
Submitted:
29 August 2024
Posted:
02 September 2024
You are already at the latest version
Abstract
The landscape of clinical management for metastatic melanoma (MM) and other solid tumors has been modernized by the advent of immune checkpoint inhibitors (ICI), including programmed cell death-1 (PD-1), programmed cell death-ligand 1 (PD-L1), and cytotoxic T lymphocyte antigen 4 (CTLA-4) inhibitors. While these agents demonstrate efficacy in suppressing tumor growth, they also lead to immune-related adverse events (irAEs), resulting in exacerbation of autoimmune diseases such as Rheumatoid arthritis (RA), Ulcerative colitis (UC), and Crohn’s disease (CD). The immune checkpoint inhibitors offer promising advancements in the treatment of melanoma and other cancers, but they also present significant challenges related to irAEs and autoimmune diseases. Ongoing research is crucial to better understand these challenges and develop strategies for mitigating adverse effects while maximizing therapeutic benefits. In this manuscript, we addressed this challenge using network-based approaches by constructing and analyzing molecular and signaling networks associated with tumor-immune cross-talk.. Our analysis revealed that IL6 is the key regulator responsible for irAEs during ICI therapies. Furthermore, we conducted an integrative network and molecular-level analysis, including virtual screening, of drug libraries, such as the Collection of Open Natural Products (COCONUT) and the Zinc15 FDA-approved library, to identify potential IL-6 inhibitors.. Subsequently, the compound amprenavir was identified as the best molecule that may disrupt essential interactions between IL6 and IL6R responsible for initiating signaling cascades underlying irAEs in ICI therapies.
Keywords:
Melanoma metastasis
; Ulcerative Colitis
; Crohn's Disease
; Rheumatoid arthritis
; Integrated bioinformatics analysis
; Virtual screening
; Molecular docking
; Molecular dynamic simulation
1. Introduction
Metastatic melanoma is an advanced and aggressive form of skin cancer that arises from the uncontrolled growth of pigment-producing cells called melanocytes [1]. Immune checkpoint inhibitors (ICI) therapies have emerged as a revolutionary approach in the treatment of melanoma. ICI therapies focus on modulating the immune system to enhance its ability to recognize and attack cancer cells. Various ICI has been discovered to inhibit specific proteins (e.g., cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4) [2], programmed cell death protein 1 (PD-1) [3] in tumour immune environment and thereby eliminate of tumour cells [3,4,5,6]. While ICI therapies hold great promise in cancer treatment, they present a distinct challenge by potentially inducing autoimmune phenotypes [7]. The mechanisms that enable the immune system to target cancer cells are also supposed to inadvertently lead to immune-related adverse events (irAEs) responsible for the exacerbation of autoimmune diseases [8]. This dual nature of immune checkpoint therapies underscores the delicate balance that must be struck between activating the immune system to combat melanoma and preventing it from attacking the body's tissues resulting in irAEs [9,10,11,12,13,14]. Understanding the cross-talk between melanoma and autoimmune diseases is challenging due to the involvement of a large number of immune cells and is vital for the safe use of ICI therapies in the regulation of tumor growth. In this manuscript, we approached this challenge using network-based approaches by constructing and analysis molecular and signaling network associated with the tumor-immune cross-talk. Detailed work flow of research is given in Figure 1.
2. Results and Discussion
2.1. Protein-Protein Interaction Network at the Interface of Melanoma and Autoimmune Diseases
We extracted genes associated with MM (n=504), RA (n=2722), UC (n=1458), and CD (n=1382) from the DisGenNet database. We found a total of 132 common genes in all four disease phenotypes for which a protein-protein interaction (PPI) network was prepared using the STRING database (Figure 2). We considered these common genes as the connecting links between melanoma and investigated autoimmune diseases.
Out of 132 genes used for the construction of the PPI network, 25 genes did not exhibit any connections with other genes, and hence a network among the common 107 genes (Figure 3).
2.2. Identification of Hub Genes from the PPI Network Associated with the Crosstalk between Melanoma and Autoimmune Diseases
To identify key hub genes in the PPI network prepared using the common genes in melanoma and autoimmune diseases, we used the Cytoscape plugin Molecular Complex Detection (MCODE). The MCODE algorithm detects interconnected network clusters based on a k-core score that represents the maximal number of connected subgraphs with all the nodes connected by a minimum number of degrees k [15]. The MCODE algorithm detected six highly connected subnetworks represented as MCODE modules [Table 1]. For the identification of hub genes responsible for the crosstalk between melanoma and autoimmune diseases, we selected module 1, containing 16 genes with the highest MCODE score of 10.13.
2.3. Pathway Enrichment Analysis of top MCODE Cluster
To identify biological processes and pathways that might play a key role in the crosstalk of melanoma and autoimmune diseases, we performed pathway enrichment analysis of genes associated with the top MCODE module using the Reactome database 2022 in Enrichr web-based server (https://maayanlab.cloud/Enrichr/). All the enriched pathways (Figure 4), along with the pathway p-values, adjusted p-values, and gene/protein sets for each case (Supplementary Table S1).
Interleukin-4 (IL-4) and Interleukin-13 (IL-13) signaling pathways, which play key roles immune regulation and inflammation and are primarily associated with allergic responses and Th2 immunity [16], were among the top enriched pathways. Previous studies also suggest that these pathways shape the tumor microenvironment and promote tumor progression by modulating immune responses and survival pathways [17]. In autoimmune diseases like RA and UC, IL-4 and IL-13 contribute to excessive inflammation and tissue damage, fostering autoantibody production and B cell survival[18]. Besides, we also found Interleukin-6 (IL-6), Interleukin-10 (IL-10), and Interleukin-1 (IL-1) signaling cascades among the top enriched pathways that regulate melanoma [19,20] and autoimmune diseases [21], exerting diverse effects on inflammation, immune dysregulation, and disease progression. Elevated IL-6 levels in melanoma correlate with advanced disease stages and therapy resistance, while in autoimmune diseases, IL-6 drives inflammation and tissue damage [21]. Conversely, IL-10 exhibits dual roles, suppressing anti-tumor immunity in melanoma yet mitigating inflammation in autoimmune diseases. In melanoma, IL-1 orchestrates tumor growth, angiogenesis, and metastasis [22] by instigating pro-inflammatory cytokine production, fostering melanoma cell invasiveness, and modulating the tumor microenvironment's immune cell composition that may contribute to autoimmune disease pathogenesis, fueling inflammation and tissue damage in conditions like RA [23] and IBD [24] by inciting cytokine production and immune cell activation. Additionally, we also found matrix metalloproteinases (MMPs), collagen degradation, MAPK signaling, and CD163-mediated anti-inflammatory responses among the top enriched pathways that are known for regulating melanoma, and autoimmune diseases [25,26,27,28,29,30,31]. In melanoma, collagen degradation promotes tumor invasion and metastasis by MMP-mediated breakdown of the extracellular matrix (ECM), facilitating melanoma cell infiltration into surrounding tissues and distant metastasis [49,50]. Elevated levels of collagen degradation products also instigate immune-mediated tissue damage and inflammation in autoimmune diseases like RA [34]. The CD163-mediated anti-inflammatory responses potentially affect the inflammation resolution phase resulting in the progression towards chronic inflammatory phenotypes together with the excurbation of autoimmune symptoms. Understanding the intricate interplay of these signaling pathwys is crucial for developing targeted therapies that effectively modulate immune responses together with management of tumor by immune checkpoint inhibitors.
2.4. Identification of Lead Molecule and Molecular Docking
IL6 (Figure 4) was found to regulate five pathways among eight enriched pathways associated with MM and autoimmune disease. This directed network suggests that inhibition of IL6 will reduce the activity of pathways such as IL6, IL4 / IL13 signaling, collagen degradation, CD163-mediating responses, MAPK signalling, and activate IL-10 signaling. Thus, inhibition of IL-6 will not only be able to suppress melanoma metastasis but reduce autoimmune phenotypes, which may exacerbate during immune checkpoint therapies [35,36,37].
To identify potential inhibitors for the IL6 protein, we utilized the FDA-approved library using the Zinc15 Database (https://zinc15.docking.org/), which contained 1615 compounds and the natural compound library of the COCONUT database (https://coconut.naturalproducts.net/), which contained 407,270 unique compounds [38]. We filtered the COCONUT database libraries prior to the virtual screening with IL6 for Lipinski's rule of five[39] in order to consider only drug-like molecules. Only 272,001 compounds were able to pass Lipinski's rule of five filtering criteria.
The active site of IL-6 was selected considering amino acid residues Phe74, Phe78, Leu178, Arg179, and Arg182, which play a significant role (hotspot residues) in its interaction with IL6R [40]. The ‘Libdock’ tool in DS2022, which is a rigid-based docking program that calculates hotspots for the protein using a grid placed into the binding site polar and apolar probes [41,42], was used for the screening process of drug libraries. The top 20 compounds based on the libdock score were further considered for flexible docking analysis using the ‘CDOCKER’ tool in the DS2022 [Table 2]. In Flexible CDOCKER docking, the compound is allowed to adapt its conformation to fit the binding site of the receptor better, and the receptor can also undergo conformational changes to accommodate the compound. This flexibility is particularly important in accurately predicting the binding affinity and mode of interaction between a compound candidate and its target protein. Only nine compounds out of 20 could be further docked with IL6 using CDOCKER protocol.
We selected the top two compounds (Figure 5) because these have the highest CDOCKER energy value (CNP0003038: -41.6684 kcal/mol) and (ZINC03809192: -34.7136 kcal/mol), and one belongs to the natural compound library and second from the ZINC database.
We performed the literature survey for these two compounds for their toxicity, bioassays, and role in tumor / autoimmune disease regulation. We found that the compound CNP0003038, also referred to as 1,1'-ethylenebis-L-tryptophan (EBT; PubChem CID: 3905118), enhances the proliferation of EoL-3 eosinophilic leukemia cells, induces the release of eosinophil cationic protein from isolated human peripheral blood eosinophils resulting in eosinophilia-myalgia syndrome [43]. EBT is also shown to induce IL-5 production in isolated human T cells and induces inflammation, mast cell infiltration, fascia thickening, and adipose tissue fibrosis in an eosinophilia-myalgia-syndrome mouse model [44]. Due to the possible toxic effects of compound CNP0003038 on the immune system, we removed it from our further analysis.
The compound from the ZINC database ‘ZINC000003809192’, also known as amprenavir, is primarily known as a protease inhibitor and used in the treatment of HIV/AIDS. It inhibits the HIV protease enzyme, thereby blocking the cleavage of viral polyproteins into functional proteins, ultimately hindering viral replication [45,46]. Amprenavir was also included in the investigation of FDA-approved small molecule drugs through in-silico screening, assessing their potential as inhibitors of extracellular signal-regulated kinase (ERK) and apoptosis inducers in MCF-7 human breast cancer cells [47]. Based on all the above facts, we selected only Amprenavir for the molecular dynamic simulation.
We employed molecular docking and molecular dynamics simulation analyses to investigate the interaction patterns, stability, and flexibility of the docked complex, which helped us explore the interaction and stability of Amprenavir with IL-6 during the simulation. Our molecular docking studies suggest that Amprenavir forms three hydrogen bonds and four hydrophobic bonds with the IL6 amino acid residues SER176, CYS73, MET67 and, ARG179, LYS54, LYS66 (more information on the bonds is provided in the supplementary Table S2). To further check if Amprenavir interferes with the binding of IL6 with IL6R, we performed additional protein-protein docking using the HDOCK tool [48]. For this, we first used IL6 and IL6R complex (PDB ID: 1P9M) [49] and redocked the protein units using HDOCK as a control scenario. Next, we used the IL6 in complex with ‘Amprenavir’ and performed the protein-protein docking with IL6R again using the HDOCK tool. Both these scenarios were compared with each other to evaluate the effect of ‘Amprenavir’ on IL6 and IL6R interactions. We observed that the docked complex of IL6_IL6R has a higher docking energy of -398.1 kcal/mol thus more stable in comparison to the IL6_Amprenavir_IL6R complex (-262.02 kcal/mol). We further compared the impact of ‘Amprenavir’ on the number of bonds formed between IL6 and IL6R.
The residues essential for IL6 binding to IL6R, includes IL6: LYS27, GLN28, ARG30, PHE74, PHE75, PHE78, LEU178, ARG179, ALA180 and ARG182 [49] which form various bonds with IL6RA: GLU163, GLN190, PHE229, ASP253, GLU277, GLU278, PHE279, and IL6RB: LYS118, LYS119, ARG128, VAL167, TYR168, PHE169, VAL230, VAL264 residues (more information on the bonds formed between IL6 and IL6R is provided in the supplementary Table S3).
Our analysis revealed that the IL6_ Amprenavir_ IL6R complex lost 6 significant bonds that were formed between the IL6_IL6R complex. However, one new bond formed between IL6 and IL6R in the presence of amprenavir (IL6:ARG179 with IL6RA: GLU163) (supplementary Table S3). We also observed IL6 amino acid residues LYS66, SER176, ARG179, and IL6R residues GLY164 and CYS192 forming bonds with amprenavir (Figure 6 a and b) (supplementary Table S3). The energies and bond assessments of the docked complex show that the compound amprenavir functions as an inhibitor for IL6 interaction with IL6R, disrupting numerous bonds originally formed between the protein and its receptor.
2.5. Molecular Dynamics Simulation
To evaluate the IL6_amprenavir docked complex flexibility and overall stability through time-dependent molecular dynamics (MD) simulation, we used the ‘Standard Dynamics Cascade’ protocol of DS 2022. The analysis of the simulation involved the use of RMSD (Root Mean Square Deviation), RMSF (Root Mean Square Fluctuation) and Radius of gyration (Rg) (Figure 7). We observed the RMSD of the docked complex (IL6_Amprenavir) for 50 ns; and found that the complex achieved convergence and stabilizes at around 20 ns (Figure 7 b).
We comprehensively analysed residue fluctuations in RMSF, which are crucial for IL6 binding with IL6R. Residues within the hydrophilic domain (Lys27, Arg30, Phe78, Arg179, and Arg182) form salt bridges with IL6R proteins. In the molecular docking of the IL6_Amprenavir_IL6R complex, we observed the disruptions of salt bridges involving residues Lys27 and Arg182 between IL6 and IL6R. The same happens in Phe 78, disengaged from its bond with IL6R (supplementary Tables S3). All these residues take part in the binding of the amprenavir into the cavity of IL6. During the MD simulation, all these residues showed minimal fluctuations of 0.4 (Å), which indicates that these IL6 residues are tightly engaged with the drug ‘amprenavir’ and are not available for interaction with IL6R. Furthermore, no residues showed a fluctuation of more than 0.7 Å (Figure 7c). In the case of the radius of gyration, during the MD run after the binding of the Amprenavir, IL6 started to achieve a more compact structure (Figure 7d). The comparison of bonds formed between IL6_amprenavir complex before and after the MD simulation is shown in Figure 7a and in the supplementary Tables S2 and S4. The analysis of the final pose of IL6_Amprenavir complex after MD simulation indicates an increase in bonds compared to the initial docked pose. Initially, it formed three Hydrogen bonds with various IL6 residues which increased to five after a 50 ns production run. Overall, our analysis indicates that the compound amprenavir binds with IL6 and forms a stable complex at the same site that is associated with IL6R interaction.
3. Methods and Methodology
3.1. Data Collection and Protein-Protein Interaction (PPI)
The genes related to MM, RA, UC & CD were extracted from the DisGenNet database (https://www.disgenet.org/), which is one of the largest publicly available collections of genes and variants associated with human diseases [50]. The data extracted from DisGeNet was subsequently analysed to identify common genes among the above-mentioned disease phenotypes. All the common genes were further used to explore their protein-protein interactions using the STRING database (http://string-db.org) [51] with a confidence score cutoff of 0.70.
3.2. Identification of Highly Interconnected Clusters in the Tumor-Autoimmune PPI Network
We used the Cytoscape plugin Molecular Complex Detection (MCODE) [53,54,55] to identify highly connected clusters in the protein-protein interaction network of the common genes associated with tumour and selected autoimmune disease phenotypes. The degree cutoff = 2 was used to control the genes to become part of the cluster. New members are added to the cluster only if their node score deviates from the cluster’s seed nodes score by less than the set cutoff of 0.2. The k-score cutoff = 2 was used to filter out clusters that do not contain a maximally inter-connected sub-cluster of at least a degree score of 2. The max. depth of 100 was set to limit the distance from the seed node within which MCODE can search for cluster members. This approach allowed us to partition the network based on its topology, pinpointing densely connected regions within the protein-protein interaction network associated with the crosstalk between melanoma and immune checkpoint therapies inducing irAEs.
3.3. Pathway Enrichment Analysis
For the top cluster identified using MCODE analysis, we identified enriched pathways using Enrichr (https://maayanlab.cloud/Enrichr/) [52]. We specifically focused on the pathways that are present in the Reactome 2022 database with the significance threshold of p-value (0.05) for the enrichment analysis. The pathways were sorted on the basics of the combined score [53], which is described as
where c is the combined score, p is the Fisher exact test p-value, and z is the z-score for deviation from expected rank. Further, we filtered enriched pathways that were previously identified to play a role in melanoma and autoimmune diseases. From the filtered pathways, we identified nodes that are common in all the enriched pathways for future analysis.
c = log(p)*z,
3.4. 3D Structure Preparation and Screening of Lead Compounds
We conducted virtual screening and molecular docking analyses to elucidate the inhibition mechanism and identify potential lead compounds for IL6 inhibition. To perform these analyses, we use a 3D model of IL6 (PDB ID: 1ALU) from the RCSB PDB database (https://www.rcsb.org/) [54]. The model was subjected to the ‘Prepare Protein’ protocols of the Biovia Discovery Studio 2022 (DS2022) [55] using the CHARMm force field [56]. The structure was further optimized using the ‘Smart Minimiser’ algorithm to achieve a stable state through energy minimization. The minimization process was done in 2000 steps, with an RMS gradient tolerance set at 0.1. With the help of the ‘Receptor-ligand Interaction’ tool of DS2022, we defined the IL6 binding site based on the amino acid residues that play a key role in its interaction with IL6R [54]. To identify potential inhibitors for IL6 proteins, we conducted a virtual screening of natural products using the Collection of Open Natural Products (COCONUT), an extensive and well-annotated resource for natural products [38], Additionally, we included an FDA-approved drug library from the ZINC 15 database[57] for the virtual screening using the ‘LibDock’ protocol of the DS2022. All the docked compounds were further subjected to the Flexible ‘CDOCKER’ program in the DS2022 [55,58]. Finally, we performed the protein-protein docking between IL6R and IL6 in complex with the screened drug molecules using the HDOCK tool [48].
3.5. Molecular Dynamic Simulation
We assessed the binding affinity of the IL6 inhibitor docked at the IL6R interaction site, we performed MD simulation using DS2022. The MD simulation was performed in an implicit solvent environment to investigate the stability, conformational changes, and dynamic behavior of the inhibitor in the binding cavity by examining the formation of diverse electrostatic interactions. For the MD simulation, the complex was subjected to an initial minimization phase consisting of 1000 steps using the steepest descent algorithm, followed by an additional 2000 steps employing the conjugate gradient method with the CHARMM force field [56]. After minimization, the systems underwent a heating phase where the initial temperature was incrementally increased from 50 K to 300 K in 50 ps intervals. Subsequently, an equilibration step lasting 100 ps was performed. The adjusted velocity frequency was configured at 50 for the heating and equilibration phases. Subsequently, a production run of 50 ns was conducted within an NVT assembly (maintaining normal volume and temperature) at a constant temperature of 300 K, with results being saved at intervals of 0.02 ns. For the entire simulation run, we analyzed trajectories consisting of 25,000 conformations. Various properties, including root-mean-square deviation (RMSD), root-mean-square fluctuation (RMSF), and the radius of gyration (ROG), were examined using the 'Analyze Trajectory Protocol' of DS2022
4. Conclusions
The introduction of ICI, such as PD-1, PD-L1, and CTLA-4 inhibitors, has revolutionized the clinical management of metastatic melanoma (MM). These inhibitors have shown remarkable efficacy in controlling the growth of tumors. However, their use also led to irAEs, resulting in exacerbating autoimmune diseases such as RA, UC, and CD in melanoma patients. Our research has explored the interface between MM and autoimmune diseases, aiming to identify potential druggable targets and understand the cross-talk between these conditions for the safe use of ICI in MM management. Using an integrative network approach, IL6 was identified as a promising target at the interface between MM and autoimmune diseases. Through structure biology approaches, lead compounds capable of inhibiting IL6, such as Amprenavir, have been identified. However, further laboratory experiments are needed to validate the efficacy of Amprenavir together with ICI.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Funding
This research was funded by the German Federal Ministry of Education and Research (BMBF) e: Med-MelAutim 01ZX2205B.
Authors’ Contributions
KPS conceived and designed the study, collected and analyzed the data, and drafted the manuscript. AS contributed to the study design, conducted experiments, and provided critical revisions to the manuscript. SG provided valuable intellectual input throughout the project and drafted the manuscript. OW participated in the study design, and provided technical expertise. All authors have read and approved the final version of the manuscript.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data is contained within the article and Supplementary Materials.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Lowe L 2023 Metastatic melanoma and rare melanoma variants: a review Pathology 55 236–44. [CrossRef] [PubMed]
- Rausch M P and Hastings K T 2017 Immune Checkpoint Inhibitors in the Treatment of Melanoma: From Basic Science to Clinical Application Cutaneous Melanoma: Etiology and Therapy 121–42.
- Shah V, Panchal V, Shah A, Vyas B, Agrawal S and Bharadwaj S 2024 Immune checkpoint inhibitors in metastatic melanoma therapy (Review) Medicine International 4.
- Rausch M P and Hastings K T 2017 Immune Checkpoint Inhibitors in the Treatment of Melanoma: From Basic Science to Clinical Application Cutaneous Melanoma: Etiology and Therapy 121–42.
- Shiravand Y, Khodadadi F, Kashani S M A, Hosseini-Fard S R, Hosseini S, Sadeghirad H, Ladwa R, O’byrne K and Kulasinghe A 2022 Immune Checkpoint Inhibitors in Cancer Therapy Current Oncology 29 3044.
- Shiravand Y, Khodadadi F, Kashani S M A, Hosseini-Fard S R, Hosseini S, Sadeghirad H, Ladwa R, O’byrne K and Kulasinghe A 2022 Immune Checkpoint Inhibitors in Cancer Therapy Current Oncology 29 3044.
- Motofei I G 2019 Melanoma and autoimmunity: spontaneous regressions as a possible model for new therapeutic approaches Melanoma research 29 231–6. [CrossRef] [PubMed]
- Ibis B, Aliazis K, Cao C, Yenyuwadee S and Boussiotis V A 2023 Immune-related adverse effects of checkpoint immunotherapy and implications for the treatment of patients with cancer and autoimmune diseases Frontiers in Immunology 14.
- Darvin P, Toor S M, Sasidharan Nair V and Elkord E 2018 Immune checkpoint inhibitors: recent progress and potential biomarkers Experimental & Molecular Medicine 2018 50:12 50 1–11.
- Khan S and Gerber D E 2020 Autoimmunity, Checkpoint Inhibitor Therapy and Immune-related Adverse Events: A Review Seminars in cancer biology 64 93.
- Hassel J C 2021 Checkpoint blocker induced autoimmunity as an indicator for tumour efficacy in melanoma British Journal of Cancer 2021 126:2 126 163–4. [CrossRef] [PubMed]
- Portenkirchner C, Kienle P and Horisberger K 2021 Checkpoint inhibitor-induced colitis—a clinical overview of incidence, prognostic implications and extension of current treatment options Pharmaceuticals 14 367.
- Lo C H, Khalili H, Lochhead P, Song M, Lopes E W, Burke K E, Richter J M, Chan A T and Ananthakrishnan A N 2021 Immune-mediated disease and Risk of Crohn’s Disease or Ulcerative Colitis: A Prospective Cohort Study Alimentary pharmacology & therapeutics 53 598.
- Jeurling S and Cappelli L C 2020 Treatment of immune checkpoint inhibitor-induced inflammatory arthritis Current opinion in rheumatology 32 315. [CrossRef] [PubMed]
- Bader G D and Hogue C W V 2003 An automated method for finding molecular complexes in large protein interaction networks BMC bioinformatics 4.
- Iwaszko M, Biały S and Bogunia-Kubik K 2021 Significance of Interleukin (IL)-4 and IL-13 in Inflammatory Arthritis Cells 10.
- Eddy K, Shah R and Chen S 2021 Decoding Melanoma Development and Progression: Identification of Therapeutic Vulnerabilities Frontiers in Oncology 10 626129.
- Napolitano M, di Vico F, Ruggiero A, Fabbrocini G and Patruno C 2023 The hidden sentinel of the skin: An overview on the role of interleukin-13 in atopic dermatitis Frontiers in Medicine 10.
- Hoejberg L, Bastholt L and Schmidt H 2012 Interleukin-6 and melanoma Melanoma research 22 327–33.
- Nemunaitis J, Fong T, Shabe P, Martineau D and Ando D 2001 Comparison of serum interleukin-10 (IL-10) levels between normal volunteers and patients with advanced melanoma Cancer investigation 19 239–47.
- Tanaka T, Narazaki M and Kishimoto T 2014 IL-6 in inflammation, immunity, and disease Cold Spring Harbor perspectives in biology 6.
- Voronov E, Carmi Y and Apte R N 2014 The role IL-1 in tumor-mediated angiogenesis Frontiers in physiology 5.
- Iwakura Y 2002 Roles of IL-1 in the development of rheumatoid arthritis: Consideration from mouse models Cytokine and Growth Factor Reviews 13 341–55.
- Silva F A R, Rodrigues B L, Ayrizono M D L S and Leal R F 2016 The Immunological Basis of Inflammatory Bowel Disease Gastroenterology research and practice 2016.
- Hofmann U B, Westphal J R, Van Muijen G N P and Ruiter D J 2000 Matrix metalloproteinases in human melanoma The Journal of investigative dermatology 115 337–44.
- Cabral-Pacheco G A, Garza-Veloz I, Rosa C C D La, Ramirez-Acuña J M, Perez-Romero B A, Guerrero-Rodriguez J F, Martinez-Avila N and Martinez-Fierro M L 2020 The Roles of Matrix Metalloproteinases and Their Inhibitors in Human Diseases International journal of molecular sciences 21 1–53.
- Lakatos G, Hritz I, Varga M Z, Juhász M, Miheller P, Cierny G, Tulassay Z and Herszényi L 2012 The impact of matrix metalloproteinases and their tissue inhibitors in inflammatory bowel diseases Digestive diseases (Basel, Switzerland) 30 289–95.
- Guo Y, Pan W, Liu S, Shen Z, Xu Y and Hu L 2020 ERK/MAPK signalling pathway and tumorigenesis Experimental and therapeutic medicine 19.
- Wei Z and Liu H T 2002 MAPK signal pathways in the regulation of cell proliferation in mammalian cells Cell research 12 9–18.
- Skytthe M K, Graversen J H and Moestrup S K 2020 Targeting of CD163+ Macrophages in Inflammatory and Malignant Diseases International journal of molecular sciences 21 1–31.
- Etzerodt A and Moestrup S K 2013 CD163 and inflammation: biological, diagnostic, and therapeutic aspects Antioxidants & redox signaling 18 2352–63.
- Miskolczi Z, Smith M P, Rowling E J, Ferguson J, Barriuso J and Wellbrock C 2018 Collagen abundance controls melanoma phenotypes through lineage-specific microenvironment sensing Oncogene 37 3166–82.
- Van Kempen L C L T, Rijntjes J, Mamor-Cornelissen I, Vincent-Naulleau S, Gerritsen M J P, Ruiter D J, Van Dijk M C R F, Geffrotin C and Van Muijen G N P 2008 Type I collagen expression contributes to angiogenesis and the development of deeply invasive cutaneous melanoma International journal of cancer 122 1019–29.
- Gencoglu H, Orhan C, Sahin E and Sahin K 2020 Undenatured Type II Collagen (UC-II) in Joint Health and Disease: A Review on the Current Knowledge of Companion Animals Animals : an open access journal from MDPI 10.
- Jarlborg M and Gabay C 2022 Systemic effects of IL-6 blockade in rheumatoid arthritis beyond the joints Cytokine 149 155742. [CrossRef] [PubMed]
- Rašková M, Lacina L, Kejík Z, Venhauerová A, Skaličková M, Kolář M, Jakubek M, Rosel D, Smetana K and Brábek J 2022 The Role of IL-6 in Cancer Cell Invasiveness and Metastasis-Overview and Therapeutic Opportunities Cells 11.
- Shahini A and Shahini A 2023 Role of interleukin-6-mediated inflammation in the pathogenesis of inflammatory bowel disease: focus on the available therapeutic approaches and gut microbiome Journal of Cell Communication and Signaling 17 55. [CrossRef] [PubMed]
- Sorokina M, Merseburger P, Rajan K, Yirik M A and Steinbeck C 2021 COCONUT online: Collection of Open Natural Products database Journal of Cheminformatics 13 1–13.
- Lipinski C A 2004 Lead- and drug-like compounds: The rule-of-five revolution Drug Discovery Today: Technologies 1 337–41.
- Tran Q H, Nguyen Q T, Vo N Q H, Mai T T, Tran T T N, Tran T D, Le M T, Trinh D T T and Minh Thai K 2022 Structure-based 3D-Pharmacophore modeling to discover novel interleukin 6 inhibitors: An in silico screening, molecular dynamics simulations and binding free energy calculations PloS one 17.
- Balasubramaniyan S, Irfan N, Umamaheswari A and Puratchikody A 2018 Design and virtual screening of novel fluoroquinolone analogs as effective mutant DNA GyrA inhibitors against urinary tract infection-causing fluoroquinolone resistant Escherichia coli RSC Advances 8 23629–47.
- Pal S, Kumar V, Kundu B, Bhattacharya D, Preethy N, Reddy M P and Talukdar A 2019 Ligand-based Pharmacophore Modeling, Virtual Screening and Molecular Docking Studies for Discovery of Potential Topoisomerase I Inhibitors Computational and Structural Biotechnology Journal 17 291–310.
- Love L A, Rader J I, Crofford L J, Raybourne R B, Principato M A, Page S W, Trucksess M W, Smith M J, Dugan E M, Turner M L, Zelazowski E, Zelazowski P and Sternberg E M 1993 Pathological and immunological effects of ingesting L-tryptophan and 1,1’-ethylidenebis (L-tryptophan) in Lewis rats The Journal of clinical investigation 91 804–11.
- Yamaoka K A, Miyasaka N, Inuo G, Saito I, Kolb J P, Fujita K and Kashiwazaki S 1994 1,1′-Ethylidenebis(tryptophan) (peak E) induces functional activation of human eosinophils and interleukin 5 production from T lymphocytes: Association of eosinophilia-myalgia syndrome with a l-tryptophan contaminant Journal of Clinical Immunology 14 50–60.
- Polli J W, Jarrett J L, Studenberg S D, Humphreys J E, Dennis S W, Brouwer K R and Woolley J L 1999 Role of P-glycoprotein on the CNS disposition of amprenavir (141W94), an HIV protease inhibitor Pharmaceutical research 16 1206–12.
- Yu Y X, Wang W, Sun H B, Zhang L L, Wu S L and Liu W T 2021 Insights into effect of the Asp25/Asp25’ protonation states on binding of inhibitors Amprenavir and MKP97 to HIV-1 protease using molecular dynamics simulations and MM-GBSA calculations SAR and QSAR in environmental research 32 615–41.
- Jiang W, Li X, Li T, Wang H, Shi W, Qi P, Li C, Chen J, Bao J, Huang G and Wang Y 2017 Repositioning of amprenavir as a novel extracellular signal-regulated kinase-2 inhibitor and apoptosis inducer in MCF-7 human breast cancer International journal of oncology 50 823–34.
- Yan Y, Tao H, He J and Huang S Y 2020 The HDOCK server for integrated protein–protein docking Nature Protocols 2020 15:5 15 1829–52.
- Boulanger M J, Chow D chone, Brevnova E E and Garcia K C 2003 Hexameric structure and assembly of the interieukin-6/IL-6 α-receptor/gp130 complex Science 300 2101–4.
- Piñero J, Bravo Á, Queralt-Rosinach N, Gutiérrez-Sacristán A, Deu-Pons J, Centeno E, García-García J, Sanz F and Furlong L I 2017 DisGeNET: a comprehensive platform integrating information on human disease-associated genes and variants Nucleic Acids Research 45 D833–9.
- Szklarczyk D, Gable A L, Lyon D, Junge A, Wyder S, Huerta-Cepas J, Simonovic M, Doncheva N T, Morris J H, Bork P, Jensen L J and Von Mering C 2019 STRING v11: protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets Nucleic Acids Research 47 D607.
- Kuleshov M V., Jones M R, Rouillard A D, Fernandez N F, Duan Q, Wang Z, Koplev S, Jenkins S L, Jagodnik K M, Lachmann A, McDermott M G, Monteiro C D, Gundersen G W and Maayan A 2016 Enrichr: a comprehensive gene set enrichment analysis web server 2016 update Nucleic Acids Research 44 W90.
- Chen E Y, Tan C M, Kou Y, Duan Q, Wang Z, Meirelles G V., Clark N R and Ma’ayan A 2013 Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool BMC bioinformatics 14.
- Somers W, Stahl M and Seehra J S 1997 1.9 A crystal structure of interleukin 6: implications for a novel mode of receptor dimerization and signaling The EMBO journal 16 989–97.
- Gogoi B, Chowdhury P, Goswami N, Gogoi N, Naiya T, Chetia P, Mahanta S, Chetia D, Tanti B, Borah P and Handique P J 2021 Identification of potential plant-based inhibitor against viral proteases of SARS-CoV-2 through molecular docking, MM-PBSA binding energy calculations and molecular dynamics simulation Molecular Diversity 25 1963.
- Vanommeslaeghe K, Hatcher E, Acharya C, Kundu S, Zhong S, Shim J, Darian E, Guvench O, Lopes P, Vorobyov I, Mackerell A D and Jr. 2010 CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. Journal of computational chemistry 31 671–90.
- Sterling T and Irwin J J 2015 ZINC 15 - Ligand Discovery for Everyone Journal of Chemical Information and Modeling 55 2324–37. [CrossRef] [PubMed]
- Yang W, Wang S, Zhang X, Sun H, Zhang M, Chen H, Cui J, Li J, Peng F, Zhu M, Yu B, Li Y, Yang L, Min W, Xue M, Pan L, Zhu H, Wu B and Gu Y 2023 New natural compound inhibitors of PDGFRA (platelet-derived growth factor receptor α) based on computational study for high−grade glioma therapy Frontiers in Neuroscience 16 1060012.
Figure 1.
This workflow outlines the process for identifying a lead compound for melanoma and autoimmune disease. The methods utilized are enhanced with various filters. Initially, gene-related information for all diseases is obtained using DisGeNET. The common genes identified are then analyzed through a protein-protein interaction (PPI) molecular map using the STRING database. The resulting PPI network is further analyzed in Cytoscape for cluster identification with MCODE. The most promising cluster undergoes enrichment analysis and a network-based approach to identify the target. Virtual screening and molecular docking are employed to find the best compound. Finally, the stability of the lead compound (amprenavir) is assessed through molecular dynamics (MD) simulation.
Figure 1.
This workflow outlines the process for identifying a lead compound for melanoma and autoimmune disease. The methods utilized are enhanced with various filters. Initially, gene-related information for all diseases is obtained using DisGeNET. The common genes identified are then analyzed through a protein-protein interaction (PPI) molecular map using the STRING database. The resulting PPI network is further analyzed in Cytoscape for cluster identification with MCODE. The most promising cluster undergoes enrichment analysis and a network-based approach to identify the target. Virtual screening and molecular docking are employed to find the best compound. Finally, the stability of the lead compound (amprenavir) is assessed through molecular dynamics (MD) simulation.
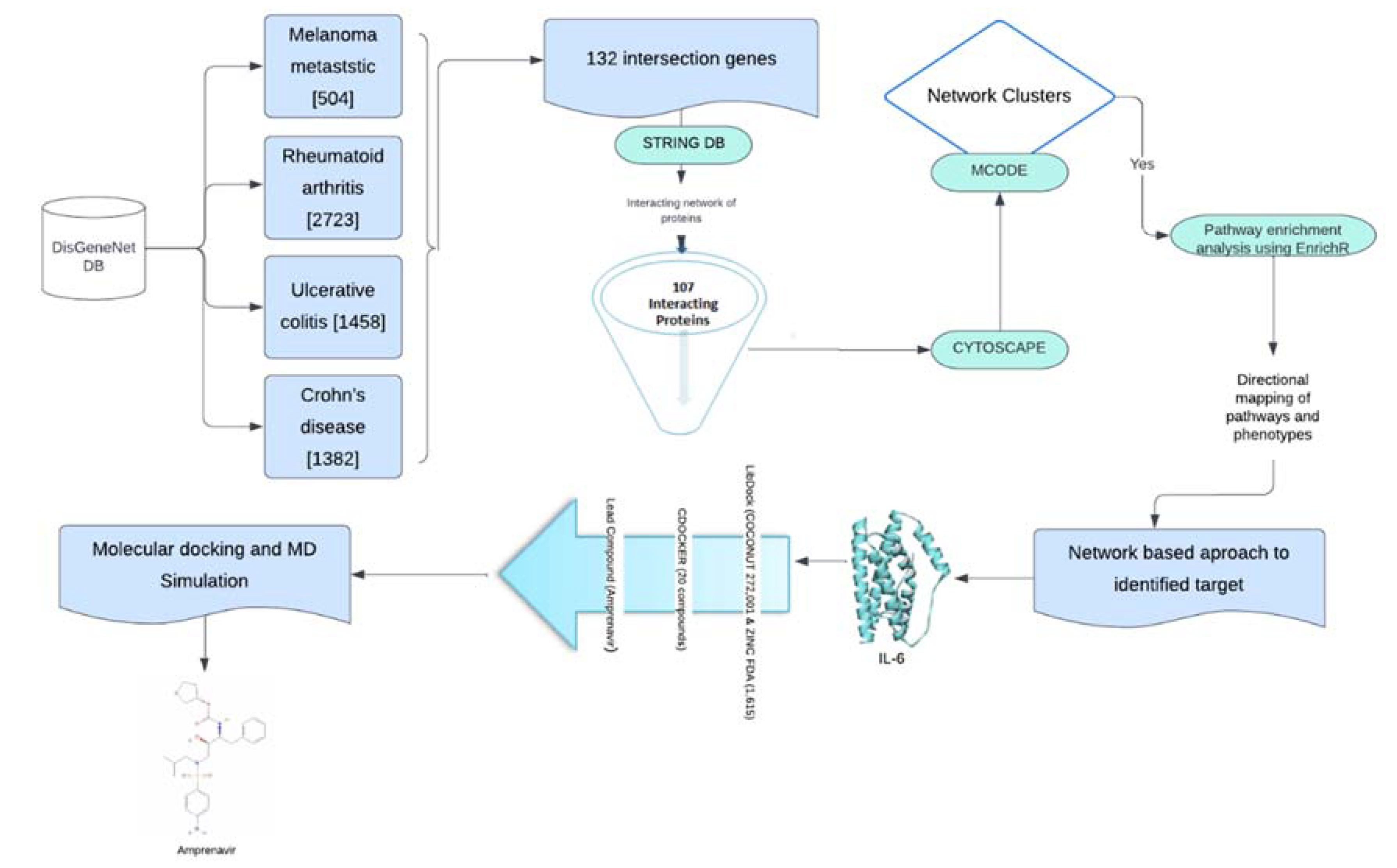
Figure 2.
Venn diagram highlighting overlapping genes between rheumatoid arthritis (RA), ulcerative colitis (UC), Crohn's Disease (CD), and melanoma metastasis (MM). A total of 132 genes were shared among all disease phenotypes.
Figure 2.
Venn diagram highlighting overlapping genes between rheumatoid arthritis (RA), ulcerative colitis (UC), Crohn's Disease (CD), and melanoma metastasis (MM). A total of 132 genes were shared among all disease phenotypes.
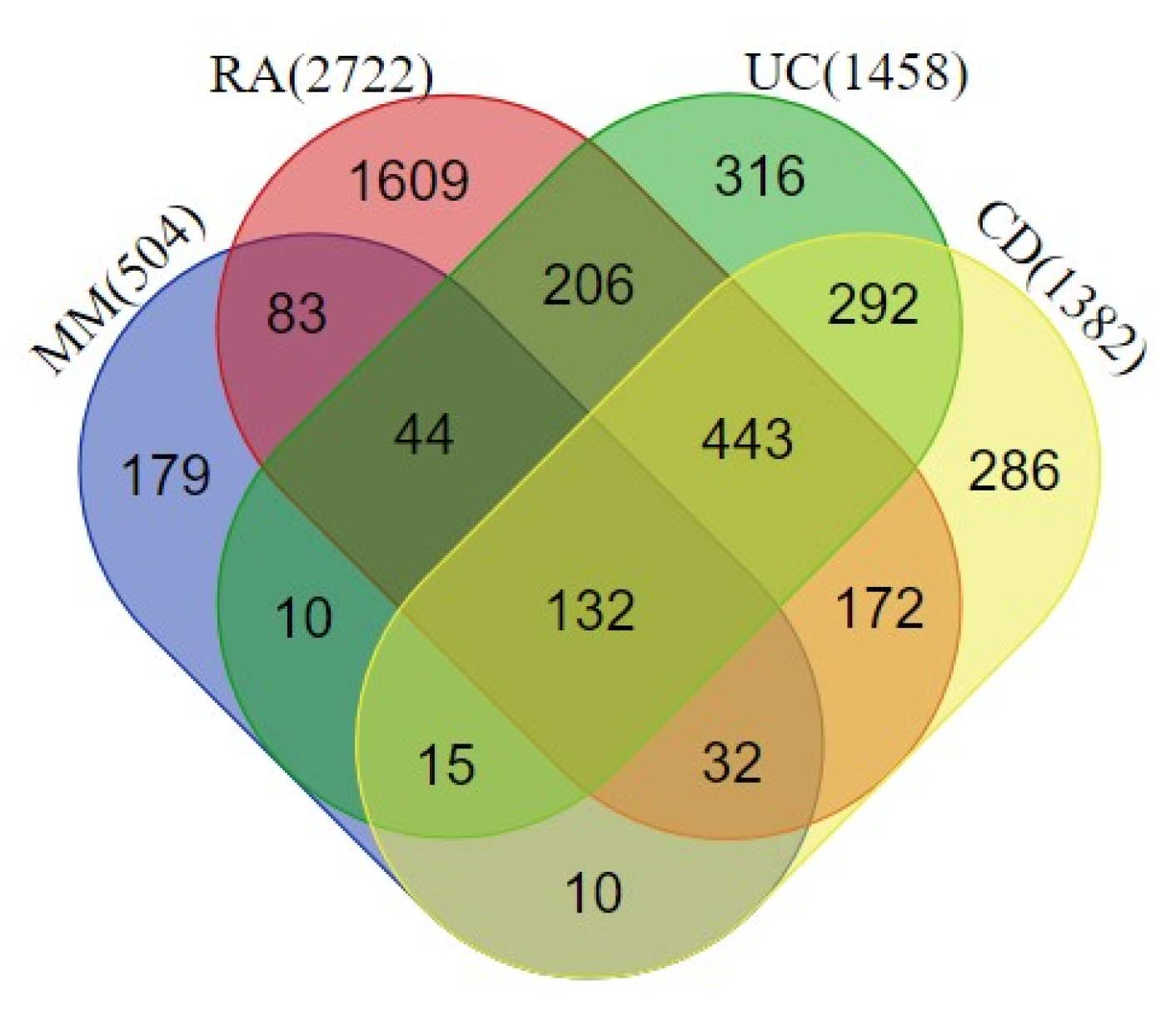
Figure 3.
A network of 107 common genes associated with the investigated four disease phenotypes. The network is prepared using the String database with connections between the nodes above the 0.7 confidence score cutoff.
Figure 3.
A network of 107 common genes associated with the investigated four disease phenotypes. The network is prepared using the String database with connections between the nodes above the 0.7 confidence score cutoff.
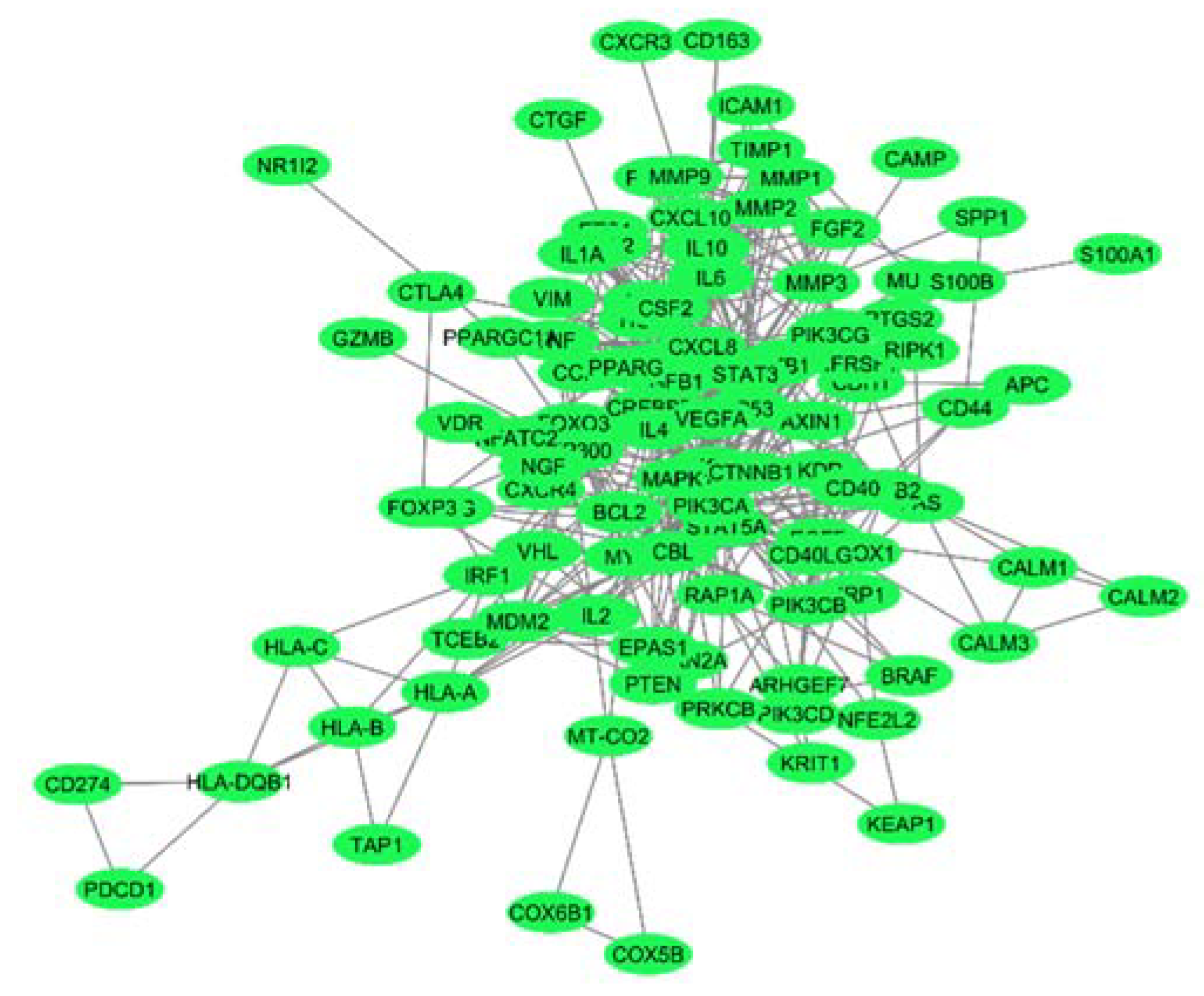
Figure 4.
A network of top enriched pathways associated with the genes present in the best cluster was identified through the MCODE analysis. The enriched pathways are shown in green rectangle boxes, genes are shown as colored ovals, and disease phenotypes (MM and Autoimmune diseases) are shown as circular nodes. The impact of genes on the pathways (dashed lines) and to melanoma and autoimmune disease phenotypes (dotted lines) is shown where the pointed arrowhead indicates activation and the blunt-end arrowhead indicate suppression.
Figure 4.
A network of top enriched pathways associated with the genes present in the best cluster was identified through the MCODE analysis. The enriched pathways are shown in green rectangle boxes, genes are shown as colored ovals, and disease phenotypes (MM and Autoimmune diseases) are shown as circular nodes. The impact of genes on the pathways (dashed lines) and to melanoma and autoimmune disease phenotypes (dotted lines) is shown where the pointed arrowhead indicates activation and the blunt-end arrowhead indicate suppression.
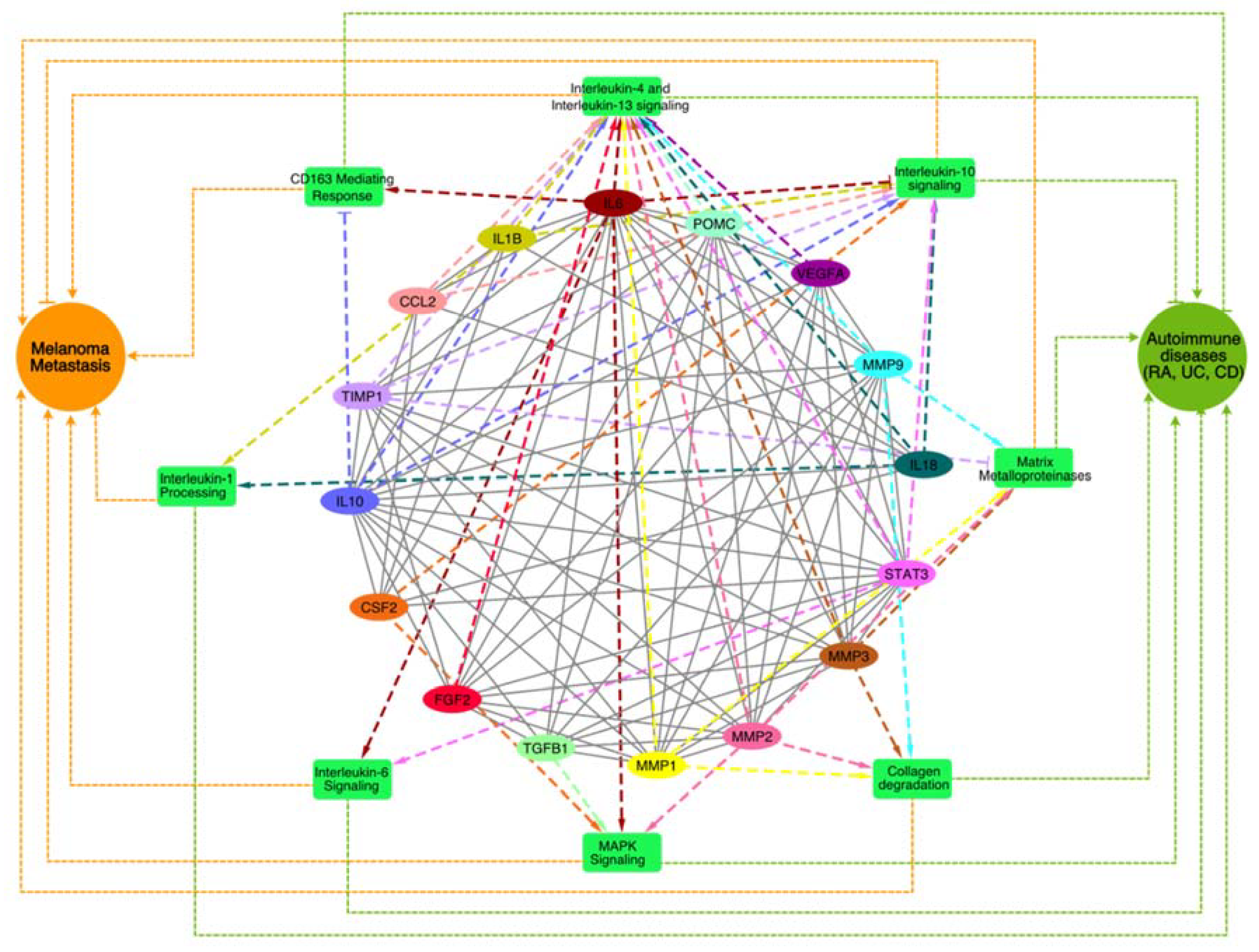
Figure 5.
2D representation of the top two compounds (ZINC000003809192 & CNP0003038), which were extracted after the virtual screening and molecular docking using the DS20222.
Figure 5.
2D representation of the top two compounds (ZINC000003809192 & CNP0003038), which were extracted after the virtual screening and molecular docking using the DS20222.
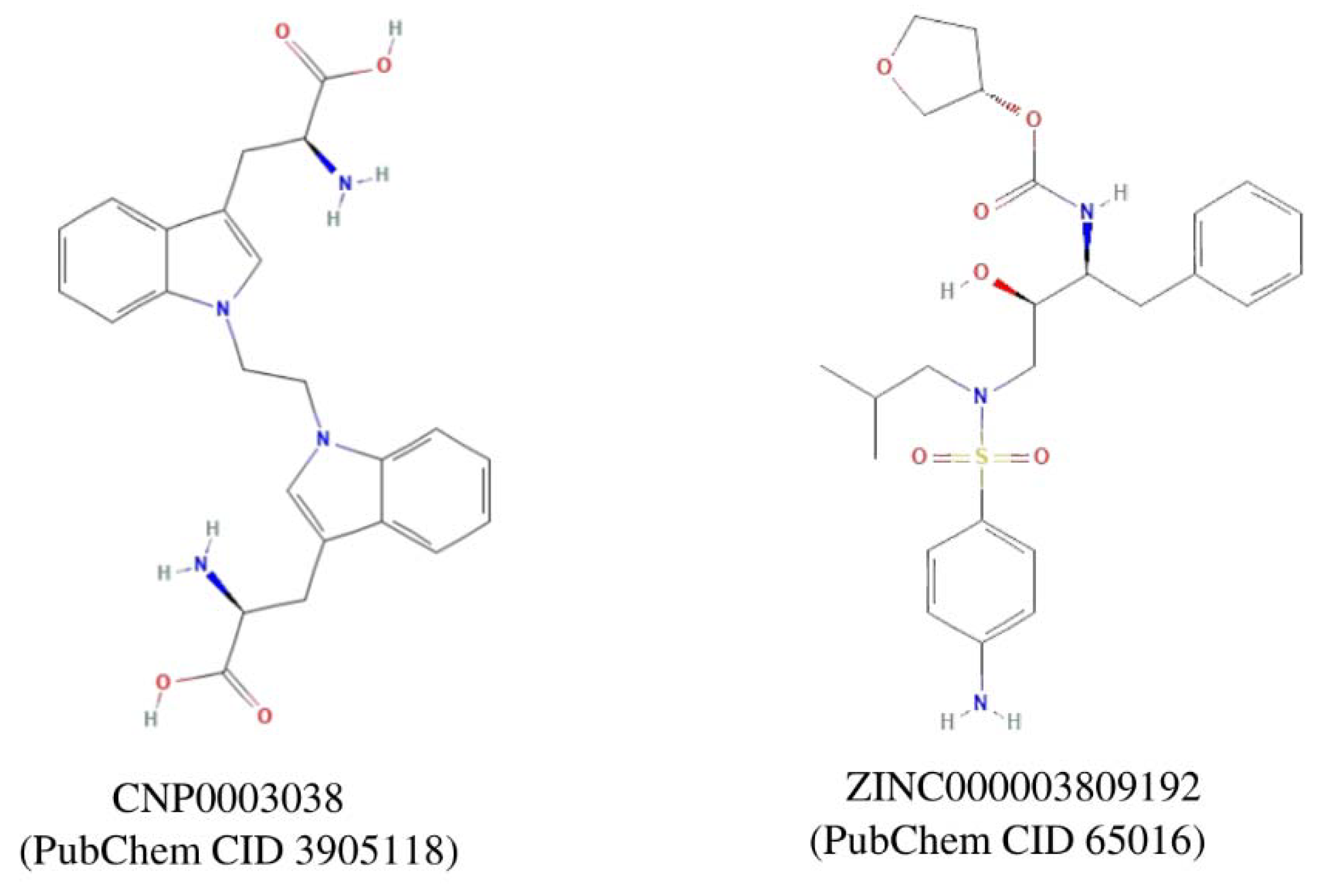
Figure 6.
The docked poses obtained from the HDOCK docking tool depict the interactions between IL6R and IL6 with amprenavir. In this illustration, IL6R is represented by two different color bases on separate chains (alpha in light green and beta in dark green), while IL6 is shown in blue. The first frame of figure (a) showcases the surface representation of IL6R and IL6 proteins and their interactions. (b) the surface representation of IL6R and IL6 proteins with amprenavir. Additionally, the frame provides a zoomed-in version of the surface, highlighting the interaction between IL6R, IL6, and amprenavir.
Figure 6.
The docked poses obtained from the HDOCK docking tool depict the interactions between IL6R and IL6 with amprenavir. In this illustration, IL6R is represented by two different color bases on separate chains (alpha in light green and beta in dark green), while IL6 is shown in blue. The first frame of figure (a) showcases the surface representation of IL6R and IL6 proteins and their interactions. (b) the surface representation of IL6R and IL6 proteins with amprenavir. Additionally, the frame provides a zoomed-in version of the surface, highlighting the interaction between IL6R, IL6, and amprenavir.
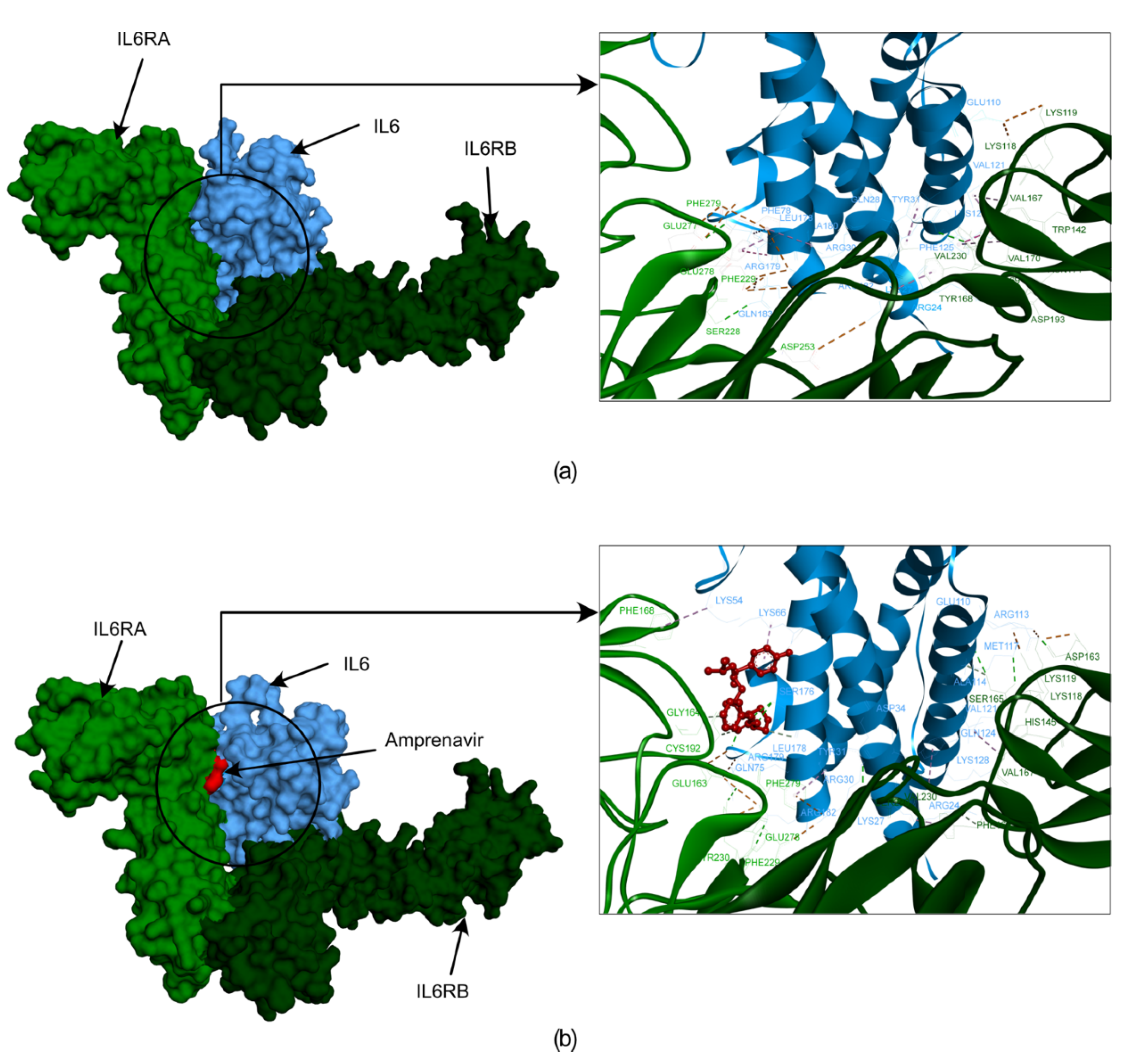
Figure 7.
MD simulation analysis of IL6_Amprenavir complex. (a) Hydrogen bonds and hydrophobic interactions between IL6 and Amprenavir are shown before and after the MD simulation. After the MD simulation, Amprenavir formed two additional hydrogen bonds with IL6 compared to the initial docked pose, while the hydrophobic bonds remained unchanged. The colored arrow indicates the nature of the bonds. All IL6 amino acid residues involved in the bond formation are labelled. (b) Root mean square deviation (RMSD) graph of the IL6 from the docked complex over a simulation period of 50 nanoseconds (ns). (c) Root mean square fluctuation (RMSF) graph of IL6 interaction site associated with IL6R. The IL6 amino acid residues that directly interact with IL6R are labelled. (d) Radius of gyration (Rg) graph of IL6 from the IL6_Amprenavir complex. The graph suggests that IL6 attain more compact confirmation after binding with the drug.
Figure 7.
MD simulation analysis of IL6_Amprenavir complex. (a) Hydrogen bonds and hydrophobic interactions between IL6 and Amprenavir are shown before and after the MD simulation. After the MD simulation, Amprenavir formed two additional hydrogen bonds with IL6 compared to the initial docked pose, while the hydrophobic bonds remained unchanged. The colored arrow indicates the nature of the bonds. All IL6 amino acid residues involved in the bond formation are labelled. (b) Root mean square deviation (RMSD) graph of the IL6 from the docked complex over a simulation period of 50 nanoseconds (ns). (c) Root mean square fluctuation (RMSF) graph of IL6 interaction site associated with IL6R. The IL6 amino acid residues that directly interact with IL6R are labelled. (d) Radius of gyration (Rg) graph of IL6 from the IL6_Amprenavir complex. The graph suggests that IL6 attain more compact confirmation after binding with the drug.
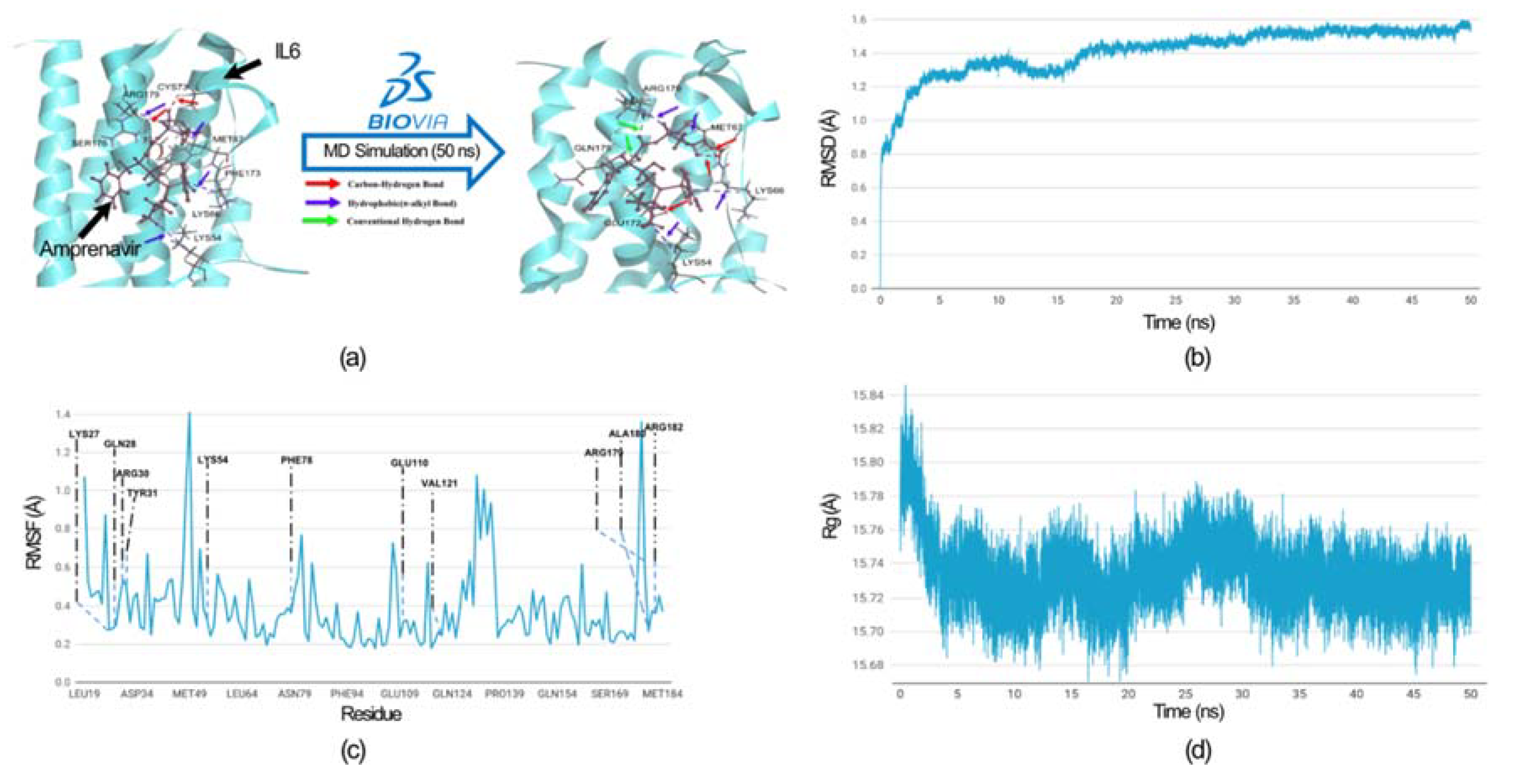

Table 1.
Network modules generated by MCODE, with their score, number of nodes and interactions, along with the associated gene names.
Table 1.
Network modules generated by MCODE, with their score, number of nodes and interactions, along with the associated gene names.
| Modules | Nodes | Interaction | MCODE Score | Genes |
|---|---|---|---|---|
| 1 | 16 | 76 | 10.133 | CCL2,CSF2,FGF2,IL10,IL18,IL1B,IL6,MMP1,MMP2,MMP3,MMP9,POMC,STAT3,TGFB1,TIMP1,VEGFA |
| 2 | 7 | 18 | 6 | CREBBP,EP300,FOXO3,HIF1A,MAPK1,MDM2,TP53 |
| 3 | 4 | 6 | 4 | AKT1,CD40,CD40LG,PIK3CG |
| 4 | 8 | 13 | 3.71 | CTNNB1,CXCL10,CXCL8,IL1A,IL4,MYC,NFKB1,TNF |
| 5 | 7 | 10 | 3.33 | CALM1,CALM2,CALM3,CXCR4,FAS,PIK3CB,STAT5A |
| 6 | 3 | 3 | 3 | HLA-B,HLA-C,HLA-DQB1 |
Table 2.
List of top 20 compounds identified after the virtual screening of IL6 binding site responsible for interacting with IL6R.
Table 2.
List of top 20 compounds identified after the virtual screening of IL6 binding site responsible for interacting with IL6R.
| S.No. | Compounds ID | Database | Compounds Name | Libdock score | -CDOCKER Energy (kcal/mol) |
|---|---|---|---|---|---|
| 1 | CNP0003841 | Coconut | N-[(3-methoxyphenyl)methyl]-3-({5-[(4-phenylpiperazin-1-yl)methyl]-1,2-oxazol-3-yl}methyl)oxetan-3-amine | 127.506 | NA |
| 2 | CNP0004058 | Coconut | 2-chloro-5-hydroxy-N-{[4-hydroxy-5-(hydroxymethyl)-3-{4-[3-(trifluoromethyl)phenyl]piperazin-1-yl}oxolan-2-yl]methyl}benzamide | 126.919 | NA |
| 3 | CNP0004582 | Coconut | 2-{[({3-methyl-4-[(7-methyl-1H-1,3-benzodiazol-2-yl)methyl]-6-(propan-2-yl)cyclohex-2-en-1-yl}methyl)carbamoyl]methoxy}acetic acid | 122.508 | 18.4652 |
| 4 | CNP0004629 | Coconut | 2-{[({3-methyl-4-[(1-methyl-1H-1,3-benzodiazol-2-yl)methyl]-6-(propan-2-yl)cyclohex-2-en-1-yl}methyl)carbamoyl]methoxy}acetic acid | 121.359 | 13.0031 |
| 5 | CNP0000288 | Coconut | 7-methoxy-2-(4-methoxyphenyl)-4-[2-(4-methoxyphenyl)ethyl]-3,4-dihydro-2H-1-benzopyran | 120.936 | 25.4486 |
| 6 | ZINC03809192 | ZINC | [(3S)-oxolan-3-yl] N-[(2S,3R)-4-[(4-aminophenyl)sulfonyl-(2-methylpropyl)amino]-3-hydroxy-1-phenylbutan-2-yl]carbamate | 120.668 | 34.7136 |
| 7 | CNP0004224 | Coconut | 4-(dimethylamino)-N-[5-hydroxy-7a-(2-{[2-(1H-indol-3-yl)ethyl]carbamoyl}ethyl)-3,3,5-trimethyl-octahydro-1H-inden-1-yl]benzamide | 118.314 | NA |
| 8 | CNP0004392 | Coconut | 4-[(2-{3-[2-(pyrrolidin-1-yl)pyridin-4-yl]-1,2,4-oxadiazol-5-yl}pyrrolidin-1-yl)methyl]benzoic acid | 118.034 | NA |
| 9 | CNP0003909 | Coconut | 3-[4-(4-methoxyphenyl)-1H-imidazol-2-yl]-4-[(4-methylphenyl)methyl]morpholine | 117.757 | 17.5344 |
| 10 | ZINC03955219 | ZINC | [(3aS,4R,6aR)-2,3,3a,4,5,6a-hexahydrofuro[2,3-b]furan-4-yl] N-[(2S,3R)-4-[(4-aminophenyl)sulfonyl-(2-methylpropyl)amino]-3-hydroxy-1-phenylbutan-2-yl]carbamate | 117.727 | 18.3056 |
| 11 | CNP0004686 | Coconut | 4-cyano-N-{2,3-dihydroxy-5-[6-(morpholin-4-yl)pyridin-3-yl]cyclopentyl}benzamide | 117.281 | NA |
| 12 | CNP0004257 | Coconut | N-[(2H-1,3-benzodioxol-5-yl)methyl]-3-({5-[(dimethylamino)methyl]-1,2-oxazol-3-yl}methyl)oxetan-3-amine | 116.072 | NA |
| 13 | CNP0003888 | Coconut | 3-[4-(4-chlorophenyl)-1H-imidazol-2-yl]-4-[(1-methyl-1H-imidazol-2-yl)methyl]morpholine | 115.838 | 16.6375 |
| 14 | CNP0004329 | Coconut | N-[(2H-1,3-benzodioxol-4-yl)methyl]-3-({5-[(4-phenylpiperazin-1-yl)methyl]-1,2-oxazol-3-yl}methyl)oxetan-3-amine | 115.688 | NA |
| 15 | CNP0004277 | Coconut | (5-{[(3-{[5-(pyridin-2-yl)-1,2-oxazol-3-yl]methyl}oxetan-3-yl)amino]methyl}furan-2-yl)methanol | 115.539 | NA |
| 16 | CNP0004720 | Coconut | 2-{[(3-{[5-(4-methoxyphenyl)-1,2-oxazol-3-yl]methyl}oxetan-3-yl)amino]methyl}phenol | 115.352 | NA |
| 17 | CNP0003796 | Coconut | N-[(4-methoxyphenyl)methyl]-3-{[5-(pyridin-2-yl)-1,2-oxazol-3-yl]methyl}oxetan-3-amine | 113.167 | NA |
| 18 | CNP0004058 | Coconut | 2-chloro-5-hydroxy-N-{[4-hydroxy-5-(hydroxymethyl)-3-{4-[3-(trifluoromethyl)phenyl]piperazin-1-yl}oxolan-2-yl]methyl}benzamide | 112.619 | NA |
| 19 | CNP0003038 | Coconut | 2-amino-3-(1-{1-[3-(2-amino-2-carboxyethyl)-1H-indol-1-yl]ethyl}-1H-indol-3-yl)propanoic acid | 111.568 | 41.6684 |
| 20 | CNP0005022 | Coconut | 4-({3-[4-(pyridin-4-yl)-1H-imidazol-2-yl]morpholin-4-yl}methyl)benzoic acid | 111.381 | 22.286 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



