
Submitted:
25 August 2024
Posted:
26 August 2024
You are already at the latest version
Abstract
Antiphospholipid syndrome (APS) otherwise known as Hughes syndrome is a systemic autoimmune disease typically linked with characteristic features like thrombosis and pregnancy morbidity with the presence of antiphospholipid antibodies (aPLs). Although anticoagulation is the current flagship treatment for APS, there are reports indicating a considerable failure following this regimen in a significant number of obstetric APS and thrombotic APS cases. Antiphospholipid antibodies (aPLs) bind anionic phospholipids or protein-phospholipid complexes within the cell membrane. Here, we gave a general overview of the biology of APS and the human gut microbiome, our current understanding of the gut bacteria-immune axis in autoimmunity focus on the potential link to APS, highlighting the implicated microorganisms in this context, and proposing recommendations on future therapeutic development.
Keywords:
Gut Microbiome
; Antiphospholipid syndrome
; Antiphospholipid antibodies
; Autoimmunity
; Therapeutics
1.0. Introduction
Antiphospholipid syndrome (APS) is an autoimmune disease characterized by the presence of persistent antiphospholipid antibodies, leading to vascular thrombosis and/or pregnancy-related complications [1]. Typical features of this syndrome are venous or arterial thrombosis, miscarriage, and thrombocytopenia [2]. In addition, APS is considered a thrombo-inflammatory disease that occurs in approximately a third of systemic lupus erythematosus (SLE) cases and is often associated with increasing organ damage over time [3]. Other clinical manifestations of this disease include chorea, livedo reticularis, autoimmune hemolytic anemia, transverse myelitis, migraine, epilepsy, Raynaud’s phenomenon, and heart valve lesions [4]. The classification of APS is based on specific clinical criteria and circumstances; the first criteria include three clinical aspects: arterial or venous thrombosis, obstetric loss, or thrombocytopenia [5] with other diagnostic parameters including lupus anticoagulant and anticardiolipin antibodies [6]. Further classification identifies APS as primary when not accompanied by other autoimmune diseases and secondary when associated with autoimmune diseases, such as an autoimmune disease, infection, drug-related factor, or cancer, with the strongest correlation observed in systemic lupus erythematosus [7]; other non-criteria manifestations such as thrombocytopenia, APS-associated nephropathy, valvular heart disease, livedo reticularis, and cognitive impairment has also been reported [7]. Another form of APS, known as catastrophic APS, is characterized by the formation of thrombi in multiple and numerous small vascular beds, resulting in organ failure and a high mortality rate in this specific group of APS patients [8,9]. Antiphospholipid antibodies are a diverse group of autoantibodies that play a crucial role in the pathogenesis of APS by interacting with various plasma proteins, including beta-2 glycoprotein 1, prothrombin, thrombomodulin, plasminogen, antithrombin III, Protein C, Protein S, Annexin II and Annexin V [10,11]. These antibodies trigger prothrombotic mechanisms by activating endothelial cells, monocytes, platelets, coagulation factors, and complement proteins, leading to disrupted fibrinolysis, coagulation pathways, inflammation, and placental injury [12,13,14,15,16]. Antiphospholipid antibodies (apL) target either anionic phospholipids directly or protein-phospholipid complexes within the cell membrane. Categorically, they can be subdivided into three main types: anti-β2-glycoprotein-1 (anti-β2GPI), anticardiolipin, and lupus anticoagulant [17,18,19,20,21]. Notably, the specificity of antiphospholipid antibodies for APS is limited because they may also be present in healthy individuals with a history of thrombosis, pregnancy-related morbidity, or individuals with autoimmune disorders [22]. Here, we gave a general overview of the biology and our current understanding of the gut bacteria-immune axis in autoimmunity with emphasis and focus on the potential link to APS, highlighting the implicated microorganisms in this context, and proposing recommendations on future therapeutic development.
2.0. Pathophysiology of APS
Infections, especially those caused by bacterial or viral pathogens, are believed to be the primary triggers for developing antiphospholipid antibodies; this is evident in the case of anti-β2GPI [22]. This process, known as molecular mimicry, involves similarities between the amino acid sequences of infectious agents (bacteria or viruses) and that of amino acid sequences from beta-2 glycoprotein-1 (β2GPI), which contributes to the formation of autoantibodies [23]. While the pathophysiology of APS depends on the diverse action of antibodies on their myriad antigenic target sites in various patients [24], the etiology of these pathogenic autoantibodies is likely a result of an intertwined intricacy of different environmental factors in individuals who carry genetic markers further increasing disease susceptibility [25,26], Although the mechanism is not fully understood, the APS has been reported to disrupt the homeostatic regulation of blood coagulation [9]. The precise mechanism of thrombosis isn’t yet defined. Still, a hypothesis is that there is a deficiency in cellular apoptosis, exposing membrane phospholipids and instigating antiphospholipid antibody formation [27]. In the APS, pathogenic antiphospholipid autoantibodies have been shown to cause various thrombotic events that encourage coagulation while preventing fibrinolysis [9]. This is achieved by autoantibodies that target phospholipid-binding proteins that cause blood clots in veins and arteries and bolster thrombosis through a diverse array of means by disrupting the function of vital phospholipid-binding proteins crucial to blood clot regulation and activate platelets, pivotal to clot formation [28].
Additionally, they activate endothelial cells, which in turn causes the expression of coagulation-related molecules hindering the activity of naturally occurring anticoagulants like protein C and protein S, which usually prevent the formation of clots, further, interfering with the fibrinolytic system and prevent the clot from dissolving [28]. Overall, APS represents a complex interplay between autoantibodies and physiological processes that predispose people to thrombotic events and pregnancy complications. In the context of APS, these antibodies play a diverse role in pregnancy loss by inducing thrombosis in the placenta, which leads to placental insufficiency and fetal death [29]. Also, the triggering of the activation of endothelial cells, monocytes, and platelets leads to the overproduction of tissue factor and thromboxane A2, ultimately leading to placental damage and fetal loss [30].
3.0. Clinical Features of APS
APS affects various tissues and organs in the body, including blood vessels, kidneys, brain, heart, lungs, skin, liver, and gastrointestinal tract, resulting in various clinical symptoms and complications. These clinical features are venous, arterial, or small vessel thrombosis, fetal loss, and thrombocytopenia. Deep vein thrombosis is the most common manifestation, while cerebrovascular accidents represent the predominant form of arterial thrombosis. Early fetal loss, preterm birth, and preeclampsia are the most common fetal and obstetric manifestations [31]. Additional clinical features such as cognitive dysfunction or demyelination may be associated with interactions between phospholipid antibodies and cells, possibly due to a disrupted blood-brain barrier or increased intrathecal synthesis of phospholipid antibodies [32]. This disease has various clinical manifestations, which differ in their respective frequencies. In over 20% of cases, patients may experience venous thromboembolism, thrombocytopenia, miscarriage or fetal loss, stroke or transient ischemic attack, migraine, and livedo reticularis. Less commonly occurring in 10-20% of cases, the APS may manifest as heart valve disease, pre-eclampsia or eclampsia, premature birth, hemolytic anemia, and coronary artery disease. Rare manifestations, occurring in less than 10% of cases, include epilepsy, vascular dementia, chorea, retinal artery, or venous thrombosis, amaurosis fugax, pulmonary hypertension, leg ulcers, digital gangrene, osteonecrosis, antiphospholipid nephropathy, and mesenteric ischemia. Occurring in less than 1% of cases, APS may lead to adrenal hemorrhage, transverse myelitis, and Budd-Chiari Syndrome [33]. This spectrum of clinical manifestations underscores the complexity and heterogeneity of APS, necessitating thorough evaluation and tailored management approaches for affected individuals.
4.0. Current Treatments of APS
Despite the volume of knowledge so far, there are still uncertainties in treating APS, with some aspects requiring further evidence [34]. The primary goals of treatment for individuals with APS include preventing thrombosis in individuals without prior incidents (i.e., primary thromboprophylaxis), effectively treating acute thrombosis if it occurs, and averting the recurrence of thrombosis in individuals with established APS (secondary thromboprophylaxis) [35]. Treatment strategies for APS include a variety of anticoagulant approaches. For instance, vitamin K antagonists such as warfarin have historically been the cornerstone of treatment for thrombotic APS [36]. In addition, low-dose aspirin is recommended for people who have abnormal antiphospholipid antibodies without a history of blood clots [37,38]. Also, heparin and direct oral anticoagulants (DOACs) such as rivaroxaban are considered in cases of warfarin intolerance, allergy, or inadequate anticoagulant control [36]. Lastly, emerging evidence suggests possible adjunctive therapies such as hydroxychloroquine, rituximab, and statins, although further research is needed [37,39]. Long-term management goals in APS revolve around preventing recurrent thrombosis while minimizing anticoagulation-related side effects, especially for Catastrophic APS, where early intervention is crucial and typically involves a combination of anticoagulants to address severe manifestations [40,41].
5.0. Involvement of Microorganisms in APS
It is well known that persistent pathogenic autoantibodies targeted at membrane phospholipids and/or their linked plasma proteins play a role in establishing APS [42]. This is the major characteristic of APS, where two mechanisms are forwarded to cause the presence of aPLs [43]. One reason explains the occurrence of clinical events such as vascular thrombosis and/or pregnancy morbidity with the persistence of aPLs such as lupus anticoagulant (LA), anticardiolipin antibodies(aCL), and anti-β2 glycoprotein 1 antibody (anti-β2GP1) [1]. However, the thrombosis event in the pathogenesis of APS rarely occurs, indicating the involvement of other determinants that modulate the thrombotic milieu. This brought about the conception of a second hit that focuses on exploring innate immunity like inflammation, infection, or surgery essential to precipitate the thrombotic event in aPL carriers, with a line of thought of infective agents being one of the mechanisms increasing the aPL exposure [44,45,46]. Research has shown that microbial pathogens can provoke the production of aPLs in an antigen-dependent manner, such as the previously mentioned molecular mimicry, or through an antigen-independent manner, which includes the breakdown of immune tolerance due to an inflammatory response [44]. Molecular mimicry is one of the most relevant mechanisms to explain the association between infections and clinical manifestations linked with aPLs in APS because it justifies the generation of cross-reactive T and B lymphocyte cells that recognize antigens from pathogens but cross-react to autoantigens [47,48].
An example is rheumatic fever, a human autoimmune disease believed to have originated from cross-reactivity with protein or carbohydrate structures from pathogens. Further research shows several similarities between rheumatic fever and APS due to the bond and cross-reactivity of streptococcal proteins with β2GP1 [49], with homologies between proteins of microorganisms and peptides generated from β2GP1 that contribute to T and B cell activation [50]. Various animal models are used to understand the potential pathogenic effect of infections exhibiting surface components analogous to the major immunogenic epitopes targeted by anti-β2GP1 antibodies. From this research, it is seen that upon immunization, high titers of antipeptide anti-β2GP1 antibodies were detected in murine immunized with Haemophilus influenza, Neisseria gonorrhea, Candida albicans, and tetanus toxoid and showed APS features like thrombocytopenia with an increased risk of fetal loss. A succinct link between gut microbiome and APS highlights the intestine as a potential chronic trigger in patients with APS. Roseburia intestinalis (a bacteria abundant in the human gut) contains amino acid sequences homologous to those found in B cell and T cell epitopes within [52,53,54]. Significantly, when R. intestinalis is administered orally using a mouse model of APS, it leads to the development of anti-human-β2GP1 antibodies with the occurrence of morbidity and mortality associated with APS [55]. Some studies demonstrate that fermented milk contains a probiotic bacterial strain that can modify aPLs in non-autoimmune animals , which further suggests that gut microbes may be able to regulate the development of pathogenic autoantibodies and APS.
Furthermore, an association between APS and other infectious agents, including hepatitis C virus (HCV) and HIV, cytomegalovirus, Epstein Barr virus, and herpes simplex virus have been reported (Table 1). Other reports have also shown the induction of APS by parvovirus unique region (VP1u) [56]. During the COVID-19 pandemic, APS severe acute respiratory syndrome coronavirus 2 (SARS-CoV2) with eight types of aPL antibodies were discovered in the plasma of over 50% of hospitalized COVID-19 patients [36,59]. Similarly, aPLs have been associated with some bacterial infections, caused by Coxiella burnetii, Helicobacter pylori, Mycoplasma pneumonia, Streptococci, Borrelia burgdorferi, and Mycobacterium tuberculosis (Table 1). For instance, syphilis patients show aCL antibodies that could have been provoked by the cross-reactivity of syphilis antibodies with treponemal cardiolipins [60]. Likewise, patients with leprosy also present aPL and β2GP1-dependent binding [44]. However, there is a paucity of data on the ability of parasitic or fungal infections to trigger aPLs [44].
6.0. Gut Microbiome
6.1. Overview
The development of the human gut microbiota begins early, even before birth, because it is essential in maintaining the host organism’s normal physiological processes [61]. This microbiota can synthesize diverse metabolic compounds that can exert beneficial and detrimental effects on human health through interactions with the host due to its ability to proliferate along the intestinal surfaces, establishing a resilient system that serves as a barrier against the intrusion of pathogenic microorganisms [61].
6.2. Gut Microbiome - Metabolic and Protective Role
The gut microbiota plays a pivotal role in metabolizing dietary components, transforming indigestible carbohydrates like cellulose, hemicelluloses, resistant starch, pectin, oligosaccharides, and lignin into short-chain fatty acids (SCFAs), including acetic, propionic, and butyric acids- produced by Firmicutes, Bacteroidetes, and certain anaerobic gut microorganisms. The gut microbiota also contributes to the host’s well-being by facilitating the synthesis of essential vitamins such as biotin, thiamine, cobalamin, riboflavin, nicotine, pantothenic acids, as well as vitamins B and K. It’s worth noting that the gut microbiota also can produce neurochemicals, including gamma-aminobutyric acid (GABA), an important inhibitory neurotransmitter in the brain; dysregulation of GABA has been linked to various neuropsychiatric disorders. Additionally, the microbiota generates various compounds such as carbohydrates, branched-chain amino acids, amines, phenols, indoles, and phenylacetic acid. Moreover, it plays a role in synthesizing bile acids, cholesterol, and conjugated fatty acids. In summary, the microbiota’s metabolic activities are multifaceted and encompass transforming dietary components, vitamin synthesis, neurochemical production, and generating various bioactive compounds [62]. The protective role of the microbiota is to occupy intestinal surfaces and create stability in the system that prevents the invasion of pathogenic microorganisms. The production of SCFAs serves as a significant energy source for intestinal epithelial cells. It strengthens the mucosal barrier, which is regarded as a tumor suppressor due to its promising anti-inflammatory and chemo-preventive properties [63]. The microbiota plays a protective role by colonizing intestinal surfaces and establishing a stable environment that acts as a barrier against the intrusion of pathogenic microorganisms. Moreover, producing short-chain fatty acids (SCFAs) is a crucial energy source for intestinal epithelial cells, reinforcing the mucosal barrier. SCFAs are also recognized as tumor suppressors, owing to their notable anti-inflammatory and chemo-preventive properties [64].
6.3. Microbiota and the Gut-Brain Axis
The brain-gut-microbiota axis is a bidirectional system enabling gut microorganisms to communicate with the central nervous system (CNS) and the CNS with the gut. The mechanisms of signal transmission are complex and not fully understood but include neural, endocrine, immune, and metabolic pathways which influence the arrays of factors affecting the microbiome-gut-brain axis such as diet, genetics, drugs, environment, exercise, cognitive behavior, stress, social interactions, and fear [65]. Gut microbes can produce most neurotransmitters in the human brain and their precursors. However, some neurotransmitters, like glutamate, GABA, dopamine, and serotonin, cannot cross the blood-brain barrier and must be synthesized in the brain from local pools of precursor neurotransmitters [66]. These precursors mostly comprise amino acids, e.g., food-derived neurotransmitters (tyrosine and tryptophan) that pass through the blood-brain barrier. They are absorbed by the corresponding cells in the brain that produce neurotransmitters [66].
The precursors are subsequently transformed via several intermediate processes utilizing different host enzymes into functional neurotransmitters, such as dopamine, norepinephrine, and serotonin; therefore, by controlling the metabolism of neurotransmitter precursors, the gut microbiota can affect host behavior due to the dietary origin of these precursors. For instance, probiotic bifidobacterial therapy can raise tryptophan levels necessary for serotonin synthesis. Certain Lactobacilli species change how gamma-aminobutyric acid (GABA) is metabolized and how the GABA receptor expresses itself and behaves in the brain [66]. Escherichia, Bacillus, and Saccharomyces spp. can produce GABA, as Lactobacillus and Bifidobacterium species can produce enterococcus, streptococcus, Escherichia, and norepinephrine can create dopamine, acetylcholine, and serotonin; Bacillus can produce both [67]. Outside the brain, dopamine production has been detected in Staphylococcus in the human intestine, which can take up the precursor l-3,4-dihydroxy-phenylalanine (l-DOPA) and convert it into dopamine by staphylococcal aromatic amino acid decarboxylase (SadA) expressed by these bacteria. More than 50% of dopamine in the human body is synthesized in the gut. Dopamine and its receptors are widely distributed in the intestinal tract and affect gastric secretion, motility, and mucosal blood flow [66,67].
6.4. Gut Microbiome in Health and Diseases
The gut microbiome refers to the diverse community of microorganisms inhabiting the human gastrointestinal tract, including bacteria, viruses, fungi, and other microbes. It plays a crucial role in maintaining overall health and is implicated in various diseases when its composition and function are disrupted as shown in Figure 1. The gut microbiota has the largest quantities of microorganisms and the most species compared to other body parts. They consist of thousands of microorganisms, including bacteria, viruses, and some eukaryotes, that colonize the digestive tract just after birth. The microbial composition of the gut microbiota varies across the digestive tract. In the stomach and small digestive tract, relatively few species of bacteria are present. The intestinal microbiota consists of more than 1500 species distributed in more than 50 different phyla. The colon contains a densely populated microbial ecosystem with up to 1012 cells for every gram of intestinal substance [68]. The most dominant bacterial phyla in the human gut are Firmicutes, Bacteroidetes, Actinobacteria, and Proteobacteria, and the most recorded bacterial genera are Bacteroides, Clostridium, Peptococcus, Bifidobacterium, Eubacterium, Ruminococcus, Faecalibacterium and Peptostreptococcus [69]. It was reported that Bacteroidetes and Firmicutes, followed by Proteobacteria, Fusobacteria, Tenericutes, Actinobacteria, and Verrucomicrobia were the most dominant phyla, making up to 90% of the total microbial population in humans [70]. Among these, Bacteroides are the most abundant, comprising about 30% of bacteria in the gut, suggesting they are particularly significant in the functioning of the host organism. Most gut bacteria (99%) are anaerobes; however, high densities of aerobic microbes are recorded in the cecum. Fungi, protists, archaea, and viruses are also present in the gut flora; however, less is known about their activities [68]. Several factors can change the gut microbiota composition and function. These factors include host genetics, diet, age [71], antibiotics use, and diseases [72]. The gut microbiota has a lot of significant functions in the human body, including supporting protection from pathogens by colonizing mucosal surfaces, creating different antimicrobial substances, enhancing the immune system, playing a vital role in digestion and metabolism, controlling epithelial cell proliferation and differentiation, modifying insulin resistance and affecting its secretion influencing brain-gut communication and thus affecting the mental and neurological functions of the host; hence, the gut microbiota plays a significant role in maintaining normal gut physiology and health [68].
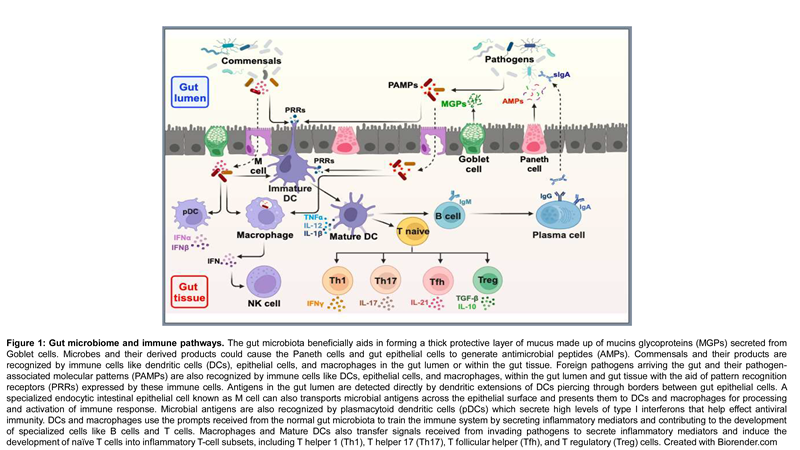 |
6.5. Gut Microbiome and Age
Maternal milk, which is the optimal food for infants, meets all their nutrition and physiologic requirements and protects against infections due to the presence of immune effectors, such as immunoglobulin A (IgA); this natural mode of feeding contributes to the maturation of the infant’s immune system and modulates the development of its gut microbiota. Human milk, which is not sterile, contains protein, fat, carbohydrate, immunoglobulins, and endocannabinoids. It also contains as many as 600 different species of bacteria, including beneficial Bifidobacterium breve, Bifidobacterium adolescentis, Bifidobacterium longum, Bifidobacterium bifidum, and Bifidobacterium dentium [73]. Moreover, the carbohydrate component of human milk has oligosaccharides, which makes up the third largest solid component of the entire food source. Human milk oligosaccharides are indigestible polymers formed by a few monosaccharides that serve as prebiotics by selectively stimulating the growth of members of the genus Bifidobacterium [74]. They have been linked with strengthening gut mucosal protection through activities against pathogens, also increasing the production of immunoglobulin A, which is correlated with modulation of the intestinal immune system [75,76]. It is noted that the Bifidobacterium-dominated microbiota of the infant changes over time into the Bacteroidetes and Firmicutes-dominated microbiota of the adult. This distribution remains stable throughout adulthood without perturbations, such as long-term dietary changes, repeated antibiotic usage, or disease. Declines in dentition, salivary function, digestion, and intestinal transit time may affect the gut microbiota upon aging. There are notable differences in the microbiota in elderly people compared with young adults, with relative proportions of Bacteroidetes predominating in elderly people compared with higher proportions of Firmicutes in young adults [77].
6.6. Gut microbiome and Environmental influences – Focus on APS
There is a research gap and a lack of a detailed scoping review on the environmental factors influencing the gut microbiome shape, structure, functions, and association with APS. The gut microbiome is extremely dynamic, and its variations are linked to various health outcomes, including neurological diseases, inflammatory bowel disease, respiratory illnesses, obesity, arthritis, depression, cardiovascular diseases, chronic liver diseases, and pancreatic disorders as stated above. These variations are often influenced by environmental factors such as environmental contaminants and medication use [78,82]. The impact of environmental factors on gut microbiome can differ based on demography and level of exposure. The interactions between the environment and gut microbiome are complex, with diverse environments playing crucial roles in shaping gut microbiome [92]. This becomes particularly important during early life stages, as differences in hygiene and diet among households and communities significantly contribute to gut colonization by various bacterial species, predisposing individuals to different health conditions [92]. The gut microbiome changes throughout life, starting from neonatal development. The nurturing surroundings where a child grows up can notably impact the development of specific gut microbiome species. This could affect their vulnerability to APS because various healthy microbiomes support a varied and balanced array of microorganisms and their activities, which is vital for a strong immune system [87].
Environmental factors such as the dissemination of contaminants, including pesticides, heavy metals, and microplastics, pose significant threats to the human gut microbiome and are highly associated with various diseases resulting from changes in the composition, diversity, and metabolic activity of gut microbiome community [80]. Most contaminants remain persistent and bioactive in the environment. Upon ingestion, contaminants are metabolized by the gut microbiome, and the bioactive metabolites, i.e., residues, affect the homeostasis balance of the gut microbiome, resulting in severe health complications. Indeed, bioactive metabolites’ effects on the gut microbiome were found to interfere with neurotransmitters, leading to cognitive impairments and mental health disorders [94]. The assimilation of glyphosate, a pesticide, has been shown to cause neurological disorders, including autism, through the modification of gene expression, DNA replication, and immunomodulation of gut microbial communities [86]. Heavy metals such as mercury, copper, lead, and cadmium in food, soil, and water have been shown to impose selective pressure on the gut microbiota composition, thereby increasing the vulnerability of the host to different types of cancers, immune response alteration, and inflammatory cytokine increments [93]. The most adverse effects of heavy metals are inhibiting gut microbiome proliferation, interfering with the composition and metabolic prowess of the gut bacterial community, and distorting the oxidative and immune status of the host [80].
Microplastics are reservoirs of organic and inorganic contaminants as well as antibiotics. Such microplastics as virgin polystyrene absorb polyaromatic hydrocarbons. Also, the concentrations of copper and zinc were determined to be highly correlated with the amounts of virgin polystyrene bead and polyvinyl chloride, respectively. These were studied to be widely distributed in the environment [79] and cause several health issues, including intestinal tract infections, mucosa damage, and increasing permeability [83]. Microplastics are home to diverse microbial communities, including pathogens [83,84]. These can displace gut microbiota, leading to the dominance and dissemination of bacterial pathogens in the gut. This impacted cytokine secretion and the proportion of Th17 and Treg cells within CD4+ cells, leading to inflammation of the intestines [88].
Medication use, including antibiotics and their effects on gut microbiota, has been extensively studied. Antibiotics are substances that kill or inhibit bacterial growth but also target commensal bacteria in the gut, causing various collateral damage [89]. Antibiotics can either be bactericidal or bacteriostatic by killing bacteria or inhibiting bacterial proliferation. Antibiotic exposure induces long-term changes in gut microbiome structure and increases the susceptibility of the host to immunological, inflammatory, metabolic, gastrointestinal, and allergic consequences [81]. Antibiotic exposure during pregnancy results in an imbalanced gut microbiome, which in turn predisposes pregnant women to severe risk of depressive symptoms [90]. After childbirth, antibiotic consumption increases postpartum depression within six months [90]. Different antibiotics target different bacterial cells, but some are broad-spectrum, with activities directly affecting the gut microbiome, leading to dysbiosis, resulting in a dysfunctional immune system which is a major factor facilitating the predisposition of the host to APS [91].
7.0. Recommendations and Perspectives: A Need for a Holistic Treatment Approach
Understanding the link between infectious disease agents, gut microbiota, and APS is necessary for curating and advancing alternative approaches to treating APS [95]. The complex, intricate relationships between the gut microbiota, the neuronal mechanisms and the hormonal pathway of the gut needs to be further studied to be able to interpret molecular-level interactions useful in animal models, healthy individuals, and patients to yield novel methods for diagnosing, preventing, and managing APS and other associated autoimmune conditions [96]. Rather than the conventional treatment approaches, which aim to eliminate venous thrombosis, there is a need for an evidence-based treatment regimen that manages the syndrome with concise consideration for older adults and pregnant mothers, due to the complications of their gut microbiota proliferation which has been found to influence the production of aPLs [95]. Also, considering all the prospects that may promote the proliferation of the intestinal microbiome leading to the release of various aCL antibodies or their molecular mimicry could hinder the established treatment regimen and cause failure and delayed recovery, further aggravating the autoimmune disease syndrome [96].
Furthermore, beyond anticoagulation, which is regarded as the cornerstone of APS treatment, current studies are progressively focused on a few biologics, largely because these biologics like such as rituximab and eculizumab, are generally indicated for refractory catastrophic APS [97] with high specificity and sensitivity, albeit with some limitations so far. Ultimately, more studies are needed to design useful safe and effective vaccines and therapy for APS.
Author Contributions
Conceptualization, O.C.A.; figures and tables, O.C.A., R.O.A., O.G.B., C.E.U., writing—original and final draft preparation, O.C.A., J.A., R.O.A., O.G.B., A.A., B.O., C.E.U, I.I., V.A., P.B., Q.A.A, O.O, S.S. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Ethics Approval and Consent to Participate
Not applicable.
Conflicts of Interest
The authors declare no competing interests in this work.
References
- Miyakis, S.; Lockshin, M.D.; Atsumi, T.; Branch, D.W.; Brey, R.L.; Cervera, R.; Derksen, R.H.W.M.; De Groot, P.G.; Koike, T.; Meroni, P.L.; et al. International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS). J. Thromb. Haemost. 2006, 4, 295–306. [Google Scholar] [CrossRef] [PubMed]
- Espinosa, G.; Cervera, R. Antiphospholipid syndrome. Arthritis Research & Therapy 2008, 10, 230. [Google Scholar]
- Riancho-Zarrabeitia, L.; Martínez-Taboada, V.; Rúa-Figueroa, I.; Alonso, F.; Galindo-Izquierdo, M.; Ovalles, J.; Olivé-Marqués, A.; Fernández-Nebro, A.; Calvo-Alén, J.; Menor-Almagro, R.; et al. Antiphospholipid syndrome (APS) in patients with systemic lupus erythematosus (SLE) implies a more severe disease with more damage accrual and higher mortality. Lupus 2020, 29, 1556–1565. [Google Scholar] [CrossRef] [PubMed]
- Torres-Jimenez, A.-R.; Ramirez-Nova, V.; Cespedes-Cruz, A.I.; Sanchez-Jara, B.; Velazquez-Cruz, A.; Bekker-Méndez, V.C.; Guerra-Castillo, F.X. Primary antiphospholipid syndrome in pediatrics: beyond thrombosis. Report of 32 cases and review of the evidence. Pediatr. Rheumatol. 2022, 20, 1–9. [Google Scholar] [CrossRef]
- Harris, E.N. Syndrome of the black swan. British Journal of Rheumatology 1987, 26, 324–326. [Google Scholar] [CrossRef] [PubMed]
- Eby, C. Antiphospholipid Syndrome Review. Clin. Lab. Med. 2009, 29, 305–319. [Google Scholar] [CrossRef] [PubMed]
- Corban, M.T.; Duarte-Garcia, A.; McBane, R.D.; Matteson, E.L.; Lerman, L.O.; Lerman, A. Antiphospholipid syndrome: role of vascular endothelial cells and implications for risk stratification and targeted therapeutics. Journal of the American College of Cardiology 2017. [Google Scholar] [CrossRef]
- Cervera, R.; Bucciarelli, S.; Plasín, M.A.; Gómez-Puerta, J.A.; Plaza, J.; Pons-Estel, G.; Shoenfeld, Y.; Ingelmo, M.; Espinos, G. Catastrophic antiphospholipid syndrome (CAPS): Descriptive analysis of a series of 280 patients from the “CAPS Registry”. J. Autoimmun. 2009, 32, 240–245. [Google Scholar] [CrossRef]
- Arreola-Diaz, R.; Majluf-Cruz, A.; Sanchez-Torres, L.; Hernandez-Juarez, J. The Pathophysiology of The Antiphospholipid Syndrome: A Perspective From The Blood Coagulation System. Clin. Appl. Thromb. 2022, 28. [Google Scholar] [CrossRef]
- Zuo, Y.; Shi, H.; Li, C.; Knight, J.S. Antiphospholipid syndrome: a clinical perspective. Chin. Med J. 2020, 133, 929–940. [Google Scholar] [CrossRef]
- Zhang, J.; McCrae, K.R. Annexin A2 mediates endothelial cell activation by antiphospholipid/anti-beta2 glycoprotein I antibodies. Blood 2005. [Google Scholar]
- Sorice, M.; Longo, A.; Capozzi, A.; Garofalo, T.; Misasi, R.; Alessandri, C.; Valesini, G. Anti-beta2-glycoprotein I antibodies induce monocyte release of tumor necrosis factor alpha and tissue factor by signal transduction pathways involving lipid rafts. Arthritis & Rheumatism 2007, 56, 2687–2697. [Google Scholar]
- Shi, T.; Giannakopoulos, B.; Yan, X.; Yu, P.; Berndt, M.C.; Andrews, R.K.; Rivard, G.E. Anti-beta2-glycoprotein I antibodies in complex with beta2-glycoprotein I can activate platelets in a dysregulated manner via glycoprotein Ib-IX-V. Arthritis & Rheumatism 2006, 54, 2558–2567. [Google Scholar]
- Pierangeli, S.S.; Girardi, G.; Vega-Ostertag, M.; Liu, X.; Espinola, R.G.; Salmon, J. Requirement of activation of complement C3 and C5 for antiphospholipid antibody–mediated thrombophilia. Arthritis Rheum. 2005, 52, 2120–2124. [Google Scholar] [CrossRef] [PubMed]
- Fischetti, F.; Durigutto, P.; Pellis, V.; Debeus, A.; Macor, P.; Bulla, R.; Tedesco, F. Thrombus formation induced by antibodies to beta2-glycoprotein I is complement dependent and requires a priming factor. Blood 2005, 106, 2340–2346. [Google Scholar] [CrossRef]
- Girardi, G.; Redecha, P.; E Salmon, J. Heparin prevents antiphospholipid antibody–induced fetal loss by inhibiting complement activation. Nat. Med. 2004, 10, 1222–1226. [Google Scholar] [CrossRef]
- Gómez-Puerta, J.A.; Espinosa, G.; Cervera, R. Antiphospholipid Antibodies: From General Concepts to Its Relation with Malignancies. Antibodies 2016, 5, 18. [Google Scholar] [CrossRef]
- Vassalo, J.; Spector, N.; de Meis, E.; Rabello, L.S.; Rosolem, M.M.; Brasil, P.E.D.; Salluh, J.I.; Soares, M. Antiphospholipid antibodies in critically ill patients with cancer: A prospective cohort study. J. Crit. Care 2014, 29, 533–538. [Google Scholar] [CrossRef]
- Sciascia, S.; Radin, M.; Bazzan, M.; Roccatello, D. Novel diagnostic and therapeutic frontiers in thrombotic anti-phospholipid syndrome. Intern. Emerg. Med. 2017, 12, 1–7. [Google Scholar] [CrossRef]
- Virachith, S.; Saito, M.; Watanabe, Y.; Inoue, K.; Hoshi, O.; Kubota, T. Anti-beta2-glycoprotein I antibody with DNA binding activity enters living monocytes via cell surface DNA and induces tissue factor expression. Clinical and Experimental Immunology 2019, 195, 167–178. [Google Scholar] [CrossRef]
- Islam, M.A. Antiphospholipid antibodies and antiphospholipid syndrome in cancer: Uninvited guests in troubled times. Seminars in Cancer Biology 2020, 64, 108–113. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, K.; Sciascia, S.; de Groot, P.G.; Devreese, K.; Jacobsen, S.; Ruiz-Irastorza, G.; Hunt, B.J. Antiphospholipid syndrome. Nature Reviews Disease Primers 2018, 4, 17103. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Tapias, P.; Blank, M.; Anaya, J.-M.; Shoenfeld, Y. Infections and vaccines in the etiology of antiphospholipid syndrome. Curr. Opin. Rheumatol. 2012, 24, 389–393. [Google Scholar] [CrossRef] [PubMed]
- Harris, E.N.; Asherson, R.A.; Hughes, G.R. Antiphospholipid antibodies–autoantibodies with a difference. Annual Review of Medicine 1988, 39, 261–271. [Google Scholar] [CrossRef]
- Gharavi, A.; Pierangeli, S. Origin of antiphospholipid antibodies: Induction of aPL by viral peptides. Lupus 1998, 7, 52–54. [Google Scholar] [CrossRef] [PubMed]
- Sherer, Y.; Blank, M.; Shoenfeld, Y. Antiphospholipid syndrome (APS): where does it come from? Best Practice & Research Clinical Rheumatology 2007, 21, 1071–1078. [Google Scholar]
- Movva, S.; Belilos, E.; Carsons, S. (2022). Antiphospholipid Syndrome. In: StatPearls.
- Bustamante, J.G.; Goyal, A.; Singhal, M. (2023). Antiphospholipid Syndrome. [Updated 7th February 2023].
- Salmon, J.E.; Girardi, G. Antiphospholipid antibodies and pregnancy loss: a disorder of inflammation. J. Reprod. Immunol. 2007, 77, 51–56. [Google Scholar] [CrossRef]
- Prima, F.A.F.D.; Valenti, O.; Hyseni, E.; Giorgio, E.; Faraci, M.; Renda, E.; De Domenico, R.; Monte, S. Antiphospholipid Syndrome during pregnancy: the state of the art. J. Prenat. Med. 2011, 5, 41–53. [Google Scholar]
- Cervera, R. Lessons from the ‘Euro-Phospholipid’ project. Autoimmunity Reviews 2008, 7, 174–178. [Google Scholar] [CrossRef]
- Martínez-Cordero, E.; García, B.E.R.; León, D.E.A. Anticardiolipin antibodies in serum and cerebrospinal fluid from patients with systemic lupus erythematosus. J. Investig. Allergol. Clin. Immunol. 1998, 7, 596–601. [Google Scholar]
- Ruiz-Irastorza, G.; Crowther, M.; Branch, W.; Khamashta, M.A. Antiphospholipid syndrome. The Lancet 2010, 376, 1498–1509. [Google Scholar] [CrossRef]
- Espinosa, G.; Cervera, R. Current treatment of antiphospholipid syndrome: lights and shadows. Nat. Rev. Rheumatol. 2015, 11, 586–596. [Google Scholar] [CrossRef]
- Rumsey, D.G.; Myones, B.; Massicotte, P. Diagnosis and treatment of antiphospholipid syndrome in childhood: A review. Blood Cells, Mol. Dis. 2017, 67, 34–40. [Google Scholar] [CrossRef]
- Zuo, Y.; Shi, H.; Li, C.; Knight, J.S. Antiphospholipid syndrome: a clinical perspective. Chin. Med J. 2020, 133, 929–940. [Google Scholar] [CrossRef] [PubMed]
- Hubben, A.; McCrae, K.R. Emerging Therapies in Antiphospholipid Syndrome. Transfus. Med. Rev. 2022, 36, 195–203. [Google Scholar] [CrossRef]
- American College of Rheumatology. (2023). Antiphospholipid Syndrome.
- Knight, J.S.; Branch, D.W.; Ortel, T.L. Antiphospholipid syndrome: advances in diagnosis, pathogenesis, and management. BMJ 2023, 380, e069717. [Google Scholar] [CrossRef]
- Tumian, N.R.; Hunt, B.J. Clinical Management of Thrombotic Antiphospholipid Syndrome. J. Clin. Med. 2022, 11, 735. [Google Scholar] [CrossRef] [PubMed]
- Garcia, D.; Erkan, D. Diagnosis and Management of the Antiphospholipid Syndrome. N. Engl. J. Med. 2018, 378, 2010–2021. [Google Scholar] [CrossRef] [PubMed]
- Misasi, R.; Longo, A.; Recalchi, S.; Caissutti, D.; Riitano, G.; Manganelli, V.; Garofalo, T.; Sorice, M.; Capozzi, A. Molecular Mechanisms of “Antiphospholipid Antibodies” and Their Paradoxical Role in the Pathogenesis of “Seronegative APS”. Int. J. Mol. Sci. 2020, 21, 8411. [Google Scholar] [CrossRef]
- Hughes, G. Thrombosis, abortion, cerebral disease, and the lupus anticoagulant. British Medical Journal (Clinical research ed.) 1983, 287, 1088. [Google Scholar] [CrossRef] [PubMed]
- Mendoza-Pinto, C.; García-Carrasco, M.; Cervera, R. (2023). Microorganisms in the Pathogenesis and Management of Anti-phospholipid Syndrome (Hughes Syndrome) Role of Microorganisms in Pathogenesis and Management of Autoimmune Diseases: Volume II: Kidney, Central Nervous System, Eye, Blood, Blood Vessels & Bowel (pp. 341-357): Springer.
- Meroni, P.L.; Borghi, M.O.; Grossi, C.; Chighizola, C.B.; Durigutto, P.; Tedesco, F. Obstetric and vascular antiphospholipid syndrome: same antibodies but different diseases? Nature Reviews Rheumatology 2018, 14, 433–440. [Google Scholar] [CrossRef] [PubMed]
- Van Os, G.; Meijers, J.; Agar, C.; Seron, M.; Marquart, J.; Åkesson, P. . Mörgelin, M. Induction of anti-β2-glycoprotein I autoantibodies in mice by protein H of Streptococcus pyogenes. Journal of Thrombosis and Haemostasis 2011, 9, 2447–2456. [Google Scholar] [CrossRef] [PubMed]
- Albert, L.J.; Inman, R.D. Molecular Mimicry and Autoimmunity. New Engl. J. Med. 1999, 341, 2068–2074. [Google Scholar] [CrossRef]
- Oldstone, M.B. Molecular Mimicry: Its Evolution from Concept to Mechanism as a Cause of Autoimmune Diseases. Monoclon. Antibodies Immunodiagn. Immunother. 2014, 33, 158–165. [Google Scholar] [CrossRef]
- Blank, M.; Krause, I.; Magrini, L.; Spina, G.; Kalil, J.; Jacobsen, S.; Thiesen, H.J.; Cunningham, M.W.; Guilherme, L.; Shoenfeld, Y. Overlapping humoral autoimmunity links rheumatic fever and the antiphospholipid syndrome. Rheumatology 2006, 45, 833–841. [Google Scholar] [CrossRef] [PubMed]
- Martirosyan, A.; Aminov, R.; Manukyan, G. Environmental Triggers of Autoreactive Responses: Induction of Antiphospholipid Antibody Formation. Front. Immunol. 2019, 10, 1609. [Google Scholar] [CrossRef]
- Blank, M.; Krause, I.; Fridkin, M.; Keller, N.; Kopolovic, J.; Goldberg, I. . Shoenfeld, Y. Bacterial induction of autoantibodies to β2-glycoprotein-I accounts for the infectious etiology of antiphospholipid syndrome. The Journal of clinical investigation 2002, 109, 797–804. [Google Scholar] [CrossRef]
- Ruff, W.E.; Dehner, C.; Kim, W.J.; Pagovich, O.; Aguiar, C.L.; Yu, A.T.; Roth, A.S.; Vieira, S.M.; Kriegel, C.; Adeniyi, O.; et al. Pathogenic Autoreactive T and B Cells Cross-React with Mimotopes Expressed by a Common Human Gut Commensal to Trigger Autoimmunity. Cell Host Microbe 2019, 26, 100–113. [Google Scholar] [CrossRef]
- Ruff, W.E.; Vieira, S.M.; Kriegel, M.A. The Role of the Gut Microbiota in the Pathogenesis of Antiphospholipid Syndrome. Curr. Rheumatol. Rep. 2014, 17, 1–10. [Google Scholar] [CrossRef]
- van Mourik, D.J.M.; Salet, D.M.; Middeldorp, S.; Nieuwdorp, M.; van Mens, T.E. The role of the intestinal microbiome in antiphospholipid syndrome. Front. Immunol. 2022, 13, 954764. [Google Scholar] [CrossRef]
- Vieira, S.M.; Yu, A.; Pagovich, O.E.; Tiniakou, E.; Sterpka, J.; Kriegel, M.A. (2013). Depletion Of The Gut Microbiota Prevents beta (2)-Glycoprotein I Antibody Production and Mortality In a Model Of Antiphospholipid Syndrome. Paper presented at the ARTHRITIS AND RHEUMATISM.
- Abdel-Wahab, N.; Talathi, S.; A Lopez-Olivo, M.; E Suarez-Almazor, M. Risk of developing antiphospholipid antibodies following viral infection: a systematic review and meta-analysis. Lupus 2017, 27, 572–583. [Google Scholar] [CrossRef]
- Mendoza-Pinto, C.; García-Carrasco, M.; Cervera, R. Role of Infectious Diseases in the Antiphospholipid Syndrome (Including Its Catastrophic Variant). Curr. Rheumatol. Rep. 2018, 20, 62. [Google Scholar] [CrossRef] [PubMed]
- Palomo, I.; Alarcón, M.; Sepulveda, C.; Pereira, J.; Espinola, R.; Pierangeli, S. Prevalence of antiphospholipid and antiplatelet antibodies in human immunodeficiency virus (HIV)-infected Chilean patients. J. Clin. Lab. Anal. 2003, 17, 209–215. [Google Scholar] [CrossRef]
- Bowles, L.; Platton, S.; Yartey, N.; Dave, M.; Lee, K.; Hart, D.P.; MacDonald, V.; Green, L.; Sivapalaratnam, S.; Pasi, K.J.; et al. Lupus Anticoagulant and Abnormal Coagulation Tests in Patients with Covid-19. New Engl. J. Med. 2020, 383, 288–290. [Google Scholar] [CrossRef]
- Noakes, D.; Evans, K.; Pathansali, R. The return of a former foe: syphilis with antiphospholipid syndrome as a cause of acute stroke. JRSM Open 2017, 8. [Google Scholar] [CrossRef]
- Miko, E.; Csaszar, A.; Bodis, J.; Kovacs, K. The Maternal–Fetal Gut Microbiota Axis: Physiological Changes, Dietary Influence, and Modulation Possibilities. Life 2022, 12. [Google Scholar] [CrossRef] [PubMed]
- Markowiak-Kopeć, P.; Śliżewska, K. The Effect of Probiotics on the Production of Short-Chain Fatty Acids by Human Intestinal Microbiome. Nutrients 2020, 12, 1107. [Google Scholar] [CrossRef] [PubMed]
- Iacob, S.; Iacob, D.G.; Luminos, L.M. Intestinal Microbiota as a Host Defense Mechanism to Infectious Threats. Front. Microbiol. 2019, 9, 3328. [Google Scholar] [CrossRef]
- Hou, K.; Wu, X.; Chen, Y.; Wang, Q.; Zhang, D.; Xiao, C.; Zhu, D.; Koya, J.B.; Wei, L.; Li, J.; Chen, S. Microbiota in health and diseases. Signal Transduction and Targeted Therapy 2022, 7. [Google Scholar] [CrossRef]
- Carabotti, M.; Scirocco, A.; Maselli, M.A.; Severi, C. The gut-brain axis: Interactions between enteric microbiota, central and enteric nervous systems. Annals of Gastroenterology: Quarterly Publication of the Hellenic Society of Gastroenterology 2015, 28, 203–209. [Google Scholar]
- Chen, Y.; Xu, J.; Chen, Y. Regulation of Neurotransmitters by the Gut Microbiota and Effects on Cognition in Neurological Disorders. Nutrients 2021, 13. [Google Scholar] [CrossRef]
- Bendriss, G.; MacDonald, R.; McVeigh, C. Microbial Reprogramming in Obsessive–Compulsive Disorders: A Review of Gut–Brain Communication and Emerging Evidence. Int. J. Mol. Sci. 2023, 24, 11978. [Google Scholar] [CrossRef]
- Gomaa, E.Z. Human gut microbiota/microbiome in health and diseases: a review. Antonie van Leeuwenhoek 2020, 113, 2019–2040. [Google Scholar] [CrossRef] [PubMed]
- Shapira, M. Gut Microbiotas and Host Evolution: Scaling Up Symbiosis. Trends Ecol. Evol. 2016, 31, 539–549. [Google Scholar] [CrossRef] [PubMed]
- Jethwani, P.; Grover, K. Gut microbiota in health and diseases—a review. Int. J. Curr. Microbiol. Appl. Sci. 2019, 8, 1586–1599. [Google Scholar] [CrossRef]
- Odamaki, T.; Kato, K.; Sugahara, H.; Hashikura, N.; Takahashi, S.; Xiao, J.-Z.; Abe, F.; Osawa, R. Age-related changes in gut microbiota composition from newborn to centenarian: a cross-sectional study. BMC Microbiol. 2016, 16, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Hasan, N.; Yang, H. Factors affecting the composition of the gut microbiota, and its modulation. PeerJ 2019, 7, e7502. [Google Scholar] [CrossRef]
- Martín, R.; Jiménez, E.; Heilig, H.; Fernández, L.; Marín, M.L.; Zoetendal, E.G.; Rodríguez, J.M. Isolation of Bifidobacteria from Breast Milk and Assessment of the Bifidobacterial Population by PCR-Denaturing Gradient Gel Electrophoresis and Quantitative Real-Time PCR. Appl. Environ. Microbiol. 2009, 75, 965–969. [Google Scholar] [CrossRef]
- German, J.B.; Freeman, S.L.; Lebrilla, C.B.; Mills, D.A. Human milk oligosaccharides: evolution, structures and bioselectivity as substrates for intestinal bacteria. Nestle Nutr Workshop Ser Pediatr Program. 2008, 62, 205–222. [Google Scholar]
- Fukuda, S.; Toh, H.; Hase, K.; Oshima, K.; Nakanishi, Y.; Yoshimura, K.; Tobe, T.; Clarke, J.M.; Topping, D.L.; Suzuki, T.; et al. Bifidobacteria can protect from enteropathogenic infection through production of acetate. Nature 2011, 469, 543–547. [Google Scholar] [CrossRef]
- Ouwehand, A.; Isolauri, E.; Salminen, S. The role of the intestinal microflora for the development of the immune system in early childhood. Eur. J. Nutr. 2002, 41, 1–1. [Google Scholar] [CrossRef]
- Zwielehner, J.; Liszt, K.; Handschur, M.; Lassl, C.; Lapin, A.; Haslberger, A.G. Combined PCR-DGGE fingerprinting and quantitative PCR indicates shifts in fecal population sizes and diversity of Bacteroides, Bifidobacteria and Clostridium cluster IV in institutionalized elderly. Exp Gerontol. 2009, 44, 440–446. [Google Scholar] [CrossRef] [PubMed]
- Tektonidou, M.G.; Andreoli, L.; Limper, M.; Amoura, Z.; Cervera, R.; Costedoat-Chalumeau, N.; Cuadrado, M.J.; Dörner, T.; Ferrer-Oliveras, R.; Hambly, K.; et al. EULAR recommendations for the management of antiphospholipid syndrome in adults. Ann. Rheum. Dis. 2019, 78, 1296–1304. [Google Scholar] [CrossRef] [PubMed]
- Brennecke, D.; Duarte, B.; Paiva, F.; Caçador, I.; Canning-Clode, J. Microplastics as vector for heavy metal contamination from the marine environment. Estuarine, Coast. Shelf Sci. 2016, 178, 189–195. [Google Scholar] [CrossRef]
- Campana, A.M.; Laue, H.E.; Shen, Y.; Shrubsole, M.J.; Baccarelli, A.A. Assessing the role of the gut microbiome at the interface between environmental chemical exposures and human health: Current knowledge and challenges. Environ. Pollut. 2022, 315, 120380–120380. [Google Scholar] [CrossRef] [PubMed]
- Cox, L.M.; Yamanishi, S.; Sohn, J.; Alekseyenko, A.V.; Leung, J.M.; Cho, I.; Kim, S.G.; Li, H.; Gao, Z.; Mahana, D.; et al. Altering the Intestinal Microbiota during a Critical Developmental Window Has Lasting Metabolic Consequences. Cell 2014, 158, 705. [Google Scholar] [CrossRef] [PubMed]
- Dempsey, J.L.; Little, M.; Cui, J.Y. Gut microbiome: An intermediary to neurotoxicity. NeuroToxicology 2019, 75, 41–69. [Google Scholar] [CrossRef]
- Deng, Yongfeng, Zehua Yan, Ruqin Shen, Meng Wang, Yichao Huang, Hongqiang Ren, Yan Zhang, and Bernardo Lemos. 2020.
- Cause Aggravated Adverse Effects in the Mouse Gut.” Environment International 143: 105916.
- Giambò, F.; Costa, C.; Teodoro, M.; Fenga, C. Role-Playing Between Environmental Pollutants and Human Gut Microbiota: A Complex Bidirectional Interaction. Front. Med. 2022, 9, 810397. [Google Scholar] [CrossRef]
- Giambò, F.; Leone, G.M.; Gattuso, G.; Rizzo, R.; Cosentino, A.; Cinà, D.; Teodoro, M.; Costa, C.; Tsatsakis, A.; Fenga, C. Genetic and Epigenetic Alterations Induced by Pesticide Exposure: Integrated Analysis of Gene Expression, MicroRNA Expression, and DNA Methylation Datasets. International Journal of Environmental Research and Public Health 2021, 18, 8697. [Google Scholar] [CrossRef]
- Gombart, A.F.; Pierre, A.; Maggini, S. A Review of Micronutrients and the Immune System–Working in Harmony to Reduce the Risk of Infection. Nutrients 2020, 12, 236. [Google Scholar] [CrossRef]
- Li, B.; Ding, Y.; Cheng, X.; Sheng, D.; Xu, Z.; Rong, Q.; Wu, Y.; Zhao, H.; Ji, X.; Zhang, Y. Polyethylene microplastics affect the distribution of gut microbiota and inflammation development in mice. Chemosphere 2019, 244, 125492. [Google Scholar] [CrossRef] [PubMed]
- Maier, L.; Goemans, C.V.; Wirbel, J.; Kuhn, M.; Eberl, C.; Pruteanu, M.; Müller, P.; Garcia-Santamarina, S.; Cacace, E.; Zhang, B.; et al. Unravelling the collateral damage of antibiotics on gut bacteria. Nature 2021, 599, 120–124. [Google Scholar] [CrossRef] [PubMed]
- Murphy, J.R.; Paul, S.; Dunlop, A.L.; Corwin, E.J. Maternal peripartum antibiotic exposure and the risk of postpartum depression. Res. Nurs. Heal. 2018, 41, 369–377. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, C.F.A.; Silveira, G.G.d.O.S.; Cândido, E.d.S.; Cardoso, M.H.; Carvalho, C.M.E.; Franco, O.L. Effects of Antibiotic Treatment on Gut Microbiota and How to Overcome Its Negative Impacts on Human Health. ACS Infect. Dis. 2020, 6, 2544–2559. [Google Scholar] [CrossRef]
- Tavalire, H.F.; Christie, D.M.; Leve, L.D.; Ting, N.; Cresko, W.A.; Bohannan, B.J.M. Shared Environment and Genetics Shape the Gut Microbiome after Infant Adoption. mBio 2021, 12. [Google Scholar] [CrossRef]
- Tikka, C.; Manthari, R.K.; Ommati, M.M.; Niu, R.; Sun, Z.; Zhang, J.; Wang, J. Immune disruption occurs through altered gut microbiome and NOD2 in arsenic induced mice: Correlation with colon cancer markers. Chemosphere 2020, 246, 125791. [Google Scholar] [CrossRef]
- Verma, H.; Phian, S.; Lakra, P.; Kaur, J.; Subudhi, S.; Lal, R.; Rawat, C.D. Human Gut Microbiota and Mental Health: Advancements and Challenges in Microbe-Based Therapeutic Interventions. Indian J. Microbiol. 2020, 60, 405–419. [Google Scholar] [CrossRef]
- Lim, W.; Crowther, M.A.; Eikelboom, J.W. Management of antiphospholipid antibody syndrome: a systematic review. JAMA 2006, 295, 1050–1057. [Google Scholar] [CrossRef]
- Garabatos, N.; Santamaria, P. Gut Microbial Antigenic Mimicry in Autoimmunity. Front. Immunol. 2022, 13, 873607. [Google Scholar] [CrossRef]
- Yun, Z.; Duan, L.; Liu, X.; Cai, Q.; Li, C. An update on the biologics for the treatment of antiphospholipid syndrome. Front. Immunol. 2023, 14. [Google Scholar] [CrossRef]

Table 1.
List of key microorganisms involved in aPL generation and thrombosis.
| Microorganism | aCL | anti-β2GP1 | LA | Thrombosis |
|---|---|---|---|---|
| Viral infection | ||||
| CMV | + | + | + | + |
| EBV | + | + | + | + |
| HIV | + | + | + | + |
| SARS-Cov-2 | + | + | + | + |
| Varicella zoster virus | + | + | + | + |
| Parvovirus B19 | + | + | + | + |
| Hepatitis A | + | - | - | + |
| Hepatitis B | + | + | + | - |
| Hepatitis C | + | + | + | + |
| Hepatitis D | + | + | + | - |
| Mumps | + | - | - | - |
| Rubella | + | - | - | - |
| Adenovirus | + | + | - | - |
| HTLV | + | + | - | - |
| Influenza A | + | - | - | + |
| Bacterial infections | ||||
| Mycobacterium leprae | + | + | + | + |
| Borrelia burgdorferi | + | + | + | + |
| Salmonella spp. | + | + | + | + |
| Streptococcus spp. | + | + | + | + |
| M. tuberculosis | + | + | - | + |
| Escherichia coli | + | + | - | + |
| Coxiella burnetii | + | - | + | - |
| Helicobacter pylori | + | + | - | - |
| Klebsiella spp. | + | - | - | - |
| Chlamydia | + | + | - | - |
| Mycoplasma pneumonia | + | + | - | + |
| Treponema pallidum | + | + | - | - |
| Parasitic infections | ||||
| Plasmodium malariae | + | + | - | + |
| Leishmania | + | + | + | - |
| Leptospira spp. | + | + | - | - |
| Plasmodium falciparum | + | - | - | - |
| Toxoplasmosis | + | - | - | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



