
Submitted:
29 August 2024
Posted:
30 August 2024
You are already at the latest version
Abstract
More than 90% of patients affected by mastocytosis are characterized by a somatic point mutation of c-KIT, which induces ligand-independent activation of the receptor and downstream signal triggering, ultimately leading to mast cell proliferation and survival. The most frequent mutation is c-KIT p.D816V, but other rarer mutations can also be found. These mutations often have a very low variant allele frequency (VAF), well below the sensitivity of common next-generation sequencing (NGS) methods used in routine diagnostic panels. Highly sensitive methods are developing for detecting mutations. This review summarizes the current indications on the recommended methods and on how to manage and interpret molecular data for the diagnosis and follow-up of patients with mastocytosis.
Keywords:
c-KIT
; gene mutations
; Systemic Mastocytosis
; digital PCR
1. Introduction
Mastocytosis is a rare clonal disease characterized by an accumulation of mast cells in various organs including skin, bone, gastrointestinal tract, lymph nodes, and spleen [1,2]. It can affect any age, with a male-to-female ratio of about 1:1. The reported prevalence of mastocytosis in the adult population ranges from 9-13 per 10,000 adults. There are no prevalence data for the pediatric forms. Systemic mastocytosis almost always arises in adulthood, generally between the ages of 20 and 50. In children, the onset of mastocytosis occurs in 75-80% of cases within the first two years of life. Most cases are sporadic, but rare familial forms have been described [1,2,3]. From a molecular point of view mastocytosis is mostly associated with a somatic point mutation in the gene encoding the KIT receptor for stem cell factor (SCF). The most frequent c-KIT gene mutation is D816V, present in approximately 85-90% of adult cases and about 50% of pediatric cases [4,5,6]. Mastocytosis is associated with a wide variety of clinical manifestations, generally due to the inappropriate release of mediators by mast cells. Among these the most frequent are itching, urticaria, angioedema, flushing, nausea, vomiting, abdominal pain, diarrhea, anaphylaxis, cardiovascular symptoms like syncope or presyncope, tachycardia, osteopenia or osteoporosis. In addition, the presence of skin involvement is frequent with diffuse red-brown lesions, especially on the trunk and limbs with the characteristic Darier's sign when rubbed [7].In the aggressive forms, mastocytosis also presents signs of organ dysfunction related to mast cell infiltration including hypersplenism, pathological fractures, ascites, malabsorption and cytopenias. The rarity of this condition and the multiplicity of clinical manifestations necessitate the integration of various disciplines for accurate diagnosis and appropriate therapeutic management [7].
About 60% of systemic mastocytosis are associated with another hematologic neoplasm, primarily chronic myelomonocytic leukemia (CMML) and chronic myeloproliferative neoplasms (MPN) and myelodysplastic syndromes (MDS) [8,9].
The WHO classification 2022 defines precise criteria for the diagnosis of systemic mastocytosis [8]. The major criterion is based on the presence of multifocal dense infiltrates of mast cells with more than 15 mast cells aggregated in bone marrow (BM) and/or other extracutaneous organs. Minor criteria include i) the presence of more than 25% of spindle-shaped or atypical mast cells in mast cell infiltrates detected in bone marrow or other extracutaneous organ histological sections, or more than 25% of mast cells with immature or atypical appearance in bone marrow smears; ii) Presence of the Asp816Val mutation in the c-KIT gene or other c-KIT mutation in bone marrow, peripheral blood, or other extracutaneous organs; iii) Expression of CD25 and/or CD2 and/or CD30 in mast cells in the bone marrow, peripheral blood, or other extracutaneous organs; and iv) persistent serum tryptase levels >20 ng/mL. WHO classification divides systemic mastocytosis into six different entities: Bone Marrow Mastocytosis (BMM), Indolent Systemic Mastocytosis (ISM), Smoldering Systemic Mastocytosis (SSM), Aggressive Systemic Mastocytosis (ASM), Systemic Mastocytosis with an Associated Hematological Neoplasm (SM-AHN) and Mast Cell Leukemia (MCL) (Table 1).
Table1: This table presents the World Health Organization (WHO) classification for systemic mastocytosis, outlining the various subtypes and diagnostic criteria. The criteria are based on clinical, histological, and genetic factors, which aid in the accurate diagnosis and management of the disease.
According to the WHO 2022 classification [8], the diagnosis of systemic mastocytosis (SM) is based on the integration of laboratory parameters (serum tryptase levels, blood chemistry tests), bone marrow (BM) morphological parameters (BM biopsy, BM blood smear), genetic parameters (presence of c-KIT mutations, cytogenetic alterations, or other markers of clonality), cytometric/immunohistochemical parameters (aberrant expression of membrane antigens), radiological and clinical parameters. In the case of patients with suspected SM but with normal or slightly elevated tryptase, highly sensitive techniques of molecular biology, such as allele-specific qPCR (ASOqPCR) and digital droplet PCR (ddPCR), are necessary [10]. The diagnostic process starts from clinical suspicion and varies according to the detected tryptase level [11]. If the tryptase level is >25 ng/mL, a complete bone marrow evaluation is immediately indicated, whereas if the level is <15 ng/mL, it is suggested to perform periodic monitoring over time. If the level is between 15-25 ng/mL, the decision to proceed with a bone marrow evaluation is based on additional important parameters, such as a REMA score ≥2 [12], the presence of the c-KIT p.D816V mutation in peripheral blood (PB), or the presence of additional symptoms suggestive of the disease.
Symptoms can be variable in type, number, location and intensity; in some cases, they are very mild or even absent, while in others, they are severe or life-threatening. Symptoms due to mediator release are also variable and include cutaneous, allergic, gastrointestinal, neuropsychiatric, osteoarticular symptoms and many others [1,2].
Anaphylaxis is certainly the most severe clinical manifestation of ISM. Its frequency is reported between 22% and 49% in adults and between 6% and 9% in children [13,14]. The triggers are numerous, with hymenoptera stings being the most frequent cause, followed by reactions to foods and drugs [15]. Idiopathic anaphylaxis can account for up to 39% of anaphylactic cases in adults. As in the general population, cofactors such as alcohol, physical exercise, and sudden temperature changes can play a significant role, being determinants in about 26% of anaphylactic reactions. A preferential association between hymenoptera venom allergy and mastocytosis is well established. The prevalence of hymenoptera venom allergy in the European adult population is about 3%, whereas it rises to 20-30% in patients with clonal mast cell disorders [13,14,15,16].
2. KIT/CD117
KIT/CD117 is a type III tyrosine kinase (TK) transmembrane receptor which binds stem cell factor [17]. C-KIT gene, located on chromosome 4q12, consists of 21 exons and codes for a glycoprotein of 145 kD and 976 amino acids. The structure of the receptor is very similar to other TK receptors with a N-terminal extracellular domain that binds SCF, and a C-terminal intracellular domain [18]. The two domains are linked by a transmembrane domain. The sequences linked to kinase activity are all within the intracellular domain which can be divided into the juxtamembrane domain, with an inhibitory function of TK activity, and the TK domain. The extracellular domain (ECD) includes five immunoglobulin-like domains important for ligand binding and receptor dimerization [17,18,19].
There are several isoforms of KIT generated by alternative splicing, including two isoforms that arise from the presence or absence of a tetrapeptide sequence: glycine-asparagine-asparagine-lysine (GNNK) [20,21,22]. Although both isoforms have the ability to bind SCF, the GNNK-negative form has a faster receptor phosphorylation capability and, therefore, a more robust downstream signal. This isoform in mice has revealed oncogenic potential. A third isoform results from the loss of a serine residue in the kinase domain. Finally, a fourth isoform results from a truncated transcript that gives rise to a truncated protein lacking kinase activity [20,21].
c-KIT is expressed in various cell types [23]. The signal activated by the binding between the receptor and SCF is involved in many biological processes, primarily cell survival, proliferation, and migration. In normal bone marrow, c-KIT is expressed in hematopoietic stem cells and plays an important role in self-renewal and differentiation into various types of mature cells. c-KIT is gradually downregulated during differentiation and remains expressed only in mast cells, natural killer (NK) cells, and dendritic cells (DCs), suggesting its role in immunity and inflammation [24,25]. In mice, homozygous loss of c-KIT in hematopoietic cells causes a defect in stem cells leading to death from severe anemia [26]. Finally, c-KIT is not only expressed in hematopoietic stem cells but also in the prostate, liver, and heart, indicating its role in the stem cells of these organs as well [27,28,29]. KIT has also been shown to be crucial in regulating oogenesis and spermatogenesis, thus playing a crucial role in fertility.
3. Downstream Signaling of KIT
The downstream signaling of KIT involves the activation of MAPK/ERK, PI3K/AKT, PLCγ, and JAK/STAT pathways [30]. The MAPK/ERK pathway is activated by the phosphorylation of Y703 and Y936 and plays a key role in transcription activation and cell proliferation [31]. The phosphorylation of Y721 activates the PI3K/AKT pathway, which promotes cell survival and escape from apoptosis [32]. In addition, the activation of the Src family of kinases (SFK) can occur resulting in cell proliferation and survival through Akt phosphorylation and cell migration through the phosphorylation of focal adhesion kinase [31,32,33]. Finally, it has been shown that phosphorylation of some KIT tyrosine residues leads to JAK/STAT activation with the translocation of STAT proteins into the nucleus where they target specific genes [32,33] (Figure 1). There are also negative feedback mechanisms to control the signal once the KIT receptor is activated. Among these, ubiquitination is the main mechanism. E3 ubiquitin-protein ligase c-CBL binds directly to activated KIT receptor trough Y568 and Y936 or indirectly through Grb2 to Y703 and Y936 or through the p85 subunit of PI3K [34,35]. Additional mechanisms include dephosphorylation, and PKC-dependent serine phosphorylation. Finally, signal inactivation can occur through phosphatases such as Src homology region 2 domain-containing phosphatase 1 which causes KIT dephosphorylation.
4. KIT Is Activated in Many Tumors
The majority of KIT alterations found in cancer are associated with gain of function mutations leading to a constitutive activation of c-KIT independently from SCF. Gain of function mutations of c-KIT are detectable in many cancers including gastrointestinal stromal tumor (GIST) [36,37], melanoma [38], seminoma [39], mastocytosis [40], acute myeloid leukemia (AML) [41].
More than 90% of mastocytosis cases are characterized by a somatic point mutation of c-KIT, which induces ligand-independent activation of the receptor and downstream signal activation, ultimately leading to mast cell proliferation and survival [42,43]. Different types of mutations have been described in mastocytosis [42,43,44]. A picture of c-KIT mutations is represented in Figure 2. Mutations located in the extracellular domain impact on KIT dimerization and mutations in the phosphotransferase domain impact on the kinase activity [45].
In children, c-KIT mutations often affect the extracellular domain of the receptor, while in adults the most frequent activating mutation is the c-KIT p.D816V found in the phosphotransferase domain encoded by exon 17 [46]. Other activating mutations, such as c-KIT V560G in the juxtamembrane domain and c-KIT D419del in the extracellular domain, are less common but found in patients with aggressive systemic mastocytosis (ASM), mast cell leukemia (MCL), and mast cell sarcoma [47,48,49]. Unlike adults, children with mastocytosis show the c-KIT p.D816V mutation in about 30% of skin biopsy cases, with other c-KIT-activating mutations in the extracellular domain present in about 40% of cases [46]. A recent study found no significant link between the progression (spontaneous regression or persistence into adolescence) and the type of c-KIT mutation in pediatric cases [50]. Finally, there are rare cases, not exceeding 5%, that do not present c-KIT mutations. Among these, we find almost all cases of mast cell sarcoma and cases with well-differentiated mast cells. However, there are also rare cases of classic systemic mastocytosis without c-KIT mutation [51].
It is also interesting to note that studies of c-KIT mutations performed on cells other than mast cells show that about 30% of individuals with mastocytosis have a multilineage myeloid and/or lymphoid involvement of hematopoiesis [52,53,54,55]. Unlike what happens in the majority of patients where the c-KIT mutation is present only in the mast cell compartment, in these patients, the mutation is also found in basophils, eosinophils, neutrophils, as well as in B and T lymphocytes. This evidence suggests that the acquisition of the mutation occurs early in a multipotent hematopoietic progenitor and that the clonal origin of mastocytosis and the associated hematologic neoplasm is common. Multilineage involvement is more frequent in AdvSM or SM forms and less frequent in ISM forms, but when present in the latter, it is associated with a worse prognosis with a higher risk of disease progression [54]. This suggests that the earlier the mutation occurs hierarchically, the higher the risk of acquiring additional mutations and disease progression. In these patients the c-KIT mutant allele burden is a surrogate for the measurement of the multilineage involvement [56].
5. Methods for the Detection of c-KIT Mutations
The frequency of the c-KIT p.D816V mutation varies depending on the sensitivity of the method used to detect it, ranging from 30% to 95% of cases [57]. Although the presence of c-KIT mutations is a diagnostic criterion for systemic mastocytosis, there is currently no globally accepted standardized method for its detection.
Various non-quantitative (or semi-quantitative) methods have been employed in the past to detect the c-KIT p.D816V mutation. These methods include RT-PCR combined with restriction fragment length polymorphism, nested RT-PCR followed by D-HPLC of PCR amplicons, and peptide nucleic acid-mediated PCR [58]. Generally, these methods are known for their relatively low sensitivity and/or higher likelihood of false-negative results, particularly in cases with low mast cell burden and lack of multilineage involvement.
However, there is currently a consensus that the search for c-KIT mutations should be performed using highly sensitive methods. The limit of detection (LOD) shall be less than or equal to 0.01% VAF [51].
This level of sensitivity is reached by ASO PCR or by droplet digital PCR (ddPCR) [59].
This recommendation stems from the fact that the allelic burden of c-KITp.D816V can be very low because the mast cell component is often poorly represented in both PB and BM.
Kristensen and colleagues developed a highly sensitive method based on mutation specific q-PCR for the detection of c-KIT mutation in mastocytosis [57]. This assay has a sensitivity ranging from 0,001% to 0,03% positive alleles and a specificity of 100%. They demonstrated for the first time that by using a highly sensitive and specific assay it is possible to detect c-KIT mutation in blood in nearly all adult patients with mastocytosis [57]. Although qPCR is characterized by an adequate level of sensitivity, it also has the disadvantage that it requires a calibration material, and there is no commonly accepted c-KIT calibrator. Currently, plasmid dilutions are used as a calibrator, but differences in the material used for calibration have made it difficult to standardize the method. To overcome this problem, attention has recently shifted to droplet digital PCR (ddPCR) [59].
Droplet Digital PCR employs the distribution of diluted target nucleic acids across numerous reactions (partitions) to precisely quantify DNA molecules without the need for calibration material [60,61]. As a result, ddPCR has emerged as a new benchmark for quantifying mutant alleles at low variant allele fractions (VAF) [60].
Greiner and colleagues validated ddPCR assay for c-KIT p.D816V [59]. They compared ddPCR to ASO PCR demonstrating a high level of concordance between the two methods. Basing on these data the EU-US Cooperative Group considers both methods suitable for the quantification of c-KIT p.D816V allelic burden from DNA [51]. ddPCR assays have the advantage that can be interlaboratory standardized since it does not require internal calibrator material.
Finally, although ultrasensitive NGS assays have been developed for detecting c-KIT p.D816V, there are still limited data. It is important to underline that standard NGS techniques used for myeloid neoplasms workup have an insufficient limit of detection, usually ranging between 1% and 5% VAF which is not adequate for c-KIT p.D816V detection in the majority of SM and particularly in patients with ISM.
6. Genomic DNA (gDNA) and mRNA: Which Is Better?
mRNA based measurement of Expressed Allelic Burden (EAB) is not interchangeable with DNA-based results. Recently Naumann and colleagues [62], by analyzing a large number of samples, demonstrated that in patients with indolent systemic mastocytosis there is a high level of concordance between c-KIT p.D816V VAF and EAB but in patients with AdvSM this correlation is weak. This paper also suggests another clinically relevant point that is represented by the significance of the transcriptional activity of c-KIT p.D816V. Since the authors demonstrated that EAB/VAF ratio of more than 2 is predictive for an advanced phenotype and a significantly inferior overall survival (OS), they conclude that the transcriptional activity of mutated c-KIT probably plays an important role in the pathophysiology of systemic mastocytosis.
In 2016 Kristensen and colleagues [63] compared gDNA and mRNA in BM and PB. They found that depending on the expression level of c-KIT p.D816V, mRNA is potentially more sensitive than gDNA. However, gDNA based assay is usually superior to mRNA in PB. The conclusion of this study is that mRNA-based analysis is the best tool to be used in BM and gDNA based assay is preferred over mRNA in PB. Needless to remark that the absence of a complete concordance between gDNA and mRNA-based assays should be considered when interpreting the mutant allele burden results.
7. What to Do when the Mutational Test Is Negative but the Clinical Suspicion Is Strong
In 5-10% of SM the mutational analysis for c-KIT p.D816V results negative [51].
The EU-US Cooperative Group has considered these data and provided three scenarios that align with this possibility. The first is that the mutational load is below the detection limit, making it a false negative. The second is that it is truly negative. The third is that the patient may have a c-KIT mutation different from D816V that cannot be detected with the chosen method [51].
If the result was obtained with a method characterized by low sensitivity, the recommendation in these cases is to use a highly sensitive method to address scenario 1.
If the negative result was obtained on PB sample, it must be considered that a negative result on PB does not necessarily indicate that the patient is negative. The analysis should be repeated on a high-quality BM sample.
Furthermore, it is described that if an analysis on PB of gDNA results borderline, typically results positive when reanalyzed using increased amount of DNA. The quantity of DNA analyzed can therefore represent a limit.
If the result is negative in BM and PB despite using a sensitive technique and the mast cell infiltrate is evident, then it is appropriate to look for other c-KIT mutations different from D816V. Whole gene sequencing is recommended. In these cases, the use of standard NGS employed in many centers is useful, while still considering the limited sensitivity of this method.
8. What Material Should Be Used for the Mutational Analysis of c-KIT? Differences between BM or PB
For the diagnosis of systemic mastocytosis different types of biological material can be used, commonly BM or PB but also tissue biopsies including skin or gastrointestinal biopsies.
In cases of a reasonable suspicion of SM, the recommended material for diagnosis is BM aspirate collected in an EDTA tube, preferably fresh, although frozen material can also be used [57].
Although mature mast cells are generally not detected in PB, the use of PB for the analysis of c-KIT mutations is possible because an increased number of mast cell precursors are detectable in PB from SM patients. These circulating mast cell precursors usually carry c-KIT mutations but the expression level of CD117 is low therefore low levels of mRNA are usually detectable in these cells [64].
PB can therefore be used for the screening but always keeping in mind that when using PB at diagnosis a negative result on PB does not necessarily mean the absence of a mutation. In case of a negative result, it is essential to proceed with the examination of BM. A high VAF in PB generally is suggestive of a multilineage involvement. Both tests using gDNA and those using mRNA on BM produce reliable results, with the particularity that if there are low levels of mutation, using mRNA produces more reliable results.
9. How Frequent c-KIT p.D816V Allelic Burden Should Be Monitored?
The EU-US Cooperative Group believes that the frequency of evaluation depends on the type of disease, the stage and the therapy. For example, for patients with ISM the evaluation must be made at diagnosis and then is no longer considered necessary except in cases where progression is suspected. In AdvSM, especially in those under specific therapies, the VAF measurement should be repeated to monitor the treatment. VAF measurement must be repeated in all patients with SM who present signs of progression [51].
10. Significance of c-KIT p.D816V Allelic Burden
It is now clear that that c-KIT p.D816V mutant allele burden is a surrogate for the extent of multilineage involvement. In many patients with ISM the mutation can be detected in many mature cells including basophils, eosinophils, neutrophils, B and T lymphocytes [52,53,54,55].
Many data confirm that multilineage involvement in ISM is one of the most important prognostic factors that impacts on the probability of disease progression. Kristensen and colleagues reported a significant correlation between c-KIT p.D816V mutant allele burden, serum tryptase levels and the percentage of neoplastic mast cells [63]. These findings were confirmed by Erben and colleagues who, not only confirmed these data, but demonstrated the correlation with the allelic burden and the type of disease and therefore with the overall survival [58]. Recently, Greiner and colleagues by making use of ddPCR established that c-KIT p.D816V mutant allele burden correlates with MC infiltration, especially in tissues. They also found that the allele burden is higher in AdvSM compared to ISM and is an independent predictor of survival [64].
11. c-KIT p.D816V as a Marker of Response to Therapy
c-KIT p.D816V mutant allele burden has been used as a biomarker to evaluate the response to treatment. The possibility of using VAF as a biomarker of response was described many years ago [65] following treatments with agents such as hydroxyurea, cladribine, and interferon alpha. In the same year, Erben suggested the use of VAF to monitor the disease after allogeneic transplant [58]. With the development of molecular therapies targeting KIT, the ability to monitor the response becomes critical. In this regard, emerging data confirm the utility of c-KIT p.D816V VAF as a biomarker. Specifically, the study by Jawhar and colleagues demonstrated that in patients treated with midostaurin, a 25% reduction in the expressed allelic burden after 6 months of treatment is an independent prognostic factor for survival [66].
12. Additional Molecular and Cytogenetic Lesions
Patients affected by mastocytosis are often characterized by additional mutations beyond c-KIT [67,68,69,70,71]. These mutations are frequently associated with a worse prognosis. The additional mutations are mainly of two types. First, they can be mutations affecting driver genes associated with specific diseases, such as JAK2 or BCR::ABL and, second, they can be mutations of genes that do not define a specific hematologic neoplasm, generally myeloid lineage associated mutations. Among these, for example, we find CBL, KRAS, RUNX1, ASXL1, DNMT3, EZH2, TET2, SRSF2, SF3B1, U2AF1 and SETD2 [70]. These mutations are frequently found in myeloid neoplasms, although they are not associated with a particular disease phenotype. The mutations of the first type, those associated with defined pathologies, are typical of mastocytosis associated with another hematologic neoplasm. For example, mastocytosis with JAK2 mutation defines mastocytosis associated with a chronic myeloproliferative disease. The mutations not associated with a specific subtype of hematologic neoplasm are often found in the advanced forms, mainly in forms associated with other hematologic diseases, and less frequently in mast cell leukemias.
Many studies have sought to give prognostic significance to these additional mutations. In particular, what is defined as the S/A/R panel, i.e., the presence and number of SRSF2/ASXL1/RUNX1 mutations, represent a negative prognostic factor [71].
However, additional mutations do not always represent an unfavorable factor. Some studies published in the literature have clearly indicated that the presence of both c-KIT and JAK2 mutations often characterizes a disease with two independent clones. In these patients, in the absence of S/A/R mutations, the 5-year overall survival rate is 77%, which is significantly different from that of patients with S/A/R mutations. In these patients, the allele burden of both mutations, c-KIT and the additional one, also has clinical and prognostic significance. Indeed, a disease with minimal mast cell infiltration, a VAF of c-KIT p.D816V lower than 5%, and relatively low tryptase levels has a significantly different clinical behavior compared to a disease with the same c-KIT allele burden but many monocytes and a 50% of VAF in one of S/A/R genes [71].
Finally, some cytogenetic abnormalities detectable from karyotype examination have shown a prognostic role. In particular, abnormalities such as monosomy 7 and complex karyotype are associated with progression to very aggressive forms like acute mast cell leukemia. For this reason, cytogenetic investigation is strongly recommended in all advanced forms to evaluate clonal evolution and leukemic transformation [72,73].
13. Summary and Conclusions
Although there is still no standardization of methods to detect and measure c-KIT mutations, it is currently recommended to use highly sensitive techniques such as ASO-PCR and digital PCR.
Both BM and PB can be used for analysis, although the significance of allele burden differs between the two materials.
A negative result in PB does not exclude the presence of a mutation in BM; if PB is negative, it is recommended to perform the analysis on BM as well.
The test can be conducted on either gDNA or RNA, with the precautions mentioned above.
c-KIT mutation can be used as a biomarker for therapy response.
NGS analysis with a myeloid panel to detect additional mutations is useful for establishing prognosis, as mutations in the SAR genes (SRSF2/ASXL1/RUNX1) are associated with a poorer prognosis.
Author Contributions
Conceptualization, DC; methodology DC, BM, ACD; validation, DC, AS, VB, CF; resources DC; writing—original draft preparation DC; writing—review and editing, DC, BM, AS, ACD, VB, CF; All authors have read and agreed to the published version of the manuscript.”
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
The authors thank Dr Silvia Marini for technical support.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Heybeli, C. Mast Cells, Mastocytosis, and Related Disorders. N Engl J Med 2015, 373, 1885. [CrossRef]
- Tzankov, A.; Duncavage, E.; Craig, F.E.; Kelemen, K.; King, R.L.; Orazi, A.; Quintanilla-Martinez, L.; Reichard, K.K.; Rimsza, L.M.; Wang, S.A.; et al. Mastocytosis. American Journal of Clinical Pathology 2021, 155, 239–266. [CrossRef]
- Valent, P.; Akin, C.; Hartmann, K.; Nilsson, G.; Reiter, A.; Hermine, O.; Sotlar, K.; Sperr, W.R.; Escribano, L.; George, T.I.; et al. Mast Cells as a Unique Hematopoietic Lineage and Cell System: From Paul Ehrlich’s Visions to Precision Medicine Concepts. Theranostics 2020, 10, 10743–10768. [CrossRef]
- Kanakura, Y.; Furitsu, T.; Tsujimura, T.; Butterfield, J.H.; Ashman, L.K.; Ikeda, H.; Kitayama, H.; Kanayama, Y.; Matsuzawa, Y.; Kitamura, Y. Activating Mutations of the C-Kit Proto-Oncogene in a Human Mast Cell Leukemia Cell Line. Leukemia 1994, 8 Suppl 1, S18-22.
- Metcalfe, D.D.; Mekori, Y.A. Pathogenesis and Pathology of Mastocytosis. Annu Rev Pathol 2017, 12, 487–514. [CrossRef]
- De Matteis, G.; Zanotti, R.; Colarossi, S.; De Benedittis, C.; Garcia-Montero, A.; Bonifacio, M.; Sartori, M.; Aprili, F.; Caruso, B.; Paviati, E.; et al. The Impact of Sensitive KIT D816V Detection on Recognition of Indolent Systemic Mastocytosis. Leukemia Research 2015, 39, 273–278. [CrossRef]
- Valent, P.; Akin, C.; Hartmann, K.; Alvarez-Twose, I.; Brockow, K.; Hermine, O.; Niedoszytko, M.; Schwaab, J.; Lyons, J.J.; Carter, M.C.; et al. Updated Diagnostic Criteria and Classification of Mast Cell Disorders: A Consensus Proposal. HemaSphere 2021, 5, e646. [CrossRef]
- Khoury, J.D.; Solary, E.; Abla, O.; Akkari, Y.; Alaggio, R.; Apperley, J.F.; Bejar, R.; Berti, E.; Busque, L.; Chan, J.K.C.; et al. The 5th Edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms. Leukemia 2022, 36, 1703–1719. [CrossRef]
- Arber, D.A.; Orazi, A.; Hasserjian, R.P.; Borowitz, M.J.; Calvo, K.R.; Kvasnicka, H.-M.; Wang, S.A.; Bagg, A.; Barbui, T.; Branford, S.; et al. International Consensus Classification of Myeloid Neoplasms and Acute Leukemias: Integrating Morphologic, Clinical, and Genomic Data. Blood 2022, 140, 1200–1228. [CrossRef]
- Arock, M.; Hoermann, G.; Sotlar, K.; Hermine, O.; Sperr, W.R.; Hartmann, K.; Brockow, K.; Akin, C.; Triggiani, M.; Broesby-Olsen, S.; et al. Clinical Impact and Proposed Application of Molecular Markers, Genetic Variants, and Cytogenetic Analysis in Mast Cell Neoplasms: Status 2022. Journal of Allergy and Clinical Immunology 2022, 149, 1855–1865. [CrossRef]
- Valent, P.; Hoermann, G.; Bonadonna, P.; Hartmann, K.; Sperr, W.R.; Broesby-Olsen, S.; Brockow, K.; Niedoszytko, M.; Hermine, O.; Chantran, Y.; et al. The Normal Range of Baseline Tryptase Should Be 1 to 15 Ng/mL and Covers Healthy Individuals With HαT. The Journal of Allergy and Clinical Immunology: In Practice 2023, 11, 3010–3020. [CrossRef]
- Alvarez-Twose, I.; González-de-Olano, D.; Sánchez-Muñoz, L.; Matito, A.; Jara-Acevedo, M.; Teodosio, C.; García-Montero, A.; Morgado, J.M.; Orfao, A.; Escribano, L. Validation of the REMA Score for Predicting Mast Cell Clonality and Systemic Mastocytosis in Patients with Systemic Mast Cell Activation Symptoms. Int Arch Allergy Immunol 2012, 157, 275–280. [CrossRef]
- Golden, D.B.K.; Wang, J.; Waserman, S.; Akin, C.; Campbell, R.L.; Ellis, A.K.; Greenhawt, M.; Lang, D.M.; Ledford, D.K.; Lieberman, J.; et al. Anaphylaxis: A 2023 Practice Parameter Update. Annals of Allergy, Asthma & Immunology 2024, 132, 124–176. [CrossRef]
- Gulen, T. Management of Mediator Symptoms, Allergy, and Anaphylaxis in Mastocytosis. Immunology and Allergy Clinics of North America 2023, 43, 681–698. [CrossRef]
- Giannetti, M.P.; Nicoloro-SantaBarbara, J.; Godwin, G.; Middlesworth, J.; Espeland, A.; Douvas, J.L.; Castells, M.C. Challenges in Drug and Hymenoptera Venom Hypersensitivity Diagnosis and Management in Mastocytosis. Diagnostics 2024, 14, 123. [CrossRef]
- Valent, P. Risk Factors and Management of Severe Life-threatening Anaphylaxis in Patients with Clonal Mast Cell Disorders. Clin Experimental Allergy 2014, 44, 914–920. [CrossRef]
- Roskoski, R. Structure and Regulation of Kit Protein-Tyrosine Kinase—The Stem Cell Factor Receptor. Biochemical and Biophysical Research Communications 2005, 338, 1307–1315. [CrossRef]
- Blechman, J.M.; Lev, S.; Givol, D.; Yarden, Y. Structure-Function Analyses of the Kit Receptor for the Steel Factor. STEM CELLS 1996, 11, 12–21. [CrossRef]
- Giebel, L.B.; Strunk, K.M.; Holmes, S.A.; Spritz, R.A. Organization and Nucleotide Sequence of the Human KIT (Mast/Stem Cell Growth Factor Receptor) Proto-Oncogene. Oncogene 1992, 7, 2207–2217.
- Chan, E.C.; Bai, Y.; Bandara, G.; Simakova, O.; Brittain, E.; Scott, L.; Dyer, K.D.; Klion, A.D.; Maric, I.; Gilfillan, A.M.; et al. KIT GNNK Splice Variants: Expression in Systemic Mastocytosis and Influence on the Activating Potential of the D816V Mutation in Mast Cells. Experimental Hematology 2013, 41, 870-881.e2. [CrossRef]
- Lennartsson, J.; Jelacic, T.; Linnekin, D.; Shivakrupa, R. Normal and Oncogenic Forms of the Receptor Tyrosine Kinase Kit. STEM CELLS 2005, 23, 16–43. [CrossRef]
- Crosier, P.S.; Ricciardi, S.T.; Hall, L.R.; Vitas, M.R.; Clark, S.C.; Crosier, K.E. Expression of Isoforms of the Human Receptor Tyrosine Kinase C-Kit in Leukemic Cell Lines and Acute Myeloid Leukemia. Blood 1993, 82, 1151–1158.
- Lennartsson, J.; Rönnstrand, L. Stem Cell Factor Receptor/c-Kit: From Basic Science to Clinical Implications. Physiological Reviews 2012, 92, 1619–1649. [CrossRef]
- Reber, L.; Da Silva, C.A.; Frossard, N. Stem Cell Factor and Its Receptor C-Kit as Targets for Inflammatory Diseases. European Journal of Pharmacology 2006, 533, 327–340. [CrossRef]
- Ray, P.; Krishnamoorthy, N.; Oriss, T.B.; Ray, A. Signaling of C-kit in Dendritic Cells Influences Adaptive Immunity. Annals of the New York Academy of Sciences 2010, 1183, 104–122. [CrossRef]
- Waskow, C.; Terszowski, G.; Costa, C.; Gassmann, M.; Rodewald, H.-R. Rescue of Lethal C-KitW/W Mice by Erythropoietin. Blood 2004, 104, 1688–1695. [CrossRef]
- Leong, K.G.; Wang, B.-E.; Johnson, L.; Gao, W.-Q. Generation of a Prostate from a Single Adult Stem Cell. Nature 2008, 456, 804–808. [CrossRef]
- Laurson, J.; Selden, C.; Clements, M.; Mavri-Damelin, D.; Coward, S.; Lowdell, M.; Hodgson, H.J.F. Putative Human Liver Progenitor Cells in Explanted Liver. Cells Tissues Organs 2007, 186, 180–191. [CrossRef]
- Zaruba, M.-M.; Soonpaa, M.; Reuter, S.; Field, L.J. Cardiomyogenic Potential of C-Kit + –Expressing Cells Derived From Neonatal and Adult Mouse Hearts. Circulation 2010, 121, 1992–2000. [CrossRef]
- Liang, J.; Wu, Y.-L.; Chen, B.-J.; Zhang, W.; Tanaka, Y.; Sugiyama, H. The C-Kit Receptor-Mediated Signal Transduction and Tumor-Related Diseases. Int. J. Biol. Sci. 2013, 9, 435–443. [CrossRef]
- Thömmes, K.; Lennartsson, J.; Carlberg, M.; Rönnstrand, L. Identification of Tyr-703 and Tyr-936 as the Primary Association Sites for Grb2 and Grb7 in the c-Kit/Stem Cell Factor Receptor. Biochem J 1999, 341 ( Pt 1), 211–216.
- Chian, R.; Young, S.; Danilkovitch-Miagkova, A.; Rönnstrand, L.; Leonard, E.; Ferrao, P.; Ashman, L.; Linnekin, D. Phosphatidylinositol 3 Kinase Contributes to the Transformation of Hematopoietic Cells by the D816V C-Kit Mutant. Blood 2001, 98, 1365–1373. [CrossRef]
- Hashimoto, K.; Matsumura, I.; Tsujimura, T.; Kim, D.-K.; Ogihara, H.; Ikeda, H.; Ueda, S.; Mizuki, M.; Sugahara, H.; Shibayama, H.; et al. Necessity of Tyrosine 719 and Phosphatidylinositol 3′-Kinase–Mediated Signal Pathway in Constitutive Activation and Oncogenic Potential of c-Kit Receptor Tyrosine Kinase with the Asp814Val Mutation. Blood 2003, 101, 1094–1102. [CrossRef]
- Sun, J.; Pedersen, M.; Rönnstrand, L. Gab2 Is Involved in Differential Phosphoinositide 3-Kinase Signaling by Two Splice Forms of c-Kit. Journal of Biological Chemistry 2008, 283, 27444–27451. [CrossRef]
- Masson, K.; Heiss, E.; Band, H.; Rönnstrand, L. Direct Binding of Cbl to Tyr568 and Tyr936 of the Stem Cell Factor Receptor/c-Kit Is Required for Ligand-Induced Ubiquitination, Internalization and Degradation. Biochemical Journal 2006, 399, 59–67. [CrossRef]
- Hirota, S.; Isozaki, K.; Moriyama, Y.; Hashimoto, K.; Nishida, T.; Ishiguro, S.; Kawano, K.; Hanada, M.; Kurata, A.; Takeda, M.; et al. Gain-of-Function Mutations of c- Kit in Human Gastrointestinal Stromal Tumors. Science 1998, 279, 577–580. [CrossRef]
- Duensing, A.; Heinrich, M.C.; Fletcher, C.D.M.; Fletcher, J.A. Biology of Gastrointestinal Stromal Tumors: KIT Mutations and Beyond. Cancer Investigation 2004, 22, 106–116. [CrossRef]
- Curtin, J.A.; Busam, K.; Pinkel, D.; Bastian, B.C. Somatic Activation of KIT in Distinct Subtypes of Melanoma. JCO 2006, 24, 4340–4346. [CrossRef]
- Sakuma, Y.; Sakurai, S.; Oguni, S.; Hironaka, M.; Salto, K. Alterations of the C-kit Gene in Testicular Germ Cell Tumors. Cancer Science 2003, 94, 486–491. [CrossRef]
- Longley, B.J.; Metcalfe, D.D.; Tharp, M.; Wang, X.; Tyrrell, L.; Lu, S.; Heitjan, D.; Ma, Y. Activating and Dominant Inactivating C- KIT Catalytic Domain Mutations in Distinct Clinical Forms of Human Mastocytosis. Proc. Natl. Acad. Sci. U.S.A. 1999, 96, 1609–1614. [CrossRef]
- Beghini, A.; Ripamonti, C.B.; Cairoli, R.; Cazzaniga, G.; Colapietro, P.; Elice, F.; Nadali, G.; Grillo, G.; Haas, O.A.; Biondi, A.; et al. KIT Activating Mutations: Incidence in Adult and Pediatric Acute Myeloid Leukemia, and Identification of an Internal Tandem Duplication. Haematologica 2004, 89, 920–925.
- Chantran, Y.; Valent, P.; Arock, M. KIT Mutations and Other Genetic Defects in Mastocytosis. Immunology and Allergy Clinics of North America 2023, 43, 651–664. [CrossRef]
- Navarro-Navarro, P.; Álvarez-Twose, I.; Pérez-Pons, A.; Henriques, A.; Mayado, A.; García-Montero, A.C.; Sánchez-Muñoz, L.; González-López, O.; Matito, A.; Caldas, C.; et al. KIT D816V Mutation in Blood for the Diagnostic Screening of Systemic Mastocytosis and Mast Cell Activation Syndromes. Allergy 2023, 78, 1347–1359. [CrossRef]
- Nedoszytko, B.; Arock, M.; Lyons, J.; Bachelot, G.; Schwartz, L.; Reiter, A.; Jawhar, M.; Schwaab, J.; Lange, M.; Greiner, G.; et al. Clinical Impact of Inherited and Acquired Genetic Variants in Mastocytosis. IJMS 2021, 22, 411. [CrossRef]
- Bibi, S.; Langenfeld, F.; Jeanningros, S.; Brenet, F.; Soucie, E.; Hermine, O.; Damaj, G.; Dubreuil, P.; Arock, M. Molecular Defects in Mastocytosis. Immunology and Allergy Clinics of North America 2014, 34, 239–262. [CrossRef]
- Bodemer, C.; Hermine, O.; Palmérini, F.; Yang, Y.; Grandpeix-Guyodo, C.; Leventhal, P.S.; Hadj-Rabia, S.; Nasca, L.; Georgin-Lavialle, S.; Cohen-Akenine, A.; et al. Pediatric Mastocytosis Is a Clonal Disease Associated with D816V and Other Activating C-KIT Mutations. Journal of Investigative Dermatology 2010, 130, 804–815. [CrossRef]
- Nakagomi, N.; Hirota, S. Juxtamembrane-Type c-Kit Gene Mutation Found in Aggressive Systemic Mastocytosis Induces Imatinib-Resistant Constitutive KIT Activation. Laboratory Investigation 2007, 87, 365–371. [CrossRef]
- Georgin-Lavialle, S.; Aguilar, C.; Guieze, R.; Lhermitte, L.; Bruneau, J.; Fraitag, S.; Canioni, D.; Chandesris, M.-O.; Suarez, F.; Grandpeix-Guyodo, C.; et al. Mast Cell Sarcoma: A Rare and Aggressive Entity—Report of Two Cases and Review of the Literature. JCO 2013, 31, e90–e97. [CrossRef]
- Valent, P.; Berger, J.; Cerny-Reiterer, S.; Peter, B.; Eisenwort, G.; Hoermann, G.; Müllauer, L.; Mannhalter, C.; Steurer, M.; Bettelheim, P.; et al. Chronic Mast Cell Leukemia (MCL) with KIT S476I: A Rare Entity Defined by Leukemic Expansion of Mature Mast Cells and Absence of Organ Damage. Ann Hematol 2015, 94, 223–231. [CrossRef]
- Meni, C.; Georgin-Lavialle, S.; Le Saché De Peufeilhoux, L.; Jais, J.P.; Hadj-Rabia, S.; Bruneau, J.; Fraitag, S.; Hanssens, K.; Dubreuil, P.; Hermine, O.; et al. Paediatric Mastocytosis: Long-Term Follow-up of 53 Patients with Whole Sequencing of KIT . A Prospective Study. Br J Dermatol 2018, 179, 925–932. [CrossRef]
- Hoermann, G.; Sotlar, K.; Jawhar, M.; Kristensen, T.; Bachelot, G.; Nedoszytko, B.; Carter, M.C.; Horny, H.-P.; Bonadonna, P.; Sperr, W.R.; et al. Standards of Genetic Testing in the Diagnosis and Prognostication of Systemic Mastocytosis in 2022: Recommendations of the EU-US Cooperative Group. The Journal of Allergy and Clinical Immunology: In Practice 2022, 10, 1953–1963. [CrossRef]
- Akin, C. Multilineage Hematopoietic Involvement in Systemic Mastocytosis. Leukemia Research 2003, 27, 877–878. [CrossRef]
- Garcia-Montero, A.C. KIT Mutation in Mast Cells and Other Bone Marrow Hematopoietic Cell Lineages in Systemic Mast Cell Disorders: A Prospective Study of the Spanish Network on Mastocytosis (REMA) in a Series of 113 Patients. Blood 2006, 108, 2366–2372. [CrossRef]
- Escribano, L.; Álvarez-Twose, I.; Sánchez-Muñoz, L.; Garcia-Montero, A.; Núñez, R.; Almeida, J.; Jara-Acevedo, M.; Teodósio, C.; García-Cosío, M.; Bellas, C.; et al. Prognosis in Adult Indolent Systemic Mastocytosis: A Long-Term Study of the Spanish Network on Mastocytosis in a Series of 145 Patients. Journal of Allergy and Clinical Immunology 2009, 124, 514–521. [CrossRef]
- Sotlar, K.; Colak, S.; Bache, A.; Berezowska, S.; Krokowski, M.; Bültmann, B.; Valent, P.; Horny, H. Variable Presence of KIT D816V in Clonal Haematological Non-mast Cell Lineage Diseases Associated with Systemic Mastocytosis (SM–AHNMD). The Journal of Pathology 2010, 220, 586–595. [CrossRef]
- Jawhar, M.; Schwaab, J.; Schnittger, S.; Sotlar, K.; Horny, H.-P.; Metzgeroth, G.; Müller, N.; Schneider, S.; Naumann, N.; Walz, C.; et al. Molecular Profiling of Myeloid Progenitor Cells in Multi-Mutated Advanced Systemic Mastocytosis Identifies KIT D816V as a Distinct and Late Event. Leukemia 2015, 29, 1115–1122. [CrossRef]
- Kristensen, T.; Vestergaard, H.; Bindslev-Jensen, C.; Møller, M.B.; Broesby-Olsen, S.; on Behalf of the Mastocytosis Centre Odense University Hospital (MastOUH) Sensitive KIT D816V Mutation Analysis of Blood as a Diagnostic Test in Mastocytosis. American J Hematol 2014, 89, 493–498. [CrossRef]
- Erben, P.; Schwaab, J.; Metzgeroth, G.; Horny, H.-P.; Jawhar, M.; Sotlar, K.; Fabarius, A.; Teichmann, M.; Schneider, S.; Ernst, T.; et al. The KIT D816V Expressed Allele Burden for Diagnosis and Disease Monitoring of Systemic Mastocytosis. Ann Hematol 2014, 93, 81–88. [CrossRef]
- Greiner, G.; Gurbisz, M.; Ratzinger, F.; Witzeneder, N.; Simonitsch-Klupp, I.; Mitterbauer-Hohendanner, G.; Mayerhofer, M.; Müllauer, L.; Sperr, W.R.; Valent, P.; et al. Digital PCR: A Sensitive and Precise Method for KIT D816V Quantification in Mastocytosis. Clinical Chemistry 2018, 64, 547–555. [CrossRef]
- Hindson, B.J.; Ness, K.D.; Masquelier, D.A.; Belgrader, P.; Heredia, N.J.; Makarewicz, A.J.; Bright, I.J.; Lucero, M.Y.; Hiddessen, A.L.; Legler, T.C.; et al. High-Throughput Droplet Digital PCR System for Absolute Quantitation of DNA Copy Number. Anal. Chem. 2011, 83, 8604–8610. [CrossRef]
- Huggett, J.F.; Cowen, S.; Foy, C.A. Considerations for Digital PCR as an Accurate Molecular Diagnostic Tool. Clinical Chemistry 2015, 61, 79–88. [CrossRef]
- Naumann, N.; Lübke, J.; Baumann, S.; Schwaab, J.; Hoffmann, O.; Kreil, S.; Dangelo, V.; Reiter, L.; Bugert, P.; Kristensen, T.; et al. Adverse Prognostic Impact of the KIT D816V Transcriptional Activity in Advanced Systemic Mastocytosis. IJMS 2021, 22, 2562. [CrossRef]
- Kristensen, T.; Broesby-Olsen, S.; Vestergaard, H.; Bindslev-Jensen, C.; Møller, M.B.; the Mastocytosis Centre Odense University Hospital (MastOUH) Comparison of gDNA -based versus mRNA -based KIT D816V Mutation Analysis Reveals Large Differences between Blood and Bone Marrow in Systemic Mastocytosis. Br J Haematol 2017, 178, 330–332. [CrossRef]
- Greiner, G.; Gurbisz, M.; Ratzinger, F.; Witzeneder, N.; Class, S.V.; Eisenwort, G.; Simonitsch-Klupp, I.; Esterbauer, H.; Mayerhofer, M.; Müllauer, L.; et al. Molecular Quantification of Tissue Disease Burden Is a New Biomarker and Independent Predictor of Survival in Mastocytosis. Haematologica 2020, 105, 366–374. [CrossRef]
- Hoermann, G.; Gleixner, K.V.; Dinu, G.E.; Kundi, M.; Greiner, G.; Wimazal, F.; Hadzijusufovic, E.; Mitterbauer, G.; Mannhalter, C.; Valent, P.; et al. The KIT D 816 V Allele Burden Predicts Survival in Patients with Mastocytosis and Correlates with the WHO Type of the Disease. Allergy 2014, 69, 810–813. [CrossRef]
- Jawhar, M.; Schwaab, J.; Naumann, N.; Horny, H.-P.; Sotlar, K.; Haferlach, T.; Metzgeroth, G.; Fabarius, A.; Valent, P.; Hofmann, W.-K.; et al. Response and Progression on Midostaurin in Advanced Systemic Mastocytosis: KIT D816V and Other Molecular Markers. Blood 2017, 130, 137–145. [CrossRef]
- Tefferi, A.; Levine, R.L.; Lim, K.-H.; Abdel-Wahab, O.; Lasho, T.L.; Patel, J.; Finke, C.M.; Mullally, A.; Li, C.-Y.; Pardanani, A.; et al. Frequent TET2 Mutations in Systemic Mastocytosis: Clinical, KITD816V and FIP1L1-PDGFRA Correlates. Leukemia 2009, 23, 900–904. [CrossRef]
- Traina, F.; Visconte, V.; Jankowska, A.M.; Makishima, H.; O’Keefe, C.L.; Elson, P.; Han, Y.; Hsieh, F.H.; Sekeres, M.A.; Mali, R.S.; et al. Single Nucleotide Polymorphism Array Lesions, TET2, DNMT3A, ASXL1 and CBL Mutations Are Present in Systemic Mastocytosis. PLoS ONE 2012, 7, e43090. [CrossRef]
- Soucie, E.; Hanssens, K.; Mercher, T.; Georgin-Lavialle, S.; Damaj, G.; Livideanu, C.; Chandesris, M.O.; Acin, Y.; Létard, S.; De Sepulveda, P.; et al. In Aggressive Forms of Mastocytosis, TET2 Loss Cooperates with c-KITD816V to Transform Mast Cells. Blood 2012, 120, 4846–4849. [CrossRef]
- Schwaab, J.; Schnittger, S.; Sotlar, K.; Walz, C.; Fabarius, A.; Pfirrmann, M.; Kohlmann, A.; Grossmann, V.; Meggendorfer, M.; Horny, H.-P.; et al. Comprehensive Mutational Profiling in Advanced Systemic Mastocytosis. Blood 2013, 122, 2460–2466. [CrossRef]
- Jawhar, M.; Schwaab, J.; Schnittger, S.; Meggendorfer, M.; Pfirrmann, M.; Sotlar, K.; Horny, H.-P.; Metzgeroth, G.; Kluger, S.; Naumann, N.; et al. Additional Mutations in SRSF2, ASXL1 and/or RUNX1 Identify a High-Risk Group of Patients with KIT D816V+ Advanced Systemic Mastocytosis. Leukemia 2016, 30, 136–143. [CrossRef]
- Naumann, N.; Jawhar, M.; Schwaab, J.; Kluger, S.; Lübke, J.; Metzgeroth, G.; Popp, H.D.; Khaled, N.; Horny, H.; Sotlar, K.; et al. Incidence and Prognostic Impact of Cytogenetic Aberrations in Patients with Systemic Mastocytosis. Genes Chromosomes & Cancer 2018, 57, 252–259. [CrossRef]
- Kluin-Nelemans, H.C.; Jawhar, M.; Reiter, A.; Van Anrooij, B.; Gotlib, J.; Hartmann, K.; Illerhaus, A.; Oude Elberink, H.N.G.; Gorska, A.; Niedoszytko, M.; et al. Cytogenetic and Molecular Aberrations and Worse Outcome for Male Patients in Systemic Mastocytosis. Theranostics 2021, 11, 292–303. [CrossRef]
Figure 1.
Illustration of KIT receptor activation by Stem Cell Factor (SCF), triggering downstream signaling pathways: JAK/STAT, PLCg MAPK/ERK, and PI3K/AKT, which collectively contribute to cell survival, proliferation, and migration.
Figure 1.
Illustration of KIT receptor activation by Stem Cell Factor (SCF), triggering downstream signaling pathways: JAK/STAT, PLCg MAPK/ERK, and PI3K/AKT, which collectively contribute to cell survival, proliferation, and migration.
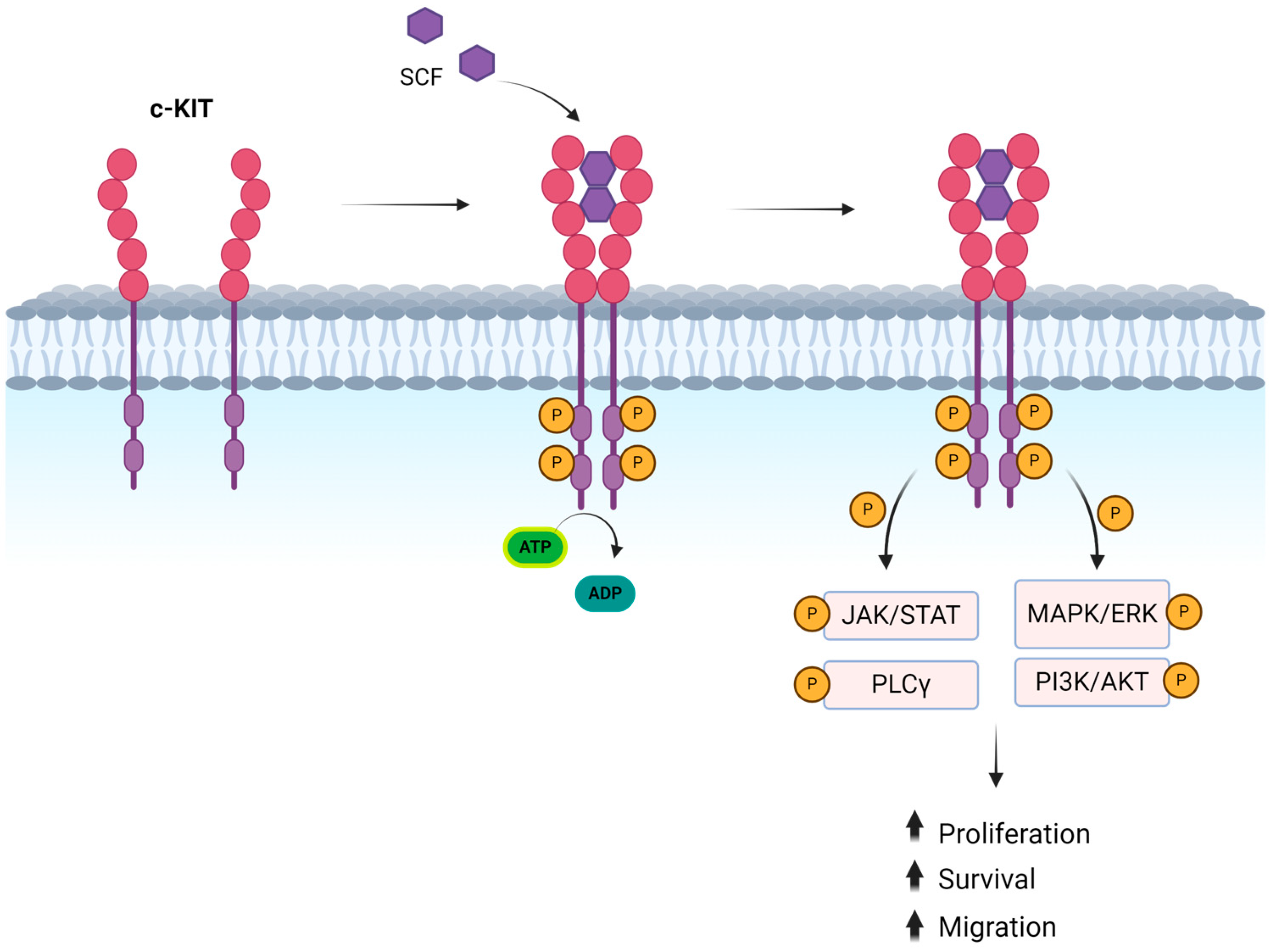
Figure 2.
Entire spectrum of c-KIT mutations. On the left, the protein structure is shown with its various domains. In the center, the corresponding gene structure is depicted, highlighting the different exons that encode each domain. On the right, the mutations identified in the respective exons are indicated. TMD: trans-membrane domain. JMD juxtamembrane domain. TK1: tyrosine kinase domain 1. TK2: tyrosine kinase domain 2.
Figure 2.
Entire spectrum of c-KIT mutations. On the left, the protein structure is shown with its various domains. In the center, the corresponding gene structure is depicted, highlighting the different exons that encode each domain. On the right, the mutations identified in the respective exons are indicated. TMD: trans-membrane domain. JMD juxtamembrane domain. TK1: tyrosine kinase domain 1. TK2: tyrosine kinase domain 2.
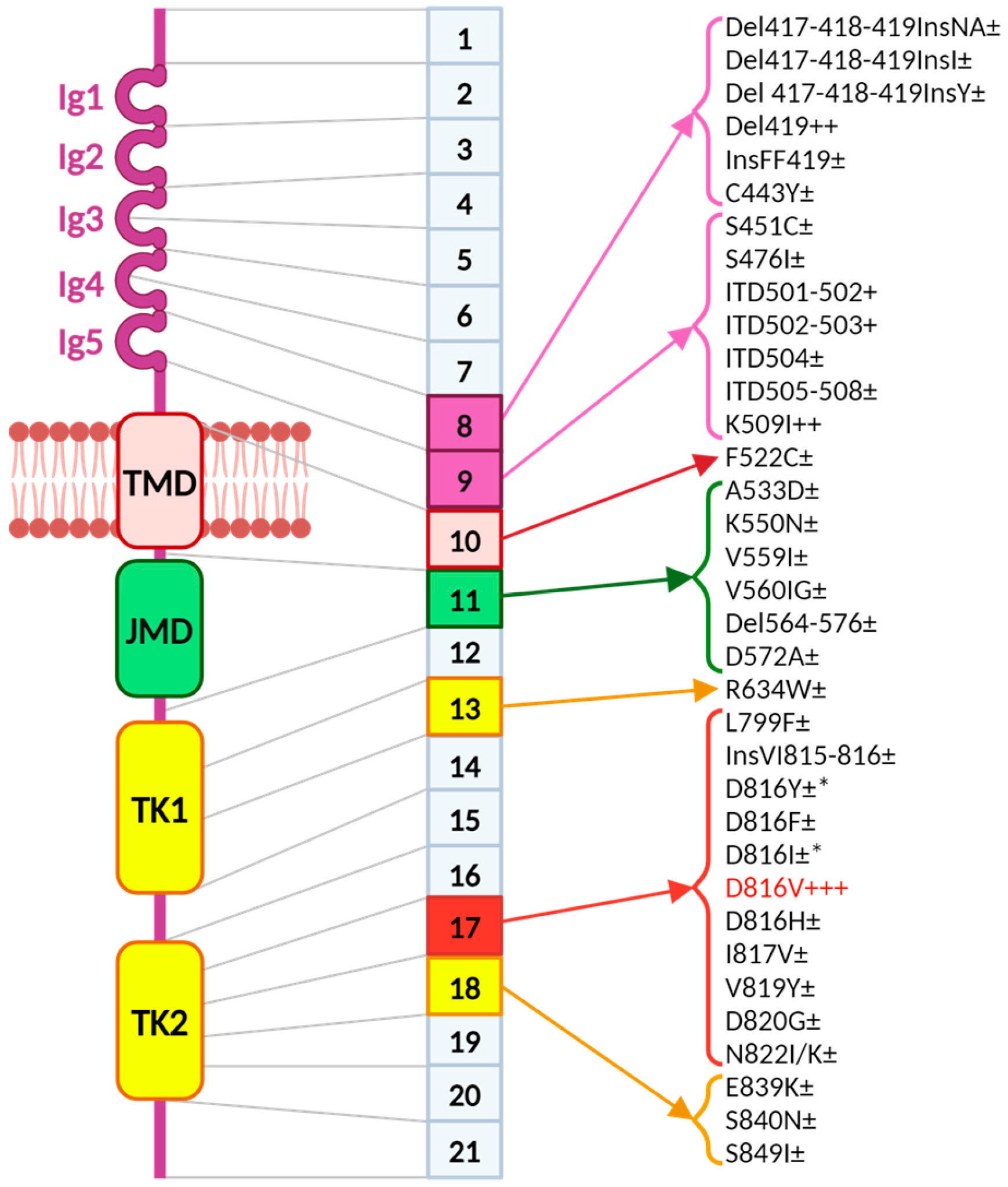

Table 1.
WHO classification of systemic mastocytosis (2022).
| Bone Marrow Mastocytosis (BBM) | |
| Indolent Systemic Mastocytosis (ISM): | Bone marrow infiltration <20% without associated hematologic disease, absence of C findings, and absence or presence of 1 B finding |
| Smoldering Systemic Mastocytosis (SSM): | Bone marrow infiltration <20% without associated hematologic disease, absence of C findings, and presence of 2 or more B findings |
| Aggressive Systemic Mastocytosis (ASM): | Bone marrow infiltration <20% without associated hematologic disease, presence of at least 1 C finding |
| Systemic Mastocytosis with Associated Hematologic Neoplasm (SM-AHN): | Mast cell percentage <20% and bone marrow histology conclusive for hematologic disease (myelodysplastic syndrome or myeloproliferative syndrome). |
| Mast Cell Leukemia (MCL): | Patients with bone marrow infiltration >20% without signs of other hematologic diseases. Typically presents with weight loss, fatigue, anemia, and other C findings, such as hypotension, flushing, diarrhea, and coagulopathy. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



