
Submitted:
17 August 2024
Posted:
20 August 2024
You are already at the latest version
Abstract
Background. Respiratory distress syndrome (RDS) is the primary cause of respiratory failure in preterm infants, but it also affects 5-7% of term infants. Dysfunctions in pulmonary surfactant metabolism, resulting from mutations of the lung surfactant genes, are rare diseases, ranging from fatal neonatal RDS to interstitial lung disease, associated with increased morbidity and mortality. This study aims to clarify the clinical significance of ABCA3 variants found in a specific family case, as existing data in the literature is inconsistent. Material and methods: A family case report was conducted; targeted panel genetic testing identified a variant of the SFTPB gene and two variants of ABCA3 genes. Comprehensive research involving a systematic review of PubMed, Google Scholar databases, and genome browsers was used to clarify the pathogenicity of the two ABCA3 variants found in the index patient. Advanced prediction tools were employed to assess the pathogenicity of the two ABCA3 variants, ensuring the validity and reliability of our findings. Results: The index case exhibited fatal neonatal RDS. Genetic testing revealed the presence of the SFTPB p.Val267Ile variant, not previously reported, a benign variant based on family genetic testing and history. Additionally, two ABCA3 gene variants were identified: c.697C>T, not yet reported, and c.838C>T. These variants were found to affect ABCA3 protein function and were likely associated with neonatal RDS. Prediction tools and data from nine other cases in the literature supported this conclusion. Conclusions: Based on in silico predictors, analysis of the presented family, and cases described in the literature, it is reasonable to consider reclassifying the two ABCA3 variants identified in the index case as pathogenic/pathogenic. Reclassification will improve genetic counseling accuracy and facilitate correct diagnosis.
Keywords:
ABCA3 c.838C>T
; ABCA3 p.Arg280Cys
; ABCA3 R280C
; ABCA3 c.697C>T
; ABCA3 p.Gln233Ter
; ABCA3 Q233X
; SFTPB p.Val267Ile
; neonatal RDS
; interstitial lung disease
; genetic testing
1. Introduction
Lung surfactant is a complex surface-active mixture of proteins (surfactant protein A, B, C, and D, 1-2%) and lipids (80-85% phospholipids and around 10% neutral lipids, mainly cholesterol) [1], a film covering the alveolar surface, with a crucial role in reducing the alveolar surface tension, maintaining normal gas exchanges and preventing end-expiratory alveolar collapse [2,3,4]. Clinically, respiratory distress syndrome (RDS) in neonates due to surfactant deficiency is suggested by grunting, tachypnea, nasal flaring, and thoracic retractions, and if gas exchange is significantly impaired, cyanosis may also occur. RDS is the leading cause of respiratory failure in preterm infants [5], one of the main reasons for admission in neonatal intensive care units (NICUs) [6], mainly occurring in preterm infants due to surfactant quantitative deficiencies – surfactant insufficient production or inactivation in the undeveloped lungs of the preterm infants below 37 weeks of gestation, surfactant qualitative deficiencies, or sepsis [7,8,9,10,11]; RDS is also diagnosed in 5-7% of the term infants [2,7,12]. In comparison, in late preterm (gestational age 34-36 weeks) and term infants (gestational age ≥37 weeks), RDS is mainly associated with delayed transition to extrauterine life (transient tachypnea of the newborn), meconium aspiration syndrome, sepsis/pneumonia, congenital cardiac defects, persistent pulmonary hypertension, air leak syndromes, and diaphragmatic hernia [2,3,6,7,8,9,12,13]. Less often, perinatal asphyxia, hypothermia, oropharyngeal, airway, and lung congenital abnormalities, and cystic fibrosis may present as RDS in near-term and term infants during the immediate neonatal period [2,3,6,13,14,15,16].
Pathogenic gene variants can lead to pulmonary surfactant metabolism dysfunctions, also known as surfactant dysfunction disorders. These disorders are particularly observed in near-term and term neonates or cases of unexpectedly severe RDS in preterm infants, considering their gestational age or associated perinatal risk factors [3,10]. Dysfunctions in pulmonary surfactant metabolism, a group of rare diseases, can be caused by variants in the genes responsible for surfactant biosynthesis [2]: SFTPA1, SFTPA2, SFTPA3, SFTPB, SFTPC, SFTPD, ABCA3, NKX2.1 [2,13,17,18,19]. The impact of these variants can lead to both qualitative and quantitative surfactant deficiencies [20], characterized by a broad spectrum of clinical manifestations, ranging from fatal neonatal respiratory failure to interstitial lung disease (ILD) in older children and even adults [8,21,22]. Consequently, these deficiencies significantly increase the neonatal and pediatric morbidity and mortality [15,22]. There are several inherited surfactant disorders known.
Surfactant Protein B (SFTPB)
Hereditary deficiency of surfactant protein B was the first genetic defect identified as genetic surfactant deficiency (OMIM 178640), presenting as severe neonatal RDS [23].
Over 50 mutations of the SFTPB gene are reported in the literature [15,24]; the estimated prevalence of SFTPB deficiency in the USA is 1 per 1 million live births [1,25]. Among these, one frameshift mutation – c.397delCinsGAA - represents 50-70% of the pathogenic variants [25,26]. The almost complete absence of SFTPB mRNA and SFTPB occurs due to unstable transcription [15,27]. Bi-allelic mutations of the SFTPB gene cause SFTPB deficiency associated with severe RDS with clinical and radiological aspects similar to RDS secondary to surfactant qualitative deficiency seen in preterm infants [10,15,17,23,28]. Survival was reported in cases with at least one allele that allows residual SFTPB protein production (partial deficiency) [29,30]. No major effects are seen in heterozygous carriers of SFTPB variants; these mutations, most probably, allow partial synthesis of functional SFTPB protein [15,24,29,30,31]. In SFTPB deficiency, symptoms occur a short time after delivery, usually in term newborns, and progress to refractory respiratory failure and death or need for lung transplantation in the first months of life [15,16,26,32,33].
Surfactant Protein C (SFTPC)
Over 40 pathogenic variants of the SFTPC gene are linked to surfactant deficiencies [1,26,33,34,35,36]. Variants of the SFTPC gene are associated with severe, fatal neonatal lung disease and ILD in older children (surfactant, pulmonary-associated protein C; SFTPC—OMIM 178620) [23,26,32,33,34,35,37].
Surfactant Proteins A and D (SFTPA and SFTPD)
Surfactant proteins A1, A2, and D are hydrophilic proteins involved in lung innate host defense; SFTPA, SFTPA1, and SFTPD proteins have the ability to opsonize and enhance the killing of bacteria, viruses, and fungi [38,39,40]. A role in maintaining surfactant lipids homeostasis was also described [41]. Polymorphisms of the SFTPA and SFTPD genes were described in association with increased susceptibility to RDS and bronchopulmonary dysplasia in preterm infants but not in term infants [1,42,43] and infections due to respiratory syncytial virus [44,45].
NKX2.1 Gene
The NKX2.1 gene encodes the thyroid transcript factor 1 (TTF 1), a factor with a critical role in regulating the expression of over 1300 genes, including the essential genes involved in thyroid development, lung development, homeostasis, and its expression, through feedback [6,10,15,25,46]. Variants of the NKX2.1 gene are associated with severe multisystemic disease (brain-lung-thyroid syndrome), characterized by autosomal dominant inheritance and variable penetrance, with no described genotype/phenotype correlation. This condition often manifests as severe neonatal RDS or ILD [6,8,15,24,47,48,49].
ABCA3 Gene
ABCA3 (ATP Binding Cassette Family A Member 3) belongs to the ATP binding cassette transporter superfamily of proteins that uses energy derived from ATP hydrolysis for substrate translocation through biological membranes [11,50]. ABCA3 phospholipid glycoprotein is localized in the lamellar bodies and is essential for surfactant biosynthesis and central regulation of the lung surfactant balance [51,52]. ABCA3 gene is expressed intensely in the alveolar epithelial cell II (AEC II), the same cells where surfactant is produced [15].
Variants of the ABCA3 gene are the most commonly reported in primary surfactant deficiency (surfactant metabolism dysfunction, OMIM 601615, OMIM 61092), with more than 400 variants documented in the literature [14,53,54]. Most reported cases of surfactant deficiencies associated with ABCA3 gene variants manifest as mild to severe, unexplained, or fatal RDS in near-term and term neonates or as ILD [14,15,21,22].
This review will focus on ABCA3’s role in surfactant biosynthesis, its central regulation, and the function of lung surfactant balance. Additionally, we will address the need to clarify the clinical significance of ABCA3 variants, focusing on ABCA3:c.838C>T and ABCA3:c.697C>T variants, by presenting a family case study and a literature review on these mutations.
2. Materials and Methods
2.1. Molecular Analysis
We present a case study of a family with an early-term infant with fatal neonatal RDS. The infant was found to be a compound heterozygous for the ABCA3 gene, as determined through molecular analysis using Next Generation Sequencing (NGS) with a targeted panel conducted by Laborärzte Singen in Singen (Hohentwiel), Germany. The variants confirmation was performed by capillary sequencing. Genomic DNA isolation from a buccal swab was performed using a Prepito NA kit (Perkin Elmer, Baesweiler, Germany); AmpliTaq Gold 360 Master Mix (ThermoFisher Scientific, Darmstadt, Germany) was used for PCR amplification, and a Rapid PCR Cleanup Enzyme set (New England Biolabs, Frankfurt, Germany) was used for PCR products purification. Probe sequencing was performed using the BigDye Terminator v3.1 Cycle Sequencing Kit (ThermoFisher Scientific, Darmstadt, Germany); electrophoresis of the sequenced products was performed using Applied Biosystems 3500Dx Genetic Analyzed (ThermoFisher Scientific, Darmstadt, Germany).
2.2. Literature Systematic Review
Given the lack of consensus on the clinical significance of ABCA3 gene variants identified in our patient, we conducted a systematic literature review to clarify existing knowledge gaps and compile the best available data.
We conducted an extensive and systematic search of the PubMed and Google Scholar databases up to July 2024, using the keywords for ABCA3 gene: “c.838C>T”, “p.Arg280Cys“, “R280C”, “c.697C>T”, “p.Gln233Ter”, ” Q233X ”, and ” Q233*”, alone or in association with the keyword ”mutation”/”variant”. Additionally, we performed further searches on the Varsome, ClinVar, and Ensembl genome browsers, concentrating on variant classification and related literature. All types of articles were analyzed. After excluding papers without relevance for the ABCA3 gene and duplicates, we analyzed the bibliographies of the selected articles to identify additional relevant publications aligned with the objectives of this paper. A thorough review of the full texts of each included article was conducted, focusing on identifying case reports involving the two ABCA3 variants found in the presented patient. We evaluated 332 publications for relevance and inclusion in our study; of these, only 7 papers were relevant to our review (Figure 1).
To predict the pathogenicity of amino acid substitutions and their molecular mechanisms, we employed advanced tools such as MutPred2, MutPred-LOF, and PolyPhen-2. These standalone and web applications are developed to classify amino acid substitutions as pathogenic or benign in humans, providing accurate and reliable predictions.
3. Results
3.1. Case Report
3.1.1. Clinical Aspects
The index case is a male newborn delivered by C-section in a level I maternity hospital due to pregnancy-induced hypertension, at 37 weeks gestational age, with a birth weight of 2600 g (10-25th percentile), length 47 cm (10-25th percentile), cranial circumference 32 cm (25th percentile), Apgar score 9/10. Persistent tachypnea and mild intercostal retractions with onset after 4 h of life were interpreted initially as transient tachypnea of the newborn, and the infant was submitted to our unit. At arrival, at 36 h of life, mild generalized cyanosis, tachypnea (60-70 breaths/minute), mild intercostal retractions, increased anterior-posterior thoracic diameter, and irritability were noted. Oxygen on nasal cannula, intravenous fluids, and prophylactic antibiotic therapy (Penicillin and Amikacin) were started while the first investigations were performed. Thoracic radiography showed inhomogeneous opacification of both lungs (Figure 2, A); mild persistence of fetal circulation (patent ductus arteriosus and foramen ovale, mild tricuspid valve regurgitation) was noted on Doppler cardiac ultrasound. The results of all other tests – biochemistry, microbiology, immunology, hematology – were all in normal limits except mild anemia (hemoglobin 11,8 g/dL, hematocrit 38%) and hypoxemia on blood gas analysis (arterial partial oxygen pressure, PaO2 of 42.8 mmHg). The persistent respiratory effort associated with an increased need for oxygen (paO2 41.7 mmHg) imposed increased respiratory support at 48 h, and heated, humidified high flow nasal (HHHFNC) cannula was started for two days, followed by intubation and mechanical ventilation from the fourth day of life (DOL), Assist/Control mode for the first 48 h, switched to synchronized intermittent mandatory ventilation (SIMV) for the next 72 h. The infant was extubated on HHHNFC at 9 DOL after reaching normal blood gases without oxygen supplementation (lung x-ray is presented in Figure 2, B). Still, hypoxemia reoccurred (PaO2 43.6 mmHg), associated with recurrence of respiratory distress signs (tachypnea, intercostal retractions), and in the absence of any other laboratory abnormalities; repeated Doppler heart ultrasound showed only persistent foramen ovale. Persistently low oxygen saturations, hypoxemia on blood gases (PaO2 36 mmHg), aggravation of the respiratory distress on HHHFNC on 100% oxygen, and the presence of bilateral focal interstitial opacities on the thoracic x-ray prompted re-intubation (Figure 2, C) and mechanical ventilation. Synchronized intermittent positive pressure ventilation (SIPPV) was used initially. However, due to a persistent increased need for oxygen (up to 100% oxygen), the infant was switched to high-frequency oscillation ventilation (HFOV) the moment the lung x-ray showed homogenous opacification of both lungs (Figure 2, D). Persistent hypoxemia (PaO2 between 25.6-51.5 mmHg) was noted despite continuous ventilator setting adjustments and different invasive respiratory support strategies trials during the following weeks.
The unusual clinical aspect of the respiratory distress of the infant suggested even from the first days of life that the initial diagnosis of transient tachypnea of the newborn was very unlikely, and we continued the investigations to clarify the etiology and to adjust and optimize the treatment. Other potential causes were eliminated after reviewing the maternal history, pregnancy, and delivery outcome, as well as the results of the blood tests, thoracic X-rays, and cardiac and cerebral ultrasounds, leading to a diagnosis by exclusion. A genetic disease of surfactant metabolism was suspected, and genetic testing was performed using one NGS panel. The results highlighted that the patient was compound heterozygous for ABCA3 c.838C>T (p.Arg280Cys, R280C, rs201299260), and c.697C>T (p.Gln233Ter, Q233X, Q233*). In addition, the patient was found to be heterozygous for SFTPB p.Val267Ile. No variants were described in the protein-coding exons, intron regions flanking the exons, and within 250 nucleotides 5′ of the start codon of the SFTPC gene.
As a result, methylprednisolone was administered in pulse therapy for three days, followed by prednisolone administration. Also, hydroxychloroquine, daily, and azithromycin, three days per week, completed the therapeutic protocol; continuing invasive respiratory support as other advanced life support, such as nitric oxide and extracorporeal membrane oxygenation, were not available at that time. No improvements in respiratory function were noted, and the severe respiratory failure led to death on the 77th DOL.
3.1.2. Autopsy and Histology
The autopsy revealed bilateral lung atelectasis, dilated pulmonary artery, and right ventricular hypertrophy secondary to severe and prolonged respiratory failure.
Lung fragments collected at autopsy were analyzed using microscopy. Chronic infantile pneumonitis, with reduced alveolarization, diffuse marked widening of the alveolar interstitium, lobular remodeling, diffuse marked AEC II hyperplasia, large number of intra alveolar macrophages, foamy cells, and few giant cells focally accompanied by cholesterol clefts, focal alveolar proteinosis, and extended areas of desquamative interstitial pneumonia seen on microscopy were also suggestive for lung damage associated with surfactant protein deficiencies (Figure 3). No signs of persistent pulmonary hypertension were seen.
3.1.3. Pathogenic Prediction
According to ClinVar (hosted by the National Center for Biotechnology Information (NCBI)), for ABCA3 c.838C>T (p.Arg280Cys, R280C) variant there are conflicting classifications of pathogenicity (https://www.ncbi.nlm.nih.gov/clinvar/variation/318566/) [55] while Ensemble reports it as likely pathogenic (https://www.ensembl.org) [56]. The Clinvar variation ID is 318566 and is reported to disrupt the ABCA3 function and in association with autosomal recessive interstitial lung disease; the PolyPhen-2 predicts this variant is probably damaging with a high score of 0.989, suggesting also/again a deleterious effect on protein function. [(http://genetics.bwh.harvard.edu/pph2/)].
We used the MutPred2 software [57] to evaluate the pathogenicity of the ABCA3 c.838C>T (p.Arg280Cys, R280C) substitution in our patient. The MutPred2 score of 0.543 suggests a moderate probability that the variant is deleterious to protein function. As regards the molecular mechanism, the MutPred2 predicts loss of ADP-ribosylation and altered transmembrane protein (probability of 0.22 and 0.1, respectively, with p-value=0.03).
The ABCA3 c.697C>T (p.Gln233Ter, Q233X) variant was not identified in databases, such as the Genome Aggregation Database, 2022. Due to mutation in codon 233 (Glutamine, CAG), a stop codon (TAG) is generated, leading to a shortened transcript and thus causing truncated protein. Nonsense variants of the ABCA3 gene are classified into “null” mutations. To predict the effect of nonsense mutation, we used MutPred-LOF software [58]. The MutPred-LOF score was 0.369, indicating a moderate probability of a loss-of-function effect on the protein caused by this variant. Additionally, it enabled us to assess the functional consequences of this variant on the protein, as follows: catalytic site(p=0); PPI hotspot(p=0); iron binding(p=0); sulfation(p=0.0001); proteolytic cleavage(p=0.0002); data suggesting high confidence in the prediction of a catalytic site, protein-protein interaction (PPI) hotspot and iron-binding site in the protein and significant chance of a sulfation site and proteolytic cleavage site being present.
3.1.4. Genetic Counseling
Furthermore, we initiated a familial investigation through targeted sequencing, which revealed that the mother is a carrier of the ABCA3 c.697C>T (p.Gln233Ter) variant. The father was identified as a carrier of both the ABCA3 c.838C>T (p.Arg280Cys, R280C) and SFTPB p.Val267Ile variant. The parents are non-consanguineous, in good health, and have no family history of respiratory conditions. Almost two years later, a second pregnancy occurred, and prenatal diagnosis was conducted through chorionic villus sampling. The sequencing results showed the presence of the ABCA3 p.Gln233Ter and SFTPB p.Val267Ile variants. The pregnancy proceeded to term, and the child had no respiratory symptoms during the neonatal period despite being a carrier of the ABCA3 p.Gln233Ter and SFTPB p.Val267Ile. The second child in the family was born at 39 weeks gestation, with a birth weight of 3670 g, had an uneventful neonatal course, and had no noticeable respiratory illnesses up to the age of 6 years (see complete pedigree in Figure 4).
Given the three variants discovered in the patient and the results of the genetic testing performed for the rest of the family, we tried to find the variant(s) responsible for the patient’s clinical picture. SFTPB p.Val267Ile, with uncertain significance, has not been reported in the literature so far. Considering that this variant was also identified in the healthy brother and father, this alteration may be considered a benign variant, a polymorphism that does not cause SFTPB deficiency.
ABCA3 gene variants identified in our patients affect the protein function and may be associated with a surfactant defect or deficiency. Considering the in silico predictors, family pedigree, and clinical manifestation, despite the current classification of these variants as uncertain, it is reasonable to consider that these variants explain the symptomatology and need to be reclassified as probably pathogenic/pathogenic. Reclassifying these variants will provide more accurate genetic counseling and ensure a correct diagnosis, appropriate treatment and optimal outcomes.
The unique characteristics of this family highlight the urgent need for reclassification of the clinical significance of these variants and underscore the importance for clinicians to carefully identify compound heterozygotes in similar cases and all autosomal recessive disorders. This is particularly critical given that bi-allelic mutations in the ABCA3 gene are the most frequently reported cause of primary surfactant deficiency (OMIM 601615) [8,14,53].
3.2. Review of the Literature on ABCA3 c.838C>T (p.Arg280Cys, R280C)
A final analysis of our patient’s symptoms and clinical course, imaging, histology, familial history, and genetic testing results for the proband and his family suggested that the early neonatal onset of the unexplained RDS on the proband could be explained by his compound heterozygous status for the mentioned ABCA3 variants (Q233X of maternal origin and R280C of paternal origin).
We reviewed the literature to identify similar cases and clarify the pathogenicity of ABCA3 c.838C>T (p.Arg280Cys, R280C) and c.697C>T (p.Gln233Ter, Q233X, Q233*) variants. The variant ABCA3 R280C allele frequency is 0.00019 in the large population dataset of the Genome Aggregation Database (gnomAD). As previously mentioned, conflicting pathogenicity classifications exist despite several reported cases and predictive software indicating the variants are probably pathogenic or pathogenic.
Of the 333 articles found in PubMed and Google Scholar, 329 were excluded as the variants described were related to other genes than ABCA2; 7 papers were found after searching the genome browsers, 4 of the duplicates of the articles selected from PubMed and Google Scholar. No other articles or mentions were found after a careful evaluation of the references of the selected papers. Finally, we identified 9 patients with ABCA3 c.838C>T (p.Arg280Cys, R280C). Their anthropometric, clinical, radiological and histological characteristics, genetic variants, family history and patients’ outcome are presented in Table 1. No references were discovered regarding ABCA3 c.697C>T (p.Gln233Ter, Q233X, Q233*).
4. Discussion
4.1. Neonatal Respiratory Distress Syndrome in Near-Term and Term Infants
Identifying the etiology of RDS is crucial for adequate management and outcome optimization. When differential diagnosis with initial investigations fails to identify the etiology of a persistent, unexplained neonatal RDS, a genetic defect in surfactant metabolism should be evaluated. Data in the literature suggests that genetic defects in genes encoding surfactant have an important role in the etiology of unexplained nRDS [14,59,68,69]. In the presented case, the initial diagnosis of transient tachypnea of the newborn (suggested by C-section delivery in the absence of labor, early-term birth, mild respiratory distress, and hypoxemia on blood gas analysis, normal blood biochemistry, and inflammation markers) seemed improbable after the first days of life with persistent hypoxemia and increased need for oxygen and respiratory support. Lung x-rays, Doppler echocardiography, brain ultrasound, repeated blood tests, and a review of parental and pregnancy history alone did not elucidate the etiology of RDS etiology in our case. However, by integrating these findings with comprehensive molecular testing for the entire family, we were able to gain a clearer understanding of the significance of the genetic variants identified in our patient and the necessity for reclassification of those ABCA3 variants.
The genes involved in lung surfactant metabolism encode the surfactant components and facilitate the assembly, organization, and folding of the surfactant phospholipids inside the LBs and the phospholipids uptake on the lung alveolar surface [17,70]. Surfactant dysfunction disorders are associated with insufficient lung surfactant production, disruption of the surfactant metabolism, and secondary damage to AEC II [15]. Their clinical picture is characterized by a wide range of clinical manifestations, from fatal neonatal respiratory failure to ILD in older children and even adults[8,15,17,22,25,54,70,71] with increased morbidity and mortality[6,22].
Unfortunately, in our case, despite maximal respiratory support and various therapies suggested in the literature – prednisone, hydroxychloroquine, and azithromycin – a fatal outcome occurred after 77 days. No specific treatment or guideline exists for surfactant metabolic diseases [4,72], with early aggressive treatment or innovative approaches being recommended in homozygous and compound heterozygous patients [14]. Supportive treatment of respiratory distress includes oxygen, surfactant, non-invasive and invasive respiratory support, nitric oxide therapy, and ECMO, but severe cases are refractory to all types of respiratory support [3,11,13,59]. Lung transplantation has limited results as it is still associated with increased morbidity and mortality [8,11,15,54]. Some authors report improvements with steroid therapy, which are explained by the anti-inflammatory effect of increased expression of ABCA3 in the AEC II [2,11,15,72,73]. An anti-inflammatory effect and changes in intracellular metabolism with variable clinical response were reported using hydroxycholoquine[11,15,72,74]. Others have reported improvements using azithromycin, which are explained by the suppressed production of cytokine and inflammatory mediators involved in interstitial lung fibrosis progression [15,34,71,72]. Experts expect that precision medicine may achieve significant improvement or even cure for these conditions in the future [10]. Various vectors, compounds that can improve cellular ABCA3 trafficking or function (similar to those used in cystic fibrosis), and gene editing strategies are also under study [10,15,75]. Mutations in glycosylation loci may benefit in the future from therapy with proteasome inhibitors [76].
4.2. Surfactant Protein B Variants
SFTPB deficiency is characterized by inactive surfactant and abnormal LBs, with multiple vacuoles and disorganized lipid membranes; SFTPC cannot be synthesized from pro-SFTPC precursor [1,15,25]. Along with mature SFTPB and SFTPC deficiency, the accumulation of non-tensioactive intermediary products inhibits further surfactant function[15,77]. In most cases, SFTPB deficiency is associated with congenital alveolar proteinosis with an accumulation of granular, eosinophilic, PAS-positive, lipo-proteinaceous material in the alveolar spaces and frequent desquamated AEC II and foamy macrophages; the aspect of desquamative interstitial pneumonitis is less frequently seen[1]. The presence of hyperplastic alveolar epithelia with prominent AEC II, thickened alveolar walls, and limited or absent inflammatory cell infiltrates tend to distinguish SFTPB deficiency from other conditions, including damage induced by mechanical ventilation or oxygen or other conditions with abnormal lung development in which alveolar architecture is preserved[1,78]. Also, the LBs are either large and disorganized at electron microscopy or appear as irregular multivesicular structures in AEC II cytoplasm and alveolar spaces[1,25]. Incompletely processed pro-SFTPC in bronchoalveolar lavage or the lung tissue may suggest SFTPB deficiency[28].
The clinical course of our patient – early onset, unexplained nRDS in a term infant, evolving to severe respiratory failure despite maximal respiratory support, lung histology – marked AEC II hyperplasia, numerous macrophages in the alveolar lumen, focal alveolar proteinosis, extended areas of desquamative interstitial pneumonia, and the results of SFTPB sequencing – suggested a possible surfactant metabolism defect due to SFTPB deficiency. Considering that SFTPB p.Val267Ile substitution (previously not reported in the literature) was identified in the healthy brother of the patient, we speculate that this alteration is a polymorphism that does not affect protein function. Also, Polyphen-2 (v2.2.3r406) predicted this variant as benign.
4.3. ABCA3 Deficiency
4.3.1. Adenosine Triphosphate-Binding Cassette Family A Member 3 (ABCA3) Protein
The biology of ABCA3 protein is very complex. ABCA3 (Adenosine Triphosphate-binding Cassette Family A Member 3) protein belongs to proteins’ ATP binding cassette transporter superfamily. ABCA3 is a phospholipidic glycoprotein of 1704 amino acids localized in the external limiting membrane of the lamellar bodies [10,11,50,52,73,79]. Six transmembrane structures mediate ABCA3 function by forming an ATP channel for lipids (disaturated-phosphatidylcholine, phosphatidylglycerol, phosphatidylethanolamine, and cholesterol) transportation from the cytosol into the LBs [1,15,52,64,80,81,82]. ABCA3 is also involved in lung surfactant transcription and assembly, SFTPB and SFTPC translation, lung surfactant structural transformation and production in AEC II, and epithelial lung cell apoptosis [4]. A possible role of ABCA3 in the metabolism of lung surfactant phospholipids was also described [79]. Intracellular metabolism of cholesterol may also be influenced by ABCA3[15,81]. The decreased pool of mature SFTPB and SFTPB aggregates into the LBs, accumulation of large quantities of pro-SFTPB in LBs with leaks in the alveolar spaces, and abnormal processing of SFTPC were described in association with ABCA3 deficiency [71].
Consequently, ABCA3 deficiency is characterized by abnormal structure LBs, abnormal lipid composition of the lung surfactant, and abnormal processing of surfactant proteins B and C [68,71,75,83,84]. Reduced ability to decrease surface alveolar tension was demonstrated in patients with ABCA3 deficiency[11]. Accumulation of dysfunctional, inefficient lung surfactant is associated with compromised gas exchanges through reduced diffusion barrier and increased discordance between ventilation and perfusion, decreased activity of the macrophages, and secondary lung lesions [1,17,64,72]. The biological mechanism of the lung lesions associated with ABCA3 deficiency is unknown [15,85,86]. However, AEC II lesions are the final pathway to the associated lung disease, as AEC II represents the key factor for alveolar maintenance and repair[24]. Inadequate lung surfactant production leads to recurrent atelectasis and hypoxemia followed by secondary chronic inflammation; coupled with abnormal intracellular surfactant metabolism, these changes lead to chronic AEC II lesions [87]. Based on in vitro mechanistic studies, three classes of ABCA3 gene variants can be identified: type 1 - ABCA3 trafficking variants, characterized by abnormal protein folding, abnormal intracellular localization and trafficking; type 2 - complete deficiency of lipid transportation and variants affecting only the lipid transport due to deficient ATP hydrolysis, with normal trafficking and localization of the ABCA3 protein [3,60,88,89,90,91]; type 3 – a compound heterozygous of type 1 and 2, often associated with a more severe phenotype, early onset, neonatal RDS and neonatal death [54].
On lung histology, ABCA3 deficiency is characterized by AEC II hyperplasia, variable degrees of interstitial thickening, prominent macrophages and proteinaceous material in the alveolar spaces, aspect frequently described as chronic or desquamative or non-specific interstitial pneumonitis or alveolar lung proteinosis; lung fibrosis is associated in fatal cases; however, these changes are frequently seen in other surfactant metabolism conditions, including SFTPB and SFTPC deficiency [11].
As the clinical picture of ABCA3 deficiency is undistinguishable from SFTPB and SFTPC deficiency, even with lung histology [13,15], electron microscopy may help. The absence of normal LBs strongly suggests ABCA3 deficiency [11,25,51]. Small, markedly abnormal LBs with dense phospholipidic membranes are characteristic of ABCA3 deficiency, as compared to SFTPB deficiency, which is characterized by disorganized LBs, with multiple vesicular inclusions dispersed in AEC II cytoplasm and alveolar lumen [1,24].
ABCA3 is expressed not only in the AEC II but also in the brain, kidney, and platelets [22]. ABCA3 expression occurs in normal fetuses at 26-27 weeks of gestation [4], even at 23-24 weeks, associated with lung inflammation [71], and is developmentally regulated. It increases with the gestational age to reach a peak around term [11,13] under the influence of steroids and TTF 1 [11,71,92,93,94].
4.3.2. Adenosine Triphosphate-Binding Cassette Family A Member 3 (ABCA3) Gene
ABCA3 synthesis is encoded by the ABCA3 gene, intensely expressed in AEC II [8,15,51]. ABCA3 gene variants are the most common cause of congenital lung surfactant defects [2,3,11,22,25,54,72,75,85]. The first cases of ABCA3 deficiency, secondary to a bi-allelic loss of function ABCA3 variant, were reported by Shulenin et al. [51] in 2004, in full-term infants with unexplained severe RDS. Most ABCA3 variants are challenging to interpret as few of them have been studied in vitro to identify their intracellular expression and function [15,54,62,75]. According to Wambach et al. [95], the incidence of ABCA3 variants is estimated between 1:4.400 and 1:20.000 in European and African descendants, most of them compound heterozygous [72]. The incidence is probably overestimated as not all missense variants are pathogenic, and the annual number of cases identified is lower than expected according to the prediction calculations [8,95]. It is also possible that mild cases may not be recognized [54]. In humans, the ABCA3 gene has 80 kb, is located on chromosome 16 (16p.13.3) [85], and comprises 33 exons [11,96]. Only 0.15% of the 274 ABCA3 gene variants reported in the genome Aggregation Database (gnomAD) are classified as pathogenic, 0.21% as likely pathogenic, and 92.62% were VUS [70]. Twenty-five pathogenic or likely pathogenic of the ABCA3 variants were reported in the ClinVar database, 11 of these with loss of function of the protein; 47 disease-causing ABCA3 variants are reported in the in silico tool SIFT, 46 of these being also included in Polyphen-2, while 49 pathogenic mutations can be found in MutationTaster2 [52]. Most reported variants are located on the exons or at the limit between introns and exons.
The expression of the ABCA3 gene is directly proportional to gestational age [17,71]. Most ABCA3 variants have an autosomal recessive inheritance [8,85,97], but uniparental disomy was also reported [98]. The most frequent pathogenic ABCA3 variant is p.Glu292Val (E292V), representing 10% of the reported pathogenic alleles [99]. This variant occurs in gnomAD with 0.23% allelic frequency [52]. Pathogenic ABCA3 variants are reported anywhere on the gene [17]. NGS – WES, WGS - identifies new variants, most of them with unknown significance (VUS). In this situation, a correlation genotype-phenotype is impossible [22,75]. Therefore, the effect cannot be predicted accurately for all the reported variants, complicating the clinical decision, patient management, and familial counseling [17,52,75]. Interpretation and counseling are also difficult in patients exhibiting a unique ABCA3 variant on one allele [11,13,22,62,64,75]. Recently, it was suggested that ABCA3 variants may be responsible for the increased severity of RDS in preterm infants compared to as expected according to their gestational age [25,50,54,83,100]. In 2004, Shulenin et al. [51] reported 12 different causative variants of the ABCA3 gene in 16/21 neonates with severe, unexplained RDS. A study comprising 68 preterm infants with a gestational age <32 weeks of gestation with unusually severe RDS identified 24/68 heterozygous for previously described rare or new ABCA3, SFTPB, and SFTPC variants, all VUS; 21 ABCA3 variants were found in 18 of the patients; 11 deaths were noted between 2 and 6 months of age and one infant presented histological aspects suggestive for ABCA3 deficiency [69].
According to Peca et al. [71], the ABCA3 deficiency phenotype depends on the residual function of the ABCA3 protein, mutation type and severity, the activity of the intracellular stress pathways, general individual aspects, other associated mutations, and modifiable environmental factors. Variable genotype-phenotype correlation has been associated with ABCA3 variants [8,22,70,75]; diverse symptoms, severity, and outcomes are associated with ABCA3 variants [63]. However, interactions with other variants (as, for example, variants of SFTPB or SFTPC) [22,62,71,95,101,102,103] or with external, environmental factors (for example, respiratory infections or smoking) [22,50,64,79,104] may induce changes of the phenotype. Similar mechanisms were suggested for mono-allelic variants of the ABCA3 gene [71,95]. According to Yang et al. [105], loss of over 50% of the ABCA3 protein function is associated with increased morbidity and mortality. A critical level for ABCA3 protein function of 20-30% was estimated by Wambach et al. [19]. Usually, bi-allelic mutations of the ABCA3 gene are associated with loss-of-function of ABCA3 protein and severe RDS with neonatal onset. Missense mutations, insertions, and small deletions are typically associated with the residual function of the protein [52,64]. Both bi-allelic and mono-allelic variants may present with RDS [51,63,95,101,103]. Null/null mutations – nonsense and frameshift – result in a truncated, non-functional ABCA3 protein [54,71] associated with a more severe phenotype, the neonatal onset of RDS, death before one year of age, need for lung transplantation, or death even with lung transplantation [9,52,62]. Late preterm and term infants with homozygous and compound heterozygous ABCA3 variants were associated with earlier presentation of severe RDS, higher radiological scores, and increased mortality rates (all p<0.05) as compared to infants with single mutations or no genetic abnormalities identified [14]. Beers et al. [106] suggested a synergistic additive effect for compound heterozygous in the cis region of the gene. Lack of gene expression, decreased expression, abnormal intracellular protein trafficking inside LBs, abnormal phospholipid folding, and functional defects, including ATP hydrolysis, were described as consequences depending on the ABCA3 locus [1]. Fatal cases of ABCA3 deficiency were described in association with abnormal trafficking, while defects in phosphatidylcholine were correlated with less severe lung disease [88,89].
4.3.3. ABCA3 c.838C>T (p.Arg280Cys, R280C) Variant
In vitro functional studies performed by Weichert et al. [86] have suggested that this variant can lead to partial retention of the ABCA3 protein in the endoplasmic reticulum and is involved in epithelial lung cells apoptosis at least one pathway, altering ABCA3 protein function (type 1, trafficking/folding defect based on in vitro studies). ABCA3 protein retention in the endoplasmic reticulum may increase reticulum endoplasmic stress and its susceptibility to stress; an adverse effect of R280C on LBs biogenesis and induced presence of apoptotic markers (glutathione on caspase 4 pathway) were demonstrated in the experimental epithelial lung cells in Weichert et al. experiments [86], also suggestive for functional impairment on ABCA3 protein and lung disease pathogenesis. These experiments confirmed previous studies by Matsumura et al. [88,89] that defined the c.838C>T (p.Arg280Cys, R280C) variant as disruptive of ABCA3 folding and trafficking. Based on increased retention of ABCA3 protein in the endoplasmic reticulum, ABCA3 variants F1203del, N124Q, N140Q, and R280C may be classified as potentially pathogenic, according to other experts [105].
Furthermore, multiple computational predictive in silico tools and conservational analysis also indicate the negative impact of the c.838C>T (p.Arg280Cys, R280C) variant on the ABCA3 protein function [55,65,66]. Nevertheless, currently, the c.838C>T (p.Arg280Cys, R280C) variant is listed as VUS, considering the existent evidence insufficient to define the mutation’s pathogenicity. Pros and cons arguments on c.838C>T (p.Arg280Cys, R280C) pathogenicity are presented in Table 2.
The initial lung imaging – usually resembling that seen in preterm infants with RDS [3,22,25,59,71] – evolves throughout the disease and may vary over time, as it happened also in our patient [8]. Later in the course of the disease, a nodular and consolidation pattern may be observed [96]. Different lung imaging aspects are expected as even identical variants of the ABCA3 gene may present with different phenotypes [112]. The same observation applies to lung histology. All these aspects are described in the literature in association with ABCA3, SFTPB, and SFTPC deficiency [13,15]. Electron microscopy of the lung tissue was reported only in the patient presented by Jackson et al. [64].
Familial history had no relevance for ABCA3 deficiency in the several reviewed cases, as in our family. This is not unexpected as ABCA3 deficiencies are rare diseases with autosomal recessive inheritance. Most patients were compound heterozygous for the ABCA3 c.838C>T (p.Arg280Cys, R280C) variant, which, together with the association of various other ABCA3 variants, may explain the different phenotypes of the subjects (Table 1). Additionally, there is an urgent need for functional studies to quantify the impact of each variant on protein function, expressed as a percentage.
Our case, a compound heterozygous for ABCA3 c.838C>T (p.Arg280Cys, R280C), and ABCA3 c.697C>T (p.Gln233Ter, Q233X, Q233*) variants, presented with fatal RDS with neonatal onset. Most probably, a cumulative damaging effect of the c.697C>T (p.Gln233Ter, Q233X, Q233*) variant (in silico predicted as probably pathogenic) significantly contributed to the severe phenotype of our patient.
We support the recommendation that in cases of unexplained, early onset, severe neonatal RDS with persistent radiological and clinical symptoms persistent over one week, evolving to hypoxemic respiratory failure despite maximal conventional therapy and with transient response to surfactant administration in term and near-term infants, genetic surfactant metabolism dysfunctions should be suspected [6,8,9,13,15,79,86]. NGS offers a crucial role in molecular medicine, a step forward to individualized medicine, as this genetic testing precisely detects single nucleotide variants [70]. NGS, WES and WGS, can help the index case and their family identify the genetic defect and provide genetic counseling. Moreover, extended genetic sequencing may replace the information offered by lung biopsy, an invasive investigation previously recommended in assessing inherited surfactant metabolism disorders [4,6,9,13,71], as a rapid and precise diagnosis is of utmost importance for genetic counseling [10,96]. These techniques may identify VUS that need predictive tools for clarifying the impact on protein function and pathogeny and further genetic counseling [15,70,110].
Our study has several additional limitations. First, the presented patient and his family were investigated by an NGS panel that included a limited number of genes, thus not providing a comprehensive analysis of their genome. Additionally, we could not perform functional studies to quantify the effect of the ABCA3 variants, which collectively impacted protein function and led to RDS. Second, we did not assess gene expression or measure the concentration of ABCA3 protein. However, the clinical, paraclinical, imaging, and histological findings, predictive tools, and the results of previously reported studies strongly suggest the pathogenicity of the associated variants in our patient and the need to reclassify these ABCA3 variants.
5. Conclusions
Our study highlighted the necessity, importance, and timing of genetic testing for a step-by-step diagnosis. NGS techniques are considered the gold standard for the genetic investigation of RDS. However, these comprehensive techniques may identify VUS, as in our case, presenting challenges in deciphering their clinical relevance, underscoring the need for continued comprehensive research and review to characterize these variants accurately.
In our case, the patient phenotype, clinical aspects, course, and outcome, along with literature data and computational predictions from in silico probability tools, indicate that the ABCA3 c.838C>T (R280C, p.Arg280Cys) and c.697C>T (p.Gln233Ter, Q233X, Q233*) variants had a cumulative effect and should be reclassified as probably pathogenic/pathogenic variants. Data from the extensive review of the literature also support our conclusions.
Author Contributions
Conceptualization, M.L.O., M.A.C., and M.C.; methodology, M.L.O., B.W.K., R.G.; software, M.D.S., A.S., and F.G.; validation, C.B., B.W.K., M.L.O., and M.C.; formal analysis, F.G., R.G., M.D.S., and A.S.; investigation, M.L.O., M.A.C., and C.B.; resources, A.S., M.D.S., and R.G.; data curation, M.L.O.; writing—original draft preparation, M.L.O., M.A.C., C.B., and R.G.; writing—review and editing, M.L.O., M.A.C., M.C., and B.W.K.; visualization, A.S., F.G., and R.G.; supervision, M.L.O., B.W.K., and M.C.; project administration, M.L.O., and M.A.C. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki, and approved by the Ethics, Medical Deontology, and Discipline Committee of the Clinical County Emergency Hospital Sibiu, Romania, according to decision 20171/12.08.2024.
Informed Consent Statement
Informed consent was obtained from the parents.
Data Availability Statement
All data generated or analyzed during this study is included in this article.
Acknowledgments
The authors gratefully acknowledge the invaluable advice and assistance provided by Florin Tripon, Ph.D., during the completion of this work. Additionally, we wish to express our gratitude to the parents of the index case for their willingness to share their family genetic testing results and for granting consent for the publication of this family study.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Wert, S.E., Whitsett, J.A., Nogee, L.M. Genetic disorders of surfactant dysfunction. Pediatr Dev Pathol. 2009; 12(4):253-74. [CrossRef] [PubMed]
- Lei, C., Wan, C., Liu, C. ABCA3 mutation-induced congenital pulmonary surfactant deficiency: A case report. Medicine (Baltimore). 2024;103(13):e37622. [CrossRef] [PubMed]
- Winter, J., Essmann, S., Kidszun, A., Aslanidis, C., Griese, M., Poplawska, K., Bartsch, M., Schmitz, G., Mildenberger, E. Neonatal respiratory insufficiency caused by an (homozygous) ABCA3-stop mutation: a systematic evaluation of therapeutic options. Klin Padiatr. 2014;226(2):53-8. [CrossRef] [PubMed]
- Wei, M., Fu, H., Han, A., Ma, L. A Term Neonatal Case With Lethal Respiratory Failure Associated With a Novel Homozygous Mutation in ABCA3 Gene. Front Pediatr. 2020;8:138. [CrossRef] [PubMed]
- Dyer, J. Neonatal Respiratory Distress Syndrome: Tackling A Worldwide Problem. P T. 2019;44(1):12-14. [PubMed]
- Magnani, J.E., Donn, S.M. Persistent Respiratory Distress in the Term Neonate: Genetic Surfactant Deficiency Diseases. Curr Pediatr Rev. 2020;16(1):17-25. [CrossRef] [PubMed]
- Abdel-Latif, M.E., Tan, O., Fiander, M., Osborn, D.A. Non-invasive high-frequency ventilation in newborn infants with respiratory distress. Cochrane Database Syst Rev. 2024;5(5):CD012712. [CrossRef] [PubMed]
- Zhang, W., Liu, Z., Lin, Y., Wang, R., Xu, J., He, Y., Zhang, F., Wu, L., Chen. D. A novel synonymous ABCA3 variant identified in a Chinese family with lethal neonatal respiratory failure. BMC Med Genomics. 2021;14(1):256. [CrossRef] [PubMed]
- Gupta, N.P., Batra, A., Puri, R., Meena. V. Novel homozygous missense mutation in ABCA3 protein leading to severe respiratory distress in term infant. BMJ Case Rep. 2020;13(10):e235520. [CrossRef] [PubMed]
- Anciuc-Crauciuc, M., Cucerea, M.C., Tripon, F., Crauciuc, G.A., Bănescu, C.V. Descriptive and Functional Genomics in Neonatal Respiratory Distress Syndrome: From Lung Development to Targeted Therapies. Int J Mol Sci. 2024;25(1):649. [CrossRef] [PubMed]
- Bullard, J.E., Wert, S.E., Nogee, L.M. ABCA3 deficiency: neonatal respiratory failure and interstitial lung disease. Semin Perinatol. 2006;30(6):327-34. [CrossRef] [PubMed]
- Reuter, S., Moser, C., Baack, M. Respiratory distress in the newborn. Pediatr Rev. 2014;35(10):417-28; quiz 429. [CrossRef] [PubMed]
- Rogulska, J., Wróblewska-Seniuk, K., Śmigiel, R., Szydłowski, J., Szczapa, T. Diagnostic Challenges in Neonatal Respiratory Distress-Congenital Surfactant Metabolism Dysfunction Caused by ABCA3 Mutation. Diagnostics (Basel). 2022;12(5):1084 Mutation. [CrossRef] [PubMed]
- Wang, J., Fan, J., Zhang, Y., Huang, L., Shi, Y. ABCA3 gene mutations shape the clinical profiles of severe unexplained respiratory distress syndrome in late preterm and term infants. Transl Pediatr. 2021;10(2):350-358. [CrossRef] [PubMed]
- Nogee, L.M. Genetic causes of surfactant protein abnormalities. Curr Opin Pediatr. 2019;31(3):330-339. [CrossRef] [PubMed]
- Bush, A., Cunningham, S., de Blic, J., Barbato, A., Clement, A., Epaud, R., Hengst, M., Kiper, N,, Nicholson, A.G., Wetzke, M., Snijders, D., Schwerk, N., Griese, M.; chILD-EU Collaboration. European protocols for the diagnosis and initial treatment of interstitial lung disease in children. Thorax. 2015;70(11):1078-84. [CrossRef] [PubMed]
- Oltvai, Z.N., Smith, E.A., Wiens, K., Nogee, L.M., Luquette, M., Nelson, A.C., Wikenheiser-Brokamp, K.A. Neonatal respiratory failure due to novel compound heterozygous mutations in the ABCA3 lipid transporter. Cold Spring Harb Mol Case Stud. 2020;6(3):a005074. [CrossRef] [PubMed]
- Hallman, M. The surfactant system protects both fetus and newborn. Neonatology. 2013;103(4):320-6. [CrossRef] [PubMed]
- Wambach, J.A., Yang, P., Wegner, D.J., Heins, H.B., Kaliberova, L.N., Kaliberov, S.A., Curiel, D.T., White, F.V., Hamvas, A., Hackett, B.P., Cole, F.S. Functional Characterization of ATP-Binding Cassette Transporter A3 Mutations from Infants with Respiratory Distress Syndrome. Am J Respir Cell Mol Biol. 2016;55(5):716-721. [CrossRef] [PubMed]
- Malý, J., Navrátilová, M., Hornychová, H., Looman, A.C. Respiratory failure in a term newborn due to compound heterozygous ABCA3 mutation: the case report of another lethal variant. J Perinatol. 2014;34(12):951-3. [CrossRef] [PubMed]
- Griese, M., Haug, M., Brasch, F., Freihorst, A., Lohse, P., von Kries, R., Zimmermann, T., Hartl, D. Incidence and classification of pediatric diffuse parenchymal lung diseases in Germany. Orphanet J Rare Dis. 2009;4:26. [CrossRef] [PubMed]
- Mitsiakos, G., Tsakalidis, C., Karagianni, P., Gialamprinou, D., Chatziioannidis, I., Papoulidis, I., Tsanakas, I., Soubasi, V. A New ABCA3 Gene Mutation c.3445G>A (p.Asp1149Asn) as a Causative Agent of Newborn Lethal Respiratory Distress Syndrome. Medicina (Kaunas). 2019;55(7):389. [CrossRef] [PubMed]
- Nogee, L.M., de Mello, D.E., Dehner, L.P., Colten, H.R. Brief report: deficiency of pulmonary surfactant protein B in congenital alveolar proteinosis. N Engl J Med. 1993;328(6):406-10. [CrossRef] [PubMed]
- van Moorsel, C.H.M., van der Vis, J.J., Grutters, J.C. Genetic disorders of the surfactant system: focus on adult disease. Eur Respir Rev. 2021;30(159):200085. [CrossRef] [PubMed]
- Hamvas, A. Evaluation and management of inherited disorders of surfactant metabolism. Chin Med J (Engl). 2010;123(20):2943-7. [PubMed]
- Nogee, L.M., Garnier, G., Dietz, H.C., Singer, L., Murphy, A.M., deMello, D.E., Colten, H.R. A mutation in the surfactant protein B gene responsible for fatal neonatal respiratory disease in multiple kindreds. J Clin Invest. 1994;93(4):1860-3. [CrossRef] [PubMed]
- Beers, M.F., Hamvas, A., Moxley, M.A., Gonzales, L.W., Guttentag, S.H., Solarin, K.O., Longmore, W.J., Nogee, L.M., Ballard, P.L. Pulmonary surfactant metabolism in infants lacking surfactant protein B. Am J Respir Cell Mol Biol. 2000;22(3):380-91. [CrossRef] [PubMed]
- Nogee, L.M., Wert, S.E., Proffit, S.A., Hull, W.M., Whitsett, J.A. Allelic heterogeneity in hereditary surfactant protein B (SP-B) deficiency. Am J Respir Crit Care Med. 2000;161(3 Pt 1):973-81. [CrossRef] [PubMed]
- Dunbar, A.E. 3rd, Wert, S.E., Ikegami, M., Whitsett, J.A., Hamvas, A., White, F.V., Piedboeuf, B., Jobin, C., Guttentag, S., Nogee, L.M. Prolonged survival in hereditary surfactant protein B (SP-B) deficiency associated with a novel splicing mutation. Pediatr Res. 2000;48(3):275-82. [CrossRef] [PubMed]
- López-Andreu, J.A., Hidalgo-Santos, A.D., Fuentes-Castelló, M.A., Mancheño-Franch, N., Cerón-Pérez, J.A., Esteban-Ricós, M.J., Pedrola-Vidal, L., Nogee, L.M. Delayed Presentation and Prolonged Survival of a Child with Surfactant Protein B Deficiency. J Pediatr. 2017;190:268-270.e1. [CrossRef] [PubMed]
- Kurland, G., Deterding, R.R., Hagood, J.S., Young, L.R., Brody, A.S., Castile, R.G., Dell, S., Fan, L.L., Hamvas, A., Hilman, B.C., Langston, C., Nogee, L.M., Redding, G.J.; American Thoracic Society Committee on Childhood Interstitial Lung Disease (chILD) and the chILD Research Network. An official American Thoracic Society clinical practice guideline: classification, evaluation, and management of childhood interstitial lung disease in infancy. Am J Respir Crit Care Med. 2013;188(3):376-94. [CrossRef] [PubMed]
- Klein, J.M., Thompson, M.W., Snyder, J.M., George, T.N., Whitsett, J.A., Bell, E.F., McCray, P.B. Jr, Nogee, L.M. Transient surfactant protein B deficiency in a term infant with severe respiratory failure. J Pediatr. 1998;132(2):244-8. [CrossRef] [PubMed]
- Ballard, P.L., Nogee, L.M., Beers, M.F., Ballard, R.A., Planer, B.C., Polk, L., deMello, D.E., Moxley, M.A., Longmore, W.J. Partial deficiency of surfactant protein B in an infant with chronic lung disease. Pediatrics. 1995;96(6):1046-52. [PubMed]
- Lawson, W.E., Grant, S.W., Ambrosini, V., Womble, K.E., Dawson, E.P., Lane, K.B., Markin, C., Renzoni, E., Lympany, P., Thomas, A.Q., Roldan, J., Scott, T.A., Blackwell, T.S., Phillips, J.A. 3rd, Loyd, J.E., du Bois, R.M. Genetic mutations in surfactant protein C are a rare cause of sporadic cases of IPF. Thorax. 2004;59(11):977-80. [CrossRef] [PubMed]
- Tredano, M., Griese, M., Brasch, F., Schumacher, S., de Blic, J., Marque, S., Houdayer, C., Elion, J,, Couderc, R., Bahuau, M. Mutation of SFTPC in infantile pulmonary alveolar proteinosis with or without fibrosing lung disease. Am J Med Genet A. 2004;126A(1):18-26. [CrossRef] [PubMed]
- Pachajoa, H., Ruiz-Botero, F., Meza-Escobar, L.E., Villota-Delgado, V,A,, Ballesteros, A., Padilla, I., Duarte, D. Fatal respiratory disease due to a homozygous intronic ABCA3 mutation: a case report. J Med Case Rep. 2016;10(1):266. [CrossRef] [PubMed]
- Kröner, C., Reu, S., Teusch, V., Schams, A., Grimmelt, A.C., Barker, M., Brand, J., Gappa, M., Kitz, R., Kramer, B.W., Lange, L., Lau, S., Pfannenstiel, C., Proesmans, M., Seidenberg, J., Sismanlar, T., Aslan, A.T., Werner, C., Zielen, S., Zarbock, R., Brasch, F., Lohse, P., Griese, M. Genotype alone does not predict the clinical course of SFTPC deficiency in paediatric patients. Eur Respir J. 2015;46(1):197-206. [CrossRef] [PubMed]
- Wright, J.R. Host defense functions of pulmonary surfactant. Biol Neonate. 2004;85(4):326-32. [CrossRef] [PubMed]
- Kingma, P.S., Whitsett, J.A. In defense of the lung: surfactant protein A and surfactant protein D. Curr Opin Pharmacol. [CrossRef] [PubMed]
- Haagsman, H.P., Hogenkamp, A., van Eijk, M., Veldhuizen, E.J. Surfactant collectins and innate immunity. Neonatology. [CrossRef] [PubMed]
- Boggaram, V. Regulation of lung surfactant protein gene expression. Front Biosci. 2003;8:d751-64. [CrossRef] [PubMed]
- Rämet, M., Haataja, R., Marttila, R., Floros, J., Hallman, M. Association between the surfactant protein A (SP-A) gene locus and respiratory-distress syndrome in the Finnish population. Am J Hum Genet. 2000;66(5):1569-79. [CrossRef] [PubMed]
- Hilgendorff, A., Heidinger, K., Bohnert, A., Kleinsteiber, A., König, I.R., Ziegler, A., Lindner, U., Frey, G., Merz, C., Lettgen, B., Chakraborty, T., Gortner, L., Bein, G. Association of polymorphisms in the human surfactant protein-D (SFTPD) gene and postnatal pulmonary adaptation in the preterm infant. Acta Paediatr. 2009;98(1):112-7. [CrossRef] [PubMed]
- Lahti, M., Lofgren, J., Marttila, R., Renko, M., Klaavuniemi, T., Haataja, R., Ramet, M., Hallman, M. Surfactant protein D gene polymorphism associated with severe respiratory syncytial virus infection. Pediatr Res. 2002;51(6):696-9. [CrossRef] [PubMed]
- Löfgren, J., Rämet, M., Renko, M., Marttila, R., Hallman, M. Association between surfactant protein A gene locus and severe respiratory syncytial virus infection in infants. J Infect Dis. 2002;185(3):283-9. [CrossRef] [PubMed]
- Wang, Y., Kuan, P.J., Xing, C., Cronkhite, J.T., Torres, F., Rosenblatt, R.L., DiMaio, J.M., Kinch, L.N., Grishin, N.V., Garcia, C.K. Genetic defects in surfactant protein A2 are associated with pulmonary fibrosis and lung cancer. Am J Hum Genet. 2009;84(1):52-9. [CrossRef] [PubMed]
- Nattes, E., Lejeune, S., Carsin, A., Borie, R., Gibertini, I., Balinotti, J., Nathan, N., Marchand-Adam, S., Thumerelle, C., Fauroux, B., Bosdure, E., Houdouin, V., Delestrain, C., Louha, M., Couderc, R., De Becdelievre. A., Fanen, P., Funalot, B., Crestani, B., Deschildre, A., Dubus, J.C., Epaud, R. Heterogeneity of lung disease associated with NK2 homeobox 1 mutations. Respir Med. 2017;129:16-23. [CrossRef] [PubMed]
- Thorwarth, A., Schnittert-Hübener, S., Schrumpf, P., Müller, I., Jyrch, S., Dame, C., Biebermann, H., Kleinau, G., Katchanov, J., Schuelke, M., Ebert, G., Steininger, A., Bönnemann, C., Brockmann, K., Christen, H.J., Crock, P., deZegher, F., Griese, M., Hewitt, J., Ivarsson, S., Hübner, C., Kapelari. K., Plecko, B., Rating, D., Stoeva, I., Ropers, H.H., Grüters, A., Ullmann, R., Krude, H. Comprehensive genotyping and clinical characterisation reveal 27 novel NKX2-1 mutations and expand the phenotypic spectrum. J Med Genet. 2014;51(6):375-87. [CrossRef] [PubMed]
- Nevel, R.J., Garnett, E.T., Worrell, J.A., Morton, R.L., Nogee, L.M., Blackwell, T.S., Young, L.R. Persistent Lung Disease in Adults with NKX2.1 Mutation and Familial Neuroendocrine Cell Hyperplasia of Infancy. Ann Am Thorac Soc. 2016;13(8):1299-304. [CrossRef] [PubMed]
- Beers, M.F., Mulugeta, S. The biology of the ABCA3 lipid transporter in lung health and disease. Cell Tissue Res. 2017;367(3):481-493. [CrossRef] [PubMed]
- Shulenin, S., Nogee, L.M., Annilo, T., Wert, S.E., Whitsett, J.A., Dean, M. ABCA3 gene mutations in newborns with fatal surfactant deficiency. N Engl J Med. 2004;350(13):1296-303. [CrossRef] [PubMed]
- Onnée, M., Fanen, P., Callebaut, I., de Becdelièvre, A. Structure-Based Understanding of ABCA3 Variants. Int J Mol Sci. 2021;22(19):10282. [CrossRef] [PubMed]
- Gonçalves, J.P., Pinheiro, L., Costa, M., Silva, A., Gonçalves, A., Pereira, A. Novel ABCA3 mutations as a cause of respiratory distress in a term newborn. Gene. 2014;534(2):417-20. [CrossRef] [PubMed]
- Jouza, M., Jimramovsky, T., Sloukova, E., Pecl, J., Seehofnerova, A., Jezova, M., Urik, M., Kunovsky, L., Slaba, K., Stourac, P., Klincova, M., Hubacek, J.A., Jabandziev, P. A Newly Observed Mutation of the ABCA3 Gene Causing Lethal Respiratory Failure of a Full-Term Newborn: A Case Report. Front Genet. 2020;11:568303. [CrossRef] [PubMed]
- National Center for Biotechnology Information. ClinVar; [VCV000318566.16]. Available online: https://www.ncbi.nlm.nih.gov/clinvar/variation/ (accessed on 17 June 2024).
- Ensembl (2022). Available online: https://www.ensembl.org/ (accessed on 17 June 2024).
- Pejaver, V., Urresti, J., Lugo-Martinez, J., Pagel, K.A., Lin, G.N., Nam, H.J., Mort, M., Cooper, D.N., Sebat, J., Iakoucheva, L.M., Mooney, S.D., Radivojac, P. Inferring the molecular and phenotypic impact of amino acid variants with MutPred2. Nat Commun. 2020;11(1):5918. [CrossRef] [PubMed]
- Pagel, K.A., Pejaver, V., Lin, G.N., Nam, H.J., Mort, M., Cooper, D.N., Sebat, J., Iakoucheva, L.M., Mooney, S.D., Radivojac, P. When loss-of-function is loss of function: assessing mutational signatures and impact of loss-of-function genetic variants. Bioinformatics. 2017;33(14):i389-i398. [CrossRef] [PubMed]
- Somaschini, M., Nogee, L.M., Sassi, I., Danhaive, O., Presi, S., Boldrini, R., Montrasio, C., Ferrari, M., Wert, S.E., Carrera, P. Unexplained neonatal respiratory distress due to congenital surfactant deficiency. J Pediatr. 2007;150(6):649-53, 653.e1. [CrossRef] [PubMed]
- Turcu, S., Ashton, E., Jenkins, L., Gupta, A., Mok, Q. Genetic testing in children with surfactant dysfunction. Arch Dis Child. 2013;98(7):490-5. [CrossRef] [PubMed]
- Williamson, M., Wallis, C. Ten-year follow up of hydroxychloroquine treatment for ABCA3 deficiency. Pediatr Pulmonol. 2014;49(3):299-301. [CrossRef] [PubMed]
- Wambach, J.A., Casey, A.M., Fishman, M.P., Wegner, D.J., Wert, S.E., Cole, F.S., Hamvas, A., Nogee, L.M. Genotype-phenotype correlations for infants and children with ABCA3 deficiency. Am J Respir Crit Care Med. 2014;189(12):1538-43. [CrossRef] [PubMed]
- Xu, K.K., Wegner, D.J., Geurts, L.C., Heins, H.B., Yang, P., Hamvas, A., Eghtesady, P., Sweet, S.C., Sessions Cole, F., Wambach, J.A. Biologic characterization of ABCA3 variants in lung tissue from infants and children with ABCA3 deficiency. Pediatr Pulmonol. 2022;57(5):1325-1330. [CrossRef] [PubMed]
- Jackson, T., Wegner, D.J., White, F.V., Hamvas, A., Cole, F.S., Wambach, J.A. Respiratory failure in a term infant with cis and trans mutations in ABCA3. J Perinatol. 2015;35(3):231-2. [CrossRef] [PubMed]
- Wang, K., Li, M., Hakonarson, H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38(16):e164. [CrossRef] [PubMed]
- Klay, D., Platenburg, M.G.J.P., van Rijswijk, R.H.N.A.J., Grutters, J.C., van Moorselm C.H.M. ABCA3 mutations in adult pulmonary fibrosis patients: a case series and review of literature. Curr Opin Pulm Med. 2020;26(3):293-301. [CrossRef] [PubMed]
- Gjeta, I., Sala, D., Bakalli, I., Celaj, E., Kola, E. Surfactant Deficiency Causing Severe Pneumonia in a Child. Curr Health Sci J. 2023;49(1):134-138. [CrossRef] [PubMed]
- Brasch, F., Schimanski, S., Mühlfeld, C., Barlage, S., Langmann, T., Aslanidis, C., Boettcher, A., Dada, A., Schroten, H., Mildenberger, E., Prueter, E., Ballmann, M., Ochs, M., Johnen, G., Griese, M., Schmitz, G. Alteration of the pulmonary surfactant system in full-term infants with hereditary ABCA3 deficiency. Am J Respir Crit Care Med. 2006;174(5):571-80. [CrossRef] [PubMed]
- Somaschini, M., Presi, S., Ferrari, M., Vergani, B., Carrera, P. Surfactant proteins gene variants in premature newborn infants with severe respiratory distress syndrome. J Perinatol. 2018;38(4):337-344. [CrossRef] [PubMed]
- Anciuc-Crauciuc, M., Cucerea, M.C., Crauciuc, G.A., Tripon, F., Bănescu, C.V. Evaluation of the Copy Number Variants and Single-Nucleotide Polymorphisms of ABCA3 in Newborns with Respiratory Distress Syndrome-A Pilot Study. Medicina (Kaunas). 2024;60(3):419. [CrossRef] [PubMed]
- Peca, D., Cutrera, R., Masotti, A., Boldrini, R., Danhaive, O. ABCA3, a key player in neonatal respiratory transition and genetic disorders of the surfactant system. Biochem Soc Trans. 2015;43(5):913-9. [CrossRef] [PubMed]
- Xiao, G.L., Gao, Y., Hao, H., Wei, T., Hong, C., Wang, Y., Lin, Y.Y., Chi, X.F., Liu, Y., Gao, H.Y., Nie. C. Novel insights into congenital surfactant dysfunction disorders by in silico analysis of ABCA3 proteins. World J Pediatr. 2023;19(3):293-301. [CrossRef] [PubMed]
- Ciantelli, M., Ghirri, P., Presi, S., Sigali, E., Vuerich, M., Somaschini, M., Ferrari, M., Boldrini, A., Carrera, P. Fatal respiratory failure in a full-term newborn with two ABCA3 gene mutations: a case report. J Perinatol. 2011;31(1):70-2. [CrossRef] [PubMed]
- Braun, S., Ferner, M., Kronfeld, K., Griese, M. Hydroxychloroquine in children with interstitial (diffuse parenchymal) lung diseases. Pediatr Pulmonol. 2015;50(4):410-9. [CrossRef] [PubMed]
- AlAnazi, A., Epaud, R., Heena, H., de Becdelievre, A., Miqdad, A.M., Fanen, P. The most frequent ABCA3 nonsense mutation -p.Tyr1515* (Y1515X) causing lethal neonatal respiratory failure in a term neonate. Ann Thorac Med. 2017;12(3):213-215. [CrossRef] [PubMed]
- Beers, M.F., Zhao, M., Tomer, Y., Russo, S.J., Zhang, P., Gonzales, L.W., Guttentag, S.H., Mulugeta, S. Disruption of N-linked glycosylation promotes proteasomal degradation of the human ATP-binding cassette transporter ABCA3. Am J Physiol Lung Cell Mol Physiol. 2013;305(12):L970-80. [CrossRef] [PubMed]
- Li, J., Ikegami, M., Na, C.L., Hamvas A,., Espinassous, Q., Chaby, R., Nogee, L.M., Weaver, T.E., Johansson, J. N-terminally extended surfactant protein (SP) C isolated from SP-B-deficient children has reduced surface activity and inhibited lipopolysaccharide binding. Biochemistry. 2004;43(13):3891-8. [CrossRef] [PubMed]
- Clark, J.C., Wert, S.E., Bachurski, C.J., Stahlman, M.T., Stripp, B.R., Weaver, T.E., Whitsett, J.A. Targeted disruption of the surfactant protein B gene disrupts surfactant homeostasis, causing respiratory failure in newborn mice. Proc Natl Acad Sci U S A. 1995;92(17):7794-8. [CrossRef] [PubMed]
- Kitazawa, H., Moriya, K., Niizuma, H., Kawano, K., Saito-Nanjo, Y., Uchiyama, T., Rikiishi, T., Sasahara, Y., Sakamoto, O., Setoguchi, Y., Kure, S. Interstitial lung disease in two brothers with novel compound heterozygous ABCA3 mutations. Eur J Pediatr. 2013;172(7):953-7. [CrossRef] [PubMed]
- Chen, F., Xie, Z., Zhang, V.W., Chen, C., Fan, H., Zhang, D., Jiang, W., Wang, C., Wu, P. Case Report: Report of Two Cases of Interstitial Lung Disease Caused by Novel Compound Heterozygous Variants in the ABCA3 Gene. Front Genet. 2022;13:875015. [CrossRef] [PubMed]
- Zarbock, R., Kaltenborn, E., Frixel, S., Wittmann, T., Liebisch, G., Schmitz, G., Griese, M. ABCA3 protects alveolar epithelial cells against free cholesterol induced cell death. Biochim Biophys Acta. 2015;1851(7):987-95. [CrossRef] [PubMed]
- Ban, N., Matsumura, Y., Sakai, H., Takanezawa, Y., Sasaki, M., Arai, H., Inagaki, N. ABCA3 as a lipid transporter in pulmonary surfactant biogenesis. J Biol Chem. 2007;282(13):9628-9634. [CrossRef] [PubMed]
- Garmany, T.H., Wambach, J.A., Heins, H.B., Watkins-Torry. J.M., Wegner, D.J., Bennet, K., An, P., Land, G., Saugstad, O.D., Henderson, H., Nogee, L.M., Cole, F.S., Hamvas, A. Population and disease-based prevalence of the common mutations associated with surfactant deficiency. Pediatr Res. 2008;63(6):645-9. [CrossRef] [PubMed]
- Cheong, N., Zhang, H., Madesh, M., Zhao, M., Yu, K., Dodia, C., Fisher, A.B., Savani, R.C., Shuman, H. ABCA3 is critical for lamellar body biogenesis in vivo. J Biol Chem. 2007;282(33):23811-7. [CrossRef] [PubMed]
- Flamein, F., Riffault, L., Muselet-Charlier, C., Pernelle, J., Feldmann, D., Jonard, L., Durand-Schneider, A.M., Coulomb, A., Maurice, M., Nogee, L.M., Inagaki, N., Amselem, S., Dubus, J.C., Rigourd, V., Brémont, F., Marguet, C., Brouard, J., de Blic, J., Clement, A., Epaud, R., Guillot, L. Molecular and cellular characteristics of ABCA3 mutations associated with diffuse parenchymal lung diseases in children. Hum Mol Genet. 2012;21(4):765-75. [CrossRef] [PubMed]
- Weichert, N., Kaltenborn, E., Hector, A., Woischnik, M., Schams, A., Holzinger, A., Kern, S., Griese, M. Some ABCA3 mutations elevate ER stress and initiate apoptosis of lung epithelial cells. Respir Res. 2011;12(1):4. [CrossRef] [PubMed]
- Nathan, N., Borensztajn, K., Clement, A. Genetic causes and clinical management of pediatric interstitial lung diseases. Curr Opin Pulm Med. 2018;24(3):253-259. [CrossRef] [PubMed]
- Matsumura, Y., Ban, N., Ueda, K., Inagaki, N. Characterization and classification of ATP-binding cassette transporter ABCA3 mutants in fatal surfactant deficiency. J Biol Chem. 2006;281(45):34503-14. [CrossRef] [PubMed]
- Matsumura, Y., Ban, N., Inagaki, N. Aberrant catalytic cycle and impaired lipid transport into intracellular vesicles in ABCA3 mutants associated with nonfatal pediatric interstitial lung disease. Am J Physiol Lung Cell Mol Physiol. 2008;295(4):L698-707. [CrossRef] [PubMed]
- Kinting, S., Höppner, S., Schindlbeck, U., Forstner, M.E., Harfst, J., Wittmann, T., Griese, M. Functional rescue of misfolding ABCA3 mutations by small molecular correctors. Hum Mol Genet. 2018;27(6):943-953. [CrossRef] [PubMed]
- Cheong, N., Madesh, M., Gonzales, L.W., Zhao, M., Yu, K., Ballard, P.L., Shuman, H. Functional and trafficking defects in ATP binding cassette A3 mutants associated with respiratory distress syndrome. J Biol Chem. 2006;281(14):9791-800. [CrossRef] [PubMed]
- Yoshida, I., Ban, N., Inagaki, N. Expression of ABCA3, a causative gene for fatal surfactant deficiency, is up-regulated by glucocorticoids in lung alveolar type II cells. Biochem Biophys Res Commun. 2004;323(2):547-55. [CrossRef] [PubMed]
- Stahlman, M.T., Besnard, V., Wert, S.E., Weaver, T.E., Dingle, S., Xu, Y., von Zychlin, K., Olson, S.J., Whitsett, J.A. Expression of ABCA3 in developing lung and other tissues. J Histochem Cytochem. 2007;55(1):71-83. [CrossRef] [PubMed]
- Kolla, V., Gonzales, L.W., Gonzales, J., Wang, P., Angampalli, S., Feinstein, S.I., Ballard, P.L. Thyroid transcription factor in differentiating type II cells: regulation, isoforms, and target genes. Am J Respir Cell Mol Biol. 2007;36(2):213-25. [CrossRef] [PubMed]
- Wambach, J.A., Wegner, D.J., Depass, K., Heins, H., Druley, T.E., Mitra, R.D., An, P., Zhang, Q., Nogee, L.M., Cole, F.S., Hamvas, A. Single ABCA3 mutations increase risk for neonatal respiratory distress syndrome. Pediatrics. 2012;130(6):e1575-82. [CrossRef] [PubMed]
- Kröner, C., Wittmann, T., Reu, S., Teusch, V., Klemme, M., Rauch, D., Hengst, M., Kappler, M., Cobanoglu, N., Sismanlar, T., Aslan, A.T., Campo, I., Proesmans, M., Schaible, T., Terheggen-Lagro, S., Regamey, N., Eber, E., Seidenberg, J., Schwerk, N., Aslanidis, C., Lohse, P., Brasch, F., Zarbock, R., Griese, M. Lung disease caused by ABCA3 mutations. Thorax. 2017;72(3):213-220. [CrossRef] [PubMed]
- Henderson, L.B., Melton, K., Wert, S., Couriel, J., Bush, A., Ashworth, M., Nogee, L.M. Large ABCA3 and SFTPC deletions resulting in lung disease. Ann Am Thorac Soc. 2013;10(6):602-7. [CrossRef] [PubMed]
- Hamvas, A., Nogee, L.M., Wegner, D.J., Depass, K., Christodoulou, J., Bennetts, B., McQuade, L.R., Gray, P.H., Deterding, R.R., Carroll, T.R., Kammesheidt, A., Kasch, L.M., Kulkarni, S., Cole, F.S. Inherited surfactant deficiency caused by uniparental disomy of rare mutations in the surfactant protein-B and ATP binding cassette, subfamily a, member 3 genes. J Pediatr. 2009;155(6):854-859.e1. [CrossRef] [PubMed]
- Hartl, D., Griese, M. Interstitial lung disease in children -- genetic background and associated phenotypes. Respir Res. 2005;6(1):32. [CrossRef] [PubMed]
- Karjalainen, M.K., Haataja, R., Hallman, M. Haplotype analysis of ABCA3: association with respiratory distress in very premature infants. Ann Med. 2008;40(1):56-65. [CrossRef] [PubMed]
- Bullard, J.E., Wert, S.E., Whitsett, J.A., Dean, M., Nogee, L.M. ABCA3 mutations associated with pediatric interstitial lung disease. Am J Respir Crit Care Med. 2005;172(8):1026-31. [CrossRef] [PubMed]
- Crossno, P.F., Polosukhin, V.V., Blackwell, T.S., Johnson, J.E., Markin, C., Moore, P.E., Worrell, J.A., Stahlman, M.T., Phillips, J.A. 3rd, Loyd, J.E., Cogan, J.D., Lawson, W.E. Identification of early interstitial lung disease in an individual with genetic variations in ABCA3 and SFTPC. Chest. 2010;137(4):969-73. [CrossRef] [PubMed]
- Wittmann, T., Frixel, S., Höppner, S., Schindlbeck, U., Schams, A., Kappler, M., Hegermann, J., Wrede, C., Liebisch, G., Vierzig, A., Zacharasiewicz, A., Kopp, M.V., Poets, C.F., Baden, W., Hartl, D., van Kaam, A.H., Lohse, P., Aslanidis, C., Zarbock, R., Griese, M. Increased Risk of Interstitial Lung Disease in Children with a Single R288K Variant of ABCA3. Mol Med. 2016;22:183-191. [CrossRef] [PubMed]
- Kaltenborn, E., Kern, S., Frixel, S., Fragnet, L., Conzelmann, K.K., Zarbock, R., Griese, M. Respiratory syncytial virus potentiates ABCA3 mutation-induced loss of lung epithelial cell differentiation. Hum Mol Genet. 2012;21(12):2793-806. [CrossRef] [PubMed]
- Yang, X., Rapp, C.K., Li, Y., Forstner, M., Griese, M. Quantifying Functional Impairment of ABCA3 Variants Associated with Interstitial Lung Disease. Int J Mol Sci. 2023;24(8):7554. [CrossRef] [PubMed]
- Beers, M.F., Hawkins, A., Shuman, H., Zhao, M., Newitt, J.L., Maguire, J.A., Ding, W., Mulugeta, S. A novel conserved targeting motif found in ABCA transporters mediates trafficking to early post-Golgi compartments. J Lipid Res. 2011;52(8):1471-82. [CrossRef] [PubMed]
- Wambach, J.A., Yang, P., Wegner, D.J., Heins, H.B., Luke, C., Li, F., White, F.V., Cole, F.S. Functional Genomics of ABCA3 Variants. Am J Respir Cell Mol Biol. 2020;63(4):436-443. [CrossRef] [PubMed]
- Karbassi, I., Maston, G.A., Love, A., DiVincenzo, C., Braastad, C.D., Elzinga, C.D., Bright, A.R., Previte, D., Zhang, K., Rowland, C.M., McCarthy, M., Lapierre, J.L., Dubois, F., Medeiros, K.A., Batish, S.D., Jones, J., Liaquat, K., Hoffman, C.A., Jaremko, M., Wang, Z., Sun, W., Buller-Burckle, A., Strom, C.M., Keiles, S.B., Higgins, J.J. A Standardized DNA Variant Scoring System for Pathogenicity Assessments in Mendelian Disorders. Hum Mutat. 2016;37(1):127-34. [CrossRef] [PubMed]
- Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., Grody, W.W., Hegde, M., Lyon, E., Spector, E., Voelkerding, K., Rehm, H.L.; ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405-24. [CrossRef] [PubMed]
- Duzkale, H., Shen, J., McLaughlin, H., Alfares, A., Kelly, M.A., Pugh, T.J., Funke, B.H., Rehm, H.L., Lebo, M.S. A systematic approach to assessing the clinical significance of genetic variants. Clin Genet. 2013;84(5):453-63. [CrossRef] [PubMed]
- Thusberg, J., Olatubosun, A., Vihinen, M. Performance of mutation pathogenicity prediction methods on missense variants. Hum Mutat. 2011;32(4):358-68. [CrossRef] [PubMed]
- Thavagnanam, S., Cutz, E., Manson, D., Nogee, L.M., Dell, SD. Variable clinical outcome of ABCA3 deficiency in two siblings. Pediatr Pulmonol. 2013;48(10):1035-8. [CrossRef] [PubMed]
Figure 1.
The flow chart of the systematic review on ABCA3 c.838C>T (p.Arg280Cys, R280C) and c.697C>T (p.Gln233Ter, Q233X, Q233*).
Figure 1.
The flow chart of the systematic review on ABCA3 c.838C>T (p.Arg280Cys, R280C) and c.697C>T (p.Gln233Ter, Q233X, Q233*).
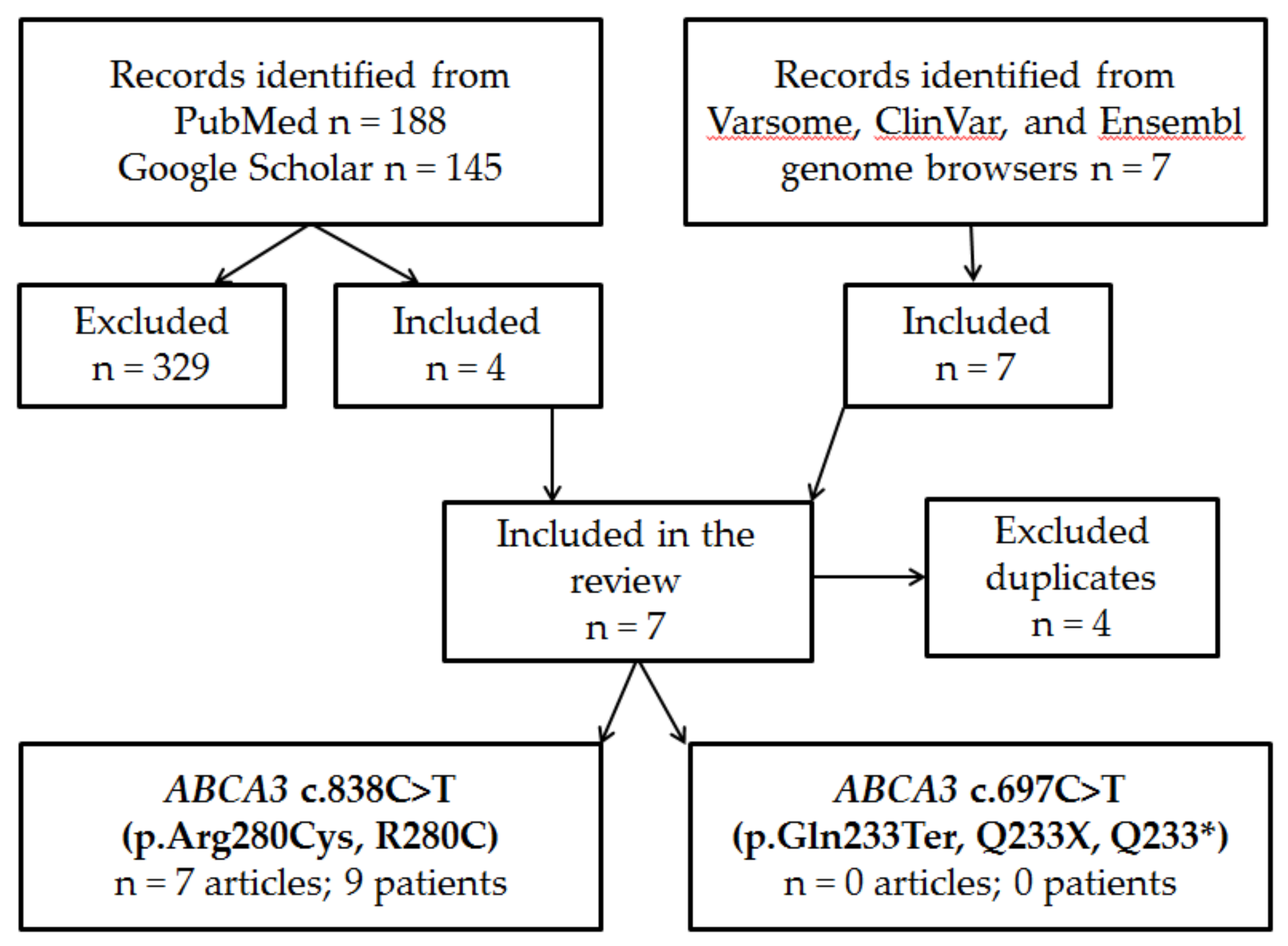
Figure 2.
Lung x-ray of index patient. A. DOL 1. B. DOL 5. C DOL 40. D DOL 60 (DOL – day of life).
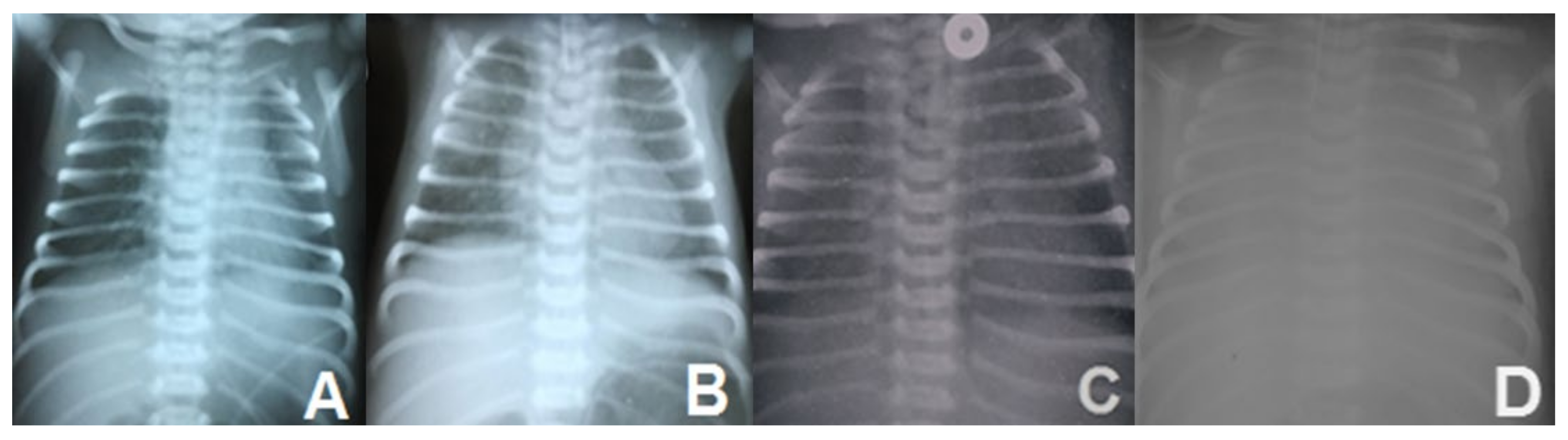
Figure 3.
Lung histology: A – Alveolar proteinosis (X 100); B – Thickened alveolar septae and vascular stasis (X 200); C – Alveoli and bonchiole filled with eosinophylic material (X 200); D – Alveoli with pseudostratified AEC II (X 200); E, F – Alveolar proteinosis (x100); A-D - hematoxylin eosin staining; E, F – periodic-acid Schiff (PAS) staining.
Figure 3.
Lung histology: A – Alveolar proteinosis (X 100); B – Thickened alveolar septae and vascular stasis (X 200); C – Alveoli and bonchiole filled with eosinophylic material (X 200); D – Alveoli with pseudostratified AEC II (X 200); E, F – Alveolar proteinosis (x100); A-D - hematoxylin eosin staining; E, F – periodic-acid Schiff (PAS) staining.
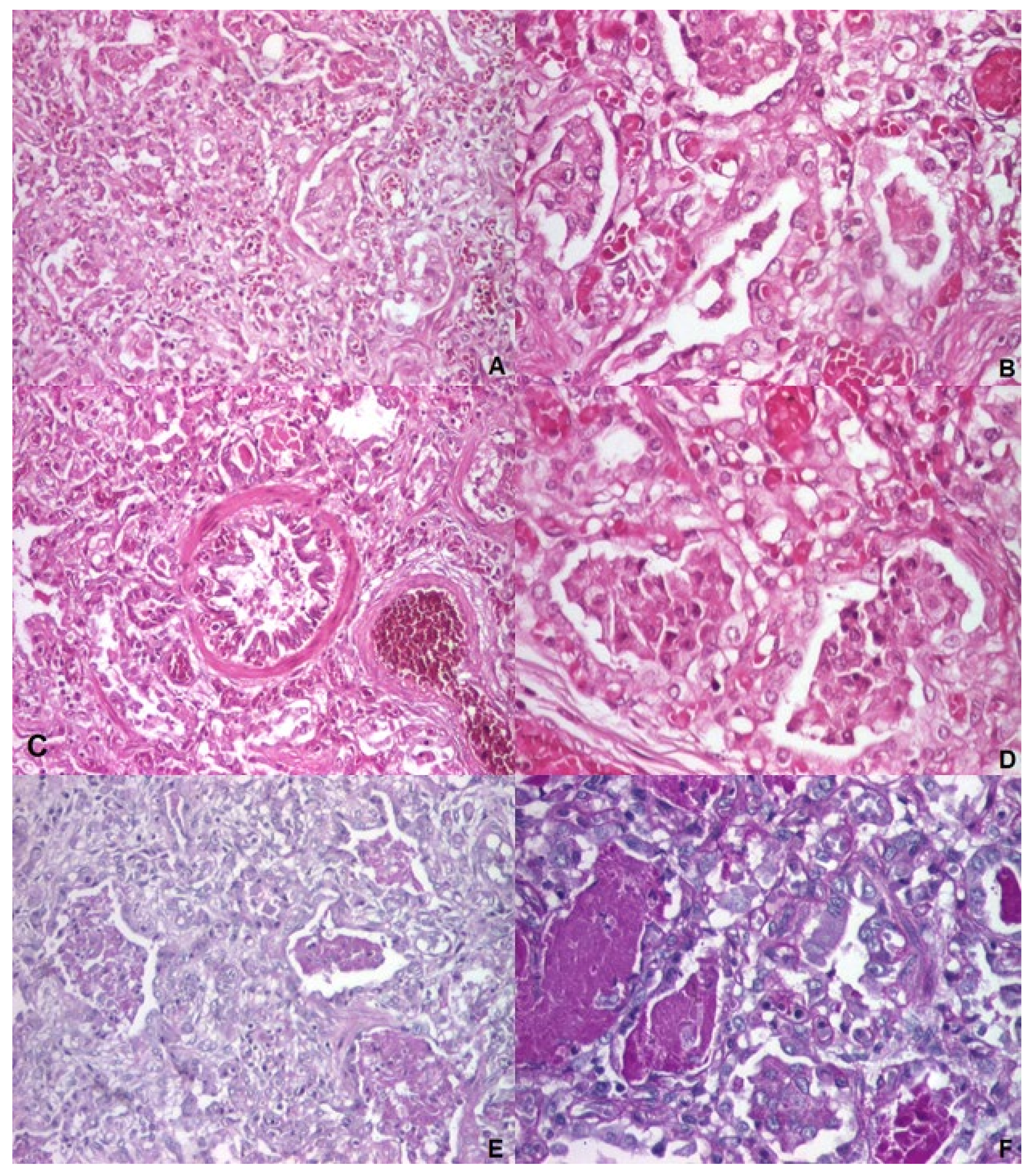
Figure 4.
Pedigree of the family studied: squares represent males, and circles represent females; the arrow points to the index case; slashed symbols indicate deceased individuals; black-filled symbols denote individuals with fatal neonatal respiratory distress syndrome, half-filled symbols represent carriers.
Figure 4.
Pedigree of the family studied: squares represent males, and circles represent females; the arrow points to the index case; slashed symbols indicate deceased individuals; black-filled symbols denote individuals with fatal neonatal respiratory distress syndrome, half-filled symbols represent carriers.

Table 1.
Anthropometric, clinical, radiological and histological characteristics, genetic variants, family history and patients outcome reported in the literature with ABCA3 c.838C>T (p.Arg280Cys, R280C) variant.
Table 1.
Anthropometric, clinical, radiological and histological characteristics, genetic variants, family history and patients outcome reported in the literature with ABCA3 c.838C>T (p.Arg280Cys, R280C) variant.
| Pacient number | Reference | Gender | GA | BW | Respiratory disease | Imaging aspects | Lung histology | EM | ABCA3 variant - alelle 1/parental origin | Other associated ABCA3 variant(s)/parental origin/Allele 2 | Variants of other surfactant genes/parental origin | Familial history | Outcome | Comments |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1. | Somaschini et al., 2007 [59] | Male | 39 weeks | 2850 g | Severe nRDS (mechanical ventilation) | N/A | N/A | N/A | R280C/wt/ | Most probably the second mutation was missed due to restrictive genetic testing protocol | Tested, none reported | No | Died at 2 days | Patient 7 in a case series of 17 cases with inherited deficiency of pulmonary surfactant |
| 2. | Turcu et al., 2013 [60] | Not reported | Term | Not reported | nRDS – Surfactant administration, ventilation, hydroxychloroquine | Interstitial changes on lung CT | CIP | N/A | c.838C>T (p.Arg280Cys)/heterozygous/parental origin not reported | c.2069A>G (p.Glu690Gly)/heterozygous/parental origin not reported | None reported | no | Alive at 13 years | Patient reported in a case serie of 323 cases analyzed for inherited deficiency of pulmonary surfactant |
| 3. | Williamson & Wallis, 2014 [61] | Female | Term | Not reported | nRDS – surfactant, mechanical ventilation, multiple corticosteroids courses, oxygen dependent at 2 years; treated with hydroxychloroquine afterwards | Extensive patchy ground glass opacification and cystic airway changes, predominantly in the upper lobes at 2 years on thoracic CT |
DIP at 2 years |
N/A | c.838C>T/heterozygous/parental origin not reported | c.2069A>G/heterozygous/parental origin not reported | Tested, no variants discovered | N/A | Final evaluation at 13 years, stable under hydroxychloroquine treatment | Case report |
| 4. | Wambach, 2014 [62] | Female | N/A | N/A | nRDS | N/A | N/A | N/A | Q1589X R280C (homozygous) |
Q1589X R280C (homozygous) |
N/A | N/A | Died < 3 months | Based on unpublished data |
| 5. | Male | N/A | N/A | nRDS | N/A | N/A | N/A | c.3997_3998delAG (null) | R280C | N/A | N/A | Alive | Based on unpublished data | |
| 6. | Female | N/A | N/A | nRDS | N/A | N/A | NA | Q1589X (p.Gln1589) R280C |
c.4195G>A (V1399M)(p.Val1399Met) | N/A | N/A | Lung transplantation at 10 months | Based on unpublished data; Reported also by Xu et al., 2022 [63] | |
| 7. | Jackson et al., 2015 [64] | Female | Term | 2870 g | Severe nRDS treated with advanced respiratory support (HFOV, iNO), weaned for respiratory support at 1 year | N/A | Diffuse interstitial fibrosis with alveolar remodeling and prominent AEC II hyperplasia | Small, dense lamellar bodies and occasional fused lamellar bodies | p.R280C (c.838C>T)/paternal (cis) | p.Val1399Met(c.4195G>A)/maternal p.Q1589X (C.4765C>T)/paternal (cis) |
Tested, none discovered | No (parents and one previous sibling without – healthy) | Alive at 3 years, mild speech and motor delay | Case report; p.V1399M is rare and has been previously reported in symptomatic infants [62]; p.Q1589X is predicted to result in a truncated protein [64] Both p.R280C and p.V1399M are predicted to be damaging to ABCA3 protein function by the majority of in silico prediction programs in ANNOVAR [65] |
| 8. | Klay et al., 2020 [66] | Female | Not reported | Not reported | ILD - onset at 19 years with dyspnea; high resolution chest CT: diffuse ground glass opacities with emphysema located in the apical regions, progressing to severe lung fibrosis |
N/A | Diffuse fibrosis with chronic inflammation, unusual interstitial pneumonia, fibroblast foci or granulomas. Mild AEC II hyperplasia, accumulation of alveolar macrophages |
N/A | c.838C>T (p.R280C, rs201299260) (trans)/one parent c.875A>T (p.E292V, rs149989682). (trans)/the other parent |
- | No report on other genetic tests | No | Proposed for lung transplantation at 25 years | Case report; Initially diagnosed with drug-induced ILD |
| 9. | Gjeta et al., 2023 [67] | Female | Not reported | Not reported | Onset at 2 years and 7 months with severe pediatric ARDS (requiring invasive respiratory support, prednisolone and azithromycin treatment), after 2 episodes of upper respiratory tract infections | Chest X-ray showed bilateral opacification suggesting interstitial bilateral pneumonia. chest CT scan showed bilateral ground-glass opacities |
N/A | N/A | c.4261 G>A p. (Gly1421Arg)/heterozygous/paternal | c.838C>T p. (Arg280Cys)/heterozygous/maternal | Tested, none reported | N/A | Alive, after prolonged oxygen therapy and on treatment with oral hydroxychloroquine and fluticasone propionate inhalations | Case report |
| 10. | Ognean et al., 2024 (this study) | Male | 37 weeks | 2700 g | Severe nRDS – advanced respiratory support (HFOV), prednison, azithromycin, hydroxychloroquine treatment | Thoracic x-ray – ground glass homogeneous opacity | CPI pattern with lobular remodeling, prominent AEC II hyperplasia, focal PAP pattern and extensive DIP-like areas, alveolar proteinosis | N/A | p.Arg280Cys (R280C, c.838C>T, rs201299260)/heterozygous/paternal origin | p.Gln233ter (Q233X, Q233*)heterozygous/maternal origin | SFTPB p.Val267Ile | No, healthy parents, one healthy sibling despite carrying p.Gln233ter and SFTPB p.Val267Ile variants | Died at 77 days of life | Current case report |
Legend: GA, gestational age; BW, birth weight; EM, electron microscopy; nRDS, neonatal respiratory distress syndrome; AEC II, alveolar epithelial type II cells; CIP, chronic interstitial pneumonitis; DIP, desquamative interstitial pneumonitis; ARDS, acute respiratory distress syndrome; ILD, interstitial lung disease; PAP, pulmonary alveolar proteinosis; MV, mechanical ventilation; HFOV, high-frequency oscillatory ventilation; iNO, inhaled nitric oxide; CT, computed tomography; ANNOVAR, ANNOtate VARiation software tool; N/A, not available.
Table 2.
Pros and cons arguments on ABCA3 c.838C>T (p.Arg280Cys, R280C) pathogenicity.
| Pros | Cons | |
| Clinical aspects | R280C mutations were reported in association with neonatal RDS and ILD, with clinical picture and course suggestive of surfactant metabolism dysfunction [59,61,62,64,66,107] | Variable phenotype, usually in association with other mutations with survival varying between 2 days and over 25 years [66]; severe phenotype was associated in 4 of the 10 cases reported in the literature (Table 1) |
| In vitro experiments | R280C mutations result in folding and trafficking defects, increased endoplasmic reticulum stress, and apoptosis induction in lung epithelial cells in vitro [86] | In vitro experiments cannot accurately represent biological functions [86] Functional impact on ABCA3 protein is less important as compared to other ABCA3 mutations [66] |
| DNA analysis | Mutations reported are associated with respiratory disease in the subjects [59,61,62,64,66,107] | Most reported inducing-disease variants are associated with other mutations in ABCA3 or SFTPB genes, some of these known as pathogenic (ex. Q1589X) [62]; co-occurrence with these mutations may suggest R280C as benign variant; Incomplete genetic testing in some patients (for example, Somaschini et al., 2007 [59] |
| Estimated allele frequency in the population | Rare, suggestive of pathogenicity (surfactant metabolic dysfunction produced by ABCA3 mutations) [108,109] | Frequency higher than for other mutations causing ABCA3 deficiency, not offering unequivocal evidence for pathogenicity [55] |
| Computational predictive tools and conservational analysis | Most indicate a damaging effect of the mutation on ABCA3 protein [55]. Calibrated prediction (examples):
|
Insufficient power for predicting pathogenicity Calibrated prediction (examples)
Bioinformatic prediction tools do not determine and or exclude pathogenicity [110]. Most of the current prediction algorithms have a 65-80% predictive accuracy for known pathogenic variants and a tendency to low specificity [111] Predictive bioinformatics seems insufficient for defining the pathogenicity of ABCA3 deficiencies [52,105] |
Legend: RDS, neonatal respiratory distress syndrome; ILD, interstitial lung disease; ABCA3 gene, ATP binding cassette subfamily A member 3 gene; SFTPB gene, surfactant Protein B gene; SIFT, Sorting Intolerant From Tolerant program, DANN, Deleterious Annotation of genetic variants using Neural Networks Computational Tool.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



