
Submitted:
02 September 2024
Posted:
03 September 2024
You are already at the latest version
Abstract
Glioblastoma (GBM) is a prevalent type of malignancy within the central nervous system (CNS), associated with a poor prognosis. The standard treatment for GBM includes surgical resection of the tumor, followed by radiotherapy and chemotherapy; yet despite these interventions, overall treatment outcomes remain suboptimal. The blood-brain barrier (BBB), which plays a crucial role in maintaining the stability of brain tissue under normal physiological conditions of the CNS,also poses a significant obstacle to the effective delivery of therapeutic agents to GBM. Recent preclinical studies have demonstrated that nanomedicine delivery systems (NDDS) offer promising results, demonstrating both effective GBM targeting and safety, thereby presenting a potential solution for targeted drug delivery. This review explores the various strategies employed in preclinical studies to overcome the BBB for drug delivery. Subsequently, the results of clinical translation of NDDS has been summarized, highlighting the progress made. Finally, we discuss potential strategies for advancing NDDS development and accelerating their translational research through well-designed clinical trials in GBM therapy.
Keywords:
Glioblastoma
; Blood-brain barrier
; Drug delivery
; Nanomedicine delivery system
; Nanoparticles
1. Introduction
Glioblastoma (GBM) is the most prevalent malignant primary brain tumor (accounting for 13% of all tumors and 50.1% of all malignant tumors) with a 5-year survival rate of less than 7% [1,2]. Despite progress in multimodal treatments encompassing surgery, radiotherapy, chemotherapy, and targeted therapy, the overall prognosis remains unfavorable, with a median survival duration of merely 8 months [1]. Figure 1 illustrates seminal milestones in GBM research and treatment. In 2021, GBM, classified as IDH-wildtype and defined as a WHO grade 4, specifically refers to diffusely infiltrative gliomas exhibiting classic histopathological features or molecular alterations [3]. Due to its heterogeneity, invasiveness, and difficulties in defining malignant tissue margins, compared to other solid tumors, along with the BBB, leading to rapid disease progression in up to 70% of patients within one year of diagnosis. Chemotherapy is one of the standardized steps in GBM treatment [4]. However, non-targeted administration often leads to low efficacy and systemic toxicity, while the presence of BBB demands higher dosages from chemotherapeutic drugs. Developing brain-targeting drugs or formulations that can cross this barrier are urgently needed for effective treatment while minimizing toxic side effects.
The blood-brain barrier (BBB) is a natural defense mechanism between the bloodstream and brain tissue. The intact BBB effectively segregates brain tissue from peripheral blood and plays a pivotal biological role in upholding the fundamental stability of the internal environment of brain tissue and the normal physiological state of the central nervous system (CNS) (Figure 2a). The BBB restricts nearly all large molecules and over 98% of small molecule candidate drugs from permeating into it [5,6,7]. Therefore, the key issue in drug therapy for GBM is to design and develop targeted nanomedicine delivery systems (NDDS) that can effectively cross the BBB and ensure the drugs reach their intended sites in the brain. Two main approaches to delivering chemotherapy drugs to GBM involve either crossing or bypassing the BBB (Figure 2b).
Biomaterials, encompassing both natural and synthetic sources, are materials that possess excellent biocompatibility and physicochemical properties, utilized in drug delivery. These materials can either be directly conjugated with drugs to enhance their pharmacokinetic properties or serve as excipients, leveraging nanotechnology to encapsulate drugs within nanocarriers (10-500nm) or attach/adsorb them onto the carrier surface, thereby forming NDDS that achieve sustained release, controlled release and targeted delivery of drugs [8]. The development of NDDS represents a promising avenue for GBM treatment, albeit not without its challenges. A significant bottleneck lies in the transition from preclinical research to clinical trials. The heterogeneity of GBM presents a challenge as it may lead to varied therapeutic responses. Moreover, the BBB further complicates drug delivery. Despite notable advancements made by NDDS in overcoming the BBB, translating these preclinical successes into clinical practice has been hindered by issues related to scalability, regulatory pathways, and clinical validation.
Recent reviews have delved into the application of Nanomedicine in brain tumor pharmacotherapy, highlighting the significance of nanotechnology in overcoming the BBB [9,10,11]. Nevertheless, amidst the heterogeneity of GBM and the constraints imposed by the BBB on drug delivery to tumor sites, the clinical advancements of nano-drug delivery systems remain largely uncharted territories. This review aims to systematically classify strategies for crossing and bypassing the BBB, highlight the key research accomplishments from the past 5 years using these systems, and discuss the strengths and weaknesses of different BBB-overcoming strategies. Finally, we offer our perspectives on the future developments and challenges in this field, aiming to provide more comprehensive and in-depth guidance for the practical application of nanomedicine in the treatment of GBM.
2. Strategies for NDDS to Cross the BBB
The BBB is a formidable obstacle for drug delivery to the brain due to its tightly regulated selective permeability. However, several strategies have been developed to enhance the delivery of nanomedicines across the BBB (Table 1).
2.1. Receptor-Mediated Transport
This is a pivotal approach in the nanomedicine delivery system for crossing the BBB, as it exploits the natural transport mechanisms of the brain's protective interface. The existence of nutrient transport receptors on BBB endothelial cell membranes is crucial for sustaining normal brain function in humans, with some overexpressed on GBM cell membranes. A tailored nanomedicine delivery system enhances BBB permeability and GBM cell targeting by binding to these receptors[43] (Figure 3).
2.1.1. Transferrin Receptors and Lactoferrin Receptors
The overexpression of transferrin receptors (Tf) and lactoferrin receptors (Lf) on BBB endothelial cells and glioma cells presents an opportunity for targeted drug delivery. Tf-functionalized nanoparticles (Tf-NPs) crossing BBB and targeting GBM in intracranial orthotopic models, reducing tumor burden and extending survival [12]. Lf, an iron-binding glycoprotein, surpasses Tf and OX-26 in BBB permeation [44]. Dong et al. developed a glioma-targeted drug delivery system utilizing biodegradable periodic mesoporous organosilica nanoparticles modified with Lf ligands, enhancing drug delivery to brain gliomas [13]. Taskeen et al. introduced an Lf receptor-mediated nanomedicine, USLP-NH2-PEG-TMZ-Lf, with enhanced BBB permeability and cytotoxicity for GBM treatment [14].
2.1.2. Acetylcholine Receptors
Nicotinic acetylcholine receptors are widely distributed on the surface of the BBB, serving as a crucial target for drug delivery. 2-methacryloyloxyethyl phosphorylcholine (MPC)-based protein nanocapsules enhance CNS protein transport. Chen et al. engineered an NDDS containing TMZ (TMZ@RVG-Zein NPs), penetrating BBB via receptor-mediated endocytosis. This NDDS exhibits exceptional biocompatibility and is capable of penetrating into GBM (U87) cell lines, thereby facilitating the release of TMZ for therapeutic effect [15].
2.1.3. Folate Receptors
BBB and glioma cells express folate receptors (FR), crucial for tumor DNA replication. Folate-modified NPs are internalized by tumor FR via receptor-mediated endocytosis [45]. Xiang Yang Zhong et al. engineered folate/iRGD-modified NPs, enhancing targeting and uptake, delivering TMZ to nuclei, inhibiting tumor proliferation. FR can integrate with other drugs for novel delivery systems, e.g., WGA/FA-MPEG-PCL NPs loaded with ETO, BCNU, DOX, targeting BBB to inhibit GBM growth [16].
2.1.4. Low-Density Lipoprotein Receptors
Members of the LDL receptor superfamily exhibit binding affinity towards a diverse range of ligands, including lipoproteins, proteases, and protease inhibitor complexes, facilitating their transport into the cell nucleus. Specifically, apolipoprotein E demonstrates selective binding to LDL receptors, while solid lipid nanoparticles functionalized with apolipoprotein E have been demonstrated to enhance BBB delivery by 1.5 times compared to non-functionalized nanoparticles [46]. Jiantang Liang and colleagues designed the versatile biomimetic nanoplatform L-D-I/NPs to selectively target GBM by binding to low density lipoprotein receptacle-associated protein-1 (LRP1) and crossing the BBB. Effectively inhibited the progression of in orthotopic GBM and significantly prolonged survival [47]. Angiopep-2 modified nanoparticles selectively target LRP1. Compared with the control group,intravenous injection of Ti@FeAu-Ang nanoparticles resulted in a 10-fold reduction in tumor volume [17]. The Au-DOX@PO-ANG NPs designed by Chen He and colleagues also significantly reduced the size of GBM in mice[18].
2.1.5. Epidermal Growth Factor Receptors
Epidermal growth factor receptors (EGFR), an ErbB family tyrosine kinase receptor, governs GBM angiogenesis and determines high proliferation and drug resistance. EGFR-targeted drugs like Cetuximab, panitumumab, nimotuzumab, and necitumumab have been clinically approved [48]. EGFR overexpression occurs in 40-70% GBM patients, with EGFRvIII, a cancer-specific deletion, present in 25-50% pleomorphic GBMs [48]. EGFRvIII's absence in normal tissues makes it a prime target for GBM-specific receptor therapies. The antitumor efficacy of Panitumumab-conjugated and TMZ-loaded poly (lactic-co-glycolic acid) nanoparticles (PmAb-TMZ-PLGA-NPs) in GBM cells with overexpressed EGFR is significantly enhanced through the inhibition of caspase-mediated autophagy by this novel NDDS, thereby promoting apoptotic cell death [19].
2.1.6. Human Insulin-like Growth Factor-1 Receptors
Insulin-like growth factor-1 receptors (IGF1R), a receptor tyrosine kinase, is prominently expressed in brain endothelial cells, brain microvessels, and specific neuronal regions. It mediates endocytosis upon high-affinity binding with IGF1, making it a suitable target for brain drug delivery [20,49]. High-affinity anti-IGF1R monoclonal antibodies penetrate the BBB after in situ brain perfusion (ISBP) administration [20]. Additionally, IGF1R5, effectively binds to IGF1R on the BBB, enhancing drug transport [49].
2.1.7. Integrins
The αvβ3/αvβ5 integrins are upregulated in angiogenic sites and GBM, underpinning the reliance on their BBB endothelial and GBM cell expression for GBM-targeted drug delivery systems [50]. Studies confirm that cyclic-Arg-Gly-Asp (cRGD)-installed micelles enhance the targeted delivery and therapeutic efficacy of epirubicin in orthotopic GBM models [51]. Additionally, α5β1 integrin receptor overexpression in GBM cells facilitates the use of RGDk-lipid nanoparticles for concurrent delivery of chemotherapeutics and siRNA, significantly inhibiting tumor growth in mouse models [21]. These discoveries underscore the efficacy of integrin-mediated carriers for drug delivery in aggressive GBM.
2.1.8. CD13
CD13 is widely overexpressed on glioma neovasculature endothelial cells and glioma cell surfaces [52]. Asn-Gly-Asp (NGR), a structural motif for endothelial cells and tumor neovasculature, functions as an effective tumor-targeting drug carrier. Circular iNGR in the blood can specifically and rapidly bind to CD13, demonstrating strong tumor vascular targeting ability [53]. Sai An et al. engineered PEGylated iNGR-modified RNAi nanoparticles that exhibit superior tumor accumulation and penetration, offering a novel avenue for GBM's targeted therapy [52].
2.1.9. Neuropilin-1
Neuropilin-1 (NRP-1), a transmembrane glycoprotein, is overexpressed in GBM cells and serves as a co-receptor for semaphorin3A and vascular endothelial growth factor, contributing significantly to tumor angiogenesis, growth, and metastasis [54]. The CPT-S-S-PEG-iRGD@IR780 nanoparticles were modified by iRGD peptide and photosensitizer IR780, which can be used as drug delivery system. Leveraging αvβ integrin and NRP-1-mediated transport, this system efficiently traverses the BBB to target GBM, enhancing anti-tumor efficacy in combined therapies [22].
2.1.10. Heat Shock Protein 70
Heat shock protein 70 (Hsp70) is selectively expressed on GBM cell membranes, serves as a precise target for GBM therapy, enhancing binding efficiency [23]. Acid-triggered gold nanoparticles (D-A-DA/TPP) selectively deliver DOX to glioma tissue by Hsp70 targeting, with TPP (a peptide that binds to Hsp70 on the membrane of glioma cells) facilitating cellular uptake. Under the weakly acidic tumor microenvironment, D-A-DA/TPP aggregation prolongs circulation, augments binding, and triggers pH-responsive DOX release [23]. Jianfen Zhou et al. devised pHA-AOHX-VAP-DOX, a nanodrug system traversing the BBB via dopamine and GRP78 receptors, a heat shock protein family member, to extend survival in nude mice with intracranial U87 glioma [24].
These examples underscore the versatility and potential of RMT in nanomedicine, particularly for GBM therapy. By binding to specific receptors overexpressed on the BBB and tumor cells, nanoparticles can be guided to their target, enhancing drug delivery efficiency while minimizing systemic side effects. The continued exploration and refinement of RMT strategies hold great promise for improving the prognosis and treatment of brain-related diseases.
2.2. Transporter-Mediated Transport
Membrane transport proteins in neurovascular units, comprising 10-15% of their structure [55], are essential for transmembrane movement of diverse substrates, including small water-soluble molecules. These proteins, categorized into four types based on their mechanisms, undergo conformational changes to facilitate transport and can be targeted by nanoparticles for drug delivery [56]. Their strategic distribution and unique mechanisms on brain capillary endothelial cells (BCECs) enhance the targeting and uptake of nanomedicines in brain tissue (Figure 4).
2.2.1. Glucose Transporters
Glucose is the brain's primary energy source, with glucose transporter-1 (GLUT1) being the predominant transporter on BCEC membranes, facilitating its rapid translocation into brain tissue [57]. Leveraging post-fasting hyperglycemia, nanocarriers can enhance drug delivery across the BBB via GLUT1 [58]. Glycosylated derivatives of heptapeptide ATWLPPR (A7R) have shown improved serum stability and enhanced BCEC uptake via GLUT1-mediated transcytosis, thereby improving BBB penetration and glioma cell absorption [59]. Zhang et al. developed a 2-DG nanocapsule system that exploits GLUT1 overexpression for targeted delivery to the GBM tumor microenvironment [25].
2.2.2. Choline Transporters
The choline transporters (ChTs) protein, prominently expressed on cerebral capillary endothelial cell lumens, efficiently traverses the BBB to transport acetylcholine and choline analogs into the CNS [60]. Li's team engineered a cholinergic-derivative-modified delivery system that significantly increased glioma drug accumulation, inducing apoptosis and enhancing therapeutic outcomes [61]. MPC-n (IVIg) utilizes high-affinity ChT1 overexpression for targeted brain tissue drug delivery, reducing therapeutic doses [62]. Hairong Wang's team devised a universal BBB-permeable smart polymer, pMPC-co-(anti-PD-L1-pPEGMA), that crosses the BBB via choline transporters to effectively treat malignant gliomas [26].
2.2.3. Amino Acid Transporters
L-type amino acid transporter 1 (LAT1) is overexpressed in both BBB and GBM cells [63], facilitating targeted brain drug delivery and minimizing peripheral exposure [64]. MeHg-L-cys harnesses LAT1 to enhance malignant glioma cell targeting and mitigate normal brain tissue toxicity [65]. Amphi-DOPA-loaded wp1066 nano-carriers significantly improved survival in orthotopic mouse GBM models [27].
2.2.4. Vitamin Transporters
The sodium-dependent vitamin C transporter (SVCT2), expressed in choroid plexus epithelial cells and brain tumor cell lines, emerges as a promising nanomedicine target [66]. Ascorbic acid's (AA) CNS penetration, facilitated by its reversible oxidation to dehydroascorbic acid (DHAA), leverages the SVCT2-mediated transport mechanism, enhancing DHAA CNS levels and offering a strategy for drug delivery systems with potential in Alzheimer's therapy [66,67]. Yao Peng's research introduced a glucose-vitamin C modified liposome for paclitaxel delivery to the brain, demonstrating superior targeting efficacy over unmodified or singly modified formulations [28].
2.2.5. Organic Cation Transporters
Organic cation/carnitine transporter 2 (OCTN2, SLC22A5), a member of the OCTN family, is implicated in drug BBB permeation [68] and is upregulated in GBM [69]. Kou's team engineered L-Carnitine-conjugated nanoparticles (LC-PLGA NPs) that exploit OCTN2 overexpression on brain endothelial and glioma cells for enhanced BBB permeability and targeted glioma cell internalization [29].
2.2.6. Organic Anion Transporters
Organic anion-transporting polypeptides (OATPs), a family of multi-specific transporters, include OATP1A2, which facilitates CNS drug uptake at the human BBB [70]. Statins illustrate this by crossing the BBB via the Oatp1a4 transporter, underscoring OATPs' utility in targeted CNS drug delivery. Brain-specific anion transporter 1 (BSAT1), exclusively present in brain microvascular endothelial cells, is pivotal in targeted drug delivery within tumors and their surrounding areas. Research demonstrates platinum-nanogel conjugates with Cx43 and BSAT1 antibodies effectively shrink tumors [30]. Given OATP substrate selectivity, current research prioritizes stroke therapy, with glioma treatment often considered a secondary or dual-targeted approach.
2.2.7. Monocarboxylate Transporters
Monocarboxylate transporter (MCT) family members mediate the cellular translocation of monocarboxylic acids like lactate and pyruvate across various tissues. MCT1-4, integral to the plasma membrane, facilitate the bidirectional exchange of short-chain monocarboxylic acids and protons in mammalian cells [71]. Glioma cells uptake β-Hydroxy-β-methylbutyrate through H+-coupled MCTs [71], and the overexpression of MCT1 and MCT4 on their surfaces correlates with poor prognosis, indicating their diagnostic and therapeutic potential [72]. Huber's research demonstrated a link between MCTs and the brain penetration of cyclic C5-curcuminoids, emphasizing their role in BBB traversal [73].
2.3. Adsorptive-Mediated Transport
Nanocarrier size and surface charge are pivotal for BBB endocytosis, with cationic systems like PEG enhancing drug solubility and cellular uptake through electrostatic interactions with the endothelial cell membrane [74]. Tumor blood vessels' and cells' upregulation of negatively charged glycoproteins increases nanocarrier accumulation in brain tumors [75] (Figure 5). While common transport receptors like albumin and cell-penetrating peptides (CPPs) improve brain penetration [76], the lack of selectivity in electrostatic targeting limits its systemic drug administration use, necessitating combination with specific ligands for enhanced targeting.
2.3.1. Cationic Albumin
Electrostatic interactions between cationic and anionic microdomains on BBB endothelial membranes initiate AMT, facilitating brain drug delivery, particularly for macromolecules [77]. Cationized albumin (pI>8) accumulates in tumor cells via AMT [75], can be integrated with anti-glioma drugs into a nano-drug delivery system to induce tumor cell death and retard growth [78]. Cationized immunoglobulin, monoclonal antibodies, and histones possess brain-targeting properties through similar mechanisms. Given AMT's non-selectivity, it is often combined with other approaches to enhance BBB permeation of nanoborne drug systems [79].
2.3.2. Cell-Penetrating Peptides
CPPs encompass natural proteins (e.g., TAT, penetration peptide, Syn-B carrier) and synthetic cationic, highly hydrophilic proteins like polyarginine peptides [80]. Coupling CPPs with nanobodies enhances brain penetration [76]. TAT exhibits BBB-targeting potential, with brain targeting efficacy correlating positively with its bound ligand's positive charge [81]. TAT-modified gold NPs outperform free doxorubicin in BBB traversal, GBM targeting, and circulation time [31]. However, CPPs' non-specific electrostatic membrane interactions lack cell selectivity, elevating side effects. Researchers have devised dual/multi-ligand systems by conjugating CPPs with specific targeting ligands to optimize BBB traversal, impart cell selectivity, and enhance drug delivery efficiency [31,82].
2.4. Cell-Mediated Transport
The unique structures, mechanical properties, and surface ligands of human cells dictate their diverse physiological functions, fostering the development of cell-based targeted drug delivery systems [83,84]. Cells serve as drug delivery vectors, encapsulating or attaching drugs and utilizing their homing mechanisms for efficient targeted delivery [85]. Promising candidates include erythrocytes, leukocytes, and stem cells. Furthermore, synthetic nano-drug delivery systems utilize native cell membranes to enhance BBB penetration and active targeting, broadening the range of cells for drug delivery applications [83].
2.4.1. Erythrocytes
Erythrocytes, owing to their abundance, distinctive biconcave morphology, and prolonged circulation lifespan of 110-120 days in circulation, have garnered attention as promising nanodrug carriers [86]. Their structural characteristics, notably the biconcave shape and absence of a nucleus, enhance drug encapsulation efficiency by up to 67% [87]. However, direct drug integration into or onto erythrocyte membranes can inflict irreparable damage, hastening clearance by the reticuloendothelial system and diminishing circulatory persistence [86]. To circumvent this, CPPs are utilized for drug loading, safeguarding membrane integrity and function [88]. Despite these merits, erythrocytes as drug carriers encounter limitations such as uncontrolled drug release and absence of specific targeting receptors. To address these, red blood cell (RBC)-NPs, nanoparticles coated with RBC membranes (RBCms), combine membrane functionalities with nanoparticle physicochemical properties, enhancing drug-loading capacity, stability, biocompatibility, and prolonged retention [89]. Furthermore, RBC-NPs exhibit refined BBB penetration and tumor-targeting abilities, facilitated by surface- modified ligands [90]. The novel nano-drug Ang-RBCm@NM-(Dox/Lex), functionalized with the surface of Angiopep-2-modified RBCms, exhibits prolonged circulation time, superior BBB penetration, and enhanced tumor accumulation, effectively suppressing tumor growth and significantly extending the median survival time of orthotopic U87MG human GBM tumor-bearing nude mice [33]. Dong Luo and colleagues developed a NDDS using the tumor-penetrating peptide iRGD (CRGDK/RGPD/EC) derived from the RBC membrane as a carrier to overcome the BBB, enhance drug targeting, and increase the 30-day survival rate from 0% to 100% [34]. Mingming Song et al. devised LMP RFA NPs, a biomimetic nanodrug delivery system for targeted GBM therapy. This system encapsulates lomitapide (LMP)-loaded tetrahedral DNA nanocages within a folate-modified erythrocyte-cancer cell-macrophage hybrid membrane (FRUR) shell, demonstrating high BBB permeability, precise tumor targeting, low side effects, and extended survival in tumor-bearing mice [32].
2.4.2. Leukocytes
Leukocytes migrate to disease sites, traverse the BBB, and penetrate hypoxic tumor regions, facilitating targeted drug delivery to challenging areas [84]. These cells, including T cells, neutrophils, monocytes/macrophages, and dendritic cells, exhibit innate affinity towards inflammation and are recruited to lesion sites by inflammatory factors[91].
Chimeric antigen receptor (CAR)-T cells are genetically modified autologous T cells that can be employed for GBM-targeted drug delivery [92,93]. Despite genetic modification of T cells, limited transport across the BBB remains one of the primary challenges CAR-T cell therapy encounters. Focused ultrasound (FUS) has been utilized to open the BBB and enhanced survival rates by 129% compared to CAR-T cell therapy alone [94]. Gloria B. Kim and her team have developed a combined selective NDDS that utilizes high-affinity TQM-13 CAR T cells as drug carriers, integrating with nanoparticles and DOX to enhance drug bioavailability while mitigating systemic toxicity [35]. Overall, T cells remain a very promising delivery vehicle for GBM treatment.
Neutrophils (NEs), the predominant immune cells with rapid responsiveness to inflammatory stimuli, are recruited to lesion sites upon activation and exhibit the ability to traverse the BBB, accessing inflamed brain tumor tissues [36,89]. A recent study harnessed this property by formulating a NEs-exosomes (NEs-Exos) system for GBM therapy, enabling the effective loading and intravenous delivery of DOX, significantly suppressed tumor growth and prolonged survival [36]. Inspired by CAR-T cell therapy, researchers have engineered CAR-neutrophils to specifically deliver tumor microenvironment-responsive nanodrugs to GBM, combining chemotherapy with immunotherapy to minimize off-target effects and extend the lifespan of tumor-bearing mice [37]. Utilizing neutrophil membrane as a biomimetic drug delivery system interacts with adherent proteins to target endothelial cell membranes within the BBB for crossing [95].
Monocytes/macrophages, recruited to lesion sites by chemotactic factors [89], exhibit robust migratory capacity towards GBM, rendering them ideal carriers for cell-mediated brain drug delivery [96]. Macrophage-derived delivery systems are tumor-targeted, releasing therapeutics for efficacy [89]. Inflammatory macrophage membranes enhance targeting, and their integration with nanoparticles augments BBB traversal and brain-targeting [97]. Xiao et al. achieved prolonged circulation, improved BBB penetration, and enhanced chemo/chemodynamic therapeutic efficacy for orthotopic C6 glioma in a mouse model through coating hybrid nanogels with macrophage membranes [38] (Figure 6).
Dendritic cells, specialized antigen-presenting cells, stimulate and regulate innate and adaptive immune responses, crucial for cytotoxic T cell activation and proliferation. Dendritic cell membrane proteins facilitate BBB traversal. Xiaoyue Ma et al. devised aDCM@PLGA/RAPA, a nano-platform coated with dendritic cell membranes, to effectively traverse the BBB, promoting tumor immune response and synergistically augmenting GBM eradication in conjunction with rapamycin (RAPA) [39].
Despite their potent anti-tumor and immune regulatory capabilities, natural killer (NK) cells have been underutilized as carriers in research. NK cells inherently possess direct cytotoxic effects, acting as effective "anti-tumor agents" [84]. They can induce target cell apoptosis by releasing perforin and granzymes, which may cause explosive release of tumor drugs and hinder the penetration of tumor drugs.
2.4.3. Stem Cells
Stem cells, such as mesenchymal stem cells (MSCs), neural stem cells (NSCs) and adipose-derived stem cells (ADSCs), are ideal carriers for glioma drug delivery due to their self-renewal, low immunogenicity and easy differentiation. These stem cells can penetrate the BBB, migrate to tumors, and are used to treat hypoxia, necrosis, inflammatory tissue and glioma, and even CNS diseases and other tumors [84,89]. MSCs and ADSCs, with their immunosuppressive properties, can be engineered to deliver therapeutic agents to tumors. Viral transfection of stem cells has been shown to induce immunoactive cytokine expression, thereby enhancing mouse survival rates [98]. ADSCs deliver apoptosation-inducing ligands (TRAIL) to brain tumors via viral transfection [99], and non-viral nanoparticles are also used to transfect stem cells targeting GBM in vitro [40]. The cancer homing ability of ADSCs and the therapeutic potential of TRAIL can induce apoptosis of primary tumor and microsatellite cells and prolong the survival time of GBM xenografts [40]. Transfection of ADSCs with bone morphogenetic protein 4 induces differentiation of brain cancer stem cells, preventing tumor recurrence, and their intranasal or intravenous administration has been shown to improve survival rates in F98 rats [100]. Furthermore, ADSCs can be engineered to express suicide genes like HSV-tk, and Malik et al. utilized PLL-PEI in conjunction with TRAIL and suicide gene strategies to extend survival in C6 glioma rats [41]. Integrating NSCs with nanoparticles in brain GBM models hastens tumor targeting and reduces nanoparticle clearance [101]. MSCs cross the BBB of Wistar rats under the action of chemokines and migrate to the tumor region, making MSCs become carriers for the delivery of GBM therapeutic drugs. The researchers also observed that MSCs may promote tumor growth by releasing exosomes [102]. However, advances in cell membrane bionic carrier technology can address the shortcomings of natural cells as carriers. MSC and NSC membranes are employed as drug delivery vehicles, retaining protocell tumor-homing and BBB-crossing capabilities while enhancing their crossing and tumor targeting through specific modifications [89].
2.5. Passive Diffusion
Small, fat-soluble molecules can form transient pores in the phospholipid bilayer, allowing passive diffusion across the BBB [103], a process influenced by molecular properties that typically restrict the penetration of the majority of molecules, with only a small subset of drugs effectively treating CNS diseases [104]. These hydrophobic molecules exploit the enhanced permeability and retention (EPR) effect for intercellular delivery and passive accumulation in cancer tissues, with passive administration across the BBB occurring via paracellular and transcellular routes tailored to drugs with specific physicochemical properties [105]. Strategies to modulate BBB permeability are essential for passive endocytosis, as demonstrated by the efficacy of fluoroethyl-modified tyrosine kinase inhibitors [106] and the ARTPC nanoplatform loaded with TMZ and surface functionalized with ApoE (ApoE-ARTPC@TMZ), which utilizes LDLRs-mediated transcytosis for enhanced BBB permeation and anti-GBM activity in vivo [42]. In vivo studies have shown that targeted liposomes demonstrate enhanced circulation time, superior BBB permeation, and GBM accumulation, leading to significant anti-GBM effects and prolonged survival in an intracranial U251-TR mouse model [42] (Figure 7). Cationic liposomes selectively target tumor vasculature due to their affinity for the negatively charged BBB endothelial cells [107]. Conversely, non-targeted lipophilic systems may fail to penetrate the BBB due to its tight junctions. The use of drugs ( such as Mannitol ) or focused ultrasound (FUS) can disrupt BBB connections, thereby enhancing drug penetration [105]. Intravenous administration of microbubbles followed by ultrasound exposure induces localized BBB disruption, facilitating transient drug access to the brain parenchyma [108]. Furthermore, microbubble-mediated FUS has been shown to improve the targeting of drugs to brain tumors [109], with enhanced efficacy when combined with other delivery methods [108].
3. Strategies for NDDS to Bypass the BBB
While crossing the BBB is essential, there are alternative strategies to bypass it entirely, offering potential solutions for drug delivery to the brain.
3.1. Intranasal Administration
By administering drugs via the nasal route, it is possible to circumvent the BBB, evade first-pass metabolism, and achieve rapid onset of action [110]. ABC and SLC transporters in the nasal cavity facilitate neurotherapeutic efficacy [111]. This approach highlights intranasal delivery of diverse drugs and biopharmaceuticals [112]. Nanoparticles target brain tumors via olfactory and trigeminal nerves, crossing the olfactory epithelium [113]. In vivo studies show reduced peripheral distribution and prolonged survival in brain tumor rats [114]. Chitosan-manganese/gold nanoparticle hybrids enhance RNA delivery to brain regions [115]. However, nasal administration's limited targeted volume and specificity can cause toxicity. Combining routes, like microbubble-mediated FUS with nasal delivery, significantly increases targeted drug delivery in tumors [108] , promising more efficient drug delivery to the brain.
3.2. Convection-Enhanced Delivery
Convection-enhanced delivery (CED), a local administration method during neurosurgery [116] , which involves placing a catheter at the tumor site and using an external pump to establish a pressure gradient for direct drug delivery to the target tissue [117]. The infusion dose does not depend on molecular size and weight, improving drug spatial distribution and reducing systemic toxicity, potentially enhancing patient survival rates at 24 and 36 months [118] and median survival time [119]. Phase I trials for recurrent high-grade gliomas confirmed the feasibility and safety of intracerebral CED of carboplatin [120]. However, in a multicenter phase III study involving 276 patients with recurrent GBM, there was no significant difference in median survival when treating CED with Cintredekin besudotox (IL13-PE38QQR) and Gliadel wafers (a carmustine implant) [121]. The rich tumor vasculature, interstitial fluid pressure within tumors, limitations in catheter technology, and imaging for drug delivery hinder the reliability and reproducibility of this technique. Drug excretion/absorption and catheter placement issues affect tumor exposure [105]. With technological advancements, CED could become a glioma drug delivery technique, but further trials are needed to confirm its efficacy.
3.3. Intracavitary/Intrathecal Drug Administration
Intrathecal drug administration, bypassing the BBB to access the ventricular system, elevates brain drug concentrations but risks tissue damage [122,123]. Compared to intraventricular injection, it's less harmful, extensively studied for delivering large molecules in stroke and neurodegenerative models [124], and a promising avenue for future anti-tumor nanomedicine delivery.
4. Progress in Clinical Trials
Preclinical research is a pivotal step in the drug development process, providing a solid data foundation for clinical trials and drug marketing by comprehensively assessing the safety, efficacy, and pharmacokinetic properties of drugs in animal models and in vitro systems. This phase of research not only aids in optimizing drug candidates and reducing the risks associated with clinical trials but also ensures that new drugs meet stringent safety and efficacy standards before marketing and comply with regulatory requirements. This chapter will focus on the progress of clinical trials of nanomedicine drug delivery systems in the treatment of GBM, systematically analyzing key data on drug safety, PK, and pharmacodynamics, and discussing the advantages and disadvantages of different drug delivery systems, as well as challenges and solutions encountered in clinical trials.
Building upon the robust foundation laid by preclinical research, the field of nanomedicine has made inroads into clinical applications, as evidenced by the favorable outcomes of Phase I clinical trials. Table 2 offers a comprehensive overview of pivotal trials that have utilized NDDS based on liposomal formulations, polymeric nanoparticles, and inorganic nanoparticles. These nanocarriers have been the subject of considerable interest and investigation [125]. Notably, the clinical progression of nanoliposomal irinotecan, assessed in trials NCT00734682, NCT02022644, NCT03086616, and NCT03119064 since 2008, underscores the extensive exploration of nanomedicine in GBM treatment. A notable Phase I trial among recurrent GBM patients aimed to ascertain the maximum tolerated dose and preliminary therapeutic effects of pegylated nanoliposomal irinotecan in conjunction with TMZ. Despite the study's premature conclusion due to unmet efficacy expectations in an interim analysis [126], it yielded critical insights for the development of advanced treatment modalities. Another Phase I trial investigated the safety, PK, and maximum tolerated dose of intravenously administered nal-IRI, considering the UGT1A1*28 genotype. The findings suggest that intravenous nal-IRI is well-tolerated and that genotype does not significantly alter the drug's PK or maximum tolerated dose [127]. The next step involves exploring the efficacy and safety of nal-IRI under CED though the outcomes of this trial have yet to be published (NCT02022644). The results of these clinical trials are anticipated to provide crucial information for the clinical translation of nanomedicine therapies for GBM. Furthermore, Phase II clinical trials are ongoing to investigate the efficacy and safety of other liposomal NDDS. For instance, the combination of pegylated liposomal doxorubicin (PLD) and TMZ has demonstrated superior outcomes compared to radiotherapy alone in preliminary studies [128]. Although the sample size was limited and lacked randomized controls, this finding warrants further investigation.
To enhance the targeting efficiency of NDDS, chemical modification has emerged as a strategy aimed at precisely targeting GBM cells, thereby bolstering the safety and efficacy of treatment. For instance, Kasenda et al. reported the effective drug delivery capability of anti-EGFR doxorubicin-loaded immunoliposomes in tumor tissues with a compromised BBB [131]. EGFR-Erbitux receptor EnGeneIC Dream Vector with mitoxantrone (EEDVsMit), an innovative nanocellular therapy, has demonstrated good tolerability and no dose-limiting toxicity observed in clinical trials [133] (Evans et al., 2024). This advancement offers novel therapeutic strategies for pediatric refractory tumors, particularly those with positive EGFR expression. Additionally, glutathione, as a targeting ligand, has been conjugated to polyethylene glycol liposomes, a strategy designed to enhance drug delivery efficiency to the brain. Glutathione pegylated liposomal doxorubicin (2B3-101) completed Phase I/II clinical trials in 2014 (NCT01386580).
In the research on strategies to enhance the targeting of NDDS, utilizing biological vectors, particularly human cells, has become a key innovation for achieving precise localization of GBM cells and enhancing the safety and efficiency of treatment. Genetically engineered NSCs that express cytosine deaminase (CD) can efficiently convert 5-fluorocytosine (5-FC) into 5-fluorouracil (5-FU) in the tumor microenvironment. Portnow et al.'s study first demonstrated the safety of single intracranial injection of CD-NSCs after oral administration of 5-FC in recurrent high-grade GBM patients [134]. Subsequent studies further evaluated the safety of repeated intracranial injection of CD-NSCs and explored the appropriate dose when combined with Leucovorin, laying the foundation for Phase II clinical trials [135]. This strategy significantly enhances the targeting of treatment by bypassing the BBB, optimizing the concentration of drugs in the affected area, and reducing systemic side effects. NSCs as an ideal biological vector demonstrate the potential to deliver a variety of anti-cancer therapies to the tumor region. For example, in clinical trials using NSCs to deliver oncolytic adenovirus (NSC-CRAd-S-pk7) to newly diagnosed malignant GBM patients, good safety was observed in the dose-escalation phase, and partial tumor volume reduction and other positive responses were observed in some patients [136]. Another clinical trial uses hCE1m6-NSCs expressing carboxylesterase and irinotecan to treat recurrent high-grade GBM to inhibit tumor growth (NCT02192359), aiming to determine the recommended II phase dose, with the results yet to be released. Additionally, monocyte antigen carrier cells have shown potential in the treatment of newly diagnosed GBM with an unmethylated MGMT gene promoter, such as the MT-201-GBM vaccine, which is made from a patient's own cells loaded with CMV pp65-LAMP mRNA and has been in clinical trials since August 2023. To determine its maximum tolerated dose (NCT04741984). Meanwhile, antigen-MRNA-loaded dendritic cells (DCS) have also entered clinical trials using autologous DC cells as mRNA delivery carriers to target Survivin for GBM of the brain (NCT06524063). This trial is currently in progress and the results have not yet been published. Therefore, combining the dual strategy of genetic engineering technology and biological vectors is expected to further promote the development of the field of GBM treatment and bring hope to more patients.
Lastly, inorganic nanoparticles composed of magnetically responsive materials, such as superparamagnetic iron oxide nanoparticles (NanoTherm®), have shown potential in prolonging overall survival in patients with recurrent GBM [129]. In 2013, NanoTherm® became the first nanotechnology-based cancer treatment device to receive CE mark approval. Furthermore, a new clinical trial (NCT06271421) is evaluating the efficacy and tolerability of the NanoTherm treatment system in recurrent GBM via Cyclic hyperthermia. Meanwhile, other types of inorganic nanoparticles, like NU-0129 (NCT03020017), a nanomedicine delivery system centered on gold nanoparticles and covalently conjugated with small interfering RNA (siRNA) oligonucleotides, target the BCL2L12 gene in GBM cells. This system is being explored through clinical trials for its potential as a brain-penetrating precision therapy [130]. Additionally, a Phase I/II clinical trial investigating AGuIX nanoparticles combined with radiotherapy and temozolomide in newly diagnosed GBM patients has shown that AGuIX nanoparticles significantly enhance the clinical potential of GBM treatment when used in conjunction with radiotherapy and temozolomide [132].
Since the clinical application of Nanomedicine delivery system over a decade ago, numerous Phase I/II clinical trials have been conducted to comprehensively evaluate their safety and efficacy, yet the outcomes of most of these trials remain unpublished. Among the published results, the strategy of encapsulating drugs within liposomes and delivering them via passive diffusion mechanisms has demonstrated more frequent application compared to other administration routes. Despite summarizing eight strategies to overcome the BBB from preclinical studies, this preference may be attributed to the generally good liposolubility of first-line drugs commonly used in GBM treatment, coupled with the unique advantages of liposomes as carriers, including high stability, low leakage rates, drug sustained-release properties, and excellent biocompatibility. Building upon this foundation, scientists have further delved into introducing ligands into liposomal formulations to enhance drug targeting. Based on the outcomes of two reported clinical trials, targeted immunoliposomes have shown potential in delivering cytotoxic drugs and potentially immunomodulatory molecules to GBM tissues, albeit without achieving the anticipated significant clinical improvement. However, the exceptional long-term remissions observed in individual patients provide positive signals for this novel therapeutic strategy, hinting at its potential positive impact on treatment outcomes [131]. The utilization of cells or cell membranes as drug delivery vehicles constitutes a prominent option in clinical translation, with notable achievements particularly evident in studies employing neural stem cells (NSCs) as carriers, which have yielded published clinical outcomes. The dual-pronged strategy of combining gene engineering technologies with NSCs as biological vectors has yielded more abundant clinical translation results compared to other cell types. This may stem from the unique advantages possessed by NSCs that are not shared by other cell types. For instance, NSCs exhibit superior BBB permeability, precise migration and tracking capabilities towards tumor regions, and differentiation potential that aids in the repair and regeneration of brain tissue, alongside relatively low immunogenicity. In contrast, the use of red blood cells as drug carriers is hindered by limitations such as burst release characteristics and the absence of specific receptors for targeted delivery.
Given the challenges faced in GBM chemotherapy, the ongoing proliferation of clinical trial projects signifies the gradual progress in the clinical translation of nanomedicines. In the future, the key to nanomedicine development will lie in optimizing drug delivery systems, enhancing drug targeting and efficacy, and actively exploring combination strategies with other treatment modalities (such as immunotherapy and radiotherapy) to achieve more pronounced therapeutic effects.
5. Technical Challenges and New Strategies
The BBB, comprising endothelial cells, pericytes, astrocytes, basement membrane, tight junctions, and adherens junctions among endothelial cells [104,137] (Figure 2a). The intact BBB safeguards the brain from toxins while restricting drug entry. Only small, lipophilic molecules (< 500 daltons) can cross it [138]. The coordinated action of three cell types constructs a robust "city wall" for brain protection, utilizing membrane receptors, ion channels, and transport proteins as "delivery channels". To deliver drugs, leveraging these channels is key. However, efflux pumps on cell membranes pose a challenge. In gliomas, BBB integrity deteriorates with tumor progression, influenced by malignancy grade [83]. Elevated malignancy correlates with heightened metabolic demands, yet nutrient and oxygen scarcity induce local hypoxia, fostering abnormal angiogenesis and BBB dysfunction [139]. Despite variable BBB integrity, local disruptions do not markedly affect drug concentrations in tumor tissues [140,141]. Consequently, BBB penetration is essential for effective anti-glioma drug design.
The main barriers for therapeutic drugs to penetrate the BBB encompass four aspects: "physical barrier", "transportation barrier", "metabolic barrier", and "immune barrier".
- (1)
- Physical barrier". This encompasses the anatomical and functional features of BBB endothelial cells, forming a vital anatomical gateway for targeted brain drug delivery. The lipid bilayer membrane of these cells exhibits lipophilicity and hosts receptors, carrier proteins, and other components that regulate molecular trafficking from the bloodstream to brain tissue. High molecular weight drugs (>500 daltons) often fail to traverse this barrier [140]. Tight and adherens junctions between endothelial cells maintain BBB integrity, preventing unrestricted substance exchange.
- (2)
- "Transportation barrier". Functionally, the surface of the BBB endothelial cells is negatively charged,, impeding negatively charged compounds from entering neurons. Endothelial membranes express specific transporters regulating substrate influx/efflux, preventing unauthorized bloodstream substances from crossing [142]. Pericytes and astrocytes encapsulate BBB endothelial cells, creating resistance that allows only small molecules (e.g., water, gases, lipids) to diffuse passively. Large, charged, polar, hydrophilic molecules (amino acids, glucose, drugs) rely on luminal membrane transport proteins/receptors [137]. ATP-driven efflux pumps (P-glycoprotein) limit toxin/drug permeability, reducing CNS exposure [104,143], impacting drug efficacy, exacerbating side effects, and challenging drug action in brain tissue.
- (3)
- "Metabolic barrier". Various drug-metabolizing enzymes, such as CYP450 enzymes, have been documented within the endothelial cells of the brain [104].
"Immune barrier". Neurovascular units, comprising pericytes and astrocytes, regulate tight junctions, waste clearance, vascular function, and neuroimmune responses, forming an 'immune barrier' that constitutes the BBB.
The main challenges in BBB drug delivery involve physical, transportation, metabolic, and immune barriers. Rational design of drug delivery systems, particularly nanocarriers, can overcome metabolic and immune barriers by reducing enzymatic reactions, phagocytosis, and immune clearance, enhancing drug stability [144]. From the perspective of morphology and function of the BBB, there are two types of coping strategies to overcome the BBB: across nd bypassing BBB. This review emphasizes how nanodrug delivery systems penetrate the BBB, focusing on overcoming physical and transportation obstacles.
6. Conclusions and Perspective
The BBB plays a pivotal role in influencing the efficacy of GBM treatment agents. NDDS have ushered in a new era for brain drug delivery, potentially overcoming the BBB to improve the treatment outcome of GBM patients. Preclinical studies reveal that, compared to drug substance, NDDS, through rational design, exhibit diverse delivery modalities and superior delivery efficiency. Encapsulation and specific chemical modifications enhance drug stability and targeting, thereby significantly mitigating toxicity and side effects, advancing towards clinical translation. Nevertheless, the number of NDDS transitioning into clinical practice remains limited. In contrast, emerging therapies such as tumor immunotherapy and cellular therapy are progressively being explored, posing additional challenges to the widespread clinical application of NDDS. Consequently, continued technological innovation, aimed at markedly improving the clinical efficacy of NDDS, is imperative for achieving their broad clinical transformation. Another challenge that lies ahead is the complexity and heterogeneity of GBM, which further requires researchers to integrate knowledge from multiple disciplines and undergo a paradigm shift in the strategic development of NDDS-specific to drive NDDS from the laboratory to the clinic [145].
In clinical trials, in order to improve the clinical efficacy of the NDDS, researchers tend to adopt the strategy of combination therapy [132], aiming to achieve a synergistic effect in three primary avenues, thereby attaining a "1+1>2" augmentation: 1) Theranostic NDDS. Endowing nanomedicines with dual functions of therapy and imaging provides a robust tool for early tumor diagnosis, treatment monitoring, and efficacy assessment. A feasible approach being the co-assembly of magnetic resonance imaging contrast agents and therapeutic drugs within NDDS, enabling real-time navigation for multimodal theranostics [146]. 2) Combination of therapeutic modalities. For instance, utilizing CAR-T cells and CAR-neutrophils for targeted drug delivery, the integration of chemotherapy and immunotherapy mitigates off-target effects and prolongs the survival of tumor-bearing mice [37,92,93]. 3) Design of multi-drug synergistic NDDS. The combined delivery of sicPLA2 and metformin based on exosomes selectively targets GBM energy metabolism for antitumor effects, demonstrating potential for personalized treatment in GBM patients [147]. However, these strategies are currently in preclinical research. Despite the aforementioned challenges, recent clinical trials of GBM using nanomedicine as a therapeutic modality indicate the gradual expansion of nanochemotherapy's clinical application in GBM treatment.
Looking ahead, further advancements in NDDS will require interdisciplinary collaboration across fields such as nanotechnology, neurobiology, and clinical medicine. Future research should focus on optimizing nanoparticle design for specific therapeutic applications, improving patient-specific targeting, and refining drug release mechanisms. Additionally, well-designed clinical trials are crucial for evaluating the safety, efficacy, and long-term outcomes of these innovative delivery systems. By addressing these challenges, NDDS can move closer to realizing their full potential in GBM therapy, ultimately leading to more effective treatments with fewer side effects and better patient outcomes.
Author Contributions
Mengyun Duan: Writing - original draft, Methodology, Data curation. Ruina Cao: Data curation. Yuan Yang: Visualization, Formal analysis. Xiaoguang Chen: Software, Methodology. Lian Liu: Visualization, Formal analysis. Boxu Ren: Supervision, Funding acquisition, Conceptualization. Lingzhi Wang and Boon-Cher Goh: Review & editing, Supervision, Conceptualization. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Singapore Ministry of Health's National Medical Research Council (NMRC/STaR/MOH-00070900; CIRG/MOH-00006400), the National Research Foundation Singapore and the Singapore Ministry of Education under its Research Centres of Excellence initiatives (Boon-Cher Goh), and NUS Center for Cancer Research (N2CR), Cancer Programme under Translational Research Programmes (TRPs), Yong Loo Lin School of Medicine, NUS (NUHSRO/2020/122/MSC/07/Cancer) (L Wang/BC Goh), Nature Science Foundation of Hubei Province (No. 2023AFB839), Hubei provincial teaching reform research project (No. 2023278).
Data Availability Statement
No new data were created or analyzed in this study. Data sharing is not applicable to this article.
Acknowledgments
The figure 2 was drawn by Figdraw.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Ostrom, Q.T.; Price, M.; Neff, C.; Cioffi, G.; Waite, K.A.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2016-2020. Neuro Oncol 2023, 25, iv1–iv99. [Google Scholar] [CrossRef] [PubMed]
- Liang, T.; Gu, L.; Kang, X.; Li, J.; Song, Y.; Wang, Y.; Ma, W. Programmed cell death disrupts inflammatory tumor microenvironment (TME) and promotes glioblastoma evolution. Cell communication and signaling : CCS 2024, 22, 333. [Google Scholar] [CrossRef] [PubMed]
- Horbinski, C.; Berger, T.; Packer, R.J.; Wen, P.Y. Clinical implications of the 2021 edition of the WHO classification of central nervous system tumours. Nat Rev Neurol 2022, 18, 515–529. [Google Scholar] [CrossRef] [PubMed]
- Smerdi, D.; Moutafi, M.; Kotsantis, I.; Stavrinou, L.C.; Psyrri, A. Overcoming Resistance to Temozolomide in Glioblastoma: A Scoping Review of Preclinical and Clinical Data. Life (Basel, Switzerland) 2024, 14. [Google Scholar] [CrossRef]
- Banks, W.A. From blood-brain barrier to blood-brain interface: new opportunities for CNS drug delivery. Nature reviews. Drug discovery 2016, 15, 275–292. [Google Scholar] [CrossRef]
- Pardridge, W.M. Blood-brain barrier delivery. Drug discovery today 2007, 12, 54–61. [Google Scholar] [CrossRef]
- Pardridge, W.M. The blood-brain barrier: bottleneck in brain drug development. NeuroRx : the journal of the American Society for Experimental NeuroTherapeutics 2005, 2, 3–14. [Google Scholar] [CrossRef]
- Tiwari, S.B.; Amiji, M.M. A review of nanocarrier-based CNS delivery systems. Current drug delivery 2006, 3, 219–232. [Google Scholar] [CrossRef]
- Quader, S.; Kataoka, K.; Cabral, H. Nanomedicine for brain cancer. Adv Drug Deliv Rev 2022, 182, 114115. [Google Scholar] [CrossRef]
- Song, X.; Qian, H.; Yu, Y. Nanoparticles Mediated the Diagnosis and Therapy of Glioblastoma: Bypass or Cross the Blood-Brain Barrier. Small (Weinheim an der Bergstrasse, Germany) 2023, 19, e2302613. [Google Scholar] [CrossRef]
- Khan, I.; Baig, M.H.; Mahfooz, S.; Imran, M.A.; Khan, M.I.; Dong, J.J.; Cho, J.Y.; Hatiboglu, M.A. Nanomedicine for glioblastoma: Progress and future prospects. Semin Cancer Biol 2022, 86, 172–186. [Google Scholar] [CrossRef] [PubMed]
- Lam, F.C.; Morton, S.W.; Wyckoff, J.; Vu Han, T.L.; Hwang, M.K.; Maffa, A.; Balkanska-Sinclair, E.; Yaffe, M.B.; Floyd, S.R.; Hammond, P.T. Enhanced efficacy of combined temozolomide and bromodomain inhibitor therapy for gliomas using targeted nanoparticles. Nature communications 2018, 9, 1991. [Google Scholar] [CrossRef]
- Dong, M.; Liu, Y.; Liu, B.; Peng, J.; Tang, Y.; Lu, G.; Shi, H.; Zhu, F. Enhanced anti-glioma efficacy of biodegradable periodic mesoporous organosilica nanoparticles through target delivery of chemotherapeutics. Journal of materials science. Materials in medicine 2023, 34, 48. [Google Scholar] [CrossRef]
- Janjua, T.I.; Cao, Y.; Ahmed-Cox, A.; Raza, A.; Moniruzzaman, M.; Akhter, D.T.; Fletcher, N.L.; Kavallaris, M.; Thurecht, K.J.; Popat, A. Efficient delivery of Temozolomide using ultrasmall large-pore silica nanoparticles for glioblastoma. Journal of controlled release : official journal of the Controlled Release Society 2023, 357, 161–174. [Google Scholar] [CrossRef]
- Chen, H.; Wang, Y.; Wang, H.; Zhang, K.; Liu, Y.; Li, Q.; Li, C.; Wen, Z.; Chen, Z. Biomimetic nanocarriers loaded with temozolomide by cloaking brain-targeting peptides for targeting drug delivery system to promote anticancer effects in glioblastoma cells. Heliyon 2024, 10, e28256. [Google Scholar] [CrossRef] [PubMed]
- Zhong, X.; Wei, G.; Liu, B.; Wang, C.; Wang, J.; Lu, Y.; Cui, W.; Guo, H. Polyhedral Oligomeric Silsesquioxane-Based Nanoparticles for Efficient Chemotherapy of Glioblastoma. Small (Weinheim an der Bergstrasse, Germany) 2023, 19, e2207248. [Google Scholar] [CrossRef] [PubMed]
- Thirumurugan, S.; Dash, P.; Liu, X.; Tseng, Y.Y.; Huang, W.J.; Li, Y.; Zhao, G.; Lin, C.; Murugan, K.; Dhawan, U.; et al. Angiopep-2-decorated titanium-alloy core-shell magnetic nanoparticles for nanotheranostics and medical imaging. Nanoscale 2022, 14, 14789–14800. [Google Scholar] [CrossRef]
- He, C.; Zhang, Z.; Ding, Y.; Xue, K.; Wang, X.; Yang, R.; An, Y.; Liu, D.; Hu, C.; Tang, Q. LRP1-mediated pH-sensitive polymersomes facilitate combination therapy of glioblastoma in vitro and in vivo. Journal of nanobiotechnology 2021, 19, 29. [Google Scholar] [CrossRef]
- Banstola, A.; Duwa, R.; Emami, F.; Jeong, J.H.; Yook, S. Enhanced Caspase-Mediated Abrogation of Autophagy by Temozolomide-Loaded and Panitumumab-Conjugated Poly(lactic-co-glycolic acid) Nanoparticles in Epidermal Growth Factor Receptor Overexpressing Glioblastoma Cells. Molecular pharmaceutics 2020, 17, 4386–4400. [Google Scholar] [CrossRef]
- Alata, W.; Yogi, A.; Brunette, E.; Delaney, C.E.; van Faassen, H.; Hussack, G.; Iqbal, U.; Kemmerich, K.; Haqqani, A.S.; Moreno, M.J.; et al. Targeting insulin-like growth factor-1 receptor (IGF1R) for brain delivery of biologics. FASEB journal : official publication of the Federation of American Societies for Experimental Biology 2022, 36, e22208. [Google Scholar] [CrossRef]
- Vangala, V.; Nimmu, N.V.; Khalid, S.; Kuncha, M.; Sistla, R.; Banerjee, R.; Chaudhuri, A. Combating Glioblastoma by Codelivering the Small-Molecule Inhibitor of STAT3 and STAT3siRNA with α5β1 Integrin Receptor-Selective Liposomes. Molecular pharmaceutics 2020, 17, 1859–1874. [Google Scholar] [CrossRef]
- Lu, L.; Zhao, X.; Fu, T.; Li, K.; He, Y.; Luo, Z.; Dai, L.; Zeng, R.; Cai, K. An iRGD-conjugated prodrug micelle with blood-brain-barrier penetrability for anti-glioma therapy. Biomaterials 2020, 230, 119666. [Google Scholar] [CrossRef] [PubMed]
- Xie, R.; Wang, Y.; Tong, F.; Yang, W.; Lei, T.; Du, Y.; Wang, X.; Yang, Z.; Gong, T.; Shevtsov, M.; et al. Hsp70-Targeting and Size-Tunable Nanoparticles Combine with PD-1 Checkpoint Blockade to Treat Glioma. Small (Weinheim an der Bergstrasse, Germany) 2023, e2300570. [Google Scholar] [CrossRef]
- Zhou, J.; Meng, N.; Lu, L.; Lu, J.; Wu, S.; Ding, Y.; Wu, S.; Bao, Y.; Xu, Q.; Chen, R.; et al. A novel peptide-drug conjugate for glioma-targeted drug delivery. Journal of controlled release : official journal of the Controlled Release Society 2024, 369, 722–733. [Google Scholar] [CrossRef]
- Zhang, Y.; Ren, Y.; Xu, H.; Li, L.; Qian, F.; Wang, L.; Quan, A.; Ma, H.; Liu, H.; Yu, R. Cascade-Responsive 2-DG Nanocapsules Encapsulate aV-siCPT1C Conjugates to Inhibit Glioblastoma through Multiple Inhibition of Energy Metabolism. ACS applied materials & interfaces 2023, 15, 10356–10370. [Google Scholar] [CrossRef]
- Wang, H.; Chao, Y.; Zhao, H.; Zhou, X.; Zhang, F.; Zhang, Z.; Li, Z.; Pan, J.; Wang, J.; Chen, Q.; et al. Smart Nanomedicine to Enable Crossing Blood-Brain Barrier Delivery of Checkpoint Blockade Antibody for Immunotherapy of Glioma. ACS nano 2022, 16, 664–674. [Google Scholar] [CrossRef]
- Bhunia, S.; Vangala, V.; Bhattacharya, D.; Ravuri, H.G.; Kuncha, M.; Chakravarty, S.; Sistla, R.; Chaudhuri, A. Large Amino Acid Transporter 1 Selective Liposomes of l-DOPA Functionalized Amphiphile for Combating Glioblastoma. Molecular pharmaceutics 2017, 14, 3834–3847. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Zhao, Y.; Chen, Y.; Yang, Z.; Zhang, L.; Xiao, W.; Yang, J.; Guo, L.; Wu, Y. Dual-targeting for brain-specific liposomes drug delivery system: Synthesis and preliminary evaluation. Bioorganic & medicinal chemistry 2018, 26, 4677–4686. [Google Scholar] [CrossRef]
- Kou, L.; Hou, Y.; Yao, Q.; Guo, W.; Wang, G.; Wang, M.; Fu, Q.; He, Z.; Ganapathy, V.; Sun, J. L-Carnitine-conjugated nanoparticles to promote permeation across blood-brain barrier and to target glioma cells for drug delivery via the novel organic cation/carnitine transporter OCTN2. Artificial cells, nanomedicine, and biotechnology 2018, 46, 1605–1616. [Google Scholar] [CrossRef]
- Baklaushev, V.P.; Nukolova, N.N.; Khalansky, A.S.; Gurina, O.I.; Yusubalieva, G.M.; Grinenko, N.P.; Gubskiy, I.L.; Melnikov, P.A.; Kardashova, K.; Kabanov, A.V.; et al. Treatment of glioma by cisplatin-loaded nanogels conjugated with monoclonal antibodies against Cx43 and BSAT1. Drug delivery 2015, 22, 276–285. [Google Scholar] [CrossRef]
- Lu, L.; Wang, L.; Zhao, L.; Liao, J.; Zhao, C.; Xu, X.; Wang, F.; Zhang, X. A Novel Blood-Brain Barrier-Penetrating and Vascular-Targeting Chimeric Peptide Inhibits Glioma Angiogenesis. Int J Mol Sci 2023, 24. [Google Scholar] [CrossRef] [PubMed]
- Song, M.; Tian, J.; Wang, L.; Dong, S.; Fu, K.; Chen, S.; Liu, C. Efficient Delivery of Lomitapide using Hybrid Membrane-Coated Tetrahedral DNA Nanostructures for Glioblastoma Therapy. Adv Mater 2024, 36, e2311760. [Google Scholar] [CrossRef]
- Zou, Y.; Liu, Y.; Yang, Z.; Zhang, D.; Lu, Y.; Zheng, M.; Xue, X.; Geng, J.; Chung, R.; Shi, B. Effective and Targeted Human Orthotopic Glioblastoma Xenograft Therapy via a Multifunctional Biomimetic Nanomedicine. Adv Mater 2018, 30, e1803717. [Google Scholar] [CrossRef] [PubMed]
- Luo, D.; Chen, Z.; Peng, Y.; Liu, C. IRGD-modified erythrocyte membrane biomimetic temozolomide nanodots for the treatment of glioblastoma. Nanotechnology 2024, 35. [Google Scholar] [CrossRef]
- Kim, G.B.; Aragon-Sanabria, V.; Randolph, L.; Jiang, H.; Reynolds, J.A.; Webb, B.S.; Madhankumar, A.; Lian, X.; Connor, J.R.; Yang, J.; et al. High-affinity mutant Interleukin-13 targeted CAR T cells enhance delivery of clickable biodegradable fluorescent nanoparticles to glioblastoma. Bioactive materials 2020, 5, 624–635. [Google Scholar] [CrossRef]
- Wang, J.; Tang, W.; Yang, M.; Yin, Y.; Li, H.; Hu, F.; Tang, L.; Ma, X.; Zhang, Y.; Wang, Y. Inflammatory tumor microenvironment responsive neutrophil exosomes-based drug delivery system for targeted glioma therapy. Biomaterials 2021, 273, 120784. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.; Cai, X.; Syahirah, R.; Yao, Y.; Xu, Y.; Jin, G.; Bhute, V.J.; Torregrosa-Allen, S.; Elzey, B.D.; Won, Y.Y.; et al. CAR-neutrophil mediated delivery of tumor-microenvironment responsive nanodrugs for glioblastoma chemo-immunotherapy. Nature communications 2023, 14, 2266. [Google Scholar] [CrossRef]
- Xiao, T.; He, M.; Xu, F.; Fan, Y.; Jia, B.; Shen, M.; Wang, H.; Shi, X. Macrophage Membrane-Camouflaged Responsive Polymer Nanogels Enable Magnetic Resonance Imaging-Guided Chemotherapy/Chemodynamic Therapy of Orthotopic Glioma. ACS nano 2021, 15, 20377–20390. [Google Scholar] [CrossRef]
- Ma, X.; Kuang, L.; Yin, Y.; Tang, L.; Zhang, Y.; Fan, Q.; Wang, B.; Dong, Z.; Wang, W.; Yin, T.; et al. Tumor-Antigen Activated Dendritic Cell Membrane-Coated Biomimetic Nanoparticles with Orchestrating Immune Responses Promote Therapeutic Efficacy against Glioma. ACS nano 2023, 17, 2341–2355. [Google Scholar] [CrossRef]
- Jiang, X.; Fitch, S.; Wang, C.; Wilson, C.; Li, J.; Grant, G.A.; Yang, F. Nanoparticle engineered TRAIL-overexpressing adipose-derived stem cells target and eradicate glioblastoma via intracranial delivery. Proceedings of the National Academy of Sciences of the United States of America 2016, 113, 13857–13862. [Google Scholar] [CrossRef]
- Malik, Y.S.; Sheikh, M.A.; Xing, Z.; Guo, Z.; Zhu, X.; Tian, H.; Chen, X. Polylysine-modified polyethylenimine polymer can generate genetically engineered mesenchymal stem cells for combinational suicidal gene therapy in glioblastoma. Acta biomaterialia 2018, 80, 144–153. [Google Scholar] [CrossRef] [PubMed]
- Ismail, M.; Yang, W.; Li, Y.; Chai, T.; Zhang, D.; Du, Q.; Muhammad, P.; Hanif, S.; Zheng, M.; Shi, B. Targeted liposomes for combined delivery of artesunate and temozolomide to resistant glioblastoma. Biomaterials 2022, 287, 121608. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.; Mu, N.; Jia, B.; Guo, Q.; Pan, L.; Zhu, M.; Zhang, W.; Zhang, K.; Li, W.; Li, M.; et al. Targeting radiation-tolerant persister cells as a strategy for inhibiting radioresistance and recurrence in glioblastoma. Neuro Oncol 2022, 24, 1056–1070. [Google Scholar] [CrossRef]
- Ji, B.; Maeda, J.; Higuchi, M.; Inoue, K.; Akita, H.; Harashima, H.; Suhara, T. Pharmacokinetics and brain uptake of lactoferrin in rats. Life sciences 2006, 78, 851–855. [Google Scholar] [CrossRef]
- Kuplennik, N.; Lang, K.; Steinfeld, R.; Sosnik, A. Folate Receptor α-Modified Nanoparticles for Targeting of the Central Nervous System. ACS applied materials & interfaces 2019, 11, 39633–39647. [Google Scholar] [CrossRef]
- Neves, A.R.; Queiroz, J.F.; Weksler, B.; Romero, I.A.; Couraud, P.O.; Reis, S. Solid lipid nanoparticles as a vehicle for brain-targeted drug delivery: two new strategies of functionalization with apolipoprotein E. Nanotechnology 2015, 26, 495103. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Li, L.; Tian, H.; Wang, Z.; Liu, G.; Duan, X.; Guo, M.; Liu, J.; Zhang, W.; Nice, E.C.; et al. Drug Repurposing-Based Brain-Targeting Self-Assembly Nanoplatform Using Enhanced Ferroptosis against Glioblastoma. Small (Weinheim an der Bergstrasse, Germany) 2023, 19, e2303073. [Google Scholar] [CrossRef]
- Tashima, T. Brain Cancer Chemotherapy through a Delivery System across the Blood-Brain Barrier into the Brain Based on Receptor-Mediated Transcytosis Using Monoclonal Antibody Conjugates. Biomedicines 2022, 10. [Google Scholar] [CrossRef]
- Yogi, A.; Hussack, G.; van Faassen, H.; Haqqani, A.S.; Delaney, C.E.; Brunette, E.; Sandhu, J.K.; Hewitt, M.; Sulea, T.; Kemmerich, K.; et al. Brain Delivery of IGF1R5, a Single-Domain Antibody Targeting Insulin-like Growth Factor-1 Receptor. Pharmaceutics 2022, 14. [Google Scholar] [CrossRef]
- Miura, Y.; Takenaka, T.; Toh, K.; Wu, S.; Nishihara, H.; Kano, M.R.; Ino, Y.; Nomoto, T.; Matsumoto, Y.; Koyama, H.; et al. Cyclic RGD-linked polymeric micelles for targeted delivery of platinum anticancer drugs to glioblastoma through the blood-brain tumor barrier. ACS nano 2013, 7, 8583–8592. [Google Scholar] [CrossRef]
- Quader, S.; Liu, X.; Chen, Y.; Mi, P.; Chida, T.; Ishii, T.; Miura, Y.; Nishiyama, N.; Cabral, H.; Kataoka, K. cRGD peptide-installed epirubicin-loaded polymeric micelles for effective targeted therapy against brain tumors. Journal of controlled release : official journal of the Controlled Release Society 2017, 258, 56–66. [Google Scholar] [CrossRef] [PubMed]
- An, S.; Jiang, X.; Shi, J.; He, X.; Li, J.; Guo, Y.; Zhang, Y.; Ma, H.; Lu, Y.; Jiang, C. Single-component self-assembled RNAi nanoparticles functionalized with tumor-targeting iNGR delivering abundant siRNA for efficient glioma therapy. Biomaterials 2015, 53, 330–340. [Google Scholar] [CrossRef]
- Huang, N.; Cheng, S.; Zhang, X.; Tian, Q.; Pi, J.; Tang, J.; Huang, Q.; Wang, F.; Chen, J.; Xie, Z.; et al. Efficacy of NGR peptide-modified PEGylated quantum dots for crossing the blood-brain barrier and targeted fluorescence imaging of glioma and tumor vasculature. Nanomedicine : nanotechnology, biology, and medicine 2017, 13, 83–93. [Google Scholar] [CrossRef]
- Wang, J.; Lei, Y.; Xie, C.; Lu, W.; Wagner, E.; Xie, Z.; Gao, J.; Zhang, X.; Yan, Z.; Liu, M. Retro-inverso CendR peptide-mediated polyethyleneimine for intracranial glioblastoma-targeting gene therapy. Bioconjugate chemistry 2014, 25, 414–423. [Google Scholar] [CrossRef] [PubMed]
- Enerson, B.E.; Drewes, L.R. The rat blood-brain barrier transcriptome. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism 2006, 26, 959–973. [Google Scholar] [CrossRef] [PubMed]
- Gyimesi, G.; Hediger, M.A. Transporter-Mediated Drug Delivery. Molecules 2023, 28. [Google Scholar] [CrossRef] [PubMed]
- Min, H.S.; Kim, H.J.; Naito, M.; Ogura, S.; Toh, K.; Hayashi, K.; Kim, B.S.; Fukushima, S.; Anraku, Y.; Miyata, K.; et al. Systemic Brain Delivery of Antisense Oligonucleotides across the Blood-Brain Barrier with a Glucose-Coated Polymeric Nanocarrier. Angew Chem Int Ed Engl 2020, 59, 8173–8180. [Google Scholar] [CrossRef]
- Anraku, Y.; Kuwahara, H.; Fukusato, Y.; Mizoguchi, A.; Ishii, T.; Nitta, K.; Matsumoto, Y.; Toh, K.; Miyata, K.; Uchida, S.; et al. Glycaemic control boosts glucosylated nanocarrier crossing the BBB into the brain. Nature communications 2017, 8, 1001. [Google Scholar] [CrossRef]
- Wang, H.; Wang, X.; Xie, C.; Zhang, M.; Ruan, H.; Wang, S.; Jiang, K.; Wang, F.; Zhan, C.; Lu, W.; et al. Nanodisk-based glioma-targeted drug delivery enabled by a stable glycopeptide. Journal of controlled release : official journal of the Controlled Release Society 2018, 284, 26–38. [Google Scholar] [CrossRef]
- Li, J.; Zhou, L.; Ye, D.; Huang, S.; Shao, K.; Huang, R.; Han, L.; Liu, Y.; Liu, S.; Ye, L.; et al. Choline-derivate-modified nanoparticles for brain-targeting gene delivery. Adv Mater 2011, 23, 4516–4520. [Google Scholar] [CrossRef]
- Li, J.; Guo, Y.; Kuang, Y.; An, S.; Ma, H.; Jiang, C. Choline transporter-targeting and co-delivery system for glioma therapy. Biomaterials 2013, 34, 9142–9148. [Google Scholar] [CrossRef] [PubMed]
- Jin, W.; Wu, Y.; Chen, N.; Wang, Q.; Wang, Y.; Li, Y.; Li, S.; Han, X.; Yang, E.; Tong, F.; et al. Early administration of MPC-n(IVIg) selectively accumulates in ischemic areas to protect inflammation-induced brain damage from ischemic stroke. Theranostics 2021, 11, 8197–8217. [Google Scholar] [CrossRef]
- Puris, E.; Gynther, M.; Auriola, S.; Huttunen, K.M. L-Type amino acid transporter 1 as a target for drug delivery. Pharm Res 2020, 37, 88. [Google Scholar] [CrossRef]
- Montaser, A.B.; Järvinen, J.; Löffler, S.; Huttunen, J.; Auriola, S.; Lehtonen, M.; Jalkanen, A.; Huttunen, K.M. L-Type Amino Acid Transporter 1 Enables the Efficient Brain Delivery of Small-Sized Prodrug across the Blood-Brain Barrier and into Human and Mouse Brain Parenchymal Cells. ACS chemical neuroscience 2020, 11, 4301–4315. [Google Scholar] [CrossRef] [PubMed]
- Wei, Z.Y.; Feng, M.M.; Yang, B.; Yan, Z.Y.; Wang, B.Q.; Bu, X.Y. Methylmercury-l-Cysteine targeting L-type amino acid transporter conjugate cytotoxicity on C6 glioma cells. Journal of biological regulators and homeostatic agents 2018, 32, 147–151. [Google Scholar] [PubMed]
- Salmaso, S.; Pappalardo, J.S.; Sawant, R.R.; Musacchio, T.; Rockwell, K.; Caliceti, P.; Torchilin, V.P. Targeting glioma cells in vitro with ascorbate-conjugated pharmaceutical nanocarriers. Bioconjugate chemistry 2009, 20, 2348–2355. [Google Scholar] [CrossRef]
- Quéléver, G.; Kachidian, P.; Melon, C.; Garino, C.; Laras, Y.; Pietrancosta, N.; Sheha, M.; Louis Kraus, J. Enhanced delivery of gamma-secretase inhibitor DAPT into the brain via an ascorbic acid mediated strategy. Organic & biomolecular chemistry 2005, 3, 2450–2457. [Google Scholar] [CrossRef]
- Kou, L.; Sun, R.; Ganapathy, V.; Yao, Q.; Chen, R. Recent advances in drug delivery via the organic cation/carnitine transporter 2 (OCTN2/SLC22A5). Expert opinion on therapeutic targets 2018, 22, 715–726. [Google Scholar] [CrossRef]
- Fink, M.A.; Paland, H.; Herzog, S.; Grube, M.; Vogelgesang, S.; Weitmann, K.; Bialke, A.; Hoffmann, W.; Rauch, B.H.; Schroeder, H.W.S.; et al. L-Carnitine-Mediated Tumor Cell Protection and Poor Patient Survival Associated with OCTN2 Overexpression in Glioblastoma Multiforme. Clinical cancer research : an official journal of the American Association for Cancer Research 2019, 25, 2874–2886. [Google Scholar] [CrossRef]
- Bakos, É.; Német, O.; Patik, I.; Kucsma, N.; Várady, G.; Szakács, G.; Özvegy-Laczka, C. A novel fluorescence-based functional assay for human OATP1A2 and OATP1C1 identifies interaction between third-generation P-gp inhibitors and OATP1A2. The FEBS journal 2020, 287, 2468–2485. [Google Scholar] [CrossRef]
- Higuchi, K.; Sivaprakasam, S.; Sennoune, S.R.; Ogura, J.; Bhutia, Y.D.; Rueda, R.; Pereira, S.L.; Ganapathy, V. A Proton-Coupled Transport System for β-Hydroxy-β-Methylbutyrate (HMB) in Blood-Brain Barrier Endothelial Cell Line hCMEC/D3. Nutrients 2021, 13. [Google Scholar] [CrossRef] [PubMed]
- Sadeghzadeh, M.; Wenzel, B.; Gündel, D.; Deuther-Conrad, W.; Toussaint, M.; Moldovan, R.P.; Fischer, S.; Ludwig, F.A.; Teodoro, R.; Jonnalagadda, S.; et al. Development of Novel Analogs of the Monocarboxylate Transporter Ligand FACH and Biological Validation of One Potential Radiotracer for Positron Emission Tomography (PET) Imaging. Molecules 2020, 25. [Google Scholar] [CrossRef]
- Huber, I.; Pandur, E.; Sipos, K.; Barna, L.; Harazin, A.; Deli, M.A.; Tyukodi, L.; Gulyás-Fekete, G.; Kulcsár, G.; Rozmer, Z. Novel cyclic C(5)-curcuminoids penetrating the blood-brain barrier: Design, synthesis and antiproliferative activity against astrocytoma and neuroblastoma cells. European journal of pharmaceutical sciences : official journal of the European Federation for Pharmaceutical Sciences 2022, 173, 106184. [Google Scholar] [CrossRef] [PubMed]
- Klibanov, A.L.; Maruyama, K.; Torchilin, V.P.; Huang, L. Amphipathic polyethyleneglycols effectively prolong the circulation time of liposomes. FEBS letters 1990, 268, 235–237. [Google Scholar] [CrossRef] [PubMed]
- Sim, T.M.; Tarini, D.; Dheen, S.T.; Bay, B.H.; Srinivasan, D.K. Nanoparticle-Based Technology Approaches to the Management of Neurological Disorders. Int J Mol Sci 2020, 21. [Google Scholar] [CrossRef]
- Herce, H.D.; Schumacher, D.; Schneider, A.F.L.; Ludwig, A.K.; Mann, F.A.; Fillies, M.; Kasper, M.A.; Reinke, S.; Krause, E.; Leonhardt, H.; et al. Cell-permeable nanobodies for targeted immunolabelling and antigen manipulation in living cells. Nature chemistry 2017, 9, 762–771. [Google Scholar] [CrossRef]
- Hervé, F.; Ghinea, N.; Scherrmann, J.M. CNS delivery via adsorptive transcytosis. The AAPS journal 2008, 10, 455–472. [Google Scholar] [CrossRef]
- Lu, W.; Sun, Q.; Wan, J.; She, Z.; Jiang, X.G. Cationic albumin-conjugated pegylated nanoparticles allow gene delivery into brain tumors via intravenous administration. Cancer research 2006, 66, 11878–11887. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, W.; Sun, Y.; Dong, X. Transthyretin-Penetratin: A Potent Fusion Protein Inhibitor against Alzheimer's Amyloid-β Fibrillogenesis with High Blood Brain Barrier Crossing Capability. Bioconjugate chemistry 2024, 35, 419–431. [Google Scholar] [CrossRef]
- Zhang, Y.; Guo, P.; Ma, Z.; Lu, P.; Kebebe, D.; Liu, Z. Combination of cell-penetrating peptides with nanomaterials for the potential therapeutics of central nervous system disorders: a review. Journal of nanobiotechnology 2021, 19, 255. [Google Scholar] [CrossRef]
- Qin, Y.; Zhang, Q.; Chen, H.; Yuan, W.; Kuai, R.; Xie, F.; Zhang, L.; Wang, X.; Zhang, Z.; Liu, J.; et al. Comparison of four different peptides to enhance accumulation of liposomes into the brain. J Drug Target 2012, 20, 235–245. [Google Scholar] [CrossRef]
- Liu, L.; Guo, K.; Lu, J.; Venkatraman, S.S.; Luo, D.; Ng, K.C.; Ling, E.A.; Moochhala, S.; Yang, Y.Y. Biologically active core/shell nanoparticles self-assembled from cholesterol-terminated PEG-TAT for drug delivery across the blood-brain barrier. Biomaterials 2008, 29, 1509–1517. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Z.; Yu, Z.; Xu, T.; Wang, L.; Meng, N.; Jin, H.; Xu, B. Novel Nano-Drug Delivery System for Brain Tumor Treatment. Cells 2022, 11. [Google Scholar] [CrossRef]
- Ayer, M.; Klok, H.A. Cell-mediated delivery of synthetic nano- and microparticles. Journal of controlled release : official journal of the Controlled Release Society 2017, 259, 92–104. [Google Scholar] [CrossRef] [PubMed]
- Choi, A.; Javius-Jones, K.; Hong, S.; Park, H. Cell-Based Drug Delivery Systems with Innate Homing Capability as a Novel Nanocarrier Platform. Int J Nanomedicine 2023, 18, 509–525. [Google Scholar] [CrossRef] [PubMed]
- Stephan, M.T.; Irvine, D.J. Enhancing Cell therapies from the Outside In: Cell Surface Engineering Using Synthetic Nanomaterials. Nano Today 2011, 6, 309–325. [Google Scholar] [CrossRef]
- Yu, H.; Yang, Z.; Li, F.; Xu, L.; Sun, Y. Cell-mediated targeting drugs delivery systems. Drug delivery 2020, 27, 1425–1437. [Google Scholar] [CrossRef]
- He, H.; Ye, J.; Wang, Y.; Liu, Q.; Chung, H.S.; Kwon, Y.M.; Shin, M.C.; Lee, K.; Yang, V.C. Cell-penetrating peptides meditated encapsulation of protein therapeutics into intact red blood cells and its application. Journal of controlled release : official journal of the Controlled Release Society 2014, 176, 123–132. [Google Scholar] [CrossRef]
- Wu, H.; Zhang, T.; Li, N.; Gao, J. Cell membrane-based biomimetic vehicles for effective central nervous system target delivery: Insights and challenges. Journal of controlled release : official journal of the Controlled Release Society 2023, 360, 169–184. [Google Scholar] [CrossRef]
- Chai, Z.; Ran, D.; Lu, L.; Zhan, C.; Ruan, H.; Hu, X.; Xie, C.; Jiang, K.; Li, J.; Zhou, J.; et al. Ligand-Modified Cell Membrane Enables the Targeted Delivery of Drug Nanocrystals to Glioma. ACS nano 2019, 13, 5591–5601. [Google Scholar] [CrossRef]
- Karp, J.M.; Leng Teo, G.S. Mesenchymal stem cell homing: the devil is in the details. Cell stem cell 2009, 4, 206–216. [Google Scholar] [CrossRef] [PubMed]
- Elmadany, N.; Alhalabi, O.T.; Platten, M.; Bunse, L. Site-Specific Considerations on Engineered T Cells for Malignant Gliomas. Biomedicines 2022, 10. [Google Scholar] [CrossRef]
- Wang, D.; Starr, R.; Chang, W.C.; Aguilar, B.; Alizadeh, D.; Wright, S.L.; Yang, X.; Brito, A.; Sarkissian, A.; Ostberg, J.R.; et al. Chlorotoxin-directed CAR T cells for specific and effective targeting of glioblastoma. Science translational medicine 2020, 12. [Google Scholar] [CrossRef] [PubMed]
- Sabbagh, A.; Beccaria, K.; Ling, X.; Marisetty, A.; Ott, M.; Caruso, H.; Barton, E.; Kong, L.Y.; Fang, D.; Latha, K.; et al. Opening of the Blood-Brain Barrier Using Low-Intensity Pulsed Ultrasound Enhances Responses to Immunotherapy in Preclinical Glioma Models. Clinical cancer research : an official journal of the American Association for Cancer Research 2021, 27, 4325–4337. [Google Scholar] [CrossRef]
- Chen, H.; Ji, J.; Zhang, L.; Chen, T.; Zhang, Y.; Zhang, F.; Wang, J.; Ke, Y. Inflammatory responsive neutrophil-like membrane-based drug delivery system for post-surgical glioblastoma therapy. Journal of controlled release : official journal of the Controlled Release Society 2023, 362, 479–488. [Google Scholar] [CrossRef] [PubMed]
- Tong, H.I.; Kang, W.; Shi, Y.; Zhou, G.; Lu, Y. Physiological function and inflamed-brain migration of mouse monocyte-derived macrophages following cellular uptake of superparamagnetic iron oxide nanoparticles-Implication of macrophage-based drug delivery into the central nervous system. Int J Pharm 2016, 505, 271–282. [Google Scholar] [CrossRef] [PubMed]
- Long, Y.; Xiang, Y.; Liu, S.; Zhang, Y.; Wan, J.; Ci, Z.; Cui, M.; Shen, L.; Li, N.; Guan, Y. Macrophage membrane modified baicalin liposomes improve brain targeting for alleviating cerebral ischemia reperfusion injury. Nanomedicine : nanotechnology, biology, and medicine 2022, 43, 102547. [Google Scholar] [CrossRef]
- Yuan, X.; Hu, J.; Belladonna, M.L.; Black, K.L.; Yu, J.S. Interleukin-23-expressing bone marrow-derived neural stem-like cells exhibit antitumor activity against intracranial glioma. Cancer research 2006, 66, 2630–2638. [Google Scholar] [CrossRef]
- Li, M.; Sun, S.; Dangelmajer, S.; Zhang, Q.; Wang, J.; Hu, F.; Dong, F.; Kahlert, U.D.; Zhu, M.; Lei, T. Exploiting tumor-intrinsic signals to induce mesenchymal stem cell-mediated suicide gene therapy to fight malignant glioma. Stem cell research & therapy 2019, 10, 88. [Google Scholar] [CrossRef]
- Mangraviti, A.; Tzeng, S.Y.; Gullotti, D.; Kozielski, K.L.; Kim, J.E.; Seng, M.; Abbadi, S.; Schiapparelli, P.; Sarabia-Estrada, R.; Vescovi, A.; et al. Non-virally engineered human adipose mesenchymal stem cells produce BMP4, target brain tumors, and extend survival. Biomaterials 2016, 100, 53–66. [Google Scholar] [CrossRef]
- Mooney, R.; Weng, Y.; Tirughana-Sambandan, R.; Valenzuela, V.; Aramburo, S.; Garcia, E.; Li, Z.; Gutova, M.; Annala, A.J.; Berlin, J.M.; et al. Neural stem cells improve intracranial nanoparticle retention and tumor-selective distribution. Future oncology (London, England) 2014, 10, 401–415. [Google Scholar] [CrossRef] [PubMed]
- Pavon, L.F.; Sibov, T.T.; de Souza, A.V.; da Cruz, E.F.; Malheiros, S.M.F.; Cabral, F.R.; de Souza, J.G.; Boufleur, P.; de Oliveira, D.M.; de Toledo, S.R.C.; et al. Tropism of mesenchymal stem cell toward CD133(+) stem cell of glioblastoma in vitro and promote tumor proliferation in vivo. Stem cell research & therapy 2018, 9, 310. [Google Scholar] [CrossRef]
- Liu, X.; Tu, M.; Kelly, R.S.; Chen, C.; Smith, B.J. Development of a computational approach to predict blood-brain barrier permeability. Drug metabolism and disposition: the biological fate of chemicals 2004, 32, 132–139. [Google Scholar] [CrossRef] [PubMed]
- Kadry, H.; Noorani, B.; Cucullo, L. A blood-brain barrier overview on structure, function, impairment, and biomarkers of integrity. Fluids Barriers CNS 2020, 17, 69. [Google Scholar] [CrossRef]
- Haumann, R.; Videira, J.C.; Kaspers, G.J.L.; van Vuurden, D.G.; Hulleman, E. Overview of Current Drug Delivery Methods Across the Blood-Brain Barrier for the Treatment of Primary Brain Tumors. CNS drugs 2020, 34, 1121–1131. [Google Scholar] [CrossRef] [PubMed]
- Radaram, B.; Pisaneschi, F.; Rao, Y.; Yang, P.; Piwnica-Worms, D.; Alauddin, M.M. Novel derivatives of anaplastic lymphoma kinase inhibitors: Synthesis, radiolabeling, and preliminary biological studies of fluoroethyl analogues of crizotinib, alectinib, and ceritinib. European journal of medicinal chemistry 2019, 182, 111571. [Google Scholar] [CrossRef]
- Fulton, M.D.; Najahi-Missaoui, W. Liposomes in Cancer Therapy: How Did We Start and Where Are We Now. Int J Mol Sci 2023, 24. [Google Scholar] [CrossRef]
- Ye, D.; Zhang, X.; Yue, Y.; Raliya, R.; Biswas, P.; Taylor, S.; Tai, Y.C.; Rubin, J.B.; Liu, Y.; Chen, H. Focused ultrasound combined with microbubble-mediated intranasal delivery of gold nanoclusters to the brain. Journal of controlled release : official journal of the Controlled Release Society 2018, 286, 145–153. [Google Scholar] [CrossRef]
- Burgess, A.; Hynynen, K. Noninvasive and targeted drug delivery to the brain using focused ultrasound. ACS chemical neuroscience 2013, 4, 519–526. [Google Scholar] [CrossRef]
- Costa, C.P.; Barreiro, S.; Moreira, J.N.; Silva, R.; Almeida, H.; Sousa Lobo, J.M.; Silva, A.C. In Vitro Studies on Nasal Formulations of Nanostructured Lipid Carriers (NLC) and Solid Lipid Nanoparticles (SLN). Pharmaceuticals (Basel, Switzerland) 2021, 14. [Google Scholar] [CrossRef]
- Van Woensel, M.; Wauthoz, N.; Rosière, R.; Mathieu, V.; Kiss, R.; Lefranc, F.; Steelant, B.; Dilissen, E.; Van Gool, S.W.; Mathivet, T.; et al. Development of siRNA-loaded chitosan nanoparticles targeting Galectin-1 for the treatment of glioblastoma multiforme via intranasal administration. Journal of controlled release : official journal of the Controlled Release Society 2016, 227, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.R.; Liu, M.; Khan, M.W.; Zhai, G. Progress in brain targeting drug delivery system by nasal route. Journal of controlled release : official journal of the Controlled Release Society 2017, 268, 364–389. [Google Scholar] [CrossRef] [PubMed]
- Van Woensel, M.; Mathivet, T.; Wauthoz, N.; Rosière, R.; Garg, A.D.; Agostinis, P.; Mathieu, V.; Kiss, R.; Lefranc, F.; Boon, L.; et al. Sensitization of glioblastoma tumor micro-environment to chemo- and immunotherapy by Galectin-1 intranasal knock-down strategy. Scientific reports 2017, 7, 1217. [Google Scholar] [CrossRef]
- Chu, L.; Wang, A.; Ni, L.; Yan, X.; Song, Y.; Zhao, M.; Sun, K.; Mu, H.; Liu, S.; Wu, Z.; et al. Nose-to-brain delivery of temozolomide-loaded PLGA nanoparticles functionalized with anti-EPHA3 for glioblastoma targeting. Drug delivery 2018, 25, 1634–1641. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Ramos, J.; Song, S.; Kong, X.; Foroutan, P.; Martinez, G.; Dominguez-Viqueria, W.; Mohapatra, S.; Mohapatra, S.; Haraszti, R.A.; Khvorova, A.; et al. Chitosan-Mangafodipir nanoparticles designed for intranasal delivery of siRNA and DNA to brain. Journal of drug delivery science and technology 2018, 43, 453–460. [Google Scholar] [CrossRef]
- Jahangiri, A.; Chin, A.T.; Flanigan, P.M.; Chen, R.; Bankiewicz, K.; Aghi, M.K. Convection-enhanced delivery in glioblastoma: a review of preclinical and clinical studies. Journal of neurosurgery 2017, 126, 191–200. [Google Scholar] [CrossRef]
- Mehta, A.M.; Sonabend, A.M.; Bruce, J.N. Convection-Enhanced Delivery. Neurotherapeutics : the journal of the American Society for Experimental NeuroTherapeutics 2017, 14, 358–371. [Google Scholar] [CrossRef]
- Desjardins, A.; Gromeier, M.; Herndon, J.E., 2nd; Beaubier, N.; Bolognesi, D.P.; Friedman, A.H.; Friedman, H.S.; McSherry, F.; Muscat, A.M.; Nair, S.; et al. Recurrent Glioblastoma Treated with Recombinant Poliovirus. The New England journal of medicine 2018, 379, 150–161. [Google Scholar] [CrossRef] [PubMed]
- Séhédic, D.; Chourpa, I.; Tétaud, C.; Griveau, A.; Loussouarn, C.; Avril, S.; Legendre, C.; Lepareur, N.; Wion, D.; Hindré, F.; et al. Locoregional Confinement and Major Clinical Benefit of (188)Re-Loaded CXCR4-Targeted Nanocarriers in an Orthotopic Human to Mouse Model of Glioblastoma. Theranostics 2017, 7, 4517–4536. [Google Scholar] [CrossRef]
- Wang, J.L.; Barth, R.F.; Cavaliere, R.; Puduvalli, V.K.; Giglio, P.; Lonser, R.R.; Elder, J.B. Phase I trial of intracerebral convection-enhanced delivery of carboplatin for treatment of recurrent high-grade gliomas. PLoS One 2020, 15, e0244383. [Google Scholar] [CrossRef]
- Kunwar, S.; Chang, S.; Westphal, M.; Vogelbaum, M.; Sampson, J.; Barnett, G.; Shaffrey, M.; Ram, Z.; Piepmeier, J.; Prados, M.; et al. Phase III randomized trial of CED of IL13-PE38QQR vs Gliadel wafers for recurrent glioblastoma. Neuro Oncol 2010, 12, 871–881. [Google Scholar] [CrossRef] [PubMed]
- Fowler, M.J.; Cotter, J.D.; Knight, B.E.; Sevick-Muraca, E.M.; Sandberg, D.I.; Sirianni, R.W. Intrathecal drug delivery in the era of nanomedicine. Adv Drug Deliv Rev 2020, 165-166, 77–95. [Google Scholar] [CrossRef] [PubMed]
- Zheng, F.; Pang, Y.; Li, L.; Pang, Y.; Zhang, J.; Wang, X.; Raes, G. Applications of nanobodies in brain diseases. Front Immunol 2022, 13, 978513. [Google Scholar] [CrossRef]
- Calias, P.; Banks, W.A.; Begley, D.; Scarpa, M.; Dickson, P. Intrathecal delivery of protein therapeutics to the brain: a critical reassessment. Pharmacology & therapeutics 2014, 144, 114–122. [Google Scholar] [CrossRef]
- Barenholz, Y. Doxil®--the first FDA-approved nano-drug: lessons learned. Journal of controlled release : official journal of the Controlled Release Society 2012, 160, 117–134. [Google Scholar] [CrossRef]
- Elinzano, H.; Toms, S.; Robison, J.; Mohler, A.; Carcieri, A.; Cielo, D.; Donnelly, J.; Disano, D.; Vatketich, J.; Baekey, J.; et al. Nanoliposomal Irinotecan and Metronomic Temozolomide for Patients With Recurrent Glioblastoma: BrUOG329, A Phase I Brown University Oncology Research Group Trial. American journal of clinical oncology 2021, 44, 49–52. [Google Scholar] [CrossRef]
- Clarke, J.L.; Molinaro, A.M.; Cabrera, J.R.; DeSilva, A.A.; Rabbitt, J.E.; Prey, J.; Drummond, D.C.; Kim, J.; Noble, C.; Fitzgerald, J.B.; et al. A phase 1 trial of intravenous liposomal irinotecan in patients with recurrent high-grade glioma. Cancer chemotherapy and pharmacology 2017, 79, 603–610. [Google Scholar] [CrossRef]
- Beier, C.P.; Schmid, C.; Gorlia, T.; Kleinletzenberger, C.; Beier, D.; Grauer, O.; Steinbrecher, A.; Hirschmann, B.; Brawanski, A.; Dietmaier, C.; et al. RNOP-09: pegylated liposomal doxorubicine and prolonged temozolomide in addition to radiotherapy in newly diagnosed glioblastoma--a phase II study. BMC cancer 2009, 9, 308. [Google Scholar] [CrossRef]
- Maier-Hauff, K.; Ulrich, F.; Nestler, D.; Niehoff, H.; Wust, P.; Thiesen, B.; Orawa, H.; Budach, V.; Jordan, A. Efficacy and safety of intratumoral thermotherapy using magnetic iron-oxide nanoparticles combined with external beam radiotherapy on patients with recurrent glioblastoma multiforme. Journal of neuro-oncology 2011, 103, 317–324. [Google Scholar] [CrossRef]
- Kumthekar, P.; Ko, C.H.; Paunesku, T.; Dixit, K.; Sonabend, A.M.; Bloch, O.; Tate, M.; Schwartz, M.; Zuckerman, L.; Lezon, R.; et al. A first-in-human phase 0 clinical study of RNA interference-based spherical nucleic acids in patients with recurrent glioblastoma. Science translational medicine 2021, 13. [Google Scholar] [CrossRef]
- Kasenda, B.; König, D.; Manni, M.; Ritschard, R.; Duthaler, U.; Bartoszek, E.; Bärenwaldt, A.; Deuster, S.; Hutter, G.; Cordier, D.; et al. Targeting immunoliposomes to EGFR-positive glioblastoma. ESMO open 2022, 7, 100365. [Google Scholar] [CrossRef]
- Thivat, E.; Casile, M.; Moreau, J.; Molnar, I.; Dufort, S.; Seddik, K.; Le Duc, G.; De Beaumont, O.; Loeffler, M.; Durando, X.; et al. Phase I/II study testing the combination of AGuIX nanoparticles with radiochemotherapy and concomitant temozolomide in patients with newly diagnosed glioblastoma (NANO-GBM trial protocol). BMC cancer 2023, 23, 344. [Google Scholar] [CrossRef] [PubMed]
- Evans, L.; Walker, R.; MacDiarmid, J.; Brahmbhatt, H.; Anazodo, A.; McCowage, G.; Gifford, A.J.; Kavallaris, M.; Trahair, T.; Ziegler, D.S. A Phase 1 Study of Intravenous EGFR-ErbituxEDVsMIT in Children with Solid or CNS Tumours Expressing Epidermal Growth Factor Receptor. Targeted oncology 2024, 19, 333–342. [Google Scholar] [CrossRef] [PubMed]
- Portnow, J.; Synold, T.W.; Badie, B.; Tirughana, R.; Lacey, S.F.; D'Apuzzo, M.; Metz, M.Z.; Najbauer, J.; Bedell, V.; Vo, T.; et al. Neural Stem Cell-Based Anticancer Gene Therapy: A First-in-Human Study in Recurrent High-Grade Glioma Patients. Clinical cancer research : an official journal of the American Association for Cancer Research 2017, 23, 2951–2960. [Google Scholar] [CrossRef] [PubMed]
- Portnow, J.; Badie, B.; Suzette Blanchard, M.; Kilpatrick, J.; Tirughana, R.; Metz, M.; Mi, S.; Tran, V.; Ressler, J.; D'Apuzzo, M.; et al. Feasibility of intracerebrally administering multiple doses of genetically modified neural stem cells to locally produce chemotherapy in glioma patients. Cancer gene therapy 2021, 28, 294–306. [Google Scholar] [CrossRef]
- Fares, J.; Ahmed, A.U.; Ulasov, I.V.; Sonabend, A.M.; Miska, J.; Lee-Chang, C.; Balyasnikova, I.V.; Chandler, J.P.; Portnow, J.; Tate, M.C.; et al. Neural stem cell delivery of an oncolytic adenovirus in newly diagnosed malignant glioma: a first-in-human, phase 1, dose-escalation trial. Lancet Oncol 2021, 22, 1103–1114. [Google Scholar] [CrossRef]
- Wu, D.; Chen, Q.; Chen, X.; Han, F.; Chen, Z.; Wang, Y. The blood-brain barrier: structure, regulation, and drug delivery. Signal transduction and targeted therapy 2023, 8, 217. [Google Scholar] [CrossRef]
- Pardridge, W.M. CNS drug design based on principles of blood-brain barrier transport. Journal of neurochemistry 1998, 70, 1781–1792. [Google Scholar] [CrossRef]
- Song, X.; Qian, H.; Yu, Y. Nanoparticles Mediated the Diagnosis and Therapy of Glioblastoma: Bypass or Cross the Blood-Brain Barrier. Small (Weinheim an der Bergstrasse, Germany) 2023, e2302613. [Google Scholar] [CrossRef]
- Gao, H. Progress and perspectives on targeting nanoparticles for brain drug delivery. Acta Pharm Sin B 2016, 6, 268–286. [Google Scholar] [CrossRef]
- van Tellingen, O.; Yetkin-Arik, B.; de Gooijer, M.C.; Wesseling, P.; Wurdinger, T.; de Vries, H.E. Overcoming the blood-brain tumor barrier for effective glioblastoma treatment. Drug Resist Updat 2015, 19, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Chu, C.; Li, J.Y.; Boado, R.J.; Pardridge, W.M. Blood-brain barrier genomics and cloning of a novel organic anion transporter. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism 2008, 28, 291–301. [Google Scholar] [CrossRef] [PubMed]
- Fromm, M.F. Importance of P-glycoprotein at blood-tissue barriers. Trends in pharmacological sciences 2004, 25, 423–429. [Google Scholar] [CrossRef]
- Peer, D.; Karp, J.M.; Hong, S.; Farokhzad, O.C.; Margalit, R.; Langer, R. Nanocarriers as an emerging platform for cancer therapy. Nature nanotechnology 2007, 2, 751–760. [Google Scholar] [CrossRef]
- Balasubramanian, V.; Liu, Z.; Hirvonen, J.; Santos, H.A. Bridging the Knowledge of Different Worlds to Understand the Big Picture of Cancer Nanomedicines. Advanced healthcare materials 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Zhang, X.; Huang, J.; Feng, K.; Zhang, Y.; Wu, J.; Ma, L.; Zhu, A.; Di, L. Bio-fabricated nanodrugs with chemo-immunotherapy to inhibit glioma proliferation and recurrence. Journal of controlled release : official journal of the Controlled Release Society 2023, 354, 572–587. [Google Scholar] [CrossRef]
- Zhan, Q.; Yi, K.; Cui, X.; Li, X.; Yang, S.; Wang, Q.; Fang, C.; Tan, Y.; Li, L.; Xu, C.; et al. Blood exosomes-based targeted delivery of cPLA2 siRNA and metformin to modulate glioblastoma energy metabolism for tailoring personalized therapy. Neuro Oncol 2022, 24, 1871–1883. [Google Scholar] [CrossRef]
Figure 1.
Timeline of important events in the history of GBM research and treatment.
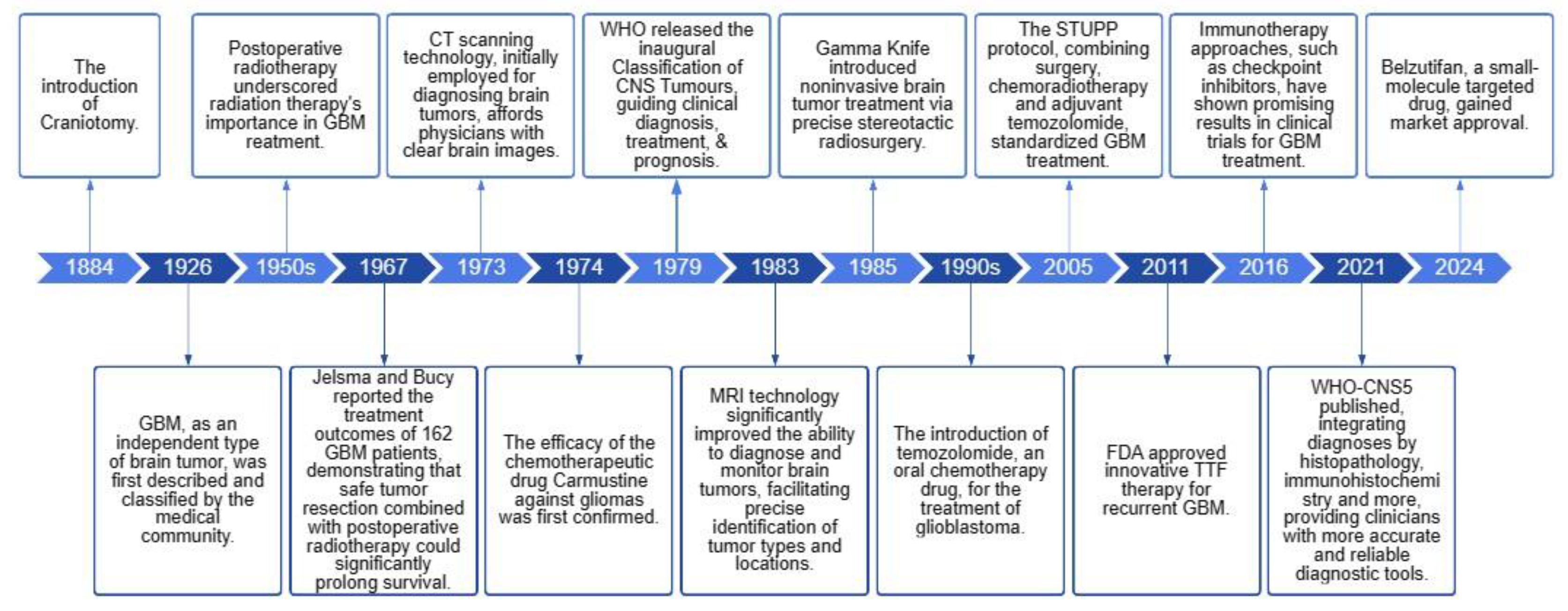
Figure 2.
Schematic diagram of the BBB structure and the mechanism of drug entry and exit from the BBB. a) Schematic cross-section of the BBB. b) Schematic diagram of different mechanisms for BBB crossing. From left to right is receptor-mediated transport, transporter -mediated transport, adsorptive-mediated transport, cell-mediated transport, Liphophilic pathway (Passive diffusion), Paracellular aqueous pathway (Passive diffusion), FUS reversibly opens the BBB (Passive diffusion), Drugs reversibly open the BBB (Passive diffusion), Active efflux.
Figure 2.
Schematic diagram of the BBB structure and the mechanism of drug entry and exit from the BBB. a) Schematic cross-section of the BBB. b) Schematic diagram of different mechanisms for BBB crossing. From left to right is receptor-mediated transport, transporter -mediated transport, adsorptive-mediated transport, cell-mediated transport, Liphophilic pathway (Passive diffusion), Paracellular aqueous pathway (Passive diffusion), FUS reversibly opens the BBB (Passive diffusion), Drugs reversibly open the BBB (Passive diffusion), Active efflux.
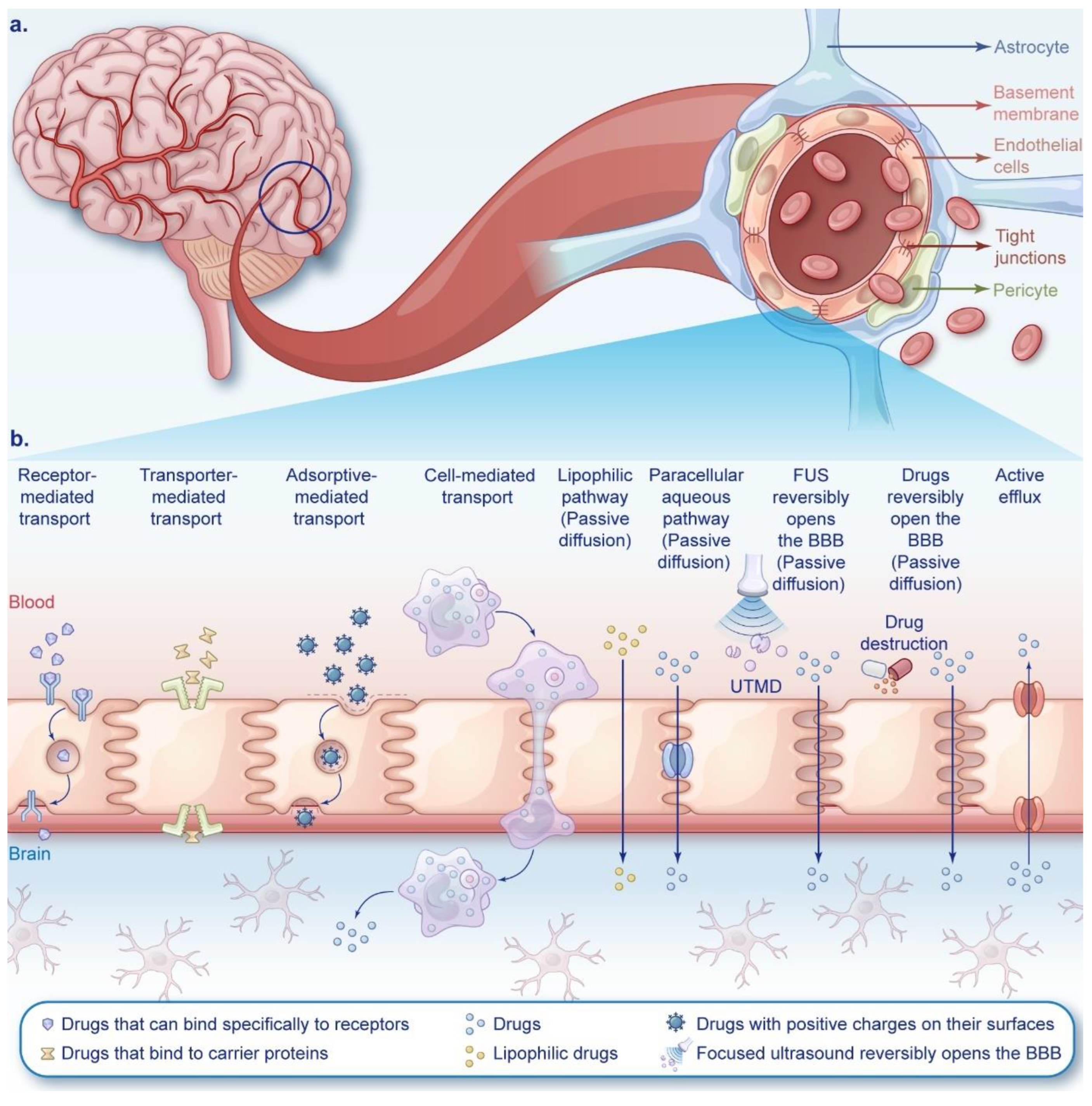
Figure 3.
Receptor-mediated transport. RTP-targeting Tf-modified nanoparticles reverse refractory GBM and improve radiosensitivity. (A) Schematic of a PEGylated agomirloaded nanoparticles that can be functionalized to enhance transport across the BBB and target RTP cells. (B) Immunofluorescence staining demonstrating the time-dependent intracellular uptake of Tf-NPs in P2 cells. Scale bar: 10 μm. (C) Tf-NPs entering a GSC spheroid were monitored by 3D confocal laser microscopy Scale bar: 20 μm. (D) P2 cells were treated with IR in the presence or absence of Tf-NPs, and γ-H2AX foci formation was investigated. Scale bar = 10 μm. (E) In vitro limiting dilution assay. (F and G) In vivo real-time NIR fluorescence imaging of P2 tumor-bearing mice after administration of Tf-NPs for the indicated time periods. (H) Schematic diagram showing the experimental time course and details of the Tf-NP and IR treatment courses. (I) In vivo bioluminescence images of P2 tumor cells in orthotopic mice intravenously injected with Tf-NPs. (J) Statistical analysis of orthotopic tumor growth from P2 cells. (K) Quantification of tumor sizes. The data were obtained from H&Estained brain sections of 8 mice per group. (L) Kaplan–Meier survival curves of mice intracranially injected with P2 cells. [43] Copyright 2022, Oxford University Press.
Figure 3.
Receptor-mediated transport. RTP-targeting Tf-modified nanoparticles reverse refractory GBM and improve radiosensitivity. (A) Schematic of a PEGylated agomirloaded nanoparticles that can be functionalized to enhance transport across the BBB and target RTP cells. (B) Immunofluorescence staining demonstrating the time-dependent intracellular uptake of Tf-NPs in P2 cells. Scale bar: 10 μm. (C) Tf-NPs entering a GSC spheroid were monitored by 3D confocal laser microscopy Scale bar: 20 μm. (D) P2 cells were treated with IR in the presence or absence of Tf-NPs, and γ-H2AX foci formation was investigated. Scale bar = 10 μm. (E) In vitro limiting dilution assay. (F and G) In vivo real-time NIR fluorescence imaging of P2 tumor-bearing mice after administration of Tf-NPs for the indicated time periods. (H) Schematic diagram showing the experimental time course and details of the Tf-NP and IR treatment courses. (I) In vivo bioluminescence images of P2 tumor cells in orthotopic mice intravenously injected with Tf-NPs. (J) Statistical analysis of orthotopic tumor growth from P2 cells. (K) Quantification of tumor sizes. The data were obtained from H&Estained brain sections of 8 mice per group. (L) Kaplan–Meier survival curves of mice intracranially injected with P2 cells. [43] Copyright 2022, Oxford University Press.
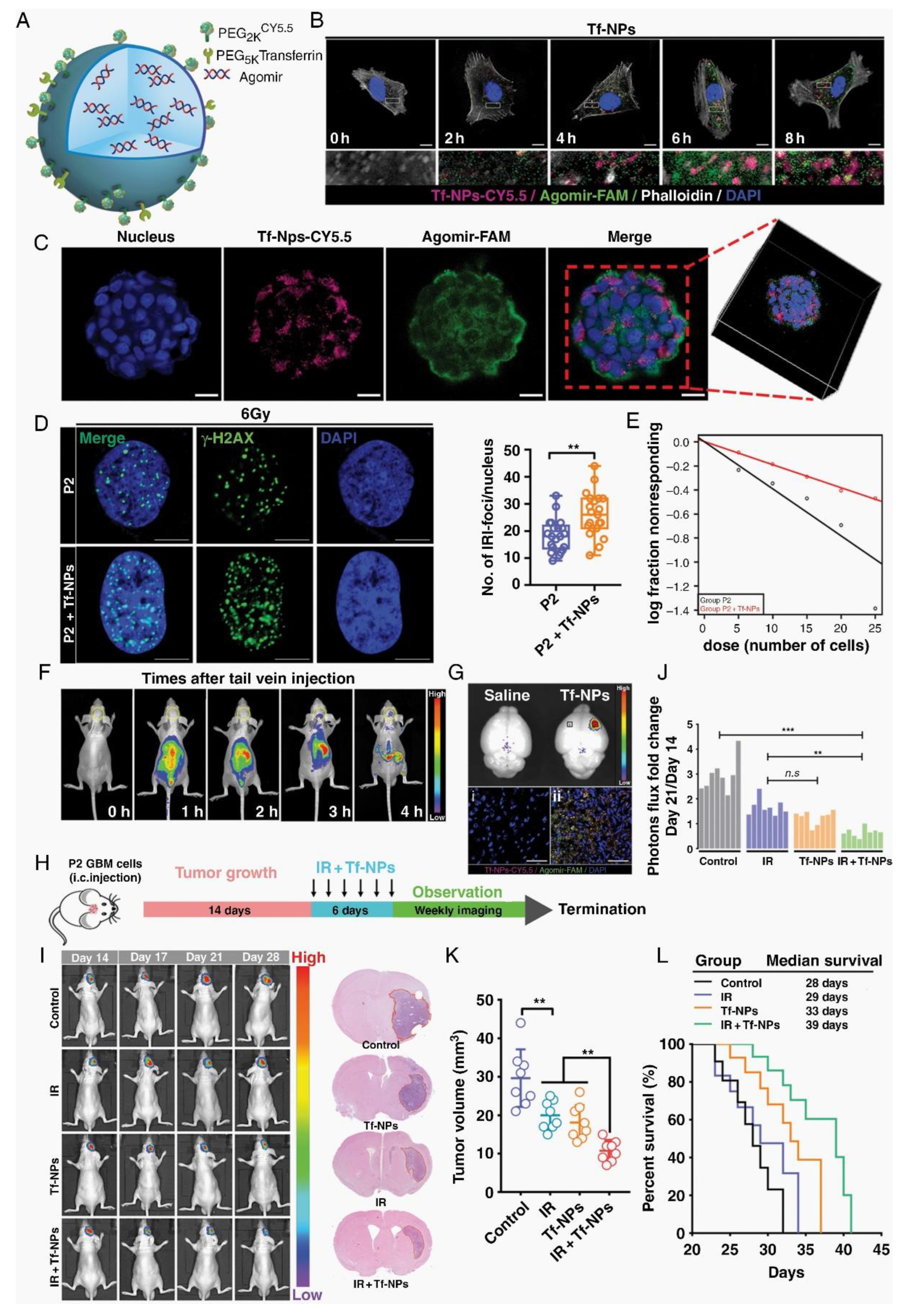
Figure 4.
Transporter-mediated transport. (A-C) The 2-DG/aV-siCPT1C NC traverses the BBB via GLUT1 receptors to enter the CNS, thereby achieving starvation therapy for GBM. (D) Biofluorescence imaging of U87-Luci glioma mice with different treatments. [25] Copyright 2023, American Chemical Society.
Figure 4.
Transporter-mediated transport. (A-C) The 2-DG/aV-siCPT1C NC traverses the BBB via GLUT1 receptors to enter the CNS, thereby achieving starvation therapy for GBM. (D) Biofluorescence imaging of U87-Luci glioma mice with different treatments. [25] Copyright 2023, American Chemical Society.
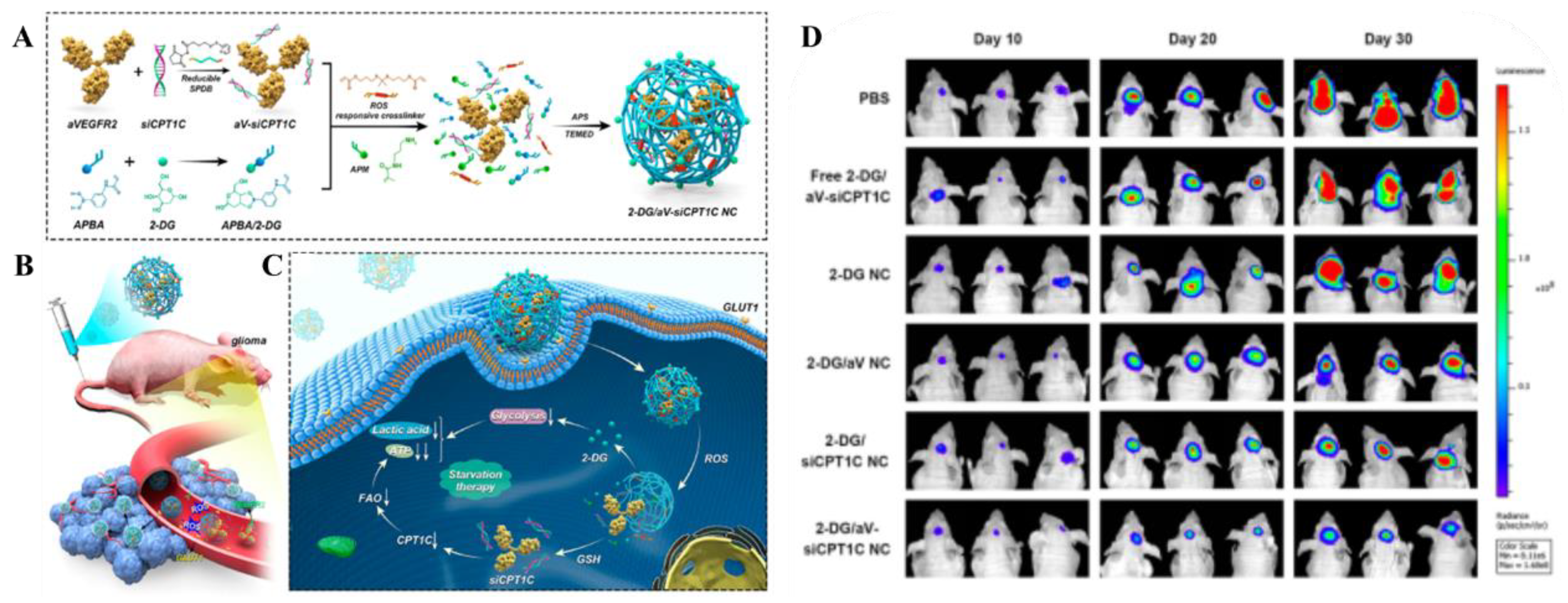
Figure 5.
Adsorptive-mediated transport. (A-B) TAT-AT7 targeted glioma blood vessels in the intracranial glioma tissue of nude mice. (C-H) TAT-AT7 inhibited the growth of glioma in nude mice [31]. Copyright 2023, MDPI.
Figure 5.
Adsorptive-mediated transport. (A-B) TAT-AT7 targeted glioma blood vessels in the intracranial glioma tissue of nude mice. (C-H) TAT-AT7 inhibited the growth of glioma in nude mice [31]. Copyright 2023, MDPI.
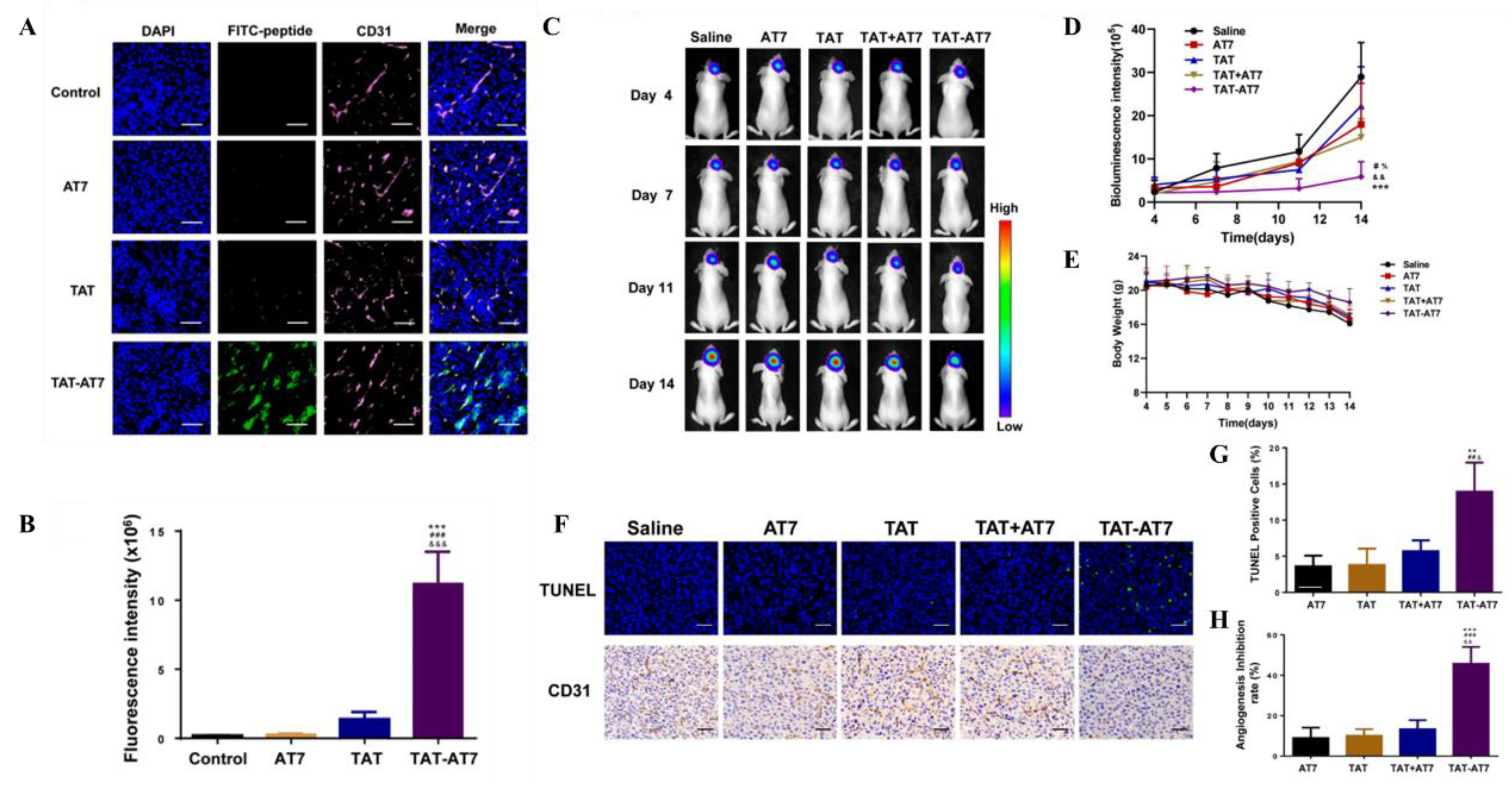
Figure 6.
Cell-mediated transport. (A) Preparation of macrophage membrane-based biomimetic vehicles and Mechanism of the Biomimetic MPM@P NGs across BBB to Penetrate Brain Parenchyma for MR Imaging-Guided Combination Chemotherapy/Chemodynamic Therapy of Orthotopic Glioma. (B) The targeted MR imaging of orthotopic glioma verified that macrophage membrane decoration facilitated the traversal of MPM@P NGs across the BBB. (C-E) Changes in C6 glioma-bearing mice tumor volume and mouse body weight after different treatments. [38] Copyright 2021, American Chemical Society.
Figure 6.
Cell-mediated transport. (A) Preparation of macrophage membrane-based biomimetic vehicles and Mechanism of the Biomimetic MPM@P NGs across BBB to Penetrate Brain Parenchyma for MR Imaging-Guided Combination Chemotherapy/Chemodynamic Therapy of Orthotopic Glioma. (B) The targeted MR imaging of orthotopic glioma verified that macrophage membrane decoration facilitated the traversal of MPM@P NGs across the BBB. (C-E) Changes in C6 glioma-bearing mice tumor volume and mouse body weight after different treatments. [38] Copyright 2021, American Chemical Society.
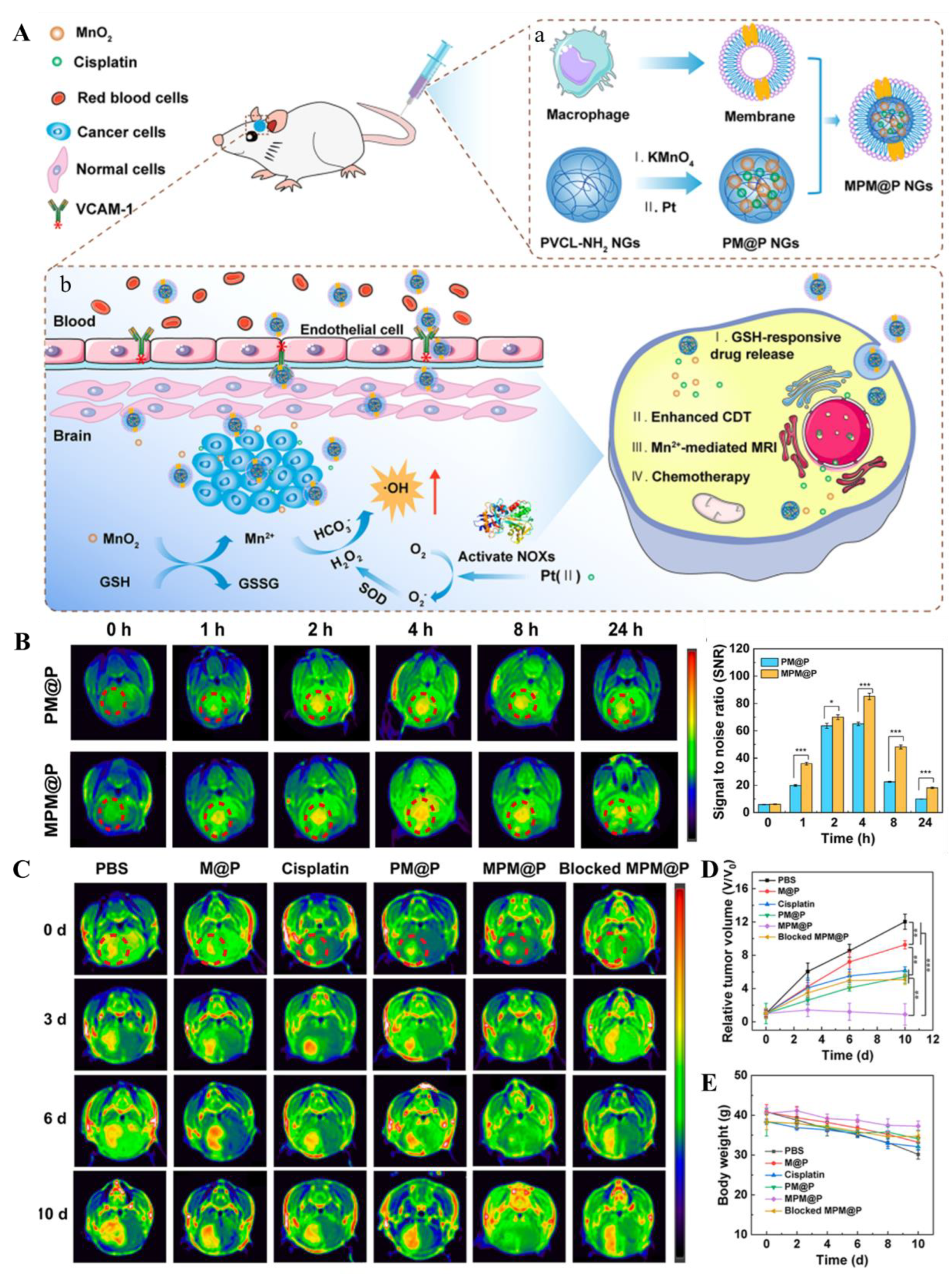

Table 1.
Preclinical research achievements of NDDS in the treatment of GBM.
| Strategies to cross the BBB | Targeting | NDDS | Drug | Combination therapy (if any) | Administration route | Administration model |
Safety Evaluation | Pharmacokinetics (PK) | Pharmacodynamics | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|
| RMT | Tf | Tf-NPs | TMZ | bromodomain inhibitor | tail vein injection | the human U87MG and murine GL261 glioma models | compare with the control group, mice treated with drug-loaded Tf-NPs maintained their body weight and body conditioning scores, and had significantly more stable WBC and PLT levels over the treatment course. | 24 h absorption efficiency was higher (13% vs > 1%). Accumulation and retention of Tf-NPs on the tumor surface, while non-functional NPs did not. NPs loaded with TMZ demonstrated an attenuated release profile with ~90% release by 48 h. | Tf-NPs bind to GBM tumors, enhance DNA damage & apoptosis, reduce tumor burden, improve survival, protect from systemic toxicity. | [12] |
| RMT | Lf | PMO-Lf@Dox | DOX | - | co-incubation | C6 cells | PMO concentration is 200 μg/mL, the cell survival rate is 90%,the hemolysis rate is< 2%. one week after tail vein injection of 60 mg/kg PMO, no obvious changes such as heart, liver, spleen, lung and kidney are seen. |
PMO-Lf@Dox-treated C6 cells showed significantly higher uptake after 24 hours than PMO@Dox-treated cells. | Lf-modified PMO enhances the inhibitory effect of Dox on C6 cells. | [13] |
| RMT | Lf | USLP-NH2-PEG-TMZ-Lf | TMZ | - | tail vein injection | transwell model with hCMEC/D3 cells; Balb/C mice |
Safety is evaluated by H&E staining of mouse organs (brain, kidney, lung, liver) post-nano-formulation IV injection, with no toxicity observed in 24 hours. | USLP-NH2-Cy5-PEG-Lf accumulated rapidly in the brain and reached its peak at 1 h after administration, and the USLP formula significantly reduces the external alignment. | In vitro apoptosis studies on GBM cell lines U87 and GL261 showed improved TMZ-induced apoptosis with USLP formulations compared to pure TMZ. | [14] |
| RMT | acetylcholine receptor | TMZ@RVG-Zein NPs | TMZ | - | co-incubation | U87 cell lines; BBB model with bEnd.3 cells | The TMZ@RVG-Zein NPs exhibited excellent stability, without causing significant side effects. | The TMZ@RVG-Zein NPs had an encapsulation efficiency (EE) of 77.9 ± 4.7% and a loading efficiency (LE) of 66.7 ± 2.9 mg/g. | The TMZ@RVG-Zein NPs have cytotoxic effects on U87 cells, induce apoptosis, and show enhanced cellular uptake compared to TMZ alone. | [15] |
| RMT | FR | iRPPFFA@TMZ | TMZ | - | tail vein injection | subcutaneous and orthotopic xenograft tumor models | The study evaluated the biocompatibility of POSS nanoparticles through a CCK-8 assay, hemolysis rate tests, and fluorescence imaging, showing low toxicity and effective cellular uptake for biomedical applications. | The multifunctional POSS nanoparticles demonstrated high stability with no significant fluorescence intensity change over 8 weeks, while the half-life of iRPPFFA nanoparticles was estimated at approximately 16 weeks. | In vivo studies showed that TMZ-loaded POSS nanoparticles significantly improved the survival of GBM-bearing mice, indicating enhanced therapeutic efficacy compared to monotherapy. | [16] |
| RMT | LRP-1 receptors | Ti@FeAu–Ang nanoparticles | Magnetic nanoparticles as therapeutic agents | - | tail vein injection | Rat GBM model | Preliminary safety analysis highlighted no toxicity to the hematological system after Ti@FeAu–Ang nanoparticle-induced hyperthermia treatment. Immunohistochemical analysis showed no significant organ damage and biological changes in vital organs such as the heart, liver, spleen, lungs, and kidneys. | The paper does not provide detailed pharmacokinetic data, but it does mention that the nanoparticles were prepared at various concentrations for the temperature elevation test and the in vivo assessment of tumor growth. | The Ti@FeAu–Ang nanoparticles demonstrated a temperature elevation of up to 12°C upon magnetic stimulation, indicating potential applications in MRI and hyperthermia-based cancer therapy. In vitro, the nanoparticles showed improved cytotoxicity up to 85% due to hyperthermia produced by a magnetic field. In vivo findings showed a 10-fold decrement in tumor volume compared to the control group. | [17] |
| RMT | LRP-1 receptors | Au-DOX@PO-ANG | DOX | radiotherapy | tail vein injection | U87-MG human GBM xenografts in nude mice | In vivo, blood biochemical indicators (CK-MB, AST, and Scr) were measured, and no significant pathological changes were observed in the main organs (heart, liver, spleen, lung, and kidney) of the Au-DOX@PO-ANG group compared to the PBS group. In vitro, the cytotoxicity of modified AuNPs was evaluated using CCK-8 assays, showing more than 80% cell viability at concentrations up to 10 mg/L, indicating non-toxicity to GBM cells. | The storage stability of the modified AuNPs in PBS (pH 7.4) at 4 °C showed little change in particle size over 4 weeks, indicating good storage ability and potential stability. | In vitro, the antitumor activity of Au-DOX@PO-ANG was evaluated using the CCK-8 assay, showing significant antitumor effects when combined with radiotherapy. In vivo, the therapeutic effect was observed through MRI, with the tumor volume rate of increase slowing in the Au-DOX@PO-ANG + RT group, and a significant increase in cell apoptosis was observed in this group, consistent with MRI data. | [18] |
| RMT | EGFR | PmAb-TMZ-PLGA-NPs | TMZ | Panitumumab | co-incubation | U-87MG and LN229 GBM cell lines. | In Vitro Cytotoxicity: Assessed using the Live/Dead assay kit and fluorescence-activated cell sorting (FACS). Immunoreactivity: Evaluated using EGFR-overexpressed U-87MG cells. |
In Vitro Drug Release: Studied using phosphate-buffered saline (PBS) at acidic (pH 5.0) and neutral (pH 7.4) conditions. Release Profile: Determined by UV-vis spectroscopy at various time points. |
Enhanced in U-87MG cells due to high EGFR expression compared to LN229 cells. | [19] |
| RMT | IGF1R | IGF1R4-mFc | Galanin Peptide | - | ISBP | Rat Model; Hargreaves Pain Model | No specific safety evaluation was described in detail. However, the use of sdAbs, which are known for their low immunogenicity and high solubility/stability, suggests potential safety advantages. | Brain Uptake: IGF1R4-mFc showed significant brain uptake compared to the negative control A20.1-mFc, with ~25% of the total amount accumulating in the brain parenchymal fraction post-ISBP. Linear Accumulation Plateau: The concentration curve for IGF1R4-mFc demonstrated a linear accumulation plateauing at approximately 400 µg (~1 µM), suggesting a saturable mechanism of transport. | Analgesic Effect: Systemic administration of IGF1R4-mFc fused with galanin induced a dose-dependent suppression of thermal hyperalgesia in the Hargreaves pain model, indicating pharmacological effectiveness of the brain-delivered cargo. | [20] |
| RMT | α5β1 integrin receptor | RGEK-lipopeptide containing coencapsulated STAT3siRNA and WP1066 | WP1066; STAT3siRNA | tail vein injection | intracranial orthotopic GL261 GBM model in C57BL/6J male mice | No Direct Safety Data: The document did not provide direct safety evaluation data. However, the use of integrin receptor-selective liposomes and their preferential accumulation in tumor tissue suggests potential for reduced systemic toxicity. | Preferential Accumulation: NIR-dye-labeled α5β1 integrin receptor-selective liposomes were found to accumulate preferentially in mouse brain tumor tissue after intravenous administration. Encapsulation Efficiency: Entrapment efficiency (%EE) for WP1066 was measured using analytical HPLC. |
Inhibition of GBM Growth: Coadministration of WP1066 and STAT3siRNA within RGDK-lipopeptide-based liposomes led to significant inhibition (>350% compared to untreated mice group) of orthotopically growing mouse GBM. | [21] | |
| RMT | αv β integrin and NRP-1 | CPT-S-S-PEG-iRGD@IR780 micelles | Camptothecin (CPT) | photodynamic therapy | tail vein injection | Intracranial orthotopic U87MG gliomas tumor model in Balb/c nude mice | The micelles were shown to be stable with controlled drug release under physiological conditions. Toxicity studies in vivo assessed mice's survival, body weight, and histopathology. | Information on the pharmacokinetic behavior of the micelles is not explicitly detailed in the abstract. However, the micelle design enables sustained release of CPT upon exposure to high glutathione levels in glioma cells. | The CPT-S-S-PEG-iRGD@IR780 micelles displayed significantly enhanced antitumor effects with laser irradiation, as compared to controls. Micelles with iRGD demonstrated favorable targeting ability to glioma cells and deep tumor penetration. | [22] |
| RMT | Hsp70 | D-A-DA/TPP | DOX | PD-1 Checkpoint Blockade | tail vein injection | C6-luc tumor-bearing mice | In vivo Toxicity Assessment: Safety evaluation is likely conducted through monitoring of animal health, blood chemistry, and tissue histology post-treatment. However, specific details are not provided. | Biodistribution and Clearance: The biodistribution and clearance of D-A-DA/TPP nanoparticles in vivo are not explicitly discussed in the excerpt. These would typically involve measuring nanoparticle concentrations in blood, tumor, and other organs over time. | Efficacy Evaluation: The efficacy of D-A-DA/TPP nanoparticles in inducing glioma apoptosis and prolonging median survival time is demonstrated through in vivo studies. Immune Response Activation: Combination with PD-1 checkpoint blockade is shown to further activate T cells and provoke an anti-tumor immune response. |
[23] |
| RMT | dopamine receptor and GRP78 receptor | pHA-AOHX-VAP-DOX | DOX | - | tail vein injection | intracranial U87 gliomabearing nude mice model | The maximum tolerated dose (MTD) was determined in healthy BALB/c mice. Hematoxylin-eosin (H&E) staining of major organs and blood samples were collected to measure blood routine and biochemical parameters. | The study evaluated the biodistribution of the conjugates in the tumor and normal tissues. The accumulation of DOX in the tumor site was assessed, and the conjugation with peptides reduced the accumulation of DOX in normal tissues. | The anti-GBM efficacy was evaluated by prolonging the survival time of mice and assessing tumor cell apoptosis through TUNEL staining. The inhibition of tumor angiogenesis was evaluated using CD31 immunofluorescence staining. | [24] |
| TMT | GLUT1 | 2-DG/aV-siCPT1C NC | 2-DG | siCPT1C | tail vein injection | Orthotopic U87-Luci cells xenograft tumor model in BALB/c nude mice | The safety of the nanocapsules was evaluated through histopathologic studies and blood biochemical tests, including ALT, AST, BUN, and CREA levels. No significant toxicity or side effects were observed on normal cells and tissues. | The nanocapsules showed an extended half-life compared to free siRNA, with the 2-DG/aV-Cy5-siCPT1C NC having an elimination half-life (t1/2) of approximately 1.2 hours. | The nanocapsules effectively targeted GBM, inhibited energy metabolism, and showed a significant inhibitory effect on the growth of GBM. The combination of 2-DG, aV, and siCPT1C resulted in decreased lactic acid levels and reduced ATP production in tumor tissues, indicating effective metabolic pathway inhibition. | [25] |
| TMT | ChTs | pMPC-co-(anti-PD-L1-pPEGMA) | anti-PD-L1 | - | intravenous injection | LCPN orthotopic glioma tumor model | The safety and biocompatibility of the delivery system were evaluated through in vitro cytotoxicity studies using bEnd. 3 cells and LCPN cells, as well as histological examination of major organs from mice. | The PK of the Cy5.5-labeled IgG in the anti-PD-L1-MP-3 formulation was studied, showing prolonged blood circulation time compared to non-choline containing controls. | The system demonstrated pH-responsive protein release in vitro, with accelerated release at pH 6.0 simulating the acidic tumor microenvironment. In vivo studies showed significant tumor growth suppression and prolonged animal survival, indicating the activation of antitumor immune responses. | [26] |
| TMT | LAT1 | WP1066-loaded liposomes of Amphi-DOPA | wp1066 | DNA vaccine | tail vein injection | orthotopic GL261 tumor model in famale C57BL/6J mice | The safety and toxicity of the liposomal formulation are evaluated through in vivo serum toxicity profiles. No significant changes in biochemical and hematological parameters suggest the system is well-tolerated. | Preferential Accumulation in Brain Tissue: Intravenously administered NIR-dye labeled Amphi-DOPA liposomes showed preferential accumulation of the dye in brain tissue, indicating successful BBB penetration. | Enhanced Overall Survivability: WP1066-loaded Amphi-DOPA Liposomes Alone: Enhanced overall survivability of C57BL/6J mice bearing orthotopically established mouse GBM by ~60% compared to untreated mice. Combination Therapy: Further enhanced overall survivability (>300% compared to untreated mice) when combining WP1066-loaded Amphi-DOPA liposomes with in vivo DC-targeted DNA vaccination using a survivin-encoded DNA vaccine. |
[27] |
| TMT | SVCT2 | PTX-Glu-Vc-Lip | PTX | - | tail vein injection | intracranial C6 glioma bearing mice | The safety of the ligand-modified liposomes is demonstrated through hemolysis assays, showing no significant increase in hemocompatibility even at high concentrations of phospholipids. | The study evaluates the plasma concentration-time profiles and brain distribution of paclitaxel after intravenous injection of different liposome formulations. The pharmacokinetic parameters, including AUC(0-t), MRT, Tmax, Cmax, and t1/2, are reported for each formulation. | The cellular uptake of the liposomes is evaluated in GLUT1- and SVCT2-overexpressed C6 cells, showing higher uptake for the Glu-Vc-Lip compared to other formulations. The in vivo imaging of DiD-loaded liposomes demonstrates the targeting efficiency to the brain tumor site. | [28] |
| TMT | OCTN2 | LC-1000-PLGA NPs | PTX | - | tail vein injection | 2 D and 3 D tumor growth models using the glioma cell line T98G. | The specific dose is not explicitly mentioned in the provided text. However, the studies involve the use of different concentrations of paclitaxel in the in vitro cytotoxicity assays and varying lengths of PEG spacers in the nanoparticles. | Biodistribution: LC-PLGA NPs showed high accumulation in the brain as indicated by biodistribution and imaging assays in mice. In Vitro Release: Paclitaxel-loaded LC-PLGA NPs showed sustained release of paclitaxel compared to Taxol. | The pharmacodynamic evaluation includes in vitro cytotoxicity assays in T98G cells, demonstrating increased toxicity with LC-PLGA NPs compared to Taxol and paclitaxel-loaded PLGA NPs, and in vivo biodistribution studies showing enhanced brain accumulation of paclitaxel with LC-1000-PLGA NPs. | [29] |
| TMT | Cx43 and BSAT1 | Cx43-NG/CDDP and BSAT1-NG/CDDP | cisplatin | - | tail vein injection | intracranial implantation of rat GBM 101/8 in female Wistar rats | Safety is evaluated by monitoring body weight changes, general condition of the animals, and comparing the median survival rates of the different treatment groups. | The PK of the nanogels is assessed by monitoring the tumor volume changes over time using MRI and comparing the median survival times of the treated groups with the control group. | The antitumor efficacy of the targeted nanogels is evaluated by comparing the glioma volume and the survival rate of rats treated with targeted nanogels conjugated with specific mAbs against Cx43 and BSAT1 to those treated with non-targeted nanogels or free cisplatin. The study demonstrates significantly reduced tumor growth and increased lifespan in animals treated with targeted nanogels. | [30] |
| AMT | TAT | TAT-AT7-modified PEI nanocomplex | secretory endostatin gene | - | intravenous injection | orthotopic U87-glioma-bearing nude mice model | TAT-AT7 showed no obvious hemolysis at concentrations ranging from 2.5 to 640 µmol/L, indicating good biosafety. | The study evaluated the cellular uptake of TAT-AT7 in bEnd3 cells (mouse brain microvascular endothelial cells) and its distribution in an intracranial glioma model, demonstrating high uptake efficiency and penetration capability. | TAT-AT7 exhibited significant inhibitory effects on HUVECs' proliferation, migration, invasion, and tubular structure formation. It also promoted apoptosis in HUVECs and inhibited zebrafish embryo angiogenesis. In vivo, TAT-AT7 significantly suppressed glioma growth, induced apoptosis of glioma cells, and inhibited angiogenesis. | [31] |
| CMT+ RMT | RF + active targeting | LMP tFNA NPs | LMP | - | tail vein injection | orthotopic U87-glioma-bearing nude mice model | In vitro and in vivo safety evaluations showed that RFA NPs had no significant cytotoxic effects on primary hepatocytes, astrocytes, or GBM cells, and no obvious immune side effects were observed in vivo. | Prolonged Circulation Time: Biomimetic NPs encapsulated with natural cell membranes prolonged the circulation time of the drug in vivo. Efficient BBB Permeability: The hybrid membrane coating facilitated efficient crossing of the BBB. |
Inhibition of GBM Growth: LMP loaded RFA NPs exhibited superior and specific anti-GBM activities in vitro and in vivo. Apoptosis/Pyroptosis Induction: LMP induced apoptosis and pyroptosis in GBM cells, reducing tumor growth. Reduced Off-Target Delivery: The RFA NPs demonstrated reduced off-target drug delivery, ensuring specificity. |
[32] |
| CMT | Active Targeting | Ang-RBCm@NM-(Dox/Lex) | Dox | Lexiscan (Lex) | Intravenous injection | orthotopic U87MG human GBM tumor-bearing nude mice | Cell Viability Studies: The nanomedicine was evaluated for cytotoxicity using in vitro cell viability assays. In vivo Toxicity Studies: While specific toxicity data are not detailed in the abstract, in vivo studies assessed the therapeutic efficacy and survival outcomes, which indirectly reflect safety. | The nanomedicine demonstrates a prolonged blood circulation time, with an elimination half-life (t1/2,β) of 9.3 hours for Ang-RBCm@NM-(Dox/Lex), which is longer than the RBCm@NM-(Dox/Lex) counterpart (t1/2,β = 7.8 hours). | Superior BBB Penetration: The angiopep-2 functionalization and Lex-mediated BBB opening facilitated superb penetration across the BBB. Effective Tumor Suppression: Treatment with Ang-RBCm@NM-(Dox/Lex) resulted in effective suppression of tumor growth and significantly improved median survival time in orthotopic U87MG human GBM tumor-bearing nude mice. |
[33] |
| CMT | Active Targeting | iRGD-EM:TNDs | TMZ | - | tail vein injection | orthotopic U87MG human GBM tumor-bearing nude mice | Safety is evaluated through hematoxylin and eosin (H&E) pathological staining of major organs, monitoring of body weight, and detection of IgE levels associated with hypersensitivity reactions. The results suggest low systemic toxicity and good biocompatibility of the iRGD-EM:TNDs. | The EM-coated nanodots demonstrate a longer elimination half-life, suggesting reduced degradation in vivo compared to traditional PEG stealth motifs. | The iRGD-EM:TNDs show enhanced cellular uptake, improved penetration in multicellular tumor spheroids, and increased transport ratios across the BBB in vitro and in vivo. The treatment with iRGD-EM:TNDs results in a 100% survival rate after 30 days post-tumor implantation and induces the highest level of cell apoptosis. | [34] |
| CMT | IL13Rα2 | T cells + BPLP-PLA-NPs (clicked) | DOX | CAR T cell therapy | tail vein injection | intracranial xenograft model using female immunodeficient nude mice | The safety evaluation includes assessing the cytotoxic effects of the nanoparticles and T cells on GBM cells in vitro and observing the behavior of T cells in vivo without causing observable side effects. | The study evaluates the retention of nanoparticles on T cells for at least 8 days, indicating the stability of the linkage for a suitable time window for in vivo delivery. | The system demonstrates enhanced cytotoxic effects in vitro with T cells clicked with doxorubicin-loaded nanoparticles compared to bare T cells. In vivo, T cells expressing TQM-13 serve as delivery shuttles for nanoparticles, significantly increasing the number of nanoparticles reaching brain tumors compared to nanoparticles alone. | [35] |
| CMT | inflammatory tumor microenvironment | NEs-Exos/DOX | DOX | - | intravenous injection | C6-Luc glioma-bearing mice models | The safety evaluation of the NEs-Exos/DOX system is not detailed in the provided text. The focus is on the efficacy of the system in crossing the BBB and targeting glioma cells. | The study does not provide specific pharmacokinetic data for the NEs-Exos/DOX system. However, it does mention that the NEs-Exos can rapidly penetrate the BBB and migrate into the brain, suggesting favorable pharmacokinetic properties for brain tumor targeting. | The pharmacodynamics of the NEs-Exos/DOX system are demonstrated through in vitro and in vivo assays, showing that the system can improve the anticancer efficacy of DOX, reduce mortality, and effectively suppress tumor growth while prolonging survival time in a glioma mouse model. | [36] |
| CMT | domain CLTX, and IgG4 hinge | CAR-neutrophils@RSiO2-TPZ | tirapazamine (TPZ) | - | intravenous injection | mouse xenograft model of GBM | Biocompatibility: CAR-neutrophils exhibited high biocompatibility with normal cells (SVG p12 glial cells, hPSCs, and hPSC-derived cells). Toxicity: Necrosis was not observed in major organs of experimental mice. However, concerns regarding off-target tissue toxicity or systemic toxicity were mentioned. | Nanoparticle delivery efficiency: CAR-neutrophils delivered >20% of administered nanodrugs to brain tumors compared to 1% by free nanodrugs. | In vitro cytotoxicity: CAR-neutrophils presented enhanced anti-tumor cytotoxicity compared to peripheral blood (PB) neutrophils. In vivo tumor growth inhibition: CAR-neutrophils loaded with TPZ-loaded SiO2 nanoparticles significantly inhibited tumor growth and prolonged animal survival in GBM xenograft models. Mechanism: Combination of CAR-enhanced direct cytolysis and chemotherapeutic-mediated tumor-killing via cellular uptake and glutathione (GSH)-induced degradation of nanoparticles within tumor cells. |
[37] |
| CMT | Active Targeting | MPM@P NGs | cisplatin | chemodynamic therapy (CDT) | intravenous injection | orthotopic C6 glioma in a mouse model | The safety of the nanogels is evaluated through in vitro hemolysis assays, in vivo hematological indices, blood biochemical analysis, and histopathological examination of major organs. | The PK of the nanogels is assessed by tracking the platinum (Pt) content in blood after intravenous injection. The half-life of MPM@P NGs is determined to be longer than that of PM@P NGs without membrane coating, indicating improved blood circulation time. | The in vivo antitumor activity of the nanogels is evaluated by monitoring tumor volume changes using T1-weighted MR imaging. The MPM@P NGs demonstrate the smallest tumor size and most efficient therapeutic effect among all groups due to the combination of enhanced CDT and chemotherapy, as well as improved BBB crossing and glioma targeting ability. | [38] |
| CMT | Active Targeting | aDCM@PLGA/RAPA | RAPA | immunotherapeutic activation | tail vein injection | orthotopic C6 tumor model | Biocompatibility Tests: Studies investigating the biocompatibility and potential toxicity of aDCM@PLGA/RAPA in vivo and in vitro were mentioned but specific details were not provided. | Stability Studies: aDCM@PLGA/RAPA demonstrated good colloidal stability in PBS and plasma, suggesting the potential for prolonged circulation time in the blood. | Immune Activation: aDCM@PLGA/RAPA effectively activated T cells and NK cells, modifying the tumor microenvironment to an immune-supportive state. Antitumor Efficacy: The combined immunotherapeutic and chemotherapeutic effects led to significant inhibition of glioma growth and induced glial differentiation. | [39] |
| CMT | Active Targeting | nanoparticle-engineered TRAIL-expressing hADSCs | TRAIL | - | local intracranial delivery | mouse intracranial xenograft model of patient-derived GBM cells | Cell Viability: MTS assay revealed no significant change in viability of hADSCs transfected with nanoparticle-laden TRAIL DNA compared to controls, indicating safety. The in vivo safety is assessed by monitoring the survival and weight changes of the mice. | The PK of the TRAIL-overexpressing hADSCs is evaluated by observing the migration and infiltration of the cells towards GBM tumors in vivo and by measuring the levels of TRAIL protein expression in vivo. | The study demonstrates that TRAIL-overexpressing hADSCs induce significant apoptosis in GBM cells in vitro and in vivo, with negligible apoptotic activity in normal brain cells. The therapeutic effects include inhibition of tumor growth, extension of animal survival, and reduction of tumor mass and microsatellites occurrence. | [40] |
| CMT | Active Targeting | PEI-PLL-transfected MSCs | suicidal genes, namely, HSV-TK and TRAIL | ganciclovir | intratumoral injection | SD rats were used with C6 glioma cells injected intracranially | Cell Viability: Cell viability of MSCs transfected with the PEI-PLL copolymer was evaluated using the MTT assay. Cell viability was more than 90% at a pDNA/polymer ratio of 1:1.5 for the PEI-PLL copolymer. In vivo Toxicity: While not explicitly stated in the abstract or methods section, the use of nonviral vectors like PEI-PLL copolymer is generally considered safer than viral vectors due to their low immunogenicity and reduced risk of oncogenicity. |
The PK of the system is assessed by monitoring the survival rates and tumor growth in the animal model after treatment with the PEI-PLL-transfected MSCs. | The study demonstrates that the combination of HSV-TK and TRAIL genes in MSCs leads to a significant decrease in cell viability and an increase in apoptosis in glioma cells, both in vitro and in vivo. The reduction in cell proliferation markers Ki67 and angiogenesis marker VEGF, along with the TUNEL assay results, indicate the therapeutic effectiveness of the MSCs in inducing apoptosis in GBM cells. | [41] |
| CMT | Active Targeting | ApoE-ARTPC@TMZ | ART | TMZ | tail vein injection | orthotopic U251-TR GBM mouse model | Conducted in vivo experiments to assess the safety of the nanoplatform, including examination of body weight, blood cell counts (RBC, WBC, PLT), and histological examination of major organs and brain tissue. | Evaluated the circulation time of the liposomes in the bloodstream using FITC-Dex as a marker, showing prolonged circulation times for ApoE-ARTPC@FITC-Dex and ARTPC@FITC-Dex compared to free FITC-Dex. | Assessed the induction of apoptosis, DNA damage, and inhibition of MGMT expression and Wnt/β-catenin signaling. The combination therapy showed enhanced cytotoxicity, increased ROS generation, and significant induction of apoptosis in vivo. | [42] |
Table 2.
Clinical trials of glioma treatment using NDDS: ongoing and completed.
| Methods to overcome the BBB | Drug |
Combination therapy (if any) |
Research Phases | NDDS | Administration route |
Safety Evaluation | PK | Primary Efficacy Endpoints | ClinicalTrials.Gov Identifier | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|
| Passive diffusion | DOX | TMZ + radiotherapy |
Phase Ⅱ | Caelyx™, PEG-Dox | Intravenous infusion + Oral Administration |
The treatment was well-tolerated, with most AEs classified as grade 1-2. However, some grade 3-4 AEs were also reported. | PK of PEG-Dox and Temozolomide were not specifically detailed in the document summary provided. However, the improved BBB penetration of PEG-Dox suggests enhanced PK compared to conventional doxorubicin. | Progression Free Survintravenous infusional at 12 Months (PFS-12): 30.2% in all patients. Median Overall Survival (mOS): 17.6 months in all patients including those from Phase I. Comparison to Historical Control: The endpoints did not differ significantly from the EORTC26981/NCIC-CE.3 data in a post-hoc statistical comparison. |
NCT00944801 | [128] |
| intratumoral injection | magnetic iron-oxide nanoparticles |
fractionated stereotactic radiotherapy | phase II | Magnetic Iron-Oxide Nanoparticles | Intratumoral Injection | Side Effects: Acute side effects during thermotherapy were classified according to the Common Toxicity Criteria (CTC) version 2.0. Common side effects included sweating (50.0%), general sensation of warmth (47.0%), and thermal stress with body temperature exceeding 38°C in 6 patients (9.1%). No serious complications were observed. | The study does not provide specific pharmacokinetic data. However, it mentions that no indication of iron being released from the intratumoral deposits or being metabolized was observed, suggesting the nanoparticles remained stable post-administration. | Overall Survival After First Tumor Recurrence (OS-2): The median OS-2 was 13.4 months (95% CI: 10.6–16.2 months) among the 59 patients with recurrent glioblastoma. Only tumor volume at study entry was significantly correlated with ensuing survival (P < 0.01). Overall Survival After Primary Tumor Diagnosis (OS-1): Median OS-1 was 23.2 months with a 95% confidence interval of 17.2–29.2 months. |
- | [129] |
| EPR | Irinotecan | - | phase I | nal-IRI | Intravenous infusion | The maximum tolerated dose (MTD) was determined for both WT and HT cohorts. Dose-limiting toxicities included diarrhea, dehydration, and fatigue. The study concluded that nal-IRI had no unexpected toxicities and that UGT1A1 genotype did not correlate with toxicity. | PK results were comparable to those seen in other PK studies of nal-IRI. The study analyzed PK parameters including maximum plasma concentrations (Cmax), areas under the plasma concentration-time curve (AUC0−t), and terminal half-life (t1/2) for total irinotecan, SN-38, and SN-38G. UGT1A1*28 genotype did not affect PK parameters. | The primary efficacy endpoint was PFS-6. The study reported PFS-6 as 2.9% for the intent-to-treat cohort, with a median PFS of 42 days and a median overall survival of 107 days. | - | [127] |
| Passive diffusion | Irinotecan | TMZ | phase I | nal-IRI | Intravenous infusion + Oral Administration | The study evaluates safety through monitoring dose-limiting toxicities (DLTs) which include grade 4 neutropenia, grade 3 diarrhea, hypokalemia, fatigue, anorexia, and other grade 3 or 4 nonhematologic toxicities. The MTD for nal-IRI was determined to be 50 mg/m^2 every 2 weeks with TMZ. | The study does not provide specific pharmacokinetic data. Enhanced BBB Penetration: Nanoliposomal encapsulation of irinotecan improved its ability to cross the BBB, as demonstrated in preclinical animal models. Tissue Analysis: In preclinical studies, nal-IRI showed a 10.9-fold increase in tumor area under the curve compared to free irinotecan and 35-fold selectivity for tumor versus normal tissue exposure. |
The primary efficacy endpoints are the assessment of response rate (complete or partial response as defined by Macdonald criteria) and PFS. The study was terminated after an interim analysis showed no activity (0% response rate) and a median PFS of 2 months. |
NCT03119064 | [126] |
| EPR + active transport | NU-0129 | - | phase 0 | siBcl2L12-SNAs | Intravenous infusion | The safety assessment revealed no significant treatment-related toxicities. The study monitored vital signs, blood chemistry, and adverse events in patients, with only two treatment-related severe adverse events (lymphopenia and hypophosphatemia) noted, which were considered 'possibly' related to the treatment. | PK analysis showed that siRNA was rapidly eliminated from plasma with a mean half-life of 0.09 hours, while gold (Au), used as a marker for the SNAs, had a much slower elimination with a mean half-life of 17 hours. The study also detailed the clearance rates and volume of distribution for both siRNA and Au. | The primary efficacy endpoints were the intratumoral accumulation of SNAs and the suppression of the Bcl2L12 gene. The study reported that NU-0129 uptake into glioma cells correlated with a significant reduction in tumor-associated Bcl2L12 protein expression. Additionally, the presence of Au in the tumor tissue was confirmed, indicating that the SNAs reached the patient tumors. | NCT03020017 | [130] |
| RMT | DOX | - | phase I | anti-EGFR ILs-dox | Intravenous infusion | The study reports safety data from the application of anti-EGFR ILs-dox in patients with relapsed glioblastoma. No grade 4 or 5 adverse events occurred. One case of severe pneumonitis was reported, which resolved with treatment. Other adverse events included febrile neutropenia in two patients, which was managed without sequelae. | The pharmacokinetic analysis showed that the mean plasma concentration of doxorubicin 24 hours after administration was 15,805 ng/ml. DOX concentrations in CSF were below 1 ng/ml in all patients, indicating that anti-EGFR ILs-dox do not cross the BBB at clinically relevant levels. However, significant levels of doxorubicin were detected in glioblastoma tissue 24 hours after application, suggesting that the disrupted BBB in high-grade gliomas may enable liposome delivery into tumor tissue. | The primary efficacy endpoints were the concentration of anti-EGFR ILs-dox in plasma, CSF, and glioblastoma tissue. The median PFS was 1.5 months, and the median OS was 8 months. One patient had a very long remission, suggesting that neoadjuvant administration may positively affect the outcome. | NCT03603379 | [131] |
| EPR | AGuIX nanoparticles |
standard of care for glioblastoma | phase I/Ⅱ | AGuIX nanoparticles | Intravenous infusion | Toxicity Assessment: DLT defined as any grade 3–4 NCI Common Terminology Criteria for Adverse Events (CTCAE) toxicity, except for alopecia, nausea, vomiting, or fever. Adverse Event Reporting: According to CTCAE (version 5.0). Neurological Status Evaluation: Clinical assessment and Mini-Mental State Examination (MMSE). |
PK parameters of AGuIX nanoparticles, including AUC, Tmax, and Cmax, will be measured on blood samples and urinary excretion during phase I of the study. | For phase I, the primary endpoint is the RP2D of AGuIX nanoparticles, with DLT defined as any grade 3–4 toxicity. For phase II, the primary endpoint is the 6-month progression-free survival rate, which will be estimated using the Kaplan–Meier method. | NCT04881032 | [132] |
| EPR + RMT | Mitoxantrone | - | phase I | EGFR-Erbitux EDVsMIT |
Intravenous infusion | The safety evaluation of EEDVsMit showed that it was well tolerated, with no dose-limiting toxicities observed. The most common drug-related adverse events were grade 1–2 fever, nausea, vomiting, rash, lymphopenia, and mildly deranged liver function tests. | The study does not provide detailed pharmacokinetic data within the summary. However, it mentions that the EDV selectively targets the cancer cell via the bispecific antibody and releases the cytotoxic drug into the tumor cell after macropinocytosis and breakdown in lysosomes. | The primary efficacy endpoints were safety and tolerability, with the study also aiming to preliminarily define the antitumor activity of mitoxantrone-containing EDVs. The best antitumor response observed was a mixed response in one patient, and all other patients had progressive disease. | NCT02687386 | [133] |
| CED | Irinotecan | - | phase I | nal-IRI | intratumoral injection | Safety is evaluated by monitoring adverse events, scheduled laboratory assessments, vital sign measurements, and physical examinations. Toxicities are graded according to the NCI CTCAEv4.0. | The study assesses the distribution of gadolinium, used in conjunction with real-time imaging, to model the drug distribution and evaluate the effectiveness of CED in delivering nal-IRI to the tumor site. | The primary efficacy endpoint is OS at 12 months (OS12), which will be considered evaluable for clinical efficacy. | NCT03086616 | - |
| CED | Irinotecan | - | phase I | Liposomal-Irinotecan | intratumoral injection | Safety is evaluated by monitoring DLTs within 30 days post-infusion, as well as any neurological or systemic grade-3 or higher toxicities. | PK measurements are taken at pre-dose, 1 day after drug administration, and 1 week post-op to estimate drug distribution and concentration in the brain. | The primary efficacy endpoints include the determination of the maximum tolerated dose and the assessment of PFS at 6 months and 10 years, as well as OS at 12 months and 10 years. | NCT 02022644 | - |
| Actively target | MT-201-GBM | standard radiation therapy and TMZ chemotherapy | phase I | MT-201-GBM | Intravenous administration | The study aims to assess the safety and tolerability of MT-201-GBM by measuring: Primary Safety Endpoint: Percentage of patients with a DLT during the DLT observation period within each dose level. Other Safety Measures: Changes in laboratory parameters (e.g., hemoglobin, creatinine, AST, bilirubin) and monitoring for adverse events. |
The study does not provide specific details on pharmacokinetic assessments. However, it does include immune response measurements, such as changes from baseline in IFN gamma, granzyme-B, and fluorospot, which may provide insights into the vaccine's biological activity. | The primary efficacy endpoints include the MTD of MT-201-GBM and immune response measurements after the second and third infusions compared to baseline. Secondary outcome measures include OS, PFS, and changes in immune response indicators. | NCT04741984 | - |
| Actively target and bypass the BBB | 5-FC | - | phase I | CD-NSCs | Intracranial Injection + Oral Administration | The study evaluated safety through the monitoring of adverse events, assessment of possible NSC migration into the systemic circulation, and testing for the presence of replication-competent retrovirus (RCR). No DLT was observed due to the CD-NSCs, and there was no development of anti-CD-NSC antibodies. | Intracerebral microdialysis was used to measure brain levels of 5-FC and 5-FU, showing CD-NSCs produced 5-FU locally in a 5-FC dose-dependent manner. Collected Blood Samples to assess systemic drug concentrations and perform immunologic correlative studies. | The primary efficacy endpoints included assessing the feasibility of treating recurrent high-grade glioma patients with intracranially administered CD-NSCs followed by oral 5-FC. Secondary objectives included assessing possible CDNSC immunogenicity, secondary tumorigenicity, and evaluating proof-of-concept regarding CD-NSC migration to tumor foci and localized conversion of 5-FC to 5-FU. | NCT01172964 | [134] |
| Actively target and bypass the BBB | 5-FC | Leucovorin | Phase I | CD-NSCs | Intracranial Injection | Safety is evaluated through monitoring of adverse events, assessment of possible NSC migration into the systemic circulation using qPCR, and testing for the presence of replication-competent retrovirus (RCR). No clinical signs of immunogenicity were observed, and only three patients developed anti-NSC antibodies. | Neuropharmacokinetic data were collected using intracerebral microdialysis to confirm continuous production of 5-FU in the brain during the course of 5-FC administration. The primary pharmacokinetic parameters of interest were the Cmax and AUC of 5-FC and 5-FU, measured in both dialysate samples and plasma. | The primary efficacy endpoints included assessing the feasibility of serially administering CD-NSCs and determining the recommended dosing for phase II testing. Secondary objectives included characterizing the relationship between intracerebral and systemic concentrations of 5-FC and 5-FU, assessing CD-NSC immunogenicity, and describing clinical activity. The best response observed was stable disease in three participants, with a range of 4 to 5 months. | NCT02015819 | [135] |
| Actively target and bypass the BBB | Irinotecan Hydrochloride | - | phase I | hCE1m6-NSCs | Intracranial administration + Intravenous administration |
Safety is evaluated through the grading of toxicities using the NCI CTCAE version 4.0, with specific attention to DLTs during the first treatment cycle. | PK data are collected via intracerebral microdialysis to measure SN-38 concentrations in the brain interstitium and plasma levels of irinotecan and SN-38. | The primary efficacy endpoints include the assessment of clinical benefit, defined as stable disease, partial response, or complete response, as measured by RANO criteria, for up to 6 months. | NCT02192359 | - |
| Actively target and bypass the BBB | Oncolytic Adenovirus (CRAd-S-pk7) | standard temozolomide chemotherapy and radiotherapy | phase I | NSC-CRAd-S-pk7 | Intracranial Injection | The trial demonstrated that the treatment was safe, with no formal dose-limiting toxicity reached. One patient developed viral meningitis (grade 3) due to an inadvertent injection into the lateral ventricle. | PK assessments were performed through medical history, imaging, blood samples, and chemistry values collected at screening and follow-up. The study did not detail specific pharmacokinetic parameters but focused on the safety and presence of the treatment. | The primary endpoints were safety and tolerability. Secondary endpoints included estimation of survival outcomes and evaluation of immune response correlation with clinical outcomes. The median progression-free survival was 9.05 months, and overall survival was 18.4 months. | NCT03072134 | [136] |
| CMT | Survivin DC cell injection | radiotherapy and TMZ chemotherapy | phase I | Survivin-loaded dendritic cell injection | Intradermal Injection + Intravenous Infusion | Safety is evaluated through the monitoring of adverse events that occur within 28 days after the first to the third dose, as judged by the investigator to be related to the study drug. DLT is a primary outcome measure. | Not specifically detailed in the document. However, the administration schedule (days 0, 14, and 28) and route of administration (ID and IV) indicate PK will be influenced by the absorption, distribution, metabolism, and excretion of the Survivin-loaded dendritic cells. | The primary efficacy endpoints include PFS measured over 2 years and OS also measured over 2 years. Secondary Outcome Measures include immune effect assessments such as Anti-Survivin antibody test, cytokine detection, and specific T-cell responses, as well as DC cell activity and in vivo processes, measured at specific time points. | NCT06524063 | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



