
Submitted:
25 August 2024
Posted:
27 August 2024
You are already at the latest version
Abstract
Colorectal cancer (CRC) is the third most commonly diagnosed type of cancer worldwide and is responsible for numerous deaths. 5-fluorouracil (5-FU) is an effective chemotherapy drug commonly used in the treatment of CRC, either as monotherapy or in combination with other drugs. However, half of CRC cases are resistant to 5-FU-based therapies. To contribute to the understanding the mechanisms underlying CRC resistance or recurrence after 5-FU-based therapies, we performed a comprehensive study integrating in silico, in vitro and in vivo approaches. We identified differentially expressed genes and enrichment of pathways associated with recurrence after 5-FU-based therapies. Using these bioinformatic data as a starting point, we selected a group of drugs that restored 5-FU sensitivity to 5-FU resistant cells. Interestingly, treatment with the novel Rac1 inhibitor, 1A-116, reversed morphological changes associated with 5-FU resistance back to a control-like status. Moreover, our in vivo studies have shown that 1A-116 affected tumor growth and the development of metastasis. All our data allowed us to postulate that targeting Rac1 represents a promising avenue for the development of new therapies for patients with CRC resistant to 5-FU-based therapies.
Keywords:
colorectal cancer
; resistance
; small GTPases
; Rac1
; repositioning
1. Introduction
Colorectal cancer (CRC) is the third most commonly diagnosed type of cancer worldwide, accounting for ten percent of all malignant diagnoses and responsible for numerous deaths, ranking third in terms of incidence, but second in terms of mortality [1] with estimations of increasing incidence for the next years [2]. Increased risk of CRC is normally associated with age, western lifestyle, diet, personal history and chronic intestinal diseases [3].
Most CRCs arise from previous adenomatous polyps. Although it remains uncertain the time it takes for an early adenoma to progress to CRC, the detection and removal of precancerous lesions before progressing to malignancy and metastasis provides a prevention strategy [1,4].
CRC treatment depends on factors such as patient’s health, tumor size and location, and the presence of metastasis, but surgical removal is the most common option [5] followed by administration of adjuvant chemotherapy when there is local-regional or distant invasion.
Since its discovery in 1957, chemotherapies based on 5-fluorouracil (5-FU, a fluorinated uracil analog) have remained the mainstay of adjuvant and palliative therapies for CRC patients [6]. 5-FU interferes with nucleosides metabolism by inhibiting the action of thymidylate synthase and missincorporating metabolites into DNA and RNA, leading to cytotoxicity and cell death [6,7,8]. Despite the clinical benefits and the extended use of 5-FU, response rates to 5-FU monotherapy are below 20% as most patients do not completely eliminate tumor cells, and tumor recurrence leads to poor outcomes [9]. To counteract this, various strategies have been developed to increase 5-FU effectiveness through the modulation of its intracellular and biochemical metabolism, mainly combining 5-FU with other cytotoxic drugs with different mechanisms of action. Combined chemotherapies FOLFOX (leucovorin+5-FU+xaliplatin), FOLFIRI (leucovorin+5-FU+irinotecan) and FOLFOXIRI (leucovorin+5-FU+oxaliplatin+irinotecan) are the most used in the clinic and have been used as the standard therapy for advanced CRC, increasing response rates up to 40-50% [10,11]. Unfortunately, despite the increased response CRC patient’s disease-free survival has not been efficiently extended [12,13], and half of CRCs are resistant to 5-FU-based therapies. Therefore, there is a need for studies focusing on characterizing resistance-mediators biological factors or identification of biomarkers to define which CRC population is most likely to respond to 5-FU-based therapies [14,15,16].
One of the main causes of failure in cancer treatment is the development of drug resistance by cancer cells. This is a very serious phenomenon since it causes the recurrence of the disease or even death [17]. Therapy resistance occurs when diseases become tolerant to pharmaceutical treatments. This concept was first considered when bacteria became resistant to certain antibiotics, but similar mechanisms have since been found in other diseases, including cancer. Some resistance mechanisms are specific to each pathology, while others, such as drug efflux can be observed at the microbial level and also in tumors, making them evolutionarily conserved [18]. Although many types of tumors are initially susceptible to chemotherapy, over time they may develop resistance to through this and other mechanisms, such as DNA mutations and metabolic changes that promote the inhibition and degradation of drugs [19]. Tumor resistance is not limited to conventional chemotherapeutic drugs, but also appears associated with the use of target therapies or biotherapies [20,21]. Consequently, numerous efforts are focused on exploring biomarkers that can predict or indicate the success of therapy.
In the case of CRC, 5-FU-based therapies increased response rates up to 40-50%, therefore strategies to improve clinical outcomes are required. To address this, a deeper understanding of the mechanisms associated with resistance or recurrence after 5-FU-based therapies is imperative.
On the other hand, Rac1 (Ras-related C3 botulinum toxin substrate 1) is a key member of the Rho GTPases family. It is well known that Rac1 is a regulator of actin-based cytoskeletal dynamics, modulating cell adhesion, morphology, and movement. Rac1 is highly expressed in different tumor types, and related to poor prognosis [22]. In tumors, it was described that Rac1 modulates cell cycle, apoptosis, proliferation, invasion, migration and angiogenesis. Rac1 also plays a key role in anti-tumor therapy and participates in immune escape mediated by the tumor microenvironment [22]. Increasingly, studies are reporting the role of Rac1 as a potential target for tumor therapy [23].
To contribute to understanding the mechanisms underlying CRC resistance to therapies, we have conducted a study integrating in silico, in vitro and in vivo approaches. First, we compared microarray gene expression data from 5-FU-treated CRC patients with and without recurrence after 5-FU monotherapy. Then, we extended the analysis to 5-FU-based therapies. Comparisons uncovered common enrichment pathways associated to chemotherapy resistance that allowed us to select drugs to overcome this phenomenon. Interestingly, inhibition of Rac1 by 1A-116 compound decreases the growth of 5-FU-resistant CRC, sensitizes cells to 5-FU therapy and prevents metastasis development, suggesting that therapies based on Rac1 inhibition could be of benefit to overcome therapy resistance.
2. Materials and Methods
2.1. Gene Expression Data Collection
Gene expression profiles of datasets GSE81653, GSE39582, and GSE72970 were downloaded from the NCBI Gene Expression Omnibus (GEO) database (https://www.ncbi.nlm.nih.gov/gds) using the GEOquery package [24,25]. These datasets contain gene expression profiling of tumor clinical samples from patients after the exposure to 5-FU alone and 5-FU based combined chemotherapy. Sample inclusion criteria for the analysis were: 1) not proceeding from studies of patients with familial hereditary polyposis, 2) including clinical information detailing the type of chemotherapy provided, and 3) presenting clinical information about tumor recurrence status. The expression values downloaded were normalized using RMA (Robust Multichip Average) [26] method from the affy or oligo packages depending on the chip model. Probe annotation was performed using the manufacturer-supplied annotation package: “hugene20sttranscriptcluster.db” [27] for the GSE81653 series and “hgu133plus2.db” [28] for the GSE39582 and GSE72970 series (a general flowchart of this work is presented in Figure 1).
If publicly available, information on clinicopathologic characteristics of patients was retrieved to explore their association with recurrent condition using Fisher's exact test, a significant association was considered for p-value < 0.05.
2.2. Identification of Differentially Expressed Genes in Patients Treated with 5-FU Monotherapy
Data analysis was carried out using the facilities of the CCT-Rosario Computational Center, member of the High Performance Computing National System (SNCAD, MincyT- Argentina) where data were introduced into the R/Bioconductor environment. Differentially expressed genes (DEGs) were identified in GSE39582 and GSE81653 datasets independently. Each dataset was divided in two groups: 1) patients treated with 5-FU monotherapy with tumoral recurrence after surgical tumor resection and 2) patients treated in the same way but without tumor recurrence. Detection platforms and sample sizes are shown in Table S1. A more detailed description of the selected samples is described in Table S2.
Four methods were used to obtain DEGs groups; two exploratory methods: fold change (“FC”) and expression in unusual proportion; and two non-parametric algorithms: the Significance Analysis of Microarrays (SAM) [29] and RankProd (RP) [30]. For DEGs selection, we used as cutoff criterion |log2fold change| > 1 for FC method. For UR, any observation far from the mean by more than two standard deviations was considered atypical. For the non-parametric hypothesis tests, genes with |log2fold change| > 1 and FDR ≤ 0.01 were considered.
2.3. Enrichment Analysis
Reactome signaling pathway enrichment analysis for DEGs was performed using an R package “ReactomePA” [31]. ReactomePA uses the hypergeometric model to assess whether the number of selected genes associated with a Reactome pathway is larger than expected. Pathways with an FDR < 0.05 were considered significantly enriched and were visualized using the dotplot tool. We generated an enrichment map and pre-clustered network using the “enchmap” and “cnetplot” tools to visualize relationships between pathways, and highlight genes related to the more significant terms.
2.4. Identification of Gene Expression Profile-Reversing Compounds
A computational drug repositioning analysis was performed using CLUE (Connectivity map Linked User Environment: https://clue.io/query; Broad Institute, Cambridge, MA; data version 1.1.1.2; software version 1.1.1.42), a cloud-based analysis platform that catalogs 473,647 expression signatures of human cell lines treated with 25,200 perturbagens [32]. CLUE computes a CMap score (tau) that measures the similarity of a queried gene set of up to 150 upregulated and 150 downregulated genes to existing drug-matched reference gene sets, from least similar or inverse (‒100) to most similar (100). From an initial list of 426 DEGs, we selected 150 upregulated genes by screening for genes detected as DEGs by three or more methods in both data sets, as well as those associated with pathways with the highest enrichment score in the recurrent phenotype. Since the number of down-regulated DEGs was lower than 150, all of them were used. The reversing drugs of interest were selected using a negative threshold of CMap score.
2.5. Integrative Meta-Analysis
To explore whether the enriched pathways between patients with and without tumor recurrence after 5-FU treatment were maintained when patients treated with other types of chemotherapy were added to the expression matrix, we performed a meta-analysis. This technique is particularly useful for combining several datasets from the same disease when these are limited in size, therefore improving their statistical power. This time, instead of analyzing the data series independently, a large matrix was constructed merging patient’s gene expression data from GSE81653, GSE39582, and GSE72970 series. In addition to the patients treated with 5-FU already used in the previous analysis, others treated with FOLFIRI and FOLFOX were included. Data merging was performed using the empirical Bayes methods (ComBat, from Sva package) [33] to reduce confounding factors due to non-biological variations between the studies. Data about platforms and sample sizes are shown in Table S3. A more detailed description of the selected samples added to the analysis is found in Table S4.
2.6. Feature Selection
Feature selection is described as a process in which a subset of relevant features is selected from a larger data set. These features can be used for several purposes like model construction, differential analysis, enrichment exploration, etc. To select the most explanatory genes among the phenotypes from the combined gene expression matrix, we used the sigFeature package [34]. This package employs a combination of Vector Support Machine and Recursive Feature Elimination (SVM-RFE) algorithms to produce a ranked list of genes [35]. The SVM-RFE is a backward feature elimination technique that iteratively removes features based on SVM classifier weights. In each iteration, an SVM model is built based on the current features subset “F”, and the weight of each feature in “F” is calculated. The features are then ranked based on weight and the bottom-ranked features are removed from F until it is empty. The top-ranked features that are discarded in the last iteration are considered the most informative between the phenotypes. The number of features retained in the analysis depends on their particular future use. In this work, we selected the top 5000 more informative features to improve their potential to predict the biological signature of the data set.
2.7. Gene Set Enrichment Analysis (GSEA)
GSEA is a computational method used to determine whether an a priori defined set of genes shows concordant and statistically significant differences between two biological states [36]. We employed the java GSEA Desktop Application v4.2.3 to perform the GSEA analysis on 5000 more informative features selected. The gene sets available from The Molecular Signatures Database 3.0 (MSigDB) were employed [37]; genesets composed of less than 15 or more than 500 genes were excluded. The phenotype label was set as “recurrent” vs “non-recurrent”. The t-statistic mean of the genes was computed in each gene set using a permutation type test with 1000 replications. Up-regulated gene sets were defined by a normalized enrichment score (NES) > 0 and down-regulated by NES < 0. Gene sets with an FDR-P value ≤ 0.05 were chosen as significantly enriched.
2.8. Detection of Motifs and Transcriptional Factors
For the discovery of transcription factor binding sites (motifs) in the promoters of co-regulated genes, we used the Cytoscape plug-in iRegulon [38,39]. A collection of 9713 position weight matrices (PWMs) was applied to analyze 10 kb centered around the transcription start site. DNA logos corresponding to each motif were extracted and the main transcriptional factors binding to them and their sets of direct targets (metatargetnoma) were screened. Cut-off criteria used in the analysis were enrichment score threshold = 5, ROC threshold for AUC calculation = 0.03, rank threshold = 5000, minimal identity between orthologous genes = 0.05, FDR > 0.001, and normalized enrichment (NES) > 3.
2.9. Predictor Genes Detection
Important predictors genes from DEGs list and “reactome_signaling_by_rho_gtpases” gene set (https://www.gsea-msigdb.org/gsea/msigdb/cards/REACTOME_SIGNALING_BY_RHO_GTPASES) were selected via least absolute shrinkage and selection operator (LASSO) logistic regression [40] in individuals GEO series and in the merged matrix using the glmnet R package. To carry out the analysis, patients from each dataset were divided into training and validation sets in an 80-20 ratio, respectively. Optimal values for the penalty parameter λ were determined through 10-fold cross-validations. For model construction, we employed the lambda value giving minimal mean cross-validated error (lambda.min). The LASSO coefficient for recurrence predictors genes for each gene list was extracted. Receiver operating characteristic (ROC) curves were plotted to validate the prediction efficiency of the model using the ROCR and pROC R packages [41,42]. We employed survival package [43] to perform a univariate Cox regression to assess the effect of the change in expression of individual predictor gene on the survival of patients in each dataset; genes with a LogRank < 0.01 were retained.
2.10. PPI Network Construction and Hub Gene Identification
The PPI network was constructed using STRING public online database [44] and exported to Cytoscape (version 3.9.1) for viewing and analysis [39]. Cytoscape plug-in CytoHubba (version 0.1) [45] was employed to retrieve the top 20 hub genes based on maximal clique centrality (MCC) algorithm. To expand the network, main interactor genes were rescued using the network expansion function of STRING plug-in. Top 20 interactors with a selectivity of 0.5 were included in the network
2.11. Drugs
For each drug, a 10 mM stock solution was prepared and stored at −20°C. Ivermectin (Parafarm) was dissolved in 50% DMSO solution, the concentration of DMSO in the final dilutions did not exceed 0.03%. Amitriptyline (Parafarm) was dissolved in sterile distilled water. Commercial 20mg/ml irinotecan solution (Kemex) and 50mg/ml 5-FU solution (Fada Pharma) were diluted in sterile distilled water to reach final stocks concentration. 1A-116 [46] was kindly provided by Dr. Georgina Cardama (Laboratorio de Oncología Molecular, Universidad Nacional de Quilmes). 1A-116 stock was prepared by dissolving the drug in acidic water (sterile distilled water brought to pH 1-2 using a 100 mM HCl solution). Once dissolved, pH of the final solution was adjusted to 5.5-6 with 100 mM NaOH and filtered. Unless indicated otherwise, doses used of drugs were 5µM 5-FU; 20µM amitriptyline; 30µM irinotecan, 15µM ivermectin and 20µM 1A-116.
2.1. Cell Culture
5-FU resistant cell lines CT265FUR, HCT1165FUR or HT295FUR were produced in our laboratory as described before [47] from CT26 (chemically induced BALB/c mice-derived colorectal carcinoma), HCT116 (male human colon adenocarcinoma) and HT29 (female human colon adenocarcinoma) cells respectively. Cells were cultured in DMEM (HCT116 and HT29) or RPMI (CT26) media, supplemented with 10% fetal bovine serum (FBS; Natocor, Argentina), penicillin (10 μg/ml), streptomycin (100 μg/ml) and L-glutamine (2 mM, DMEM medium). Cells were maintained at 37°C in a 5% CO2 atmosphere and routinely tested for mycoplasma.
2.13. Cells Immunostaining Techniques
7× 103 cells were cultured in coverslips for 24 h, treated for the 36 h, fixed in 4% paraformaldehyde and permeabilized with a 0.5% PBS-Triton and blocked with BSA (2%, Sigma). Cells were first incubated α-Tubulin (Sigma, 1:1000 dilution) and Beta-Catenin (Santa Cruz, sc-59737; 1/50 dilution) or E-Cadherin (67A4) (Santa Cruz, sc-21791; 1/50 dilution), and then with Alexa Fluor 488-conjugated secondary antibody (Invitrogen, 1:500 dilution), and counterstained with DAPI (Sigma, 1:10000 dilution) and phalloidin-rhodamine conjugate (Invitrogen, 1:2000 dilution) to observe cell nucleus and actin skeleton, respectively. Coverslips were fixed to a slide with Mowiol and observed under a Nikon Ti2E fluorescent microscope or and Zeiss LSM880 confocal microscope. Cell and nuclear areas were measured with the ImageJ software.
2.14. Viability Assays
For in vitro cell viability assays 1x104 CT265FUR, HCT1165FUR or HT295FUR cells were seeded on 96-well plates and incubated for 36 h at 37°C at a 5% CO2 atmosphere with increasing doses of the individual drugs and combinations of selected doses with 5 µM 5-FU. Controls were treated in the same way with corresponding vehicle to each drug. After the incubation time, treatments were removed and an MTT assay (Sigma Aldrich) was performed as described before [47]. Viability was expressed as the percentage of control untreated samples. The concentration of drugs that decreased cell proliferation by 50% (IC50) as compared to controls was calculated with GraphPad Prism v8.0 (GraphPad Sofware, La Jolla, CA, USA). When indicated, viable cells were counted using Trypan Blue (0,4%; Sigma), and viability was expressed as the percentage of control untreated samples.
2.15. Immunoblotting
CRC cells were seeded in 60 mm plates until they achieved 80% confluence. Cells were lysed with RIPA buffer containing protease inhibitor cocktail (Roche Diagnostics, Mannheim, Germany) and then detached from the plates to collect protein extracts. Protein levels were quantified using the Lowry assay, and 40 µg of protein per sample was denatured for 5 minutes at 95 °C in SDS-PAGE sample buffer before being loaded onto gels. Samples were resolved on 8% SDS–polyacrylamide gels and transferred to PVDF membranes (Amersham Hybond P, GE Healthcare Life Sciences) for 90 minutes with constant current at 4 °C. Membranes were blocked with 1% (w/v) BSA in TBS-Tween (50 mM Tris, 150 mM NaCl, 0.05% Tween, pH 7.5) for 60 minutes at room temperature and then incubated with the specified primary antibody overnight at 4 °C. The primary antibodies used included β-catenin (BD Biosciences, 1:1000 dilution) and E-cadherin (BD Biosciences, 1:1000 dilution). After three washes with TBS-Tween to remove the primary antibodies, membranes were incubated with the appropriate peroxidase-conjugated secondary antibody (BioRad, 1:5000 dilution) for one hour at room temperature. Detection was carried out using chemiluminescence (Bio-Lumina; Kalium Technologies, Argentina) and imaged with a Licor C-Digit Blot Scanner (LI-COR Biosciences) according to the manufacturer's instructions. Quantification was performed by densitometry using Image J software. Cropped images are displayed in the main figures, with full-length membranes shown in the supplementary information.
2.16. GTP-Rac1 Pull-Down Assay
GST-Pak1 pull-down experiment to determine Rac1 activity in cells was performed as indicated [48]. Briefly, exponentially growing cells were harvested and resuspended in lysis buffer containing 20 mM Tris- HCl [pH 7.5], 150 mM NaCl, 5 mM MgCl2, 0.5% Triton X-100, 10 mM beta-glycerophosphate, 1 mM DTT, Cømplete (Roche), and 10 µg of GST fusion protein containing the Pak1 Rac1 binding domain (GST-Pak1 RBD, bacterially-expressed). After incubations for 10 min on ice, cell lysates were pre-cleared by centrifugation at 14,000 rpm for 10 min at 4°C and supernatants incubated with glutathione-Sepharose beads (GE Healthcare Life Biosciences) for 1h at 4°C under gentle rotation. After extensive washes in lysis buffer, protein complexes were released by boiling in SDS-PAGE sample buffer, separated electrophoretically, transferred onto nitrocellulose filters, and analyzed by immunoblotting using an anti-Rac1 antibody (BD Biosciences, 1:1000). GTP-Rac1 levels were quantified with the ImageJ analysis software using as normalizing control the total levels of Rac1 found in each cell lysate.
2.17. Animal Studies
8 weeks-old BALB/c female mice were obtained from the School of Veterinary Sciences at the National University of La Plata and treated in accordance with the Canadian Council on Animal Care and ARRIVE guidelines. Animals were maintained in the CIPReB facilities (Centro de Investigación y Producción de Reactivos Biológicos, Medicine School, National University of Rosario). 1x106 CT265FUR viable cells were resuspended in PBS (100 μl) and injected subcutaneously into the right flank of each animal. For all experiments, mice were distributed and treated as follows: control, consist in a daily intraperitoneal (ip) administration of vehicle (1% absolute alcohol solution); ivermectin, ip administration of 2 mg/kgBW/day (dissolved in absolute alcohol and then diluted in water to the final concentration); 1A-116, ip administration of 5 mg/kg BW/day in sterile water; 5-FU, ip administration 20 mg/kgBW/week in sterile water; iver + 5-FU, ip administration of ivermectin and 5-FU treatments; 1A-116+5-FU, ip administration of 1A-116 and 5-FU treatments.
Animals were periodically weight and checked for changes in skin, fur, eyes, secretions, excretions and autonomic activity (lacrimation, pilo-erection, unusual respiratory pattern, movement, etc).
Tumor volumes were calculated as V = 0.4ab2, where a is the measurement of the tumor along its longest axis and b its shortest. When any of the groups reached the ethically permitted tumor volume, the animals were euthanized. Lungs, spleens and tumors were removed, fixed and stained with hematoxylin-eosin for histological evaluation and detection and counting of micrometastases in an Olympus BX40 microscope.
For intrasplenic inoculation of cells, mice were anesthetized by intraperitoneal injection of acepromazine/ketamine/midazolam (50mg/kg, 100mg/kg and 50mg/kg, respectively). A small incision was made to access the spleen and allow injection of 1x106 CT265FUR viable cells resuspended in PBS (100 μl). Two days after surgery, animals were distributed in groups as described and treatments were initiated. Three weeks after injection, animals were sacrificed, for collection and weighting of spleens and livers.
For subcutaneous tumor development, 2 independent experimental rounds were performed. For the first round N = 4/group; for the second round, N=6/group. For intrasplenic injection the number of animals used was N=6/group.
2.18. Statistical Analysis
Statistical analyzes were carried out using the GraphPad Prism 8.0 software (GraphPad Software, Inc., La Jolla, CA, USA). Single comparisons between two groups were performed with the Student’s-test, whereas for multiple comparisons the ANOVA followed by Tukey’s multiple comparisons post-test was employed. Correlation between the two variables was assessed with Spearman’s rank correlation coefficient (r). For differential gene expression analysis, statistical significance was tested with the Student’s t-test followed by a False Discovery Rate (FRD) correction with Benjamini–Hochberg procedure. Survival analysis was implemented according to the Kaplan–Meier analysis and log-rank test. Overall survival (OS) was defined as the time between the date of surgery and the date of death or the date of the last follow-up. In all cases, p-values less than 0.05 were considered statistically significant and were marked with an asterisk as follows: *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001.
3. Results
3.1. Identification of Differentially Expressed Genes and Key Pathways in Recurrent CRC after 5-FU Monotherapy
To identify DEGs and key pathways associated with 5-FU resistance, raw data of the datasets GSE39582 and GSE81653 were downloaded from GEO database. Each dataset was normalized using the RMA method (Figure S1A,B). Dataset GSE39582 contained a total of 585 samples, 82 of which met the requirements described in Section 2.1. Dataset GSE81653 contained 593 samples, 192 of which met the requirements. The selected patients from each study were divided into patients with and without tumor relapse groups. Before performing the analysis, probes with intensity values close to chip background were filtered and discarded (the proportion of probes removed is shown in Figure S1C,D).
In order to explore the putative association between recurrence and clinical parameters, correlation was assessed using Fisher's exact test. The results indicated that 5-FU resistance was not associated to age, TNM stage (Tumor, Node, Metastasis staging system), or the main mutations described for CRC (KRAS, TP53 and BRAF; Table S5). As no correlation was noted, we performed enrichment analysis of gene sets associated to recurrent and non-recurrent phenotypes for both datasets. Through this approach, we observed that coordinate expression of genes grouped in categories linked to cell adhesion and migration was associated to a recurrent phenotype, while a good response to 5-FU monotherapy correlated with immune system and complement activation (Figure 2A,B and Tables S6-S7).
In parallel, we aimed to identify DEGs associated to the 5-FU-resistant phenotype. To select DEGs in each dataset independently we employed four methods, two exploratory (fold change [FC] and unusual ratio [UR]) and two non-parametric hypothesis tests (RankProd [RP] and the Significance Analysis of Microarrays [SAM]). Cut-off criterions for DEGs selection were described in Materials and Methods. DEGs lists were compared to extract common genes detected by at least two methods in both studies. The number of genes obtained by each method in each dataset was represented by a Venn diagram (Figure 2C,E). A total of 388 upregulated and 39 downregulated common DEGs were selected (Table S8). In order to detect pathways enriched in recurrent phenotype, we performed an over-representation analysis (ORA). The results of this analysis indicated that the main upregulated pathways were associated with signaling mediated by Rho GTPase proteins and their effectors (Figure 2D), while among the downregulated pathways were found those related to cytokine receptors, receptors binding to peptide ligands and the immune response (Figure 2F).
Transcription factors are key regulators of biological processes that function by binding to genes regulatory regions. Each transcription factor recognizes a collection of DNA sequences or binding sites that can be represented as motifs. Motif characterization is important for understanding the regulatory functions of transcription factors shaping gene regulatory networks. To go further in characterizing expression changes associated to resistance, we look for common transcriptional factors binding sites present at the promotor regions of upregulated genes, finding that the most relevant factors include Serum Response Factor (SRF), Myocyte Enhancer Factor 2C and 2A (MEF2C, MEF2A), Msh Homeobox 2 (MSX2), and common motives were found to FOXO1, HMGA2, HMGA1, FOXA2. FOXA1. SOX10, FOS, JUN, HLTF and RUNX3 (Figure 3A). Genes specifically regulated by each transcription factor were extracted to perform a functional enrichment analysis (P < 0.05), through a word cloud graph, which indicates that the most relevant cellular processes modulated are related to signaling, GTPases and E-cadherin (Figure 3B).
To visualize the most relevant relationships between pathways and functions associated to the 5-FU-resistant phenotype, we constructed an enrichment map that indicates over-representation of two particular clusters: Rho GTPases and TGFbeta signaling (Figure 3C,D). 5-FU-resitance associated DEGs are highly connected genes by 20 hub genes, as visualize in Figure 3E.
3.2. Determinants of Resistance to 5-FU-Based Therapies
To go further in the search for determinants to resistance in CRC, we decided to extend our approach to databases of patients receiving therapies based on 5-FU, including FOLFOX and FOLFIRI. This approach allowed us to increase our samples to 567 to perform more robust analyses. As in the case of 5-FU monotherapy, no evident correlation was observed between resistance and available clinical parameters (Table S9). Unbiased GSEAs associated with the transcriptome of recurrent phenotype following 5-FU-based chemotherapies using Reactome, GO, and KEGG databases indicated a positive enrichment for Rho GTPases activate PKNs and Formation of beta-catenin:TCF transactivating complex, and negative enrichment for immunoregulatory interactions between lymphoid and non-lymphoid cell and complement cascade (Figure 4A, Table S10). Moreover, after selecting the 5000 most explanatory genes in the recurrent phenotype after 5-FU-based chemotherapies by the Recursive Feature Elimination algorithm (RFE), and a chemical and genetic perturbations database (MSigDB) we found a positive enrichment of gene sets associated with resistance to other therapies generally used to treat tumors other than CRC such as gefitinib, tamoxifen, gemcitabine and radiation (Figure 4B, Table S11), indicating that some of the identified pathways could be orchestrating general resistance mechanisms. Surprisingly, gene sets more present in this 5000 subset were related to neutrophil degranulation and Rho GTPases, among which Rac1 seems to be the most relevant (Figure 4C). Consistently, through pull-down experiments in CRC cells, we observed a significant increase in Rac1 activity associated with 5-FU resistance (Figure 4D). As it was the case for 5-FU monotherapy, we found binding motifs for SRF, JUN and FOS transcription factors present at the promotor regions of upregulated genes (Figure 4E) associated to 5-FU-based therapies resistance.
3.3. Selection of Drugs to Overcome 5-FU Resistance in CRC
In order to overcome 5-FU resistance, we search for compounds with the ability to reverse the expression of genes and pathways identified in our previous analysis by LINCS. This tool allows entering 150 upregulated and 150 downregulated genes. For 5-FU monotherapy, we used all common downregulated genes as they were less than 150. To reduce the number of upregulated genes to 150, we selected DEGs detected by three or more statistical methods (in both databases) and those with the higher association (with a lower P value) with overrepresented pathways in functional enrichment analysis (Table S12). The platform detected similar and opposite expression profiles in nine cell lines, including HT29.
Through this approach we obtained a list of compounds that could potentially reverse 5-FU resistant phenotype (Table S13). Some of these compounds are currently used in the clinic combined to 5-FU, such as irinotecan [49,50] or to treat other types of recurrent cancers such as topotecan [51,52]; but we also found other compounds such as a norepinephrine inhibitor (amitriptyline), MEK inhibitors, a COX inhibitor, ivermectin and the Rho-associated kinase inhibitor (Rockout) among others. Accordingly, cell perturbations that could reverse the expression profile associated to 5-FU resistance determined by the same platform include inhibition of Raf, MEK, PKC or Src and loss of function of Rho GTPases activating proteins (Table S14).
3.4. Ivermectin and the Rac1 Inhibitor 1A-116 Restore 5-FU Sensitivity to 5-FU Resistant Cells
As drug repositioning provides cheaper, effective, and safe drugs with fewer side effects and fastens drug development, we focused on irinotecan, amitriptyline and ivermectin to confirm by cell studies their potential to sensitize 5-FU-resistant CRC cells. In parallel, as our studies highlighted a prominent role of the Rho GTPase Rac1 on 5-FU-resistance modulation, we decided to explore the effects of 1A-116, a novel Rac1 inhibitor previously reported to be promising to treat other cancer types [46,53,54,55]. To characterize the effect of these drugs on resistance, we used three 5-FU resistant cells lines generated as described before [47]. IC50 values for each selected drug were calculated on sensitive and resistant cell lines (Table S15).
To evaluate the potential effect of 1A-116 and selected repurposing drugs on reversing resistance of CRC cells, we treated resistant cells with 5-FU in the presence of each drug. For all the assays, we selected doses for each drug with a minimal statistical effect on cell viability as evaluated by MTT-based assays (Figure S2 and Table S15). We found that incubation of cells with irinotecan, ivermectin and 1A-116 improved responses to 5-FU in all cell lines tested (Figure 5A), while amitriptyline only had a partial effect on HT29 and CT26 resistant cells. For 1A-116 treatment, we confirmed the inhibition of Rac1 activation by this compound and its effect on reversing 5-FU resistance by counting viable cells after treatment (Figure S3A,B).
In vitro generation of 5-FU-resistance is associated to cell morphological changes as it was previously described for several cell lines including CRC [56,57,58,59]. Previous reports also described that resistance to 5-FU in CRC cells promotes the loss of epithelial markers [58,59]. To confirm the link between resistance acquisition and EMT, we analyzed the expression of epithelial markers in CRC cells, validating the loss of E-Cadherin and beta-catenin expression in resistance cells (Figure 5B,C). To assess morphological changes associated to 5-FU resistance we performed nuclear, cytoskeletal and microtubular staining of control and 5-FU-resistant cells (Figure S3D,E). In order to quantify changes in morphology, we measured nuclear and cell areas for each condition, noting a significant increase in both parameters for resistant cells (Figure 5E-G). Indeed, the relationship between nucleus/cytoplasm decreased in resistant cells and cytoskeletal architecture changes became evident. Interestingly, treatment with the Rac1 inhibitor 1A-116 was enough to reverse the expression of epithelial markers (Figure 5B,D) and morphological changes back to control-like values (Figure 5E-G and S3B,C) suggesting a critical role for Rac1 activation on triggering events leading to 5-FU-resistance in CRC.
3.5. Rac1 Inhibitor 1A-116 Reduces the Growth of CRC Resistant cells, Sensitizes Them to 5-FU and Prevents Metastasis Development
Once confirmed by in vitro assays the possibility to reverse resistance to 5-FU we moved to an in vivo assay with CT265FUR cells. For this experiment, we selected one drug in repositioning, ivermectin, and the Rac1 inhibitor 1A-116. Tumor growth kinetics indicated that 5-FU or ivermectin alone had not statistical effect on resistant CRC cells growing (Figure 6A,B). Surprisingly, Rac1 inhibitor 1A-116 administration statistically affected the growth evolution of tumor cells. Interestingly, both ivermectin and 1A-116 were able to induce sensitivity to 5-FU in resistant CT265FUR cells (Figure 6A,B and S4A,B). No signs of toxicity were associated with treatments as evaluated by general animal behavior and weight (Figure 6C and S4C) and the measurement of metabolic parameters (Figure S4D).
At the end of the experiment, we collected tumor, spleen, liver and lungs for histological observation. We did not observe histological differences between tumors by H&E staining (Figure S5), but it was evident that animals treated with 1A-116 or combination of 1A-116 with 5-FU did not develop splenomegaly (Figure 6D and Table 1). Microscopic visualization of histological sections indicated that combined treatment with 1A-116 and 5-FU reduced liver and lung metastasis development in mice (Figure 6E,F and Table 1) suggesting a role for Rac1 in CRC tumor growth, resistance and metastatic dissemination. To further confirm the antimetastatic potential of Rac1 inhibition on 5-FU resistant CRC we performed an intrasplenic cells injection, noting that 1A-116 treatment drastically reduced spleen tumor formation and resistant cells dissemination to the liver (Figure 6G-J).
3.6. Generation of Prognostic Signatures to Predict 5-FU-Based Therapies Resistance
Finally, we used our lists of genes associated to resistance to obtain by LASSO regression analysis a minimal group of predictive genes that could anticipate responses to both 5-FU monotherapy and 5-FU-based therapies (Figure 7A and Figure S6) and evaluated their predictive efficiency by generation of ROC curves (Figure 7B). Unfortunately, there were not many common genes between the different predictive lists, but all of them displayed an acceptable predictive efficiency.
As it was shown that Rho GTPases were important markers associated to resistance, we selected by LASSO the most important Rho GTPases-related genes to predict recurrence in the 5-FU monotherapy datasets and combined treatments merged matrix, and then analyzed their recurrence predictive efficiency for each signature by generation of ROC curves (Figure 7C). ROC curve analysis demonstrated the good performance of the established model, with similar predictive efficiency to those based on DEGs. Interestingly, some of these predictive genes were associated to overall CRC patients’ survival (Figure 7D).
To understand the relationship between genes predicting resistance, we constructed a protein-protein interaction network. Using the STRING database, we identified the main interacting proteins shared among these genes (Figure 7E) and extracted the top ten hub genes from this network (Figure 7F). Interestingly, top ten genes are GTPases Rac1, RhoA, and RhoB, their GEFs (Guanine nucleotide Exchange Factors Vav1, Vav2, Vav3 and Tiam1), their effector Pak1, phosphoinositide-3-kinase regulatory subunit (PIK3R2) and beta-catenin (CTNNB1). Taken together, these data indicate a prominent role for Rho GTPases, particularly Rac1, in modulating resistance to 5-FU-based therapies in CRC.
4. Discussion
CRC is the third most commonly diagnosed type of cancer worldwide. Early diagnosis, adenomas removal during screenings and improved CRC treatments have reduced morbidity and mortality rates. However, CRC incidence is increasing in middle- and low-income countries, and early-onset CRC is also emerging, positioning CRC as a growing global public health challenge [60].
Since their discovery, 5-FU-based chemotherapies have been commonly used to treat CRC. However, resistance to treatment has greatly affected 5-FU clinical use. As CRC is a heterogeneous disease characterized by different genetic scenarios [61], a CRC molecular comprehensive characterization could shed light on the understanding of resistance mechanisms and improve therapies.
In this work, we addressed the identification of genes and pathways associated with development of resistance to 5-FU and 5-FU-based treatments in CRC. Comparison of gene expression datasets revealed the enrichment of pathways previously reported to be associated with resistance such as “TGFbeta signaling” and “SMAD2/3 activation by TGFbeta” [62,63,64]. TGFbeta is a cytokine involved both in physiological and pathological processes. Canonical signaling is mediated by SMAD transcription factors, but TGFbeta “non-canonical” pathway involves activation of proteins such as MAPK, PI3K/AKT and small GTPases as Rac1 and RhoA [65]. Categories and genes related to cell stemness [66,67], EGFR signaling [68,69], apoptosis [70], DNA repair [71], and interaction between tumor cells and the immune system [72] were also present in our analysis.
When we looked for potential compounds reversing 5-FU resistance-associated gene expression, we identified molecules belonging to different categories. Some of them were involved in modulation of hormonal messages (dopamine and serotonin receptor agonists and antagonists), selective estrogen receptor modulation (this category includes tamoxifen and raloxifene, widely used in breast cancer) and adrenergic receptor inhibition such as phentolamine. Additionally, we found anti-inflammatory compounds, mainly COX inhibitors. We focused on irinotecan, amitriptyline and ivermectin, three drugs under repositioning and selected ivermectin to our in vivo approach. As predicted by our analysis, ivermectin was able to re-sensitize CRC cells to 5-FU monotherapy, but no significant metastasis prevention was associated to this treatment. Noteworthy, the antitumor mechanism of action proposed for ivermectin is associated to P21(Rac1)-Activated Kinase 1 (PAK1) function [73,74,75].
Interestingly, the most represented pathways in all datasets were directly or indirectly related to Rho GTPases activation. Indeed, activation of Rho GTPases may be under control of the Serum Response Factor as many upregulated genes in recurrent phenotype contained serum response elements in their promoter sequences. Many studies have identified SRF as a central agent in the development of multiple types of cancer, which has classified it as a potential biomarker and therapeutic target, especially for cancers with a poor prognosis. SRF controls the expression of cytoskeleton, morphogenesis and cell migration genes, but also SRF-MRTF complex activity also responds to Rho GTPase-induced actin changes, thereby coupling cytoskeletal gene expression to cytoskeletal dynamics [76,77,78]. Accordingly, it was recently described that active Rac1 modulates SRF/MRTF, which initiates a switch to a mesenchymal-like state characterized by therapy resistance in melanoma [79].
Rho-GTPases regulate a variety of important cellular activities, such as cytoskeletal remodeling, cell adhesion, cell movement, vesicle transport, angiogenesis, and cell cycle regulation [76,80,81,82,83]. Rho-GTPases are generally described as "molecular switches" because they fluctuate between their active conformation attached to GTP and the inactive conformation bound to GDP. The activation of the “molecular switch” is controlled by guanine nucleotide exchange factors (GEFs), which stimulate the release of GDP bound to the inactive form and promote combination with GTP [84]. The inactive state of Rho-GTPase is maintained by inhibitory molecules of the guanine nucleotide dissociation (GDIs) and GTPase activity activating proteins (GAPs) [84]. Through changes in Rho-GTPases protein levels, their activity or their effector proteins, abnormal signaling could contribute to different steps of cancer progression, including proliferation, survival, invasion and metastasis [22,85].
Rac1, RhoA and Cdc42 are the three classical members of Rho-GTPase family, being Rac1 the one that has received increased attention [82]. Rac1 is widely expressed in tissues, and is considered a regulatory factor related to cell movement and invasion. Rac1 is highly expressed and overactivated in many tumor types and it had lately been related to resistance to therapy in several reports [79,86,87,88,89,90].
Based on our data and the literature, Rac1 could be engaging cellular mechanisms leading to 5-FU-based resistance in CRC. To confirm our hypothesis, we treated 5-FU-resistant CRC cells with Rac1 inhibitor 1A-116 noting that doses not compromising viability of CRC cells were enough to overt morphological changes associated to resistance, and re-sensitize resistant cells to 5-FU. Moreover, Rac1 inhibition restored the expression of epithelial markers to 5-FU-resistant cells, previously characterized in a mesenchymal-like state associated with therapy resistance. Administration of Rac1 inhibitor to mice bearing CRC tumors reduced tumor growth, sensitized resistant tumors to 5-FU monotherapy, and decreased metastasis development. Interestingly, tumors derived from animals treated with Rac1 inhibitor showed an increased number of immune cell infiltrates (data not shown), suggesting that Rac1 inhibition could be acting through different mechanisms, including immune escape mediated by tumor microenvironment [91].
Additionally, our data suggest that Rac1 inhibition could be an important strategy to overcome resistance to therapy in different cancer types. Indeed, Rac1 modulated pathways could be playing essential roles in developing resistance to different therapeutic approaches such as endocrine therapies, targeted therapies and radiotherapy, as our enrichment analysis indicate common profiles between resistance to 5-FU-based therapies and gemcitabine and gefitinib in non-small cell lung cancer [92,93], dasatinib for breast, lung, and ovarian tumors [94], tamoxifen in estrogen receptor-positive breast cancer [95], and postradiation tumor escape of CRC [96].
Altogether our data point to Rac1 as a potential target to overcome therapy-resistance for CRC and other types of tumors, and suggest that Rac1 inhibitor 1A-116 could represent a good therapeutic agent to overt CRC resistance for 5-FU-based therapies.
5. Conclusions
CRC is the one of the third most diagnosed type of cancer worldwide, and the second in terms of mortality. Therapies to treat CRC are often associated with the development of resistance, so initially responding tumors became resistant to treatment. Therapies based on 5-FU have been used in the clinics since 1950, but almost half of patients develop therapy-resistance.
Our findings indicate that inhibition of Rac1 activation by the compound 1A-116 reverses 5-FU resistance in CRC cells, decreases tumor growth and prevents metastasis development, suggesting a therapeutic application of 1A-116 for the treatment of therapy-resistant CRC. Further studies are needed to fully understand Rac1's role in CRC progression and therapy resistance.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Figure S1: Dataset normalization and probes intensity filters; Figure S2: Cell viability assays; Figure S3: Rac1 activity inhibition in 5-FU resistant cells; Figure S4: Inhibition of Rac1 activity modulates in vivo CRC growth; Figure S5: Tumor histology; Figure S6: LASSO regression model; Table S1: 5-FU monotherapy detection platforms and sample sizes used in this study; Table S2: Detailed description of 5-FU monotherapy samples selected for analysis; Table S3: 5-FU-based therapies platforms and sample sizes used in this study; Table S4: Detailed description of 5-based therapies samples selected for analysis; Table S5: Available clinical data of patients treated with 5-FU monotherapy; Table S6: GSEA analysis between 5-FU treated recurrent and non-recurrent phenotypes for the GSE39582 dataset; Table S7: GSEA analysis between 5-FU treated recurrent and non-recurrent phenotypes for the GSE81653 dataset; Table S8: Common DEGs identified in patient with tumor recurrence after 5-FU therapy; Table S9: Available clinical data of patients treated with 5-FU-based therapies; Table S10: GSEA analysis between 5-FU, FOLFIRI and FOLFLOX treated recurrent and non-recurrent phenotypes for the merged matrix; Table S11: GSEA analysis between 5-FU, FOLFIRI and FOLFLOX treated recurrent and non-recurrent phenotypes for the 5000 more explicative genes selected by RFE algorithm form merged matrix; Table S12: Selected DEGs to perform CLUE Query (v.1.1) analysis; Table S13: CLUE Query (v.1.1) analysis for selected up-regulated and downregulated genes; Table S14: Perturbations CLUE Query (v.1.1) analysis for selected up-regulated and downregulated genes; Table S15: IC50 values of selected drugs were chosen from in silico studies on 5-FU sensitive and resistant cell lines.
Author Contributions
L.E.A participated in all experimental work, analyzed data, and contributed to both artwork and figure design, and manuscript writing; F.M. carried out cell culture assays; A.A. analyzed clinical data and performed statistical studies; N.C.L. and L.C.Z. contribute to design of figures and data processing and cell culture assays; M.M. performed Rac1-GTP pull-down and cell culture assays; C.F. contribute to perform animal experimental procedures; M.M-M. conceived the work, analyzed data, wrote the manuscript, and performed the final editing of figures. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by grants to M.M-M from Instituto Nacional del Cáncer (INC, Asistencia Financiera III, 2015), Agencia Nacional de Promoción Científica y Tecnológica (ANPCyT, PICT2018-0900 and PICT2019-0801), Agencia Santafesina de Ciencia, Tecnología e Innovación (PEICID-202-106) and Fundación Florencio Fiorini.
Institutional Review Board Statement
Authors declare that all experimental protocols involving animals were approved by the “Committee for the Care and Use of Laboratory Animals (CICUAL)” of the Rosario Medical School (RS 4702/2022 and 4704/2022). Mice were treated in accordance with the Canadian Council on Animal Care and ARRIVE 2.0 guidelines.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data presented in this study are available upon request from the corresponding author.
Acknowledgments
We thank G. Chapo and all CIPReB personnel for their help with mouse housing and monitoring, M. Armando and M. Derio for technical support, and A. Tomé and R. Vena for microscopy assistance. MM-M thanks Dr. Cardama for providing 1A-116 and Dr. Xosé Bustelo for continuous support.
References
- F. Bray, M. Laversanne, H. Sung, et al. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 2024, 74, 229–263. [CrossRef] [PubMed]
- L. Rahib, B.D. Smith, R. Aizenberg, et al. Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef] [PubMed]
- M. S. Hossain, H. Karuniawati, A.A. Jairoun, et al. Colorectal cancer: A review of carcinogenesis, global epidemiology, current challenges, risk factors, preventive and treatment strategies. Cancers (Basel) 2022, 14, 1732. [Google Scholar] [CrossRef]
- J. A. Sninsky, B.M. Shore, G. V. Lupu, et al. Risk factors for colorectal polyps and cancer. Gastrointest Endosc Clin N Am. 2022, 32, 195–213. [Google Scholar] [CrossRef] [PubMed]
- NCI, Colon Cancer Treatment - National Cancer Institute, PDQ Cancer Information Summaries. 2023. https://www.cancer.gov/types/colorectal/hp/colon-treatment-pdq (accessed 2 Nov 2023).
- D. B. Longley, D.P. Harkin, P.G. Johnston. 5-Fluorouracil: Mechanisms of action and clinical strategies. Nat Rev Cancer. 2003, 3, 330–338. [Google Scholar] [CrossRef]
- P. Noordhuis, U. Holwerda, C.L. Van der Wilt, et al. 5-Fluorouracil incorporation into RNA and DNA in relation to thymidylate synthase inhibition of human colorectal cancers. Annals of Oncology. 2004, 15, 1025–1032. [Google Scholar] [CrossRef]
- D. M. Thomas, J.R. Zalcberg. 5-Fluorouracil: A pharmacological paradigm in the use of cytotoxics. Clin Exp Pharmacol Physiol. 1988, 25, 887–895. [Google Scholar] [CrossRef]
- S. Giacchetti, B. Perpoint, R. Zidani, et al. Phase III multicenter randomized trial of oxaliplatin added to chronomodulated fluorouracil-leucovorin as first-line treatment of metastatic colorectal cancer. Journal of Clinical Oncology. 2000, 18, 136–47. [Google Scholar] [CrossRef]
- B. Gustavsson, G. Carlsson, D. MacHover, et al. A review of the evolution of systemic chemotherapy in the management of colorectal cancer. Clin Colorectal Cancer. 2015, 14, 1–10. [CrossRef]
- J. Gu, Z. Li, J. Zhou, et al. Response prediction to oxaliplatin plus 5-fluorouracil chemotherapy in patients with colorectal cancer using a four-protein immunohistochemical model. Oncol Lett. 2019, 18, 2091–2101. [CrossRef]
- R. M. Goldberg, D.J. Sargent, R.F. Morton, et al. A randomized controlled trial of fluorouracil plus leucovorin, irinotecan, and oxaliplatin combinations in patients with previously untreated metastatic colorectal cancer. Journal of Clinical Oncology 2004, 22, 23–30. [Google Scholar] [CrossRef] [PubMed]
- J. Y. Douillard, D. Cunningham, A.D. Roth, et al. Irinotecan combined with fluorouracil compared with fluorouracil alone. as first-line treatment for metastatic colorectal cancer: a multicentre randomised trial. Lancet 2000, 355, 1041–1047. [Google Scholar] [CrossRef] [PubMed]
- P. J. Maxwell, D.B. Longley, T. Latif, et al. Identification of 5-fluorouracil-inducible target genes using cDNA microarray profiling. Cancer Res. 2003, 63, 4602–6. [Google Scholar]
- S.M. Offer, N.J. Wegner, C. Fossum, et al. Phenotypic profiling of DPYD variations relevant to 5-fluorouracil sensitivity using real-time cellular analysis and in vitro measurement of enzyme activity. Cancer Res 2013, 73, 1958–1968. [CrossRef] [PubMed]
- A. Sadanandam, C.A. Lyssiotis, K. Homicsko, et al. A colorectal cancer classification system that associates cellular phenotype and responses to therapy. Nat Med. 2013, 19, 619–625. [Google Scholar] [CrossRef] [PubMed]
- D. B. Longley, P.G. Johnston. Molecular mechanisms of drug resistance. Journal of Pathology. 2005, 205, 275–292. [Google Scholar] [CrossRef]
- R. Kabra, N. Chauhan, A. Kumar, et al. Efflux pumps and antimicrobial resistance: Paradoxical components in systems genomics. Prog Biophys Mol Biol. 2019, 141, 15–24. [Google Scholar] [CrossRef]
- M. Nikolaou, A. Pavlopoulou, A.G. Georgakilas, et al. The challenge of drug resistance in cancer treatment: a current overview. Clin Exp Metastasis. 2018, 35, 309–318. [Google Scholar] [CrossRef]
- M. Russo, G. Crisafulli, A. Sogari, et al. Adaptive mutability of colorectal cancers in response to targeted therapies. Science. 2019, 366, 1473–1480. [Google Scholar] [CrossRef]
- N. Vasan, J. Baselga, D.M. Hyman. A view on drug resistance in cancer. Nature. 2019, 575, 299–309. [Google Scholar] [CrossRef]
- F. M. Vega, A.J. Ridley. Rho GTPases in cancer cell biology. FEBS Lett. 2008, 582, 2093–2101. [Google Scholar] [CrossRef] [PubMed]
- C. Bailly, J. Beignet, G. Loirand, et al. Rac1 as a therapeutic anticancer target: Promises and limitations. Biochem Pharmacol. 2022, 203, 115180. [Google Scholar] [CrossRef]
- D. Sean, P.S. Meltzer. GEOquery: A bridge between the Gene Expression Omnibus (GEO) and BioConductor. Bioinformatics. 2007, 23, 1846–1847. [Google Scholar] [CrossRef] [PubMed]
- T. Barrett, T.O. Suzek, D.B. Troup, et al. NCBI GEO: Mining millions of expression profiles - Database and tools. Nucleic Acids Res. 2005, 33, D562–D566. [Google Scholar] [CrossRef]
- R. A. Irizarry, B. Hobbs, F. Collin, et al. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003, 4, 249–264. [Google Scholar] [CrossRef]
- James, W. MacDonald. Affymetrix hugene20 annotation data. 2017. [Google Scholar]
- Carlson, M. Affymetrix Affymetrix HG-U133_Plus_2 Array annotation data (chip hgu133plus2). 2016.
- V. G. Tusher, R. Tibshirani, G. Chu. Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci U S A. 2001, 98, 5116–5121. [Google Scholar] [CrossRef] [PubMed]
- F. Hong, R. Breitling, C.W. McEntee, et al. RankProd: A bioconductor package for detecting differentially expressed genes in meta-analysis. Bioinformatics. 2006, 22, 2825–2827. [Google Scholar] [CrossRef]
- G. Yu, Q.Y. He. ReactomePA: An R/Bioconductor package for reactome pathway analysis and visualization. Mol Biosyst. 2016, 12. [Google Scholar] [CrossRef]
- A. Subramanian, R. Narayan, S.M. Corsello, et al. A Next Generation Connectivity Map: L1000 Platform and the First 1,000,000 Profiles. Cell. 2017, 171, 1437–1452. [Google Scholar] [CrossRef]
- J. T. Leek, W.E. Johnson, H.S. Parker, et al. The SVA package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics. 2012, 28, 882–883. [Google Scholar] [CrossRef] [PubMed]
- P. Das, A. Roychowdhury, S. Das, et al. sigFeature: Novel Significant Feature Selection Method for Classification of Gene Expression Data Using Support Vector Machine and t Statistic. Front Genet. 2020, 11, 247. [Google Scholar] [CrossRef] [PubMed]
- I. Guyon, J. Weston, S. Barnhill, et al. Gene selection for cancer classification using support vector machines. Mach Learn. 2002, 46, 389–422. [Google Scholar] [CrossRef]
- A. Subramanian, P. Tamayo, V.K. Mootha, et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005, 102, 15545–15550. [CrossRef] [PubMed]
- A. Liberzon, A. Subramanian, R. Pinchback, et al. Molecular signatures database (MSigDB) 3.0. Bioinformatics. 2001, 27, 1739–1740. [Google Scholar] [CrossRef]
- R. Janky, A. Verfaillie, H. Imrichová, B. et al. iRegulon: From a gene list to a gene regulatory network using large motif and track collections. PLoS Comput Biol. 2014, 10, e1003731. [Google Scholar] [CrossRef]
- P. Shannon, A. Markiel, O. Ozier, et al. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Z. Y. Algamal, M.H. Lee. Penalized logistic regression with the adaptive LASSO for gene selection in high-dimensional cancer classification. Expert Syst Appl. 2015, 42, 9326–9332. [Google Scholar] [CrossRef]
- T. Sing, O. Sander, N. Beerenwinkel, et al. ROCR: Visualizing classifier performance in R. Bioinformatics. 2005, 21, 3940–3941. [Google Scholar] [CrossRef]
- X. Robin, N. Turck, A. Hainard, et al. pROC: An open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinformatics. 2011, 12. [Google Scholar] [CrossRef]
- T.M. Therneau, T. Lumley. Package ‘survival’. R Topics Documented. 2015.
- D. Szklarczyk, J.H. Morris, H. Cook, et al. The STRING database in 2017: Quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res 2017, 45, D362–D368. [CrossRef] [PubMed]
- C. H. Chin, S.H. Chen, H.H. Wu, et al. cytoHubba: Identifying hub objects and sub-networks from complex interactome. BMC Syst Biol. 2014, 8. [Google Scholar] [CrossRef]
- G. Cardama, M. Comin, L. Hornos, et al. Preclinical development of novel Rac1-GEF signaling inhibitors using a rational design approach in highly aggressive breast cancer cell lines. Anticancer Agents Med Chem. 2013, 14, 840–851. [Google Scholar] [CrossRef]
- L. E. Anselmino, M. V. Baglioni, F. Malizia, et al. Repositioning metformin and propranolol for colorectal and triple negative breast cancers treatment. Sci Rep. 2021, 11, 8091. [Google Scholar] [CrossRef]
- A. Castro-Castro, V. Ojeda, M. Barreira, et al. Coronin 1A promotes a cytoskeletal-based feedback loop that facilitates Rac1 translocation and activation. EMBO Journal. 2011, 30, 3913–3927. [Google Scholar] [CrossRef] [PubMed]
- L. Saltz, Y. Shimada, D. Khayat. CPT-11 (irinotecan) and 5-fluorouracil: A promising combination for therapy of colorectal cancer. European Journal of Cancer Part A. 1996, 32, S24–S31. [Google Scholar] [CrossRef] [PubMed]
- H. Satake, A. Tsuji, M. Nakamura, et al. Phase I study of primary treatment with 5-FU, oxaliplatin, irinotecan, levofolinate, and panitumumab combination chemotherapy in patients with advanced/recurrent colorectal cancer involving the wild-type RAS gene: the JACCRO CC-14 study. Int J Clin Oncol. 2018, 23, 490–496. [Google Scholar] [CrossRef]
- M. W. Lee, H. Ryu, I.C. Song, et al. Efficacy of cisplatin combined with topotecan in patients with advanced or recurrent ovarian cancer as second- or higher-line palliative chemotherapy. Medicine. 2020, 99, e19931. [Google Scholar] [CrossRef]
- H. S. Kim, S.Y. Park, C.Y. Park, et al. A multicentre, randomised, open-label, parallel-group Phase 2b study of belotecan versus topotecan for recurrent ovarian cancer. Br J Cancer. 2021, 124, 375–382. [Google Scholar] [CrossRef]
- G. A. Cardama, J. Maggio, L. Valdez Capuccino, et al. Preclinical efficacy and toxicology evaluation of RAC1 inhibitor 1A-116 in human glioblastoma models. Cancers (Basel). 2022, 14, 4810. [Google Scholar] [CrossRef]
- A. L. Hemsing, K.P. Rye, K.J. Hatfield, et al. NPM1-mutated patient-derived AML cells are more vulnerable to Rac1 inhibition. Biomedicines. 2022, 10, 1881. [Google Scholar]
- M. Cabrera, E. Echeverria, F.R. Lenicov, et al. Pharmacological Rac1 inhibitors with selective apoptotic activity in human acute leukemic cell lines. Oncotarget. 2017, 8, 98509–98523. [Google Scholar] [CrossRef] [PubMed]
- M. de C. Filgueiras, A. Morrot, P.M.G. Soares, et al. Effects of 5-fluorouracil in nuclear and cellular morphology, proliferation, cell cycle, apoptosis, cytoskeletal and caveolar distribution in primary cultures of smooth muscle cells. PLoS One. 2013, 8, e63177. [Google Scholar] [CrossRef]
- A. Długosz-Pokorska, M. Pięta, T. Janecki, et al. New uracil analogs as downregulators of ABC transporters in 5-fluorouracil-resistant human leukemia HL-60 cell line. Mol Biol Rep. 2019, 46, 5831–5839. [Google Scholar] [CrossRef] [PubMed]
- A. Y. Kim, J.H. Kwak, N.K. Je, et al. Epithelial-mesenchymal transition is associated with acquired resistance to 5-fluorocuracil in HT-29 colon cancer cells. Toxicol Res. 2015, 31, 151–156. [Google Scholar] [CrossRef] [PubMed]
- A. C. M. Sousa-Squiavinato, D. A. Arregui Ramos, M. S. Wagner, et al. Long-term resistance to 5-fluorouracil promotes epithelial-mesenchymal transition, apoptosis evasion, autophagy and reduced proliferation rate in colon cancer cells. Eur J Pharmacol. 2022, 933, 175253. [Google Scholar] [CrossRef]
- Y. Xi, P. Xu. Global colorectal cancer burden in 2020 and projections to 2040. Transl Oncol. 2021, 14, 101174. [Google Scholar] [CrossRef]
- J. Guinney, R. Dienstmann, X. Wang, et al. The consensus molecular subtypes of colorectal cancer. Nat Med. 2015, 21, 1350–1356. [Google Scholar] [CrossRef]
- G. Romano, L. Santi, M.R. Bianco, et al. The TGF-β pathway is activated by 5-fluorouracil treatment in drug resistant colorectal carcinoma cells. Oncotarget. 2016, 7, 22077–22091. [Google Scholar] [CrossRef]
- M. Zhang, Y.Y. Zhang, Y. Chen, et al. TGF-β signaling and resistance to cancer therapy. Front Cell Dev Biol. 2021, 9, 786728. [Google Scholar] [CrossRef]
- Y. Wang, Q. Wei, Y. Chen, et al. Identification of hub genes associated with sensitivity of 5-fluorouracil based chemotherapy for colorectal cancer by integrated bioinformatics analysis. Front Oncol. 2021, 11, 604315. [Google Scholar] [CrossRef] [PubMed]
- A. Bertrand-Chapel, C. Caligaris, T. Fenouil, et al. SMAD2/3 mediate oncogenic effects of TGF-β in the absence of SMAD4. Commun Biol. 2022, 5, 1068. [Google Scholar] [CrossRef]
- Y. H. Cho, E.J. Ro, J.S. Yoon, et al. 5-FU promotes stemness of colorectal cancer via p53-mediated WNT/β-catenin pathway activation. Nat Commun. 2020, 11, 5321. [Google Scholar] [CrossRef] [PubMed]
- C. Denise, P. Paoli, M. Calvani, et al. 5-Fluorouracil resistant colon cancer cells are addicted to OXPHOS to survive and enhance stem-like traits. Oncotarget. 2015, 6, 41706–41721. [Google Scholar] [CrossRef] [PubMed]
- M. Zhou, P. Yu, K. Hou, et al. Effect of RAS status on anti-EGFR monoclonal antibodies + 5-FU infusion-based chemotherapy in first-line treatment of metastatic colorectal cancer: A meta-analysis. Meta Gene. 2016, 9, 110–119. [Google Scholar] [CrossRef] [PubMed]
- S. J. Gao, S.N. Ren, Y.T. Liu, et al. Targeting EGFR sensitizes 5-Fu-resistant colon cancer cells through modification of the lncRNA-FGD5-AS1-miR-330-3p-Hexokinase 2 axis. Mol Ther Oncolytics. 2021, 23. [Google Scholar] [CrossRef]
- S. Blondy, V. David, M. Verdier, et al. 5-Fluorouracil resistance mechanisms in colorectal cancer: From classical pathways to promising processes. Cancer Sci. 2020, 111, 3142–3154. [Google Scholar] [CrossRef]
- Sethy, C.N. Kundu. 5-Fluorouracil (5-FU) resistance and the new strategy to enhance the sensitivity against cancer: Implication of DNA repair inhibition. Biomedicine and Pharmacotherapy. 2021, 137, 111285. [Google Scholar] [CrossRef]
- X. Huang, K. Ke, W. Jin, et al. Identification of genes related to 5-fluorouracil based chemotherapy for colorectal cancer. Front Immunol. 2022, 13, 887048. [CrossRef]
- Q. Dou, H.N. Chen, K. Wang, et al. Ivermectin induces cytostatic autophagy by blocking the PAK1/Akt Axis in breast cancer. Cancer Res. 2016, 76, 4457–4469. [Google Scholar] [CrossRef]
- K. Wang, W. Gao, Q. Dou, et al. Ivermectin induces PAK1-mediated cytostatic autophagy in breast cancer. Autophagy. 2016, 12, 2498–2499. [Google Scholar] [CrossRef] [PubMed]
- L. Chen, S. Bi, Q. Wei, et al. Ivermectin suppresses tumour growth and metastasis through degradation of PAK1 in oesophageal squamous cell carcinoma. J Cell Mol Med. 2020, 24, 5387–5401. [Google Scholar] [CrossRef] [PubMed]
- J. K. Westwick, Q.T. Lambert, G.J. Clark, et al. Rac regulation of transformation, gene expression, and actin organization by multiple, PAK-independent pathways. Mol Cell Biol. 1997, 17, 1324–1335. [Google Scholar] [CrossRef] [PubMed]
- R. Treisman, A.S. Alberts, E. Sahai. Regulation of SRF activity by Rho family GTPases. in: Cold Spring Harb Symp Quant Biol. 1998, 63, 643–651. [Google Scholar] [CrossRef]
- S. Montaner, R. Perona, L. Saniger, et al. Activation of serum response factor by RhoA is mediated by the nuclear factor-κB and C/EBP transcription factors. Journal of Biological Chemistry. 1999, 274, 8506–8515. [Google Scholar] [CrossRef] [PubMed]
- D. A. Lionarons, D.C. Hancock, S. Rana, et al. RAC1P29S induces a mesenchymal phenotypic switch via Serum Response Factor to promote melanoma development and therapy resistance. Cancer Cell. 2019, 36, 68–83. [Google Scholar] [CrossRef] [PubMed]
- A. Hall. Rho GTpases and the actin cytoskeleton. Science. 1998, 279, 509–514. [Google Scholar] [CrossRef]
- S. A. Benitah, P.F. Valerón, L. Van Aelst, et al. Rho GTPases in human cancer: An unresolved link to upstream and downstream transcriptional regulation. Biochim Biophys Acta Rev Cancer. 2004, 1705, 121–132. [Google Scholar] [CrossRef]
- E. E. Bosco, J.C. Mulloy, Y. Zheng. Rac1 GTPase: A “Rac” of all trades. Cellular and Molecular Life Sciences. 2009, 66, 370. [Google Scholar] [CrossRef]
- L. Van Aelst, C. D’Souza-Schorey. Rho GTPases and signaling networks. Genes Dev. 1997, 11, 2295–2322. [Google Scholar] [CrossRef]
- J. Cherfils, M. Zeghouf. Regulation of small GTPases by GEFs, GAPs, and GDIs. Physiol Rev. 2013, 93, 269–309. [Google Scholar] [CrossRef] [PubMed]
- R. B. Haga, A.J. Ridley. Rho GTPases: Regulation and roles in cancer cell biology. Small GTPases. 2016, 7, 207–221. [Google Scholar] [CrossRef] [PubMed]
- G. A. Cardama, D.F. Alonso, N. Gonzalez, et al. Relevance of small GTPase Rac1 pathway in drug and radio-resistance mechanisms: Opportunities in cancer therapeutics. Crit Rev Oncol Hematol. 2018, 124, 29–36. [Google Scholar] [CrossRef] [PubMed]
- H.L. Goel, B. Pursell, L.D. Shultz, et al. P-Rex1 promotes resistance to VEGF/VEGFR-targeted therapy in prostate cancer. Cell Rep 2016, 14, 2193–2208. [CrossRef] [PubMed]
- T. Zhang, N. Wang. MiR-135a confers resistance to gefitinib in non-small cell lung cancer cells by upregulation of RAC1. Oncol Res. 2018, 26, 1191–1200. [Google Scholar] [CrossRef] [PubMed]
- M. Dokmanovic, D.S. Hirsch, Y. Shen, et al. Rac1 contributes to trastuzumab resistance of breast cancer cells: Rac1 as a potential therapeutic target for the treatment of trastuzumab-resistant breast cancer. Mol Cancer Ther 2009, 8, 1557–1569. [CrossRef]
- P. De, B.J. Rozeboom, J.C. Aske, et al. Active rac1 promotes tumorigenic phenotypes and therapy resistance in solid tumors. Cancers (Basel) 2020, 12, 1551. [CrossRef]
- J. Liang, L. Oyang, S. Rao, et al. Rac1, A potential target for tumor therapy. Front Oncol. 2021, 11, 674426. [Google Scholar] [CrossRef]
- P. Tooker, W.C. Yen, S.C. Ng, et al. Bexarotene (LGD1069, Targretin), a selective retinoid X receptor agonist, prevents and reverses gemcitabine resistance in NSCLC cells by modulating gene amplification. Cancer Res. 2007, 67, 4425–4433. [Google Scholar] [CrossRef]
- C. D. Coldren, B.A. Helfrich, S.E. Witta, et al. Baseline gene expression predicts sensitivity to gefitinib in non-small cell lung cancer cell lines. Molecular Cancer Research. 2006, 4, 521–528. [Google Scholar] [CrossRef]
- F. Huang, K. Reeves, X. Han, et al. Identification of candidate molecular markers predicting sensitivity in solid tumors to dasatinib: Rationale for patient selection. Cancer Res. 2007, 67, 2226–2238. [Google Scholar] [CrossRef] [PubMed]
- S. Massarweh, C.K. Osborne, C.J. Creighton, et al. Tamoxifen resistance in breast tumors is driven by growth factor receptor signaling with repression of classic estrogen receptor genomic function. Cancer Res 2008, 68, 826–33. [CrossRef] [PubMed]
- Y. Monnier, P. Farmer, G. Bieler, et al. CYR61 and αVβ5 Integrin Cooperate to Promote Invasion and Metastasis of Tumors Growing in Preirradiated Stroma. Cancer Res. 2008, 68, 7323–7331. [Google Scholar] [CrossRef] [PubMed]
- Author 1, A.B.; Author 2, C.D. Title of the article. Abbreviated Journal Name Year, Volume, page range.
Figure 1.
General flowchart of the experimental approach. In this work, three data series were selected from the GEO database. Studies contained gene expression data from tumors of CRC patients treated with different chemotherapies. Patients were followed up to record tumor relapse or death after treatment. First, we selected patients treated only with 5-FU (with and without tumor relapse) and used different methods to obtain DEGs in each data series. Then, we performed an overrepresentation analysis of pathways (ORA) based on common identified genes. These genes were also used for screening of potential drugs able to reverse the expression profile associated with recurrence by the L1000 project of ConnectivityMap. Then we extended our studies by incorporating patients treated with other 5-FU-based chemotherapies constructing a gene expression matrix by merging data from different studies. Using the SigFeature package that applies the SVM-REF algorithm, genes were ordered based on their informative capacity among the phenotypes. With the 5000 most informative genes, a GSEA analysis was carried out. Based on this analysis, we selected a set of drugs to test in vitro and in vivo.
Figure 1.
General flowchart of the experimental approach. In this work, three data series were selected from the GEO database. Studies contained gene expression data from tumors of CRC patients treated with different chemotherapies. Patients were followed up to record tumor relapse or death after treatment. First, we selected patients treated only with 5-FU (with and without tumor relapse) and used different methods to obtain DEGs in each data series. Then, we performed an overrepresentation analysis of pathways (ORA) based on common identified genes. These genes were also used for screening of potential drugs able to reverse the expression profile associated with recurrence by the L1000 project of ConnectivityMap. Then we extended our studies by incorporating patients treated with other 5-FU-based chemotherapies constructing a gene expression matrix by merging data from different studies. Using the SigFeature package that applies the SVM-REF algorithm, genes were ordered based on their informative capacity among the phenotypes. With the 5000 most informative genes, a GSEA analysis was carried out. Based on this analysis, we selected a set of drugs to test in vitro and in vivo.
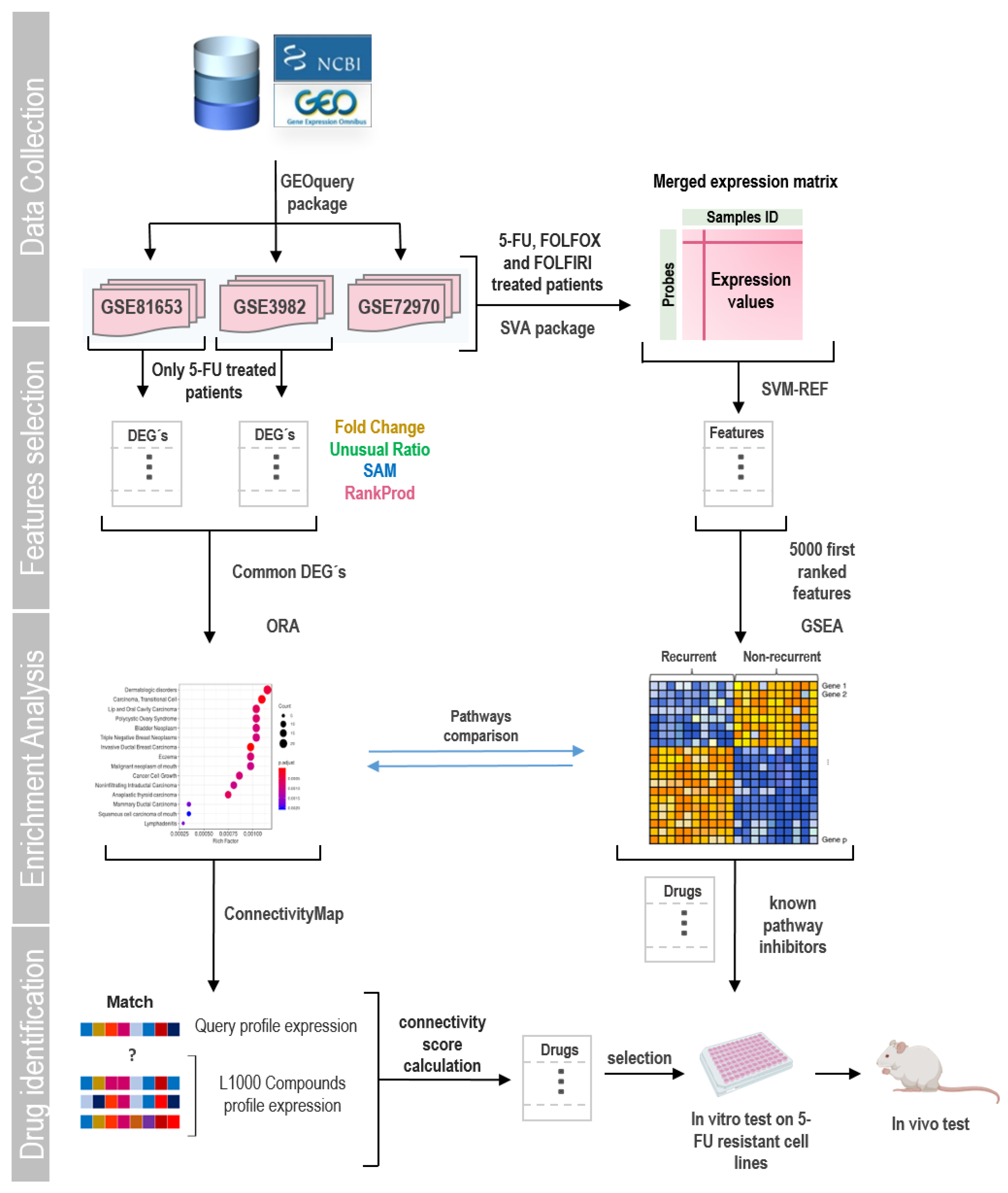
Figure 2.
Gene sets enrichments and DEGs associated to 5-FU monotherapy resistance. (A,B) Representative upregulated (left) and downregulated (right) gene sets in the recurrent phenotype according to Gene Set Enrichment Analysis (GSEA) for selected 5-FU treated patient form GEO dataset (A) GSE39582 and (B) GSE81653. NES and FDR values are indicated within each graph. Positive and negative enrichments are indicated with upward- and downward-pointing arrows, respectively. (C-F) DEGs analysis. Venn diagrams of overexpressed (C) and underexpressed (E) genes identified by four statistical methods (FC: fold change; RP: RankProd package; SAM: Significance Analysis of Microarray; UR: unusual ratio) for series GSE39582 (left diagram) and GSE81653 (right diagram); intersections indicate genes detected by two or more methods. (D) Reactome functional classification of upregulated (D) and downregulated (F) genes detected as DEGs in both datasets. Dot size is proportional to the number of genes associated with a term. Dot color intensity indicates the adjusted p-value resulting from the Over-representation analysis. This graph displays only significant terms (p<0.05).
Figure 2.
Gene sets enrichments and DEGs associated to 5-FU monotherapy resistance. (A,B) Representative upregulated (left) and downregulated (right) gene sets in the recurrent phenotype according to Gene Set Enrichment Analysis (GSEA) for selected 5-FU treated patient form GEO dataset (A) GSE39582 and (B) GSE81653. NES and FDR values are indicated within each graph. Positive and negative enrichments are indicated with upward- and downward-pointing arrows, respectively. (C-F) DEGs analysis. Venn diagrams of overexpressed (C) and underexpressed (E) genes identified by four statistical methods (FC: fold change; RP: RankProd package; SAM: Significance Analysis of Microarray; UR: unusual ratio) for series GSE39582 (left diagram) and GSE81653 (right diagram); intersections indicate genes detected by two or more methods. (D) Reactome functional classification of upregulated (D) and downregulated (F) genes detected as DEGs in both datasets. Dot size is proportional to the number of genes associated with a term. Dot color intensity indicates the adjusted p-value resulting from the Over-representation analysis. This graph displays only significant terms (p<0.05).
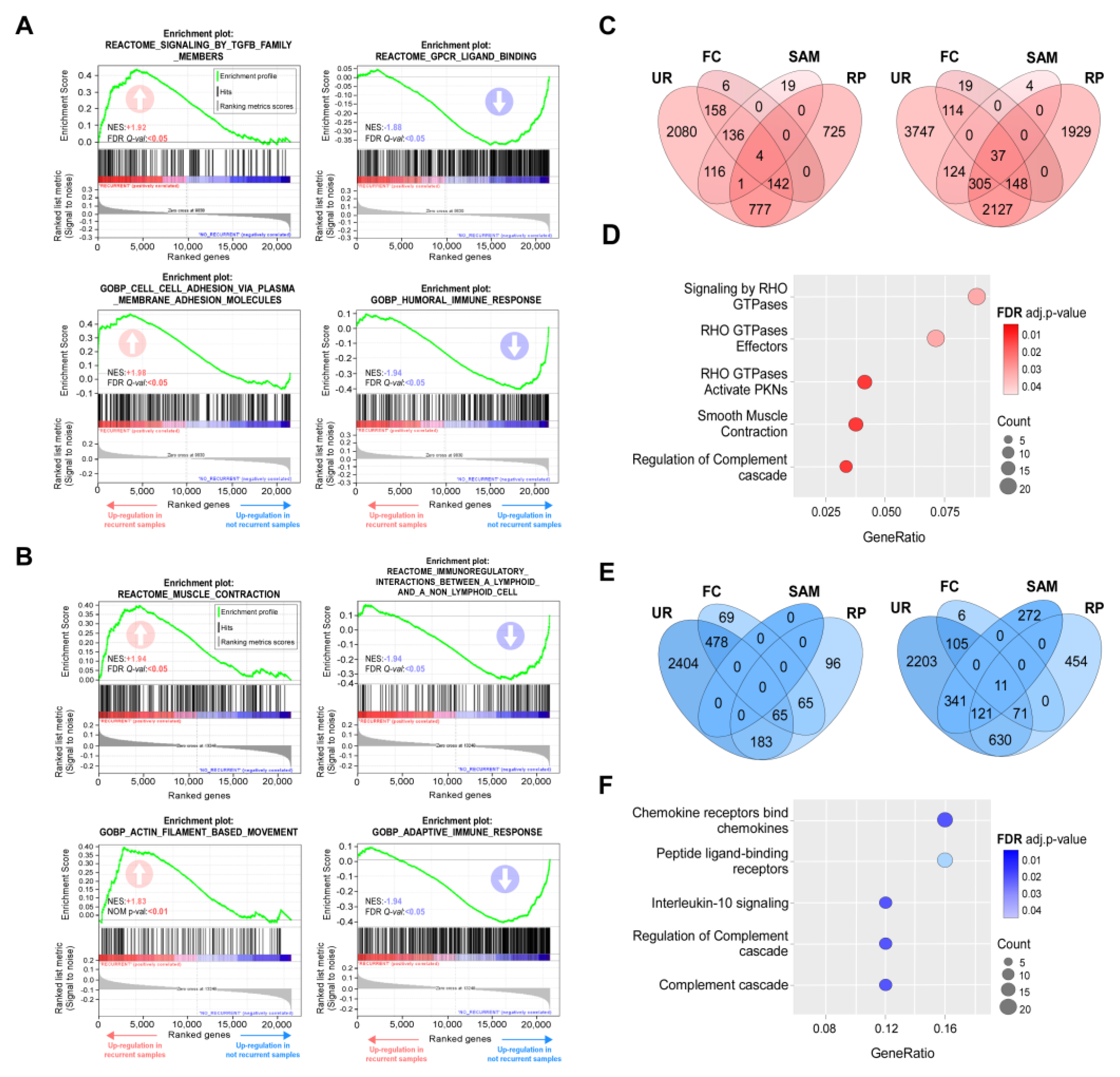
Figure 3.
Regulation of DEGs associated to 5-FU resistant phenotype. (A) Transcription factor binding sites found enriched in the promoter regions of upregulated genes identify in the 5-FU recurrent transcriptome. NES and percentage of regulated genes (targets) are indicated. (B) Word cloud showing the most frequent terms in the enrichment analysis result for each group of genes controlled by a transcriptional factor. The size of the word is proportional to the frequency the term appeared in the Over-representation analysis (p>0.05). (C) Enrichment map for visualizing the pathway/function relationships of DEGs associated to recurrence. Dot size is proportional to the number of genes associated with a term. Dot color intensity indicates the adjusted p-value resulting from the Over-representation analysis. The thickness of the gray lines represents the level of connection between pathways. This graph displays only significant terms (p<0.05). (D) Cnetplot showing the DEGs related with RHO GTPases pathway; dot color indicates the fold change for the recurrent condition. (E) Top 20 hub genes identified by CytoHubba Cytoscape plugin. A greater red color intensity is associated whit highest number of connections in the total DEGs PPI network.
Figure 3.
Regulation of DEGs associated to 5-FU resistant phenotype. (A) Transcription factor binding sites found enriched in the promoter regions of upregulated genes identify in the 5-FU recurrent transcriptome. NES and percentage of regulated genes (targets) are indicated. (B) Word cloud showing the most frequent terms in the enrichment analysis result for each group of genes controlled by a transcriptional factor. The size of the word is proportional to the frequency the term appeared in the Over-representation analysis (p>0.05). (C) Enrichment map for visualizing the pathway/function relationships of DEGs associated to recurrence. Dot size is proportional to the number of genes associated with a term. Dot color intensity indicates the adjusted p-value resulting from the Over-representation analysis. The thickness of the gray lines represents the level of connection between pathways. This graph displays only significant terms (p<0.05). (D) Cnetplot showing the DEGs related with RHO GTPases pathway; dot color indicates the fold change for the recurrent condition. (E) Top 20 hub genes identified by CytoHubba Cytoscape plugin. A greater red color intensity is associated whit highest number of connections in the total DEGs PPI network.
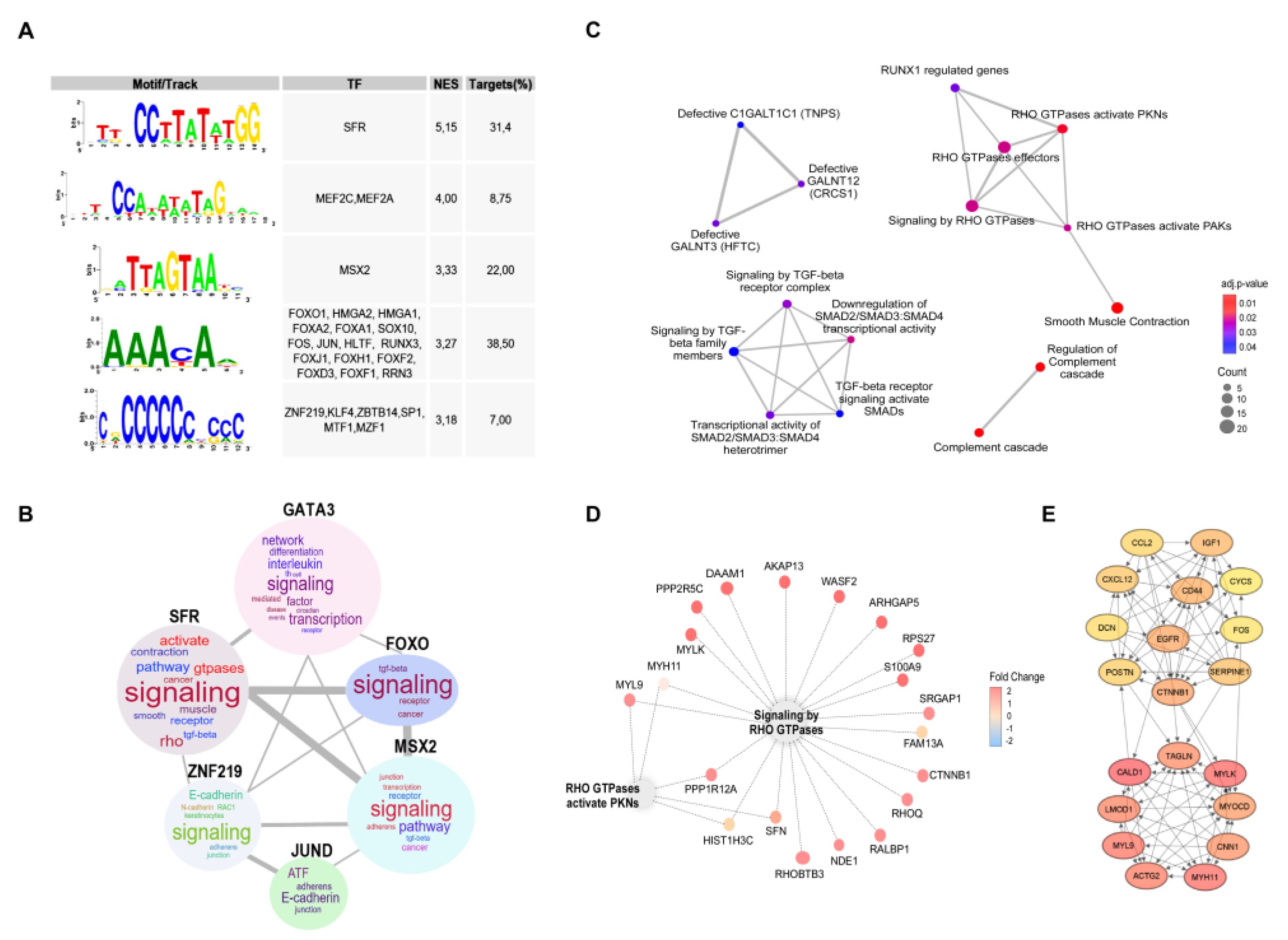
Figure 4.
Gene sets enrichments associated with resistance to 5-FU-based therapies. (A) Gene Set Enrichment Analysis (GSEA) associated with the transcriptome of recurrent phenotype following 5-FU-based chemotherapies using the Reactome database. (B) GSEA associated with the 5000 most explanatory genes in the recurrent phenotype after 5-FU-based chemotherapies selected by the Recursive Feature Elimination algorithm (RFE), gene sets from ‘c2.cgp.v2023.1.Hs.symbols.gmt’ database were employed to perform the analysis. The normalized enrichment scores (NES) and false discovery rate q–values (FDR q–val) are indicated within each graph. Positive and negative enrichments are specified by upward– and downward–pointing arrows, respectively. (C) Over-representation analysis (ORA) of the 5000 most explanatory selected by RFE. Dot size is proportional to the number of genes associated with a term. Dot color indicates the p-value resulting from the ORA analysis. (D) Rac1 activation in control (CT26) and 5-FU resistant (CT265FUR) cells determined by pull-down assays. Levels of Rac1-GTP and total Rac1 were analyzed by Western blot (upper panel) and quantified by ImageJ (lower panel). (E) Transcription factor binding sites found enriched in the promoter regions of 5000 RFE retained genes. The NES and percentage of regulated genes (targets) are also indicated.
Figure 4.
Gene sets enrichments associated with resistance to 5-FU-based therapies. (A) Gene Set Enrichment Analysis (GSEA) associated with the transcriptome of recurrent phenotype following 5-FU-based chemotherapies using the Reactome database. (B) GSEA associated with the 5000 most explanatory genes in the recurrent phenotype after 5-FU-based chemotherapies selected by the Recursive Feature Elimination algorithm (RFE), gene sets from ‘c2.cgp.v2023.1.Hs.symbols.gmt’ database were employed to perform the analysis. The normalized enrichment scores (NES) and false discovery rate q–values (FDR q–val) are indicated within each graph. Positive and negative enrichments are specified by upward– and downward–pointing arrows, respectively. (C) Over-representation analysis (ORA) of the 5000 most explanatory selected by RFE. Dot size is proportional to the number of genes associated with a term. Dot color indicates the p-value resulting from the ORA analysis. (D) Rac1 activation in control (CT26) and 5-FU resistant (CT265FUR) cells determined by pull-down assays. Levels of Rac1-GTP and total Rac1 were analyzed by Western blot (upper panel) and quantified by ImageJ (lower panel). (E) Transcription factor binding sites found enriched in the promoter regions of 5000 RFE retained genes. The NES and percentage of regulated genes (targets) are also indicated.
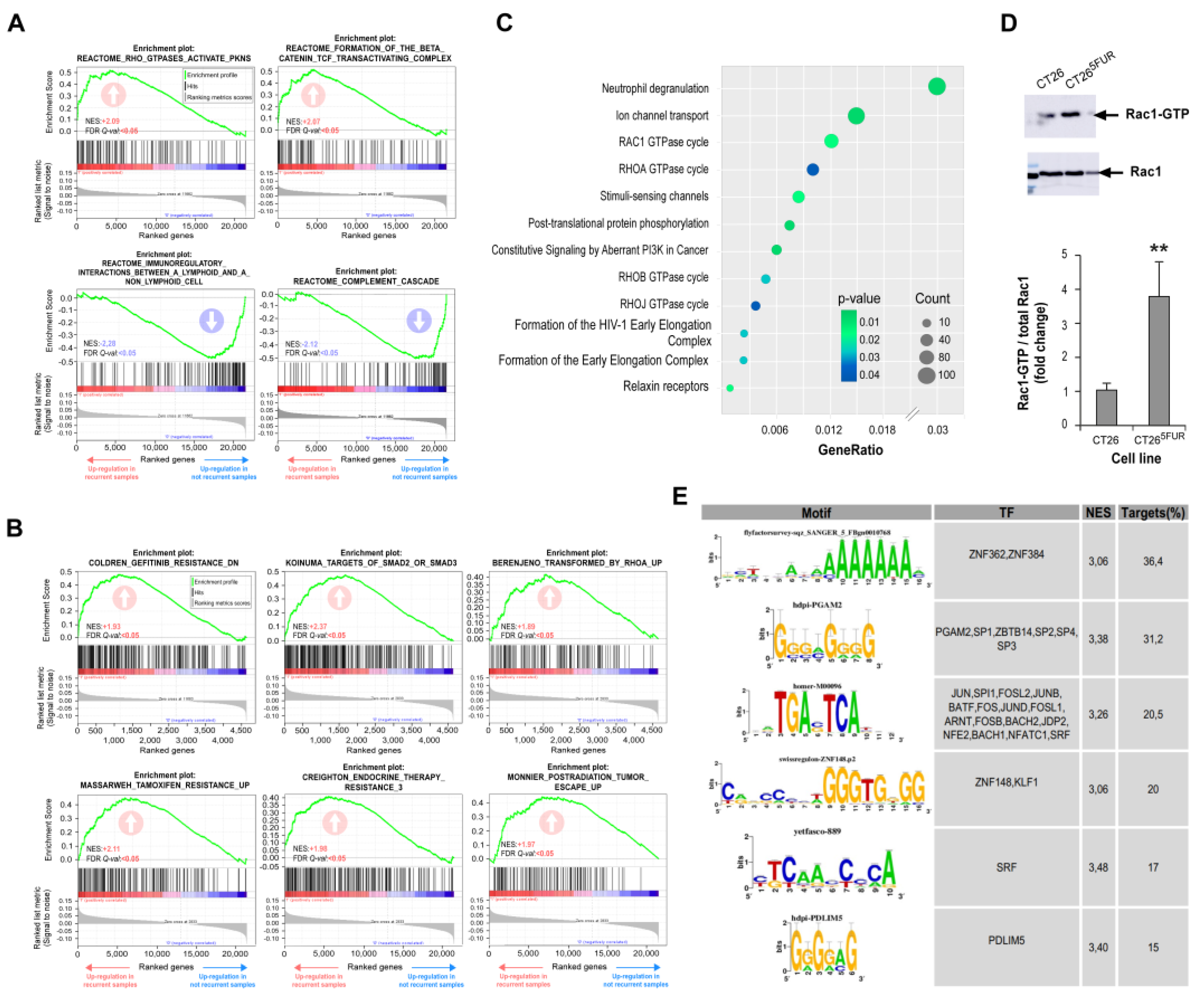
Figure 5.
Characterization of 5-FU resistant cells and viability assays. (A) Viability of CT265FUR, HCT1165FUR and HT295FUR cells treated with selected drugs individually and in combination with 5µM 5-FU (n = 3). (B) Representative immunoblots showing modulation of epithelial markers expression associated to generation of resistance (left panel) and reversion of the expression after Rac1 inhibition (right panel). For loading control, we used the abundance of endogenous beta-actin (E-Cad: E-Cadherin; B-Cat: beta-catenin; B-Act: beta-actin). (C,D) Confocal images for immunofluorescent detection of E-Cadherin (green color, left panel) and beta-catenin (green color, right panel) in CRC cells counterstained with F-actin and nuclei (red and blue colors respectively, scale bar=10μm). (E-G) Cells area, nuclei area and nucleus/cell areas relationship were quantified for each cell line (n > 50 per treatment).
Figure 5.
Characterization of 5-FU resistant cells and viability assays. (A) Viability of CT265FUR, HCT1165FUR and HT295FUR cells treated with selected drugs individually and in combination with 5µM 5-FU (n = 3). (B) Representative immunoblots showing modulation of epithelial markers expression associated to generation of resistance (left panel) and reversion of the expression after Rac1 inhibition (right panel). For loading control, we used the abundance of endogenous beta-actin (E-Cad: E-Cadherin; B-Cat: beta-catenin; B-Act: beta-actin). (C,D) Confocal images for immunofluorescent detection of E-Cadherin (green color, left panel) and beta-catenin (green color, right panel) in CRC cells counterstained with F-actin and nuclei (red and blue colors respectively, scale bar=10μm). (E-G) Cells area, nuclei area and nucleus/cell areas relationship were quantified for each cell line (n > 50 per treatment).
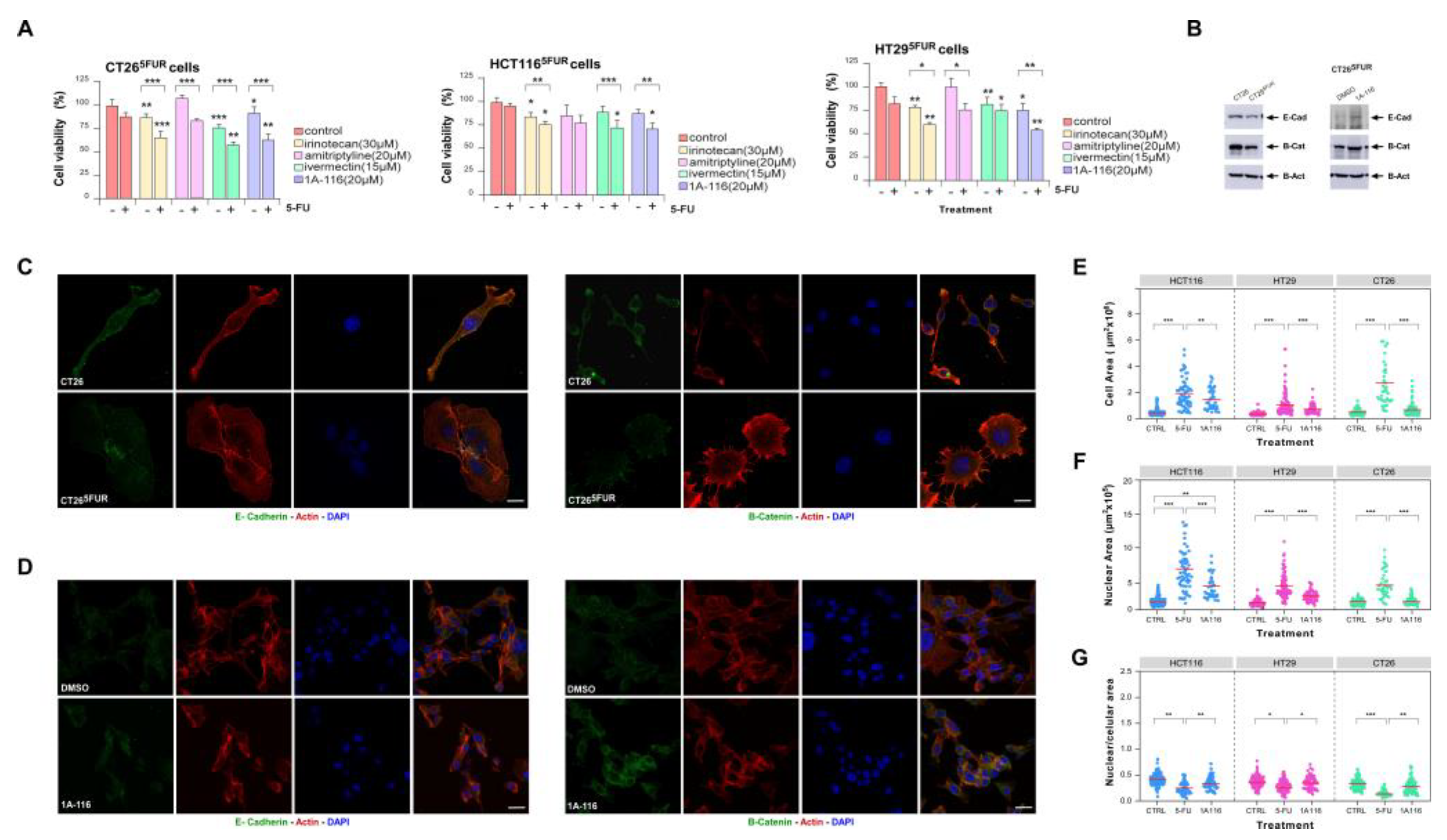
Figure 6.
Ivermectin and 1A-116 in vivo studies. (A) BALB/c mice were subcutaneously challenged with CT265FUR cells. Ten days later tumors become evident and animals were randomly distributed in groups for treatment: control, 5-FU, ivermectin (IVER), ivermectin plus 5-FU (IVER+5-FU), 1A116 and 1A116+5FU (n = 4 per treatment). The tumor size was measured biweekly with a caliper and volume estimated (time = 0 indicates the beginning of treatment). (B) At the end of the experiment, animals were sacrificed and tumors of each group were removed and weighed. (C) Body weight of the animals was measured at the beginning and at the end of the treatments to evaluate signs of its toxicity. (D) Representative images of tumor and spleen for each treatment group. (E) Haematoxylin and eosin lung staining images showing representative micrometastases (indicated with yellow arrows) for control, 5-FU, 1A116 and 1A116+5-FU groups. (F) Haematoxylin and eosin liver staining images showing representative micrometastases (indicated with yellow arrows). (G-J) Analysis of Rac1 inhibition on experimental metastasis development. After intrasplenic injection tumor containing spleens (G) and livers (H) were collected and weight (I). Representative images of spleens (upper panel) and livers (lower panel). (J) Metastasis were counted under a magnifying glass.
Figure 6.
Ivermectin and 1A-116 in vivo studies. (A) BALB/c mice were subcutaneously challenged with CT265FUR cells. Ten days later tumors become evident and animals were randomly distributed in groups for treatment: control, 5-FU, ivermectin (IVER), ivermectin plus 5-FU (IVER+5-FU), 1A116 and 1A116+5FU (n = 4 per treatment). The tumor size was measured biweekly with a caliper and volume estimated (time = 0 indicates the beginning of treatment). (B) At the end of the experiment, animals were sacrificed and tumors of each group were removed and weighed. (C) Body weight of the animals was measured at the beginning and at the end of the treatments to evaluate signs of its toxicity. (D) Representative images of tumor and spleen for each treatment group. (E) Haematoxylin and eosin lung staining images showing representative micrometastases (indicated with yellow arrows) for control, 5-FU, 1A116 and 1A116+5-FU groups. (F) Haematoxylin and eosin liver staining images showing representative micrometastases (indicated with yellow arrows). (G-J) Analysis of Rac1 inhibition on experimental metastasis development. After intrasplenic injection tumor containing spleens (G) and livers (H) were collected and weight (I). Representative images of spleens (upper panel) and livers (lower panel). (J) Metastasis were counted under a magnifying glass.
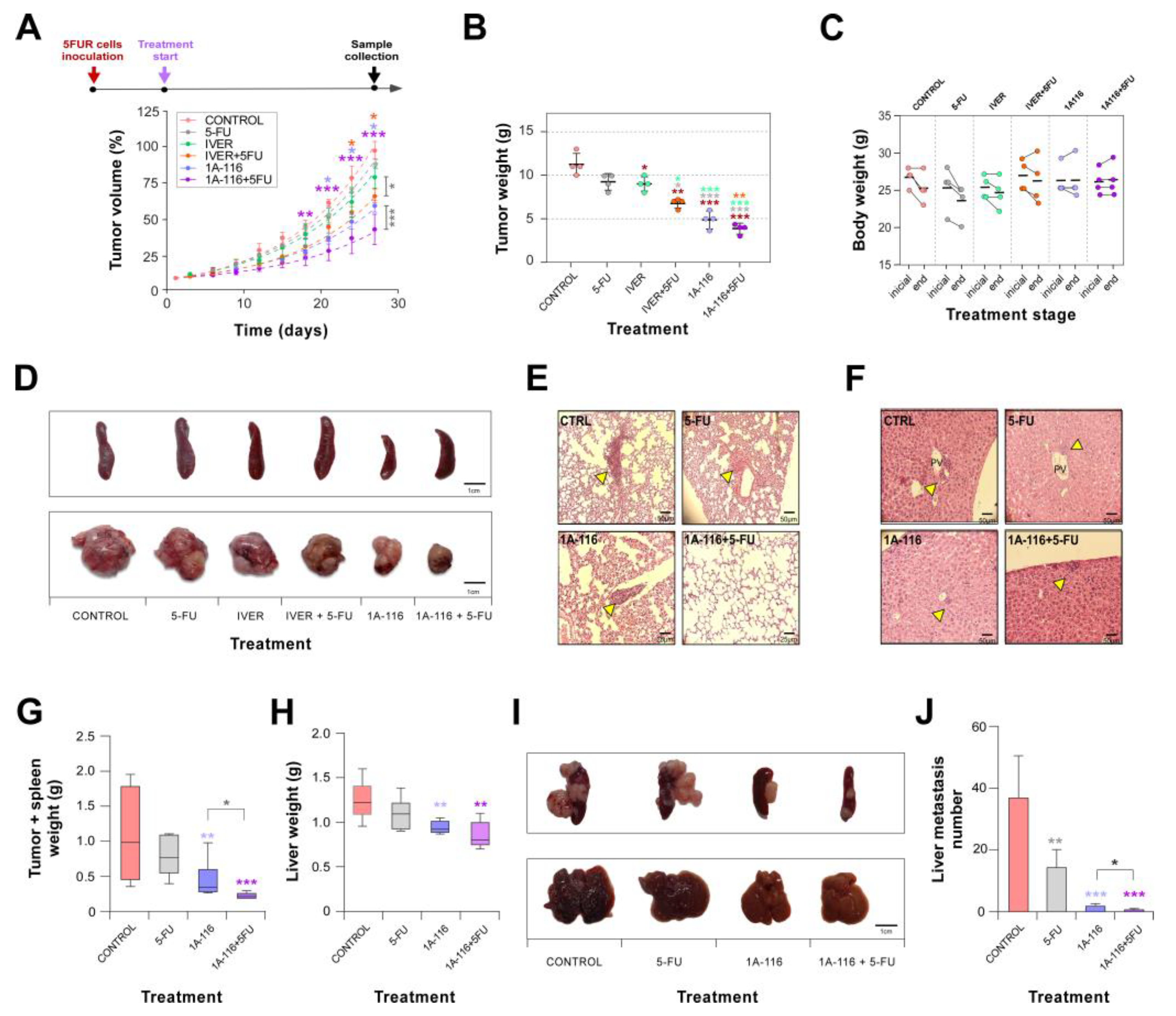
Figure 7.
Generation of recurrence predictor signatures. (A) LASSO coefficient profiles showing the most important DEGs predictors of recurrence selected by LASSO regression analysis (left, DEGs predictors in data sets of patients treated with 5-FU monotherapy; right, DEGs predictors in pooled matrix of patients treated with 5-FU, FOLFIRI, or FOLFOX). (B) Receiver operating characteristic (ROC) curves showing the predictive efficiency of recurrence for each DEGs signature on its respective data set (training + validation). (C) LASSO coefficient profiles showing the most important genes predictors of recurrence from Rho GTPases gene set selected by LASSO regression analysis in the 5-FU monotherapy datasets and combined treatments merged matrix. (D) ROC curves showing the recurrence predictive efficiency for each Rho GTPases genes signature on its respective data set (training + validation). In both (A) and (C), the predictors common to more than one dataset or that appeared as predictors even when different gene sets were used to perform the analysis are highlighted with a green color. (E) Heatmap showing the relationship between recurrence predictors genes expression and the overall survival. Patients were divided into groups with high and low mRNA predictor gene expression at median. Each heatmap cell corresponds to a predictor gene log10 HR (hazard ratio) for the respective dataset detailed in column name. Colors in the red range indicate HR > 0, while colors in the blue range indicate a HR < 0. The graph only displays the predictor genes with significance level p < 0.05 in log-rank test. (F) Expanded PPI network of the set of all recurrence predictor genes (marked in colour), the main common interactor proteins were obtained by STRING Cytoscape StringApp and are shown in grey (selectivity of interactors = 0,5). (G) Top ten hub genes extracted from the predictors expanded PPI network, a greater red color intensity is associated whit highest number of connections in the network. AUC, area under the curve.
Figure 7.
Generation of recurrence predictor signatures. (A) LASSO coefficient profiles showing the most important DEGs predictors of recurrence selected by LASSO regression analysis (left, DEGs predictors in data sets of patients treated with 5-FU monotherapy; right, DEGs predictors in pooled matrix of patients treated with 5-FU, FOLFIRI, or FOLFOX). (B) Receiver operating characteristic (ROC) curves showing the predictive efficiency of recurrence for each DEGs signature on its respective data set (training + validation). (C) LASSO coefficient profiles showing the most important genes predictors of recurrence from Rho GTPases gene set selected by LASSO regression analysis in the 5-FU monotherapy datasets and combined treatments merged matrix. (D) ROC curves showing the recurrence predictive efficiency for each Rho GTPases genes signature on its respective data set (training + validation). In both (A) and (C), the predictors common to more than one dataset or that appeared as predictors even when different gene sets were used to perform the analysis are highlighted with a green color. (E) Heatmap showing the relationship between recurrence predictors genes expression and the overall survival. Patients were divided into groups with high and low mRNA predictor gene expression at median. Each heatmap cell corresponds to a predictor gene log10 HR (hazard ratio) for the respective dataset detailed in column name. Colors in the red range indicate HR > 0, while colors in the blue range indicate a HR < 0. The graph only displays the predictor genes with significance level p < 0.05 in log-rank test. (F) Expanded PPI network of the set of all recurrence predictor genes (marked in colour), the main common interactor proteins were obtained by STRING Cytoscape StringApp and are shown in grey (selectivity of interactors = 0,5). (G) Top ten hub genes extracted from the predictors expanded PPI network, a greater red color intensity is associated whit highest number of connections in the network. AUC, area under the curve.
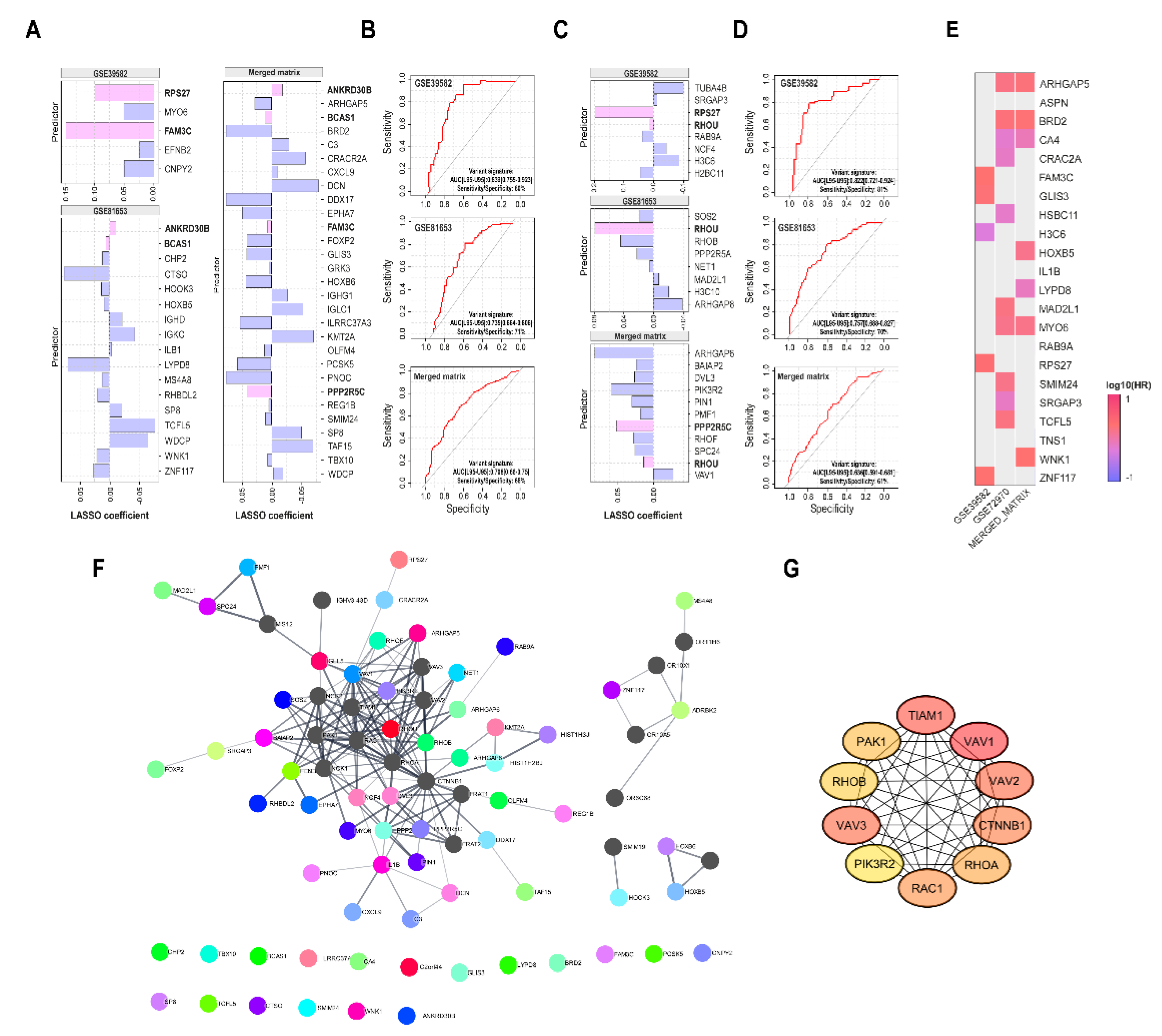

Table 1.
Analysis of metastasis. Mean of metastatic nodes detected per mouse by H&E in lung and liver. At the end of the experiment, the spleens of the mice were removed and measured with a caliper to detect signs of splenomegaly. The mean spleen lengths for each treatment are shown. (n=4).
Table 1.
Analysis of metastasis. Mean of metastatic nodes detected per mouse by H&E in lung and liver. At the end of the experiment, the spleens of the mice were removed and measured with a caliper to detect signs of splenomegaly. The mean spleen lengths for each treatment are shown. (n=4).
| Organ | control | 5-FU | iver | iver+5-FU | 1A-116 | 1A-116+5-FU |
|---|---|---|---|---|---|---|
| Lung (nodes/mouse) | 2±1 | 1.33±1.15 | 1±1 | 0.33±0.58 | 0.67±0.58 | 0±0* |
| Liver (nodes/mouse) | 1.33±0.58 | 1.33±1.53 | 1.33±1.53 | 1.33±1.53 | 0.67±0.58 | 0.67±0.58 |
| Splenomegaly** (spleen length mm) | 24.25±2.06 | 25.75±2.06 | 24.75±1.50 | 26±1.41 | 20.50±1.91* | 23.25±1.50 |
| *p<0.05 ordinary one-way ANOVA vs. control | ||||||
| **Normal length reference: 15–20 mm | ||||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



