
Submitted:
27 August 2024
Posted:
28 August 2024
You are already at the latest version
Abstract
Pigeon Newcastle disease, caused by pigeon paramyxovirus type 1 (PPMV-1), is a significant infectious disease in pigeons that can result in substantial mortality and poses a severe threat to the pigeon industry. Rapid and accurate onsite diagnosis of pigeon disease is crucial for timely diagnosis and the implementation of effective prevention and control measures. In this study, we established a rapid detection method for PPMV-1 based on recombinase-aided amplification (RAA) and CRISPR/Cas12a. The RAA primers target the conserved regions of the L gene for preamplification in clinical nucleic acid samples, followed by CRISPR/Cas12a detection of the target gene. Visualization could be achieved by combination with a lateral flow dipstick (LFD). This method demonstrated high specificity, showing no cross-reactivity with non-PPMV-1 samples. The sensitivity of the method assessed by fluorescence analysis reached 100 copy/µL, and when it was combined with an LFD, the sensitivity was 103 copies/µL. The constructed RAA-CRISPR/Cas12a-LFD visual detection method was applied to clinical sample testing and was found to enable rapid and accurate detection of swab samples and tissue specimens. Its sensitivity was consistent with the current gold standard, quantitative real-time PCR results. The RAA-CRISPR/Cas12a-LFD detection method we developed provides a novel approach for the rapid, simple, precise, and specific onsite diagnosis of pigeon Newcastle disease.
Keywords:
pigeon Newcastle disease
; RAA
; CRISPR/Cas12a
; detection
1. Introduction
Newcastle disease (ND) is an important contagious disease caused by Newcastle disease virus (NDV), which affects a majority of avian species and causes significant economic losses to the poultry industry worldwide[1]. Pigeon paramyxovirus type 1 (PPMV-1) is an antigenic variant strain of NDV that primarily infects pigeons, leading to Newcastle disease in pigeons and posing a significant threat to the pigeon breeding industry [2,3,4]. Currently, PPMV-1 is prevalent in many countries and regions worldwide. In China, several provinces have reported the presence of PPMV-1 infections in pigeon flocks, causing severe harm to both meat pigeons and racing pigeons[5,6].
Diagnostic methods for Newcastle disease in pigeons, specifically PPMV-1 infection, include virus isolation, hemagglutination (HA) and hemagglutination inhibition (HI), and reverse transcription‒polymerase chain reaction (RT‒PCR) [5,7,8]. However, these detection methods have limitations such as long processing times, the requirement for expensive equipment, and inconvenience for onsite testing. Therefore, a rapid, precise, and specific PPMV-1 detection method that is convenient for onsite application is highly important for the prevention and control of pigeon Newcastle disease.
The clustered regularly interspaced short palindromic repeats (CRISPR) system was first discovered in the 1980s and has emerged as a preferred tool for genome editing [9,10,11]. In recent years, the CRISPR/Cas system has been widely developed for onsite rapid pathogen detection due to its exceptional specificity and sensitivity. This method relies on the nonspecific nuclease activity elicited by the Cas12a or Cas13a protein upon binding to the target gene sequence guided by gRNA[12,13,14]. To date, the combination of isothermal amplification technology (IAT) and CRISPR/Cas12a has been applied to detect pathogens such as SARS-CoV-2, Monkey B virus, human norovirus and ASFV [15,16,17,18]. In this study, we developed a novel detection method that combines CRISPR/Cas12a with recombinase-aided amplification (RAA) for the identification of PPMV-1. This assay involves three steps: (1) RNA extraction and RT-RAA reaction at 42°C for 30 minutes; (2) CRISPR/Cas12a reaction at 37°C for 10 minutes; and (3) rapid detection within 2 minutes via a fluorescence analyzer or lateral flow dipstick (LFD). Our novel molecular detection approach (RAA-CRISPR/Cas12a-LFD) provides a rapid, precise, and specific way to detect PPMV-1 without the need for sophisticated equipment. It is well suited for swift screening and diagnosis of clinical samples, offering vital technical assistance for prompt confirmation and efficient prevention and control of pigeon Newcastle disease.
2. Materials and Methods
2.1. Virus and Clinical Samples
Avian influenza virus (AIV), pigeon circovirus (PiCV) and avian infectious bronchitis virus (IBV) strains were preserved by the Laboratory for Prevention and Control of Major Poultry Diseases (Shanxi Agricultural University, China). In this study, swab samples were collected from the throat and cloaca of meat pigeons at live bird markets in Jinzhong city, Shanxi Province, China. Viral nucleic acids were extracted using the 8-Minute Superfast Virus DNA/RNA Extraction Kit (Zhongshi Gene Technology (Tianjin) Co., Ltd.), with the operation process conducted with strict adherence to the manufacturer’s instructions. The viral cDNA used for qPCR and PCR was generated via reverse transcription of the viral genomic RNA using the ReverTra AceTM qPCR RT Kit (TOYOBO Co., Ltd.). The viral genomic RNA/cDNA was stored at -80°C until use.
2.2. Reagents and Instruments
The HiScribe™ T7 Quick High Yield RNA Synthesis Kit, En Gen® Lba Cas12a and Buffer 2.1 were purchased from New England Biolabs (Massachusetts, USA). The Spin Column RNA Cleanup & Concentration Kit was acquired from Sangon Biotech (Shanghai Co., Ltd.). PrimeSTAR Max Premix (2X) and the reporter probe were obtained from Takara Bio (Dalian, China). The RT-RAA Basic Nucleic Acid Amplification Kit was from Hangzhou ZC Bio-Sci & Tech Co. Ltd. (Hangzhou, China). CRISPR Cas12/13 HybriDetect strips were purchased from Warbio Biotech (Nanjing, China). The Universal DNA Purification Kit was purchased from Tiangen Biotech Co., Ltd. (Beijing, China). RT‒qPCR was conducted in a QuantStudio 5 (Applied Biosystems, Massachusetts, USA).
2.3. Design and Synthesis of crRNA and Target DNA Primers
The full-genome sequences of 28 PPMV-1 strains were obtained from the NCBI database and aligned using MegAlign software. On the basis of the nucleotide recognition properties of CRISPR/Cas12a (5′-TTTN-3′) and the results of genome sequence alignment, a PAM sequence was chosen at the relatively conserved L gene of PPMV-1. A pair of crRNA transcription template strands were designed, incorporating the T7 promoter, the scaffold sequence of LbCas12a, and the 20 bp sequence adjacent to the PAM sequence as essential reference elements. A portion of the gene sequence that contained the conserved region of the L gene was selected as the target gene sequence. The crRNA transcription template strands and target DNA primers are presented in Table 1. The two synthetic crRNA transcription template strands were gradually annealed to form a double strand following denaturation. In vitro transcription and purification were subsequently performed using the HiScribe™ T7 Quick High Yield RNA Synthesis Kit and a column-based RNA rapid concentration and purification kit, respectively.
2.4. ssDNA Reporter Probe Design and Validation
The reporter probe, consisting of a labeled fluorescent group (FAM), a quenching group (BHQ1/Biotin), and the sequence TTATTATT, was synthesized by TaKaRa Bio (Dalian, China). The synthesized reporter probe was diluted with enzyme-free water to a concentration of 200 μM to prepare a stock solution. The stock solution was further diluted to a final concentration of 1 μM in a light-protected tube to form the working solution, with the remainder stored at -20°C until use.
To evaluate the trans-cleavage activity of CRISPR/Cas12a, a PCR tube was prepared with a total volume of 20 μL, containing 2 μL of Cas12a (1 μM), 1 μL of crRNA (100 ng/μL), 2 μL of buffer, 1 μL of RNasin (50 U), 10 μL of RNase-free ddH2O, 3 μL of target DNA, and 1 μL of Reporter I (1 μM) (5′ FAM-TTATTATT-BHQ1 3′). The mixture was subsequently analyzed via an QuantStudio 5 instrument to detect the trans-cleavage activity of CRISPR/Cas12a
2.5. Primer Design and Recombinase-Aided Amplification (RAA)
A set of primers was designed on the basis of the detection site of PPMV-1 and the principles of RAA primer design. After a series of RAA primers were tested, the one with the best amplification effect was chosen for subsequent experiments. The sequences of the primers used were as follows: PPMV-1-RAA-F: AACCTCAACTAACCGCCTCTTGATAGAGTTT; and PPMV-1-RAA-R: CTGCCATTACCTGGCAGTTTCTTAATCT. The lyophilized primers were diluted to 10 μM with enzyme-free water to prepare the working solution, and RAA was carried out according to the instructions of the RT-RAA Basic Nucleic Acid Amplification Kit.
2.6. RAA-CRISPR/Cas12a Sensitivity Analysis
The amplified and recovered product of the target DNA was diluted to concentrations ranging from 100 to 1010 copies/μL, and these dilutions were then used as templates for subsequent reactions after amplification with the RT-RAA Basic Nucleic Acid Amplification Kit. Sensitivity analysis was conducted by using the QuantStudio5 FAM channel to collect fluorescence signals every 2 minutes, with three replicates performed for each reaction. The reaction protocol consisted of a prereaction step at 37 °C for 1 minute followed by fluorescence signal acquisition at 37 °C for 2 minutes (30 cycles).
2.7. RAA-CRISPR/Cas12a Specificity Analysis
To evaluate the specificity of the established RAA-CRISPR/Cas12a method for targeting PPMV-1, we selected AIV, IBV and PiCV for testing. Following viral RNA amplification via RT-RAA, the reaction mixture was prepared with 2 μL of Cas12a (1 μM), 1 μL of crRNA (100 ng/μL), 2 μL of buffer, 1 μL of RNasin (50 U), 10 μL of RNase-free ddH2O, 1 μL of Reporter I (1 μM), and 3 μL of the RAA-amplified reaction mixture containing viral RNAs or ddH2O as negative control. The fluorescence signals were detected at 2-minute intervals via the QuantStudio 5 FAM channel, with each reaction performed in triplicate.
2.8. RAA-CRISPR/Cas12a-LFD
To visualize the analysis results, we established an LFD-based RAA-CRISPR/Cas12a assay. The reaction mixture was assembled with 2 μL of Cas12a (1 μM), 1 μL of crRNA (100 ng/μL), 2 μL of buffer, 1 μL of RNasin (50 U), 10 μL of RNase-free ddH2O, 1 μL of Reporter II (5′ FAM-TTATTATT-biotin 3′) (1 μM), and 3 μL of the recovered target DNA product after RAA. The reaction mixture was incubated at 37°C for 10 minutes, and then the volume was adjusted to 60 μL with RNase-free water, followed by thorough mixing. The dedicated Cas12/13 nucleic acid test strips were subsequently inserted into the reaction tube. The strips were allowed to saturate the entire reading area, which typically took 1–2 minutes. The results were directly read on the basis of the coloration of the test strips. The presence of both a red band on the control line (C line) and the test line (T line) indicated a positive result, as did the scenario where the control line did not develop coloration while the test line did. Conversely, a negative result was determined when only a red band appeared on the control line, with no coloration on the test line.
2.9. Clinical Sample Testing
Sixty pigeon throat and cloacal swab samples were collected from live bird markets and pigeon farms and preserved in PBS. Three positive tissue samples were pulverized using liquid nitrogen and subsequently suspended in PBS. Both the 60 pigeon swab samples and the 3 positive tissue samples were homogenized via a vortex mixer and then centrifuged at 10,000 r/min for 10 minutes at 4°C. Nucleic acids were extracted from the samples via an 8-minute ultrafast virus DNA/RNA dual extraction kit. Detection of PPMV-1 was performed via the RAA-CRISPR/Cas12a-LFD assay, with real-time quantitative PCR (qPCR) and conventional PCR also being employed for comparative analysis.
2.10. Data Analysis
The significance of differences among various groups was analyzed via Student’s t test in GraphPad Prism software, with p<0.05 indicating a statistically significant difference between the two groups (*p<0.05, **p<0.01, ***p<0.001).
3. Results
3.1. Schematic of the RAA-CRISPR/Cas12a System for the Detection of PPMV-1
In this study, we established a novel strategy for the detection of PPMV-1. As depicted in Figure 1, the detection method relying on RAA-CRISPR/Cas12a involves the following steps: nucleic acid extraction and amplification, specific gene detection, and result output and visualization. During sample preprocessing and amplification, nucleic acids are swiftly extracted from the samples and utilized as templates for RAA amplification. In the detection phase, specifically transcribed and purified crRNA (Figure 1B) is designed to direct Cas12a to identify the target DNA that is complementary to the crRNA. This initiates the cis-cleavage activity of Cas12a toward the target gene and the trans-cleavage activity toward the reporter gene, facilitating subsequent visual detection and onsite testing.
3.2. crRNA-Guided CRISPR/Cas12a Cleavage Activity and System Optimization
On the basis of the PPMV-1 virus sequence alignment results and the design principles of the CRISPR/Cas12a system, a conserved region within the L gene was selected as the target sequence for crRNA design. The strand sequences used for crRNA transcription are shown in Table 1. After the synthesized crRNA strand sequences were annealed, transcription was performed via an in vitro transcription kit. The transcription products were then purified and verified via agarose gel electrophoresis, and the results revealed distinct bright bands (Figure 2A). This finding indicates that the crRNA was successfully transcribed in vitro.
Further analysis was conducted to assess whether the crRNA could guide Cas12a to cleave the PPMV-1 target gene. In this CRISPR/Cas12a system, the crRNA effectively guides the Cas12a protein to perform specific recognition and activate both cis- and trans-cleavage activities. By using PPMV-1 target DNA as a template, specific fluorescence signals can be detected, and the fluorescence signal exhibited a significant difference when compared to the negative control group (Figure 2B). Cas12a and crRNA are key components of this detection system, and their concentration ratio has a significant impact on the detection results. To optimize the CRISPR/Cas12a reaction system, five different concentrations of Cas12a/crRNA were tested. The optimal reaction conditions were achieved at a concentration ratio of Cas12a protein to crRNA of 1:2, with concentrations of 50 nM for Cas12a and 100 nM for crRNA (Figure 2C). Under these conditions, the Cas12a/crRNA reactions were analyzed using a real-time fluorescence instrument, resulting in the observation of the strongest fluorescence signal.
3.3. Specificity and Sensitivity of the RAA-CRISPR/Cas12a System
To evaluate the specificity of the RAA-CRISPR/Cas12 system developed in this study, the optimized reaction system was used to detect common pathogens in poultry, including AIV (avian influenza virus), IBV (infectious bronchitis virus), PiCV (pigeon circovirus), and PPMV-1. The fluorescence detection results indicated that specific fluorescence signals were detected only when PPMV-1 was used as the template, and there were no reactions with the genomes of the other pathogens (Figure 3A). These findings demonstrate that the detection method established for PPMV-1 in this study has good specificity.
To assess the sensitivity of detection via the RAA-CRISPR/Cas12a system, serial dilutions of target DNA (ranging from 100 to 1010 copies) were tested. These templates were first amplified via RAA and then analyzed for sensitivity via the CRISPR/Cas12a detection system. The fluorescence signals were collected every 2 minutes, with three replicates per reaction. Fluorescence intensity analysis revealed that the target DNA could be effectively detected at a concentration of 100 copies/μL (Figure 3B), which suggests that the RAA-CRISPR/Cas12 system has excellent sensitivity for detecting PPMV-1.
To determine the sensitivity of the established RAA-CRISPR/Cas12a system combined with a lateral flow dipstick for detecting PPMV-1, diluted target DNA (with concentrations ranging from 100 to 1010 copies/µL) was used as the template. Following RAA amplification and the CRISPR/Cas12a reaction, the CRISPR Cas12/13 HybriDetect test strip was immediately inserted. The results after 2 minutes indicated that a clear T line could be observed when the target DNA concentration was 103 copies/μL (Figure 3C). These findings demonstrate that the RAA-CRISPR/Cas12a-LFD system can be effectively used for the detection of PPMV-1.
3.4. On-Site Detection Capability of RAA-CRISPR/Cas12a-LFD
To establish the RAA-CRISPR/Cas12a-LFD-based PPMV-1 detection method, this approach was applied to clinical sample detection. Sixty pigeon throat and cloacal swabs from Shanxi Province (China) were collected and tested for PPMV-1 infection. The results obtained with RAA-CRISPR/Cas12a-LFD in the 60 clinical swab samples are shown in Figure 4; 14 out of the 60 samples were positive for PPMV-1 (Figure 4, Table 2).
To assess the effectiveness and reliability of the RAA-CRISPR/Cas12a-LFD method in clinical applications, the results were compared with those obtained via traditional PCR and real-time quantitative PCR (qPCR) methods by using the same clinical swab samples. A total of 14 samples were found to be positive by both the RAA-CRISPR/Cas12a-LFD method and qPCR, and there was a total of 8 positive samples that were also detected with traditional PCR (Table 2). These results indicate that, compared with conventional PCR, the RAA-CRISPR/Cas12a-LFD method has superior detection performance in complex clinical swabs. The positive detection rate of the RAA-CRISPR/Cas12a-LFD method was consistent with that of qPCR. The RAA-CRISPR/Cas12a-LFD method was also applied to tissue samples positive for PPMV-1. All three PPMV-1-positive tissue samples tested yielded positive results via the RAA-CRISPR/Cas12a-LFD method (Figure 5A). This finding fully demonstrates the accuracy and reliability of the RAA-CRISPR/Cas12a-LFD method in detecting PPMV-1 in tissue samples.
In terms of detection time, the established RAA-CRISPR/Cas12a-LFD method requires only 50 minutes to complete the entire detection process (Figure 5B). This rapid turnaround time highlights the method’s potential utility for timely and efficient PPMV-1 detection in clinical settings, offering a reliable and efficient alternative to traditional PCR and qPCR methods.
4. Discussion
PPMV-1, an antigenic variant of NDV, has a clear host preference for pigeons and has caused substantial economic losses in the poultry industry worldwide[6,19]. While most PPMV-1 isolates do not cause significant clinical manifestations in chickens, they demonstrate high pathogenicity in pigeons [20,21,22]. Some studies have reported that NDV strains originating from pigeons can also exhibit high pathogenicity in chickens, highlighting the necessity for prompt monitoring and pathogenicity assessment of pigeon-derived NDV strains [23,24]. Although PPMV-1 infection is endemically prevalent in domestic pigeons, it can also occur in wild doves, other bird species, and even humans, leading to severe clinical symptoms and potentially fatal outcomes [25,26]. Therefore, rapid diagnosis and control of PPMV-1 are crucial for the pigeon industry and public health safety.
Currently, the commonly used detection methods for PPMV-1 include virus isolation and identification, HA-HI testing, ELISA testing, RT‒PCR, and real-time PCR [7,8,27]. However, these methods have limitations such as long detection times, the requirement for expensive equipment, complex operation procedures, and the need for specialized technical personnel. Loop-mediated isothermal amplification (LAMP) has been developed for the rapid detection of NDV and offers significant advantages in terms of convenience, speed, and sensitivity [28,29]. However, LAMP has several drawbacks, including the complexity of primer design, the risk of contamination, and the possibility of nonspecific amplification. Owing to the small-scale nature of most pigeon or racing pigeon breeding farms, with limited detection conditions, there is an urgent need for a rapid, easy-to-use, highly specific, and sensitive detection method for PPMV-1.
Recently, a blocking lateral flow assay (bLFA) strip has been developed for the rapid clinical detection of NDV antibodies, and this method allows for the detection of Newcastle disease-specific antibodies at room temperature in 10 minutes, and it demonstrates high specificity and sensitivity[30]. However, rapid and precise detection of pathogens is also of significant importance for the prevention and control of Newcastle disease in pigeons. In the present study, we developed an RAA-CRISPR/Cas12a-LFD assay, which is simple, rapid, and accurate, for the detection of PPMV-1. Unlike LAMP and qRT‒PCR, our method does not require high temperatures or expensive equipment, and the reaction can be completed in just 50 minutes. Furthermore, the integration of CRISPR technology enhances the specificity of our method because of its precise gene-editing capabilities. Moreover, the combined utilization of LFD transforms the detection method into a simple and visual assay, making it ideal for convenient onsite clinical testing.
5. Conclusions
We developed a novel method that combines RAA-CRISPR/Cas12a with LFD for the detection of PPMV-1 in pigeons. This method offers several advantages, including ease of operation, cost-effective equipment, short experimental time, and high specificity and sensitivity. This method shows significant potential for clinical applications and can be used for onsite testing and routine monitoring of PPMV-1 in pigeon flocks.
Author Contributions
Conceptualization, L.L., D. W., and J.L.; methodology, L.L. and D.W.; software, D.W.; validation, L.L., D.W., and Z.G.; formal analysis, L. L., D.W., Z.G. and J. L.; investigation, L.L., D.W., Z.G., J. T.; resources, Y.W., S. G. and X. W.; data curation, D.W., Z. G., J.T., X.L. and P. R.; writing—original draft preparation, L.L. and D.W.; writing—review and editing, L.L. and J.L.; visualization, D.W. and Y.G.; supervision, J.L.; project administration, L.L. and J.L.; funding acquisition, L.L. and J.L. All authors have read and agreed to the published version of this manuscript.
Funding
This work was supported by the Special Research Fund of Shanxi Agricultural University for High-level Talents, China (grant No.2021XG004), the National Natural Science Foundation of China (grant No. 32202788), the Fundamental Research Program of Shanxi Province (grant No. 202103021224156), the State Key Laboratory for Animal Disease Control and Prevention Foundation (grant No. SKLADCPKFKT202403), the special fund for Science and Technology Innovation Teams of Shanxi Province (grant No.202304051001041), the Science and Technology Innovation Program of Shanxi Agricultural University (2021BQ78), the earmarked fund for Modern Agro-industry Technology Research System of Shanxi Province, China (2023CYJSTX15-13).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Conflicts of Interest
The authors declare no conflicts of interest. The funders had no role in the design of this study, the collection, analyses, or interpretation of the data; the writing of the manuscript; or the decision to publish the results
References
- Dimitrov, K. M.; Ramey, A. M.; Qiu, X.; Bahl, J.; Afonso, C. L., Temporal, geographic, and host distribution of avian paramyxovirus 1 (Newcastle disease virus). Infect. Genet. Evol. 2016, 39, 22-34. [CrossRef]
- Esmaeelzadeh-Dizaji, R.; Molouki, A.; Hosseini, H.; Fallah-Mehrabadi, M. H.; Ziafati-Kafi, Z.; Takalou, A.; Eram, N.; Kumar, N.; Ashuri, A.; Sadri, N.; Ghalyanchi-Langeroudi, A., Molecular characterization of a pigeon paramyxovirus type 1 virus isolated from Eurasian collared doves in Iran, 2017. J .Vet. Sci. 2022, 23, (3), e29. [CrossRef]
- Molini, U.; Aikukutu, G.; Khaiseb, S.; Cattoli, G.; Dundon, W. G., Phylogenetic Analysis of Pigeon Paramyxoviruses Type-1 Identified in Mourning Collared-doves ( Streptopelia decipiens) in Namibia, Africa. J. Wildl. Dis. 2018, 54, (3), 601-606. [CrossRef]
- Zhan, T.; Lu, X.; He, D.; Gao, X.; Chen, Y.; Hu, Z.; Wang, X.; Hu, S.; Liu, X., Phylogenetic analysis and pathogenicity assessment of pigeon paramyxovirus type 1 circulating in China during 2007-2019. Transbound Emerg. Dis. 2022, 69, (4), 2076-2088. [CrossRef]
- Yu, X.; Luo, Y.; Wang, J.; Shu, B.; Jiang, W.; Liu, S.; Li, Y.; Li, J.; Hou, G.; Peng, C.; Wang, S.; Yuan, L.; Yu, J.; Liu, H.; Wang, Z., A molecular, epidemiological and pathogenicity analysis of pigeon paramyxovirus type 1 viruses isolated from live bird markets in China in 2014-2021. Virus. Res. 2022, 318, 198846. [CrossRef]
- Zhan, T.; He, D.; Lu, X.; Liao, T.; Wang, W.; Chen, Q.; Liu, X.; Gu, M.; Wang, X.; Hu, S.; Liu, X., Biological Characterization and Evolutionary Dynamics of Pigeon Paramyxovirus Type 1 in China. Front. Vet. Sci. 2021, 8, 721102. [CrossRef]
- Sheng, W.; Wang, K.; Gui, Y.; Qi, X.; Shen, L.; Zhang, Y.; Tang, C.; Li, X.; Tao, J.; Cao, C.; Qian, W.; Liu, J., Molecular characteristics and phylogenetic analysis of pigeon paramyxovirus type 1 isolates from pigeon meat farms in Shanghai (2009-2012). Sci. Rep. 2024, 14, (1), 10741. [CrossRef]
- Zhang, Y.; Wang, W.; Li, Y.; Liu, J.; Wang, W.; Bai, J.; Yang, Z.; Liu, H.; Xiao, S., A pigeon paramyxovirus type 1 isolated from racing pigeon as an inactivated vaccine candidate provides effective protection. Poult. Sci. 2022, 101, (10), 102097. [CrossRef]
- Cong, L.; Ran, F. A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P. D.; Wu, X.; Jiang, W.; Marraffini, L. A.; Zhang, F., Multiplex genome engineering using CRISPR/Cas systems. Science 2013, 339, (6121), 819-23. [CrossRef]
- Hsu, P. D.; Lander, E. S.; Zhang, F., Development and applications of CRISPR-Cas9 for genome engineering. Cell 2014, 157, (6), 1262-1278. [CrossRef]
- Ishino, Y.; Shinagawa, H.; Makino, K.; Amemura, M.; Nakata, A., Nucleotide sequence of the iap gene, responsible for alkaline phosphatase isozyme conversion in Escherichia coli, and identification of the gene product. J. Bacteriol. 1987, 169, (12), 5429-33. . [CrossRef]
- Chen, J. S.; Ma, E.; Harrington, L. B.; Da Costa, M.; Tian, X.; Palefsky, J. M.; Doudna, J. A., CRISPR-Cas12a target binding unleashes indiscriminate single-stranded DNase activity. Science 2018, 360, (6387), 436-439. [CrossRef]
- Ma, L.; Zhu, M.; Meng, Q.; Wang, Y.; Wang, X., Real-time detection of Seneca Valley virus by one-tube RPA-CRISPR/Cas12a assay. Front. Cell Infect. Microbiol. 2023, 13, 1305222. [CrossRef]
- Singh, M.; Misra, C. S.; Bindal, G.; Rangu, S. S.; Rath, D., CRISPR-Cas12a assisted specific detection of mpox virus. J. Med. Virol. 2023, 95, (8), e28974. [CrossRef]
- Chen, Y.; Zong, N.; Ye, F.; Mei, Y.; Qu, J.; Jiang, X., Dual-CRISPR/Cas12a-Assisted RT-RAA for Ultrasensitive SARS-CoV-2 Detection on Automated Centrifugal Microfluidics. Anal. Chem. 2022, 94, (27), 9603-9609. [CrossRef]
- Liu, L.; Duan, J. J.; Wei, X. Y.; Hu, H.; Wang, Y. B.; Jia, P. P.; Pei, D. S., Generation and application of a novel high-throughput detection based on RPA-CRISPR technique to sensitively monitor pathogenic microorganisms in the environment. Sci. Total. Environ. 2022, 838, (Pt 2), 156048. [CrossRef]
- Qian, W.; Huang, J.; Wang, T.; Fan, C.; Kang, J.; Zhang, Q.; Li, Y.; Chen, S., Ultrasensitive and visual detection of human norovirus genotype GII.4 or GII.17 using CRISPR-Cas12a assay. Virol. J. 2022, 19, (1), 150. [CrossRef]
- Chen, X. L., Liu, C..C, Li F.X., Zhou, J.H., Huang, Z.H., Zhang, H.L., Wang, H.L., Huang, P., Cao, Z.G., Chiu, S.D. Rapid and Visual Detection of Monkey B Virus Based on Recombinase Polymerase Amplification. Zoonoses 2023, 3:39. [CrossRef]
- Wang, H. Y.; Wu, M. C.; Chen, H. W.; Lai, Y. C.; Huang, W. H.; Chang, H. W.; Jeng, C. R.; Cheng, C. H.; Wang, P. J.; Lai, Y. H.; Chang, Y. C., Isolation, full sequence analysis, and in situ hybridization of pigeon paramyxovirus-1 genotype VI.2.1.1.2.2 from oriental turtle doves (Streptopelia orientalis). Poult. Sci. 2023, 102, (10), 102974. [CrossRef]
- Guo, H.; Liu, X.; Xu, Y.; Han, Z.; Shao, Y.; Kong, X.; Liu, S., A comparative study of pigeons and chickens experimentally infected with PPMV-1 to determine antigenic relationships between PPMV-1 and NDV strains. Vet. Microbiol. 2014, 168, (1), 88-97. [CrossRef]
- Hossain, I.; Parvin, R.; Rahman, M. M.; Begum, J. A.; Chowdhury, E. H.; Islam, M. R.; Diel, D. G.; Nooruzzaman, M., Comparative pathogenicity of a genotype XXI.1.2 pigeon Newcastle disease virus isolate in pigeons and chickens. Microb. Pathog. 2023, 178, 106068. [CrossRef]
- Wang, X.; Ren, S.; Wang, X.; Wang, C. Y.; Fan, M.; Jia, Y.; Gao, X.; Liu, H.; Xiao, S.; Yang, Z., Genomic characterization of a wild-bird-origin pigeon paramyxovirus type 1 (PPMV-1) first isolated in the northwest region of China. Arch. Virol. 2017, 162, (3), 749-761. [CrossRef]
- Nooruzzaman, M.; Barman, L. R.; Mumu, T. T.; Chowdhury, E. H.; Dimitrov, K. M.; Islam, M. R., A Pigeon-Derived Sub-Genotype XXI.1.2 Newcastle Disease Virus from Bangladesh Induces High Mortality in Chickens. Viruses 2021, 13, (8):1520. [CrossRef]
- Kommers, G. D.; King, D. J.; Seal, B. S.; Carmichael, K. P.; Brown, C. C., Pathogenesis of six pigeon-origin isolates of Newcastle disease virus for domestic chickens. Vet. Pathol. 2002, 39, (3), 353-62. [CrossRef]
- Hurley, S.; Eden, J. S.; Bingham, J.; Rodriguez, M.; Neave, M. J.; Johnson, A.; Howard-Jones, A. R.; Kok, J.; Anazodo, A.; McMullan, B.; Williams, D. T.; Watson, J.; Solinas, A.; Kim, K. W.; Rawlinson, W., Fatal Human Neurologic Infection Caused by Pigeon Avian Paramyxovirus-1, Australia. Emerg. Infect. Dis. 2023, 29, (12), 2482-2487. [CrossRef]
- Liu, M.; Qu, Y.; Wang, F.; Liu, S.; Sun, H., Genotypic and pathotypic characterization of Newcastle disease virus isolated from racing pigeons in China. Poult. Sci. 2015, 94, (7), 1476-1482. [CrossRef]
- Tian, Y.; Xue, R.; Yang, W.; Li, Y.; Xue, J.; Zhang, G., Characterization of ten paramyxovirus type 1 viruses isolated from pigeons in China during 1996-2019. Vet. Microbiol. 2020, 244, 108661. [CrossRef]
- Liang, R.; Liang, L.; Ren, X.; Jia, Y.; Han, K.; Zhao, J.; Song, C.; Cui, S., Development of a TaqMan loop-mediated isothermal amplification assay for the rapid detection of pigeon paramyxovirus type 1. Arch. Virol. 2021, 166, (6), 1599-1605. [CrossRef]
- Song, H. S.; Kim, H. S.; Kim, J. Y.; Kwon, Y. K.; Kim, H. R., The Development of Novel Reverse Transcription Loop-Mediated Isothermal Amplification Assays for the Detection and Differentiation of Virulent Newcastle Disease Virus. Int. J. Mol. Sci. 2023, 24, (18). [CrossRef]
- Lv, R.; Guo, J.; Zhang, Y.; Wang, X.; Li, G.; Meng, Z.; Wang, L.; Chai, S.; Li, Q.; Zhang, G., Development and Evaluation of a Blocking Lateral Flow Assay Strip for Detection of Newcastle Disease Virus Antibodies. Vet. Sci. 2023, 10, 152. [CrossRef]
Figure 1.
Schematic diagram of RAA-CRISPR/Cas12a-LFD detection and crRNA positioning. (A) Schematic diagram of the detection workflow of the RAA-CRISPR/Cas12a-LFD system. (B) Schematic diagram of the position of crRNA on the PPMV-1 genome. The crRNA is designed based on the conserved positions of the L gene in the PPMV-1 genome.
Figure 1.
Schematic diagram of RAA-CRISPR/Cas12a-LFD detection and crRNA positioning. (A) Schematic diagram of the detection workflow of the RAA-CRISPR/Cas12a-LFD system. (B) Schematic diagram of the position of crRNA on the PPMV-1 genome. The crRNA is designed based on the conserved positions of the L gene in the PPMV-1 genome.

Figure 2.
Transcription of crRNA and the detection and optimization of crRNA-mediated CRISPR/Cas12a cleavage activity. (A) Electrophoresis results of the in vitro transcribed crRNA. The crRNA template was transcribed in vitro using a T7 transcription kit, followed by purification and electrophoretic analysis. (B) Validation of cleavage activity in the CRISPR/Cas12a system. The negative control (NC) utilized nuclease-free water as the template, while the PPMV-1 sample employed PPMV-1 target DNA plasmid as the template. The prepared mixtures were incubated at 37 °C for 10, 20, 30, and 40 minutes, respectively. A strong fluorescent signal could be detected after 10 minutes, and the result was presented based on the 10-minute reaction. (C) Fluorescence intensity of the CRISPR/Cas12a system was measured using various ratios of Cas12a protein and crRNA. The negative control (NC) in this case did not include any template. For (B) and (C), each sample group contains three replicates. The Student’s t test was used for statistical analysis in GraphPad Software Prism 8. *, p<0.5; **, p<0.01; ***, p<0.001.
Figure 2.
Transcription of crRNA and the detection and optimization of crRNA-mediated CRISPR/Cas12a cleavage activity. (A) Electrophoresis results of the in vitro transcribed crRNA. The crRNA template was transcribed in vitro using a T7 transcription kit, followed by purification and electrophoretic analysis. (B) Validation of cleavage activity in the CRISPR/Cas12a system. The negative control (NC) utilized nuclease-free water as the template, while the PPMV-1 sample employed PPMV-1 target DNA plasmid as the template. The prepared mixtures were incubated at 37 °C for 10, 20, 30, and 40 minutes, respectively. A strong fluorescent signal could be detected after 10 minutes, and the result was presented based on the 10-minute reaction. (C) Fluorescence intensity of the CRISPR/Cas12a system was measured using various ratios of Cas12a protein and crRNA. The negative control (NC) in this case did not include any template. For (B) and (C), each sample group contains three replicates. The Student’s t test was used for statistical analysis in GraphPad Software Prism 8. *, p<0.5; **, p<0.01; ***, p<0.001.
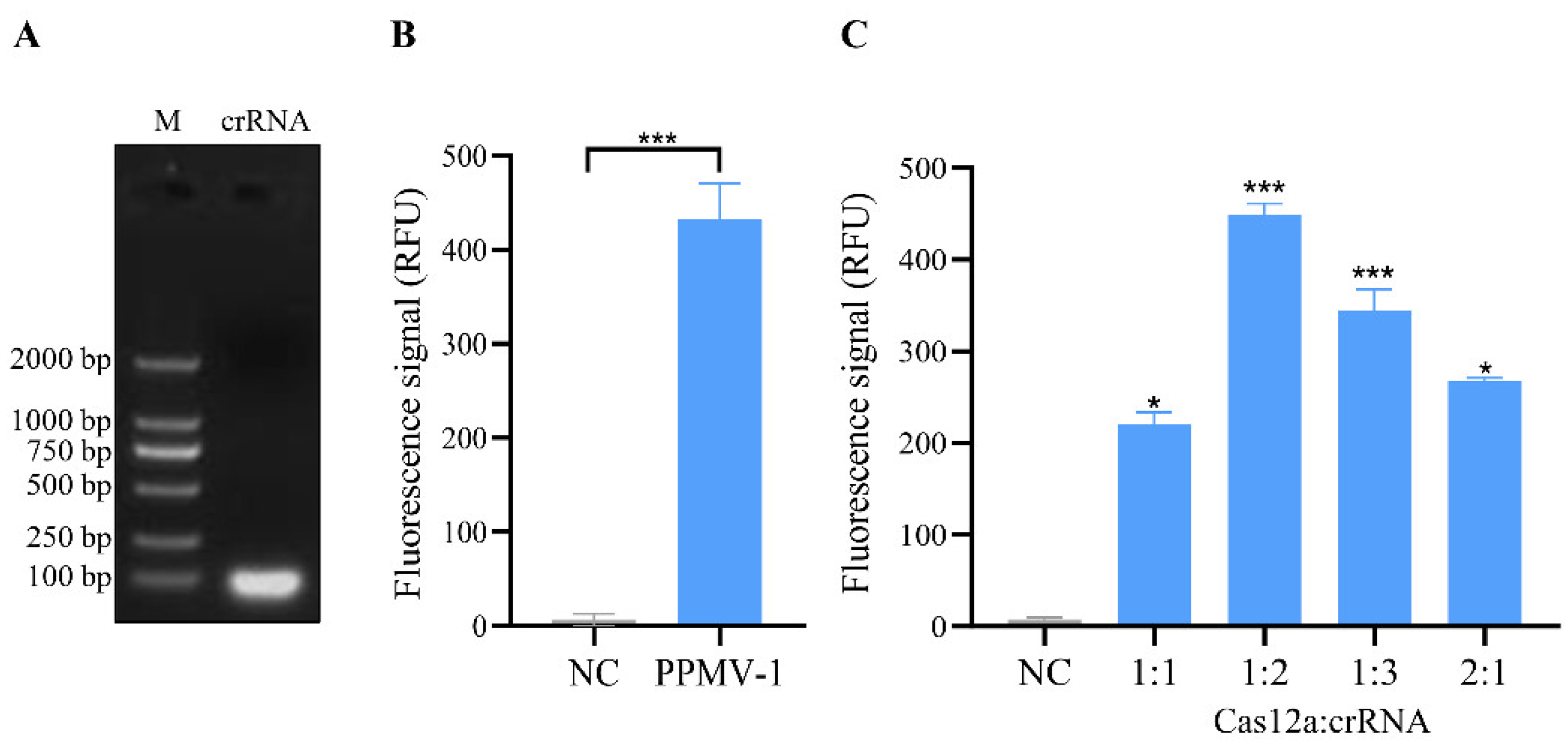
Figure 3.
Specificity and sensitivity analysis of the RAA-CRISPR/Cas12a system for PPMV-1 detection. (A) Specificity analysis of RAA-CRISPR/Cas12a system. Fluorescence intensity responding to IBV, AIV, PiCV and PPMV-1 in the RAA-CRISPR/Cas12a system; NC, negative control (no template). (B) Sensitivity analysis of RAA-CRISPR/Cas12a system. Products of RAA-amplified, serially diluted target DNA were used as templates for the RAA-CRISPR/Cas12a sensitivity analysis. (C) Sensitivity analysis of RAA-CRISPR/Cas12a combined with LFD. Data is representative of three independent experiments. The Student’s t-test was used for statistical analysis in GraphPad Software Prism 8. *, p<0.5; **, p<0.01; ***, p<0.001.
Figure 3.
Specificity and sensitivity analysis of the RAA-CRISPR/Cas12a system for PPMV-1 detection. (A) Specificity analysis of RAA-CRISPR/Cas12a system. Fluorescence intensity responding to IBV, AIV, PiCV and PPMV-1 in the RAA-CRISPR/Cas12a system; NC, negative control (no template). (B) Sensitivity analysis of RAA-CRISPR/Cas12a system. Products of RAA-amplified, serially diluted target DNA were used as templates for the RAA-CRISPR/Cas12a sensitivity analysis. (C) Sensitivity analysis of RAA-CRISPR/Cas12a combined with LFD. Data is representative of three independent experiments. The Student’s t-test was used for statistical analysis in GraphPad Software Prism 8. *, p<0.5; **, p<0.01; ***, p<0.001.
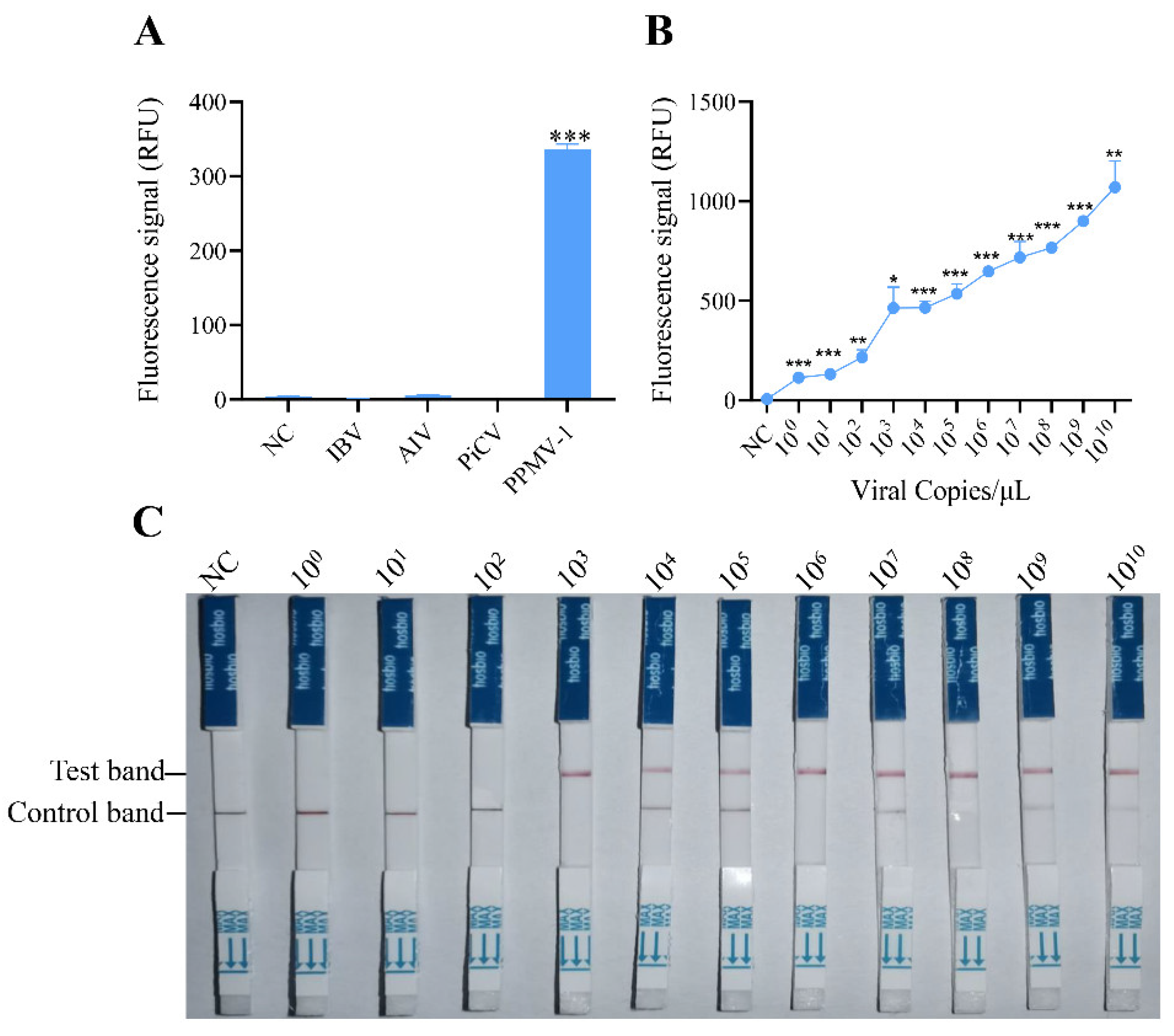
Figure 4.
Detection of clinical pigeon throat and cloacal swab samples using the RAA-CRISPR/Cas12a-LFD technique. RNA extracted from 60 throat and cloacal swab samples of pigeons was used as the template for RAA-CRISPR/Cas12a-LFD detection.
Figure 4.
Detection of clinical pigeon throat and cloacal swab samples using the RAA-CRISPR/Cas12a-LFD technique. RNA extracted from 60 throat and cloacal swab samples of pigeons was used as the template for RAA-CRISPR/Cas12a-LFD detection.
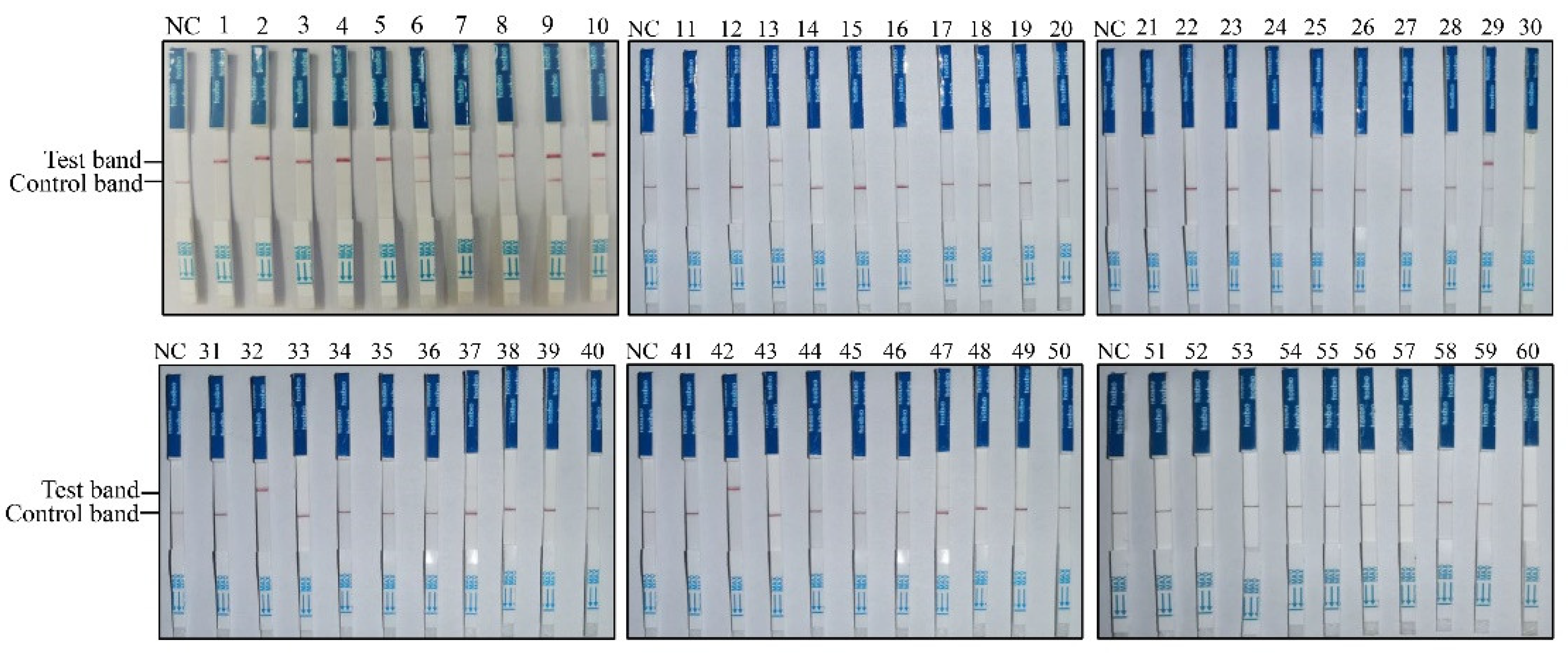
Figure 5.
Detection of tissue samples in diseased pigeons and the timeline for the RAA-CRISPR/Cas12a-LFD assay. (A) Detection results of lung tissue homogenates from three diseased pigeons. (B) Schematic diagram of the total time and duration of each step for detection using RAA-CRISPR/Cas12a-LFD.
Figure 5.
Detection of tissue samples in diseased pigeons and the timeline for the RAA-CRISPR/Cas12a-LFD assay. (A) Detection results of lung tissue homogenates from three diseased pigeons. (B) Schematic diagram of the total time and duration of each step for detection using RAA-CRISPR/Cas12a-LFD.

Table 1.
The crRNA, primers and probes used in this study.
| Primer name | sequence (5′→3′) |
|---|---|
| PPMV-1-crRNA-F | GAAATTAATACGACTCACTATAGGGTAATTTCTACTAAGTGTAGATTAGAATCAAATGATTTTGAT |
| PPMV-1-crRNA-R | ATCAAAATCATTTGATTCTAATCTACACTTAGTAGAAATTACCCTATAGTGAGTCGTATTAATTTC |
| PPMV-1-Target DNA-F | CCATGGGAAGAAGAATTCAGGT |
| PPMV-1-Target DNA -R | CTCGAGTCATGATTGCGGTTT |
| PPMV-1-RAA-F | AACCTCAACTAACCGCCTCTTGATAGAGTTT |
| PPMV-1-RAA-R Reporter I Reporter II |
CTGCCATTACCTGGCAGTTTCTTAATCT FAM-TTATTATT-BHQ1 FAM-TTATTATT-Biotin |
Table 2.
Comparison of clinical sample detection results between RAA-CRISPR/Cas12-LFD and conventional PCR and qPCR.
Table 2.
Comparison of clinical sample detection results between RAA-CRISPR/Cas12-LFD and conventional PCR and qPCR.
| Detection results | PCR (Positive/Total) |
qPCR (Positive/Total) |
RAA-CRISPR/Cas12a-LFD (Positive/Total) |
|---|---|---|---|
| Number of Positive samples | 8/60 | 14/60 | 14/60 |
| Positive rate | 13% | 23% | 23% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



