
Submitted:
29 August 2024
Posted:
02 September 2024
You are already at the latest version
Abstract
According to evolutionary cancer cell biology ECCB, cancer stem cells do not arise from normal stem cells, but rather from the functional ACD phenotype of the germline. This phenotype proliferates through asymmetric cell division, producing self-renewing cells and non-proliferating daughter cells with cancer stem cell qualities (CSCs). ECCB posits that cancer stem cells themselves do not proliferate. Similar to protists, there is a close reciprocal relationship between both sister cells that collectively form a germ and stem cell lineage. These sister cells share functional roles: the proliferating cancer germline cells generate stem cells, while the non-proliferating cancer stem cells accumulate progenitor cells for new germline clones. ECCB distinguishes between primary CSCs, which are associated with carcinogenesis and primary tumors, and secondary CSCs, which are linked to metastases. This unicellular stem cell system is homologous to that of parasitic protists, such as amoebae. Both cancer stem cells and amoebae stem cells are generated by an oxygen-sensitive germline and are vulnerable to damage when oxygen levels exceed 6.0% (germline hyperoxia). Oxygen concentrations above this threshold can damage the germline genome. Dysfunctional germline cells do not undergo senescence; instead, they continue to cycle through defective symmetric cell cycles However, to regain functionality, the germline genome requires repair by hyperpolyploid giant cells known as native PGCCs, which are homologous to the multinucleated genome repair structures MGRS found in protists. Native PGCCs differ in some respects from genotoxic-induced PGCCs
Keywords:
Cancer
; Entamoeba
; germline
; asymmetric cell division
; CSCs
; loss of function
; hyperpolyploidy
1. Introduction
According to the most common definitions today, stem cells and cancer stem cells are either (i) undifferentiated or partially differentiated cells that can differentiate into various types of cells and cycle indefinitely to produce more stem cells, or (ii) undifferentiated cells that continuously divide to produce some offspring that remain as stem cells and others that are destined to differentiate. However recent findings have shown that these definitions are no longer adequate.
Researchers have long sought a model analogous to the cancer cell system to understand the origins and nature of cancer and cancer stem cells (CSCs). Such a model could potentially explain why cancer occurs not only in humans and vertebrates but also in invertebrates and primitive metazoans. In the absence of this model, cancer has often been described as the uncontrolled growth of cells driven by various extrinsic and intrinsic factors [1]. Consequently, the origin of CSCs has been traced back to other stem cells such as normal adult stem cells (ASCs) or embryonic stem cells (ESCs).
This work explores the origin of CSCs and cancer through the lens of Evolutionary Cancer Cell Biology (ECCB), demonstrating why current CSC hypotheses are not evolutionarily valid. ECCB uncovers the deep homologous relationship between CSCs and the Urgermline of the common ancestor of amoebozoans, metazoans, and fungi (AMF), highlighting the shared characteristics of cancer and CSCs with unicellularity [1]. However, before delving into this perspective, a brief overview of current CSC research outside the ECCB framework will be provided.
Current CSCs hypotheses
In recent years, both empirical and molecular research on cancer stem cells (CSCs) has made significant progress, leading to the collection of substantial new data. However, due to a lack of evolutionary insight, the origin and development of CSCs could not be fully understood.
Several studies conducted over the past three to four years have underscored the limitations and shortcomings of earlier cancer stem cell concepts. Researchers have identified CSCs as a small subset of cells that play a fundamental role in cancer development, progression, metastasis, and treatment resistance [2]. The similarities between cancer stem cells (CSCs) and normal stem cells (NSCs) have been well-documented, but the differences could not be adequately explained.
As reported by Rossi et al. in 2020 [3], there is ongoing controversy regarding the origin of CSCs from normal or embryonic stem cells. While some researchers support this hypothesis, others oppose it and propose alternative origins, such as cell-cell fusion, gene transfer, or mutations [4]. Additionally, some findings suggest that CSCs may arise from normal stem cells that fail to regulate their proliferation under abnormal conditions [5,6].
According to Tweedell [7], NSC populations typically consist of a mixture of quiescent stem cells, active stem cells, and progenitor cells at various stages of differentiation. Part of the stem cell progeny is sequestered within tissue niches during the different stages of organ development and differentiation. This concept has been applied to CSCs and their niche, which regulates both stem and progenitor cells, serving as a specific topographical and functional site. [8,9,10] In summary, the origin of CSCs remains controversial, with ongoing debate about whether CSCs arise from normal stem cells, progenitor cells, or dysfunctional progenitor cells present in tissue [1].
CSCs cannot be born from NSCs
There are far too many differences that allow the origin of the CSCs to be left in normal, adult, or embryonic stem cells. CSCs exhibit unlimited proliferation, excessive self-renewal, drug resistance, and the ability to generate heterogeneous populations with offspring (bulk tumor cells) that cannot replicate the tumor or form metastases. These divergent characteristics and mechanisms suggest a genome distinct from that of normal stem cells (NSCs). Most notably, CSCs lack the regulatory systems present of adult stem cells that prevent uncontrolled proliferation [4,11] and possess hyperpolyploid genome repair systems absent in NSCs [12]. On the other hand, from an evolutionary perspective, it is evident that the existing similarities between CSCs and NSCs reflect their shared relationship with the AMF ancestor and its Urgermline. [1].
1.1. Requirements for a Modern Stem Cell Concept
According to most researchers, further efforts are needed to clarify the origin of CSCs to enhance existing therapies and develop new clinically relevant cancer treatments. Recently, Loh and Ma [13] discussed the need to re-evaluate the origin, hallmarks, and characteristics of cancer stem cells. They argue that substantial evidence suggests cancer cells possess a plastic state influenced by the interplay of stressors and the environment, known as the CSC niche. The researchers propose that the features acquired through de-differentiation require a re-assessment of the basic attributes of the CSC state. Cellular plasticity allows various cancer cells to adopt a stem-like state, enabling tumor cells to enhance their malignant properties, including resistance to therapy and metastasis.
The author of this work also advocates for a reevaluation of the cancer stem cell (CSC) concept concerning germline progenitors and germline plasticity, as proposed by Evolutionary Cancer Cell Biology. From the ECCB perspective, current CSC definitions are imprecise and require adjustment. Many inconsistencies could be clarified and better explained through this lens. ECCB posits that CSCs are “born” from dysfunctional cells that have lost their capacity for asymmetric cell division (ACD), with cancer representing a fundamental transition to a lower cell organization system of unicellular imprinting. Both this transition and the subsequent evolution of the cancer cell system are governed by an ancestral gene regulatory network (aGRN). The genome of the ancestral cell system, including the aGRN, is contained within the ancestral genome compartment of all metazoans and humans and can be reactivated at any time by intrinsic and extrinsic factors. These switching mechanisms, which originated during the transition from unicellularity to multicellularity [1,12], are crucial for cancer initiation.
1.2. Proliferating Germline and Non-Proliferating CSCs
Like the ancestral cell system of the AMF ancestor, the unicellular system of cancer consists of a germline and a somatic cell line [1]. During cancer evolution, this unicellular cancer cell system generates heterogeneous germline clones that give rise to numerous stem cell lineages. All CSCs are germline cells produced by more or less evolved ACD clones, which cycle through asymmetric cell division, resulting in two unequal daughter cells: one that self-renews and another that exits the cell cycle (Figure 1). The exiting cells may differentiate environmental dependant into either non-committed CSCs (quiescent or temporarily CSCs) or committed CSCs for cyst-like amplification, both of which are non-proliferative. Transitory, quiescent stem cells have the potential to revert to a self-renewing state, producing proliferating ACD clones. Under optimal environmental conditions, they can also commit to becoming genuine CSCs.
In summary, CSCs and the ACD germline form a reciprocal, inseparable unit: proliferating germline cells generate CSCs, while non-proliferating CSCs may generate proliferating germline clones. This inseparable cell pair is referred to as an ACD-lineage for self-renewing and stem cell production
2. The Unicellular Model of Cancer and Cancer Stem Cells
As already mentioned, cancer is a complex and highly atypical disease that cannot be fully explained by genetic alterations or the accumulation of mutated genes over a lifetime. Rather, it represents a systemic shift from a multicellular cell system to a more primitive unicellular system, driven by hyperoxic shock above 6.0% O2 (germline and stem cell hyperoxia), genome dysfunctionalities, and cells seeking opportunities for genome repair. In the past, cancer has even been likened to a parasitic multistage disease caused by a highly transformative pathogen [14].
2.1. The Germ and Soma Model of Parasitic Amoebae
ECCB is a new oncological discipline that emerged from the comparative analysis of the life cycles of cancer and protists, along with their stem cells [1]. It reveals that the stem cells of cancer and protists are evolutionary siblings and that parasitic protists possess a cellular system remarkably similar to the germ and soma cell systems found in cancer. One of the most striking similarities between the two systems is the presence of a cyst-like polyploid stage followed by a hyperpolyploid stage that has been observed both in the multinucleated genome repair syncytia (MGRS) of amoebae and the polyploid giant cancer cells (PGCC). These impressive unicellular features, observed exclusively in cancer and amoebae, strongly suggest a common evolutionary origin and indicate that native CSCs arise from the cancer germline and not from embryogenic or normal stem cell lineages. Both stem cell systems bear the inherent mechanisms of the evolutionary Urgermline and its stemness.
The history of the ECCB began with the development of oxygen-consuming culture media for parasitic amoebae. Contrary to the prevailing trend of cultivating entamoebae in synthetic media, the author of this work successfully established oxygen-consuming cell cultures using metabolically suppressed bacteria. These oxygen-consuming bacteria (OCB) effectively mimicked the hypoxic conditions of the intestine, characterized by an oxygen gradient ranging from 0.1 to 5.7% O2. In 2013, the author reviewed and updated these findings [15,16].
The in vitro results obtained with OCB media underline the remarkable plasticity of the germline and reveal that hypoxia is the natural physioxic environment for all germline and stem cell activities. The hypoxic environment provided by OCB sediment cultures facilitates several key processes: (i) asymmetric cell division and the formation of cancer stem cells (CSCs), (ii) the germ-to-soma transition (GST) processes and soma-to-germ transition, which generate new germline clones, (iii) cyst-like accumulation of genome copies through polyploidization and depolyploidization cycles, (iii) the production of fusogens in dysfunctional phenotypes that proliferate by symmetric cell division (DSCD), (iv) homotypic cell fusion and formation of multinucleated genome repair syncytia (MGRS) (v) genome repair via the MGRS repair pathway [1].
In contrast, studies using synthetic media with an oxygen content above 6.0% O2 (germline hyperoxia) showed that: (i) hyperoxic conditions damage the germline genome, (ii) the functional ACD germline phenotype, which normally gives rise to germline stem cells (GSCs, CSCs), is irreversibly replaced by a dysfunctional DSCD phenotype, (iii) DSCD cells proliferate indefinitely through defective symmetric cell division, resulting in tetraploidy and aberrant mitosis, and (iv) these dysfunctional genomes can only be repaired through homotypic cell fusion and hyperpolyploidy [17,18,19,20].
In 2023 Hazra et al. [20] wrote “ In Entamoeba, a protozoan parasite that causes amoebic dysentery and liver abscesses in humans, the formation of MGCs (multinucleated giant cells) is a unique phenomenon and not been reported in any other protozoa. Accordingly, the formation of MGCs in Entamoeba is thought to be a survival strategy to cope with adverse conditions. This organism forms MGCs through cell aggregation and fusion in response to osmotic and heat stress. The MGCs in Entamoeba are thought to have increased resistance to various stresses and can survive under adverse conditions. The authors hypothesized that the increased survival ability could provide redundancy in case of DNA damage or mutations. Additionally, MGCs may play a role in the virulence of Entamoeba as they are found in the inflammatory foci of amoebic liver abscesses and other infections caused by Entamoeba“. The researchers do not realize that MGCs serve to repair the dysfunctional DSCD phenotype that they have grown in hyperoxic cultures.
Parallel to advancements in studying the hypoxic cell biology of parasitic protists [16,21,22], cancer researchers have successfully progressed into the hypoxic biology of cancer stem cell lineages. However, the unicellular origin of ACD-lineages was not recognized due to a lack of evolutionary understanding. Despite this, the phenomena of polyploidy and hyperpolyploidy were accurately characterized, as was the genotoxically induced PGCC process following radiation and chemotherapy treatments [23,24,25]. Additionally, cancer researchers have detailed the various aspects of epithelial-mesenchymal transition (EMT) in cancer [26,27,28,29,30] and have shown how oxygen-sensitive germ cells disseminate into the blood and tissues, forming surviving cell clusters with oxygen-resistant cells [31,32,33,34,35,36].
2.2. The Age of the Unicellular Germ and Soma Cell System Adopted by Cancer
According to the ECCB, the evolutionary origin of cancer stem cells dates back approximately 2300 million years ago (Mya), predating the emergence of metazoans [1,37]. During this period, the common AMF ancestor evolved the dual life cycle consisting of a non-gametogenic oxygen-sensitive germline (Urgermline) and a somatic oxygen-resistant cell line. The non-gametogenic Urgermline, which had the capability of generating unipotent germline stem cells (GSCs aka CSCs), served as an ancestral blueprint for all modern self-renewing and stem cell lineage (ACD-lineages). It also plays a central role in cancer cell biology, largely reflecting the AMF heritage has also been transferred to the parasitic protists of humans and other metazoans [16]. In contrast, somatic cancer cells which are oxygen resistant, are not harmed by excess oxygen and contribute to the reconstruction of functional germline clones and CSCs, both by soma-to-germ transition (SGT) and by circulating cancer cell clusters.
2.3. The Unicellular Germ and Soma Cell System during the Transition Period to Multicellularity
Around 1,750 million years ago, with the onset of multicellularity, evolutionary pressures led to the suppression of the unicellular AMF life cycle. However, as early multicellular organisms became unstable and dysfunctional, they reverted to the stable AMF life cycle, and its associated repair mechanisms. From this point onwards, all early and later metazoans incorporated the AMF genome into their ancestral genome compartment, preserving it in a latent state that can be activated when necessary.
This evolutionarily conserved strategy of switching between the multicellular and phylogenetic genome compartments was a recurring phenomenon during the transition to multicellularity that allowed early and later metazoans to toggle between different genomic states. Genes associated with this back-and-forth strategy remained in a constant standby mode, enabling transitions from multicellularity to unicellularity even today, particularly when multicellular genome errors need to be repaired using unicellular mechanisms, as observed in cancer [1,12].
In this context, cancer mimics the alternative lifestyles of organisms from the transitional period to multicellularity. Just as the early multicellular organisms that oscillated between impaired multicellular evolution and a stable unicellular lifestyle, cancer cell system alternate between a restricted DSCD phenotype, which lacks stemness and differentiation capacities, and a functional ACD phenotype, which is capable of stemness and differentiation.
Suppressor and anti suppressor genes from this transition period were preserved in the ancestral genome compartment of all metazoans. Over time, these genes evolved into tumor suppressor genes (TSGs) and oncogenes. Additionally, genes from early multicellular dead ends were retained as supplementary reservoir genes, which could be repurposed during metazoan evolution.
2.4. Ancient Hyperoxic Ranges of More Than 6.0 % O2, in Tissue and Bloodstream, Damage ACD-Lineages of Cancer
All modern-day germlines, including progenitor and stem cells, originate from the AMF Urgermline and are predominantly hypoxic, with normoxic ranges below 6.0% O2 (germline physioxia). Oxygen levels above 6.0% O2 (germline hyperoxia), as found in tissues and the bloodstream, far from the hypoxic niche, are harmful to germ and stem cells and cause significant damage across all animal cell systems including cancer. This sensitivity is consistent across humans, mammals, vertebrates, invertebrates, and protists such as parasitic amoebae. When exposed to hyperoxic conditions, germlines reduce the gene activity associated with homologous recombination (HR) repair. This reduction leads to irreparable DNA double-strand breaks (DNA DSBs) and subsequent loss of function [1,12].
3. Non-Proliferating CSCs and Cyst-like Amplification Cycles
As shown in Figure 1, CSCs and the productive germline form a reciprocal, inseparable unit: proliferating germline cells can generate non-proliferating CSCs committed for differentiation. These CSCs accumulate precursor cells for a new CSC cell generation through amitotic cyst-like polyploidization-depolyploidization cycles. Under favorable environmental conditions, committed stem cells, which are capable of polyploid amplification, can differentiate cysts (as in Entamoeba) or cyst-like structures (as in cancer). During this process, they amplify their DNA content and accumulate progenitors for new germline clones. During cancer evolution, this unicellular cancer cell system continuously generates a large number of heterogeneous germline clones and subclones, which give rise to further CSCs and stem cell lineages. According to the ECCB, the functional ACD cancer phenotype producing primary CSCs arises from a dysfunctional non-cancerous phenotype, which cycles through unlimited tetraploid cell cycles and defective symmetric cell divisions (DSCD phenotype) [1,12].
Cyst-like polyploidization and depolyploidization cycles of cancer and protists involve four distinct phases (Figure 2 right): (i) committed genome copying and polyploidization, (ii) reductive nuclear division, (iii) haploidization of the daughter nuclei, and (iv) diploidization to germline cells. Polyploidization is more efficient than proliferation through slower ACD cell cycling and ensures accurate DNA replication through associated homologous recombination (HR).
In the past, current CSC biology assumes that stem cells “proliferate indefinitely“ to produce more identical stem cells”. This view fails to acknowledge that CSCs are stem cells of unicellular origin, fundamentally different from the more evolved multicellular stem cells found in humans and metazoans. Moreover, current CSC research often overlooks critical distinctions, such as the difference between proliferating ACD germline cells and non-proliferating CSCs, as well as between committed and not committed CSCs. Additionally, it tends to confuse CSC amplification through polyploidization and reductive nuclear division with mitotic proliferation. However, it is important to recognize that committed stem cells, like those in amoebae and cancer, are not inherently proliferative. Instead, uncommitted, quiescent CSCs can revert to proliferative, self-renewing germline cells, thereby reinforcing the germline that originally produced them (Figure 2 left).
4. Functional and Dysfunctional Germline States
As mentioned earlier, all germlines and clones are sensitive to oxygen. They can be damaged by germline hyperoxia, which occurs when oxygen levels exceed 6.0%, as typically found in blood and tissue [1]. Under g/hyperoxia conditions, the germline irreversibly loses its asymmetric cell division and stem cell potential due to severe DNA double-strand breaks (DNA DSB), and alterations in homologous recombination (HR) genes, leading to an overall genome dysfunctionality. Hyperoxic damage occurs when oxygen-sensitive germline cells and CSCs leave the hypoxic niche and migrate via the bloodstream into well-oxygenated tissues [12,23]. The increased oxygen pressure in these tissues and the bloodstream transforms ACD germ cells into an irreparably dysfunctional DSCD cell state, resulting in the cessation of CSC production.
4.1. The “Life Cycle of Stemness“
Naturally occurring DSCD phenotypes in cancer and protists, which lose function, do not initiate apoptosis programs. Instead, they cycle throug aberrant cell cycles and defective symmetric cell division. These DSCD phenotypes are tetraploid and exhibit cytokinetic failure due to both mature and immature nuclei in the same cell. They undergo depolyploidization and re-polyploidization cycles, alternating between tetraploidy and diploidy (4n > 2n > 4n) [1,12]. To restore functionality, these cells require repair via the MGRS/PGCC pathway.
Loss of stemness and irreparable germline dysfunctionality occur cyclically, both in tumorigenesis and in amoebiasis, the infectious disease caused by parasitic amoebae. The “life cycle of stemness“ (Figure 3) is a constant alternation between three distinct phases: a functional ACD germline phase from which stem cells arise, a cyst-free dysfunctional DSCD germline phase, and between an MGRS/PGCC repair phase, which reconstructs the functional germline genome with stemness, ACD potential, and CSC production. The „stemness cycle“ results in amoebiasis in cyst-positive and cyst-negative phases, during which the disease may be detectable or not by coprological analysis. In cancer, a similar alternation occurs between ACD phenotypes generating CSCs and DSCD phenotypes lacking stemness [1,12].
4.2. Contradictions to the“Life Cycle of Cancer”
The concept of the „life cycle of stemness” contradicts the “cancer life cycle“ hypothesis put forth by genotoxic PGCC research, which understands the cancer life cycle as an alternation of somatic cell cycles and polyploidization cycles by the germline (Figure 4).
According to the ECCB, the direct transition from mitotic cell cycles to germline ploidy cycles and hyperpolyploidization (Figure 4) does not occur from somatic cells [38] but exclusively in germline cells, either from committed CSCs, which then enter cyst-like polyploidization cycles or (ii) from defective DSCD cells that required genomic repair via the polyploid/hyperpolyploid MGRS/PGCC pathway. Cells that enter the MGRS/ PGCC pathway are already germline DSCD cells, but not necessarily senescent or arrested in mitosis, as claimed by genotoxic PGCC research [38].
In a recent reassessment of their concept of the “cancer life cycle,” the above researchers agreed that “mitotic cancer cells enter accelerated senescence caused by oncogenic, genotoxic, or oxidative stress” [39] and hypothesized that “DNA damage associated with senescence activates the DNA damage response (DDR)”, leading to germline-related ploidy cycles. ECCB’s perspective diverges from this view. First, it argues that DSCD cells requiring MGRS/PGCC repair, possess the capability for unlimited proliferation without triggering senescence programs. These cells can proliferate indefinitely through symmetric cell cycles and aberrant mitoses, resulting in tetraploidy, binucleation, and immature nuclei. Therefore, DDR-induced MGRS/PGCC repair is less associated with senescence, but primarily with the intrinsic abnormality of DSCD cells that transforms them into fusogenic cells.
5. Heterogeneity in Tumors: Heterogenous ACD-Lineages and Heterogenous CSCs
As previously mentioned, ECCB distinguishes between the primary CSCs of the native ACD-lineage and secondary CSCs from metastatic ACD-lineages.
The notion of “ACD-lineages“ was introduced as a replacement for “CSC-lineages“ because it more clearly indicates that CSCs appear as non-proliferative products of germline sublines and clones and are the differentiation products of productive ACD germline phenotypes. This also allows for better distinction between the primary ACD lineage of the primary tumor and the multitude of secondary ACD lineages in later tumors and metastases.
In contrast to the primary ACD-lineage, secondaryACD-lineages arise from the heterotypic fusion of cancer cells with non-cancerous somatic cells, followed by fractal processes of soma-to-germ transitions (SGT), commonly referred to as epithelial-mesenchymal transition (EMT). By differentiating between primary ACD-lineagen and primary CSCs, secondaryACD-lineagen, and secondary CSCs - as outlined in Table 1 - ECCB offers a clearer understanding of the heterogeneity and origin of cancer stem cells and cancer stem-like cells.
5.1. The Primary ACD-Lineage, Primary CSCs and Replacement CSCs
According to ECCB, the primary ACD-lineage of cancer originates from a dysfunctional non-cancerous DSCD cell that requires repair through the unicellular MGRS/PGCC pathway. This atypical repair process results in the formation of spores with carcinogenic potential, which act as progenitors for the native ACD-lineage, ultimately giving rise to the primary tumor. The primary ACD germline proliferates through ACD cell cycles, generating primary CSCs (pCSCs) as long as environmental factors support its hypoxic proliferation (Figure 3).
When the primary ACD phenotype becomes dysfunctional and transits to a to a DSCD phenotype, the depletion of CSCs and associated signaling factors trigger a soma-to-germ transition (SGT/EMT) from the primary somatic cell line. This transition regenerates germline sublines from the oxygen-resistant somatic sister line that has preserved the primary germline genome. Primary SGT processes, which form replacement sublines for the now dysfunctional maternal germline, are essential to ensure the production of CSCs.
5.2. Secondary ACD-Lineages and Secondary CSCs
As cancer progresses, primary somatic cells can fuse with non-cancerous host cells, such as macrophages, enriching the primary germline genome with active multicellular genes (MGs) acquired through heterotypic cell fusion. This fusion process leads to the formation of secondary sublines and clones through secondary SGT/EMT. In amoebae, SGT processes occur as a continuum, whereas in cancer, EMT can be fractal, forming multiple germline sublines and clones and multiple secondary ACD-lineages with very different secondary CSCs.
Alongside the existing primary ACD germline and its sublines, which continue to produce primary CSCs, multiple secondary ACD-lineages give rise to numerous secondary CSCs fractions and subpopulations that exhibit enhanced anti-host capabilities. These alternative mechanisms of CSC formation could contribute to a better understanding of the heterogeneous nature of cancer stem cells and stem-like cell generations as the so-called “somatic cell dedifferentiation” process. [13]. Each instance of heterotypic cell fusion with non-cancerous host cells and fractal SGT/EMT processes potentially contributes to the expansion of the germline genome, increasing CSC heterogeneity, potency, and resistance to antitumor therapies.
6. The Two Phases of the MGRS/PGCC Repair Process
As previously described, dysfunctional tetraploid DSCD germlines express stemness and differentiation potential, traits exclusively exhibited by the “healthy” diploid ACD phenotype. Instead, non-senescent DSCD cell lines persist in aberrant symmetric cell proliferation and accumulate DSCD populations. This symmetrical cell accumulation increases the likelihood of homotypic cell fusion, which triggers the biphasic MGRS/PGCC repair process forming viable spores, which give rise to new functional ACD-lineages[1,12].
6.1. Phase 1: It Is the Phase of Multiple Defective Cyst-like Polyploidization- Depolyploidization Cycles
Following cell fusion, each of the defective DSCD nuclei within the MGRS/PGCC structure undergoes a cyst-like polyploidization-depolyploidization cycle, which serves to increase the DNA content within the MGRS/PGCC structure [12,23]. Unlike the functional polyploidization-depolyploidization cycle observed in cysts of Entamoeba (Figure 5), which ultimately produces 8-16 functional daughter nuclei capable of generating germlines, germline clones, and stem cells, the daughter nuclei resulting from this first MGRS/PGCC phase remain dysfunctional and require further repair. Conventional repair mechanisms, such as homologous recombination (HR), are insufficient to correct the DNA double-strand break (DNADSB) damage present in the embedded DSCD nuclei [12,23].
6.2. Phase 2: Is the Phase of Defective Nuclear Fusion and the Formation of Hyperpolyploid Giant Nuclei
In this phase, the defective nuclear progeny fuse, leading to the formation of high-grade hyperpolyploid nuclei with an extensive DNA mass. These giant nuclei possess the capability to eliminate DNA DSB fragments and to reconstitute the functional germline genome architecture, and HR genes (homologous recombination gene) function. The MGRS structures themselves acquire stemness potential and pass it on to the viable spores they generate.
7. Native PGCCs –the Multinucleated MGRS’ of Cancer
Native PGCCs resulting from cell-to-cell fusion are integral to the life cycle of stem cells, which are in constant flux within cancer and tumors (Figure 3). Homologous to the MGRS of protists, PGCCs play a crucial role in reconstructing the functional genome of the germline and regenerating the ACD germline phenotype, which, in turn, generates CSCs.
There is limited information available about native PGCCs, which arise during malignancy, early carcinogenesis, and within primary tumors, yet they are hard to detect until cancer is already suspected. Native PGCCs play a crucial role in cancer progression by continually repairing newly emerging DSCD cells induced by germline hyperoxia in blood and tissue, thereby contributing to new cancer stem cells (CSCs).
For a long time, the homology between amoeba MGRS and the native PGCC structures of cancer was not known. Therefore, the only viable method for gaining insight into the role and function of PGCCs - occasionally observed in tissue and cancer cell cultures - has been through induction using irradiation, chemotherapeutic agents, and stress factors such as cobalt chloride (CoCl2) [40,41,42]. In particular, CoCl2 appears to create hypoxic conditions that induce oxygen-damaged DSCD cells to enter the MGRS/PGCC pathway
However, the results and characteristics of genotoxically induced mononucleated PGCCs can only be extrapolated to native PGCCs to a limited extent. First, the effects of genotoxic treatments are significantly more severe than the “natural“ hyperoxic stress caused by oxygen levels exceeding 6.0% outside the hypoxic niche. Secondly, the native PGCCs remain largely unexplored and have not been adequately categorized within the cancer development process. These limitations have led to assumptions that may not always be accurate.
There are many unresolved questions regarding native PGCCs. One critical area of inquiry is whether a causal relationship exists between native PGCCs and aneuploidy. While significant evidence suggests a link, many aspects remain unclear. Key questions include: (i) Could some DSCD cells, which have suffered hyperoxic DNAD DSB damage, be predisposed to aneuploidy? (ii) Could some genomic defects, which cannot be fully repaired by the PGCC process, result in the formation of aneuploid-favoring spores and clones, thereby perpetuating aneuploid cell cycling? Or (iii) could PGCCs themselves generate aneuploidy? These questions are crucial for understanding the possible role of native PGCCs and the development of cancer aneuploidy.
Further questions include: Do native PGCCs arise solely from homotypic DSCD cell fusions, or can they also arise from different primary and secondary germline cells? Understanding whether native PGCCs can also emerge from such heterotypic fusions would provide deeper insights into the role and genesis of native PGCCs in cancer development. Such hypotheses need to be rigorously tested using biopsy samples from untreated cancers.
8. Uninucleated, Genotoxic- Induced PGCCs
Unlike native PGCCs, which naturally arise during carcinogenesis and cancer progression, genotoxic induced uninucleated PGCC structures resulting from irradiation and chemotherapeutics require significantly more time to initiate polyploidization and depend on the assistance of nursing cells [23].
8.1. Consequences of the Genotoxic Damage: A Unique Amplification Cycle and High Ranges of Hyperpolydization
In contrast to native PGCCs, which contain multiple DSCD nuclei and undergo several polyploidization-depolyploidization cycles, genotoxic induced PGCCs undergo a singular, cyst-like polyploidization cycle. By the end of phase 1, the number of daughter nuclei and the overall DNA mass are significantly lower than those in native PGCCs. The daughter nuclei produced at the end of phase 1 are homogenous to the damaged parent nucleus, thus continuing to harbor the same DNA defects.
During the subsequent phase 2, genotoxic induced PGCCs achieve extreme levels of hyperpolyploidy (up to 380n) and accumulate the critical DNA mass necessary for genome repair. However, the outcomes of this PGCC repair process remain genomically unclear. Numerous studies suggest that the final spore progeny are heterogeneous, with persisting genomic damage, such as aneuploidy.
8.2. Accelerated PGCC Senescence and Senescent DSCD Cells
The few cancer cells that survive genotoxic insults appear to undergo a prolonged adaptation, contrasting with the rapid MGRS structures observed in amoebae. In amoebae, the MGRS process occurs from tetraploid or multinucleated DSCD cells grown in synthetic, nutrient-deficient media [18,19]. It can be induced in young and middle-aged DSCD cells, but never in senescent DSCDs [17].
In contrast, the delay in polyploidization observed in living, genotoxically treated cancer cells has been interpreted by some researchers as a phase of accelerated senescence. This interpretation is based on the assumption that cells capable of genome repair through ploidy cycles and hyperpolyploidization must bypass senescence, with PGCCs potentially shortening and accelerating the senescence process [38,39]. However, according to ECCB, this statement is not applicable. The delay is more likely due to the extent of cell damage that must be resolved before the cyst-like amplification cycle (phase 1) can be initiated.
8.3. Recent Statements from Genotoxic Cancer Cell Research
Despite the limited understanding of the natural MGRS/PGCC pathway, recent years have seen the publication of numerous excellent reviews by the research group of Jekaterina Erenpreisa and even a book by Brazilian researchers that comprehensively describes the results of genotoxic-induced PGCC research. [43].
Over the years, genotoxic research on cancer cells has shown that PGCCs are responsible for extensive genomic restructuring, leading to tumor-initiating cells in response to stress. In 2016, Niu et al. [44] proposed that the giant cell cycle is a source for mitotically competent tumor-initiating cell production and provides genome instability. In 2018-2019, researchers emphasized that giant cancer cells are unrecognized triggers of tumorigenesis, metastasis, and therapy resistance [45,46,47].
According to a review by Ammend et al. [45], PGCCs occurred in the hypoxic environment of primary tumors as a response to therapy. The progeny of these PGCCs exhibit CSC properties and can repopulate the tumor. The proportion of giant cells can significantly increase in response to genotoxic stress, and PGCCs can develop heightened metastatic and invasive potential. The clinicopathological significance of PGCCs has been examined by many researchers [48,49], as well as their role as circulating cancer cells [50], which is associated with tumor grading and metastasis [46]. PGCCs occurs by stress [40,51], and the PGCC phenotypes that emerge during cancer development are various [52,53].
9. Convergences and Controversies between ECCB and Current Cancer Research
Obviously, some recent statements from genotoxic PGCC research are consistent with the findings of the ECCB, but others are not. In particular, ECCB considerations on the compartmentalized genome, ancestral genome modules, and regulation by hypoxic/hyperoxic environments are now shared by both research fields. The author of this paper, who reported on the progress of the emerging ECCB field in 2018/2019, contributed to this alignment by revealing the that all eukaryotes, from protists to mammals, retain the genome of their unicellular common AMF ancestor within their ancestral genome compartments [54,55,56]. This ancestral genome can be reactivated under harmful environmental conditions, such as oxygenic stress and germline hyperoxia
The primary controversy between the ECCB (which advocates for cancer as a unicellular cell system), and genotoxic-driven PGCC research revolves around whether the cancer cell system is fundamentally a unicellular cell system or a result of aberrations within the multicellular cell system. While it is well-established that polyploidy, hyperpolyploidy, and PGCCs are not associated with multicellularity, current PGCC research asserts that CSCs originate from multicellular adult stem cells or embryonic cell stages. Current opinion attributes CSC formation to epigenetic changes, mutations, DNA damage, and altered multicellular gene activity. In contrast, ECCB posits that the potential unicellular cancer genome is preserved within the ancestral genome compartment of humans and metazoans, existing in a reactivatable silent state. According to this view, the reactivation of this genome originates from non-cancerous DSCD cells. [56,57].
Recent hypotheses emerging from genotoxic-induced PGCC research continue to view cancer as a disease within the multicellular framework, suggesting that it does not necessarily involve a transition to a complete unicellular cell system. In contrast, the ECB relies on extensive evidence and proposes that spontaneous solid cancers are due to an irreversible switch to an extensive pre-metazoan cell system that evolved through the common AMF ancestor. According to the ECCB, this transition to a unicellular lifestyle occurs through the loss of asymmetric cell division capacity and stemness potential, cessation of CSC production, and activation of a natural DSCD program within the multicellular cell system.
In contrast to the ECCB, the conclusions drawn from current cancer research remain ambiguous. Although they describe various evolutionary links between cancer origins and processes such as mammalian embryogenesis, sporulation in protozoa, gametogenesis and embryogenesis in insects, or PGCC reproduction [38], these connections primarily underscore the deep homology of cancer to a common ancestor and its evolutionary branches. These homologies remember distant cousins who have inherited certain traits from a shared ancestor but are no longer closely related.
As a result, current statements on cancer origins do not offer a viable alternative to the ECCB framework. They fall short of providing a deeper understanding of cancer, do not clarify how or from what cancer originates, and fail to illuminate what cancer fundamentally is.
10. Is Cancer Unicellular or Multicellular?
The debate about whether cancer takes over a unicellular cell system or remains multicellular is of central importance for the understanding of cancer. The multicellular cancer concept views carcinogenesis as a series of aberrations within the multicellular system, linking it to processes such as cell plasticity, epigenomic alterations, disrupted differentiation, and the rewiring of gene regulatory networks (GRNs) [38]. In this view, the multicellular GRN functions as a regulator that strives for equilibrium with its environment, which can adopt a stable, pre-programmed configuration via a “pre-programmed attractor” [38,58]. This “attractor” can be likened to a software application, programmed during early phylo-ontogenetic evolution, which can be activated under stress to reprogram the genome. In this sense, cancer ultimately represents an imbalance in multicellular gene expression: older genes of unicellular origin tend to be overexpressed, while newer genes from more recent phylostates, which are responsible for multicellular evolution, are underexpressed. These changes in gene expression patterns cause cancers to adopt phenotypes that otherwise only occur in unicellular organisms.
The hypothesis above is supported by phylostratigraphy and phylogenetic studies that trace the origins of cancer genes to the transitional period from unicellularity to multicellularity. However, critics argue that this perspective focuses on a time that is far too late to fully explain the fundamental nature of cancer [59,60,61,62,63,64]. Older concepts emphasize the regulation of genome expression and the self-organization of gene expression during tissue and organ differentiation [38,39]. This hypothesis suggests that changes in cell fate, such as those occurring during the differentiation of highly specialized cell types, can also lead to the formation of dysfunctional tumors under unusual environmental stress.
Recently, Erenpreisa et al. came even closer to the ECCB framework when they said that “cancer cells can adapt to unforeseen environmental challenges through exploration by trial and error, existing at the edge between order and chaos”. [65] When confronted with potentially lethal damage, cells scan their gene networks, revisiting hidden transcriptional configurations preserved in the mammalian genome, reflecting 3.5 billion years of cellular evolution“. This view draws an analogy to the ECCB in its approach to carcinogenesis, particularly in the interplay between unicellularity and multicellularity during the transition period. Erenpreisa and colleagues [66] describe this period as involving “vestigial transcriptional programs” or “predetermined chaos,” which may be the most effective strategy for facilitating the rare escape of “lucky” survivors from near-lethal damage. The forward motion is uncertainty, fluctuations, and a duality of opposites engaged in an intensive “dialogue with the environment”.
The underlying question remains: Is cancer complete access to the unicellularity of the ancestral genome compartment, or is it an intrinsic flaw within the multicellular system itself? ECCB provides substantial arguments supporting a complete change to a unicellular cell system.
11. Conclusions and Perspectives
From an evolutionary perspective, cancer represents a transition to a lower system of cellular organization, marking a switch from multicellularity to unicellularity. This switch occurred frequently during the transition period to multicellularity, as early multicellular organisms oscillated between both alternatives in response to intrinsic or extrinsic factors. All multicellular organisms, including humans and mammals, maintain this unicellular cell system - derived from the AMF ancestor - in their ancestral genome compartment, in a state of responsiveness. This explains why cancer has a different stem cell biology than the host organism.
According to the ECCB framework, cancer stem cells (CSCs) are non-proliferative cells that result from asymmetric cell division but are sister cells of self-renewing germline cells. ECCB distinguishes between two types of CSCs: non-committed quiescent CSCs, which have the potential to revert into self-renewing germline cells, and committed CSCs (genuine CSCs), which are capable of amplification through cyst-like polyploidization – depolyploidization cycles (but not for proliferation).
The ECCB considers self-renewing germline and cancer stem cells (CSCs) to be an interchangeable entity termed ACD-lineage. In this model, self-renewing ACD cells generate CSCs, and CSCs generate germline sublines and clones either by self-renewing conversion or through an accumulation of progenitor cells This dynamic interplay is of fundamental importance for an understanding of the role and behavior of CSCs in cancer cell biology.
The multitude of secondary ACD-lineages including secondary CSCs in older tumors and metastases can better explain the heterogeneity of the stem cell population than the current notion of CSC-lineages. It shows that the generation of heterogenous non-proliferative CSCs lineages depends on the numerous germline sublines and clones, which in turn rely on the variety of heterotypic cell-to-cell fusions with non-cancerous somatic cells and subsequent fractal SGT/EMT processes.”
Stemness is a hallmark of each ACD-lineage. It can be lost but regained through the MGRS/PGCC repair pathway. The DSCD phenotype, which requires repair, does not exhibit stemness. This distinction highlights the unique capacity of the MGRS/PGCC pathway to reestablish stemness.
Multinucleated MGRS’ and native PGCCs are hyperpolyploid genome repair structures formed through homotypic cell fusion. These structures are distinct from genotoxically induced PGCCs, which arise following irradiation or treatment with chemotherapeutic agents. MGRS were first observed in protists as early as 1906 [67]. Unaware of these early findings, the term “neosis”, was introduced in 2008 to describe PGCCs as a new type of asymmetric cell division [68,69]. Unfortunately, this was an error: PGCCs are not proliferative and do not undergo either proliferation or asymmetric cell division. Instead, they produce spores through cyst-like genome accumulation and hyperpolyploidization.
The ECCB framework further distinguishes between primary CSCs and secondary CSCs, i.e. primary ACD-lineages and secondary ACD-lineages. Primary CSCs are both carcinogenic and tumorigenic, arising either post-malignancy from the primary ACD-lineage or within tumors through native PGCCs processes. Secondary ACD-lineages with secondary CSCs, on the other hand, are initiated by heterotypic somatic cell fusion and fractal SGT/EMT processes, as before related. The resulting secondary CSCs incorporate functional multicellular genes into the unicellular cancer genome, forming new germline clones capable of producing metastatic secondary CSCs with higher invasive potential. This distinction highlights the different origins and roles of primary and secondary CSCs in cancer development and progression, with secondary CSCs playing a critical role in enhancing the metastatic capabilities of cancer.
In summary, a deeper understanding of cancer stem cell biology is crucial for advancing experimental cancer research. The novel insights into the biology of cancer stem cells described in the present study could reveal new molecular targets in the fight against cancer. This is particularly important for improving our understanding of the surface antigenicity of the cell of origin and other cells closely associated with transformation and carcinogenesis, such as non-cancerous DSCD cells and tumor DSCDs. Investigating the ACD-DSCD-MGRS sequence for antigenicity, carcinogenic potential, and surface markers could provide valuable new research data. These findings may ultimately contribute to the development of an anti-cancer vaccine.
Abbreviations
ACD, asymmetric cell division; aCLS,amplifying cyst-like structure; aGRN, ancestral gene regulatory network; AMF, amoebozoans, metazoans and fungi; ACD, asymmetric cell division, CSCs, cancer stem cells; DSCD, dysfunctional symmetric cell division;; MGRS, multinucleated genome repair syncytia; PGCC, polyploid giant cancer cell; SGT, soma-to-germ transition;
References
- Niculescu, V.F. Understanding cancer from an evolutionary perspective: high-risk reprogramming of genome-damaged stem cells. Academia Medicine. 2024, 2. [Google Scholar] [CrossRef]
- Stem cells and cancer stem cells: therapeutic applications inn isease and injury. 2013, 9 Hayat MA ed. Springer. eISSN: 2215-1001.
- Rossi, F.; Noren, H.; Jove, R.; et al. Differences and similarities between cancer and somatic stem cells: therapeutic implications. Stem Cell Res Ther 2020, 11, 489. [Google Scholar] [CrossRef] [PubMed]
- Nimmakayala, R.K.; Batra, S.K.; Ponnusamy, M.P. Unraveling the journey of cancer stem cells from origin to metastasis. Biochim Biophys Acta Rev Cancer. 2019, 1871, 50–63. [Google Scholar] [CrossRef] [PubMed]
- Rycaj, K.; Tang, D.G. Cell-of-origin of cancer versus cancer stem cells: assays and interpretations. Cancer Res. 2015, 75, 4003–4011. [Google Scholar] [CrossRef] [PubMed]
- Shah, M.; Allegrucci, C. Stem cell plasticity in development nd cancer: epigenetic origin of cancer stem cells. Subcell Biochem. 2013, 61, 545–565. [Google Scholar] [CrossRef]
- Tweedell, K.S. The Adaptability of Somatic Stem Cells: A Review. J Stem Cells Regen Med. 2017, 13, 3–13. [Google Scholar] [CrossRef]
- Spradling, A.; Drummond-Barbosa, D.; Kai, T. Stem cells find their niche. Nature. 2001, 414, 98–104. [Google Scholar] [CrossRef]
- Sell, S. Stem cell origin of cancer and differentiation therapy. Crit Rev Oncol Hematol. 2004, 51, 1–28. [Google Scholar] [CrossRef]
- Scadden, D.T. The stem-cell niche as an entity of action. Nature. 2006, 441, 1075–1079. [Google Scholar] [CrossRef]
- Spillane, J.B.; Henderson, M.A. Cancer stem cells: a review. ANZ J Surg. 2007, 77, 464–468. [Google Scholar] [CrossRef] [PubMed]
- Niculescu, V.F.; Niculescu, E.R. The Enigma of Cancer Polyploidy as Deciphered by Evolutionary Cancer Cell Biology (ECCB). Academia Medicine. 2024. [CrossRef]
- Loh, J.J.; Ma, S. Hallmarks of cancer stemness. Cell stem cell. 2024, 31, 617–639. [Google Scholar] [CrossRef]
- Niculescu, V.F. aCLS cancers: Genomic and epigenetic changes transform the cell of origin of cancer into a tumorigenic pathogen of unicellular organization and lifestyle. Gene. 2020, 726, 144174. [Google Scholar] [CrossRef]
- Niculescu, V.F. Growth of Entamoeba invadens in sediments with metabolically repressed bacteria leads to multicellularity and redefinition of the amoebic cell system. Roum Arch Microbiol Immunol. 2013, 72, 25–48. [Google Scholar] [PubMed]
- Niculescu, V.F. The stem cell biology of the protist pathogen Entamoeba invadens in the context of eukaryotic stem cell evolution. Stem Cell Biology and Research 2015. [Google Scholar] [CrossRef]
- Krishnan, D.; Ghosh, S.K. Cellular Events of Multinucleated Giant Cells Formation During the Encystation of Entamoeba invadens. Front Cell Infect Microbiol 2018, 8, 262. [Google Scholar] [CrossRef]
- Mukherjee, C.; Clark, C.G.; Lohia, A. Entamoeba shows reversible variation in ploidy under different growth conditions and between life cycle phases. PLoS Negl Trop Dis. 2008, 2, e281. [Google Scholar] [CrossRef]
- Mukherjee, C.; Majumder, S.; Lohia, A. Inter-cellular variation in DNA content of Entamoeba histolytica originates from temporal and spatial uncoupling of cytokinesis from the nuclear cycle. PLoS Negl Trop Dis. 2009, 3, e409. [Google Scholar] [CrossRef] [PubMed]
- Hazra, S.; Kalyan Dinda, S.; Kumar Mondal, N.; Hossain, S.R.; Datta, P.; et al. Giant cells: multiple cells unite to survive. Front Cell Infect Microbiol 2023, 13, 1220589. [Google Scholar] [CrossRef]
- Niculescu, V.F. The cell system of Giardia lamblia in the light of the protist stem cell biology. Stem Cell Biol Res. 2014, 1, 3. [Google Scholar] [CrossRef]
- Niculescu, V.F. Evidence for asymmetric cell fate and hypoxia induced differentiation in the facultative pathogen protist Colpoda cucullus. Microbiol Discov. 2014, 2, 3. [Google Scholar] [CrossRef]
- Salmina, K.; Huna, A.; Kalejs, M.; Pjanova, D.; Scherthan, H.; et al. The Cancer Aneuploidy Paradox: In the Light of Evolution. Genes. 2019, 10, 83. [Google Scholar] [CrossRef] [PubMed]
- Illidge, T.; Cragg, M.; Fringe, B.; Olive, P.; Erenpreisa, J.E. Polyploid giant cells pro- vide a survival mechanism for p53 mutant cells after DNA damage. Cell Biol Int. 2000, 24, 621e33. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, A.; Cragg, M.S.; Erenpreisa, J.; Emzinsh, D.; Lukman, H.; Illidge, T.M. Endo- polyploid cells produced after severe genotoxic damage have the potential to repair DNA double strand breaks. J Cell Sci. 2003, 11, 4095e106. [Google Scholar] [CrossRef]
- Chen, Q.; Zou, J.; He, Y.; Pan, Y.; Yang, G.; et al. A narrative review of circulating tumor cells clusters: A key morphology of cancer cells in circulation promote hematogenous metastasis. Front. Oncol. 2022, 12, 944487. [Google Scholar] [CrossRef]
- Gema, M–B.; Héctor, P.; Patricia, M.; David, O.; Eva, C.; Vanesa, S.; et al. The morphological and molecular features of the epithelial–to–mesenchymal transition. Nat Protoc 2009, 4, 1591–1613. [Google Scholar] [CrossRef]
- Yang, J.; Antin, P.; Berx, G.; Blanpain, C.; Brabletz, T; et al. Author correction: Guidelines and definitions for research on epithelial–mesenchymal transition. Nat Rev Mol Cell Biol. 2021, 22, 834. [Google Scholar] [CrossRef]
- Hapach, L.; Carey, S.; Schwager, S.; Taufalele, P.; Wang, W.; et al. Phenotypic heterogeneity and metastasis of breast cancer cells. Cancer Res. 2021, 81, 3649–3663. [Google Scholar] [CrossRef]
- Derynck, R.; Turley, S.; Akhurst, RJ. TGFb biology in cancer progression and immunotherapy. Nat Rev Clin Oncol 2021, 18, 9–34. [Google Scholar] [CrossRef]
- Aceto, N.; Bardia, A.; Miyamoto, D.; Donaldson, M.; Wittner, B; et al. Circulating tumor cell clusters are oligoclonal precursors of breast cancer metastasis. Cell. 2014, 158, 1110–1122. [Google Scholar] [CrossRef]
- Amintas, S.; Bedel, A.; Moreau–Gaudry, F.; Boutin, J.; Buscail, L; et al. Circulating tumor cell clusters: United we stand divided we fall. Int J Mol Sci Int J Mol Sci. 2020, 21, 2653. [Google Scholar] [CrossRef]
- Murlidhar, V.; Reddy, R.; Fouladdel, S.; Zhao, L.; Ishikawa, M; et al. Poor prognosis indicated by venous circulating tumor cell clusters in early–stage lung cancers. Cancer Res. 2017, 77, 5194–5206. [Google Scholar] [CrossRef] [PubMed]
- Aceto, N. Bring along your friends: Homotypic and heterotypic circulating tumor cell clustering to accelerate metastasis. BioMed J. 2020, 43, 18–23. [Google Scholar] [CrossRef]
- Vetter, M.; Landin, J.; Szczerba, BM.; Castro–Giner, F.; Gkountela, S; et al. Denosumab treatment is associated with the absence of circulating tumor cells in patients with breast cancer. Breast Cancer Res. 2018, 20, 141. [Google Scholar] [CrossRef]
- Ting, D.; Wittner, B.; Ligorio, M.; Vincent Jordan, N.; Shah, A; et al. Single–cell RNA sequencing identifies extracellular matrix gene expression by pancreatic circulating tumor cells. Cell Rep 2014, 8, 1905–1918. [Google Scholar] [CrossRef] [PubMed]
- Niculescu, V.F. Introduction to Evolutionary Cancer Cell Biology (ECCB) and Ancestral Cancer Genomics. 2023. [Google Scholar] [CrossRef]
- Erenpreisa, J.; Salmina, K.; Anatskaya, O.; Cragg, M.S. Paradoxes of cancer: Survival at the brink. Semin Cancer Biol. 2022, 81, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Erenpreisa, J.; Giuliani, A.; Yoshikawa, K.; Falk, M.; Hildenbrand, G; et al. Spatial-Temporal Genome Regulation in Stress-Response and Cell-Fate Change. Int J Mol Sci. 2658; ;24. [Google Scholar] [CrossRef]
- Lopez-Sánchez, LM.; Jimenez, C.; Valverde, A.; Hernandez, V.; Peñarando, J; et al. CoCl2, a mimic of hypoxia, induces formation of polyploid giant cells with stem characteristics in colon cancer. PLoS One. 2014, 9, e99143. [Google Scholar] [CrossRef]
- Liu, Y.; Shi, Y.; Wu, M.; Liu, J.; Wu, H; et al. Hypoxia-induced polypoid giant cancer cells in glioma promote the transformation of tumor-associated macrophages to a tumor-supportive phenotype. CNS Neurosci Ther. 2022, 28, 1326–1338. [Google Scholar] [CrossRef]
- Liu Zhang, S.; Zhang, D.; Yang, Z.; Zhang, X. Tumor Budding, Micropapillary Pattern, and Polyploidy Giant Cancer Cells in Colorectal Cancer: Current Status and Future Prospects. Stem Cells Int. 2016, 4810734. [Google Scholar] [CrossRef]
- PGCC: uma resposta complexa do câncer. Casotti- Correia M, Louro- Drumond I, Dummer-Meira D, editors. Atena Editura, 2023. [CrossRef]
- Niu, N.; Zhang, J.; Zhang, N.; et al. Linking genomic reorganization to tumor initiation via the giant cell cycle. Oncogenesis 2016, 5, e281. [Google Scholar] [CrossRef]
- Amend, SR.; Torga, G.; Lin, KC.; Kostecka, LG.; de Marzo, A; et al. Polyploid giant cancer cells: Unrecognized actuators of tumorigenesis, metastasis, and resistance. Prostate, 1489. [Google Scholar] [CrossRef]
- Mirzayans, R.; Andrais, B.; Murray, D. Roles of Polyploid/Multinucleated Giant Cancer Cells in Metastasis and Disease Relapse Following Anticancer Treatment. Cancers (Basel). 2018, 10, 118. [Google Scholar] [CrossRef]
- Fei, F.; Zhang, D.; Yang, Z.; Wang, S.; Wang, X; et al. The number of polyploid giant cancer cells and epithelial-mesenchymal transition-related proteins are associated with invasion and metastasis in human breast cancer. J Exp Clin Cancer Res. 2015, 34, 158. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Yang, X.; Yang, Z.; Fei, F.; Li, S; et al. Daughter Cells and Erythroid Cells Budding from PGCCs and Their Clinicopathological Significances in Colorectal Cancer. J Cancer. 2017, 8, 469–478. [Google Scholar] [CrossRef]
- Saini, G.; Joshi, S.; Garlapati, C.; Li, H.; Kong, J; et al. Polyploid giant cancer cell characterization: New frontiers in predicting response to chemotherapy in breast cancer. Semin Cancer Biol. 2022, 81, 220–231. [Google Scholar] [CrossRef] [PubMed]
- Ali, AM.; BenMohamed, F.; Decina, A.; Mukherjee, S.; Levi, S; et al. Circulating cancer giant cells with unique characteristics frequently found in patients with myelodysplastic syndromes (MDS). Med Oncol. 2023, 40, 204. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Zhao, Y.; Deng, Z.; Zhao, R.; Huang, Q. Stress-Induced Polyploid Giant Cancer Cells: Unique Way of Formation and Non-Negligible Characteristics. Front Oncol. 2021, 11, 724781. [Google Scholar] [CrossRef]
- Xuan, B.; Ghosh, D.; Dawson, M.R. Contributions of the distinct biophysical phenotype of polyploidal giant cancer cells to cancer progression. Semin Cancer Biol. ;81. [CrossRef]
- White-Gilbertson, S.; Voelkel-Johnson, C. Giants and monsters: Unexpected characters in the story of cancer recurrence. Adv Cancer Res. 2020, 148, 201–232. [Google Scholar] [CrossRef]
- Niculescu, V.F. Carcinogenesis: Recent Insights in Protist Stem Cell Biology Lead To a Better Understanding of Atavistic Mechanisms Implied in Cancer Development. MOJ Tumor Res 2018, 1, 00004. [Google Scholar] [CrossRef]
- Niculescu, V.F. The cancer stem cell family: atavistic origin and tumorigenic development. MOJ Tumor Res. 2018, 1, 70–73. [Google Scholar] [CrossRef]
- Niculescu, V.F. Cancer Stem Cells and the Unicellular Life Cycle of Cancer. Nov Appro in Can Study. NACS, 2019; 3. [Google Scholar] [CrossRef]
- Niculescu, V.F. The reproductive life cycle of cancer: Hypotheses of cell of origin, TP53 drivers and stem cell conversions in the light of the atavistic cancer cell theory. Med Hypotheses, 2019; 123, 19–23. [Google Scholar] [CrossRef]
- Huang, S. Reprogramming cell fates: reconciling rarity with robustness. Bioessays, 2009; 31, 546–560. [Google Scholar] [CrossRef]
- Domazet-Lošo, T.; Tautz, D. An ancient evolutionary origin of genes associated with human genetic diseases. Mol Biol Evol. 2008, 25, 2699–2707. [Google Scholar] [CrossRef] [PubMed]
- Domazet-Loso, T.; Tautz, D. Phylostratigraphic tracking of cancer genes suggests a link to the emergence of multicellularity in metazoa. BMC Biol 2010, 8. [Google Scholar] [CrossRef]
- Trigos, A.S.; Pearson, R.B.; Papenfuss, A.T.; Goode, D.L. Altered interactions between unicellular and multicellular genes drive hallmarks of transformation in a diverse range of solid tumors, Proc. Natl. Acad. Sci. U. S. A. 2017, 114, 6406–6411. [Google Scholar] [CrossRef] [PubMed]
- Trigos, A.S.; Pearson, R.B.; Papenfuss, A.T.; Goode, D.L. How the evolution of multicellularity set the stage for cancer, Br. J. Cancer 2018, 118, 145–152. [Google Scholar] [CrossRef]
- Trigos, A.S.; Pearson, R.B.; Papenfuss, A.T.; Goode, D.L. Somatic mutations in early metazoan genes disrupt regulatory links between unicellular and multicellular genes in cancer, Elife. 2019. [Google Scholar] [CrossRef]
- Lineweaver, C.H.; Davies, P. Comparison of the atavistic model of cancer to somatic mutation theory: phylostratigraphic analyses support the atavistic model, in: B.S. Gerstman (Ed.), The Physics of Cancer: Research Advances, World Scientific, 2020, pp. 243–261. [CrossRef]
- Erenpreisa, J.; Giuliani, A. Resolution of complex issues in genome regulation and Cancer Requires non-linear and network-based thermodynamics, Int. J. Mol. Sci. 2019, 21. [Google Scholar] [CrossRef]
- Erenpreisa, J.; Salmina, K.; Anatskaya, O.; Alexander Vinogradov, A.; Cragg, M.S. The Enigma of Cancer Resistance to Treatment. Organisms. 2022, 5, 2. [Google Scholar] [CrossRef]
- Craig, C.F. Studies upon the amebae in the intestine of man. J Infect Dis. 1908, 5, 324–377. [Google Scholar] [CrossRef]
- Sundaram, M.; Guernsey, D.L.; Rajaraman, M.M.; Rajaraman, R. Neosis: a novel type of cell division in cancer. Cancer Biol. Ther. 2004, 3, 207–218. [Google Scholar] [CrossRef]
- Rajaraman, R.; Guernsey, D.L.; Rajaraman, M.M.; Rajaraman, S.R. Neosis – a parasexual somatic reduction division in Cancer. Int. J. Hum. Genet. 2007, 7, 29–48. [Google Scholar] [CrossRef]
Figure 1.
The germ and soma life cycle of cancer and protists (Entamoeba) consists of a germline (green) and a somatic cell line (blue). The oxgen sensitive germline proliferates by asymmeric cell division (ACD) giving rise to unequal daughter cells: a cell that self-renews and a cell that exits the cell cycle and gives rise to committed and uncommitte stem cells (germline stem cells, cancer stem cells). Committed stem cells (precysts), which are capable differentiation and form cysts (Entamoeba) or cyst-like structures ( cancer), amplify their DNA content (cyst- or cyst-like ploidization) and form progenitor cells for new germline clones or subclones by reductive nuclear division and cellularization (depolyploidization). During cancer evolution, the unicellular cancer cell system generates clones and subclones via polyploidization-depolyploidization cycles, which give rise to a large number heterogeneous germline clones. In turn, that give rise to a large number of CSCs lineages. All CSCs are germline cells generated by more or less evolved ACD germline clones.
Figure 1.
The germ and soma life cycle of cancer and protists (Entamoeba) consists of a germline (green) and a somatic cell line (blue). The oxgen sensitive germline proliferates by asymmeric cell division (ACD) giving rise to unequal daughter cells: a cell that self-renews and a cell that exits the cell cycle and gives rise to committed and uncommitte stem cells (germline stem cells, cancer stem cells). Committed stem cells (precysts), which are capable differentiation and form cysts (Entamoeba) or cyst-like structures ( cancer), amplify their DNA content (cyst- or cyst-like ploidization) and form progenitor cells for new germline clones or subclones by reductive nuclear division and cellularization (depolyploidization). During cancer evolution, the unicellular cancer cell system generates clones and subclones via polyploidization-depolyploidization cycles, which give rise to a large number heterogeneous germline clones. In turn, that give rise to a large number of CSCs lineages. All CSCs are germline cells generated by more or less evolved ACD germline clones.
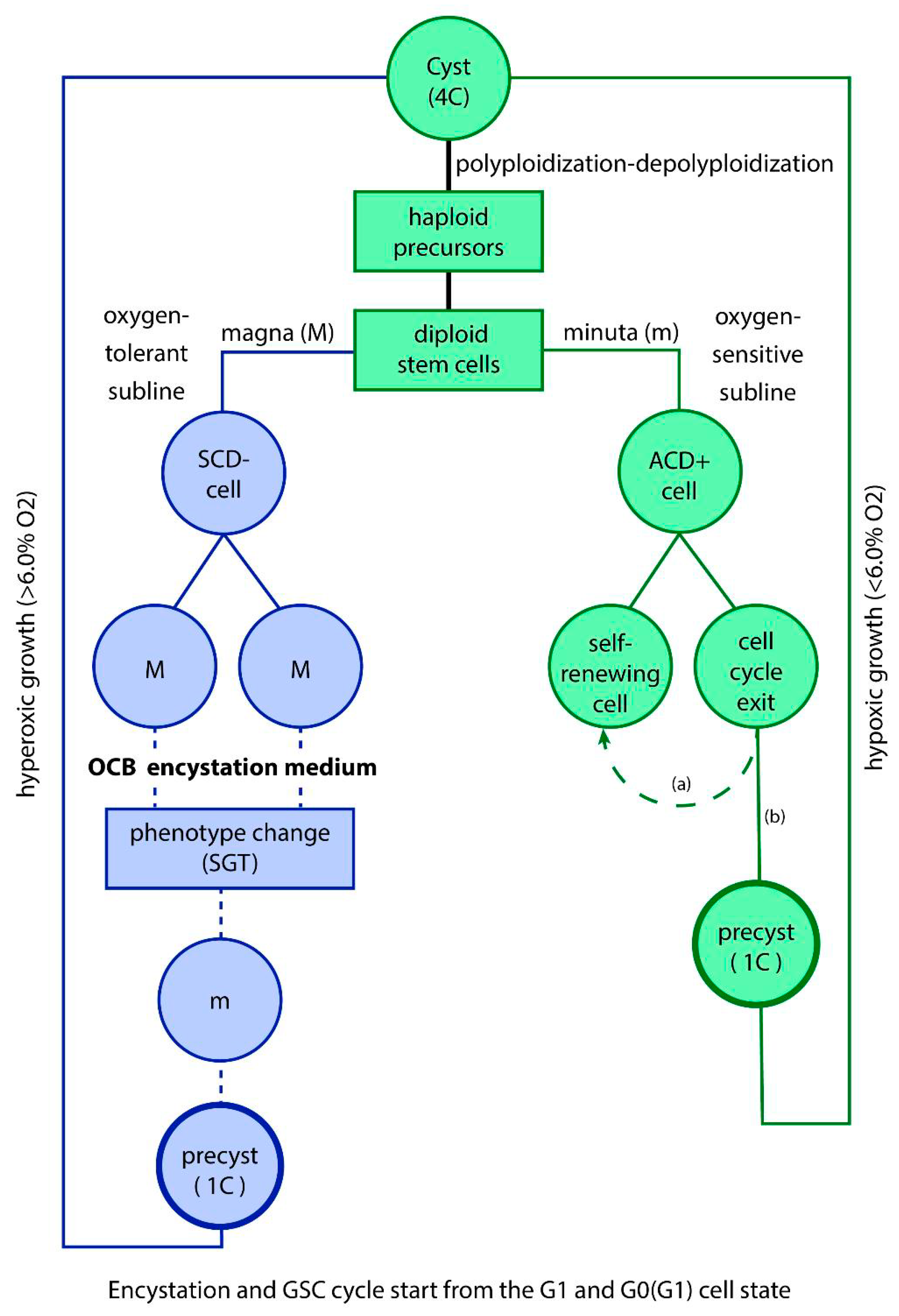
Figure 2.
Amplification by cyst-like polyploidization cycles. Both in cancer and protists, polyploidization and depolyploidization cycles have four distinct phases: (i) committed genome copying and polyploidization, (ii) reductive nuclear division, (iii) haploidization of the daughter nuclei, and (iv) diploidization to germline cells. Polyploidization is more effective than propagation by slowly proliferating ACD cell cycles. In Entamoeba histolytica / E. invadens it forms 4 polyploid nuclei, which give rise during ex-cystation to 16 haploid germline cell progenitors, and in E.coli to 8 polyploid nuclei, and 32 progenitors. (From V.F. Niculescu, Cancer genes and cancer stem cells in tumorigenesis: Evolutionary deep homology and controversies, Genes & Diseases, https://doi.org/10.1016/j.gendis.2022.03.010 (CC BY-NC-ND license).
Figure 2.
Amplification by cyst-like polyploidization cycles. Both in cancer and protists, polyploidization and depolyploidization cycles have four distinct phases: (i) committed genome copying and polyploidization, (ii) reductive nuclear division, (iii) haploidization of the daughter nuclei, and (iv) diploidization to germline cells. Polyploidization is more effective than propagation by slowly proliferating ACD cell cycles. In Entamoeba histolytica / E. invadens it forms 4 polyploid nuclei, which give rise during ex-cystation to 16 haploid germline cell progenitors, and in E.coli to 8 polyploid nuclei, and 32 progenitors. (From V.F. Niculescu, Cancer genes and cancer stem cells in tumorigenesis: Evolutionary deep homology and controversies, Genes & Diseases, https://doi.org/10.1016/j.gendis.2022.03.010 (CC BY-NC-ND license).
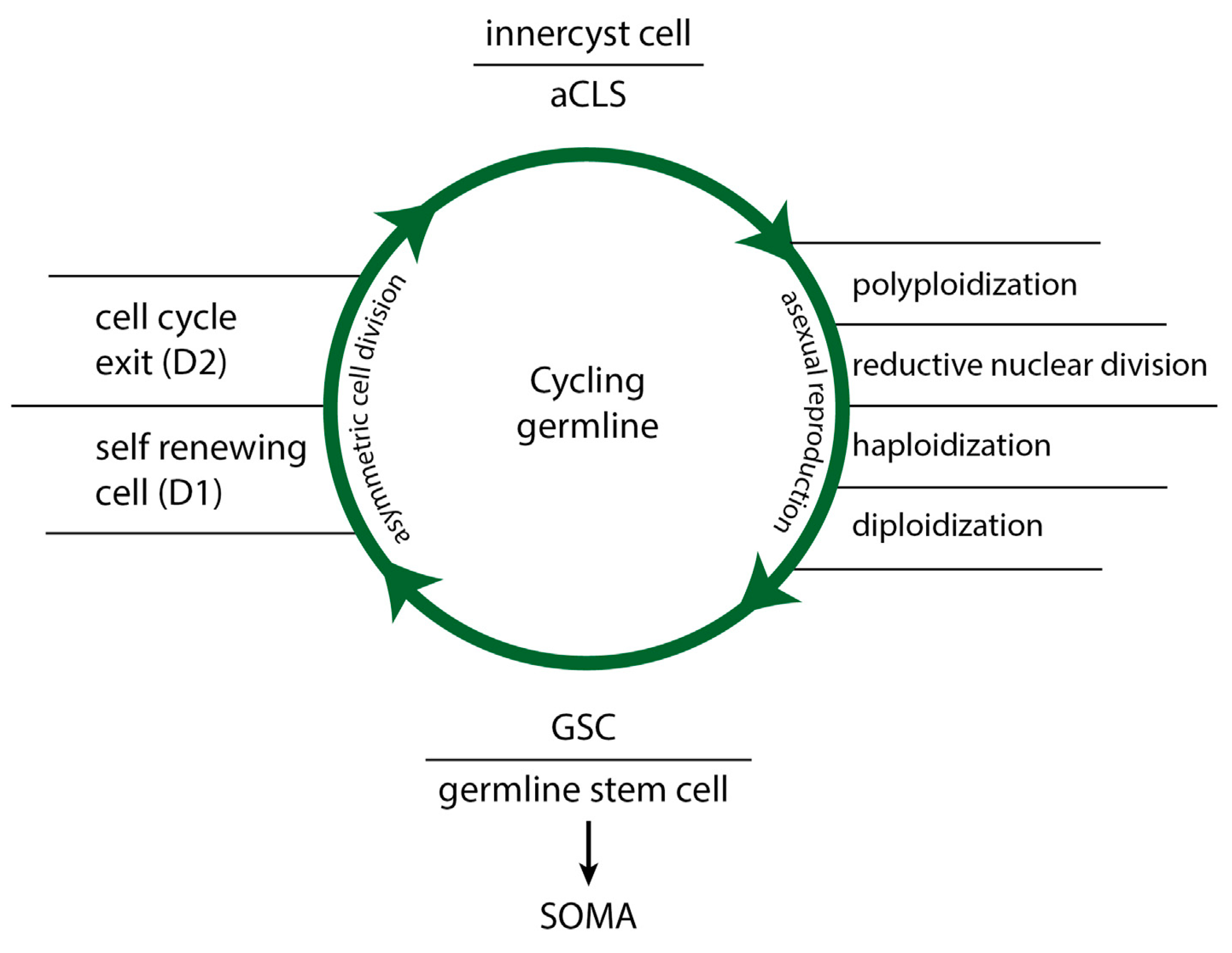
Figure 3.
The life cycle of stemness (stemness cycle) in cancer and protists: alternation of ACD and DSCD phenotypes and repair through the MGRS/PGCC pathway.
Figure 3.
The life cycle of stemness (stemness cycle) in cancer and protists: alternation of ACD and DSCD phenotypes and repair through the MGRS/PGCC pathway.
Figure 4.
The “cancer life cycle“) as an alternation of (i) mitotic cell cycles by the somatic cell line and (ii) polyploid amitotic cycles by the germline PGCC research (from Erenpreisa et al. doi: 10.1016/j.semcancer.2020.12.009 ;This figure was originally published under CC-BY.).
Figure 4.
The “cancer life cycle“) as an alternation of (i) mitotic cell cycles by the somatic cell line and (ii) polyploid amitotic cycles by the germline PGCC research (from Erenpreisa et al. doi: 10.1016/j.semcancer.2020.12.009 ;This figure was originally published under CC-BY.).
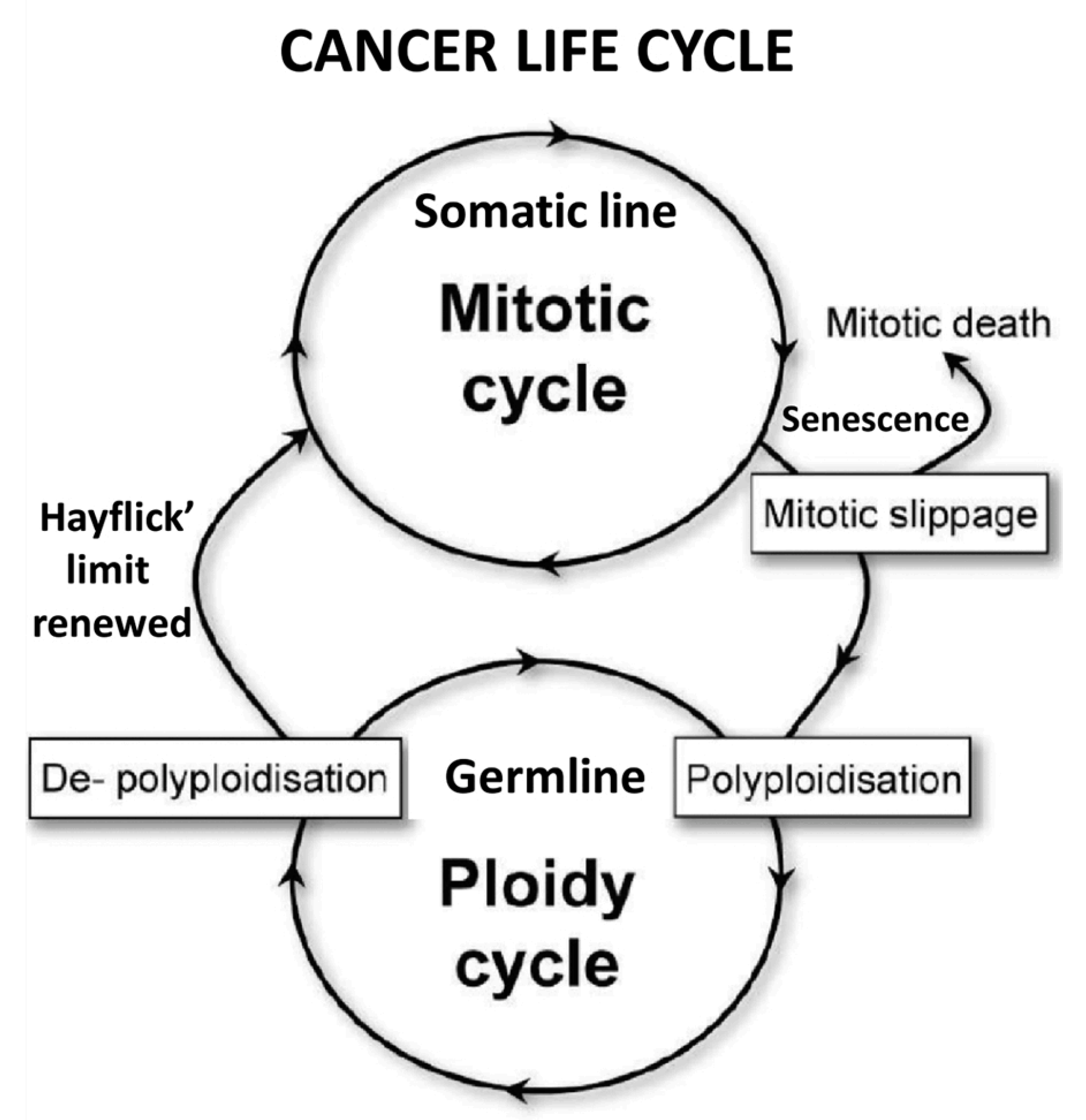
Figure 5.
Cyst of Entamoeba (left, A) with four functional tetraploid nuclei (10-20 mm) and a PGCC structure (right, B) , up to 10 times larger than the regular cancer cells capable of hyperpolyploidization up to 380n. https://media01.stockfood.com/largepreviews/ NDE4NzQxODYy/13507802-Cysts-of-Entamoeba-histolytica-protozoan-illustration.jpg; Credit: Science photo Library, Kon, Katerina (according to StockFood emailfrom 2024, August 13. Credit: National Cancer Institute , https://www.cancer.gov/types/breast/research.
Figure 5.
Cyst of Entamoeba (left, A) with four functional tetraploid nuclei (10-20 mm) and a PGCC structure (right, B) , up to 10 times larger than the regular cancer cells capable of hyperpolyploidization up to 380n. https://media01.stockfood.com/largepreviews/ NDE4NzQxODYy/13507802-Cysts-of-Entamoeba-histolytica-protozoan-illustration.jpg; Credit: Science photo Library, Kon, Katerina (according to StockFood emailfrom 2024, August 13. Credit: National Cancer Institute , https://www.cancer.gov/types/breast/research.

Table 1.
Functional and dysfunctional germline cels and stem cells in cancer and protists.
| Germline phenotypes | ||
| functional | dysfunctional | |
| ACD-phenotype | DSCD phenotype | |
| Proliferatiion | asymmetric cell division | dysfunctional symmetric cell division |
| Progeny | inequal daughter cells | equal daughter cells |
| (D1, self renewing, D2, stem cell) | (no stem cells) | |
| Commited stem cells | ||
| capable of differentiation or/and | ||
| cell amplification through | ||
| Stem cells | polyploidization-depolyploidization cycles; | |
| (non proliferating) | form haploid progenitors for | |
| new germline clones and stem cells (CSC) | ||
| Non-commited quiescent stem cells | ||
| capable to transform | ||
| into self renewing germline cells, | ||
| which continue ACD proliferation | ||
| tetraploidy, multinucleation, | ||
| Cell cycle | no aberrations | mature and immature nuclei, |
| characteristics | cytokinesis failure, mitotic defects | |
| homotypic cell fusion | ||
| Cell fusion | no fusion | forms multinucleated syncytia (MGRS’, PGCCs) that produce spores, |
| germline clones and stem cells (CSCs) | ||
| Germ to soma transition (GST) and | The MGRS/PGCC pathway for | |
| Cell conversion | soma-to-germ transition (SGT, EMT) ; | genome repair and function regain: |
| (plasticity) | Somatic cells maintain germline genome | It generates viable spores that in turn form |
| integrity; SGT generate new functional | new functional germline clones and new stem | |
| gemline clones and stem cells (CSCs) | cells (CSCs) | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



