
Submitted:
18 September 2024
Posted:
19 September 2024
You are already at the latest version
Abstract
As with all new fields of discovery, work on the biological role of G-quadruplexes (GQ) has produced a number of results that at first glance are quite baffling, sometimes because they do not fit well together, but mostly because they are different from commonly held expectations. Like other classes of flipons, those that form G-quadruplexes have a repeat sequence motif that enables the fold. The canonical DNA motif (G3N1-7)3G3, where N is any nucleotide and G is guanine, a feature that is under active selection in avian and mammalian genomes. The involvement of G-flipons in genome maintenance traces back to the invertebrate Caenorhabditis elegans and to ancient DNA repair pathways. A role for GQ in transcription is supported by the observation that yeast Rap1 protein binds both B-DNA, in a sequence-specific manner, and GQ, in a structure-specific manner, through the same helix. Other sequence-specific transcription factors (TF) also engage both conformations to actuate cellular transactions. Noncoding RNAs can also modulate GQ formation in a sequence-specific manner and engage the same cellular machinery as localized by TF, linking the ancient RNA world with the modern protein world. The coevolution of noncoding RNAs and sequence-specific proteins is supported by studies of early embryonic development, where the transient formation of G-quadruplexes coordinates the epigenetic specification of cell fate.
Keywords:
Flipons
; G-Quadruplex
; Triplex
; Z-DNA
; Proliferation
; Transcription
; Translation
; Repair
; Repression
; Sister chromatids
; CTCG
; Chromatin Loops
; Class Switch Recombination
; HIV
Introduction
The idea that the repetitive genome encodes genetic information by shape rather than by sequence is relatively new. The unit of information is the flipon, a genomic element that can adopt alternative structures under physiological conditions. The conformation formed depends on the repeat sequence involved. The classic example is provided by left-handed Z-DNAs and Z-RNAs (collectively called ZNAs) that are formed by runs of alternating guanosine and cytosine [1,2]. Collectively, the repetitive genome comprises over 50% of the human sequence, compared to 2.5% for protein coding genes.
Flipons in the B-DNA conformation have little informational value as the repeats are frequent in the genome. They also lack the complexity of codons, so do not contribute directly to the Watson and Crick genetics that focuses on protein variation. Instead, flipons alter the readout of genetic information by localizing structure-specific complexes to genomic loci able to power the flip from a right-handed B-DNA or A-RNA helix to an alternative DNA or RNA fold. The readout of RNAs then varies dynamically with flipon structure. Here the focus is on G-flipons that form G-quadruplexes (GQ) in DNA (dGQ), RNA (rGQ) or DNA/RNA hybrids (hGQ). GQ are inherently more stable than ZNA helices. Consequently, G-flipons can actuate biological processes that are quite distinct from those modulated by Z-flipons.
GQ forming sequences are defined by the canonical DNA motif (G3N1-7)3G3, where G is guanine and N is any nucleotide. Four G-bases hydrogen bond to each other to form a tetrad that then folds into a four stranded structure (Figure 1A). In place of the Watson-Crick base-pairing scheme, the rather unconventional Hoogsteen hydrogen bonds stabilize the interaction (Figure 1B, highlighted by colored shading). The G-tetrad was first observed in X-ray diffraction studies of 5'-GMP and 3'-GMP gels, each stacking the tetrads on top of another in a different manner [3]. The preferred helical arrangement of GQ crystalline fibers was later revealed by structural studies of polyinosinic and polyguanylic RNAs [4].
It was once widely believed that GQ did not exist in cells. If present, then the GQ formed predisposed to genetic instability and to disease [5]. There was much excitement when the Tetrahymena telomere sequence repeats [6] were shown to form GQ [7]. In contrast, later work revealed that telomeres in vivo were more likely to form a different type of structure called a T-loop [8]. Closure of the loop lead to formation of a three-stranded DNA structure that incorporated the single stranded telomeric end and a subtelomeric segment. This structure was protected by a shelterin protein complex. The T-loop model seemingly ruled out a role for GQ in telomere maintenance (but see below). The prevailing view that GQ were bad was reinforced by the many loss-of-function (LOF) helicase variants that were associated with human mendelian diseases. The failure of these variants to resolve GQ was considered causal for the genomic instability, even though the helicases also resolve other non-B structures, such as cruciforms and the Holliday junctions (HJs) that form during recombination [9]. Further, a role for GQ in pathology was suggested by an analysis of repeat expansion diseases. In some cases, the sequences involved were predicted to freeze in the GQ conformation, thereby interfering with a variety of cellular functions, including DNA replication, transcription and RNA processing [10].
However, there was evidence that GQ played an essential biological role in the adaptive immune system. The GQ were associated with class switch recombination of immunoglobulin heavy chain (IgH) genes. Of interest were the noncoding switch (S) regions in the IgH gene that underwent transcription to produce R-loops. The non-template strand was G-rich and 2 to 10 kb in length. When displaced by RNA transcript, the single-stranded G-rich DNA was able to fold back on itself to form GQ [11]. The targeting of the AID cytosine deaminase protein to the GQ structure by the helicase DDX1 was essential for both class switching and immunoglobulin somatic hypermutation that is critical for antibody affinity maturation [11,12,13,14]. The cytosine to uridine substitution catalyzed by the cytidine deaminase was not only mutagenic, but also recruited the repair machinery required for DNA recombination. In other contexts, GQ formation in G-rich DNA due to R-loop formation was proposed as pathogenic [15].
Other experimental approaches to unraveling the biology of GQ were complicated by the equilibrium that exists between different flipon conformations, with the transition occurring in unmodified DNA and without requiring any strand cleavage [1]. Early experiments using dimethyl sulfoxide footprinting of RNA failed to show the protection of guanine bases expected if a GQ had formed inside a cell [16]. These results were interpreted to show that GQ were not biologically relevant. However, there was a problem with the experimental design: chemical modification of any G-quadruplexes that unfolded during the time course of the experiment would prevent the structure from reforming [17]. In other words, the longer the experiment ran, the less chance there was of detecting the presence of GQ in a cell. Nevertheless, the study highlighted the possibility that GQ were formed dynamically in cells and that they were rapidly resolved to reform B-DNA.
There were also limitations to other experimental approaches designed to detect GQ. Tools designed to detect GQ in cells were able to induce their formation. This risk of artefact increased when assays were performed on cell extracts. Here various factors came into play, such as the buffers used, and the loss of proteins that might otherwise restrain the B-DNA flip to GQ. Even well accepted ChIP-seq protocols to map protein interactions potentially mislead, as recently shown by a stringent analysis of the GQ binding substrates of PRC2 (Polycomb Repressor Complex 2) interactions [18]. Combined, these uncertainties limited the widespread acceptance of G-flipons as important components of the genetic repertoire. The repetitive genome was just considered “junk” [19].
The intent of this review is to integrate information from a wide range of research papers, including some whose significance has been long overlooked and are not mentioned in many recent GQ reviews [20,21,22,23,24,25,26,27,28]. The initial focus is on the genetic evidence that speaks to an early evolutionary role for G-flipons in maintaining genomic stability and on the proteins that localize the machinery required for nucleotide and base excision repair (NER, BER respectively) by inducing GQ formation. Different classes of helicase then power the resolution of GQ to reform B-DNA, completing the flipon cycle. By changing the readout of genetic information, flipons dynamically reprogram a cell in response to environmental perturbations.
I will then discuss roles for G-flipons in transcription that emerged later in evolution. This feature reflects a change in how GQ recognition occurs, from interactions involving single-stranded loops and modified bases, to those mediated by proteins that bind both B-DNA and G-quadruplexes through a different face of the same helix.
Biophysical and Computational Studies of the G-Quadruplex
The basic building block for a G-quadruplex is a guanine tetrad formed by Hoogsteen hydrogen bonding, [4] (Figure 1B) between bases [29]. Interestingly, the parallel nature of these bonds contributes to sigma bonds that increases the stability of the G-tetrads relative to those formed by xanthine where the bonding is anti-parallel [30]. A recent review describes 48 different possible GQ folds, reflecting whether the four strands are parallel, anti-parallel or a mix, made from one to 4 different strands with lateral, diagonal or propeller loop topology [31] (Figure 1). Further, the guanosine residues may be either in the syn or anti conformation (with the guanine base either lying over the sugar or pointed away from it) [32]. The GQ can also be left-handed [33]. The folds are stabilized by a central metal, with a potassium ion preferred over the smaller sodium and lithium ions for parallel strand GQ. The metal preference for other GQ folds varies and depends on whether they are made from RNA or DNA [34]. Non-consecutive guanosines can form tetrads with the extra residue everted from the stack to form a bulge. In the case of GGA repeats, the adenine bases that are excluded from the quadruplex can interact with the tetrads to produce a heptad structure [35,36].
The stability of GQ is also affected by the loop composition, decreasing with loop length, and varying with the loop nucleotide sequence [37]. With long runs of G repeats, defined as over 500 bases in length, the loops can basepair to give even higher order structures. Of the 299 such long G runs reported, over 67% are located within 6M bp (base pairs) of telomeres. [38]. Interestingly, loop length and sequence variation has increased during evolution, especially in mammals, as has GQ length, number, and density in the genome [39]. G-flipons are also more frequent on the non-template strand of coding genes [40,41].
Besides GQ formation by neighboring G3 repeats, it has been proposed that GQ are formed by a pair of G3 repeats in an enhancer and a pair of G3 repeats from a promoter [42]. Further, a hybrid GQ can form between a pair of DNA G3 repeats in the non-template strand and a matching RNA G3 pair in the nascent transcript [43]. The GQ formed by strands that are not physically connected to each other also show structural variation. The GQ can assemble by stacking tetrads, one on top of the other or by pairing bases from the separate strands l to form a G-wire [44,45]. G-wires were originally proposed to explain the alignment of homologous chromosomes during meiosis [46]. Tetrads missing the fourth base can incorporate into the vacant space a guanine provided in trans, potentially acting as a sensor for a local change in concentration of the replacement nucleotide [47].
RNA tetrads only form parallel rGQ when G-repeats are contiguous [34]. A variety of different rGQ folds are stabilized by pairing schemes involving G bases that are separated by other nucleotides. [48]. rGQ composed of only 2 tetrads have been reported [49] and are stabilized by the 2′-hydroxyl group present in RNA [34]. In contrast, there are many possible variations of dGQ composed of 3 or more tetrads, making it difficult to computationally predict from sequence alone those flipons that actually form dGQ in vivo. A database that combines results from a variety of experimental methods now overcomes this problem by providing a set of well validated G-flipons detected in many different studies using a variety of approaches [50]. The mappings show that in the human genome, dGQ forming sequences are enriched in transcription start sites (TSS), in introns and at transcription termination sites (TTS) [39].
GQ Binding Proteins
The plethora of different dGQ topologies allows for different modes of protein recognition (Figure 2 and Figure 3). Strategies to confirm these interactions and the specificity of binding to GQ include those that synthesize control oligonucleotides containing the 8-aza-7-deazaguanosine base (Figure 1C) that will not form the Hoogsteen hydrogen bonds necessary to stabilize a GQ (Figure 1B, crimson shading), despite having the same chemical composition as guanine [51]. In these studies, different modes of docking to GQ have been identified, including binding to loop sequences or to 5' and 3' single-strand extensions that give the helicases something to pull on so that they can unwind the structure. Proteins can bind to loops formed when adducted bases such as 8-oxo-G prevent the incorporation of a DNA strand into a GQ, or to the everted bases across from an apurinic/apyrimidinic (AP) site. Proteins also dock to the planar tetrad surfaces that form the GQ endplate (Figure 2). Specific binding to RNA rather than DNA GQ is favored by intrinsically disordered regions (IDR) enriched in arginine, glycine repeats, as recently reviewed [52], and visualized in the FMR crystal structure of the Fragile X Mental Retardation Protein bound to an RNA GQ [53]. In principle, the preformed GQ site for docking IDR lowers the entropic cost of binding.
The stability of GQ and strength of their interaction with proteins can vary with the loop length and the loop sequence composition [54,55], as revealed by studies of nucleolin and the 2E4 Darpin [56,57]. Further, the latching of a single base by the REV1 polymerase [58], and the docking to an AP site by APE1 (AP endonuclease 1) [59], can create a surface that induces GQ folding. As we will discuss, the use of SANT (Swi3, Ada2, N-Cor, and TFIIIB) domains to recognize parallel-strand GQ is of particular interest as the domains can use the same helix to bind B-DNA in a sequence-specific manner (Figure 2). In total, 50 GQ-peptide structures are present in the Protein Data Base (PDB) showing a variety of interactions [26,57]. A subset of validated GQ interacting proteins is given in Figure 3. Listings of additional proposed GQ binding proteins can be found in recent publications ([51,He, 2023 #3109, 60] and online at the G4IPBD database (http://people.iiti.ac.in/~amitk/bsbe/ipdb/index.php, accessed 15th September, 2024) [61] and the QUADRatlas database (https://rg4db.cibio.unitn.it/, accessed) 15th September, 2024 [62].
The Accumulating Evidence for the Biological Importance of G-Quadruplexes
Despite the numerous challenges to studying the cellular functions of high energy and dynamic flipon conformations, much progress has been made. There are two key aspects to the biology: first the events that promote and resolve the formation of the alternative flipon structures and second, the transactions that the alternative flipon conformations modulate. There are well validated proteins that can induce the flip to GQ and many helicases capable of their resolution (Figure 3, up and down arrows). Although GQ formation does not inherently require any change, modification, or cleavage of DNA or RNA, such events may change the propensity of G-flipons to flip from one conformation to another. The GQ formed in these processes differ in topology. The structured loops they form are recognized by specific sets of proteins, as are the GQ endplates (Figure 3, top). The outcomes depend on which cellular machinery is localized to a particular GQ. The complexes formed enable cells to reprogram their responses to environmental perturbations.
The trans actions occurring between GQ formed at different sites are also important in understanding their cellular functions. The complexes nucleated by one GQ have the potential to associate with other G4-anchored structures to form membraneless condensates (Figure 4) [64,65]. These complexes can be quite large and visible by light microscopy [64]. The interactions enable the sequencing and timing of events within the cell (Figure 4A). Pairing of promoters GQ with GQ formed at enhancers, splice sites and polyadenylation sites then generate production lines for the processing of transcripts. Anchoring of the lines to the nuclear scaffold to form factories [66,67] that enable the transcriptional bursts associated with gene expression [68]. The pliability of these production lines is revealed by the constant updates to nuclear architecture [69].
The GQ Architecture of Retroviruses
The simplest example of GQ mediated integration may be provided by retroviruses, such as the human immunodeficiency virus 1 (HIV-1). These viruses encode G-flipons in the long terminal repeat that is present at either end of their 9.6 kb genomic insert [70] (Figure 4B). The arrangement enables the formation of chromatin loops that separates the viral protein coding genome from that of the host. In this state, the virus is likely latent. Nevertheless, the virus is poised to replicate on removal of the loop restraint (Figure 4B). The HIV-1 plus strand mRNA also contains 11 potential G-quadruplexes with 9 in the coding sequence. The topologies are mixed, raising the possibility that particular pairings affect the splicing, stability, recombination, and repair of transcripts [71]. Long Interspersed elements (LINEs) are another class of retrotransposons of similar length to retroviruses that have a G-flipon conserved in their 3'UTR (untranslated region). The pairing pf the LINE GQ with GQ in cellular enhancers has the potential to form a loop that controls their expression in a tissue-specific manner [72]. Conversely, the 5’UTR G-flipons that LINE families acquire during evolution can themselves act as tissue specific enhancers of cellular genes [73].
G-flipon functions within the cell and their modulation by G-flipon cycles are described below,
Cell Division
Interestingly, the first evidence hinting at a biological role for GQ came from the round worm Caenorhabditis elegans. Sequences with the G-quadruplex motifs underwent deletion in strains with dog-1 (deletions of guanine-rich DNA) LOF variants, but not those sequences with only 3 G3 repeats that are unable to form GQ [74]. Mutant strains of dog-1 lacking the trans-lesion polymerases (TLS) polymerases, POL eta and POL kappa had significantly more G-tract deletions than dog-1 by itself [75]. Interestingly, the combined deletion of dog-1 and the spindle-checkpoint component mdf-1 enabled long term survival [76], even though a high incidence of lethal mutations in this strain was revealed by the use of balancer chromosomes. In total, 126 (13%) of the 954 mono-G/C tracts larger than 14 bp, were deleted over 470 generations when both genes were absent. A role for GQ in sister chromatid alignment by the cohesin proteins during mitosis was suggested by effects of dog-1 LOF on the spindle checkpoint. The absence of other phenotypes also supported the consensus that GQ had only a limited role in normal cell biology, not only in C. elegans, but also in other organisms.
Epigenetic Maintenance. The dog-1 homolog in the DT40 chicken lymphoblastoid cell line, the 5' FANCJ (Fanconi Anemia Complementation Group J) helicase (a member of the Fe-S superfamily 2 (SF2)) [77] also was found to prevent deletion of guanine repeats (G-repeats) with the potential to form GQ. Effects of the mutation were enhanced by loss of the REV1 polymerase that localizes TLS to sites of polymerase stalling. Interestingly, REV1 catalytic activity was not necessary to prevent deletion, although the LOF variant did enhance the rate of G-repeat loss. Also, in the FANCJ model, the combined deletion of the Werner and Bloom Syndrome 3' helicases (RecQ SF2) [78] also increased G-repeat deletion, likely because of GQ accumulation [77].
Of interest was that the TLS pathway was required to maintain the epigenetic state of dividing cells, as monitored by cell-surface expression of a protein with an intronic G-flipon that regulated gene expression. Whereas in the wildtype cell, the histone modifications associated with this G-flipon were maintained, they were lost following rev1 deletion. Instead, resolution of the GQ formed during DNA replication was through the gap-fill repair pathway. The subsequent incorporation of unmodified histones led to diminished gene transcription and surface marker expression. This rev1-dependent phenotype could be reverted by re-expression of human FANCJ helicase [77]. The opposite effect was observed when a G-flipon was experimentally inserted into a repressed locus. In this case, rev1 deletion led to depression of the segment, consistent with the replacement of repressive histone with unmodified histones that were permissive to gene expression [79]. These results support a model where the formation of GQ by G-flipons during periods of cell proliferation helps in transmitting the current epigenetic state to progeny, an important biological outcome.
DNA replication and Sister Chromatid Conformation. The involvement of GQ in cell proliferation is further supported by other evidence. During assembly of the DNA polymerase complex at the origin of replication (OOR), the MTBP protein assists in the loading of CDC45 into the replicative helicase. The C-terminal domain of MTBP binds GQ in vitro [80]. Notably, G-flipons are enriched in OOR. Indeed, in chicken DT20 cells a minimal, functional OOR consists of a 90 bp fragment that has two G-flipons on the same strand [81]. These constructs establish the nucleosome depleted region (NDR) bounded by histone H2A.Z that is typical of the OOR. Collectively, the results suggest a model in which the MTBP binds GQ at the OOR to initiate the assembly of the replication complex.
Another potential role for GQ during proliferation and transmission of epigenetic state is to align sister chromatids, as mapping of intra- and inter chromatin interactions between homologous chromosomes reveals a high degree of symmetry in the architecture of topologically associated domains (TADs), and in the loops formed within TADs [82]. In this regard, a recent report suggests that G-flipons are enriched near sites bound by the CTCF (CCCTC-binding factor), a protein associated with loop formation. Interestingly, the strand orientation of the G-flipons mirrors the inverse orientation of the two CTCF sites that associate with each other to form the base of the loop [83]. CTCF however is not known to bind GQ [51].
DNA Repair
G-flipons in nucleotide excision repair (NER) . The REV1 pathway also plays a role in NER that is triggered by UV irradiation and the formation of DNA crosslinks. In this situation, loading of the repair pathway proteins such as XPCC and RAD23 is triggered by the protein ZRF1 and its yeast homolog Zuo1 that recognizes the lesion and induces GQ formation [84]. Triggering of this pathway by cytosine deaminases can result in single base substitutions at a sequence tagged site (STS) with a C to G transversion resulting from the preferential insertion of cytidine into the lesion by REV1 [85]. This mutation (STS13) is prevalent in cancers [86].
NER in the transcription coupled repair pathway (TCR) depends on the Cockayne Syndrome B (CSB) helicase (encoded by ERCC6) that binds GQ [87]. On sensing a lesion, CSB displaces DSIF (DRB Sensitivity Inducing Factor) from the RNA polymerase 2 (RNAP2) complex, inducing a conformational switch that halts transcriptional elongation and initiates TCR [88]. LOF variants of CSB are associated with premature aging phenotypes[87].
G-flipons in base excision repair (BER) . APE1 plays a similar role in stabilizing GQ formed by AP DNA, but not unmodified DNA, to initiate BER pathway [59]. The pathway removes oxidized bases, such as 8-oxo-G. It is proposed that regulation of the APE1 by acetylation coordinates the expression of genes involved in cellular pathways that respond to oxidative damage. Interestingly, the GQ involved are formed from G-flipons with a “spare tire” (Figure 1F). The extra runs of G-repeats allow formation of a GQ despite damage to one of the other repeats [89].
The 8-oxoG modification can arise due to toxins in the environment. The adduct is also generated during the flavin-dependent LSD1 (lysine demethylase 1A, encoded by KDM1A) demethylation of H3K9me2, where hydrogen peroxide is a product of the reaction. The LSD1 enzyme is activated during the induction of BCL2 gene expression by estrogen [59]. The repair of the lesion through the BER pathways depends on GQ formation. Before the involvement of GQ in this process was known, the finding, it was proposed that DNA strand breaks were a general mechanism for initiating gene transcription [90].
Hemin and Oxidative damage. Another cause of oxidative damage is due to the production of highly reactive oxidative species catalyzed by hemin, an iron-containing porphyrin that is present at high concentration in the cell [91]. Hemin binds with high affinity (Kd ~ 10 nM) to GQ, an interaction that was initially highlighted for its ability to increase production of superoxide [92]. However, it appears that in cells that this reaction is squelched, presumably by proteins that bind to GQ [91]. In such cases, GQ may act as a sink for free hemin and trigger the rapid repair through the BER pathway of any damage hemin causes. In such cases, GQ protect rather than damage the genome.
GQ and Telomeres
The formation of T-loops by telomeres described above does not rule out a role for GQ formation in telomere protection. Indeed, the TRF2/RAP1 complex protects telomeres from homologous recombination by repressing PARP1 localization to telomeres and by inhibiting the SLX4 resolvase that binds to HJs. Loss of TRF2 and RAP1 in both humans and mice leads to rapid telomere attrition, with increased rates of telomere deletion and fusion[93]. TRF2 preferentially docks to rGQ rather than dGQ. The protein binds rGQ formed by the noncoding Telomeric Repeat-Containing RNA (TERRA) telomere transcript through an RG rich domain [94]. Interestingly, the HIV retrovirus may form a dGQ to cap the DNA flap sequence produced during the pre-integration phase of reverse transcription, potentially protecting the end in much the same way as proposed for telomeres [95].
Resolution of G-Quadruplexes
Implicit in the G-flipon cycle is the need to reset flipons to a resting state. As shown in Figure 3, many helicases enable this outcome. The most studied example is the ATP dependent DEAH box SF2 helicase DDX36 (RHAU), a highly specific GQ resolvase that unwinds parallel dGQ. The enzyme makes helical contacts with the GQ end plate [96,97]. Binding by the helix alone has a relatively high Kd of 1 μM. The additional engagement of a 3' single-stranded dGQ tail by other residues accounts for the nM affinity of the enzyme for its substrate. Using a ratchet mechanism, the helicase disassembles the dGQ, one guanine at a time. The chemical energy derived from ATP is converted into a pulling force by rotation of the C-terminal domain. The twist provides access to the helicase core [97]. In the absence of nucleotide, or in the ADP bound state, D. melanogaster DDX36 stabilizes the GQ [98].
The cocrystal structure of dGQ with the SF1 Thermus oshimai 5′-3′ Pif1 helicase shows the enzyme in an unwinding state with engagement of a single-stranded thymine repeat [99]. The related yeast helicases PiF1 and Rrm3 cooperate to unfold a wide range of dGQ topologies, including those formed not only by telomeres, but also by centromeres and tRNA repeat sequences [100,101]. The enzyme unfolds dGQ in an ATP-dependent manner, unwinding both parallel and antiparallel dGQ [99]. The interaction of the Pif1 with the parallel stranded dGQ differs from that of DDX36. The contact is mediated by a cluster of amino acids, including two arginine/lysine cation-π interactions at either end of the dGQ, plus ionic contacts with the phosphate backbone. The SF2 RecQ BLM helicase also can unfold a range of dGQ folds through a variety of different mechanisms [102]. Collectively, the helicases play key but distinct roles in flipping dGQ back to the B-DNA conformation.
G-Quadruplexes and Gene Expression
The SANT domain, and gene expression. The widely held assumption is that a crystal structure of a protein engaged with B-DNA precludes an interaction with any other DNA conformation, especially if the substrate is bound with nM affinity. Of course, crystal structures by their nature represent a low energy state. The example of Rap1 is therefore instructive (Figure 2). Prior to its role in telomere protection, Rap1 was characterized as a sequence-specific transcription factor that bound to a UAS (upstream activating site) in yeast [103]. The base-specific interaction with B-DNA was confirmed by crystallographic study of a telomeric sequence (Figure 2A)[104]. Only later did crystal structures show that Rap1 also docked to GQ. Surprisingly, both DNA interactions involved the same helix, but a different face [105] (Figure 2B). The GQ contacts were hydrophobic, with the helix lying on the planar surface of the terminal tetrad, while the B-DNA contacts were consistent with those found for the UAS. Both interactions have a Kd≈20-30 nM [105], yielding a switch that has two stable states (Figure 2C). The switch state then depends on the context and the availability of helicases. The example illustrates the potential of flipons to switch the readout of genetic information from a genome by changes to their conformation [106].
While this finding might seem anomalous, many subsequent studies have demonstrated the ability of proteins to bind specifically to a cognate B-DNA sequence, and also to a GQ. In both cases, the affinity is often nanomolar. This finding is true for binding of the SP1 transcription factor to the c-MYC parallel GQ [107] and for a range of other proteins that bind GQ and a B-DNA motif[21]. Interestingly, like Rap1, many of the GQ binding proteins include a SANT/Myb domain such as ZRF1 [108] and TRF2 [109,110]. Interestingly, the yeast Zuo1 protein has replaced the SANT domain with a highly hydrophobic helix that could well interact with the endplate of a GQ [108]. SANT domain proteins are found in multiple chromatin-modifying and remodeling complexes, although their interactions with GQ are not yet reported [111].
GQ and transcription complexes. Given the enrichment of G-flipons in promoters, a key question was how do the GQ stabilizing and resolving proteins impact transcription. GQ binding proteins like YY1 (Yin Yang 1) are known to form homodimers that promote enhancer-promoter contacts [51,112,113]. So do transcription factors that bind GQ. One of the surprises of the ENCODE project was the identification of HOT (high occupancy target) loci where upwards of a 100 TF bound, even to sites lacking their sequence-specific binding motif. The findings were initially dismissed as methodological artefacts [114], but were later shown not to be so [115,116]. The primary studies focused on the sequence-specificity of TFs, not the GQs that were also formed at promoters. The ability of TF to bind both B-DNA and GQ offered a resolution to this HOT dilemma [51]. Indeed, recent findings suggest that it is GQ formation that recruits TF to transcriptional hubs [117]. In this new model, as described here, TFs play a different role. Through the complexes they anchor, TF localize helicases to resolve the GQ formed by promoters. A specific helicase might recognize a particular GQ fold, a GQ loop of particular length or composition, or display a preference for a 5' or 3' single-stranded flanking sequence. The biological outcomes then depend on the GQ topology and the helicase involved. The model explains the diversity of functions enabled by the G-flipon cycle (Figure 3).
G-Quadruplexes and Transcriptional Bursts
One extension of this model is that docking of TF to GQ maintains a transcription state following its initiation by the binding of a sequence-specific TF to B-DNA. Consequently, there would be no need for any further sequence-specific interactions with the promoter. However, this possibility is not consistent with the observed rapid reset of promoters that occurs after each round of transcription [118,119]. The fast disassembly of the transcriptional complexes following each round of transcription is mirrored by the abrupt dissolution of promoter condensates triggered by the high levels of nascent RNAs produced [120]. The evidence suggests that transcription occurs in bursts followed by a reset rather than by a preset level of expression.
Earlier experiments based on single molecule FISH suggest that the transcriptional burst frequency, but not the burst size, depends on the rate of promoter reset [118]. One contribution to burst size is the frequency with which sister chromatids are transcribed. Curiously, only one allele is active at a time, rather than both undergoing simultaneous transcription [118]. The localization of many different helicases to the locus may allow one allele to reload a sequence-specific TF to reform an initiation complex while the other one fires. Such coordinated activity is consistent with the symmetrical chromatin architecture observed for sister chromatids, as described above [82]. The lack of co-bursting by maternal and paternal chromosomes is consistent with recent single cell studies of allele-specific transcription [125].
G-Quadruplexes and Promoter Pausing
How then do GQ modulate transcriptional bursting? The formation of GQ at promoters often is detectable before the initiation of transcription [126]. In such situations, there is no preference for which strand forms a GQ, further suggesting that gene transcription is not required to induce the flip from B-DNA [107]. Further, the GQ flip is not modulated by human topoisomerase I (TOP1), even though the enzyme is enriched at these sites [127]. Instead, TOP1 is inhibited by GQ, with an IC50 ~ 100 nM [128].
It is possible that the GQ formed at promoters engage RPOL2, but prevent elongation by holding the enzyme in a poised state (Figure 5A). The YY1 mediated looping between promoter and enhancer GQ could further freeze RPOL2 in place through the condensate formed [51,112,113]. Locking down RPOL2 then provides time to properly position other GQ anchored condensates required to correctly splice and polyadenylate the pre-mRNA produced. Without such an arrangement to coordinate downstream events, an elongating RPOL2 appear to terminate transcription prematurely, then detach from the DNA template (Figure 4) [129]. Once RPOL2 is released from the TSS, the enhancer-promoter condensate disassembles, allowing the promoter to reset for another transcriptional burst [120].
G- and Z-flipons in Promoter Reset
This scenario provides a different perspective on why TF engagement of promoter GQ is important in regulating gene expression. In the new scheme and as described above, TF do not directly drive gene expression by binding a cognate motif. Instead, they engage GQ, localizing complexes that contain the helicases required to rapidly resolve GQ. The reset then allows the DNA duplex to reform and B-DNA sequence-specific proteins to seed formation of a new pre-initiation complex.
For this to happen, it is also necessary to clear the existing pre-initiation complex (PIC) used previously to dock RPOL2 at the transcription bubble (Figure 5). This process often involves Z-DNA forming sequences near the TSS. Indeed, many of the promoters regulating embryonic and neurological development contain both G- and Z-flipons that have been validated experimentally [130]. Many other factors affect promoter reset. For example, the capture by Z-flipons of the energy released by an elongation RNAP2 is thought to power the re-engagement of the RNAP2 complex [131,132]. In these cases, Z-DNA formation is initiated by the negative supercoiling generated following release of RPOL2 from the pause site. The energy stored in Z-DNA is then used to power dissociation of the existing PIC (Figure 5B). The reset of Z-flipons then actuates removal of the PIC from the promoter (Figure 5C)[132]. There is preliminary evidence that the Z-DNA formed nay also engages GTFE (General Transcriptional Factor E), leading to redocking of the RPOL2 at newly formed transcription bubble [132,133].
The reengagement of the RPOL2 complex also depends on reformation of the PIC that is promoted by sequence-specific TF binding to B-DNA. The positive supercoiling induced by the PIC [134] likely plays a role in both steps as it leads to unwinding of DNA on either side of the PIC [131]. The uncoiling of DNA is permissive to GQ formation in the promoter region, signaling that the PIC is engaged, while the unwinding assists the docking of RNAP2 by further opening the transcription bubble to allow engagement the RPOL2 catalytic center (Figure 5C).
This mechanism is quite flexible and adaptable. The insertion of flipons during evolution into promoters by retrotransposons provides a mechanism for modulating gene expression. In humans, the copying and pasting of the ALU family of SINEs (Short Interspersed Nuclear Elements) throughout the genome has greatly enhanced this type of genetic variation [106]. An extreme example of the alternative outcomes enabled by the insertion of flipons into promoter regions is provided by the experimental observation that some flipons can form either GQ or Z-DNA [130,135]. The particular structure adopted may depend on which TSS is used to initiate gene expression. The formation of Z-DNA downstream would be driven by transcription, while the flip upstream to GQ would be driven by TF engagement.
The involvement of G-flipons in both polymerase pausing and in promoter reset may produce some paradoxical results when ligands that stabilize GQ are employed experimentally. The outcome then depends on the step in the G-flipon cycle that is most affected. The immediate effect of disrupting the GQ dependent enhancer-promoter condensate is the release of RPOL2 from the promoter and a transcriptional burst. The failure to reset the promoter will maintain the NDR and increase the chance of DNA damage, leading to decreased transcription initiation and eventually to cell death. It is also possible that a GQ stabilizing ligand may disrupt the reset of GQ at genomic sites other than the promoter, leading to premature transcript termination due to the disruption of downstream RNA processing events.
Gene Repression
GQ and Gene Repression. The promoter reset occurs in competition with complexes that suppress gene expression. These competitors include the PRC2 complex that engages the GQ formed at promoters through the SANT domain of the EZH2 (enhancer of zeste 2) component. For active genes, binding of PRC2 the GQ formed by a nascent RNA likely prevents engagement of the GQ formed by the single-stranded promoter DNA [136]. However, in other situations, binding of a small RNA to the coding strand would promote GQ formation by the promoter DNA without the transcription of a GQ RNA competitor. In this situation, proteins, such as PRC2, that are localized to the site by the small RNA, would enhance formation of a repressive complex at the promoter. In these situations, the small RNA could be produced from a locus elsewhere in the genome [130]. Indeed, the small RNAs direct the hiwi (human ortholog of piwi) mediated repression of human endogenous retroelements in early development are produced from over 6000 clusters [137,138,139]. By localizing a different set of proteins to the site, small RNAs acting in trans could also promote transcriptional activation (Figure 4A). Such a role has been proposed for the other piwi-related agonaute family member complexes [140,141].
R Loop Resolution
A number of mechanisms exist to regulate dGQ formation by R-loops (Figure 3). For example, helicases such as SETX, and RTEL1 can facilitate the flip of GQ back to B-DNA through the resolution of RNA:DNA hybrids [142,143]. Nucleases that digest the RNA strand of hybrids, such as RNaseH1, play an important role in their removal [144]. Other proteins such as ATRX prevent R-loop formation at telomeres by sequestering RNA. Deletion of ATRX leads to increased formation of GQ at telomeres [145].
Transactional Chromatin Looping and Transcript elongation
In cellulo studies reveal that delays in RNAP2 transcript elongation occur at the CTCF binding sites involved in chromatin loop formation. CCTF binds to the large subunit of RNAP2 and the interaction is also associated with cohesin recruitment [146,147,148]. Conversely, CTCF binding to DNA increases, following deletion of the DNA methylase DNMT1.
These findings are consistent with a model where stalling of the polymerase by CTCF results in an R-loop that promotes GQ formation at the site. The GQ structure produced then inhibits DNMT1, preventing DNA methylation of the locus by trapping the enzyme. The trap works as the binding affinity of DNMT1 is higher for GQ than to either duplex, hemi-methylated or single-stranded DNA [149]. The resolution of the GQ by helicases then allows redocking of CTCF to the original DNA site, leading to reinstatement of the chromatin loop formed with the promoter (Figure 4). The CTCF binding sites necessary for this transaction lie in reverse orientation to each other. They are then fully aligned at the base of the loop and held in that state until the next splicing event [83]. After the splicing complex is assembled, the flipon cycle then resets the DNA locus to await splicing of the next transcript.
DNA G-Quadruplexes and Splicing
How GQ formation by DNA affects splicing is therefore of considerable interest. Pausing of RNAP2 is associated with alternative splicing (reviewed in [150]). The sites at which RNAP2 pauses have been investigated at nucleotide resolution. Careful in vivo measurements show dependence of pause sites on the structure of the RNA:DNA hybrid produced, but not on the canonical DNA motifs that form GQ [151]. The lack of direct involvement of dGQ may reflect the action of the FACT (Facilitates Chromatin Transcription) complex in maintaining the existing epigenetic state by removing nucleosomes in front of the RNAP2 and replacing them behind the enzyme. This mechanism prevents the net accumulation of local DNA supercoiling that might otherwise change flipon conformation[152]..
However, CTCF mediated looping is associated with alternative splicing and may allow dGQ to play an indirect role in splicing by maintaining CTCF sites methylation free. The role for CTCF is well substantiated. There is evidence that the DNA loops formed between promoter and the spliceosome mediate the transfer of various splicing factors that initially accumulate in promoter regions [153,154]. There is also ancillary evidence that R-loop formation at promoter sites promotes splicing [155], consistent with a role for GQ in forming promoter/spliceosome condensates.
Alternative splicing is also associated with demethylated DNA, consistent with a role of CTCF anchored loops in splicing. The deletion of DNMT1 enhances the alternative splicing of the CD45 transcript, as does inducing DNA demethylation by increasing expression of TET1 (tet methylcytosine dioxygenase 1) and TET2 enzymes [156,157].. Interestingly, the complement of the degenerate RPOL2 pause motif given by Gajos et al, has a weak match to a CTCF motif (the orientation is inverted relative to those enriched at TSS). In this case, the inhibition of DNA methylation by GQ may provide a partial explanation for how this conformation can indirectly influence the selection of splice sites [40].
The CTCF-dependent mechanism of connecting promoters with RNA processing condensates involved in splicing is quite flexible. For example, the multiple alternative splices of the protocadherin Pcdh gene family connect the production of each isoform with a different active promoter [158,159]. Similar dependence on promoter selection is reported for other RNA processing steps in which the polyadenylation of transcripts occurs at different sites [160,161] (Figure 4). In both outcomes, GQs potentially prevent the loss of CTCF binding sites by inhibiting DNA methylation of the locus. The GQ also localizes proteins with roles in the splicing and polyadenylation. The many proposed GQ binding proteins involved are listed in [60], in the G4IPBD database and QUADRatlas databases, with a validated subset given in [51,He, 2023 #3109]).
RNA G-Quadruplexes and Splicing
rGQ can also form in the RNA transcripts produced, including those with only two tetrads [34] and those folded with non-contiguous G nucleotides [48]. These structures have the potential to alter RPOL2 elongation rate and the RNA processing performed [40,41].}. For example, the splicing factors U2AF65 and SRSF1 bind to GQ RNA with nanomolar affinity, each showing specificity for different GQ substrates [162]. The small molecule cephaeline and the related compound emetine are both reported to impair the formation of GQ by RNA. Both compounds globally disrupt alternative RNA splicing [163].
GQ formation may also alter the co-transcriptional N6-methyladenosine (m6A) modification of RNA. It has been proposed that this epigenetic mark can affect splice site selection, but that issue is unresolved [164,165,166]. The involvement of rGQ in m6A modification is also controversial. Interestingly, the methyltransferase METTL3/METTL14 heterodimer that writes m6A within the consensus DRACH motif (D = A, G, or U; R = A or G; H = A, C, or U) binds to rG4 structures preferentially through its RGG domain [167,168]. Also, the RBM15 protein that also binds rG4 localizes METl3 to certain transcripts and to a subset of H3K36me3 marks [51,166,169]. The mapping of GQ and m6A to splice junctions is dependent on the methods used. Over 81% of GQ that map in HeLa cells are formed from only 2 tetrads that can stably fold into rGQ [164]. The mapping frequency also depends on the m6A detection protocol employed and the cell line studied, varying from 14% in HeLa cells to 40% in HEK cells [164]. More recent methods are even more sensitive than those used in the earlier analysis, but reproducibility across studies remains a problem [170]. Current mappings do not reveal any enrichment of the DRACH motif in GQ loops, suggesting that rGQ might localize METl3 to modify sequences in their neighborhood [164]. Alternatively, m6A modification may inhibit rGQ formation, as seen for GGA repeats [171]. Interestingly, m6A bases are read by heterogeneous ribonucleic acid proteins (hnRNPs) involved in alternative splicing, such as hnRNP C and hnRNP A2B1 [172{Ye, 2024 #3182].
The role of m6A in splicing was also investigated in genetically modified animals. The expression of a hypomorphic METTL3 allele in mouse embryonic stem cells did not appear to change splicing patterns, although there was slower turnover of many of the wildtype m6A modified RNAs [165], Further, in wildtype cells, the distribution of m6A in processed nuclear mRNAs was similar to that found in cytoplasmic mRNAs. Around 70% of the observed m6A sites were in terminal exons, with ~70% in the 3' UTR. With chromatin associated RNAs that were not completely processed, ∼93% of the m6As in the partially spliced transcripts were in exons and only ~10% of m6As were within 50 nucleotides of 5' or 3' splice sites. Notably, methylation was mostly performed before splicing [173].
Rather than working with a genomic knockout, another group examined the immediate effects of acute depletion of METTL3 protein. This approach was designed to minimize the downstream effects on the expression of other genes resulting from METTL3 loss. Around 6%–10% of high-confidence m6A regions were mapped to introns, mainly in protein coding genes, either around stop-codon regions or at the beginning of the 3′ UTR. The loss of METTL3 disrupted inclusion of alternative introns/exons in the nascent transcriptome, particularly at those 5' splice sites proximal to m6A peaks, suggesting that the sites were occluded or the isoforms were protected by proteins bound to m6A. Among those genes showing altered splicing were those encoding proteins for m6A modification (Wtap, Ythdc1, Ythdf1, and Spen), suggesting a negative feedback regulatory mechanism that would be absent in cells with METTL3 deleted from the germline [166]. Overall, the different results for GQ RNA formation at splice sites and METTL3 deficiency are consistent with a model where rGQ folding in introns can promote m6A modification of exons, with rapid degradation of splicing isoforms with retained introns marked by m6A.
G-Quadruplexes and Translation
GQ and ribosome assembly. rGQs appear to play an important role in ribosome structure and maturation, with ribosomal RNAs enriched for G-flipons [174]. Many ribosomal proteins have been identified as rGQ ligands in different screens [62,162]. Further, rGQ binding and resolving proteins such as nucleolin and nucleophosmin help structure the nucleolar condensates that guide ribosome assembly [56,175,176,177].
rGQ and translation. rGQ formation by mRNA is the subject of much interest, especially in the untranslated regions that regulate translation. These exons contain alternative translation initiation sites and microRNA (miR) binding sites that affect the production of different protein isoforms. The complexities involved are described in a number of recent reviews. The articles provide examples of how rGQ in the 5’UTRs can switch the use of start codons to produce completely different protein products, which rGQ in the 3’UTR can modulate the translation of mRNAs and interactions with small regulatory RNAs such as miRNA [178,179,180,181]. Analysis of G-flipons in 5'- and 3'UTR provides evidence of positive selection, which can alter the alternative splicing of these exons. Single nucleotide variants in both 5'- and 3'UTR are associated with quantitative trait loci [182]. Bioinformatic approaches have also been used to identify G-flipon RNA binding protein, as annotated in the QUADRatlas database.
By modulating mRNA translation RNAs, rGQs contribute in many ways to phenotypic pliability [28]. Here helicases such as DHX36 and CCHC-type zinc finger nucleic acid-binding protein (CNBP/ZNF9 play a central role in promoting mRNA translation by resolving rGQ [183,184]. The m6A modifications of RNA that are associated with rGQ formation during transcription (as described above) also impact translation. The removal of these marks from the 5' UTR near the start codon by the m6A erasers AlkB homolog H5 (ALKBH5) and fat mass and obesity (FTO) decreases ribosome translational pausing, increasing protein synthesis [185]. Such m6A modifications also dynamically regulate heat shock responses by enhancing N7-methylguanosine cap-independent translation [186]. Further, the class I cytoplasmic m6A readers, YTHDF1 and YTHDF3, promote the degradation of target transcripts [187], potentially eliminating partially processed transcripts with retained introns. The endogenous repeat elements present in these introns, such as ALU SINE inverted repeats, might otherwise activate dsRNA and Z-RNA dependent immune responses [132]. The potential of rGQ to enhance m6A modifications provides additional mechanistic insight into how G-flipons increase phenotype pliability by regulating RNA dependent epigenetic outcomes.
G-Quadruplexes and Development.
Pioneering Factors. Other mechanisms exist for the induction of alternative flipon conformations. Sequence-specific pioneering transcription factors, such as HNF4 and GATA4, can dock to their motifs on nucleosome bound DNA. The master regulators of embryonic development then localize complexes that evict histone octamers from the locus, generating a negatively supercoiled NDR at the site [188,189]. The energy released by removal of a nucleosome is sufficient to induce a number of different alternative DNA conformations [190]. The relaxation of these structures to B-DNA is sufficient to power the assembly of the different biological machines that actuate alternative cellular responses (Figure 3).
GQs are able to facilitate a number of different processes in the cell that are directed by sequence-specific TF. Small noncoding RNAs, such as those used in the piwi system to regulate endogenous retroelements [191], provide another means by which GQ formation can be regulated in a sequence-specific manner. In both cases, the alternative flipon conformations engage the same structure-specific cellular machinery. The question arises as to two these two different systems for sequence-specific regulation of gene expression and RNA translation are used to coordinate development, especially during early embryogenesis. To explore the role of small RNAs in this process, the sequence-specific match between experimentally confirmed flipons and miR highly conserved in eutherian mammals was explored. Intriguingly, promoters with miR matches to G- and Z-flipons were highly enriched in developmental genes (FDR > 10-100), consistent with a role in early development [130].
Notably, GQ are enriched in human embryonic stem cells (hESC). About 18,000 GQ were mapped to NDR as defined by ATAC seq. Following differentiation into neural stem cells and cranial neural crest cells, the number of detectable GQ was reduced by 25-50%, with findings differing by lineage [192]. In hESC, GQ were mapped to ~50% of bivalent promoters that contain both active H3K4me1 and repressive H3K27me3 marks and are lowly transcribed. The GQ in hESC overlapped sites bound by CTCF (~36 %), the cohesin component RAD51 (~50%) and RING1B that mediates repression by recruiting PRC1 to R-loops (~55%) [193]. Differentiation was associated with the loss of bivalent promoters reflecting the potential of GQ to localize either activating or repressive protein complexes during lineage specification. Collectively the results are consistent with a model where small RNAs bootstrap development, much in the same way a computer loads an initial program to specify the inputs and outputs that are necessary for an operating system to run. Here, the programming of flipon conformation by small RNAs would establish epigenetic marks to template tissue differentiation by sequence-specific B-DNA binding proteins. The bootstrapping by small RNAs that occurs after the erasure of existing parental epigenetic marks early in development could potentially involve miR transmitted by either maternal or paternal gametes [194,195,196,197]. Further research is needed to address such mechanisms.
Summary and Outlook
Flipons are genetically encoded elements that dynamically change their conformation under physiological conditions without requiring strand cleavage or a change in sequence. They vary by the non-B-DNA structure they form. Z-flipons flip rapidly, with an in vitro relaxation time of 100 ms and have ancient, well documented roles in self-recognition and immunity through the structure-specific interaction with Zα domain [132]. G-flipons are much more stable, with higher melting temperatures than their B-DNA structure. Yet, like Z-flipons, GQ are formed and resolved dynamically to perform a number of important biological roles (Figure 3). Flipons that form triplexes are also likely to influence gene expression and development [198,199], with examples related to the hemoglobin locus [200], stabilization of by histone H3 tails [201,202], and by binding of the Drosophila GAGA protein triplex-DNA through the same domain that engages B-DNA in a sequence-specific manner [203]. Triplex forming sequences are also enriched in repeat elements, such as ALU SINEs (short interspersed nuclear element) that form part of the repetitive genome [106]. Their biology may reflect the RNA motifs they deliver to a locus that engage both sequence- and structure-specific proteins that scaffold formation of various chromatin modifying complexes [204].
Based on a dynamic form of encoding, flipon biology can be best visualized as a cycle that exchanges energy for information. The flip to an alternative conformation is regulated both genetically and by environmental events, by base modifications that enhance or suppress the transition and depend upon proteins and noncoding RNAs that modulate the formation or resolution of the alternative conformation. These modulators are also subject to modification to tune the cycle. Other factors also affect the equilibrium by binding in a sequence-specific manner to the right-handed B-DNA conformations or to single-stranded RNA. While it has been usual to consider the effects of evolution on the individual components involved in cellular processes, the optimization of so many different parameters represents a combinatorically challenging calculation full of cascading complexity, similar in logic to the epicycles once used to predict planetary orbits in a bygone era. Instead, flipons offer a simpler alternative to optimize context-specific responses that allow rapid adjustments of cellular state. By programming and refreshing epigenetic state, flipons facilitate the formation and maintenance of cellular memory [2]. Here, the various ways in which G-flipons impact a wide variety of biological processes is described, with a focus on recent experimental validation of GQ and descriptions of the current unknowns.
References
- Herbert, A., A Genetic Instruction Code Based on DNA Conformation. Trends Genet 2019, 35, 887–890. [CrossRef] [PubMed]
- Herbert, A., Flipons and the logic of soft-wired genomes. 1st ed.; CRC Press: Boca Raton, 2024.
- Gellert, M.; Lipsett, M. N.; Davies, D. R., Helix Formation by Guanylic Acid. Proceedings of the National Academy of Sciences 1962, 48, (12), 2013-2018. [CrossRef]
- Arnott, S.; Chandrasekaran, R.; Marttila, C. M., Structures for polyinosinic acid and polyguanylic acid. Biochem J 1974, 141, (2), 537-43. [CrossRef] [PubMed]
- Sauer, M.; Paeschke, K., G-quadruplex unwinding helicases and their function in vivo. Biochem Soc Trans 2017, 45, (5), 1173-1182. [CrossRef]
- Blackburn, E. H.; Gall, J. G., A tandemly repeated sequence at the termini of the extrachromosomal ribosomal RNA genes in Tetrahymena. J Mol Biol 1978, 120, (1), 33-53. [CrossRef]
- Sundquist, W. I.; Klug, A., Telomeric DNA dimerizes by formation of guanine tetrads between hairpin loops. Nature 1989, 342, (6251), 825-9. [CrossRef] [PubMed]
- Griffith, J. D.; Comeau, L.; Rosenfield, S.; Stansel, R. M.; Bianchi, A.; Moss, H.; de Lange, T., Mammalian telomeres end in a large duplex loop. Cell 1999, 97, (4), 503-14. [CrossRef]
- Huber, M. D.; Duquette, M. L.; Shiels, J. C.; Maizels, N., A conserved G4 DNA binding domain in RecQ family helicases. J Mol Biol 2006, 358, (4), 1071-80. [CrossRef]
- Maizels, N., G4-associated human diseases. EMBO Rep 2015, 16, (8), 910-22. [CrossRef]
- Duquette, M. L.; Handa, P.; Vincent, J. A.; Taylor, A. F.; Maizels, N., Intracellular transcription of G-rich DNAs induces formation of G-loops, novel structures containing G4 DNA. Genes Dev 2004, 18, (13), 1618-29. [CrossRef]
- Okazaki, I. M.; Kinoshita, K.; Muramatsu, M.; Yoshikawa, K.; Honjo, T., The AID enzyme induces class switch recombination in fibroblasts. Nature 2002, 416, (6878), 340-5. [CrossRef] [PubMed]
- Qiao, Q.; Wang, L.; Meng, F. L.; Hwang, J. K.; Alt, F. W.; Wu, H., AID Recognizes Structured DNA for Class Switch Recombination. Mol Cell 2017, 67, (3), 361-373 e4.
- Ribeiro de Almeida, C.; Dhir, S.; Dhir, A.; Moghaddam, A. E.; Sattentau, Q.; Meinhart, A.; Proudfoot, N. J., RNA Helicase DDX1 Converts RNA G-Quadruplex Structures into R-Loops to Promote IgH Class Switch Recombination. Mol Cell 2018, 70, (4), 650-662 e8.
- Richard, P.; Manley, J. L., R Loops and Links to Human Disease. J Mol Biol 2017, 429, (21), 3168-3180. [CrossRef] [PubMed]
- Guo, J. U.; Bartel, D. P., RNA G-quadruplexes are globally unfolded in eukaryotic cells and depleted in bacteria. Science 2016, 353, (6306).
- Di Antonio, M.; Ponjavic, A.; Radzevicius, A.; Ranasinghe, R. T.; Catalano, M.; Zhang, X.; Shen, J.; Needham, L. M.; Lee, S. F.; Klenerman, D.; Balasubramanian, S., Single-molecule visualization of DNA G-quadruplex formation in live cells. Nat Chem 2020, 12, (9), 832-837. [CrossRef]
- Guo, J. K.; Blanco, M. R.; Walkup, W. G. t.; Bonesteele, G.; Urbinati, C. R.; Banerjee, A. K.; Chow, A.; Ettlin, O.; Strehle, M.; Peyda, P.; Amaya, E.; Trinh, V.; Guttman, M., Denaturing purifications demonstrate that PRC2 and other widely reported chromatin proteins do not appear to bind directly to RNA in vivo. Mol Cell 2024, 84, (7), 1271-1289 e12.
- Doolittle, W. F., Is junk DNA bunk? A critique of ENCODE. Proc Natl Acad Sci U S A 2013, 110, (14), 5294-300. [CrossRef]
- Varshney, D.; Spiegel, J.; Zyner, K.; Tannahill, D.; Balasubramanian, S., The regulation and functions of DNA and RNA G-quadruplexes. Nat Rev Mol Cell Biol 2020, 21, 459–474. [CrossRef]
- Spiegel, J.; Adhikari, S.; Balasubramanian, S., The Structure and Function of DNA G-Quadruplexes. Trends Chem 2020, 2, (2), 123-136. [CrossRef]
- Yadav, P.; Kim, N.; Kumari, M.; Verma, S.; Sharma, T. K.; Yadav, V.; Kumar, A., G-Quadruplex Structures in Bacteria: Biological Relevance and Potential as an Antimicrobial Target. J Bacteriol 2021, 203, (13), e0057720. [CrossRef]
- Wang, E.; Thombre, R.; Shah, Y.; Latanich, R.; Wang, J., G-Quadruplexes as pathogenic drivers in neurodegenerative disorders. Nucleic Acids Res 2021, 49, (9), 4816-4830. [CrossRef] [PubMed]
- Lejault, P.; Mitteaux, J.; Sperti, F. R.; Monchaud, D., How to untie G-quadruplex knots and why? Cell Chem Biol 2021, 28, (4), 436-455. [CrossRef]
- Sato, K.; Knipscheer, P., G-quadruplex resolution: From molecular mechanisms to physiological relevance. DNA Repair (Amst) 2023, 130, 103552. [CrossRef]
- Troisi, R.; Sica, F., Structural overview of DNA and RNA G-quadruplexes in their interaction with proteins. Curr Opin Struct Biol 2024, 87, 102846. [CrossRef] [PubMed]
- Sahayasheela, V. J.; Sugiyama, H., RNA G-quadruplex in functional regulation of noncoding RNA: Challenges and emerging opportunities. Cell Chem Biol 2024, 31, (1), 53-70. [CrossRef] [PubMed]
- Cammas, A.; Desprairies, A.; Dassi, E.; Millevoi, S., The shaping of mRNA translation plasticity by RNA G-quadruplexes in cancer progression and therapy resistance. NAR Cancer 2024, 6, (2), zcae025. [CrossRef]
- Sen, D.; Gilbert, W., A sodium-potassium switch in the formation of four-stranded G4-DNA. Nature 1990, 344, (6265), 410-4. [CrossRef] [PubMed]
- Fonseca Guerra, C.; Zijlstra, H.; Paragi, G.; Bickelhaupt, F. M., Telomere Structure and Stability: Covalency in Hydrogen Bonds, Not Resonance Assistance, Causes Cooperativity in Guanine Quartets. Chemistry – A European Journal 2011, 17, (45), 12612-12622.
- Sundaresan, S.; Uttamrao, P. P.; Kovuri, P.; Rathinavelan, T., The entangled world of DNA quadruplex folds. BioRxiv 2024, 2024. [CrossRef]
- Marusic, M.; Sket, P.; Bauer, L.; Viglasky, V.; Plavec, J., Solution-state structure of an intramolecular G-quadruplex with propeller, diagonal and edgewise loops. Nucleic Acids Res 2012, 40, (14), 6946-56. [CrossRef]
- Roschdi, S.; Yan, J.; Nomura, Y.; Escobar, C. A.; Petersen, R. J.; Bingman, C. A.; Tonelli, M.; Vivek, R.; Montemayor, E. J.; Wickens, M.; Kennedy, S. G.; Butcher, S. E., An atypical RNA quadruplex marks RNAs as vectors for gene silencing. Nat Struct Mol Biol 2022, 29, (11), 1113-1121. [CrossRef]
- Fay, M. M.; Lyons, S. M.; Ivanov, P., RNA G-Quadruplexes in Biology: Principles and Molecular Mechanisms. J Mol Biol 2017, 429, (14), 2127-2147. [CrossRef]
- Matsugami, A.; Okuizumi, T.; Uesugi, S.; Katahira, M., Intramolecular higher order packing of parallel quadruplexes comprising a G:G:G:G tetrad and a G(:A):G(:A):G(:A):G heptad of GGA triplet repeat DNA. J Biol Chem 2003, 278, (30), 28147-53. [CrossRef]
- Palumbo, S. L.; Memmott, R. M.; Uribe, D. J.; Krotova-Khan, Y.; Hurley, L. H.; Ebbinghaus, S. W., A novel G-quadruplex-forming GGA repeat region in the c-myb promoter is a critical regulator of promoter activity. Nucleic Acids Res 2008, 36, (6), 1755-69. [CrossRef]
- Piazza, A.; Adrian, M.; Samazan, F.; Heddi, B.; Hamon, F.; Serero, A.; Lopes, J.; Teulade-Fichou, M. P.; Phan, A. T.; Nicolas, A., Short loop length and high thermal stability determine genomic instability induced by G-quadruplex-forming minisatellites. Embo J 2015, 34, (12), 1718-34. [CrossRef] [PubMed]
- Williams, J. D.; Houserova, D.; Johnson, B. R.; Dyniewski, B.; Berroyer, A.; French, H.; Barchie, A. A.; Bilbrey, D. D.; Demeis, J. D.; Ghee, K. R.; Hughes, A. G.; Kreitz, N. W.; McInnis, C. H.; Pudner, S. C.; Reeves, M. N.; Stahly, A. N.; Turcu, A.; Watters, B. C.; Daly, G. T.; Langley, R. J.; Gillespie, M. N.; Prakash, A.; Larson, E. D.; Kasukurthi, M. V.; Huang, J.; Jinks-Robertson, S.; Borchert, G. M., Characterization of long G4-rich enhancer-associated genomic regions engaging in a novel loop:loop 'G4 Kissing' interaction. Nucleic Acids Res 2020, 48, (11), 5907-5925. [CrossRef]
- Wu, F.; Niu, K.; Cui, Y.; Li, C.; Lyu, M.; Ren, Y.; Chen, Y.; Deng, H.; Huang, L.; Zheng, S.; Liu, L.; Wang, J.; Song, Q.; Xiang, H.; Feng, Q., Genome-wide analysis of DNA G-quadruplex motifs across 37 species provides insights into G4 evolution. Commun Biol 2021, 4, (1), 98. [CrossRef] [PubMed]
- Lee, C. Y.; McNerney, C.; Ma, K.; Zhao, W.; Wang, A.; Myong, S., R-loop induced G-quadruplex in non-template promotes transcription by successive R-loop formation. Nat Commun 2020, 11, (1), 3392. [CrossRef]
- Georgakopoulos-Soares, I.; Parada, G. E.; Wong, H. Y.; Medhi, R.; Furlan, G.; Munita, R.; Miska, E. A.; Kwok, C. K.; Hemberg, M., Alternative splicing modulation by G-quadruplexes. Nat Commun 2022, 13, (1), 2404. [CrossRef]
- Hegyi, H., Enhancer-promoter interaction facilitated by transiently forming G-quadruplexes. Sci Rep 2015, 5, 9165. [CrossRef]
- Zheng, K. W.; Xiao, S.; Liu, J. Q.; Zhang, J. Y.; Hao, Y. H.; Tan, Z., Co-transcriptional formation of DNA:RNA hybrid G-quadruplex and potential function as constitutional cis element for transcription control. Nucleic Acids Res 2013, 41, (10), 5533-41. [CrossRef]
- Varizhuk, A. M.; Protopopova, A. D.; Tsvetkov, V. B.; Barinov, N. A.; Podgorsky, V. V.; Tankevich, M. V.; Vlasenok, M. A.; Severov, V. V.; Smirnov, I. P.; Dubrovin, E. V.; Klinov, D. V.; Pozmogova, G. E., Polymorphism of G4 associates: from stacks to wires via interlocks. Nucleic Acids Res 2018, 46, (17), 8978-8992. [CrossRef] [PubMed]
- Kolesnikova, S.; Curtis, E. A., Structure and Function of Multimeric G-Quadruplexes. Molecules 2019, 24, (17).
- Sen, D.; Gilbert, W., Formation of parallel four-stranded complexes by guanine-rich motifs in DNA and its implications for meiosis. Nature 1988, 334, (6180), 364-6. [CrossRef]
- Li, X.-m.; Zheng, K.-w.; Zhang, J.-y.; Liu, H.-h.; He, Y.-d.; Yuan, B.-f.; Hao, Y.-h.; Tan, Z., Guanine-vacancy–bearing G-quadruplexes responsive to guanine derivatives. Proceedings of the National Academy of Sciences 2015, 112, (47), 14581-14586. [CrossRef]
- Banco, M. T.; Ferre-D'Amare, A. R., The emerging structural complexity of G-quadruplex RNAs. Rna 2021, 27, (4), 390-402. [CrossRef]
- Lavezzo, E.; Berselli, M.; Frasson, I.; Perrone, R.; Palu, G.; Brazzale, A. R.; Richter, S. N.; Toppo, S., G-quadruplex forming sequences in the genome of all known human viruses: A comprehensive guide. PLoS Comput Biol 2018, 14, (12), e1006675. [CrossRef] [PubMed]
- Qian, S. H.; Shi, M. W.; Xiong, Y. L.; Zhang, Y.; Zhang, Z. H.; Song, X. M.; Deng, X. Y.; Chen, Z. X., EndoQuad: a comprehensive genome-wide experimentally validated endogenous G-quadruplex database. Nucleic Acids Res 2024, 52, (D1), D72-D80. [CrossRef] [PubMed]
- Spiegel, J.; Cuesta, S. M.; Adhikari, S.; Hansel-Hertsch, R.; Tannahill, D.; Balasubramanian, S., G-quadruplexes are transcription factor binding hubs in human chromatin. Genome Biol 2021, 22, (1), 117. [CrossRef] [PubMed]
- Brazda, V.; Cerven, J.; Bartas, M.; Mikyskova, N.; Coufal, J.; Pecinka, P., The Amino Acid Composition of Quadruplex Binding Proteins Reveals a Shared Motif and Predicts New Potential Quadruplex Interactors. Molecules 2018, 23, (9).
- Vasilyev, N.; Polonskaia, A.; Darnell, J. C.; Darnell, R. B.; Patel, D. J.; Serganov, A., Crystal structure reveals specific recognition of a G-quadruplex RNA by a β-turn in the RGG motif of FMRP. Proceedings of the National Academy of Sciences 2015, 112, (39), E5391-E5400. [CrossRef]
- Guedin, A.; Gros, J.; Alberti, P.; Mergny, J. L., How long is too long? Effects of loop size on G-quadruplex stability. Nucleic Acids Res 2010, 38, (21), 7858-68. [CrossRef]
- Zhang, A. Y.; Bugaut, A.; Balasubramanian, S., A sequence-independent analysis of the loop length dependence of intramolecular RNA G-quadruplex stability and topology. Biochemistry 2011, 50, (33), 7251-8. [CrossRef]
- Saha, A.; Duchambon, P.; Masson, V.; Loew, D.; Bombard, S.; Teulade-Fichou, M. P., Nucleolin Discriminates Drastically between Long-Loop and Short-Loop Quadruplexes. Biochemistry 2020, 59, (12), 1261-1272. [CrossRef]
- Ngo, K. H.; Liew, C. W.; Heddi, B.; Phan, A. T., Structural Basis for Parallel G-Quadruplex Recognition by an Ankyrin Protein. J Am Chem Soc 2024, 146, (20), 13709-13713. [CrossRef]
- Weaver, T. M.; Cortez, L. M.; Khoang, T. H.; Washington, M. T.; Agarwal, P. K.; Freudenthal, B. D., Visualizing Rev1 catalyze protein-template DNA synthesis. Proc Natl Acad Sci U S A 2020, 117, (41), 25494-25504. [CrossRef]
- Roychoudhury, S.; Pramanik, S.; Harris, H. L.; Tarpley, M.; Sarkar, A.; Spagnol, G.; Sorgen, P. L.; Chowdhury, D.; Band, V.; Klinkebiel, D.; Bhakat, K. K., Endogenous oxidized DNA bases and APE1 regulate the formation of G-quadruplex structures in the genome. Proc Natl Acad Sci U S A 2020, 117, (21), 11409-11420. [CrossRef] [PubMed]
- Pipier, A.; Devaux, A.; Lavergne, T.; Adrait, A.; Coute, Y.; Britton, S.; Calsou, P.; Riou, J. F.; Defrancq, E.; Gomez, D., Constrained G4 structures unveil topology specificity of known and new G4 binding proteins. Sci Rep 2021, 11, (1), 13469. [CrossRef]
- Mishra, S. K.; Tawani, A.; Mishra, A.; Kumar, A., G4IPDB: A database for G-quadruplex structure forming nucleic acid interacting proteins. Sci Rep 2016, 6, 38144. [CrossRef] [PubMed]
- Bourdon, S.; Herviou, P.; Dumas, L.; Destefanis, E.; Zen, A.; Cammas, A.; Millevoi, S.; Dassi, E., QUADRatlas: the RNA G-quadruplex and RG4-binding proteins database. Nucleic Acids Res 2023, 51, (D1), D240-D247. [CrossRef]
- Fleming, A. M.; Zhou, J.; Wallace, S. S.; Burrows, C. J., A Role for the Fifth G-Track in G-Quadruplex Forming Oncogene Promoter Sequences during Oxidative Stress: Do These "Spare Tires" Have an Evolved Function? ACS Cent Sci 2015, 1, (5), 226-233. [CrossRef] [PubMed]
- Handwerger, K. E.; Cordero, J. A.; Gall, J. G., Cajal bodies, nucleoli, and speckles in the Xenopus oocyte nucleus have a low-density, sponge-like structure. Mol Biol Cell 2005, 16, (1), 202-11. [CrossRef] [PubMed]
- Shin, Y.; Brangwynne, C. P., Liquid phase condensation in cell physiology and disease. Science 2017, 357, (6357).
- Iborra, F. J.; Pombo, A.; Jackson, D. A.; Cook, P. R., Active RNA polymerases are localized within discrete transcription "factories' in human nuclei. J Cell Sci 1996, 109 ( Pt 6), 1427-36. [CrossRef]
- Jackson, D. A., The amazing complexity of transcription factories. Brief Funct Genomic Proteomic 2005, 4, (2), 143-57. [CrossRef]
- Chubb, J. R.; Trcek, T.; Shenoy, S. M.; Singer, R. H., Transcriptional pulsing of a developmental gene. Curr Biol 2006, 16, (10), 1018-25. [CrossRef]
- Marshall, W. F.; Straight, A.; Marko, J. F.; Swedlow, J.; Dernburg, A.; Belmont, A.; Murray, A. W.; Agard, D. A.; Sedat, J. W., Interphase chromosomes undergo constrained diffusional motion in living cells. Curr Biol 1997, 7, (12), 930-9. [CrossRef]
- Ruggiero, E.; Tassinari, M.; Perrone, R.; Nadai, M.; Richter, S. N., Stable and Conserved G-Quadruplexes in the Long Terminal Repeat Promoter of Retroviruses. ACS Infect Dis 2019, 5, (7), 1150-1159. [CrossRef] [PubMed]
- Amrane, S.; Jaubert, C.; Bedrat, A.; Rundstadler, T.; Recordon-Pinson, P.; Aknin, C.; Guedin, A.; De Rache, A.; Bartolucci, L.; Diene, I.; Lemoine, F.; Gascuel, O.; Pratviel, G.; Mergny, J. L.; Andreola, M. L., Deciphering RNA G-quadruplex function during the early steps of HIV-1 infection. Nucleic Acids Res 2022, 50, (21), 12328-12343. [CrossRef]
- Sahakyan, A. B.; Murat, P.; Mayer, C.; Balasubramanian, S., G-quadruplex structures within the 3' UTR of LINE-1 elements stimulate retrotransposition. Nat Struct Mol Biol 2017, 24, (3), 243-247. [CrossRef] [PubMed]
- Sakamoto, M.; Ishiuchi, T., YY1-dependent transcriptional regulation manifests at the morula stage. MicroPubl Biol 2024, 2024.
- Kruisselbrink, E.; Guryev, V.; Brouwer, K.; Pontier, D. B.; Cuppen, E.; Tijsterman, M., Mutagenic capacity of endogenous G4 DNA underlies genome instability in FANCJ-defective C. elegans. Curr Biol 2008, 18, (12), 900-5. [CrossRef]
- Jones, M.; Rose, A., A DOG's View of Fanconi Anemia: Insights from C. elegans. Anemia 2012, 2012, 323721. [CrossRef]
- Tarailo-Graovac, M.; Wong, T.; Qin, Z.; Flibotte, S.; Taylor, J.; Moerman, D. G.; Rose, A. M.; Chen, N., Spectrum of variations in dog-1/FANCJ and mdf-1/MAD1 defective Caenorhabditis elegans strains after long-term propagation. BMC Genomics 2015, 16, (1), 210. [CrossRef]
- Sarkies, P.; Murat, P.; Phillips, L. G.; Patel, K. J.; Balasubramanian, S.; Sale, J. E., FANCJ coordinates two pathways that maintain epigenetic stability at G-quadruplex DNA. Nucleic Acids Res 2012, 40, (4), 1485-98. [CrossRef]
- Liu, Y.; Zhu, X.; Wang, K.; Zhang, B.; Qiu, S., The Cellular Functions and Molecular Mechanisms of G-Quadruplex Unwinding Helicases in Humans. Front Mol Biosci 2021, 8, 783889. [CrossRef]
- Sarkies, P.; Reams, C.; Simpson, L. J.; Sale, J. E., Epigenetic instability due to defective replication of structured DNA. Mol Cell 2010, 40, (5), 703-13. [CrossRef]
- Kumagai, A.; Dunphy, W. G., MTBP, the partner of Treslin, contains a novel DNA-binding domain that is essential for proper initiation of DNA replication. Mol Biol Cell 2017, 28, (22), 2998-3012. [CrossRef]
- Poulet-Benedetti, J.; Tonnerre-Doncarli, C.; Valton, A. L.; Laurent, M.; Gerard, M.; Barinova, N.; Parisis, N.; Massip, F.; Picard, F.; Prioleau, M. N., Dimeric G-quadruplex motifs-induced NFRs determine strong replication origins in vertebrates. Nat Commun 2023, 14, (1), 4843. [CrossRef] [PubMed]
- Mitter, M.; Gasser, C.; Takacs, Z.; Langer, C. C. H.; Tang, W.; Jessberger, G.; Beales, C. T.; Neuner, E.; Ameres, S. L.; Peters, J. M.; Goloborodko, A.; Micura, R.; Gerlich, D. W., Conformation of sister chromatids in the replicated human genome. Nature 2020, 586, (7827), 139-144. [CrossRef]
- Hou, Y.; Li, F.; Zhang, R.; Li, S.; Liu, H.; Qin, Z. S.; Sun, X., Integrative characterization of G-Quadruplexes in the three-dimensional chromatin structure. Epigenetics 2019, 14, (9), 894-911. [CrossRef] [PubMed]
- De Magis, A.; Gotz, S.; Hajikazemi, M.; Fekete-Szucs, E.; Caterino, M.; Juranek, S.; Paeschke, K., Zuo1 supports G4 structure formation and directs repair toward nucleotide excision repair. Nat Commun 2020, 11, (1), 3907. [CrossRef] [PubMed]
- Ketkar, A.; Smith, L.; Johnson, C.; Richey, A.; Berry, M.; Hartman, J. H.; Maddukuri, L.; Reed, M. R.; Gunderson, J. E. C.; Leung, J. W. C.; Eoff, R. L., Human Rev1 relies on insert-2 to promote selective binding and accurate replication of stabilized G-quadruplex motifs. Nucleic Acids Res 2021, 49, (4), 2065-2084. [CrossRef]
- Sondka, Z.; Dhir, N. B.; Carvalho-Silva, D.; Jupe, S.; Madhumita; McLaren, K.; Starkey, M.; Ward, S.; Wilding, J.; Ahmed, M.; Argasinska, J.; Beare, D.; Chawla, M. S.; Duke, S.; Fasanella, I.; Neogi, A. G.; Haller, S.; Hetenyi, B.; Hodges, L.; Holmes, A.; Lyne, R.; Maurel, T.; Nair, S.; Pedro, H.; Sangrador-Vegas, A.; Schuilenburg, H.; Sheard, Z.; Yong, S. Y.; Teague, J., COSMIC: a curated database of somatic variants and clinical data for cancer. Nucleic Acids Res 2024, 52, (D1), D1210-D1217. [CrossRef]
- Liano, D.; Chowdhury, S.; Di Antonio, M., Cockayne Syndrome B Protein Selectively Resolves and Interact with Intermolecular DNA G-Quadruplex Structures. J Am Chem Soc 2021, 143, (49), 20988-21002.
- Kokic, G.; Wagner, F. R.; Chernev, A.; Urlaub, H.; Cramer, P., Structural basis of human transcription-DNA repair coupling. Nature 2021, 598, (7880), 368-372. [CrossRef] [PubMed]
- Fleming, A. M.; Zhu, J.; Ding, Y.; Esders, S.; Burrows, C. J., Oxidative Modification of Guanine in a Potential Z-DNA-Forming Sequence of a Gene Promoter Impacts Gene Expression. Chem Res Toxicol 2019, 32, (5), 899-909. [CrossRef]
- Ju, B. G.; Lunyak, V. V.; Perissi, V.; Garcia-Bassets, I.; Rose, D. W.; Glass, C. K.; Rosenfeld, M. G., A topoisomerase IIbeta-mediated dsDNA break required for regulated transcription. Science 2006, 312, (5781), 1798-802. [CrossRef]
- Gray, L. T.; Puig Lombardi, E.; Verga, D.; Nicolas, A.; Teulade-Fichou, M. P.; Londono-Vallejo, A.; Maizels, N., G-quadruplexes Sequester Free Heme in Living Cells. Cell Chem Biol 2019, 26, (12), 1681-1691 e5.
- Li, Y.; Geyer, C. R.; Sen, D., Recognition of anionic porphyrins by DNA aptamers. Biochemistry 1996, 35, (21), 6911-22. [CrossRef] [PubMed]
- Rai, R.; Chen, Y.; Lei, M.; Chang, S., TRF2-RAP1 is required to protect telomeres from engaging in homologous recombination-mediated deletions and fusions. Nat Commun 2016, 7, 10881. [CrossRef] [PubMed]
- Mei, Y.; Deng, Z.; Vladimirova, O.; Gulve, N.; Johnson, F. B.; Drosopoulos, W. C.; Schildkraut, C. L.; Lieberman, P. M., TERRA G-quadruplex RNA interaction with TRF2 GAR domain is required for telomere integrity. Sci Rep 2021, 11, (1), 3509. [CrossRef]
- Lyonnais, S.; Hounsou, C.; Teulade-Fichou, M. P.; Jeusset, J.; Le Cam, E.; Mirambeau, G., G-quartets assembly within a G-rich DNA flap. A possible event at the center of the HIV-1 genome. Nucleic Acids Res 2002, 30, (23), 5276-83.
- Heddi, B.; Cheong, V. V.; Martadinata, H.; Phan, A. T., Insights into G-quadruplex specific recognition by the DEAH-box helicase RHAU: Solution structure of a peptide-quadruplex complex. Proc Natl Acad Sci U S A 2015, 112, (31), 9608-13. [CrossRef]
- Chen, M. C.; Tippana, R.; Demeshkina, N. A.; Murat, P.; Balasubramanian, S.; Myong, S.; Ferre-D'Amare, A. R., Structural basis of G-quadruplex unfolding by the DEAH/RHA helicase DHX36. Nature 2018, 558, (7710), 465-469. [CrossRef] [PubMed]
- You, H.; Lattmann, S.; Rhodes, D.; Yan, J., RHAU helicase stabilizes G4 in its nucleotide-free state and destabilizes G4 upon ATP hydrolysis. Nucleic Acids Res 2017, 45, (1), 206-214. [CrossRef] [PubMed]
- Dai, Y. X.; Guo, H. L.; Liu, N. N.; Chen, W. F.; Ai, X.; Li, H. H.; Sun, B.; Hou, X. M.; Rety, S.; Xi, X. G., Structural mechanism underpinning Thermus oshimai Pif1-mediated G-quadruplex unfolding. EMBO Rep 2022, 23, (7), e53874. [CrossRef]
- Muellner, J.; Schmidt, K. H., Yeast Genome Maintenance by the Multifunctional PIF1 DNA Helicase Family. Genes (Basel) 2020, 11, (2).
- Varon, M.; Dovrat, D.; Heuze, J.; Tsirkas, I.; Singh, S. P.; Pasero, P.; Galletto, R.; Aharoni, A., Rrm3 and Pif1 division of labor during replication through leading and lagging strand G-quadruplex. Nucleic Acids Res 2024, 52, (4), 1753-1762. [CrossRef]
- Wu, W. Q.; Hou, X. M.; Li, M.; Dou, S. X.; Xi, X. G., BLM unfolds G-quadruplexes in different structural environments through different mechanisms. Nucleic Acids Res 2015, 43, (9), 4614-26. [CrossRef]
- Huet, J.; Cottrelle, P.; Cool, M.; Vignais, M. L.; Thiele, D.; Marck, C.; Buhler, J. M.; Sentenac, A.; Fromageot, P., A general upstream binding factor for genes of the yeast translational apparatus. Embo J 1985, 4, (13A), 3539-3547. [CrossRef]
- König, P.; Giraldo, R.; Chapman, L.; Rhodes, D., The Crystal Structure of the DNA-Binding Domain of Yeast RAP1 in Complex with Telomeric DNA. Cell 1996, 85, (1), 125-136. [CrossRef]
- Traczyk, A.; Liew, C. W.; Gill, D. J.; Rhodes, D., Structural basis of G-quadruplex DNA recognition by the yeast telomeric protein Rap1. Nucleic Acids Res 2020, 48, (8), 4562-4571. [CrossRef] [PubMed]
- Herbert, A., ALU non-B-DNA conformations, flipons, binary codes and evolution. Royal Society Open Science 2020, 7, (6), 200222. [CrossRef] [PubMed]
- Esain-Garcia, I.; Kirchner, A.; Melidis, L.; Tavares, R. C. A.; Dhir, S.; Simeone, A.; Yu, Z.; Madden, S. K.; Hermann, R.; Tannahill, D.; Balasubramanian, S., G-quadruplex DNA structure is a positive regulator of MYC transcription. Proc Natl Acad Sci U S A 2024, 121, (7), e2320240121. [CrossRef]
- Shrestha, O. K.; Sharma, R.; Tomiczek, B.; Lee, W.; Tonelli, M.; Cornilescu, G.; Stolarska, M.; Nierzwicki, L.; Czub, J.; Markley, J. L.; Marszalek, J.; Ciesielski, S. J.; Craig, E. A., Structure and evolution of the 4-helix bundle domain of Zuotin, a J-domain protein co-chaperone of Hsp70. PLoS One 2019, 14, (5), e0217098. [CrossRef]
- Biffi, G.; Tannahill, D.; Balasubramanian, S., An intramolecular G-quadruplex structure is required for binding of telomeric repeat-containing RNA to the telomeric protein TRF2. J Am Chem Soc 2012, 134, (29), 11974-6. [CrossRef]
- Sharma, S.; Mukherjee, A. K.; Roy, S. S.; Bagri, S.; Lier, S.; Verma, M.; Sengupta, A.; Kumar, M.; Nesse, G.; Pandey, D. P.; Chowdhury, S., Human telomerase is directly regulated by non-telomeric TRF2-G-quadruplex interaction. Cell Rep 2021, 35, (7), 109154. [CrossRef]
- Boyer, L. A.; Latek, R. R.; Peterson, C. L., The SANT domain: a unique histone-tail-binding module? Nat Rev Mol Cell Biol 2004, 5, (2), 158-63. [CrossRef]
- Weintraub, A. S.; Li, C. H.; Zamudio, A. V.; Sigova, A. A.; Hannett, N. M.; Day, D. S.; Abraham, B. J.; Cohen, M. A.; Nabet, B.; Buckley, D. L.; Guo, Y. E.; Hnisz, D.; Jaenisch, R.; Bradner, J. E.; Gray, N. S.; Young, R. A., YY1 Is a Structural Regulator of Enhancer-Promoter Loops. Cell 2017, 171, (7), 1573-1588 e28.
- Li, L.; Williams, P.; Ren, W.; Wang, M. Y.; Gao, Z.; Miao, W.; Huang, M.; Song, J.; Wang, Y., YY1 interacts with guanine quadruplexes to regulate DNA looping and gene expression. Nat Chem Biol 2021, 17, (2), 161-168. [CrossRef]
- Wreczycka, K.; Franke, V.; Uyar, B.; Wurmus, R.; Bulut, S.; Tursun, B.; Akalin, A., HOT or not: examining the basis of high-occupancy target regions. Nucleic Acids Res 2019, 47, (11), 5735-5745. [CrossRef]
- Ramaker, R. C.; Hardigan, A. A.; Goh, S. T.; Partridge, E. C.; Wold, B.; Cooper, S. J.; Myers, R. M., Dissecting the regulatory activity and sequence content of loci with exceptional numbers of transcription factor associations. Genome Res 2020, 30, (7), 939-950. [CrossRef] [PubMed]
- Partridge, E. C.; Chhetri, S. B.; Prokop, J. W.; Ramaker, R. C.; Jansen, C. S.; Goh, S. T.; Mackiewicz, M.; Newberry, K. M.; Brandsmeier, L. A.; Meadows, S. K.; Messer, C. L.; Hardigan, A. A.; Coppola, C. J.; Dean, E. C.; Jiang, S.; Savic, D.; Mortazavi, A.; Wold, B. J.; Myers, R. M.; Mendenhall, E. M., Occupancy maps of 208 chromatin-associated proteins in one human cell type. Nature 2020, 583, (7818), 720-728. [CrossRef] [PubMed]
- Lago, S.; Nadai, M.; Cernilogar, F. M.; Kazerani, M.; Dominiguez Moreno, H.; Schotta, G.; Richter, S. N., Promoter G-quadruplexes and transcription factors cooperate to shape the cell type-specific transcriptome. Nat Commun 2021, 12, (1), 3885. [CrossRef] [PubMed]
- Bartman, C. R.; Hsu, S. C.; Hsiung, C. C.; Raj, A.; Blobel, G. A., Enhancer Regulation of Transcriptional Bursting Parameters Revealed by Forced Chromatin Looping. Mol Cell 2016, 62, (2), 237-247. [CrossRef]
- Hasegawa, Y.; Struhl, K., Promoter-specific dynamics of TATA-binding protein association with the human genome. Genome Res 2019, 29, (12), 1939-1950. [CrossRef]
- Henninger, J. E.; Oksuz, O.; Shrinivas, K.; Sagi, I.; LeRoy, G.; Zheng, M. M.; Andrews, J. O.; Zamudio, A. V.; Lazaris, C.; Hannett, N. M.; Lee, T. I.; Sharp, P. A.; Cisse, II; Chakraborty, A. K.; Young, R. A., RNA-Mediated Feedback Control of Transcriptional Condensates. Cell 2021, 184, (1), 207-225 e24.
- De Nicola, B.; Lech, C. J.; Heddi, B.; Regmi, S.; Frasson, I.; Perrone, R.; Richter, S. N.; Phan, A. T., Structure and possible function of a G-quadruplex in the long terminal repeat of the proviral HIV-1 genome. Nucleic Acids Res 2016, 44, (13), 6442-51. [CrossRef]
- Butovskaya, E.; Heddi, B.; Bakalar, B.; Richter, S. N.; Phan, A. T., Major G-Quadruplex Form of HIV-1 LTR Reveals a (3 + 1) Folding Topology Containing a Stem-Loop. J Am Chem Soc 2018, 140, (42), 13654-13662. [CrossRef]
- Krafcikova, P.; Demkovicova, E.; Halaganova, A.; Viglasky, V., Putative HIV and SIV G-Quadruplex Sequences in Coding and Noncoding Regions Can Form G-Quadruplexes. J Nucleic Acids 2017, 2017, 6513720. [CrossRef]
- Pathak, R., G-Quadruplexes in the Viral Genome: Unlocking Targets for Therapeutic Interventions and Antiviral Strategies. Viruses 2023, 15, (11).
- Ramskold, D.; Hendriks, G. J.; Larsson, A. J. M.; Mayr, J. V.; Ziegenhain, C.; Hagemann-Jensen, M.; Hartmanis, L.; Sandberg, R., Single-cell new RNA sequencing reveals principles of transcription at the resolution of individual bursts. Nat Cell Biol 2024. [CrossRef]
- Shen, J.; Varshney, D.; Simeone, A.; Zhang, X.; Adhikari, S.; Tannahill, D.; Balasubramanian, S., Promoter G-quadruplex folding precedes transcription and is controlled by chromatin. Genome Biol 2021, 22, (1), 143. [CrossRef]
- Baranello, L.; Wojtowicz, D.; Cui, K.; Devaiah, B. N.; Chung, H. J.; Chan-Salis, K. Y.; Guha, R.; Wilson, K.; Zhang, X.; Zhang, H.; Piotrowski, J.; Thomas, C. J.; Singer, D. S.; Pugh, B. F.; Pommier, Y.; Przytycka, T. M.; Kouzine, F.; Lewis, B. A.; Zhao, K.; Levens, D., RNA Polymerase II Regulates Topoisomerase 1 Activity to Favor Efficient Transcription. Cell 2016, 165, (2), 357-71. [CrossRef] [PubMed]
- Marchand, C.; Pourquier, P.; Laco, G. S.; Jing, N.; Pommier, Y., Interaction of Human Nuclear Topoisomerase I with Guanosine Quartet-forming and Guanosine-rich Single-stranded DNA and RNA Oligonucleotides. Journal of Biological Chemistry 2002, 277, (11), 8906-8911. [CrossRef] [PubMed]
- Schwalb, B.; Michel, M.; Zacher, B.; Fruhauf, K.; Demel, C.; Tresch, A.; Gagneur, J.; Cramer, P., TT-seq maps the human transient transcriptome. Science 2016, 352, (6290), 1225-8. [CrossRef]
- Herbert, A.; Pavlov, F.; Konovalov, D.; Poptsova, M., Conserved microRNAs and Flipons Shape Gene Expression during Development by Altering Promoter Conformations. Int J Mol Sci 2023, 24, (5).
- Herbert, A., Flipons and small RNAs accentuate the asymmetries of pervasive transcription by the reset and sequence-specific microcoding of promoter conformation. J Biol Chem 2023, 299, (9), 105140. [CrossRef]
- Herbert, A., The ancient Z-DNA and Z-RNA specific Zα fold has evolved modern roles in immunity and transcription through the natural selection of flipons. Royal Society Open Science 2024, 11, (6).
- Beknazarov, N.; Konovalov, D.; Herbert, A.; Poptsova, M., Z-DNA formation in promoters conserved between human and mouse are associated with increased transcription reinitiation rates. Sci Rep 2024, 14, (1), 17786. [CrossRef] [PubMed]
- Le, S. N.; Brown, C. R.; Harvey, S.; Boeger, H.; Elmlund, H.; Elmlund, D., The TAFs of TFIID Bind and Rearrange the Topology of the TATA-Less RPS5 Promoter. Int J Mol Sci 2019, 20, (13).
- Kouzine, F.; Wojtowicz, D.; Baranello, L.; Yamane, A.; Nelson, S.; Resch, W.; Kieffer-Kwon, K. R.; Benham, C. J.; Casellas, R.; Przytycka, T. M.; Levens, D., Permanganate/S1 Nuclease Footprinting Reveals Non-B DNA Structures with Regulatory Potential across a Mammalian Genome. Cell Syst 2017, 4, (3), 344-356. [CrossRef]
- Song, J.; Gooding, A. R.; Hemphill, W. O.; Love, B. D.; Robertson, A.; Yao, L.; Zon, L. I.; North, T. E.; Kasinath, V.; Cech, T. R., Structural basis for inactivation of PRC2 by G-quadruplex RNA. Science 2023, 381, (6664), 1331-1337. [CrossRef]
- Watanabe, T.; Totoki, Y.; Toyoda, A.; Kaneda, M.; Kuramochi-Miyagawa, S.; Obata, Y.; Chiba, H.; Kohara, Y.; Kono, T.; Nakano, T.; Surani, M. A.; Sakaki, Y.; Sasaki, H., Endogenous siRNAs from naturally formed dsRNAs regulate transcripts in mouse oocytes. Nature 2008, 453, (7194), 539-43. [CrossRef]
- Ha, H.; Song, J.; Wang, S.; Kapusta, A.; Feschotte, C.; Chen, K. C.; Xing, J., A comprehensive analysis of piRNAs from adult human testis and their relationship with genes and mobile elements. BMC Genomics 2014, 15, (1), 545. [CrossRef]
- Ozata, D. M.; Yu, T.; Mou, H.; Gainetdinov, I.; Colpan, C.; Cecchini, K.; Kaymaz, Y.; Wu, P. H.; Fan, K.; Kucukural, A.; Weng, Z.; Zamore, P. D., Evolutionarily conserved pachytene piRNA loci are highly divergent among modern humans. Nat Ecol Evol 2020, 4, (1), 156-168.
- Li, L. C.; Okino, S. T.; Zhao, H.; Pookot, D.; Place, R. F.; Urakami, S.; Enokida, H.; Dahiya, R., Small dsRNAs induce transcriptional activation in human cells. Proc Natl Acad Sci U S A 2006, 103, (46), 17337-42. [CrossRef] [PubMed]
- Matsui, M.; Chu, Y.; Zhang, H.; Gagnon, K. T.; Shaikh, S.; Kuchimanchi, S.; Manoharan, M.; Corey, D. R.; Janowski, B. A., Promoter RNA links transcriptional regulation of inflammatory pathway genes. Nucleic Acids Res 2013, 41, (22), 10086-109. [CrossRef] [PubMed]
- Leonaite, B.; Han, Z.; Basquin, J.; Bonneau, F.; Libri, D.; Porrua, O.; Conti, E., Sen1 has unique structural features grafted on the architecture of the Upf1-like helicase family. Embo J 2017, 36, (11), 1590-1604. [CrossRef] [PubMed]
- Lansdorp, P.; van Wietmarschen, N., Helicases FANCJ, RTEL1 and BLM Act on Guanine Quadruplex DNA in Vivo. Genes (Basel) 2019, 10, (11).
- Nguyen, H. D.; Yadav, T.; Giri, S.; Saez, B.; Graubert, T. A.; Zou, L., Functions of Replication Protein A as a Sensor of R Loops and a Regulator of RNaseH1. Mol Cell 2017, 65, (5), 832-847 e4.
- Yan, Q.; Wulfridge, P.; Doherty, J.; Fernandez-Luna, J. L.; Real, P. J.; Tang, H. Y.; Sarma, K., Proximity labeling identifies a repertoire of site-specific R-loop modulators. Nat Commun 2022, 13, (1), 53. [CrossRef]
- Chernukhin, I.; Shamsuddin, S.; Kang, S. Y.; Bergstrom, R.; Kwon, Y. W.; Yu, W.; Whitehead, J.; Mukhopadhyay, R.; Docquier, F.; Farrar, D.; Morrison, I.; Vigneron, M.; Wu, S. Y.; Chiang, C. M.; Loukinov, D.; Lobanenkov, V.; Ohlsson, R.; Klenova, E., CTCF interacts with and recruits the largest subunit of RNA polymerase II to CTCF target sites genome-wide. Mol Cell Biol 2007, 27, (5), 1631-48. [CrossRef]
- Gomes, N. P.; Espinosa, J. M., Gene-specific repression of the p53 target gene PUMA via intragenic CTCF-Cohesin binding. Genes Dev 2010, 24, (10), 1022-34. [CrossRef] [PubMed]
- Nanavaty, V.; Abrash, E. W.; Hong, C.; Park, S.; Fink, E. E.; Li, Z.; Sweet, T. J.; Bhasin, J. M.; Singuri, S.; Lee, B. H.; Hwang, T. H.; Ting, A. H., DNA Methylation Regulates Alternative Polyadenylation via CTCF and the Cohesin Complex. Mol Cell 2020, 78, (4), 752-764 e6.
- Mao, S. Q.; Ghanbarian, A. T.; Spiegel, J.; Martinez Cuesta, S.; Beraldi, D.; Di Antonio, M.; Marsico, G.; Hansel-Hertsch, R.; Tannahill, D.; Balasubramanian, S., DNA G-quadruplex structures mold the DNA methylome. Nat Struct Mol Biol 2018, 25, (10), 951-957. [CrossRef]
- Alharbi, A. B.; Schmitz, U.; Bailey, C. G.; Rasko, J. E. J., CTCF as a regulator of alternative splicing: new tricks for an old player. Nucleic Acids Res 2021, 49, (14), 7825-7838. [CrossRef]
- Gajos, M.; Jasnovidova, O.; van Bommel, A.; Freier, S.; Vingron, M.; Mayer, A., Conserved DNA sequence features underlie pervasive RNA polymerase pausing. Nucleic Acids Res 2021, 49, (8), 4402-4420. [CrossRef]
- Ehara, H.; Kujirai, T.; Shirouzu, M.; Kurumizaka, H.; Sekine, S. I., Structural basis of nucleosome disassembly and reassembly by RNAPII elongation complex with FACT. Science 2022, 377, (6611), eabp9466. [CrossRef]
- Cramer, P.; Pesce, C. G.; Baralle, F. E.; Kornblihtt, A. R., Functional association between promoter structure and transcript alternative splicing. Proc Natl Acad Sci U S A 1997, 94, (21), 11456-60. [CrossRef] [PubMed]
- Cramer, P.; Caceres, J. F.; Cazalla, D.; Kadener, S.; Muro, A. F.; Baralle, F. E.; Kornblihtt, A. R., Coupling of transcription with alternative splicing: RNA pol II promoters modulate SF2/ASF and 9G8 effects on an exonic splicing enhancer. Mol Cell 1999, 4, (2), 251-8. [CrossRef]
- He, X.; Yuan, J.; Gao, Z.; Wang, Y., Promoter R-Loops Recruit U2AF1 to Modulate Its Phase Separation and RNA Splicing. J Am Chem Soc 2023, 145, (39), 21646-21660. [CrossRef] [PubMed]
- Shukla, S.; Kavak, E.; Gregory, M.; Imashimizu, M.; Shutinoski, B.; Kashlev, M.; Oberdoerffer, P.; Sandberg, R.; Oberdoerffer, S., CTCF-promoted RNA polymerase II pausing links DNA methylation to splicing. Nature 2011, 479, (7371), 74-9. [CrossRef]
- Marina, R. J.; Sturgill, D.; Bailly, M. A.; Thenoz, M.; Varma, G.; Prigge, M. F.; Nanan, K. K.; Shukla, S.; Haque, N.; Oberdoerffer, S., TET-catalyzed oxidation of intragenic 5-methylcytosine regulates CTCF-dependent alternative splicing. Embo J 2016, 35, (3), 335-55. [CrossRef]
- Guo, Y.; Monahan, K.; Wu, H.; Gertz, J.; Varley, K. E.; Li, W.; Myers, R. M.; Maniatis, T.; Wu, Q., CTCF/cohesin-mediated DNA looping is required for protocadherin alpha promoter choice. Proc Natl Acad Sci U S A 2012, 109, (51), 21081-6. [CrossRef]
- Monahan, K.; Rudnick, N. D.; Kehayova, P. D.; Pauli, F.; Newberry, K. M.; Myers, R. M.; Maniatis, T., Role of CCCTC binding factor (CTCF) and cohesin in the generation of single-cell diversity of protocadherin-alpha gene expression. Proc Natl Acad Sci U S A 2012, 109, (23), 9125-30. [CrossRef]
- Lamas-Maceiras, M.; Singh, B. N.; Hampsey, M.; Freire-Picos, M. A., Promoter-Terminator Gene Loops Affect Alternative 3'-End Processing in Yeast. J Biol Chem 2016, 291, (17), 8960-8. [CrossRef]
- Tan-Wong, S. M.; Zaugg, J. B.; Camblong, J.; Xu, Z.; Zhang, D. W.; Mischo, H. E.; Ansari, A. Z.; Luscombe, N. M.; Steinmetz, L. M.; Proudfoot, N. J., Gene loops enhance transcriptional directionality. Science 2012, 338, (6107), 671-5. [CrossRef]
- von Hacht, A.; Seifert, O.; Menger, M.; Schutze, T.; Arora, A.; Konthur, Z.; Neubauer, P.; Wagner, A.; Weise, C.; Kurreck, J., Identification and characterization of RNA guanine-quadruplex binding proteins. Nucleic Acids Res 2014, 42, (10), 6630-44. [CrossRef]
- Zhang, J.; Harvey, S. E.; Cheng, C., A high-throughput screen identifies small molecule modulators of alternative splicing by targeting RNA G-quadruplexes. Nucleic Acids Res 2019, 47, (7), 3667-3679. [CrossRef] [PubMed]
- Jara-Espejo, M.; Fleming, A. M.; Burrows, C. J., Potential G-Quadruplex Forming Sequences and N(6)-Methyladenosine Colocalize at Human Pre-mRNA Intron Splice Sites. ACS Chem Biol 2020, 15, (6), 1292-1300. [CrossRef]
- Darnell, R. B.; Ke, S.; Darnell, J. E., Jr., Pre-mRNA processing includes N(6) methylation of adenosine residues that are retained in mRNA exons and the fallacy of "RNA epigenetics". Rna 2018, 24, (3), 262-267. [CrossRef] [PubMed]
- Wei, G.; Almeida, M.; Pintacuda, G.; Coker, H.; Bowness, J. S.; Ule, J.; Brockdorff, N., Acute depletion of METTL3 implicates N (6)-methyladenosine in alternative intron/exon inclusion in the nascent transcriptome. Genome Res 2021, 31, (8), 1395-1408. [CrossRef] [PubMed]
- Fleming, A. M.; Nguyen, N. L. B.; Burrows, C. J., Colocalization of m(6)A and G-Quadruplex-Forming Sequences in Viral RNA (HIV, Zika, Hepatitis B, and SV40) Suggests Topological Control of Adenosine N (6)-Methylation. ACS Cent Sci 2019, 5, (2), 218-228. [CrossRef]
- Yoshida, A.; Oyoshi, T.; Suda, A.; Futaki, S.; Imanishi, M., Recognition of G-quadruplex RNA by a crucial RNA methyltransferase component, METTL14. Nucleic Acids Res 2022, 50, (1), 449-457. [CrossRef]
- Patil, D. P.; Chen, C. K.; Pickering, B. F.; Chow, A.; Jackson, C.; Guttman, M.; Jaffrey, S. R., m(6)A RNA methylation promotes XIST-mediated transcriptional repression. Nature 2016, 537, (7620), 369-373. [CrossRef] [PubMed]
- Ye, H.; Li, T.; Rigden, D. J.; Wei, Z., m6ACali: machine learning-powered calibration for accurate m6A detection in MeRIP-Seq. Nucleic Acids Res 2024, 52, (9), 4830-4842. [CrossRef]
- Iwasaki, Y.; Ookuro, Y.; Iida, K.; Nagasawa, K.; Yoshida, W., Destabilization of DNA and RNA G-quadruplex structures formed by GGA repeat due to N(6)-methyladenine modification. Biochem Biophys Res Commun 2022, 597, 134-139. [CrossRef]
- Shi, H.; Wei, J.; He, C., Where, When, and How: Context-Dependent Functions of RNA Methylation Writers, Readers, and Erasers. Mol Cell 2019, 74, (4), 640-650. [CrossRef]
- Ke, S.; Pandya-Jones, A.; Saito, Y.; Fak, J. J.; Vagbo, C. B.; Geula, S.; Hanna, J. H.; Black, D. L.; Darnell, J. E., Jr.; Darnell, R. B., m(6)A mRNA modifications are deposited in nascent pre-mRNA and are not required for splicing but do specify cytoplasmic turnover. Genes Dev 2017, 31, (10), 990-1006. [CrossRef] [PubMed]
- Mestre-Fos, S.; Penev, P. I.; Suttapitugsakul, S.; Hu, M.; Ito, C.; Petrov, A. S.; Wartell, R. M.; Wu, R.; Williams, L. D., G-Quadruplexes in Human Ribosomal RNA. J Mol Biol 2019, 431, (10), 1940-1955. [CrossRef]
- Scognamiglio, P. L.; Di Natale, C.; Leone, M.; Poletto, M.; Vitagliano, L.; Tell, G.; Marasco, D., G-quadruplex DNA recognition by nucleophosmin: new insights from protein dissection. Biochim Biophys Acta 2014, 1840, (6), 2050-9. [CrossRef] [PubMed]
- Okuwaki, M.; Saotome-Nakamura, A.; Yoshimura, M.; Saito, S.; Hirawake-Mogi, H.; Sekiya, T.; Nagata, K., RNA-recognition motifs and glycine and arginine-rich region cooperatively regulate the nucleolar localization of nucleolin. J Biochem 2021, 169, (1), 87-100. [CrossRef] [PubMed]
- Santos, T.; Salgado, G. F.; Cabrita, E. J.; Cruz, C., Nucleolin: a binding partner of G-quadruplex structures. Trends Cell Biol 2022, 32, (7), 561-564. [CrossRef]
- Tian, B.; Manley, J. L., Alternative polyadenylation of mRNA precursors. Nat Rev Mol Cell Biol 2017, 18, (1), 18-30. [CrossRef]
- Leppek, K.; Das, R.; Barna, M., Functional 5' UTR mRNA structures in eukaryotic translation regulation and how to find them. Nat Rev Mol Cell Biol 2018, 19, (3), 158-174. [CrossRef]
- Schuster, S. L.; Hsieh, A. C., The Untranslated Regions of mRNAs in Cancer. Trends Cancer 2019, 5, (4), 245-262. [CrossRef]
- Mayr, C., What Are 3' UTRs Doing? Cold Spring Harb Perspect Biol 2019, 11, (10).
- Lee, D. S. M.; Ghanem, L. R.; Barash, Y., Integrative analysis reveals RNA G-quadruplexes in UTRs are selectively constrained and enriched for functional associations. Nat Commun 2020, 11, (1), 527. [CrossRef]
- Sauer, M.; Juranek, S. A.; Marks, J.; De Magis, A.; Kazemier, H. G.; Hilbig, D.; Benhalevy, D.; Wang, X.; Hafner, M.; Paeschke, K., DHX36 prevents the accumulation of translationally inactive mRNAs with G4-structures in untranslated regions. Nat Commun 2019, 10, (1), 2421. [CrossRef]
- Benhalevy, D.; Gupta, S. K.; Danan, C. H.; Ghosal, S.; Sun, H. W.; Kazemier, H. G.; Paeschke, K.; Hafner, M.; Juranek, S. A., The Human CCHC-type Zinc Finger Nucleic Acid-Binding Protein Binds G-Rich Elements in Target mRNA Coding Sequences and Promotes Translation. Cell Rep 2017, 18, (12), 2979-2990. [CrossRef] [PubMed]
- Dong, L.; Mao, Y.; Zhou, A.; Liu, X. M.; Zhou, J.; Wan, J.; Qian, S. B., Relaxed initiation pausing of ribosomes drives oncogenic translation. Sci Adv 2021, 7, (8).
- Zhou, J.; Wan, J.; Gao, X.; Zhang, X.; Jaffrey, S. R.; Qian, S. B., Dynamic m(6)A mRNA methylation directs translational control of heat shock response. Nature 2015, 526, (7574), 591-4. [CrossRef] [PubMed]
- Zaccara, S.; Jaffrey, S. R., A Unified Model for the Function of YTHDF Proteins in Regulating m(6)A-Modified mRNA. Cell 2020, 181, (7), 1582-1595 e18.
- Cirillo, L. A.; Lin, F. R.; Cuesta, I.; Friedman, D.; Jarnik, M.; Zaret, K. S., Opening of compacted chromatin by early developmental transcription factors HNF3 (FoxA) and GATA-4. Mol Cell 2002, 9, (2), 279-89. [CrossRef]
- Zaret, K. S., Pioneer Transcription Factors Initiating Gene Network Changes. Annu Rev Genet 2020, 54, 367-385. [CrossRef] [PubMed]
- Herbert, A., Nucleosomes and flipons exchange energy to alter chromatin conformation, the readout of genomic information, and cell fate. Bioessays 2022, 44, (12), e2200166. [CrossRef]
- Czech, B.; Munafo, M.; Ciabrelli, F.; Eastwood, E. L.; Fabry, M. H.; Kneuss, E.; Hannon, G. J., piRNA-Guided Genome Defense: From Biogenesis to Silencing. Annu Rev Genet 2018, 52, 131-157. [CrossRef]
- Zyner, K. G.; Simeone, A.; Flynn, S. M.; Doyle, C.; Marsico, G.; Adhikari, S.; Portella, G.; Tannahill, D.; Balasubramanian, S., G-quadruplex DNA structures in human stem cells and differentiation. Nat Commun 2022, 13, (1), 142. [CrossRef]
- Skourti-Stathaki, K.; Torlai Triglia, E.; Warburton, M.; Voigt, P.; Bird, A.; Pombo, A., R-Loops Enhance Polycomb Repression at a Subset of Developmental Regulator Genes. Mol Cell 2019, 73, (5), 930-945 e4.
- Yang, Q.; Lin, J.; Liu, M.; Li, R.; Tian, B.; Zhang, X.; Xu, B.; Liu, M.; Zhang, X.; Li, Y.; Shi, H.; Wu, L., Highly sensitive sequencing reveals dynamic modifications and activities of small RNAs in mouse oocytes and early embryos. Sci Adv 2016, 2, (6), e1501482. [CrossRef]
- Zhang, Y.; Zhang, X.; Shi, J.; Tuorto, F.; Li, X.; Liu, Y.; Liebers, R.; Zhang, L.; Qu, Y.; Qian, J.; Pahima, M.; Liu, Y.; Yan, M.; Cao, Z.; Lei, X.; Cao, Y.; Peng, H.; Liu, S.; Wang, Y.; Zheng, H.; Woolsey, R.; Quilici, D.; Zhai, Q.; Li, L.; Zhou, T.; Yan, W.; Lyko, F.; Zhang, Y.; Zhou, Q.; Duan, E.; Chen, Q., Dnmt2 mediates intergenerational transmission of paternally acquired metabolic disorders through sperm small non-coding RNAs. Nat Cell Biol 2018, 20, (5), 535-540. [CrossRef]
- Paloviita, P.; Hyden-Granskog, C.; Yohannes, D. A.; Paluoja, P.; Kere, J.; Tapanainen, J. S.; Krjutskov, K.; Tuuri, T.; Vosa, U.; Vuoristo, S., Small RNA expression and miRNA modification dynamics in human oocytes and early embryos. Genome Res 2021, 31, (8), 1474-1485. [CrossRef]
- Tomar, A.; Gomez-Velazquez, M.; Gerlini, R.; Comas-Armangue, G.; Makharadze, L.; Kolbe, T.; Boersma, A.; Dahlhoff, M.; Burgstaller, J. P.; Lassi, M.; Darr, J.; Toppari, J.; Virtanen, H.; Kuhnapfel, A.; Scholz, M.; Landgraf, K.; Kiess, W.; Vogel, M.; Gailus-Durner, V.; Fuchs, H.; Marschall, S.; Hrabe de Angelis, M.; Kotaja, N.; Korner, A.; Teperino, R., Epigenetic inheritance of diet-induced and sperm-borne mitochondrial RNAs. Nature 2024, 630, (8017), 720-727. [CrossRef]
- Maldonado, R.; Langst, G., The chromatin - triple helix connection. Biol Chem 2023, 404, (11-12), 1037-1049.
- Leisegang, M. S.; Warwick, T.; Stotzel, J.; Brandes, R. P., RNA-DNA triplexes: molecular mechanisms and functional relevance. Trends Biochem Sci 2024, 49, (6), 532-544. [CrossRef] [PubMed]
- Zhou, Z.; Giles, K. E.; Felsenfeld, G., DNA.RNA triple helix formation can function as a cis-acting regulatory mechanism at the human beta-globin locus. Proc Natl Acad Sci U S A 2019, 116, (13), 6130-6139. [CrossRef] [PubMed]
- Maldonado, R.; Schwartz, U.; Silberhorn, E.; Langst, G., Nucleosomes Stabilize ssRNA-dsDNA Triple Helices in Human Cells. Mol Cell 2019, 73, (6), 1243-1254 e6.
- Kohestani, H.; Wereszczynski, J., The effects of RNA.DNA-DNA triple helices on nucleosome structures and dynamics. Biophys J 2023, 122, (7), 1229-1239.
- Jimenez-Garcia, E.; Vaquero, A.; Espinas, M. L.; Soliva, R.; Orozco, M.; Bernues, J.; Azorin, F., The GAGA factor of Drosophila binds triple-stranded DNA. J Biol Chem 1998, 273, (38), 24640-8. [CrossRef] [PubMed]
- Leisegang, M. S.; Bains, J. K.; Seredinski, S.; Oo, J. A.; Krause, N. M.; Kuo, C. C.; Gunther, S.; Senturk Cetin, N.; Warwick, T.; Cao, C.; Boos, F.; Izquierdo Ponce, J.; Haydar, S.; Bednarz, R.; Valasarajan, C.; Fuhrmann, D. C.; Preussner, J.; Looso, M.; Pullamsetti, S. S.; Schulz, M. H.; Jonker, H. R. A.; Richter, C.; Rezende, F.; Gilsbach, R.; Pfluger-Muller, B.; Wittig, I.; Grummt, I.; Ribarska, T.; Costa, I. G.; Schwalbe, H.; Brandes, R. P., HIF1alpha-AS1 is a DNA:DNA:RNA triplex-forming lncRNA interacting with the HUSH complex. Nat Commun 2022, 13, (1), 6563. [CrossRef]
Figure 1.
G-quadruples fold in many different ways. A. The core four-stranded structure formed by stacking the guanine tetrads shown in B. with Hoogsteen hydrogen bonds highlighted in yellow and crimson. The four strands may form from G repeats on the same molecule or arise from different molecules or from either RNA or DNA. C. The base 8-aza-7-deazaguanosine retains the same molecular composition as guanosine, but with the red ring nitrogen in a different position, preventing the formation of the Hoogsteen hydrogen bonds shown with crimson shading. Control oligonucleotides incorporating this nucleotide will not form GQ. In intramolecular GQ, the stands may be parallel (D, G), anti-parallel (E, H), or hybrid (F, I). The topology of the connecting loops is shown in blue and can be propeller (D), lateral (E) or diagonal (F). M+ indicates a metal ion located at the core of the tetrad. K+ promotes GQ formation while Li+ does not. The dotted strand in F labeled 5 indicates that many sequences capable of forming GQ contain a “spare” tire that can maintain the fold when one of the other repeats is damaged [63]. The cartons in G,H, I show the phosphate backbone as a ribbon and the bases as sticks. PDB codes are given below the structures.
Figure 1.
G-quadruples fold in many different ways. A. The core four-stranded structure formed by stacking the guanine tetrads shown in B. with Hoogsteen hydrogen bonds highlighted in yellow and crimson. The four strands may form from G repeats on the same molecule or arise from different molecules or from either RNA or DNA. C. The base 8-aza-7-deazaguanosine retains the same molecular composition as guanosine, but with the red ring nitrogen in a different position, preventing the formation of the Hoogsteen hydrogen bonds shown with crimson shading. Control oligonucleotides incorporating this nucleotide will not form GQ. In intramolecular GQ, the stands may be parallel (D, G), anti-parallel (E, H), or hybrid (F, I). The topology of the connecting loops is shown in blue and can be propeller (D), lateral (E) or diagonal (F). M+ indicates a metal ion located at the core of the tetrad. K+ promotes GQ formation while Li+ does not. The dotted strand in F labeled 5 indicates that many sequences capable of forming GQ contain a “spare” tire that can maintain the fold when one of the other repeats is damaged [63]. The cartons in G,H, I show the phosphate backbone as a ribbon and the bases as sticks. PDB codes are given below the structures.
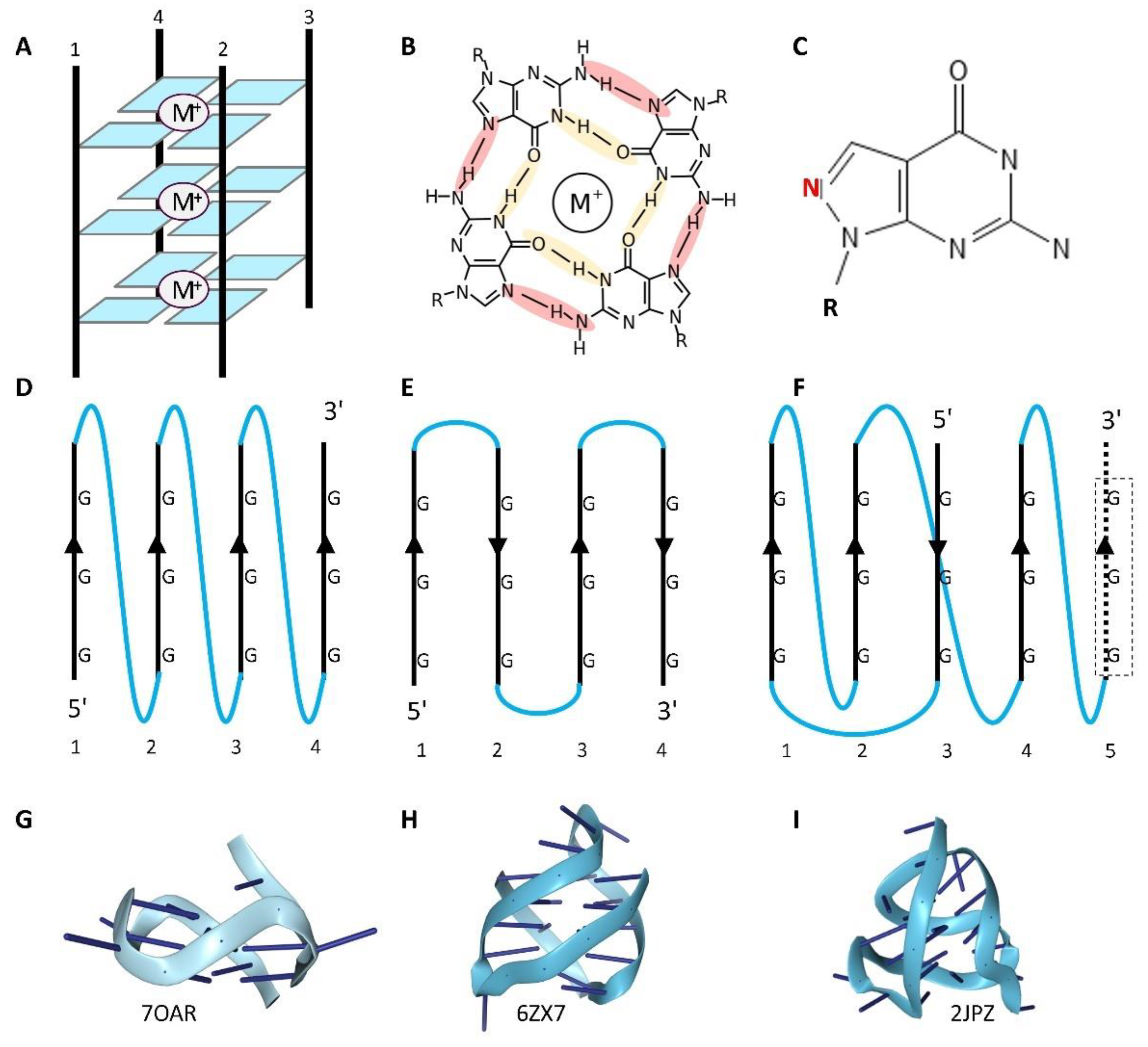
Figure 2.
Some proteins bind both B-DNA and GQ. The yeast Rap1 protein binds both B-DNA in a sequence-specific manner A [104] and to GQ in a structure-specific manner B. [105] through different faces of the same helix in the SANT/Myb1 domain (Images by Daniela Rhodes) C. The Flipon cycle creates a two-state switch that arises due to the similar affinities of Rap1for B-DNA and GQ (i.e. Kb ≈ Kg ≈ ~20-30 nM).
Figure 2.
Some proteins bind both B-DNA and GQ. The yeast Rap1 protein binds both B-DNA in a sequence-specific manner A [104] and to GQ in a structure-specific manner B. [105] through different faces of the same helix in the SANT/Myb1 domain (Images by Daniela Rhodes) C. The Flipon cycle creates a two-state switch that arises due to the similar affinities of Rap1for B-DNA and GQ (i.e. Kb ≈ Kg ≈ ~20-30 nM).

Figure 3.
The G-flipon cycle. Many factors induce and resolve GQ to modulate specific outcomes. Other proteins prevent their formation, such as nucleosomes and repressor proteins. Transcription factors can dock to G-flipons in either a sequence-specific manner to their right-handed conformation or to the GQ structure. Once formed, the resolution of GQ can be coupled to the different outcomes shown, with both activation and inhibition of gene expression. The inhibition of enzymes like DNMT1 and TOP1 favors maintenance of an unmethylated, nucleosome depleted state that is necessary to rapidly reprogram cellular responses to environmental inputs.(BER: base excision repair; NER: nucleotide excision repair). References for each gene are given in the text.
Figure 3.
The G-flipon cycle. Many factors induce and resolve GQ to modulate specific outcomes. Other proteins prevent their formation, such as nucleosomes and repressor proteins. Transcription factors can dock to G-flipons in either a sequence-specific manner to their right-handed conformation or to the GQ structure. Once formed, the resolution of GQ can be coupled to the different outcomes shown, with both activation and inhibition of gene expression. The inhibition of enzymes like DNMT1 and TOP1 favors maintenance of an unmethylated, nucleosome depleted state that is necessary to rapidly reprogram cellular responses to environmental inputs.(BER: base excision repair; NER: nucleotide excision repair). References for each gene are given in the text.
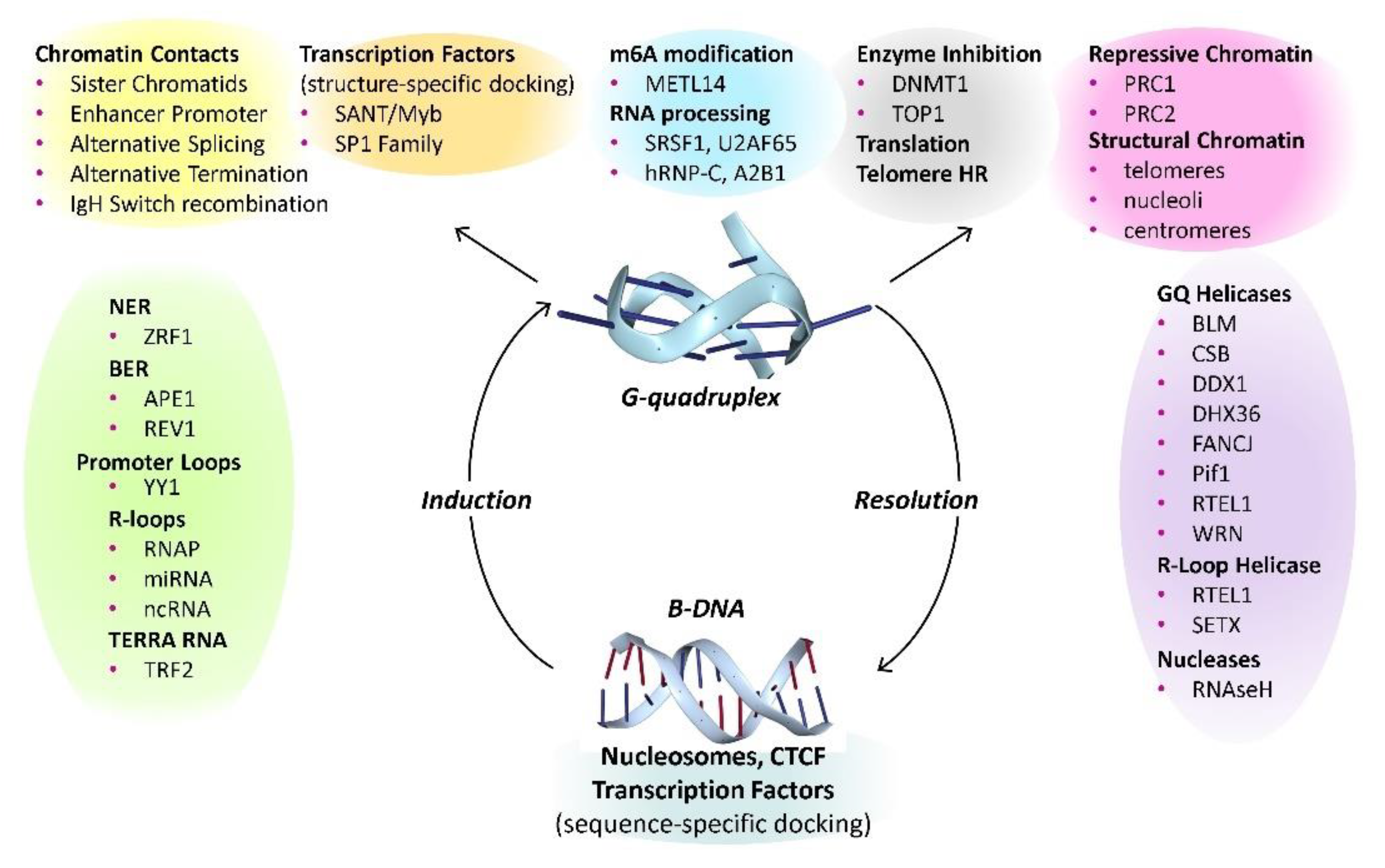
Figure 4.
G-flipons nucleate condensates. In the scheme presented. GQ formation seeds condensates that promote the interaction between different genomic regions. This arrangement can maintain G-flipons in an active, but poised state, locked and loaded. A. The sequential contacts between GQ formed at different chromosomal sites ensure that RNA processing occurs in the correct temporal order. The splice and polyadenylation events may vary with the specific promoter used to seed the condensate. The GQ variant formed by a promoter may favor selection of a particular splice or polyadenylation site. The dotted lines indicate that both DNA and RNA GQ can participate in these reactions. B. Retroviruses are enriched for G-flipons in their LTR that can adopt different conformations as seen in the NMR structures PDB:2N47 [121] and PDB:6HiK [122]. Formation of GQ by both retroviral long terminal repeats may anchor chromosomal loops that are stabilized by a condensate at their base. In this state, the viral genome is poised, but not actively transcribed. Dissolution of the condensate then release of RPOL2 to initiate transcription. Eleven potential and conserved GQ on the viral plus strand with different folds exist in HIV mRNA, 9 of which are in protein coding regions. Two potential GQ were detected on the negative strand [71,123,124]. The number of potential G-flipons in the LTR differs between retroviruses [70]. Whether more GQ increases viral virulence or whether the G-flipons enable different processing events is not currently known.
Figure 4.
G-flipons nucleate condensates. In the scheme presented. GQ formation seeds condensates that promote the interaction between different genomic regions. This arrangement can maintain G-flipons in an active, but poised state, locked and loaded. A. The sequential contacts between GQ formed at different chromosomal sites ensure that RNA processing occurs in the correct temporal order. The splice and polyadenylation events may vary with the specific promoter used to seed the condensate. The GQ variant formed by a promoter may favor selection of a particular splice or polyadenylation site. The dotted lines indicate that both DNA and RNA GQ can participate in these reactions. B. Retroviruses are enriched for G-flipons in their LTR that can adopt different conformations as seen in the NMR structures PDB:2N47 [121] and PDB:6HiK [122]. Formation of GQ by both retroviral long terminal repeats may anchor chromosomal loops that are stabilized by a condensate at their base. In this state, the viral genome is poised, but not actively transcribed. Dissolution of the condensate then release of RPOL2 to initiate transcription. Eleven potential and conserved GQ on the viral plus strand with different folds exist in HIV mRNA, 9 of which are in protein coding regions. Two potential GQ were detected on the negative strand [71,123,124]. The number of potential G-flipons in the LTR differs between retroviruses [70]. Whether more GQ increases viral virulence or whether the G-flipons enable different processing events is not currently known.
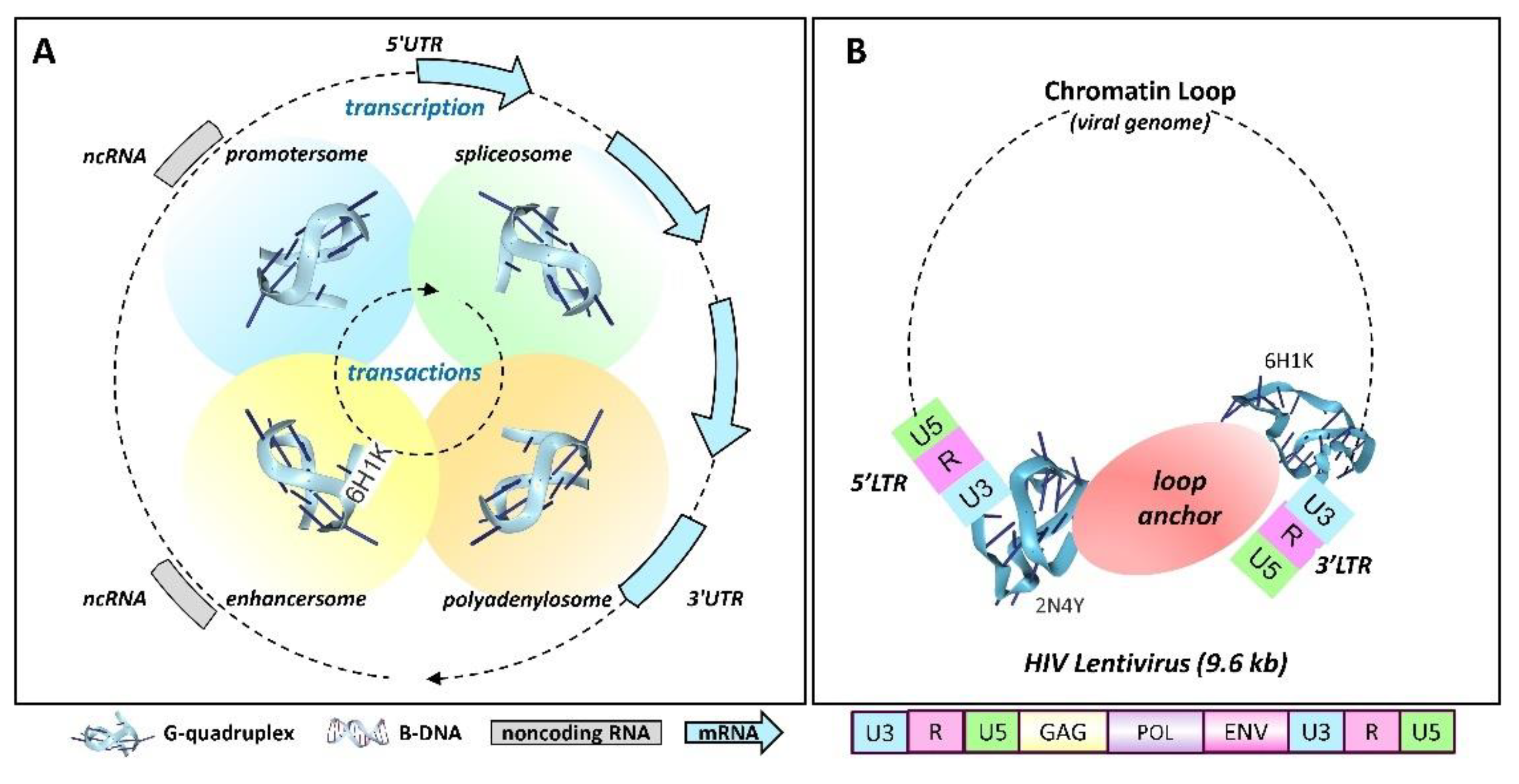
Figure 5.
Flipons Cycle Promoters. The reset and reinitiation of transcription complexes is actuated by G- and Z-flipons. A. In this model, a condensate is anchored by GQ formed by enhancer and promoter sequences. The condensate stabilizes an enhancer-promoter loop and holds the RNA polymerase (RPOL2) in a poised state. B. Breakdown of the condensate triggers transcript elongation. The negative supercoiling 5' to RNAP2and induces Z-DNA formation that actuates removal of the pre-initiation complex (PIC). C. Resolution of the promoter GQ by helicases then enables rebinding of TF (transcription factors) and reassembly of the PIC. The separation of strands indicates the partial unwinding that is necessary to form the transcription bubble and a GQ.
Figure 5.
Flipons Cycle Promoters. The reset and reinitiation of transcription complexes is actuated by G- and Z-flipons. A. In this model, a condensate is anchored by GQ formed by enhancer and promoter sequences. The condensate stabilizes an enhancer-promoter loop and holds the RNA polymerase (RPOL2) in a poised state. B. Breakdown of the condensate triggers transcript elongation. The negative supercoiling 5' to RNAP2and induces Z-DNA formation that actuates removal of the pre-initiation complex (PIC). C. Resolution of the promoter GQ by helicases then enables rebinding of TF (transcription factors) and reassembly of the PIC. The separation of strands indicates the partial unwinding that is necessary to form the transcription bubble and a GQ.
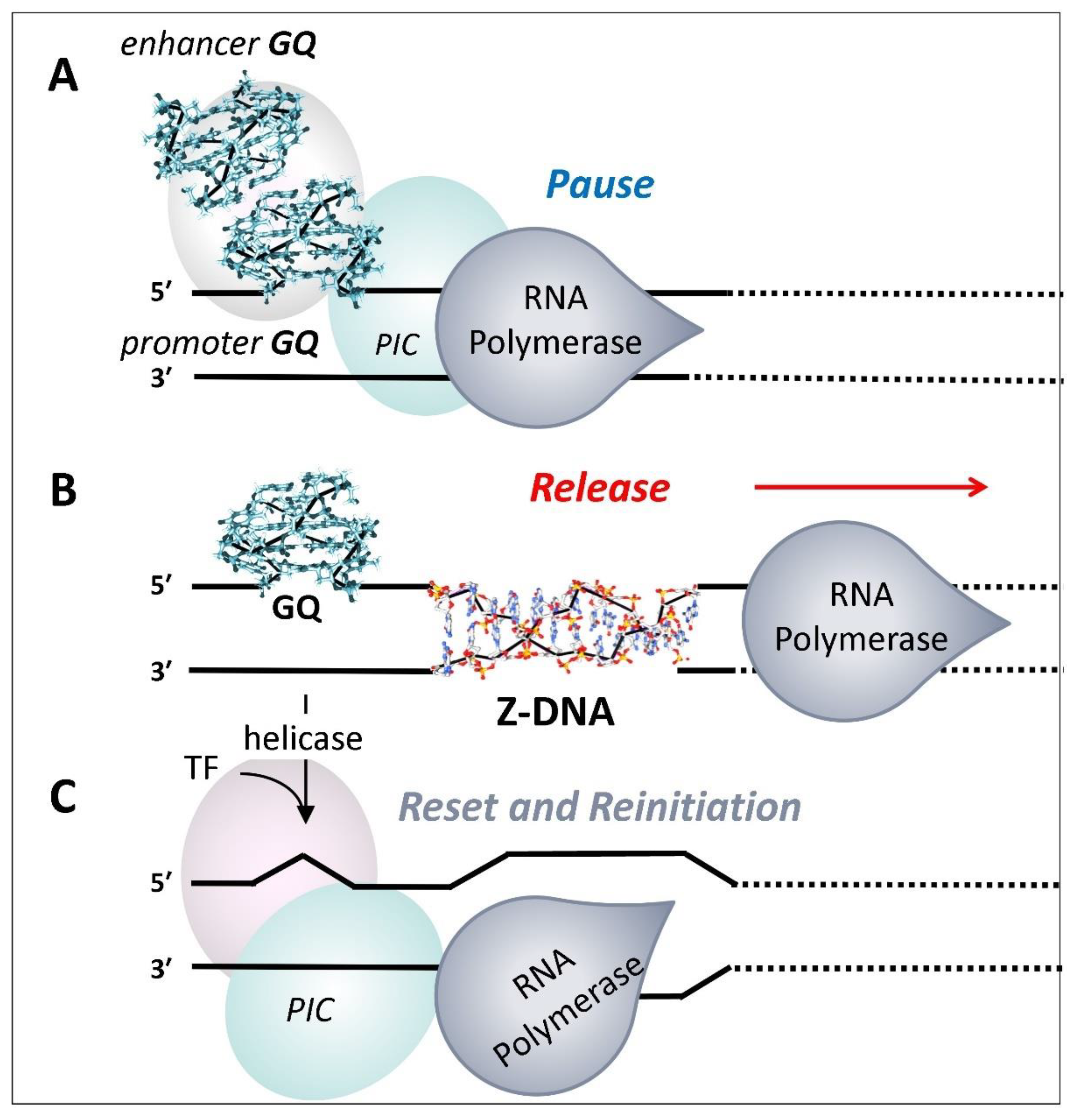
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



