
Submitted:
22 August 2024
Posted:
22 August 2024
You are already at the latest version
Abstract
Decidualization of the uterine endometrium is a critical process for embryo implantation in mammals, primarily occurring on gestational day 8 in pregnant mice. However, the interplay between the maternal gut microbiome, metabolism, and the uterus at this specific time point remains poorly understood. This study employed a multi-omics approach to investigate the metabolic, gut microbiome, and transcriptomic changes associated with early pregnancy (gestational day 8 (E8)) in mice. Serum metabolomics revealed a distinct metabolic profile in E8 compared to controls, with differential metabolites primarily enriched in amino acid metabolism pathways. Gut microbial composition showed that E8 mice exhibited higher alpha-diversity and a significant shift of beta-diversity. Specifically, the E8 group displayed a decrease in pathogenic Proteobacteria and an increase in beneficial Bacteroidetes and S24-7 taxa. Transcriptomics identified myriads of distinct gene between E8 and control mice. The differentially expressed genes were enriched in pathways involved in Alanine, Aspartate and Glutamate metabolism, PI3K-Akt signaling, and PPAR signaling pathway. Integrative analysis of the multi-omics data uncovered potential mechanistic relationships among the differential metabolites, gut microbiota, and uterine gene expression changes. Notably, the gene Asns showed strong correlations with specific gut S24-7 and metabolite L-Aspartatic acid, suggesting its potential role in mediating the crosstalk between the maternal environment and embryo development during early pregnancy. These findings provide valuable insights into the complex interplay between the maternal metabolome, gut microbiome, and uterine transcriptome in the context of early pregnancy, which may contribute to our understanding of the underlying mechanisms of embryo implantation and development.
Keywords:
Gestational day 8
; Gut microbiota
; Serum metabolomics
; Uterine transcriptome
; Asparagine synthetase
1. Introduction
Mammalian pregnancy is a critical biological process that involves intricate interactions between the maternal and fetal units. Two key events during early pregnancy are embryo implantation and placental development, which are crucial for fetal health and survival(Hemberger, Hanna, & Dean, 2020; Huang et al., 2023). In particular, gestation day 8 (GD8) in mice is considered a critical period for these processes, involving complex molecular and cellular events, including immune cell migration, cytokine production, and placental tissue formation. Embryo implantation is the process by which the blastocyst attaches to the uterine wall and initiates the formation of the placenta, which serves as the interface between the maternal and fetal tissues(Akaeda, Aikawa, & Hirota, 2024; Red-Horse et al., 2004). This event is tightly regulated by a delicate balance between maternal and embryonic signaling pathways, involving various hormones, growth factors, and cytokines(Boldeanu et al., 2020; Gurner, Truong, Harvey, & Gardner, 2020). Disruption of this process can lead to implantation failure or abnormal placentation, both of which can have severe consequences for fetal development and pregnancy outcomes. The placenta is a crucial organ that facilitates the exchange of nutrients, gases, and waste products between the mother and the fetus(Gude, Roberts, Kalionis, & King, 2004). During early placental development, specialized cells called trophoblasts proliferate, differentiate, and invade the maternal decidua, establishing the foundation for the placental structure(Burton & Fowden, 2015; Cindrova-Davies & Sferruzzi-Perri, 2022). This process is regulated by a complex interplay between maternal and fetal signals, including immunological factors, angiogenic molecules, and transcription factors.
The role of the gut microbiome in maternal health and fetal development has gained increasing attention in recent years. The gut microbiota not only impacts the host’s immune system, but may also influence the interactions between the mother and the fetus through various mechanisms(Fuhler, 2020; Macpherson, de Aguero, & Ganal-Vonarburg, 2017; Nyangahu & Jaspan, 2019). For instance, alterations in the gut microbiome have been associated with preterm birth, low birth weight, and other pregnancy-related complications(Giannella et al., 2023; Groer, Gregory, Louis-Jacques, Thibeau, & Walker, 2015; Siena et al., 2021). The gut microbiome can regulate host metabolism, immune responses, and endocrine function, all of which are critical for a healthy pregnancy(Koren et al., 2012; Nuriel-Ohayon, Neuman, & Koren, 2016). During pregnancy, the maternal gut microbiome undergoes dynamic changes, which may have downstream effects on embryo implantation and placental development(Kuang et al., 2017). For example, the gut microbiome can modulate the production of metabolites, such as short-chain fatty acids, that can influence uterine receptivity and placental function(Moylan, Nguyen-Ngo, Lim, & Lappas, 2020; Siena et al., 2021). Furthermore, the maternal gut microbiome-host interactions may also involve changes in gene expression patterns in the maternal organs, such as the uterus and placenta, which are crucial for supporting fetal growth and development(Antony et al., 2015; Lu, Shi, Jiang, & Zhang, 2024). Understanding the interplay between the maternal gut microbiome, metabolome, and transcriptome during critical stages of pregnancy, such as embryo implantation and placental development, may provide important insights into the mechanisms underlying maternal-fetal crosstalk and inform the development of new pregnancy intervention strategies.
Metabolomics, the study of the comprehensive profile and dynamic changes of all small molecules (e.g., amino acids, fatty acids, carbohydrates, etc.) within a biological system, has emerged as a powerful tool to investigate the interactions between the gut microbiome and host metabolism(Lamichhane, Sen, Dickens, Oresic, & Bertram, 2018; Xie, Zhang, Zheng, & Jia, 2013). Changes in the maternal metabolome may reflect alterations in the gut microbiome composition and function, which in turn can impact the health of the mother and the developing fetus(Jasarevic & Bale, 2019; Vuong et al., 2020). By integrating gut microbiome, metabolomic, and transcriptomic analyses, researchers can gain a more comprehensive understanding of the complex interplay between the maternal gut-metabolism-transcriptome axis and its influence on critical stages of pregnancy, such as embryo implantation and placental development.
Given the considerations outlined above, the present study aims to investigate the potential associations between maternal gut microbiome, metabolome, and transcriptome during the critical gestational D8 in mice. Our goal is to elucidate the interrelationships between these factors and explore their underlying biological mechanisms, with the ultimate aim of providing a theoretical foundation for future clinical interventions.
2. Materials and Methods
2.1. Ethics and Consent
All the animals were raised in compliance with the care and use guidelines of experimental animals according to the Animal Welfare legislation of China. This study was approved by the Ethics Committee of Anhui Medical University.
2.2. Animals and Samples Collection
A total of 12 female C57BL/6J mice purchased from the VitonLihua Ltd. (Beijing, China) were applied in this study. Animals were housed under specific pathogen-free conditions with a 12-hour light/12-hour dark cycle, temperature (24 ± 2°C) and humidity (50% ± 10%). Serum samples were collected and centrifuged for metabolomics. Following serum collection, colon luminal contents were carefully collected from mice and transferred to sterile tubes, whereafter they were immediately stored in a -80 °C refrigerator following swift freezing with liquid nitrogen to be utilized in gut microbial diversity analyses. A portion of uterus tissue was collected and rapidly into the cryopreserved tubes and stored in a -80 °C refrigerator for subsequent transcriptome sequencing.
2.3. RNA-Seq and Differential Gene Expression Analysis
RNA sequencing (RNA-seq) and differential gene expression analysis were performed according to previously published research (Y. Zhu et al., 2021). Briefly, total RNA was isolated from uterus tissues using a Trizol kit (Invitrogen, USA) following the manufacturer’s protocol. cDNA libraries were then constructed from the extracted RNA using the NEBNext® Ultra™ Directional RNA Library Prep Kit for Illumina® (NEB, United States). The prepared libraries were subjected to paired-end sequencing on a HiSeq 4,000 platform (Illumina, USA) by Novogene (Novogene, Beijing, China), generating approximately 46.2 million 150-bp paired-end reads. Differential gene expression analysis, over-represented gene ontology (GO) terms, pathways, and biological processes were subsequently identified from the sequencing data.
2.4. Microbial DNA Extraction and 16S rRNA Gene Sequencing
Following previously published methodology (Liu et al., 2023), microbial DNA was extracted and the 16S rRNA gene was subsequently sequenced. Sequencing was conducted on an Illumina MiSeq platform in accordance with the manufacturer’s instructions (PANOMIX Bio-Pharm Technology Co., Ltd., Soochow, China). The resulting data underwent splicing and filtering using Trimmomatic (version 0.35) (Bolger, Lohse, & Usadel, 2014), yielding a set of high-quality sequences for downstream analysis. The reads were then clustered into operational taxonomic units (OTUs) at a similarity threshold of 97.0% using USEARCH (version 10.0) (Edgar, 2010). Species classification information was obtained by annotating feature sequences with a Naive Bayes classifier based on the reference database provided by Silva (http://www.arb-silva.de) (Quast et al., 2013). Finally, alpha diversity indices of Chao1 and Shannon, as well as beta diversity calculated using the Bray-Curtis dissimilarity matrix, were performed using Quantitative Insights into Microbial (QIIME) (version 1.9.1). Additionally, principal component analysis (PCA) based on a Bray-Curtis matrix was carried out using the vegan package.
2.5. Nontargeted Serum Metabolite Profiling and Analysis
The metabolite profiles of serum were detected by an LC-MS/MS system consisting of an ultra-high-performance liquid chromatography (UHPLC) system (Vanquish, Thermo Fisher Scientific, Waltham, MA, USA) coupled to a Q Exactive (QE) HFX mass spectrometer (Orbitrap MS, Thermo Fisher Scientific, Waltham, MA, USA). The UHPLC system was equipped with a UPLC BEH Amide column (2.1 mm × 100 mm, 1.7 μm) and was used for the separation of biochemicals. The mobile phase consisted of 25 mmol/L ammonium acetate (Sigma-Aldrich, Darmstadt, Germany) and 25 mmol/L ammonium hydroxide (Fisher Chemical, Waltham, MA, USA) in water (pH = 9.75) (solvent A) and acetonitrile (solvent B). The autosampler temperature was maintained at 4 °C, and the injection volume was 3 μL. The QE HFX mass spectrometer was operated in information-dependent acquisition (IDA) mode to obtain MS/MS spectra, under the control of the Xcalibur acquisition software (Thermo Fisher Scientific, Waltham, MA, USA). In this mode, the acquisition software continuously evaluates the full-scan MS spectrum. The electrospray ion (ESI) conditions were as follows: sheath gas flow rate of 30 Arb, auxiliary gas flow rate of 25 Arb, capillary temperature of 350 °C, full MS resolution of 60,000, MS/MS resolution of 7500, collision energy of 10/30/60 in negative ion mode (NCE), and spray voltage of 3.6 kV (positive) or −3.2 kV (negative).Next, the original LC-MS/MS data were converted to the mzXML format using ProteoWizard and processed with an in-house program, which was developed using R and based on XCMS, for peak detection, extraction, alignment, and integration. Subsequently, for further multivariate analysis, the raw data were pretreated. Pretreatment included de-noising based on the relative standard deviation (RSD), filling the missing data via half of the minimum value, and normalization by the internal standard normalization method. The final dataset contained the information of peak number, sample name, and normalized peak area and was imported to SIMCA16.0.2 software package (Sartorius Stedim Data Analytics AB, Umea, Västerbotten, Sweden). Firstly, we performed Principal Component Analysis (PCA), an unsupervised analysis, to reduce the dimensions of the data. The 95% confidence interval (95% CI) in the PCA score plot was used as the threshold to identify potential outliers in the dataset. Secondly, in order to visualize the group separation and find significantly changed metabolites, we conducted the supervised orthogonal projections to latent structure discriminate analysis (OPLS-DA) and acquired the value of variable importance in projection (VIP). A 7-fold cross-validation test was performed to evaluate the goodness of fit of the OPLS-DA model using the values of R2Y and Q2. R2Y indicates how well the variation of a variable is explained, and Q2indicates how well a variable can be predicted. A 200-time permutation test was then conducted to assess the robustness of the model. The precursor molecule passed the combined criteria: 1) absolute log2 fold change (|log2FC|) great than 0.26, 2) VIP > 1, and 3) coarse P value < 0.05 (nonparametric Wilcoxon t-test) were considered significantly changed between mice in E8 and Sham groups. For clustering heatmaps, the data were normalized using z-scores of the intensity areas of differential metabolites and were plotted by Pheatmap package in R language. Further, after scanning for the differential metabolite features, the fragment information obtained from the MS/MS model was then further matched with the annotations in HMDB, Metlin, LipidMaps, and in-house standards database (PPM < 10) to obtain accurate metabolite information. Finally, commercial databases, including Kyoto Encyclopedia of Genes and Genomes (KEGG) (http://www.genome.jp/kegg/ (accessed on 15 June 2021) and MetaboAnalyst (http://www.metaboanalyst.ca/ (accessed on 15 June 2021), were used for pathway enrichment analysis, when P value of metabolic pathway < 0.05, metabolic pathway were considered as statistically significant enrichment.
2.6. Statistical Analysis
The present study adhered to rigorous statistical methods to ensure the validity and reliability of the results. Specifically, the standard error of the mean (SEM) was used to represent error bars. Statistical significance was assigned at P values of less than 0.05, which were computed using R software. The Shapiro-Wilk test was utilized to determine the sample distribution type. For two groups, the F test was employed to test homoscedasticity, and significance was calculated by means of the two-sided Mann-Whitney test or unpaired. To control for the false discovery rate (FDR), multiple testing was corrected using the Benjamini-Hochberg method, considering FDR-corrected P value of less than 0.05 as statistically significant. The correlation analysis was investigated using the Spearman’s test (two-sided).
3. Results
3.1. E8 Mice Show a Distinct Serum Metabolic Profile Compared with Mice in Sham group
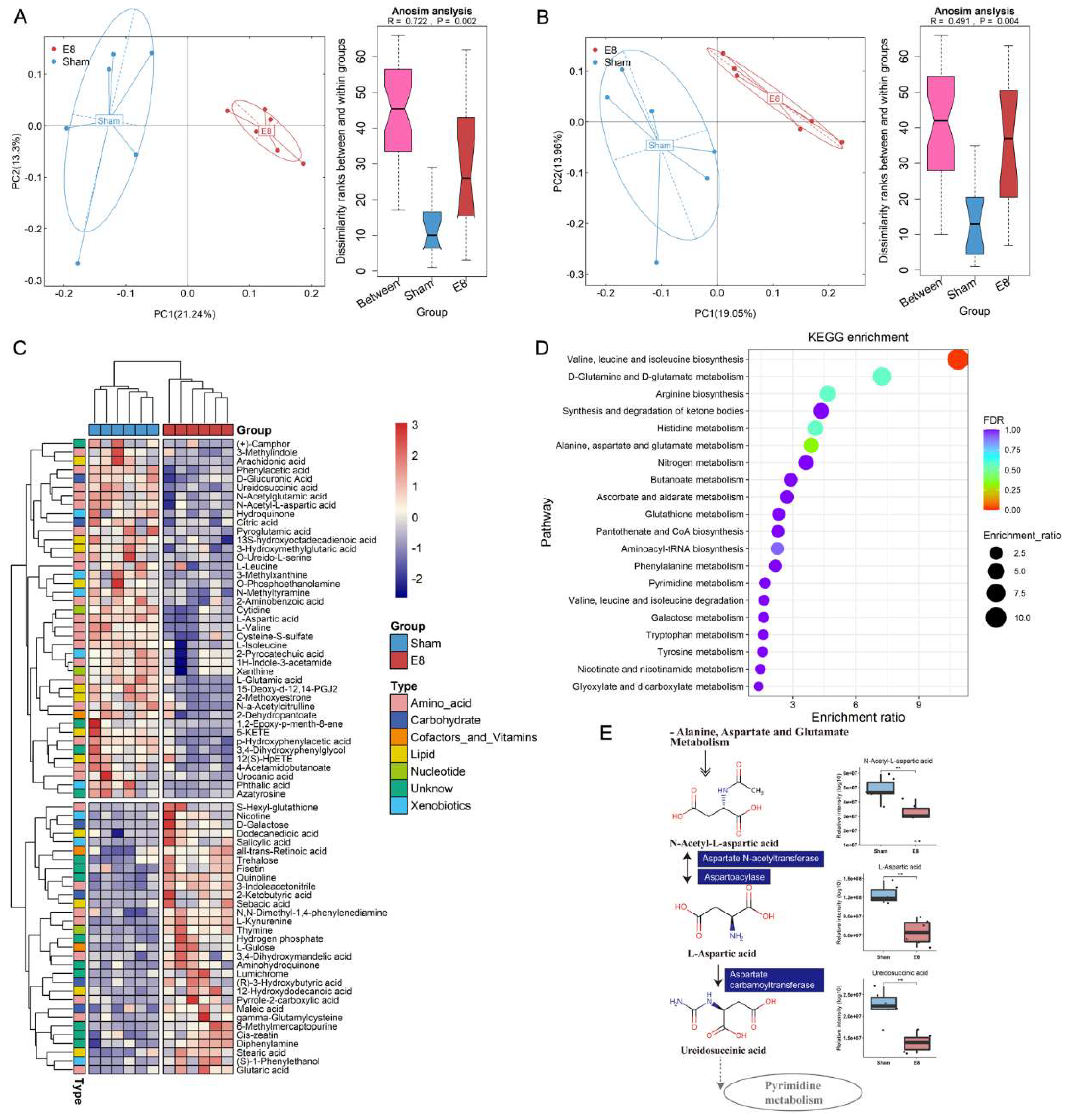
To identify the metabolic signature associated with the stimulant introduced by early pregnancy, an untargeted global metabolic profiling analysis was performed on serum samples collected from six mice at eight days post-pregnancy (E8) and six control (Sham) mice. Typical extracted ion chromatograms (Base Peak Chromatogram) from two ESI modes are displayed in Figure S1A, B. In the principal component analysis (PCA) score plots of both ESI+ and ESI-, the quality control (QC) samples were observed to cluster tightly together, indicating the reliability of the present study. Additionally, the coefficients of variation of peak distribution in the QC samples confirmed that the analysis was stable and repeatable (Figure S1C, D). The metabolomic analysis revealed a distinct serum metabolic profile in E8 mice compared to the Sham group, as demonstrated by the PCA score plots in positive and negative ion modes (Figure 1A, B). Of the 351 annotated metabolites, a total of 72 were found to be significantly altered between the two groups, as revealed by Welch’s two-sample t-test. Among the altered metabolites, 31 (43.06%) were enriched in E8 mice, while 41 (56.94%) were down-regulated (Figure 1C, Table S1). To further delineate potential correlations with early pregnancy, individual serum metabolites were subjected to in-depth analyses. A Random Forest (RF) analysis was performed, which ranked the altered 72 serum metabolites by their contribution to the group separation. The top 19 differentiating metabolites with a Mean Decrease in Gini index greater than 0.1 are shown in Figure S1E. Besides, KEGG enrichment analysis based on the differential metabolites was conducted to reveal altered biological process and demonstrated the functional differences of the metabolites. The differential metabolites induced by pregnancy were mainly enriched in “Valine, leucine and isoleucine biosynthesis”, “Arginine biosynthesis” and “Alanine, aspartate and glutamate metabolism” (Figure 1D). Taken together, these findings show a sub-metabolic pathway from N-Acetyl-L-aspartic acid to Ureidosuccinic acid within Alanine, aspartate and glutamate biosynthesis pathways and showed that Aspartate N-acetyltransferase, Aspartoacylase, and Aspartate carbamoyltransferase are required (Figure 1E). These findings suggest significant metabolic alterations in E8 mice, potentially reflecting changes in key biological processes and metabolic pathways.
Figure 1.
Serum metabolites were significantly changed in E8 group as compared to Sham group. (A-B) Principal Coordinate Analysis (PCoA) based on Bray-Curtis dissimilarity calculated by metabolite features in ESI+ and ESI- mode indicating an obvious shift in mice of E8 group from that in Sham group. (C) Heatmap clustering of distinct serum metabolites from the comparison between E8 and Sham groups. (D) KEGG pathway analysis was performed based on differential expressed metabolites between E8 and Sham groups. The color of bubbles represents the value of adjusted P value, and the size of bubbles represents the number of counts (sorted by enrichment ratio). (E) Schematic diagram of 3 E8-depleted metabolites participating in the Alanine, Aspartate and Glutamate Metabolism KEGG pathways.
Figure 1.
Serum metabolites were significantly changed in E8 group as compared to Sham group. (A-B) Principal Coordinate Analysis (PCoA) based on Bray-Curtis dissimilarity calculated by metabolite features in ESI+ and ESI- mode indicating an obvious shift in mice of E8 group from that in Sham group. (C) Heatmap clustering of distinct serum metabolites from the comparison between E8 and Sham groups. (D) KEGG pathway analysis was performed based on differential expressed metabolites between E8 and Sham groups. The color of bubbles represents the value of adjusted P value, and the size of bubbles represents the number of counts (sorted by enrichment ratio). (E) Schematic diagram of 3 E8-depleted metabolites participating in the Alanine, Aspartate and Glutamate Metabolism KEGG pathways.
3.2. Gut Microbiota Composition Was Altered in E8 Mice
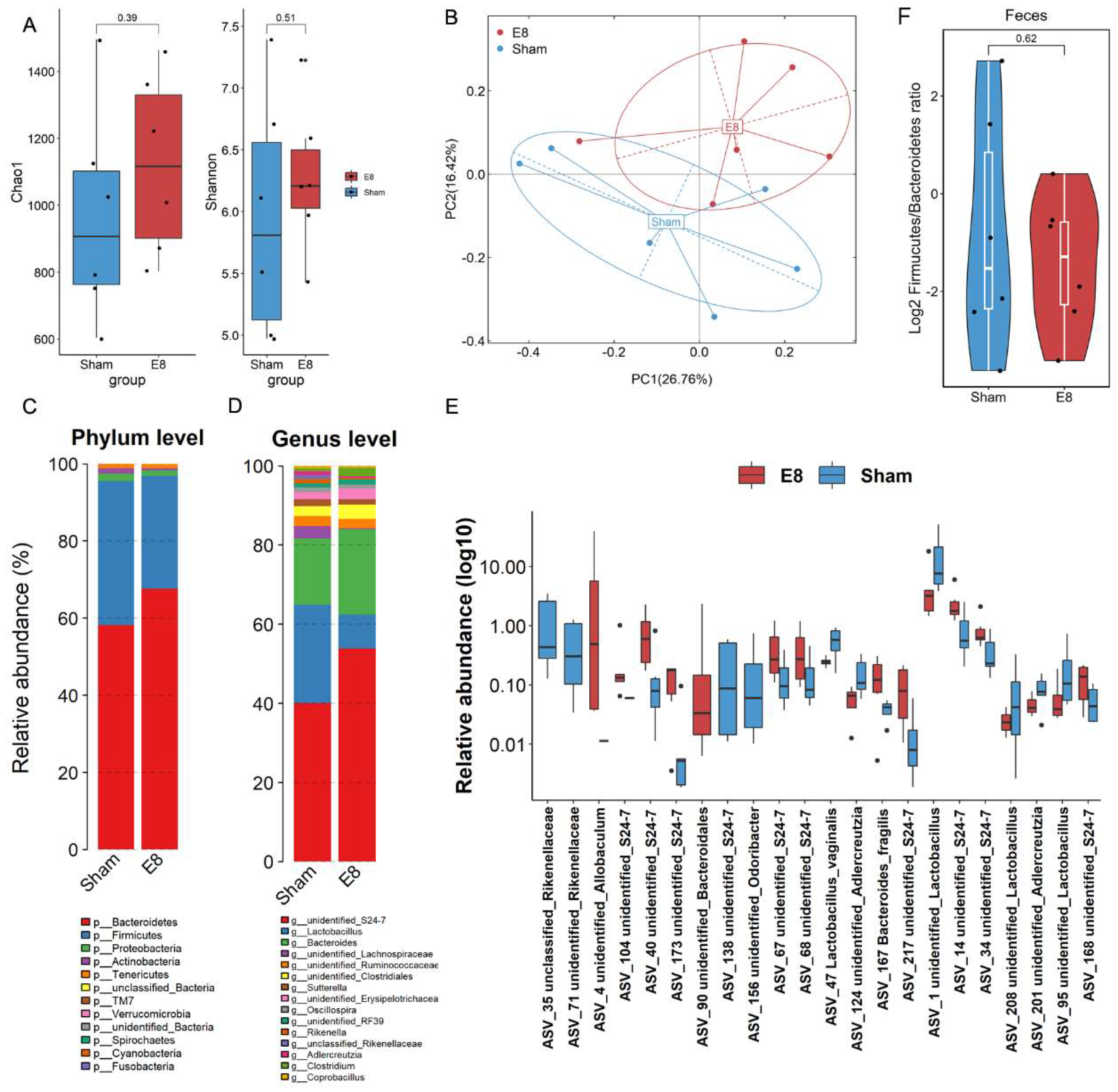
We compared the gut microbial composition of cecum contents from pregnant mice in E8 group versus sham C57BL/6J mice using 16S rRNA gene sequencing. E8 mice demonstrated with relatively higher α-diversity, while exhibited a marked shift in microbial composition when compared to sham mice, as evidenced by a significant difference in β-diversity (p = 0.002) (Figure 2A, B). The relative abundance of pathogenic Proteobacteria at phylum level and Rikenella at genus level were decreased in E8 group. In addition, the relative abundance of Bacteroidetes and S24-7 were enriched in E8 mice (Figure 2C, D). At the ASV level, we detected 22 bacteria showed significantly different between E8 and Sham groups. 9 unidentified S24-7 (ASV104, ASV40, ASV173, ASV67-68, ASV217, ASV14, ASV34, and ASV168), while unidentified Rikenellaceae (ASV35, and ASV71), and Lactobacillus vaginalis were decreased (Figure 2E, Table S2). Subsequently, RF analysis was performed to examine the ability to discriminate samples from E8 or Sham mice based on the differential gut microbiota. A sum of thirteen bacteria was showed in Figure S2, among which the ASVs that belonged to S24-7 could be a prominent microbial biomarker that associated with the early pregnancy of mice. Furthermore, we compared the Firmicutes/Bacteroidetes ratio, a measure of the relative abundance of Firmicutes and Bacteroidetes bacteria in the gut microbiome which has been suggested to indicate overall gut health. The results exhibited a lower Firmicutes abundance in subjects of E8 group (Figure 2F), indicating the pivotal role of embryo implantation and placental development in shape host gut microbial composition.
Figure 2.
Compositional analysis of gut microbiota in mice between Sham and E8 groups. (A) Faecal alpha diversity analysis estimated by Chao1 and Shannon indexes between mice in Sham and E8 groups. (B) Principal coordination analysis (PCoA) and analysis of similarities (anosim) were calculated based on bray-curtis matrix. (C-D) The taxonomic composition distribution between two groups on phylum- and genus-level of gut microbiota. (E) Log10-transformed relative abundance of significantly different ASVs between Sham and E8. (F) The ratio of Firmicutes compared to Bacteroidota in E8 mice compared with Sham mice.
Figure 2.
Compositional analysis of gut microbiota in mice between Sham and E8 groups. (A) Faecal alpha diversity analysis estimated by Chao1 and Shannon indexes between mice in Sham and E8 groups. (B) Principal coordination analysis (PCoA) and analysis of similarities (anosim) were calculated based on bray-curtis matrix. (C-D) The taxonomic composition distribution between two groups on phylum- and genus-level of gut microbiota. (E) Log10-transformed relative abundance of significantly different ASVs between Sham and E8. (F) The ratio of Firmicutes compared to Bacteroidota in E8 mice compared with Sham mice.
3.3. The Transcriptome of Gravid Uterus in E8 Mice Was Distinct Compared to Un-Pregnant Mice
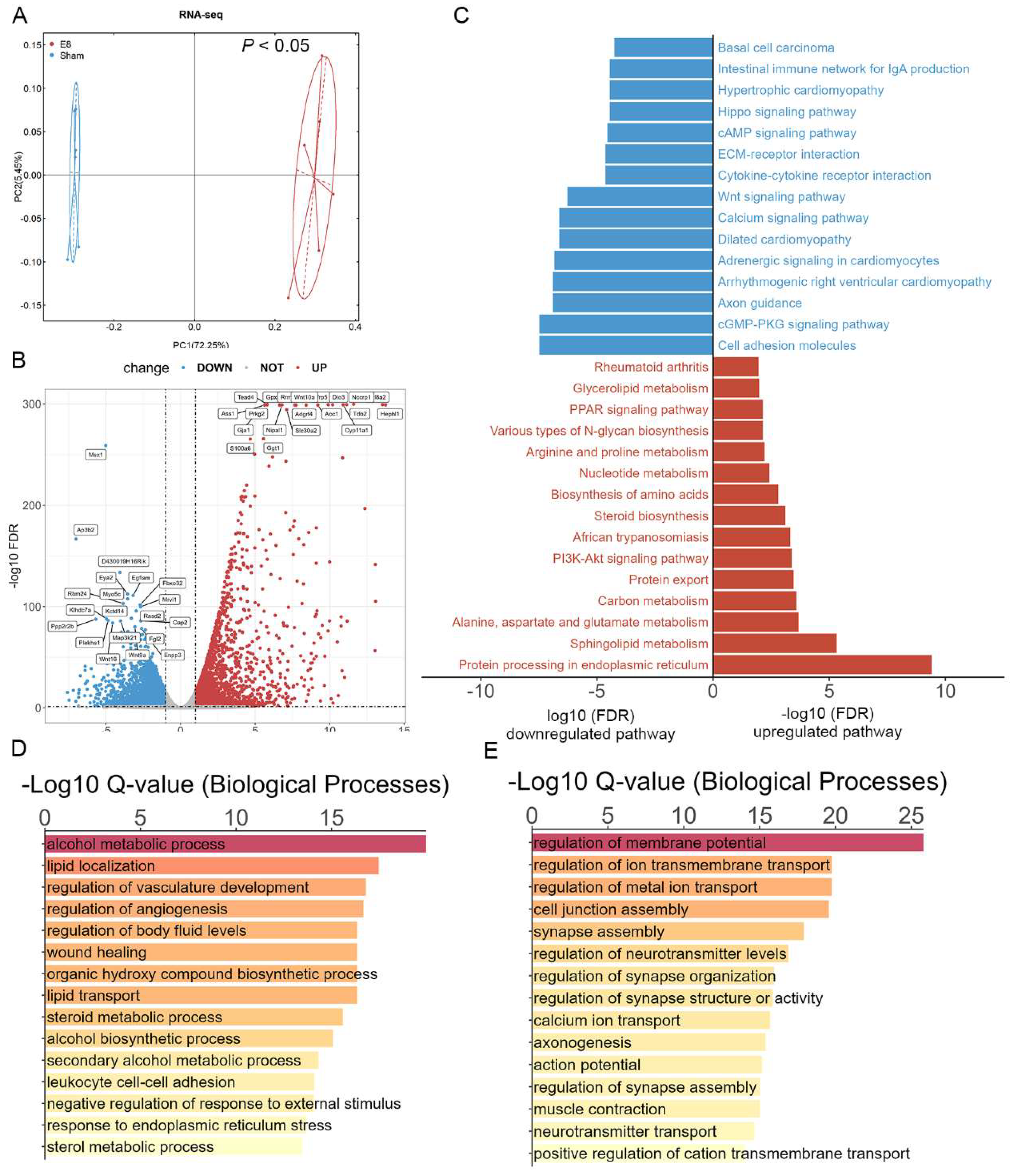
To clarify the specific mechanism of early pregnant in reshaping the gene expression profile from the perspective of transcriptome, we performed RNA-seq of uterus from mice in E8, and Sham groups. Firstly, the PCA result showed that samples were clearly separated by groups, which indicated the reliability and validity of our data, laying the foundation for further analysis (Figure 3A). The differential expressed genes (DEGs) between E8 and Sham individuals were then identified with the criterion |log2 Fold Change | ≥ 1 and P value < 0.05 (Table S3). we labeled top 20 significantly up-regulated and down-regulated DEGs in the indicated groups (Figure 3B). Next, KEGG enrichment analyses were employed and interestingly found that the DEGs between E8 and Sham were significantly enriched in the “Protein processing in endoplasmic reticulum”, “PI3K-Akt signaling pathway”, and “PPAR signaling pathway” (Figure 3C, Table S4). Interestingly, GO enrichment analysis found that most of the upregulated genes induced by embryo implantation fall into the broad categories of regulation of vasculature development, regulation of angiogenesis (Figure 3D-E, Table S5). Taken together, the results above revealed that embryo implantation may mediated the development of embryo through modulation of gene expression in uterus of the pregnant mice.
Figure 3.
Analysis of differential expressed genes in E8 and Sham groups. (A) PCA score plot indicating an obvious separation of the transcriptomic profile between E8 and Sham groups. (B) Volcano plot showing differential genes (top 20 upregulated and top 20 down-regulated genes in E8 and Sham mice). Criteria for significant differences (VIP > 1, adjusted P < 0.05 and fold change ≥ 2). (C) KEGG enrichment analysis of the DEGs in the E8 versus Sham comparison. The red column represents the top 15 KEGG pathways in E8 and blue column represents the top 15 down-regulated KEGG pathways. (D-E) The bar plot for the top 15 significant enrichment annotations for biological process (BP) of all upregulated genes (D) and down-regulated genes (E).
Figure 3.
Analysis of differential expressed genes in E8 and Sham groups. (A) PCA score plot indicating an obvious separation of the transcriptomic profile between E8 and Sham groups. (B) Volcano plot showing differential genes (top 20 upregulated and top 20 down-regulated genes in E8 and Sham mice). Criteria for significant differences (VIP > 1, adjusted P < 0.05 and fold change ≥ 2). (C) KEGG enrichment analysis of the DEGs in the E8 versus Sham comparison. The red column represents the top 15 KEGG pathways in E8 and blue column represents the top 15 down-regulated KEGG pathways. (D-E) The bar plot for the top 15 significant enrichment annotations for biological process (BP) of all upregulated genes (D) and down-regulated genes (E).
3.4. The Putative Mechanistic Correlations among Serum Metabolites, Gut Microbiota and Uterus Transcriptome
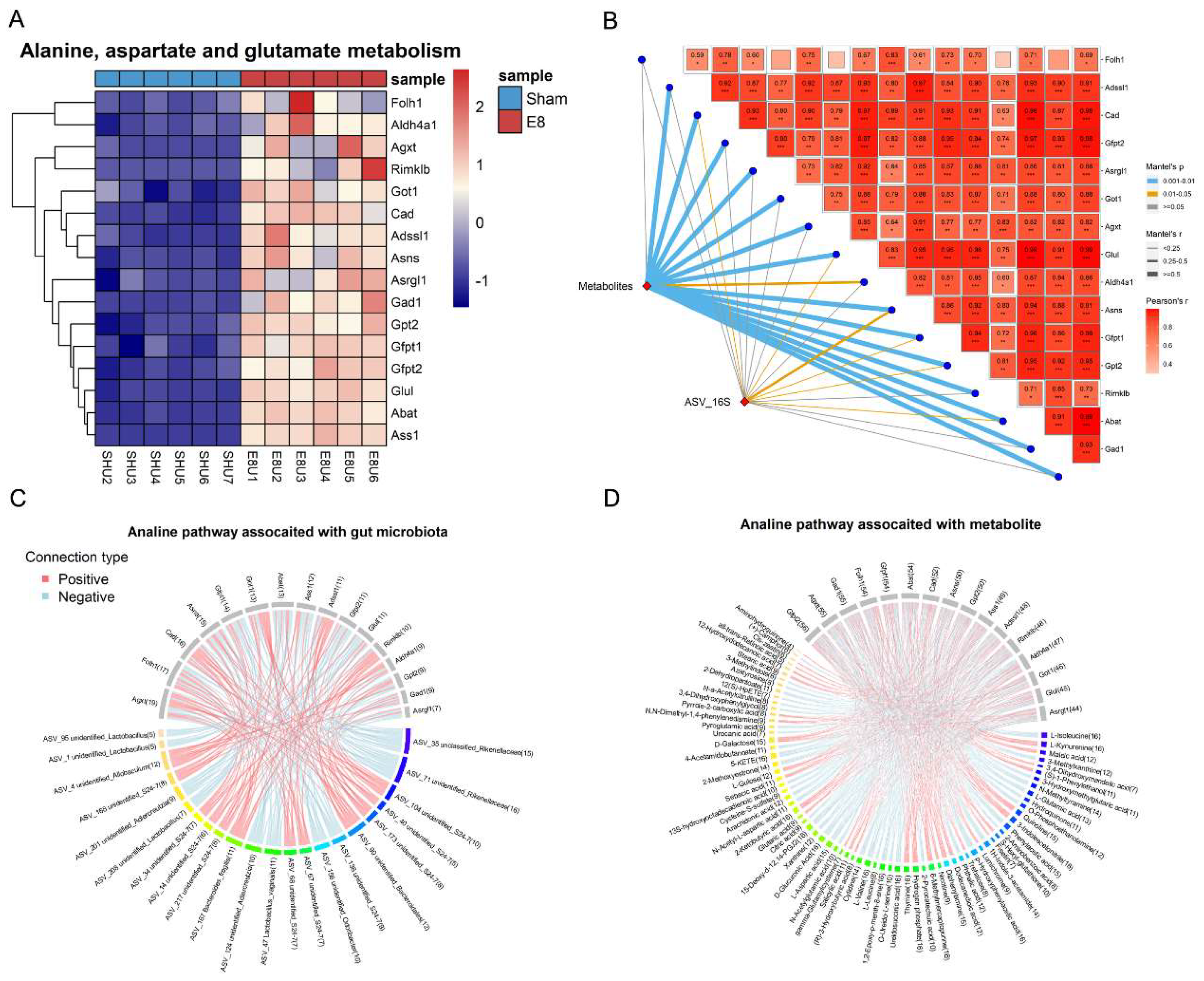
The multi-omics nature of this dataset provided an excellent opportunity to discover the association among metabolites, microbes and genes. Spearman’s correlations were conducted for the differentially abundant bacteria, metabolites, and genes, focusing on the E8 and Sham groups. Consequently, 533 associations between the species and the metabolites (Figure 4A, Table S6) were found to be significantly different (p < 0.05). WGCNA analysis identified three modules based on differential metabolites and found E8-depleted unclassified_Rikenellaceae were positively correlated with modules blue and brown, while anti-associated with module turquoise (Figure 4B, Table S7). To shed light on the core interaction among gene, metabolite and gut microbiota plays pivotal role in clarifying the molecular connection upon early embryo implantation in uterus. An integrated analysis of multi-omics data including transcriptomics, metabolomics and colon microbiota was employed. To derive insights on whether there is a distinct gene aligned with the distinct metabolite and microbe contributing to the potential mechanism of embryo development, we adopted Mantel Test to perform integrative analysis of multi-omics using DEGs from Alanine, aspartate and glutamate metabolism (Figure 5A), the significantly altered metabolites from metabolomics (Figure 1C) and significantly differed genera from colon metagenomics (Figure 2E). We initially correlated distance-corrected dissimilarities of metabolites and genera with those of genes (Figure 5B). Overall, Asns exhibited strongest correlations with microbiota and metabolites. Specifically, we adopted chord diagram to present one-to-one interactions between genes and gut microbiota (Figure 5C), genes and metabolites (Figure 5D). Interestingly, we detected Asns showed positively correlation with S24-7, and they both anti-correlated L-Aspartatic acid (Figure S3A-C).
Figure 4.
Associations between differential expressed metabolites and gut microbiota. (A) Chord diagram display the significant associations between differential expressed metabolites and ASVs. The associated metabolites are colored gray, and the associated ASVs are colored successive. Each line indicates a significant correlation between a bacterium and a metabolite, with the red color corresponding to a positive association (P value < 0.05) and the blue color representing a negative association (P value < 0.05). (B) Correlations between metabolite modules and ASVs. The absolute correlation coefficient (|r|) corresponds to the size of the circle, and P value is indicated by asterisk (“*”, P < 0.05; “**”, P < 0.01).
Figure 4.
Associations between differential expressed metabolites and gut microbiota. (A) Chord diagram display the significant associations between differential expressed metabolites and ASVs. The associated metabolites are colored gray, and the associated ASVs are colored successive. Each line indicates a significant correlation between a bacterium and a metabolite, with the red color corresponding to a positive association (P value < 0.05) and the blue color representing a negative association (P value < 0.05). (B) Correlations between metabolite modules and ASVs. The absolute correlation coefficient (|r|) corresponds to the size of the circle, and P value is indicated by asterisk (“*”, P < 0.05; “**”, P < 0.01).
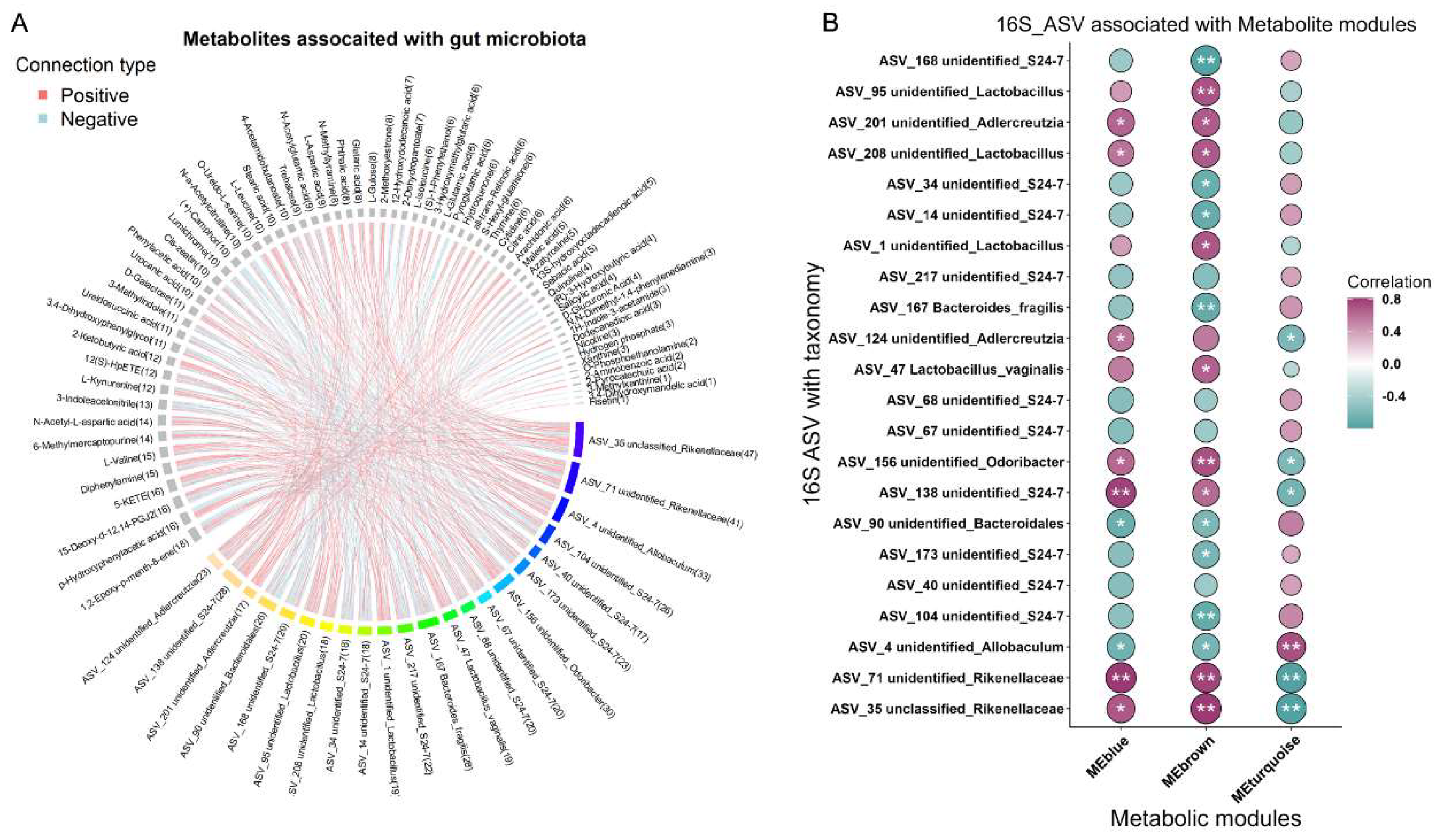
Figure 5.
Variations of serum metabolites and gut microbiota have extensive relationship with pregnancy related KEGG pathways. (A) Heatmap shows differential expressed genes in the Alanine, aspartate and glutamate metabolism pathway. (B) 16S rRNA gene sequencing based ASVs composition and metabolites composition were related to each of 16 significantly differential genes in the Alanine, aspartate and glutamate metabolism pathway using Mantel-test analysis. Edge width corresponds to the Mantel’s r statistic for the corresponding distance correlations, and edge color denotes the statistical significance. Pairwise comparisons of genes are shown, with a color gradient denoting Spearman’s correlation coefficient. (C) Chord diagram displays the significant associations between each ASV and gene in the Alanine, aspartate and glutamate metabolism pathway. The associated genes are colored gray, and the associated ASVs are colored successive. Each line indicates a significant correlation between a bacterium and a metabolite, with the red color corresponding to a positive association (P value < 0.05) and the blue color representing a negative association (P value < 0.05). (D) Chord diagram displays the significant associations between each metabolite and gene in the Alanine, aspartate and glutamate metabolism pathway. The associated genes are colored gray, and the associated metabolites are colored successive. Each line indicates a significant correlation between a bacterium and a metabolite, with the red color corresponding to a positive association (P value < 0.05) and the blue color representing a negative association (P value < 0.05).
Figure 5.
Variations of serum metabolites and gut microbiota have extensive relationship with pregnancy related KEGG pathways. (A) Heatmap shows differential expressed genes in the Alanine, aspartate and glutamate metabolism pathway. (B) 16S rRNA gene sequencing based ASVs composition and metabolites composition were related to each of 16 significantly differential genes in the Alanine, aspartate and glutamate metabolism pathway using Mantel-test analysis. Edge width corresponds to the Mantel’s r statistic for the corresponding distance correlations, and edge color denotes the statistical significance. Pairwise comparisons of genes are shown, with a color gradient denoting Spearman’s correlation coefficient. (C) Chord diagram displays the significant associations between each ASV and gene in the Alanine, aspartate and glutamate metabolism pathway. The associated genes are colored gray, and the associated ASVs are colored successive. Each line indicates a significant correlation between a bacterium and a metabolite, with the red color corresponding to a positive association (P value < 0.05) and the blue color representing a negative association (P value < 0.05). (D) Chord diagram displays the significant associations between each metabolite and gene in the Alanine, aspartate and glutamate metabolism pathway. The associated genes are colored gray, and the associated metabolites are colored successive. Each line indicates a significant correlation between a bacterium and a metabolite, with the red color corresponding to a positive association (P value < 0.05) and the blue color representing a negative association (P value < 0.05).
4. Discussion
Growing evidence suggests that the dynamic changes in the maternal gut microbiome during pregnancy are crucial for fetal development(Amato, Pradhan, Mallott, Shirola, & Lu, 2024; Di Simone et al., 2020; Pronovost et al., 2023; Sun et al., 2023). Alterations in maternal gut microbiome diversity have been observed in early pregnancy, accompanied by concomitant changes in the maternal blood metabolome. These changes may influence early embryo implantation and normal fetal development through the modulation of host immunity, inflammatory responses, and metabolic states. Increased abundances of certain bacterial genera (e.g., Lactobacillus, Bifidobacterium) in the maternal gut during pregnancy have been associated with healthy fetal development, birth weight, and immune function. Conversely, the overgrowth of pathogenic bacteria has been linked to pregnancy complications, such as early pregnancy loss and preeclampsia. Furthermore, the changes in the maternal gut metabolome, such as altered levels of short-chain fatty acids and amino acids, may also regulate the intrauterine environment by influencing maternal endocrine and energy metabolism, thereby impacting embryonic and fetal development. However, the existing research has primarily focused on late pregnancy, while the changes in the gut microbiome, metabolome, and uterine transcriptome during early gestation, as well as the molecular interactions between these factors, remain relatively unexplored. Therefore, it is crucial to identify the key gut bacteria and circulating metabolites that influence embryo implantation during early pregnancy. Elucidating these factors has the potential to provide further insights into the underlying mechanisms by which the gut microbiome, metabolism, and gene expression interact to influence the process of early embryo establishment. In the present study, we discovered a great disturbance in the serum metabolome, gut microbiota and gene expression of E8 mice as compared with control mice. The key findings were that serum L-aspartic acid levels were significantly decreased in the E8 group mice, and there was a significant enrichment of the amplicon sequence variant (ASV) corresponding to the gut microbiome family S24-7. The differentially abundant metabolites and genes were concurrently enriched in the Alanine, Aspartate and Glutamate Metabolism pathway. Specifically, the Asns gene within this pathway was significantly negatively correlated with L-aspartic acid levels, but significantly positively correlated with the abundance of S24-7.
Previous researches have proposed that the dominant changed serum metabolites, such as classified into amino acids and lysophosphatidylcholines (LPCs), were induced by early pregnancy(Zhao et al., 2021). Particularly, in normal pregnancy, the levels of most serum amino acids increased, which may play a pivotal role in contributing fetal health and growth(Barchitta et al., 2020). In this study, we found that the majority of serum metabolites increased or decreased in the E8 mice were fell into amino acids. L-Aspartic acid and Phenylacetic acid were detected with decreased trend in mice pregnant at 8 days. The significantly reduced levels of L-Aspartic acid in the serum of early pregnant mice are, to the best of our knowledge, a novel finding. This could represent an active self-protective mechanism by the mother, as L-Aspartic acid has been associated with Gestational Diabetes Mellitus (GDM), which can have adverse effects on both the mother and the fetus(Yang et al., 2023). Additionally, L-Aspartic acid has been implicated in causing neurotoxicity in newborn mice(Stegink, 1976), prompting recommendations against supplementation during pregnancy and lactation(Holecek, 2023). Therefore, we have reason to believe that the decreased serum levels of L-Aspartic acid in early pregnant mice may be an innate protective mechanism employed by the gestating females to safeguard themselves and their offspring.
The research findings indicate that the depletion of the maternal gut microbiome significantly impacts the growth and development of the placenta(Pronovost et al., 2023), suggesting that a healthy maternal gut microbiome is crucial for the mother’s gestational period and fetal development. However, studies on the changes in the maternal gut microbiome during early pregnancy remain limited. This study, therefore, investigated the composition of the maternal gut microbiome in pregnant mice at gestational day 8. The most prominent change observed was in the abundance of the family S24-7. Bacteria within family S24-7 (phylum Bacteroidetes) are dominant in the mouse gut microbiota and detected in the intestine of other animals. Recently, the name Muribaculaceae is used to substitute S24-7(Lagkouvardos et al., 2019). Previous research indicated that the increased abundance of S24-7 was negatively correlated with the production of inflammatory cytokines(Dong et al., 2022). In addition, research has proposed that the S24-7 is a major utilizer of mucin glycans, while many enteric pathogens require sugars as a nutrient source. Therefore, the increased abundance of S24-7 during early pregnancy may effectively inhibit the growth of pathogenic bacteria by occupying their ecological niche, which could be crucial for the early stages of pregnancy(Pereira et al., 2020).
In the current study, a number of differential metabolites and differentially expressed genes were found to be enriched in the same KEGG pathway: Alanine, aspartate and glutamate metabolism. Among the genes in this pathway, Asns gene showed strongest correlation with metabolomics and 16S rRNA amplicon data. Asns is a key enzyme in convertion L-Aspartic acid into asparagine(Lomelino, Andring, McKenna, & Kilberg, 2017). To data Asns deficiency or mutation in the parents has been reported can cause a number of pediatric neurological and developmental abnormalities, such as intellectual disability, developmental delays, and asparagine synthetase deficiency (ASD)(Schleinitz et al., 2018; Wang et al., 2020; L. Zhu et al., 2023). Hence, increased expression of the Asns gene and decreased levels of L-Aspartic acid have been observed, suggesting the existence of a reciprocal regulation between maternal genetics and metabolism during early pregnancy. The upregulation of Asns may help prevent the occurrence of adverse pregnancy outcomes and ensure normal fetal development. Furthermore, the maternal reduction in L-Aspartic acid levels mediated through Asns appears to serve a protective function by modulating systemic metabolism.
5. Conclusions
In summary, changes in the gut microbiome composition, serum metabolic profiles, and uterine gene expression have been detected in early pregnant mice. Among these multiomic alterations, the significantly increased abundance of the S24-7 genus and Asns gene expression were found to be inversely correlated with L-Aspartic acid levels. These findings suggest that S24-7, Asns, and L-Aspartic acid may represent potential targets for the traditional Chinese medicine-based intervention of pregnancy-related diseases and fetal developmental disorders, although the underlying mechanisms require further detailed investigation.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org
Author contributions
MH and SL conceived and designed the experiments and revised the manuscript; MH, SL, YL, and JG performed the experiments, analyzed the data, and wrote the manuscript; YY and RD collected the samples and performed the experiments. All authors read and approved the final manuscript.
Ethics statement
All procedures were conducted according to the Regulations for the Administration of Affairs Concerning Experimental Animals (Ministry of Science and Technology, China, revised in March 2017), and approved by the Institutional Animal Care and Use Committee at the Experimental Animal Center of Anhui Medical University (AHMUASY-20230622).
Data Availability Statement
Data are available on reasonable request. Data can be obtained from a third party and are not publicly available. Annotated metabolomic data of serum is available from supplementary Table S1.
Acknowledgements
This work was supported by National Natural Science Foundation of China (82302568, 82301917), Anhui Provincial National Science Foundation (2308085QH283, 2308085QH252), Basic and Clinical Collaboration Enhancement Program Foundation of Anhui Medical University (2022xkjT012) and Funding Program for Young Backbone Teachers’ Domestic Visiting and Seminar Program of Anhui Provincial Department of Education (gxjnfx2023003).
Disclosure statement
No potential conflict of interest was reported by the author(s).
References
- Akaeda, S., Aikawa, S., & Hirota, Y. (2024). Spatial and molecular anatomy of the endometrium during embryo implantation: a current overview of key regulators of blastocyst invasion. FEBS J. [CrossRef]
- Amato, K. R., Pradhan, P., Mallott, E. K., Shirola, W., & Lu, A. (2024). Host-gut microbiota interactions during pregnancy. Evol Med Public Health, 12(1), 7-23. [CrossRef]
- Antony, K. M., Ma, J., Mitchell, K. B., Racusin, D. A., Versalovic, J., & Aagaard, K. (2015). The preterm placental microbiome varies in association with excess maternal gestational weight gain. Am J Obstet Gynecol, 212(5), 653 e651-616. [CrossRef]
- Barchitta, M., Maugeri, A., Magnano San Lio, R., Favara, G., La Mastra, C., La Rosa, M. C., & Agodi, A. (2020). Dietary Folate Intake and Folic Acid Supplements among Pregnant Women from Southern Italy: Evidence from the “Mamma & Bambino” Cohort. Int J Environ Res Public Health, 17(2). [CrossRef]
- Boldeanu, L., Dijmarescu, A. L., Radu, M., Silosi, C. A., Popescu-Driga, M. V., Poenariu, I. S., . . . Novac, L. V. (2020). The role of mediating factors involved in angiogenesis during implantation. Rom J Morphol Embryol, 61(3), 665-672. [CrossRef]
- Bolger, A. M., Lohse, M., & Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics, 30(15), 2114-2120. [CrossRef]
- Burton, G. J., & Fowden, A. L. (2015). The placenta: a multifaceted, transient organ. Philos Trans R Soc Lond B Biol Sci, 370(1663), 20140066. [CrossRef]
- Cindrova-Davies, T., & Sferruzzi-Perri, A. N. (2022). Human placental development and function. Semin Cell Dev Biol, 131, 66-77. [CrossRef]
- Di Simone, N., Santamaria Ortiz, A., Specchia, M., Tersigni, C., Villa, P., Gasbarrini, A., . . . D’Ippolito, S. (2020). Recent Insights on the Maternal Microbiota: Impact on Pregnancy Outcomes. Front Immunol, 11, 528202. [CrossRef]
- Dong, L., Du, H., Zhang, M., Xu, H., Pu, X., Chen, Q., . . . Gao, F. (2022). Anti-inflammatory effect of Rhein on ulcerative colitis via inhibiting PI3K/Akt/mTOR signaling pathway and regulating gut microbiota. Phytother Res, 36(5), 2081-2094. [CrossRef]
- Edgar, R. C. (2010). Search and clustering orders of magnitude faster than BLAST. Bioinformatics, 26(19), 2460-2461. [CrossRef]
- Friedman, J., & Alm, E. J. (2012). Inferring correlation networks from genomic survey data. PLoS Comput Biol, 8(9), e1002687. [CrossRef]
- Fuhler, G. M. (2020). The immune system and microbiome in pregnancy. Best Pract Res Clin Gastroenterol, 44-45, 101671. [CrossRef]
- Giannella, L., Grelloni, C., Quintili, D., Fiorelli, A., Montironi, R., Alia, S., . . . Ciavattini, A. (2023). Microbiome Changes in Pregnancy Disorders. Antioxidants (Basel), 12(2). [CrossRef]
- Groer, M. W., Gregory, K. E., Louis-Jacques, A., Thibeau, S., & Walker, W. A. (2015). The very low birth weight infant microbiome and childhood health. Birth Defects Res C Embryo Today, 105(4), 252-264. [CrossRef]
- Gude, N. M., Roberts, C. T., Kalionis, B., & King, R. G. (2004). Growth and function of the normal human placenta. Thromb Res, 114(5-6), 397-407. [CrossRef]
- Gurner, K. H., Truong, T. T., Harvey, A. J., & Gardner, D. K. (2020). A combination of growth factors and cytokines alter preimplantation mouse embryo development, foetal development and gene expression profiles. Mol Hum Reprod, 26(12), 953-970. [CrossRef]
- Hemberger, M., Hanna, C. W., & Dean, W. (2020). Mechanisms of early placental development in mouse and humans. Nat Rev Genet, 21(1), 27-43. [CrossRef]
- Holecek, M. (2023). Aspartic Acid in Health and Disease. Nutrients, 15(18). [CrossRef]
- Huang, C. C., Hsueh, Y. W., Chang, C. W., Hsu, H. C., Yang, T. C., Lin, W. C., & Chang, H. M. (2023). Establishment of the fetal-maternal interface: developmental events in human implantation and placentation. Front Cell Dev Biol, 11, 1200330. [CrossRef]
- Jasarevic, E., & Bale, T. L. (2019). Prenatal and postnatal contributions of the maternal microbiome on offspring programming. Front Neuroendocrinol, 55, 100797. [CrossRef]
- Koren, O., Goodrich, J. K., Cullender, T. C., Spor, A., Laitinen, K., Backhed, H. K., . . . Ley, R. E. (2012). Host remodeling of the gut microbiome and metabolic changes during pregnancy. Cell, 150(3), 470-480. [CrossRef]
- Kuang, Y. S., Lu, J. H., Li, S. H., Li, J. H., Yuan, M. Y., He, J. R., . . . Qiu, X. (2017). Connections between the human gut microbiome and gestational diabetes mellitus. Gigascience, 6(8), 1-12. [CrossRef]
- Lagkouvardos, I., Lesker, T. R., Hitch, T. C. A., Galvez, E. J. C., Smit, N., Neuhaus, K., . . . Clavel, T. (2019). Sequence and cultivation study of Muribaculaceae reveals novel species, host preference, and functional potential of this yet undescribed family. Microbiome, 7(1), 28. [CrossRef]
- Lamichhane, S., Sen, P., Dickens, A. M., Oresic, M., & Bertram, H. C. (2018). Gut metabolome meets microbiome: A methodological perspective to understand the relationship between host and microbe. Methods, 149, 3-12. [CrossRef]
- Liu, Q., He, M., Zeng, Z., Huang, X., Fang, S., Zhao, Y., . . . Chen, C. (2023). Extensive identification of serum metabolites related to microbes in different gut locations and evaluating their associations with porcine fatness. Microb Biotechnol. [CrossRef]
- Lomelino, C. L., Andring, J. T., McKenna, R., & Kilberg, M. S. (2017). Asparagine synthetase: Function, structure, and role in disease. J Biol Chem, 292(49), 19952-19958. [CrossRef]
- Lu, X., Shi, Z., Jiang, L., & Zhang, S. (2024). Maternal gut microbiota in the health of mothers and offspring: from the perspective of immunology. Front Immunol, 15, 1362784. [CrossRef]
- Macpherson, A. J., de Aguero, M. G., & Ganal-Vonarburg, S. C. (2017). How nutrition and the maternal microbiota shape the neonatal immune system. Nat Rev Immunol, 17(8), 508-517. [CrossRef]
- Moylan, H. E. C., Nguyen-Ngo, C., Lim, R., & Lappas, M. (2020). The short-chain fatty acids butyrate and propionate protect against inflammation-induced activation of mediators involved in active labor: implications for preterm birth. Mol Hum Reprod, 26(6), 452-468. [CrossRef]
- Nuriel-Ohayon, M., Neuman, H., & Koren, O. (2016). Microbial Changes during Pregnancy, Birth, and Infancy. Front Microbiol, 7, 1031. [CrossRef]
- Nyangahu, D. D., & Jaspan, H. B. (2019). Influence of maternal microbiota during pregnancy on infant immunity. Clin Exp Immunol, 198(1), 47-56. [CrossRef]
- Pereira, F. C., Wasmund, K., Cobankovic, I., Jehmlich, N., Herbold, C. W., Lee, K. S., . . . Berry, D. (2020). Rational design of a microbial consortium of mucosal sugar utilizers reduces Clostridiodes difficile colonization. Nat Commun, 11(1), 5104. [CrossRef]
- Pronovost, G. N., Yu, K. B., Coley-O’Rourke, E. J. L., Telang, S. S., Chen, A. S., Vuong, H. E., . . . Hsiao, E. Y. (2023). The maternal microbiome promotes placental development in mice. Sci Adv, 9(40), eadk1887. [CrossRef]
- Quast, C., Pruesse, E., Yilmaz, P., Gerken, J., Schweer, T., Yarza, P., . . . Glockner, F. O. (2013). The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res, 41(Database issue), D590-596. [CrossRef]
- Red-Horse, K., Zhou, Y., Genbacev, O., Prakobphol, A., Foulk, R., McMaster, M., & Fisher, S. J. (2004). Trophoblast differentiation during embryo implantation and formation of the maternal-fetal interface. J Clin Invest, 114(6), 744-754. [CrossRef]
- Schleinitz, D., Seidel, A., Stassart, R., Klammt, J., Hirrlinger, P. G., Winkler, U., . . . Hirrlinger, J. (2018). Novel Mutations in the Asparagine Synthetase Gene (ASNS) Associated With Microcephaly. Front Genet, 9, 245. [CrossRef]
- Shannon, P., Markiel, A., Ozier, O., Baliga, N. S., Wang, J. T., Ramage, D., . . . Ideker, T. (2003). Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res, 13(11), 2498-2504. [CrossRef]
- Siena, M., Laterza, L., Matteo, M. V., Mignini, I., Schepis, T., Rizzatti, G., . . . Gasbarrini, A. (2021). Gut and Reproductive Tract Microbiota Adaptation during Pregnancy: New Insights for Pregnancy-Related Complications and Therapy. Microorganisms, 9(3). [CrossRef]
- Stegink, L. D. (1976). Absorption, utilization, and safety of aspartic acid. J Toxicol Environ Health, 2(1), 215-242. [CrossRef]
- Sun, Z., Lee-Sarwar, K., Kelly, R. S., Lasky-Su, J. A., Litonjua, A. A., Weiss, S. T., & Liu, Y. Y. (2023). Revealing the importance of prenatal gut microbiome in offspring neurodevelopment in humans. EBioMedicine, 90, 104491. [CrossRef]
- Vuong, H. E., Pronovost, G. N., Williams, D. W., Coley, E. J. L., Siegler, E. L., Qiu, A., . . . Hsiao, E. Y. (2020). The maternal microbiome modulates fetal neurodevelopment in mice. Nature, 586(7828), 281-286. 7828. [CrossRef]
- Wang, C., He, G., Ge, Y., Li, R., Li, Z., & Lin, Y. (2020). A novel compound heterozygous missense mutation in ASNS broadens the spectrum of asparagine synthetase deficiency. Mol Genet Genomic Med, 8(6), e1235. [CrossRef]
- Xie, G., Zhang, S., Zheng, X., & Jia, W. (2013). Metabolomics approaches for characterizing metabolic interactions between host and its commensal microbes. Electrophoresis, 34(19), 2787-2798. [CrossRef]
- Yang, J., Wu, J., Tekola-Ayele, F., Li, L. J., Bremer, A. A., Lu, R., . . . Zhang, C. (2023). Plasma Amino Acids in Early Pregnancy and Midpregnancy and Their Interplay With Phospholipid Fatty Acids in Association With the Risk of Gestational Diabetes Mellitus: Results From a Longitudinal Prospective Cohort. Diabetes Care, 46(4), 722-732. [CrossRef]
- Zhao, R., An, Z., Sun, Y., Xia, L., Qiu, L., Yao, A., . . . Liu, L. (2021). Metabolic profiling in early pregnancy and associated factors of folate supplementation: A cross-sectional study. Clin Nutr, 40(9), 5053-5061. [CrossRef]
- Zhu, L., Sun, Y., Xu, Y., Jin, P., Ding, H., & Dong, M. (2023). Case report: A compound heterozygous mutations in ASNS broadens the spectrum of asparagine synthetase deficiency in the prenatal diagnosis. Front Pediatr, 11, 1273789. [CrossRef]
- Zhu, Y., Mao, H., Peng, G., Zeng, Q., Wei, Q., Ruan, J., & Huang, J. (2021). Effect of JAK-STAT pathway in regulation of fatty liver hemorrhagic syndrome in chickens. Anim Biosci, 34(1), 143-153. [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



