
Submitted:
30 August 2024
Posted:
03 September 2024
You are already at the latest version
Abstract
Pancreatic ductal adenocarcinoma (PDAC) remains one of the most lethal malignancies, characterized by its aggressive progression and dismal prognosis. Advances in epigenetic profiling, specifically DNA methylation analysis, have significantly deepened our understanding of PDAC pathogenesis. This review synthesizes findings from recent genome-wide DNA methylation studies, notably those by Espinet et al. and Wang et al., which have delineated a complex DNA methylation landscape differentiating between normal and cancerous pancreatic tissues, as well as across various stages and molecular subtypes of PDAC. These studies identified specific differentially methylated regions (DMRs) that not only enhance our grasp of the epigenetic drivers of PDAC but also offer potential biomarkers for early diagnosis and prognosis, enabling the customization of therapeutic approaches. The review further explores how DNA methylation profiling could facilitate the development of subtype-tailored therapies, potentially improving treatment outcomes based on precise molecular characterizations. Overall, leveraging DNA methylation alterations as functional biomarkers holds promise for advancing our understanding of disease progression and refining PDAC management strategies, which could lead to improved patient outcomes and a deeper comprehension of the disease’s underlying biological mechanisms.
Keywords:
DNA methylation
; pancreatic ductal adenocarcinoma
; pancreatic cancer
; whole-genome bisulfite sequencing
; metastasis
Introduction
Pancreatic cancer is the third leading cause of cancer-related deaths in the United States and is projected to take the place of colorectal cancer as the second leading cause by 2030 [1,2]. It is relatively uncommon but exceptionally lethal, characterized by the lowest five-year survival rate among common cancers at just 13% [1]. The most prevalent and most malignant form of pancreatic cancer is pancreatic ductal adenocarcinoma (PDAC), accounting for 90% of pancreatic cancer cases and resulting in nearly 50,000 deaths annually in the US alone [3]. Late diagnosis often due to asymptomatic early stages, frequent metastasis, and lack of effective treatment contributes to the poor survival outcomes of the disease. Less than 20% of patients are eligible for surgical resection of tumors, and even then, resistance to chemotherapy and radiotherapy, in part due to the characteristic dense stroma and desmoplastic reaction, limits the effectiveness of treatment [4]. Next-generation sequencing (NGS) has facilitated the discovery as well as the characterization of genetic aberrations that contribute to PDAC disease initiation and progression [5]. More specifically, 90-92% of PDAC patients have gain-of-function mutations in the KRAS proto-oncogene in exocrine pancreas cells. These oncogenic mutations lead to the initiation of disease and the formation of precancerous lesions known as pancreatic intraepithelial neoplasia (PanIN) [6,7,8]. As these lesions progress, inactivating mutations or deletions of classic tumor suppressor genes such as CDKN2A, TP53, and SMAD4 further promote disease progression [5,9,10]. Despite a well-understood genetic mutational landscape, therapeutic applications remain limited, and understanding the mechanism of disease progression, namely metastasis, continues to pose a significant challenge.
Over the years, PDAC research has shifted to include the effects of epigenetic mechanisms on cancer progression [11,12,13,14]. It has become increasingly evident that epigenetic mechanisms play a role in disease pathology. Through their mechanism of transcription regulation, epigenetic alterations have profound effects on gene expression and genome stability and can equally promote oncogenic pathways. DNA methylation is one epigenetic mechanism that has widely been found dysregulated in many cancers, PDAC included [15,16,17,18]. DNA methylation predominately takes place at cytosines followed by guanines, or CpG regions. It is known to be negatively associated with gene expression at promoter and enhancer regions while being positively associated with gene expression at gene body regions, although developments in methylome analysis and further studies have suggested the relationship between methylation and gene expression is more complex than previously anticipated [19,20]. Advanced methods of investigating the methylome have enabled greater coverage of CpG sites and even non-CpG sites with higher resolution, allowing for more comprehensive studies on the role of DNA methylation to be conducted. However, further exploration is constrained by the accessibility and quality of the tumor samples. The dense stromal contents within the tumor microenvironment leads to stromal interference, posing the problem of low cellularity when attempting to isolate neoplastic epithelial cells for investigating their molecular mechanisms. These epithelial cells are the minority among the large diversity of cell types that comprise the surrounding stroma, complicating the isolation of pure neoplastic populations. As a result, extensive efforts are being made toward understanding disease characterization and progression, along with the development of original techniques to effectively isolate tumor cells.
In this review, we will discuss the current status of studies investigating the role of DNA methylation in PDAC progression [14]. We will equally highlight two recently published comprehensive DNA methylome studies in PDAC patients each using unique techniques to circumvent the universal challenge of stromal infiltration faced when studying this disease, and mention potential future directions [21,22].
Regulation of DNA Methylation
DNA methylation is a key epigenetic modification mechanism involving the transfer of a methyl group from S-adenosyl-L-methionine (SAM) to the 5’ carbon of cytosine, resulting in the formation of 5-methylcytosine [23]. This process primarily occurs within CpG islands—genomic regions characterized by a high frequency of cytosine-guanine dinucleotides [24]. DNA methylation plays a crucial role in regulating gene expression without altering the DNA sequence, thereby contributing to the complexity and diversity of the genome. It also serves as a stable epigenetic marker that can be inherited across multiple cell divisions and even generations, making it vital for transcriptional regulation, development, differentiation, genomic imprinting, stability, and the onset of various diseases [25,26]. Recent advances in epigenetics have shed light on the diverse mechanisms governing DNA methylation, with the majority of regulators being classified into three main categories: writers, erasers, and readers [27].
Writers include the DNA methyltransferase (DNMT) family, which catalyzes the methylation process. The DNMT family consists of DNMT1, DNMT2, DNMT3a, DNMT3b, DNMT3L, and their respective isoforms. Notably, DNMT3a and DNMT3b, often referred to as de novo methyltransferases, establish methylation patterns during development by methylating unmethylated cytosines [28]. DNMT1, on the other hand, is essential for maintaining the DNA methylome, as it preferentially targets hemimethylated DNA during replication to ensure the transmission of methylation patterns from the parent to the daughter strand, thereby functioning as a cellular memory system [29,30].
Erasers are responsible for demethylation through either active or passive mechanisms. Active demethylation can occur through several processes: 1) enzymatic removal of the methyl group; 2) nucleotide or base excision repair, which removes methylated nucleotides; 3) oxidation of 5-methylcytosine to 5-hydroxymethylcytosine by the TET protein family, which prevents DNMT1 recognition and maintenance [31,32].
The three categories of readers are the methyl-CpG-binding domain (MBD) family (MeCP2, MBD1, 2, 3 & 4), SRA domain-containing proteins, and Methyl-CpG binding zinc fingers. Readers interpret methylation signals to regulate gene transcription, with their effects varying across different genomic regions [33]. In promoter and enhancer regions, DNA methylation is generally associated with reduced gene expression [34,35,36,37,38]. Methylation at the promoter CpGs can silence gene expression through several mechanisms: 1) physical obstruction of transcription factor binding by the methyl group in the DNA major groove; 2) failure of transcription factors to recognize methylated promoter CpG sequences; 3) recruitment of MBD proteins to methylated DNA, which in turn attract histone deacetylases and chromatin remodelers, leading to the formation of compact, inactive heterochromatin [35,36,38] (Figure 1). Conversely, DNA methylation in intergenic regions and gene bodies is positively correlated with transcription elongation [39,40]. While studies have demonstrated that DNA methylation in these regions plays a role in maintaining genome stability, the regulatory mechanisms behind this process remain underexplored [41,42].
Aberrant DNA Methylation in PDAC
Dysregulation of DNA methylation is found to be closely associated with cancers. Numerous studies have highlighted that DNA methylation patterns deviate significantly from their normal states across cancer types [15,16,43] (Figure 1).
To detect these aberrant methylation patterns in PDAC, several sodium bisulfite-based techniques have been employed. These methods are based on the principle that sodium bisulfite treatment selectively converts unmethylated cytosine residues into uracil, while 5-methylcytosine residues remain unchanged, allowing for the differentiation between methylated and unmethylated sites [44,45]. One of the earliest techniques developed was bisulfite modification followed by methylation-specific PCR (MSP). Despite being easy to conduct, MSP has limitations: 1) It requires prior knowledge of the candidate genes, as only a limited number of CpG sites can be analyzed per primer pair [46]; 2) PCR bias leads to more efficient amplification of T-rich unmethylated sequences, compromising the accuracy of DNA methylation quantification [47]; 3) The high concentration of bisulfite during conversion can degrade up to 90% of the DNA, leading to PCR amplification failures [48]. Microarray sequencing offers a more efficient alternative to MSP, enabling the simultaneous analysis of thousands of regions of interest within the DNA methylome. However, this method still covers only a small fraction of the methylome, often missing key regulatory and non-coding sequences.
Advancements in NGS have facilitated the development and widespread application of reduced-representation bisulfite sequencing (RRBS) and whole-genome bisulfite sequencing (WGBS). RRBS uses methylation-sensitive restriction enzymes to enrich the genomic DNA for CpG-rich regions, enabling the sequencing of methylated regions that were previously difficult to analyze with conventional bisulfite-based methods, such as repeated sequences [49,50]. WGBS, on the other hand, is the only method that provides a comprehensive profile of the DNA methylome at single base-pair resolution, identifying a significantly larger number of differentially methylated regions (DMRs) [51]. However, despite the vast number of DMRs identified through WGBS, most studies rely on association-based methods to identify candidate genes and pathways, often without establishing causality versus mere correlation in DNA methylation patterns. Further in vitro and in vivo functional studies are needed to unravel the mechanisms by which these altered DMRs drive pancreatic cancer progression and correlate with patient survival outcomes.
Using the aforementioned methods, researchers were able to identify multiple gene promoters that are aberrantly methylated within PDAC, contributing to cancer aggressiveness (Table 1). Similar to many other types of cancers, promoter CpGs of DNA repair and tumor suppressor genes are frequently hypermethylated in PDAC, resulting in repression of these genes [52]. Conversely, oncogene promoters often exhibit hypomethylation, which drives tumor proliferation [53]. These aberrant methylation patterns are shown to be strongly associated with increased cell proliferation, invasion, metastasis, poorer prognosis, and reduced survival rates in PDAC patients (Figure 1).
Cellular pathways regulated by differential DNA methylation have also been identified. For example, Mishra et al. performed a differential methylation analysis on level-3 pancreatic cancer data from TCGA, identifying 23,688 CpG sites with differential methylation between tumor and normal samples—13,501 of these were hypermethylated, and 10,187 were hypomethylated [8,54]. They mapped 4,252 genes to these differentially methylated CpGs, highlighting aberrant methylation in methylation writers, erasers, and readers, as well as homeobox genes, and genes involved in pancreatic development and signaling. Subsequent pathway and gene ontology (GO) enrichment analyses revealed key enriched pathways including cancer-related pathways, MAPK signaling, calcium signaling, focal adhesion, regulation of the actin cytoskeleton, and gap junction pathways. Using Ingenuity Pathway Analysis (IPA), they identified top canonical pathways such as axonal guidance signaling, G-protein-coupled receptor signaling, hepatic fibrosis/hepatic stellate cell activation, cancer molecular mechanisms, and Sertoli cell junction pathways. Unsupervised hierarchical clustering of the data revealed three distinct patient clusters, each characterized by unique patterns of hyper- or hypomethylated CpGs, suggesting the presence of three potential pancreatic cancer subtypes within TCGA. In another study from an Indian cohort, hyper- and hypomethylation are shown to regulate 91 genes, many involved in ion transport regulation, interferon alpha/beta signaling, morphogenesis, development, and transcriptional dysregulation pathways [55]. Additionally, Bailey et al., using data from the Australian Pancreatic Cancer Genome Initiative (APGI), identified four stable PDAC subtypes: squamous, pancreatic progenitor, immunogenic, and aberrantly differentiated endocrine exocrine (ADEX) [56]. Each subtype was distinguished by distinct gene expression and DNA methylation patterns, as well as specific enriched pathways.
Collectively, these studies reveal that the DNA methylome undergoes significant rearrangement during PDAC progression, altering gene transcription patterns and pathways involved in cancer cell invasion and migration. These changes can be used to predict survival outcomes, tumor grades, and histopathological stages in PDAC patients. The strong association between methylation status and patient outcomes, particularly in genes like PD-L1 (Table 1), highlights the active role of epigenetic modifications in cancer dynamics, suggesting that DNA methylation markers could improve prognostic assessments and therapeutic targeting of pancreatic cancer.
One major challenge these above studies faced while profiling PDAC patient samples has been how to isolate or enrich neoplastic populations in given tumor samples to ensure tumor purity. PDAC is characterized by low cellularity and intense desmoplasia, resulting in a low signal-to-noise ratio that complicates the study of the molecular and cellular mechanisms of PDAC tissues. The neoplastic cellularity of the tumor samples used in the TCGA dataset ranged from 0% to 53%, with a median of 18% based on their pathology review. The tumor purity estimates from whole exome sequencing data ranged from 9% to 89%, with a median of 33% [8]. In the Bailey et al. study, among the pancreatic cancer samples analyzed, 179 samples exhibited a cellularity greater than 40%, estimated by a combination of qPure analysis of high-density SNP profiles and KRAS amplicon sequencing [56]. These samples were subsequently utilized for transcriptomic analyses to balance the stromal gene expression. However, a significant portion of the samples (n = 204) displayed lower cellularity, ranging from 12% to 40%, leading to the exclusion of these patient samples from the downstream analyses. This substantial variance in cellularity, particularly the prevalence of low-cellularity samples, underscores the urgent need for innovative methods to enhance signal-to-noise ratios or to isolate pure neoplastic populations for more accurate downstream assays and analyses.
Recent WGBS studies on PDAC patients
To overcome this issue, Espinet et al. recently attempted to isolate EpCAM+ epithelial cells and exclude cells with CD45, an immune cell marker from surgically resected primary tumor tissues using FACS [22]. They then performed WGBS with these sorted epithelial cells. While the authors showed only a small percentage of EpCAM−/CD45− cells had KRAS mutant reads in RNA-seq data, this approach could potentially neglect tumor cell heterogeneity that could account for epithelial PDAC cells that have lost EpCAM expression. In addition, it should be noted that only early-stage PDAC tumors were profiled in this study likely due to ineligibility for surgical resection in advanced-stage PDAC patients. Nonetheless, Espinet et al. identified 56,177 DMRs with an overall predominant trend of hypomethylation in the PDAC-derived samples compared to those derived from normal pancreas. Principal component analysis (PCA) divided the tumor-derived samples into 2 distinct clusters based on DMRs. These two clusters were found to be differentially enriched in an interferon (IFN) signaling signature, as well as signatures for the two most common molecular subtypes, progenitor-like and basal-like/squamous. High expression of this interferon signature was correlated with poor prognosis and was more prevalent in tumors classified as the aggressive squamous subtype. Within the more aggressive IFNhigh cluster, a trend of hypomethylation was found at expected partially methylated domains (PMDs), as well as in other genomic locations, notably outside of CpG islands. In particular, hypomethylation at repetitive elements (LTR/ERV, LINEs, and SINEs) was identified and associated with higher expression of LTR/ERV and LINE-derived transcripts, enrichment of gene signatures linked to dsRNA response, and increased IFN signaling. Their functional in vitro/in vivo experiments suggested that the interaction of PDAC cells with high IFN signaling reprograms and activates stromal cells in a cell-intrinsic process, creating a pro-tumorigenic microenvironment. Moreover, this IFN signaling was shown to be mediated by STAT1, as a JAK/STAT inhibitor impaired the tumor growth in this context. Given the close association between IFN signaling, hypomethylation at repeat elements, dsRNA response, and more aggressive cancer phenotypes, the hypomethylation of repetitive elements was suggested to activate tumoral IFN signaling, which in turn reshapes TME into a pro-tumorigenic environment, ultimately resulting in aggressive cancer phenotypes (Figure 1).
The analysis of surgically resected tumors with the aforementioned method can potentially cause sample-bias, neglecting late-stage PDAC patient samples. Importantly, this issue is not limited to the study above; extensive datasets like TCGA and many other studies also exhibit similar sample biases [8]. Utilization of patient-derived organoid models can be an alternative way to complement this deficiency [57,58,59,60,61]. The ability to expand organoid cultures from a small amount of tissue such as fine needle aspiration (FNA) biopsy allows us to grow cells from primary tumors, metastatic lesions, pleural effusion, and ascites of PDAC patients [57,62,63,64,65]. This method significantly enhances the representation of late-stage PDAC patients in research, providing a more comprehensive understanding of the disease’s progression. In the 3D pancreatic organoid cultures, only epithelial cells can grow and form organoids in the presence of growth factors and WNT agonists. This method effectively reduces stromal contamination in the samples, as well as ensures capture/expansion of all relevant epithelial cells, making it suitable for any omics studies.
To better represent various stages of PDAC patients’ epigenome, our group utilized a panel of patient-derived organoids (PDOs, n = 35) including human normal ductal organoids (hNs, n = 4), early-stage (hTs, n = 12), and late-stage PDAC (hTs, n = 19) [21]. DMR analysis between hNs and hTs identified 374 significant DMRs, with the majority (87%) being hypermethylated in PDAC organoids. These DMRs tended to be enriched in bivalent transcription start sites (TSS) and enhancer regions, suggesting that developmentally poised genes are more prone to methylation changes during PDAC progression. Subsequent GO analysis suggested that substantial epigenetic alterations occur as PDAC develops from normal pancreatic ducts, influencing the genes related to transcription, ion channels, membranes, and DNA binding. Comparative DMR analysis between early-stage and late-stage PDAC organoids further revealed 5,374 DMRs, with the vast majority being hypomethylated in late-stage PDAC PDOs. These regions are significantly enriched in genes associated with cell adhesion, synapse signaling, and neurotransmitter activity, indicating a shift in cellular function and interaction as the disease progresses. We further employed DMR analysis to distinguish two major molecular subtypes of PDAC—progenitor and squamous—identifying methylation signatures that could potentially enhance PDAC subtyping and prognosis predictions. [8,56,62,66,67]. The progenitor subtype exhibits a favorable prognosis characterized by the expression of transcription factors required for pancreatic endoderm cell fate determination, such as HNF4A and GATA6 [56,68,69]. Conversely, the squamous subtype, associated with a worse prognosis, shows loss of these endoderm specification TFs. Previously, DNA methylation has been implicated in the silencing of these genes [8,56]. Our study highlighted a potential role of GATA6 TF in maintaining the hypomethylated status of progenitor subtype-associated genes for their expression, likely through recruiting TET enzymes to its binding sites.
Overall, WGBS profiling of PDAC PDOs, which had not yet been explored, provides a unique opportunity to explore the DNA methylome in PDAC tumors and identify novel diagnostic markers. While it remains to be addressed whether organoid culture conditions such as growth factors and WNT signaling alter epigenomes of organoid cultures, it is obvious that PDOs provide a promising window for exploring the DNA methylome in PDAC tumors. It is important to note that there was a difference in which groups of samples had DMRs that were hypermethylated or hypomethylated across the two studies, as well as the genomic region of these DMRs. Nonetheless, both methods were proficient in segregating tumor cells from their surrounding stroma, offering new insight and direction for studying the DNA methylation landscape of PDAC.
Conclusion
The genome-wide profiling of DNA methylation of PDAC specimens in a single base pair resolution through studies such as those conducted by Espinet et al. and Wang et al. has provided an invaluable resource for understanding the epigenetic alterations that characterize this aggressive cancer. These studies reveal a complex landscape of differential methylation that not only distinguishes between normal and cancerous pancreatic tissues but also between different stages and molecular subtypes of the disease. The identification of specific DMRs offers potential biomarkers that could be critical for early diagnosis, prognosis, and the development of targeted therapies.
Looking to the future, the application of methylated cell-free DNA (cfDNA) circulating in the blood as a non-invasive diagnostic tool holds promise for the detection and management of PDAC. Methylation analysis of cfDNA serves as liquid-biopsy-based biomarkers, offering a practical approach for routine screening and monitoring of PDAC, which potentially enables more precise tracking of disease progression and treatment response. As the field advances, integrating findings from detailed methylome analyses with cfDNA technologies could lead to significant improvements in the specificity and sensitivity of PDAC diagnostics [70].

Table 1.
Summary of gene promoters that are differentially methylated in PDAC compared to normal pancreatic tissues and their impacts on PDAC progression. ↑ denotes increase and ↓ denotes decrease.
Table 1.
Summary of gene promoters that are differentially methylated in PDAC compared to normal pancreatic tissues and their impacts on PDAC progression. ↑ denotes increase and ↓ denotes decrease.
| Gene Promoter | Hypermethylated or Hypomethylated in PDAC | Affected Gene Expression Level | Molecular Function of the Gene | Functional Phenotype in PDAC | Method | Reference |
| PD-L1 promoter | Hypomethylated | PD-L1 ↑ | Programmed cell death ligand | Aggressive clinical phenotypes ↑, immune cell infiltration ↓, expression of immune checkpoint genes ↓, overall survival ↓ | Microarray | [71] |
| HNF4A promoter | Hypermethylated |
HNF4A ↓ | Controls genes that are especially important for development and function of beta cells in the pancreas; regulate cell proliferation through the Wnt/beta-Catenin Pathway | Patient survival ↓, cell growth, colony formation, and invasiveness ↑, expression decreases during early PDAC stages | Targeted bisulfite sequencing | [72] |
| BEND4 promoter | Hypermethylated | BEND4 ↓ | BEN domain-containing protein 4 involves in non-homologous end-joining signaling by interacting with Ku80 and promotes DNA damage repair; inhibits cell growth, migration and invasion | Prognosis ↓, synthetic lethality towards ATM inhibitor | MSP | [73] |
|
NPY and FAIM2 promoters |
Hypermethylated | NPY ↓, FAIM2 ↓ | NPY is involved in cell motion, invasion, and proliferation, and its hypermethylation is observed in certain cancers. FAIM2 is associated with apoptosis inhibition. | Prognosis ↓, progress to later stages ↑ | Microarray | [55] |
| CDH13 promoter | Hypermethylated | CDH13 ↓ | Cadherin, involved in cell-cell adhesion and contact inhibition of cell growth | Invasion and metastasis ↑ | MSP | [74] |
| Cyclin D2 promoter | Hypermethylated | Cyclin D2 ↓ | Involved in cell cycle regulation, cell differentiation, and neoplastic transformation | Unknown in PDAC context | MSP | [75] |
| SPARC promoter | Hypermethylated | SPARC ↓ | A multifunctional glycoprotein, involved in cell proliferation, cell spreading, adhesion, motility, and invasion | Proliferation ↑ | MSP, microarray | [76] |
| DKK1 promoter | Hypermethylated | DKK1 ↓ | Regulates Wnt signaling by binding to the Wnt coreceptor lipoprotein-related protein-5 (LRP5)/Arrow | Proliferation ↑, migration & invasion ↑ | MSP | [77] |
| MUC Promoter | Hypomethylated | Not tested | Mucins, bind to pathogens as part of the immune system; renewal and differentiation of the epithelium; involved in cell adhesion and signaling | Unknown in PDAC context | Microarray | [78] |
Moreover, the DMRs identified in these studies could serve as a critical foundation for developing cfDNA-based diagnostics. By leveraging such epigenetic signatures, researchers and clinicians might be able to distinguish between benign conditions and malignant transformations within the pancreas more effectively, overcoming current diagnostic challenges. This approach not only promises to enhance our diagnostic capabilities but also opens new avenues for personalized medicine in PDAC, where treatment strategies can be tailored based on specific epigenetic profiles. The ability to identify molecular subtypes through methylation profiling significantly enhances the potential for subtype-tailored therapies, which could improve treatment outcomes. For instance, patients identified with the classical (progenitor) subtype of PDAC might benefit specifically from tailored treatments such as FOLFIRINOX, a regimen known for its efficacy in this group.
In conclusion, the identification of recurrent DMRs in PDAC underscores the functional significance of epigenetic alterations in the progression of this disease. Utilizing these DNA methylation alterations as functional biomarkers offers a promising avenue for advancing our understanding of disease progression and molecular subtyping. This approach not only enhances the precision of diagnostic and prognostic models but also opens possibilities for the development of targeted therapies tailored to specific epigenetic profiles. By integrating these epigenetic insights, we can significantly improve the strategic management of PDAC, potentially leading to better patient outcomes and a deeper comprehension of the disease’s underlying mechanisms.
Acknowledgments
We would like to thank all members of the C.-I.H. laboratory for helpful discussions and suggestions for the manuscript. P.L. is supported by the Charles and Nanci Cooper Undergraduate Research Fellowship at University of California Davis. C.-I.H. is supported by the National Cancer Institute (NCI) K22CA226037 and R37CA249007, University of California Research Initiatives, Cancer Research Coordinating Committee (CRCC) C21CR2020, and the UC Davis Comprehensive Cancer Center Pilot Grant (NCI P30CA093373).
References
- Siegel, R.L., A.N. Giaquinto, and A. Jemal, Cancer statistics, 2024. CA Cancer J Clin, 2024. 74(1): p. 12-49.
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Zheng, Y.; Yang, F.; Zhu, L.; Zhu, X.-Q.; Wang, Z.-F.; Wu, X.-L.; Zhou, C.-H.; Yan, J.-Y.; Hu, B.-Y.; et al. The Molecular Biology of Pancreatic Adenocarcinoma: Translational Challenges and Clinical Perspectives. Signal Transduct. Target. Ther. 2021, 6, 1–23. [Google Scholar] [CrossRef]
- Zeng, S., et al., Chemoresistance in Pancreatic Cancer. Int J Mol Sci, 2019. 20(18).
- Waddell, N.; Pajic, M.; Patch, A.-M.; Chang, D.K.; Kassahn, K.S.; Bailey, P.; Johns, A.L.; Miller, D.; Nones, K.; Quek, K.; et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature 2015, 518, 495–501. [Google Scholar] [CrossRef] [PubMed]
- Kanda, M.; Matthaei, H.; Wu, J.; Hong, S.M.; Yu, J.; Borges, M.; Hruban, R.H.; Maitra, A.; Kinzler, K.; Vogelstein, B.; et al. Presence of somatic mutations in most early-stage pancreatic intraepithelial neoplasia. Gastroenterology 2012, 142, 730–733. [Google Scholar] [CrossRef] [PubMed]
- Waters, A.M.; Der, C.J. KRAS: The Critical Driver and Therapeutic Target for Pancreatic Cancer. Cold Spring Harb. Perspect. Med. 2018, 8, a031435. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network. Electronic address, a.a.d.h.e. and N. Cancer Genome Atlas Research, Integrated Genomic Characterization of Pancreatic Ductal Adenocarcinoma. Cancer Cell, 2017. 32(2): p. 185-203 e13.
- Halbrook, C.J.; Lyssiotis, C.A.; di Magliano, M.P.; Maitra, A. Pancreatic cancer: Advances and challenges. Cell 2023, 186, 1729–1754. [Google Scholar] [CrossRef]
- Jones, S.; Zhang, X.; Parsons, D.W.; Lin, J.C.-H.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Kamiyama, H.; Jimeno, A.; et al. Core Signaling Pathways in Human Pancreatic Cancers Revealed by Global Genomic Analyses. Science 2008, 321, 1801–1806. [Google Scholar] [CrossRef]
- Xu, J.; Roe, J.; Lee, E.; Tonelli, C.; Ji, K.Y.; Younis, O.W.; Somervile, T.D.; Yao, M.; Milazzo, J.P.; Tiriac, H.; et al. Engrailed-1 Promotes Pancreatic Cancer Metastasis. Adv. Sci. 2023, 11, e2308537. [Google Scholar] [CrossRef]
- Roe, J.-S.; Hwang, C.-I.; Somerville, T.D.; Milazzo, J.P.; Lee, E.J.; Da Silva, B.; Maiorino, L.; Tiriac, H.; Young, C.M.; Miyabayashi, K.; et al. Enhancer Reprogramming Promotes Pancreatic Cancer Metastasis. Cell 2017, 170, 875–888.e20. [Google Scholar] [CrossRef]
- Kim, H.-R.; Yim, J.; Yoo, H.-B.; Lee, S.E.; Oh, S.; Jung, S.; Hwang, C.-I.; Shin, D.-M.; Kim, T.; Yoo, K.H.; et al. EVI1 activates tumor-promoting transcriptional enhancers in pancreatic cancer. NAR Cancer 2021, 3, zcab023. [Google Scholar] [CrossRef]
- Wang, S.S.; Xu, J.; Ji, K.Y.; Hwang, C.-I. Epigenetic Alterations in Pancreatic Cancer Metastasis. Biomolecules 2021, 11, 1082. [Google Scholar] [CrossRef] [PubMed]
- Saghafinia, S.; Mina, M.; Riggi, N.; Hanahan, D.; Ciriello, G. Pan-Cancer Landscape of Aberrant DNA Methylation across Human Tumors. Cell Rep. 2018, 25, 1066–1080.e8. [Google Scholar] [CrossRef] [PubMed]
- Paz, M.F.; Fraga, M.F.; Avila, S.; Guo, M.; Pollan, M.; Herman, J.G.; Esteller, M. A systematic profile of DNA methylation in human cancer cell lines. Cancer Res. 2003, 63, 1114–1121. [Google Scholar] [PubMed]
- Bergman, Y. and H. Cedar, DNA methylation dynamics in health and disease. Nat Struct Mol Biol, 2013. 20(3): p. 274-81.
- Lomberk, G.; Blum, Y.; Nicolle, R.; Nair, A.; Gaonkar, K.S.; Marisa, L.; Mathison, A.; Sun, Z.; Yan, H.; Elarouci, N.; et al. Distinct epigenetic landscapes underlie the pathobiology of pancreatic cancer subtypes. Nat. Commun. 2018, 9, 1978. [Google Scholar] [CrossRef]
- Spainhour, J.C.; Lim, H.S.; Yi, S.V.; Qiu, P. Correlation Patterns Between DNA Methylation and Gene Expression in The Cancer Genome Atlas. Cancer Informatics 2019, 18. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.; Sen, S.; Weeks, R.J.; Eccles, M.R.; Chatterjee, A. Promoter DNA Hypermethylation and Paradoxical Gene Activation. Trends Cancer 2020, 6, 392–406. [Google Scholar] [CrossRef]
- Wang, S.S.; Hall, M.L.; Lee, E.; Kim, S.-C.; Ramesh, N.; Lee, S.H.; Jang, J.-Y.; Bold, R.J.; Ku, J.-L.; Hwang, C.-I. Whole-genome bisulfite sequencing identifies stage- and subtype-specific DNA methylation signatures in pancreatic cancer. iScience 2024, 27, 109414. [Google Scholar] [CrossRef] [PubMed]
- Espinet, E.; Gu, Z.; Imbusch, C.D.; Giese, N.A.; Büscher, M.; Safavi, M.; Weisenburger, S.; Klein, C.; Vogel, V.; Falcone, M.; et al. Aggressive PDACs Show Hypomethylation of Repetitive Elements and the Execution of an Intrinsic IFN Program Linked to a Ductal Cell of Origin. Cancer Discov. 2020, 11, 638–659. [Google Scholar] [CrossRef]
- Robertson, K.D. DNA methylation and human disease. Nat. Rev. Genet. 2005, 6, 597–610. [Google Scholar] [CrossRef] [PubMed]
- Jabbari, K.; Bernardi, G. Cytosine methylation and CpG, TpG (CpA) and TpA frequencies. Gene 2004, 333, 143–149. [Google Scholar] [CrossRef]
- Li, E. and Y. Zhang, DNA methylation in mammals. Cold Spring Harb Perspect Biol, 2014. 6(5): p. a019133.
- Takahashi, Y.; Valencia, M.M.; Yu, Y.; Ouchi, Y.; Takahashi, K.; Shokhirev, M.N.; Lande, K.; Williams, A.E.; Fresia, C.; Kurita, M.; et al. Transgenerational inheritance of acquired epigenetic signatures at CpG islands in mice. Cell 2023, 186, 715–731.e19. [Google Scholar] [CrossRef]
- Biswas, S.; Rao, C.M. Epigenetic tools (The Writers, The Readers and The Erasers) and their implications in cancer therapy. Eur. J. Pharmacol. 2018, 837, 8–24. [Google Scholar] [CrossRef] [PubMed]
- Okano, M.; Bell, D.W.; Haber, D.A.; Li, E. DNA Methyltransferases Dnmt3a and Dnmt3b Are Essential for De Novo Methylation and Mammalian Development. Cell 1999, 99, 247–257. [Google Scholar] [CrossRef] [PubMed]
- Bestor, T.H.; Ingram, V.M. Two DNA methyltransferases from murine erythroleukemia cells: purification, sequence specificity, and mode of interaction with DNA. Proc. Natl. Acad. Sci. 1983, 80, 5559–5563. [Google Scholar] [CrossRef]
- Probst, A.V.; Dunleavy, E.; Almouzni, G. Epigenetic inheritance during the cell cycle. Nat. Rev. Mol. Cell Biol. 2009, 10, 192–206. [Google Scholar] [CrossRef]
- Wu, S.C. and Y. Zhang, Active DNA demethylation: many roads lead to Rome. Nat Rev Mol Cell Biol, 2010. 11(9): p. 607-20.
- Zhou, X.; Zhuang, Z.; Wang, W.; He, L.; Wu, H.; Cao, Y.; Pan, F.; Zhao, J.; Hu, Z.; Sekhar, C.; et al. OGG1 is essential in oxidative stress induced DNA demethylation. Cell. Signal. 2016, 28, 1163–1171. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Yang, M.; Wang, S.; Abbas, S.J.; Zhang, J.; Li, Y.; Shao, R.; Liu, Y. An Overview of Epigenetic Methylation in Pancreatic Cancer Progression. Front. Oncol. 2022, 12, 854773. [Google Scholar] [CrossRef]
- Angeloni, A.; Bogdanovic, O. Enhancer DNA methylation: implications for gene regulation. Essays Biochem. 2019, 63, 707–715. [Google Scholar] [CrossRef] [PubMed]
- Brenet, F.; Moh, M.; Funk, P.; Feierstein, E.; Viale, A.J.; Socci, N.D.; Scandura, J.M. DNA Methylation of the First Exon Is Tightly Linked to Transcriptional Silencing. PLOS ONE 2011, 6, e14524. [Google Scholar] [CrossRef]
- Du, Q. , et al., Methyl-CpG-binding domain proteins: readers of the epigenome. Epigenomics, 2015. 7(6): p. 1051-73.
- Skoulidis, F.; Cassidy, L.D.; Pisupati, V.; Jonasson, J.G.; Bjarnason, H.; Eyfjord, J.E.; Karreth, F.A.; Lim, M.; Barber, L.M.; Clatworthy, S.A.; et al. Germline Brca2 Heterozygosity Promotes KrasG12D -Driven Carcinogenesis in a Murine Model of Familial Pancreatic Cancer. Cancer Cell 2010, 18, 499–509. [Google Scholar] [CrossRef]
- Watt, F.; Molloy, P.L. Cytosine methylation prevents binding to DNA of a HeLa cell transcription factor required for optimal expression of the adenovirus major late promoter. Genes Dev. 1988, 2, 1136–1143. [Google Scholar] [CrossRef]
- Ball, M.P. , et al., Targeted and genome-scale strategies reveal gene-body methylation signatures in human cells. Nat Biotechnol, 2009. 27(4): p. 361-8.
- Jones, P.A. The DNA methylation paradox. Trends Genet. 1999, 15, 34–37. [Google Scholar] [CrossRef]
- Neri, F.; Rapelli, S.; Krepelova, A.; Incarnato, D.; Parlato, C.; Basile, G.; Maldotti, M.; Anselmi, F.; Oliviero, S. Intragenic DNA methylation prevents spurious transcription initiation. Nature 2017, 543, 72–77. [Google Scholar] [CrossRef]
- Yoder, J.A.; Walsh, C.P.; Bestor, T.H. Cytosine methylation and the ecology of intragenomic parasites. Trends Genet. 1997, 13, 335–340. [Google Scholar] [CrossRef]
- Ibrahim, J. , et al., Genome-wide DNA methylation profiling and identification of potential pan-cancer and tumor-specific biomarkers. Mol Oncol, 2022. 16(12): p. 2432-2447.
- Frommer, M.; E McDonald, L.; Millar, D.S.; Collis, C.M.; Watt, F.; Grigg, G.W.; Molloy, P.L.; Paul, C.L. A genomic sequencing protocol that yields a positive display of 5-methylcytosine residues in individual DNA strands. Proc. Natl. Acad. Sci. 1992, 89, 1827–1831. [Google Scholar] [CrossRef]
- Wang, R.Y.-H.; Gehrke, C.W.; Ehrlich, M. Comparison of bisulfite modification of 5-methyldeoxycytidine and deoxycytidine residues. Nucleic Acids Res. 1980, 8, 4777–4790. [Google Scholar] [CrossRef] [PubMed]
- Umer, M.; Herceg, Z. Deciphering the Epigenetic Code: An Overview of DNA Methylation Analysis Methods. Antioxidants Redox Signal. 2013, 18, 1972–1986. [Google Scholar] [CrossRef] [PubMed]
- Warnecke, P.M.; Stirzaker, C.; Melki, J.R.; Millar, D.S.; Paul, C.L.; Clark, S.J. Detection and measurement of PCR bias in quantitative methylation analysis of bisulphite-treated DNA. Nucleic Acids Res. 1997, 25, 4422–4426. [Google Scholar] [CrossRef]
- Ehrich, M.; Zoll, S.; Sur, S.; Boom, D.v.D. A new method for accurate assessment of DNA quality after bisulfite treatment. Nucleic Acids Res. 2007, 35, e29–e29. [Google Scholar] [CrossRef] [PubMed]
- Meissner, A.; Gnirke, A.; Bell, G.W.; Ramsahoye, B.; Lander, E.S.; Jaenisch, R. Reduced representation bisulfite sequencing for comparative high-resolution DNA methylation analysis. Nucleic Acids Res. 2005, 33, 5868–5877. [Google Scholar] [CrossRef]
- Harrison, A.; Parle-McDermott, A. DNA Methylation: A Timeline of Methods and Applications. Front. Genet. 2011, 2, 74. [Google Scholar] [CrossRef] [PubMed]
- Weichenhan, D. , et al., Tagmentation-Based Library Preparation for Low DNA Input Whole Genome Bisulfite Sequencing. Methods Mol Biol, 2018. 1708: p. 105-122.
- Jin, B. and K.D. Robertson, DNA methyltransferases, DNA damage repair, and cancer. Adv Exp Med Biol, 2013. 754: p. 3-29.
- Das, P.M. and R. Singal, DNA methylation and cancer. J Clin Oncol, 2004. 22(22): p. 4632-42.
- Mishra, N.K.; Guda, C. Genome-wide DNA methylation analysis reveals molecular subtypes of pancreatic cancer. Oncotarget 2017, 8, 28990–29012. [Google Scholar] [CrossRef]
- Chatterjee, A. , et al., DNA methylome in pancreatic cancer identified novel promoter hyper-methylation in NPY and FAIM2 genes associated with poor prognosis in Indian patient cohort. Cancer Cell Int, 2022. 22(1): p. 334.
- Bailey, P.; Chang, D.K.; Nones, K.; Johns, A.L.; Patch, A.-M.; Gingras, M.-C.; Miller, D.K.; Christ, A.N.; Bruxner, T.J.C.; Quinn, M.C.; et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016, 531, 47–52. [Google Scholar] [CrossRef]
- Boj, S.F.; Hwang, C.-I.; Baker, L.A.; Chio, I.I.C.; Engle, D.D.; Corbo, V.; Jager, M.; Ponz-Sarvise, M.; Tiriac, H.; Spector, M.S.; et al. Organoid Models of Human and Mouse Ductal Pancreatic Cancer. Cell 2015, 160, 324–338. [Google Scholar] [CrossRef] [PubMed]
- Boj, S.F.; Hwang, C.-I.; A Baker, L.; Engle, D.D.; A Tuveson, D.; Clevers, H. Model organoids provide new research opportunities for ductal pancreatic cancer. Mol. Cell. Oncol. 2015, 3, e1014757. [Google Scholar] [CrossRef]
- Hwang, C.; Boj, S.F.; Clevers, H.; A Tuveson, D. Preclinical models of pancreatic ductal adenocarcinoma. J. Pathol. 2015, 238, 197–204. [Google Scholar] [CrossRef]
- Romero-Calvo, I.; Weber, C.R.; Ray, M.; Brown, M.; Kirby, K.; Nandi, R.K.; Long, T.M.; Sparrow, S.M.; Ugolkov, A.; Qiang, W.; et al. Human Organoids Share Structural and Genetic Features with Primary Pancreatic Adenocarcinoma Tumors. Mol. Cancer Res. 2019, 17, 70–83. [Google Scholar] [CrossRef] [PubMed]
- Sharick, J.T.; Jeffery, J.J.; Karim, M.R.; Walsh, C.M.; Esbona, K.; Cook, R.S.; Skala, M.C. Cellular Metabolic Heterogeneity In Vivo Is Recapitulated in Tumor Organoids. Neoplasia 2019, 21, 615–626. [Google Scholar] [CrossRef] [PubMed]
- Tiriac, H.; Belleau, P.; Engle, D.D.; Plenker, D.; Deschenes, A.; Somerville, T.D.D.; Froeling, F.E.M.; Burkhart, R.A.; Denroche, R.E.; Jang, G.H.; et al. Organoid Profiling Identifies Common Responders to Chemotherapy in Pancreatic Cancer. Cancer Discov. 2018, 8, 1112–1129. [Google Scholar] [CrossRef]
- Chen, S.; Wang, M.; Liu, L.; Wang, G.; Wang, L.; Zhong, C.; Gao, C.; Wu, W.; Li, L. Ultrasound-guided fine-needle aspiration/biopsy-based pancreatic organoids establishment: an alternative model for basic and preclinical research. Gastroenterol. Rep. 2022, 11, goad019. [Google Scholar] [CrossRef]
- Lee, J.H.; Kim, H.; Ku, J.-L.; Chun, J.W.; Seo, H.Y.; Kim, S.C.; Paik, W.H.; Ryu, J.K.; Lee, S.K.; Lowy, A.M.; et al. Establishment of Patient-Derived Pancreatic Cancer Organoids from Endoscopic Ultrasound-Guided Fine-Needle Aspiration Biopsies. Gut Liver 2021, 16, 625–636. [Google Scholar] [CrossRef] [PubMed]
- Vilgelm, A.E.; Bergdorf, K.; Wolf, M.; Bharti, V.; Shattuck-Brandt, R.; Blevins, A.; Jones, C.; Phifer, C.; Lee, M.; Lowe, C.; et al. Fine-Needle Aspiration-Based Patient-Derived Cancer Organoids. iScience 2020, 23, 101408. [Google Scholar] [CrossRef] [PubMed]
- Collisson, E.A.; Sadanandam, A.; Olson, P.; Gibb, W.J.; Truitt, M.; Gu, S.; Cooc, J.; Weinkle, J.; Kim, G.E.; Jakkula, L.; et al. Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nat. Med. 2011, 17, 500–503. [Google Scholar] [CrossRef]
- Collisson, E.A.; Bailey, P.; Chang, D.K.; Biankin, A.V. Molecular subtypes of pancreatic cancer. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 207–220. [Google Scholar] [CrossRef]
- Brunton, H.; Caligiuri, G.; Cunningham, R.; Upstill-Goddard, R.; Bailey, U.-M.; Garner, I.M.; Nourse, C.; Dreyer, S.; Jones, M.; Moran-Jones, K.; et al. HNF4A and GATA6 Loss Reveals Therapeutically Actionable Subtypes in Pancreatic Cancer. Cell Rep. 2020, 31, 107625–107625. [Google Scholar] [CrossRef]
- O’Kane, G.M.; Grünwald, B.T.; Jang, G.-H.; Masoomian, M.; Picardo, S.; Grant, R.C.; Denroche, R.E.; Zhang, A.; Wang, Y.; Lam, B.; et al. GATA6 Expression Distinguishes Classical and Basal-like Subtypes in Advanced Pancreatic Cancer. Clin. Cancer Res. 2020, 26, 4901–4910. [Google Scholar] [CrossRef]
- García-Ortiz, M.V.; Cano-Ramírez, P.; Toledano-Fonseca, M.; Aranda, E.; Rodríguez-Ariza, A. Diagnosing and monitoring pancreatic cancer through cell-free DNA methylation: progress and prospects. Biomark. Res. 2023, 11, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Chen, X. , et al., Analysis of PD-L1 promoter methylation combined with immunogenic context in pancreatic ductal adenocarcinoma. Cancer Immunol Immunother, 2024. 73(8): p. 149.
- Hatziapostolou, M. , et al., Promoter Methylation Leads to Hepatocyte Nuclear Factor 4A Loss and Pancreatic Cancer Aggressiveness. Gastro Hep Adv, 2024. 3(5): p. 687-702.
- Yao, Y.; Lv, H.; Zhang, M.; Li, Y.; Herman, J.G.; Brock, M.V.; Gao, A.; Wang, Q.; Fuks, F.; Zhang, L.; et al. Epigenetic silencing of BEND4, a novel DNA damage repair gene, is a synthetic lethal marker for ATM inhibitor in pancreatic cancer. Front. Med. 2024, 18, 721–734. [Google Scholar] [CrossRef]
- Sakai, M.; Hibi, K.; Koshikawa, K.; Inoue, S.; Takeda, S.; Kaneko, T.; Nakao, A. Frequent promoter methylation and gene silencing of CDH13 in pancreatic cancer. Cancer Sci. 2004, 95, 588–591. [Google Scholar] [CrossRef]
- Matsubayashi, H.; Sato, N.; Fukushima, N.; Yeo, C.J.; Walter, K.M.; Brune, K.; Sahin, F.; Hruban, R.H.; Goggins, M. Methylation of cyclin D2 is observed frequently in pancreatic cancer but is also an age-related phenomenon in gastrointestinal tissues. Clin Cancer Res 2003, 9, 1446–52. [Google Scholar]
- Sato, N.; Fukushima, N.; Maehara, N.; Matsubayashi, H.; Koopmann, J.; Su, G.H.; Hruban, R.H.; Goggins, M. SPARC/osteonectin is a frequent target for aberrant methylation in pancreatic adenocarcinoma and a mediator of tumor–stromal interactions. Oncogene 2003, 22, 5021–5030. [Google Scholar] [CrossRef] [PubMed]
- Weng, Y.-C.; Ma, J.; Zhang, J.; Wang, J.-C. Long non-coding RNA LINC01133 silencing exerts antioncogenic effect in pancreatic cancer through the methylation of DKK1 promoter and the activation of Wnt signaling pathway. Cancer Biol. Ther. 2018, 20, 368–380. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, S.; Iwaya, H.; Akahane, T.; Hamada, T.; Higashi, M.; Hashimoto, S.; Tanoue, S.; Ohtsuka, T.; Ido, A.; Tanimoto, A. Sequential evaluation of MUC promoter methylation using next-generation sequencing-based custom-made panels in liquid-based cytology specimens of pancreatic cancer. Diagn. Cytopathol. 2022, 50, 499–507. [Google Scholar] [CrossRef] [PubMed]
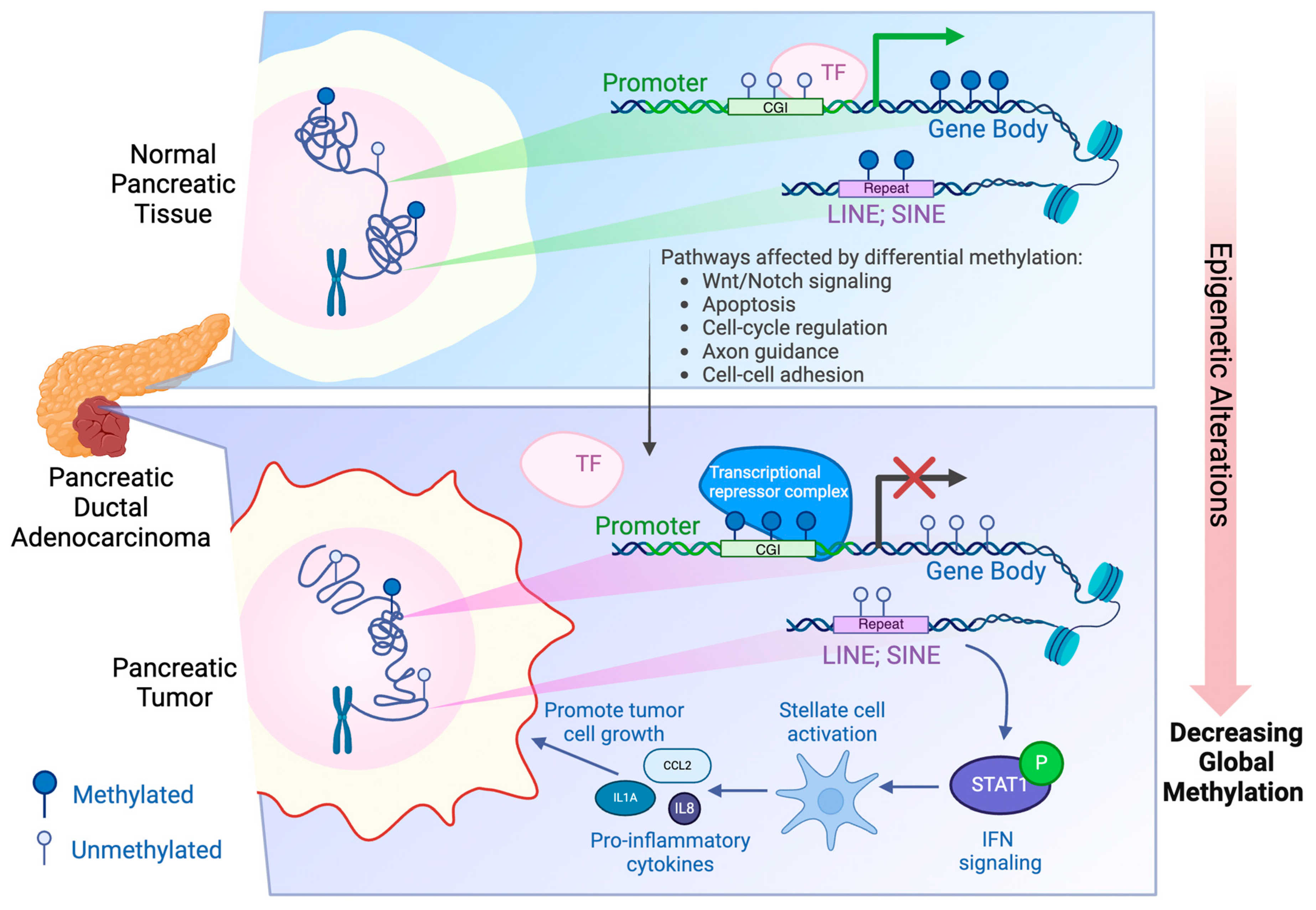
Figure 1.
Schematic illustration of the epigenetic alterations, specifically DNA methylation, during PDAC progression. Compared to normal pancreatic tissues, PDAC has promoter CpG islands (CGI) that are hypermethylated, resulting in a closed heterochromatin state as well as gene silencing. In addition, gene bodies and intergenic regions such as repeat sequences LINE and SINE are often hypomethylated in PDAC, leading to a decreased global methylation. Hypomethylation of LINEs and SINEs activates the Interferon signaling, creating a pro-inflammatory phenotype that promotes tumor cell growth and aggressiveness [22]. Created with BioRender.com.
Figure 1.
Schematic illustration of the epigenetic alterations, specifically DNA methylation, during PDAC progression. Compared to normal pancreatic tissues, PDAC has promoter CpG islands (CGI) that are hypermethylated, resulting in a closed heterochromatin state as well as gene silencing. In addition, gene bodies and intergenic regions such as repeat sequences LINE and SINE are often hypomethylated in PDAC, leading to a decreased global methylation. Hypomethylation of LINEs and SINEs activates the Interferon signaling, creating a pro-inflammatory phenotype that promotes tumor cell growth and aggressiveness [22]. Created with BioRender.com.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



