
Submitted:
20 August 2024
Posted:
22 August 2024
You are already at the latest version
Abstract
Acinar cell carcinoma (ACC) accounts for about 1% of pancreatic cancers. The molecular and clinical features of ACC are less characterized than pancreatic ductal adenocarcinoma. We retrospectively evaluated the clinical and molecular features of ACC patients who underwent germline and/or somatic molecular testing at The University of Texas MD Anderson Cancer Center from 2008 to 2022. Patient information was extracted from our institutional database with the approval of Institutional Review Board. We identified 16 patients with available molecular testing results. Fourteen patients had metastatic disease, 1 had borderline resectable disease, and 1 had localized resectable disease at diagnosis. Fifteen patients were wild type for KRAS (1 patient had unknown KRAS status). Somatic/germline mutations of DNA damage repair genes (BRCA1/2, PALB2, and ATM) were present in 5 of 12 patients tested for these genes. One patient was found to have RET fusion and responded favorably to selpercatinib for over 42 months. The median overall survival (OS) was 24 months for patients with metastatic disease. One of the additional two cases which had BostonGene tests was found to have NTRK1 fusion. RNA and TME analysis by BostonGene of the two cases reported immune desert features and relatively lower RNA levels of CEACAM5, CD47, CD74 and MMP1 and higher CDH6 compared with PDAC.
Keywords:
KRAS
; immunohistochemistry
; acinar cell carcinoma
; pancreatic
; OS
1. Introduction
Acinar cell carcinoma (ACC) is rare, accounting for about 1-2% of pancreatic cancers, and it has distinct clinical and molecular features [1].Because of low ACC case numbers, treatment guidelines for this cancer are limited. Authors have reported better survival and greater chemosensitivity of ACC compared to pancreatic adenocarcinoma [2]. A retrospective analysis demonstrated longer survival in pancreatic cancer patients harboring actionable molecular alterations who underwent molecularly matched therapies in comparison with pancreatic cancer patients who did not have actionable molecular alterations [3]. Furthermore, the clinical and molecular features of ACC differ from those of pancreatic adenocarcinoma [4]. However, these features of ACC are not well understood. With the development of precision oncology and increased uptake of tumor molecular profiling, including next-generation sequencing (NGS), understanding the mutations associated with ACC is critical for development of targeted therapy and novel treatment strategies.
In this study, we present a case series of 16 patients treated for ACC at The University of Texas MD Anderson Cancer Center (MD Anderson). We evaluated the molecular and clinical characteristics of these patients to improve our knowledge of the clinical and molecular features of ACC and suggest possible indications for treatment. We also included additional two cases who had Boston Gene tests including genomic alterations, RNA expressions and tumor microenvironment (TME) features.
2. Materials and Methods
Sixteen patients with ACC who underwent molecular testing at MD Anderson from 2008 to 2022 were identified retrospectively. Tumor genomic alteration information, including KRAS mutation information, along with demographic, clinical, molecular, and pathological information was extracted from the institutional Palantir Foundry data science platform using natural language processing, with the approval of the MD Anderson Institutional Review Board (IRB). The information was validated via chart review, which included a review of each patient’s age at diagnosis, sex, presenting symptoms at diagnosis, tumor site, pathology and cytology results, epidemiological data, treatment details (surgery, radiation therapy, and/or chemotherapy), and status at last follow-up visit. Overall survival (OS) duration was calculated from the date of initial diagnosis to the date of death or last follow-up visit. The Kaplan-Meier method was used to estimate the median OS and the Gehan-Breslow-Wilcoxon test was used to compare OS.
We also included additional two cases who had Boston Gene tests including genomic alterations, RNA expressions and tumor microenvironment (TME) features under another IRB in collaborative efforts with Boston Gene. The Boston Gene Molecular Functional Portrait™ (MF Portrait™) integrates data obtained using Comprehensive Genomic Profile (CGP) and shows the composition and activity of both the malignant and the TME components. Gene signature profiling of >10,000 cancer patients identified four distinct tumor microenvironment subtypes conserved across 20 different solid cancers: Immune-Enriched, Fibrotic (IE/F); Immune-Enriched, Non-Fibrotic (IE); Fibrotic (F); and Immune Desert (D). The IE subtype is associated with a superior response to immune checkpoint inhibitors (ICI) in cutaneous melanoma, bladder cancer, lung adenocarcinoma, and gastric cancer. The D subtype and IE/F subtype with low tumor mutational burden (TMB) are associated with poor response to ICI in cutaneous melanoma and bladder cancer [5]. Visualization of tumor and microenvironment components in projection to a cohort of patients with a similar diagnosis, as well as the most significant alterations was done based on RNA expression profiling, which may differ slightly from DNA data, and RNA expression levels of selective genes were compared with PDAC reference cohort cases.
3. Results
3.1. Demographic characteristics
In this case series of 16 pancreatic ACC patients who underwent molecular profiling at MD Anderson, 14 patients had pure ACC, 1 patient had ACC mixed with neuroendocrine tumor, and 1 patient had ACC with ductal adenocarcinoma. The demographic features of these patients are shown in Table 1.
3.2. Clinical and Molecular Characteristics of ACC
The primary tumor was in the head of the pancreas in 50% of the patients (N = 8), the body of the pancreas in 31% of the patients (N = 5), and in the tail of the pancreas in 13% of the patients (N = 2). The tumor involved both the head and body of the pancreas in 1 patient. The most common symptom was abdominal pain or discomfort, which occurred in 12 patients; 3 patients presented with obstructive jaundice. At the time of initial diagnosis,14 patients had metastatic disease, 1 had borderline resectable disease, and 1 had localized resectable disease (Table 1). Ten patients had liver metastasis at diagnosis. Five patients had regional lymph node and vascular involvement, making their tumors unresectable at the time of diagnosis.
The immunohistochemical staining profiles for the 16 study patients was summarized in Table 2. ACC demonstrated positivity for markers associated with acinar cell differentiation, such as trypsin (N = 9/9) and chymotrypsin (N = 2/2), epithelial origin, such as cytokeratin 7 (N = 8/9), and neuroendocrine differentiation, such as synaptophysin (N = 5/12), alpha 1-antichymotrypsin(N=2/2), and chromogranin (N = 3/13). Additionally, expression of other markers, such as BCL10 (N = 2/2) and BCL2 (N = 2/2), alpha 1 antitrypsin (N=1/2) was observed.
3.3. Genetic alterations in ACC
Fifteen patients (94%) were wild type (WT) for KRAS, with 1 patient having an unknown KRAS status. Somatic and/or germline mutations of DNA damage repair genes (BRCA1/2, PALB2, and ATM) were present in 5 of 12 patients (42%) tested for at least some of these genes. The most frequent genomic alteration was the mutation of NF1, followed by CTNNB1 and SMAD4. Of note, one of the patients had SAT-B1-RET fusion. Due to the rare nature of pancreatic ACC and the extended period of study, the available genetic alterations of 14 patients is shown as an Oncoplot in Figure 1.
3.4. Treatment
The details of treatment received by our patients are shown in Table 3. Six patients underwent first-line treatment with 5-fluorouracil, irinotecan, and oxaliplatin (FOLFIRINOX). Three of these patients had partial responses, 2 had stable disease, and 1 had progressive disease. Also, 5 patients received treatment with 5-fluorouracil, and oxaliplatin (FOLFOX), with 1 patient having a response, 2 having stable disease, and 2 having progressive disease. Furthermore, 5 patients underwent treatment with a gemcitabine-based regimens. Only 1 patient had a response after receiving gemcitabine/nab-paclitaxel combined with cisplatin. The other 4 patients who received gemcitabine-based regimens did not respond.
Fourteen patients had metastatic disease, 1 had borderline resectable disease, and 1 had localized resectable disease at initial diagnosis. The 1 patient who had resectable disease underwent the Whipple procedure without neoadjuvant or adjuvant chemotherapy and 2 years later presented with liver metastasis, which was treated with extended partial hepatectomy. The patient then received gemcitabine and capecitabine in the adjuvant setting for 3 months, which was discontinued because of toxicity. The patient underwent active surveillance for 5 years after liver resection and had a recurrence, with a surveillance scan showing left hilar lymph node enlargement and biopsy confirmed pancreatic ACC. The patient received three cycles of FOLFOX followed by two cycles of gemcitabine and nab-paclitaxel (Abraxane). Afterwards the patient’s condition worsened, with progression of disease, peritoneal carcinomatosis, and ultimately death. This patient’s OS was 92 months.
Also of note is that one of our patients was positive for SAT-B1-RET fusion. Before the fusion was identified, this patient underwent treatment with FOLFIRINOX. Since the fusion has been identified, she has been receiving treatment with selpercatinib, which was approved by the U.S. Food and Drug Administration for treatment of any cancer with RET fusion/gene rearrangement [6]. At the time of writing, patient’s disease is still responding favorably to selpercatinib, but the patient did experience the development of chylous ascites, which occurs in about 7% of patients taking selpercatinib [7].
At the time of ACC diagnosis,12 patients had metastasis to the liver, the most common site of metastasis (N=12/14). Two patients had distant metastasis to the lungs, pleura, and supraclavicular lymph nodes. Only 1 patient underwent liver-directed therapy, which was a partial hepatectomy. This was the same patient who underwent the Whipple procedure and presented with liver metastasis 2 years later. As mentioned above, patient went on active surveillance for 5 years and eventually had progression of disease. Fifteen patients were deemed to have unresectable ACC at the time of diagnosis. Another patient, who received FOLFIRINOX in the neoadjuvant setting, later underwent surgical resection of the primary tumor. His ACC progressed during FOLFIRINOX administration and was treated with gemcitabine and abraxane, but he died because of disease progression. The median OS was 24 months for the patients with metastatic disease in our study. The median OS in the gemcitabine- based treatment group was 20.5 months, and that in the FOLFOX based group was 26 months. The OS between the above 2 treatment groups was compared with Gehan-Breslow-Wilcoxon test. The obtained p-value was 0.3346 (data not show).
We also included additional two cases who had Boston Gene tests including genomic alterations, RNA expressions and tumor microenvironment (TME) analysis. The features were summarized in Figure 2. The two cases included one case with mixed ACC and adenocarcinoma, which was found to have NTRK1 fusion (case 17#, Figure 2A) and another case with pure ACC on histology (#18). Both cases showed immune desert features (Figure 2A). Cell compositions and RNA signatures showed few T cells, T cells trafficking, and B cells were observed in the tumors (Figure 2B,C). CEACAM5 RNA expressions were low in both cases compared with PDAC and CDH6 RNA expression levels were higher than PDAC (Figure 2D). Both CD 47, MMP1 and CD74 RNA levels showed lower trend than the reference PDAC data (Figure 2D). The FAP expression showed opposite trend in the two cases: with once sample lower than PDAC (18#, pure ACC) and the other one higher than PDAC references (17#, mixed ACC-adenocarcinoma, Figure 2D).
4. Discussion
In this case series, we summarized the clinical and molecular features of 16 ACC patients. We found that all the patients who had tested for KRAS mutation were negative (15/15) and 5 of 12 (42%) of the patients who underwent DNA damage repair gene testing had germline and/or somatic mutations in DNA damage repair genes (BRCA1/2, PALB2, and ATM). One patient had RET fusion and has been on a RET inhibitor for more than 42 months. One of the additional two cases who had Boston Gene test also identified NTRK1 fusion in one patient. Our study suggests the importance of molecular testing for ACC to identify actionable genomic alterations.
ACC is typically seen in individuals above the age of 60, and predominantly occurs in male patients, with a male-to-female patient ratio of 3.6 [1,8]. Our population demographics were similar, and most of the patients were male (81%, Table 1). Other authors have described lipase hypersecretion syndrome, a type of paraneoplastic syndrome that makes up the Schmid triad with multifocal fat necrosis and polyarthralgia, in about 10-15% of patients with pancreatic ACC [1,9]. Most of our patients presented with abdominal pain (N=12, 75%) or obstructive jaundice (N=3, 19%) and was an incidental finding in the remaining one patient. We did not observe the paraneoplastic syndrome in our limited population. Consistent with other report, liver metastasis is the most common metastatic site in our patients (N=10/14) [10]. The role of liver directed therapy in ACC is uncertain. No significant survival was found with tumor debulking surgery (N=7) in patients with metastatic disease [11]. However, long-term survival after aggressive surgery of primary tumor and liver metastases resection in pancreatic ACC was reported [12]. One of our patients presented a significant OS of 92 months. This patient underwent the Whipple procedure and removed metastatic liver lesions. Even if no conclusions can be drawn from a single case, this patient’s cancer treatment history potentially points to improved progression-free survival after surgery in selected ACC patients who undergo resection of metastatic lesions. The selection of such patients who could benefit from aggressive surgery regarding clinical, biological, and molecular features remain undefined.
Because of the rarity of ACC and its morphological resemblance to normal pancreatic acini, diagnosis can be challenging. ACC also shares histological features with neuroendocrine carcinoma, pancreatoblastoma, and mixed tumors [13]. Mixed acinar-endocrine carcinomas are histologically similar to the pure ACCs but have more than 25% endocrine cells [14]. Immunohistochemical staining is often required to confirm the diagnosis of ACC, as it helps to differentiate this tumor from other pancreatic neoplasms with similar morphological features [15]. Trypsin, chymotrypsin, lipase, amylase, and carboxyl ester lipase are some of the exocrine pancreatic enzymes used to differentiate ACC from other pancreatic tumors [16]. These enzymes are expressed at varying degrees in pancreatic ACC, so they may not be expressed in all these tumors. Antibodies against trypsin and BCL10, which identify carboxyl ester lipase, have been most sensitive in immunohistochemical staining of ACCs [17]. ACC was reported to be strongly positive for trypsin and chymotrypsin and negative or focally positive for synaptophysin and chromogranin [14]. In our study, all patients tested were positive for trypsin (N = 9/9) and chymotrypsin (N = 2/2). 5/12 patients were positive for synaptophysin and 3/13 were positive for chromogranin. One patient had mixed ACC with NET. 2 patients were tested positive for BCL-10 (2/2) and BCL-2 (2/2).
Identification of the genetic alterations in ACC is key to further exploration of targetable mutations for treatment. KRAS is mutated in the majority of pancreatic adenocarcinoma cases, and researchers recently showed that targeting KRAS in treatment of pancreatic cancer produced promising results [18,19]. None of our 16 patients tested positive for KRAS mutation (15 tested negative, and 1 was not tested for it). This finding is consistent with earlier reports of a lower KRAS mutation rate for ACC (13.6%) than for pancreatic adenocarcinoma (85.1%) [4,20,21,22,23]. As described by Kim and Knepper [24], KRAS WT tumors are well known to have higher actionable mutation and gene fusion rates than KRAS mutated tumors. These two authors reported high gene fusion rates in patients with KRAS WT pancreatic ductal adenocarcinoma [24]. Their analysis of tumor tissue from almost 2,500 patients with pancreatic cancer revealed that those lacking mutations of the KRAS gene (KRAS WT) frequently harbored mutations of genes associated with various critical cellular functions. These include DNA damage repair genes such as ATM, BAP1, BRCA2, FANCE, PALB2, and RAD50. In another study, Singh et al. [25] described the oncogenic drivers for KRAS WT pancreatic cancer. Of the 795 samples of pancreatic cancer, mostly PDAC, they analyzed, including samples from five cases of ACC, 9.2% were WT for KRAS. The mitogen-activated protein kinase pathway was a key oncogenic driver for KRAS WT pancreatic cancers. Also, the researchers identified BRAF mutations and receptor tyrosine kinase fusions to be possible targetable mutations in this population.
Of note, the highest mutation percentage we identified in our patient population was that for NF1 and RB1 (28%, Figure 1). Ramakrishnan et al. showed that for pancreatic ductal adenocarcinoma patients who were tested wild type for KRAS, inactivation of the NF1 gene played a vital role in oncogenesis by triggering acinar-to-ductal metaplasia and pancreatic cancer development in situ [26]. The role of the NF1 gene in pancreatic ACC needs to be explored further. Also, La Rosa et al. [27] described the role of TP53 mutation in pancreatic cancer. Their study revealed TP53 positivity in 13% of primary ACCs and 31% of metastases. They concluded that TP53 positivity may be associated with tumor progression and shortened survival. In our study, only 1 patient presented TP53 positivity. It should be noted that not all patients were tested for all possible gene mutations by the limited gene panels, suggesting that the number presented may be an underestimation.
In our patients, somatic and/or germline mutations of DNA damage repair genes (BRCA1/2, PALB2, and ATM) were present in 5 of 12 patients (42%) tested for at least some of these genes (Figure 1). High germline and somatic DNA damage repair gene mutations have been well documented in ACC with mutation rates up to 45% by whole exome sequencing [2,4,22,23,28,29]. Pure pancreatic ACCs had higher prevalence of germline BRCA1, BRCA2, and PALB2 pathogenic variants (42%, N=13/31) than ACCs with mixed with ductal or neuroendocrine histology (11%, N=2/18) based on a recent report with mostly BRCA2 (35%, N=11/31) [29]. Others also reported high rates of homologous recombination–related gene mutations in ACC cases compared to pancreatic ductal adenocarcinoma (PDAC), including mutations of BRCA2 (13.6%), BRCA1 (2.3%), and ATM (11.4%); they also reported that 25% had mutation of at least one of five genes (ATM, ATR, BRCA1, BRCA2, and PALB2) [4]. Furthermore, these authors reported a higher response rate for FOLFIRINOX (53.8%) than for gemcitabine and nab-paclitaxel (23.5%) in ACC patients according to a search of the Japanese Nationwide Comprehensive Genomic Profiling database [4]. A previous retrospective analysis demonstrated higher response rates for platinum-containing (40%) and irinotecan-containing (29%) regimens than for gemcitabine-containing (7%) regimens [30]. It also demonstrated a higher response rate for monotherapy with tegafur/gimeracil/oteracil (17%) than for monotherapy with gemcitabine (3%) [30]. Other researchers also observed higher response rates for treatment with platinum-based regimens in ACC patients with homologous recombination–related gene mutations in several studies [31]. As seen in the comparative analysis of treatment and outcome (Table 3), the improved response of our patients to 5-FOLFOX–based regimens (median OS 26 months) than to gemcitabine-based regimens (median OS 20.5 months) was not statistically significant (p=0.3346). This might be due to our relatively small sample size. A larger study by Sakakida et al. of patients with rare subtypes of pancreatic cancer, including 44 patients with ACC, demonstrated a higher overall response rate for FOLFORINOX than for gemcitabine-based treatment (61.5% vs. 23.5%; p = 0.06) and a significantly longer median time to treatment failure for the former than for the latter (42.3 weeks vs. 21.0 weeks; p = 0.004) [4]. Only one patient had a response after receiving gemcitabine/nab-paclitaxel combined with cisplatin and all the other four patients who received gemcitabine-based regimens did not respond. The results implied the preference of FOLFIRINOX/platinum chemotherapy in ACC.
Among our patients, one had RET fusion (specifically, SAT-B1-RET fusion). This patient is undergoing treatment with selpercatinib for more than 42 months. Targetable fusions like RET fusion may open more avenues for treatment in the ACC patient population [7,32,33]. Gene rearrangements such as BRAF and CRAF fusion were reported in 23% of patients in a 44 patients case series including 16 pure ACC, 14 mixed acinar/neuroendocrine, and other histology [28]. The study revealed the diversity of the BRAF breakpoints and fusion partners. Others also reported up to 30% fusion genes affecting BRAF, CRAF, RET, and NTRK1/2/3 in ACC [29]. Interesting, the RAF gene fusions were mutually exclusive with the inactivation of DNA repair genes (45%) and the BRAF oncogenic alterations were exclusively found in non-DNA damage repair gene mutated pure ACCs [28,29]. The studies suggested the importance of MAPK pathways and DNA damage repair genes. Authors have reported chromosome 11p loss to be the most frequent genomic alteration in ACC patients (50% (6/12)), with APC/β-catenin pathway gene mutation occurring frequently (23.5% (4/17)) [34]. Also, investigators found CTNNB1 mutations in 13.6% of ACC cases [4] and mismatch repair deficiency in 14% of ACC cases according to immunohistochemistry [35]. We did not find chromosome 11p loss nor CTNNB1 mutation, likely due to limited sample size.
In the additional two cases who underwent a comprehensive molecular profiling using Boston Gene test including DNA and RNA sequencing and TME analysis, one of the patients was found to have NTRK1 fusion (Figure 2A). The results described above demonstrate the importance of molecular profiling and gene fusion testing of ACC patients to create more personalized treatment plans. National Comprehensive Cancer Network guidelines currently recommend tumor genomic profiling (e.g., NGS) to guide precision-based and targeted approaches to cancer treatment. The U.S. Food and Drug Administration has approved DNA-based assays for mutation analysis. These assays are effective in evaluating mutation landscapes but accurately identifying gene fusions and exon-skipping events, especially novel ones, which remains a challenge [36]. Whereas NGS reliably detects single-nucleotide variants and small insertions/deletions, it is less reliable for detecting larger structural variants such as chromosomal rearrangements, including fusions and copy-number variations. Fusion breakpoints occurring within introns or repetitive regions in DNA negatively impact assay performance, because of size limitations of hybridization-capture probes [36]. NGS often requires relatively sizeable quantities of high-quality DNA or RNA, which can be challenging to obtain, especially from archival or degraded samples for RNA based assays. RNA-based assays are more effective in identifying gene fusions and exon skipping. This poses a unique challenge, however, as RNA is unstable, has varying expression levels, and lacks a double-stranded context [37]. The other layer of complexities includes the diversity of breakpoints of partners of gene fusion which the limited gene panels used in clinical testing may not be able to capture the full spectrum of tumor somatic gene rearrangements without whole exome sequencing or whole genome sequencing. Advancement of clinical testing platforms is warranted. Being able to identify targetable fusions in molecular testing is of the utmost importance, as it provides the opportunity for individualized treatment approaches for ACC and other KRAS wildtype tumors based on molecular profiles. Hence, we recommend both DNA and RNA based fusion panel tests to increase the chance of identifying targetable genomic alterations, especially gene fusions, in ACC. This approach may lead to more effective therapies tailored to the specific genetic alterations driving ACC.
The features of the two cases which had genomic analysis, RNA expressions and tumor microenvironment (TME) analysis were summarized in Figure 2. One case was mixed ACC and adenocarcinoma (NTRK1 fusion positive, #17) and the other case was pure ACC on histology (#18, Figure 2A). Both tumor tissues showed immune desert features with very few immune cells such as T cells and B cells (Figure 2A, 2B and 2C). Both CD47, MMP1 and CD74 RNA levels showed lower trends than the reference PDAC data (Figure 2D). PDAC is well known for immune suppressive TME with overexpression of CD74, CD47, and stromal MMPs signals [38,39,40]. Effective therapeutics to overcome the immune resistance in PDAC remains challenging [41,42]. Profiling patients’ samples using comprehensive platforms including TME analysis in clinical practice and correlative studies under clinical trials could be critical to understand the resistance mechanisms [43]. The CEACAM5 gene encodes the tumor marker carcinoembryonic antigen (CEA) and CEACAM5 is commonly overexpressed in PDAC and associated with epithelial-mesenchymal transition and poor prognosis [44]. Targeting CEACAM5 is under investigation in gastrointestinal cancer, and it is role in pancreatic cancer remains unclear [45]. CEACAM5 RNA expressions were lower in both cases compared with PDAC (Figure 2D). It was reported previously that CEA is not a sensitive tumor marker for ACC and only 15% of cases had elevated CEA levels [46]. CDH6 RNA expression levels were higher than PDAC (Figure 2D). CDH6 was expressed in cholangiocarcinoma, gastric cancer, renal cell carcinoma, ovarian cancer, and other cancers but less studied in pancreatic cancer including ACC [47,48,49]. The FAP expression showed opposite trend in the two cases: with one sample lower than PDAC (#18, pure ACC) and the other one higher than PDAC references (#17, mixed ACC-adenocarcinoma, Figure 2D). FAP is highly expressed in cancer-associated fibroblasts (CAFs) and critical in PDAC [50]. Less is known about the roles of the gene expression in pancreatic ACC. The sample sizes here are small and more studies at the RNA level and TME analysis in ACC would be helpful to understand the molecular and immune features of ACC.
Limitations of the present study include that it is a single-institution case series with potential population bias and limited numbers of patients. The molecular testing panels, which evolved over the years, included expanded gene panels over the years in different testing platforms and variations in gene fusion panels. Thus, different genes were tested among our patients. For example, KRAS mutation testing was not routinely performed in one case diagnosed in 2009, which was early in the era of mutation testing for pancreatic cancer and not all patients were tested by the DNA and/or RNA based gene fusion panel. Only two patients had RNA sequencing and TME analysis using the BostonGene testing platform.
5. Conclusions
Herein we describe the molecular and clinical course of pancreatic ACC in 16 patients. No patients had KRAS mutations, HRR gene mutation rates were high (5/12, 42%), and one patient had SAT-B1-RET fusion treated with RET inhibitor had OS more than 42 months. We also observed a tendency towards better OS with FOLFIRINOX than with other treatment regimens (median OS duration, 26 months versus 20.5, p=0.3346). In summary, our study shows the clinical and molecular features of ACC and suggests the use of molecular profiling and gene fusion panels in the treatment of ACC.
Author Contributions
Conceptualization and design, D.Z.; methodology, A.B.P., M.Y and D.Z.; software (Microsoft excel, GraphPad prism, Foundry), A.B.P., M.Y. A. K., K.K., A.T., K. K., and D.Z.; data analysis and validation, A.B.P., A.Y. M. Y, A. K., K.K., A.T., K. K., and D.Z.; Resources, data analysis and interpretation, K.A.-M., B.G.S., J.W., M.L., R.A.W., S.P., M.H., A.M., H.W., M.H.G.K., L.R.P., J.P.S. and S.K.; writing—original draft preparation, A.B.P., L.F.C and D.Z.; writing—review and editing, A.B.P., L.F.C, A., C., A. K., K.K., A.T., K. K., and D.Z.; data visualization, A.B.P. and D.Z.; supervision, D.Z.. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki and approved by the Institutional Review Board MD Anderson Cancer Center, protocol number 2023-0091.
Informed Consent Statement
Patient consent was waived as per IRB guidelines.
Data Availability Statement
Individual patient level data is not publicly available to maintain compliance with HIPPA regulations and IRB protocol. Anonymized data are available for non-commercial use from corresponding author upon request pending data usage agreement and/or IRB-approved collaboration.
Acknowledgments
Authors would like to thank Donald Norwood and David Farris for providing expertise with scientific editing.
Conflicts of Interest
Brandon Smaglo: Consulting for Ipsen. Shubham Pant: Advisory for Zymeworks, Ipsen, Novartis, Janssen, Boehringer Ingelheim, and AskGene Pharma; and he receives research funding from Mirati Therapeutics, Lilly, Xencor, Novartis, Rgenix, Bristol-Myers Squibb, Astellas Pharma, Framewave, 4D Pharma, Boehringer Ingelheim, NGM Biopharmaceuticals, Janssen, Arcus Biosciences, Elicio Therapeutics, Bionte, Ipsen, Zymeworks, Pfizer, ImmunoMET, Imuneering, and Amal Therapeutics. Anirban Maitra: Consultant for Tezcat Biosciences, is listed as an inventor on a patent licensed to Thrive Earlier Detection (an Exact Sciences Company) relevant to early detection of pancreatic cancer. Andrey Kravets: Stock option of BostonGene. John Paul Shen: Grant/Research support/ Collaboration: Celsius Therapeutics, BostonGene, Caris Life Sciences, Natera, Xilis, Palantir, Genentech. Consulting/Stock ownership: Engine Biosciences, NaDeNo Nanoscience. Dan Zhao: Research support/Clinical trials: hMirati/BMS, CARsgen, TriSalus, and Affini-T. Consulting: Ipsen. The other authors declare no conflicts of interest.
References
- Calimano-Ramirez, L.F.; Daoud, T.; Gopireddy, D.R.; Morani, A.C.; Waters, R.; Gumus, K.; Klekers, A.R.; Bhosale, P.R.; Virarkar, M.K. Pancreatic acinar cell carcinoma: A comprehensive review. World J. Gastroenterol. 2022, 28, 5827–5844. [Google Scholar] [CrossRef]
- Lowery, M.A.; Klimstra, D.S.; Shia, J.; Yu, K.H.; Allen, P.J.; Brennan, M.F.; O’Reilly, E.M. Acinar Cell Carcinoma of the Pancreas: New Genetic and Treatment Insights into a Rare Malignancy. Oncol. 2011, 16, 1714–1720. [Google Scholar] [CrossRef]
- Pishvaian, M.J.; Blais, E.M.; Brody, J.R.; Lyons, E.; DeArbeloa, P.; Hendifar, A.; Mikhail, S.; Chung, V.; Sahai, V.; Sohal, D.P.S.; et al. Overall survival in patients with pancreatic cancer receiving matched therapies following molecular profiling: a retrospective analysis of the Know Your Tumor registry trial. Lancet Oncol. 2020, 21, 508–518. [Google Scholar] [CrossRef] [PubMed]
- Sakakida, T.; Ishikawa, T.; Doi, T.; Morita, R.; Kataoka, S.; Miyake, H.; Yamaguchi, K.; Moriguchi, M.; Sogame, Y.; Yasuda, H.; et al. Genomic landscape and clinical features of rare subtypes of pancreatic cancer: analysis with the national database of Japan. J. Gastroenterol. 2023, 58, 575–585. [Google Scholar] [CrossRef]
- Bagaev, A.; Kotlov, N.; Nomie, K.; Svekolkin, V.; Gafurov, A.; Isaeva, O.; Osokin, N.; Kozlov, I.; Frenkel, F.; Gancharova, O.; et al. Conserved pan-cancer microenvironment subtypes predict response to immunotherapy. Cancer Cell 2021, 39, 845–865. [Google Scholar] [CrossRef] [PubMed]
- Subbiah V, Wolf J, Konda B, et al. Tumour-agnostic efficacy and safety of selpercatinib in patients with RET fusion-positive solid tumours other than lung or thyroid tumours (LIBRETTO-001): a phase 1/2, open-label, basket trial. The Lancet Oncology. 2022;23(10):1261-1273.
- Fricke, J.; Wang, J.; Gallego, N.; Mambetsariev, I.; Kim, P.; Babikian, R.; Chen, B.T.; Afkhami, M.; Subbiah, V.; Salgia, R. Selpercatinib and Pralsetinib Induced Chylous Ascites in RET-Rearranged Lung Adenocarcinoma: A Case Series. Clin. Lung Cancer 2023, 24, 666–671. [Google Scholar] [CrossRef]
- Zong, Y.; Qi, C.; Peng, Z.; Shen, L.; Zhou, J. Patients With Acinar Cell Carcinoma of the Pancreas After 2005 A Large Population Study. Pancreas 2020, 49, 781–787. [Google Scholar] [CrossRef]
- Xing-Mao, Z.; Hong-Juan, Z.; Qing, L.; Qiang, H. Pancreatic acinar cell carcinoma—case report and literature review. BMC Cancer 2018, 18, 1083. [Google Scholar] [CrossRef]
- Lowery, M.A.; Klimstra, D.S.; Shia, J.; Yu, K.H.; Allen, P.J.; Brennan, M.F.; O’Reilly, E.M. Acinar Cell Carcinoma of the Pancreas: New Genetic and Treatment Insights into a Rare Malignancy. Oncol. 2011, 16, 1714–1720. [Google Scholar] [CrossRef] [PubMed]
- Sridharan, V.; Mino-Kenudson, M.; Cleary, J.M.; Rahma, O.E.; Perez, K.; Clark, J.W.; Clancy, T.E.; Rubinson, D.A.; Goyal, L.; Bazerbachi, F.; et al. Pancreatic acinar cell carcinoma: A multi-center series on clinical characteristics and treatment outcomes. Pancreatology 2021, 21, 1119–1126. [Google Scholar] [CrossRef]
- Yamada, S.; Motegi, H.; Kurihara, Y.; Shimbo, T.; Kikuchi, I.; Wakabayashi, T.; Sato, T. A resected case of acinar cell carcinoma of the pancreas with liver metastasis following chemotherapy using modified FOLFIRINOX. Surg. Case Rep. 2023, 9, 1–10. [Google Scholar] [CrossRef]
- Skacel, M.; Ormsby, A.H.; Petras, R.E.; McMahon, J.T.; Henricks, W.H. Immunohistochemistry in the Differential Diagnosis of Acinar and Endocrine Pancreatic Neoplasms. Appl. Immunohistochem. Mol. Morphol. 2000, 8, 203–209. [Google Scholar] [CrossRef] [PubMed]
- Holen, K.D.; Klimstra, D.S.; Hummer, A.; Gonen, M.; Conlon, K.; Brennan, M.; Saltz, L.B. Clinical Characteristics and Outcomes From an Institutional Series of Acinar Cell Carcinoma of the Pancreas and Related Tumors. J. Clin. Oncol. 2002, 20, 4673–4678. [Google Scholar] [CrossRef] [PubMed]
- Said, S.; Kurtin, P.J.; Nasr, S.H.; Graham, R.P.; Dasari, S.; Vrana, J.A.; Yasir, S.; Torbenson, M.S.; Zhang, L.; Mounajjed, T.; et al. Carboxypeptidase A1 and regenerating islet-derived 1α as new markers for pancreatic acinar cell carcinoma. Hum. Pathol. 2020, 103, 120–126. [Google Scholar] [CrossRef] [PubMed]
- Ishimoto-Namiki U, Ino Y, Esaki M, Shimada K, Saruta M, Hiraoka N. Novel Insights Into Immunohistochemical Analysis For Acinar Cell Neoplasm of The Pancreas: Carboxypeptidase A2, Carboxypeptidase A1, and Glycoprotein 2. The American journal of surgical pathology. 2023;47(5):525-534.
- La Rosa S, Adsay V, Albarello L, et al. Clinicopathologic study of 62 acinar cell carcinomas of the pancreas: insights into the morphology and immunophenotype and search for prognostic markers. The American journal of surgical pathology. 2012;36(12):1782-1795.
- Strickler JH, Satake H, George TJ, et al. Sotorasib in KRAS p.G12C–Mutated Advanced Pancreatic Cancer. The New England journal of medicine. 2023;388(1):33-43.
- Yousef, A.; Yousef, M.; Chowdhury, S.; Abdilleh, K.; Knafl, M.; Edelkamp, P.; Alfaro-Munoz, K.; Chacko, R.; Peterson, J.; Smaglo, B.G.; et al. Impact of KRAS mutations and co-mutations on clinical outcomes in pancreatic ductal adenocarcinoma. npj Precis. Oncol. 2024, 8, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Hoorens, A.; Lemoine, N.R.; McLellan, E.; Morohoshi, T.; Kamisawa, T.; Heitz, P.U.; Stamm, B.; Rüschoff, J.; Wiedenmann, B.; Klöppel, G. Pancreatic acinar cell carcinoma. An analysis of cell lineage markers, p53 expression, and Ki-ras mutation.. 1993, 143, 685–98. [Google Scholar]
- Florou, V.; Elliott, A.; Bailey, M.H.; Stone, D.; Affolter, K.; Soares, H.P.; Nevala-Plagemann, C.; Scaife, C.; Walker, P.; Korn, W.M.; et al. Comparative Genomic Analysis of Pancreatic Acinar Cell Carcinoma (PACC) and Pancreatic Ductal Adenocarcinoma (PDAC) Unveils New Actionable Genomic Aberrations in PACC. Clin. Cancer Res. 2023, 29, 3408–3417. [Google Scholar] [CrossRef]
- Liu, Y.; Raimondo, M.; Wallace, M.B.; Mody, K.; Stauffer, J.A.; Zhang, L.; Ji, B.; Bi, Y. Exome Sequencing of Pancreatic Acinar Carcinoma Identified Distinctive Mutation Patterns. Pancreas 2021, 50, 1007–1013. [Google Scholar] [CrossRef]
- Furukawa, T.; Sakamoto, H.; Takeuchi, S.; Ameri, M.; Kuboki, Y.; Yamamoto, T.; Hatori, T.; Yamamoto, M.; Sugiyama, M.; Ohike, N.; et al. Whole exome sequencing reveals recurrent mutations in BRCA2 and FAT genes in acinar cell carcinomas of the pancreas. Sci. Rep. 2015, 5, srep08829. [Google Scholar] [CrossRef]
- Dae Won Kim MMP, MD; and Todd Knepper, PharmD. KRAS Wild-Type and KRAS Mutant Pancreatic Ductal Adenocarcinoma: Are These One in the Same or Separate Entities? ASCO Daily News. July 19, 2023.
- Singh, H.; Keller, R.B.; Kapner, K.S.; Dilly, J.; Raghavan, S.; Yuan, C.; Cohen, E.F.; Tolstorukov, M.; Andrews, E.; Brais, L.K.; et al. Oncogenic Drivers and Therapeutic Vulnerabilities in KRAS Wild-Type Pancreatic Cancer. Clin. Cancer Res. 2023, 29, 4627–4643. [Google Scholar] [CrossRef]
- Ramakrishnan, G.; Parajuli, P.; Singh, P.; Friend, C.; Hurwitz, E.; Prunier, C.; Razzaque, M.S.; Xu, K.; Atfi, A. NF1 loss of function as an alternative initiating event in pancreatic ductal adenocarcinoma. Cell Rep. 2022, 41, 111623–111623. [Google Scholar] [CrossRef] [PubMed]
- La Rosa, S.; Bernasconi, B.; Frattini, M.; Tibiletti, M.G.; Molinari, F.; Furlan, D.; Sahnane, N.; Vanoli, A.; Albarello, L.; Zhang, L.; et al. TP53 alterations in pancreatic acinar cell carcinoma: new insights into the molecular pathology of this rare cancer. Virchows Arch. 2015, 468, 289–296. [Google Scholar] [CrossRef] [PubMed]
- Chmielecki, J.; Hutchinson, K.E.; Frampton, G.M.; Chalmers, Z.R.; Johnson, A.; Shi, C.; Elvin, J.; Ali, S.M.; Ross, J.S.; Basturk, O.; et al. Comprehensive Genomic Profiling of Pancreatic Acinar Cell Carcinomas Identifies Recurrent RAF Fusions and Frequent Inactivation of DNA Repair Genes. Cancer Discov. 2014, 4, 1398–1405. [Google Scholar] [CrossRef] [PubMed]
- Mandelker, D.; Marra, A.; Zheng-Lin, B.; Selenica, P.; Blanco-Heredia, J.; Zhu, Y.; Gazzo, A.; Wong, D.; Yelskaya, Z.; Rai, V.; et al. Genomic Profiling Reveals Germline Predisposition and Homologous Recombination Deficiency in Pancreatic Acinar Cell Carcinoma. J. Clin. Oncol. 2023, 41, 5151–5162. [Google Scholar] [CrossRef]
- Takahashi H, Ikeda M, Shiba S, et al. Multicenter Retrospective Analysis of Chemotherapy for Advanced Pancreatic Acinar Cell Carcinoma: Potential Efficacy of Platinum- and Irinotecan-Containing Regimens. Pancreas. 2021;50(1):77-82.
- Sunami, T.; Yamada, A.; Kondo, T.; Kanai, M.; Nagai, K.; Uchida, Y.; Yokode, M.; Matsumori, T.; Uza, N.; Murakami, H.; et al. Exceptional Response of Pancreatic Acinar Cell Carcinoma and Bile Duct Cancer to Platinum-Based Chemotherapy in a Family With a Germline BRCA2 Variant. Pancreas 2022, 51, 1258–1262. [Google Scholar] [CrossRef]
- Duke, E.S.; Bradford, D.; Marcovitz, M.; Amatya, A.K.; Mishra-Kalyani, P.S.; Nguyen, E.; Price, L.S.L.; Zirkelbach, J.F.; Li, Y.; Bi, Y.; et al. FDA Approval Summary: Selpercatinib for the Treatment of Advanced RET Fusion-Positive Solid Tumors. Clin. Cancer Res. 2023, 29, 3573–3578. [Google Scholar] [CrossRef]
- Chou, A.; Brown, I.S.; Kumarasinghe, M.P.; Perren, A.; Riley, D.; Kim, Y.; Pajic, M.; Steinmann, A.; Rathi, V.; Jamieson, N.B.; et al. RET gene rearrangements occur in a subset of pancreatic acinar cell carcinomas. Mod. Pathol. 2019, 33, 657–664. [Google Scholar] [CrossRef]
- Abraham SC, Wu TT, Hruban RH, et al. Genetic and immunohistochemical analysis of pancreatic acinar cell carcinoma: frequent allelic loss on chromosome 11p and alterations in the APC/beta-catenin pathway. Am J Pathol. 2002;160(3):953-962.
- Liu W, Shia J, Gönen M, Lowery MA, O’Reilly EM, Klimstra DS. DNA mismatch repair abnormalities in acinar cell carcinoma of the pancreas: frequency and clinical significance. Pancreas. 2014;43(8):1264-1270.
- Chen, H.; Wang, B.; Zhang, Y.; Shu, Y.; Dong, H.; Zhao, Q.; Yang, C.; Li, J.; Duan, X.; Zhou, Q. A unified DNA- and RNA-based NGS strategy for the analysis of multiple types of variants at the dual nucleic acid level in solid tumors. J. Clin. Lab. Anal. 2023, 37, e24977. [Google Scholar] [CrossRef]
- Heydt, C.; Wölwer, C.B.; Camacho, O.V.; Wagener-Ryczek, S.; Pappesch, R.; Siemanowski, J.; Rehker, J.; Haller, F.; Agaimy, A.; Worm, K.; et al. Detection of gene fusions using targeted next-generation sequencing: a comparative evaluation. BMC Med Genom. 2021, 14, 1–14. [Google Scholar] [CrossRef]
- Oh, K.; Yoo, Y.J.; Torre-Healy, L.A.; Rao, M.; Fassler, D.; Wang, P.; Caponegro, M.; Gao, M.; Kim, J.; Sasson, A.; et al. Coordinated single-cell tumor microenvironment dynamics reinforce pancreatic cancer subtype. Nat. Commun. 2023, 14, 1–13. [Google Scholar] [CrossRef]
- Hong, W.C.; Lee, D.E.; Kang, H.W.; Kim, M.J.; Kim, M.; Kim, J.H.; Fang, S.; Kim, H.J.; Park, J.S. CD74 Promotes a Pro-Inflammatory Tumor Microenvironment by Inducing S100A8 and S100A9 Secretion in Pancreatic Cancer. Int. J. Mol. Sci. 2023, 24, 12993. [Google Scholar] [CrossRef] [PubMed]
- Michaels, A.D.; Newhook, T.E.; Adair, S.J.; Morioka, S.; Goudreau, B.J.; Nagdas, S.; Mullen, M.G.; Persily, J.B.; Bullock, T.N.J.; Slingluff, C.L.; et al. CD47 Blockade as an Adjuvant Immunotherapy for Resectable Pancreatic Cancer. Clin. Cancer Res. 2018, 24, 1415–1425. [Google Scholar] [CrossRef] [PubMed]
- Hilmi, M.; Delaye, M.; Muzzolini, M.; Nicolle, R.; Cros, J.; Hammel, P.; Cardot-Ruffino, V.; Neuzillet, C. The immunological landscape in pancreatic ductal adenocarcinoma and overcoming resistance to immunotherapy. Lancet Gastroenterol. Hepatol. 2023, 8, 1129–1142. [Google Scholar] [CrossRef] [PubMed]
- Bear, A.S.; Vonderheide, R.H.; O’Hara, M.H. Challenges and Opportunities for Pancreatic Cancer Immunotherapy. Cancer Cell 2020, 38, 788–802. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Tandurella, J.A.; Gai, J.; Zhu, Q.; Lim, S.J.; Thomas, D.L.; Xia, T.; Mo, G.; Mitchell, J.T.; Montagne, J.; et al. Multi-omic analyses of changes in the tumor microenvironment of pancreatic adenocarcinoma following neoadjuvant treatment with anti-PD-1 therapy. Cancer Cell 2022, 40, 1374–1391. [Google Scholar] [CrossRef]
- Gebauer, F.; Wicklein, D.; Horst, J.; Sundermann, P.; Maar, H.; Streichert, T.; Tachezy, M.; Izbicki, J.R.; Bockhorn, M.; Schumacher, U. Carcinoembryonic Antigen-Related Cell Adhesion Molecules (CEACAM) 1, 5 and 6 as Biomarkers in Pancreatic Cancer. PLOS ONE 2014, 9, e113023–e113023. [Google Scholar] [CrossRef]
- Kopetz, S.; Boni, V.; Kato, K.; Raghav, K.P.S.; Pallis, A.; Habermehl, C.; Galipelli, S.; Courlet, P.; Rivera, I.R. First-in-human trial of M9140, an anti-CEACAM5 antibody drug conjugate (ADC) with exatecan payload, in patients (pts) with metastatic colorectal cancer (mCRC). J. Clin. Oncol. 2024, 42, 3000–3000. [Google Scholar] [CrossRef]
- Zhou, W.; Han, X.; Fang, Y.; Han, S.; Cai, Y.; Kuang, T.; Lou, W.; Wang, D. Clinical Analysis of Acinar Cell Carcinoma of the Pancreas: A Single-Center Experience of 45 Consecutive Cases. Cancer Control. 2020, 27. [Google Scholar] [CrossRef]
- Goeppert, B.; Ernst, C.; Baer, C.; Roessler, S.; Renner, M.; Mehrabi, A.; Hafezi, M.; Pathil, A.; Warth, A.; Stenzinger, A.; et al. Cadherin-6 is a putative tumor suppressor and target of epigenetically dysregulated miR-429 in cholangiocarcinoma. Epigenetics 2016, 11, 780–790. [Google Scholar] [CrossRef]
- Casal JI, Bartolomé RA. Beyond N-Cadherin, Relevance of Cadherins 5, 6 and 17 in Cancer Progression and Metastasis. Int J Mol Sci. 2019;20(13).
- Luo, S.; Lin, R.; Liao, X.; Li, D.; Qin, Y. Identification and verification of the molecular mechanisms and prognostic values of the cadherin gene family in gastric cancer. Sci. Rep. 2021, 11, 1–21. [Google Scholar] [CrossRef]
- Lo, A.; Li, C.-P.; Buza, E.L.; Blomberg, R.; Govindaraju, P.; Avery, D.; Monslow, J.; Hsiao, M.; Puré, E. Fibroblast activation protein augments progression and metastasis of pancreatic ductal adenocarcinoma. J. Clin. Investig. 2017, 2. [Google Scholar] [CrossRef]
Figure 1.
Oncoplot of genomic alterations identified in the study patients. Del, deletion.
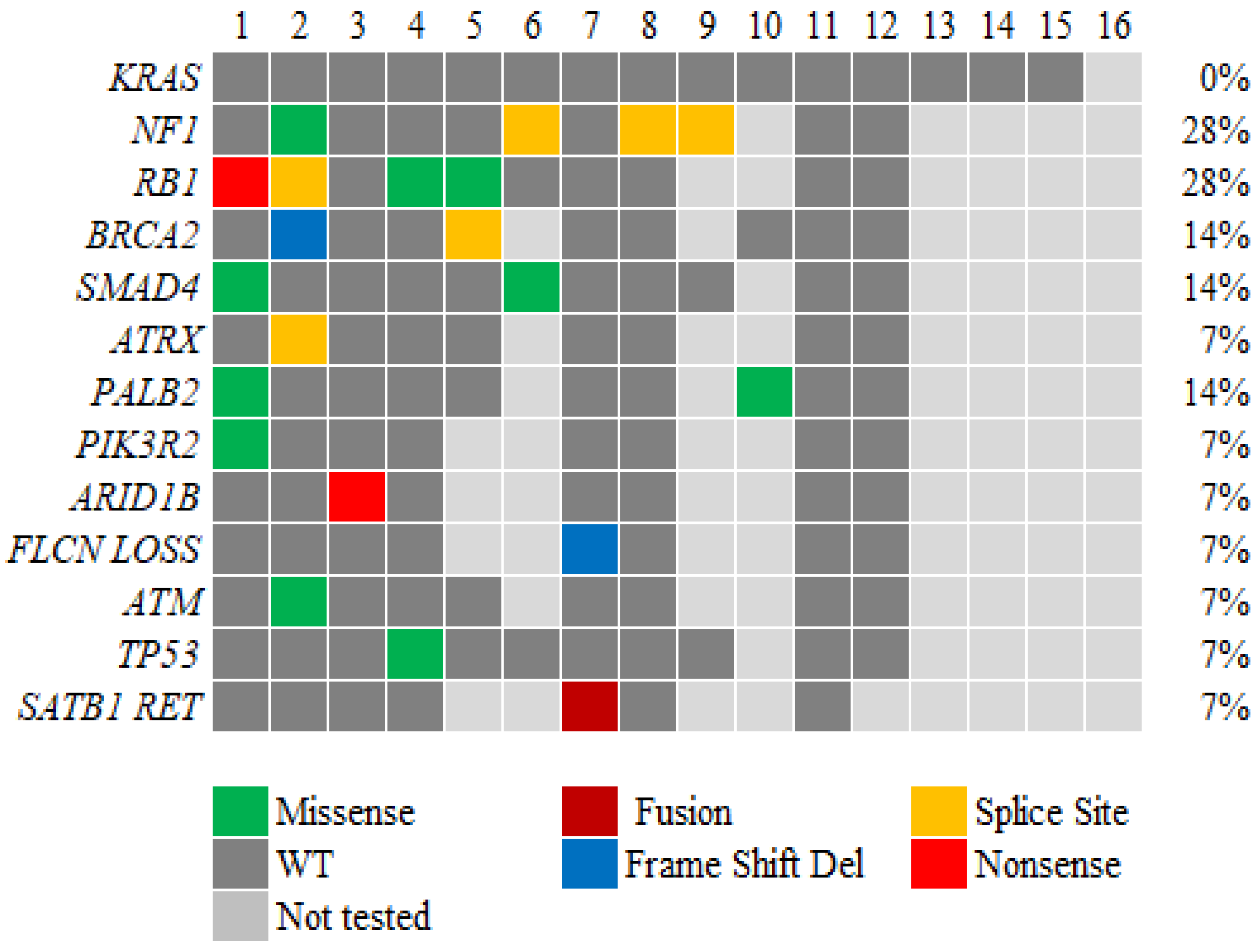
Figure 2.
RNA expression and TME features for the two cases tested by BostonGene. (#17, mixed ACC with adenocarcinoma, found NTRK1 fusion) and #18 (pure ACC). A: TME features of both cases revealed immune desert type. B and C, RNA expression signatures and % of cells composition. D, Relative expression of selective genes (dashed lines for reference ranges of PDAC).
Figure 2.
RNA expression and TME features for the two cases tested by BostonGene. (#17, mixed ACC with adenocarcinoma, found NTRK1 fusion) and #18 (pure ACC). A: TME features of both cases revealed immune desert type. B and C, RNA expression signatures and % of cells composition. D, Relative expression of selective genes (dashed lines for reference ranges of PDAC).
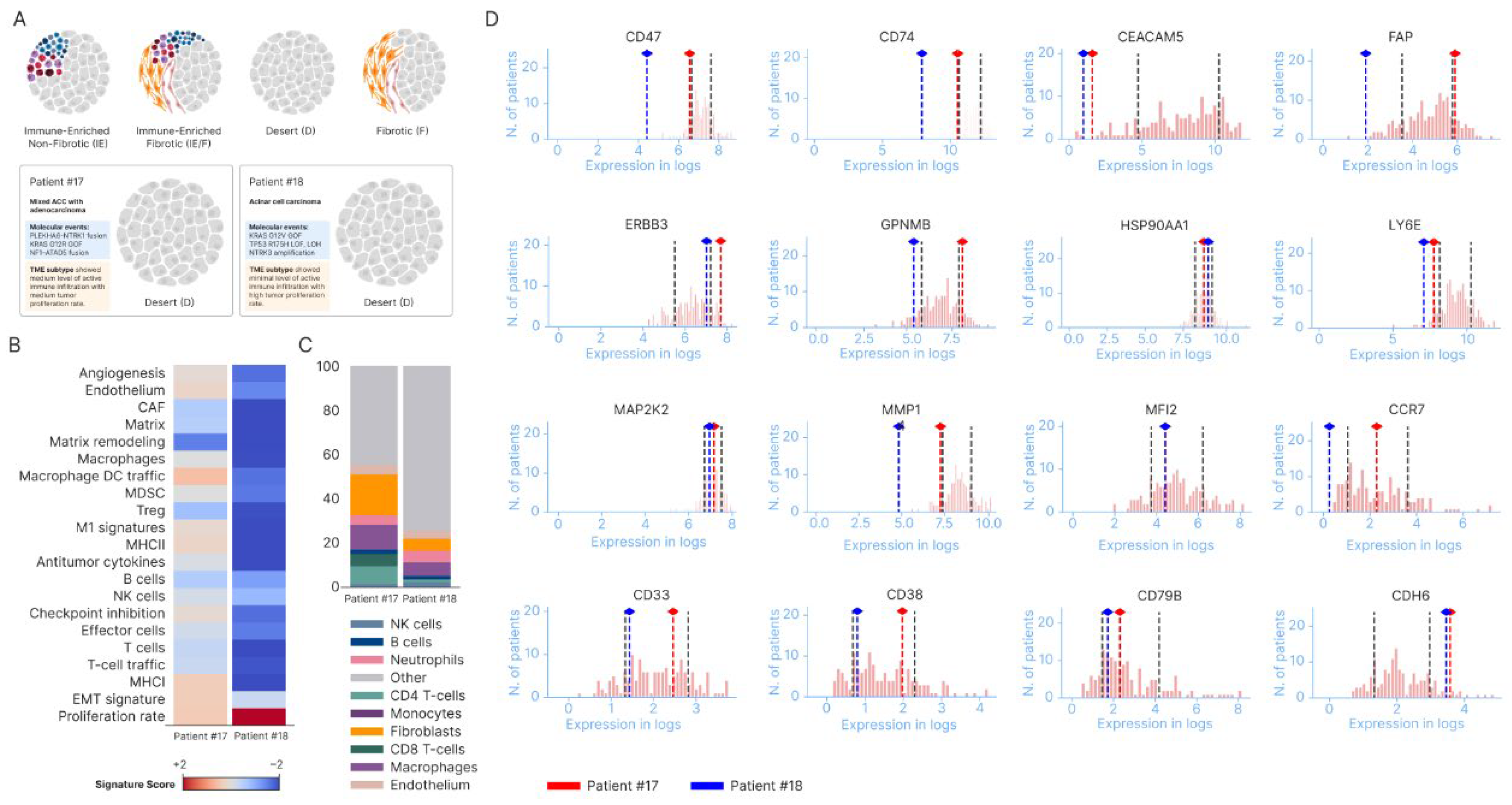

Table 1.
Demographic characteristics of the 16 study patients.
| Characteristics | Number of patients (%) |
|---|---|
| Sex | |
| Male Female |
13 (81) 3 (19) |
| Median age at diagnosis, years | 62.5 |
| Race White Hispanic Asian |
14 (88) 1 (6) 1 (6) |
| Primary tumor location Head of pancreas Body of pancreas Tail of pancreas Body and head of pancreas |
8 (50) 5 (31) 2 (13) 1 (6) |
| Tumor status at initial diagnosis Resectable/borderline resectable Metastatic |
2 (12) 14 (88) |
| Smoking status (current/former smoker) | 9 (56) |
| Alcohol status (current/former drinker) | 10 (63) |
Table 2.
Histology and immunohistochemistry results for the 16 study patients.
| Characteristic | Number of patients |
|---|---|
| Histology Pure acinar cell carcinoma Mixed acinar-neuroendocrine Mixed acinar and adenocarcinoma |
14 1 1 |
| Marker positivity Trypsin Chymotrypsin Synaptophysin BCL-10 Cytokeratin 7 Alpha 1-antichymotrypsin Chromogranin Alpha 1 antitrypsin BCL-2 |
(N of positive/tested) 9/9 2 /2 5 /12 2 /2 8 /9 2 /2 3/13 1 /2 2 /2 |
Table 3.
Treatment Response, POD-progression of disease, DOD- died of disease, rx-treatment, OS-overall survival, mo-months, yrs-years, rxed-treated, XRT-radiation.
Table 3.
Treatment Response, POD-progression of disease, DOD- died of disease, rx-treatment, OS-overall survival, mo-months, yrs-years, rxed-treated, XRT-radiation.
| Age/Sex | First | Second | Third and beyond | OS (months) | |
|---|---|---|---|---|---|
| 1 | 59/ M | Xelox 3 cycles with stable disease, rx switch due to loss of vascular access | Gemcitabine+Xeloda->3 cycles and POD | Xeloda 1 month with POD, then FOLFIRI 4 cycles with POD, followed by Phase I | 17 |
| 2 | 62/M | Gemcitabine +Cisplatin,3 months with POD | Gemcitabine/Erlotinib 2 mo. with POD | Phase I trial | 17 |
| 3 | 53/M | Folfox 2 cycles with POD | Folfirinox2cycles POD | Gem/Abraxane,3 mo. With stable d/s. Next line Gem/Abraxane/Xeloda with continued response till 8 months, then rx break f/u POD and then Phase I | 29 |
| 4 | 70/F | Whipple surgery, recurrence in 3 yrs with liver mets rxed with partial hepatectomy | Xeloda/Gemcitabine in adj setting for 4 mo. and surveillance for 5 yrs. | Recurrence rxed with Folfox+Avastin for 3 mo. with POD followed by Gem/Abraxane, 1 mo. & DOD | 92 |
| 5 | 66/M | Folfirinox 5mo with stability, rx changed due to toxicity | Gemcitabine +Tazarotene + Xeloda for 6mo with stable d/s for 12 mo. followed by d/s POD | 33 | |
| 6 | 52/M | Folfox 2months with POD | Xeloda XRT 1month | Folfirinox 2 mo. with POD,then Gem/Abraxane 8 mo. with mixed response, then Erlotinib/Avastin 3 mo. with progression | 24 |
| 7 | 55/M | Neoadjuvant Gem/Abraxane+ Cisplatin (2 mo.) | Folfirinox 2mo with POD | Gem/Abraxane/5 FU/Cisplatin 4 mo. with mixed response, the Gem/Abraxane/5FU/Cisplatin/ Erlotinib 4 mo. mixed response and then Phase II trial | 24 |
| 8 | 67/M | Gem/Abraxane/Xeloda with response for 2 mo. followed by stable d/s for 2 mo. then POD | 7 | ||
| 9 | 72/M | Folfirinox 6 cycles with partial response | 15 | ||
| 10 | 65/F | Gem/Cis 8 cycles mixed response | Folfox 4 cycles with POD | Gem/Abraxane for 9 mo. with response | 39 |
| 11 | 49/M | Folfirinox 12 cycles with response | Xeloda maintenance with response for 17 mo. | 31 | |
| 12 | 58/F | Folfirinox 1 mo. with POD | Referred to Phase I | 26 | |
| 13 | 70/M | Folfox 10 cycles with stable response | Xelox mo. with mixed response | Folfox 2 mo. with POD then Everolimus for 6mo with POD followed by Gem/Abraxane 1 mo. with POD | 46 |
| 14 | 61/M | Folfirinox 15 mo. with response | Gem/Abraxane 2mo POD | Folfiri 2 mo. POD &Lost to f/u | 24 |
| 15 16 |
66/M 59/M |
Folfirinox 4 mo. with stable Capecitabine &Oxaliplatin for 6mo with response |
Xeloda+ XRT 1 mo. with response followed by surgery. Lost to follow up |
Adj Folfirinox 2mo f/u POD Gem/Abraxane 2 mo with POD followed by Phase I | 21 6 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



