, B.; Irani, S.R.; Joubert, B.; Leypoldt, F.; et al. Updated Diagnostic Criteria for Paraneoplastic Neurologic Syndromes. Neurol.-Neuroimmunol. Neuroinflammation 2021, 8. [Google Scholar] [CrossRef]
- Hadjivassiliou, M.; Manto, M.; Mitoma, H. Rare Etiologies in Immune-Mediated Cerebellar Ataxias: Diagnostic Challenges. Brain Sci. 2022, 12, 1165. [Google Scholar] [CrossRef]
- Flanagan, E.P. Paraneoplastic Disorders of the Nervous System. Contin. Lifelong Learn. Neurol. 2020, 26, 1602–1628. [Google Scholar] [CrossRef]
- Dalmau, J.; Rosenfeld, M.R. Paraneoplastic syndromes of the CNS. Lancet Neurol. 2008, 7, 327–340. [Google Scholar] [CrossRef] [PubMed]
- Peterson, K.; Rosenblum, M.K.; Kotanides, H.; Posner, J.B. Paraneoplastic cerebellar degeneration: I. A clinical analysis of 55 anti-Yo antibody-positive patients. Neurology 2011, 77, 1690–1690. [Google Scholar] [CrossRef]
- Small, M.; Treilleux, I.; Couillault, C.; Pissaloux, D.; Picard, G.; Paindavoine, S.; Attignon, V.; Wang, Q.; Rogemond, V.; Lay, S.; et al. Genetic alterations and tumor immune attack in Yo paraneoplastic cerebellar degeneration. Acta Neuropathol. 2018, 135, 569–579. [Google Scholar] [CrossRef]
- Titulaer, M.J.; Lang, B.; Verschuuren, J.J. Lambert–Eaton myasthenic syndrome: From clinical characteristics to therapeutic strategies. Lancet Neurol. 2011, 10, 1098–1107. [Google Scholar] [CrossRef]
- Hülsbrink, R.; Hashemolhosseini, S. Lambert-Eaton myasthenic syndrome - diagnosis, pathogenesis and therapy. Clin. Neurophysiol. 2014, 125, 2328–2336. [Google Scholar] [CrossRef]
- Fukuda, T.; Motomura, M.; Nakao, Y.; Shiraishi, H.; Yoshimura, T.; Iwanaga, K.; Tsujihata, M.; Dosaka-Akita, H.; Eguchi, K. Reduction of P/Q-type calcium channels in the postmortem cerebellum of paraneoplastic cerebellar degeneration with Lambert-Eaton myasthenic syndrome. Ann. Neurol. 2002, 53, 21–28. [Google Scholar] [CrossRef]
- Shimizu, F.; Takeshita, Y.; Sano, Y.; Hamamoto, Y.; Shiraishi, H.; Sato, T.; Yoshimura, S.; Maeda, T.; Fujikawa, S.; Nishihara, H.; et al. GRP78 antibodies damage the blood–brain barrier and relate to cerebellar degeneration in Lambert-Eaton myasthenic syndrome. Brain 2019, 142, 2253–2264. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.S. Glucose-regulated proteins in cancer: Molecular mechanisms and therapeutic potential. Nat. Rev. Cancer 2014, 14, 263–276. [Google Scholar] [CrossRef] [PubMed]
- Jakobsen, C.G.; Rasmussen, N.; Laenkholm, A.-V.; Ditzel, H.J. Phage Display–Derived Human Monoclonal Antibodies Isolated by Binding to the Surface of Live Primary Breast Cancer Cells Recognize GRP78. Cancer Res. 2007, 67, 9507–9517. [Google Scholar] [CrossRef]
- Mintz, P.J.; Kim, J.; Do, K.A.; et al. Fingerprinting the circulating repertoire of antibodies from cancer patients. Nat. Biotechnol. 2003, 21, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Amaresan, R.; Gopal, U. Cell surface GRP78: A potential mechanism of therapeutic resistant tumors. Cancer Cell Int. 2023, 23, 100. [Google Scholar] [CrossRef]
- Tanigawa; Tsunemi, S. ; Nakanishi, T.; Fujita, Y.; Bouras, G.; Miyamoto, Y.; Miyamoto, A.; Nomura, E.; Takubo, T.; Tanigawa, N. Proteomics-based identification of a tumor-associated antigen and its corresponding autoantibody in gastric cancer. Oncol. Rep. 2010, 23, 949–956. [Google Scholar] [CrossRef]
- Raiter, A.; Vilkin, A.; Gingold, R.; Levi, Z.; Halpern, M.; Niv, Y.; Hardy, B. The Presence of Anti-GRP78 Antibodies in the Serum of Patients with Colorectal Carcinoma: A Potential Biomarker for Early Cancer Detection. Int. J. Biol. Markers 2014, 29, 431–435. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Gronow, M.; Pizzo, S.V. Physiological Roles of the Autoantibodies to the 78-Kilodalton Glucose-Regulated Protein (GRP78) in Cancer and Autoimmune Diseases. Biomedicines 2022, 10, 1222. [Google Scholar] [CrossRef]
- Shahmohammadi, S.; Doosti, R.; Shahmohammadi, A.; Azimi, A.; Sahraian, M.A.; Fattahi, M.-R.; Moghadasi, A.N. Neuromyelitis optica spectrum disorder (NMOSD) associated with cancer: A systematic review. Mult. Scler. Relat. Disord. 2021, 56, 103227. [Google Scholar] [CrossRef]
- Srichawla, B.S.; Doshi, K.; Cheraghi, S.N.; Sivakumar, S. The temporal relationship of paraneoplastic aquaporin-4-IgG seropositive neuromyelitis optica spectrum disorder (NMOSD) and breast cancer: A systematic review and meta-analysis. Neurol. Sci. 2023, 44, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Duan, Y.; Wang, X.; Duan, X.; Gao, H.; Ji, X.; Xiao, X.; Zhu, F.; Xue, Q. Neuromyelitis optica spectrum disorders associated with AQP4-positive-cancer—A case series. Front. Neurol. 2022, 13, 1071519. [Google Scholar] [CrossRef]
- Minomo, S.; Ichijo, M.; Shimizu, F.; Sato, R.; Kanda, T.; Takai, Y.; Misu, T.; Sakurai, Y.; Amino, T.; Kamata, T. Paraneoplastic Neuromyelitis Optica Spectrum Disorder Related to Glucose-regulated Protein 78 (GRP78) Autoantibodies in a Patient with Lynch Syndrome-associated Colorectal Cancer. Intern. Med. 2023, 62, 1653–1657. [Google Scholar] [CrossRef]
Figure 1.
Structure of the blood-brain barrier (BBB). (A) The blood-brain barrier (BBB) consists of endothelial cells, pericytes, astrocytes and basement membrane. The neurovascular unit (NVU) is composed of endothelial cells, pericytes and astrocytes of the BBB, neurons, oligodendrocytes and microglia, which closely communicate with each other in order to regulate brain homeostasis. (B) Tight junctions (claudin-5, occludin, ZO-1, ZO-2 and ZO-3) and adhesion junctions (JAMs and VE-cadherin) between BBB-endothelial cells (BBB-ECs) develop the BBB. (C) Transcellular migration of lymphocytes involves the following 4 steps: 1) in the rolling process, activated lymphocytes slow their flow speed due to the interaction of VLA-4 from the surface of lymphocyte with vascular cell adhesion molecules 1 (VCAM-1) on BBB-ECs; 2) in adhesion pathways, the lymphocytes adhere to endothelial cells and transverse the BBB by coupling the VLA-4 and LFA-1 expressed on lymphocytes with the endothelial cell receptor (VCAM-1 and intracellular adhesion molecules (ICAM-1); 3) in adhesion, interaction between VCAM-1 and ICAM on BBB-ECs and their ligands (LFA-1 and VLA-4) on leukocytes induces the arrest of immune cells from the blood at the brain endothelial cells; 4) interaction between ICAM-1 and ICAM-2 and their ligands (LFA-1 and LFA-2) is involved in crawling and migration.
Figure 1.
Structure of the blood-brain barrier (BBB). (A) The blood-brain barrier (BBB) consists of endothelial cells, pericytes, astrocytes and basement membrane. The neurovascular unit (NVU) is composed of endothelial cells, pericytes and astrocytes of the BBB, neurons, oligodendrocytes and microglia, which closely communicate with each other in order to regulate brain homeostasis. (B) Tight junctions (claudin-5, occludin, ZO-1, ZO-2 and ZO-3) and adhesion junctions (JAMs and VE-cadherin) between BBB-endothelial cells (BBB-ECs) develop the BBB. (C) Transcellular migration of lymphocytes involves the following 4 steps: 1) in the rolling process, activated lymphocytes slow their flow speed due to the interaction of VLA-4 from the surface of lymphocyte with vascular cell adhesion molecules 1 (VCAM-1) on BBB-ECs; 2) in adhesion pathways, the lymphocytes adhere to endothelial cells and transverse the BBB by coupling the VLA-4 and LFA-1 expressed on lymphocytes with the endothelial cell receptor (VCAM-1 and intracellular adhesion molecules (ICAM-1); 3) in adhesion, interaction between VCAM-1 and ICAM on BBB-ECs and their ligands (LFA-1 and VLA-4) on leukocytes induces the arrest of immune cells from the blood at the brain endothelial cells; 4) interaction between ICAM-1 and ICAM-2 and their ligands (LFA-1 and LFA-2) is involved in crawling and migration.
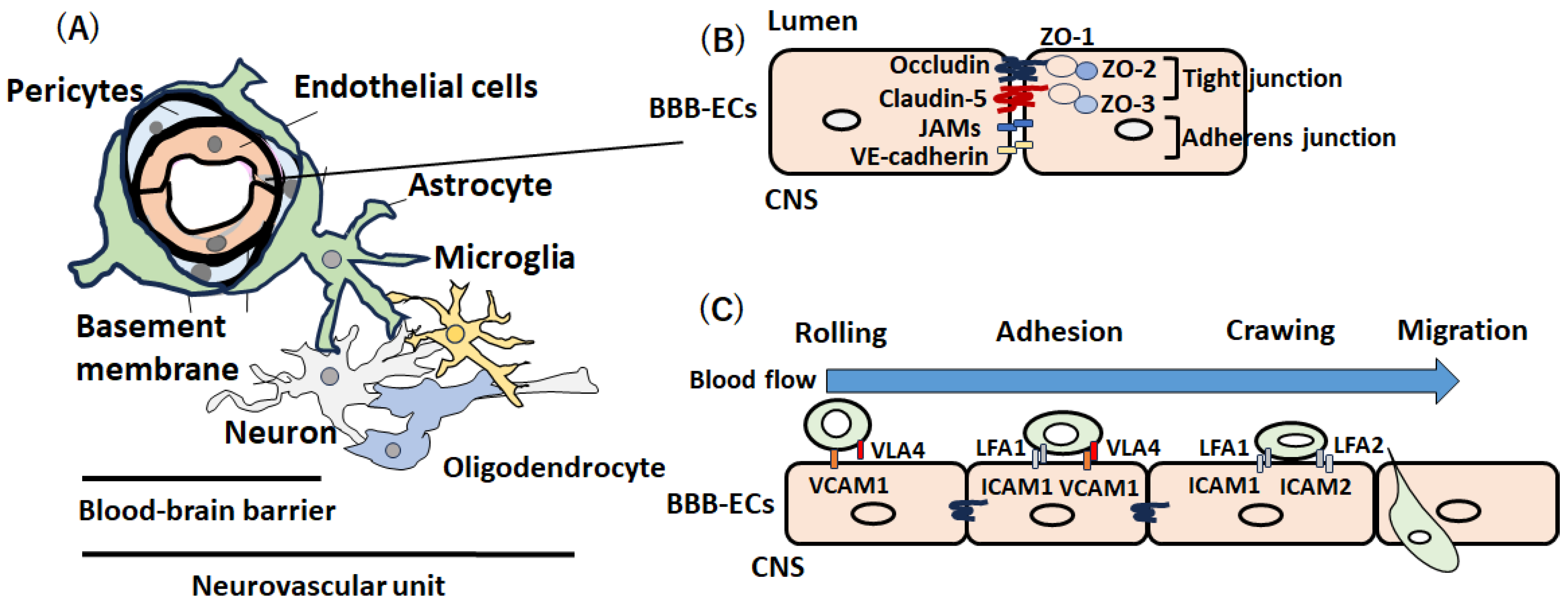
Figure 2.
Flow of lymphocytes in multiple sclerosis (MS). After T and B lymphocytes are activated in the periphery, they infiltrate the CNS and trigger the central autoimmune response, leading to myelin and axonal damage during onset or remission in MS. The cell adhesion molecules (CAMs) expressed on the surface of inflamed endothelial cells, including vascular cell adhesion molecule 1 (VCAM-1), intracellular adhesion molecules (ICAM-1), melanoma cell adhesion molecules (MCAMs) and activated leukocyte cell adhesion molecules (ALCAMs), play a role in trans-endothelial immune cell infiltration. After activated lymphocytes disrupt the BBB, massive numbers of lymphocytes enter the CNS, leading to relapse in MS. Lymphocytes in the CNS aggregate in the meninges in progressive MS.
Figure 2.
Flow of lymphocytes in multiple sclerosis (MS). After T and B lymphocytes are activated in the periphery, they infiltrate the CNS and trigger the central autoimmune response, leading to myelin and axonal damage during onset or remission in MS. The cell adhesion molecules (CAMs) expressed on the surface of inflamed endothelial cells, including vascular cell adhesion molecule 1 (VCAM-1), intracellular adhesion molecules (ICAM-1), melanoma cell adhesion molecules (MCAMs) and activated leukocyte cell adhesion molecules (ALCAMs), play a role in trans-endothelial immune cell infiltration. After activated lymphocytes disrupt the BBB, massive numbers of lymphocytes enter the CNS, leading to relapse in MS. Lymphocytes in the CNS aggregate in the meninges in progressive MS.
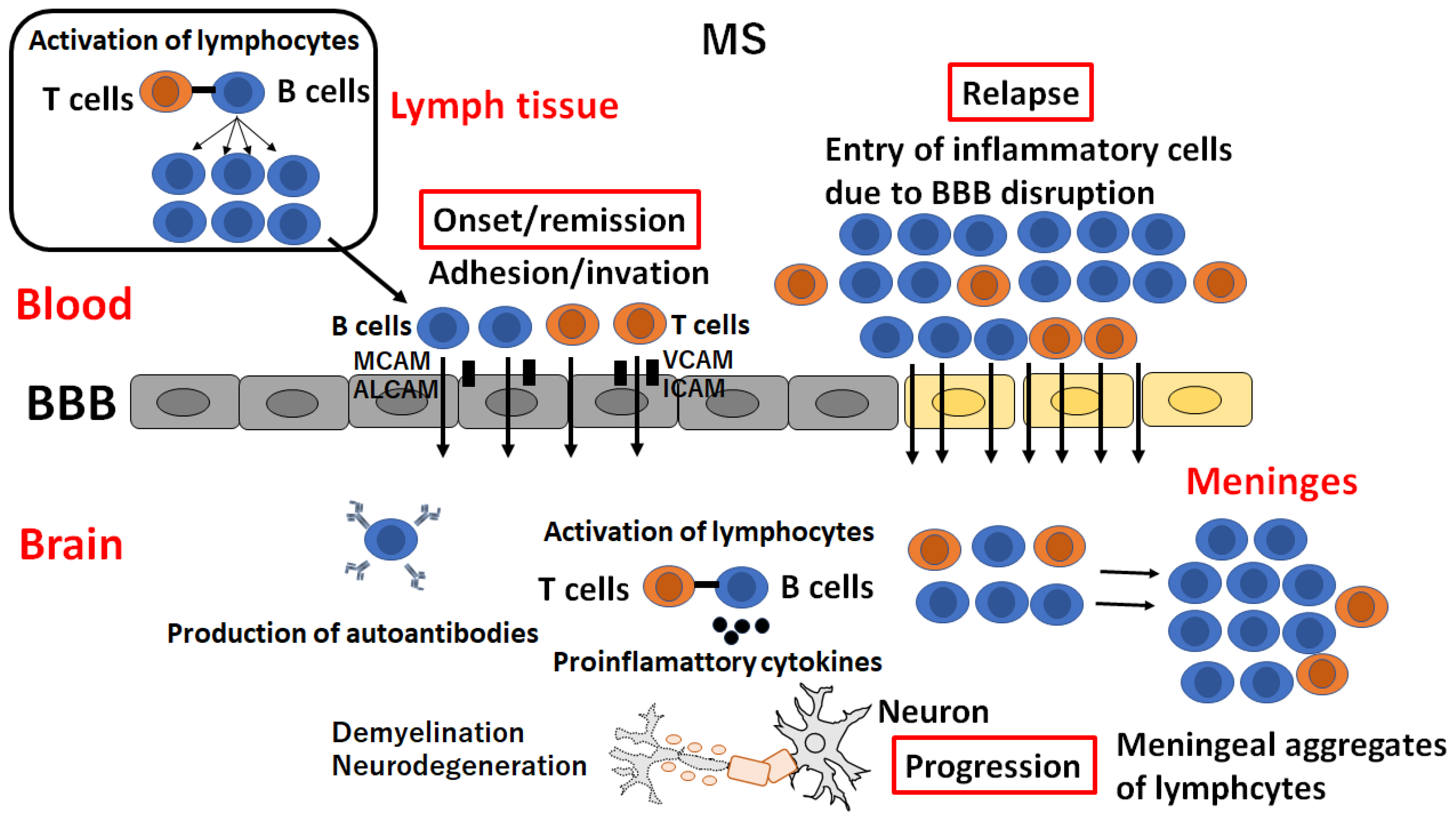
Figure 3.
Possible mechanism of BBB breakdown in NMOSD. GRP78 autoantibodies in NMO-IgG bind to GRP78 on the blood-brain barrier (BBB)-endothelial cells (ECs) and activate nuclear factor-kappa B (NF-κB) signaling in BBB-ECs, leading to enhanced permeability via degradation of tight junctions. Infiltration of AQP4 autoantibodies in NMO-IgG into the CNS causes binding of AQP4 autoantibodies to AQP4 on the endfeet of astrocytes, thus giving rise to complement-dependent astrocyte cytotoxicity. IL-6 secreted by astrocytes induces inflammation, which further mediates BBB breakdown.
Figure 3.
Possible mechanism of BBB breakdown in NMOSD. GRP78 autoantibodies in NMO-IgG bind to GRP78 on the blood-brain barrier (BBB)-endothelial cells (ECs) and activate nuclear factor-kappa B (NF-κB) signaling in BBB-ECs, leading to enhanced permeability via degradation of tight junctions. Infiltration of AQP4 autoantibodies in NMO-IgG into the CNS causes binding of AQP4 autoantibodies to AQP4 on the endfeet of astrocytes, thus giving rise to complement-dependent astrocyte cytotoxicity. IL-6 secreted by astrocytes induces inflammation, which further mediates BBB breakdown.
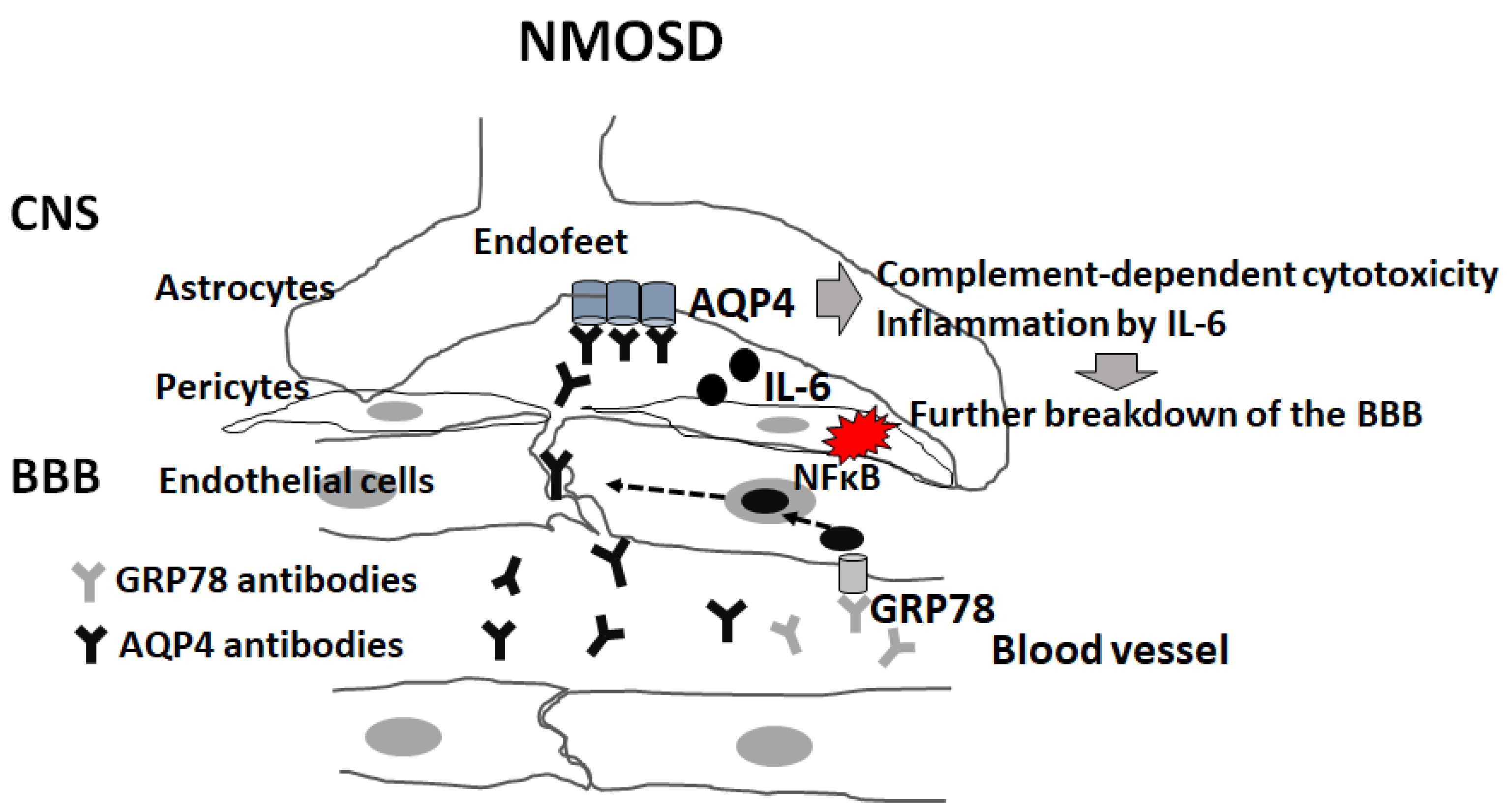
Figure 4.
Possible mechanism of BBB breakdown in PCD-LEMS. Glucose-regulated protein (GRP) 78 autoantibodies and P/Q-type voltage-gated calcium channel (VGCC) autoantibodies are produced by cross-reactivity with tumor cells, as both GRP78 and P/Q-type VGCC are expressed on the surface of tumor cells (1). After GRP78 autoantibodies bind to GRP78 on endothelial cells, permeability of the BBB is increased via the activation of nuclear factor-kappa B (NF-κB) (2), thereby inducing the infiltration of P/Q-type VGCC autoantibodies into the CNS space (3). Binding of P/Q-type VGCC autoantibodies to P/Q-type VGCC causes injury to Purkinje and granule cells (4).
Figure 4.
Possible mechanism of BBB breakdown in PCD-LEMS. Glucose-regulated protein (GRP) 78 autoantibodies and P/Q-type voltage-gated calcium channel (VGCC) autoantibodies are produced by cross-reactivity with tumor cells, as both GRP78 and P/Q-type VGCC are expressed on the surface of tumor cells (1). After GRP78 autoantibodies bind to GRP78 on endothelial cells, permeability of the BBB is increased via the activation of nuclear factor-kappa B (NF-κB) (2), thereby inducing the infiltration of P/Q-type VGCC autoantibodies into the CNS space (3). Binding of P/Q-type VGCC autoantibodies to P/Q-type VGCC causes injury to Purkinje and granule cells (4).
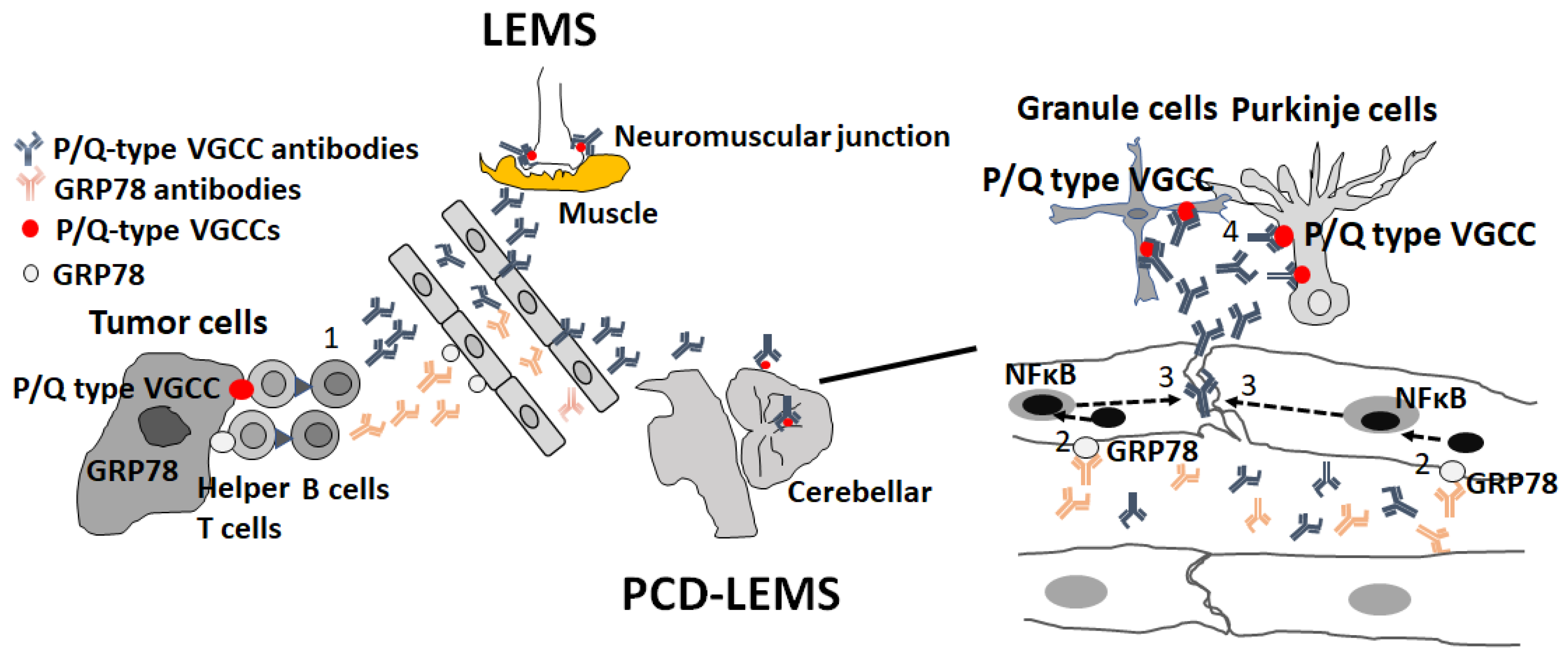

Table 1.
Effect of DMT on the BBB-ECs.
| DMT | Effects on the BBB-ECs | Cross the BBB? |
|---|---|---|
| Steroid | ↑Tight junction (occludin, claudin-5) ↑BBB function ↑TIMP-1 ↓MMP-9 |
Yes |
| Interferon-β | ↑BBB function ↓VCAM-1, ICAM-1, E selectin ↓Trans-endothelial migration of lymphocyte |
Yes, slightly |
| Anti-α4 integrin Ab Natalizumab |
↓↓Trans-endothelial migration of lymphocyte (Block the interaction between α4 integrin and VCAM1) |
None |
| Anti-CD20 Abs Ofatumumab, Ocrelizumab, Rituximab, |
Indirect effect due to depletion of B cells ↓proinflammatory cytokines ↓complement activity ↓autoantibody |
None |
| Fingolimod | ↓S1P1/S1P3 on the surface of BMECs ↓Trans-endothelial migration of lymphocyte ↓VCAM-1 ↑Tight junction (claudin-5) |
Yes |
| Dimethyl fumarate | ↓Trans-endothelial migration of lymphocyte ↓VCAM-1, ICAM-1 ↑BBB function via Nrf2-activity |
Yes |
| Cladribine | ↓ICAM-1, E-selectin ↓MMP-2, MMP-9 ↓CXCL8, CCL5 |
Yes |
| Anti-IL6 Ab Satralizumab |
↑BBB function ↓CCL2, CXCL8 ↓Trans-endothelial migration of lymphocyte |
None |
| Anti-CD19 Ab Inebilizumab |
Indirect effect due to depletion of B cells ↓proinflammatory cytokines ↓complement activity ↓autoantibody |
None |
| Anti-complement Ab Eculizumab Ravulizumab |
Unknown | Unknown |
DMT=disease-modifying therapies, BBB-ECs=blood-brain barrier-endothelial cells, TIMP=tissue inhibitor of metalloproteinases-1, MMP=matrix metalloproteinases, VCAM=vascular cell adhesion molecule, ICAM=Intercellular adhesion molecule, CXCL=C-X-C motif chemokine ligand, CCL=chemokine (C-C motif) ligand.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



