
Submitted:
28 August 2024
Posted:
29 August 2024
You are already at the latest version
Abstract
Radiotherapy remains a cornerstone in cancer treatment, leveraging ionizing radiation to eradicate malignant cells. Its efficacy, however, is frequently challenged by the heterogeneous sensitivity of tumors and surrounding tissues to radiation. Therefore, understanding the molecular mechanisms underlying radiosensitivity is crucial for improving treatment outcomes. Among the myriad of molecular players involved, the tumor suppressor protein p53 stands out as a central regulator with significant implications for radiosensitivity. Known as the “guardian of the genome,” p53 plays a pivotal role in maintaining genomic stability and orchestrating cellular responses such as cell cycle arrest, DNA repair, apoptosis, and senescence in response to various stress signals, including radiation-induced DNA damage. Activation of p53 triggers the transcription of target genes involved in DNA repair pathways, such as p21, MDM2, and GADD45, facilitating the repair of radiation-induced DNA damage or the elimination of irreparably damaged cells. This, in turn, influences the overall radiosensitivity of tissues. Mutations in the TP53 gene, which encodes p53, are among the most frequent genetic alterations in human cancers. Loss or dysfunction of p53 can compromise the cellular response to radiation, leading to increased resistance to therapy and poorer clinical outcomes. Conversely, intact p53 function is associated with enhanced radiosensitivity due to its ability to promote cell cycle arrest and apoptosis in response to radiation-induced DNA damage. In conclusion, elucidating the molecular mechanisms by which p53 influences radiosensitivity is essential for advancing our understanding of radiation response in cancer cells and developing more effective therapeutic approaches in cancer treatment. This review provides a comprehensive overview of the multifaceted role of p53 in modulating cellular responses to radiation, emphasizing its influence on radiosensitivity.
Keywords:
radiation
; p53
; radiosensitivity
; cancer therapy
; DNA damage and repair
; Apoptosis
1. Introduction
Discovered in 1979, p53 is a protein of approximately 53 kDa known as the “Guardian of the Genome.” It is the most commonly mutated tumor suppressor gene in human cancers, performing multiple regulatory functions by receiving, modulating, and relaying information through various downstream signals such as cellular senescence, cell metabolism, inflammation, autophagy, and other biological processes that control the survival and death of abnormal cells [1,2]. p53 also plays a crucial role in determining cellular responses to various stresses, such as DNA damage, nutrient deficiency, and hypoxia, by inducing gene transcription that regulates the cell cycle and programmed cell death (apoptosis). This regulation is associated with sensitivity to radiotherapy and chemotherapy of malignant tumors [3,4].
Radiotherapy is one of the most common cancer treatments, often used in combination with chemotherapy or surgery. However, radioresistance is a key factor limiting radiotherapeutic efficacy. Several cellular signaling pathways and factors, including the p53-p21 and p53-Bcl-2 pathways, are involved in radioresistance where the p53 is considered the most crucial regulator [5,6] for example, the control of G1 and G2 checkpoints, especially G1 arrest, which is considered closely associated with p53 status; p53 affects the radiosensitivity of cancer cells by regulating cell cycle arrest, apoptosis and DNA repair signaling pathways.
For the present review, we reviewed the multifaceted roles of p53, its regulation of radiosensitivity, factors involved in radiosensitivity, and the role of p53 in cancer treatment. And furthermore highlighted the importance of understanding the mechanisms involved in p53 signaling.
2. p53 Signal Transduction Pathway
The p53 signal transduction pathway involves a series of steps that respond to cellular stress and regulate the activity of the p53 protein. Upon receiving cellular stress signals, such as DNA damage, hypoxia, or oncogenic stress, sensor proteins like ATM (Ataxia-telangiectasia mutated) and ATR (ATM- and Rad3-related) kinases activate the p53 signaling pathway. This activation triggers a cascade of phosphorylation events and other posttranslational modifications (PTMs), which lead to the stabilization of p53 and prevent its degradation by MDM2 (Mouse double minute 2 homolog). Stabilized p53 accumulates in the cell nucleus, where it exerts its transcriptional regulatory functions by binding to specific DNA sequences known as p53 response elements in the promoters of target genes. This binding results in the expression of p53 target genes involved in cell-cycle arrest (e.g., p21, 14-3-3σ), DNA repair (e.g., p53R2, DDB2), autophagy (e.g., DRAM), senescence (e.g., PAI-1), or apoptosis (e.g., BAX, PUMA, NOXA) if the damage is irreparable, ultimately leading to the suppression of tumor growth (Figure 1) [7,8]. In unstressed cells, p53 protein levels are tightly regulated via a negative-feedback loop, whereby p53 induces the transcription of its own negative regulator, MDM2. In conjunction with its homolog MDMX, MDM2 ubiquitinates p53, resulting in its nuclear export and proteasomal degradation [9].
Conversely, upon DNA damage, wild-type (WT) p53 restrains the process of cell replication until the damage is repaired, thus preventing the propagation of DNA-defective cells and the acquisition of a cancer phenotype (Figure 1). However, TP53 mutations disrupt this process, causing cells to lose control of cell proliferation, leading to propagation of damaged DNA into their progeny, which may then transform into cancerous cells [10].
Beyond its role in responding to DNA damage, p53 mediates responses to various cellular stresses and is involved in radio- and chemosensitivity of malignant tumors. It is known that many cancers possess one or more inactivated components of the p53 pathway [11,12].
To maintain low levels of p53 protein in healthy, unstressed cells, the primary p53 inhibitor, E3 ubiquitin ligase MDM2, directs its constitutive proteasomal degradation. In turn, p53 stimulates MDM2 expression, establishing a feedback loop between p53 and MDM2. Amplification of the MDM2 gene is present in approximately 17% of tumors, resulting in decreased p53 levels and poor patient prognosis. Thus, the MDM2-p53 interaction is a crucial target for cancer treatment [13,14,15,16].
3. Functions of p53
p53 activation is primarily mediated through post-translational modifications, such as phosphorylation and acetylation, which stabilize the protein and enhance its transcriptional activity. Key kinases involved in these modifications include ATM, ATR, CHK1 (Checkpoint kinase 1), and CHK2, which are activated by DNA damage and other stress signals. Once activated, p53 functions as a transcription factor, binding to specific DNA sequences and regulating the expression of a wide array of target genes involved in cell cycle arrest, DNA repair, apoptosis, and senescence.
3.1. Cell Cycle Arrest
The cell cycle is composed of four distinct phases: G1, S, G2, and M. Progression through these phases is tightly regulated by cyclins, CDKs, and their inhibitors. p53 plays a key role in regulating the cell cycle, particularly at the G1/S and G2/M checkpoints, ensuring that cells with damaged DNA do not proceed to replicate their genome or divide. p53 inhibits cell cycle progression through various mechanisms. The primary methods are through G1/S checkpoint and G2/M checkpoint activation. G1/S checkpoint activation is achieved via both upregulation of p21 (CDKN1A), a potent CDK inhibitor, and retinoblastoma protein (RB) pathway. Upon activation by stress signals, p53 transcriptionally activates the expression of p21. The p21 protein binds to and inhibits the activity of cyclin CDK2 or cyclin CDK4 complexes, causing the cell cycle to arrest in G1 phase. p21 also stabilizes the RB in its hypo-phosphorylated form. Hypo-phosphorylated RB binds to E2F transcription factors, preventing them from activating genes required for S phase entry, thereby enforcing G1 arrest. G2/M checkpoint activation is achieved via induction of 14-3-3σ and expression of GADD45 [5]. p53 upregulates the expression of 14-3-3σ, a protein that sequesters CDK1-cyclin B complexes in the cytoplasm. This prevents their activation and entry into the nucleus, thereby inhibiting the transition from G2 to M phase. p53 also induces the expression of GADD45, which interacts with CDK1, inhibiting its kinase activity and contributing to G2 arrest [17].
Besides, mutations in the TP53 gene can cause cell cycle dysregulation. These mutations often result in the loss of p53’s ability to induce cell cycle arrest, leading to unchecked cell proliferation despite the presence of DNA damage. This contributes to genomic instability and tumor progression.
The role of p53 in inducing cell cycle arrest in response to stress is fundamental to its function as a tumor suppressor. By halting cell cycle progression, p53 allows cells to repair damaged DNA or undergo apoptosis if the damage is irreparable, thus preventing the propagation of genetic mutations. Understanding the molecular mechanisms underlying p53-mediated cell cycle arrest provides critical insights into cancer biology and highlights potential therapeutic targets for cancer treatment [18].
3.2. DNA Repair
To facilitate DNA repair, p53 both directly activates repair mechanisms and halts the cell cycle. As the “guardian of the genome,” p53 constantly monitors the genome for signs of DNA damage, such as double-strand breaks (DSBs). p53 is involved in a variety of DNA repair processes, including nonhomologous end-joining (NHEJ), homologous recombination (HR), base excision repair (BER), mismatch repair (MMR), and nucleotide excision repair (NER). DSBs induced by IR are repaired either by NHEJ or HR, with DNA helicases playing diverse roles in genome maintenance during recombination and replication [19].
p53 has been demonstrated to play a role in regulating HR through both transcriptional-activation-dependent and -independent mechanisms, separate from its involvement in cell-cycle checkpoint control. Research indicates that p53 can influence the repair of DSBs by affecting transcriptional regulation, as evidenced by its direct interaction with the RAD51 promoter, a key protein for the strand invasion step of HR, leading to changes in RAD51 expression. Besides affecting RAD51 expression, p53 seems to directly interact with RAD51 and RAD54 proteins to modulate HR. Additionally, p53 impacts the expression and activity of genes such as BRCA1 and BRCA2, which are essential for the HR repair process. On the other hand, NHEJ repair pathway, consisting of a catalytic subunit DNA-dependent protein kinase (DNA-PKcs) and regulatory components (Ku proteins). p53 can impact the expression and function of DNA-PKcs and Ku proteins, thereby influencing the effectiveness of the NHEJ pathway. p53 interacts with the Ku70/Ku80 heterodimer, which is mandatory for binding to the broken DNA end and recruiting additional proteins to aid in processing and sealing the break during NHEJ, and this interaction can affect how efficiently NHEJ repairs DNA. While p53’s interactions with NHEJ components have known effects on cellular survival, cell-cycle regulation, and DNA repair, the precise molecular mechanisms underlying these interactions are still not well understood or remain unclear [19,20].
3.3. Apoptosis
In mammalian cells, there are two separate but ultimately merging pathways to apoptosis: the BCL-2-regulated pathway, also known as the intrinsic, mitochondrial, or stress pathway, which is triggered by stressors like cytokine deprivation, ER stress, or DNA damage; and the death receptor pathway, also known as the extrinsic pathway, which is triggered by binding of some ligands to the members of the tumor necrosis factor receptor (TNFR) family containing an intracellular death domain. p53 activates a broad range of signals through both the apoptosis pathways [17].
In intrinsic pathway, p53 interacts with specific DNA sequences to control the expression of important pro-apoptotic genes, including BAX (Bcl-2-associated X protein), PUMA (p53 upregulated modulator of apoptosis), and NOXA (also known as PMAIP1, which encodes a pro-apoptotic Bcl-2 family protein). The proteins produced by these genes induce apoptosis by causing mitochondrial dysfunction. Additionally, p53 enhances mitochondrial outer membrane permeabilization (MOMP) by boosting the levels of pro-apoptotic Bcl-2 family members (like BAX and PUMA) and suppressing anti-apoptotic ones (such as BCL-2 and BCL-XL). This process results in the release of cytochrome c and other pro-apoptotic factors from the mitochondria into the cytosol. Cytochrome c in the cytosol helps to form the apoptosome, a complex that activates caspases, crucial enzymes for carrying out apoptosis [21].
On the other hand, p53 can regulate the expression of death receptors on the cell surface, such as Fas (CD95/Apo-1) and DR5 (Death Receptor 5), thereby increasing the cell’s sensitivity to extrinsic apoptotic signals. It can also influence the expression of ligands that bind to these death receptors. For instance, p53 may elevate the expression of Fas ligand (FasL) in certain situations, which binds to the Fas receptor on target cells and initiates apoptosis. Furthermore, p53 integrates signals from both intrinsic and extrinsic apoptotic pathways. Activation of death receptors recruits adaptor proteins and initiator caspases, like caspase-8, which can then interact with components of the intrinsic pathway. p53 enhances the effects of this extrinsic signaling by promoting mitochondrial dysfunction and strengthening the apoptotic signal [17,18].
3.4. Senescence
Senescence is a permanent halt in the cell cycle that plays a critical role in aging and serves as a potent natural defense against tumors by countering oncogenic threats. Through its control over senescence, p53 significantly contributes to tumor suppression, with its effectiveness closely tied to its expression levels and the specific cellular environment (Figure 1). p53-mediated senescence is triggered by various internal or external stressors causing DNA damage. This activates pathways involving p53 and/or p16INK4A. In the latter pathway, p16INK4A inhibits Cdk4/6, leading to phosphorylation of Rb, and inhibition of E2F transcription, leading to senescence. In the first pathway, DNA damage activates ATM/ATR, which in turn activates Chk1/Chk2 kinases, leading to activation of p53 and p21CIP1, resulting in senescence. In addition, elevated levels of p21CIP1 can inhibit CDK4/6, and contribute to senescence. Furthermore, p53 directly induces senescence by stabilizing fibrinogen activator inhibitor-1, a marker of senescent cells. p53 also promotes the transcription of genes encoding promyelocytic leukemia protein, contributing to cellular senescence [22].
Over 50% of cancers have partial or whole loss of function for TP53, the gene encoding p53, making it the most frequently altered gene in human cancer. Tumor cells with Tp53 mutations gain a selective advantage, enabling them to bypass cell cycle checkpoints, apoptosis, and senescence, and to proliferate in conditions that are inhospitable to normal cells [23].
4. p53 Regulates Radiosensitivity
Radiosensitivity is a complex, multidimensional concept encompassing various biological and physical factors. Biologically, multiple processing, including the initiation of apoptosis, cell cycle progression, DNA damage repair, gene mutations, oncogene accumulation, and loss of tumor suppressor gene function, all can influence cellular radiosensitivity.
p53 has been extensively investigated as a potential target for radiosensitization and radioprotection. Various posttranslational modifications impact the p53 protein, including phosphorylation, acetylation, methylation, ubiquitination, sumoylation, and neddylation [24,25]. The mechanisms by which p53 mediates radiosensitivity are outlined below:
4.1. The p53 Pathway Protects Cells from p53-Independent Apoptosis Caused by Radiation
The p53 pathway plays a critical role in protecting cells from radiation-induced p53-independent apoptosis. Due to the cell-type specificity and complexity of the p53-mediated response to ionizing radiation (IR), the effects of p53 loss on radiation sensitivity vary among different cell types [26]. When cells survive apoptosis following severe, irreversible radiation damage, they face two options: mitotic catastrophe followed by physical death, or irreversible growth arrest. The latter is dependent on p53 and involves the p53-responsive CDKN1A gene, which encodes the CDK inhibitor p21CIP1. At borderline lethal levels of IR, mice can die from p53-dependent apoptosis within the hematopoietic compartment. However, the deletion of p53 in these mice has been shown to reduce cell death and enhance survival [27]. This indicates that the p53 pathway is integral to the cellular response to radiation, mediating not only apoptosis but also other cell fate decisions that contribute to overall radiosensitivity. Understanding the dual role of p53 in promoting both cell death and survival depending on the context and level of radiation exposure provides valuable insights into designing therapeutic strategies. These strategies can aim to modulate p53 activity to protect normal cells during radiotherapy or enhance the radiosensitivity of tumor cells.
4.2. Mutations in p53 Increase Resistance to Radiation in Cancer Cells
p53 is the most frequently mutated gene in human cancers, with alterations identified in approximately half of all malignancies. These mutations often result in the inactivation of p53’s tumor-suppressing functions. For instance, in ovarian cancer cells, PA-1, which expresses WT p53, is highly susceptible to irradiation and shows significant p53 accumulation post-irradiation. Conversely, Caov-3 and SK-OV-3 cell lines, which harbor mutant p53, exhibit varying levels of radioresistence and do not show p53 accumulation upon irradiation [28].
In experiments with SCC-61 (wild-type p53) and SQ-20B (mutant p53) human squamous cell carcinoma cell lines, both showed hyper-radiosensitivity (HRS) at modest radiation doses. However, SQ-20B cells were more resistant to radiation and a chemotherapeutic agent paclitaxel compared to SCC-61 cells. Partial restoration of p53 function in these cells led to increased radiosensitivity, supporting the idea that mutant p53 contributes to radiation resistance (Figure 2) [29].
Mutant p53 also promotes adaptive responses to cancer-related stress conditions, thereby supporting tumor progression. This adaptive response includes promoting cell survival and proliferation under conditions of metabolic stress, hypoxia, and DNA damage, which are common in the tumor microenvironment. These findings suggest that p53 mutations not only contribute to cancer development but also affect the efficacy of radiotherapy. Tumor cells with mutant p53 can evade the typical cell death pathways induced by radiation, leading to increased survival and proliferation despite treatment. Understanding the role of p53 mutations in radiation resistance is crucial for developing more effective cancer therapies that can either restore p53 function or bypass the resistance mechanisms induced by its mutation [30].
4.3. MDM2-p53 Interaction Regulates Radiosensitivity
Several studies have shown that small-molecule inhibitors of MDM2 can increase the expression of p53 and p21, leading to cell cycle arrest at the G1/S and/or G2/M phase and inducing apoptosis. In some cancer cells, p53 retains WT status but its function is inhibited by MDM2. Inhibitors of MDM2-p53 interaction in such cancer cells can restore p53 activity, potentially enhancing the efficacy of radiation therapy [31,32]. Previous research indicates that correcting the balance between p53 and MDM2 can improve radiosensitivity. Inhibitors of MDM2-p53 interaction are considered promising agents in cancer therapy because they can activate p53, resulting in increased radiosensitivity in certain cancer types [33]. Thus, balancing the p53-MDM2 interaction can enhance radiosensitivity, suggesting that inhibitors of MDM2-p53 interaction could be beneficial in cancer therapy by activating p53 and boosting radiosensitivity in specific cancer types.
4.4. Ribosomal Protein-MDM2 Interaction in Radiosensitivity
Mice lacking Rps27l on a heterozygous Tp53 background have demonstrated increased sensitivity to radiation. This heightened sensitivity can be attributed to firstly, the absence of Rps27l induces ribosomal stress, which stabilizes Mdm2 and disrupts the balance between Mdm2 and Mdm4, thereby reducing Mdm2’s capacity to degrade p53. This leads to elevated p53 levels, particularly under radiation-induced stress, resulting in increased apoptosis. Secondly, elevated levels of Mdm2 enhance its interaction with Nbs1, which diminishes Nbs1’s binding to Atm, thereby impairs Atm activation. This impairment in the DNA damage response further contributes to heightened sensitivity to radiation (Figure 3). Overall, these findings underscore the physiological role of RPS27L in regulating the MDM2-p53 axis and MDM2-MRN-ATM pathways, which are pivotal for maintaining the DNA damage response and providing radioprotection [33].
4.5. Translationally Controlled Tumor Protein (TCTP)-MDM2 Mediated Radiosensitivity
Translationally controlled tumor protein (TCTP) is a highly conserved protein present in all eukaryotic cells. It binds to MDM2 and prevents MDM2 from auto-ubiquitination, thereby facilitating MDM2-mediated degradation of p53. Consistent with previous studies, down-regulation of TCTP led to increased levels of p53 during radiation treatment due to MDM2 instability. In experiments where p53 small interfering RNA or silencing RNA (siRNA) was administered to TCTP-silenced A549 cells (a lung cancer cell line), the radiosensitizing effect of TCTP down-regulation was nullified, confirming the involvement of p53 [34,35]. Furthermore, TCTP down-regulation has been shown to decrease the proliferative potential of tumor cells in breast cancer, prostate cancer, lung cancer, and squamous cell carcinoma [36,37]. Therefore, intracellular levels of TCTP play a crucial role in determining radiosensitivity in cancer cells. By reducing TCTP levels, A549 cells become more sensitive to radiation through enhanced activation of p53 and increased DNA damage. In conclusion, down-regulation of TCTP represents a potential strategy to enhance the susceptibility of cancer cells to radiation therapy, potentially improving treatment efficacy while minimizing adverse effects [38].
4.6. MicroRNAs (miRs) in Radiosensitivity
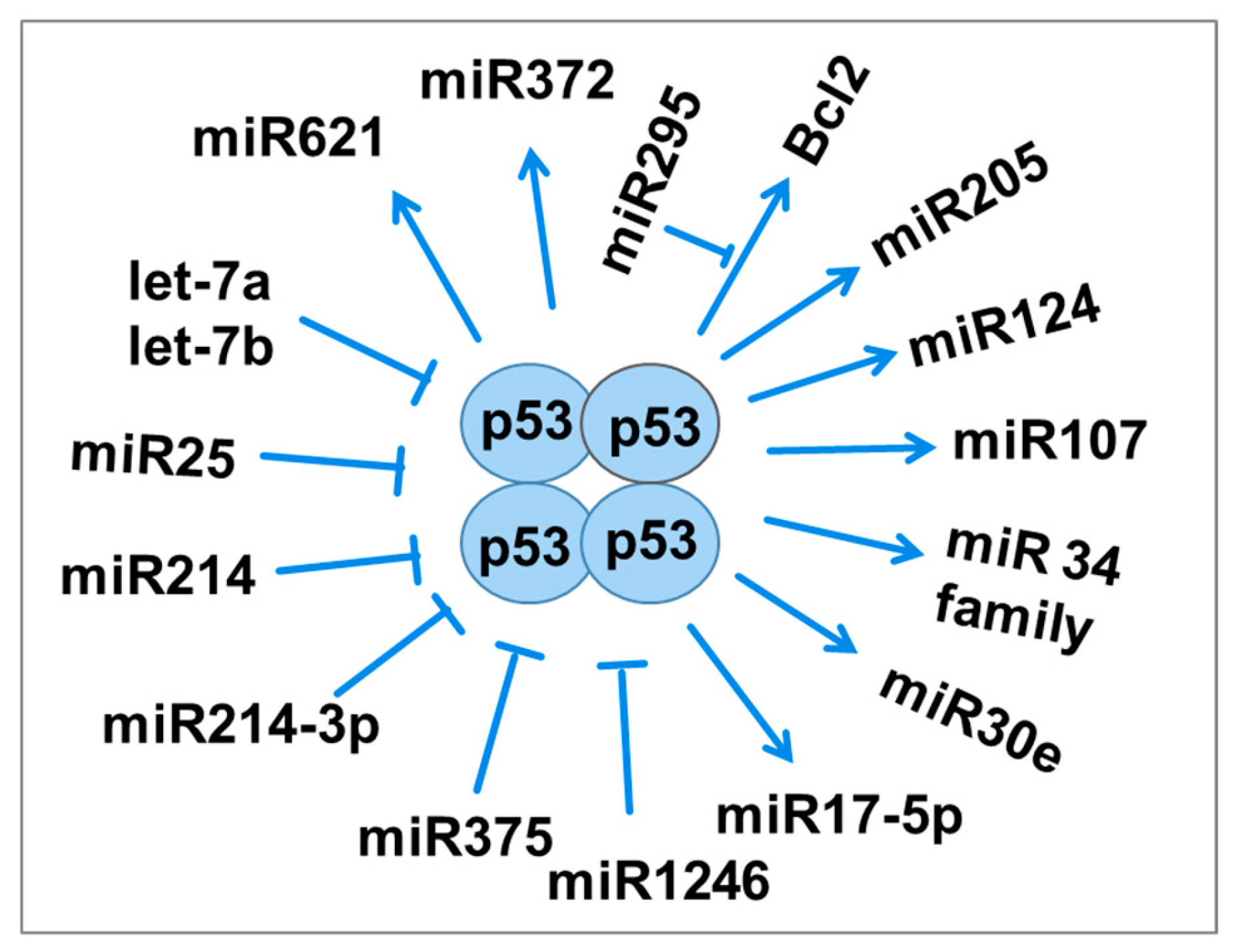
MicroRNAs (miRNAs) are single-stranded RNA molecules that play important roles in post-transcriptional gene regulation, influencing processes such as cell division, differentiation, apoptosis, metabolism, and carcinogenesis [39]. In response to radiation-induced DNA damage, the p53 gene activates the expression of miR-34. Overexpression of miR-34b has been shown to enhance radiosensitivity [40].
MiR-34a remains stable in serum after irradiation, suggesting it may serve as a novel biomarker, mediator, and therapeutic target for radiation injury, radiosensitivity, and radiation protection [41]. Glioblastoma cells exhibit increased expression of miR-34a following exposure to 60Gy of radiation, which correlates with decreased p53 expression. Even radioresistant cells can undergo apoptosis due to miR-34a regulation [42,43].
MiR-295 is another miRNA directly regulated by p53, binding to Bcl2 to potentially inhibit apoptosis and promote radioresistance in tumor cells. Increased expression of miR-375 has been associated with recurrent gastric cancer, where it downregulates p53 protein expression, thereby promoting radioresistance by preventing cell cycle arrest and apoptosis (Figure 4) [44,45,46,47].
4.7. Cell Division Cycle Protein 20 (CDC20) Regulates Radiosensitivity of p53 Mutant HCC Cells
CDC20 is one of the key downstream signaling molecules in the MDM2-p53 pathway.
There is a strong correlation between CDC20 expression and the prognosis of hepatocellular carcinoma (HCC). Inhibiting CDC20 has been shown to increase the radiosensitivity of HCC cells, identifying CDC20 as a potential biomarker and therapeutic target for a number of cancer types. WT p53, when activated by ionizing radiation (IR), triggers the expression of p21 and inhibits CDC20, which results in apoptosis and arrest in the G2/M phase of the cell cycle. On the other hand, mutant p53 fails to effectively regulate CDC20 and is less capable of inducing growth arrest and apoptosis. These results highlight that blocking CDC20 can enhance the sensitivity to radiotherapy and suggest that CDC20 could serve as a new target and prognostic marker for hepatocellular carcinoma (HCC), particularly in the presence of p53 mutations [48]. Patients with HCC exhibit a worse prognosis when CDC20 expression is high. However, when CDC20 is inhibited, HCC tumor cells become more radiosensitive [49]. Tumor development and cellular senescence are strongly linked to cell cycle arrest. Using siRNA to transfect and inhibit CDC20 expression in mantle cell lymphoma (MCL) cells, flow cytometry demonstrated a marked reduction in MCL cell proliferation and enhanced cell cycle arrest at the G2/M phase in MCL cells [50]. This suggests that targeting CDC20 could improve the efficacy of radiation therapy in treating HCC and MCL which is related to p53 status.
4.8. Deficiency of 53BP1 Inhibits the Radiosensitivity
Tumor suppressor p53-binding protein 1 (53BP1) is a significant member of the DNA damage response (DDR) pathway and a recently identified cancer suppressor gene. Involved in DDR, 53BP1 maintains genomic stability and, along with p53 and ATM, regulates apoptosis. The failure of 53BP1 to effectively anchor damaged chromosome ends can lead to chromosome aberrations. Research has shown that 53BP1 deletion occurs to varying degrees during the initiation and progression of many malignancies. This deletion is strongly associated with tumor staging, malignancy grading, and even resistance to therapy [51].
Elevated 53BP1 expression can influence the expression of specific DDR pathway proteins to enhance DDR, which is linked to increased sensitivity to therapy. Recent studies have identified the ATM-CHK2-p53 pathway as the primary apoptotic mechanism involved in DDR repair; the reduced expression of 53BP1 closely correlates with reduced expression of the essential components of this pathway, ATM, CHK2 and p53, leading to radiation resistance [52,53,54]. In conclusion, 53BP1 plays a significant role in determining radiosensitivity. Its deficiency inhibits the DDR, leading to reduced radiosensitivity and potentially contributing to tumor resistance to radiation therapy.
5. Targeting p53 in Cancer Treatment
Targeting p53 in cancer treatment is an area of intense research and interest. Efforts to restore p53 functionality in tumors as a therapeutic strategy to induce cancer cell death began decades ago, but these efforts have mostly met with limited success. Few p53 drug development initiatives have reached advanced clinical trial phases [55]. Nevertheless, several promising approaches towards p53-based therapy have emerged in recent years, giving hope that TP53 alterations will finally become ‘actionable’ targets. These approaches include:
5.1. Restoration of WT Activity
Several molecules have been identified that can reverse the oncogenic properties of mutant p53. Among these, the most widely investigated are PRIMA-1 (2,2-bis(hydroxymethyl)-1-azabicyclo(2,2,2)octan-3-one) and PRIMA-1MET. PRIMA-1 was originally discovered by screening a library of low molecular weight molecules for their ability to restore the tumor suppressive properties of mutant p53 [56]. This molecule can restore sequence-specific DNA binding capability to mutant p53 proteins and demonstrate in vivo tumor suppressive activity [57,58]. Therefore, restoring p53 function or mimicking its activity through pharmacological agents is an area of active research aimed at developing novel cancer therapies.
Some small molecules can reactivate the WT functions of mutant p53. PhiKan083 is a carbazole derivative discovered through in silico screening of the crystal structure of p53. By binding mutated p53, PhiKan083 raises the melting temperature of the protein, resulting in the reactivation of its function [59]. CP-31398 is another small molecule that can restore the protein folding of mutated p53 to a more natural conformation, allowing for WT function [60,66].
5.2. Blocking Interaction of WT p53 with MDM2/MDM4: Stabilizing WT p53
5.2.1. Low Molecular Weight Compounds
Although p53 mutations are common in cancer, they are not universal. In many cancers retaining WT p53, the protein can be inactivated through interaction with cellular proteins such as MDM2 or MDM4. Blocking the binding of these proteins to p53 using low molecular weight compounds, such as Nutlin-3 and MI-219 (specific inhibitors of the MDM2-p53 interaction), inhibits its degradation and restores WT function [61].
5.2.2. Stapled Peptides
Compared with low molecular weight inhibitors of the MDM2-p53 interaction, small peptides are potentially more specific in their mode of action and can be produced more cheaply. Stapling involves the formation of a hydrocarbon bond between 2 non-adjacent amino acids in the synthesized peptide. The stapled peptide PM2 shows potential in preventing MDM2 from inhibiting WT p53, making it a promising candidate for combination therapy with radiation. Furthermore, some of these peptides have been shown to promote apoptosis and suppress cancer cell growth both in vitro and in xenograft models [62].
5.2.3. Reactivating Suppressed p53 Functions
In some cancers, the TP53 gene remains intact, but the tumor suppressor p53 is inhibited through various mechanisms. MDM2 is the primary negative regulator of p53. It prevents p53 from entering the nucleus, inhibits its DNA binding, and promotes its proteasomal degradation [63]. Genetic amplification is the most frequent genomic alteration of MDM2, initially discovered in soft-tissue sarcoma. Notably, amplification and overexpression of MDM2 are mutually exclusive with p53 mutation [64]. Therefore, inhibiting MDM2 in cancers with WT 53 is a promising therapeutic strategy that has been successfully applied in clinical settings [65,66].
MDM2 is an E3 ubiquitin ligase that controls p53 degradation. Many tumors overexpress MDM2, even those without p53 mutations. Targeting MDM2 for p53 stabilization seems promising, and numerous reports have been published on this approach. For example, nutlins are cis-imidazoline compounds that act as antagonists of the MDM2-p53 interaction. Analysis of the crystal structure showed that nutlin binds in the pocket of MDM2, preventing the p53-MDM2 interaction [66].
5.3. Degradation of Mutant p53 with “Gain-of-Function”
Inhibiting mutant p53 by promoting its protein degradation is an effective strategy for treating cancers with mutant p53 with “gain-of-function”. Mutant p53 proteins are inherently unstable in normal cells [67] but are stabilized in cancer cells, for example, by HSP90 (heat shock protein 90), which is often overexpressed in cancer [68]. The discovery of several HSP90 inhibitors has made the ablation of mutant p53 a feasible anti-cancer approach [58].
5.4. Tumor Immunotherapy
Tumor-associated antigen-specific cytotoxic T lymphocytes can mediate the host immune response against cancer in vivo. The p53 protein, especially when targeting missense mutations, can serve as a tumor antigen. Some cancer patients have antibodies against p53, though the frequency and clinical significance are still under debate [66,69].
5.5. Gene Therapy with WT p53
The earliest attempts with WT p53 gene replacement for cancer treatment involved direct administration of a retroviral p53 expression vector into an orthotopic human lung tumor model, inducing tumor regression in most treated mice. Due to the advantages of adenoviral vectors (which do not integrate into the host genome), most subsequent trials used replication-defective recombinant p53 virus (Ad5CMV-p53). Although a few trials reached phase III, final FDA approval has not yet been granted [66,70].
Despite significant progress, targeting p53 in cancer treatment faces several challenges, including off-target effects, drug resistance, and the complexity of p53 signaling pathways. However, ongoing research efforts continue to uncover new insights into p53 biology and develop novel therapeutic strategies to exploit its potential as a target for cancer therapy.
6. Conclusions
In summary, the WT form of p53 plays a crucial role in maintaining genome stability and preventing cancer development. The TP53 gene, encoding p53, is extensively mutated in approximately half of human cancers. Functioning as a tumor suppressor, p53 safeguards genome integrity and inhibits tumor formation by inducing cell cycle arrest, promoting DNA repair, cellular senescence, and apoptosis. Additionally, it regulates autophagy and cancer cell metabolism. Despite its protective role, cancer cells employ various strategies to neutralize p53 to enhance their survival and proliferation. The primary method of disabling p53 is through mutations in the TP53 gene. Therefore, developing new approaches to reactivate mutant p53 is essential and could offer valuable insights for treating malignant cancers.
From a clinical perspective, TP53 mutations are not only prevalent but also diverse, significantly contributing to cancer progression across various tissues. This diversity highlights the challenge in effectively targeting mutant p53. Recent advances in cancer research have identified promising strategies for targeting mutant p53, such as small molecule compounds and gene therapy aimed at restoring WT p53 function. In addition to its canonical functions, p53’s role in regulating cellular metabolism, including glycolysis and oxidative phosphorylation, underscores its multifaceted role in cancer biology. Reactivating mutant p53 holds great promise in personalized cancer therapies, potentially improving treatment outcomes and patient survival. This approach could revolutionize current cancer treatment paradigms. However, overcoming resistance mechanisms developed by cancer cells to evade p53 reactivation remains a critical hurdle. Novel therapeutic modalities that circumvent these mechanisms are actively being explored. The global burden of cancer emphasizes the urgent need for innovative therapies targeting mutant p53, with potential implications for public health worldwide. These perspectives underscore both the challenges and potential avenues for therapeutic intervention in cancer biology, emphasizing the importance of ongoing research and development.
Author Contributions
Tusher -Al-Arafat, Aihong Mao and Bing Wang conceptualized, conceived and designed the study. Tusher -Al-Arafat prepared the first draft of the manuscript. Aihong Mao, Takanori Katsube and Bing Wang reviewed, edited and finalized the manuscript. All authors have read and approved the final manuscript.
Funding
Tusher -Al-Arafat was a fellow of the Nuclear Researchers Exchange Programme 2023 supported by the Ministry of Education, Culture, Sport, Sciences and Technology (MEXT) and the Nuclear Safety Research Association of Japan. Aihong Mao was a fellow of The Japan-China Sasakawa Medical Fellowship (fiscal year 2020) from The Japan China Medical Association supported by The Nippon Foundation, Japan.
Acknowledgments
The authors thank Ms. Aya Arakawa and Ms. Hiromi Arai (QST-NIRS, Japan), for their expert technical assistance and administrative support. Thanks are also due to the anonymous peer reviewers for providing the constructive comments that strengthened the presentation of this work.
Conflicts of Interest
The authors declare no conflicts of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
References
- Mireștean, C.C.; Iancu, R.I.; Iancu, D.P.T. p53 Modulates Radiosensitivity in Head and Neck Cancers—From Classic to Future Horizons. Diagnostics 2022, 12, 3052. [Google Scholar] [CrossRef] [PubMed]
- Feroz, W.; Sheikh, A.M.A. Exploring the multiple roles of guardian of the genome: P53. Egypt. J. Med. Hum. Genet. 2020, 21, 49. [Google Scholar] [CrossRef]
- Kong, X.; Yu, D.; Wang, Z.; Li, S. Relationship between p53 status and the bioeffect of ionizing radiation (Review). Oncol. Lett. 2021, 22, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Chen, J. The Cell-Cycle Arrest and Apoptotic Functions of p53 in Tumor Initiation and Progression. Cold Spring Harb. Perspect. Med. 2016, 6, a026104. [Google Scholar] [CrossRef] [PubMed]
- Engeland, K. Cell cycle arrest through indirect transcriptional repression by p53: I have a DREAM. Cell Death Differ. 2017, 25, 114–132. [Google Scholar] [CrossRef]
- Middleton, F.K.; Pollard, J.R.; Curtin, N.J. The Impact of p53 Dysfunction in ATR Inhibitor Cytotoxicity and Chemo- and Radiosensitisation. Cancers 2018, 10, 275. [Google Scholar] [CrossRef]
- Joerger, A. C., & Fersht, A. R. (2016). The p53 pathway: origins, inactivation in cancer, and emerging therapeutic approaches. Annual review of biochemistry, 85, 375-404.
- Stegh, A. H. (2012). Targeting the p53 signaling pathway in cancer therapy–the promises, challenges and perils. Expert opinion on therapeutic targets, 16(1), 67-83.
- Carr, M.I.; Jones, S.N. Regulation of the Mdm2-p53 signaling axis in the DNA damage response and tumorigenesis. Transl. Cancer Res. 2016, 5, 707–724. [Google Scholar] [CrossRef]
- Marei, H.E.; Althani, A.; Afifi, N.; Hasan, A.; Caceci, T.; Pozzoli, G.; Morrione, A.; Giordano, A.; Cenciarelli, C. p53 signaling in cancer progression and therapy. Cancer Cell Int. 2021, 21, 1–15. [Google Scholar] [CrossRef]
- Zhu, G.; Pan, C.; Bei, J.-X.; Li, B.; Liang, C.; Xu, Y.; Fu, X. Mutant p53 in Cancer Progression and Targeted Therapies. Front. Oncol. 2020, 10. [Google Scholar] [CrossRef]
- Patil, M.R.; Bihari, A. A comprehensive study of p53 protein. J. Cell. Biochem. 2022, 123, 1891–1937. [Google Scholar] [CrossRef]
- Hassin, O.; Oren, M. Drugging p53 in cancer: one protein, many targets. Nat. Rev. Drug Discov. 2023, 22, 127–144. [Google Scholar] [CrossRef] [PubMed]
- Meek, D.W. Regulation of the p53 response and its relationship to cancer. Biochem. J. 2015, 469, 325–346. [Google Scholar] [CrossRef] [PubMed]
- Graves, B.; Thompson, T.; Xia, M.; Janson, C.; Lukacs, C.; Deo, D.; Di Lello, P.; Fry, D.; Garvie, C.; Huang, K.-S.; et al. Activation of the p53 pathway by small-molecule-induced MDM2 and MDMX dimerization. Proc. Natl. Acad. Sci. 2012, 109, 11788–11793. [Google Scholar] [CrossRef] [PubMed]
- Rivlin, N.; Brosh, R.; Oren, M.; Rotter, V. Mutations in the p53 Tumor Suppressor Gene: Important Milestones at the Various Steps of Tumorigenesis. Genes Cancer 2011, 2, 466–474. [Google Scholar] [CrossRef]
- Varna, M.; Bousquet, G.; Plassa, L.-F.; Bertheau, P.; Janin, A. TP53 Status and Response to Treatment in Breast Cancers. J. Biomed. Biotechnol. 2011, 2011, 1–9. [Google Scholar] [CrossRef]
- Ozaki, T.; Nakagawara, A. Role of p53 in Cell Death and Human Cancers. Cancers 2011, 3, 994–1013. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Williams, A. B., & Schumacher, B. (2016). p53 in the DNA-damage-repair process. Cold Spring Harbor perspectives in medicine, 6(5), a026070.
- Rodriguez-Pastrana, I.; Birli, E.; Coutts, A.S. p53-dependent DNA repair during the DNA damage response requires actin nucleation by JMY. Cell Death Differ. 2023, 30, 1636–1647. [Google Scholar] [CrossRef]
- Pemberton, J.M.; Pogmore, J.P.; Andrews, D.W. Neuronal cell life, death, and axonal degeneration as regulated by the BCL-2 family proteins. Cell Death Differ. 2021, 28, 108–122. [Google Scholar] [CrossRef]
- Mijit, M.; Caracciolo, V.; Melillo, A.; Amicarelli, F.; Giordano, A. Role of p53 in the Regulation of Cellular Senescence. Biomolecules 2020, 10, 420. [Google Scholar] [CrossRef]
- Olivier, M., Hollstein, M., & Hainaut, P. (2010). TP53 mutations in human cancers: origins, consequences, and clinical use. Cold Spring Harbor perspectives in biology, 2(1), a001008.
- Habash, M.; Bohorquez, L.C.; Kyriakou, E.; Kron, T.; Martin, O.A.; Blyth, B.J. Clinical and Functional Assays of Radiosensitivity and Radiation-Induced Second Cancer. Cancers 2017, 9, 147. [Google Scholar] [CrossRef]
- Zhang, J.; Shen, L.; Sun, L.-Q. The regulation of radiosensitivity by p53 and its acetylation. Cancer Lett. 2015, 363, 108–118. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Liu, X.; Zhao, T.; Li, F.; Wang, Q.; Zhang, P.; Hirayama, R.; Chen, W.; Jin, X.; Zheng, X.; et al. Comparable radiation sensitivity in p53 wild-type and p53 deficient tumor cells associated with different cell death modalities. Cell Death Discov. 2021, 7, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Al Bitar, S., & Gali-Muhtasib, H. (2019). The role of the cyclin dependent kinase inhibitor p21cip1/waf1 in targeting cancer: Molecular mechanisms and novel therapeutics. Cancers, 11(10), 1475.
- Concin, N.; Zeillinger, C.; Stimpfel, M.; Schiebel, I.; Tong, D.; Wolff, U.; Reiner, A.; Leodolter, S.; Zeillinger, R. p53-dependent radioresistance in ovarian carcinoma cell lines. Cancer Lett. 2000, 150, 191–199. [Google Scholar] [CrossRef] [PubMed]
- Spring, P.M.; Arnold, S.M.; Shajahan, S.; Brown, B.; Dey, S.; Lele, S.M.; Valentino, J.; Jones, R.; Mohiuddin, M.; Ahmed, M.M. Low Dose Fractionated Radiation Potentiates the Effects of Taxotere in Nude Mice Xenografts of Squamous Cell Carcinoma of Head and Neck. Cell Cycle 2004, 3, 477–483. [Google Scholar] [CrossRef]
- Alvarado-Ortiz, E.; de la Cruz-López, K.G.; Becerril-Rico, J.; Sarabia-Sánchez, M.A.; Ortiz-Sánchez, E.; García-Carrancá, A. Mutant p53 Gain-of-Function: Role in Cancer Development, Progression, and Therapeutic Approaches. Front. Cell Dev. Biol. 2021, 8. [Google Scholar] [CrossRef]
- Stewart-Ornstein, J.; Iwamoto, Y.; Miller, M.A.; Prytyskach, M.A.; Ferretti, S.; Holzer, P.; Kallen, J.; Furet, P.; Jambhekar, A.; Forrester, W.C.; et al. p53 dynamics vary between tissues and are linked with radiation sensitivity. Nat. Commun. 2021, 12, 1–11. [Google Scholar] [CrossRef]
- Shangary, S.; Wang, S. Targeting the MDM2-p53 Interaction for Cancer Therapy. Clin. Cancer Res. 2008, 14, 5318–5324. [Google Scholar] [CrossRef]
- Zhao, Y.; Tan, M.; Liu, X.; Xiong, X.; Sun, Y. Inactivation of ribosomal protein S27-like confers radiosensitivity via the Mdm2-p53 and Mdm2-MRN-ATM axes. Cell Death Dis. 2018, 9, 1–11. [Google Scholar] [CrossRef]
- Amson, R.; Pece, S.; Lespagnol, A.; Vyas, R.; Mazzarol, G.; Tosoni, D.; Colaluca, I.; Viale, G.; Rodrigues-Ferreira, S.; Wynendaele, J.; et al. Reciprocal repression between P53 and TCTP. Nat. Med. 2012, 18, 91–99. [Google Scholar] [CrossRef]
- Kaarbø, M.; Storm, M.L.; Qu, S.; Wæhre, H.; Risberg, B.; Danielsen, H.E.; Saatcioglu, F. TCTP Is an Androgen-Regulated Gene Implicated in Prostate Cancer. PLOS ONE 2013, 8, e69398. [Google Scholar] [CrossRef]
- Wu, D.; Guo, Z.; Min, W.; Zhou, B.; Li, M.; Li, W.; Luo, D. Upregulation of TCTP expression in human skin squamous cell carcinoma increases tumor cell viability through anti-apoptotic action of the protein. Exp. Ther. Med. 2011, 3, 437–442. [Google Scholar] [CrossRef] [PubMed]
- Gu, X.; Yao, L.; Ma, G.; Cui, L.; Li, Y.; Liang, W.; Zhao, B.; Li, K. TCTP promotes glioma cell proliferation in vitro and in vivo via enhanced beta-catenin/TCF-4 transcription. Neuro Oncol. 2014, 16, 217–227. [Google Scholar] [CrossRef]
- Jung, J.; Lee, J.-S.; Lee, Y.-S.; Lee, K. Radiosensitivity of Cancer Cells Is Regulated by Translationally Controlled Tumor Protein. Cancers 2019, 11, 386. [Google Scholar] [CrossRef]
- Bajan, S.; Hutvagner, G. RNA-Based Therapeutics: From Antisense Oligonucleotides to miRNAs. Cells 2020, 9, 137. [Google Scholar] [CrossRef] [PubMed]
- Balça-Silva, J.; Neves, S.S.; Gonçalves, A.C.; Abrantes, A.M.; Casalta-Lopes, J.; Botelho, M.F.; Sarmento-Ribeiro, A.B.; Silva, H. Effect of miR-34b overexpression on the radiosensitivity of non-small cell lung cancer cell lines. Anticancer Res. 2012, 32, 1603–1609. [Google Scholar]
- Mert, U.; Özgür, E.; Tiryakioglu, D.; Dalay, N.; Gezer, U. Induction of p53-inducible microRNA miR-34 by gamma radiation and bleomycin are different. Front. Genet. 2012, 3, 33413. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, A.; Udaka, Y.; Tsunoda, Y.; Yamamoto, G.; Tsuji, M.; Oyamada, H.; Oguchi, K.; Mizutani, T. Analysis of p53 and miRNA expression after irradiation of glioblastoma cell lines. Anticancer Res. 2012, 32, 4709–4713. [Google Scholar] [PubMed]
- Wang, B.; Li, D.; Kovalchuk, O. p53 Ser15 phosphorylation and histone modifications contribute to IR-induced miR-34a transcription in mammary epithelial cells. Cell Cycle 2013, 12, 2073–2083. [Google Scholar] [CrossRef]
- Liu, Y.; Xing, R.; Zhang, X.; Dong, W.; Zhang, J.; Yan, Z.; Li, W.; Cui, J.; Lu, Y. miR-375 targets the p53 gene to regulate cellular response to ionizing radiation and etoposide in gastric cancer cells. DNA Repair 2013, 12, 741–750. [Google Scholar] [CrossRef]
- Wu, S.-Y.; Wu, A.T.; Liu, S.-H. MicroRNA-17-5p regulated apoptosis-related protein expression and radiosensitivity in oral squamous cell carcinoma caused by betel nut chewing. Oncotarget 2016, 7, 51482–51493. [Google Scholar] [CrossRef]
- Shao, Y.; Song, X.; Jiang, W.; Chen, Y.; Ning, Z.; Gu, W.; Jiang, J. MicroRNA-621 Acts as a Tumor Radiosensitizer by Directly Targeting SETDB1 in Hepatocellular Carcinoma. Mol. Ther. 2019, 27, 355–364. [Google Scholar] [CrossRef] [PubMed]
- Okazaki, R. Role of p53 in Regulating Radiation Responses. Life 2022, 12, 1099. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S., Zhang, Y., Lu, X., Ding, H., Han, B., Song, X.,... & Wang, J. (2021). CDC20 regulates the cell proliferation and radiosensitivity of P53 mutant HCC cells through the Bcl-2/Bax pathway. International journal of biological sciences, 17(13), 3608.
- Yang, G.; Wang, G.; Xiong, Y.; Sun, J.; Li, W.; Tang, T.; Li, J. CDC20 promotes the progression of hepatocellular carcinoma by regulating epithelial-mesenchymal transition. Mol. Med. Rep. 2021, 24, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Yang, P.; Wang, J.; Gao, S.; Xiao, S.; Zhang, W.; Zhu, M.; Wang, Y.; Ke, X.; Jing, H. p53 directly downregulates the expression of CDC20 to exert anti-tumor activity in mantle cell lymphoma. Exp. Hematol. Oncol. 2023, 12, 1–23. [Google Scholar] [CrossRef]
- Abuetabh, Y.; Wu, H.H.; Chai, C.; Al Yousef, H.; Persad, S.; Sergi, C.M.; Leng, R. DNA damage response revisited: the p53 family and its regulators provide endless cancer therapy opportunities. Exp. Mol. Med. 2022, 54, 1658–1669. [Google Scholar] [CrossRef]
- Han, X., Zhang, L., Chung, J., Mayca Pozo, F., Tran, A., Seachrist, D. D., ... & Zhang, Y. (2014). UbcH7 regulates 53BP1 stability and DSB repair. Proceedings of the National Academy of Sciences, 111(49), 17456-17461.
- Yao, J.; Huang, A.; Zheng, X.; Liu, T.; Lin, Z.; Zhang, S.; Yang, Q.; Zhang, T.; Ma, H. 53BP1 loss induces chemoresistance of colorectal cancer cells to 5-fluorouracil by inhibiting the ATM–CHK2–P53 pathway. J. Cancer Res. Clin. Oncol. 2016, 143, 419–431. [Google Scholar] [CrossRef]
- Xiao, Y.; Zheng, X.; Huang, A.; Liu, T.; Zhang, T.; Ma, H. Deficiency of 53BP1 inhibits the radiosensitivity of colorectal cancer. Int. J. Oncol. 2016, 49, 1600–1608. [Google Scholar] [CrossRef]
- Duffy, M.J.; Synnott, N.C.; Crown, J. p53 in cancer: ready for therapeutic targeting? Transl. Cancer Res. 2016, 5, 627–631. [Google Scholar] [CrossRef]
- Duffy, M.J.; Synnott, N.C.; McGowan, P.M.; Crown, J.; O’connor, D.; Gallagher, W.M. p53 as a target for the treatment of cancer. Cancer Treat. Rev. 2014, 40, 1153–1160. [Google Scholar] [CrossRef]
- Bykov, V.J.; Issaeva, N.; Shilov, A.; Hultcrantz, M.; Pugacheva, E.; Chumakov, P.; Bergman, J.; Wiman, K.G.; Selivanova, G. Restoration of the tumor suppressor function to mutant p53 by a low-molecular-weight compound. Nat. Med. 2002, 8, 282–288. [Google Scholar] [CrossRef]
- Zhou, X.; Hao, Q.; Lu, H. Mutant p53 in cancer therapy—the barrier or the path. J. Mol. Cell Biol. 2018, 11, 293–305. [Google Scholar] [CrossRef] [PubMed]
- F. M. Boeckler, A. C. Joerger, G. Jaggi, T. J. Rutherford, D. B. Veprintsev, and A. R. Fersht, “Targeted rescue of a destabilized mutant of p53 by an in silico screened drug,” Proceedings of the National Academy of Sciences of the United States of America, vol. 105, no. 30, pp. 10360–10365, 2008.
- Rippin, T.M.; Bykov, V.J.N.; Freund, S.M.V.; Selivanova, G.; Wiman, K.G.; Fersht, A.R. Characterization of the p53-rescue drug CP-31398 in vitro and in living cells. Oncogene 2002, 21, 2119–2129. [Google Scholar] [CrossRef] [PubMed]
- Shangary, S.; Wang, S. Targeting the MDM2-p53 Interaction for Cancer Therapy. Clin. Cancer Res. 2008, 14, 5318–5324. [Google Scholar] [CrossRef] [PubMed]
- Mortensen, A.C.L.; Spiegelberg, D.; Brown, C.J.; Lane, D.P.; Nestor, M. The Stapled Peptide PM2 Stabilizes p53 Levels and Radiosensitizes Wild-Type p53 Cancer Cells. Front. Oncol. 2019, 9, 923. [Google Scholar] [CrossRef] [PubMed]
- Freedman, D.A.; Wu, L.; Levine, A.J. Functions of the MDM2 oncoprotein. Cell. Mol. Life Sci. 1999, 55, 96–107. [Google Scholar] [CrossRef]
- Oliner JD, Saiki AY, Caenepeel S. The role of MDM2 amplifcation and overexpression in tumorigenesis. Cold Spring Harb Perspect Med. 2016;6:a026336.
- Hu, J.; Cao, J.; Topatana, W.; Juengpanich, S.; Li, S.; Zhang, B.; Shen, J.; Cai, L.; Cai, X.; Chen, M. Targeting mutant p53 for cancer therapy: direct and indirect strategies. J. Hematol. Oncol. 2021, 14, 1–19. [Google Scholar] [CrossRef]
- Suzuki, K.; Matsubara, H. Recent Advances in p53 Research and Cancer Treatment. BioMed Res. Int. 2011, 2011, 978312. [Google Scholar] [CrossRef]
- Mantovani, F.; Collavin, L.; Del Sal, G. Mutant p53 as a guardian of the cancer cell. Cell Death Differ. 2018, 26, 199–212. [Google Scholar] [CrossRef]
- Trepel, J.; Mollapour, M.; Giaccone, G.; Neckers, L. Targeting the dynamic HSP90 complex in cancer. Nat. Rev. Cancer 2010, 10, 537–549. [Google Scholar] [CrossRef]
- Vojtesek, B.; Kovarik, J.; Dolezalova, H.; Nenutil, R.; Havlis, P.; Brentani, R.; Lane, D. Absence of p53 autoantibodies in a significant proportion of breast cancer patients. Br. J. Cancer 1995, 71, 1253–1256. [Google Scholar] [CrossRef]
- Zhang, S. Y., Lu, Y. Y., & Peng, Z. H. (2012). 10—Recombinant Adenoviral-p53 Agent (Gendicine®): Quality Control, Mechanism of Action, and Its Use for Treatment of Malignant Tumors. Recent advances in cancer research and therapy, 215-243.
Figure 1.
The tetrameric transcription factor p53 is activated upon cellular stress through multiple phosphorylation events. Depending on the type of stress, this activation results in upregulation or repression of genes involved in cell-cycle arrest, senescence, DNA repair, apoptosis, and autophagy. In unstressed cells, p53 protein levels are tightly regulated via a negative-feedback loop mediated by MDM2. Abbreviations: Atg10, Autophagy-related 10; Bax, BCL-2-associated X protein; Btg2, B cell translocation gene-2, Cdkn1α, Cyclin-dependent kinase inhibitor-1α; Ddb2, Damage-specific DNA-binding protein-2; Foxo3, Forkhead box O3; Gadd45α, Growth arrest and DNA-damage-inducible 45α; Noxa, NADPH oxidase activator; Pai1, Plasminogen activator inhibitor-1; Pig3, p53-inducible protein-3; Pml, Promyelocytic leukemia; P53r2, p53-inducible ribonucleotide reductase; Puma, p53 upregulated modulator of apoptosis; Tpp1, Tripeptidyl peptidase-1; Tsc2, Tuberous sclerosis-2.
Figure 1.
The tetrameric transcription factor p53 is activated upon cellular stress through multiple phosphorylation events. Depending on the type of stress, this activation results in upregulation or repression of genes involved in cell-cycle arrest, senescence, DNA repair, apoptosis, and autophagy. In unstressed cells, p53 protein levels are tightly regulated via a negative-feedback loop mediated by MDM2. Abbreviations: Atg10, Autophagy-related 10; Bax, BCL-2-associated X protein; Btg2, B cell translocation gene-2, Cdkn1α, Cyclin-dependent kinase inhibitor-1α; Ddb2, Damage-specific DNA-binding protein-2; Foxo3, Forkhead box O3; Gadd45α, Growth arrest and DNA-damage-inducible 45α; Noxa, NADPH oxidase activator; Pai1, Plasminogen activator inhibitor-1; Pig3, p53-inducible protein-3; Pml, Promyelocytic leukemia; P53r2, p53-inducible ribonucleotide reductase; Puma, p53 upregulated modulator of apoptosis; Tpp1, Tripeptidyl peptidase-1; Tsc2, Tuberous sclerosis-2.
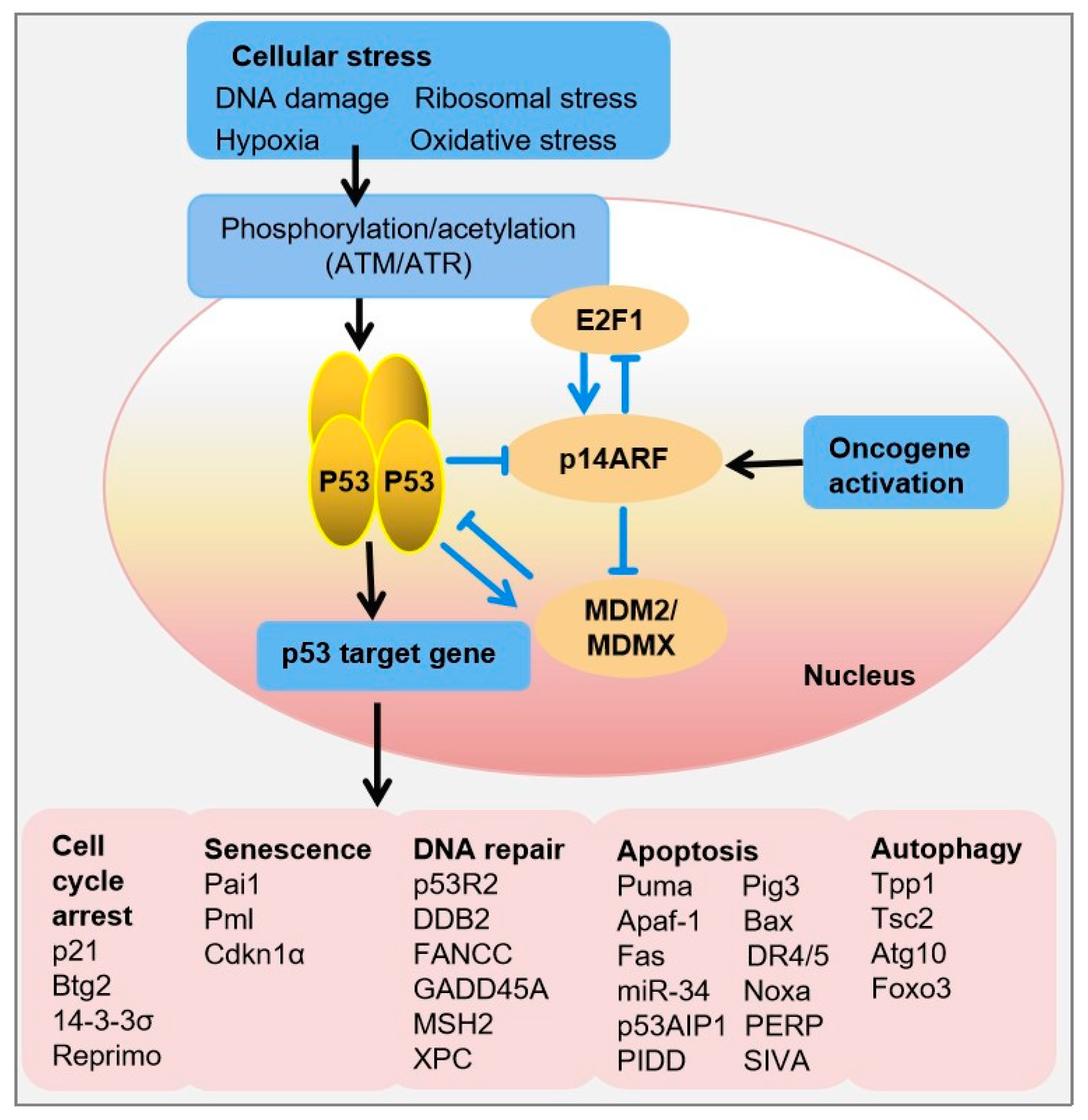
Figure 2.
Mutant p53 promotes adaptive responses to cancer-related stress conditions to support tumor progression. Abbreviations: NOX4, NADPH oxidase 4; ROS, reactive oxygen species; Akt/mTOR, protein kinase B (Akt)/mammalian target of rapamycin (mTOR); DDR, DNA damage response; UCP2, Uncoupling protein 2; PGC-1α, Peroxisome proliferator-activated receptor gamma coactivator 1-alpha; SESNs, Sestrin; AMPK, AMP-activated protein kinase.
Figure 2.
Mutant p53 promotes adaptive responses to cancer-related stress conditions to support tumor progression. Abbreviations: NOX4, NADPH oxidase 4; ROS, reactive oxygen species; Akt/mTOR, protein kinase B (Akt)/mammalian target of rapamycin (mTOR); DDR, DNA damage response; UCP2, Uncoupling protein 2; PGC-1α, Peroxisome proliferator-activated receptor gamma coactivator 1-alpha; SESNs, Sestrin; AMPK, AMP-activated protein kinase.
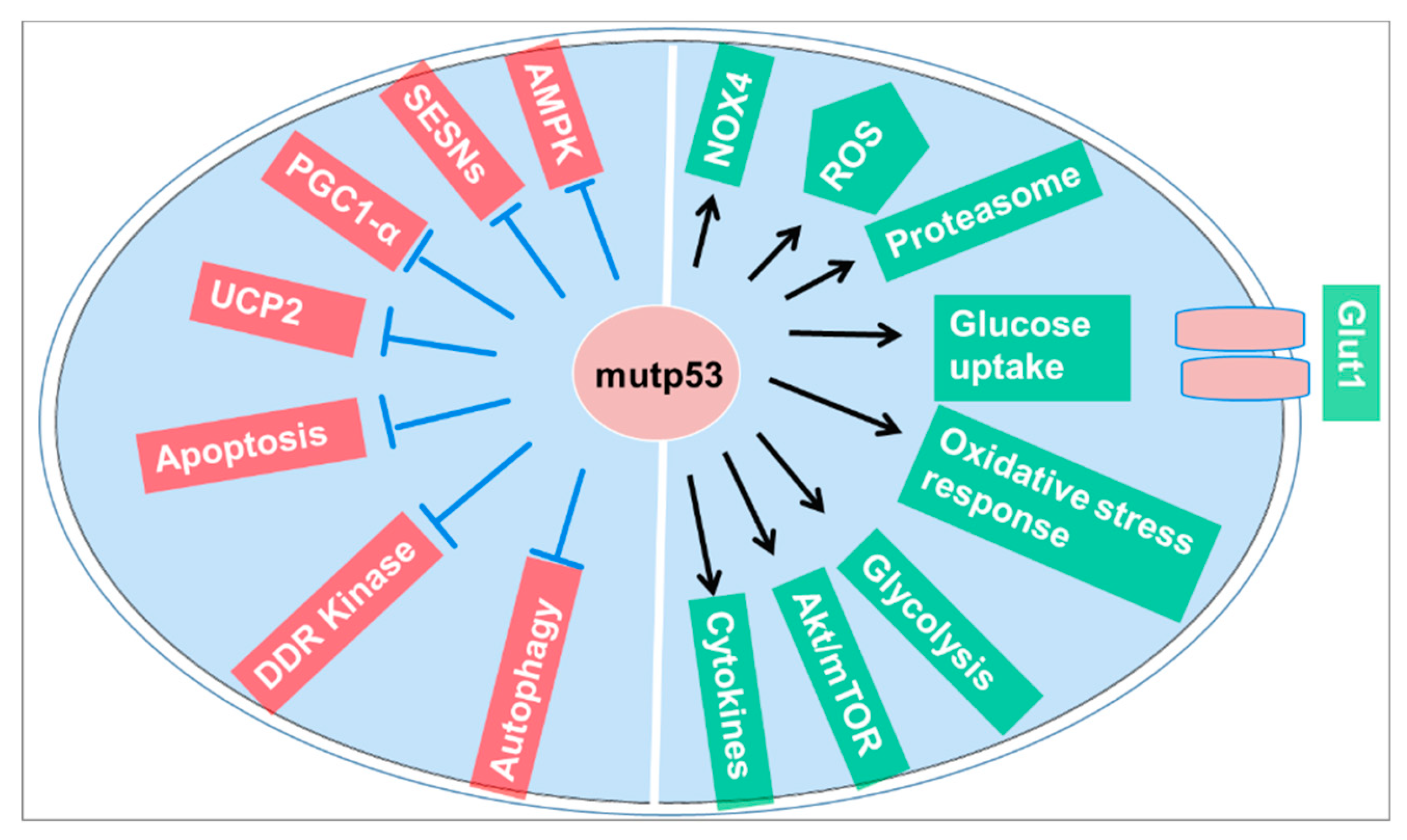
Figure 3.
A model for radiosensitivity upon inactivation of ribosomal protein S27l (Red circle represents inactivated S271) mediated by the MDM2-p53 and MDM2–MRN–ATM axes.
Figure 3.
A model for radiosensitivity upon inactivation of ribosomal protein S27l (Red circle represents inactivated S271) mediated by the MDM2-p53 and MDM2–MRN–ATM axes.
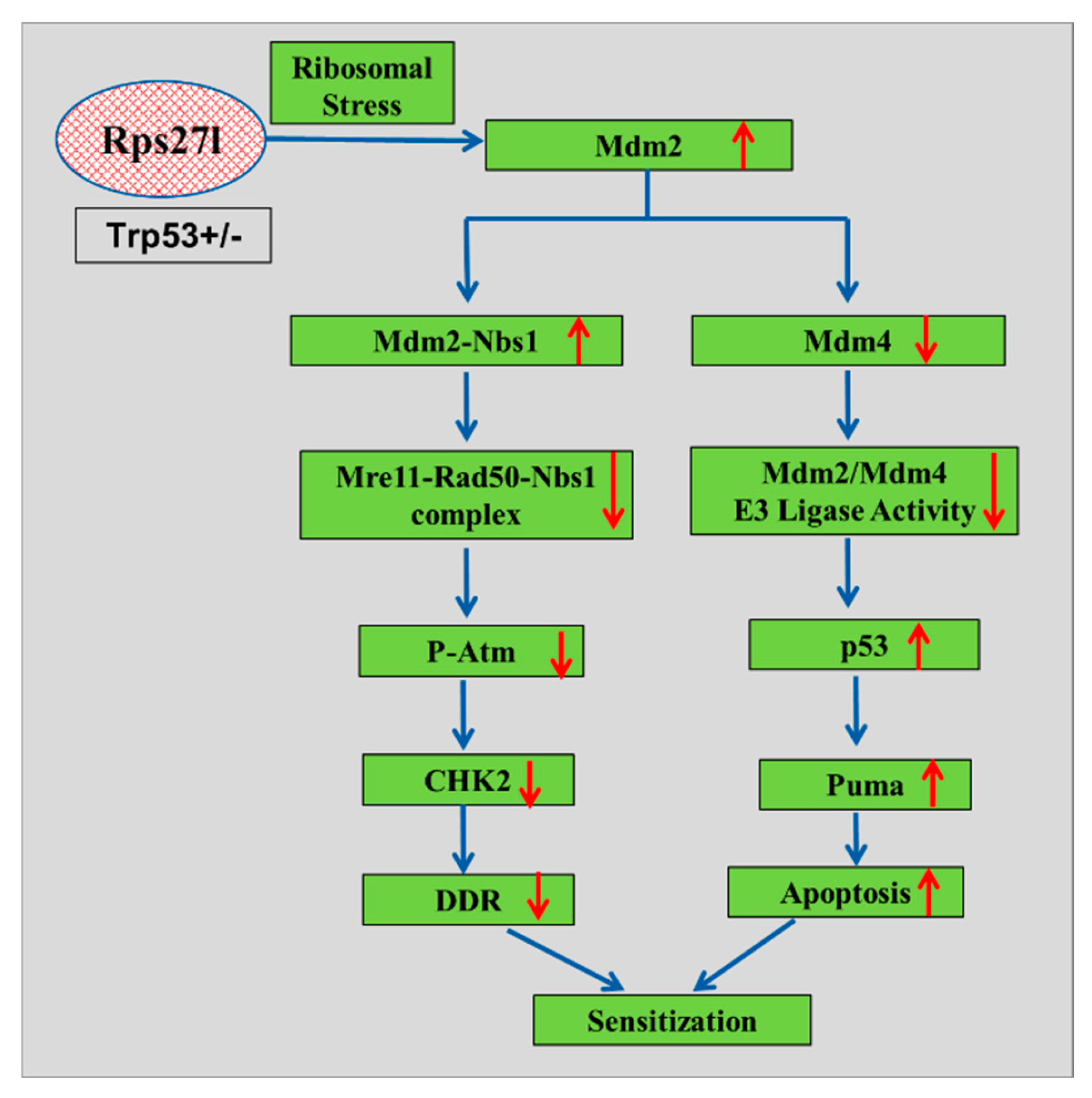
Figure 4.
The relationship between p53 and miRNAs in response to radiation involves miRNAs either activating p53 or inhibiting its function.
Figure 4.
The relationship between p53 and miRNAs in response to radiation involves miRNAs either activating p53 or inhibiting its function.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



