
Submitted:
21 August 2024
Posted:
26 August 2024
You are already at the latest version
Abstract
A genome represents the complete collection of genetic material in an organism or cell. This genetic information is encoded in nucleic acids, which can be either single or double-stranded and arranged in linear or circular forms. DNA, the most common type of genetic material, consists of just four nucleotides: adenine, guanine, thymine, and cytosine. These nucleotides are organized in a double helix structure, a discovery made by Watson and Crick. The sequence of these nucleotides forms the basic blueprint of a gene or genome. The process of determining this sequence is known as genome sequencing. The field of genome sequencing has evolved dramatically over time, advancing from sequencing short DNA fragments to analyzing millions of base pairs. Initially, efforts were focused on determining the sequence of individual genes, but now whole genome sequencing is both rapid and widely accessible. Recent improvements in sequencing technology emphasize faster, more accurate results, lower costs, and better data analysis. Sanger sequencing, which became the gold standard for DNA sequencing for about thirty years, was instrumental in completing various genome sequences, including the human genome. It laid the groundwork for newer sequencing methods. Today, these newer methods, known collectively as “Next-Generation Sequencing” (NGS), include several generations of technology. Second-generation methods, such as Illumina, Pyrosequencing, ABI/SOLiD, and Ion Torrent Sequencing, and third-generation methods like PacBio and Helicos Sequencing, have advanced the field further. Fourth-generation technology, such as Nanopore sequencing, is also emerging. Most modern research now relies on NGS for analyzing biological sequences, and understanding these technologies offers insight into current methods and future developments.
Keywords:
Genome
; DNA sequencing
; Next Generation Sequencing
1. Introduction
A genome, serving as the blueprint of life, encompasses an organism's complete DNA sequence, encoding genetic instructions.
Nucleic acids, whether single or double-stranded, store this genetic information in either linear or circular arrangements. Nucleotides, including adenine, guanine, thymine, and cytosine, serve as the building blocks of DNA, formed in a double helix structure that was identified in 1953 by Watson and Crick [1]. The most fundamental level of understanding about a gene or genome is its nucleotide sequence. DNA sequencing provides information about the nucleotide sequence of a DNA molecule, which aids in our understanding of the genetic code that all life on Earth is based on. Additionally, it aids in the diagnosis and treatment of genetic disorders [2].
The advancement from Frederick Miescher's first isolation of DNA in 1869 to the development of next-generation sequencing was achieved through ongoing efforts within the scientific community.
Sanger sequencing and Maxam & Gilbert sequencing were initial sequencing technologies created by Sanger et al. [3] in 1977 and Maxam et al [4] respectively. Their discovery enabled the exploration of the genetic codes of living organisms, motivating scientists to innovate faster and more efficient sequencing technologies.
Fleischmann et al. [5] (1995) reported on the Sanger method's genome sequencing of Haemophilus influenza, the first free-living microorganism with complete functionality. Sanger sequencing has emerged as the prevailing choice among sequencing methods due to its high efficiency and was regarded as gold standard DNA sequencing technology around 3 decades. Since then, numerous approaches have been created from enhancements made to Sanger's methodology as whole genome sequencing (WGS) using this technology is exceedingly costly, labour-intensive, and yields low accuracy and output [6]. NGS (Next Generation Sequencing) encompasses a range of techniques categorized into second, third, and fourth generation sequencing.
The beginning of next-generation sequencing (NGS) platforms, employing diverse sequencing chemistries and advanced instrumentation, has significantly streamlined the sequencing process. These technologies not only simplify sequencing but also generate sequencing data at much higher volumes compared to traditional methods. Consequently, the time required to sequence an entire genome at the desired depth has been reduced to days or weeks, a stark improvement from the prolonged timelines of Sanger sequencing [7]. Progress in DNA sequencing technology has led to significant advancements, which have made sequencing faster and more affordable, have led to a broad spectrum of new applications. These include research in transcriptomics, epigenomics, cancer [8,9], human genetics [10], infectious diseases [11], personal genomics [12,13,14], ecology [15,16] and the analysis of ancient DNA [17,18].
2. Historical Background:
In 1953, following the landmark revelation of double helical structure of DNA by James. D. Watson and Francis Crick [1], extensive effort was undertaken to sequence DNA. Subsequently, in 1965, Robert Holley accomplished the sequencing of initial tRNA molecule, an achievement that earned him the Nobel Prize in 1986 [19].
In 1972, Walter Fries achieved a significant milestone by sequencing DNA of a complete gene, specifically gene responsible for encoding the MS2 bacteriophage coat protein. Fries employed RNAses to break down virus RNA and separate oligonucleotides, which were subsequently separated by chromatography or electrophoresis [20]. Concurrently, Frederick Sanger pursued an alternative DNA sequencing approach and in 1977, he introduced the pioneering "chain termination method." This method involved radiolabelled partially digested fragments.
Sanger's method quickly became the dominant technique in the field of sequencing for the following three decades. Frederick Sanger is revered as a luminary in genomics, earning him Nobel Prize in 1980, second Nobel Prize; his first was awarded in 1958 for groundbreaking studies on insulin's structure. In 1977, Sanger's method reached another milestone when he applied it to sequence entire genome of E. coli-infecting bacteriophage PhiX174. This accomplishment propelled PhiX174 to become the standard DNA positive control in laboratories worldwide [21]. Concurrently, in same year, Maxam and Gilbert presented a different approach to sequence DNA, which relied on chemical alteration of DNA. Their approach involved utilizing chemicals to induce breaks in the DNA sequence at specific bases [4]. Unlike Sanger sequencing, Maxam-Gilbert sequencing did not depend on DNA polymerase. However, both Sanger sequencing and Maxam-Gilbert sequencing shared a common challenge: they lacked automation and were labor-intensive and time-consuming. Despite these limitations, the scientific community recognized the immense potential of Sanger sequencing. Consequently, numerous research groups worked on automation of the process.
The Human Genome Project, initiated in 1990 to decode human genome consisting of 3.2 billion nucleotide bp for medical advancements, spurred the development of faster sequencing technologies. In 2005, Roche 454 Life Sciences introduced pyrosequencing, marking the beginning of the "next-generation sequencing" era [22]. Thousands to millions of short sequencing reads could be identified using this high-throughput technique in a single machine cycle, eliminating the necessity for cloning.
3. First Generation Sequencing:
The technologies known as First-Generation Sequencing were Maxam-Gilbert and Sanger sequencing [23].
3.1. Sanger Sequencing
Known also as the "chain termination method," Sanger sequencing" is a technique used to determine the sequence of nucleotides in DNA.
The Sanger Sequence, named after its creator Nobel Laureate Frederick Sanger and his associates, has become widely adopted due to its efficiency and minimal radioactivity [24]. In 1980, the phiX174 genome, spanning 5374 base pairs [25] and the bacteriophage λ genome, spanning 48501 base pairs [26] were the first genomes to undergo sequencing employing sanger technique. Subsequently, Sanger sequencing was essential in numerous sequencing endeavors, with its most notable achievement being the deciphering of the human genome [27].
Principle: It involves a modification of the DNA replication process, relying on the specific incorporation by DNA polymerase of chain-terminating dideoxynucleotides (ddNTPs) during in vitro DNA replication.
Procedure: This process modifies the DNA replication mechanism through the selective integration of chain-terminating chemically modified nucleotides known as dideoxynucleotides (ddNTPs) by DNA polymerase. The procedure involves the utilization of reagents such as ddNTPs, DNA polymerase, dNTPs, primers and template DNA that needs to be sequenced. During annealing process, DNA polymerase integartes both ddNTPs and dNTPs in a random manner for chain extension. Due to the absence of the 3' hydroxyl group required for DNA chain elongation, ddNTPs are unable to attach to the 5' phosphate of the next dNTP [25]. Once ddNTPs are integrated into a growing DNA strand, they hinder further elongation, causing extension to halt [28]. This procedure yields fragments of different lengths, which are then separated by size using gel electrophoresis and can be seen with an imaging device, like an X-ray or UV light source [29,30]. DNA samples are introduced into one end of a gel matrix and an electric current is applied during the gel electrophoresis process. Due to its negative charge, DNA migrates towards positive electrode positioned at opposite end of gel.
Size is the main factor influencing how quickly oligonucleotides migrate as every DNA fragment has the same charge per mass unit. Fragments which are small, encounter less resistance and thus travel more swiftly through the gel. As a result, the gel is interpreted with the oligonucleotides organized in increasing size order, starting from the bottom. Because DNA polymerase only synthesizes DNA in the 5' to 3' orientation, beginning with a primer supplied, each terminal ddNTP corresponds to a particular nucleotide in the original sequence. For instance, the first nucleotide from the 5' end is where the shortest segment ends, the second nucleotide from the 5' end is where the second-shortest fragment ends, and so on. This method of interpreting the gel bands allows us to infer the 5' to 3' sequencing of the original DNA strand.
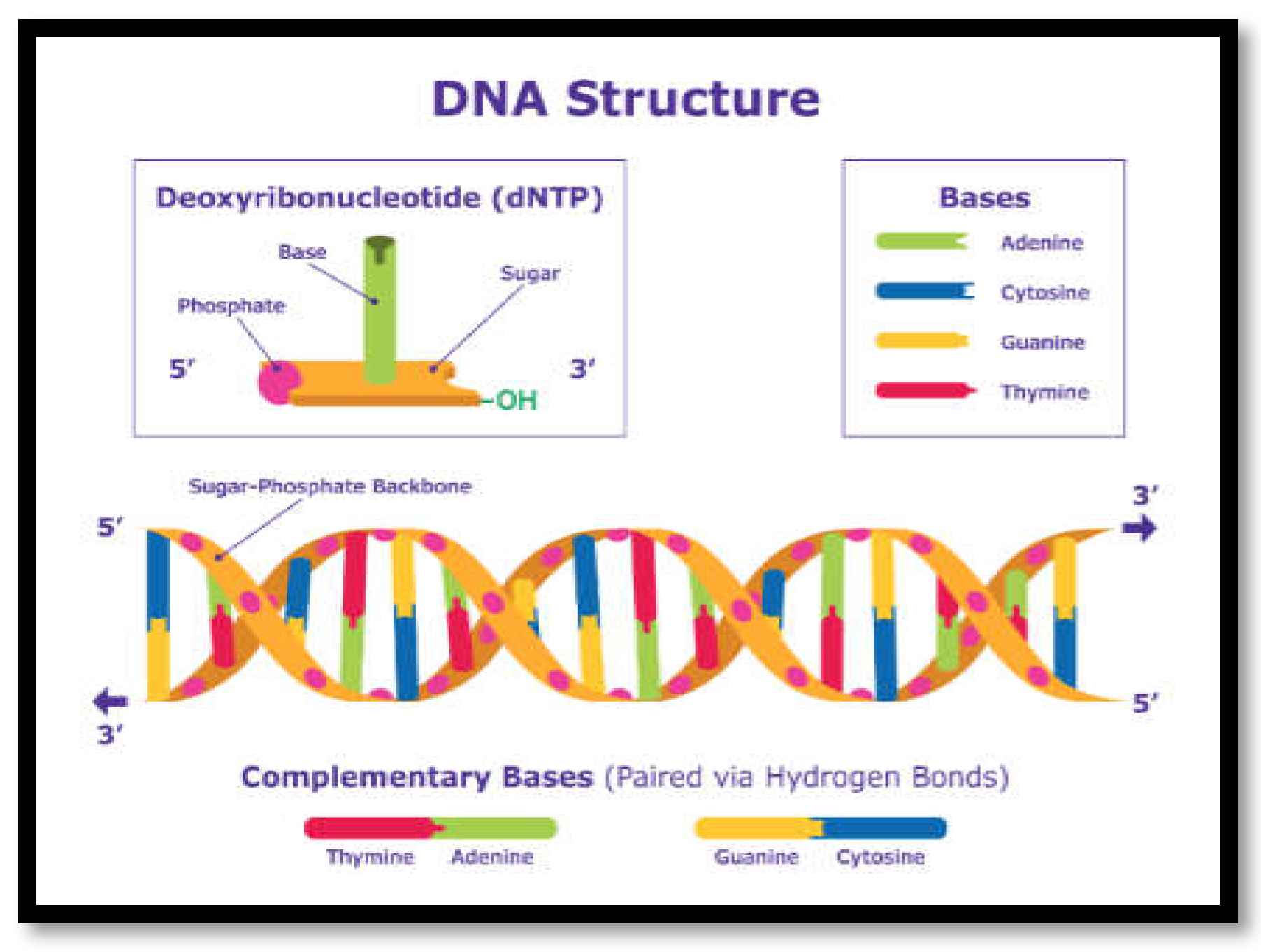
Figure 1.
Schematic structure of DNA. Source: https://www.sigmaaldrich.com.
Figure 1.
Schematic structure of DNA. Source: https://www.sigmaaldrich.com.
The first automatic DNA sequencer, developed by Leroy Hood's laboratory in association with Applied Biosystems (ABI) at Caltech in 1986, marked a significant milestone in the field of genetics [31]. This technology revolutionized DNA sequencing by automating the process, making it faster and more efficient than traditional methods. Today, automatic DNA sequencers are ubiquitous in genetics research and clinical diagnostics, holding a pivotal position in an extensive array of scientific and medical applications. This method utilizes fluorescently tagged ddNTPs and capillary gel electrophoresis is used to separate the fragments. Fluorescence detection generates a chromatogram of the sequence.
Advantages:
Sanger sequencing offers a safer alternative to the Maxam and Gilbert method, with reduced toxicity and lower radioactivity, providing incredibly precise long sequence reads of approximately 700 bp. It is widely applied in sequence-based research.
Disadvantages:
Sanger sequencing generates short DNA fragments (300-1000 nucleotides), with poor quality at sequence ends due to primer binding. Electrophoretic separation is time-consuming. Cloning is necessary, risking vector inclusion in reads. It struggles with accurate readings for similar-length DNA fragments and is costly per base, impractical for whole genome projects.
3.2. Maxam And Gilbert Sequencing
This technique, also referred to as chemical sequencing, necessitates either double-stranded DNA (dsDNA) or single-stranded DNA (ssDNA) labeled with radioactivity, typically at 5' end. Chemical agents are employed to break DNA strand at particular base pairs. The sequence is then determined by dividing distinct fragments based on their sizes.
Principle: It relies on chemical cleavage, where denatured DNA undergoes various chemical processes customized to specific bases. This results in fragments terminating precisely at that particular base. These labeled fragments are then subjected to high-resolution gel electrophoresis and autoradiography for detection.
Procedure: The first step in the procedure is to separate the double-stranded sample DNA into homogenous single-stranded DNA. This single-stranded DNA is labeled with radioactivity at the 5' end, typically achieved through a kinase reaction using gamma-32P ATP. The template DNA is divided into four aliquots, each treated with a distinct chemical. These chemical treatments induce breaks at a small proportion of one or two of the four nucleotide bases in each reaction (G, A+G, C, C+T). For instance, formic acid is used to depurinate purines (A+G), guanines are methylated with dimethyl sulphate, and hydrazine is employed to hydrolyze pyrimidines (C+T). In the C-only reaction, the reaction involving thymine is hindered by adding sodium chloride to hydrazine reaction. Subsequently, hot piperidine is applied at location of modified base to cleave modified DNAs. In order to separate the sizes of the fragments produced by the four processes, they are electrophoresed side by side in denaturing acrylamide gels [30]. The fragments are visible on the gel after autoradiography using X-ray film. This process produces a series of dark bands, each of which represents the position of a single radiolabeled DNA molecule. Reading the sequence from bottom to top (5' - 3') is then performed. The appearance of a band in a specific lane identifies the corresponding nucleotide: Guanine (G) in G and G+A lanes, Cytosine (C) in the C+T and C lanes, Thymine (T) in the C+T lane, and Adenine (A) in the G+A lane.
Advantages: Capable of directly reading purified DNA, utilized for analyzing DNA-protein interactions, employed in the analysis of epigenetic modifications and nucleic acid structure.
Disadvantages: Limited to sequencing short DNA fragments, employs hazardous and radioactive chemicals [32], incapable of reading DNA fragments longer than 500 base pairs.
4. Next Generation Sequencing
Compared to first-generation sequencing techniques, next-generation sequencing (NGS) technologies provide faster and more cost-effective nucleotide sequencing with higher throughput data, enabling population-scale genome research. They represent a fundamentally different approach to sequencing, sparking a revolution in genomic research and yielding numerous groundbreaking discoveries by providing countless insights into the genome, transcriptome, and epigenome of various species. While NGS shares the basic principle of capillary electrophoresis-based Sanger sequencing, it enhances this method by facilitating large-scale parallel sequencing, accurately deciphering millions of fragments of DNA from one sample. NGS can sequence extensive stretches of DNA base pairs, producing in a single sequencing run hundreds of gigabases of data. NGS, also known as Massive Parallel Sequencing [33,34,35,36], has marked a revolutionary advancement in genomics and molecular biology by enabling the rapid sequencing of entire genomes within a single day. NGS technologies present three key advancements over first-generation sequencing methods [37].
- i)
- NGS methods do not require bacterial cloning; instead, they prepare libraries for sequencing in a system without cells.
- ii)
- NGS systems process millions of sequencing processes in parallel and concurrently.
- iii)
- Bases are found in a parallel, cyclical manner. These significant advancements enable scientists to execute whole genome sequencing quickly and inexpensively.
There are four main NGS technology platforms as of 2024.
Roche/454 sequencing: The latest iteration of Roche/454 sequencing, formerly known as 454, is the GS FLX+ sequencer from Roche Diagnostics. This sequencer produces approximately 1,000,000 reads, each containing 700 base pairs (bp). It stands as the initial commercially available NGS platform utilizing pyrosequencing, which follows the notion of sequencing by synthesis. Each time a nucleotide is added to DNA strand, the release of pyrophosphate sets off a sequence of chemical reactions that produce light, making it possible to identify nucleotide that has been incorporated [38].
ABI/SOLiD sequencing: The SOLiD platform, pioneered by Life Technologies, functions based on principle of Sequencing by Oligonucleotide Ligation and Detection (SOLiD) approach [39] leveraging DNA ligase's mismatch sensitivity to determine nucleotide sequence. In this method, fluorescently labeled oligo probes are designed to complement the target DNA strand. When there is a match between the sequence of these probes and the strand being sequenced, DNA ligation occurs. This ligation event produces a fluorescent signal, which is then used to deduce the sequence information.
Ion torrent/ proton sequencing: It is based on semiconductor technology, also adopts the sequencing by synthesis method. As dNTPs are incorporated into a developing DNA strand, hydrogen ions are emitted and captured. This sequence of events transpires within a semiconductor chip. Release of hydrogen ions generates a notably positive voltage within the microenvironment, detectable by a transistor-based apparatus, and subsequently converted into a voltage signal.
Illumina sequencing: It currently stands as the dominant NGS platform in the market, responsible for over 90% of the sequencing data generated globally. Operating on the sequencing by synthesis approach, it detects fluorescence signals as fluorescently labeled dNTPs are added to an evolving DNA chain. Images are captured following each dNTP incorporation reaction, and high-quality sequence data is derived by analyzing and processing these images. Illumina acquired Solexa in 2007 [40] and supplies a majority of NGS platforms worldwide. Illumina's HiSeq X TEN system, comprising 10 HiSeq X units, represents the latest high-throughput sequencer available.
Next Generation Sequencing Workflow
Instead of sequencing directly, the NGS adds few steps such as:
Construction of the library: The DNA that has to be sequenced is broken up into different lengths of suitable size (50–500 NT) followed by adapter ligation [37].
Clonal amplification: To increase the observable signal from each target during sequencing, the DNA library must first be clonally amplified and attached to a solid substrate. Each distinct DNA molecule in the library is attached to the surface of a bead or flow cell and amplified by PCR to generate a precise set of clones.
Sequence library: Using a sequencing equipment, the full DNA content of the library is sequenced simultaneously. Despite variances among NGS methodologies, they all use an adaptation of sequencing by synthesis technique. This technique reads individual bases along a polymerized strand as they lengthen. The process of starting a new cycle involves identifying the integrated base, synthesizing bases on single-stranded DNA, and removing reactants.
Data analysis: Countless complex data points made up of brief DNA reads are generated by each NGS experiment. Three steps make up the analysis process: primary, secondary, and tertiary analysis. Converting instrument detector raw signals into digital data or base calls is known as primary analysis. These raw data are gathered with every cycle of sequencing. Files comprising base calls arranged into sequencing reads (FASTQ files) and their matching quality ratings (Phred quality score) are produced by primary analysis. After quality-based read trimming and filtering, read alignment to a reference genome or read assembly for new genomes, including variant calling, constitute secondary analysis. A BAM file with aligned readings is one of the stage's main outputs. Tertiary analysis, which entails interpreting results and deriving significant insights from the data, is the most difficult stage.
5. Second Generation Sequencing
The initial wave of sequencing, particularly Sanger sequencing, dominated the market for thirty years, but time and cost were a significant barrier. To overcome the shortcomings of the first generation of sequencers, a new generation has emerged in 2005 and the years that have followed. The essential characteristics of sequencing technology of the second generation comprise:
- i)
- Several millions of short reads are generated simultaneously in parallel.
- ii)
- The sequencing procedure is completed faster than it was in the first generation.
- iii)
- The inexpensive nature of sequencing.
- iv)
- There is no need for electrophoresis since the sequencing output is directly detected.
Second generation sequencing comprises of: Illumina sequencing, ABI/SOLiD Sequencing, Pyrosequencing, Ion Torrent Sequencing.
5.1. Pyrosequencing
Pyrosequencing, also known as emulsion PCR, relies on real-time DNA synthesis monitoring. Bioluminescence is used by this four-enzyme DNA sequencing technique to identify DNA synthesis.
Principle:Pyrosequencing adheres to the "sequencing by synthesis" approach, which identifies the nucleotide incorporation by a DNA polymerase. Named Pyrosequencing due to its reliance on light emission triggered by a chain reaction upon the release of pyrophosphate.
Procedure:
The single-stranded DNA targeted for sequencing is fragmented into approximately 800-1000 base pair fragments [7]. These fragments are added with adapters, creating a library which are then linked to beads. Bead-bound DNA populations undergo emulsion PCR (emPCR) in a water-in-oil emulsion microreactors to encapsulate each bead with a clonal DNA population [41]. The ideal scenario is for one DNA molecule to land on a single bead, with each bead amplifying inside a separate emulsion droplet. Subsequently, beads coated with DNA are transferred onto a picoliter reaction plate, with one bead fitting into each well. In the next step, adenosine 5' phosphosulfate and luciferin substrates are added to the DNA fragments in the wells and they are exposed to enzymes such as ATP sulfurylase, DNA polymerase, and apyrase. DNA polymerase starts nucleotide incorporation onto the single-strand DNA template at the 3' end upon adding one of the four types of nucleotides to the wells, producing pyrophosphate. The enzyme ATP sulfurylase changes the released pyrophosphate into adenosine triphosphate (ATP) when adenosine 5' phosphosulfate is present. Then, ATP contributes to the luciferase-mediated transformation of luciferin into oxyluciferin. A detector captures the light emitted during this process, which correlates with the amount of ATP involved in the conversion [42]. Apyrase breaks down unused nucleotides and ATP, enabling the cycle to restart with a different nucleotide. This cycle repeats, with each nucleotide added sequentially until synthesis is complete. A detector notices the light that is released and utilized to ascertain the type and quantity of nucleotides provided. For instance, the light emitted from identical DNA fragments will be three times brighter if three consecutive cytosine nucleotides are added to it compared to DNA fragments with only one cytosine nucleotide added. The lack of light emitted after adding cytosine indicates that the next base in the single-stranded DNA template must be one of the other three nucleotides.
To prevent false signals from premature luciferase reactions, Deoxyadenosine α-thio triphosphate is used in place of deoxyadenosine triphosphate (A) among the four nucleotides used in pyrosequencing.
Advantages: faster than sanger sequencing, it utilized natural nucleotides rather than extensively modified dNTPs as in chain termination methods, it allowed real-time observation rather than relying on lengthy electrophoresis processes [43,44].
Disadvantages: When the same nucleotide is included simultaneously in homopolymer repetitions, it becomes difficult to sequence them since the light produced cannot be precisely discriminated for longer than six base pairs [45], Low bases per run.
Illumina Sequencing
For quick and accurate sequencing, Illumina NGS technology uses clonal amplification and sequencing by synthesis (SBS) chemistry. In the process, DNA bases are recognized and concurrently incorporated into a chain of nucleic acids.
Principle:
The idea is based on the chemistry of sequencing-by-synthesis, using novel reversible terminator nucleotides for the four bases [46]. A specific fluorescent dye is applied to each nucleotide, and the integration of these nucleotides is done by means of a DNA polymerase enzyme.
Procedure: First, random fragmentation of DNA materials into sequences is performed. Next, adapters are ligated to both ends of each sequence. These adapters bind themselves to their corresponding complementary adapters, which are affixed to a slide containing various versions of complementary adapters. The adapters are bonded to their corresponding complementary adapters, which are attached to a slide that has different complementary adapter versions on a flat surface. In the subsequent phase, "PCR bridge amplification" is utilized to replicate each sequence attached to the solid plate, resulting in multiple identical copies of each sequence [47,48]. A cluster represents a group of sequences originating from the same parent sequence, contains roughly a million copies of the initial sequence in each cluster [49]. To conclude, the identification of each nucleotide in the sequences takes place. A mixture of DNA polymerases, sequencing primers, and the four modified nucleotides is hybridized to the sequences using Illumina's reversible terminators sequencing by synthesis method [35]. These modified nucleotides are then used to elongate the primers via polymerases. Each nucleotide type is labeled with a specific fluorescent tag to ensure uniqueness. The nucleotides have a 3’ hydroxyl group that is inactive, guaranteeing the incorporation of only single nucleotide. A coupled-charge device camera detects the unique light signal that each nucleotide in a cluster emits when it is stimulated by a laser. These signals are converted into a nucleotide sequence by computer systems. The cycle is completed by removing the fluorescently labeled terminator and starting a new cycle with a new incorporation [49].
Compared to other technologies, base calls are directly derived from measurements of signal strength at each cycle, significantly reducing raw error rates. The final outcome is highly accurate base-by-base sequencing that mitigates errors inherent to sequence context. This makes strong base calling possible throughout the genome, particularly in areas with homopolymers and repetitive sequences [35]. This sequencing approach exhibits a 1% total error rate.
Advantages: produces large data at low cost per base. Illumina platforms have a 99.9% accuracy rate [50].
Limitation: longer run time. Only short length is sequenced. the data analysis sometimes can be complex and challenging. Nucleotide substitutions are the most common type of mistake in this technique [51].
5.2. Ion Torrent Sequencing:
It depends on the detection of hydrogen ions generated during DNA polymerization [52]. Ion semiconductor sequencing does not require optical detection or altered nucleotides, in contrast to previous sequencing-by-synthesis methods. It is also known by various names such as silicon sequencing, semiconductor sequencing, pH-mediated sequencing
Principle:
According to the "sequencing by synthesis" theory, ion semiconductor sequencing creates a complementary DNA strand by sequencing a template strand. In the process of adding additional nucleotides to the expanding DNA template, hydrogen ions are produced as a byproduct. An ion sensor, which functions similarly to a pH meter, can detect and measure the voltage caused by this release, which changes the pH of the solution.
Procedure: DNA fragmentation and adaptor attachment to the ends of the pieces of DNA signals start of the process. These adapted DNA libraries are linked to beads via adapter sequences. A clonal population of DNA is encapsulated in each bead during emulsion PCR (emPCR), which is carried out in a water-in-oil emulsion. After emPCR, beads containing clonally amplified DNA templates are transferred to a chip with millions of microwells, where each microwell typically accommodates only one bead. One kind of unmodified deoxyribonucleotide triphosphate (dNTP) is provided for each microwell [53]. The dNTP is integrated into the developing complementary strand when it aligns with the template strand's leading nucleotide in the microwell. This incorporation releases a hydrogen ion [54,55], causing variation in pH that activates ISFET (ion-sensitive field-effect transistor) sensor within complementary metal-oxide-semiconductor (CMOS) sequencing chip [52]. The ISFET sensor detects these pH changes as signals indicating successful nucleotide incorporation during DNA sequencing. Both incorporation and a biological reaction will not occur if the dNTP is not complementary. Before introducing the next nucleotide, any unincorporated nucleotides are removed through washing. Voltage changes occur upon the incorporation of correct nucleotide. There is a voltage doubling when two neighboring nucleotides integrate the same nucleotide, releasing two hydrogen ions. There is no necessity of intermediate signal processing because the sequence of electrical pulses that is sent from the device to a computer is instantly translated into a DNA sequence. It is not necessary to use tagged nucleotides or optical measurements because nucleotide incorporation events can be directly detected by electronics. Software can then be used for DNA assembly and signal processing.
Advantages: short run time of this technique allows for repeated runs to generate more data within short period of time, incorporate significantly greater read lengths at a reduced cost [56].
Disadvantages: Ion Torrent's sequencing technology falls in between short and long read length NGS technologies, with a read length of 200 bp. Massive data generation benefits short read technologies, however Ion Torrent lags behind in terms of overall data output. As such, Ion Torrent needs to prove that it is a stand-alone sequencing method appropriate for large-scale de novo sequencing initiatives.
5.3. ABI/Solid Sequencing
Life Technologies created detection and oligonucleotide ligation sequencing devices [39], which have been commercially accessible since 2006.With this next-generation technology, 108–109 short sequence reads can be produced simultaneously. Two base encoding is used to decode the raw data generated by the sequencing platform into sequence data.
Principle: This method relies on the Sequencing by Oligonucleotide Ligation and Detection technique, which uses DNA ligase's sensitivity to mismatches to determine the nucleotide sequence. Fluorescence-tagged oligo probes ligate DNA when their sequences match the strand to be sequenced, resulting in a fluorescence signal that may be utilized to show the sequence information.
Procedure: The DNA that has to be sequenced is broken up into pieces and are attached to a universal P1 adapter sequence, ensuring each fragment has an identical and known starting sequence. These library of DNA fragments are attached to a magnetic bead, which undergoes emulsion PCR resulting in clonal populations where only one type of fragment is present per bead. Emulsion PCR occurs in microreactors containing all the essential PCR reagents.
In the sequencing process, beads containing the final PCR products are placed onto a glass slide. DNA fragments are then methodically joined to 8-mers that have a fluorescent label at the end, and the color that the label emits is then recorded. The information is then displayed in color space, with the nucleotide sequence represented by four fluorescent hues, which correspond to 16 possible combinations of two bases. 2 Every time the sequencer performs this ligation cycle, the complimentary strand is removed, and a new sequencing cycle is started from position n-1 of the template. The cycle continues until each base has been sequenced twice [57].
Advantages: Sequencing methods that utilize successive offset primers, which are spaced less than one base pair apart, yield more precise data compared to other techniques. The accuracy percentage is 94.94% [58]. This heightened accuracy is primarily attributed to the sequencing of each nucleotide on the template twice. Consequently, miscalling a single nucleotide polymorphism (SNP) requires two adjacent colors to be inaccurately identified, a scenario that is rare.
Disadvantages: This sequencing method typically generates lower data output and shorter read lengths, often requiring around 6 to 7 days to complete a full run, particularly for larger genomes. Additionally, palindromic sequences pose challenges for accurate sequencing when employing the machine's ligation-based technique [59]. The substitution error is the most common one.
6. Third Generation Sequencing
The advent of the second generation of sequencing technology has transformed DNA analysis, becoming the preferred method over the first generation. However, PCR amplification is frequently required for second-generation sequencing (SGS) methods, which is expensive and time-consuming. It also became evident that genomes are extremely complex, with many repeating areas that are unresolvable using SGS methods. Moreover, the comparatively short reads exacerbate challenges of genome assembly. To overcome these limitations, scientists have introduced "third-generation sequencing," a novel approach that offers streamlined sample preparation and cost-effective sequencing that does not require PCR amplification. It includes Pac Bio Sequencing, Helicos Sequencing.
6.1. Pac Bio Sequencing
Among the frequently employed third-generation sequencing methods is Pacific Biosystems (PacBio), which utilizes Single Molecule Real Time (SMRT) sequencing [37,60]. In contrast to the preceding two generations, PacBio's long-read sequencing facilitated by SMRT Sequencing technology eliminates the necessity for PCR amplification, while boasting read lengths that are 100 times longer than those achieved with Next-Generation Sequencing (NGS). This real-time sequencing technique by PacBio eradicates the need for interruptions between reading processes [61].
Principle:
Zero mode waveguides (ZMWs), constructed from thin metallic films as subwavelength optical nanostructures, serve as potent analytical tools [62]. Their capability to confine excitation volumes to attoliters facilitates the isolation of individual molecules, allowing optical examination at physiologically significant concentrations of fluorescently labeled biomolecules. In the context of PacBio SMRT sequencing, the concept revolves around incorporating arrays of these nanostructures into devices for real-time analysis of numerous single-molecule reactions or binding events.
Procedure:
Pacific Biosciences has developed Single Molecule Real Time DNA Sequencing (SMRT) technology and offers the PacBio RS II sequencer [61]. The SMRT sequencer utilizes Zero Mode Waveguides (ZMWs), each containing approximately 150,000 ultra-microwells. Individual DNA polymerase molecules are attached to the underside of these wells using the biotin-streptavidin technique. A, C, G, and T are fluorescently labeled nucleotides that are incorporated by DNA polymerase during sequencing. These nucleotides are identified by their distinct colors and bind to the single-stranded template DNA that has been immobilized inside the well. As phosphodiester bonds form, resulting in nucleotide incorporation, the emitted fluorescence ceases as the dye diffuses out. ZMWs are continuously observed with CCD cameras, which record a series of pulses that are then transformed into single molecular traces that correspond to the template sequence. The unique quality of this platform lies in its ability to simultaneously add and measure all four nucleotides in real time, resulting in accelerated genome assembly compared to alternative methods. A read length of 900 base pairs yields a reported accuracy of 99.3% [63].
Advantages: Since it operates on single molecule real-time sequencing principles, there's no need for PCR amplification. Quicker runtime (usually a few hours).
Disadvantages: Each SMRT cell produced by this approach yields between 70 and 140 MB of data, with variations based on the GC concentration of the template DNA, a notably lower output compared to alternative methods. Additionally, it experiences random insertion and deletion errors, as noted by Mardis (2011). Moreover, the machine is relatively sizable and comes with a hefty price tag [64].
6.2. Helicos Sequencing
Helicos Single Molecule Sequencing (SMS) provides precise quantity and sequence data by unbiased direct sequencing of cellular nucleic acids, providing a unique viewpoint on genome biology. This was the pioneering technology that enabled DNA sequencing without the need for amplification, effectively bypassing any biases and errors typically associated with amplification processes [24,61].
Principle:
DNA polymerase is used in Helicos single-molecule sequencing, which combines sequencing by hybridization and sequencing by synthesis. In this procedure, individual nucleic acid molecules are broken down, melted into single strands, and then poly-A tailed in the case of genomic DNA.
Procedure:
After being broken up into single strands, the DNA destined for sequencing is given a poly-A tail at the 3' end. These ready-made DNA strands are adhered to a glass Helicos Flow Cell that has oligo-dT-50 oligonucleotide coating applied. To finish the nucleotides complementary to the poly-A tail, dTTP and polymerase are next added to the flow cell. Following this fill-in step, sequencing by synthesis is initiated by the addition of fluorescently labeled dCTP, dGTP, and dATP reversible terminator nucleotides, which are referred to as virtual terminators [65] one at a time. These terminator nucleotides bind as a single complementary nucleotide and block further extension until the terminator is cleaved. With the assistance of a DNA polymerase, nucleotide incorporation occurs at the corresponding position in each of the separate developing DNA strands.
Unincorporated nucleotides are washed through the flow cell after inclusion. Following laser illumination of the flow-cell surface, inclusion is identified by the fluorescent light emission. To ensure accurate conversion of images into individual DNA molecules and capture A, T, G, or C nucleotide sequence data, the HeliScope Sequencer employs a CCD camera. This camera captures images, recording details such as strands that have integrated a nucleotide, positional information, and cycle details. The terminator moiety of the inserted nucleotide is broken after the pictures are taken, enabling the addition of the subsequent complimentary nucleotide. The fluorescent label separates from the nucleotide after hundreds of photos representing every channel in the flow cell have been taken, enabling the device to move on to the subsequent nucleotide in the addition cycle. The HeliScope Sequencer does 120 cycles of distinct nucleotide additions in a typical run. After real-time image processing is finished, researchers can get sequence file and start aligning it to relevant reference transcriptome or genomic sequences. This file encapsulates both the position of the DNA strand and the addition of nucleotide strings.
Advantages: Produces data at a low cost. Facilitates direct sequencing of RNA molecules from cells, eliminating the necessity for reverse transcription or amplification, as well as mitigating biases and inaccuracies induced by cDNA synthesis and PCR amplification.
Disadvantages:
high error rates and short read lengths, which can be mitigated by repeating the sequencing process but eventually raise the cost per base for a certain degree of precision.
7. Fourth Generation Sequencing:
7.1. Nanopore Sequencing:
Since Oxford Nanopore Technologies (ONT) unveiled the MinION sequencer in 2014, nanopore sequencing technology has developed dramatically, becoming one of the most powerful sequencing technologies available today and able to detect single molecules. There are numerous possible applications for it, including the study of ions, medicines, polymers, proteins, DNA, RNA, peptides and macromolecules. The single molecule nanopore technology field is on the brink of DNA sequencing technology advancement, marking the culmination of three generations of progress. Particles that are marginally smaller than the pore size are directed through the pore by applying an external voltage. There are two main categories into which nanopore technologies can be classified: solid-state and biological.
Transmembrane protein channels, another name for biological nanopores, are frequently integrated into substrates like polymer films, liposomes, and planar lipid bilayers. Of these, a-Hemolysin (a-HL), also known as a-toxin, is the most prominent and widely used biological nanopore, significantly improving DNA sequencing capabilities. Biological nanopores include Bacteriophage phi29 [66] and Mycobacterium smegmatis porin A (MspA)[67]. Solid-state nanopores includes silicon dioxide (SiO2)[68], silicon nitride (Si3N4)[69], boron nitride (BN)[70], aluminium oxide (Al2O3)[71], graphene [72] and polymer membranes [73] with microfabrication technologies.
Utilizing nanopores in ultra-thin membranes offers several advantages, as the minimum thickness of 0.335 nm aligns perfectly with the distance in a DNA chain between two nucleotides [74]. This setup presents significant benefits for DNA sequencing, potentially achieving exceptionally high spatial resolution with single-layer membranes. Solid-state nanopores have become a cutting-edge method in several domains, such as molecular translocation processes, DNA sequencing, protein detection, and disease diagnosis. Furthermore, it has been suggested that hybrid nanopores combine the advantages of solid-state and biological nanopores.
Principle: Applying voltage across the membrane and tracking the ionic current flowing through the nanopore are the key components of the detecting process. Particles that are only a little bit smaller than the pore size interrupt the current level when the nanopore is at the molecular scale, producing a discernible signal.
Procedure:
This technique utilizes a "nanopore," a minute protein pore that serves as a biosensor and is embedded in an electrically resistive polymer membrane. Sequencing DNA involves directing DNA fragments through a protein nanopore, which can be either a synthetic material or a protein-made nanoscale aperture [75]. An electric current flows through the protein pore as DNA is directed through it during this procedure. A voltage blockage happens as the DNA passes through the pore with the help of a secondary motor protein, which modifies the current that passes through the pore. Nanopore sequencing relies on the principle that as individual nucleotides pass through the pore, they alter the ionic current, generating signals that are time-specific and examined in real-time [76]. An ionic current produced by constant voltage in an electrolytic solution propels single-stranded DNA or RNA molecules, which carry negative charges, through the nanopore from the negatively charged "cis" side to the positively charged "trans" side. A motor protein moves the nucleic acid molecule through the nanopore gradually to control the rate of translocation. The specific type of molecule traversing the pore influences the amount of ion current flow [77]. Computer methods that analyze changes in ionic current during translocation to match the nucleotide sequence within the sensing area enable real-time single-molecule sequencing. The motor protein also exhibits helicase activity, which controls translocation speed and allows double-stranded DNA or RNA-DNA duplexes to unwind into single-stranded molecules that pass through the nanopore. The efficiency of nanopore sequencing based platforms is determined by the stability, pore shape, process speed, and signal detecting system properties.
Advantages: Sequencing single molecules in real-time comes at a low cost, typically ranging from $25 to $40 per gigabase of sequence. This technology is capable of reading exceptionally long DNA molecules in a single read.
Disadvantages: DNA sequencing using nanopores at the single-molecule level faces challenges due to limited sequencing accuracy, with an error rate of approximately 12%. This error rate is distributed, with around 3% mismatches, 4% insertions, and 5% deletions [78]. Achieving ultra-precise and high-speed DNA detection beyond the capabilities of current optical as well as electrical technologies presents a major challenge for this sequencing [79].

Table 1.
Summary of various sequencing technologies.
| Sequencing types | Year | Sequencing Chemistry |
Read lengths | Sequencing run time | Data generated | Advantages | Disadvantages |
|---|---|---|---|---|---|---|---|
| Sanger sequencing | 1977 | Chain termination | 700bp | 20 minutes – 3 hrs | 1.9-84 Kb/run | Gold standard method | Limited throughput Not cost effective |
| Maxam gilbert sequencing | 1977 | Chemical sequencing | 500bp | NA | NA | Capable to read purified DNA | Uses hazardous and radioactive chemicals |
| Pyrosequencing |
2005 | Sequencing by synthesis | 700-1000bp | 23 hrs | 700 MB/run | Real time sequencing Longer read length | Homopolymer reads |
| Illumina | 2006 | Reversible dye terminators | 50-250bp | 11-14 days | 600 GB/run |
Large data Low cost per base |
Longer run time Short read length |
| Ion torrent | 2010 | H+ ion detection | 200bp – 600bp | 2.5-4 hrs | 10-1000MB (depends upon chip used) | Short run time | Short read length |
| ABI/Solid | 2007 | Sequencing by ligation | 75bp | 6-7 days | 30 GB/ run | Higher accuracy | Less data output Longer run time |
| Pacbio | 2011 | ZMW single molecule | 15-25 kb | 12-30 hrs | 70-140 MB/ cell | Real time sequencing Long read length |
Less data output |
| Helicos | 2007 | Helicos single molecule | 25bp | 5-10 days | 21-35 GB/ run | Generates big data Low cost |
High error rate |
| Nanopore | 2019 | Nanopore with DNA transistor | 13-20 kb | 3 days | Several gigabytes | Real time sequencing Low cost |
Practically to be proven |
8. Conclusion
The emergence of NGS (Next-Generation Sequencing) has transformed the study of biological systems to unprecedented levels. Approximately 50 years ago, the first sequencing technology emerged, laying the groundwork for subsequent advancements. Since the inception of NGS in 2005, these technologies have continually evolved. Notably, the transition from the initial Sanger technique to modern NGS methods, with Illumina's technology leading the way, has been remarkable for its rapid DNA and RNA sequencing capabilities. Over the past decade, new sequencing technologies have made significant strides. Second-generation sequencing has significantly transformed DNA analysis compared to earlier methods and is now extensively employed.
However, the polymerase chain reaction (PCR) amplification steps inherent in second-generation sequencing are both time-consuming and costly, and the short read lengths they produce can complicate genome assembly. Third-generation sequencing approaches have addressed some of these limitations by obviating the need for amplification during sequencing, thereby reducing errors and enabling single-molecule sequencing. These methods facilitate cost-effective assembly of billions of sequences with high throughput, supporting numerous research endeavors in biological sequence analysis.
Despite advancements, challenges persist with Illumina-NGS technologies, such as data acquisition and storage issues, as well as complexities in data analysis and interpretation. As newer NGS platforms emerge, they are anticipated to generate even larger datasets, necessitating the development of advanced techniques and software for efficient data processing. Though the Sanger method remains a traditional approach in genome studies, the utilization of NGS methods has been steadily increasing alongside technological advancements.
Continuous technological improvements and decreasing costs are making NGS more accessible and ubiquitous, enabling its seamless incorporation into everyday clinical, research, agricultural, and environmental applications. The future of NGS looks bright, promising to explore new realms of understanding and drive progress with significant implications for human health, agriculture, environmental preservation, and beyond.
References
- Watson, J.D.; Crick, F.H. Molecular structure of nucleic acids: a structure for deoxyribose nucleic acid. Nature 1953, 171, 737-738. [CrossRef]
- Le Tourneau, Christophe, and Maud Kamal, eds. Pan-cancer integrative molecular portrait towards a new paradigm in precision medicine. Springer International Publishing 2015, 12306.
- anger, F.; Nicklen, S.; Coulson, A. R. DNA sequencing with chain-terminating inhibitors. Proceedings of the National Academy of Sciences 1977, 74, 5463-5467.
- Maxam, A.M.; Gilbert, W.A. A new method for sequencing DNA. Proceedings of the National Academy of Sciences 1977, 74, 560-564.
- Fleischmann, R.D.; Adams, M.D.; White, O.; Clayton, R.A.; Kirkness, E.F.; Kerlavage, A.R. Whole-genome random sequencing and assembly of Haemophilus influenzae Rd. Science 1995, 269, 496-512.
- Tripp, S.; Grueber, M. Economic impact of the human genome project. Battelle Memorial Institute 2011, 58, 1-58.
- Gupta, A.K.; Gupta, U.D. Next generation sequencing and its applications. Animal Biotechnology 2020, 2 nd Edn., Academic Press, 395-421.
- Thomas, R. K.; Nickerson, E.; Simons, J. F.; Jänne, P. A.; Tengs, T.; Yuza, Y.; Meyerson, M. Sensitive mutation detection in heterogeneous cancer specimens by massively parallel picoliter reactor sequencing. Nature Medicine 2006, 12, 852-855. [CrossRef]
- Ley, T. J.; Mardis, E. R.; Ding, L.; Fulton, B.; McLellan, M. D.; Chen, K.; Wilson, R. K. DNA sequencing of a cytogenetically normal acute myeloid leukaemia genome. Nature 2008, 456, 66-72.
- Ng, S. B.; Buckingham, K. J.; Lee, C.; Bigham, A. W.; Tabor, H. K.; Dent, K. M.; Bamshad, M. J. Exome sequencing identifies the cause of a mendelian disorder. Nature Genetics 2010, 42, 30-35. [CrossRef]
- Andries, K.; Verhasselt, P.; Guillemont, J.; Göhlmann, H. W.; Neefs, J. M.; Winkler, H.; Jarlier, V. A diarylquinoline drug active on the ATP synthase of Mycobacterium tuberculosis. Science 2005, 307, 223-227. [CrossRef]
- Wheeler, D. A.; Srinivasan, M.; Egholm, M.; Shen, Y.; Chen, L.; McGuire, A.; Rothberg, J. M. The complete genome of an individual by massively parallel DNA sequencing. Nature 2008, 452, 872-876. [CrossRef]
- Lupski, J. R.; Reid, J. G.; Gonzaga-Jauregui, C.; Rio Deiros, D.; Chen, D. C.; Nazareth, L.; Gibbs, R. A. Whole-genome sequencing in a patient with Charcot–Marie–Tooth neuropathy. New England Journal of Medicine 2010, 362, 1181-1191.
- Schuster, S. C.;Miller, W.; Ratan, A.; Tomsho, L. P.; Giardine, B.; Kasson, L. R.; Hayes, V. M. Complete Khoisan and Bantu genomes from southern Africa. Nature 2010, 463, 943-947. [CrossRef]
- Venter, J. C.; Remington, K.; Heidelberg, J. F.; Halpern, A. L.; Rusch, D.; Eisen, J. A.; Smith, H. O. Environmental genome shotgun sequencing of the Sargasso Sea. Science 2004, 304, 66-74. [CrossRef]
- Sogin, M. L.; Morrison, H. G.; Huber, J. A.; Welch, D. M.; Huse, S. M.; Neal, P. R.; Herndl, G. J. Microbial diversity in the deep sea and the underexplored “rare biosphere”. Proceedings of the National Academy of Sciences 2006, 103, 12115-12120.
- Noonan, J. P.; Hofreiter, M.; Smith, D.; Priest, J. R.; Rohland, N.; Rabeder, G.; Rubin, E. M. Genomic sequencing of Pleistocene cave bears. Science 2005, 309, 597-599. [CrossRef]
- Green, R. E.; Krause, J.; Ptak, S. E.; Briggs, A. W.; Ronan, M. T.; Simons, J. F.; Pääbo, S. Analysis of one million base pairs of Neanderthal DNA. Nature 2006, 444, 330-336. [CrossRef]
- Holley, R.W.; Apgar, J.; Everett, G.A.; Madison, J.T.; Marquisee, M.; Merrill, S.H.; Penswick, J.R.; Zamir, A. Structure of a ribonucleic acid. Science 1965, 147,1462-1465.
- Jou, W.M.; Haegeman, G.; Ysebaert, M.; Fiers, W. Nucleotide Sequence of the Gene Coding for the Bacteriophage MS2 Coat Protein. Nature 1972, 237, 82-88. [CrossRef]
- Sanger, F.; Air, G. M.; Barrell, B. G.; Brown, N. L.; Coulson, A. R.; Fiddes, J. C.; Smith, M. Nucleotide sequence of bacteriophage φX174 DNA. Nature 1977, 265, 687-695. [CrossRef]
- Martin, A.C.; Goldstein, B. Apical constriction: themes and variations on a cellular mechanism driving morphogenesis. Development 2014, 141, 1987-1998. [CrossRef]
- Thudi, M.; Li, Y.; Jackson, S.A.; May, G.D.; Varshney, R.K. Current State-of-Art of Sequencing Technologies for Plant Genomics Research. Briefings In Functional Genomics 2012, 11, 3-11. [CrossRef]
- Pareek, C.S.; Smoczynski, R.; Tretyn, A. Sequencing technologies and genome sequencing Journal of applied genetics 2011, 52, 413–435.
- Sanger, F.; Coulson, A.R. A rapid method for determining sequences in DNA by primed synthesis with DNA polymerase. Journal of Molecular Biology 1975, 94, 441-448. [CrossRef]
- Sanger, F.; Coulson, A.R.; Barrell, B.G.; Smith, A.J.; Roe, B.A. Cloning in single stranded bacteriophage as an aid to rapid dna sequencing. Journal of Molecular Biology 1980, 143, 161-178. [CrossRef]
- Durbin, R.M. A map of human genome variation from population scale sequencing. Nature 2010, 467, 1061-73.
- Chidgeavadze, Z.G.; Beabealashvilli, R.S.; Atrazhev, A.M.; Kukhanova, M.K.; Azhayev, A.V.; Krayevsky, A.A. 2', 3'-Dideoxy-3'aminonucleoside 5'-triphosphates are the terminators of DNA synthesis catalyzed by DNA polymerases. Nucleic Acids Research 1984, 12, 1671–1686.
- Masoudi-Nejad, A.; Narimani, Z.; Hosseinkhan, N. Next generation sequencing and sequence assembly: methodologies and algorithms. Springer Science & Business Media 2013, Vol. 4.
- El-Metwally, S.; Ouda, O. M.; Helmy, M. Next generation sequencing technologies and challenges in sequence assembly. Springer Science & Business 2014, Vol. 7.
- Smith, L.M.; Sanders, J.Z.; Kaiser, R.J.; Hughes, P.; Dodd, C.; Connell, C.R.; Heiner, C.; Kent, B.H.; Hood, L.E. Fluorescence detection in automated DNA sequence analysis. Nature 1986, 321, 674 – 679. [CrossRef]
- Moti, T. B.; Guyassa, C.; Dima, C.; Home, M. R. I. Illumina sequencing technology review. Image, 2022, 10.
- Margulies, M.; Egholm, M.; Altman, W. E.; Attiya, S.; Bader, J. S.; Bemben, L. A.; Rothberg, J. M. Genome sequencing in microfabricated high-density picolitre reactors. Nature 2005, 437, 376-380. [CrossRef]
- Shendure, J., Porreca, G. J., Reppas, N. B., Lin, X., McCutcheon, J. P., Rosenbaum, A. M.; Church, G. M. Accurate multiplex polony sequencing of an evolved bacterial genome. Science 2005, 309, 1728-1732. [CrossRef]
- Bentley, D. R.; Balasubramanian, S.; Swerdlow, H. P.; Smith, G. P.; Milton, J.; Brown, C. G.; Roe, P. M. Accurate whole human genome sequencing using reversible terminator chemistry. Nature 2008, 456, 53-59. [CrossRef]
- Drmanac, R.; Sparks, A. B.; Callow, M. J.; Halpern, A. L.; Burns, N. L.; Kermani, B. G.; Reid, C. A. Human genome sequencing using unchained base reads on self-assembling DNA nanoarrays. Science 2010, 327, 78-81. [CrossRef]
- Van Dijk, E.L.; Auger, H.; Jaszczyszyn, Y.; Thermes, C. Ten years of next-generation sequencing technology. Trends in Genetics 2014, 30, 418-426. [CrossRef]
- Nyrén, P.; Pettersson, B.; Uhlén, M. Solid phase DNA minisequencing by an enzymatic luminometric inorganic pyrophosphate detection assay. Analytical Biochemistry 1993, 208, 171-175. [CrossRef]
- Shendure, J.; Ji, H. Next-generation DNA sequencing. Nature Biotechnology 2008, 26, 1135-1145.
- Balasubramanian, S. Solexa sequencing: Decoding genomes on a population scale. Clinical Chemistry 2015, 61, 21-24. [CrossRef]
- Tawfik, D.S.; Griffiths, A.D. Man-made cell-like compartments for molecular evolution. Nature Biotechnology 1998, 16, 652-656. [CrossRef]
- Nyrén, P.; Lundin, A. Enzymatic method for continuous monitoring of inorganic pyrophosphate synthesis. Analytical Biochemistry 1985, 151, 504-509. [CrossRef]
- Ronaghi, M.; Karamohamed, S.; Pettersson, B.; Uhlén, M.; Nyrén, P. Real-time DNA sequencing using detection of pyrophosphate release. Analytical biochemistry 1996, 242, 84-89. [CrossRef]
- Ronaghi, M.; Uhlén, M.; Nyrén, P. A sequencing method based on real-time pyrophosphate. Science 1998, 281, 363-365. [CrossRef]
- Huse, S. M.; Huber, J. A.; Morrison, H. G.; Sogin, M. L.; Welch, D. M. Accuracy and quality of massively parallel DNA pyrosequencing. Genome Biology 2007, 8, 1-9. [CrossRef]
- Goodwin, S.; McPherson, J. D.; McCombie, W. R. Coming of age: ten years of next-generation sequencing technologies. Nature Reviews Genetics 2016, 17, 333-351. [CrossRef]
- Voelkerding, K. V.; Dames, S. A.; Durtschi, J. D. Next-generation sequencing: from basic research to diagnostics. Clinical Chemistry 2009, 55, 641-658. [CrossRef]
- Adessi, C.; Matton, G.; Ayala, G.; Turcatti, G.; Mermod, J.J.; Mayer, P.; Kawashima, E. Solid phase DNA amplification: characterisation of primer attachment and amplification mechanisms. Nucleic Acids Research 2000, 28, 87-e87.
- Chen, R.; Im, H.; Snyder, M. Whole-exome enrichment with the Agilent sure select human all exon platform. Cold Spring Harbor Protocols 2015, 7, 3659. [CrossRef]
- Morey, M.; Fernández-Marmiesse, A.; Castiñeiras, D.; Fraga, J. M.; Couce, M. L.; Cocho, J. A. A glimpse into past, present, and future DNA sequencing. Molecular Genetics and Metabolism 2013, 110, 3-24.
- Dohm, J.C.; Lottaz, C.; Borodina, T.; Himmelbauer, H. Substantial biases in ultra-short read data sets from high-throughput DNA sequencing. Nucleic Acids Research 2008, 36, 105. [CrossRef]
- Rothberg, J.M.; Hinz, W.; Rearick, T.M.; Schultz, J.; Mileski, W.; Davey, M.; Leamon, J.H.; Johnson, K.; Milgrew, M.J.; Edwards, M.; Hoon, J. An integrated semiconductor device enabling non-optical genome sequencing. Nature 2011, 475, 348-352. [CrossRef]
- Pennisi, E. Semiconductors Inspire New Sequencing Technologies. Science 2010, 327, 1190. [CrossRef]
- Alberts, B.; Bray, D.; Lewis, J.; Raff, M.; Roberts, K.; Watson, J. D. Molecular biology of the cell, 3 rd Edn.; Garland Publishers: New York, 1994; pp. 966-996.
- Purushothaman, S.; Toumazou, C.; Ou, C.P. Protons and single nucleotide polymorphism detection: A simple use for the Ion Sensitive Field Effect Transistor. Sensors and Actuators B: Chemical 2006, 114, 964-968. [CrossRef]
- Diekstra. A.; Bosgoed, E.; Rikken, A.; van Lier, B.; Kamsteeg, E.J.; Tychon, M.; Derks, R.C.; van Soest, R.A.; Mensenkamp, A.R.; Scheffer, H.; Neveling, K.; Nelen, M.R. Translating sanger-based routine DNA diagnostics into generic massive parallel ion semiconductor sequencing. Clinical Chemistry 2015, 61,154-62. [CrossRef]
- Mardis, E.R. Next-generation DNA sequencing methods. Annual Review of Genomics and Human Genetics 2008, 9, 387-402.
- Liu, L.; Li, Y.; Li, S.; Hu, N.; He, Y.; Pong, R.; Lin, D.; Lu, L.; Law, M. Comparison of next-generation sequencing systems. Journal of Biomedicine and Biotechnology, 2012, 251364. [CrossRef]
- Huang, Y. F.; Chen, S. C.; Chiang, Y. S.; Chen, T. H.; Chiu, K. P. Palindromic sequence impedes sequencing-by-ligation mechanism. In BMC Systems Biology 2012, 6,1-7. [CrossRef]
- Eid. J.; Fehr, A.; Gray, J.; Luong, K.; Lyle. J.; Otto, G.; Peluso, P.; Rank, D.; Baybayan, P.; Bettman, B. Real-time DNA sequencing from single polymerase molecules. Science 2009, 323, 133–138. [CrossRef]
- Schadt, E.E.; Turner, S.; Kasarskis, A. A Window into Third-Generation Sequencing. Human Molecular Genetics 2010, 19, 227-240. [CrossRef]
- Levene, M. J.; Korlach, J.; Turner, S. W.; Foquet, M.; Craighead, H. G.; Webb, W. W. Zero-mode waveguides for single-molecule analysis at high concentrations. Science, 2003, 299, 682-686. [CrossRef]
- Metzker, M.L. Sequencing technologies—the next generation. Nature Reviews Genetics 2010, 11, 31-46.
- Pavlovic, S.; Klaassen, K.; Stankovic, B.; Stojiljkovic, M.; Zukic, B. Next-generation sequencing: The enabler and the way ahead. Microbiomics, 2020, 16, 175-200.
- Bowers, J.; Mitchell, J.; Beer, E.; Buzby, P.R.; Causey, M.; Efcavitch, J.W.; Jarosz, M.; Krzymanska-Olejnik, E.; Kung, L.; Lipson, D.; Lowman, G.M. Virtual terminator nucleotides for next-generation DNA sequencing. Nature Methods, 2009, 6, 593-595. [CrossRef]
- Wendell, D.; Jing, P.; Geng, J.; Subramaniam, V.; Lee, T. J.; Montemagno, C.; Guo, P. Translocation of double-stranded DNA through membrane-adapted phi29 motor protein nanopores. Nature Nanotechnology 2009, 4, 765-772. [CrossRef]
- Branton, D.; Deamer, D.W.; Marziali, A.; Bayley, H.; Benner, S.A.; Butler, T.; Schloss, J. A. The potential and challenges of nanopore sequencing Nature Biotechnology 2008, 26, 1146–53.
- Storm, A.J.; Chen, J.H.; Ling, X.S.; Zandbergen, H.W.; Dekker, C. Fabrication of solid-state nanopores with single–nanometre precision. Nature Materials 2003, 2, 537–40. [CrossRef]
- Heng, J. B.; Ho, C.; Kim, T.; Timp, R.; Aksimentiev, A.; Grinkova, Y. V.; Timp, G. Sizing DNA using a nanometer-diameter pore. Biophysical Journal 2004, 87, 2905-2911. [CrossRef]
- Liu, S.; Lu, B.; Zhao, Q.; Li, J.; Gao, T.; Chen, Y.; Yu, D. Boron nitride nanopores: highly sensitive DNA single-molecule detectors. Advanced Materials 2013, 25, 4549-4554. [CrossRef]
- Venkatesan, B.M.; Bashir R. Nanopore sensors for nucleic acid analysis. Nature Nanotechnology 2011, 6, 615–24.
- Garaj, S.; Hubbard, W.; Reina, A.; Kong, J.; Branton, D.; Golovchenko, J.A. Graphene as a subnanometre trans-electrode membrane. Nature 2010, 467, 190–3. [CrossRef]
- Menestrina, J.; Yang, C.; Schiel, M.; Vlassiouk, I.; Siwy, Z. S. Charged particles modulate local ionic concentrations and cause formation of positive peaks in resistive-pulse-based detection. The Journal of Physical Chemistry C 2014, 118, 2391-2398. [CrossRef]
- Traversi, F.; Raillon, C.; Benameur, S.M.; Liu, K.; Khlybov, S.; Tosun, M.; Detecting the translocation of DNA through a nanopore using graphene nanoribbons. Nature Nanotechnology 2013, 8,939–45. [CrossRef]
- Heo, Yun. Improving quality of high-throughput sequencing reads. Diss. University of Illinois at Urbana-Champaign, 2015.
- Bayley, H. Nanopore sequencing: from imagination to reality. Clinical chemistry 2015, 61, 25-31. [CrossRef]
- Stoddart, D.; Heron, A. J.; Mikhailova, E.; Maglia, G.; Bayley, H. Single-nucleotide discrimination in immobilized DNA oligonucleotides with a biological nanopore. Proceedings of the National Academy of Sciences. 2009, 106, 7702-7707. [CrossRef]
- Ip, C.L.; Loose, M.; Tyson, J.R.; de Cesare, M.; Brown, B.L.; Jain, M.; Leggett, R.M.; Eccles, D.A.; Zalunin, V.; Urban, J.M.; Piazza, P. MinION Analysis and Reference Consortium: Phase 1 data release and analysis. F1000Research 2015, 4, 1075. [CrossRef]
- Feng, Y.; Zhang, Y.; Ying, C.; Wang, D.; Du, C. Nanopore-based fourth-generation DNA sequencing technology. Genomics, Proteomics and Bioinformatics, 2015, 13, 4-16. [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



