
Submitted:
18 August 2024
Posted:
19 August 2024
You are already at the latest version
Abstract
Streptococcus agalactiae is a highly contagious Gram- positive bacterium that causes mastitis, has a high infectivity for mammary epithelial cells, and is challenging to treat. However, the molecular interactions between it and mammary epithelial cells remains poorly understood. This study analyzed differential gene expression in mammary epithelial cells with varying levels of S. agalactiae infection using UID- Dual Seq and bioinformatics tools. This study identified 211 differentially expressed mRNAs (DEmRNAs) and 452 differentially expressed lncRNAs (DElncRNAs) in host cells, primarily enriched in anti-inflammatory responses, immune responses, and cancer-related processes. Additionally, 854 pathogen differentially expressed mRNAs (pDEmRNAs) were identified, mainly enriched in protein metabolism, gene expression, and biosynthesis processes. Mammary epithelial cells activate pathways, such as the ERK1/2 pathway to produce, to produce reactive oxygen species (ROS) to eliminate bacteria. However, the bacteria disrupt the host's innate immune mechanisms by interfering with the alternative splicing processes of mammary epithelial cells. Specifically, the bacterial genes of tsf, prfB, and infC can interfere with lncRNAs targeting RUNX1 and BCL2L11 in mammary epithelial cells, affecting the alternative splicing of target genes and altering normal molecular regulation.
Keywords:
Mastitis
; Streptococcus agalactiae
; UID- Dual
; RNA- seq
; mammary epithelial cells
1. Introduction
Udders are vital functional organs in cows and provide a significant source of protein (milk) for humans. Milk is produced by the mammary glands and is collected into the milk ducts through physical methods such as squeezing and excreting[1]. Tyoically, after the mammary gland is infected with pathogenic microorganisms, or stimulated by physical, chemical, and other factors, inflammatory changes can occur in the mammary plasma or parenchymal tissue, resulting in mastitis. This disease is primarily caused by various pathogenic bacteria or microorganisms in the environment infecting the mammary glands via milk ducts, leading to inflammation[2]. Based on clinical symptoms, mastitis can be divided into clinical and subclinical forms [3,4]. Clinical mastitis is characterized by pronounced signs of inflammation in the udder, microbial presence, and chemical changes in milk[5]. Cows with clinical mastitis are affected by pathogenic bacteria that cause fibrosis and atrophy of the mammary gland, leading to premature culling[6]. However, cows with subclinical mastitis exhibit only mild inflammation after pathogenic bacteria enter teat duct. The somatic cell count (SSC) in milk was increases, with no other clinical manifestations[7]. Although subclinical mastitis does not pose an immediate risk, if left untreated, it will eventually develop into clinical mastitis and cause huge economic losses.
The study by Paramanandham et al. indicated that Staphylococcus aureus is the most common mastitis pathogen, Escherichia coli is the main pathogen causing clinical mastitis and Streptococcus spp. can cause both subclinical and clinical mastitis worldwide[8]. In many cases of subclinical mastitis, the inflammatory response is predominantly caused by Streptococcus agalactiae[9,10,11]. S. agalactiae is classified as group B according to the Lancefield bacterial taxonomy. The bacterium is mostly β-hemolytic, although some non-hemolytic and CAMP-positive strains have been observed[12]. Due to the variety of virulence factors, S. agalactiae can adhere to and invade host cells, inducing an inflammatory response in the mammary gland. Therefore, it is essential to analyze the transcriptional regulation of inflammatory genes during S. agalactiae invasion of the mammary gland. The mammary epithelium in first line of defense in the mammary gland. It can effectively initiate an immune response, eliminating pathogens before any abnormal changes occur in the mammary gland, which is crucial for resistance to mastitis and affects susceptibility[13]. When the pathogen successfully breaches the host's physical defenses, mammary gland epithelial detect bacteria through specific pattern recognition receptors and initiate a series of immune responses. Currently, researchers have conducted high-throughput sequencing studies on mammary infected with S. agalactiae and identified several immune-related receptors or pathways, but the findings are not consistent. However, the mechanisms underlying S. agalactiae infection in subclinical mastitis are not well understood.
Zhang et al. found that, compared with healthy cow mammary glands, a total of 129 differentially expressed genes and 144 differentially expressed proteins were identified in mammary glands infected with S. agalactiae[14]. Intramammary infection with S. agalactiae triggered a complex host innate immune response involving complement and coagulation cascades, ECM-receptor interaction, focal adhesion, and phagosome and bacterial invasion of epithelial cells pathways. Tong et al. used proteomics to discover that differentially expressed proteins included enzymes and proteins associated with various metabolic processes and cellular immunity in S. agalactiae-infected bovine mammary epithelial cells[15]. Subsequently, they used the ubiquitinome analysis to find that ubiquitinated proteins are associated with regulating cell junctions in the host[16]. Sbardella et al. infected dairy cow mammary glands with S. agalactiae via manual intervention, and screened 122 differential genes in the sequencing data using three different statistical methods, but only platelet activation showed a significant enrichment pathway[17]. Additionally, it has been found that differential genes in mammary alveolar tissue infected with S. agalactiae are mainly involved in innate immune response, inflammatory response, chemokine signaling, Wnt signaling, complement and coagulation cascades compared with normal tissues[18]. Richards et al. concluded that lactose metabolism is an important metabolic pathway for S. agalactiae to adapt to the bovine mammary environment, as determined by sequencing analysis of isolated S. agalactiae[19]. In mammary glands infected with S. agalactiae, differentially expressed miRNAs were mainly involved in the RIG-I-like receptor signaling pathway, cytosolic DNA sensing pathway, and Notch signaling pathway[20].
Most of the studies are in vivo, and research on immune changes following mammary epithelial cell infection is lacking. Therefore, studying the immune regulation of mammary epithelial cells infected with S. agalactiae is essential. The transcriptome serves as a powerful indicator of the physiological state of a cell, whether healthy or diseased. Consequently, transcriptome analysis has become a crucial tool for understanding the molecular changes that occur during bacterial infections of eukaryotic cells. Previously, transcriptomic studies were technically limited to analyzing mRNA expression changes in either the bacterial pathogen or the infected eukaryotic host cell. However, the increasing sensitivity of high-throughput RNA sequencing now enables "Dual RNA-seq" studies, simultaneously capturing all classes of coding and noncoding transcripts in both the pathogen and the host[21].
To the best of our knowledge, this study represents the first report of Dual RNA-seq on mammary epithelial cells infected with S. agalactiae. Identifying key differentially expressed genes and pathways in infected mammary epithelial cells provides a foundation for a better understanding of the central mechanisms of host defense during subclinical infections like mastitis.
2. Materials and Methods
2.1. Bacterial Strains and Growth Conditions
S. agalactiae strain ATCC27956 was inoculated onto Edwards Medium Modified (EMM) Agar and incubated at 37°C for 24 h. A single colony was randomly selected and cultured in Todd Hewitt Broth (THB) with agitation at 37°C for 12 h and the growth was monitored by measuring the OD600nm.
2.2. Cell Culture
MAC-T cells were cultured in T25 cell culture flasks with Dulbecco's modified eagle culture medium (DMEM) containing 10% fetal bovine serum (FBS) and maintained in 5% CO2 at 37°C. The cells were cultured until they reached 80% of the confluence for further experiments.
2.3. Intracellular Infection Model
The intracellular infection model followed the method described by Tong et al[16]. MAC-T cells were cultured in T75 cell culture flasks until and they reached a density of 1x106 cells/ml. Control (M Group) and MOI group (100:1) groups were then established. The MOI group was incubated for 2 hours(S Group) and 6 hours(H Group), respectively. Each group included at least three biological replicates. The cells were washed twice with phosphate-buffered saline (PBS), and DMEM containing lysozyme (20 μg/mL) and gentamicin (100 μg/mL) were added. The culture was maintained at 37 °C in 5% CO2 for 2 hours to remove the extracellular bacteria. The collected sterilized cell culture medium was applied to bacterial plates to ensure that elimination of extracellular bacteria. The MAC-T cells were rinsed three more times with PBS to remove any remaining extracellular adherent bacteria and were then culture in 10% FBS-DMEM.
2.4. RNA Extraction and cDNA Library Construction
Total RNAs were extracted from both the control and S. agalactiae-treated group using TRIzol Reagent (Invitrogen, cat. NO 15596026) following the manufacturer's instructions. DNA digestion was performed after RNA extraction using DNaseI. RNA quality was assessed by measuring the A260/A280 ratio using a Nanodrop TM OneC spectrophotometer (Thermo Fisher Scientific Inc.). RNA Integrity was confirmed using a Qsep100 (BiOptic Inc.) and a 5300 Fragment Analyzer system (Agilent). The qualified RNAs were quantified using a Qubit3.0 with the Qubit TM RNA Broad Range Assay kit (Life Technologies, Q10210).
2μg total RNA were used for stranded RNA sequencing library preparation using KC-DigitalTM Total RNA Library Prep Kit (Catalog NO. DLR08702, Wuhan Seqhealth Co., Ltd. China), Ribo- off rRNA Depletion Kit (Vazyme # N406/ N409), MICROB Express Kit, (Thermo # AM1905), Ribo- off rRNA Depletion Kit (Bacteria), (Vazyme # N407) following the manufacturer’s instruction. The library products, ranging from 200- 500bps were enriched, quantified, and sequenced using a DNBSEQ- T7 sequencer (MGI Tech Co., Ltd. China) with PE150 model. The UID- dual RNA-seq experiment, high throughput sequencing and data analysis were conducted by Seqhealth Technology Co., LTD (Wuhan, China).
2.5. RNA- Seq Data Analysis
Raw sequencing data were first filtered by using fastp (version 0.23.0), low-quality reads were discarded, and the reads contaminated with adaptor sequences were trimmed. Clean Reads were further processed using in-house scripts to eliminate duplication bias introduced during library preparation and sequencing. In brief, clean reads were first clustered based on UMI sequences, with reads sharing the same UMI sequence grouped into the same cluster. Reads within the same cluster were compared by pairwise alignment, and those with sequence identity exceeding 95% were assigned to a new sub-cluster. After all sub-clusters were generated, multiple sequence alignment was performed to obtain one consensus sequence for each sub-cluster. following these steps, any errors and biases introduced during PCR amplification or sequencing were removed.
2.6. Reads Alignment and Differential Expression Analysis of RNA-Seq
The reference genomes of the two species were merged, and then the deduplicated data were mapped to the reference genomes of Bos taurus from http://asia.ensembl.org/Bos_taurus/Info/Index. and S. agalactiae from https://bacteria.ensembl.org/ Streptococcus_agalactiae_gca_900458965/Info/Index/. using STAR software (version 2.5.3a) with default parameters. The reads mapped to the exon regions of each gene were counted using feature Counts (Subread-1.5.1; Bioconductor) and then RPKM was calculated.
Differentially expressed genes between groups were identified using the edgeR package (version 3.28.1). A p-value cutoff of 0.05 and fold-change cutoff of 1 or -1 were used to etermine the statistical significance of gene expression differences.
2.7. Analysis of the Alternative Splicing Prediction
The alternative splicing prediction were predicted by rMATS (Version 3.2.5)[22].
2.8. Prediction of Target Gene of lncRNA
2.9. GO and KEGG Pathway Analysis of DEmRNAs and Target Genes of DElncRNAs
Gene Ontology (GO) and The Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways enrichment analysis was performed using the DAVID 2021 (Dec. 2021) functional annotation tool.
2.10. Differential Gene Co-Expression Analysis
Co-expression analysis of mRNA was performed using https://www.bioinformatics.com.cn (last accessed on 20 June 2024), an online platform for data analysis and visualization[27].
3. Results
3.1. Transcriptome Assembly Profiles Evaluation
Thirteen samples were deeply sequenced, yielding 156.06 Gb of mRNA and lncRNA transcription data(Table 1). The raw reads were filtered and the clean data were analyzed downstream.
3.2. Analysis of Differentially Expressed mRNAs
Cluster pattern analysis of host DEmRNAs between the control (n= 5), S. agalactiae - S groups (n= 5), and S. agalactiae – H groups (n= 3) is illustrated in Figure 1A. A total of 3370 DEmRNAs (S_M) (Supplementary File 1) were filtered by the thresholds of p < 0.05 and |log2(fold- change)| > 1, out of which 2001 were up-regulated and 1369 were down-regulated (Figure 1B). A total of 4730 DEmRNA (H_M) were found, with 2472 were up-regulated and 2258 were down-regulated (Figure 1C) (Supplementary File 2). A total of 2085 DEmRNA (H_ S) were identified, with 1102 were up-regulated and 983 were down-regulated (Figure 1D) (Supplementary File 3).
Gene Ontology (GO) was used to classify the functions of DEGs (Figure 2). The DEGs enriched in the three comparison groups were annotated using three GO categories: biological process (BP), cellular component (CC), and molecular function (MF).
In the comparisons of S_M, H_M, and H_S, GO enrichment analysis showed that the upregulated genes in biological processes were mainly enriched in negative chemotaxis, extrinsic apoptotic signaling pathway in the absence of ligand, small GTPase mediated signal transduction, and regulation of ERK1 and ERK2 cascade. In molecular functions, they were primarily enriched in transcription factors, and in cellular components, they were mainly enriched in cell membrane structures and complexes. The downregulated genes were mainly enriched in NADH dehydrogenase (ubiquinone) activity and translation elongation factor activity in molecular functions, in the Lsm2-8 complex and nucleolus in cellular components, and in mRNA splicing via the spliceosome and translational elongation in biological processes. Additionally, they were enriched in pathways related to apoptosis.
The enrichment analysis of the KEGG pathway in DEmRNAs is depicted in Figure 2C. In the three comparison groups, signaling pathways were primarily enriched in disease-related pathways such as cancer, leukemia, and diabetes. Additionally, they were mainly enriched in environmental information processing pathways such as the MAPK signaling pathway, TGF-beta signaling pathway, and Notch signaling pathway. Pathways involved in cellular processes included the p53 signaling pathway, apoptotic signaling pathway, and cellular senescence. Pathways related to organismal systems included thermogenesis and the IL-17 signaling pathway.
3.3. Analysis of Host mRNA Alternative Splicing
Using rMATs to analyze differential AS events in S_M, H_M, and H_S, a total of 130,178 alternative splicing events were detected by target and junction reads. After setting the threshold p-value < 0.05 and |Δψ| > 0.1 for alternative splicing filtering, a total of 10,750 differentially expressed alternative splicing events were identified across the three comparison groups (Table 2).
Among the three comparison groups, the SE type was identified the most frequently, with a total of 104,687 events, followed by the MXE type with 21,901 events. The A5SS, A3SS, and RI types were relatively less frequent, with 982, 1,216, and 1,392 events, respectively. Differential analysis of AS events revealed that there were 3,980 differentially expressed SE types across the three comparison groups, of which 1,505 were upregulated and 2,475 were downregulated. The analysis identified 6,501 differentially expressed MXE types, with 3,501 upregulated and 3,000 downregulated.
In the S_M group, the numbers of differentially spliced genes identified in the SE, MXE, A5SS, A3SS, and RI events were 983, 2,065, 32, 25, and 24, respectively. In the H_M group, the numbers were 1,684, 2,505, 44, 32, and 45, respectively. In the H_S group, the numbers were 1,313, 1,931, 26, 21, and 20, respectively. Among the differentially spliced genes, the MXE type was the most common, while the A3SS type was the least common.
The differentially spliced genes in the three comparison groups are mainly enriched in biological processes including the ubiquitin-dependent protein catabolic process, positive regulation of GTPase activity, and protein polyubiquitination. The cellular components show primary enrichment in the nucleus, cytoplasm, nucleoplasm, and nuclear body. The molecular functions are predominantly associated with ATP binding, RNA binding, and GTPase activator activity (Figure 3).
The enrichment analysis of the KEGG pathway in AS DEmRNAs is depicted in Figure 4. In the three comparison groups, signaling pathways are enriched in various categories, including disease, metabolism, genetic information processing, environmental information processing, and cellular processes. Pathways enriched in metabolism include those involved in lysine degradation and fatty acid metabolism. Pathways enriched in genetic information processing include the ubiquitin-mediated proteolysis and nucleotide excision repair. The main pathway enriched in environmental information processing is the sphingolipid signaling. Pathways enriched in cellular processes include those involved in focal adhesion, tight junction, and autophagy in animals. Disease-related pathways include those associated with Yersinia infection, renal cell carcinoma, and bacterial invasion of epithelial cells.
3.4. Analysis of Differentially Expressed lncRNA
Each sample yielded novel DElncRNAs (Figure 5A), and according to the CNCI, COC, Pfam, and CPAT databases, a total of 5995 novel DElncRNAs were identified (Figure 5B). After correction, the uniformity among samples was relatively high. The heatmap showed hierarchical clustering of DElncRNAs (Figure 5C). The volcano plot illustrated the DElncRNAs between S_M, H_M, and H_S (Figure 5D-F). The S_M comparison had 4377 significantly different genes, with a total of 2798 targeted genes. The H_M comparison had 4050 significantly different genes, with a total of 376 targeted genes. The H_S comparison had 1868 significantly different genes, with a total of 189 targeted genes. A total of 452 mRNAs targeted by lncRNAs were identified as common among the three comparison groups in the final analysis (Figure 6) (Supplementary File 4).
The results of the Venn analysis showed that 211 DEmRNAs among the three comparative groups. 8 genes were common among the differentially expressed genes, alternatively spliced genes, and lncRNA-targeted mRNAs of the three control groups, namely TPM2, ARRB1, TCHP, BCL2L11, LSMEM1, RUNX1, DNAH12, and IDS (Figure 7) (Supplementary File 5).
To further understand the dynamic changes at the gene expression level during the metastatic progression, we classified all DEmRNAs into eight patterns (Cluster1, …, Cluster8) using Mfuzz (Figure 8).
The genes in Cluster 1 are upregulated in two stages in the S and H groups after the cells are infected, while the genes in Cluster 3 do not show significant differences between the M and S groups but are rapidly upregulated in the H group. The genes in Clusters 2 and 4 are upregulated in the S group and downregulated in the H group; however, the difference between the M and S groups is not significant in Cluster 2. The genes in Cluster 4 are downregulated in the H group, but their expression levels are still higher than in the M group. The genes in Clusters 5 and 8 are downregulated in both the S and M groups, but the genes in Cluster 8 show significant differences among genomes, with no significant difference between the S and M groups. The genes in Cluster 7 are downregulated in the S group and rapidly upregulated in the H group, with significant differences between the H group and both the M and S groups. Through examining each cluster, TPM2 and TCHP were found to be enriched in Cluster 5, ARRB1, BCL2L11, LSMEM1, and IDS in Cluster 3, and RUNX1 and DNAH12 in Cluster 1 (Figure 9).
Time series analysis indicates that Clusters 1, 3, and 6 exhibit an upregulation trend in gene expression, while Clusters 5 and 8 exhibit a downregulation trend. GO enrichment analysis of the five clusters reveals that in Cluster 1, RUNX1 and other genes are enriched in the biological process of positive regulation of transcription by RNA polymerase II (Figure S1A). Additionally, this cluster is enriched in both the apoptotic process and the negative regulation of the apoptotic process. RUNX1 and other genes are associated with the cellular components of the nucleus and nucleoplasm. Regarding MF, RUNX1 in Cluster 1, along with other genes, is enriched in ATP binding and DNA-binding transcription activator activity involving RNA polymerase. RUNX1 is implicated in chronic myeloid leukemia and transcriptional misregulation in cancer signaling pathways.
Cluster 3 (Figure S1B) mainly participates in BP including the inflammatory response, immune response, innate immune response, apoptotic process, positive regulation of the ERK1 and ERK2 cascade, and regulation of reactive oxygen species metabolism. BCL2L11 and other genes are enriched in the apoptotic process and the positive regulation of apoptosis. They are also associated with the cellular components of the membrane and mitochondrion. For MF, Cluster 3 is mainly enriched in transcriptional activator activity, RNA polymerase regulatory region sequence-specific binding, GTPase activator activity, and ubiquitin-protein ligase activity. KEGG analysis indicates that BCL2L11 primarily participates in pathways related to cancer, Epstein-Barr virus infection, FoxO signaling, apoptosis, and most genes are involved in the MAPK signaling pathway, metabolic pathways, IL-17 signaling pathway, NOD-like receptor signaling pathway, and TNF signaling pathway.
Cluster 6 (Figure S1D) mainly participates in biological processes including the regulation of transcription by RNA polymerase II, negative regulation of Rho protein signal transduction, ubiquitin-dependent protein catabolic process, phosphorylation, signal transduction, and small GTPase-mediated signal transduction. For molecular function (MF), most genes in Cluster 6 are involved in GTPase activator activity, ubiquitin-protein transferase activity, ubiquitin-protein ligase activity, ATP binding, protein binding, RNA polymerase II cis-regulatory region sequence-specific binding, and DNA-binding transcription activator activity. The KEGG pathways primarily involved include the MAPK signaling pathway, transcriptional misregulation in cancer, and other pathways in cancer.
Cluster 5 (Figure S1C) is primarily enriched in the BP of RNA splicing via the spliceosome and protein folding. For CC, it is enriched in the spliceosomal complex. For MF, it is enriched in RNA binding, unfolded protein binding, and transcription coactivator activity. KEGG analysis reveals that the genes are involved in the spliceosome and nucleotide excision repair pathways. Cluster 8 (Figure S1E) shares similar enriched biological processes with Cluster 5, including RNA splicing via the spliceosome and protein folding. Additionally, it is enriched in the regulation of transcription by RNA polymerase II and immune system processes. Similar to Cluster 5, it is enriched in the spliceosomal complex for cellular components, but most genes are also enriched in the nucleus and cytoplasm. For MF, in addition to being enriched in RNA binding like Cluster 5, it is also enriched in DNA-binding transcription factor activity specific to RNA polymerase II, RNA polymerase II cis-regulatory region sequence-specific DNA binding, and DNA-binding transcription repressor activity specific to RNA polymerase II.
3.5. Analysis of pDEmRNAs of Pathogens
The cluster pattern analysis of pathogenic pDEmRNAs between S. agalactiae normal-treated groups (n = 5) and S. agalactiae deeply treated groups (n = 3) is illustrated in Figure 9A. A total of 864 pDEmRNAs (Supplementary File 6) were filtered using the thresholds of p-values < 0.05 and log2(fold-change) > 1 or <-1, of which 861 were up-regulated and 3 were down-regulated (Figure 9B).
GO enrichment analysis of pDEmRNAs reveals that the enriched BP primarily involve protein metabolism, gene expression, and biosynthesis, including the organic substance biosynthetic process, cellular biosynthetic process, regulation of transcription, cellular protein metabolic process, peptide metabolic process, and gene expression (Figure 9D). The enriched CC are mainly located in the cell, cell part, intracellular, intracellular part, cytoplasm, and protein-containing complex. The enriched MF primarily include RNA-binding, catalytic activity, transferase activity, and oxidoreductase activity. KEGG pathway analysis shows that the main enrichments are in metabolism-related pathways (Figure 9C), such as the biosynthesis of amino acids, purine metabolism, glycolysis/gluconeogenesis, carbon metabolism, and fatty acid metabolism. Further enrichments are found in genetic information processing pathways, primarily ribosome, aminoacyl-tRNA biosynthesis, and protein export. The pathways related to environmental information processing include ABC transporters, the phosphotransferase system, and the two-component system. Disease-related signaling pathways include vancomycin resistance, beta-lactam resistance, and cationic antimicrobial peptide (CAMP) resistance. The D-alanine metabolism signaling pathway is involved in the construction of cell wall peptidoglycan and teichoic acid.
3.6. Gene Co-Expression Analysis Interaction Network
Using gene co-expression analysis, which targeted the lncRNAs of the eight candidate genes in Clusters 1, 3, and 5, as well as differentially expressed genes related to the transcriptional regulation of S. agalactiae, it was found that six upregulated genes of S. agalactiae were correlated with TCONS_00001766- RUNX1, TCONS_00001789- RUNX1, ENSBTAG00000048558- DNAH12, TCONS_00026618- TCHP, TCONS_00085732- IDS, TCONS_00019399- ARRB1, TCONS_00065698- LSMEM1, RNase-MRP- TPM2, and TCONS-00007590- BCL2L11. The genes tsf, prfB, and infC significantly affected these nine candidate lncRNAs (Figure 10).
3.7. The Expression Level of Candidate Genes
Statistical analysis of high-throughput sequencing data showed that TPM2, TCHP, and TCONS_00026618 were downregulated, while LSMEM1, RUNX1, IDS, DNAH12, ARRB1, BCL2L11, and lncRNAs RNase_ MRP, TCONS_00001766, TCONS_00065689, TCONS_00001789, TCONS_00085732, TCONS_00019399, TCONS_00007590, and ENSBTAG00000048558 were upregulated (Figure 11).
4. Discussion
S. agalactiae, commonly referred to as Group B Streptococcus (GBS), is a zoonotic pathogen and a highly infectious Gram-positive bacterium[28]. This pathogen can infect the mammary glands of dairy cows, leading to subclinical mastitis, which can progress to clinical mastitis if left unchecked[28]. Research has demonstrated that S. agalactiae infection in the mammary gland initiates a series of innate immune responses in the host. Concurrently, the bacteria invade mammary epithelial cells to evade host defenses and antibiotics[14]. During this infection process, various molecular interactions occur, highlighting the importance of studying the molecular changes that mediate mastitis.
Zhang et al. utilized transcriptomics and proteomics to analyze mammary tissue infected by S. agalactiae, investigating the host’s immune response to the pathogen[14]. Tong et al. conducted ubiquitination sequencing and analysis on mammary epithelial cells infected with S. agalactiae[16]. Mayara et al. perfused mammary tissue with S. agalactiae to observe transcriptional changes, focusing on the most affected biological functions and pathways during this process[18]. These studies primarily examined the resistance mechanisms of mammary tissue or the ubiquitination processes in mammary epithelial cells following S. agalactiae infection, each emphasizing different aspects. This study utilized in vitro cultured mammary epithelial cells infected with varying times of S. agalactiae and performed interaction transcriptome analysis to investigate the molecular mechanisms of host-pathogen interactions during transcriptional regulation.
The GO enrichment analysis of DEmRNAs showed enrichment in processed related to inflammation, disease occurrence, damage repair, and apoptosis regulation. Upregulated genes were enriched in exogenous apoptosis regulation processes, while downregulated genes were enriched in endogenous apoptosis signaling pathways. This could be due to the induction of exogenous apoptotic signals and the inhibition of endogenous apoptotic processes following pathogen invasion. Upregulated genes were significantly enriched in small GTPase-mediated signal transduction and regulation of the ERK1 and ERK2 cascades.
Rho-GTPase acts as a molecular switch during inflammatory cell migration by cycling between the inactive Rho-GDP and active Rho-GTP forms. It plays a crucial role in actin cytoskeleton dynamics and precise regulation of leukocyte immune functions. Existing reports indicate that dysregulation of Rho-GTPase signaling is associated with various inflammatory diseases[29]. Small GTPase can mediate ERK1/2 to enter the signal nucleus through the MAPK signaling pathway, leading to apoptosis, inflammatory stress responses, and reactive oxygen species (ROS) production[30]. ROS acts as a double-edged sword, potentially playing roles in both pro-inflammatory and anti-inflammatory processes.
Recent studies have revealed the physiological importance of ROS as signaling molecules, crucial for maintaining cellular function and homeostasis[31]. Previous research indicated that S. agalactiae infection in human endothelial cells induces ROS[32], which can persist for a week compared to Staphylococcus aureus and Escherichia coli[33]. In this study, we found that genes related to the regulation of reactive oxygen species metabolic processes were upregulated as mammary epithelial cells were increasingly infected by the pathogen. This indicates that mammary epithelial cells employ ROS signaling as a defensive measure during infection. However, ROS production further activates the ERK1/2 pathway, triggering various immune processes to eliminate cells, but also leading to apoptosis[34,35]. ROS can also induce autophagy, allowing the pathogen to survive in mammary epithelial cells[36]. These findings suggest that during S. agalactiae infection in mammary epithelial cells, the ERK1/2 pathway induces inflammatory responses through MAPK signaling, accompanied by increased ROS production.
KEGG analysis identified several crucial pathways, including transcription misregulation in cancer, IL-17 signaling pathway, P53 signaling pathway, TGF-beta signaling pathway, MAPK signaling pathway, pathways in cancer, and apoptosis. Transcription misregulation in cancer pathways is triggered by pathogen invasion, interferes with the regulation of cancer-related transcription factors. The upregulated RUNX1 can inhibit the invasiveness of most breast cancer subtypes, especially in the early stages of tumorigenesis, and prevent the epithelial-to-mesenchymal transition in breast cancer cells[32]. In time-series analysis, RUNX1 expression gradually increases with the severity of bacterial infection, likely due to bacterial interference and disruption of the cell's regulatory system. Recent studies have shown that the IL-17 signaling pathway is also involved in the occurrence of mastitis[37], particularly related to the inflammatory response in mammary epithelial cells[38]. The p53 signaling pathway and MAPK signaling pathway are enriched in exosomes from bacterial-infected cells[39]. The p53 signaling pathway is a complex cellular stress response network with various inputs and downstream outputs, related to its role as a tumor suppressor pathway[40]. The MAPK signaling pathway is involved in tumor formation, invasion, metastasis, and apoptosis[41], and its activation is also implicated in mastitis[42]. The TGF-beta signaling pathway induces apoptosis in mammary epithelial cells[43]. Moreover, TGF-beta1 can cooperate with the ERK1/2 pathway to promote Gram-positive bacterial adhesion and infection of mammary epithelial cells[44]. Additionally, the RhoA/Rho kinase signaling cascade aids in TGF-beta induced changes in cytoskeletal organization and cell permeability[45]. BCL2L11, involved in the apoptosis pathway, primarily participates in the extrinsic apoptotic signal pathway in the absence of ligand, positive regulation of cysteine-type endopeptidase activity involved in the apoptotic process, and protein kinase binding among upregulated genes. It is also involved in EGFR tyrosine kinase inhibitor resistance.
It is conceivable that upon pathogens interference, host cells initiate immune and inflammatory responses to combat bacterial infection, potentially leading to adverse effects such as progression toward cancer. Enriched signaling pathways in the upregulated clusters 1 and 3 suggest that cells may gradually transform towards a cancerous state. However, cells cannot evade the process of apoptosis. In Cluster 3, several disease-related pathways are upregulated, including the pathway in cancer. Concurrently, BCL2L11 is enriched in this pathway along with other genes.
Alternative splicing is a crucial mechanism of genetic regulation, enhancing the diversity and complexity of the transcriptome and proteome from a limited number of genes. Numerous studies suggest that alternative splicing events can lead to changes in protein expression or function during disease onset and progression[46]. In this study, variable splicing showed that DEmRNAs were mainly involved in regulating GTPase activity, protein ubiquitination, ATP binding, and RNA binding. Some studies suggest that immune-related GTPase directly mediates pathogen membrane destruction by binding to the pathogen membrane, thereby exposing the pathogen to cytoplasmic defenses[47]. Immune-related GTPase can also utilize the ubiquitination system to tag intracellular pathogens[48]. In this study, differentially spliced genes were involved in ubiquitin-dependent protein catabolic processes and protein polyubiquitination. This indicates that S. agalactiae infection in mammary epithelial cells activates the host cell's immune defense mechanisms, leading to the overactivation of the GTPase system, which collaborates with the intracellular ubiquitination system to combat intracellular bacterial damage. However, the influence of bacterial intracellular molecular regulation disrupts normal transcriptional regulation in host cells, particularly the normal alternative splicing process, potentially causing dysregulation of GTPase signaling. Differentially spliced genes were enriched in ATP binding and RNA binding functions, indicating that S. agalactiae interferes with the splicing forms of ATP binding proteins and RNA binding proteins, indirectly affecting cellular energy metabolism and transcriptional regulation.
Among the three groups of DEmRNAs, the upregulated gene RUNX1 exhibited MXE, A3SS, and SE events in the S_M group (all not significant), A3SS and SE events in the H_M group (not significant), and significant A3SS and SE events in the H_S group. KEGG analysis of the H_S group indicated that RUNX1 is primarily involved in cancer-related pathways, including cancer transcription misregulation pathways and tight junctions. GO analysis revealed involvement in nucleoplasm localization, ATP binding, and protein-containing complex processes. This suggests that as bacterial infection intensifies, the splicing process of RUNX1 in host cells is disrupted, altering its expression pattern and potentially increasing the risk of cellular transformation. Studies have demonstrated that RUNX1 is associated with RNA Pol II-transcribed protein and lncRNA genes, as well as RNA Pol I-transcribed ribosomal genes, which are crucial for the growth and phenotype maintenance of mammary epithelial cells[49]. This further implies that bacterial interference with the transcription process indirectly induce morphological changes in mammary epithelial cells, thereby contributing to carcinogenesis. Recent research indicates that RUNX1 plays a role in breast cancer migration and invasion[50].
In this study, through the analysis of DElncRNA target gene prediction, alternatively spliced genes, and DEmRNA, which identified eight candidate genes at the intersection, including RUNX1 and BCL2L11. RUNX1 is a crucial transcription factor that induces the expression of numerous genes. Among the upregulated genes, BCL2L11 is one that RUNX1 induces[51]. BCL2L11 plays a crucial dual role in disease mechanisms by inhibiting autophagy and initiating apoptosis[52]. Under normal conditions, BCL2L11 undergoes alternative splicing, forming at least 18 different isoforms[51]. In this study, however, BCL2L11 underwent a MXE event between the H and M groups. MXE results in different exon combinations that may maintain protein folding but alter the specificity and selectivity of protein functions[53]. This indicates that under continuous bacterial infection, the alternative splicing of BCL2L11 is affected, altering its splicing form and resulting in changes to BCL2L11 conformation. This splicing pattern reduces proteome diversity, leading to protein dysfunction and altered biological functions.
In the time series analysis, genes involved in BP of DNA binding transcription activator activity are upregulated in clusters 1 and 6. However, the transcription factors of the host cell are downregulated in clusters 5 and 8. This indicates that some factors from the pathogens have replaced the host's transcription factors, thereby affecting the host's transcriptional regulation. It is hypothesized that during pathogen infection of host cells, the host's spliceosome is disrupted and consequently downregulated. The host's cell nucleus is similarly affected, which interferes with the host's transcriptional regulation, particularly the activation of nucleic acid transcription factors and the specificity of RNA polymerase.
Interestingly, in the GO enrichment analysis of DEmRNAs in the host cell transcriptome, both spliceosome and mRNA splicing processes were downregulated, indicating that bacterial interference affects the normal gene splicing process of the mammary epithelial cell. Conversely, some proteins secreted by S. agalactiae are involved. Combined with the differential expression analysis of the pathogenic bacteria, multiple genes are upregulated during infection, suggesting an influence on the host cell molecular transcriptional regulation and their interaction during infection. For example, nusB, rimN, yhbY, infC, prfB, and tsf are involved. Studies have shown that the nusB is transcribed in opposition to the eukaryotic system and can be a potential antibacterial target[54]. Additionally, it utilizes biological coupling of transcription and translation to control downstream gene expression[55]. The protein YrdC translated from rimN is involved in tRNA modification and preferentially binds to RNA[56,57]. yhbY is involved in ribosome assembly and exhibits RNA binding activity[58]. infC guides transcriptional regulation and is upregulated in bacterial infection in the animal liver[59,60]. The RF2 protein encoded by prfB is required for recognizing stop codons during bacterial translation termination[61]. This suggests that bacteria proliferate extensively and express proteins capable of invading host cells. tsf can contribute to the production of biologically active bacterial keratinase[62], and this site can also confer strong antibiotic resistance[63], making it a potential therapeutic target. To adapting to the host system, bacteria employ various strategies, including the production of virulence factors and the formation of biofilms, to evade the host immune system and resist antibiotics[64].
Bacteria can manipulate the host signaling pathways by regulating the host lncRNAs to escape immune clearance. Therefore, bacteria also can induce significant alteration in the cell transcriptome and develop various strategies to modify immune signaling for its survival[65]. Currently, lncRNAs have been shown to play a crucial role in regulating alternative splicing in response to various stimuli or diseases[66]. Additionally, increasing evidence indicates that lncRNAs are important in the regulatory circuits controlling innate and adaptive immune responses to bacterial pathogens[67]. In the co-expression network analysis, these three bacterial genes (tsf, prfB, and infC) have the most significant effect on the differential targeting of lncRNAs to RUNX1 and BCL2L11. Both RUNX1 and BCL2L11 undergo abnormal alternative splicing during the infection process. It is hypothesized that during the infection of bovine mammary epithelial cells by S. agalactiae, genes such as tsf, prfB, and infC, which are involved in RNA binding, infiltrate host cells and disrupt the targeting of lncRNAs to RUNX1 and BCL2L11. This affects the host's normal alternative splicing process, disrupting normal cell proliferation regulation and apoptosis.
5. Conclusions
This study analyzed the infection of bovine mammary epithelial cells by S. agalactiae using absolute quantitative interaction transcriptome sequencing. Analysis of the results revealed that when S. agalactiae infection triggers both immune and inflammatory responses in mammary epithelial cells, as well as induces cell carcinogenesis and apoptosis. Furthermore, to evade cellular immune defenses, S. agalactiae interferes with normal alternative splicing processes by generating lncRNAs that disrupt the regulation of apoptosis and disease-related pathways, thus achieving immune evasion.
Supplementary Materials
The following supporting information can be downloaded at: Preprints.org, Figure S1: GO and KEGG analysis of different clusters in the three comparison groups. The Y- axis on the left represents GO and KEGG terms of genes, including biological process (BP), cellular component (CP), molecular function (MF) and KEGG pathways, the X- axis indicates p-values, The red bars represent upregulated genes. The green bars represent downregulated genes. The area of shapes represents gene numbers.; Supplement File1: Differentially expressed genes between the S and M groups; Supplement File2: Differentially expressed genes between the H and M groups; Supplement File3: Differentially expressed genes between the H and S groups; Supplement File4: Genes targeted by lncRNA; Supplement File5: Venn analysis of alternatively spliced genes, lncRNA, and differentially expressed genes; Supplement File6: Differentially expressed genes of pathogens infecting cells at different times.
Author Contributions
Conceptualization, J.G. and Y.M.; methodology, J.G. and T.L.; resources, J.G., J.X. and X.C.; data curation, X.X.; writing—original draft preparation, J.G.; writing—review and editing, J.M.; visualization, Y.L.; project administration, J.G. and Y.M.; funding acquisition, J.G. and Y.M. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Postdoctoral Fund of the Gansu Provincial Department of Human Resources and Social Security (NO. 03824006), the Discipline Team Project of Gansu Agricultural University (GAU-XKTD-2022-20).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data is contained within the article or supplementary material.
Acknowledgments
MAC-T cells were provided by the team of Professor Zhang from the College of Life Science and Technology, Gansu Agricultural University.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Ouamba, A.J.K.; Gagnon, M.; LaPointe, G.; Chouinard, P.Y.; Roy, D. Graduate Student Literature Review: Farm management practices: Potential microbial sources that determine the microbiota of raw bovine milk. Journal of dairy science 2022, 105, 7276–7287. [Google Scholar] [CrossRef] [PubMed]
- Kurban, D.; Roy, J.P.; Kabera, F.; Fréchette, A.; Um, M.M.; Albaaj, A.; Rowe, S.; Godden, S.; Adkins, P.R.F.; Middleton, J.R.; et al. Diagnosing Intramammary Infection: Meta-Analysis and Mapping Review on Frequency and Udder Health Relevance of Microorganism Species Isolated from Bovine Milk Samples. Animals : an open access journal from MDPI 2022, 12, 3288. [Google Scholar] [CrossRef]
- Wang, Y.; Nan, X.; Zhao, Y.; Jiang, L.; Wang, H.; Zhang, F.; Hua, D.; Liu, J.; Yao, J.; Yang, L.; et al. Dietary Supplementation of Inulin Ameliorates Subclinical Mastitis via Regulation of Rumen Microbial Community and Metabolites in Dairy Cows. Microbiology spectrum 2021, 9, e0010521. [Google Scholar] [CrossRef]
- Hoekstra, J.; Zomer, A.L.; Rutten, V.; Benedictus, L.; Stegeman, A.; Spaninks, M.P.; Bennedsgaard, T.W.; Biggs, A.; De Vliegher, S.; Mateo, D.H.; et al. Genomic analysis of European bovine Staphylococcus aureus from clinical versus subclinical mastitis. Scientific reports 2020, 10, 18172. [Google Scholar] [CrossRef]
- Khan, M.Z.; Wang, J.; Ma, Y.; Chen, T.; Ma, M.; Ullah, Q.; Khan, I.M.; Khan, A.; Cao, Z.; Liu, S. Genetic polymorphisms in immune- and inflammation-associated genes and their association with bovine mastitis resistance/susceptibility. Frontiers in immunology 2023, 14, 1082144. [Google Scholar] [CrossRef] [PubMed]
- Kan, X.; Hu, G.; Liu, Y.; Xu, P.; Huang, Y.; Cai, X.; Guo, W.; Fu, S.; Liu, J. Mammary Fibrosis Tendency and Mitochondrial Adaptability in Dairy Cows with Mastitis. Metabolites 2022, 12, 1035. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Richoux, R.; Boutinaud, M.; Martin, P.; Gagnaire, V. Role of somatic cells on dairy processes and products: a review. Dairy science & technology 2014, 94, 517–538. [Google Scholar] [CrossRef]
- Krishnamoorthy, P.; Suresh, K.P.; Jayamma, K.S.; Shome, B.R.; Patil, S.S.; Amachawadi, R.G. An Understanding of the Global Status of Major Bacterial Pathogens of Milk Concerning Bovine Mastitis: A Systematic Review and Meta-Analysis (Scientometrics). Pathogens (Basel, Switzerland) 2021, 10, 545. [Google Scholar] [CrossRef]
- Abd El-Razik, K.A.E.; Arafa, A.A.; Fouad, E.A.; Younes, A.M.; Almuzaini, A.M.; Abdou, A.M. Isolation, identification and virulence determinants of Streptococcus agalactiae from bovine subclinical mastitis in Egypt. Journal of infection in developing countries 2021, 15, 1133–1138. [Google Scholar] [CrossRef]
- Lakew, B.T.; Fayera, T.; Ali, Y.M. Risk factors for bovine mastitis with the isolation and identification of Streptococcus agalactiae from farms in and around Haramaya district, eastern Ethiopia. Tropical animal health and production 2019, 51, 1507–1513. [Google Scholar] [CrossRef]
- Lin, L.; Huang, X.; Yang, H.; He, Y.; He, X.; Huang, J.; Li, S.; Wang, X.; Tang, S.; Liu, G.; et al. Molecular epidemiology, antimicrobial activity, and virulence gene clustering of Streptococcus agalactiae isolated from dairy cattle with mastitis in China. J Dairy Sci 2021, 104, 4893–4903. [Google Scholar] [CrossRef] [PubMed]
- Kabelitz, T.; Aubry, E.; van Vorst, K.; Amon, T.; Fulde, M. The Role of Streptococcus spp. in Bovine Mastitis. Microorganisms 2021, 9, 1497. [Google Scholar] [CrossRef]
- Yang, F.; Yuan, L.; Xiang, M.; Jiang, Q.; Zhang, M.; Chen, F.; Tong, J.; Huang, J.; Cai, Y. A Novel TLR4-SYK Interaction Axis Plays an Essential Role in the Innate Immunity Response in Bovine Mammary Epithelial Cells. Biomedicines 2022, 11, 97. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Jiang, H.; Fan, Y.; Chen, Z.; Li, M.; Mao, Y.; Karrow, N.A.; Loor, J.J.; Moore, S.; Yang, Z. Transcriptomics and iTRAQ-Proteomics Analyses of Bovine Mammary Tissue with Streptococcus agalactiae-Induced Mastitis. Journal of agricultural and food chemistry 2018, 66, 11188–11196. [Google Scholar] [CrossRef] [PubMed]
- Tong, J.; Sun, M.; Zhang, H.; Yang, D.; Zhang, Y.; Xiong, B.; Jiang, L. Proteomic analysis of bovine mammary epithelial cells after in vitro incubation with S. agalactiae: potential biomarkers. Veterinary research 2020, 51, 98. [Google Scholar] [CrossRef]
- Tong, J.; Ji, X.; Zhang, H.; Xiong, B.; Cui, D.; Jiang, L. The Analysis of the Ubiquitylomic Responses to Streptococcus agalactiae Infection in Bovine Mammary Gland Epithelial Cells. Journal of inflammation research 2022, 15, 4331–4343. [Google Scholar] [CrossRef]
- Sbardella, A.P.; Weller, M.; Fonseca, I.; Stafuzza, N.B.; Bernardes, P.A.; FF, E.S.; da Silva, M.; Martins, M.F.; Munari, D.P. RNA sequencing differential gene expression analysis of isolated perfused bovine udders experimentally inoculated with Streptococcus agalactiae. Journal of dairy science 2019, 102, 1761–1767. [Google Scholar] [CrossRef]
- Weller, M.; Fonseca, I.; Sbardella, A.P.; Pinto, I.S.B.; Viccini, L.F.; Brandão, H.M.; Gern, J.C.; Carvalho, W.A.; Guimarães, A.S.; Brito, M.; et al. Isolated perfused udder model for transcriptome analysis in response to Streptococcus agalactiae. The Journal of dairy research 2019, 86, 307–314. [Google Scholar] [CrossRef]
- Richards, V.P.; Choi, S.C.; Pavinski Bitar, P.D.; Gurjar, A.A.; Stanhope, M.J. Transcriptomic and genomic evidence for Streptococcus agalactiae adaptation to the bovine environment. BMC genomics 2013, 14, 920. [Google Scholar] [CrossRef]
- Pu, J.; Li, R.; Zhang, C.; Chen, D.; Liao, X.; Zhu, Y.; Geng, X.; Ji, D.; Mao, Y.; Gong, Y.; et al. Expression profiles of miRNAs from bovine mammary glands in response to Streptococcus agalactiae-induced mastitis. The Journal of dairy research 2017, 84, 300–308. [Google Scholar] [CrossRef] [PubMed]
- Westermann, A.J.; Barquist, L.; Vogel, J. Resolving host-pathogen interactions by dual RNA-seq. PLoS pathogens 2017, 13, e1006033. [Google Scholar] [CrossRef]
- Park, J.W.; Tokheim, C.; Shen, S.; Xing, Y. Identifying differential alternative splicing events from RNA sequencing data using RNASeq-MATS. Methods in molecular biology (Clifton, N.J.) 2013, 1038, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.J.; Yang, D.C.; Kong, L.; Hou, M.; Meng, Y.Q.; Wei, L.; Gao, G. CPC2: a fast and accurate coding potential calculator based on sequence intrinsic features. Nucleic acids research 2017, 45, W12–w16. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Park, H.J.; Dasari, S.; Wang, S.; Kocher, J.P.; Li, W. CPAT: Coding-Potential Assessment Tool using an alignment-free logistic regression model. Nucleic acids research 2013, 41, e74. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Luo, H.; Bu, D.; Zhao, G.; Yu, K.; Zhang, C.; Liu, Y.; Chen, R.; Zhao, Y. Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucleic acids research 2013, 41, e166. [Google Scholar] [CrossRef] [PubMed]
- Mistry, J.; Chuguransky, S.; Williams, L.; Qureshi, M.; Salazar, G.A.; Sonnhammer, E.L.L.; Tosatto, S.C.E.; Paladin, L.; Raj, S.; Richardson, L.J.; et al. Pfam: The protein families database in 2021. Nucleic acids research 2021, 49, D412–d419. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Chen, M.; Huang, X.; Zhang, G.; Zeng, L.; Zhang, G.; Wu, S.; Wang, Y. SRplot: A free online platform for data visualization and graphing. PloS one 2023, 18, e0294236. [Google Scholar] [CrossRef]
- Kabelitz, T.; Aubry, E.; van Vorst, K.; Amon, T.; Fulde, M. The Role of Streptococcus spp. in Bovine Mastitis. Microorganisms 2021, 9. [Google Scholar] [CrossRef]
- Dipankar, P.; Kumar, P.; Dash, S.P.; Sarangi, P.P. Functional and Therapeutic Relevance of Rho GTPases in Innate Immune Cell Migration and Function during Inflammation: An In Silico Perspective. Mediators of inflammation 2021, 2021, 6655412. [Google Scholar] [CrossRef]
- Pan, J.S.; Hong, M.Z.; Ren, J.L. Reactive oxygen species: a double-edged sword in oncogenesis. World journal of gastroenterology 2009, 15, 1702–1707. [Google Scholar] [CrossRef]
- Hirata, Y. [Reactive Oxygen Species (ROS) Signaling: Regulatory Mechanisms and Pathophysiological Roles]. Yakugaku zasshi : Journal of the Pharmaceutical Society of Japan 2019, 139, 1235–1241. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, J.S.S.; Santos, G.D.S.; Moraes, J.A.; Saliba, A.M.; Barja-Fidalgo, T.C.; Mattos-Guaraldi, A.L.; Nagao, P.E. Reactive oxygen species generation mediated by NADPH oxidase and PI3K/Akt pathways contribute to invasion of Streptococcus agalactiae in human endothelial cells. Memorias do Instituto Oswaldo Cruz 2018, 113, e140421. [Google Scholar] [CrossRef]
- Hussen, J. Bacterial species-specific modulatory effects on phenotype and function of camel blood leukocytes. BMC veterinary research 2021, 17, 241. [Google Scholar] [CrossRef]
- Ma, F.; Yang, S.; Zhou, M.; Lu, Y.; Deng, B.; Zhang, J.; Fan, H.; Wang, G. NADPH oxidase-derived reactive oxygen species production activates the ERK1/2 pathway in neutrophil extracellular traps formation by Streptococcus agalactiae isolated from clinical mastitis bovine. Veterinary microbiology 2022, 268, 109427. [Google Scholar] [CrossRef]
- Wei, C.D.; Li, Y.; Zheng, H.Y.; Tong, Y.Q.; Dai, W. Palmitate induces H9c2 cell apoptosis by increasing reactive oxygen species generation and activation of the ERK1/2 signaling pathway. Molecular medicine reports 2013, 7, 855–861. [Google Scholar] [CrossRef]
- Geng, N.; Liu, K.; Lu, J.; Xu, Y.; Wang, X.; Wang, R.; Liu, J.; Liu, Y.; Han, B. Autophagy of bovine mammary epithelial cell induced by intracellular Staphylococcus aureus. Journal of microbiology (Seoul, Korea) 2020, 58, 320–329. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Liu, L.; Augustino, S.M.A.; Duan, T.; Hall, T.J.; MacHugh, D.E.; Dou, J.; Zhang, Y.; Wang, Y.; Yu, Y. Identification of novel molecular markers of mastitis caused by Staphylococcus aureus using gene expression profiling in two consecutive generations of Chinese Holstein dairy cattle. Journal of animal science and biotechnology 2020, 11, 98. [Google Scholar] [CrossRef]
- Liu, J.; Gao, Y.; Zhang, X.; Hao, Z.; Zhang, H.; Gui, R.; Liu, F.; Tong, C.; Wang, X. Transcriptome sequencing analysis of bovine mammary epithelial cells induced by lipopolysaccharide. Animal biotechnology 2024, 35, 2290527. [Google Scholar] [CrossRef]
- Chen, Y.; Jing, H.; Chen, M.; Liang, W.; Yang, J.; Deng, G.; Guo, M. Transcriptional Profiling of Exosomes Derived from Staphylococcus aureus-Infected Bovine Mammary Epithelial Cell Line MAC-T by RNA-Seq Analysis. Oxidative medicine and cellular longevity 2021, 2021, 8460355. [Google Scholar] [CrossRef]
- Hernández Borrero, L.J.; El-Deiry, W.S. Tumor suppressor p53: Biology, signaling pathways, and therapeutic targeting. Biochimica et biophysica acta. Reviews on cancer 2021, 1876, 188556. [Google Scholar] [CrossRef]
- Asl, E.R.; Amini, M.; Najafi, S.; Mansoori, B.; Mokhtarzadeh, A.; Mohammadi, A.; Lotfinejad, P.; Bagheri, M.; Shirjang, S.; Lotfi, Z.; et al. Interplay between MAPK/ERK signaling pathway and MicroRNAs: A crucial mechanism regulating cancer cell metabolism and tumor progression. Life sciences 2021, 278, 119499. [Google Scholar] [CrossRef]
- Ząbek, T.; Semik-Gurgul, E.; Ropka-Molik, K.; Szmatoła, T.; Kawecka-Grochocka, E.; Zalewska, M.; Kościuczuk, E.; Wnuk, M.; Bagnicka, E. Short communication: Locus-specific interrelations between gene expression and DNA methylation patterns in bovine mammary gland infected by coagulase-positive and coagulase-negative staphylococci. Journal of dairy science 2020, 103, 10689–10695. [Google Scholar] [CrossRef]
- Di, H.S.; Wang, L.G.; Wang, G.L.; Zhou, L.; Yang, Y.Y. The Signaling Mechanism of TGF-β1 Induced Bovine Mammary Epithelial Cell Apoptosis. Asian-Australasian journal of animal sciences 2012, 25, 304–310. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Gao, Y.; Xia, X.; Che, Y.; Wang, Y.; Liu, H.; Sun, Y.; Ren, W.; Han, W.; Yang, J.; et al. TGF-β1 promotes Staphylococcus aureus adhesion to and invasion into bovine mammary fibroblasts via the ERK pathway. Microbial pathogenesis 2017, 106, 25–29. [Google Scholar] [CrossRef]
- Clements, R.T.; Minnear, F.L.; Singer, H.A.; Keller, R.S.; Vincent, P.A. RhoA and Rho-kinase dependent and independent signals mediate TGF-beta-induced pulmonary endothelial cytoskeletal reorganization and permeability. American journal of physiology. Lung cellular and molecular physiology 2005, 288, L294–306. [Google Scholar] [CrossRef] [PubMed]
- Park, J.E.; Ryoo, G.; Lee, W. Alternative Splicing: Expanding Diversity in Major ABC and SLC Drug Transporters. The AAPS journal 2017, 19, 1643–1655. [Google Scholar] [CrossRef] [PubMed]
- Dockterman, J.; Coers, J. How did we get here? Insights into mechanisms of immunity-related GTPase targeting to intracellular pathogens. Current opinion in microbiology 2022, 69, 102189. [Google Scholar] [CrossRef]
- Haldar, A.K.; Piro, A.S.; Finethy, R.; Espenschied, S.T.; Brown, H.E.; Giebel, A.M.; Frickel, E.M.; Nelson, D.E.; Coers, J. Chlamydia trachomatis Is Resistant to Inclusion Ubiquitination and Associated Host Defense in Gamma Interferon-Primed Human Epithelial Cells. mBio 2016, 7. [Google Scholar] [CrossRef]
- Rose, J.T.; Moskovitz, E.; Boyd, J.R.; Gordon, J.A.; Bouffard, N.A.; Fritz, A.J.; Illendula, A.; Bushweller, J.H.; Lian, J.B.; Stein, J.L.; et al. Inhibition of the RUNX1-CBFβ transcription factor complex compromises mammary epithelial cell identity: a phenotype potentially stabilized by mitotic gene bookmarking. Oncotarget 2020, 11, 2512–2530. [Google Scholar] [CrossRef] [PubMed]
- Ariffin, N.S. RUNX1 as a Novel Molecular Target for Breast Cancer. Clinical breast cancer 2022, 22, 499–506. [Google Scholar] [CrossRef] [PubMed]
- Sionov, R.V.; Vlahopoulos, S.A.; Granot, Z. Regulation of Bim in Health and Disease. Oncotarget 2015, 6, 23058–23134. [Google Scholar] [CrossRef]
- Luo, S.; Rubinsztein, D.C. BCL2L11/BIM: a novel molecular link between autophagy and apoptosis. Autophagy 2013, 9, 104–105. [Google Scholar] [CrossRef] [PubMed]
- Lam, S.D.; Babu, M.M.; Lees, J.; Orengo, C.A. Biological impact of mutually exclusive exon switching. PLoS computational biology 2021, 17, e1008708. [Google Scholar] [CrossRef]
- Qiu, Y.; Chan, S.T.; Lin, L.; Shek, T.L.; Tsang, T.F.; Zhang, Y.; Ip, M.; Chan, P.K.; Blanchard, N.; Hanquet, G.; et al. Nusbiarylins, a new class of antimicrobial agents: Rational design of bacterial transcription inhibitors targeting the interaction between the NusB and NusE proteins. Bioorganic chemistry 2019, 92, 103203. [Google Scholar] [CrossRef]
- Sherman, M.W.; Sandeep, S.; Contreras, L.M. The Tryptophan-Induced tnaC Ribosome Stalling Sequence Exposes High Amino Acid Cross-Talk That Can Be Mitigated by Removal of NusB for Higher Orthogonality. ACS synthetic biology 2021, 10, 1024–1038. [Google Scholar] [CrossRef] [PubMed]
- Teplova, M.; Tereshko, V.; Sanishvili, R.; Joachimiak, A.; Bushueva, T.; Anderson, W.F.; Egli, M. The structure of the yrdC gene product from Escherichia coli reveals a new fold and suggests a role in RNA binding. Protein science : a publication of the Protein Society 2000, 9, 2557–2566. [Google Scholar] [CrossRef] [PubMed]
- Fu, T.M.; Liu, X.; Li, L.; Su, X.D. The structure of the hypothetical protein smu.1377c from Streptococcus mutans suggests a role in tRNA modification. Acta crystallographica. Section F, Structural biology and crystallization communications 2010, 66, 771–775. [Google Scholar] [CrossRef]
- Barkan, A.; Klipcan, L.; Ostersetzer, O.; Kawamura, T.; Asakura, Y.; Watkins, K.P. The CRM domain: an RNA binding module derived from an ancient ribosome-associated protein. RNA (New York, N.Y.) 2007, 13, 55–64. [Google Scholar] [CrossRef]
- Pediconi, D.; Spurio, R.; LaTeana, A.; Jemiolo, D.; Gualerzi, C.O.; Pon, C.L. Translational regulation of infC operon in Bacillus stearothermophilus. Biochemistry and cell biology = Biochimie et biologie cellulaire 1995, 73, 1071–1078. [Google Scholar] [CrossRef]
- Xiao, Y.; Wu, L.; He, L.; Tang, Y.; Guo, S.; Zhai, S. Transcriptomic analysis using dual RNA sequencing revealed a Pathogen-Host interaction after Edwardsiella anguillarum infection in European eel (Anguilla anguilla). Fish & shellfish immunology 2022, 120, 745–757. [Google Scholar] [CrossRef]
- Kurita, D.; Abo, T.; Himeno, H. Molecular determinants of release factor 2 for ArfA-mediated ribosome rescue. The Journal of biological chemistry 2020, 295, 13326–13337. [Google Scholar] [CrossRef] [PubMed]
- Han, K.Y.; Song, J.A.; Ahn, K.Y.; Park, J.S.; Seo, H.S.; Lee, J. Enhanced solubility of heterologous proteins by fusion expression using stress-induced Escherichia coli protein, Tsf. FEMS microbiology letters 2007, 274, 132–138. [Google Scholar] [CrossRef] [PubMed]
- Che, R.X.; Xing, X.X.; Liu, X.; Qu, Q.W.; Chen, M.; Yu, F.; Ma, J.X.; Chen, X.R.; Zhou, Y.H.; God'Spower, B.O.; et al. Analysis of multidrug resistance in Streptococcus suis ATCC 700794 under tylosin stress. Virulence 2019, 10, 58–67. [Google Scholar] [CrossRef]
- Kasthuri, T.; Barath, S.; Nandhakumar, M.; Karutha Pandian, S. Proteomic profiling spotlights the molecular targets and the impact of the natural antivirulent umbelliferone on stress response, virulence factors, and the quorum sensing network of Pseudomonas aeruginosa. Frontiers in cellular and infection microbiology 2022, 12, 998540. [Google Scholar] [CrossRef]
- Wen, Y.; Chen, H.; Luo, F.; Zhou, H.; Li, Z. Roles of long noncoding RNAs in bacterial infection. Life sciences 2020, 263, 118579. [Google Scholar] [CrossRef] [PubMed]
- Romero-Barrios, N.; Legascue, M.F.; Benhamed, M.; Ariel, F.; Crespi, M. Splicing regulation by long noncoding RNAs. Nucleic acids research 2018, 46, 2169–2184. [Google Scholar] [CrossRef] [PubMed]
- Schmerer, N.; Schulte, L.N. Long noncoding RNAs in bacterial infection. Wiley interdisciplinary reviews. RNA 2021, 12, e1664. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Figure 1 Screening differently expressed mRNAs (DEmRNAs) in S. agalactiae infected mammary epithelial cells among the control (n= 5), S. agalactiae – S groups (n= 5), and S. agalactiae - H groups (n= 3). A. Gene expression level analysis in the S_M, H_M, H_S. The X-axis of the box plot represents the sample name, while the Y-axis represents log10 (RPKM). The box plots for each region correspond to five statistical measures (maximum, upper quartile, median, lower quartile, and minimum values, respectively). B. Cluster analysis of DEmRNAs in mammary epithelial cells between the control group (M1, M2, M3, M4, and M5), S groups (S1, S2, S3, S4, and S5) and H treated groups (H1, H2, and H3). Red indicates highly expressed genes, and blue indicates low expressed genes. Each column represents a sample, and each row represents a gene. On the left is the tree diagram of mRNA clustering. The closer the two mRNA branches are, the closer their expression level is. The upper part is the tree diagram of sample clustering, and the bottom is the name of each sample. The closer the two-sample branches are to each other, the closer the expression pattern of all genes in the two samples is and the trend of the more recent gene expression. C. D. E. Volcano plot of global DEmRNAs in S_M, H_M and H_S, respectively. Red dots (Up) represent significantly up-regulated genes (p-values< 0.05, log2(fold-change) >1); blue dots (Down) represent significantly down-regulated genes (p-values< 0.05, log2(fold-change) <-1); grey dots represent insignificantly differential expressed genes. F. Upset map analysis of S_M, H_M and H_S. The origin and connecting lines of the X-axis represent intersections, while the black bars represent the number of differentially expressed genes in each group; The number of differentially expressed genes at the intersection of each group on the Y-axis.
Figure 1.
Figure 1 Screening differently expressed mRNAs (DEmRNAs) in S. agalactiae infected mammary epithelial cells among the control (n= 5), S. agalactiae – S groups (n= 5), and S. agalactiae - H groups (n= 3). A. Gene expression level analysis in the S_M, H_M, H_S. The X-axis of the box plot represents the sample name, while the Y-axis represents log10 (RPKM). The box plots for each region correspond to five statistical measures (maximum, upper quartile, median, lower quartile, and minimum values, respectively). B. Cluster analysis of DEmRNAs in mammary epithelial cells between the control group (M1, M2, M3, M4, and M5), S groups (S1, S2, S3, S4, and S5) and H treated groups (H1, H2, and H3). Red indicates highly expressed genes, and blue indicates low expressed genes. Each column represents a sample, and each row represents a gene. On the left is the tree diagram of mRNA clustering. The closer the two mRNA branches are, the closer their expression level is. The upper part is the tree diagram of sample clustering, and the bottom is the name of each sample. The closer the two-sample branches are to each other, the closer the expression pattern of all genes in the two samples is and the trend of the more recent gene expression. C. D. E. Volcano plot of global DEmRNAs in S_M, H_M and H_S, respectively. Red dots (Up) represent significantly up-regulated genes (p-values< 0.05, log2(fold-change) >1); blue dots (Down) represent significantly down-regulated genes (p-values< 0.05, log2(fold-change) <-1); grey dots represent insignificantly differential expressed genes. F. Upset map analysis of S_M, H_M and H_S. The origin and connecting lines of the X-axis represent intersections, while the black bars represent the number of differentially expressed genes in each group; The number of differentially expressed genes at the intersection of each group on the Y-axis.
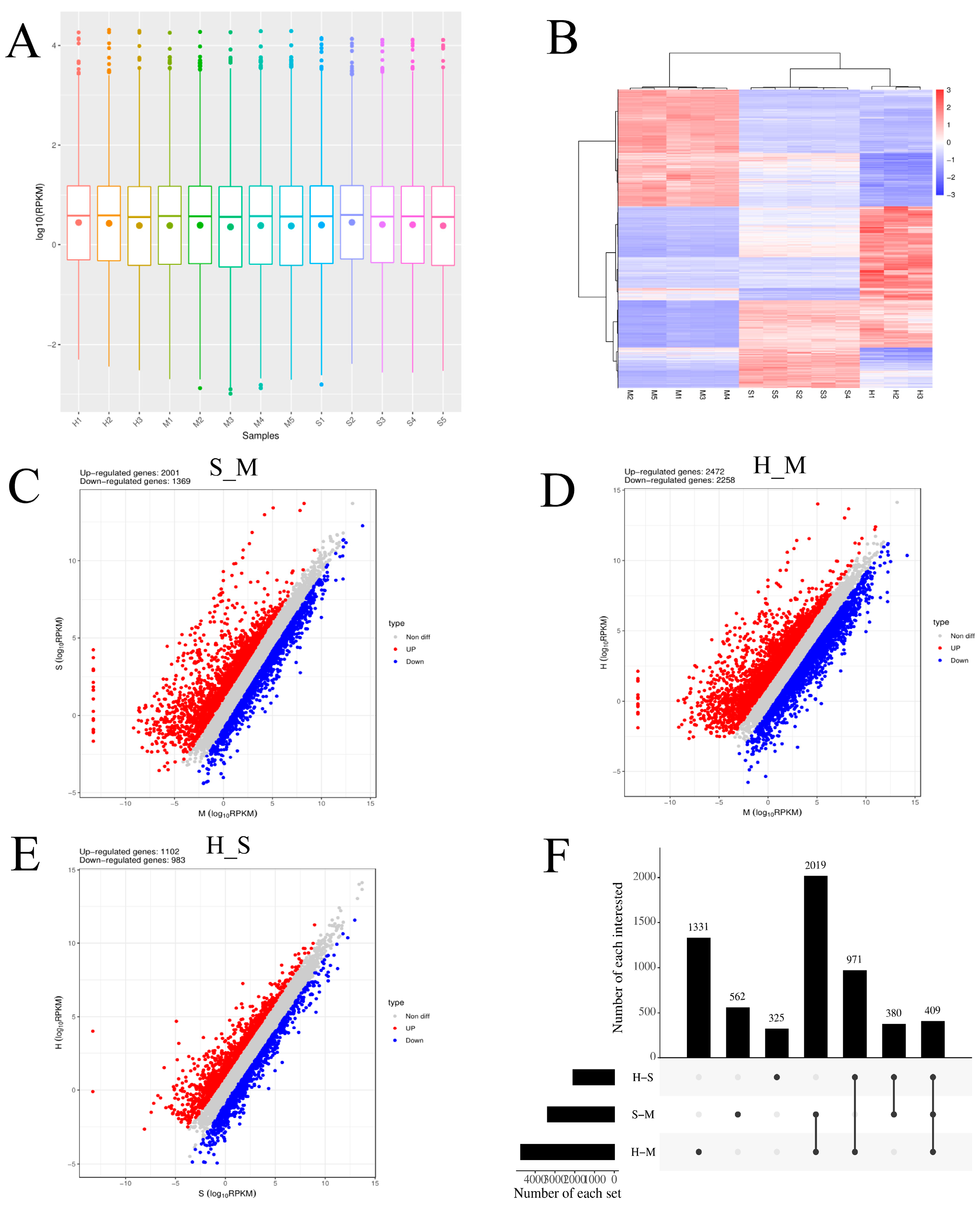
Figure 2.
GO and KEGG analysis of DEmRNAs in the S_M, H_M, H_S. A. The Y- axis on the left represents GO terms of up-regulated genes, including biological process (BP), cellular component (CP), and molecular function (MF), the X- axis indicates different comparison groups. The area of a circle represents DEGs number. Low p- values are shown in the red circle, and high p-values are shown in the blue circle. B. The Y- axis on the left represents GO terms of down-regulated genes, including biological process (BP), cellular component (CP), and molecular function (MF), the X- axis indicates different comparison groups. The area of a circle represents DEGs number. Low p-values are shown in the red circle, and high p-values are shown in the blue circle. C. The Y- axis on the left represents KEGG pathways, the X- axis indicates genes enrichment of each term. The shapes represent different groups. The area of shapes represents DEmRNAs numbers.
Figure 2.
GO and KEGG analysis of DEmRNAs in the S_M, H_M, H_S. A. The Y- axis on the left represents GO terms of up-regulated genes, including biological process (BP), cellular component (CP), and molecular function (MF), the X- axis indicates different comparison groups. The area of a circle represents DEGs number. Low p- values are shown in the red circle, and high p-values are shown in the blue circle. B. The Y- axis on the left represents GO terms of down-regulated genes, including biological process (BP), cellular component (CP), and molecular function (MF), the X- axis indicates different comparison groups. The area of a circle represents DEGs number. Low p-values are shown in the red circle, and high p-values are shown in the blue circle. C. The Y- axis on the left represents KEGG pathways, the X- axis indicates genes enrichment of each term. The shapes represent different groups. The area of shapes represents DEmRNAs numbers.
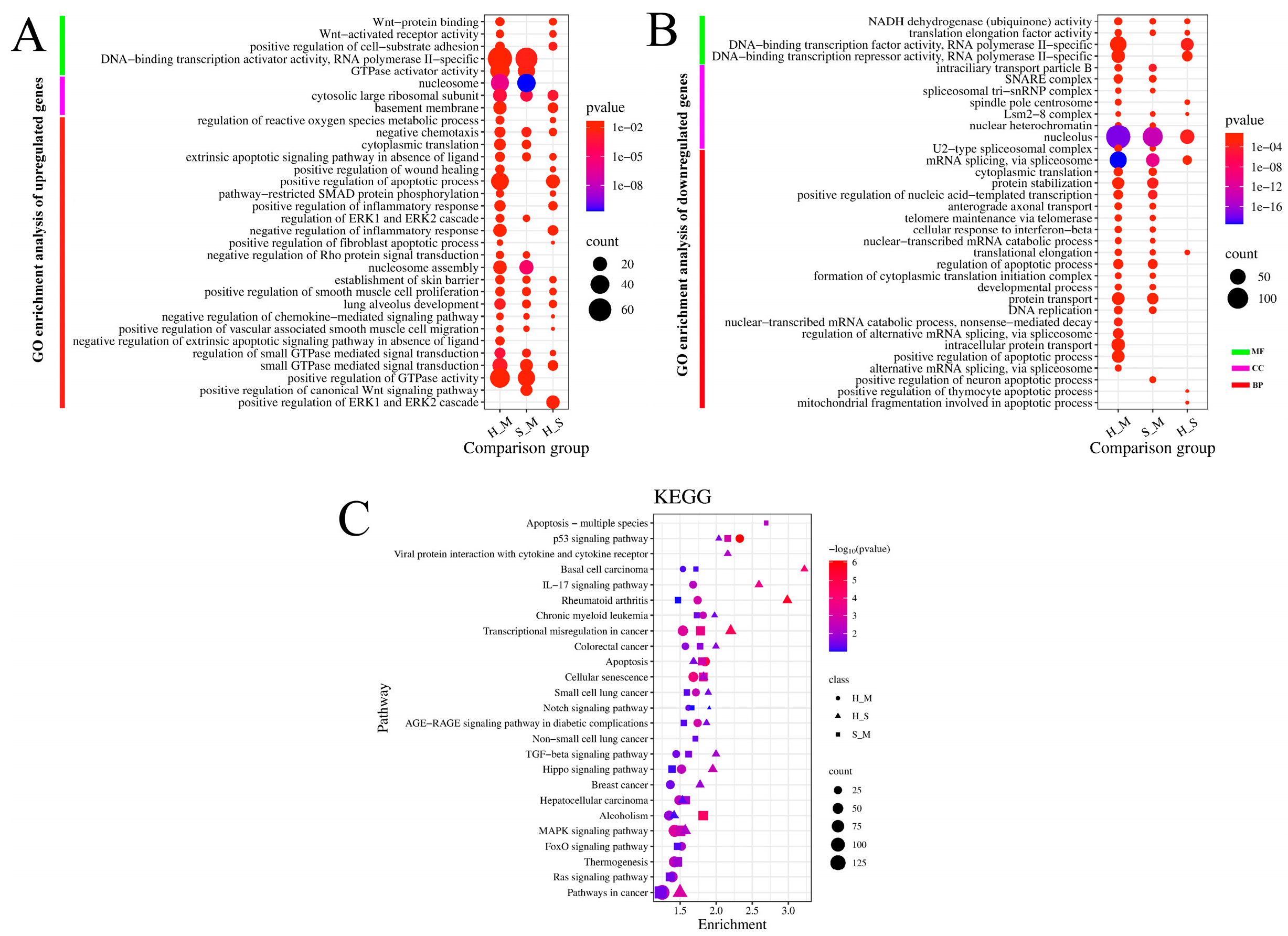
Figure 3.
GO enrichment result of differentially spliced genes in the S_M, H_M, H_S. The Y-axis on the left represents GO terms, including biological process (BP), cellular component (CP), and molecular function (MF), and the X-axis indicates gene enrichment of each term. Low p-values are shown in the red circle, and high p-values are shown in the green circle. The area of a circle represents of DEmRNAs number.
Figure 3.
GO enrichment result of differentially spliced genes in the S_M, H_M, H_S. The Y-axis on the left represents GO terms, including biological process (BP), cellular component (CP), and molecular function (MF), and the X-axis indicates gene enrichment of each term. Low p-values are shown in the red circle, and high p-values are shown in the green circle. The area of a circle represents of DEmRNAs number.
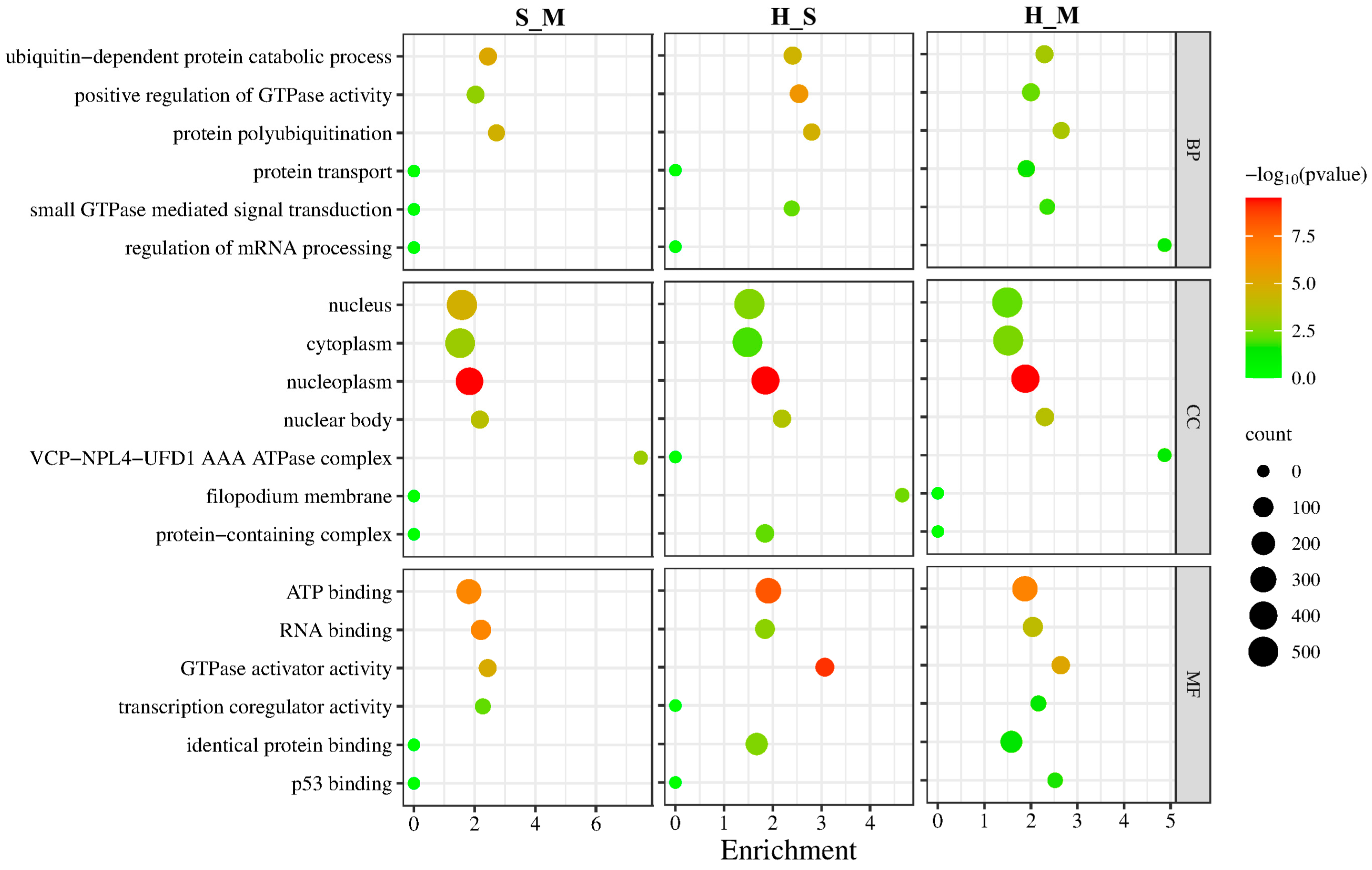
Figure 4.
The pathways of spliced genes in the S_M, H_M, H_S. The Y-axis on the left represents KEGG pathways, the Y-axis on the right represents the major category to which each pathway belongs, the X-axis indicates DEmRNAs numbers of each pathway.
Figure 4.
The pathways of spliced genes in the S_M, H_M, H_S. The Y-axis on the left represents KEGG pathways, the Y-axis on the right represents the major category to which each pathway belongs, the X-axis indicates DEmRNAs numbers of each pathway.
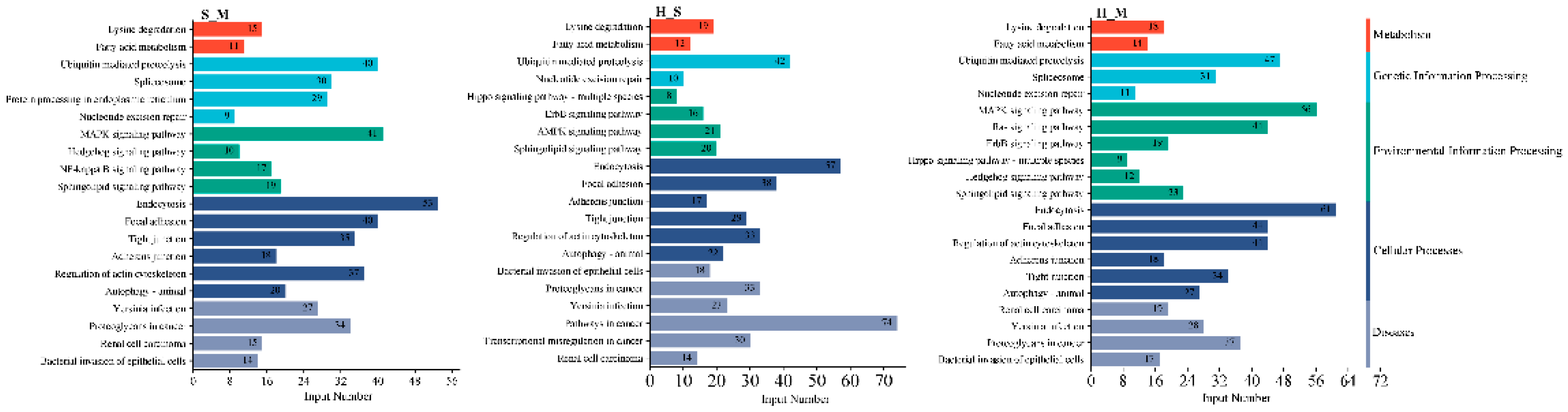
Figure 5.
Screening DElncRNAs compared between M group, S group, and H group. A. Distribution of DElncRNAs in each sample, with the Y-axis representing the number of genes and the X-axis representing different samples; B. Venn analysis of novel DElncRNAs obtained from four software programs: CNKI, COC, Pfam, and CPAT. C. Cluster analysis of DElncRNAs in mammary epithelial cells between the control group (M1, M2, M3, M4, and M5), normally treated groups (S1, S2, S3, S4, and S5) and deeply treated groups (H1, H2, and H3). Red indicates highly expressed genes, and green indicates low expressed genes. Each column represents a sample. D E F. Volcano plot of global DElncRNAs in S_M, H_M and H_S, respectively. Gradient red dots represent significantly regulated genes (p<0.05, |log2(fold-change)| >1); dark green dots represent significantly differential expressed genes.
Figure 5.
Screening DElncRNAs compared between M group, S group, and H group. A. Distribution of DElncRNAs in each sample, with the Y-axis representing the number of genes and the X-axis representing different samples; B. Venn analysis of novel DElncRNAs obtained from four software programs: CNKI, COC, Pfam, and CPAT. C. Cluster analysis of DElncRNAs in mammary epithelial cells between the control group (M1, M2, M3, M4, and M5), normally treated groups (S1, S2, S3, S4, and S5) and deeply treated groups (H1, H2, and H3). Red indicates highly expressed genes, and green indicates low expressed genes. Each column represents a sample. D E F. Volcano plot of global DElncRNAs in S_M, H_M and H_S, respectively. Gradient red dots represent significantly regulated genes (p<0.05, |log2(fold-change)| >1); dark green dots represent significantly differential expressed genes.
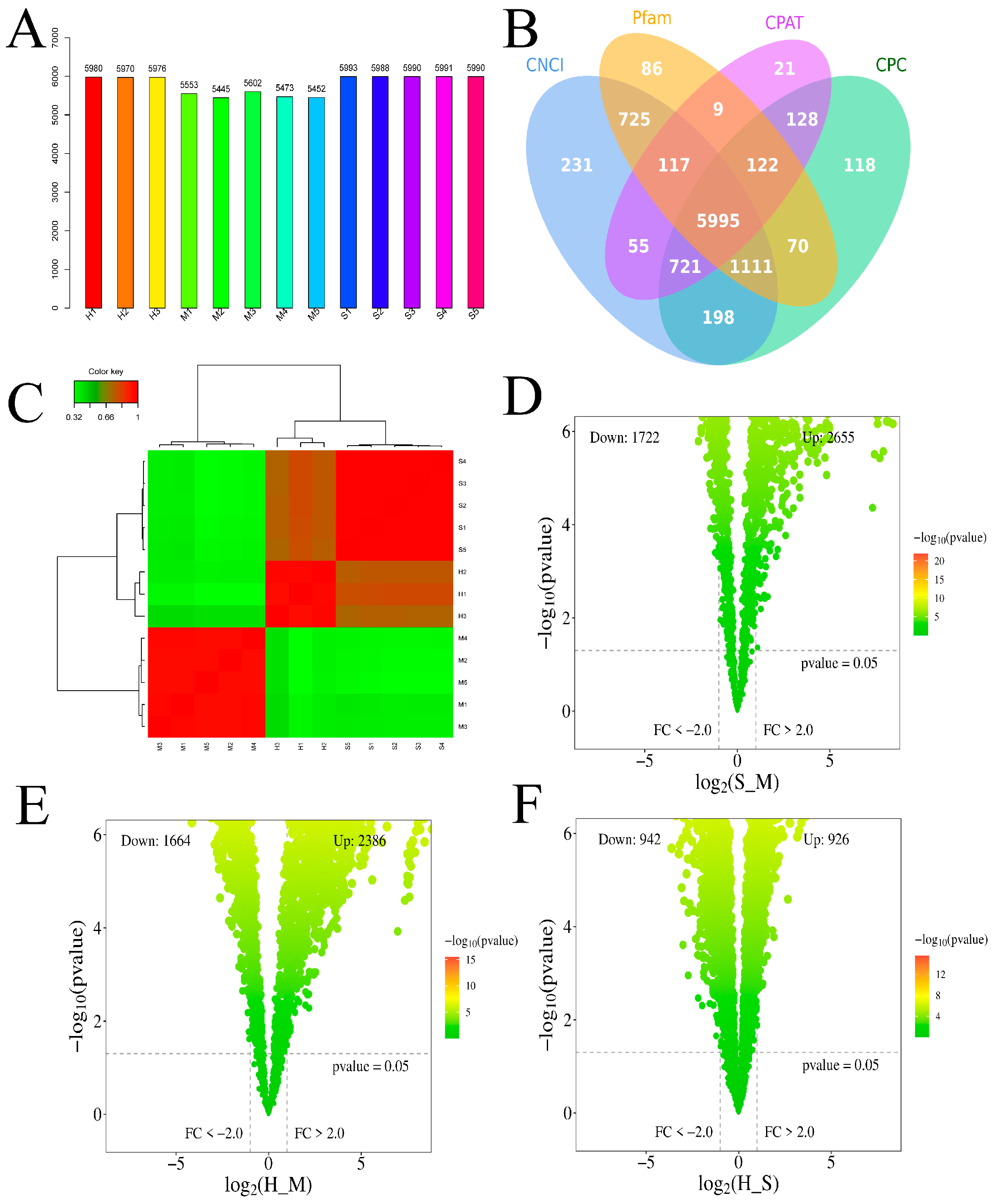
Figure 6.
Analysis of DElncRNA target genes. The red circle represents lncRNA, and the purple circle represents mRNA.
Figure 6.
Analysis of DElncRNA target genes. The red circle represents lncRNA, and the purple circle represents mRNA.
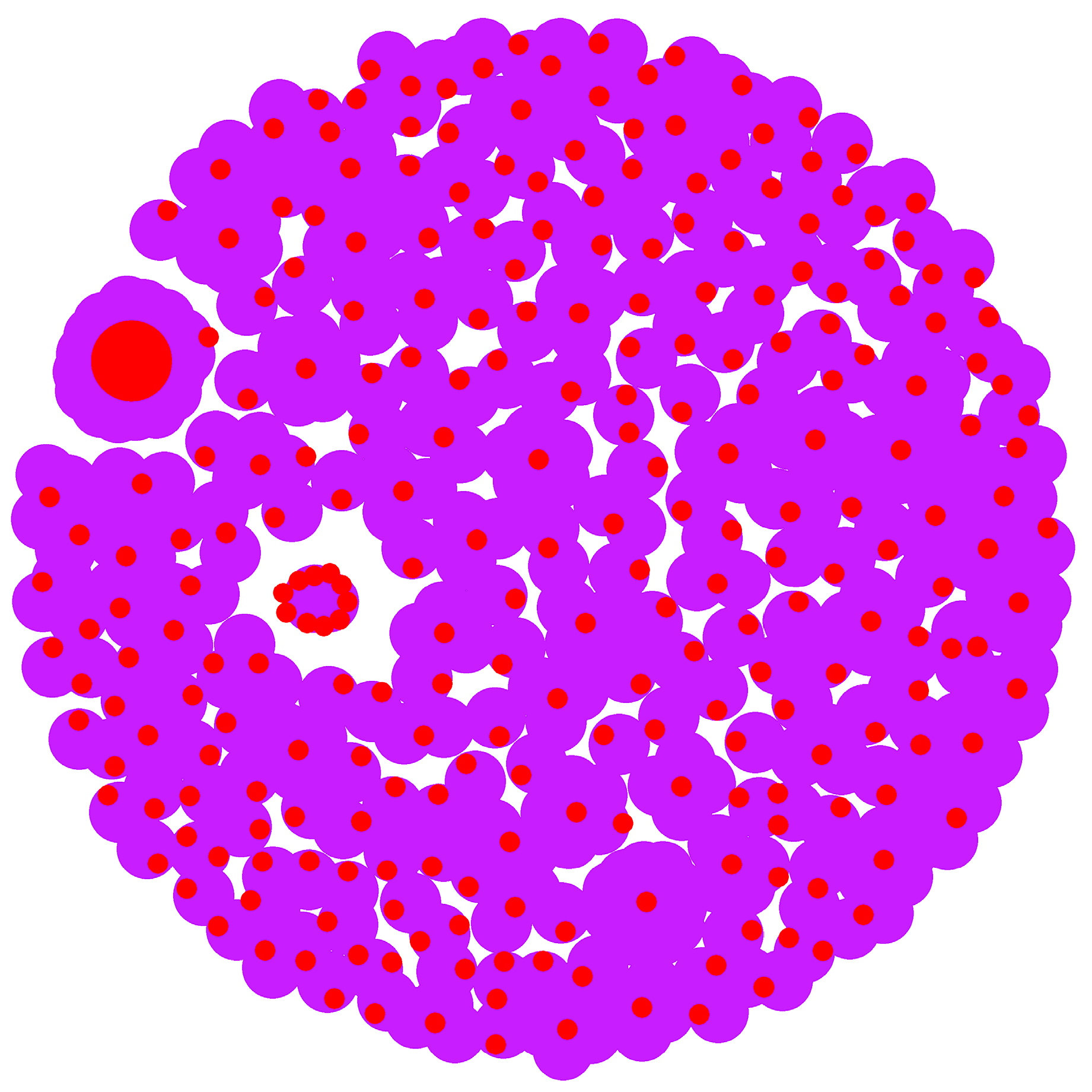
Figure 7.
Venn map analysis of DElncRNA target genes, DEmRNA and AS genes. DEG means differential expression genes; AS means Alternative splicing; S_M, H_S and H_M represent three comparison groups; Target lncRNA means three comparative groups targeting mRNA.
Figure 7.
Venn map analysis of DElncRNA target genes, DEmRNA and AS genes. DEG means differential expression genes; AS means Alternative splicing; S_M, H_S and H_M represent three comparison groups; Target lncRNA means three comparative groups targeting mRNA.
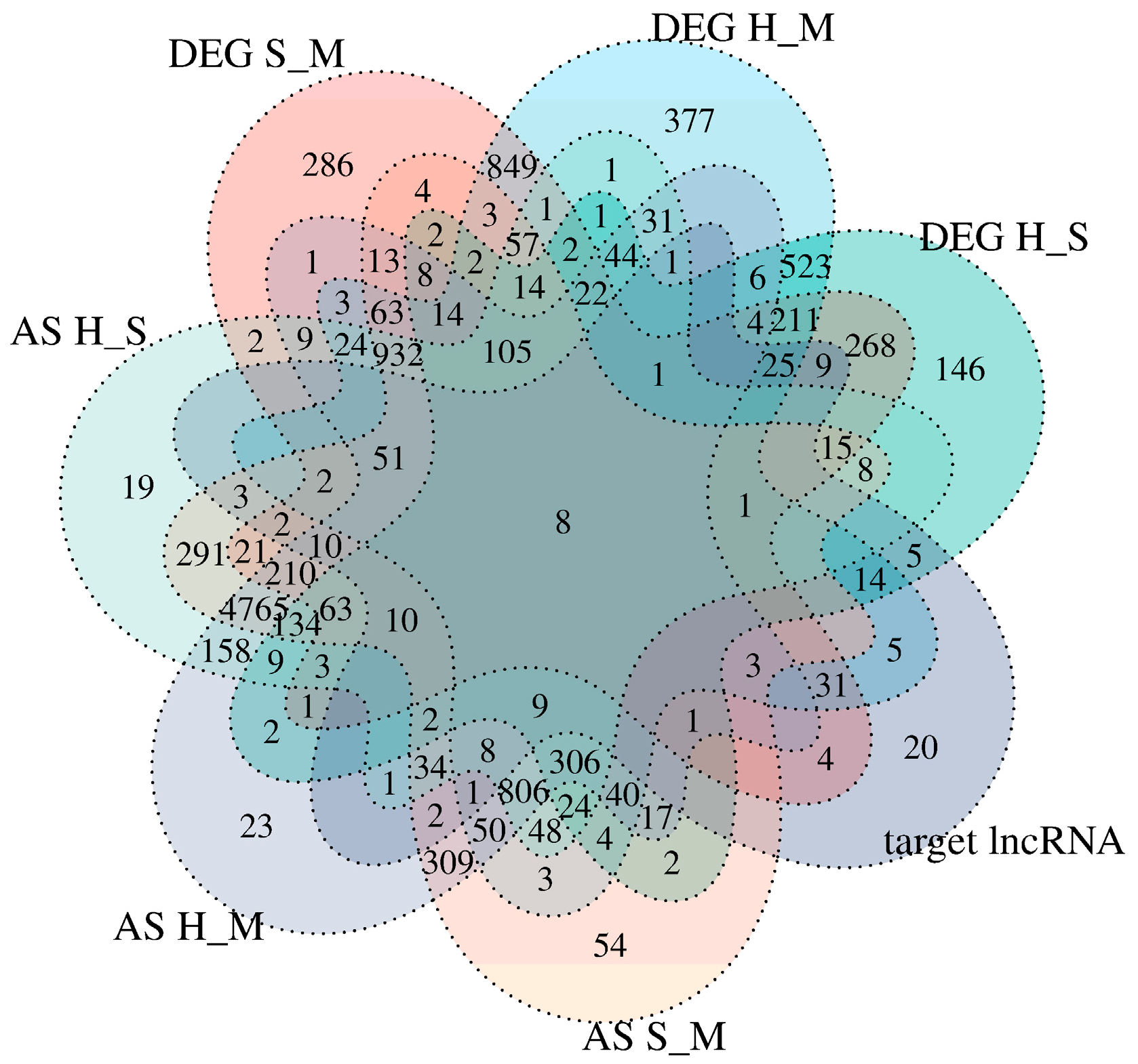
Figure 8.
Trend analysis of S. agalactiae infection in breast epithelial cells. This series of charts uses Mfuzzy to illustrate the dynamic changes in DEmRNAs during pathogen infection. The yellow and green lines represent genes with small differences in expression changes, while the red and purple lines indicate genes with large differences in expression changes.
Figure 8.
Trend analysis of S. agalactiae infection in breast epithelial cells. This series of charts uses Mfuzzy to illustrate the dynamic changes in DEmRNAs during pathogen infection. The yellow and green lines represent genes with small differences in expression changes, while the red and purple lines indicate genes with large differences in expression changes.
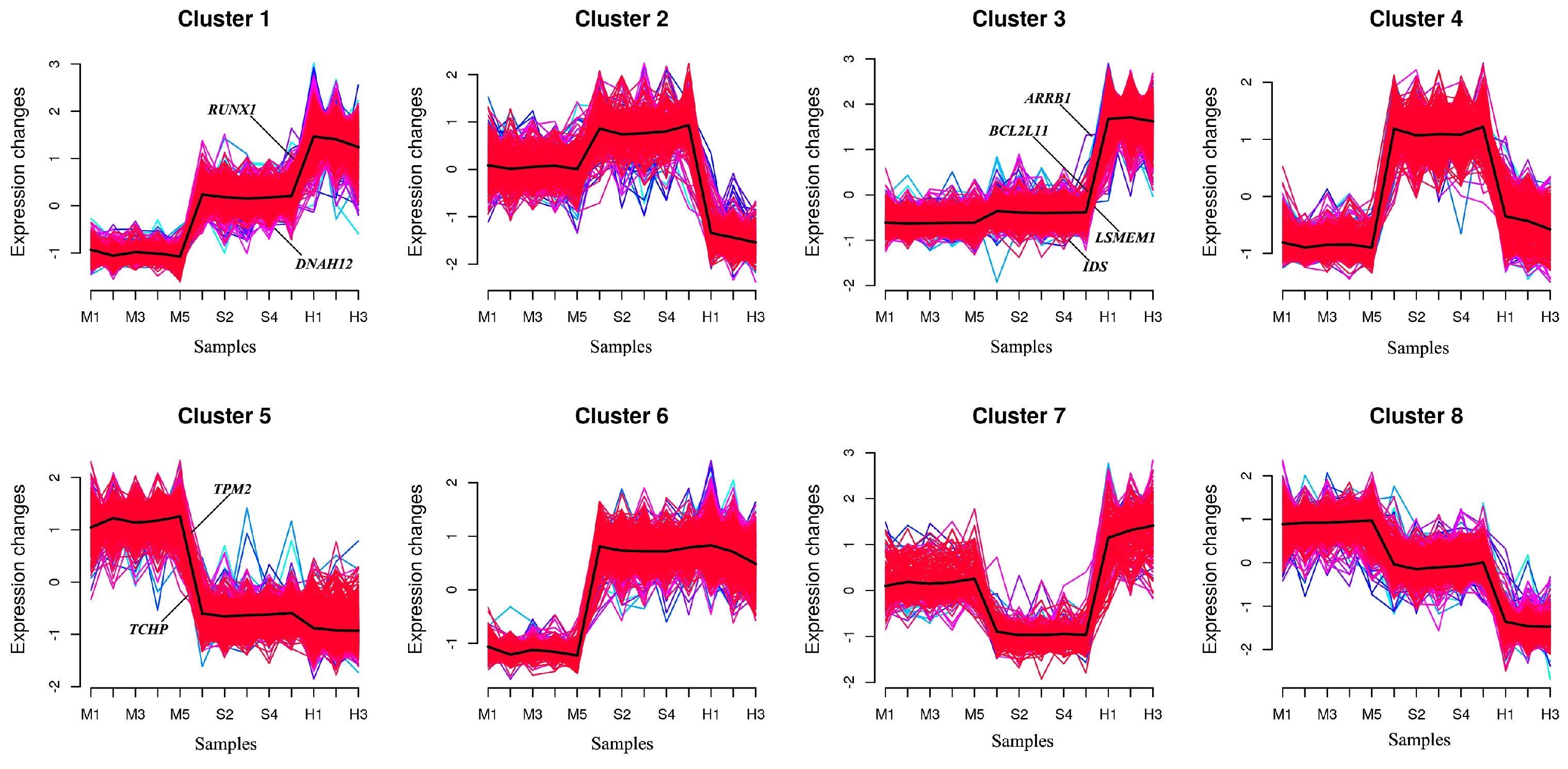
Figure 9.
Screening and enrichment analysis of pDEmRNAs of pathogens in S. agalactiae- normally treated groups (n= 5) compared with S. agalactiae- deeply treated groups. A. Cluster analysis of pDEmRNAs in S. agalactiae between normal treated group (S1, S2, S3, S4, and S5) and deeply treated group (H1, H2, and H3). Red indicates highly expressed genes, and blue indicates low expressed genes. Each column represents a sample, and each row represents a gene. On the left is the tree diagram of mRNA clustering. B. Volcano plot of global pDEmRNAs in S. agalactiae between normal treated group and deeply treated group. Red dots (Up) represent significantly up-regulated genes (p< 0.05, log2(fold-change) >1); blue dots (Down) represent significantly down-regulated genes (p< 0.05, log2(fold-change) <-1); grey dots represent insignificantly differential expressed genes. C. KEGG pathway classified annotation of pDEmRNAs in S. agalactiae. The pathway is exhibited on the left axis, and the area of the circle represents the number of genes listed on the right axis. D. Annotation of pDEmRNAs using Gene Ontology (GO) in S. agalactiae. The rich factor of mRNAs for each GO annotation is exhibited in the left axis.
Figure 9.
Screening and enrichment analysis of pDEmRNAs of pathogens in S. agalactiae- normally treated groups (n= 5) compared with S. agalactiae- deeply treated groups. A. Cluster analysis of pDEmRNAs in S. agalactiae between normal treated group (S1, S2, S3, S4, and S5) and deeply treated group (H1, H2, and H3). Red indicates highly expressed genes, and blue indicates low expressed genes. Each column represents a sample, and each row represents a gene. On the left is the tree diagram of mRNA clustering. B. Volcano plot of global pDEmRNAs in S. agalactiae between normal treated group and deeply treated group. Red dots (Up) represent significantly up-regulated genes (p< 0.05, log2(fold-change) >1); blue dots (Down) represent significantly down-regulated genes (p< 0.05, log2(fold-change) <-1); grey dots represent insignificantly differential expressed genes. C. KEGG pathway classified annotation of pDEmRNAs in S. agalactiae. The pathway is exhibited on the left axis, and the area of the circle represents the number of genes listed on the right axis. D. Annotation of pDEmRNAs using Gene Ontology (GO) in S. agalactiae. The rich factor of mRNAs for each GO annotation is exhibited in the left axis.
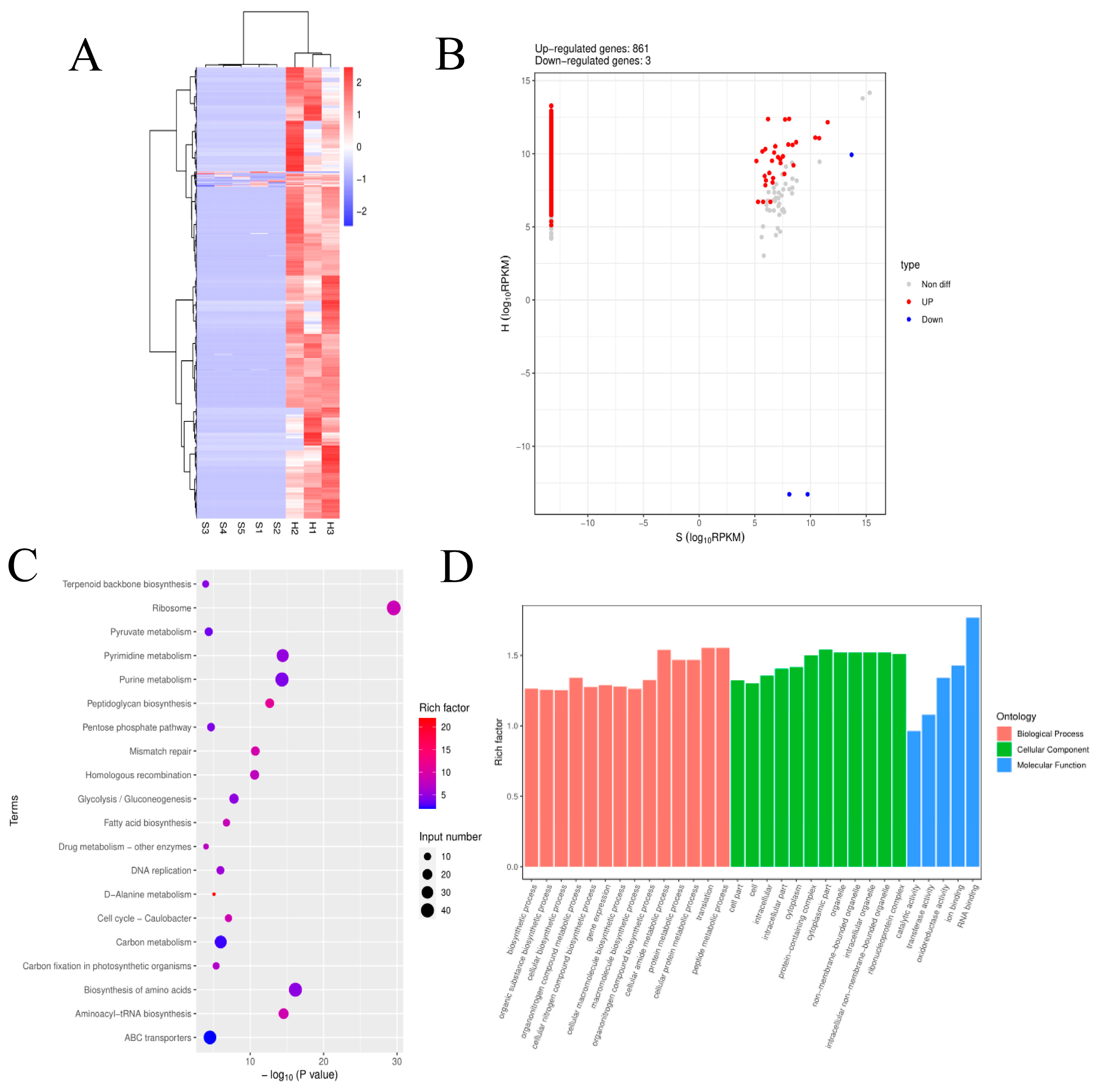
Figure 10.
Co-expression network of host cell DElncRNA, DEmRNA, and pathogen pDEmRNA. Red indicates upregulation, blue indicates downregulation, and the color intensity represents strength.
Figure 10.
Co-expression network of host cell DElncRNA, DEmRNA, and pathogen pDEmRNA. Red indicates upregulation, blue indicates downregulation, and the color intensity represents strength.
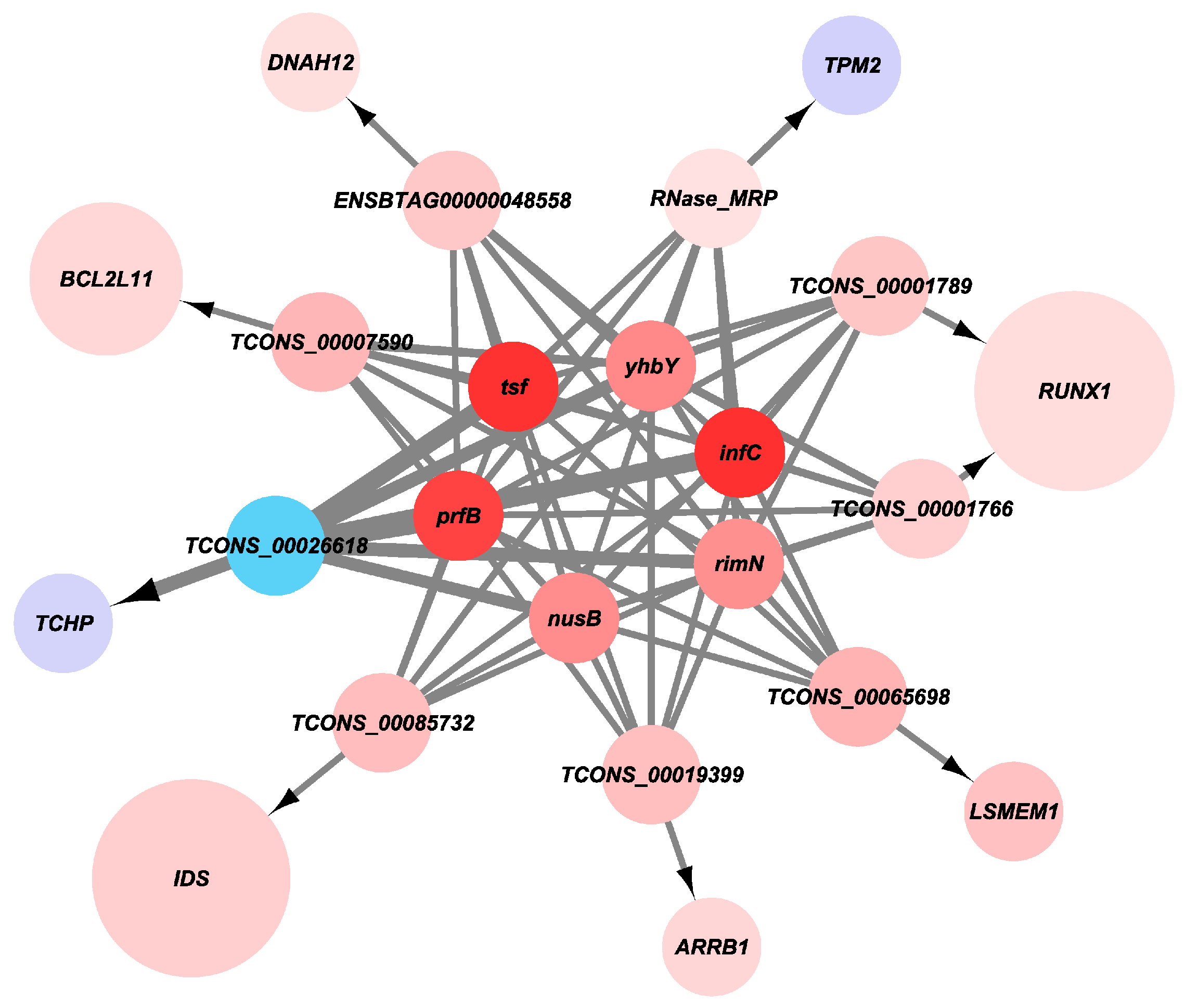
Figure 11.
The expression level of candidate gene.
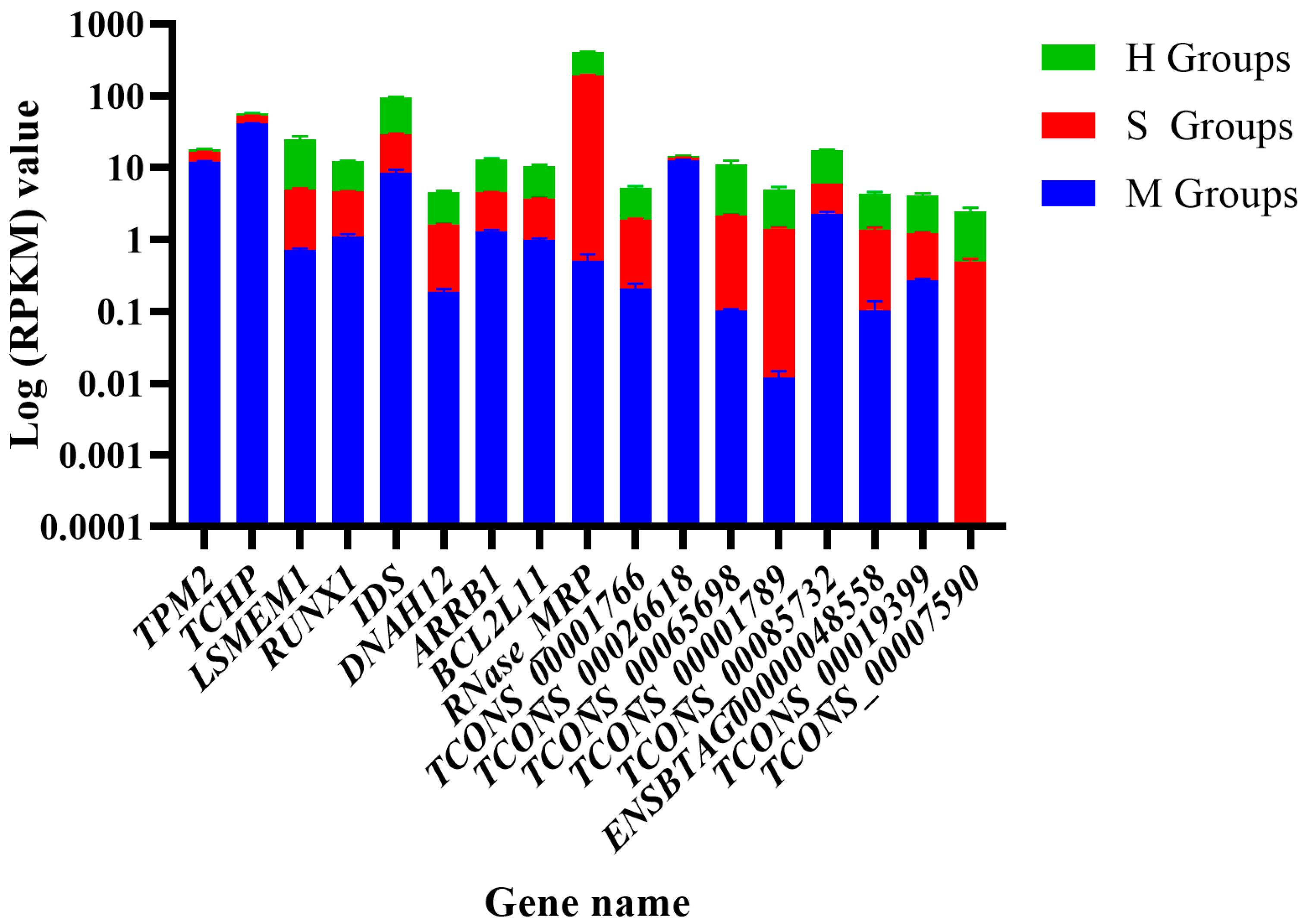

Table 1.
mRNA sequence quality.
| Sample | Group | Total raw reads | Total clean reads | Total clean base (G) | Effective Rate (%) | Reads with UIDs | Dedup Reads |
|---|---|---|---|---|---|---|---|
| M1 | Control (M Group) |
81298832 | 70041960 | 10.38 | 86.15 | 64794252(92.51%) | 61112168(87.25%) |
| M2 | 80917920 | 70388474 | 10.46 | 86.99 | 65098992(92.49%) | 60322418(85.70%) | |
| M3 | 92091998 | 79660982 | 11.81 | 86.5 | 73798986(92.64%) | 69202752(86.87%) | |
| M4 | 82288064 | 70524828 | 10.46 | 85.7 | 65226928(92.49%) | 61014132(86.51%) | |
| M5 | 91369374 | 79413762 | 11.81 | 86.92 | 73703106(92.81%) | 67255788(84.69%) | |
| S1 | Treat1 (S Group) |
102946650 | 92613194 | 13.63 | 89.96 | 86948336(93.88%) | 79863190(86.23%) |
| S2 | 71198348 | 63027196 | 9.25 | 88.52 | 59120464(93.80%) | 56144004(89.08%) | |
| S3 | 86815548 | 76934788 | 11.25 | 88.62 | 72256950(93.92%) | 68365948(88.86%) | |
| S4 | 92741594 | 82904518 | 12.12 | 89.39 | 77832968(93.88%) | 73052626(88.12%) | |
| S5 | 104732530 | 93522804 | 13.67 | 89.3 | 87853102(93.94%) | 82741910(88.47%) | |
| H1 | Treat2 (H Group) |
60552640 | 43447378 | 6.3 | 72.94 | 41488876(95.49%) | 40567390(93.37%) |
| H2 | 72552826 | 51713480 | 7.61 | 71.28 | 49400638(95.53%) | 46882558(90.66%) | |
| H3 | 83335490 | 62442120 | 9.2 | 74.93 | 59544572(95.36%) | 55991706(89.67%) |
Table 2.
Type of alternative splicing and statistics of differential alternative splicing events.
| EventType. | NumEvents. JC. only |
SigEvents. JC. only (up: down) | NumEvents. JC+ Reads On Target | SigEvents. JC+ reads On Target (up: down) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| S_ M | H_ M | H_ S | S_ M | H_M | H_ S | S_ M | H_ M | H_ S | S_ M | H_ M | H_ S | |
| SE | 36032 | 33110 | 35535 | 428:497 | 557:1034 | 434:818 | 36038 | 33113 | 35536 | 450:533 | 593:1091 | 462:851 |
| MXE | 7816 | 6661 | 7424 | 993:1090 | 1398:1132 | 1159:794 | 7816 | 6661 | 7424 | 982:1083 | 1375:1130 | 1144:787 |
| A5SS | 348 | 324 | 309 | 17:16 | 19:21 | 9:12 | 349 | 324 | 309 | 17:15 | 21:23 | 13:13 |
| A3SS | 418 | 405 | 393 | 12:12 | 18:13 | 12:10 | 418 | 405 | 393 | 12:13 | 19:13 | 12:09 |
| RI | 494 | 455 | 426 | 7:21 | 13:37 | 8:17 | 503 | 457 | 432 | 6:18 | 13:32 | 7:13 |
SE, Skipped exon, A5SS, Alternative 5’splice, A3SS, Alternative 3’splice, MXE, Mutually exclusive, RI, Retained intron.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



