
Submitted:
21 August 2024
Posted:
22 August 2024
You are already at the latest version
Abstract
Cognitive impairment is a core feature of schizophrenia, playing a pivotal role in understanding the causes, processes, and outcomes of the disorder. Cognitive impairments in schizophrenia encompass a wide range of domains, including processing speed, episodic memory, working memory, and executive function. These deficits persist throughout the course of the illness and significantly impact functional outcome and quality of life. Therefore, identifying the biological basis of cognitive deficits in schizophrenia and developing effective treatments are imperative. The role of N-methyl-D-aspartate (NMDA) receptors in synaptic transmission and plasticity has long been recognized, making them potential targets for schizophrenia treatment. This review will focus on emerging pharmacology targeting NMDA receptor, offering strategies for the prevention and treatment of cognitive deficits in schizophrenia.
Keywords:
NMDA receptor
; cognitive impairment
; excitation/inhibition balance
; schizophrenia
1. Introduction
Schizophrenia is a chronic and severe mental disorder, typically emerging during adolescence or early adulthood, with an estimated lifetime prevalence of approximately 1% [1]. It often manifests with positive symptoms, such as hallucinations and delusions, negative symptoms characterized by diminished motivation and reduced emotional expression, alongside cognitive impairments including decreased attention and memory. Importantly, cognitive impairment associated with schizophrenia (CIAS) is highly prevalent, affecting approximately 80% of individuals with the disorder. CIAS often predates the onset of positive and negative symptoms and persists throughout the course of the illness. Even after achieving "stability" in positive symptom through the usage of antipsychotic medications, many patients continue to experience cognitive symptoms, which in turn contribute to poorer functional outcomes [2,3]. Therefore, CIAS is considered the strongest predictor of long-term functional prognosis in individuals with schizophrenia.
Treating cognitive impairments is a crucial aspect of improving functional outcomes in schizophrenia. The glutamatergic system, illustrated in the Figure 1, is one of the key mechanisms underlying cognitive deficits. The glutamatergic system plays a central role in various cognitive processes, including learning, memory, and executive function [4]. Dysfunction within this neurotransmitter system has been implicated in the cognitive impairments observed in schizophrenia. Glutamate hypothesis of schizophrenia is based on the ability of N-methyl-D-aspartate (NMDA) receptor antagonists to induce schizophrenia-like symptoms, as well as emergent literature demonstrating disturbances of NMDA receptor-related gene expression and metabolic pathways in schizophrenia [5]. Therefore, it seems that NMDA receptor and synapse function may participate in the pathogenesis of schizophrenia, suggesting that modulators of NMDA receptor signaling are promising candidates for CIAS therapy.
2. The NMDA Receptor Function and Cognition
NMDA receptors are cation-selective ligand-gated ion channels, together with other ionotropic receptors (AMPA receptors, kainate receptors) and G-protein coupled receptors (metabotropic glutamate receptors, or mGluRs), mediate glutamatergic synaptic transmission throughout the central nervous system (CNS) [6]. The channels contain two obligatory GluN1 and two modulatory GluN2 (A-D) subunits or a combination of GluN2 and GluN3 subunits. Activation of the NMDA receptor is both voltage-dependent and ligand-gated, and requires the binding of two ligands, glutamate and either D-serine or glycine. Glutamate serves as the neurotransmitter, released from presynaptic terminals in an activity-dependent manner, whereas D-serine or glycine acts as a modulator, maintaining relatively constant levels in the extracellular fluid. The ion-channel integral to the NMDA receptors is voltage-dependently blocked by magnesium ions (Mg2+), and depolarization removes this block [7,8,9]. Thus, the NMDA receptor serves as a coincidence detector, linking neurotransmitter activation with the electrical properties of neurons. Sustained NMDA receptor activation triggers signaling to the nucleus and coordinated changes in gene expression, supporting the establishment of long-term synaptic plasticity (LTP), which are the basis of learning and memory [10]. However, excessive glutamate can chronically overstimulate NMDA receptors, resulting in excessive intracellular calcium and excitotoxicity, a mechanism implicated in neuronal death in various CNS disorders. Both deficiency and excess of NMDA receptor activity can be detrimental. Excitotoxicity associated with NMDA receptors has prompted the search for antagonists as potential neuroprotective agents, while their role in synaptic plasticity has inspired research into receptor potentiators for the treatment of cognitive dysfunction.
NMDA receptors are integral to the intricate processes of synaptic transmission, neuronal plasticity, and cognitive functions in the brain [11]. Their uniqueness among glutamate receptors lies in their high calcium permeability and voltage-dependent activation properties. This distinctiveness allows NMDARs to act as molecular switches that mediate critical aspects of synaptic plasticity, particularly LTP, a phenomenon widely associated with learning and memory. Specifically, activation of NMDA receptors permits the influx of calcium ions into the postsynaptic neuron. This influx of calcium serves as a pivotal signal, triggering a cascade of intracellular events that profoundly impact synaptic strength and neuronal connectivity, including the activation of protein kinases, such as calcium/calmodulin-dependent protein kinase II (CaMKII) and protein kinase C (PKC). The phosphorylation of kinases influences target proteins, leading to the changes in synaptic efficacy, including alterations in the function and density of neurotransmitter receptors at the synapse. Furthermore, the activation of NMDA receptors during LTP induction also influences gene expression within the neuron. This modulation of gene transcription and translation is mediated by calcium-dependent signaling pathways and transcription factors, such as cAMP response element-binding protein (CREB). The activation of CREB leads to the expression of genes that encode proteins involved in synaptic strengthening and the structural remodeling of synapses, thereby contributing to the consolidation of memory.
3. NMDA Receptor Hypofunction in Schizophrenia
The normal functioning of NMDA receptors is crucial for maintaining cognitive functions such as learning and memory. Dysfunction of NMDA receptors is a key mechanism underlying schizophrenia, especially in CIAS [12,13]. Early findings have shown that NMDA receptor antagonists, such as phencyclidine (PCP), ketamine, and MK801, can induce schizophrenia-like behaviors, including cognitive symptoms, in both preclinical and clinical studies [14,15]. Animal studies have confirmed that administering NMDA receptor antagonists like MK-801 leads to cognitive impairments across multiple domains in rodents [16]. NMDA receptor subunit NR1 knockdown mice exhibit behavioral abnormalities, which can be ameliorated to varying degrees with antipsychotics or psychoactive drugs [17,18,19]. Furthermore, autoimmune encephalitis linked to NMDA receptor-specific antibodies has been associated with severe psychosis [20]. Both genetic and environmental factors that precipitate NMDA receptor hypofunction have been implicated in the disease progression and symptoms of schizophrenia [21]. Specifically, NMDA receptors can affect cognitive function at the cellular and neural network levels.
3.1. Cellular and Molecular Level: Synaptic Plasticity
Synaptic plasticity is the experience-dependent change in connectivity between neurons that is believed to underlie learning and memory [22]. Patients with schizophrenia experiencing negative life events, such as uterine infection or pregnancy complications, psychosocial causes, amphetamine abuse, autoimmune disease and other brain trauma, all these activity-dependent modifications are essential for NMDA receptor function, synaptic transmission and neuronal circuits establishment. Post-mortem studies of the brains of patients with schizophrenia have revealed lower levels of expression of the obligatory NMDA receptor subunit GluN1 [23], increased expression of the endogenous NMDA receptor antagonist kynurenate [24], and a reduction in levels of the NMDA receptor co-agonist D-serine, along with reduced activity of its synthesizing enzyme, D-serine racemase [25]. Additionally, biological pathway analyses of genome-wide association studies (GWAS) data (~60,000 subjects from the Psychiatric Genetics Consortium) revealed that genetic variants associated with schizophrenia are enriched in pathways related to the postsynaptic density, the postsynaptic membrane, dendritic spines, and histone methylation [26]. Analyses of copy number variants (CNV) have linked de novo mutations in genes encoding the NMDA receptor and proteins that associated with the postsynaptic density to a higher risk of schizophrenia [26,27].
Changes in NMDA receptor expression and function involve enhancements or suppressions in synaptic transmission efficacy, for instance, LTP facilitates memory formation, long-term depression (LTD) verifies memory content and maintains a balance between memory and forgetting. Consistent with this, NMDA receptor hypofunction is implicated in the behavioral manifestations, including the social withdrawal, increased locomotor activity and cognitive impairment of schizophrenia in humans and animal models [28,29,30]. Our findings revealed that the diminished excitatory neurotransmission in the medial prefrontal cortex could be a common pathophysiology regardless of the prenatal and postnatal pathogenesis in developmental models of schizophrenia, and that might underlie the mechanism of defective working memory in those models [31]. Activation of neuregulin 1, a growth factor genetically linked to schizophrenia in humans [32], promotes internalization of NMDA receptors from the cell surface by actin-dependent mechanism in prefrontal pyramidal neurons [33]. Additionally, overactivation of the ErbB4 receptor by neuregulin suppresses NMDA receptor activity in the prefrontal cortex of patients with schizophrenia [34], eliciting schizophrenic-like symptoms.
3.2. Neural network level: Excitation/Inhibition Balance and Neural Oscillation
The excitatory synaptic transmission and inhibitory synaptic transmission are the two main synaptic transmissions in our brain [35]. The balance between excitatory and inhibitory synaptic transmission (E/I balance) is essential for normal neural development, behavior and cognition. Whereas, an E/I imbalance leads to neurological disorders, such as schizophrenia [36]. Importantly, NMDA receptors regulate α-amino-3-hydroxy-5-methylisoxazole-4-propionate receptor (AMPAR)-mediated excitatory and γ-amino- butyric acid receptor (GABAR)-mediated inhibitory synaptic transmission, suggesting that NMDA receptors play an important role in the establishment and maintenance of the E/I balance [37]. Hypofunction of NMDA receptors have found in human and animal models of schizophrenia, which can impact the balance between excitation (E) and inhibition (I) [13,38,39]. Specifically, within inhibitory neurons, reduced NMDA receptor function leads to a decrease in GABA neurotransmission. Consequently, inhibition on excitatory neurons is relieved, leading to an increase in excitatory neurotransmission. This imbalance in E/I results in abnormal neural oscillations and cognitive impairments (Figure 2) [40,41].
Neural oscillations are a crucial mechanism for establishing precise temporal coordination between neuronal responses, which is highly relevant for cognitive processes like memory, perception, and consciousness. In patients with schizophrenia, the synchronization of gamma-band activity is abnormal, suggesting a crucial role for dysfunctional oscillations in the generation of the cognitive deficits and other symptoms of this disorder. The neurocircuitry hypotheses may help explain how reduced NMDAR activity contributes to the symptoms of schizophrenia [42]. One hypothesis is that the cognitive impairments may arise from hypofunctional NMDA receptors on cortical GABA interneurons, particularly fast-spiking parvalbumin interneurons, leading to changes in cortical network oscillations [43,44]. This hypothesis is supported by the fact that patients with schizophrenia have reduced parvalbumin expression in the dorsolateral prefrontal cortex (PFC) and abnormal gamma band oscillations [43,45], which have been implicated in the synchronization of neural ensembles during working memory and attention. Moreover, NMDA receptor hypofunction on cortical interneurons could boost glutamatergic projection neurons' activity excessively, leading to hyperstimulation of GABAergic interneurons in the ventral tegmental area. This overstimulation can dampen the meso-cortical dopamine pathway, causing insufficient dopamine release in the PFC and potentially contributing to negative and cognitive symptoms [46]. In individuals with schizophrenia, there is an upregulation of the serotonin (5-HT) receptor subtypes 5-HT1A and a downregulation of 5-HT2A in the PFC [47], which are important for emotion and cognition and involved in modulating NMDA receptor activity [48,49]. Consequently, alterations in 5-HT signaling could potentially influence cognitive and negative symptoms by impacting NMDA receptor function in the PFC.
Overall, NMDA receptors are critical for synaptic plasticity, learning, and memory processes in the brain. They play a crucial role in regulating the balance between excitatory and inhibitory neurotransmission. Dysfunction in NMDA receptor signaling disrupts this balance, leading to aberrant neural circuitry function and cognitive deficits observed in schizophrenia.
4. Targeting NMDA Receptors in Schizophrenia
Given the critical role of NMDA receptors in cognition and their implication in schiz ophrenia, targeting NMDA receptors presents a promising approach for treating CIAS. Notably, strategies targeting the glycine-binding site are expected to have less adverse effects compared to modulating the glutamate-binding site [50]. Approaches to enhance NMDA receptor function directly or indirectly include modulating glycine or D-serine concentrations in the synaptic cleft and developing selective NMDA receptor modulators (Figure 3).
4.1. Direct Enhancement of NMDA Receptor Function
A wide range of compounds that directly activate NMDA receptors, such as glycine and D-serine, have been shown to be effective in clinical trials for improving cognitive function and reducing symptom severity in schizophrenia patients. These agonists act by enhancing NMDA receptor-mediated neurotransmission, thereby restoring synaptic plasticity and neural circuitry function.
4.1.1. Enhancement of NMDA Receptor Functions by Glycine
Glycine is the simplest amino acid and acts as a neurotransmitter in the brain. When glycine receptors are activated, chloride enters the neuron via ionotropic receptors, causing an inhibitory postsynaptic potential. Glycine is also a required co-agonist along with glutamate for NMDA receptors. In contrast to the inhibitory role of glycine in the spinal cord, this behavior is facilitated at the NMDA receptors which are excitatory [51]. Studies on postmortem of schizophrenic patients have revealed the increases in NMDA-associated glycine binding sites in the cerebral cortex [52]. High serum glycine levels have been reported in patients with pre-pulse inhibition deficits in chronic schizophrenia patients [53]. Similarly, rats treated with a glycine-rich diet display disturbances in sensory gating [54]. Conversely, some studies have reported lower plasma glycine levels in schizophrenia patients compared to healthy controls, and these lower glycine levels were associated with a greater severity of negative symptoms [55]. These findings suggest that glycine levels may compensate for changes in glutamate NMDA receptor transmission in patients with schizophrenia. Further large-scale studies measuring glycine concentrations in both serum and cerebrospinal fluid are needed to elucidate the complex relationship between glycine signaling and the pathogenesis of schizophrenia.
Although there is controversy over the glycine levels in patients with schizophrenia, glycine induced NMDA receptor mediated enhanced neurotransmission is considered a potential safe and feasible method for improving negative symptoms as well as CIAS. It has been reported that the potentiation of NMDA receptor function by the infusion of glycine into the prefrontal cortex ameliorated PCP-induced behavioral deficits in latent learning [56]. Glycinamide, a prodrug of glycine, can be converted to glycine in CNS and it prevented MK-801 induced deficits in a novel object recognition task in rabbits [57]. Briefly, the Table 1 summarizes the key findings from several clinical studies examining the effects of glycine supplementation on the treatment of schizophrenic symptoms. The studies varied in sample size and glycine dosage, but the majority reported significant improvements in negative and cognitive symptoms of schizophrenia with glycine treatment. However, several studies did not find any significant effects on symptoms. The lack of consistency across trials may be due to small sample sizes, various doses of glycine, different trial durations and clinical ratings. Overall, the evidence suggests that glycine may be a promising adjunctive therapy for targeting the cognitive and negative symptom domains in schizophrenia.
4.1.2. Enhancement of NMDA Receptor Functions by D-Serine
D-serine is an endogenous ligand for the glycine site of the NMDA receptor [74]. For the GluN1/N2 subunits of NMDA receptor, the binding affinity of D-serine is three-fold more potent than that of glycine [75]. Notably, D-serine is the primary coagonist of synaptic NMDA receptors, whereas glycine is the primary coagonist of extrasynaptic NMDA receptors [76]. The differential localization and coagonist preferences of synaptic versus extrasynaptic NMDA receptors have important implications for their distinct roles in neuronal signaling and synaptic function. Synaptic NMDA receptors are typically associated with excitatory neurotransmission and the induction of LTP, processes critical for learning and memory. Moreover, depletion of D-serine diminishes NMDA receptor activity [74] and LTP [77]. In contrast, extrasynaptic NMDA receptors have been linked to excitotoxicity and the propagation of pathological signals, which may contribute to the cognitive deficits observed in neuropsychiatric disorders like schizophrenia [78,79].
Accumulating evidence highlights the potential therapeutic role of D-serine in the modulation of NMDA receptor functions for the treatment of schizophrenia. Indeed, reduced levels of D-serine have been found in the serum of patients with schizophrenia compared to healthy individuals [80]. A postmortem brain study also revealed decreased D-serine in the cerebrospinal fluid (CSF) of schizophrenia patients [81]. Correspondingly, D-serine supplementation during juvenile and adolescent stages has been shown to prevent the onset of cognitive deficits, prodromal and the core symptoms of schizophrenia in adult offspring after maternal immune activation [82]. Our recent study demonstrated that chronic D-serine treatment ameliorated cognitive dysfunction in a neurodevelopmental mouse model of schizophrenia. Mechanistically, we found that D-serine restores the excitation/inhibition balance by reconstituting both synaptic and intrinsic inhibitory control of cingulate pyramidal neurons. This effect was mediated through the facilitation of parvalbumin-positive (PV) interneuron-preferential NMDA receptor function and the activation of small-conductance calcium-activated potassium (SK) channels in pyramidal neurons, respectively [29]. However, it is important to note that the rapid metabolism of D-serine by the enzyme D-amino acid oxidase (DAAO) may reduce its bioavailability, potentially posing a challenge for its therapeutic use in schizophrenia [83]. Additionally, there are concerns regarding the potential nephrotoxicity associated with high concentrations of D-serine, as observed in rats developing acute tubular necrosis with higher doses [84,85]. Nonetheless, the measurement of serum D- and L-serine levels has been proposed as a potential biological marker for schizophrenia [80]. Further research with larger sample sizes and specific controls, following the guidelines for accurate measurement and detection methods [86], is warranted to fully elucidate the therapeutic potential of modulating the D-serine/NMDA receptor axis in schizophrenia.
Building upon the same lines, D-serine has been extensively investigated, both employed alone and as an adjunct to antipsychotics for improving negative and cognitive symptoms of schizophrenia in numerous clinical studies. Key findings from these studies are summarized in Table 2. Many clinical trials have reported significant improvements in the negative and cognitive symptoms of schizophrenia with D-serine supplementation. For instance, a pilot, double-blind, placebo-controlled, randomized parallel group mechanistic proof-of-concept trial demonstrated a 35.7% improvement in negative symptoms (cognitive impairment is a common negative symptom of schizophrenia) compared with placebo in individuals at high risk of schizophrenia [87]. Similarly, a study of 31 Taiwanese schizophrenic patients receiving D-serine (30 mg/kg/day) as an adjunct to standard antipsychotics revealed significant improvements in their positive, negative, and cognitive symptoms, as well as enhanced performance on the Wisconsin Card Sorting Test (WCST) [88]. However, not all studies have yielded positive results. A multicenter, add-on randomized controlled trial indicated that the effect of low-dose D-serine (2 g/day) on the treatment of negative and cognitive symptoms appeared to be small [89]. Conversely, the first randomized, double-blind, placebo-controlled study using a higher dose of D-serine (60 mg/kg) in schizophrenia patients reported significant improvements in clinical symptoms, suggesting that a minimum daily dose of 3.6 g of D-serine may be necessary to achieve improvements in negative symptoms [90]. It is important to note that high-dose D-serine administration can lead to adverse effects, such as peripheral neuropathies, oxidative damage [91], neurotoxicity [92], and renal toxicity [93,94].
In summary, the available clinical evidence suggests that D-serine may hold promise for the treatment of negative and cognitive symptoms in schizophrenia, particularly when used as an adjunct to antipsychotic medications. However, the therapeutic benefit appears to be dose-dependent, and the potential for adverse effects, especially with higher doses, should be carefully considered in the design and implementation of future clinical trials.
4.2. Indirect Enhancement of NMDA Receptor Function
As described previously, glutamate, glycine and D-serine directly target postsynaptic NMDA receptor and activate NMDA receptor functioning. However, the beneficial effect of directly targeting the NMDA receptor with above compounds is limited due to several factors, including the need for high doses, a narrow therapeutic window, poor central nervous system (CNS) penetration, and the associated side effects. Alternatively, as illustrated in the Figure 3, indirect enhancement of NMDA receptor function by improving the availability of synaptic glycine and D-serine levels from in astrocytes provides a new approach to help meet the needs of patients in schizophrenia.
4.2.1. Enhancement of NMDA Receptor Functions by GlyT1 Inhibitors
Glycine transporter type 1 (GlyT1) is involved in the reuptake of glycine from the synaptic cleft. By inhibiting GlyT1, compounds such as sarcosine, BI 425809, and bitopertin can reduce the reuptake of glycine, increasing its concentration in the synaptic cleft [103]. GlyT1 is highly colocalized with NMDA receptors on glial cells and neurons in the cortex, hippocampus, septum and thalamus [104]. GlyT1 effectively regulates synaptic glycine reuptake and enhances NMDA receptor function by promoting the binding of glycine to the subtypes of NMDA receptors [105]. Thus, selective inhibition of astrocytic GlyT1 represents a promising therapeutic strategy to enhance synaptic glycine concentrations and boost NMDA receptor activity.
Multiple lines of evidence indicate that inhibition of GlyT1 enhances NMDA receptor functions, which holds promise for the treatment of CIAS. BI 425809 is a novel, potent and selective GlyT1 inhibitor that can increase synaptic glycine concentration, thereby enhancing NMDA receptor signaling and improving neural plasticity, which in turn ameliorates cognitive function [106,107]. A randomized controlled trial found that PF-03463275 as a GlyT1 inhibitor could enhance cognitive training and neuroplasticity in schizophrenia [108]. Glycyldodecylamide, a compound that blocks neuronal glycine uptake and which may therefore increase intrasynaptic glycine levels, inhibits PCP-induced psychosis in schizophrenia [109]. Subsequent chronic (2-week) administration of (R)-(N-[3-(4'-fluorophenyl)-3-(4'-phenylphenoxy) propyl]) sarcosine (NFPS, also known as ALX5407), a GlyT1 inhibitor, enhanced PCP-induced cognitive deficits [110,111]. Sarcosine, another GlyT1 inhibitor, has promising therapeutic potential in ameliorating cognitive deficits in both animal models [112,113,114] and patients with schizophrenia [115]. Intriguingly, it has been proven that sarcosine may enhance NMDA receptor function by more than one mechanism and may have different effects from NMDA receptor agonist like glycine [116]. In addition, other GlyT1 inhibitors, such as SSR103800 and SSR504734, have also exhibited similar beneficial effects in sensorimotor gating, learning and memory, and schizophrenia-like behaviors [117,118]. Furthermore, selective genetic disruption of GlyT1 resulted in enhancement of NMDA receptor function, memory retention and protects against an amphetamine disruption of sensory gating, suggesting that inhibition of GlyT1 might have both cognitive-enhancing and antipsychotic effects [119]. These findings indicate that GlyT1 is a promising drug target for the treatment of schizophrenia-related behaviors and cognitive deficits, even though the high binding affinity of the GlyT1 inhibitor can cause unpredictable toxicity [120,121,122].
4.2.2. Enhancement of NMDA Receptor Functions by DAAO Inhibitors
In contrast to the GlyT1 inhibitors, another promising therapeutic strategy for treating schizophrenia is to indirectly increase synaptic D-serine levels by targeting D-amino acid oxidase (DAAO). DAAO is an enzyme that degrades D-serine, by inhibiting DAAO via DAAO inhibitors, leading to increased synaptic D-serine levels and the regulation of NMDA receptor-evoked electrophysiological activity, thereby ameliorating the NMDA receptor hypofunction and cognitive deficits observed in schizophrenia. Interestingly, the expression and activity of DAAO are found to be elevated in individuals with schizophrenia, and this enhanced DAAO activity is thought to contribute to the reduced D-serine levels and subsequent impairment of NMDA receptor functioning [83,123]. Furthermore, genetic variations in DAAO and its activator have been associated with the negative symptoms and cognitive deficits observed in schizophrenic patients [124,125,126]. Adding to the therapeutic potential of DAAO inhibition, it has been reported that certain antipsychotic medications, such as chlorpromazine (a first-generation antipsychotic) and risperidone (a second-generation antipsychotic), may possess DAAO-inhibiting properties [127,128]. Indeed, elevated levels of D-serine have been observed in rodents after the administration of DAAO inhibitors [129,130]. Consistently, pre-pulse inhibition deficits and cognitive deficits relevant to schizophrenia were ameliorated after treatment with DAAO Inhibitors [130,131]. Moreover, mutant mice lacking DAAO exhibit increased NMDA receptor functions [132] and facilitate hippocampal LTP and spatial learning [133]. Other animal studies have indicated that DAAO is involved in the mechanism of D-serine nephrotoxicity [134], which also could be attenuated by DAAO Inhibitors [135]. Therefore, DAAO Inhibitors combined with D-serine or alone might be beneficial for enhancing NMDA receptor functions in schizophrenia.
4.2.3. Enhancement of NMDA Receptor Functions by other NMDAR Modulators
Several compounds that modulate NMDA receptor activity indirectly have been investigated as potential therapeutics for schizophrenia. For example, agents that target the glycine site on the NMDA receptor, such as D-cycloserine (DCS), has shown mixed results in treating negative symptoms and cognition of schizophrenia [136]. Lower doses of 50 mg/day produced persistent benefits for negative symptoms and memory deficits when added to first-generation antipsychotics [137,138], but higher doses of 100 mg/day or more worsened psychotic symptoms [139]. However, the efficacy of DCS did not achieve statistical significance in a meta-analysis of add-on trials, in contrast to the more consistently positive results with the full NMDAR co-agonist glycine [138,140]. Beyond its effects on symptoms, DCS positively modulates NMDAR-dependent forms of LTP and LTD in hippocampal brain slices of juvenile rats without affecting basal synaptic transmission [141]. The modulation of synaptic plasticity by DCS may contribute to its potential therapeutic effects, though the clinical efficacy appears to be influenced by factors like antipsychotic medication type.
Other modulators, including allosteric modulators and subtype-selective agonists, are also being explored for their therapeutic potential. These modulators bind to sites distinct from the agonist binding sites, inducing conformational changes that enhance or inhibit receptor function. Positive allosteric modulators (PAMs) can enhance NMDA receptor activity, potentially offering benefits for cognitive enhancement and treating psychiatric disorders. The relatively non-selective PAMs that alter NMDAR function independent of subunit composition, such as CAD-9303, has been studied in the treatment of the cognitive deficits and negative symptoms of schizophrenia [12]. GluN2A-selective PAMs including GNE-6901 and GNE-8324 have provided proof-of-principle for the development of allosteric modulators of NMDA receptors, however their poor pharmacokinetic properties and poor central nervous system exposures hinder their uses in vivo [142]. These PAMs also showed cell-selective functional differences in brain slice neurophysiology experiments, with GNE-6901 enhancing NMDA receptor synaptic responses on both excitatory and inhibitory neurons, whereas GNE-8324 selectively enhanced NMDA receptor response on inhibitory neurons but not excitatory neurons. The reason for this synaptic selectivity might involve differences in the microenvironment between synapses onto excitatory and inhibitory neurons that result in different susceptibility to potentiation by specific modes of PAM action [50]. Accumulating evidence suggests that GluN2B PAMs may have effects on cognitive function [50,143]. In contrast, negative allosteric modulators (NAMs) of GluN2B have been shown to induce schizophrenia-like effects and disrupt cognition, like to the actions of NMDA receptor blockers [144], suggesting GluN2B potentiation may induce an opposing effect.
Overall, targeting NMDA receptors offers a rational approach for addressing the underlying neurobiological abnormalities in schizophrenia. By restoring NMDA receptor function and synaptic plasticity, these therapeutic strategies hold the potential to improve cognitive deficits and alleviate symptoms, thereby enhancing the quality of life for individuals affected by schizophrenia.
5. Conclusions
The NMDA receptor hypofunction hypothesis provides a framework for understanding the neurobiological basis of schizophrenia, particularly its cognitive symptoms. Targeting NMDA receptor pathways represents a promising avenue for developing novel therapeutics aimed at restoring cognitive function and improving outcomes for individuals affected by schizophrenia. Data from clinical and animal studies have strongly implicated NMDA receptors as central hubs in the complex pathophysiological processes underlying schizophrenia. Accordingly, CIAS therapeutic drugs based on the regulation of NMDA receptor function are currently under active development. Importantly, several NMDAR-enhancing agents, particularly those that indirectly modulate NMDA receptor function, have demonstrated significant alleviation of schizophrenia-like behavioral deficits and cognitive dysfunctions in both animal models and patients with schizophrenia. Moreover, current findings suggest that indirectly targeting NMDA receptors appears to be more beneficial and results in fewer adverse effects than directly modulating NMDA receptor functions. Additionally, as the development of new antipsychotic drugs progresses, the establishment of comprehensive safety profiles for these potential compounds will be highly informative. This could elucidate their precise mechanisms of action and enable the evaluation of their therapeutic effects in both animal models and clinical studies.
Notably, however, this review presents an oversimplified summary of the treatment alternatives for an extremely complex psychiatric disorder. Human diseases are inherently more complex, and only certain aspects can be partially modeled in animal studies. Clinical trials remain essential and irreplaceable in the drug development process. Nonetheless, preclinical animal studies are highly valuable and indispensable for advancing our understanding of the underlying mechanisms and informing the development of new therapeutic interventions. Moving forward, integrating insights from single-cell sequencing techniques may unveil cell-type-specific NMDA receptor hypofunction in the etiology of schizophrenia, paving the way for more targeted and personalized therapeutic interventions.
Author Contributions
Ting Zhang: Writing– original draft, Data curation. Chang Liu: Writing– original draft, Data curation. Ning Zhong: Writing– review & editing. Yi-Chen Wang: Writing– review & editing. Yi-Yun Huang: Writing– review & editing. Xiao-Qin Zhang: Writing – review & editing, Supervision.
Funding
This work was supported by grants from the Natural Science Foundation of Zhejiang Province (LY24H090001), the Young Doctor Innovation Research Program of Ningbo (2022J083), the National Natural Science Foundation of China (32201322), the Municipal Key R&D Program of Ningbo (2023Z175) and the 111 Project of China (D16013).
Conflicts of Interest
The authors declare that they have no conflict of interest.
References
- Solmi, M.; Seitidis, G.; Mavridis, D.; Correll, C.U.; Dragioti, E.; Guimond, S.; Tuominen, L.; Dargél, A.; Carvalho, A.F.; Fornaro, M.; et al. Incidence, prevalence, and global burden of schizophrenia - data, with critical appraisal, from the Global Burden of Disease (GBD) 2019. Mol Psychiatry 2023, 28, 5319–5327. [Google Scholar] [CrossRef] [PubMed]
- Horan, W.P.; Catalano, L.T.; Green, M.F. An Update on Treatment of Cognitive Impairment Associated with Schizophrenia. Curr Top Behav Neurosci 2023, 63, 407–436. [Google Scholar] [CrossRef] [PubMed]
- Javitt, D.C. Cognitive Impairment Associated with Schizophrenia: From Pathophysiology to Treatment. Annu Rev Pharmacol Toxicol 2023, 63, 119–141. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, P.; Dey, A.; Gopalakrishnan, A.V.; Swati, K.; Ojha, S.; Prakash, A.; Kumar, D.; Ambasta, R.K.; Jha, N.K.; Jha, S.K.; et al. Glutamatergic neurotransmission: A potential pharmacotherapeutic target for the treatment of cognitive disorders. Ageing Res Rev 2023, 85, 101838. [Google Scholar] [CrossRef]
- Moghaddam, B.; Javitt, D. From revolution to evolution: the glutamate hypothesis of schizophrenia and its implication for treatment. Neuropsychopharmacology 2012, 37, 4–15. [Google Scholar] [CrossRef]
- Hansen, K.B.; Wollmuth, L.P.; Bowie, D.; Furukawa, H.; Menniti, F.S.; Sobolevsky, A.I.; Swanson, G.T.; Swanger, S.A.; Greger, I.H.; Nakagawa, T.; et al. Structure, Function, and Pharmacology of Glutamate Receptor Ion Channels. Pharmacol Rev 2021, 73, 298–487. [Google Scholar] [CrossRef]
- Zhou, C.; Tajima, N. Structural insights into NMDA receptor pharmacology. Biochem Soc Trans 2023, 51, 1713–1731. [Google Scholar] [CrossRef]
- Vyklicky, V.; Korinek, M.; Smejkalova, T.; Balik, A.; Krausova, B.; Kaniakova, M.; Lichnerova, K.; Cerny, J.; Krusek, J.; Dittert, I.; et al. Structure, function, and pharmacology of NMDA receptor channels. Physiol Res 2014, 63, S191–203. [Google Scholar] [CrossRef]
- Hansen, K.B.; Yi, F.; Perszyk, R.E.; Menniti, F.S.; Traynelis, S.F. NMDA Receptors in the Central Nervous System. Methods Mol Biol 2017, 1677, 1–80. [Google Scholar] [CrossRef]
- Lüscher, C.; Malenka, R.C. NMDA receptor-dependent long-term potentiation and long-term depression (LTP/LTD). Cold Spring Harb Perspect Biol 2012, 4. [Google Scholar] [CrossRef]
- Dupuis, J.P.; Nicole, O.; Groc, L. NMDA receptor functions in health and disease: Old actor, new dimensions. Neuron 2023, 111, 2312–2328. [Google Scholar] [CrossRef] [PubMed]
- Veselinović, T.; Neuner, I. Progress and pitfalls in developing agents to treat neurocognitive deficits associated with schizophrenia. CNS Drugs 2022, 36, 819–858. [Google Scholar] [CrossRef]
- Nakazawa, K.; Sapkota, K. The origin of NMDA receptor hypofunction in schizophrenia. Pharmacol Ther 2020, 205, 107426. [Google Scholar] [CrossRef]
- Coyle, J.T.; Tsai, G.; Goff, D. Converging evidence of NMDA receptor hypofunction in the pathophysiology of schizophrenia. Ann N Y Acad Sci 2003, 1003, 318–327. [Google Scholar] [CrossRef]
- Jentsch, J.D.; Roth, R.H. The neuropsychopharmacology of phencyclidine: from NMDA receptor hypofunction to the dopamine hypothesis of schizophrenia. Neuropsychopharmacology 1999, 20, 201–225. [Google Scholar] [CrossRef] [PubMed]
- Janus, A.; Lustyk, K.; Pytka, K. MK-801 and cognitive functions: Investigating the behavioral effects of a non-competitive NMDA receptor antagonist. Psychopharmacology (Berl) 2023, 240, 2435–2457. [Google Scholar] [CrossRef] [PubMed]
- Ramsey, A.J. NR1 knockdown mice as a representative model of the glutamate hypothesis of schizophrenia. Prog Brain Res 2009, 179, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Nakazawa, K.; Jeevakumar, V.; Nakao, K. Spatial and temporal boundaries of NMDA receptor hypofunction leading to schizophrenia. NPJ Schizophr 2017, 3, 7. [Google Scholar] [CrossRef]
- Bygrave, A.M.; Masiulis, S.; Nicholson, E.; Berkemann, M.; Barkus, C.; Sprengel, R.; Harrison, P.J.; Kullmann, D.M.; Bannerman, D.M.; Kätzel, D. Knockout of NMDA-receptors from parvalbumin interneurons sensitizes to schizophrenia-related deficits induced by MK-801. Transl Psychiatry 2016, 6, e778. [Google Scholar] [CrossRef]
- Dalmau, J.; Gleichman, A.J.; Hughes, E.G.; Rossi, J.E.; Peng, X.; Lai, M.; Dessain, S.K.; Rosenfeld, M.R.; Balice-Gordon, R.; Lynch, D.R. Anti-NMDA-receptor encephalitis: case series and analysis of the effects of antibodies. Lancet Neurol 2008, 7, 1091–1098. [Google Scholar] [CrossRef]
- Snyder, M.A.; Gao, W.J. NMDA receptor hypofunction for schizophrenia revisited: Perspectives from epigenetic mechanisms. Schizophr Res 2020, 217, 60–70. [Google Scholar] [CrossRef] [PubMed]
- Cotman, C.W.; Nieto-Sampedro, M. Cell biology of synaptic plasticity. Science 1984, 225, 1287–1294. [Google Scholar] [CrossRef] [PubMed]
- Catts, V.S.; Lai, Y.L.; Weickert, C.S.; Weickert, T.W.; Catts, S.V. A quantitative review of the postmortem evidence for decreased cortical N-methyl-D-aspartate receptor expression levels in schizophrenia: How can we link molecular abnormalities to mismatch negativity deficits? Biol Psychol 2016, 116, 57–67. [Google Scholar] [CrossRef]
- Schwarcz, R.; Rassoulpour, A.; Wu, H.Q.; Medoff, D.; Tamminga, C.A.; Roberts, R.C. Increased cortical kynurenate content in schizophrenia. Biol Psychiatry 2001, 50, 521–530. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, K.; Engberg, G.; Shimizu, E.; Nordin, C.; Lindström, L.H.; Iyo, M. Reduced D-serine to total serine ratio in the cerebrospinal fluid of drug naive schizophrenic patients. Prog Neuropsychopharmacol Biol Psychiatry 2005, 29, 767–769. [Google Scholar] [CrossRef]
- Psychiatric genome-wide association study analyses implicate neuronal, immune and histone pathways. Nat Neurosci 2015, 18, 199–209. [CrossRef]
- Kirov, G.; Pocklington, A.J.; Holmans, P.; Ivanov, D.; Ikeda, M.; Ruderfer, D.; Moran, J.; Chambert, K.; Toncheva, D.; Georgieva, L.; et al. De novo CNV analysis implicates specific abnormalities of postsynaptic signalling complexes in the pathogenesis of schizophrenia. Mol Psychiatry 2012, 17, 142–153. [Google Scholar] [CrossRef] [PubMed]
- Mohn, A.R.; Gainetdinov, R.R.; Caron, M.G.; Koller, B.H. Mice with reduced NMDA receptor expression display behaviors related to schizophrenia. Cell 1999, 98, 427–436. [Google Scholar] [CrossRef]
- Zhang, X.-Q.; Xu, L.; Zhu, X.-Y.; Tang, Z.-H.; Dong, Y.-B.; Yu, Z.-P.; Shang, Q.; Wang, Z.-C.; Shen, H.-W. D-serine reconstitutes synaptic and intrinsic inhibitory control of pyramidal neurons in a neurodevelopmental mouse model for schizophrenia. Nature Communications 2023, 14, 8255. [Google Scholar] [CrossRef]
- Sawa, A.; Snyder, S.H. Schizophrenia: diverse approaches to a complex disease. Science 2002, 296, 692–695. [Google Scholar] [CrossRef]
- Zhang, X.Q.; Xu, L.; Ling, Y.; Hu, L.B.; Huang, J.; Shen, H.W. Diminished excitatory synaptic transmission correlates with impaired spatial working memory in neurodevelopmental rodent models of schizophrenia. Pharmacol Biochem Behav 2021, 10.1016/j.pbb.2021.173103, 173103. [CrossRef]
- Stefansson, H.; Sigurdsson, E.; Steinthorsdottir, V.; Bjornsdottir, S.; Sigmundsson, T.; Ghosh, S.; Brynjolfsson, J.; Gunnarsdottir, S.; Ivarsson, O.; Chou, T.T.; et al. Neuregulin 1 and susceptibility to schizophrenia. Am J Hum Genet 2002, 71, 877–892. [Google Scholar] [CrossRef] [PubMed]
- Gu, Z.; Jiang, Q.; Fu, A.K.; Ip, N.Y.; Yan, Z. Regulation of NMDA receptors by neuregulin signaling in prefrontal cortex. J Neurosci 2005, 25, 4974–4984. [Google Scholar] [CrossRef] [PubMed]
- Hahn, C.G.; Wang, H.Y.; Cho, D.S.; Talbot, K.; Gur, R.E.; Berrettini, W.H.; Bakshi, K.; Kamins, J.; Borgmann-Winter, K.E.; Siegel, S.J.; et al. Altered neuregulin 1-erbB4 signaling contributes to NMDA receptor hypofunction in schizophrenia. Nat Med 2006, 12, 824–828. [Google Scholar] [CrossRef] [PubMed]
- Levinson, J.N.; El-Husseini, A. Building excitatory and inhibitory synapses: balancing neuroligin partnerships. Neuron 2005, 48, 171–174. [Google Scholar] [CrossRef]
- Sohal, V.S.; Rubenstein, J.L.R. Excitation-inhibition balance as a framework for investigating mechanisms in neuropsychiatric disorders. Mol Psychiatry 2019, 24, 1248–1257. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Sun, X.; Duan, J. NMDARs regulate the excitatory-inhibitory balance within neural circuits. Brain Science Advances 2023, 9, 3–14. [Google Scholar] [CrossRef]
- Balu, D.T. The NMDA receptor and schizophrenia: From pathophysiology to treatment. Adv Pharmacol 2016, 76, 351–382. [Google Scholar] [CrossRef]
- Gaebler, A.J.; Fakour, N.; Stöhr, F.; Zweerings, J.; Taebi, A.; Suslova, M.; Dukart, J.; Hipp, J.F.; Adhikari, B.M.; Kochunov, P.; et al. Functional connectivity signatures of NMDAR dysfunction in schizophrenia—integrating findings from imaging genetics and pharmaco-fMRI. Translational Psychiatry 2023, 13, 59. [Google Scholar] [CrossRef]
- Gonzalez-Burgos, G.; Lewis, D.A. NMDA receptor hypofunction, parvalbumin-positive neurons, and cortical gamma oscillations in schizophrenia. Schizophr Bull 2012, 38, 950–957. [Google Scholar] [CrossRef]
- Gonzalez-Burgos, G.; Fish, K.N.; Lewis, D.A. GABA neuron alterations, cortical circuit dysfunction and cognitive deficits in schizophrenia. Neural Plast 2011, 2011, 723184. [Google Scholar] [CrossRef]
- Sigurdsson, T. Neural circuit dysfunction in schizophrenia: Insights from animal models. Neuroscience 2016, 321, 42–65. [Google Scholar] [CrossRef] [PubMed]
- Jadi, M.P.; Behrens, M.M.; Sejnowski, T.J. Abnormal gamma oscillations in N-methyl-D-aspartate receptor hypofunction models of schizophrenia. Biol Psychiatry 2016, 79, 716–726. [Google Scholar] [CrossRef]
- Uhlhaas, P.J.; Singer, W. Abnormal neural oscillations and synchrony in schizophrenia. Nat Rev Neurosci 2010, 11, 100–113. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Burgos, G.; Cho, R.Y.; Lewis, D.A. Alterations in cortical network oscillations and parvalbumin neurons in schizophrenia. Biol Psychiatry 2015, 77, 1031–1040. [Google Scholar] [CrossRef]
- Ellaithy, A.; Younkin, J.; González-Maeso, J.; Logothetis, D.E. Positive allosteric modulators of metabotropic glutamate 2 receptors in schizophrenia treatment. Trends Neurosci 2015, 38, 506–516. [Google Scholar] [CrossRef] [PubMed]
- Selvaraj, S.; Arnone, D.; Cappai, A.; Howes, O. Alterations in the serotonin system in schizophrenia: a systematic review and meta-analysis of postmortem and molecular imaging studies. Neurosci Biobehav Rev 2014, 45, 233–245. [Google Scholar] [CrossRef] [PubMed]
- Yuen, E.Y.; Jiang, Q.; Chen, P.; Feng, J.; Yan, Z. Activation of 5-HT2A/C receptors counteracts 5-HT1A regulation of n-methyl-D-aspartate receptor channels in pyramidal neurons of prefrontal cortex. J Biol Chem 2008, 283, 17194–17204. [Google Scholar] [CrossRef]
- Yuen, E.Y.; Jiang, Q.; Chen, P.; Gu, Z.; Feng, J.; Yan, Z. Serotonin 5-HT1A receptors regulate NMDA receptor channels through a microtubule-dependent mechanism. J Neurosci 2005, 25, 5488–5501. [Google Scholar] [CrossRef]
- Hanson, J.E.; Yuan, H.; Perszyk, R.E.; Banke, T.G.; Xing, H.; Tsai, M.C.; Menniti, F.S.; Traynelis, S.F. Therapeutic potential of N-methyl-D-aspartate receptor modulators in psychiatry. Neuropsychopharmacology 2024, 49, 51–66. [Google Scholar] [CrossRef]
- López-Corcuera, B.; Geerlings, A.; Aragón, C. Glycine neurotransmitter transporters: an update. Mol Membr Biol 2001, 18, 13–20. [Google Scholar] [CrossRef]
- Ishimaru, M.; Kurumaji, A.; Toru, M. Increases in strychnine-insensitive glycine binding sites in cerebral cortex of chronic schizophrenics: evidence for glutamate hypothesis. Biol Psychiatry 1994, 35, 84–95. [Google Scholar] [CrossRef] [PubMed]
- Heresco-Levy, U.; Bar, G.; Levin, R.; Ermilov, M.; Ebstein, R.P.; Javitt, D.C. High glycine levels are associated with prepulse inhibition deficits in chronic schizophrenia patients. Schizophr Res 2007, 91, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Waziri, R.; Baruah, S. A hyperglycinergic rat model for the pathogenesis of schizophrenia: preliminary findings. Schizophr Res 1999, 37, 205–215. [Google Scholar] [CrossRef]
- Neeman, G.; Blanaru, M.; Bloch, B.; Kremer, I.; Ermilov, M.; Javitt, D.C.; Heresco-Levy, U. Relation of plasma glycine, serine, and homocysteine levels to schizophrenia symptoms and medication type. Am J Psychiatry 2005, 162, 1738–1740. [Google Scholar] [CrossRef] [PubMed]
- Mouri, A.; Noda, Y.; Noda, A.; Nakamura, T.; Tokura, T.; Yura, Y.; Nitta, A.; Furukawa, H.; Nabeshima, T. Involvement of a dysfunctional dopamine-D1/N-methyl-d-aspartate-NR1 and Ca2+/calmodulin-dependent protein kinase II pathway in the impairment of latent learning in a model of schizophrenia induced by phencyclidine. Mol Pharmacol 2007, 71, 1598–1609. [Google Scholar] [CrossRef]
- Hoffman, K.L.; Basurto, E. Clozapine and glycinamide prevent MK-801-induced deficits in the novel object recognition (NOR) test in the domestic rabbit (Oryctolagus cuniculus). Behav Brain Res 2014, 271, 203–211. [Google Scholar] [CrossRef]
- Waziri, R. Glycine therapy of schizophrenia. Biological psychiatry 1988, 23, 210–211. [Google Scholar] [CrossRef]
- Rosse, R.B.; Theut, S.K.; Banay-Schwartz, M.; Leighton, M.; Scarcella, E.; Cohen, C.G.; Deutsch, S.I. Glycine adjuvant therapy to conventional neuroleptic treatment in schizophrenia: an open-label, pilot study. Clinical neuropharmacology 1989, 12, 416–424. [Google Scholar] [CrossRef]
- Costa, J.; Khaled, E.; Sramek, J.; Bunney, W., Jr.; Potkin, S.G. An open trial of glycine as an adjunct to neuroleptics in chronic treatment-refractory schizophrenics. Journal of clinical psychopharmacology 1990, 10, 71–72. [Google Scholar] [CrossRef]
- Javitt, D.C.; Zylberman, I.; Zukin, S.R.; Heresco-Levy, U.; Lindenmayer, J.P. Amelioration of negative symptoms in schizophrenia by glycine. The American journal of psychiatry 1994, 151, 1234–1236. [Google Scholar]
- Leiderman, E.; Zylberman, I.; Zukin, S.R.; Cooper, T.B.; Javitt, D.C. Preliminary investigation of high-dose oral glycine on serum levels and negative symptoms in schizophrenia: An open-label trial. Biological Psychiatry 1996, 39, 213–215. [Google Scholar] [CrossRef]
- HerescoLevy, U.; Javitt, D.C.; Ermilov, M.; Mordel, C.; Horowitz, A.; Kelly, D. Double-blind, placebo-controlled, crossover trial of glycine adjuvant therapy for treatment-resistant schizophrenia. British Journal of Psychiatry 1996, 169, 610–617. [Google Scholar] [CrossRef] [PubMed]
- Heresco-Levy, U.; Javitt, D.C.; Ermilov, M.; Mordel, C.; Silipo, G.; Lichtenstein, M. Efficacy of high-dose glycine in the treatment of enduring negative symptoms of schizophrenia. Archives of General Psychiatry 1999, 56, 29–36. [Google Scholar] [CrossRef]
- Potkin, S.G.; Jin, Y.; Bunney, B.G.; Costa, J.; Gulasekaram, B. Effect of clozapine and adjunctive high-dose glycine in treatment-resistant schizophrenia. American Journal of Psychiatry 1999, 156, 145–147. [Google Scholar] [CrossRef]
- Evins, A.E.; Fitzgerald, S.M.; Wine, L.; Rosselli, R.; Goff, D.C. Placebo-controlled trial of glycine added to clozapine in schizophrenia. American Journal of Psychiatry 2000, 157, 826–828. [Google Scholar] [CrossRef] [PubMed]
- E, E.A.; M, F.S.; L, W.; R, R.; C, G.D. Placebo-controlled trial of glycine added to clozapine in schizophrenia. The American journal of psychiatry 2000, 157, 826–828. [Google Scholar]
- Heresco-Levy, U.; Ermilov, M.; Lichtenberg, P.; Bar, G.; Javitt, D.C. High-dose glycine added to olanzapine and risperidone for the treatment of schizophrenia. Biological Psychiatry 2004, 55, 165–171. [Google Scholar] [CrossRef]
- Pablo, D.; Sreenivasa, B.; M, D.S.; Bill, D. Double-blind, placebo-controlled, crossover trial of clozapine plus glycine in refractory schizophrenia negative results. Journal of clinical psychopharmacology 2005, 25, 277–278. [Google Scholar]
- Greenwood, L.-M.; Leung, S.; Michie, P.T.; Green, A.; Nathan, P.J.; Fitzgerald, P.; Johnston, P.; Solowij, N.; Kulkarni, J.; Croft, R.J. The effects of glycine on auditory mismatch negativity in schizophrenia. Schizophrenia Research 2018, 191, 61–69. [Google Scholar] [CrossRef]
- McLean Hospital, B., Massachusetts; Department of Psychiatry, H.M.S., Boston, Massachusetts; McLean Hospital, B., Massachusetts; Department of Psychiatry, H.M.S., Boston, Massachusetts; McLean Hospital, B., Massachusetts; Molecular, D.o.; Human Genetics, B.C.o.M., Houston, Texas; Department of Biostatistics, U.o.A.a.B., Birmingham, Alabama; New York State Psychiatric Institute, N.Y.; McLean Hospital, B., Massachusetts, et al. Targeted Treatment of Individuals With Psychosis Carrying a Copy Number Variant Containing a Genomic Triplication of the Glycine Decarboxylase Gene. Biological Psychiatry 2019, 86, 523–535.
- Serrita, J.; Ralevski, E.; Yoon, G.; Petrakis, I. A Pilot Randomized, Placebo-Controlled Trial of Glycine for Treatment of Schizophrenia and Alcohol Dependence. Journal of Dual Diagnosis 2019, 15, 46–55. [Google Scholar] [CrossRef] [PubMed]
- Strzelecki, D.; Kałuzyńska, O.; Józefowicz, O.; Pawełczyk, T.; Rabe-Jabłońska, J. Initial glycine serum level is not a predictor of the recovery resulting from glycine augmentation of antipsychotic treatment. Archives of Psychiatry and Psychotherapy 2011, 13, 5–11. [Google Scholar]
- Mothet, J.P.; Parent, A.T.; Wolosker, H.; Brady, R.O., Jr.; Linden, D.J.; Ferris, C.D.; Rogawski, M.A.; Snyder, S.H. D-serine is an endogenous ligand for the glycine site of the N-methyl-D-aspartate receptor. Proc Natl Acad Sci U S A 2000, 97, 4926–4931. [Google Scholar] [CrossRef] [PubMed]
- Mothet, J.P.; Le Bail, M.; Billard, J.M. Time and space profiling of NMDA receptor co-agonist functions. J Neurochem 2015, 135, 210–225. [Google Scholar] [CrossRef] [PubMed]
- Papouin, T.; Ladépêche, L.; Ruel, J.; Sacchi, S.; Labasque, M.; Hanini, M.; Groc, L.; Pollegioni, L.; Mothet, J.P.; Oliet, S.H. Synaptic and extrasynaptic NMDA receptors are gated by different endogenous coagonists. Cell 2012, 150, 633–646. [Google Scholar] [CrossRef]
- Yang, Y.; Ge, W.; Chen, Y.; Zhang, Z.; Shen, W.; Wu, C.; Poo, M.; Duan, S. Contribution of astrocytes to hippocampal long-term potentiation through release of D-serine. Proc Natl Acad Sci U S A 2003, 100, 15194–15199. [Google Scholar] [CrossRef]
- Hardingham, G.E.; Bading, H. Synaptic versus extrasynaptic NMDA receptor signalling: implications for neurodegenerative disorders. Nat Rev Neurosci 2010, 11, 682–696. [Google Scholar] [CrossRef]
- Li, S.T.; Ju, J.G. Functional roles of synaptic and extrasynaptic NMDA receptors in physiological and pathological neuronal activities. Curr Drug Targets 2012, 13, 207–221. [Google Scholar] [CrossRef]
- Hashimoto, K.; Fukushima, T.; Shimizu, E.; Komatsu, N.; Watanabe, H.; Shinoda, N.; Nakazato, M.; Kumakiri, C.; Okada, S.; Hasegawa, H.; et al. Decreased serum levels of D-serine in patients with schizophrenia: evidence in support of the N-methyl-D-aspartate receptor hypofunction hypothesis of schizophrenia. Arch Gen Psychiatry 2003, 60, 572–576. [Google Scholar] [CrossRef]
- Bendikov, I.; Nadri, C.; Amar, S.; Panizzutti, R.; De Miranda, J.; Wolosker, H.; Agam, G. A CSF and postmortem brain study of D-serine metabolic parameters in schizophrenia. Schizophr Res 2007, 90, 41–51. [Google Scholar] [CrossRef]
- Fujita, Y.; Ishima, T.; Hashimoto, K. Supplementation with D-serine prevents the onset of cognitive deficits in adult offspring after maternal immune activation. Sci Rep 2016, 6, 37261. [Google Scholar] [CrossRef] [PubMed]
- Verrall, L.; Burnet, P.W.; Betts, J.F.; Harrison, P.J. The neurobiology of D-amino acid oxidase and its involvement in schizophrenia. Mol Psychiatry 2010, 15, 122–137. [Google Scholar] [CrossRef] [PubMed]
- Carone, F.A.; Ganote, C.E. D-serine nephrotoxicity. The nature of proteinuria, glucosuria, and aminoaciduria in acute tubular necrosis. Arch Pathol 1975, 99, 658–662. [Google Scholar]
- Suzuki, M.; Gonda, Y.; Yamada, M.; Vandebroek, A.A.; Mita, M.; Hamase, K.; Yasui, M.; Sasabe, J. Serum D-serine accumulation after proximal renal tubular damage involves neutral amino acid transporter Asc-1. Sci Rep 2019, 9, 16705. [Google Scholar] [CrossRef]
- Mothet, J.P.; Billard, J.M.; Pollegioni, L.; Coyle, J.T.; Sweedler, J.V. Investigating brain d-serine: Advocacy for good practices. Acta Physiol (Oxf) 2019, 226, e13257. [Google Scholar] [CrossRef]
- Kantrowitz, J.T.; Woods, S.W.; Petkova, E.; Cornblatt, B.; Corcoran, C.M.; Chen, H.; Silipo, G.; Javitt, D.C. D-serine for the treatment of negative symptoms in individuals at clinical high risk of schizophrenia: a pilot, double-blind, placebo-controlled, randomised parallel group mechanistic proof-of-concept trial. Lancet Psychiatry 2015, 2, 403–412. [Google Scholar] [CrossRef]
- Tsai, G.; Yang, P.; Chung, L.C.; Lange, N.; Coyle, J.T. D-serine added to antipsychotics for the treatment of schizophrenia. Biol Psychiatry 1998, 44, 1081–1089. [Google Scholar] [CrossRef]
- Weiser, M.; Heresco-Levy, U.; Davidson, M.; Javitt, D.C.; Werbeloff, N.; Gershon, A.A.; Abramovich, Y.; Amital, D.; Doron, A.; Konas, S.; et al. A multicenter, add-on randomized controlled trial of low-dose d-serine for negative and cognitive symptoms of schizophrenia. J Clin Psychiatry 2012, 73, e728–734. [Google Scholar] [CrossRef]
- Kantrowitz, J.T.; Epstein, M.L.; Lee, M.; Lehrfeld, N.; Nolan, K.A.; Shope, C.; Petkova, E.; Silipo, G.; Javitt, D.C. Improvement in mismatch negativity generation during d-serine treatment in schizophrenia: Correlation with symptoms. Schizophr Res 2018, 191, 70–79. [Google Scholar] [CrossRef] [PubMed]
- da Silva Lde, B.; Leipnitz, G.; Seminotti, B.; Fernandes, C.G.; Beskow, A.P.; Amaral, A.U.; Wajner, M. D-serine induces lipid and protein oxidative damage and decreases glutathione levels in brain cortex of rats. Brain Res 2009, 1256, 34–42. [Google Scholar] [CrossRef]
- Katsuki, H.; Nonaka, M.; Shirakawa, H.; Kume, T.; Akaike, A. Endogenous D-serine is involved in induction of neuronal death by N-methyl-D-aspartate and simulated ischemia in rat cerebrocortical slices. J Pharmacol Exp Ther 2004, 311, 836–844. [Google Scholar] [CrossRef] [PubMed]
- Ganote, C.E.; Peterson, D.R.; Carone, F.A. The nature of D-serine--induced nephrotoxicity. Am J Pathol 1974, 77, 269–282. [Google Scholar]
- Kantrowitz, J.T.; Malhotra, A.K.; Cornblatt, B.; Silipo, G.; Balla, A.; Suckow, R.F.; D'Souza, C.; Saksa, J.; Woods, S.W.; Javitt, D.C. High dose D-serine in the treatment of schizophrenia. Schizophr Res 2010, 121, 125–130. [Google Scholar] [CrossRef] [PubMed]
- Tsai, G.E.; Yang, P.; Chung, L.C.; Tsai, I.C.; Tsai, C.W.; Coyle, J.T. D-serine added to clozapine for the treatment of schizophrenia. Am J Psychiatry 1999, 156, 1822–1825. [Google Scholar] [CrossRef]
- Heresco-Levy, U.; Javitt, D.C.; Ebstein, R.; Vass, A.; Lichtenberg, P.; Bar, G.; Catinari, S.; Ermilov, M. D-serine efficacy as add-on pharmacotherapy to risperidone and olanzapine for treatment-refractory schizophrenia. Biol Psychiatry 2005, 57, 577–585. [Google Scholar] [CrossRef]
- Lane, H.Y.; Lin, C.H.; Huang, Y.J.; Liao, C.H.; Chang, Y.C.; Tsai, G.E. A randomized, double-blind, placebo-controlled comparison study of sarcosine (N-methylglycine) and D-serine add-on treatment for schizophrenia. Int J Neuropsychopharmacol 2010, 13, 451–460. [Google Scholar] [CrossRef]
- Ermilov, M.; Gelfin, E.; Levin, R.; Lichtenberg, P.; Hashimoto, K.; Javitt, D.C.; Heresco-Levy, U. A pilot double-blind comparison of d-serine and high-dose olanzapine in treatment-resistant patients with schizophrenia. Schizophr Res 2013, 150, 604–605. [Google Scholar] [CrossRef] [PubMed]
- D'Souza, D.C.; Radhakrishnan, R.; Perry, E.; Bhakta, S.; Singh, N.M.; Yadav, R.; Abi-Saab, D.; Pittman, B.; Chaturvedi, S.K.; Sharma, M.P.; et al. Feasibility, safety, and efficacy of the combination of D-serine and computerized cognitive retraining in schizophrenia: an international collaborative pilot study. Neuropsychopharmacology 2013, 38, 492–503. [Google Scholar] [CrossRef]
- Heresco-Levy, U.; Durrant, A.R.; Ermilov, M.; Javitt, D.C.; Miya, K.; Mori, H. Clinical and electrophysiological effects of D-serine in a schizophrenia patient positive for anti-N-methyl-D-aspartate receptor antibodies. Biol Psychiatry 2015, 77, e27–29. [Google Scholar] [CrossRef]
- Kantrowitz, J.T.; Epstein, M.L.; Beggel, O.; Rohrig, S.; Lehrfeld, J.M.; Revheim, N.; Lehrfeld, N.P.; Reep, J.; Parker, E.; Silipo, G.; et al. Neurophysiological mechanisms of cortical plasticity impairments in schizophrenia and modulation by the NMDA receptor agonist D-serine. Brain 2016, 139, 3281–3295. [Google Scholar] [CrossRef]
- Sehatpour, P.; Iosifescu, D.V.; De Baun, H.M.; Shope, C.; Mayer, M.R.; Gangwisch, J.; Dias, E.; Sobeih, T.; Choo, T.H.; Wall, M.M.; et al. Dose-dependent augmentation of neuroplasticity-based auditory learning in schizophrenia: A double-blind, placebo-controlled, randomized, target engagement clinical trial of the NMDA glutamate receptor agonist d-serine. Biol Psychiatry 2023, 94, 164–173. [Google Scholar] [CrossRef] [PubMed]
- Sur, C.; Kinney, G.G. Glycine transporter 1 inhibitors and modulation of NMDA receptor-mediated excitatory neurotransmission. Curr Drug Targets 2007, 8, 643–649. [Google Scholar] [CrossRef] [PubMed]
- Balu, D.T.; Coyle, J.T. The NMDA receptor 'glycine modulatory site' in schizophrenia: D-serine, glycine, and beyond. Curr Opin Pharmacol 2015, 20, 109–115. [Google Scholar] [CrossRef]
- Piniella, D.; Zafra, F. Functional crosstalk of the glycine transporter GlyT1 and NMDA receptors. Neuropharmacology 2023, 232, 109514. [Google Scholar] [CrossRef] [PubMed]
- Rosenbrock, H.; Desch, M.; Wunderlich, G. Development of the novel GlyT1 inhibitor, iclepertin (BI 425809), for the treatment of cognitive impairment associated with schizophrenia. Eur Arch Psychiatry Clin Neurosci 2023, 273, 1557–1566. [Google Scholar] [CrossRef] [PubMed]
- Dudzik, P.; Lustyk, K.; Pytka, K. Beyond dopamine: Novel strategies for schizophrenia treatment. Med Res Rev 2024, 10.1002/med.22042. [CrossRef]
- Surti, T.S.; Ranganathan, M.; Johannesen, J.K.; Gueorguieva, R.; Deaso, E.; Kenney, J.G.; Krystal, J.H.; D'Souza, D.C. Randomized controlled trial of the glycine transporter 1 inhibitor PF-03463275 to enhance cognitive training and neuroplasticity in schizophrenia. Schizophr Res 2023, 256, 36–43. [Google Scholar] [CrossRef]
- Javitt, D.C.; Sershen, H.; Hashim, A.; Lajtha, A. Reversal of phencyclidine-induced hyperactivity by glycine and the glycine uptake inhibitor glycyldodecylamide. Neuropsychopharmacology 1997, 17, 202–204. [Google Scholar] [CrossRef]
- Hashimoto, K.; Fujita, Y.; Ishima, T.; Chaki, S.; Iyo, M. Phencyclidine-induced cognitive deficits in mice are improved by subsequent subchronic administration of the glycine transporter-1 inhibitor NFPS and D-serine. Eur Neuropsychopharmacol 2008, 18, 414–421. [Google Scholar] [CrossRef]
- Kopec, K.; Flood, D.G.; Gasior, M.; McKenna, B.A.; Zuvich, E.; Schreiber, J.; Salvino, J.M.; Durkin, J.T.; Ator, M.A.; Marino, M.J. Glycine transporter (GlyT1) inhibitors with reduced residence time increase prepulse inhibition without inducing hyperlocomotion in DBA/2 mice. Biochem Pharmacol 2010, 80, 1407–1417. [Google Scholar] [CrossRef]
- Chen, H.H.; Stoker, A.; Markou, A. The glutamatergic compounds sarcosine and N-acetylcysteine ameliorate prepulse inhibition deficits in metabotropic glutamate 5 receptor knockout mice. Psychopharmacology (Berl) 2010, 209, 343–350. [Google Scholar] [CrossRef]
- Kumar, A.; Akhtar, A.; Kuhad, A.; Sah, S.P. Sarcosine (glycine transporter inhibitor) attenuates behavioural and biochemical changes induced by ketamine, in the rat model of schizophrenia. Exp Brain Res 2023, 241, 451–467. [Google Scholar] [CrossRef] [PubMed]
- Pei, J.C.; Hung, W.L.; Lin, B.X.; Shih, M.H.; Lu, L.Y.; Luo, D.Z.; Tai, H.C.; Studer, V.; Min, M.Y.; Lai, W.S. Therapeutic potential and underlying mechanism of sarcosine (N-methylglycine) in N-methyl-D-aspartate (NMDA) receptor hypofunction models of schizophrenia. J Psychopharmacol 2019, 33, 1288–1302. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.H.; Lin, C.H.; Liu, C.Y.; Chen, S.J.; Lane, H.Y. Efficacy and cognitive effect of sarcosine (N-methylglycine) in patients with schizophrenia: A systematic review and meta-analysis of double-blind randomised controlled trials. J Psychopharmacol 2020, 34, 495–505. [Google Scholar] [CrossRef]
- Zhang, H.X.; Hyrc, K.; Thio, L.L. The glycine transport inhibitor sarcosine is an NMDA receptor co-agonist that differs from glycine. J Physiol 2009, 587, 3207–3220. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, H.; Inoue, T.; Izumi, T.; Nakagawa, S.; Koyama, T. SSR504734, a glycine transporter-1 inhibitor, attenuates acquisition and expression of contextual conditioned fear in rats. Behav Pharmacol 2010, 21, 576–579. [Google Scholar] [CrossRef]
- Black, M.D.; Varty, G.B.; Arad, M.; Barak, S.; De Levie, A.; Boulay, D.; Pichat, P.; Griebel, G.; Weiner, I. Procognitive and antipsychotic efficacy of glycine transport 1 inhibitors (GlyT1) in acute and neurodevelopmental models of schizophrenia: latent inhibition studies in the rat. Psychopharmacology (Berl) 2009, 202, 385–396. [Google Scholar] [CrossRef]
- Tsai, G.; Ralph-Williams, R.J.; Martina, M.; Bergeron, R.; Berger-Sweeney, J.; Dunham, K.S.; Jiang, Z.; Caine, S.B.; Coyle, J.T. Gene knockout of glycine transporter 1: characterization of the behavioral phenotype. Proc Natl Acad Sci U S A 2004, 101, 8485–8490. [Google Scholar] [CrossRef]
- Cioffi, C.L. Glycine transporter-1 inhibitors: a patent review (2011-2016). Expert Opin Ther Pat 2018, 28, 197–210. [Google Scholar] [CrossRef]
- Hashimoto, K. Glycine transporter inhibitors as therapeutic agents for schizophrenia. Recent Pat CNS Drug Discov 2006, 1, 43–53. [Google Scholar] [CrossRef]
- Hashimoto, K. Glycine transport inhibitors for the treatment of schizophrenia. Open Med Chem J 2010, 4, 10–19. [Google Scholar] [CrossRef]
- Verrall, L.; Walker, M.; Rawlings, N.; Benzel, I.; Kew, J.N.; Harrison, P.J.; Burnet, P.W. d-Amino acid oxidase and serine racemase in human brain: normal distribution and altered expression in schizophrenia. Eur J Neurosci 2007, 26, 1657–1669. [Google Scholar] [CrossRef] [PubMed]
- Jagannath, V.; Theodoridou, A.; Gerstenberg, M.; Franscini, M.; Heekeren, K.; Correll, C.U.; Rössler, W.; Grünblatt, E.; Walitza, S. Prediction analysis for transition to schizophrenia in individuals at clinical High risk for psychosis: the relationship of DAO, DAOA, and NRG1 variants with negative symptoms and cognitive deficits. Front Psychiatry 2017, 8, 292. [Google Scholar] [CrossRef]
- Hall, J.; Whalley, H.C.; Moorhead, T.W.; Baig, B.J.; McIntosh, A.M.; Job, D.E.; Owens, D.G.; Lawrie, S.M.; Johnstone, E.C. Genetic variation in the DAOA (G72) gene modulates hippocampal function in subjects at high risk of schizophrenia. Biol Psychiatry 2008, 64, 428–433. [Google Scholar] [CrossRef]
- Lin, E.; Lin, C.H.; Lai, Y.L.; Huang, C.H.; Huang, Y.J.; Lane, H.Y. Combination of G72 genetic variation and G72 protein level to detect schizophrenia: machine learning approaches. Front Psychiatry 2018, 9, 566. [Google Scholar] [CrossRef] [PubMed]
- Iwasa, S.; Tabara, H.; Song, Z.; Nakabayashi, M.; Yokoyama, Y.; Fukushima, T. Inhibition of D-amino acid oxidase activity by antipsychotic drugs evaluated by a fluorometric assay using D-kynurenine as substrate. Yakugaku Zasshi 2011, 131, 1111–1116. [Google Scholar] [CrossRef]
- Krogmann, A.; Peters, L.; von Hardenberg, L.; Bödeker, K.; Nöhles, V.B.; Correll, C.U. Keeping up with the therapeutic advances in schizophrenia: a review of novel and emerging pharmacological entities. CNS Spectr 2019, 24, 38–69. [Google Scholar] [CrossRef]
- Adage, T.; Trillat, A.C.; Quattropani, A.; Perrin, D.; Cavarec, L.; Shaw, J.; Guerassimenko, O.; Giachetti, C.; Gréco, B.; Chumakov, I.; et al. In vitro and in vivo pharmacological profile of AS057278, a selective d-amino acid oxidase inhibitor with potential anti-psychotic properties. Eur Neuropsychopharmacol 2008, 18, 200–214. [Google Scholar] [CrossRef]
- Kuo, C.Y.; Lin, C.H.; Lane, H.Y. Targeting D-amino acid oxidase (DAAO) for the treatment of schizophrenia: rationale and current status of research. CNS Drugs 2022, 36, 1143–1153. [Google Scholar] [CrossRef]
- Hashimoto, K.; Fujita, Y.; Horio, M.; Kunitachi, S.; Iyo, M.; Ferraris, D.; Tsukamoto, T. Co-administration of a D-amino acid oxidase inhibitor potentiates the efficacy of D-serine in attenuating prepulse inhibition deficits after administration of dizocilpine. Biol Psychiatry 2009, 65, 1103–1106. [Google Scholar] [CrossRef] [PubMed]
- Almond, S.L.; Fradley, R.L.; Armstrong, E.J.; Heavens, R.B.; Rutter, A.R.; Newman, R.J.; Chiu, C.S.; Konno, R.; Hutson, P.H.; Brandon, N.J. Behavioral and biochemical characterization of a mutant mouse strain lacking D-amino acid oxidase activity and its implications for schizophrenia. Mol Cell Neurosci 2006, 32, 324–334. [Google Scholar] [CrossRef] [PubMed]
- Maekawa, M.; Watanabe, M.; Yamaguchi, S.; Konno, R.; Hori, Y. Spatial learning and long-term potentiation of mutant mice lacking D-amino-acid oxidase. Neurosci Res 2005, 53, 34–38. [Google Scholar] [CrossRef] [PubMed]
- Maekawa, M.; Okamura, T.; Kasai, N.; Hori, Y.; Summer, K.H.; Konno, R. D-amino-acid oxidase is involved in D-serine-induced nephrotoxicity. Chem Res Toxicol 2005, 18, 1678–1682. [Google Scholar] [CrossRef] [PubMed]
- Williams, R.E.; Lock, E.A. Sodium benzoate attenuates D-serine induced nephrotoxicity in the rat. Toxicology 2005, 207, 35–48. [Google Scholar] [CrossRef] [PubMed]
- Goff, D.C. D-cycloserine in schizophrenia: new strategies for improving clinical outcomes by enhancing plasticity. Curr Neuropharmacol 2017, 15, 21–34. [Google Scholar] [CrossRef]
- Goff, D.C.; Tsai, G.; Manoach, D.S.; Coyle, J.T. Dose-finding trial of D-cycloserine added to neuroleptics for negative symptoms in schizophrenia. Am J Psychiatry 1995, 152, 1213–1215. [Google Scholar] [CrossRef]
- Goff, D.C.; Tsai, G.; Levitt, J.; Amico, E.; Manoach, D.; Schoenfeld, D.A.; Hayden, D.L.; McCarley, R.; Coyle, J.T. A placebo-controlled trial of D-cycloserine added to conventional neuroleptics in patients with schizophrenia. Arch Gen Psychiatry 1999, 56, 21–27. [Google Scholar] [CrossRef] [PubMed]
- van Berckel, B.N.; Hijman, R.; van der Linden, J.A.; Westenberg, H.G.; van Ree, J.M.; Kahn, R.S. Efficacy and tolerance of D-cycloserine in drug-free schizophrenic patients. Biol Psychiatry 1996, 40, 1298–1300. [Google Scholar] [CrossRef]
- Tuominen, H.J.; Tiihonen, J.; Wahlbeck, K. Glutamatergic drugs for schizophrenia: a systematic review and meta-analysis. Schizophr Res 2005, 72, 225–234. [Google Scholar] [CrossRef]
- Vestring, S.; Dorner, A.; Scholliers, J.; Ehrenberger, K.; Kiss, A.; Arenz, L.; Theiss, A.; Rossner, P.; Frase, S.; Du Vinage, C.; et al. D-Cycloserine enhances the bidirectional range of NMDAR-dependent hippocampal synaptic plasticity. Transl Psychiatry 2024, 14, 18. [Google Scholar] [CrossRef]
- Hackos, D.H.; Lupardus, P.J.; Grand, T.; Chen, Y.; Wang, T.M.; Reynen, P.; Gustafson, A.; Wallweber, H.J.; Volgraf, M.; Sellers, B.D.; et al. Positive allosteric modulators of GluN2A-containing NMDARs with distinct modes of action and impacts on circuit function. Neuron 2016, 89, 983–999. [Google Scholar] [CrossRef]
- Ladagu, A.D.; Olopade, F.E.; Adejare, A.; Olopade, J.O. GluN2A and GluN2B N-methyl-D-aspartate receptor (NMDARs) subunits: their roles and therapeutic antagonists in neurological diseases. Pharmaceuticals (Basel) 2023, 16. [Google Scholar] [CrossRef] [PubMed]
- Preskorn, S.H.; Baker, B.; Kolluri, S.; Menniti, F.S.; Krams, M.; Landen, J.W. An innovative design to establish proof of concept of the antidepressant effects of the NR2B subunit selective N-methyl-D-aspartate antagonist, CP-101,606, in patients with treatment-refractory major depressive disorder. J Clin Psychopharmacol 2008, 28, 631–637. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
A schematic overview of glutamatergic system in the brain, including glutamate synthesis and release, and subsequent glutamate activities through its receptors. EAATs, excitatory amino acid transporters; NMDARs, N-methyl-D-aspartate receptors; AMPARs, α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid receptors; BDNF, brain-derived neurotrophic factor. Figure created with BioRender.com.
Figure 1.
A schematic overview of glutamatergic system in the brain, including glutamate synthesis and release, and subsequent glutamate activities through its receptors. EAATs, excitatory amino acid transporters; NMDARs, N-methyl-D-aspartate receptors; AMPARs, α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid receptors; BDNF, brain-derived neurotrophic factor. Figure created with BioRender.com.
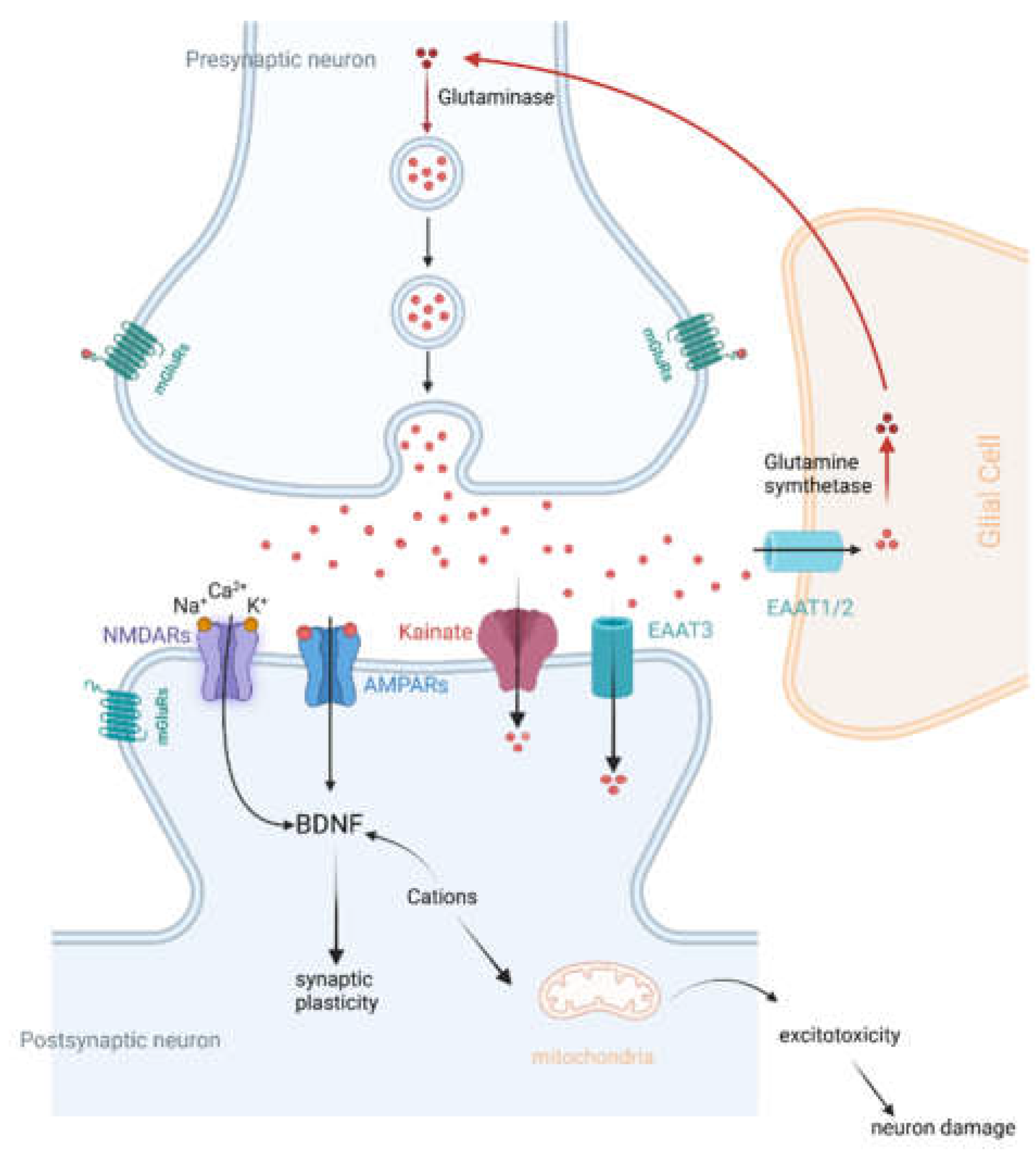
Figure 2.
The balance between excitation (E) and inhibition (I) and neural oscillations form the foundation of cognitive functions. In healthy individuals, a well-regulated E/I neural network sustains normal oscillations and cognitive functions. However, E/I imbalance leads to abnormal oscillations, consequently resulting in cognitive impairments. Figure created with BioRender.com.
Figure 2.
The balance between excitation (E) and inhibition (I) and neural oscillations form the foundation of cognitive functions. In healthy individuals, a well-regulated E/I neural network sustains normal oscillations and cognitive functions. However, E/I imbalance leads to abnormal oscillations, consequently resulting in cognitive impairments. Figure created with BioRender.com.
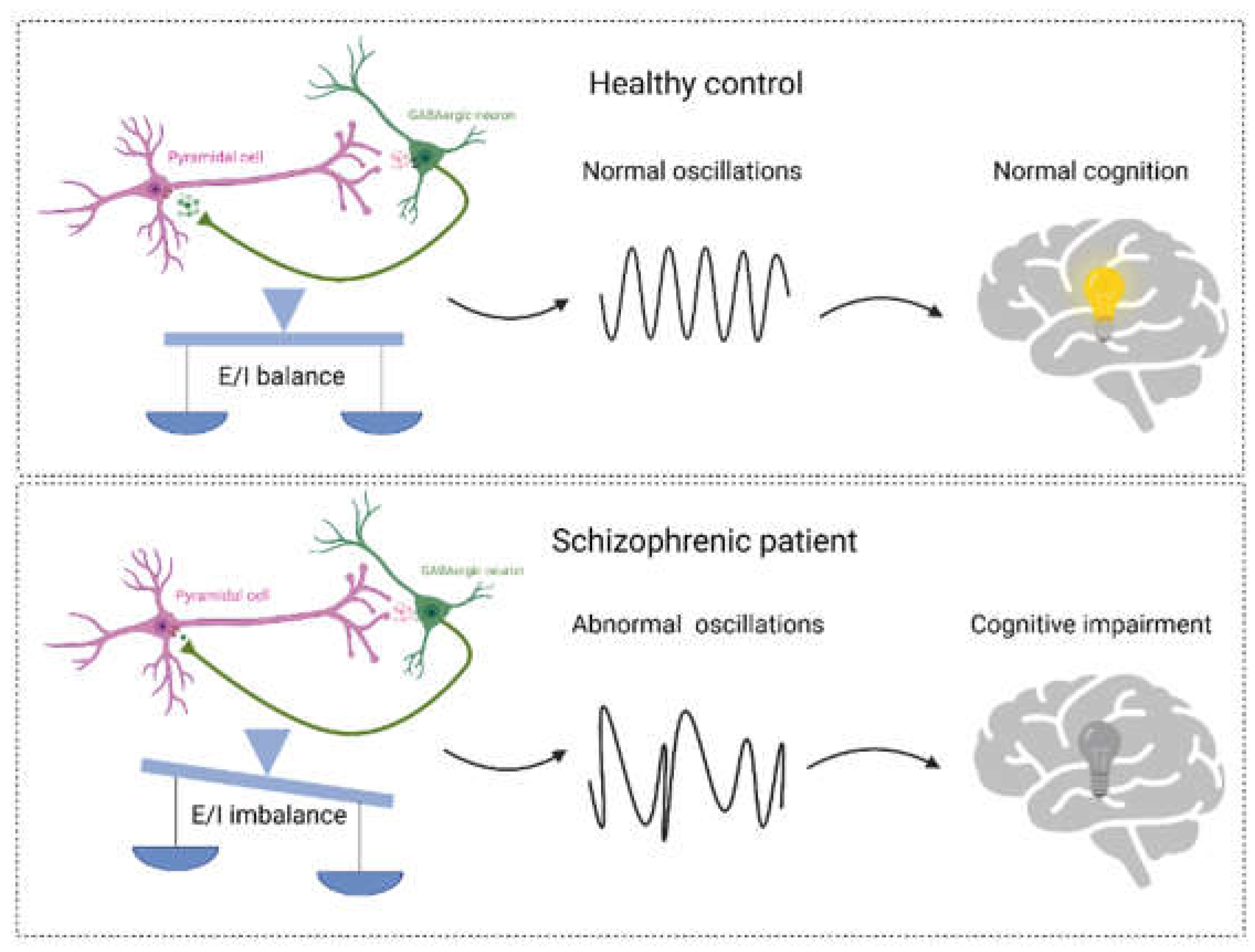
Figure 3.
An overview of the direct/indirect treatments in the regulation of NMDA recep tor function and the therapeutic effects and possible underlying mechanisms in the treatment of schizophrenia. (1) Enhancing NMDA receptor functions through direct treatments (blue: e.g., glutamate, glycine, and D-serine). (2) Boosting NMDA receptor functions via indirect treatments (red: e.g., GLT1 inhibitors, DAAO inhibitors, and GlyT1 inhibitors). Bottom panels: Effects of NMDA receptor activation on neural morphology and brain activity, which contribute to the amelioration of symptoms in schizophrenia. GLT1, Glutamate transporter; GlyT1, Glycine transporter; SR, Serine racemase; DAAO, D-amino acid oxidase. Figure created with BioRender.com.
Figure 3.
An overview of the direct/indirect treatments in the regulation of NMDA recep tor function and the therapeutic effects and possible underlying mechanisms in the treatment of schizophrenia. (1) Enhancing NMDA receptor functions through direct treatments (blue: e.g., glutamate, glycine, and D-serine). (2) Boosting NMDA receptor functions via indirect treatments (red: e.g., GLT1 inhibitors, DAAO inhibitors, and GlyT1 inhibitors). Bottom panels: Effects of NMDA receptor activation on neural morphology and brain activity, which contribute to the amelioration of symptoms in schizophrenia. GLT1, Glutamate transporter; GlyT1, Glycine transporter; SR, Serine racemase; DAAO, D-amino acid oxidase. Figure created with BioRender.com.
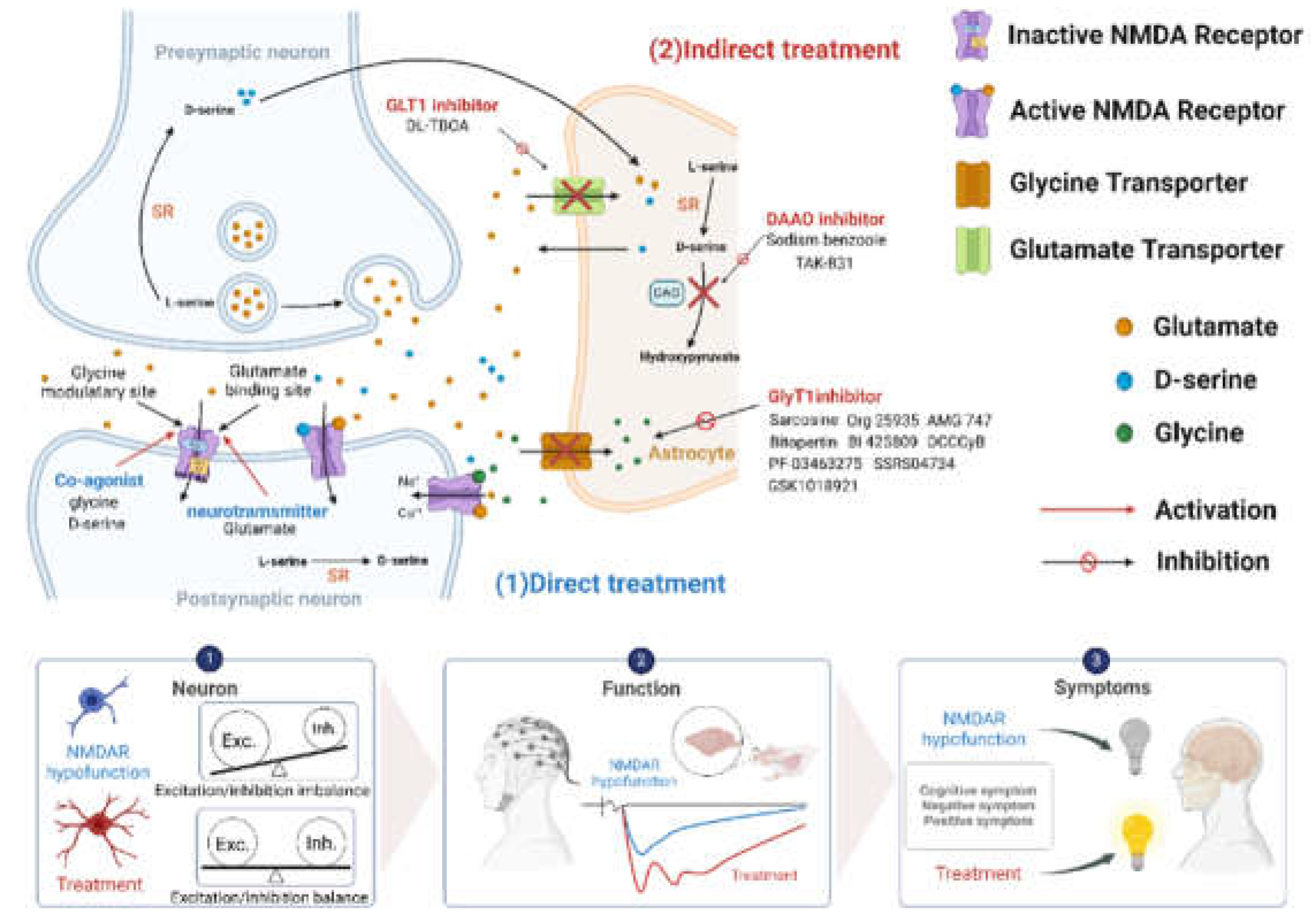

Table 1.
Effects of glycine on the treatment of schizophrenic symptoms in clinical studies.
| Compound | Sample size (placebo vs. experiment) |
Dosage | Clinical results | References |
|---|---|---|---|---|
| Glycine | 11 (no placebo) | 5-25 (g/day) | No significant effects on symptoms | [58] |
| 6 (no placebo) | 10.8 (g/day) | No significant effects on symptoms | [59] | |
| 6 (no placebo) | 15 (g/day) | No significant effects on symptoms | [60] | |
| 7 vs. 7 | 2-30 (g/day) | Significant improvements in negative symptoms | [61] | |
| 5 (no placebo) | 0.14-0.8 (g/kg/day) | Significant improvements in negative symptoms | [62] | |
| 11 vs. 11 | 0.8 (g/kg/day) | Significant improvements in depressive, cognitive and negative symptoms | [63] | |
| 22 vs. 22 | 0.8 (g/kg/day) | Significant improvements in depressive, cognitive and negative symptoms | [64] | |
| 10 vs. 9 | 30 (g/day) | No significant effects on symptoms | [65] | |
| 13 vs. 14 | 60 (g/day) | No significant effects on symptoms | [66] | |
| 6 vs. 6 | 0.2-0.8 (g/kg/day) | Significant improvements in negative symptoms | [67] | |
| 12 vs. 12 | 60 (g/day) | No significant effects on symptoms | [68] | |
| 45 (55) vs. 42 (54) | 15-60 (g/day) | No significant effects on symptoms | [69] | |
| 2 vs. 2 | 6-48 (g/day) | Significant improvements in Clinical symptoms | [70] | |
| 2 (no placebo) | 5.4-86.5 (g/day) | Significant improvements in Clinical symptoms | [71] | |
| 10 vs.10 | 0.8 (g/kg/day) | No significant effects on symptoms | [72] | |
| 29 (no placebo) | 0.8 (g/kg/day) | Significant improvements in positive and negative symptoms | [73] |
Table 2.
Effects of D-serine on the treatment of schizophrenic symptoms in clinical studies.
| Compound | Sample size (placebo vs. experiment) |
Dosage | Clinical results | References |
|---|---|---|---|---|
| D-serine | 15 vs. 14 | 30 (mg/kg/day) | Significant improvements in positive, negative, and cognitive symptoms | [88] |
| 10 vs. 10 | 30 (mg/kg/day) | No significant effects on symptoms | [95] | |
| 38 vs.37 | 20-30 (mg/kg/day) | Significant improvements in negative, positive, cognitive, and depression symptoms | [96] | |
| 16 (20)vs.16 (20) | 2 (g/day) | No significant effects on symptoms | [97] | |
| 12 vs.19 vs.16 | 30,60,120 (mg/kg/day) | Significant improvements in symptoms | [94] | |
| 69 (98)vs. 73 (97) | 2 (g/day) | No significant effects on symptoms | [89] | |
| 5 (10)vs. 3 (8) | 1.5-3 (g/day) | Significant improvements in negative symptom | [98] | |
| 23 (26)vs. 25 (27) | 30 (mg/kg/day) | No significant effects on symptoms | [99] | |
| 17 | 1.5-4 (g/day) | Significant improvements in symptoms | [100] | |
| 20 (24)vs. 15 (20) | 60 (mg/kg/day) | Significant improvements in negative symptoms | [87] | |
| 13 | 60 (mg/kg/day) | Improvements in auditory plasticity | [101] | |
| 16 vs.16 | 60 (mg/kg/day) | Significant improvements in symptoms | [90] | |
| 9 vs. 12 | 100 mg/kg | Significant plasticity improvement | [102] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



