
Submitted:
18 August 2024
Posted:
19 August 2024
You are already at the latest version
Abstract
Recently, oriented external electric fields (OEEFs) have earned much attention due to the possibility of tuning the properties of electronic systems. From a theoretical perspective, one can resort to electronic structure calculations to understand how the direction and strength of OEEFs affect the properties of electronic systems. However, for multi-reference (MR) systems, calculations employing the popular Kohn-Sham density functional theory with the traditional semilocal and hybrid exchange-correlation energy functionals can yield erroneous results. Owing to its decent compromise between accuracy and efficiency for MR systems at the nanoscale (i.e., MR nanosystems), in this study, thermally-assisted-occupation density functional theory (TAO-DFT) is adopted to explore the electronic properties of n-acenes (n = 2--10), containing n linearly fused benzene rings, in OEEFs, where the OEEFs of various electric field strengths are applied along the long axes of n-acenes. According to our TAO-DFT calculations, the ground states of n-acenes in OEEFs are singlets for all the cases examined. The effect of OEEFs is shown to be significant on the vertical ionization potentials and vertical electron affinities of ground-state n-acenes with odd-number fused benzene rings. Moreover, the MR character of ground-state n-acenes in OEEFs increases with the increase of the acene length and/or the electric field strength.
Keywords:
TAO-DFT
; OEEFs
; electronic properties
; acenes
; multi-reference character
1. Introduction
Among a wide variety of carbon materials, graphene (i.e., a two-dimensional system) has been of great interest due to its interesting properties and possible applications [1,2,3,4,5,6]. The long spin diffusion length and high carrier mobility of graphene offers attractive possibilities for graphene-based spintronics and electronics. Nevertheless, since graphene does not possess a band gap, it cannot be used for transistor applications.
To include a band gap into graphene, the carriers can be confined to quasi-one-dimensional systems, such as graphene nanoribbons (GNRs). Owing to their promising properties and potential applications, GNRs (i.e., narrow and long strips of graphene) have recently attracted a lot of attention from many researchers [7,8,9,10,11,12,13,14,15,16,17,18,19]. Because of the significant effects of edges and quantum confinement, the properties of GNRs are highly dependent on their edge shape (e.g., zigzag, armchair, or chiral) and geometrical structure (e.g., width and length). On the other hand, oriented external electric fields (OEEFs) [20,21,22,23,24,25,26,27,28,29,30,31] have also gained considerable attention due to the possibility of varying the properties of electronic systems in recent years. However, to date, there have been scarce studies on the effect of OEEFs on the properties of GNRs [26].
A good understanding of the properties of GNRs in OEEFs can benefit the relevant molecular design and potential applications. From a theoretical perspective, one can perform electronic structure calculations to know how the direction and strength of OEEFs affect the properties of GNRs. In this work, we conduct a computational study to investigate the OEEF effect on the electronic properties of the narrowest zigzag GNRs of various lengths. For finite-size models of the narrowest zigzag GNRs of various lengths, as illustrated in Figure 1, we take linear acenes (denoted as n-acenes, containing n linearly fused benzene rings), where the OEEFs of various electric field strengths F [given in atomic units (1 a.u. ≈ 51.4 V / Å)] are applied along the long axes of n-acenes. Accordingly, this study aims at exploring the electronic properties of n-acenes in OEEFs (with various values of n and F). While the electronic properties of n-acenes have been extensively studied [32,33,34,35,36,37,38,39,40,41], reports on the effect of OEEFs on the electronic properties of n-acenes are very scarce. Previous findings have shown that in the absence of OEEFs (i.e., ), the longer n-acenes have significant multi-reference (MR) character in their electronic ground states [32,33,34,35,36,37,38,39,40,41]. Accordingly, it is expected that the longer n-acenes in OEEFs can also exhibit pronounced MR character in their electronic ground states.
Over the past thirty years, Kohn-Sham density functional theory (KS-DFT) [42,43] has been the most widely used electronic structure method for exploring the ground-state properties of electronic systems at the nanoscale (i.e., nanosystems), especially for systems with single-reference (SR) character in their electronic ground states (the so-called SR systems). Nonetheless, for systems with MR character in their electronic ground states (the so-called MR systems), calculations employing KS-DFT with the traditional semilocal [44,45,46] and hybrid [47,48,49] exchange-correlation (xc) energy functionals can yield erroneous results due to the presence of strong static correlation effects in MR systems [50,51,52,53]. On the other hand, to reliably predict the ground-state properties of small MR systems, one typically resorts to ab initio MR electronic structure methods [32,34,38,54,55,56,57,58,59,60]. However, for MR nanosystems, reliably accurate MR electronic structure methods are inapplicable as the computational cost of performing these MR calculations can be prohibitively high. Therefore, it is essential to adopt an efficient and reliable electronic structure method for studying the ground-state properties of MR nanosystems (e.g., the longer n-acenes).
Thermally-assisted-occupation density functional theory (TAO-DFT) [33] has recently emerged as a cost-effective solution to the challenge of studying MR nanosystems. TAO-DFT, which has similar computational cost as KS-DFT, is suitable for studying the ground-state properties of nanosystems. For an MR system, the representability of ground-state electron density, which incorporates fractional orbital occupations generated by the Fermi-Dirac (FD) distribution function with some fictitious temperature , in TAO-DFT can be greatly improved [33,61], when compared with that in KS-DFT. The semilocal [33,36] and hybrid [39,62] exchange-correlation- (xc) energy functionals (i.e., the combined xc and -dependent energy functionals) can also be adopted in TAO-DFT. Besides, the popular dispersion correction schemes [63,64] can also be used in TAO-DFT for the efficient calculations of non-covalent interactions [36,39,65]. Simple schemes for determining the optimal system-independent [40] and system-dependent [66] values of an xc energy functional in TAO-DFT have been recently developed. Moreover, the fundamental distinction among three different electronic structure methods, such as finite-temperature density functional theory (FT-DFT) [43,67], TAO-DFT [33], and KS-DFT [42,43], has been discussed in a recent study [68].
Within the framework of TAO-DFT, a number of extensions, such as TAO-DFT with the polarizable continuum model [69], TAO-DFT-based ab initio molecular dynamics [70], and a real-time extension of TAO-DFT [68], have been recently proposed for diverse applications. Over the past few years, TAO-DFT and its extensions have been employed to explore a very wide range of properties (e.g., electronic [37,65,71,72,73,74,75,76], hydrogen storage [65], spectroscopic [70,77,78], and equilibrium thermodynamic [70] properties) of MR nanosystems.
Since TAO-DFT is a well-suited electronic structure method for studying MR nanosystems due to its decent compromise between accuracy and efficiency, in this study, we adopt TAO-DFT to obtain the electronic properties (e.g., singlet-triplet energy gaps, vertical ionization potentials, vertical electron affinities, fundamental gaps, symmetrized von Neumann entropy, active orbital occupation numbers, and real-space representation of active orbitals) of n-acenes (n = 2–10) in OEEFs of various electric field strengths F.
2. Computational Details
All calculations are performed with Q-Chem 4.3 [79], using the 6-31G(d) basis set. Calculations are carried out using TAO-LDA (i.e., TAO-DFT with the local density approximation (LDA) xc energy functional) with the recommended fictitious temperature = 7 mhartree [33] to obtain the electronic properties of n-acenes (n = 2–10) in OEEFs, where the OEEFs of various electric field strengths F = 0.000, 0.001, 0.002, 0.003, 0.004, and 0.005 a.u. are applied along the long axes of n-acenes.
3. Results and Discussion
3.1. Singlet-Triplet Energy Gap
The singlet-triplet (ST) energy gap (the so-called ST gap), i.e., the energy splitting between the lowest triplet and singlet states, of a molecule offers an understanding on its ground-state nature, which could also be helpful to explore the possible MR character of the molecule [80,81,82,83,84], and to offer useful information for photovoltaic applications [85].
In this work, the ST gap () of n-acene in an OEEF is computed using
where / is the spin-unrestricted TAO-LDA energy of the lowest triplet / singlet state of n-acene in an OEEF (evaluated at the optimized lowest triplet / singlet state molecular geometry).
As shown in Figure 2, in an OEEF, the ST gap of n-acene decreases in a monotonic manner with increasing acene length (also see Table S1 in Supplementary Information (SI)). As the electric field strength F increases, the ST gap of n-acene slightly decreases. For each case (i.e., n and F), the singlet state has lower energy than the triplet state. Therefore, for all the OEEFs considered, n-acenes (n = 2–10) possess singlet ground states. As MR systems generally have small ST gaps, the ground states of the longer n-acenes in OEEFs can have MR character.
On the other hand, the spin-symmetry constraint, which can be satisfied by an exact theory, ensures that the exact spin-unrestricted and spin-restricted calculations must yield the same energy for the lowest singlet state of an MR system. Nonetheless, such a spin-symmetry constraint can be greatly violated by KS-DFT with the traditional semilocal and hybrid xc energy functionals as well as SR electronic structure methods (e.g., the Hartree-Fock theory) [33,35,36,37,39,51,61,68,69,70], leading to strikingly different energies for the lowest singlet state of an MR system (the so-called spin-symmetry breaking). In principle, one can adopt reliably accurate MR electronic structure methods to resolve this problem. In practice, such MR methods are, however, very impractical for studying MR nanosystems (e.g., the longer n-acenes), because of the prohibitively high computational cost. Very recently, a TAO-DFT-based response theory [61] has been proposed to demonstrate that for any MR system, TAO-DFT with a sufficiently large fictitious temperature can always resolve the spin-symmetry breaking problem.
Here, we investigate whether this spin-symmetry constraint can be satisfied by TAO-LDA (i.e., with the recommended fictitious temperature = 7 mhartree) [33], and hence, carry out additional spin-restricted TAO-LDA calculations for the lowest singlet energies of n-acenes in OEEFs (evaluated at the respective optimized molecular geometries). We find that within the numerical precision considered, the spin-restricted and spin-unrestricted TAO-LDA energies for the lowest singlet states of n-acenes in OEEFs are essentially identical, implying that for all the cases examined, essentially no unphysical spin-symmetry breaking effects occur in our spin-unrestricted TAO-LDA solutions.
3.2. Vertical Ionization Potential, Vertical Electron Affinity, and Fundamental Gap
The vertical ionization potential, vertical electron affinity, and fundamental gap of a ground-state molecule are also key electronic properties. These properties are important in spectroscopy and catalyst selection, and are also essential for choosing candidate materials for electronic devices and solar cells. Based on their definitions, the vertical ionization potential is the energy change when an electron is removed from the ground-state molecule (without altering the ground-state molecular geometry), the vertical electron affinity is the energy change when an electron is added to the ground-state molecule (without altering the ground-state molecular geometry), and the fundamental gap is the difference between the vertical ionization potential and vertical electron affinity.
At the spin-unrestricted TAO-LDA optimized geometry of ground-state n-acene in an OEEF, we compute the vertical ionization potential [36,37,39]
vertical electron affinity
and fundamental gap
of ground-state n-acene in an OEEF. Here, is the total energy of the N-electron molecule (i.e., ground-state n-acene) in an OEEF, obtained with spin-unrestricted TAO-LDA.
The , , and of ground-state n-acene in an OEEF are presented in Figure 3, Figure 4, and Figure 5, respectively (also see Tables S2 to S4 in SI). For the smaller electric field strength F (e.g., a.u.), with the increase of the acene length, the monotonically decreases, the monotonically increases, and hence, the monotonically decreases, similar to those found in the absence of OEEFs (i.e., ) [36,37]. For the larger F (e.g., a.u.), the and display odd-even oscillation patterns, while the shows a similar trend as that observed for the smaller F (e.g., a.u.).
As F increases, both of the and greatly increase for n-acene with odd-number fused benzene rings, while the / slightly decreases / increases for n-acene with even-number fused benzene rings. As a consequence, the of ground-state n-acene slightly decreases with increasing F. In general, electronic systems with the fundamental gaps of 1–3 eV are suitable for photovoltaic applications. Therefore, among all the cases examined, 10-acene in OEEFs of F = 0.000, 0.001, 0.002, 0.003, 0.004, and 0.005 a.u. can be appropriate for photovoltaic applications.
3.3. Symmetrized von Neumann Entropy
We investigate the MR character of ground-state n-acene in an OEEF using the symmetrized von Neumann entropy [35,36,37,39]
Here, = ↑ or ↓ denotes the up-spin or down-spin, and (i.e., a value between 0 and 1) is the occupation number of the -spin orbital, obtained with spin-unrestricted TAO-LDA [33], approximately yielding the occupation number of the -spin natural orbital [71,86]. Based on Equation (5), the terms with = 0 or 1 give no contribution to the , while the terms with greatly deviating from 0 and 1 (e.g., close to 0.5) can lead to a large increase in the . Consequently, for an SR system, since all values of are very close to 0 or 1, the should be vanishingly small. By contrast, for an MR system, since the occupation numbers of active spin-orbitals [i.e., those with noticeable fractional occupation numbers (e.g., between 0.1 and 0.9)] are closer to 0.5, and/or the number of active spin-orbitals increases, the can be very large.
As shown in Figure 6, the of ground-state n-acene in an OEEF monotonically increases with increasing acene length (also see Table S5 in SI). This implies that the MR character of ground-state n-acene in an OEEF should generally increase with increasing acene length. Besides, as the electric field strength F increases, the (i.e., a measure of MR character) of ground-state n-acene slightly increases.
3.4. Active Orbital Occupation Numbers
According to the aforementioned spin-unrestricted TAO-DFT calculations, with increasing acene length, the of ground-state n-acene in an OEEF monotonically increases. This indicates that as n increases, the occupation numbers of active spin-orbitals are closer to 0.5, and/or the number of active spin-orbitals increases. In spin-restricted TAO-DFT, this implies that for the ground state (i.e., lowest singlet state) of n-acene (containing N electrons) in an OEEF, as n increases, the occupation numbers of active orbitals [i.e., those with noticeable fractional occupation numbers (e.g., between 0.2 and 1.8)] are closer to 1, and/or the number of active orbitals increases. Here, the orbital is defined as the HOMO (highest occupied molecular orbital), and the orbital is defined as the LUMO (lowest unoccupied molecular orbital) [33,37,39].
To demonstrate this, in Figure 7, we present the occupation numbers of active orbitals for the ground state of n-acene in an OEEF, computed using spin-restricted TAO-LDA. As shown, the shorter n-acenes (e.g., ) in an OEEF possess nonradical nature in their ground states, because all the orbital occupation numbers are very close to 0 or 2. With the increase of the acene length, the occupation numbers of active orbitals are closer to 1, and/or the number of active orbitals increases, suggesting that the longer n-acenes in an OEEF possess increasing polyradical nature in their ground states. Therefore, as the acene length increases, there is a transition from the nonradical nature of the shorter n-acenes to the increasing polyradical nature of the longer n-acenes in an OEEF. For n-acenes in an OEEF of the larger electric field strength F, the evolution of polyradical nature is more rapid.
It is worth mentioning that with increasing acene length, the occupation numbers of active orbitals exhibit a curve crossing behavior in the approach to 1. For example, in an OEEF, the orbital with the HOMO / LUMO character in the shorter n-acenes can become the LUMO / HOMO in the longer n-acenes. This curve crossing behavior has been reported by the recent studies of n-acenes in the absence of OEEFs (i.e., ), using TAO-DFT [33,37,39,40] as well as an accurate MR electronic structure method [38].
3.5. Real-Space Representation of Active Orbitals
In Figure 8 and Figure 9, we report the real-space representation of active orbitals (HOMO and LUMO) for the ground states of the longer n-acenes (n = 9 and 10) in OEEFs, obtained with spin-restricted TAO-LDA (for the cases of the shorter n-acenes (n = 2–8), see Figures S1 to S7 in SI). The edge localization of active orbitals for n-acenes in OEEFs is observed, similar to the edge localization of active orbitals previously found for n-acenes in the absence of OEEFs (i.e., ) [32,34,35,37].
As mentioned previously, the curve crossing behavior is observed, and can be easily seen here. For example, in an OEEF of the larger electric field strength F (e.g., a.u.), the orbital with the HOMO / LUMO character in the shorter n-acenes (n = 2–9) becomes the LUMO / HOMO in 10-acene. Alternatively, it can also be regarded that for 10-acene in an OEEF, the curve crossing behavior can occur when F is sufficiently large (e.g., a.u.), highlighting the role of OEEFs.
4. Conclusions
In conclusion, we have employed TAO-DFT to explore the electronic properties of n-acenes (n = 2–10) in OEEFs, where the OEEFs of various electric field strengths F = 0.000, 0.001, 0.002, 0.003, 0.004, and 0.005 a.u. are applied along the long axes of n-acenes. Since the longer n-acenes (e.g., ) in OEEFs have been shown to possess pronounced MR character in their ground states, KS-DFT with the traditional semilocal and hybrid xc energy functionals can yield incorrect results for these MR systems. On the other hand, owing to their prohibitively high computational cost, reliably accurate MR electronic structure methods are generally inapplicable for exploring the electronic properties of MR nanosystems (e.g., the longer n-acenes) in OEEFs. Therefore, TAO-DFT seems to be a promising electronic structure method for the present study due to its decent compromise between accuracy and efficiency.
According to our TAO-DFT calculations, the ST gap of n-acene in an OEEF monotonically decreases with increasing acene length. For all the OEEFs considered, n-acenes (n = 2–10) possess singlet ground states. The use of OEEFs has been shown to be significant for tuning the vertical ionization potentials and vertical electron affinities of ground-state n-acenes with odd-number fused benzene rings. For the smaller F (e.g., a.u.), as n increases, the vertical ionization potential and fundamental gap monotonically decrease, while the vertical electron affinity and symmetrized von Neumann entropy monotonically increase, similar to those found in the absence of OEEFs (i.e., ) [36,37]. For the larger F (e.g., a.u.), the vertical ionization potential and vertical electron affinity display odd-even oscillation patterns, while the fundamental gap and symmetrized von Neumann entropy show similar trends as those observed for the smaller F (e.g., a.u.). Similar to the findings of previous studies on n-acenes in the absence of OEEFs (i.e., ) [35,37], the shorter n-acenes (e.g., ) in an OEEF possess nonradical nature in their ground states, and the longer n-acenes in an OEEF possess increasing polyradical nature in their ground states. Therefore, with the increase of the acene length, there is a transition from the nonradical nature of the shorter n-acenes to the increasing polyradical nature of the longer n-acenes in an OEEF. For n-acenes in an OEEF of the larger F, the evolution of polyradical nature is more rapid.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org
Author Contributions
Conceptualization, J.-D.C.; Data curation, C.-Y.C.; Formal analysis, C.-Y.C.; Funding acquisition, J.-D.C.; Investigation, C.-Y.C.; Methodology, C.-Y.C. and J.-D.C.; Project administration, J.-D.C.; Resources, J.-D.C.; Software, J.-D.C.; Supervision, J.-D.C.; Validation, C.-Y.C. and J.-D.C.; Visualization, C.-Y.C.; Writing–original draft, C.-Y.C. and J.-D.C.; Writing–review & editing, J.-D.C. Both authors have read and agreed to the published version of the manuscript.
Funding
This research work was previously funded by the National Science and Technology Council of Taiwan (Grant Nos.: NSTC113-2112-M-002-032 and MOST110-2112-M-002-045-MY3).
Data Availability Statement
The data supporting this article have been included as part of the Supplementary Information.
Acknowledgements
This research work was previously supported by the National Science and Technology Council of Taiwan (Grant Nos.: NSTC113-2112-M-002-032 and MOST110-2112-M-002-045-MY3), and was also supported by National Taiwan University as well as the National Center for Theoretical Sciences of Taiwan.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Novoselov, K.S. et al. Electric field effect in atomically thin carbon films. Science 2004, 306, 666–669. [Google Scholar] [CrossRef]
- Novoselov, K.S. et al. Two-dimensional gas of massless Dirac fermions in graphene. Nature 2005, 438, 197–200. [Google Scholar] [CrossRef]
- Zhang, Y.; Tan, Y.; Stormer, H.L.; Kim, P. Experimental observation of the quantum Hall effect and Berry’s phase in graphene. Nature 2005, 438, 201–204. [Google Scholar] [CrossRef]
- Geim, A.K.; Novoselov, K.S. The rise of graphene. Nat. Mater. 2007, 6, 183–191. [Google Scholar] [CrossRef]
- Geim, A.K. Graphene: Status and prospects. Science 2009, 324, 1530–1534. [Google Scholar] [CrossRef] [PubMed]
- Madurani, K.A.; Suprapto, S.; Machrita, N.I.; Bahar, S.L.; Illiya, W.; Kurniawan, F. Progress in graphene synthesis and its application: History, challenge and the future outlook for research and industry. ECS J. Solid State Sci. Technol. 2020, 9, 093013. [Google Scholar] [CrossRef]
- Son, Y.W.; Cohen, M.L.; Louie, S.G. Energy gaps in graphene nanoribbons. Phys. Rev. Lett. 2006, 97, 216803. [Google Scholar] [CrossRef] [PubMed]
- Han, M.Y.; Özyilmaz, B.; Zhang, Y.; Kim, P. Energy band-gap engineering of graphene nanoribbons. Phys. Rev. Lett. 2007, 98, 206805. [Google Scholar] [CrossRef]
- Owens, F.J. Electronic and magnetic properties of armchair and zigzag graphene nanoribbons. J. Chem. Phys. 2008, 128, 194701. [Google Scholar] [CrossRef]
- Lee, H.; Ihm, J.; Cohen, M.L.; Louie, S.G. Calcium-decorated graphene-based nanostructures for hydrogen storage. Nano Lett. 2010, 10, 793–798. [Google Scholar] [CrossRef]
- Kimouche, A. et al. Ultra-narrow metallic armchair graphene nanoribbons. Nat. Commun. 2015, 6, 10177. [Google Scholar] [CrossRef] [PubMed]
- Houtsma, R.K.; de la Rie, J.; Stöhr, M. Atomically precise graphene nanoribbons: Interplay of structural and electronic properties. Chem. Soc. Rev. 2021, 50, 6541–6568. [Google Scholar] [CrossRef]
- Wang, H. et al. Graphene nanoribbons for quantum electronics. Nat. Rev. Phys. 2021, 3, 791–802. [Google Scholar] [CrossRef]
- Saraswat, V.; Jacobberger, R.M.; Arnold, M.S. Materials science challenges to graphene nanoribbon electronics. ACS Nano 2021, 15, 3674–3708. [Google Scholar] [CrossRef]
- Luo, H.; Yu, G. Preparation, bandgap engineering, and performance control of graphene nanoribbons. Chem. Mater. 2022, 34, 3588–3615. [Google Scholar] [CrossRef]
- Gu, Y.; Qiu, Z.; Müllen, K. Nanographenes and graphene nanoribbons as multitalents of present and future materials science. J. Am. Chem. Soc. 2022, 144, 11499–11524. [Google Scholar] [CrossRef]
- Friedrich, N. et al. Addressing electron spins embedded in metallic graphene nanoribbons. ACS Nano 2022, 16, 14819–14826. [Google Scholar] [CrossRef]
- Jiang, S.; Neuman, T.; Boeglin, A.; Scheurer, F.; Schull, G. Topologically localized excitons in single graphene nanoribbons. Science 2023, 379, 1049–1054. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Pratap, S.; Kumar, V.; Mishra, R.K.; Gwag, J.S.; Chakraborty, B. Electronic, transport, magnetic, and optical properties of graphene nanoribbons and their optical sensing applications: A comprehensive review. Luminescence 2023, 38, 909–953. [Google Scholar] [CrossRef]
- Aragonés, C. et al. Electrostatic catalysis of a Diels-Alder reaction. Nature 2016, 531, 88–91. [Google Scholar] [CrossRef]
- Shaik, S.; Mandal, D.; Ramanan, R. Oriented electric fields as future smart reagents in chemistry. Nat. Chem. 2016, 8, 1091–1098. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L. et al. Electrochemical and electrostatic cleavage of alkoxyamines. J. Am. Chem. Soc. 2018, 140, 766–774. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Danovich, D.; Ramanan, R.; Shaik, S. Oriented-external electric fields create absolute enantioselectivity in Diels-Alder reactions: Importance of the molecular dipole moment. J. Am. Chem. Soc. 2018, 140, 13350–13359. [Google Scholar] [CrossRef]
- Joy, J.; Stuyver, T.; Shaik, S. Oriented external electric fields and ionic additives elicit catalysis and mechanistic crossover in oxidative addition reactions. J. Am. Chem. Soc. 2020, 142, 3836–3850. [Google Scholar] [CrossRef] [PubMed]
- Shaik, S.; Danovich, D.; Joy, J.; Wang, Z.; Stuyver, T. Electric-field mediated chemistry: Uncovering and exploiting the potential of (oriented) electric fields to exert chemical catalysis and reaction control. J. Am. Chem. Soc. 2020, 142, 12551–12562. [Google Scholar] [CrossRef] [PubMed]
- Zhao, F.; Cao, T.; Louie, S.G. Topological phases in graphene nanoribbons tuned by electric fields. Phys. Rev. Lett. 2021, 127, 166401. [Google Scholar] [CrossRef]
- Cunha, L.A.; Lee, J.; Diptarka, H.; McCurdy, C.W.; Head-Gordon, M. Exploring spin symmetry-breaking effects for static field ionization of atoms: Is there an analog to the Coulson-Fischer point in bond dissociation? J. Chem. Phys. 2021, 155, 014309. [Google Scholar] [CrossRef]
- Yu, S.; Vermeeren, P.; Hamlin, T.A.; Bickelhaupt, F.M. How oriented external electric fields modulate reactivity. Chem. Eur. J. 2021, 27, 5683–5693. [Google Scholar] [CrossRef]
- Wu, J.; Long, T.; Wang, H.; Liang, J.-X.; Zhu, C. Oriented external electric fields regurating the reaction mechanism of CH4 oxidation catalyzed by Fe(IV)-Oxo-corrolazine: Insight from density functional calculations. Front. Chem. 2022, 10, 896944. [Google Scholar] [CrossRef]
- Scheele, T.; Neudecker, T. Investigating the accuracy of density functional methods for molecules in electric fields. J. Chem. Phys. 2023, 159, 124111. [Google Scholar] [CrossRef]
- Scheele, T.; Neudecker, T. Using oriented external electric fields to manipulate rupture forces of mechanophores. Phys. Chem. Chem. Phys. 2023, 25, 28070–28077. [Google Scholar] [CrossRef] [PubMed]
- Hachmann, J.; Dorando, J.J.; Aviles, M.; Chan, G.K.L. The radical character of the acenes: A density matrix renormalization group study. J. Chem. Phys. 2007, 127, 134309. [Google Scholar] [CrossRef] [PubMed]
- Chai, J.-D. Density functional theory with fractional orbital occupations. J. Chem. Phys. 2012, 136, 154104. [Google Scholar] [CrossRef]
- Mizukami, W.; Kurashige, Y.; Yanai, T. More π electrons make a difference: Emergence of many radicals on graphene nanoribbons studied by ab initio DMRG theory. J. Chem. Theory and Comput. 2013, 9, 401–407. [Google Scholar] [CrossRef]
- Rivero, P.; Jiménez-Hoyos, C.A.; Scuseria, G.E. Entanglement and polyradical nature of polycyclic aromatic hydrocarbons predicted by projected Hartree-Fock theory. J. Phys. Chem. B 2013, 117, 12750–12758. [Google Scholar] [CrossRef] [PubMed]
- Chai, J.-D. Thermally-assisted-occupation density functional theory with generalized-gradient approximations. J. Chem. Phys. 2014, 140, 18A521. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.-S.; Chai, J.-D. Electronic properties of zigzag graphene nanoribbons studied by TAO-DFT. J. Chem. Theory Comput. 2015, 11, 2003–2011. [Google Scholar] [CrossRef]
- Fosso-Tande, J.; Nguyen, T.-S.; Gidofalvi, G.; DePrince III, A.E. Large-scale variational two-electron reduced-density-matrix-driven complete active space self-consistent field methods. J. Chem. Theory Comput. 2016, 12, 2260–2271. [Google Scholar] [CrossRef]
- Chai, J.-D. Role of exact exchange in thermally-assisted-occupation density functional theory: A proposal of new hybrid schemes. J. Chem. Phys. 2017, 146, 044102. [Google Scholar] [CrossRef]
- Chen, B.-J.; Chai, J.-D. TAO-DFT fictitious temperature made simple. RSC Adv. 2022, 12, 12193–12210. [Google Scholar] [CrossRef]
- Dai, Y.; Sancho-García, J.-C.; Negri, F. Impact of di- and poly-radical characters on the relative energy of the doubly excited and La states of linear acenes and cyclacenes. Chemistry 2023, 5, 616–632. [Google Scholar] [CrossRef]
- Hohenberg, P.; Kohn, W. Inhomogeneous electron gas. Phys. Rev. 1964, 136, B864–B871. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L.J. Self-consistent equations including exchange and correlation effects. Phys. Rev. 1965, 140, A1133–A1138. [Google Scholar] [CrossRef]
- Dirac, P.A.M. Note on exchange phenomena in the Thomas atom. Proc. Cambridge Philos. Soc. 1930, 26, 376–385. [Google Scholar] [CrossRef]
- Perdew, J.P.; Wang, Y. Accurate and simple analytic representation of the electron-gas correlation energy. Phys. Rev. B 1992, 45, 13244–13249. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D. A new mixing of Hartree-Fock and local density-functional theories. J. Chem. Phys. 1993, 98, 1372–1377. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Stephens, P.J.; Devlin, F.J.; Chabalowski, C.F.; Frisch, M.J. Ab initio calculation of vibrational absorption and circular dichroism spectra using density functional force fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- Kümmel, S.; Kronik, L. Orbital-dependent density functionals: Theory and applications. Rev. Mod. Phys. 2008, 80, 3–60. [Google Scholar] [CrossRef]
- Cohen, A.J.; Mori-Sánchez, P.; Yang, W. Insights into current limitations of density functional theory. Science 2008, 321, 792–794. [Google Scholar] [CrossRef] [PubMed]
- Cohen, A.J.; Mori-Sánchez, P.; Yang, W. Challenges for density functional theory. Chem. Rev. 2012, 112, 289–320. [Google Scholar] [CrossRef] [PubMed]
- Teale, A.M. et al. DFT Exchange: Sharing perspectives on the workhorse of quantum chemistry and materials science. Phys. Chem. Chem. Phys. 2022, 24, 28700–28781. [Google Scholar] [CrossRef]
- Andersson, K.; Malmqvist, P.-Å.; Roos, B.O. Second-order perturbation theory with a complete active space self-consistent field reference function. J. Chem. Phys. 1992, 96, 1218. [Google Scholar] [CrossRef]
- Gidofalvi, G.; Mazziotti, D.A. Active-space two-electron reduced-density-matrix method: Complete active-space calculations without diagonalization of the N-electron hamiltonian. J. Chem. Phys. 2008, 129, 134108. [Google Scholar] [CrossRef]
- Gryn’ova, G.; Coote, M.L.; Corminboeuf, C. Theory and practice of uncommon molecular electronic configurations. WIREs Comput. Mol. Sci. 2015, 5, 440–459. [Google Scholar] [CrossRef]
- Goli, V.D.P.; Prodhan, S.; Mazumdar, S.; Ramasesha, S. Correlated electronic properties of some graphene nanoribbons: A DMRG study. Phys. Rev. B 2016, 94, 035139. [Google Scholar] [CrossRef]
- Hagymási, I.; Legeza, Ö. Entanglement, excitations, and correlation effects in narrow zigzag graphene nanoribbons. Phys. Rev. B 2016, 94, 165147. [Google Scholar] [CrossRef]
- Piris, M. Global method for electron correlation. Phys. Rev. Lett. 2017, 119, 063002. [Google Scholar] [CrossRef]
- van Meer, R.; Gritsenko, O.V.; Baerends, E.J. A non-JKL density matrix functional for intergeminal correlation between closed-shell geminals from analysis of natural orbital configuration interaction expansions. J. Chem. Phys. 2018, 148, 104102. [Google Scholar] [CrossRef]
- Wang, Y.-Y.; Chai, J.-D. Spin symmetry in thermally-assisted-occupation density-functional theory. Phys. Rev. A 2024, 109, 062808. [Google Scholar] [CrossRef]
- Xuan, F.; Chai, J.-D.; Su, H. Local density approximation for the short-range exchange free energy functional. ACS Omega 2019, 4, 7675–7683. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Hansen, A.; Brandenburg, J.G.; Bannwarth, C. Dispersion-corrected mean-field electronic structure methods. Chem. Rev. 2016, 116, 5105–5154. [Google Scholar] [CrossRef] [PubMed]
- Seenithurai, S.; Chai, J.-D. Effect of Li adsorption on the electronic and hydrogen storage properties of acenes: A dispersion-corrected TAO-DFT study. Sci. Rep. 2016, 6, 33081. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-Y.; Hui, K.; Chung, J.-H.; Chai, J.-D. Self-consistent determination of the fictitious temperature in thermally-assisted-occupation density functional theory. RSC Adv. 2017, 7, 50496–50507. [Google Scholar] [CrossRef]
- Mermin, N.D. Thermal properties of the inhomogeneous electron gas. Phys. Rev. 1965, 137, A1441–A1443. [Google Scholar] [CrossRef]
- Tsai, H.-Y.; Chai, J.-D. Real-time extension of TAO-DFT. Molecules 2023, 28, 7247. [Google Scholar] [CrossRef] [PubMed]
- Seenithurai, S.; Chai, J.-D. TAO-DFT with the polarizable continuum model. Nanomaterials 2023, 13, 1593. [Google Scholar] [CrossRef]
- Li, S.; Chai, J.-D. TAO-DFT-based ab initio molecular dynamics. Front. Chem. 2020, 8, 589432. [Google Scholar] [CrossRef]
- Yeh, C.-N.; Chai, J.-D. Role of Kekulé and non-Kekulé structures in the radical character of alternant polycyclic aromatic hydrocarbons: A TAO-DFT study. Sci. Rep. 2016, 6, 30562. [Google Scholar] [CrossRef] [PubMed]
- Yeh, C.-N.; Wu, C.; Su, H.; Chai, J.-D. Electronic properties of the coronene series from thermally-assisted-occupation density functional theory. RSC Adv. 2018, 8, 34350–34358. [Google Scholar] [CrossRef] [PubMed]
- Tönshoff, C.; Bettinger, H.F. Pushing the limits of acene chemistry: The recent surge of large acenes. Chem. Eur. J. 2021, 27, 3193–3212. [Google Scholar] [CrossRef] [PubMed]
- Gupta, D.; Omont, A.; Bettinger, H.F. Energetics of formation of cyclacenes from 2,3-didehydroacenes and implications for astrochemistry. Chem. Eur. J. 2021, 27, 4605–4616. [Google Scholar] [CrossRef] [PubMed]
- Nieman, R.; Carvalho, J.R.; Jayee, B.; Hansen, A.; Aquino, A.J.; Kertesz, M.; Lischka, H. Polyradical character assessment using multireference calculations and comparison with density-functional derived fractional occupation number weighted density analysis. Phys. Chem. Chem. Phys. 2023, 25, 27380–27393. [Google Scholar] [CrossRef] [PubMed]
- Somani, A.; Gupta, D.; Bettinger, H.F. Computational studies of dimerization of [n]-cyclacenes. J. Phys. Chem. A 2024, ASAP. [Google Scholar] [CrossRef] [PubMed]
- Hanson-Heine, M.W.D. Static correlation in vibrational frequencies studied using thermally-assisted-occupation density functional theory. Chem. Phys. Lett. 2020, 739, 137012. [Google Scholar] [CrossRef]
- Hanson-Heine, M.W.D. Static electron correlation in anharmonic molecular vibrations: A hybrid TAO-DFT study. J. Phys. Chem. A 2022, 126, 7273–7282. [Google Scholar] [CrossRef]
- Shao, Y. et al. Advances in molecular quantum chemistry contained in the Q-Chem 4 program package. Mol. Phys. 2015, 113, 184–215. [Google Scholar] [CrossRef]
- Su, Y. et al. Thermally controlling the singlet–triplet energy gap of a diradical in the solid state. Chem. Sci. 2016, 7, 6514–6518. [Google Scholar] [CrossRef]
- Yu, L.; Wu, Z.; Xie, G.; Zhong, C.; Zhu, Z.; Cong, H.; Ma, D.; Yang, C. Achieving a balance between small singlet-triplet energy splitting and high fluorescence radiative rate in a quinoxaline-based orange-red thermally activated delayed fluorescence emitter. Chem. Commun. 2016, 52, 11012–11015. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.B.; Michl, J. Singlet fission. Chem. Rev. 2010, 110, 6891–6936. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Liu, Q.; Feng, W.; Sun, Y.; Li, F. Upconversion luminescent materials: Advances and applications. Chem. Rev. 2015, 115, 395–465. [Google Scholar] [CrossRef]
- Romero, N.A.; Nicewicz, D.A. Organic photoredox catalysis. Chem. Rev. 2016, 116, 10075–10166. [Google Scholar] [CrossRef] [PubMed]
- Xia, J. et al. Singlet fission: Progress and prospects in solar cells. Adv. Mater. 2017, 29, 1601652. [Google Scholar] [CrossRef]
- Löwdin, P.-O.; Shull, H. Natural orbitals in the quantum theory of two-electron systems. Phys. Rev. 1956, 101, 1730–1739. [Google Scholar] [CrossRef]
Figure 1.
Structure of 5-acene, containing five linearly fused benzene rings, in an OEEF, where the arrow indicates the OEEF direction.
Figure 1.
Structure of 5-acene, containing five linearly fused benzene rings, in an OEEF, where the arrow indicates the OEEF direction.
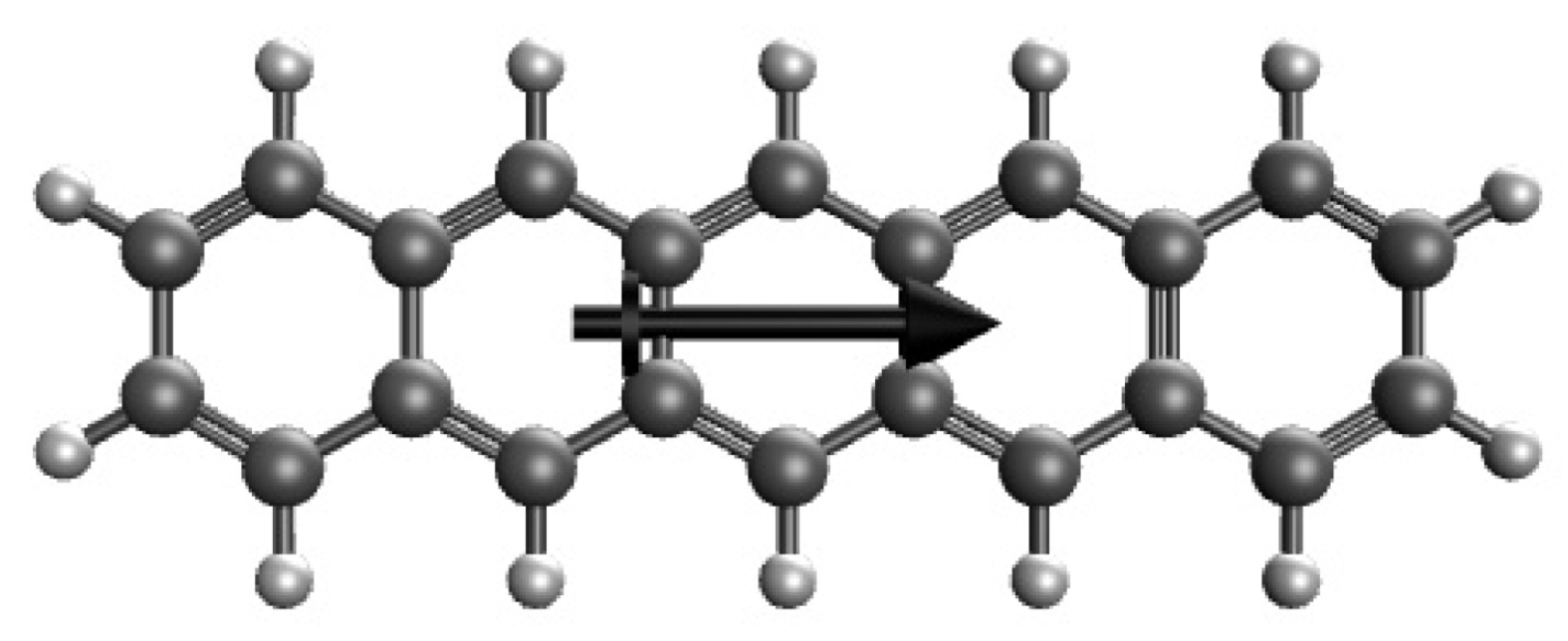
Figure 2.
Singlet-triplet energy gap of n-acene in an OEEF of the electric field strength F = 0.000, 0.001, 0.002, 0.003, 0.004, and 0.005 a.u., calculated using spin-unrestricted TAO-LDA.
Figure 2.
Singlet-triplet energy gap of n-acene in an OEEF of the electric field strength F = 0.000, 0.001, 0.002, 0.003, 0.004, and 0.005 a.u., calculated using spin-unrestricted TAO-LDA.
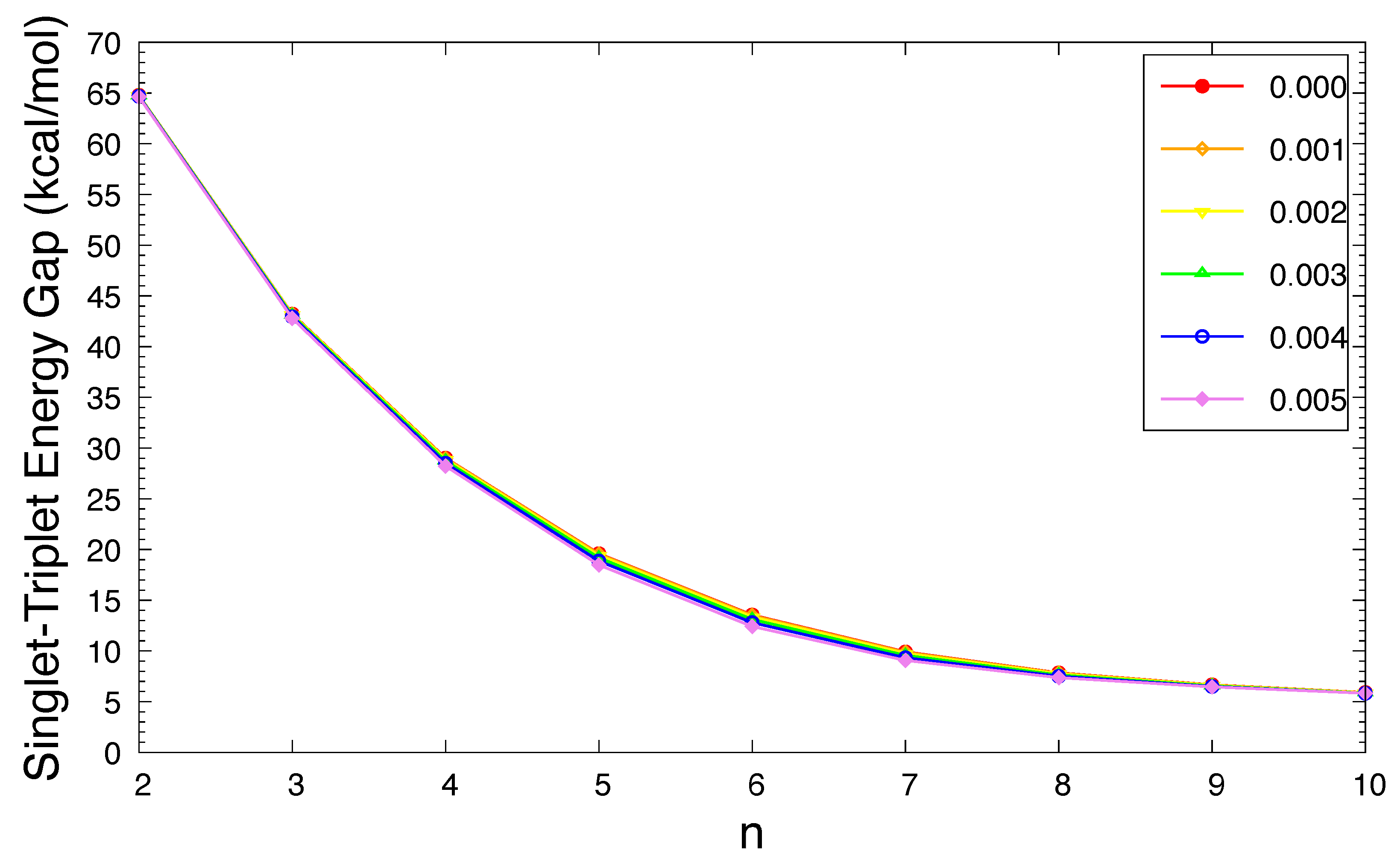
Figure 3.
Vertical ionization potential for the ground state of n-acene in an OEEF of the electric field strength F = 0.000, 0.001, 0.002, 0.003, 0.004, and 0.005 a.u., calculated using spin-unrestricted TAO-LDA.
Figure 3.
Vertical ionization potential for the ground state of n-acene in an OEEF of the electric field strength F = 0.000, 0.001, 0.002, 0.003, 0.004, and 0.005 a.u., calculated using spin-unrestricted TAO-LDA.
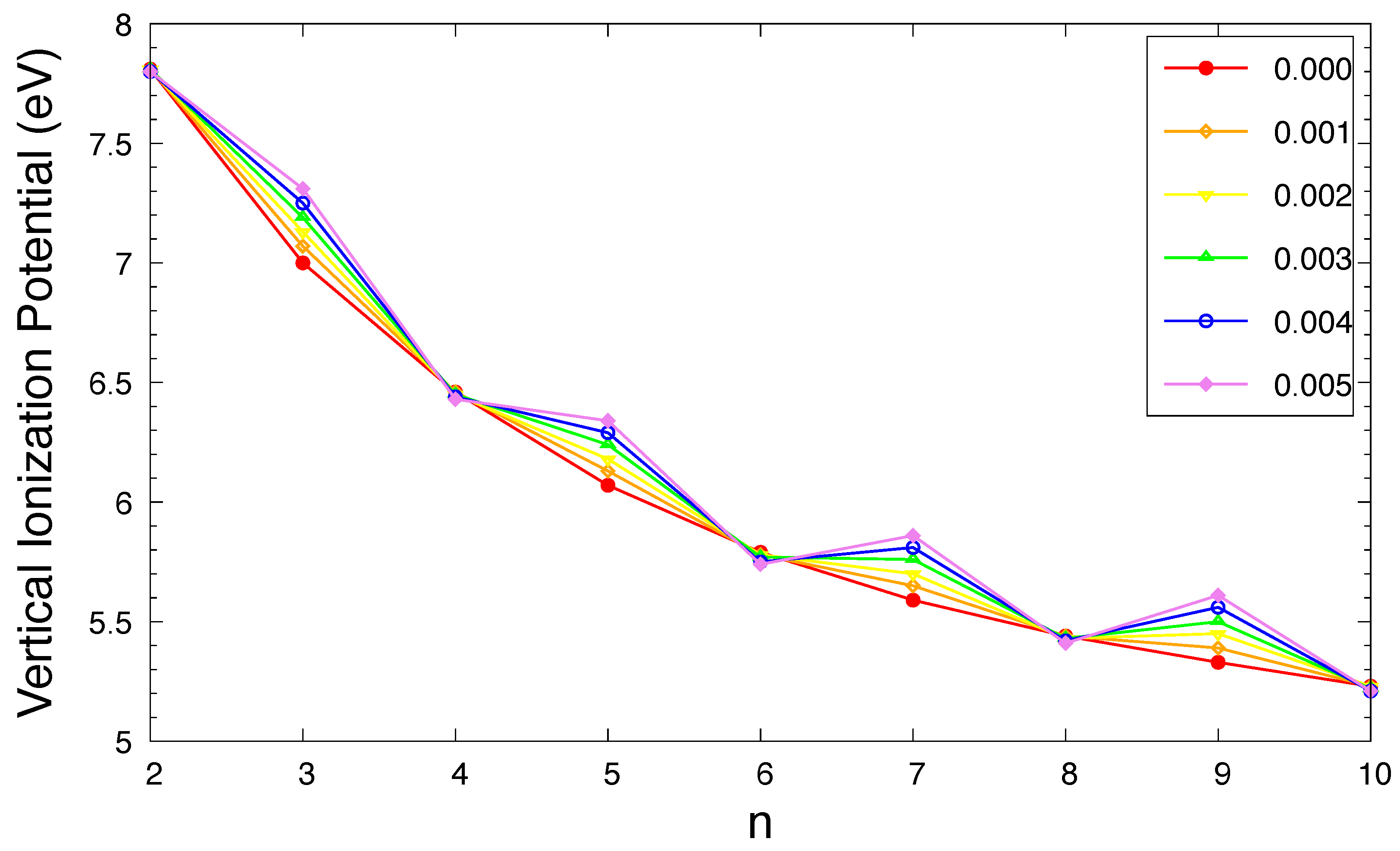
Figure 4.
Vertical electron affinity for the ground state of n-acene in an OEEF of the electric field strength F = 0.000, 0.001, 0.002, 0.003, 0.004, and 0.005 a.u., calculated using spin-unrestricted TAO-LDA.
Figure 4.
Vertical electron affinity for the ground state of n-acene in an OEEF of the electric field strength F = 0.000, 0.001, 0.002, 0.003, 0.004, and 0.005 a.u., calculated using spin-unrestricted TAO-LDA.
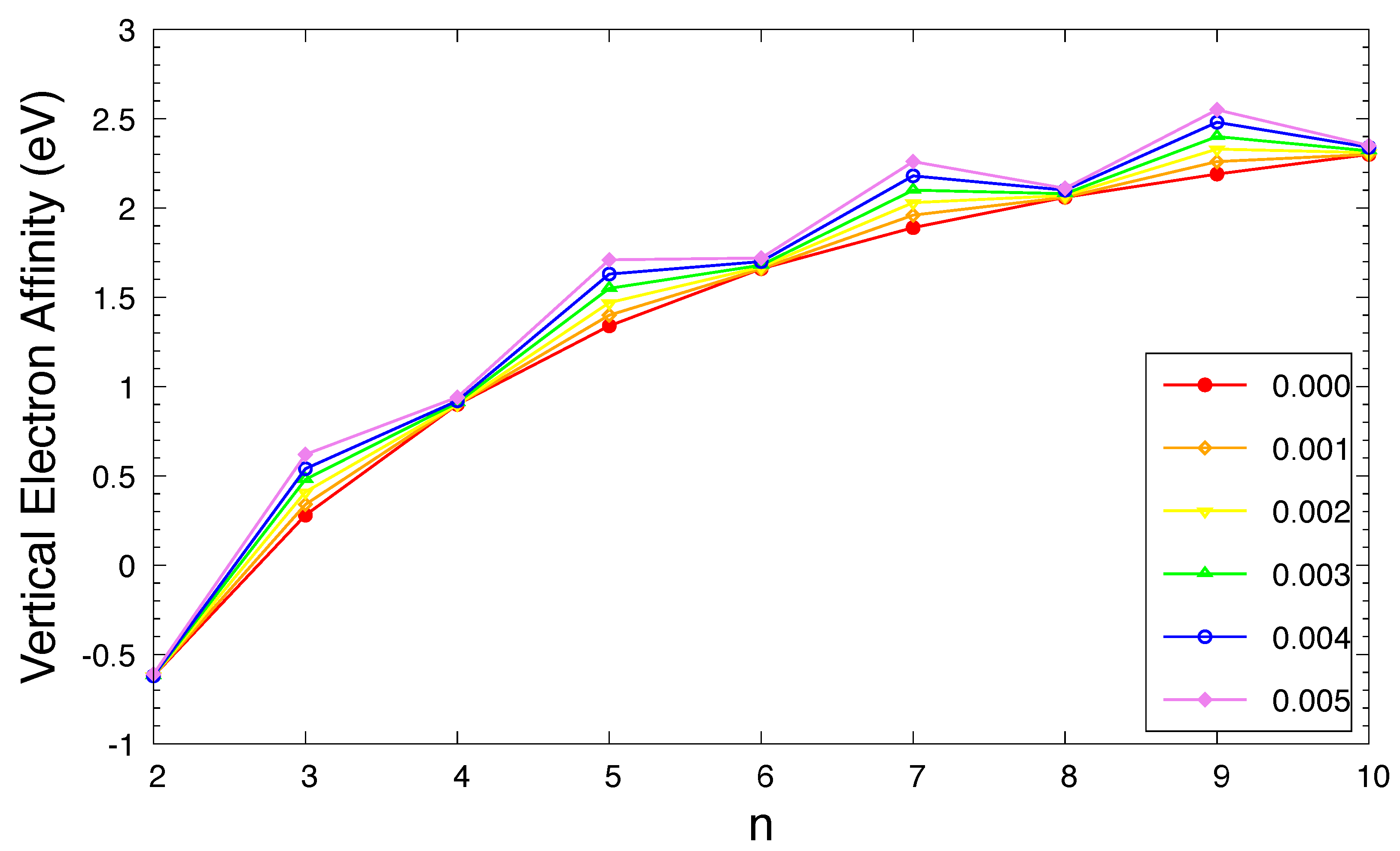
Figure 5.
Fundamental gap for the ground state of n-acene in an OEEF of the electric field strength F = 0.000, 0.001, 0.002, 0.003, 0.004, and 0.005 a.u., calculated using spin-unrestricted TAO-LDA.
Figure 5.
Fundamental gap for the ground state of n-acene in an OEEF of the electric field strength F = 0.000, 0.001, 0.002, 0.003, 0.004, and 0.005 a.u., calculated using spin-unrestricted TAO-LDA.
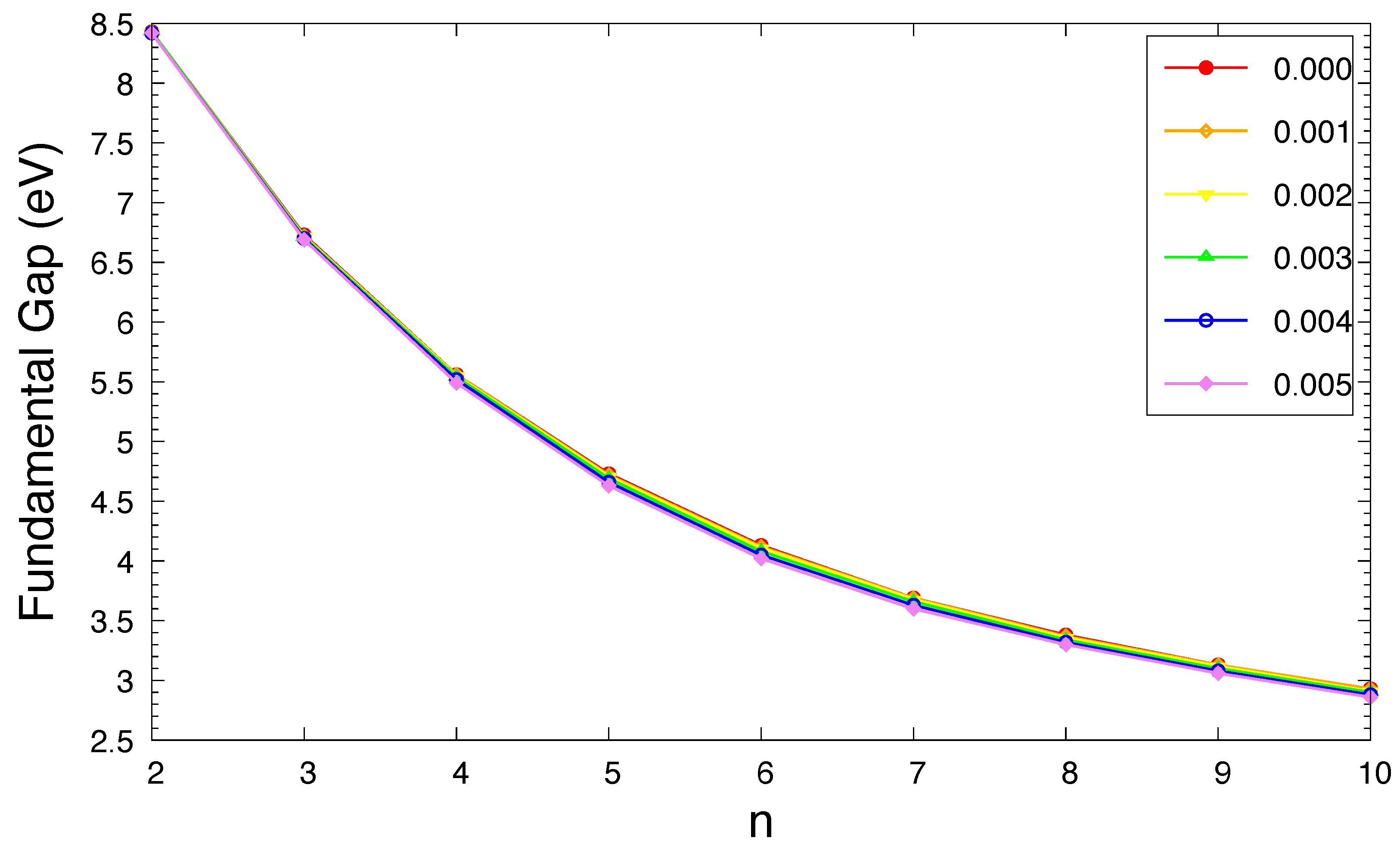
Figure 6.
Symmetrized von Neumann entropy for the ground state of n-acene in an OEEF of the electric field strength F = 0.000, 0.001, 0.002, 0.003, 0.004, and 0.005 a.u., calculated using spin-unrestricted TAO-LDA.
Figure 6.
Symmetrized von Neumann entropy for the ground state of n-acene in an OEEF of the electric field strength F = 0.000, 0.001, 0.002, 0.003, 0.004, and 0.005 a.u., calculated using spin-unrestricted TAO-LDA.
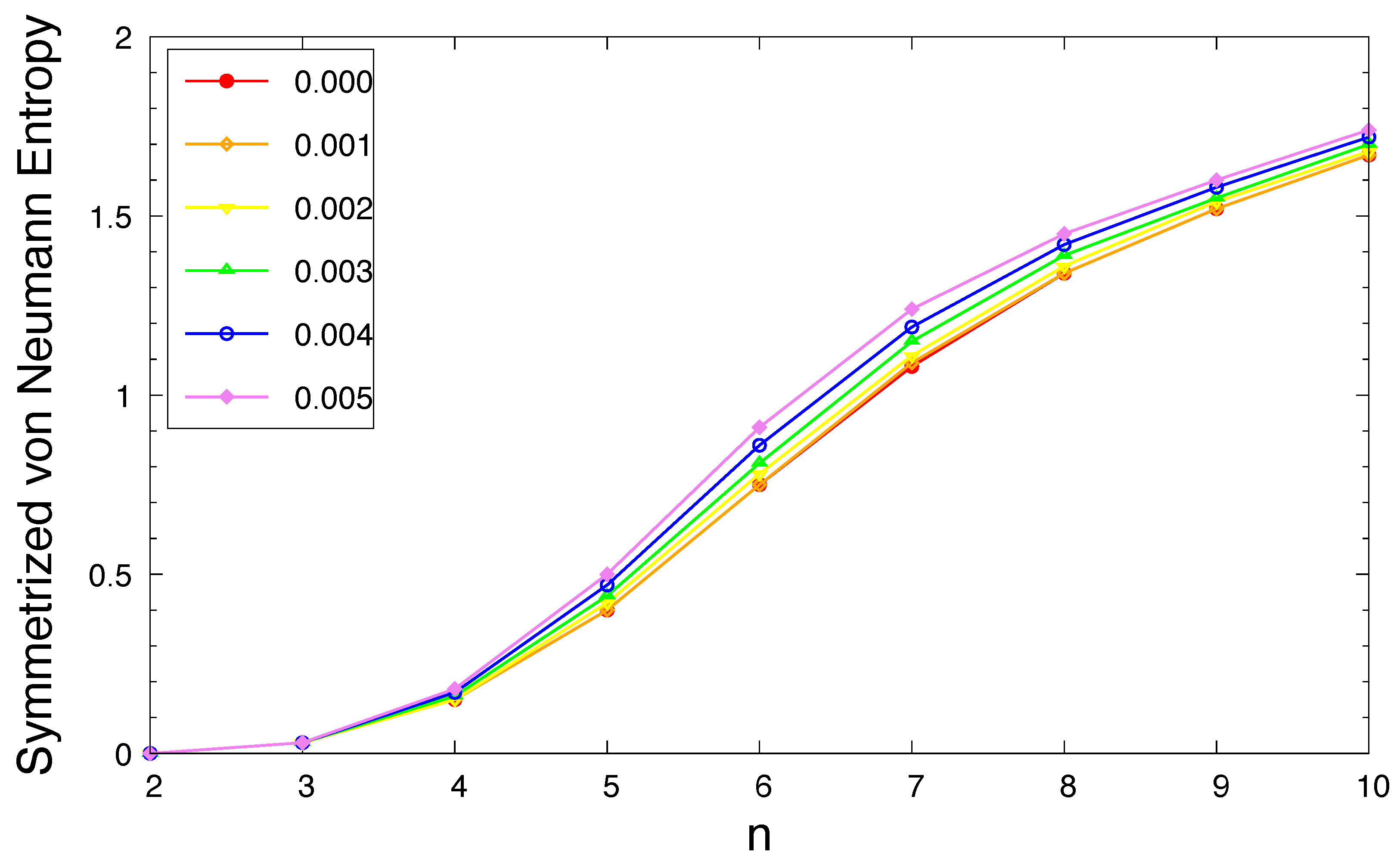
Figure 7.
Occupation numbers of active orbitals for the ground state of n-acene in an OEEF of the electric field strength F = (a) 0.000, (b) 0.001, (c) 0.002, (d) 0.003, (e) 0.004, and (f) 0.005 a.u., calculated using spin-restricted TAO-LDA. Here, the HOMO / LUMO is denoted as the H / L for brevity.
Figure 7.
Occupation numbers of active orbitals for the ground state of n-acene in an OEEF of the electric field strength F = (a) 0.000, (b) 0.001, (c) 0.002, (d) 0.003, (e) 0.004, and (f) 0.005 a.u., calculated using spin-restricted TAO-LDA. Here, the HOMO / LUMO is denoted as the H / L for brevity.
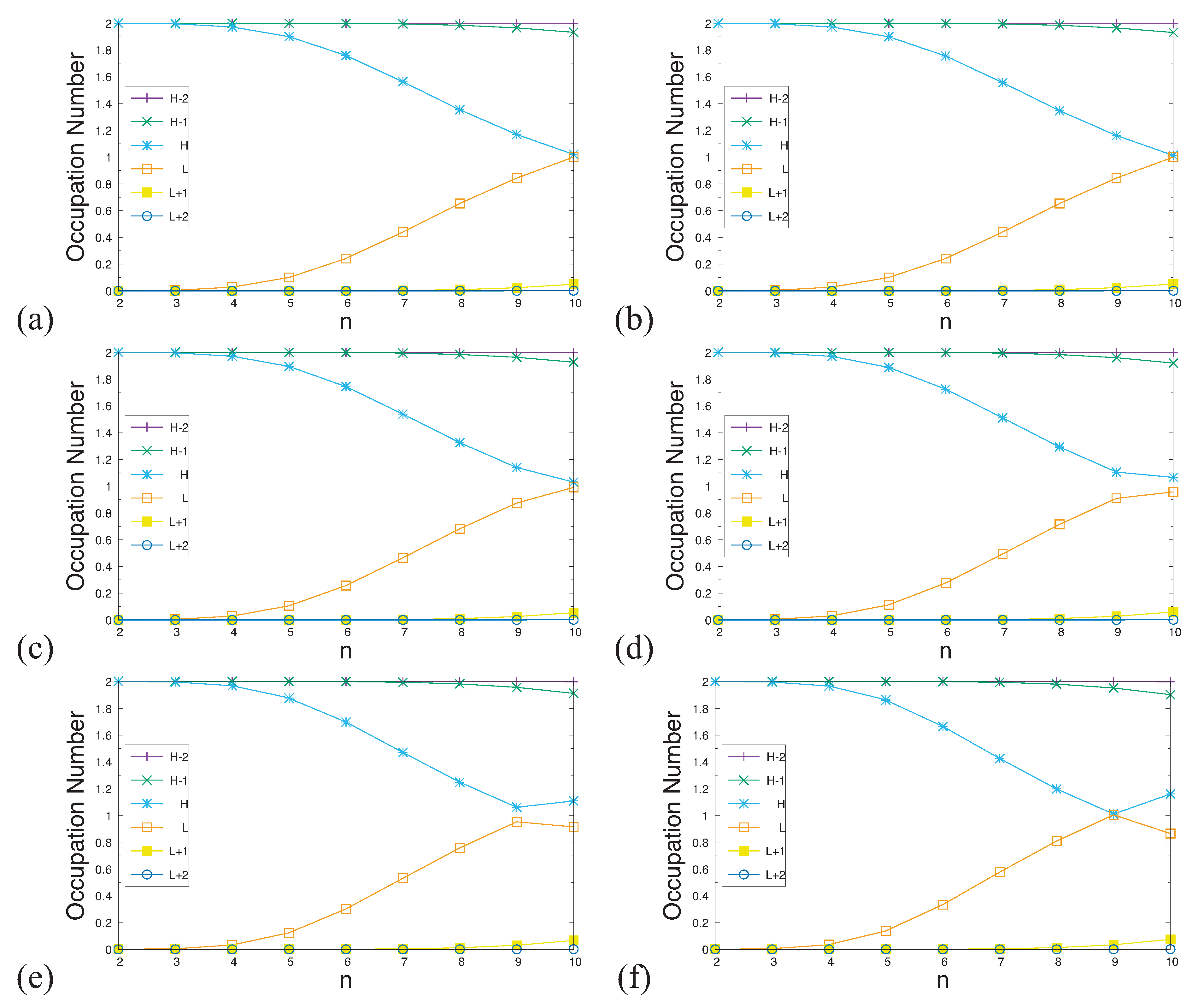
Figure 8.
Real-space representation of the HOMO (left) and LUMO (right) for the ground state of 9-acene in an OEEF of the electric field strength F = (a) 0.000, (b) 0.001, (c) 0.002, (d) 0.003, (e) 0.004, and (f) 0.005 a.u., calculated using spin-restricted TAO-LDA, at an isovalue of 0.02 e/Å3, where the orbital occupation numbers are shown in parentheses.
Figure 8.
Real-space representation of the HOMO (left) and LUMO (right) for the ground state of 9-acene in an OEEF of the electric field strength F = (a) 0.000, (b) 0.001, (c) 0.002, (d) 0.003, (e) 0.004, and (f) 0.005 a.u., calculated using spin-restricted TAO-LDA, at an isovalue of 0.02 e/Å3, where the orbital occupation numbers are shown in parentheses.
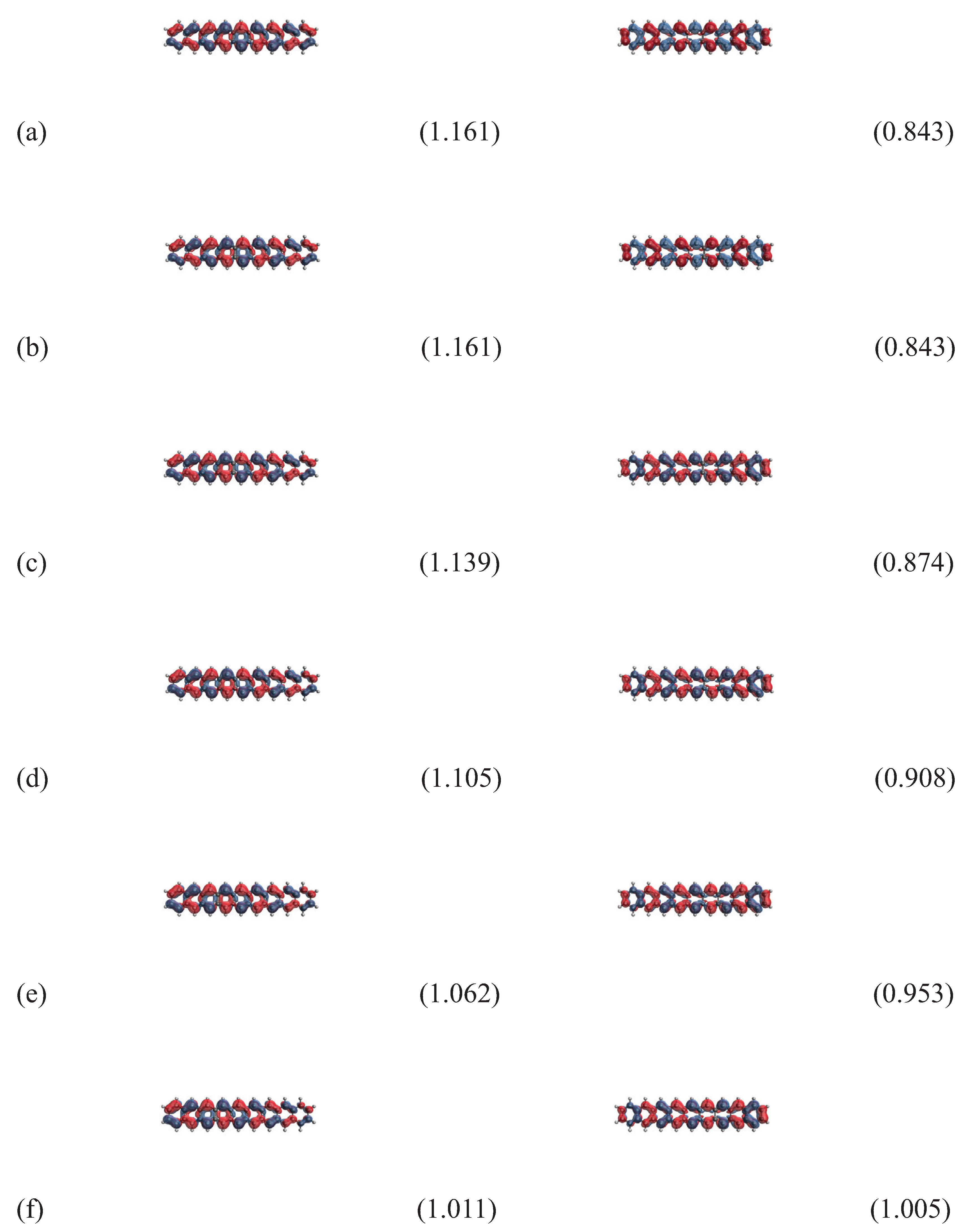
Figure 9.
Real-space representation of the HOMO (left) and LUMO (right) for the ground state of 10-acene in an OEEF of the electric field strength F = (a) 0.000, (b) 0.001, (c) 0.002, (d) 0.003, (e) 0.004, and (f) 0.005 a.u., calculated using spin-restricted TAO-LDA, at an isovalue of 0.02 e/Å3, where the orbital occupation numbers are shown in parentheses.
Figure 9.
Real-space representation of the HOMO (left) and LUMO (right) for the ground state of 10-acene in an OEEF of the electric field strength F = (a) 0.000, (b) 0.001, (c) 0.002, (d) 0.003, (e) 0.004, and (f) 0.005 a.u., calculated using spin-restricted TAO-LDA, at an isovalue of 0.02 e/Å3, where the orbital occupation numbers are shown in parentheses.

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



