
Submitted:
02 September 2024
Posted:
03 September 2024
You are already at the latest version
Abstract
This comprehensive overview of the historical milestones in cell culture underscores key breakthroughs that have shaped the field over time. It begins with Wilhelm Roux's seminal experiments in the 1880s, followed by the pioneering efforts of Ross Granville Harrison, who initiated groundbreaking experiments that fundamentally shaped the landscape of cell culture in the early 20th century. Carrel's influential contributions, notably the immortalization of chicken heart cells, mark a significant advancement in cell culture techniques. Holtfreter, Moscona, and Leighton introduced innovations such as 3D cell culture, laying the groundwork for further exploration. The advent of induced pluripotent stem cells by Takahashi and Yamanaka in 2006 was revolutionary, enabling the reprogramming of differentiated cells into a pluripotent state. Since then, recent innovations include spheroids, organoids, and organ-on-a-chip technologies, aiming to mimic the structure and function of tissues and organs in vitro, pushing the boundaries of biological modeling and disease understanding. In this review, we overview the history of cell culture shedding light on the main discoveries, pitfalls and hurdles that were overcome during the transition from 2D to 3D cell culture techniques. Finally, we discussed the future directions for cell culture research that may accelerate the development of more effective and personalized treatments.

Keywords:
Cell culture
; 2D cell culture
; 3D cell culture
; Organoids
; Spheroids
; Organs-on-a-chip
1. An Overview: The Cell Culture History
Cell culture entails a spectrum of techniques that facilitate the in vitro development of cells, whether of animal or plant origin, thereby isolating them from their native biological context. This approach seeks to partially recapitulate the physicochemical conditions prevailing in the cell’s original microenvironment. The art and science of cell culture have enjoyed a rich history, finding application across diverse realms within the biological and biomedical sciences, underscoring their profound methodological significance in dissecting cellular responses to distinct biophysical and biochemical stimuli [1,2].
The inception of cell culture finds its roots in the fields of embryology, biological development, and later, the study of cancer. The earliest documented pursuits can be traced to the late 19th and early 20th centuries. In the 1880s, Wilhelm Roux, a pioneering German experimental embryologist from the University of Halle (Germany), embarked on groundbreaking experiments involving embryonic cells extracted from avian sources. His work yielded compelling evidence that it was feasible to sustain cellular life beyond the confines of the host organism by immersing them in a saline solution [3]. Shortly thereafter, the notable legacy of Leo Loeb, a German medical practitioner who later migrated to the United States, came to the fore. His decision to leave Germany was motivated by his dissatisfaction with the nation's nationalistic and militaristic situation. At the Washington University (USA), Loeb emerged as a distinguished experimental pathologist whose groundbreaking contributions in cell culture, transplantation, and hormonal research left an indelible mark on the landscape of medical science [4,5,6,7,8,9,10]. Loeb's profound dedication to research, combined with his visionary approach to humanitarianism, firmly established him as a pivotal figure in the annals of experimental pathology. His enduring influence continues to inspire and guide scientists worldwide, providing the foundational framework for numerous scientific breakthroughs and advancements in the field.
In 1906, the researcher Ross Granville Harrison (Johns Hopkins University, USA) developed pioneering experiments that laid the foundation for cell culture as we know it today. His investigations focused on growing tissue samples in test tubes. Harrison's primary focus was on the study of developing nerve fibers in frogs, where he maintained organ fragments in test tubes containing a liquid medium composed of blood clots, saline solution, and agar [11]. Furthermore, Harrison played a crucial role in the development of the "hanging drop" technique, which involved culturing cells within plasma on the underside of glass slides, creating droplets where the cells gathered. This innovative approach, later validated, continues to be employed in contemporary research, evolving through time with refinements and adaptations [12].
Harrison's notable work culminated in his publication titled “Observations on the Development of Living Nerve Fibers”[11]. In this work, he successfully observed the in vitro development of nerve fibers from a single cell or a cluster over a defined period. However, his research faced a persistent challenge in the form of bacterial contaminations, prompting him to introduce aseptic methodologies. This included the sterilization of surgical materials and the heating of experimental glassware, enabling the conduction of experiments and cell cultivation for extended periods, up to 5 weeks [2,13].
Another luminary in the field of cell culture is Alexis Carrel (Rockefeller Institute, USA) and a Nobel laureate in Medicine in 1912, acclaimed for his introduction of sutures in surgical procedures [14]. Carrel built upon Harrison's pioneering work from 1906 by developing a method for culturing cells in hanging drops, utilizing glass plate covers. During his investigations, Carrel observed that cells proliferated beyond the confines of the tissue and could be sequentially transferred and manipulated onto new plates. These experiments led to the conception of the "Carrel Flasks," which served as the precursor to contemporary cell culture flasks [15]. Subsequently, while cultivating cardiomyocytes in chicken plasma, Carrel noted that the interaction between cells and the culture medium was directly linked to increased cell proliferation. However, he also discerned that region closer to the center of the culture exhibited a higher likelihood of necrosis development. To address this challenge, the researcher cultivated tissue fragments on silk threads saturated with plasma, creating a surface where all cells had uniform access to the available nutrients within the culture medium [16]. This groundbreaking method allowed cells to grow within a three-dimensional (3D) structure, marking the first description of 3D cell culture (Figure 1).
Also, a partnership with Charles Lindbergh should also be highlighted [14]. Lindbergh was responsible for developing methods to separate blood serum from the rest of the blood and for introducing the use of glassware known as “Pyrex Glass” for cell cultivation. The flasks had the crucial advantage as they were resistant to high temperatures, enabling sterilization in autoclaves, and maintaining temperatures between 120 and 170 ºC. Carrel consistently emphasized the need to use sterile materials [15,16], an important consideration, since these experiments were conducted before Alexander Fleming discovered the first antibiotic. Around the 1930s, Carrel and Lindbergh published studies describing technologies that supported many experiments until the 1980s, when more sophisticated growth factors, cytokines and complex culture media were introduced, characterising the technologies currently used worldwide [17,18,19,20,21,22].
One of Carrel's notable contributions was the isolation and cultivation of one of the first immortalized cell lines derived from chicken embryonic hearts [15,16]. This was only possible due to the adoption of a strict sterilization methodology and consecutive changes in culture media involving washes with Riger's solution. This strain underwent hundreds of passages and was maintained until mid-1964, when it was finalized 2 years after Carrel's death. The strain described and cultivated by Carrel generated significant interest at the time, and it was established that the cells could survive indefinitely.
The immortalization of cell cultures is a phenomenon that can occur spontaneously over time, but can also be induced by factors such as oncogenic viral infections, radiation, and carcinogenic substances, and has been observed in various cultures throughout the 40s and 60s. One notable example of immortalized cells is HeLa. These cells, which have become fundamental in scientific research, originated in 1951 when Henrietta Lacks was diagnosed with aggressive cervical adenocarcinoma at the Johns Hopkins Hospital in Baltimore (USA). After performing a cervical biopsy, the samples were sent to Dr. George Gay, Director of the Tissue Culture Laboratory [2,13]. Mary Kubicek, his assistant, noticed that the cells remained viable in a nutrient solution based on chicken plasma and cultured Henrietta Lacks' specimen, resulting in robust, rapidly dividing cell cultures. This remarkable cell line was named HeLa, abbreviated as the initial letter of the patient's name (Henrietta Lacks). It is worth mentioning that after more than 70 years since their isolation, HeLa cells still survive, which is more than twice the lifespan of Henrietta, who passed away in October 1951 at the age of 31 [2,12,23].
After Carrel's pioneering work, approximately 35 years had passed before other researchers began to investigate and improve cell culture techniques. Notable scientists such as Johannes Holtfreter, Aron Arthur Moscona and Joseph Leighton, contributed in advancing and refining of cell culture techniques [24].
In the field of developmental biology, Johannes Holtfreter, from the University of Heidelberg (Germany), described an innovative method that allowed the formation of spherical cell aggregates to prevent cells from adhering to the surface of the culture flasks, thus promoting the tridimensional development of these cells. Later, Holtfreter further refined the techniques previously used by introducing an apparatus that agitated the culture flasks. This facilitated contact between the cells and promoted diffusion of the surrounding nutrients [25,26].
Another notable researcher in the field of developmental biology, Aron Arthur Moscona (University of Chicago, USA), made several contributions to refining cell culture techniques. Initially, studies on avian embryonic cells showed that cells from distinct organs did not assemble a mixed structure [27]. Furthermore, in a subsequent investigation, Moscona designed an experiment where cells derived from the lungs of mice and chicks were cultured into contact, resulting in the formation of cell aggregates after a few days. As a result, Moscona obtained liver and cartilage tissues in vitro. This pioneering work positioned Moscona at the forefront of research on cellular chimeras [28]. In addition, Moscona introduced a technique for cultivating cells using Erlenmeyer flasks under constant agitation. The continuous shaking of the culture flasks was intended to prevent the cells from adhering to the surface while stimulating the formation of cell aggregates in a three-dimensional configuration [29].
Back in the 1950s, Joseph Leighton (University of Princeton, USA), a specialist in histology and cellular pathology, raised a crucial concern about maintaining cellular tissue architecture during development in culture flasks. He noted that, despite the remarkable importance of two-dimensional (2D) cultures, this technique had significant limitations, especially with regard to the space available for cell development, which was not in line with the natural development of these cells in vivo [30]. In one of his most innovative studies, Leighton cultivated cells and tissue fragments in a three-dimensional (3D) matrix made up of a cellulose sponge saturated with plasma obtained from bird embryos. This system was then inserted into a culture flask containing nutrients and subjected to constant agitation. As a result, the study revealed that the 3D arrangement of the cellulose sponge matrix allowed the cells to proliferate and migrate in all directions, more accurately reproducing the behaviour of these cells in their organs of origin (in vivo). In addition, these 3D cultures had a significantly larger cell surface area when compared to 2D cell cultures [31]. Based on Leighton's pioneering studies, it became clear that there was a distinction between 2D and 3D cell culture methods, with 3D cell culture systems standing out for their advantages, including greater fidelity in reproducing in vivo cellular development and behaviour [24,30].
In the early 1960s, Ernst McCulloch (University of Toronto, Canada) and James Till (Ontario Cancer Institute, Toronto, Canada) began a series of experiments involving the injection of bone marrow cells into irradiated mice. The authors observed that small nodules formed in the spleens of the mice, directly proportional to the number of bone marrow cells injected. Till and McCulloch termed these nodules “spleen colonies” and postulated that each nodule originated from a single bone marrow cell: perhaps a stem cell [32,33]. In later work, Till and McCulloch, in collaboration with Andy Becker (undergraduate student) and Lou Siminovitch, from the University of Toronto (Canada), published, in 1963, two articles that represent fundamental milestones for the consolidation of self-renewal capacity and, as a result, the formulation of the concept of bone marrow stem cells [34,35].
Another important milestone in the history of cell cultures refers to the work developed, from the 1960s onwards, by the Russian physician Alexander Friedenstein (University of Moscow, Russia) which represented cardinal contributions in the discovery and establishment of the concept of mesenchymal stromal/stem cells. From bone marrow cell cultures, Friedenstein and collaborators identified and isolated a subpopulation of non-hematopoietic cells, adherent to culture vials, with a fibroblastoid appearance, with formation of discrete colonies resulting from clonal multiplication, from a single fibroblastic colony-forming cells, the so-called “Fibroblast Colony Forming Cells” (FCFC) or “colony forming units fibroblastic” (CFU-F) [36,37,38,39,40,41]. In vivo transplantation experiments demonstrated the multipotential nature of CFU-F, since it was possible to obtain lineages of mesenchymal/mesodermal origin (osteocytes, chondrocytes and adipocytes) from a single stromal cell [42,43]. Cells of mesenchymal origin were later named marrow stromal stem cells by Maureen Owen (University of Oxford, UK) [44,45] and, subsequently, Arnold Caplan proposed, in 1991, the term mesenchymal stem cells (MSC) [46]. In 2005, the International Society for Cellular Therapy (ISCT) proposed that the scientific community adopt, in all written and oral communications, the nomenclature multipotent mesenchymal stromal cells [47], but variations in nomenclature still persist in the literature, such as: mesenchymal stem cells, mesenchymal stromal cells and mesenchymal stromal/stem cells. Finally, the work of Friedenstein and collaborators, especially the partnership established with Maureen Owen [41], represented pioneering and seminal contributions opening new perspectives in cell therapy, regenerative and translational medicine.
In 1964, Malcolm Steinberg and Stephen A. Roth, both from the University of Princeton (USA), proposed the adhesion hypothesis, which posited that cellular rearrangement was influenced by thermodynamic mediators on different adhesion surfaces [48]. However, this hypothesis gained greater significance only in later years, particularly as cells began to be isolated and studied in greater depth, with a focus on stem cells.
Research involving stem cells was accelerated from the 1980s onwards, when various researchers were able to isolate and cultivate pluripotent stem cells derived from mouse embryos [49]. In 1981, Martin Evans and Matthew Kaufman, both from the University of Cambridge (UK), reported the establishment of cell lines derived from mouse blastocysts, which could differentiate in vitro or, after inoculation into mice, give rise to tumors with cells originating from the 3 embryonic layers – teratomas [50]. Still in the same year, in december 1981, Gail R. Martin (University of California, USA) published an article in which she describes “[...] the establishment of cell lines from normal mouse embryos that form teratocarcinomas when injected into mice”. In this work, Martin used the term "embryonic stem cells" for the first time in literature [51]. It is important to highlight that the establishment of in vitro embryonic stem cell cultures allowed the modification and implantation of these cells in adult females, generating genetically modified mice [52]. Because of these works, Martin John Evans was, together with Mario Capecchi and Oliver Smithies, awarded the Nobel Prize in Medicine and Physiology in 2007.
About 17 years after the works of Evans, Kaufman, and Martin, James Thomson's team (University of Wisconsin, USA) established, for the first time, the cultivation of human embryonic stem cells obtained from the inner cell mass of blastocysts from human embryos on the 5th day after fertilization. These pluripotent cells, which had high differentiation potential across a broad range of tissues, were characterized by their normal karyotypes and high telomerase activity levels, making them useful for various applications in research and medicine [53].
A paradigm shift occurred in 2006 when Kazutoshi Takahashi and Shinya Yamanaka, from the University of Kyoto (Japan), described a method that allowed the reprogramming of already differentiated stem cells, creating the so-called "induced pluripotent stem cells" (iPSC). Researchers were able to obtain iPSC from adult fibroblasts and mouse embryonic stem cells using only specific markers and growth factors (Sox2, Oct3/4, Klf4, and c-Myc). The result was that iPSC exhibited properties and characteristics similar to embryonic stem cells, as well as expressing some of the same marker genes [54,55]. In 2007, James Thomson's team obtained pluripotent cells from differentiated adult human cells (fibroblasts). However, the authors used a different combination of genes (Oct4, Sox2, NANOG, and Lin28) compared to those used by Yamanaka's group [56]. The Figure 2 provides a chronological overview, spotlighting eminent researchers who have pioneered and refined cell culture methodologies since 1885 up to nowadays.
With the capability to generate pluripotent stem cells from adult fibroblasts, there has been a substantial increase in the availability of raw materials for research in cellular biology and development. This advancement has spurred remarkable progress and the development of previously unimaginable cell culture techniques. Additionally, as somatic cell reprogramming methodologies have become established, various cell types have been effectively utilized in generating iPSCs, expanding beyond fibroblasts. It's notable that peripheral blood cells and urinary cells offer less invasive procurement methods compared to fibroblasts, which typically necessitate skin biopsies. This advantage in accessibility and non-invasiveness underscores the significance of these alternative sources in iPSC derivation. The Table 1 showcases pivotal studies regarding iPSC derivation from diverse cell sources.
2. Two-Dimensional (2D) Cell Cultures
Two-dimensional (2D) cell cultures has been widely used in biomedical research. This technique is used to investigate the physiology of cells and tissues under conditions that partially mimic those found in vivo. These investigations cover a range of topics including cell differentiation, migration, growth, physiological mechanisms, and cellular responses to biochemical changes in the microenvironment in which they are cultured [1,64,65]. The 2D cell culture technique is based on the growth of a single cell line on flat, adherent surfaces, such as Petri dishes or culture flasks, containing a supplemented culture medium, and can be applied to a wide range of tissues and cell types [21,66,67,68]. Even for cells that do not naturally adhere to plastic or glass surfaces, such as embryonic stem cells and induced pluripotent stem cells (iPSCs), it is possible to promote adhesion using specific coatings like poly-L-lysine, Matrigel, or fibronectin [69]. This facilitates the cultivation of these cells in a monolayer configuration. Monolayer cell culture methods have some characteristics that make them attractive for research in cellular biochemistry, such as uniform access to nutrients and growth factors present in the culture medium, resulting in homogeneous cell growth and proliferation [70,71,72]. The technology of 2D cell culture has been a crucial tool in biomedical research since the early 20th century, initially focusing on understanding cellular physiological mechanisms. However, over time, various other approaches and applications for this technique have been investigated. It has been widely used in cancer-related studies, although their limitations have led to a gradual decrease in their use in this context. Nevertheless, this technique is widely used in toxicity tests. These tests are crucial for evaluating cellular viability in response to therapeutic candidates and other compounds in general. Additionally, they allow for the determination of the impact of various compounds on genetic material, including genotoxicity and mutagenesis tests [73,74,75,76,77]. Studies employing these approaches have played an important role in reducing the use of animals in research, as suggested by the "3Rs" principles (Reduction, Refinement, and Replacement), proposed by Russell and Burch in their work "The Principles of Humane Experimental Technique", published in 1959 [78].
Cellular toxicity tests have a wide application for therapeutic candidates, as any promising compound should not exhibit significant cytotoxic activity. A classic example of this is the testing of plant-derived compounds that may have potential application as phytotherapeutics. Prior to conducting tests in animal or human models, it is essential to identify potential cytotoxic effects of these substances in vitro during preclinical phases [79,80,81].
Furthermore, techniques have been developed to enable the simultaneous cultivation of multiple cell lines in a monolayer environment, commonly referred to as co-culture, which developed to mimic the in vivo microenvironment more efficiently [82]. These tests are undertaken to investigate potential cellular interactions between different cell lines or to analyse how these lines interact with the surrounding microenvironment and the extracellular matrix (ECM) [83,84]. The applications of these techniques are particularly notable in research involving nervous system cells, where the co-culture of microglial cells with neural stem cells (NSCs) can induce dopaminergic differentiation of NSCs due to the release of differentiation factors. On the other hand, the co-culture of these NSCs with astrocytes promotes their neural differentiation [83].
However, despite the significant and fundamental contribution of 2D cell culture to the advancement of knowledge in various areas of biomedical sciences, this technique also has some limitations. One of the main limitations is the lack of contact between cells and the surrounding extracellular matrix (ECM), which can lead to a low fidelity to in vivo processes. This is because cells in vivo have specific structural and morphological characteristics that play a crucial role in cellular physiology [1]. Another limitation is associated with the composition of the ECM used in the cultivation. Some cell lines require a highly complex ECM for proper in vitro proliferation, such as hepatic cell lines, which are surrounded by a highly intricate ECM in the liver. Therefore, the stabilization of these cell lines in monolayer cultures becomes a challenging task due to the complexity in reproducing the microenvironment required for the cells to perform vital functions [85,86,87]. Cellular physiology, in vivo, is influenced by cell morphology and organization, aspects that are impacted in 2D cell culture. This can affect cellular proliferation, differentiation, apoptosis, protein expression, and other cellular processes [83]. In this regard, the development of new study models that can reduce the use of animals in research, while allowing for a more faithful representation of in vivo conditions in vitro, becomes an important step forward for the advancement of therapeutic efficacy tests, pathophysiology, and tests of new drugs [21,88,89,90]. To overcome some of the inherent limitations of 2D cell culture, a more complex cell culture methodology has been increasingly explored as an alternative method to mimic in vitro the behaviour of tissues in vivo: the three-dimensional (3D) cell culture.
3. Three-Dimensional (3D) Cell Culture: Spheroids and Organoids
Three-dimensional (3D) cell culture models, such as spheroids and organoids, complement and offer some new perspectives on two-dimensional (2D) cell cultures [19,20,91,92,93,94,95,96,97,98,99,100). This technology is currently considered a highly promising alternative for use in conjunction with animal models and 2D cell culture, allowing for a reduction in the use of these models in basic research [19,21,22,97,101,102,103,104,105]. These models allow the simultaneous cultivation of different cell lines enabling the replication of both cell-cell and cell-ECM interactions. In addition, it can mimic the characteristics of the organ or tissue from which the cells are derived, including gene expression, cell proliferation, differentiation, migration and metabolic functions [86,89,106,107,108,109].
The importance of 3D cell cultures was initially highlighted in 1970 when Sutherland, from the Ontario Cancer Treatment and Research Foundation (London Clinic) and the Departments of Therapeutic Radiology and of Surgery (University of Western, London, Ontario, Canada), and his colleagues developed multicellular spheroids to recapitulate the functional phenotype of human tumour cells and their responses to radiotherapy [110,111]. In 1977, one of the first three-dimensional cultures was performed by Hamburger and Salmon (University of Arizona, USA) using a soft agar solution, demonstrating that the morphology and behaviour of cells growing in a tumour mass and under 3D conditions showed remarkable similarities [112]. In the same year, Matrigel was introduced as a basement membrane extracellular matrix extracted from mouse sarcoma tumours, containing a unique mix of ECM components and growth factors [113]. This preparation became fundamental for supporting in vitro cell cultures, enabling the growth of various cell lines in a three-dimensional conformation. Over the years, Matrigel has established itself as an indispensable tool in the development of 3D cultures, facilitating the mimicry of the complex cellular and structural interactions found in living tissues. The commercial availability of Matrigel and other similar preparations has continuously optimized the culture process, allowing for significant advancements in research.
More specifically, spheroids represent the basic units of tissue engineering capable of mimicking the events that naturally occur during embryogenesis, morphogenesis, and organogenesis. Spheroids consist of the cultivation or co-cultivation of any type of primary cell lines, adult stem cells or iPSC, that self-organize into three-dimensional architectures, either spontaneously or through external stimuli, forming small spherical cellular aggregates without the need for a predefined culture substrate for cells to adhere to (Figure 3B) [114,115].
Currently, several methods exist to produce spheroids and they can vary from the hanging drop technique to the use of magnetic levitation [98,116,117,118,119]. Spheroids are used as models in disease studies, drug screening, and the identification of potential new targets and therapeutic candidates [18,100,120,121,122,123]. The spherical structure of spheroids leads to the formation of gradients of nutrients, lactate, oxygen, carbon dioxide, and pH. These gradients significantly influence cell proliferation, with more proliferative cells found on the outer surface, quiescent or senescent cells in the middle, and apoptotic cells in the inner regions creating a necrotic core [124]. It is important to mention that necrotic core is considered a key limitation for several authors in the literature. On the other hand, this characteristic makes it an ideal model for studying various types of cancer, due to its similarity to the environment found in cancer cells [20,105,121].
In contrast, organoids are more complex 3D systems than spheroids. Currently, there are different definitions of organoids. Initial scientific interpretations defined organoids as structures derived from stem cell clusters that self-organize and self-renew through cell-cell and cell-ECM interactions to mimic organogenesis in vitro. Some studies have shown that organoids can also be created using differentiated cells [125,126].
The 3D scaffolds allow cells to develop and self-organize into a three-dimensional structure similar to that of the organ from which they are derived. Organoids can be obtained by combining iPSC and adult tissue-derived stem cells (such as adipocytes and bone marrow cells) and can be differentiated into various human cell lines, in addition to being derived from primary culture cells, isolated directly from the target tissue [127]. However, these 3D systems present characteristics such as self-organization, multicellularity, and functionality, with the potential for cell differentiation and self-organization mediated by the complexity of the culture medium, strict environmental control, and the addition of growth and differentiation factors [21,90,128]. These processes can be clearly evidenced by cerebral organoids, representing a significant advancement compared to 2D culture, where neuronal development was limited due to the lack of cell-cell interaction and deficit in neuronal self-organization. Different strategies have been adopted to generate organoids in which specific cell lines are cultivated in solid three-dimensional scaffolds derived from natural ECM or biopolymers, ceramics, and metals [114,115,129,130,131], or in suspension on bioreactors [126].
Currently, organoids are used as models in research aimed at identifying and understanding the pathophysiology of various genetic and infectious diseases (including Sars-CoV-2 infection), the mechanisms involved in the development and treatment of tumours, and for the study of new medications and therapies, such as cell therapy, for diseases that lack effective treatment or a reliable cure [21,89,93,115,132,133,134,135]. For instance, hepatic organoids have been employed in the study of cystic fibrosis, liver steatosis, Alagille syndrome, viral hepatitis, sclerosing cholangitis, and alcohol-related diseases [136,137,138].
A critical aspect to be considered is the increasing complexity achieved by organoids. Notably, the most sophisticated liver organoid was developed by Takebe and colleagues in 2013, where different stem cell lines were used to induce not only the three-dimensional formation of differentiated cellular aggregates into liver cells but also in situ vascularization [139]. The vascularization has represented a significant challenge in the development of organoids, as it seeks to achieve the highest possible fidelity to the in vivo tissue.
The cerebral organoid model was pioneered developed by the group of Juergen A. Knoblich (Institute of Molecular Biotechnology, Vienna, Austria) and it was one of the first organoids obtained from iPSC derived from a human patient. The obtained brain organoid was then used to study a specific type of microcephaly, as an alternative to the difficulty of reproducing this microcephaly in a murine model [140,141]. Since then, cerebral organoids or “mini-brains“ has been employed in studying the cellular and molecular bases of various neurological disorders, such as autism, schizophrenia, Alzheimer's and Parkinson's disease, and other similar disorders [140]. This approach provides valuable insights into the underlying mechanisms of these conditions and identifies potential therapeutic targets. Additionally, cerebral organoids enable the investigation of complex processes involved in human brain development, including cell proliferation, neuronal migration, cortical layer formation, and neural circuit establishment [141,142]. However, similar to hepatic organoids, brain organoids also have limitations in terms of model complexity. Notably, the lack of vascularization and absence of immune cells, along with the reduced spontaneous formation of astrocytes and GABAergic inhibitory circuits, pose significant challenges. In this context, current research is aimed at overcoming these limitations, with the goal of improving the fidelity of the models and their ability to accurately recapitulate human brain physiology.
In addition to brain and liver organoids, models of organs such as the uterus, fallopian tubes, ovaries, and endometrium have been used in the study of diseases related to the female reproductive system, encompassing topics such as endometriosis, endometrial hyperplasia, and carcinomas [107,143,144]. Additionally, organoids representing organs such as the intestine, lungs, and mammary glands have become a focus of investigation [91,93,145,146,147]. The Figure 3 summarizes the three cell culture methods used as experimental models.
Organoids and spheroids are promising technologies in regenerative and translational medicine, especially in personalized medicine. In particular, personalized medicine represents a promising approach that can revolutionize the treatment of various diseases, especially those of genetic origin. This area is based on the use of genetic markers, transcriptomics, proteomics, and metabolomics, aiming to individualize preventive and therapeutic methods [21,137,148,149].
Personalized medicine achieves a good standard treatment for each patient by precisely identifying markers [128]. This approach is potentiated by the use of cell culture models derived from the patient's own cells, enabling a deeper understanding of disease development and the interactions between genetic and epigenetic factors that results in personalized treatments for each patient, including everything from medication administration to the adoption of alternative therapeutic strategies [128,150].
However, despite the substantial innovations provided by 3D cell culture models in the biomedical field and the notable impact on reducing the use of animal models, these models still present limitations that render them unable to completely replace traditional study methods [90,101,114]. The primary intrinsic limitation of 3D cell culture models lies in the absence of functional vascularization (presence of blood vessels), which frequently results in immature cellular development, inefficient nutrient distribution, and the formation of necrotic areas in the central nucleus of the culture [108,151]. Despite excessive efforts, few groups have managed to create a complex model with angiogenesis induction.
In the face of these challenges, research groups have been working on exploring new strategies, such as laser ablation, the use of canalized scaffolds, and the simultaneous cultivation of vascular endothelial cells, with the aim of establishing a microvascular network within the culture scaffolds or in direct contact with the cells [152,153,154]. However, it is essential to highlight that protocols involving the cultivation of vascular endothelial cells still require additional investigations to determine the ideal cultivation conditions in order to establish a standardized method [101,129,155].
Furthermore, compared to 2D cell culture systems, the 3D methods require considerable time and significant quantities of reagents and materials for proper implementation, without allowing for precise control over the physical-chemical properties of the cellular microenvironment that influence the maintenance of the culture. In this context, research is ongoing to integrate 3D cell culture with microfluidic systems, known as organs-on-a-chip. Although these systems show promising prospects, they represent a relatively recent technology that still in its initial stages of development [96,156,157].
4. Organs-on-a-Chip
The technology of 3D cell culture known as organ-on-a-chip emerges as a promising approach to overcome the inherent limitations of conventional 2D, spheroids and organoids [128,158]. This technology is based on microfluidic systems where various 3D cell cultures are maintained in integrated systems, interacting through microtubes and microstructures (Figure 3C). These systems have some advantages over traditional cell culture methods, such as the ability to control cell adhesion, provide mechanical stimulation to cells, and allow for tissue perfusion, the creation of artificial vascularization that replicates the characteristics of blood vessels, and the uniform distribution of nutrients to cells. This enables the modelling of complex human organism characteristics in a highly controlled in vitro environment [90,96,128,159].
In 2010, researchers at Harvard University developed the first lung-on-a-chip model. In this pioneering study, Huh and colleagues conceived a microfluidic system composed of two separate microchannels separated by a porous polydimethylsiloxane (PDMS) membrane [160]. This membrane was coated with a specific extracellular matrix, and subsequently, human alveolar epithelial cells and pulmonary microvascular endothelial cells were cultured on opposite sides of the membrane. As the cells reached adequate confluence, an air-liquid interface was created by applying vacuum to the sides of the culture compartment, with the purpose of reproducing the biomechanical forces associated with respiratory movements and thus mimicking the natural functioning of the lung more precisely. The results of this study demonstrated that the system allowed for simulating cellular responses to pulmonary bacterial infections, evaluating pulmonary inflammatory responses, and conducting toxicity studies of compounds, highlighting the potential of these microfluidic systems as viable alternatives to traditional animal tests [128,160,161].
Subsequently, from a similar design, other organs-on-a-chip models were developed to evaluate the toxicity of different compounds, investigate the potential of new medications or therapeutic targets, and model diseases. A notable example is the heart-on-a-chip model developed by researchers at the University of California (USA), based on the differentiation of human iPSCs into cardiomyocytes, known as the microphysiological cardiac system (MPS). In this study, the researchers evaluated the cellular responses of the developed model in the presence of pharmacological agents with known clinical effects, such as Isoprenaline, E-4031, Verapamil, and Metoprolol, and compared the results with the pharmacological responses of these drugs in traditional cell culture models. After 24 hours of culture, it was observed that the cells in the system presented spontaneous contractions (with a frequency of 55~80 bpm), a vital characteristic for mimicking the behaviour of a natural heart. The results demonstrated that the system is highly effective as a versatile study model with various applications in the pharmaceutical industry, as well as in developmental biology studies. This study highlighted the superiority of the MPS compared to traditional 2D cell culture models used for similar purposes [162].
In the following year, a brain-on-a-chip model was developed by researchers at Johns Hopkins University (Baltimore, MD, USA) who aimed to elucidate and understand the mechanisms involved in the migration of neural progenitor cells (NPCs) in the central nervous system when stimulated by chemoattractants. Using a silicone elastomeric device, the researchers induced the differentiation of human pluripotent cells into glial and neural cells to replicate the microenvironment of the central nervous system. This model allowed for the mimicking and evaluation of cellular interactions between NPCs and the brain tissue, providing valuable information about processes still not fully understood. The authors highlighted that the brain-on-a-chip model represents a promising and convenient tool for studies related to neurological development, neural oncology, toxicology, and neural regeneration [163].
In 2018, Weber and colleagues, from the University of Washington (Seattle, WA, USA), established a kidney-on-a-chip model by culturing primary human proximal tubular epithelial cells (PTECs) in a microfluidic chip system. The researchers successfully evaluated the nephrotoxicity of polymyxin B, a polypeptide antibiotic, along with two structural analogues, NAB539 and NAB741. When the cells were exposed to polymyxin B, a significant increase in renal damage signals and cholesterol biosynthesis was observed. However, minimal changes were observed when the cells were exposed to the analogues of polymyxin, demonstrating the preclinical safety of NAB741 and NAB739 [164].
On the other hand, in 2019, Jang and colleagues developed various liver-on-a-chip models using sinusoidal endothelial cells from the liver, as well as primary human, rat, and canine hepatocytes. The researchers aimed to evaluate the hepatotoxic effects induced by bosentan, a receptor antagonist of endothelin, a compound known to cause cholestasis in humans but not in rats and dogs. Initially, it was observed that the liver-on-a-chip models produced albumin, a characteristic protein of liver cells. The results demonstrated that these systems could detect and mimicking not only hepatic toxicity phenotypes but also conditions such as steatosis, cholestasis, fibrosis, and liver cell lesions. In summary, the authors emphasized that microfluidic chip systems provide powerful tools for a better understanding and prediction of liver toxicities, lesions, and diseases compared to traditional cell culture methods [165].
Also in 2019, researchers at the Cincinnati Children’s Hospital Medical Center (Cincinnati, OH, USA) developed a pancreas-on-a-chip model by culturing epithelial cells derived from pancreatic ducts and islets in a single microfluidic system. The purpose of this study was to evaluate the functional relationship between these two types of cells in patients diagnosed with cystic fibrosis, a genetic disease associated with dysfunction of the cystic fibrosis transmembrane conductance regulator protein. Additionally, the researchers investigated the relationship between cystic fibrosis and other pancreatic dysfunctions, such as the development of diabetes. The results indicated that attenuation of the cystic fibrosis transmembrane conductance regulator protein led to a reduction of approximately 50% in the amount of insulin secreted by pancreatic islet cells, a crucial discovery for understanding the development of diabetes in these patients. The authors highlighted that the pancreas-on-a-chip model can be a valuable tool in diagnosing diseases like diabetes, identifying new therapeutic targets, and promoting personalized medicine for treating this condition [166].
5. New Perspectives for Cell Culture Methodologies
Animal models (Figure 4A), which are still the standard and minimum requirement for pre-clinical to clinical stages, have significant limitations. These models, along with more advanced in vitro models like spheroids and organoids (Figure 4B), have their own set of drawbacks. To overcome the challenge of reducing animal experimentation, the key is to find a method that can simulate interactions between biological systems without the inherent limitations of animal models.
The increasing complexity of cell culture systems aims primarily to replicate human physiology with the maximum possible fidelity, both in the context of normal development and in the pathophysiology of diseases. These advances are critically important in technical applications such as drug screening and personalized medicine. Consequently, various studies are being developed to obtain a cell culture model that mimics even more complex systems, improving the imitation of human physiology. Particularly, microfluidic technology has provided numerous possibilities, including the integration of multiple organs-on-a-chip to simulate interactions between entire systems. However, as with any technology in its initial stages of development, organs-on-a-chip models still present some limitations that need to be carefully addressed and evaluated before a standardized method for their acquisition and application can be established [88,92,94,151].
The interaction of different organs-on-a-chip systems, also known as human-on-a-chip or body-on-a-chip, represents a promising approach to addressing these challenges. This methodology aims to simulate a human organism and its complex system interactions in a single microfluidic device. In addition to incorporating essential biological barriers, these systems overcome one of the main limitations of conventional 3D cultures, which is the lack of adequate vascularization and gas exchange. In essence, the human-on-a-chip is a convergence of various organs-on-a-chip models, making the method even more complex to simulate nutrient and oxygen exchange between different organ systems (Figure 4C).
This represents a notable advancement in biomedical research, offering the ability to study systemic interactions and evaluate the impact of treatments in a more realistic context, without the need for animal experimentation. The continuous development of this technology promises to revolutionize preclinical research and accelerate progress towards more effective and personalized treatments. The main applications of experimental models in preclinical trials, as well as the main differences, are described in Table 2.
6. Conclusion Remarks
It is unquestionable that 2D cell cultures have played a critical role in the development of science. It is also important to emphasise that a large part of our knowledge about human diseases, drug development, and therapeutic is mainly due to the use of 2D cell culture alongside animal models. However, these models still present limitations that have hindered the translation of data obtained in basic research into clinical trials. Currently, various tridimensional cell culture models (3D) are being developed, evaluated, and utilized as study platforms in the fields of biotechnology and biomedicine. This technology enables the reproduction of complex biological characteristics and mechanisms in a highly controlled in vitro system showing significant potential in preclinical studies, new drug screening and therapeutic applications. In this way, 3D cell culture platforms have in recent years complemented and expanded research perspectives and enabled great advances in basic and applied research, reducing the gap between preclinical studies and their potential application in regenerative and translational medicine.
Author Contributions
Conceptualization, L.G.M., L.P.G., F.A.R.L., and J.T.R.P.; Methodology, L.G.M. and L.P.G.; Writing – Original Draft Preparation, L.G.M., L.P.G., M.V.C., M.L.D., M.F.A., J.A.R.F.; Figure elaboration, L.G.M. and L.P.G.; Writing – Review & Editing, L.G.M., L.P.G., J.T.R.P., and F.A.R.L.; Supervision, J.T.R.P, F.A.R.L.; Funding Acquisition, L.G.M., L.P.G., M.V.C, M.L.D., J.T.R.P., F.A.R.L., M.A.L.
Funding
L.G. Moro was financially supported by the National Council for Scientific and Technological Development (CNPq – Brazil – Grant 140520/2021-0); L.P. Guarnier was financially supported by the São Paulo Research Foundation (FAPESP – Brazil – Grant 2023/03687-9); M. F. Azevedo was financially supported by the São Paulo Research Foundation (FAPESP – Brazil – Grant 2023/05446-9); M. V. de Castro was financially supported by the São Paulo Research Foundation (FAPESP – Brazil – Grant 2020/09702-1). M. L. Dias was financially supported by the National Council for Scientific and Technological Development (CNPq – Brazil – Grant 170653/2023-4).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data that support the findings of this research are available from the corresponding author upon reasonable request.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Duval, K.; Grover, H.; Han, L.H.; Mou, Y.; Pegoraro, A.F.; Fredberg, J.; Chen, Z. Modeling Physiological Events in 2D vs. 3D Cell Culture. Physiology 2017, 32, 266–277. [CrossRef]
- Jedrzejczak-Silicka, M. History of Cell Culture. In New Insights into Cell Culture Technology; IntechOpen, 2017. Available online: https://www.intechopen.com/chapters/53566 (accessed on 29 August 2023).
- Sander, K. Landmarks in Developmental Biology 1883–1924: Historical Essays from Roux’s Archives; Springer Berlin Heidelberg: Berlin, Heidelberg, 1997. Available online: http://link.springer.com/10.1007/978-3-642-60492-8 (accessed on 30 August 2024).
- Loeb, L. The production of deciduomata and the relation between the ovaries and the formation of the decidua. J. Am. Med. Assoc. 1908, L, 23, 1897.
- Loeb, L. The Influence of certain Bacteria on the Coagulation of the Blood. J. Med. Res. 1903, 10, 3, 407–419.
- Loeb, L. Wounds of the pregnant uterus. Proc. Soc. Exp. Biol. Med. 1906, 4, 1, 93–94. [CrossRef]
- Loeb, L. The cyclic changes in the ovary of the guinea pig. J. Morphol. 1911, 2, 1, 37–70. [CrossRef]
- Loeb, L. On Transplantation of tumors. J. Med. Res. 1901, 6, 1, 28–38.
- Loeb, L. Further Investigations in Transplantation of tumors. J. Med. Res. 1902, 8, 1, 44–73.
- Loeb, L. On the Blood Lymph Cells and inflammatory Processes of Limulus. J. Med. Res. 1902, 7, 1, 145–158.
- Harrison, R.G. Observations on the living developing nerve fiber. Proc. Soc. Exp. Biol. Med. 1906, 4, 1, 140–143.
- Taylor, M.W. A History of Cell Culture. In Viruses and Man: A History of Interactions; Springer International Publishing: Cham, 2014, 41–52. Available online: https://doi.org/10.1007/978-3-319-07758-1_3 (accessed on 30 April 2021).
- Rodríguez-Hernandez, C.; García, S.E.; Olvera-Sandoval, C.; Ramírez Castillo, F.; Loera Muro, A.; González, F.; Guerrero-Barrera, A. Cell culture: History, Development and Prospects. International Journal of Current Research and Academic Review. 2014, 2, 188-200.
- Malinin, T.I. Remembering Alexis Carrel and Charles A. Lindbergh. Tex. Heart Inst. J. 1996, 23, 1, 28–35.
- Carrel, A.; Burrows, M.T. Cultivation of tissues in vitro and its technique. J. Exp. Med. 1911, 13, 3, 387–396. [CrossRef]
- Carrel, A.; Burrows, M.T. An addition to the technique of the cultivation of tissues in vitro. J. Exp. Med. 1911, 14, 3, 244–247. [CrossRef]
- Carrel, A.; Lindbergh, C.A. The culture of Whole Organs. Science 1935, 81, 2112, 621–623. [CrossRef]
- Caprio, N.D.; Burdick, J.A. Engineered biomaterials to guide spheroid formation, function, and fabrication into 3D tissue constructs. Acta Biomater. 2022, S1742-7061, 22, 00620-1.
- Nguyen, R.; Da Won Bae, S.; Qiao, L.; George, J. Developing liver organoids from induced pluripotent stem cells (iPSCs): An alternative source of organoid generation for liver cancer research. Cancer Lett. 2021, 508, 13–17. [CrossRef]
- Chen, G.; Liu, W.; Yan, B. Breast Cancer MCF-7 Cell Spheroid Culture for Drug Discovery and Development. J. Cancer Ther. 2022, 13, 3, 117–130. [CrossRef]
- da Silva da Costa, F.A.; Soares, M.R.; Malagutti-Ferreira, M.J.; da Silva, G.R.; Lívero, F.A.R.; Ribeiro-Paes, J.T. Three-Dimensional Cell Cultures as a Research Platform in Lung Diseases and COVID-19. Tissue Eng. Regen. Med. 2021, 18, 5, 735–745. [CrossRef]
- Hospodiuk-Karwowski, M.; Chi, K.; Pritchard, J.; Catchmark, J.M. Vascularized pancreas-on-a-chip device produced using a printable simulated extracellular matrix. Biomed. Mater. 2022, 17, 6, 065006. [CrossRef]
- Lucey, B.P.; Nelson-Rees, W.A.; Hutchins, G.M. Henrietta Lacks, HeLa Cells, and Cell Culture Contamination. Arch. Pathol. Lab. Med. 2009, 133, 9, 1463–1467.
- Byrnes, W.M. Ernest Everett Just, Johannes Holtfreter, and the origin of certain concepts in embryo morphogenesis. Mol. Reprod. Dev. 2009, 76, 10, 912–921.
- Holtfreter, J. A study of the mechanics of gastrulation. J. Exp. Zool. 1944, 95, 2, 171–212.
- Holtfreter, J. Neural induction in explants which have passed through a sublethal cytolysis. J. Exp. Zool. 1947, 106, 2, 197–222. [CrossRef]
- Moscona, A.; Moscona, H. The dissociation and aggregation of cells from organ rudiments of the early chick embryo. J. Anat. 1952, 86, 3, 287–301.
- Moscona, A. The development in vitro of chimeric aggregates of dissociated embryonic chick and mouse cells. Proc. Natl. Acad. Sci. USA 1957, 43, 1, 184–194. [CrossRef]
- Moscona, A. Rotation-mediated histogenetic aggregation of dissociated cells. A quantifiable approach to cell interactions in vitro. Exp. Cell Res. 1961, 22, 455–475. [CrossRef]
- Hoffman, R.M. 3D Sponge-Matrix Histoculture: An Overview. Methods Mol. Biol. 2018, 1760, 11–17.
- Leighton, J. The growth patterns of some transplantable animal tumors in sponge matrix tissue culture. J. Natl. Cancer Inst. 1954, 15, 2, 275–293. [CrossRef]
- McCulloch, E.A.; Till, J.E. The radiation sensitivity of normal mouse bone marrow cells, determined by quantitative marrow transplantation into irradiated mice. Radiat. Res. 1960, 13, 1, 115–135. [CrossRef]
- Till, J.E.; McCulloch, E.A. A direct measurement of the radiation sensitivity of normal mouse bone marrow cells. Radiat. Res. 1961, 14, 2, 213–222. [CrossRef]
- Becker, A.J.; McCulloch, E.A.; Till, J.E. Cytological demonstration of the clonal nature of spleen colonies derived from transplanted mouse marrow cells. Nature 1963, 197, 4866, 452–454. [CrossRef]
- Siminovitch, L.; McCulloch, E.A.; Till, J.E. The distribution of colony-forming cells among spleen colonies. J. Cell. Comp. Physiol. 1963, 62, 3, 327–336. [CrossRef]
- Friedenstein, A.J. Osteogenetic activity of transplanted transitional epithelium. Acta Anat. Basel. 1961, 45, 31–59. [CrossRef]
- Friedenstein, A.J.; Piatetzky-Shapiro, I.I.; Petrakova, K.V. Osteogenesis in transplants of bone marrow cells. J. Embryol. Exp. Morphol. 1966, 16, 381–390.
- Friedenstein, A.J.; Latzinik, N.V.; Gorskaya, Y.F.; Luria, E.A.; Moskvina, I.L. Stromal cells responsible for transferring the microenvironment of the hemopoietic tissues. Transplantation 1978, 9, 3, 267–274.
- Friedenstein, A.J.; Deriglazova, U.F.; Kulagina, N.N.; Panasuk, A.F.; Rudakowa, S.F.; Luriá, E.A; Ruadkow, I.A. Precursors for fibroblasts in different populations of hematopoietic cells as detected by the in vitro colony assay method. Exp. Hematol. 1974, 2, 83–92.
- Owen, M.E.; Cave, J.; Joyner, C.J. Clonal analysis in vitro of osteogenic differentiation of marrow CFU-F. J. Cell Sci. 1987, 87, 731–738. [CrossRef]
- Triffitt, J.T. The Collaborative Spark That Ignited the Field of Stromal Stem Cell Biology. Bioengineering 2024, 11, 652. [CrossRef]
- Friedenstein, A.J.; Chailakhjan, R.K.; Lalykina, K.S. The development of fibroblast colonies in monolayer cultures of guinea-pig bone marrow and spleen cells. Cell Tissue Kinet. 1970, 3, 393–403. [CrossRef]
- Friedenstein, A.J.; Chailakhyan, R.K.; Gerasimov, U.V. Bone marrow osteogenic stem cells: In vitro cultivation and transplantation in diffusion chambers. Cell Tissue Kinet. 1987, 20, 3, 263–272. [CrossRef]
- Owen, M. Marrow stromal stem cells. J. Cell Sci. Suppl. 1988, 10, 63–76.
- Owen, M.; Friedenstein, A.J. Stromal stem cells: Marrow-derived osteogenic precursors. Ciba Found. Symp. 1988, 136, 42–60.
- Caplan, A.I. Mesenchymal stem cells. J. Orthop. Res. 1991, 9, 641–650. [CrossRef]
- Horwitz, E.M.; Le Blanc, K.; Dominici, M.; Mueller, I.; Slaper-Cortenbach, I.; Marini, F.C.; Deans, R.J.; Krause, D.S.; Keating, A. Clarification of the Nomenclature for MSC: The International Society for Cellular Therapy Position Statement. Cytotherapy 2005, 7, 5, 393–395. [CrossRef]
- Steinberg, M.S. The Problem of Adhesive Selectivity in Cellular Interactions. Proc. Natl. Acad. Sci. USA 1964, 52, 1, 94–100.
- Evans, M. Origin of mouse embryonal carcinoma cells and the possibility of their direct isolation into tissue culture. J. Reprod. Fertil. 1981, 62, 625–631.
- Evans, M.J.; Kaufman, M.H. Establishment in culture of pluripotential cells from mouse embryos. Nature 1981, 292, 5819, 154–156. [CrossRef]
- Martin, G.R. Isolation of a pluripotent cell line from early mouse embryos cultured in medium conditioned by teratocarcinoma stem cells. Proc. Natl. Acad. Sci. USA, 1981, 78 12, 7634–7638. [CrossRef]
- Bradley, A.; Evans, M.; Kaufman, M.H.; Robertson, E. Formation of germ-line chimaeras from embryo-derived teratocarcinoma cell lines. Nature 1984, 309, 255–256. [CrossRef]
- Thomson, J.A.; Itskovitz-Eldor, J.; Shapiro, S.S.; Waknitz, M.A.; Swiergiel, J.J.; Marshall, V.S.; Jones, J.M. Embryonic stem cell lines derived from human blastocysts. Science, 1998, 282, 5391, 1145–1147. [CrossRef]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131(5), 861–872. [CrossRef]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 4, 663–676.
- Yu, J.; Vodyanik, M.; Smuga-Otto, K.; Frane, J.; Antosiewicz-Bourget, J.; Tian, S.; Nie, J.; Jonsdottir, G.A.; Ruotti, V.; Stewart, R.; Slukvin, I.I.; Thomson, J.A. Induced pluripotent stem cell lines derived from human somatic cells. Science 2007, 318, 1917–1920. [CrossRef]
- Aasen, T.; Raya, A.; Barrero, M.J.; Garreta, E.; Consiglio, A.; Gonzalez, F.; Vassena, R.; Bilić, J.; Pekarik, V.; Tiscornia, G.; Edel, M.; Boué, S.; Izpisúa Belmonte, J.C. Efficient and rapid generation of induced pluripotent stem cells from human keratinocytes. Nat. Biotechnol. 2008, 26, 11, 1276–1284. [CrossRef]
- Loh, Y.H.; Agarwal, S.; Park, I.H.; Urbach, A.; Huo, H.; Heffner, G.C.; Kim, K.; Miller, J.D.; Ng, K.; Daley, GQ. Generation of induced pluripotent stem cells from human blood. Blood 2009, 113, 22, 5476–5479. [CrossRef]
- Haase, A.; Olmer, R.; Schwanke, K.; Wunderlich, S.; Merkert, S.; Hess, C.; Zweigerdt, R.; Gruh, I.; Meyer, J.; Wagner, S.; Maier, L.S.; Han, D.W.; Glage, S.; Miller, K.; Fischer, P.; Schöler, H.R.; Martin, U. Generation of induced pluripotent stem cells from human cord blood. Cell Stem Cell 2009, 5, 4, 434–441. [CrossRef]
- Cai, J.; Li, W.; Su, H.; Qin, D.; Yang, J.; Zhu, F.; Xu, J.; He, W.; Guo, X.; Labuda, K.; Peterbauer, A.; Wolbank, S.; Zhong, M.; Li, Z.; Wu, W.; So, K.F.; Redl, H.; Zeng, L.; Esteban, M.A.; Pei D. Generation of human induced pluripotent stem cells from umbilical cord matrix and amniotic membrane mesenchymal cells. J. Biol. Chem. 2010, 285, 11227–11234. [CrossRef]
- Yan, X.; et al. Induction of pluripotent stem cells from human third molar mesenchymal stromal cells. J. Biol. Chem. 2010, 285(38), 29270–29278.
- Zhou, T.; Benda, C.; Duzinger, S.; Huang,Y.; Li, X.; Li, Y.; Guo, X.; Cao, G.; Chen, S.; Hao, L.; Chan, Y.; Kwong-Man, Ng.; Ho, J.C.; Wieser, M.; Wu, J.; Redl, H.; Tse, H.F.; Grillari, J.; Grillari-Voglauer, R. Generation of induced pluripotent stem cells from urine. J. Am. Soc. Nephrol. 2011, 22(7), 1221–1228. [CrossRef]
- Zhou, T.; Benda, C.; Dunzinger, S.; Huang, Y.; Ho, J.C.; Yang, J.; Wang, Y.; Zhang, Y.; Zhuang, Q.; Li, Y.; Bao, X.; Tse, H.F.; Grillari, J.; Grillari-Voglauer, R.; Pei, D.; Esteban, M.A. Generation of human induced pluripotent stem cells from urine samples. Nat. Protoc,. 2012, 7, 12, 2080–2089. [CrossRef]
- Huh, D.; Hamilton, G.A.; Ingber, D.E. From 3D cell culture to organs-on-chips. Trends Cell Biol. 2011, 21, 12, 745–754. [CrossRef]
- Yao, T.; Asayama, Y. Animal-cell culture media: History, characteristics, and current issues. Reprod. Med. Biol. 2017, 16, 2, 99–117.
- Bartfeld, S.; Clevers, H. Stem cell-derived organoids and their application for medical research and patient treatment. J. Mol. Med. 2017, 95, 7, 729–738. [CrossRef]
- Huch, M. Building stomach in a dish. Nat. Cell Biol. 2015, 17, 8, 966–967. [CrossRef]
- Jensen, C.; Teng, Y. Is It Time to Start Transitioning From 2D to 3D Cell Culture?. Front. Mol. Biosci. 2020, 7. [CrossRef]
- Lam, M.T.; Longaker, M.T. Comparison of several attachment methods for human iPS, embryonic, and adipose-derived stem cells for tissue engineering. J. Tissue Eng. Regen. Med. 2012, 6, 3, s80–s86. [CrossRef]
- Arruda de Faria, C.; Silva Júnior, W.A.; Caetano Andrade Coelho, K.B.; Bassi, M.; Colombari, E.; Zanette, D.L; Ribeiro-Paes, JT. Mesenchymal stromal cells-based therapy in a murine model of elastase-induced emphysema: Simvastatin as a potential adjuvant in cellular homing. Pulm. Pharmacol. Ther. 2021, 70, 102075. [CrossRef]
- Longhini-dos-Santos, N.; Barbosa-de-Oliveira, V.A.; Stessuk, T.; Sakalem, M.E.; Ribeiro-Paes, J.T. Cell therapy decreases inflammation and improves the morphology of the lung parenchyma in a murine model of cigarette smoke-induced emphysema. Int. J. New Technol. Res. 2018, 4, 1, 263154. [CrossRef]
- Passanha, F.R.; Geuens, T.; Konig, S.; van Blitterswijk, C.A.; LaPointe, V.L.S. Cell culture dimensionality influences mesenchymal stem cell fate through cadherin-2 and cadherin-11. Biomaterials 2020, 254, 120127. [CrossRef]
- Cui, T.; Liu, W.; Yu, C.; Ren, J.; Li, Y.; Shi, X.; Li, Q.; Zhang, J. Protective effects of allicin on acute myocardial infarction in rats via hydrogen sulfide-mediated regulation of coronary arterial vasomotor function and myocardial calcium transport. Front. Pharmacol. 2022, 752244. [CrossRef]
- Gillet, J.P.; Varma, S.; Gottesman, M.M. The clinical relevance of cancer cell lines. J. Natl. Cancer Inst. 2013, 105, 7, 452–458. [CrossRef]
- Imamura, Y.; Mukohara, T.; Shimono, Y.; Funakoshi, Y.; Chayahara, N.; Toyoda, M.; Kiyota, N.; Takao, S.; Kono, S.; Nakatsura, T.; Minami, H. Comparison of 2D- and 3D-culture models as drug-testing platforms in breast cancer. Oncol. Rep. 2015, 33,4, 1837–1843.
- Mukherjee, S.G.; O'Claonadh, N.; Casey, A.; Chambers, G. Comparative in vitro cytotoxicity study of silver nanoparticle on two mammalian cell lines. Toxicol. In Vitro 2012, 26, 2, 238–251. [CrossRef]
- Zhou, H.M.; Shen, Y.; Wang, Z.J.; Li, L.; Zheng, Y.F.; Häkkinen, L.; Haapasalo, M. In vitro cytotoxicity evaluation of a novel root repair material. J. Endod. 2013, 39, 4, 478–483. [CrossRef]
- Tannenbaum, J.; Bennett, B.T. Russell and Burch’s 3Rs then and now: The need for clarity in definition and purpose. J. Am. Assoc. Lab. Anim. Sci. 2015, 54, 2, 120–132.
- Gusson-Zanetoni, J.P.; da Silva, J.S.G.M.; Picão, T.B.; Cardin, L.T.; Prates, J.; Sousa, S.O. Effect of Piper cubeba total extract and isolated lignans on head and neck cancer cell lines and normal fibroblasts. J. Pharmacol. Sci. 2022, 148, 1, 93–102. [CrossRef]
- Kamaruddin, M.S.H.; Chong, G.H.; Mohd Daud, N.; Putra, N.R.; Md Salleh, L.; Suleiman, N. Bioactivities and green advanced extraction technologies of ginger oleoresin extracts: A review. Food Res. Int. 2023, 164, 112283. [CrossRef]
- Popovici, V.; Matei, E.; Cozaru, G.C.; Bucur, L.; Gîrd, C.E.; Schröder, V.; Ozon, E.A.; Musuc, A.M.; Mitu, M.A.; Atkinson, I.. In Vitro Anticancer Activity of Mucoadhesive Oral Films Loaded with Usnea barbata (L.) F. H. Wigg Dry Acetone Extract, with Potential Applications in Oral Squamous Cell Carcinoma Complementary Therapy. Antioxidants 2022, 11, 1934. [CrossRef]
- Miki, Y.; Ono, K.; Hata, S.; Suzuki, T.; Kumamoto, H.; Sasano, H. The advantages of co-culture over mono cell culture in simulating in vivo environment. J. Steroid Biochem. Mol. Biol. 2012, 131, 3, 68–75. [CrossRef]
- Liu, R.; Meng, X.; Yu, X.; Wang, G.; Dong, Z.; Zhou, Z.; Qi, M.; Yu, X.; Ji, T.; Wang, F.. From 2D to 3D Co-Culture Systems: A Review of Co-Culture Models to Study the Neural Cells Interaction. Int. J. Mol. Sci. 2022, 23, 21, 13116. [CrossRef]
- Vis, M.A.M.; Ito, K.; Hofmann, S. Impact of Culture Medium on Cellular Interactions in in vitro Co-culture Systems. Front. Bioeng. Biotechnol. 2020, 4, 8, 911. [CrossRef]
- Cheaito, K.; Bahmad, H.F.; Jalloul, H.; Hadadeh, O.; Msheik, H.; El-Hajj, A.; Mukherji, D.; Al-Sayegh, M.; Abou-Kheir, W. Epidermal Growth Factor Is Essential for the Maintenance of Novel Prostate Epithelial Cells Isolated From Patient-Derived Organoids. Front. Cell Dev. Biol. 2020, 29, 8, 571677. [CrossRef]
- Wang, Z.; Wang, L.; Su, X.; Pu, J.; Jiang, M.; He, B. Rational transplant timing and dose of mesenchymal stromal cells in patients with acute myocardial infarction: a meta-analysis of randomized controlled trials. Stem Cell Res. Ther. 2017, 1, 21. [CrossRef]
- Geurts, M.H.; van der Vaart, J.; Beumer, J.; Clevers, H. The Organoid Platform: Promises and Challenges as Tools in the Fight against COVID-19. Stem Cell Rep. 2021, 16, 3, 412–8. [CrossRef]
- Alhaque, S.; Themis, M.; Rashidi, H. Three-dimensional cell culture: from evolution to revolution. Philos. Trans. R. Soc. B Biol. Sci. 2018, 373, 1750, 20170216. [CrossRef]
- Bose, S.; Clevers, H.; Shen, X. Promises and Challenges of Organoid-Guided Precision Medicine. Med 2021, 2, 9, 1011–26. [CrossRef]
- Fetah, K.; Tebon, P.; Goudie, M.J.; Eichenbaum, J.; Ren, L.; Barros, N.; Rohollah, N.; Samad, A.; Nureddin, A.; Mehmet, R. D.; Ali,K. The emergence of 3D bioprinting in organ-on-chip systems. Prog. Biomed. Eng. 2019, 1, 1, 012001.
- van der Vaart, J.; Clevers, H. Airway organoids as models of human disease. J. Intern. Med. 2021, 289, 5, 604–13. [CrossRef]
- Sun, X.Y.; Ju, X.C.; Li, Y.; Zeng, P.M.; Wu, J.; Zhou, Y.Y.; Shen, L.B.; Dong, J.; Chen, Y.J.; Luo, Z.G. Generation of vascularized brain organoids to study neurovascular interactions. eLife, 2022, 4, 11, e76707. [CrossRef]
- Peng, L.; Gao, L.; Wu, X.; Fan, Y.; Liu, M.; Chen, J.; Song, J.; Kong, J.; Dong, Y.; Li, B.; Liu, A.; Bao, F. Lung Organoids as Model to Study SARS-CoV-2 Infection. Cells 2022, 11, 17, 2758. [CrossRef]
- Marrero, D.; Pujol-Vila, F.; Vera, D.; Gabriel, G.; Illa, X.; Elizalde-Torrent, A.; Alvarez, M.; Villa, R. Gut-on-a-chip: Mimicking and monitoring the human intestine. Biosens. Bioelectron. 2021, 181, 113156. [CrossRef]
- Criscione, J.; Rezaei, Z.; Hernandez Cantu, C.M.; Murphy, S.; Shin, S.R.; Kim, D.H. Heart-on-a-chip platforms and biosensor integration for disease modeling and phenotypic drug screening. Biosens. Bioelectron. 2023, 220, 114840. [CrossRef]
- Ko, J.; Park, D.; Lee, S.; Gumuscu, B.; Jeon, N.L. Engineering Organ-on-a-Chip to Accelerate Translational Research. Micromachines 2022, 13, 8, 1200. [CrossRef]
- Telles-Silva, K.A.; Pacheco, L.; Komatsu, S.; Chianca, F.; Caires-Júnior, L.C.; Araujo, B.H.S., Goulart, E.; Zatz, M.Applied Hepatic Bioengineering: Modeling the Human Liver Using Organoid and Liver-on-a-Chip Technologies. Front. Bioeng. Biotechnol. 2022, 10, 845360. [CrossRef]
- Gaitán-Salvatella, I.; López-Villegas, E.O.; González-Alva, P.; Susate-Olmos, F.; Álvarez-Pérez, M.A. Case Report: Formation of 3D Osteoblast Spheroid Under Magnetic Levitation for Bone Tissue Engineering. Front. Mol. Biosci. 2021, 8, 672518. [CrossRef]
- Baarsma, H.A.; Van der Veen, C.H.T.J.; Lobee, D.; Mones, N.; Oosterhout, E.; Cattani-Cavalieri, I. Epithelial 3D-spheroids as a tool to study air pollutant-induced lung pathology. SLAS Discov. 2022, 27, 3, 185–90. [CrossRef]
- Ryu, N.E.; Lee, S.H.; Park, H. Spheroid Culture System Methods and Applications for Mesenchymal Stem Cells. Cells 2019, 8, 12, 1620. [CrossRef]
- Rauth, S.; Karmakar, S.; Batra, S.K.; Ponnusamy, M.P. Recent advances in organoid development and applications in disease modeling. Biochim. Biophys. Acta BBA - Rev. Cancer, 2021, 1875, 2, 188527. [CrossRef]
- Long, C.; Wang, J.; Gan, W.; Qin, X.; Yang, R.; Chen, X. Therapeutic potential of exosomes from adipose-derived stem cells in chronic wound healing. Front. Surg. 2022, 9, 1030288. [CrossRef]
- He, C.; Lu, D.; Lin, Z.; Chen, H.; Li, H.; Yang, X.; Yang, M.; Wang, K.; Wei, X.; Zheng, S.; Xu, X. . Liver Organoids, Novel and Promising Modalities for Exploring and Repairing Liver Injury. Stem Cell Rev. Rep. 2023, 19, 2, 345–57. [CrossRef]
- Chen, J.; Na, F. Organoid technology and applications in lung diseases: Models, mechanism research and therapy opportunities. Front. Bioeng. Biotechnol. 2022, 10, 1066869. [CrossRef]
- Grimm, D.; Schulz, H.; Krüger, M.; Cortés-Sánchez, J.L.; Egli, M.; Kraus, A. The Fight against Cancer by Microgravity: The Multicellular Spheroid as a Metastasis Model. Int. J. Mol. Sci. 2022, 23, 6, 3073. [CrossRef]
- Hofer, M.; Lutolf, M.P. Engineering organoids. Nat. Rev. Mater. 2021, 6, 5, 402–20. [CrossRef]
- Alzamil, L.; Nikolakopoulou, K.; Turco, M.Y. Organoid systems to study the human female reproductive tract and pregnancy. Cell Death Differ. 2021, 28, 1, 35–51. [CrossRef]
- Brassard, J.A.; Nikolaev, M.; Hübscher, T.; Hofer, M.; Lutolf, M.P. Recapitulating macro-scale tissue self-organization through organoid bioprinting. Nat Mater. 2021, 20,1, 22–9. [CrossRef]
- Miller, A.J.; Dye, B.R.; Ferrer-Torres, D.; Hill, D.R.;Overeem, A.W., Shea, L.D.; Spence, J.R. Generation of lung organoids from human pluripotent stem cells in vitro. Nat Protoc. 2019, 14, 2, 518–40. [CrossRef]
- Sutherland, R.M.; Inch, W.R.; McCredie, J.A.; Kruuv, J. A Multi-Component Radiation Survival Curve Using an in vitro Tumour Model. Int J Radiat Biol Relat Stud Phys Chem Med. 1970, 18, 5, 491–95. [CrossRef]
- Sutherland, R.M.; McCredie, J.A.; Inch, W.R. Growth of Multicell Spheroids in Tissue Culture as a Model of Nodular Carcinomas. J Natl Cancer Inst. 1971, 46, 1, 113–20. [CrossRef]
- Hamburger, A.W.; Salmon, S.E. Primary Bioassay of Human Tumor Stem Cells. Science. 1977, 197, 4302, 461–63. [CrossRef]
- Orkin, R.W.; Gehron, P.; McGoodwin, E.B.; Martin, G.R.; Valentine, T.; Swarm, R. A Murine Tumor Producing a Matrix of Basement Membrane. J Exp Med. 1977, 145, 1, 204–20. [CrossRef]
- Garreta, E.; Kamm, R.D.; Chuva de Sousa Lopes, S.M.; Lancaster, M.A.; Weiss, R.; Trepat, X.; Hyun, I.; Montserrat, N. Rethinking organoid technology through bioengineering. Nat Mater. 2021, 20, 2, 145–55. [CrossRef]
- Clevers, H. Modeling Development and Disease with Organoids. Cell. 2016, 165, 7, 1586–97. [CrossRef]
- Jeong, Y.; Tin, A.; Irudayaraj, J. Flipped Well-Plate Hanging-Drop Technique for Growing Three-Dimensional Tumors. Front Bioeng Biotechnol. 2022, 4,10, 898699. [CrossRef]
- Kotze, L.A.; Beltran, C.G.G.; Lang, D.; Loxton, A.G.; Cooper, S.; Meiring, M.; Koegelenberg, C.F.N.; Allwood, B.W.; Malherbe, S.T.; Hiemstra, A.M.; Glanzmann, B.; Kinnear, C.; Walzl, G.; du Plessis, N. Establishment of a Patient-Derived, Magnetic Levitation-Based, Three-Dimensional Spheroid Granuloma Model for Human Tuberculosis. mSphere. 2021, 6, 4. [CrossRef]
- Rodoplu, D.; Matahum, J.S.; Hsu, C.H. A microfluidic hanging drop-based spheroid co-culture platform for probing tumor angiogenesis. Lab Chip. 2022, 22, 7, 1275–85. [CrossRef]
- Souza, G.R.; Molina, J.R.; Raphael, R. M.; Ozawa, M.G.; Stark, D.J.; Levin, C.S.; Bronk, L.F.; Ananta, J.S.; Mandelin, J.; Georgescu, M.M.; Bankson, J.A.; Gelovani, J.G.; Killian, T.C.; Arap, W.; Pasqualini, R. Three-dimensional tissue culture based on magnetic cell levitation. Nat Nanotechnol. 2010, 5,4, 291–6. [CrossRef]
- Goulart, E.; Caires-Junior, L.C.; de Telles-Silva, K.A.; Araujo, B.H.S.; Rocco, S.A.; Sforca, M.; Kobayashi, G.S.; Musso, C.M.; Assoni, A.F.; Oliveira, D.; Caldini, E.; Raia, S.; Lelkes, P.I.; Zatz, M. 3D bioprinting of liver spheroids derived from human induced pluripotent stem cells sustain liver function and viability in vitro. Biofabrication. 2019, 12, 1, 015010. [CrossRef]
- Bruns J.; Zustiak, S.P. Hydrogel-Based Spheroid Models of Glioblastoma for Drug Screening Applications. Mo Med. 2021, 118, 4, 346–51.
- Guillaume, O.; Kopinski-Grünwald, O.; Weisgrab, G.; Baumgartner, T.; Arslan, A.; Whitmore K.; Van Vlierberghe, S.; Ovsianikov, A. Hybrid spheroid microscaffolds as modular tissue units to build macro-tissue assemblies for tissue engineering. Acta Biomater, 2023, 15, 165, 72-85. [CrossRef]
- Huang, Z.; Yu, P.; Tang, J. Characterization of Triple-Negative Breast Cancer MDA-MB-231 Cell Spheroid Model. OncoTargets Ther. 2020, 13, 5395–405. [CrossRef]
- Decarli, M.C.; Vidigal De Castro, M.; Adami Nogueira, J.; Harue, T.; Nagahara, M.; Buzatto Westin, C.; Leite, R.; de Oliveira, A.; Da Silva, J.V.; Moroni, L.; Mota, C.; Moraes, Â.M. Development of a Device Useful to Reproducibly Produce Large Quantities of Viable and Uniform Stem Cell Spheroids with Controlled Diameters. Biomaterials Advances. 2022, 135, 112685.
- Marsee, A.; Roos, F.J.M.; Verstegen, M.M.A.; Organoid Consortium H.P.B.; Gehart. H; de Koning, E.; Lemaigre, F.; Forbes, S.J.; Peng, W.C.; Huch, M.; Takebe, T.; Vallier, L.; Clevers, H.; van der Laan, L.J.W.; Spee, B. Building consensus on definition and nomenclature of hepatic, pancreatic, and biliary organoids. Cell Stem Cell. 2021, 28, 5, 816–32. [CrossRef]
- Rossi, G.; Manfrin, A.; Lutolf, M.P. Progress and potential in organoid research. Nat Rev Genet. 2018, 19,11, 671–87. [CrossRef]
- Zhao, Z.; Chen, X.; Dowbaj, A.M.; Sljukic, A.; Bratlie, K.; Lin, L.; Li, F.S. E.; Gowri, M. B.; Zhaowei, C.; Soragni, A.; Meritxell, H.; Zenga, Y. A.; Wang, Q.; Yu, H. Organoids. Nat Rev Methods Primer. 2022, 2,1, 1–21.
- Sakalem, M.E.; Ribeiro-Paes, J.T. New methodologies for old problems: tridimensional gastrointestinal organoids and guts-on-a-chip. J Coloproctology Rio. 2018, 38, 90–3. [CrossRef]
- Shahabipour, F.; Ashammakhi, N.; Oskuee, R.K.; Bonakdar, S.; Hoffman, T.; Shokrgozar, M.A.; Khademhosseini, A. Key components of engineering vascularized 3-dimensional bioprinted bone constructs. Transl Res., 2020, 216, 57–76. [CrossRef]
- Schutgens, F.; Clevers, H. Human Organoids: Tools for Understanding Biology and Treating Diseases. Annu Rev Pathol Mech Dis. 2020, 15, 1. 211–34. [CrossRef]
- Gu, Q.; Tomaskovic-Crook, E.; Wallace, G.G.; Crook, J.M. 3D Bioprinting Human Induced Pluripotent Stem Cell Constructs for In Situ Cell Proliferation and Successive Multilineage Differentiation. Adv Healthc Mater. 2017, 6, 17, 1700175.
- Kim, W.; Gwon, Y.; Park, S.; Kim, H.; Kim, J. Therapeutic strategies of three-dimensional stem cell spheroids and organoids for tissue repair and regeneration. Bioact Mater. 2023, 19, 50–74. [CrossRef]
- Lee, S.Y.; Koo, I.S.; Hwang, H.J.; Lee, D.W. In Vitro three-dimensional (3D) cell culture tools for spheroid and organoid models. SLAS Discov. 2023, 28, 4, 119–37. [CrossRef]
- Fang, Z.; Li, P.; Du, F.; Shang, L.; Li, L. The role of organoids in cancer research. Exp Hematol Oncol. 2023, 12, 1, 69. [CrossRef]
- LeSavage, B.L.; Suhar, R.A.; Broguiere, N.; Lutolf, M.P.; Heilshorn, S.C. Next-generation cancer organoids. Nat Mater. 2022, 21, 2, 143–59. [CrossRef]
- Wei, J.; Zhang, W.; Zhao, B. Human liver organoid: modeling liver steatosis and beyond. Cell Regen. 2023, 12, 1, 17. [CrossRef]
- Nuciforo, S.; Heim, M.H. Organoids to model liver disease. JHEP Rep. 2021,3, 1, 100198. [CrossRef]
- Bouwmeester, M.C.; Bernal, P.N.; Oosterhoff, L.A.; van Wolferen, M.E.; Lehmann, V.; Vermaas, M.; Buchholz, M.B.; Peiffer, Q.C.; Malda, J.; van der Laan, L.J.W.; Kramer, N.I.; Schneeberger, K.; Levato, R.; Spee, B. Bioprinting of Human Liver-Derived Epithelial Organoids for Toxicity Studies. Macromol Biosci. 2021, 21, 12, 2100327. [CrossRef]
- Takebe, T.; Sekine, K.; Enomura, M.; Koike, H.; Kimura, M.; Ogaeri, T.; Zhang, R.R.; Ueno, Y.; Zheng, Y.W.; Koike, N.; Aoyama, S.; Adachi, Y.; Taniguchi, H. Vascularized and functional human liver from an iPSC-derived organ bud transplant. Nature. 2013, 499, 7459, 481–4.
- Eichmüller, O.L.; Knoblich, J.A. Human Cerebral Organoids — a New Tool for Clinical Neurology Research. Nat Rev Neurol. 2022, 18, 11, 661–80. [CrossRef]
- Lancaster, M.A.; Renner, M.; Martin, C.A.; Wenzel, D.; Bicknell, L.S.; Hurles M. E.; Homfray, T.; Penninger, M. P.; Jackson, P. A.; Knoblich, A. J. Cerebral Organoids Model Human Brain Development and Microcephaly. Nature. 2013, 501, 7467, 373–79. [CrossRef]
- Lancaster, M.A.; Knoblich, J.A. Generation of cerebral organoids from human pluripotent stem cells. Nat Protoc. 2014, 9, 10, 2329–40. [CrossRef]
- Dumont, S.; Jan, Z.; Heremans, R.; Van Gorp, T.; Vergote, I.; Timmerman D. Organoids of epithelial ovarian cancer as an emerging preclinical in vitro tool: a review. J Ovarian Res. 2019, 12, 1, 105. [CrossRef]
- Liu, H.D.; Xia, B.R.; Jin, M.Z.; Lou, G. Organoid of ovarian cancer: genomic analysis and drug screening. Clin Transl Oncol. 2020, 22, 8, 1240–51. [CrossRef]
- Hai, J.; Zhang, H.; Zhou, J.; Wu, Z.; Chen, T.; Papadopoulos, E.; Dowling, C.M.; Pyon, V.; Pan, Y.; Liu, J.B.; Bronson, R.T.; Silver, H.; Lizotte, P.H.; Deng, J.; Campbell, J.D.; Sholl, L.M.; Ng, C.; Tsao, M.S.; Thakurdin, C.; Bass, A.J.; Wong, K.K. Generation of genetically engineered mouse lung organoid models for squamous cell lung cancers allows for the study of combinatorial immunotherapy. Clin Cancer Res Off J Am Assoc Cancer Res. 2020, 26, 13, 3431–42.
- Reid, J.A.; Mollica, P.A.; Bruno, R.D.; Sachs, P.C. Consistent and reproducible cultures of large-scale 3D mammary epithelial structures using an accessible bioprinting platform. Breast Cancer Res. 2018, 20, 1, 122.
- Sprangers, J.; Zaalberg, I.C.; Maurice, M.M. Organoid-based modeling of intestinal development, regeneration, and repair. Cell Death Differ. 2021, 28, 1, 95–107. [CrossRef]
- Salgado, A.J.; Oliveira, J.M.; Martins, A.; Teixeira, F.G.; Silva, N.A.; Neves, N.M.; Sousa, N.; Reis, R.L. Tissue engineering and regenerative medicine. Int Rev Neurobiol. 2013, 108,1-33. [CrossRef]
- Tang, X.Y.; Wu, S.; Wang, D.; Chu, C.; Hong, Y.; Tao, M.; Hu, H.; Xu, M.; Guo, X.; Liu, Y. Human organoids in basic research and clinical applications. Signal Transduct Target Ther. 2022, 7, 1, 1–17. [CrossRef]
- Gilazieva, Z.; Ponomarev, A.; Rutland, C.; Rizvanov, A.; Solovyeva, V. Promising applications of tumor spheroids and organoids for personalized medicine. Cancers. 2020, 12, 10, 2727. [CrossRef]
- Hofer, M.; Lutolf, M.P. Engineering organoids. Nat Rev Mater. 2021, 6, 5, 402–20. [CrossRef]
- Voges, H.K.; Foster, S.R.; Reynolds, L.; Parker, B.L; Devilée, L.; Quaife-Ryan, G.A.; Fortuna, P.R.J.; Mathieson, E.; Fitzsimmons, R.; Lor, M.; Batho, C.; Reid, J.; Pocock, M.; Friedman, C.E.; Mizikovsky, D.; Francois, M.; Palpant, N.J.; Needham, E.J.; Peralta, M.; Monte-Nieto, G.D.; Jones, L.K.; Smyth, I.M.; Mehdiabadi, N.R; Bolk, F.; Janbandhu, V.; Yao, E.; Harvey, R.P.; Chong, J.J.H.; Elliott, D.A; Stanley, E.G.; Wiszniak, S.; Schwarz, Q.; James, D.E.; Mills, R.J.; Porrello, E.R.; Hudson, J.E. Vascular cells improve functionality of human cardiac organoids. Cell Rep. 2023, 42, 5, 112322. [CrossRef]
- Salewskij, K.; Penninger, J.M. Blood vessel organoids for development and disease. Circ Res. 2023, 132, 4, 498–510. [CrossRef]
- Shelton, SE. Vascular microphysiological systems. Curr Opin Hematol. 2024, 31, 3, 155–61. [CrossRef]
- Varani, J.; McClintock, S.D.; Aslam, M.N. Organoid culture to study epithelial cell differentiation and barrier formation in the colon: bridging the gap between monolayer cell culture and human subject research. Vitro Cell Dev Biol - Anim. 2021, 57, 2, 174–90. [CrossRef]
- Shpichka, A.; Bikmulina, P.; Peshkova, M.; Kosheleva, N.; Zurina, I.; Zahmatkesh, E.; Khoshdel-Rad, N.; Lipina, M.; Golubeva, E.; Butnaru, D.; Svistunov, A.; Vosough, M.; Timashev, P. Engineering a model to study viral infections: bioprinting, microfluidics, and organoids to defeat coronavirus disease 2019 (COVID-19). Int J Bioprinting. 2020, 6,4, 302. [CrossRef]
- Xing, Y.;Liu, J.; Guo, X.; Liu, H.; Zeng, W.; Wang, Y.; Zhang, C.; Lu, Y.; He, D.; Ma, S.; He, Y.; Xing, X.H. Engineering organoid microfluidic system for biomedical and health engineering: a review. Chin J Chem Eng. 2021, 30, 244–54. [CrossRef]
- Silva da Costa, F.A.; Soares, M.R.; Malagutti-Ferreira, M.J.; Silva, G.R.; Lívero, F.A.D.R.; Ribeiro-Paes, J.T. Three-dimensional cell cultures as a research platform in lung diseases and COVID-19. Tissue Eng Regen Med. 2021, 18, 5, 735–45. [CrossRef]
- Li, Z.; Hui, J.; Yang, P.; Mao, H. Microfluidic organ-on-a-chip system for disease modeling and drug development. Biosensors. 2022, 12, 6, 370. [CrossRef]
- Huh, D.; Matthews, B.D.; Mammoto, A; Montoya-Zavala, M.; Hsin, H.Y.; Ingber, D.E. Reconstituting organ-level lung functions on a chip. Science. 2010, 328, 5986, 1662–8. [CrossRef]
- Huh, D.; Hamilton, G.A.; Ingber, D.E. From 3D cell culture to organs-on-chips. Trends Cell Biol. 2011, 21, 12, 745–54. [CrossRef]
- Mathur, A.; Loskill, P.; Shao, K.; Huebsch, N.; Hong, S.; Marcus, S.G.; Marks, N.; Mandegar, M.; Conklin, B.R.; Lee, L.P.; Healy, K.E. Human iPSC-based cardiac microphysiological system for drug screening applications. Sci Rep. 2015, 5, 1, 8883. [CrossRef]
- Kilic, O.; Pamies, D.; Lavell, E.; Schiapparelli, P.; Feng, Y.; Hartung, T.; Bal-Price, A.; Hogberg, H.T.; Quinones-Hinojosa, A.; Guerrero-Cazares, H.; Levchenko, A . Brain-on-a-chip model enables analysis of human neuronal differentiation and chemotaxis. Lab Chip. 2016, 16, 21, 4152–62. [CrossRef]
- Weber, E.J.; Lidberg, K.A.; Wang, L.; Bammler, T.K.; MacDonald, J.W.; Li, M.J.; Redhair, M.; Atkins, W.M.; Tran, C.; Hines, K.M.; Herron, J.; Xu, L.; Monteiro, M.B.; Ramm, S.; Vaidya, V.; Vaara, M.; Vaara, T.; Himmelfarb, J.; Kelly, E.J. Human kidney on a chip assessment of polymyxin antibiotic nephrotoxicity. JCI Insight. 2018, 3, 24. [CrossRef]
- Jang, K.J.; Otieno, M.A.; Ronxhi, J.; Lim, H. K.; Ewart, L.; Kodella, K.R.; Petropolis, D.B.; Kulkarni, G.; Rubins, J.E.; Conegliano, D.; Nawroth, J.; Simic, D.; Lam, W.; Singer, M.; Barale, E.; Singh, B.; Sonee, M.; Streeter, A.J.; Manthey, C.; Jones, B.; Srivastava, A.; Andersson, L.C.; Williams, D.; Park, H.; Barrile, R.; Sliz, J.; Herland, A.; Haney, S.; Karalis, K.; Ingber, D.E.; Hamilton, G.A. Reproducing human and cross-species drug toxicities using a Liver-Chip. Sci Transl Med. 2019, 11, 517. [CrossRef]
- Mun, K.; Arora, K.; Huang, Y.; Yang, F.; Yarlagadda, S.; Ramananda, Y.; Abu-El-Haija, M.; Palermo, J.J.; Appakalai, B.N.; Nathan, J.D.; Naren, A.P. Patient-derived pancreas-on-a-chip to model cystic fibrosis-related disorders. Nat Commun. 2019, 10, 1, 3124. [CrossRef]
Figure 1.
(A) Cells growing around the silk threads impregnated with plasma, as described and demonstrated by Carrel and Burrows (Adapted from [16]); (B) Schematic representation of the available cell proliferation directions provided by the silk threads surface.
Figure 1.
(A) Cells growing around the silk threads impregnated with plasma, as described and demonstrated by Carrel and Burrows (Adapted from [16]); (B) Schematic representation of the available cell proliferation directions provided by the silk threads surface.
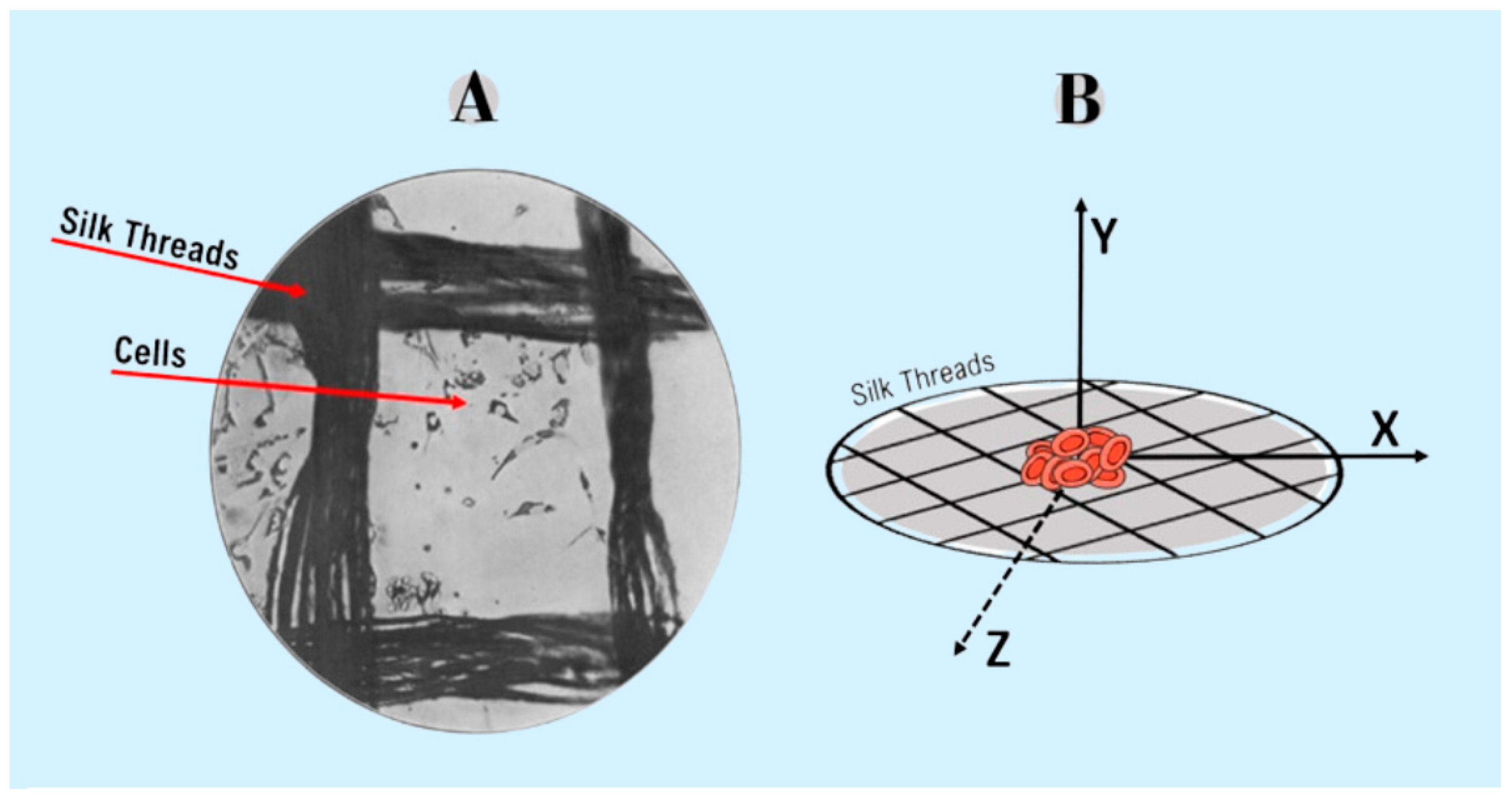
Figure 2.
Timeline’s review of researchers which contributed to scientific cell culture knowledge and techniques since 1885.
Figure 2.
Timeline’s review of researchers which contributed to scientific cell culture knowledge and techniques since 1885.

Figure 3.
Cell culture techniques used as experimental models in research. (A) two-dimensional (2D) cell culture; (B) three-dimensional (3D) cell culture; and (C) organ-on-a-chip basic design.
Figure 3.
Cell culture techniques used as experimental models in research. (A) two-dimensional (2D) cell culture; (B) three-dimensional (3D) cell culture; and (C) organ-on-a-chip basic design.
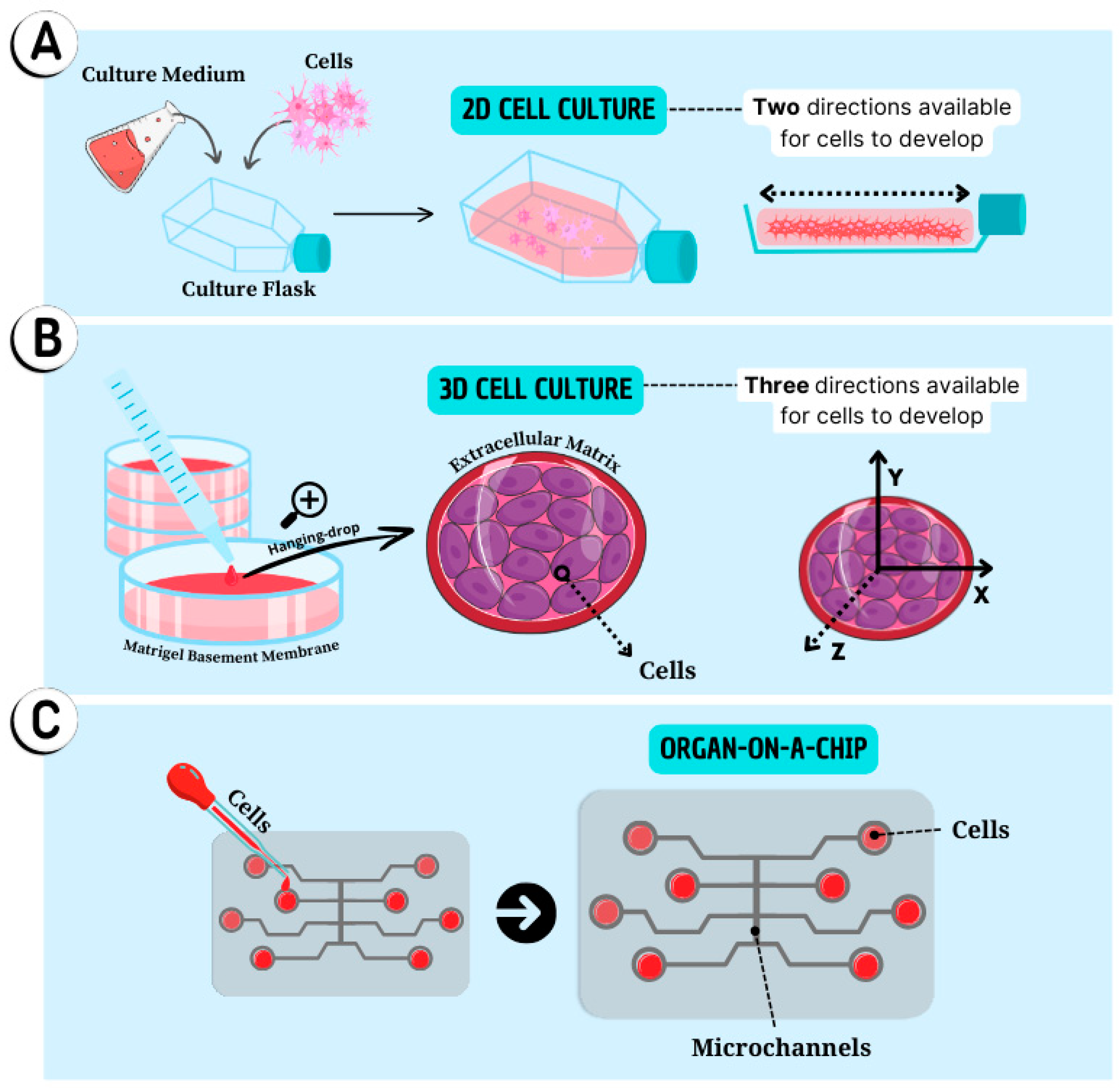
Figure 4.
(A) Traditional and (B) 3D cell culture research study models. (C) A schematic representation of the association (connection) of individual organs-on-a-chip for the development of a human-on-a-chip.
Figure 4.
(A) Traditional and (B) 3D cell culture research study models. (C) A schematic representation of the association (connection) of individual organs-on-a-chip for the development of a human-on-a-chip.
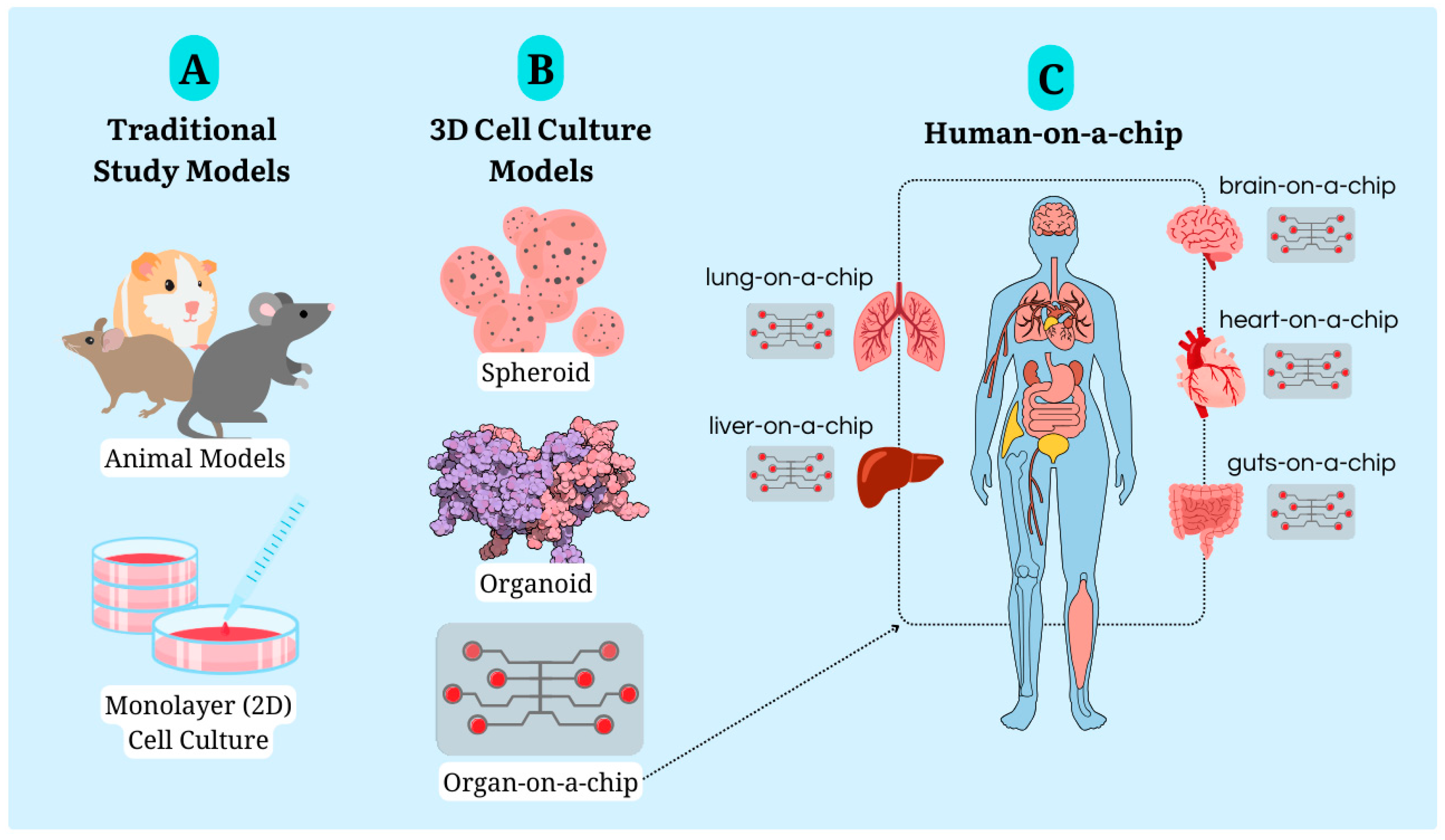

Table 1.
Key studies on the derivation of induced pluripotent stem cells (iPSCs) from various cell sources.
Table 1.
Key studies on the derivation of induced pluripotent stem cells (iPSCs) from various cell sources.
| Cell Source | Cell Lineage | Species | Factors | Methods | Reference |
|---|---|---|---|---|---|
| Embryonic/Adult Fibroblasts | MEF and TTF cells | Mice | Oct3/4, Sox2, c-Myc, and Klf4 | Retroviral vector transduction; Plat-E cells line expansion | [55] |
| Adult Fibroblasts | MSC derived from human OCT4 knock-in ES cells | Human | Oct4, Sox2, NANOG, and Lin28 | Lentiviral vector transduction 293FT cell line expansion | [56] |
| Keratinocytes | Keratinocytes foreskin derived | Human | Oct4, Sox2, c-Myc, and Klf4 | Retroviral MSCVpuro vector transduction; Phoenix Amphotropic cell line expansion | [57] |
| Peripheral Blood Cells | CD34+ mobilized human peripheral blood cells | Human | Oct4, Sox2, Klf4 and c-Myc | Retroviral vector transduction; 293T cells line expansion | [58] |
| Cord Blood Cells |
Human cord blood (CB)-derived endothelial cells (ECs) |
Human | Oct4, Sox2, NANOG, and Lin28 | Lentiviral vector Addgene transduction | [59] |
| Amniotic Membrane MSC | MSC placenta derived | Human | Sox2, Klf4, Oct4, and c-Myc | pMX-based retroviruses transduction | [60] |
| Dental MSC | MSC Human third molars derived | Human | Oct3/4, Sox2, c-Myc, and Klf4 | EcoRI site of the pMXs-based retroviruses transduction | [61] |
| Urine Cells | RPTE cells | Human | Oct4, Sox2, Klf4, and c-Myc | Adgene retroviral vector transduction, HEK293T cells expansion | [62,63] |
Table 2.
Multidimensional analysis of experimental models in biosciences.
| Animal Model | 2D Cell Culture | 3D Cell Culture | Organ-on-a-chip | |
|---|---|---|---|---|
| Ease of Maintenance | + | +++ | +++ | ++ |
| Recapitulation of Biology Development | +++ | N/A | ++ | +++ |
| Length of Experiments | ++ | +++ | +++ | +++ |
| Genetic Engineering | + | +++ | +++ | +++ |
| Physiological Complexity | +++ | N/A | + | ++ |
| Relative Cost | + | +++ | ++ | ++ |
| Recapitulation of Human Physiology | ++ | + | +++ | +++ |
Legend. (+) Good; (++) Very good; (+++) Excellent; (N/A) Not Applicable.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



