
Submitted:
31 August 2024
Posted:
02 September 2024
You are already at the latest version
Abstract
The BAFF-APRIL system is crucial for the pathogenesis of systemic lupus erythematosus (SLE) by promoting B cell survival, differentiation and the maintenance of humoral autoimmunity. Here, we investigated the relationship of BCMA expression on B cell subsets with its ligands BAFF and APRIL, together with soluble BCMA, and with clinical and serologic variables in a cohort of 100 SLE patients (86 under conventional and 14 under belimumab therapy) and 30 healthy controls (HC) using multicolor flow cytometry and ELISA. We found that BCMA expression in SLE patients was significantly increased on all B cell subsets compared to HCs, with all examined components of the BAFF-APRIL system being upregulated. BCMA expression was significantly increased on switched and unswitched memory B cells compared to naïve B cells, both in HCs and SLE. BCMA expression on B cells correlated with plasmablast frequencies, serum anti-dsDNA antibodies and complement consumption, while soluble BCMA correlated with plasmablast frequency, highlighting its potential as clinical biomarker. Belimumab treatment significantly reduced BCMA expression on most B cell subsets and soluble TACI, and contributes to the inhibition of almost the entire BAFF-APRIL system and restoration of B cell homeostasis. These results provide insights into the complex dysregulation of the BAFF-APRIL system in SLE and highlight the therapeutic potential of targeting its components, particularly BCMA, in addition to its use as biomarker for disease activity.
Keywords:
SLE
; BAFF
; BCMA
; TACI
; lupus
; B cells
; plasma cells
1. Introduction
Systemic lupus erythematosus (SLE) is a prototypic systemic autoimmune disease characterized by multiple organ manifestations and autoantibody production against a variety of nuclear antigens that strongly contribute to disease pathogenesis [1]. Although the etiology of SLE has not yet been fully clarified, antibody-secreting cells such as plasmablasts as well as long-lived plasma cells and their precursor B cells are crucial both for development and perturbation of the disease [2,3,4]. Given their role in persistent autoantibody production, long-lived plasma cells represent novel cellular targets for biologic therapies in SLE, such as proteasome inhibitors [5] and anti-CD38 monoclonal antibodies [6,7]. Since plasma cells express high levels of BCMA, it is an attractive target to deplete autoreactive plasma cells. In fact, recent studies demonstrated clinical efficacy of a dual CD19/BCMA-directed chimeric antigen receptor (CAR)-T cell therapy in SLE [8]. In addition, we recently utilized the CD3-BCMA bispecific antibody teclistamab, approved for multiple myeloma [9], in a refractory case of SLE, which resulted in a complete clinical and serologic remission, despite the discontinuation of immunosuppressive therapies. The beneficial responses were associated with depletion of plasma cells in the bone marrow and a preferential eradication of CD27+ memory B cells (Alexander et al. in press).
Among pathogenic B cell subsets, activated naïve [10,11,12,13] as well as double-negative memory B cells, particularly T-bet+ CD11c+ age-associated B cells [14,15], CD27- IgD- CXCR5- CD11c+ double negative [16,17,18,19] and CXCR5- CD19low plasmablast precursors [20], are newly identified subsets of autoreactive B cells that are expanded during flares of the disease and are implicated in humoral dysregulation of SLE. Although genetic predispositions and environmental factors have been identified in the pathogenesis of SLE, the generation and underlying molecular mechanisms of autoreactive B cells largely remain unclear. Important factors that activate B cells and stimulate them to differentiate into plasmablasts and plasma cells are soluble factors BAFF (B cell activating factor) and APRIL (a proliferation-inducing ligand). Both are ligands to the surface receptors B cell maturation antigen (BCMA) and transmembrane activator and CAML interactor (TACI) on B cells and binding to them induces proliferation and survival of B cells [21,22,23,24,25,26,27]. BAFF and APRIL have also been reported to protect plasma cells [28] and multiple myeloma cells [29] from apoptosis.
BCMA, a member of the tumor necrosis factor (TNF) receptor superfamily, is a membrane-bound surface receptor that is expressed on B cell lineage derived cells, particularly on plasmablasts and plasma cells, along with a subset of activated CD38+ memory B cells [28]. The amount of its expression represents the state of B cell activation both in healthy individuals and SLE patients [25,30]. BCMA expression can be induced by stimulation with cytokines and is essential for the survival of long-lived plasma cells in the bone marrow [31]. Furthermore, BCMA is shed by gamma-secretase and then released into the blood, where it can be measured as soluble BCMA (sBCMA) by ELISA [32,33]. We previously reported elevated serum levels of sBCMA in a cohort of untreated SLE patients compared to healthy controls, with a strong correlation to disease activity [32]. These findings were confirmed in a cohort of SLE patients under treatment, suggesting a role of sBCMA as biomarker for disease activity in SLE [34].
Previous studies investigating BCMA expression on immune cell subsets by flow cytometry demonstrated significantly increased BCMA expression on SLE B cells compared to healthy controls [25,35,36]. They also revealed correlations of BCMA expression on B cells with disease severity in SLE, yet with contradictory results. While one study reported increased BCMA expression on almost all B cell subsets and a positive correlation of BCMA expression on total B cells with disease activity [25], another study described decreased BCMA expression on almost all B cell subsets in a small cohort of SLE patients compared to healthy controls, and a negative correlation between BCMA-expressing B cells and disease activity [37].
Based on the apparent role of BCMA in SLE pathogenesis, we now aimed to analyze the expression of BCMA on freshly isolated B cell subsets in SLE patients under conventional and BAFF-targeting therapy with belimumab that confirm and extend previous results, and demonstrate that increased BCMA expression on B cells is a stable and reproducible feature of SLE. BCMA expression on B cells strongly correlates with the frequency of circulating plasmablasts and serum dsDNA antibody titers and is downregulated under belimumab treatment. We also observed higher BCMA expression levels on memory B cell subsets compared to naïve B cells, identifying BCMA as a potential therapeutic target for depleting plasma cells and their memory B cell precursors with monoclonal or bispecific antibodies, as well as BCMA-directed CAR-T cell therapies.
2. Results
2.1. B Cell Subset Distribution and their BCMA Expression
To investigate the BCMA expression on peripheral blood B cell subsets and plasmablasts, we performed multicolor flow cytometry on freshly obtained peripheral blood after red blood cell lysis in cohort of 100 SLE patients compared to 30 healthy controls (HC). Patient demographics and disease characteritics are shown in Table 1. Patients were categorized according to their treatment into those receiving standard-of-care (SLE-SOC), defined as patients without biologics (n=86), and those with belimumab treatment (SLE-BEL, n=14).
We analyzed five CD19+ B cell subsets for their surface expression of BCMA, in particular IgD+CD27- naïve (NB), Mitotracker+IgD+CD27-CD24- activated naïve (aNB), IgD-CD27- double-negative (DN), IgD+CD27+ non-switched memory (NSM), IgD-CD27+ switched memory (SM), as well as IgD-CD27highHLA-DRhigh plasmablasts (PB) and IgD-CD27highHLA-DRlow plasma cells (PC). The gating strategy of flow cytometry experiments is illustrated in Figure 1A, and representative BCMA staining examples provided in Figure 1B.
Compared to healthy controls, SLE patients under conventional therapy exhibited significantly higher frequencies of aNB (3.8% vs. 1.5%, p < 0.001), DN (8.7% vs 6.6%, p = 0.003), PB (2.5% vs. 0.5%, p < 0.001) and PC (0.4 vs. 0.1%, p < 0.001) (Figure 1C). The corresponding absolute cell counts are provided in the online supplementary Figure S1A.
When investigated for their BCMA expression, total CD19+ B cells expressed significantly higher levels in SLE compared to HCs (median MFI 84.7 vs. 57.9, p < 0.0001), while antibody secreting IgD-CD27high cells showed no statistical differences (Figure S1B in the online supplement). On a subset level, all investigated B cells in SLE expressed higher BCMA levels compared to HCs, particularly NB (MFI 74.4 vs. 46.0, p < 0.0001); aNB (MFI 85.0 vs. 65.1, p < 0.0001); DN (MFI 75.2 vs. 62.3, p < 0.0001); NSM (MFI 107.0 vs. 79.0, p < 0.0001); and SM (MFI 95.5 vs. 74.9, p < 0.0001), whereas both PB (MFI 315.5 vs. 276.0, p = 0.140) and PC (MFI 246.0 vs. 222.0, p = 0.152) displayed no significant difference (Figure 1D). We next examined whether the surface expression intensity of BCMA differed between the analyzed B cell subsets. Indeed, the highest expression levels for BCMA in HCs were found on non-switched and swiched memory B cells, with significantly higher levels compared to NB (Figure 1E). Similar findings were obtained in SLE patients, but with an additional significant increase of BCMA expression in both NSM and SM compared to DN B cells (Figure 1F). Collectively, these data demonstrate upregulation of BCMA surface expression across all B cell subsets in SLE, with increased levels on memory B cells compared to naïve B cells.
2.2. Soluble Markers of the BAFF-APRIL System and Their Correlations
We next investigated plasma concentrations of soluble BCMA, TACI, and BAFF in our cohort of 86 SLE patients under conventional therapy. Compared to HCs, we found significantly higher levels of soluble BCMA (median 36.9 vs. 27.1 p = 0.001), TACI (median 325.7 vs. 210.0, p = 0.039) and BAFF (median 742.6 vs. 552.0, p = 0.006) (Figure 2A–C). When analyzing their interactions, we detected a moderate but significant correlation between soluble BCMA and TACI in SLE patients (r = 0.38, p = 0.0007) but not in HCs (Figure 2D,E). This correlation was even stronger in SLE patients treated with belimumab (r = 0.67, p = 0.008) (Supplementary Figure S2A). In contrast, soluble BCMA and BAFF levels were negatively correlated (r = −0.33, p = 0.118), and a similar trend was observed between soluble TACI and BAFF (r = −0.34, p = 0.100), although not reaching statistical significance.
Furthermore, we sought to determine the correlation between soluble BCMA levels and surface BCMA expression on B cells. Interestingly, sBCMA in SLE significantly correlated with surface BCMA expression on plasmablasts (r = 0.35, p = 0.040) but not on B cells, possibly reflecting the shedding of BCMA from the increased frequencies of PB evident in SLE (Figure 2E). In contrast, no correlation was found between sBCMA and surface expression of BCMA on B cells or plasmablasts in HCs (Figure 2D). Additionally, surface expression of BCMA on plasmablasts in SLE negatively correlated with serum BAFF levels (r = −0.41, p = 0.002), whereas BCMA expression on B cells significantly correlated with serum BAFF concentrations (r = 0.49, p = 0.013) (Figure 2E).
Collectively, all examined soluble components of the BAFF-APRIL system were upregulated in SLE with moderate correlations between soluble BCMA and TACI, and soluble BCMA with surface BCMA on PB and soluble BAFF on B cells. Remarkably, no significant correlations were detected in healthy controls, highlighting the chronic upregulated humoral activity and disturbed immunological homeostasis in SLE.
2.3. Correlations of BCMA with Clinical and Serologic Variables and Plasmablast Frequency
We finally evaluated the complex interactions of soluble BCMA and BCMA expression on B cells and plasmablasts, with markers of clinical and serologic disease activity, as well as frequencies of circulating plasmablasts. Interestingly, no correlations were found between soluble BCMA or surface BCMA expression and clinical disease activity in SLE, as measured by clinical domains of the SLEDAI-2K (Figure 2E). In contrast, we found significant correlations between BCMA expression on B cells and serum anti-dsDNA antibody levels (r = 0.24, p = 0.031) and between soluble BCMA (r = −0.26, p = 0.017) and complement consumption for C3 (Figure 2F). More importantly, both soluble BCMA (r = 0.24, p = 0.029) and surface BCMA on B cells (r = 0.55, p < 0.0001) and plasmablasts (r = 0.46, p < 0.0001) strongly correlated with the frequency of circulating plasmablasts among CD19+ B cells, identifying these markers as surrogates for humoral activity in SLE. These findings were confirmed by using absolute cell counts (Figure S2C,D) and in patients undergoing belimumab treatment (Figure S2B).
2.4. Impact of Belimumab Treatment on the BAFF/APRIL System
Previous studies have demonstrated that the BAFF-targeting monoclonal antibody belimumab impacts the BAFF/APRIL system by inhibiting the binding of soluble BAFF to its receptors (BAFF receptor, TACI and BCMA) expressed on B cells [38]. Therefore, we aimed to evaluate BCMA expression on B cell subsets in an independent cohort of 14 SLE patients under stable treatment with intravenous belimumab (SLE-BEL), in comparison to SLE patients receiving conventional therapy (SLE-SOC). The clinical characteristics of the belimumab treated patients are provided in Table S1.
Consistent with previous findings, we noted significant reductions in the frequencies of naïve B cells (21.3% vs. 61.5%, p < 0.0001) and activated naïve B cells (1.3% vs. 3.8%, p = 0.006), along with increased frequencies of non-switched (11.0% vs. 6.1%, p = 0.015) and switched memory B cells (44.6% vs. 13.2%, p < 0.0001) in belimumab treated compared to conventionally treated SLE patients (Figure S3A) [39,40]. Surface BCMA expression levels were significantly lower in belimumab vs. conventionally treated patients on naïve B cells (74.4 vs. 65.1, p = 0.001), non-switched (107.0 vs. 86.1, p = 0.008), and switched memory B cells (95.5 vs. 82.0, p = 0.005) (Figure 3A). This resulted in a normalization of BCMA expression on memory B cell subsets in belimumab-treated SLE patients compared to HCs, but BCMA expression remained elevated on naïve and double-negative memory B cells (Figure 3B).
Finally, we prospectively followed a cohort of 9 SLE patients after initiation of belimumab treatment to assess changes in their soluble BCMA and TACI levels. The disease characteristics of these patients are provided in Table S2. After a median follow-up of 8 months (range 5–39 months), circulating plasmablasts decreased significantly (from 19.7% to 4.4% among CD19+ B cells, p = 0.004) (Figure S3B). Plasma concentrations of soluble BCMA did not show significant changes during this period (median 31.4 vs. 27.4 ng/mL, p = 0.547) (Figure 3C), whereas soluble TACI significantly decreased (median 195.3 vs. 81.2 pg/mL, p = 0.039) (Figure 3D).
3. Discussion
Given the central role of BCMA and its ligands BAFF and APRIL in the dysregulated B cell homeostasis observed in SLE, we investigated the complex interplay between BCMA expression on peripheral blood B cells subsets, soluble BCMA plasma concentrations and clinical disease variables as well as peripheral blood plasmablast expansion, a hallmark of the disease. Our data reveal significantly increased BCMA expression levels on all investigated B cell subsets in SLE, that strongly correlated with serum anti-dsDNA antibody levels and the frequency of circulating plasmablasts, the latter also correlating with soluble BCMA, identifying these markers as surrogates for disease activity as well as biomarkers for humoral activity in SLE. More importantly, we found higher BCMA expression levels on memory compared to naïve B cells, highlighting BCMA as an attractive therapeutic target for preferential depletion of memory B cell subsets along with plasmablasts and plasma cells constitutively expressing BCMA.
In contrast to BAFF receptor, BCMA is upregulated on activated B cells, reflected by increased BCMA expression along with other B cell activation markers, such as CD86 [25] and CD38 [28], and activation of B cells by CpG/IL-2/IL-15 induced BCMA expression in vitro [30]. In line with this notion, we found significantly increased surface BCMA levels on all investigated B cells subsets, but not plasmablasts, in SLE patients. The hierarchy of BCMA expression from lowest to highest was: naïve B cells, double-negative memory B cells, activated naïve B cells, switched memory B cells, non-switched memory B cells, plasma cells, and plasmablasts. Our findings are consistent with studies by Kim et al. [25] and Álvarez Gómez et al. [35], but not with earlier works by Salazar-Camarena et al. [34,37], who measured the frequency of BCMA-positive B cells rather than the expression density based on the mean fluorescent intensity (MFI) values on B cells. These studies reported lower frequencies of BCMA-positive B cells in SLE patients compared to healthy controls, with a negative correlation with the Mexican (MEX)-SLEDAI score. Additionally, Kim et al. found elevated BCMA expression on memory B cells and especially on plasmablasts and plasma cells, whereas BCMA expression on naïve B cells was relatively low [25]. Álvarez Gómez et al. reported increased BCMA expression on resting naïve B cells and switched memory B cells, but not on DN and activated naïve B cells [35], possibly due to differences in defining these subsets by using other biomarkers. Collectively, our data confirm and extend previous findings, and demonstrate that increased BCMA expression on B cells is a stable and reproducible feature of SLE, with memory B cells displaying higher expression levels compared to naïve B cell subsets. This notion could be relevant for BCMA-targeted therapies, in which naïve B-cell subsets are spared from depletion. BCMA is already an established target in the treatment of multiple myeloma, using either bispecific antibody constructs, antibody–drug conjugates, or chimeric antigen receptor (CAR)-modified T cell therapies [41], and could become an attractive target in SLE for depleting plasma cells and memory B cells. Indeed, our recent data on the CD3-BCMA bispecific antibody teclistamab demonstrated complete depletion of plasma cells in the bone marrow, together with a preferential depletion of memory over naïve B cells among the few remaining B cells after treatment, resulting in a complete remission “off-therapy” (Alexander et al., in press).
Membrane-bound BCMA can undergo γ-secretase–mediated shedding from the cell surface, leading to circulation of soluble BCMA and reduced activation of surface BCMA by BAFF and APRIL [32]. Furthermore, BAFF and APRIL are ligands of BCMA and TACI, with BAFF also binding exclusively to the BAFF receptor, which is essential for B cell proliferation and survival [42]. In our cohort of patients, plasma concentrations of BCMA, BAFF, and TACI were significantly increased compared to healthy controls, consistent with previous reports in the literature [32,33,34,43,44]. Additionally, we observed a strong correlation between sBCMA and sTACI, but not sBAFF, in SLE patients. Soluble TACI binds to BAFF, leading to its inhibition [43], potentially explaining the negative correlation between BAFF and sTACI. TACI shedding is mediated by ADAM10 independently of BAFF [45].
In vitro, concentrations of soluble BCMA shedded by γ-secretase correlated with membrane BCMA levels on B cell [32]. We therefore expected to detect a correlation between sBCMA and BCMA expressed on B cells, but this was not evident. Instead, sBCMA strongly correlated with the BCMA expression on plasmablasts, probably because they display the highest BCMA expression levels and are increased in SLE. More strikingly, sBCMA strongly correlated with the frequency of circulating plasmablasts. Thus, sBCMA levels may serve as a useful surrogate for humoral activity in SLE, similar to findings in multiple myeloma where sBCMA levels are elevated and correlate with the proportion of myeloma cells in the bone marrow [46].
BCMA expression on B cells is linked to their activation and maturation, particularly on CD27+ memory B cells and antibody-secreting cells [30]. Consequently, increased BCMA expression on B cells may result in elevated autoantibody production. Indeed, we observed a moderate correlation between BCMA expression on B cells and serum anti-dsDNA levels. In addition, negative correlations were found between serum C3 levels and both sBCMA and sTACI, suggesting that that high BCMA and TACI expression promotes autoantibody production and immune complex formation. Consistent with these findings, Laurent et al. reported a negative correlation between sBCMA and C3 levels in SLE [32], collectively identifying membrane-bound BCMA on B cells as a biomarker for disease activity.
There is ample evidence that the monoclonal antibody belimumab, which targets BAFF, can at least partially restore the impaired B-cell homeostasis in SLE by reducing B-cell activation and inhibiting the differentiation of autoreactive antibody-secreting cells through reduced binding of BAFF to its receptors BAFF receptor, TACI and BCMA [39,40,47]. Indeed, our cohort of belimumab-treated patients had lower BCMA expression on naïve and memory B cells than conventionally treated patients, with BCMA expression on non-switched and switched memory B cells reaching similar levels to healthy controls. This suggests that these B cell subgroups are preferentially inhibited by belimumab and are more dependent on BAFF signaling. These findings align with previous studies, which reported a significant decrease in BCMA MFI on total B cells after three months of belimumab treatment [38]. However, they did not analyze membrane-bound BCMA at the B cell subset level. Furthermore, intraindividual reductions in sTACI levels were observed during belimumab treatment, consistent with Piantoni et al. [48]. This indicates that belimumab reduces TACI expression on B cell membranes, as shown by Hirano et al. [38]. Piantoni et al. also reported intraindividual decreases in sBCMA during belimumab treatment [48], providing more reliable data than in our study because they included more treated patients with defined time points [49]. Taken together, these results support the notion that neutralization of BAFF by belimumab inhibits the entire BAFF-APRIL system and downregulates its receptors in the long term, leading to reduced plasmablast frequencies and clinical efficacy.
The strength of our study is the evaluation of BCMA surface expression at the B cell subset level in a large cohort of SLE patients, combined with the analysis of the soluble ligands of BCMA. However, our results may be confounded by the heterogeneity of clinical manifestations and underlying treatments among the investigated patients. Notably, the cohort of belimumab-treated patients is relatively small, and most data were obtained cross-sectionally, with a lack of longitudinal analysis of BCMA expression on B cells. Nevertheless, our study provides a detailed analysis of BCMA-expressing B cells and their correlations with clinical variables and is probably one of the most comprehensive analyses of the BAFF/APRIL system in SLE to date. Future investigations could benefit from the inclusion of transcriptomic or metabolomic data and functional analyses of BCMA-expressing B cells to complement our phenotypic evaluations.
In conclusion, we identified increased BCMA expression on B cell subsets as a prominent and reproducible feature of SLE. BCMA expression on B cells strongly correlates with the frequency of circulating plasmablasts and serum anti-dsDNA antibody titers and it is downregulated with belimumab treatment. Soluble BCMA correlates with the level of BCMA expression on plasmablasts and their frequency in peripheral blood, identifying these markers as surrogates for clinical and humoral activity that could be utilized for monitoring SLE patients in clinical routine. Furthermore, we found that BCMA-expression is significantly higher on memory B cell subsets compared to their naïve counterparts, positioning BCMA as a potential therapeutic target for depleting plasma cells and their memory B cell precursors using monoclonal or bispecific antibodies, as well as BCMA-directed CAR-T cell therapies.
4. Materials and Methods
4.1. Patient and Control Blood Samples
We recruited 100 patients with SLE according to the 2019 EULAR/ACR classification criteria [50] from the Department of Rheumatology and Clinical Immunology at Charité-Universitätsmedizin Berlin between April and November 2016. As controls, we included 30 age- and gender-matched healthy individuals. Clinical and serologic manifestations, including the SLEDAI-2K [51], and immunosuppressive medication regimes were recorded by the treating physician. Patients were subdivided according to their treatment into groups of those with (n=14) or without (n=86) receiving belimumab. Patients in the belimumab group were under intravenous belimumab therapy for a median duration of 9 months (range 5–39 months) and their clinical characteristics are provided in Table S1. All samples were collected 4 weeks after the previous belimumab infusion. From 9 out of 14 patients with belimumab treatment, plasma was collected for the analysis of soluble BCMA and TACI.
4.2. Cell Isolation and Flow Cytometry
For flow cytometry, five mL of freshly obtained heparinized blood was mixed with 50 mL of Red Blood Cell Lysis Buffer (BD Biosciences, USA) in Falcon tubes for 10 minutes to eliminate red blood cells. After centrifugation, samples were washed with phosphate-buffered saline containing bovine serum albumin and centrifuged again to generate peripheral blood mononuclear cells (PBMCs). They were incubated with MitoTracker Deep Red (MTR) at a concentration of 1:100,0000 at 37 °C for 10 minutes, washed and centrifuged. Subsequently, cells were stained in 100 μL PBS for 20 min at 4 °C with the antibodies indicated in Supplementary Table S3. DAPI was added to stain dead cells and samples were acquired on a FACSCanto II (BD Biosciences, USA). FACS analysis was performed following the guidelines for the use of flow cytometry and cell sorting in immunological studies [52]. Cytometry data were analyzed using FlowJo v10.6.2 (FlowJo, LLC, Ashland, OR, USA).
4.3. ELISA
Heparinized blood samples were centrifuged for 10 minutes to obtain plasma from each participant, which was frozen at −20 °C for subsequent analysis. ELISA was performed to analyze plasma levels of BAFF (BAFF/BLyS Quantikine), TACI (DuoSet) and BCMA (DuoSet), all R&D Systems, USA, according to the manufacturer’s protocols. SoftMax Pro V.6.5 was used to analyze ELISA results.
4.4. Statistical Analysis
Statistical analysis was conducted using SPSS Statistics version 29.0, while figures were generated with GraphPad Prism version 9.0. To compare data between different cohorts, we utilized the Mann-Whitney U test. Within the same cohort, comparisons of B cell subsets were performed using the Kruskal-Wallis test. Intraindividual changes under belimumab treatment was assessed applying the Wilcoxon signed-rank test. Spearman’s rank correlation test was employed for correlation analyses. Statistical significance was defined as a P value of less than 0.05. In the figures, statistical significance was represented by the following symbols: ns or no symbol means p > 0.05, ✱ means p < 0.05, ✱✱ means p < 0.01, ✱✱✱ means p < 0.001 and ✱✱✱✱ means p < 0.0001. In the results section, comparison values presented in brackets always represent medians.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Figure S1, Figure S2, Figure S3, Table S1, Table S2, Table S3.
Author Contributions
Conceptualization F.H., J.M., Q.C. and T.A.; methodology J.M. and Q.C.; formal analysis J.M. and Q.C.; investigation J.M., S.A.L, E.M., F.S.T. and Q.C.; resources F.H., T.A.; data curation, J.M.; writing—original draft preparation J.M. and T.A.; writing—review and editing E.M., A.E.B., G.K.; F.H., and T.A.; visualization J.M. and T.A.; supervision F.H. and T.A.; project administration F.H. and Q.C.; funding acquisition F.H. and T.A. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the DFG (SFB-TR128 TP B08 and TR130 TP15).
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki, and approved by the Ethics Committee of Charité-Universitätsmedizin Berlin (EA1/124/09).
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Conflicts of Interest
F.H. received honoria from AstraZeneca. T.A. received study support from Janssen-Cilag and honoria from AstraZeneca and GSK. Other authors declare no conflicts of interest.
References
- Tsokos, G.C. Systemic lupus erythematosus. N Engl J Med 2011, 365, 2110–2121. [Google Scholar] [CrossRef] [PubMed]
- Arbuckle, M.R.; McClain, M.T.; Rubertone, M.V.; Scofield, R.H.; Dennis, G.J.; James, J.A.; Harley, J.B. Development of autoantibodies before the clinical onset of systemic lupus erythematosus. N Engl J Med 2003, 349, 1526–1533. [Google Scholar] [CrossRef]
- Iwata, S.; Tanaka, Y. B-cell subsets, signaling and their roles in secretion of autoantibodies. Lupus 2016, 25, 850–856. [Google Scholar] [CrossRef]
- Hiepe, F.; Dörner, T.; Hauser, A.E.; Hoyer, B.F.; Mei, H.; Radbruch, A. Long-lived autoreactive plasma cells drive persistent autoimmune inflammation. Nat Rev Rheumatol 2011, 7, 170–178. [Google Scholar] [CrossRef]
- Alexander, T.; Sarfert, R.; Klotsche, J.; Kühl, A.A.; Rubbert-Roth, A.; Lorenz, H.M.; Rech, J.; Hoyer, B.F.; Cheng, Q.; Waka, A.; et al. The proteasome inhibitior bortezomib depletes plasma cells and ameliorates clinical manifestations of refractory systemic lupus erythematosus. Ann Rheum Dis 2015, 74, 1474–1478. [Google Scholar] [CrossRef]
- Ostendorf, L.; Burns, M.; Durek, P.; Heinz, G.A.; Heinrich, F.; Garantziotis, P.; Enghard, P.; Richter, U.; Biesen, R.; Schneider, U.; et al. Targeting CD38 with Daratumumab in Refractory Systemic Lupus Erythematosus. N Engl J Med 2020, 383, 1149–1155. [Google Scholar] [CrossRef] [PubMed]
- Roccatello, D.; Fenoglio, R.; Caniggia, I.; Kamgaing, J.; Naretto, C.; Cecchi, I.; Rubini, E.; Rossi, D.; De Simone, E.; Del Vecchio, G.; et al. Daratumumab monotherapy for refractory lupus nephritis. Nat Med 2023, 29, 2041–2047. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; He, S.; Zhang, W.; Zhang, H.; DeStefano, V.M.; Wada, M.; Pinz, K.; Deener, G.; Shah, D.; Hagag, N.; et al. BCMA-CD19 compound CAR T cells for systemic lupus erythematosus: A phase 1 open-label clinical trial. Ann Rheum Dis 2024. [Google Scholar] [CrossRef] [PubMed]
- Moreau, P.; Garfall, A.L.; van de Donk, N.; Nahi, H.; San-Miguel, J.F.; Oriol, A.; Nooka, A.K.; Martin, T.; Rosinol, L.; Chari, A.; et al. Teclistamab in Relapsed or Refractory Multiple Myeloma. N Engl J Med 2022, 387, 495–505. [Google Scholar] [CrossRef]
- Presley, A.D.; Fuller, K.M.; Arriaga, E.A. MitoTracker Green labeling of mitochondrial proteins and their subsequent analysis by capillary electrophoresis with laser-induced fluorescence detection. J Chromatogr B Analyt Technol Biomed Life Sci 2003, 793, 141–150. [Google Scholar] [CrossRef]
- Wirths, S.; Lanzavecchia, A. ABCB1 transporter discriminates human resting naive B cells from cycling transitional and memory B cells. Eur J Immunol 2005, 35, 3433–3441. [Google Scholar] [CrossRef]
- Tipton, C.M.; Fucile, C.F.; Darce, J.; Chida, A.; Ichikawa, T.; Gregoretti, I.; Schieferl, S.; Hom, J.; Jenks, S.; Feldman, R.J.; et al. Diversity, cellular origin and autoreactivity of antibody-secreting cell population expansions in acute systemic lupus erythematosus. Nat Immunol 2015, 16, 755–765. [Google Scholar] [CrossRef] [PubMed]
- Jacobi, A.M.; Mei, H.; Hoyer, B.F.; Mumtaz, I.M.; Thiele, K.; Radbruch, A.; Burmester, G.R.; Hiepe, F.; Dörner, T. HLA-DRhigh/CD27high plasmablasts indicate active disease in patients with systemic lupus erythematosus. Ann Rheum Dis 2010, 69, 305–308. [Google Scholar] [CrossRef] [PubMed]
- Rubtsov, A.V.; Rubtsova, K.; Fischer, A.; Meehan, R.T.; Gillis, J.Z.; Kappler, J.W.; Marrack, P. Toll-like receptor 7 (TLR7)-driven accumulation of a novel CD11c⁺ B-cell population is important for the development of autoimmunity. Blood 2011, 118, 1305–1315. [Google Scholar] [CrossRef] [PubMed]
- Sachinidis, A.; Xanthopoulos, K.; Garyfallos, A. Age-Associated B Cells (ABCs) in the Prognosis, Diagnosis and Therapy of Systemic Lupus Erythematosus (SLE). Mediterr J Rheumatol 2020, 31, 311–318. [Google Scholar] [CrossRef]
- Jenks, S.A.; Cashman, K.S.; Zumaquero, E.; Marigorta, U.M.; Patel, A.V.; Wang, X.; Tomar, D.; Woodruff, M.C.; Simon, Z.; Bugrovsky, R.; et al. Distinct Effector B Cells Induced by Unregulated Toll-like Receptor 7 Contribute to Pathogenic Responses in Systemic Lupus Erythematosus. Immunity 2018, 49, 725–739. [Google Scholar] [CrossRef]
- Wei, C.; Anolik, J.; Cappione, A.; Zheng, B.; Pugh-Bernard, A.; Brooks, J.; Lee, E.H.; Milner, E.C.; Sanz, I. A new population of cells lacking expression of CD27 represents a notable component of the B cell memory compartment in systemic lupus erythematosus. J Immunol 2007, 178, 6624–6633. [Google Scholar] [CrossRef]
- Torigoe, M.; Iwata, S.; Nakayamada, S.; Sakata, K.; Zhang, M.; Hajime, M.; Miyazaki, Y.; Narisawa, M.; Ishii, K.; Shibata, H.; et al. Metabolic Reprogramming Commits Differentiation of Human CD27(+)IgD(+) B Cells to Plasmablasts or CD27(-)IgD(-) Cells. J Immunol 2017, 199, 425–434. [Google Scholar] [CrossRef]
- Jacobi, A.M.; Reiter, K.; Mackay, M.; Aranow, C.; Hiepe, F.; Radbruch, A.; Hansen, A.; Burmester, G.R.; Diamond, B.; Lipsky, P.E.; et al. Activated memory B cell subsets correlate with disease activity in systemic lupus erythematosus: Delineation by expression of CD27, IgD, and CD95. Arthritis Rheum 2008, 58, 1762–1773. [Google Scholar] [CrossRef]
- Szelinski, F.; Stefanski, A.L.; Schrezenmeier, E.; Rincon-Arevalo, H.; Wiedemann, A.; Reiter, K.; Ritter, J.; Lettau, M.; Dang, V.D.; Fuchs, S.; et al. Plasmablast-like Phenotype Among Antigen-Experienced CXCR5-CD19(low) B Cells in Systemic Lupus Erythematosus. Arthritis Rheumatol 2022, 74, 1556–1568. [Google Scholar] [CrossRef]
- Moore, P.A.; Belvedere, O.; Orr, A.; Pieri, K.; LaFleur, D.W.; Feng, P.; Soppet, D.; Charters, M.; Gentz, R.; Parmelee, D.; et al. BLyS: Member of the tumor necrosis factor family and B lymphocyte stimulator. Science 1999, 285, 260–263. [Google Scholar] [CrossRef] [PubMed]
- Morais, S.A.; Vilas-Boas, A.; Isenberg, D.A. B-cell survival factors in autoimmune rheumatic disorders. Ther Adv Musculoskelet Dis 2015, 7, 122–151. [Google Scholar] [CrossRef]
- Yap, D.Y.; Lai, K.N. Cytokines and their roles in the pathogenesis of systemic lupus erythematosus: From basics to recent advances. J Biomed Biotechnol 2010, 2010, 365083. [Google Scholar] [CrossRef]
- Bossen, C.; Cachero, T.G.; Tardivel, A.; Ingold, K.; Willen, L.; Dobles, M.; Scott, M.L.; Maquelin, A.; Belnoue, E.; Siegrist, C.A.; et al. TACI, unlike BAFF-R, is solely activated by oligomeric BAFF and APRIL to support survival of activated B cells and plasmablasts. Blood 2008, 111, 1004–1012. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Gross, J.A.; Dillon, S.R.; Min, J.K.; Elkon, K.B. Increased BCMA expression in lupus marks activated B cells, and BCMA receptor engagement enhances the response to TLR9 stimulation. Autoimmunity 2011, 44, 69–81. [Google Scholar] [CrossRef] [PubMed]
- Schneider, P.; MacKay, F.; Steiner, V.; Hofmann, K.; Bodmer, J.L.; Holler, N.; Ambrose, C.; Lawton, P.; Bixler, S.; Acha-Orbea, H.; et al. BAFF, a novel ligand of the tumor necrosis factor family, stimulates B cell growth. J Exp Med 1999, 189, 1747–1756. [Google Scholar] [CrossRef]
- Vincent, F.B.; Morand, E.F.; Schneider, P.; Mackay, F. The BAFF/APRIL system in SLE pathogenesis. Nat Rev Rheumatol 2014, 10, 365–373. [Google Scholar] [CrossRef]
- Avery, D.T.; Kalled, S.L.; Ellyard, J.I.; Ambrose, C.; Bixler, S.A.; Thien, M.; Brink, R.; Mackay, F.; Hodgkin, P.D.; Tangye, S.G. BAFF selectively enhances the survival of plasmablasts generated from human memory B cells. J Clin Invest 2003, 112, 286–297. [Google Scholar] [CrossRef]
- Moreaux, J.; Legouffe, E.; Jourdan, E.; Quittet, P.; Rème, T.; Lugagne, C.; Moine, P.; Rossi, J.F.; Klein, B.; Tarte, K. BAFF and APRIL protect myeloma cells from apoptosis induced by interleukin 6 deprivation and dexamethasone. Blood 2004, 103, 3148–3157. [Google Scholar] [CrossRef]
- Darce, J.R.; Arendt, B.K.; Wu, X.; Jelinek, D.F. Regulated expression of BAFF-binding receptors during human B cell differentiation. J Immunol 2007, 179, 7276–7286. [Google Scholar] [CrossRef]
- O’Connor, B.P.; Raman, V.S.; Erickson, L.D.; Cook, W.J.; Weaver, L.K.; Ahonen, C.; Lin, L.L.; Mantchev, G.T.; Bram, R.J.; Noelle, R.J. BCMA is essential for the survival of long-lived bone marrow plasma cells. J Exp Med 2004, 199, 91–98. [Google Scholar] [CrossRef]
- Laurent, S.A.; Hoffmann, F.S.; Kuhn, P.H.; Cheng, Q.; Chu, Y.; Schmidt-Supprian, M.; Hauck, S.M.; Schuh, E.; Krumbholz, M.; Rübsamen, H.; et al. γ-Secretase directly sheds the survival receptor BCMA from plasma cells. Nat Commun 2015, 6, 7333. [Google Scholar] [CrossRef] [PubMed]
- Meinl, E.; Thaler, F.S.; Lichtenthaler, S.F. Shedding of BAFF/APRIL Receptors Controls B Cells. Trends Immunol 2018, 39, 673–676. [Google Scholar] [CrossRef] [PubMed]
- Salazar-Camarena, D.C.; Palafox-Sánchez, C.A.; Cruz, A.; Marín-Rosales, M.; Muñoz-Valle, J.F. Analysis of the receptor BCMA as a biomarker in systemic lupus erythematosus patients. Sci Rep 2020, 10, 6236. [Google Scholar] [CrossRef] [PubMed]
- Álvarez Gómez, J.A.; Salazar-Camarena, D.C.; Román-Fernández, I.V.; Ortiz-Lazareno, P.C.; Cruz, A.; Muñoz-Valle, J.F.; Marín-Rosales, M.; Espinoza-García, N.; Sagrero-Fabela, N.; Palafox-Sánchez, C.A. BAFF system expression in double negative 2, activated naïve and activated memory B cells in systemic lupus erythematosus. Front Immunol 2023, 14, 1235937. [Google Scholar] [CrossRef]
- Zhao, L.D.; Li, Y.; Smith, M.F., Jr.; Wang, J.S.; Zhang, W.; Tang, F.L.; Tian, X.P.; Wang, H.Y.; Zhang, F.C.; Ba, D.N.; et al. Expressions of BAFF/BAFF receptors and their correlation with disease activity in Chinese SLE patients. Lupus 2010, 19, 1534–1549. [Google Scholar] [CrossRef] [PubMed]
- Salazar-Camarena, D.C.; Ortiz-Lazareno, P.C.; Cruz, A.; Oregon-Romero, E.; Machado-Contreras, J.R.; Muñoz-Valle, J.F.; Orozco-López, M.; Marín-Rosales, M.; Palafox-Sánchez, C.A. Association of BAFF, APRIL serum levels, BAFF-R, TACI and BCMA expression on peripheral B-cell subsets with clinical manifestations in systemic lupus erythematosus. Lupus 2016, 25, 582–592. [Google Scholar] [CrossRef] [PubMed]
- Hirano, A.; Fujioka, K.; Kida, T.; Omura, S.; Sofue, H.; Sakashita, A.; Sagawa, T.; Isoda, Y.; Kasahara, A.; Sagawa, R.; et al. Association between early immunophenotypic changes and therapeutic response of belimumab in patients with systemic lupus erythematosus. Lupus 2023, 32, 63–73. [Google Scholar] [CrossRef]
- Ramsköld, D.; Parodis, I.; Lakshmikanth, T.; Sippl, N.; Khademi, M.; Chen, Y.; Zickert, A.; Mikeš, J.; Achour, A.; Amara, K.; et al. B cell alterations during BAFF inhibition with belimumab in SLE. EBioMedicine 2019, 40, 517–527. [Google Scholar] [CrossRef]
- Huang, W.; Quach, T.D.; Dascalu, C.; Liu, Z.; Leung, T.; Byrne-Steele, M.; Pan, W.; Yang, Q.; Han, J.; Lesser, M.; et al. Belimumab promotes negative selection of activated autoreactive B cells in systemic lupus erythematosus patients. JCI Insight 2018, 3. [Google Scholar] [CrossRef]
- Shah, N.; Chari, A.; Scott, E.; Mezzi, K.; Usmani, S.Z. B-cell maturation antigen (BCMA) in multiple myeloma: Rationale for targeting and current therapeutic approaches. Leukemia 2020, 34, 985–1005. [Google Scholar] [CrossRef]
- Batten, M.; Groom, J.; Cachero, T.G.; Qian, F.; Schneider, P.; Tschopp, J.; Browning, J.L.; Mackay, F. BAFF mediates survival of peripheral immature B lymphocytes. J Exp Med 2000, 192, 1453–1466. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, F.S.; Kuhn, P.H.; Laurent, S.A.; Hauck, S.M.; Berer, K.; Wendlinger, S.A.; Krumbholz, M.; Khademi, M.; Olsson, T.; Dreyling, M.; et al. The immunoregulator soluble TACI is released by ADAM10 and reflects B cell activation in autoimmunity. J Immunol 2015, 194, 542–552. [Google Scholar] [CrossRef] [PubMed]
- Ortiz-Aljaro, P.; Montes-Cano, M.A.; García-Lozano, J.R.; Aquino, V.; Carmona, R.; Perez-Florido, J.; García-Hernández, F.J.; Dopazo, J.; González-Escribano, M.F. Protein and functional isoform levels and genetic variants of the BAFF and APRIL pathway components in systemic lupus erythematosus. Sci Rep 2022, 12, 11219. [Google Scholar] [CrossRef]
- Smulski, C.R.; Kury, P.; Seidel, L.M.; Staiger, H.S.; Edinger, A.K.; Willen, L.; Seidl, M.; Hess, H.; Salzer, U.; Rolink, A.G.; et al. BAFF- and TACI-Dependent Processing of BAFFR by ADAM Proteases Regulates the Survival of B Cells. Cell Rep 2017, 18, 2189–2202. [Google Scholar] [CrossRef]
- Sanchez, E.; Li, M.; Kitto, A.; Li, J.; Wang, C.S.; Kirk, D.T.; Yellin, O.; Nichols, C.M.; Dreyer, M.P.; Ahles, C.P.; et al. Serum B-cell maturation antigen is elevated in multiple myeloma and correlates with disease status and survival. Br J Haematol 2012, 158, 727–738. [Google Scholar] [CrossRef]
- Stohl, W.; Hiepe, F.; Latinis, K.M.; Thomas, M.; Scheinberg, M.A.; Clarke, A.; Aranow, C.; Wellborne, F.R.; Abud-Mendoza, C.; Hough, D.R.; et al. Belimumab reduces autoantibodies, normalizes low complement levels, and reduces select B cell populations in patients with systemic lupus erythematosus. Arthritis Rheum 2012, 64, 2328–2337. [Google Scholar] [CrossRef] [PubMed]
- Piantoni, S.; Regola, F.; Masneri, S.; Merletti, M.; Lowin, T.; Airò, P.; Tincani, A.; Franceschini, F.; Andreoli, L.; Pongratz, G. Characterization of B- and T-Cell Compartment and B-Cell Related Factors Belonging to the TNF/TNFR Superfamily in Patients With Clinically Active Systemic Lupus Erythematosus: Baseline BAFF Serum Levels Are the Strongest Predictor of Response to Belimumab after Twelve Months of Therapy. Front Pharmacol 2021, 12, 666971. [Google Scholar] [CrossRef]
- Parodis, I.; Sjöwall, C.; Jönsen, A.; Ramsköld, D.; Zickert, A.; Frodlund, M.; Sohrabian, A.; Arnaud, L.; Rönnelid, J.; Malmström, V.; et al. Smoking and pre-existing organ damage reduce the efficacy of belimumab in systemic lupus erythematosus. Autoimmun Rev 2017, 16, 343–351. [Google Scholar] [CrossRef]
- Aringer, M.; Costenbader, K.; Daikh, D.; Brinks, R.; Mosca, M.; Ramsey-Goldman, R.; Smolen, J.S.; Wofsy, D.; Boumpas, D.T.; Kamen, D.L.; et al. 2019 European League Against Rheumatism/American College of Rheumatology Classification Criteria for Systemic Lupus Erythematosus. Arthritis Rheumatol 2019, 71, 1400–1412. [Google Scholar] [CrossRef]
- Gladman, D.D.; Ibañez, D.; Urowitz, M.B. Systemic lupus erythematosus disease activity index 2000. J Rheumatol 2002, 29, 288–291. [Google Scholar] [PubMed]
- Cossarizza, A.; Chang, H.D.; Radbruch, A.; Akdis, M.; Andrä, I.; Annunziato, F.; Bacher, P.; Barnaba, V.; Battistini, L.; Bauer, W.M.; et al. Guidelines for the use of flow cytometry and cell sorting in immunological studies. Eur J Immunol 2017, 47, 1584–1797. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Distribution of peripheral blood B cell subsets and their surface expression of BCMA in 86 SLE patients under standard-of-care (SLE-SOC) and 30 HCs. (A) Dot plots illustrate the gating strategy of flow cytometry experiments. (B) Histograms show representative examples of BCMA expression in SLE, HC and isotype control (iso-ctr). (C) Frequencies of CD19+ B cell subsets, particularly IgD+CD27- naïve B cells (NB), Mitotracker+IgD+ activated naïve B cells (aNB), IgD-CD27- double-negative B cells (DN), IgD+CD27+ non-switched memory B cells (NSM), IgD-CD27+ switched memory B cells (SM), IgD-CD27highHLA-DRhigh plasmablasts (PB) and IgD-CD27highHLA-DRlow plasma cells (PC). (D) BCMA expression on B cell subsets. (E) Comparison of BCMA expression among different B cell subsets in HC and (F) in SLE-SOC. Median values with interquartile ranges are presented. Comparison between HC and SLE-SOC was performed using the Mann-Whitney test, and comparison of BCMA expression between different B cell subsets with Kruskal-Wallis test. * p < 0.05; ** p < 0.01; *** p < 0.001; **** p < 0.0001. MFI: median fluorescence intensity.
Figure 1.
Distribution of peripheral blood B cell subsets and their surface expression of BCMA in 86 SLE patients under standard-of-care (SLE-SOC) and 30 HCs. (A) Dot plots illustrate the gating strategy of flow cytometry experiments. (B) Histograms show representative examples of BCMA expression in SLE, HC and isotype control (iso-ctr). (C) Frequencies of CD19+ B cell subsets, particularly IgD+CD27- naïve B cells (NB), Mitotracker+IgD+ activated naïve B cells (aNB), IgD-CD27- double-negative B cells (DN), IgD+CD27+ non-switched memory B cells (NSM), IgD-CD27+ switched memory B cells (SM), IgD-CD27highHLA-DRhigh plasmablasts (PB) and IgD-CD27highHLA-DRlow plasma cells (PC). (D) BCMA expression on B cell subsets. (E) Comparison of BCMA expression among different B cell subsets in HC and (F) in SLE-SOC. Median values with interquartile ranges are presented. Comparison between HC and SLE-SOC was performed using the Mann-Whitney test, and comparison of BCMA expression between different B cell subsets with Kruskal-Wallis test. * p < 0.05; ** p < 0.01; *** p < 0.001; **** p < 0.0001. MFI: median fluorescence intensity.
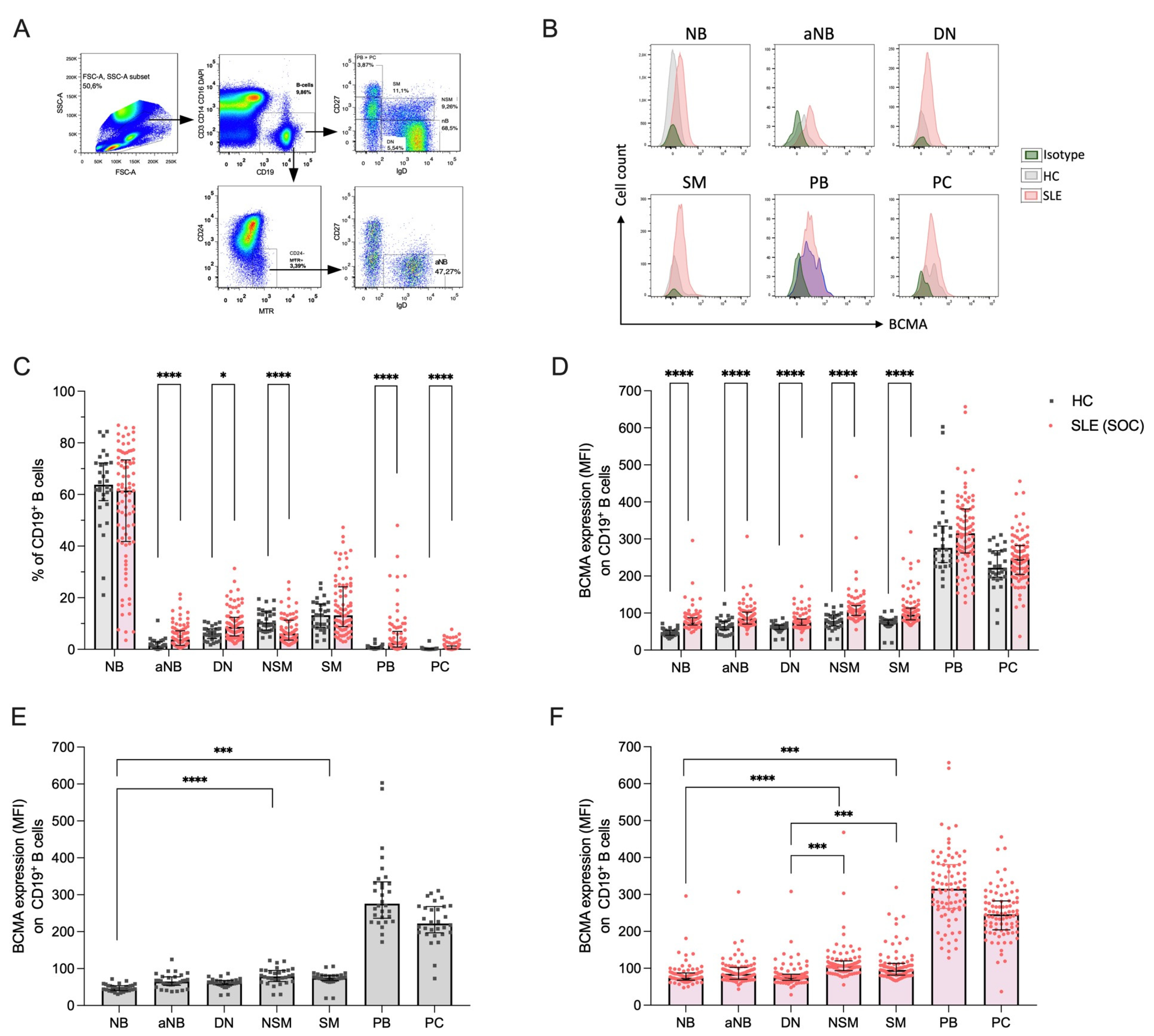
Figure 2.
Soluble markers of the BAFF-APRIL system and their correlations in 30 healthy controls (HC) and 86 SLE patients under standard-of-care (SLE-SOC). (A) Median values of soluble BCMA, (B) soluble TACI, and (C) BAFF. (D) Heatmap of correlations between sBCMA, sTACI, sBAFF, and BCMA expression on B cells (mBCMA BC) in HC, and (E) SLE-SOC patients. (F) Heatmap of correlations between BCMA expression on B cells (mBCMA BC), BCMA expression on plasmablasts (mBCMA PB), soluble BCMA, TACI, and BAFF with clinical and serologic variables, particularly clinical SLEDAI-2K (cSLEDAI) scores, levels of anti-dsDNA antibodies (anti-dsDNA), complement 3 (C3) levels and plasmablast (PB) frequency. Median values with interquartile ranges are presented. Correlation analyses were performed using the Spearman ranked test, and the Spearman’s correlation coefficient (r) was calculated. * p < 0.05; ** p < 0.01; *** p < 0.001; **** p < 0.0001.
Figure 2.
Soluble markers of the BAFF-APRIL system and their correlations in 30 healthy controls (HC) and 86 SLE patients under standard-of-care (SLE-SOC). (A) Median values of soluble BCMA, (B) soluble TACI, and (C) BAFF. (D) Heatmap of correlations between sBCMA, sTACI, sBAFF, and BCMA expression on B cells (mBCMA BC) in HC, and (E) SLE-SOC patients. (F) Heatmap of correlations between BCMA expression on B cells (mBCMA BC), BCMA expression on plasmablasts (mBCMA PB), soluble BCMA, TACI, and BAFF with clinical and serologic variables, particularly clinical SLEDAI-2K (cSLEDAI) scores, levels of anti-dsDNA antibodies (anti-dsDNA), complement 3 (C3) levels and plasmablast (PB) frequency. Median values with interquartile ranges are presented. Correlation analyses were performed using the Spearman ranked test, and the Spearman’s correlation coefficient (r) was calculated. * p < 0.05; ** p < 0.01; *** p < 0.001; **** p < 0.0001.
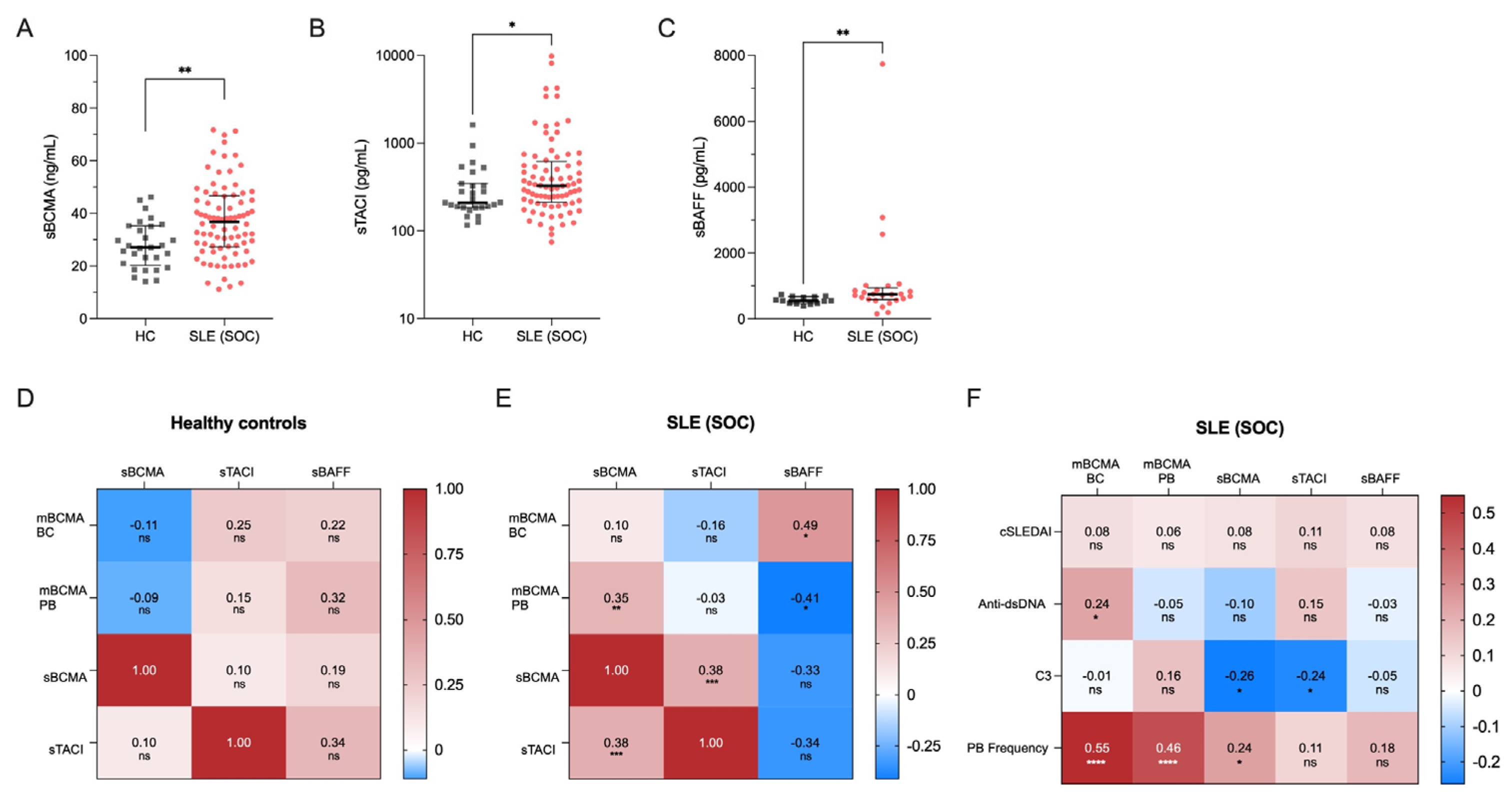
Figure 3.
Effects of belimumab treatment on BCMA expression on B cell subsets. (A) BCMA expression levels on different B cell subsets in 86 SLE patients under standard of care (SLE-SOC) compared to 14 SLE patients under belimumab treatment (SLE-BEL). (B) BCMA expression on different B cell subsets in SLE-BEL patients compared to 30 HCs. (C) Levels of soluble BCMA (sBCMA) and (D) soluble TACI (sTACI) in a cohort of 9 SLE patients before and at a median of 8 months after initiating belimumab treatment. Median values with interquartile ranges are presented. The Mann-Whitney U test was performed to compare SLE-SOC with SLE-BEL and HC. The Wilcoxon signed-rank test was used to compare sBCMA and sTACI before and during belimumab treatment. * p < 0.05; ** p < 0.01; *** p < 0.001; **** p < 0.0001.
Figure 3.
Effects of belimumab treatment on BCMA expression on B cell subsets. (A) BCMA expression levels on different B cell subsets in 86 SLE patients under standard of care (SLE-SOC) compared to 14 SLE patients under belimumab treatment (SLE-BEL). (B) BCMA expression on different B cell subsets in SLE-BEL patients compared to 30 HCs. (C) Levels of soluble BCMA (sBCMA) and (D) soluble TACI (sTACI) in a cohort of 9 SLE patients before and at a median of 8 months after initiating belimumab treatment. Median values with interquartile ranges are presented. The Mann-Whitney U test was performed to compare SLE-SOC with SLE-BEL and HC. The Wilcoxon signed-rank test was used to compare sBCMA and sTACI before and during belimumab treatment. * p < 0.05; ** p < 0.01; *** p < 0.001; **** p < 0.0001.

Table 1.
Demographics and Clinical Characteristics of Investigated SLE Patients and Controls.
| Characteristics | SLE patients (n=100) | Healthy controls (n=30) | P-Value |
|---|---|---|---|
| Age, median (range) | 38.5 (19–80) | 28.7 (22–59) | 0.11 |
| Gender, female, n (%) | 90 (90) | 26 (86.7) | 0.61 |
| Ethnicity, n (%) Caucasian Asian African Latin American |
90 (90) 1 (1) 6 (6) 3 (3) |
26 (86.7) 2 (6.7) 2 (6.7) 0 (0) |
0.61 0.07 0.89 0.34 |
| Disease duration, median years, (range) | 6.5 (6–40) | ||
| Disease activity SLEDAI-2K, median (range) Clinically active, n (%) DORIS Remission n (%) |
4 (0–26) 46 (46) 54 (54) |
||
| Active clinical manifestations at time of presentation, n (%) Musculoskeletal Mucocutaneous Polyserositis Nephritis CNS Cytopenia |
33 (33) 18 (18) 3 (3) 5 (5) 2 (2) 43 (43) |
||
| Serology Anti-dsDNA positive, n (%) C3-deficiency, n (%) |
71 (71) 71 (71) |
||
| Medication, n (%) Prednisolone Prednisolone dosage (mg/d), median Prednisolone ≥ 7.5 mg/d Hydroxychloroquine Methotrexate Azathioprine Mycophenolate mofetil Calcineurin inhibitors Belimumab |
75 (75) 5.0 30 (30) 78 (78) 11 (11) 34 (34) 14 (14) 5 (5) 14 (14) |
Abbreviations: SLEDAI-2K, Systemic Lupus Erythematosus Disease Activity-Index 2000. Statistical analysis of age differences was conducted using the Mann-Whitney test, and sex differences were analyzed with the chi-square test.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



